
CJD Mortality in Canada: 1998–2013

 Download this article as a PDF (321 KB - 10 pages)
Download this article as a PDF (321 KB - 10 pages) Published by: The Public Health Agency of Canada
Issue: Volume 41-8: Protein misfolding disorders
Date published: August 6, 2015
ISSN: 1481-8531
Submit a manuscript
About CCDR
Browse
Volume 41-8, August 6, 2015: Protein misfolding disorders
Surveillance
Creutzfeldt-Jakob disease mortality in Canada, 1998 to 2013
Coulthart MB1*, Jansen GH1,2, Connolly T1, D’Amour R1, Kruse J1, Lynch J1, Sabourin S1, Wang Z1†, Giulivi A2, Ricketts MN3, Cashman NR4
Affiliations
1 Canadian Creutzfeldt-Jakob Disease Surveillance System, Centre for Foodborne, Environmental and Zoonotic Infectious Diseases, Public Health Agency of Canada, Ottawa, ON
2 The Ottawa Hospital, University of Ottawa and Eastern Ontario Regional Laboratory Association, Ottawa, ON
3 University of Ottawa, Ottawa, ON
4 Department of Medicine (Neurology), Brain Research Centre, University of British Columbia, Vancouver, BC
†Deceased November 2014
Correspondence
DOI
https://doi.org/10.14745/ccdr.v41i08a01
The authors dedicate this paper to the memory of their friend and colleague Zheng Wang (1962–2014).
Abstract
Background: Human prion diseases, known collectively as Creutzfeldt-Jakob disease (CJD), are fatal, infectious neurodegenerative disorders that occur in all human populations.
Objective: To summarize national surveillance data for CJD in Canada between January 1, 1998, and December 31, 2013.
Methods: Detailed investigations were conducted of individual suspected CJD cases, with collaboration between Canadian health professionals and investigators affiliated with a central CJD surveillance registry operated by the Public Health Agency of Canada. Data were collected on the clinical profile, family history, and results of paraclinical and laboratory investigations, including post-mortem neuropathological examination.
Results: A total of 662 deaths from definite and probable CJD were identified in Canadian residents during the study period, comprising 613 cases of sporadic CJD (92.6%), 43 cases of genetic prion disease (6.5%), 4 cases of iatrogenic CJD (0.6%), and 2 cases of variant CJD disease (0.3%). The overall crude mortality rate for sporadic CJD was 1.18 per million per year [95% confidence interval (CI): 1.08,1.27]. Age-specific rates ranged from 0.05 [95% CI: 0.03,0.08] in persons under 50 years of age to 7.11 [95% CI: 6.20,8.11] in those aged 70 to 79. A significant net upward trend in age-adjusted rates was observed over the study period. Standardized mortality ratios, calculated for 10 individual Canadian provinces with reference to national average mortality rates, did not differ significantly from 1.0.
Conclusion: Creutzfeldt-Jakob disease remains rare in Canada, although mortality rates vary by two orders of magnitude between older and younger age groups. The upward trend in age-standardized sporadic CJD mortality rate over the study period can be better accounted for by gradually improving case ascertainment than by a real increase in incidence.
Introduction
Prion diseases are rare, lethal, degenerative brain disorders that affect humans and several other mammals. Remarkably, these diseases are transmissible by an infection-like molecular process involving a pathologically altered, self-propagating form of a host glycoprotein, the prion protein (PrP). The infective agents of these diseases are therefore referred to as prions, or proteinaceous infectious particlesFootnote 1. Despite recently recognized similarities between the molecular mechanisms of prion diseases linked with pathological behaviour of PrP and other “prion-like” diseases involving misfolding of other proteins, to date there is no evidence as yet that prion-like diseases are transmissible between individuals under natural conditionsFootnote 2Footnote 3. Thus, prion diseases pose a unique combination of challenges to public health, including long, silent incubation periods; lack of protective host immunity; resistance to decontamination; limited tools for diagnosis; and potential for both zoonotic and health care-based transmissionFootnote 4.
Prion diseases of humans—for brevity, referred to collectively here as Creutzfeldt-Jakob disease (CJD)—exhibit various clinicopathological presentations that reflect distinct endogenous and exogenous etiologic origins. Among “classic” forms of CJD the most common subtype, sporadic CJD (sCJD), is defined by characteristic prion neuropathology (particularly spongiform degeneration and deposition of pathologic PrP in brain tissue) in the absence of a discernible genetic or infectious cause. Sporadic CJD appears to occur endemically in all populations with an average mortality rate of one to two per million per year, and generally constitutes 85% to 95% of all CJD cases identified through epidemiologic surveillanceFootnote 5. Genetically caused forms (5% to 15% of classic CJD cases), which apparently also occur universally, are associated with any of over 50 different pathogenic mutations in the host gene that encodes PrP. Some of these genetic forms—in particular, Gerstmann-Sträussler-Scheinker disease and fatal familial insomnia—exhibit distinctive clinicopathologic characteristics and can usually be diagnosed as genetic in origin even when the specific underlying genetic mutation is unknownFootnote 6.
Iatrogenic cases of classic CJD (iCJD) have occurred through accidental prion transmission in the health care setting, particularly with therapeutic use of cadaveric tissues or tissue extracts presumably contaminated by donations from individuals with unrecognized sCJD. Although iCJD is very rare, epidemics have been well-documented, with 469 cases reported worldwide as of 2012Footnote 7. In addition, carefully conducted epidemiological studies have yielded new evidence that a fraction of “sporadic” CJD cases may be attributable to remote iatrogenic exposure through surgery, particularly early in lifeFootnote 8Footnote 9. Most recently, a new human prion disease, variant CJD (vCJD), emerged in the mid-1990s in the United Kingdom via zoonotic transmission to humans of a prion disease of cattle, bovine spongiform encephalopathy (BSE)Footnote 10Footnote 11Footnote 12. As of April 2015, 229 cases of vCJD have been reported in residents of 12 countries, including two imported cases in CanadaFootnote 13Footnote 14. This number includes five probable cases, all in the United Kingdom, of secondary (human-to-human) transmission of vCJD infection via the bloodborne routeFootnote 15Footnote 16Footnote 17.
Prompted by the need to better understand the epidemiology of CJD, and the responsibility to promptly and expertly address iatrogenic and zoonotic transmission risks, dedicated systems for prospective, population-based epidemiologic surveillance of CJD began to be established in many countries in the early 1990sFootnote 18. In Canada the initial motivator was a question raised by the Krever Commission on the Blood System in Canada, concerning the possibility that CJD might be transmitted by the bloodborne routeFootnote 19Footnote 20. In response, in April 1998 the Government of Canada formally initiated prospective national epidemiologic surveillance of CJD through the Canadian Creutzfeldt-Jakob Disease Surveillance System (CJDSS), now operated by the Public Health Agency of CanadaFootnote 21. Since 2000, all forms of CJD have been nationally notifiable in Canada, and are now reportable in all provinces and territories. The present article provides an overview of the findings of this national surveillance program over a 16-year period extending from January 1, 1998, to December 31, 2013.
Methods
Data collection
The CJDSS employs an internationally established methodology for epidemiologic surveillance of CJDFootnote 18. Following this methodology, after notification by a health professional of a suspected case of CJD in a Canadian resident, CJDSS investigators collaborate to facilitate diagnostic investigations, assemble and organize case files, interpret and disseminate results, support disease reporting, and analyze and manage public health issues. The central case registry and an electronic database are used by the CJDSS to track events, rates and trends of CJD occurrence in Canada. Access to CJDSS investigators for collaborating health professionals, as well as the public, is facilitated by the use of a toll-free telephone line (1-888-489-2999) and a dedicated e-mail address (cjdss@phac-aspc.gc.ca).
Case definitions
Diagnostic investigation of CJD and other subacute encephalopathies in the living patient is frequently challengingFootnote 22. Thus, high-quality epidemiologic surveillance entails collection of a variety of types of supporting information, as well as follow-up on individual cases in real time. A diagnosis of definite CJD requires neuropathologic examination; thus cranial autopsy is routinely considered. Cases lacking neuropathologic examination can be formally classified as probable CJD based on non-neuropathologic criteria, such as clinical profile, results of cerebrospinal fluid (CSF) protein immunoassays, findings of electroencephalography or magnetic resonance imaging, and genetic analysis. Where information is even more limited, it can be feasible to assign the lowest grade formal diagnostic classification of possible CJD. Surveillance case definitions employed by the CJDSS, which have been published by the Public Health Agency of Canada’s National Notifiable Diseases Surveillance System, are based on those used internationallyFootnote 18Footnote 23.
Mortality rate calculations
Although prospective national surveillance of CJD in Canada was formally initiated as of April 1, 1998, some information was also available on CJD deaths occurring in January, February and March of 1998. Thus, data presented in this report include CJD deaths in Canada recorded by the CJDSS as having occurred between January 1, 1998, and December 31, 2013. With the national scope and population-based approach of the CJDSS, for the purpose of this report the entire population of Canada was used to estimate mortality rates, which are expressed here in units of cases per million population per year. Note also that although no cases of CJD were identified from the Canadian territories (Yukon, Northwest Territories and Nunavut) during the period of surveillance, because the CJDSS was prepared to accept notifications from these regions their populations were included in denominators of national rate estimates. Canadian population and demographic data were adapted from the Statistics Canada CANSIM online socioeconomic databaseFootnote 24. On the basis of these data, over the entire period of study a nominal total of 521,794,297 person-years were included within the scope of surveillance, where a person-year represents membership of an individual in an at-risk population over a period of one yearFootnote 25.
Exact 95% confidence intervals (CIs) for mortality rate estimates were based on a Poisson model of case counts. The hypothesis of a linear trend in age-adjusted mortality rates over the period of surveillance was tested using nonparametric linear regression, in which the slope of the regression was calculated as the median of slopes connecting all pairs of data points. In addition, a two-tailed test of rank correlation between year and mortality rate was performed using Kendall’s tau coefficient, which is based on the proportion of paired observations of two variables in which the respective ranks of the two observations matchFootnote 26. Standardized mortality ratios (SMRs) for sCJD (defined as the ratio of observed total mortality rates in a study population to “expected” mortality rates in the study population derived using known age-specific rates for a reference population) were estimated for each province. National CJD mortality rates were used to calculate expected rates, again basing 95% CIs on a Poisson model.
With the low numbers of identified cases of genetic prion disease, iCJD and vCJD (see below), only limited statistical analysis was performed for these disease subtypes, with results reported here consisting primarily of case numbers and estimated crude mortality rates for genetic prion disease.
Ethics framework
Formal enrollment of a patient with the CJDSS (case referral) is done with written informed consent for each main component of data collection (autopsy, genetics, medical record review and interview) under a protocol approved by the Health Canada–Public Health Agency of Canada Research Ethics Board (Certificate REB-2009-0036). The personal information collected is registered with the Government of Canada as a personal information bank (PIB)Footnote 27.
Results
Diagnostic yield and quality
Between January 1, 1998, and December 31, 2013, the CJDSS opened 1,420 case-referral files on patients suspected of having CJD, or being at risk for CJD (i.e., family members of patients with genetic prion disease). A total of 3,689 laboratory investigations were performed by Public Health Agency of Canada laboratories, comprising 2,193 CSF protein immunoassays, 635 genetic analyses, and 861 neuropathological examinations including 828 full cranial autopsies (Table 1). During the same period the deaths of 662 Canadian residents were attributed to definite or probable CJD, comprising 613 sCJD (92.6%), 43 genetic prion disease (6.5%), 4 iCJD (0.6%), and 2 vCJD (0.3%). Among all 662 CJD diagnoses, 535 (80.8%) were definite and 127 (19.2%) were probable. In addition, 22 deaths were attributed to possible CJD, and 77 cases were unclassifiable. Of the 613 sCJD diagnoses, 498 (81.2%) were definite, and 115 (18.8%) were probable. Among 575 non-CJD diagnoses, CJD was excluded by neuropathology in 308 (53.6%) and by other criteria in 267 (46.4%).
| YearTable 1 footnote 1 | TestsTable 1 footnote 2 | ReferralsTable 1 - Footnote 3Table 1 footnote 4 | AutopsiesTable 1 footnote 5 | DeathsTable 1 footnote 6 | DefiniteTable 1 footnote 7 | ProbableTable 1 footnote 8 |
|---|---|---|---|---|---|---|
| 1998 | 50 | 44 | 24 | 24 | 20 | 4 |
| 1999 | 84 | 63 | 34 | 32 | 28 | 4 |
| 2000 | 126 | 82 | 42 | 35 | 32 | 3 |
| 2001 | 161 | 101 | 49 | 30 | 23 | 7 |
| 2002 | 158 | 103 | 54 | 36 | 32 | 4 |
| 2003 | 137 | 75 | 44 | 29 | 27 | 2 |
| 2004 | 268 | 90 | 63 | 44 | 33 | 11 |
| 2005 | 257 | 97 | 58 | 44 | 37 | 7 |
| 2006 | 234 | 80 | 57 | 44 | 37 | 7 |
| 2007 | 259 | 101 | 49 | 39 | 33 | 6 |
| 2008 | 305 | 100 | 57 | 49 | 34 | 15 |
| 2009 | 346 | 104 | 74 | 53 | 46 | 7 |
| 2010 | 281 | 76 | 46 | 38 | 31 | 7 |
| 2011 | 342 | 102 | 54 | 51 | 40 | 11 |
| 2012 | 351 | 103 | 69 | 63 | 46 | 17 |
| 2013 | 330 | 99 | 54 | 51 | 36 | 15 |
| Total | 3,689 | 1,420 | 828 | 662 | 535 | 127 |
Age and sex distributions for sCJD
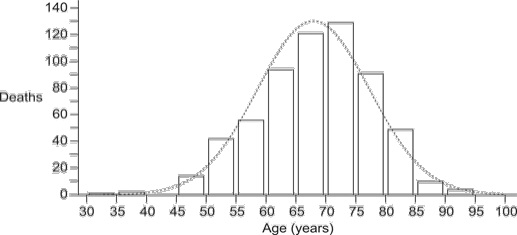
Figure 1 illustrates the distribution of ages at death for the 613 sCJD cases. The distribution departed significantly from normality, owing to leftward skewness (D’Agostino-Pearson coefficient –0.297, P = 0.012). Age at death ranged from 33 to 92 years, with a median of 69 [95% CI: 68,70] and inter-quartile range 62,75. Males numbered 291 (47.5%) [95% CI: 43.5, 51.4] and females 322 (52.4%) [95% CI: 48.6,56.5], indicating a sex ratio indistinguishable from 1. Age distributions were also very similar between sexes, with males having a median of 68 years [95% CI: 67, 70; inter-quartile range: 60,74.75], and females a median of 69 years [95% CI: 67,70; inter-quartile range: 61,75].
Figure 1: Distribution of age at death for 613 cases of sporadic CJDin Canada, 1998 to 2013 Figure 1 footnote 1Figure 1 footnote 2Figure 1 footnote 3
Text description: Figure 1
Figure 1: Distribution of age at death for 613 cases of sporadic CJDin Canada, 1998 to 2013 Figure 1 footnote 1Figure 1 footnote 2Figure 1 footnote 3
| Age Stratum (years): | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 30-34 | 35-39 | 40-44 | 45-49 | 50-54 | 55-59 | 60-64 | 65-69 | 70-74 | 75-79 | 80-84 | 85-89 | 90-94 | 95-99 | All | |
| Deaths: | 1 | 2 | 0 | 14 | 42 | 56 | 94 | 121 | 129 | 91 | 49 | 10 | 4 | 0 | 613 |
sCJD mortality
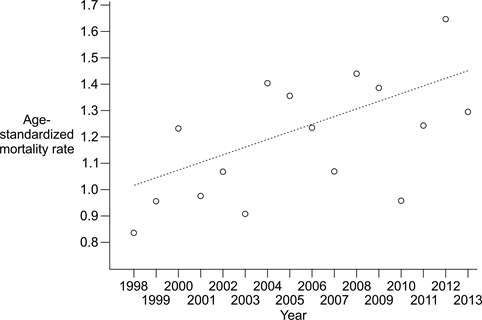
Table 2 provides age-specific case counts and mortality rates for sCJD for five age strata: ≤49 years; 50–59 years; 60–69 years; 70–79 years; and ≥80 years. Crude rates varied widely with age, ranging from 0.05 for ages ≤49 years to 7.11 for ages 70–79 years; only 17 of the 613 sCJD cases (2.8%) were identified in persons ≤49 years of age. Total crude mortality for all age strata over the entire period of surveillance was 1.18, with annual rates ranging from 0.73 in 1998 to 1.78 in 2012. Age-standardized rates were also calculated, using the 2006 Canadian census population as a reference (Table 2). These rates ranged from 0.84 for 1998 to 1.65 for 2012, with a total rate over the surveillance period of 1.20. When analyzed by nonparametric linear regression, the age-standardized rates were significantly associated with year, according to the linear function: Rate = 0.029 (Year) – 57.30 [95% CI: 0.002, 0.052 for the slope estimate] (Figure 2). Kendall’s rank-correlation coefficient tau (0.45) was significant at P = 0.017 (two-sided test).
| YearTable 2 footnote 1 | AgeTable 2 footnote 2 | MortalityTable 2 footnote 3 | ||||||
|---|---|---|---|---|---|---|---|---|
| ≤49 | 50–59 | 60–69 | 70–79 | ≥80 | All ages | CrudeTable 2 footnote 4 | Age-adjustedTable 2 footnote 5 | |
| 1998 | 0 | 3 | 5 | 12 | 2 | 22 | 0.73 [0.46,1.10] | 0.84 [0.52,1.27] |
| 1999 | 2 | 4 | 9 | 10 | 2 | 27 | 0.89 [0.58,1.29] | 0.96 [0.63,1.39] |
| 2000 | 1 | 4 | 5 | 14 | 8 | 32 | 1.04 [0.71,1.47] | 1.23 [0.84,1.75] |
| 2001 | 0 | 4 | 12 | 8 | 3 | 27 | 0.87 [0.57,1.26] | 0.98 [0.64,1.42] |
| 2002 | 2 | 5 | 6 | 14 | 4 | 31 | 0.99 [0.67,1.40] | 1.07 [0.73,1.52] |
| 2003 | 2 | 1 | 13 | 11 | 0 | 27 | 0.85 [0.56,1.24] | 0.91 [0.60,1.32] |
| 2004 | 0 | 10 | 14 | 13 | 5 | 42 | 1.31 [0.95,1.77] | 1.40 [1.01,1.90] |
| 2005 | 1 | 8 | 12 | 21 | 0 | 42 | 1.30 [0.94,1.76] | 1.36 [0.98,1.83] |
| 2006 | 0 | 5 | 13 | 17 | 4 | 39 | 1.19 [0.85,1.63] | 1.24 [0.88,1.69] |
| 2007 | 2 | 7 | 14 | 8 | 4 | 35 | 1.06 [0.74,1.48] | 1.07 [0.75,1.49] |
| 2008 | 0 | 9 | 13 | 18 | 8 | 48 | 1.44 [1.06,1.91] | 1.44 [1.06,1.91] |
| 2009 | 1 | 7 | 19 | 16 | 5 | 48 | 1.42 [1.05,1.89] | 1.39 [1.02,1.84] |
| 2010 | 2 | 6 | 19 | 4 | 4 | 35 | 1.03 [0.71,1.43] | 0.96 [0.67,1.33] |
| 2011 | 1 | 8 | 20 | 12 | 5 | 46 | 1.33 [0.98,1.78] | 1.24 [0.91,1.66] |
| 2012 | 1 | 13 | 22 | 20 | 6 | 62 | 1.78 [1.36,2.28] | 1.65 [1.26,2.11] |
| 2013 | 2 | 4 | 19 | 22 | 3 | 50 | 1.42 [1.05,1.87] | 1.30 [0.96,1.71] |
| All years | 17 | 98 | 215 | 220 | 63 | 613 | 1.18 [1.08,1.27] | 1.20 [1.11,1.30] |
| MortalityTable 2 footnote 3 | 0.05 [0.03,0.08] | 1.41 [1.14,1.72] | 4.59 [4.00,5.25] | 7.11 [6.20,8.11] | 3.44 [2.64,4.40] | 1.18 [1.08,1.27] | – | – |
Figure 2: Age-standardized mortality from sporadic CJDin Canada, 1998 to 2013 Figure 2 footnote 1Figure 2 footnote 2Figure 2 footnote 3
Text description: Figure 2
Figure 2: Age-standardized mortality from sporadic CJDin Canada, 1998 to 2013 Figure 2 footnote 1Figure 2 footnote 2Figure 2 footnote 3
| Year | Age-standardized mortality rate |
|---|---|
| 1998 | 0.84 |
| 1999 | 0.96 |
| 2000 | 1.23 |
| 2001 | 0.98 |
| 2002 | 1.07 |
| 2003 | 0.91 |
| 2004 | 1.40 |
| 2005 | 1.36 |
| 2006 | 1.24 |
| 2007 | 1.07 |
| 2008 | 1.44 |
| 2009 | 1.39 |
| 2010 | 0.96 |
| 2011 | 1.24 |
| 2012 | 1.65 |
| 2013 | 1.30 |
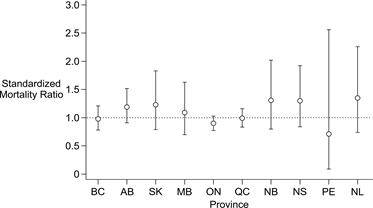
To examine the possibility that CJD mortality rates varied geographically during the study period, SMRs and 95% CIs were estimated for each province, using the estimated national age-specific mortality rates as a reference to calculate expected rates. The results are shown in Figure 3. It can be seen that both the SMR estimates and the widths of their respective 95% CIs varied considerably, with the latter expected given differing case numbers among provinces with different population sizes [range: <5 cases (PEI) to 209 cases (Ontario)]. Despite this variation, the 95% CIs of SMRs for all provinces included the value 1.0, indicating a lack of statistical support for geographic variation in mortality rates.
Figure 3: Standardized mortality ratios for sporadic CJD in Canadianprovinces, 1998 to 2013 Figure 3 footnote 1Figure 3 footnote 2Figure 3 footnote 3Figure 3 footnote 4
Text description: Figure 3
Figure 3: Standardized mortality ratios for sporadic CJD in Canadianprovinces, 1998 to 2013 Figure 3 footnote 1Figure 3 footnote 2Figure 3 footnote 3Figure 3 footnote 4
| Province | Standardized Mortality Ratio | 95% Confidence Interval |
|---|---|---|
| BC | 0.98 | 0.78-1.21 |
| AB | 1.19 | 0.91-1.52 |
| SK | 1.23 | 0.79-1.83 |
| MB | 1.09 | 0.70-1.63 |
| ON | 0.90 | 0.78-1.03 |
| QC | 0.99 | 0.84-1.16 |
| NB | 1.31 | 0.80-2.02 |
| NS | 1.30 | 0.84-1.92 |
| PE | 0.71 | 0.09-2.56 |
| NL | 1.35 | 0.74-2.26 |
Genetic prion disease, iCJD and vCJD
Basic features of the 43 identified cases of genetic prion disease are summarized in Table 3. Cases of all three known classic clinicopathologic presentations of genetic prion disease (CJD, Gerstmann-Sträussler-Scheinker disease, fatal familial insomnia) were identified. Autopsy rates were high in this group as well, with 33 of the 43 diagnoses (77%) supported by neuropathology in addition to an identified mutation in the host gene that encodes PrP. Interestingly, 9 of the 20 cases of Gerstmann-Sträussler-Scheinker disease (45%) were only discovered at autopsy and lacked an identified mutation because genetic analysis had not been requested. Clearly the total mortality rate from genetic prion diseases over the study period was much lower than that for sCJD, at 0.08 per million per year [95% CI: 0.06,0.11]. Lastly, four cases of iCJD (all with receipt of a dura mater graft as a risk factor) and two cases of vCJD (one definite and one probable) were identified.
| SubtypeTable 3 footnote 1 | Mutation identifiedTable 3 footnote 2 | Mutation not identifiedTable 3 footnote 3 | Definite (%)Table 3 footnote 4 | Probable (%)Table 3 footnote 5 | Total |
|---|---|---|---|---|---|
| CJD | 21 | 0 | 16 (76) | 5 (24) | 21 |
| GSS | 11 | 9 | 16 (80) | 4 (20) | 20 |
| FFI | 2 | 0 | 1 (50) | 1 (50) | 2 |
| All | 34 | 9 | 33 (79) | 10 (21) | 43 |
Discussion
Based on national prospective epidemiologic surveillance, a total of 662 deaths were attributed to all forms of CJD in Canada between 1998 and 2013. For the most common etiologic subtype, sCJD (92.6% of all cases), we estimated an overall average crude mortality rate of 1.18 definite and probable deaths per million per year. We also confirmed the strong relationship between crude sCJD mortality rates and age, with a very low age-specific rate of 0.05 in those under 50 years, and a maximum of 7.11 in the age stratum 70 to 79 years. Another known epidemiologic pattern confirmed by our data was the lack of detectable sex bias in mortality rates. Evidence was found for a modest net upward trend in age-standardized sCJD mortality rate over the 16-year period. However, consistent with strong national coverage, no statistical support was found for geographic deviation in rate with respect to that expected from the national rate for any of the 10 Canadian provinces in which cases were observed.
The epidemiology of CJD has been studied systematically for nearly four decadesFootnote 5Footnote 6Footnote 7Footnote 12Footnote 28Footnote 29Footnote 30. Early estimates of mortality based primarily on retrospective approaches to case identification typically fell well below one per million per yearFootnote 29. A more structured, prospective approach to case finding undertaken in France between 1978 and 1982 provided the first well-controlled demonstration of a within-country gradient of observed rates (0.56 for France, 0.86 for metropolitan Paris, and 1.19 for the city of Paris), representing a positive correlation of rates with degree of urbanization and suggesting a significant ascertainment effectFootnote 30. The combination of retrospective and prospective studies in France and later in the United Kingdom demonstrated a rise in apparent mortality with time as well, which was also attributed to improved case ascertainmentFootnote 30Footnote 31.
More recent estimates of CJD mortality rates have tended toward the range of one to two per million per yearFootnote 5. This has occurred in parallel with methodological enhancements such as establishment of national registries, application of a prospective approach, formulation of standard case definitions, recognition of disease subtypes, development and validation of diagnostic tools, and increased awareness among collaborating health professionals, all strengthening the plausibility of improved ascertainment, particularly in older patients and/or patients with less typical presentationsFootnote 5Footnote 32. In Switzerland, a temporary increase in CJD mortality in the years 2001 to 2004 was ultimately attributed to heightened awareness of the disease as a result of the vCJD epidemic in the United Kingdom; the authors went so far as to suggest that the “true” mortality rate for sCJD may exceed two per million per yearFootnote 33. In a recent study incorporating data from 10 countries, significant statistical correlations were demonstrated between observed sCJD mortality rate and several indices of surveillance intensity, including rates of case notification to the central registry as well as rates of autopsy and other laboratory investigationsFootnote 34. In light of these considerations, we believe that the observed upward trend in age-standardized sCJD mortality rates in Canada between 1998 and 2013 can be best accounted for by gradually improving case ascertainment, rather than extrinsic causes leading to a real rate elevation. Thus, although under-ascertainment could legitimately be considered a potential limitation of the present study, such a positive trend suggests that further improvements could be realized. Given the strong correlation of mortality rates with age, it is also worth keeping in mind the possibility that population aging will contribute to real increases in sCJD rates in the future.
The surveillance findings presented here confirm and extend those of earlier published studies that included either retrospective or prospective data on CJD mortality in Canada, and are consistent with those of other CJD surveillance systems internationallyFootnote 5Footnote 34Footnote 35Footnote 36. In addition to providing high-quality estimates of overall, age-specific and sex-specific mortality rates for classic forms of CJD, the Canadian CJD Surveillance System has demonstrated an ability to detect, investigate and deal with rare events, such as four identified cases of iCJD and two of vCJD. Our results indicate, therefore, that a collaborative environment has been established for robust, national prospective epidemiologic surveillance of CJD in Canada.
It is important for health professionals to remain alert to the possibility of CJD in patients. Likewise, it is important for public health professionals to use the surveillance data and the resulting epidemiologic insights to assess and mitigate any public health risks associated with the ongoing, possibly under-ascertained and potentially increasing occurrence of classic forms of CJD. Finally, there are persisting uncertainties regarding the epidemiology of vCJD and possible new zoonotic prion diseases, which warrant continued vigilance for novel CJD events or trendsFootnote 37Footnote 38.
Acknowledgements
The authors express their sincere gratitude to the many health professionals, health care institutions, service providers and, above all, affected patients and families, without whose generous collaboration, this work would not have been possible. Special thanks are also extended to the staff of the Prion Laboratory Section, National Public Health Laboratories, Public Health Agency of Canada, and of the Neuropathology Laboratory Services of the University of Ottawa for their skill and dedication in delivering high-quality laboratory diagnostic services. The Canadian population and demographic information used in this report was adapted from Statistics Canada; this does not constitute an endorsement by Statistics Canada of this publication.
Conflict of interest
None.
Funding
Funding for the Canadian CJD Surveillance System is provided through the Public Health Agency of Canada’s Prion Diseases Program.
Page details
- Date modified: