
Procédures opérationnelles pour l’Initiative de partage du travail concernant les médicaments génériques (IPTMG)
Télécharger le format de rechange (Format PDF, 750 ko, 10 pages)
Registre des modifications du document
| Version | Description du changement | Auteur | Date d'entrée en vigueur |
|---|---|---|---|
| v 1.0 | Publication originale | Groupe de travail sur les médicaments génériques du Consortium Australie–Canada–Singapour–Suisse (ACSS) | 2016-05-20 |
| v 2.0 | Mise à jour à la suite de la première demande dans le cadre de l'Initiative de partage du travail concernant les médicaments génériques (IPTMG) | Groupe de travail sur les médicaments génériques du Consortium ACSS | 2017-10-05 |
| v 3.0 | Mise à jour pour intégrer les améliorations aux procédures opérationnelles (par exemple, en tenant compte des commentaires des intervenants) | Groupe de travail sur les médicaments génériques du Consortium Access | 2022-10-30 |
Also available in English under the title:
Access Consortium: Operational procedures for the Generic Medicines Work-Sharing Initiative (GMWSI)
La présente publication peut être reproduite sans autorisation pour usage personnel ou interne seulement, dans la mesure où la source est indiquée en entier.
Cat.: H164-344/2022F-PDF
ISBN: 978-0-660-45704-8
Pub.: 220462
Table of Contents
- Introduction
- Portée
- Considérations liées à la demande
- Approche opérationnelle.
- Première ronde d'évaluation
- Deuxième ronde d'évaluation
- Troisième ronde d'évaluation (le cas échéant)
- Étapes à l'échelle nationale
- Liens connexes
Introduction
Le présent document décrit les procédures opérationnelles et les recommandations pour la planification et la mise en œuvre de l'Initiative de partage du travail concernant les médicaments génériques (IPTMG) à l'intention des organismes de réglementation du Consortium Access. Ces organismes de réglementation sont les suivants :
- Therapeutic Goods Administration (TGA), Australie
- Santé Canada (SC), Canada
- Health Sciences Authority (HSA), Singapour
- Swissmedic (SMC), Suisse
- Medicines and Healthcare products Regulatory Agency (MHRA), Royaume-Uni
L'initiative est basée sur la procédure décentralisée de l'Europe, où un organisme agissant à titre d'autorité de réglementation de référence (ARR) sera appelé à évaluer les modules 2 à 5. Chaque organisme participant agit à titre d'autorité de réglementation concernée (ARC) et, avec l'ARR, évalue son module 1 respectif. Les ARC procèdent à un examen par les pairs des rapports d'évaluation (RE) et de la liste de questions (LQ) proposée fournis par l'ARR pour les modules 2 à 5, consultent les modules au besoin et fournissent des commentaires supplémentaires au besoin.
Chaque organisme prend sa propre décision en fonction des recommandations formulées dans les RE. Si, au cours du processus, les organismes participants ne sont pas en mesure de résoudre les problèmes en lien avec les données, ils peuvent demander des renseignements supplémentaires et entreprendre un examen indépendant plus approfondi.
Portée
Pour être envisagé dans le cadre de cette initiative, le produit proposé doit être considéré comme un produit générique par tous les organismes participants. Toutes les formes pharmaceutiques (posologiques) sont admissibles.
Considérations liées à la demande
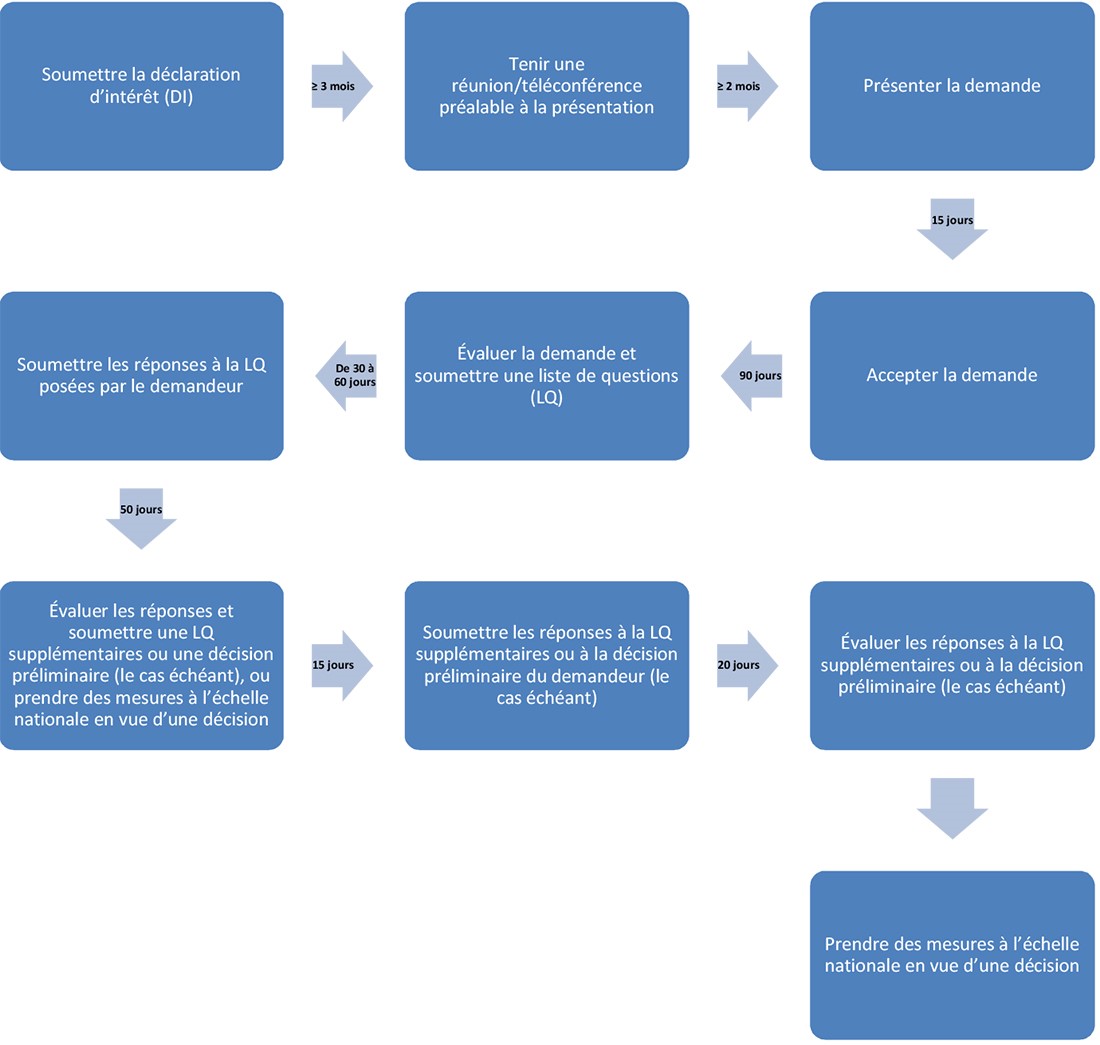
Un demandeur qui souhaite participer à cette initiative novatrice de partage du travail doit soumettre un formulaire de déclaration d'intérêt (DI) à chaque organisme proposé pour cette initiative. Le demandeur doit soumettre ce formulaire au moins 3 mois avant la date prévue de présentation.
Dans le cadre de cette initiative, il doit soumettre ses demandes à au moins 2 des membres du Consortium Access en même temps.
Il doit également présenter des modules 2 à 5 identiques à tous les organismes participant à l'initiative. Toutefois, il peut y avoir des différences mineures dans les demandes présentées aux divers organismes. Par exemple, il peut y avoir des différences dans les formats d'emballage, comme les bouteilles par rapport aux emballages-coques à dose unitaire. Les différences majeures entre les demandes peuvent compliquer le processus de partage du travail, ce qui peut retarder le processus d'évaluation. Par exemple, la présentation de plusieurs études utilisant différents produits de comparaison est considérée comme une différence majeure.
S'il y a des différences mineures entre les ensembles de données, le demandeur doit fournir un tableau intitulé « Résumé des différences ». Ce tableau fait partie du formulaire de DI. Le tableau doit décrire les différences dans la qualité et l'information des études de bioéquivalence (chaque organisme participant s'en voit attribuer une). Les organismes du Consortium Access discuteront de ces différences et détermineront si la demande convient à l'Initiative de partage du travail concernant les médicaments génériques (IPTMG).
Veuillez noter que tous les organismes du Consortium Access acceptent l'utilisation de produits de comparaison étrangers dans les études de bioéquivalence. Par conséquent, pour les présentations exigeant une équivalence clinique (qu'il s'agisse d'une bioéquivalence pharmacocinétique ou d'une équivalence thérapeutique), il peut être possible d'utiliser un seul produit de comparaison étranger en fournissant des justifications propres au pays. Veuillez consulter la section des liens connexes pour les références.
Prenez également note que le module 1 est propre à chaque pays. Par conséquent, il continuera d'être différent pour les demandes déposées dans les différentes administrations du Consortium Access (conformément aux exigences nationales).
Bien qu'un demandeur puisse proposer une ARR privilégiée, les organismes du Consortium Access choisiront ultimement l'ARR et les ARC pour toute demande. Les autorités fondent leur décision sur des facteurs tels que les besoins opérationnels du Consortium Access.
En général, un organisme (ARR) effectuera l'évaluation et les autres organismes participants agiront à titre d'ARC. Toutefois, dans certains cas, la demande peut être divisée. Dans de telles situations, plusieurs organismes effectuent l'évaluation initiale. Les rôles de chaque organisme participant seront déterminés en fonction des exigences opérationnelles.
Dans la DI, le demandeur doit indiquer le délai souhaité pour soumettre ses réponses à la LQ fournie par les organismes. Le délai doit être de 30 ou 60 jours civils.
Les demandes doivent répondre pleinement aux exigences de toutes les administrations à inclure dans la procédure. Les demandeurs doivent également reconnaître qu'ils devront travailler en collaboration avec les organismes. Bien qu'une seule demande qui traite de questions plus vastes pour plus d'un organisme puisse sembler plus onéreuse, cela permettra de réduire le fardeau réglementaire global.
Approche opérationnelle
Le processus doit être en mesure de fonctionner simultanément au sein des systèmes de réglementation des organismes participants. La présente section décrit les étapes et les questions dont on doit tenir compte lors de la mise en œuvre du processus.
Tous les échéanciers et jours sont basés sur des jours civils. Si un jalon tombe une fin de semaine ou un jour férié national, il est devancé au jour ouvrable précédent. Veuillez noter que les délais suivants sont les délais prévus et qu'ils peuvent ainsi être modifiés en fonction de la complexité de la demande.
Réunion ou téléconférence préalable à la présentation (au moins 2 mois à l'avance)
Une fois que les organismes participants auront reçu un formulaire de DI, ils travailleront ensemble pour discuter de la demande, de sa pertinence pour l'inclure dans l'initiative de partage du travail, de l'ARR et des ARC ainsi que des prochaines étapes.
Il est fortement recommandé de tenir une réunion ou une téléconférence préalable à la présentation entre le demandeur et son organisme local du Consortium Access, ou tous les organismes participants, si possible (ce qui pourrait ne pas être autorisé en raison de difficultés opérationnelles et liées aux ressources). Cette réunion sert à discuter des aspects techniques de la présentation et à confirmer la logistique et les attentes relatives aux exigences, aux échéanciers d'évaluation et au processus. La réunion donne également aux organismes l'occasion de répondre à toute question supplémentaire que pourrait avoir le demandeur.
La téléconférence doit avoir lieu au moins 2 mois avant la date de présentation de la demande convenue. De plus, le demandeur doit suivre les procédures habituelles de son organisme local lorsqu'il demande la tenue d'une réunion préalable à la présentation.
Le demandeur sera prié de soumettre ses questions au moins 2 semaines avant la téléconférence préalable à la présentation. Dans les 2 semaines suivant cette téléconférence, il devra fournir un compte rendu de la réunion résumant les points qui ont été convenus.
Présentation de la demande (moins de 15 jours)
Les demandes doivent être présentées en même temps à chacun des organismes participants ou selon ce qui a été convenu avec eux. Le processus commence dès que tous les organismes ont reçu les demandes. Il s'agit du « jour 15 » du processus.
S'il y a lieu, le dossier permanent de la substance active/fiche maîtresse des médicaments doit être soumis à chaque organisme participant avant le dépôt de la demande, accompagnée des formulaires locaux appropriés.
Acceptation de la demande (15 jours)
Une fois que les organismes participants auront reçu la demande, l'ARR et les ARC évalueront et valideront les renseignements techniques et administratifs. Elles vérifieront si la législation nationale et les exigences en matière de données (par exemple, les formulaires de demande, les frais d'utilisation) ont été respectées et s'il est possible d'accepter la demande à des fins d'évaluation.
L'ARR et les ARC informeront ensuite le demandeur si sa demande a été acceptée aux fins d'évaluation. Si tel est le cas, le demandeur recevra également un résumé des délais prévus pour chaque étape du processus. Le jour d'acceptation de la demande d'évaluation par l'ARR correspond au « jour 0 » du processus. Les ARC s'efforceront d'accepter la demande le même jour que le fera l'ARR.
Première ronde d'évaluation
Évaluation initiale par l'ARR (60 jours)
L'ARR évalue les modules 2 à 5 et prépare un RE et une LQ. En même temps, l'ARR et les ARC évaluent leur module 1 national et préparent une LQ pour ce module. L'ARR transmet ensuite le RE et la LQ des modules 2 à 5 aux ARC.
Bien qu'une LQ regroupées soit l'option privilégiée, il se peut que l'ARR envoie plutôt des questions de façon périodique au demandeur local pour obtenir des précisions pendant le processus d'évaluation. Le cas échéant, on accorde un court délai de réponse (par exemple, 5 jours) à ces questions de clarification. L'ARR communique ensuite ces réponses aux ARC.
Examen par les pairs réalisé par les ARC (25 jours)
Dans le cadre du processus d'examen par les pairs, les ARC :
- effectuent un examen par les pairs du RE et de la LQ
- consultent les modules (si nécessaire)
- font part des commentaires et des questions supplémentaires sur les modules 2 à 5 à l'ARR
Finalisation des RE et de la LQ (5 jours)
L'ARR et les ARC discutent de la LQ et de toute question supplémentaire. L'ARR prépare la LQ regroupées sur les modules 2 à 5. L'ARR et chaque ARC transmettent la LQ regroupées ainsi que leurs questions sur le module 1 (y compris les questions concernant les renseignements sur les produits et l'étiquetage), au demandeur local.
Soumission des réponses à la LQ par le demandeur (30 ou 60 jours)
Le demandeur prépare et envoie les mêmes réponses à la LQ pour les modules 2 à 5 à l'ARR et aux ARC par l'entremise des demandeurs locaux respectifs. En même temps, le demandeur local envoie les réponses aux questions du module 1 aux organismes respectifs.
Comme il a été mentionné, le demandeur doit indiquer dans la DI le délai souhaité pour soumettre ses réponses à la LQ (soit 30 ou 60 jours). Toutefois, le demandeur pourra répondre en tout temps après 30 jours et avant 60 jours.
Deuxième ronde d'évaluation
Évaluation des réponses à la LQ (30 jours)
L'ARR prépare un RE des réponses à la LQ regroupée pour les modules 2 à 5 et le transmet aux ARC. En même temps, l'ARR et les ARC préparent un RE des réponses aux questions propres à leur pays pour le module 1.
Examen par les pairs réalisé par les ARC (15 jours)
Les ARC procèdent à un examen par les pairs du RE des réponses pour les modules 2 à 5 et fournissent une rétroaction. Si nécessaire, l'ARR prépare une LQ supplémentaire, que chaque organisme envoie au demandeur local. Le délai de 15 jours comprend le temps consacré à l'examen par les pairs et à toute coordination entre l'ARR et les ARC.
Finalisation des RE et de la LQ supplémentaires (5 jours)
Si une LQ supplémentaires (en général, cela correspond à la décision préliminaire en Suisse) n'est pas exigée, chaque autorité est tenue de prendre une décision finale. Les autorités prennent également les mesures administratives nécessaires pour achever le processus dans leur pays.
Présentation des réponses à la LQ supplémentaires (le cas échéant) (15 jours)
Le demandeur prépare et envoie les réponses à la LQ supplémentaires (le cas échéant) à l'ARR et aux ARC.
Troisième ronde d'évaluation (le cas échéant)
Évaluation des réponses à la LQ supplémentaires (15 jours)
L'ARR prépare un RE des réponses à la LQ supplémentaires à la suite du processus décrit pour la deuxième ronde.
Examen par les pairs des réponses par les ARC, finalisation des RE et de la LQ supplémentaires (le cas échéant) (5 jours)
Les ARC procèdent à un examen par les pairs des réponses et fournissent une rétroaction afin que l'ARR puisse finaliser le RE.
Étapes à l'échelle nationale
Chaque organisme prend une décision finale (ou cherche à obtenir des éclaircissements sur des questions distinctes avant d'en prendre une) et entreprend les étapes administratives nécessaires pour achever le processus à l'échelle nationale. Selon le résultat de l'évaluation de chaque organisme, une lettre d'autorisation est émise ou des questions supplémentaires sont posées.
Ces communications n'ont pas nécessairement lieu en même temps.
L'une des 2 périodes maximales totales suivantes écoulées entre le moment où la demande est acceptée (jour 0) et le début des étapes à l'échelle nationale :
- De 170 à 200 jours civils (y compris le temps de réponse du demandeur et selon si une seule liste de questions est exigée)
- De 205 à 235 jours civils (si une liste de questions supplémentaires est exigée)
Veuillez adresser toute autre question au sujet de cette initiative à votre organisme de réglementation local :
- Australie : PMABinternationalevaluations@health.gov.au
- Canada : collaboration@hc-sc.gc.ca
- Singapour : HSA_TP_Enquiry@hsa.gov.sg
- Suisse : Networking@swissmedic.ch
- Royaume-Uni : Access-MHRA@mhra.gov.uk
Les communications par courriel doivent inclure « Access Consortium – Initiative de partage du travail concernant les médicaments génériques » dans la ligne de mention objet.
Liens connexes
- A survey of the regulatory requirements for the acceptance of foreign comparator products by participating regulators and organizations of the International Generic Drug Regulators Programme
- Biopharmaceutic studies (lignes directrices de l'Australie)
- Comparator products in bioequivalence/therapeutic equivalence studies (lignes directrices du Royaume-Uni)
- Ligne directrice : Utilisation d'un produit de référence étranger comme produit de référence canadien (lignes directrices du Canada)
- Guidance on therapeutic product registration in Singapore: Product interchangeability and biowaiver request for chemical generic drugs application (lignes directrices de Singapour)
- Guidance document: Authorisation of human medicinal product with known active pharmaceutical substances HMV4 (lignes directrices de Suisse)
Détails de la page
- Date de modification :