
Guide technique et sur l’assemblage adapté aux présentations fondées sur la table des matières de l’IMDRF de Santé Canada
(Version PDF, 660 Ko, 22 pages)
Sur cette page
- 1. Introduction
- 2. Guide sur l'élaboration d'une présentation fondée sur la TdM
- 3. Lignes directrices techniques
- 3.1 Structure des dossiers
- 3.2 Conventions d'appellation de dossier
- 3.3 Format et appellation des fichiers
- 3.4 Limites quant à la taille des fichiers et des présentations
- 3.5 Sécurité des documents
- 3.6 Mise en signets des fichiers PDF
- 3.7 Liens hypertextes dans les fichiers PDF
- 3.8 Règles concernant le niveau de détail
- 3.9 Pagination
- Annexe 1 : Conseils utiles
1. Introduction
1.1 Contenu du Forum international des organismes de réglementation des matériels médicaux (IMDRF)
Le contenu, y compris l'objectif et la portée du présent document, découle d'un document de l'IMDRF intitulé « Assembly and Technical Guide for IMDRF Table of Contents Submissions ». D'autres éléments exigés par Santé Canada sont indiqués dans les encadrés, comme dans l'exemple ci-dessous.
Exigences propres à Santé Canada
Exemple
1.2 Objectif/Aperçu
Le Forum international des organismes de réglementation des matériels médicaux (IMDRF) a été mis en œuvre en février 2011 sous forme de lieu permettant de discuter des futures orientations en matière d'harmonisation de la réglementation visant les instruments médicaux. Il s'agit d'un groupe formé d'organismes de réglementation des instruments médicaux du monde entier qui se réunissent volontairement dans le but de poursuivre les importants travaux préparatoires du Groupe de travail sur l'harmonisation mondiale (GTHM). Le forum accélérera l'harmonisation et la convergence de la réglementation visant les matériels médicaux à l'échelle internationale.
La présentation réglementée de produit (PRP) proposée a été adoptée par l'IMDRF à titre de nouvelle question dans le cadre de sa séance d'ouverture à Singapour (mars 2012). Jusqu'à maintenant, voici les réalisations du groupe de travail :
- établir que la norme de Health 7 (HL7) sur la présentation réglementée des produits (PRP) convient à l'échange électronique de renseignements liés aux demandes précommercialisation d'homologation des instruments médicaux;
- concevoir une table des matières (TdM) complète pour les demandes précommercialisation suivantes :
- autorisations de mise en marché d'instruments médicaux autres que les dispositifs de diagnostic in vitro (nDDIV),
- autorisations de mise en marché de dispositifs de diagnostic in vitro (DDIV).
Le présent document fournit des lignes directrices précises sur la rédaction d'une demande en vue de la PRP, y compris des lignes directrices harmonisées sur la structure des dossiers et les formats de fichiers acceptés pour les demandes fondées sur la TdM.
1.3 Portée et application
Le présent guide est destiné à l'assemblage de présentations sur la réglementation des instruments médicaux fondées sur la table des matières (TdM) de l'IMDRF faisant actuellement partie des types de présentations acceptées par Santé Canada.
2. Guide sur l'élaboration d'une présentation fondée sur la TdM
Un certain nombre de documents de référence et de guides doivent être consultés pendant l'élaboration d'une demande d'homologation d'instruments médicaux fondée sur la TdM. La présente section donne de l'information sur ces documents de référence ainsi que sur la façon de les utiliser pour produire une présentation fondée sur la TdM.
2.1 Documents de référence
Le tableau ci-dessous énumère les documents requis pour assembler une demande de réglementation fondée sur la TdM de l'IMDRF.
| Document | Description | Lieu |
|---|---|---|
IMDRF In Vitro Diagnostic Medical Device Market Authorization Table of Contents (IVD MA ToC) (en anglais seulement) [IMDRF/RPS WG/N13] ou IMDRF Non-In Vitro Diagnostic Medical Device Market Authorization Table of Contents (nIVD MA ToC) (en anglais seulement) [IMDRF/RPS WG/N9] |
Ces documents définissent les noms des titres et la structure hiérarchique de la TdM. Ils comprennent aussi des renseignements détaillés au sujet du contenu associé à chaque titre. |
www.imdrf.org (disponible en anglais seulement) |
IMDRF Assembly and Technical Guide for IMDRF Table of Content (ToC) Submissions [page web actuelles] |
Ce document fournit des renseignements au sujet des documents de référence disponibles relativement à la TdM de l'IMDRF et aux caractéristiques techniques harmonisées des présentations fondées sur la TdM. |
www.imdrf.org (disponible en anglais seulement) |
IMDRF Standard ToC Folder Structures (présenté sous forme de fichier compressé) |
Il s'agit d'une structure de dossiers fournie par l'IMDRF afin de reproduire la structure hiérarchique et les titres de la TdM. Remarque : certains titres ont été modifiés par rapport aux noms complets utilisés dans les documents IVD MA ToC et nIVD MA Toc afin de réduire la longueur du chemin d'accès. |
www.imdrf.org (disponible en anglais seulement) |
Matrices de classification régionales |
Comme les documents sur la TdM de l'IMDRF sont exhaustifs, ce ne sont pas tous les titres qui sont requis pour tous les types de présentations et toutes les régions. La matrice de classification indique, pour un certain type de présentations, si un titre est requis, non requis, facultatif, conditionnel, etc. |
Variés – consultez les sites Web régionaux pour en savoir davantage. |
Le guide technique et sur l'assemblage régional pour les présentations fondées sur la table des matières de l'IMDRF |
En plus de ce qui est indiqué dans le présent document, les régions peuvent avoir d'autres exigences ou lignes directrices régionales particulières liées à l'élaboration et à l'organisation d'une présentation fondée sur la TdM qui sont rassemblées dans un guide technique et sur l'assemblage régional (p. ex. méthode de transmission ou directives particulières pour le transfert des fichiers). |
Variés – consultez les sites Web régionaux pour en savoir davantage. |
2.2 Exemple de procédure générale pour l'élaboration d'une présentation fondée sur la TdM
La présente section donne un exemple de méthode pouvant être utilisée pour assembler une présentation fondée sur la TdM de l'IMDRF. D'autres stratégies peuvent être acceptées, y compris l'utilisation d'un logiciel de publication de présentations offert sur le marché pour générer une présentation répondant aux exigences.
Étape 1
TéléchargezNote de bas de page 1 la structure de dossiers normalisée pour la TdM de l'IMDRF convenant à la structure de TdM applicable (p. ex. DDIV ou nDDIV).
Étape 2a
Commencez à élaborer la présentation en consultant le document IMDRF Market Authorization Table of Contents approprié (IVD MA ToC ou nIVD MA ToC) ainsi que les lignes directrices régionales liées au contenu. Consultez la matrice de classification régionale pour déterminer les titres qui doivent être utilisés selon le type de présentation. Pour de plus amples renseignements concernant les matrices de classification, veuillez consulter l'Annexe 1 du présent guide. Voir la section 2.3, Considérations importantes liées à l'utilisation dans plusieurs régions, ci-dessous pour connaître les facteurs importants à prendre en compte dans ce processus.
Étape 2b
Consultez le présent document et ses équivalents régionaux pour la région concernée afin de connaître les exigences techniques liées aux présentations.
Étape 3
Consultez la matrice de classification de la région concernée pour déterminer les dossiers qui peuvent être supprimés de la structure complète en fonction du type de présentation. Voir la section 3.1, Structure des dossiers, pour de plus amples renseignements.
2.3 Considérations importantes liées à l'utilisation dans plusieurs régions
Les responsables de la mise en œuvre doivent examiner la possibilité d'utiliser un même contenu pour plusieurs régions. Même si certaines régions peuvent avoir des exigences additionnelles en ce qui a trait au contenu sous certains titresNote de bas de page 2, il peut être sage de concevoir une version de la présentation qui n'est pas propre à une région en utilisant la structure de dossier normalisée complète pour la TdM de l'IMDRFNote de bas de page 3. Il faut faire une copie de la version complète pour chaque région à laquelle vous avez l'intention de soumettre une présentation avant de supprimer les dossiers qui ne sont pas requis pour la région concernée. Il sera ensuite plus facile de concevoir des adaptations régionales à partir de la structure et du contenu de la présentation de base. Cette démarche réduit les risques :
- d'inclure du contenu régional qui n'est pas requis dans la présentation;
- d'oublier des éléments requis en raison de dossiers qui ont été supprimés, mais qui sont exigés pour d'autres régions.
Inversement, si l'approche susmentionnée ne peut être utilisée et qu'une présentation est créée à partir d'une présentation déjà soumise à une autre région, prenez soin :
- d'examiner les titres propres à la région ou qui exigent une orientation régionale et de les adapter si nécessaire;
- de vous assurer que le contenu régional qui n'est pas pertinent pour l'organisme de réglementation visé est supprimé;
- de veiller à ce que tous les dossiers qui auraient pu être supprimés pour la présentation initiale soient réexaminés aux fins d'inclusion dans la nouvelle présentation en vertu de la matrice de classification régionale pour la nouvelle région;
- de vous assurer que le contenu est à jour (p. ex. antécédents commerciaux à jour).
Lignes directrices propres à Santé Canada
Les renseignements fournis précédemment ne devraient pas être communiqués à nouveau, sauf s'ils sont touchés par un changement.
3. Lignes directrices techniques
Ces lignes directrices ont été établies pour assurer l'uniformité des exigences entre les régions. Les sections suivantes décrivent les lignes directrices de base pour la soumission d'une présentation fondée sur la TdM.
3.1 Structure des dossiers
Les documents de l'IMDRF ainsi que les documents In Vitro Diagnostic Medical Device Market Authorization Table of Contents (IVD MA ToC) et Non-In Vitro Diagnostic Device Market Authorization Table of Contents (nIVD MA ToC), définissent le contenu de chaque dossier. La structure des dossiers doit être établie conformément aux exigences de l'IMDRF. Consultez le fichier IMDRF Standard ToC Folder Structure, qui est un modèle physique sur la structure des dossiers fourni par l'IMDRF pour faciliter la préparation des demandes sous la forme prescrite par la TdM.
Lignes directrices propres à Santé Canada
Le dossier de premier niveau d'une présentation contient tous les autres dossiers et leur contenu. Le nom du dossier de premier niveau devrait être le nom de l'instrument ou le numéro de l'homologation ou de la demande. Si le numéro de l'homologation ou de la demande est disponible, il s'agit de la meilleure approche. Si le nom de l'instrument est utilisé, il ne doit pas contenir plus de 15 caractères.
Par exemple, si le nom de l'instrument est « 2000X Ultrasound », le dossier peut être nommé « 2000X » comme indiqué ci-dessous.
Le dossier racine ne devrait pas contenir de fichiers; il ne devrait contenir que les dossiers requis.
Figure 1 – Capture d'écran démontrant la hiérarchie des dossiers, incluant le dossier racine pour le 200X (instrument fictif servant d'exemple).

Équivalent textuel
La hiérarchie des dossiers avec un dossier racine nommé 200X et les sous-répertoires 1.02-Lettre d'accompagnement, 1.03 Liste de termes-acronymes, et 1.04 Formulaires-Rens admin ainsi que les fichiers PDFs correspondants.
Les matrices de classification régionales décrivent les éléments de la TdM qui sont exigés pour chaque présentation sur la réglementation concernée. Les facteurs qui influencent l'inclusion ou l'exclusion du contenu des présentations seront examinés plus attentivement ci-après.
Chaque dossier de la présentation est considéré comme étant requis ou non requis pour une présentation en particulier. Ces termes sont définis de façon précise dans la matrice de classification (p. ex. classification « requis » ou « non requis ») ou à travers l'interprétation de la classification (p. ex. en évaluant les conditionsNote de bas de page 4 pour les dossiers classés « requis conditionnellement » ou par le biais d'une décision du demandeur pour ceux qui sont classés « facultatifs »). Dans cette optique, la figure 1 ci-dessous décrit les classifications qui peuvent amener à juger qu'un dossier est « requis » ou « non requis » pour la présentation.
Tous les dossiers considérés comme étant « requis » ne devraient pas être supprimés. Le contenu doit être présenté dans ce dossier.
Tous les dossiers considérés comme étant « non requis » devraient être supprimés pour s'assurer que la présentation ne contient pas de dossiers vides. Si un dossier principal ne contient aucun contenu, il faut aussi le supprimer.
Tous les dossiers considérés comme étant « requis conditionnellement » doivent être classés par le demandeur en fonction des conditions exigées. Un dossier doit être conservé si l'on considère que son contenu est requis ou supprimé si l'on considère que son contenu est non requis.
Pour tous les dossiers considérés comme étant « facultatifs », le demandeur doit prendre la décision de le supprimer s'il n'est pas utilisé.
Il convient de souligner que certaines régions peuvent exiger une déclaration décrivant pourquoi une section n'est pas fournie. Veuillez consulter la section C – Énoncés pour les titres sans objet, de l'annexe 1 pour approfondir la discussion.
Figure 2 – Les classifications définies dans la matrice de classification (rectangles) déterminent si le contenu est requis ou non requis (ovales) pour une présentation en particulier.
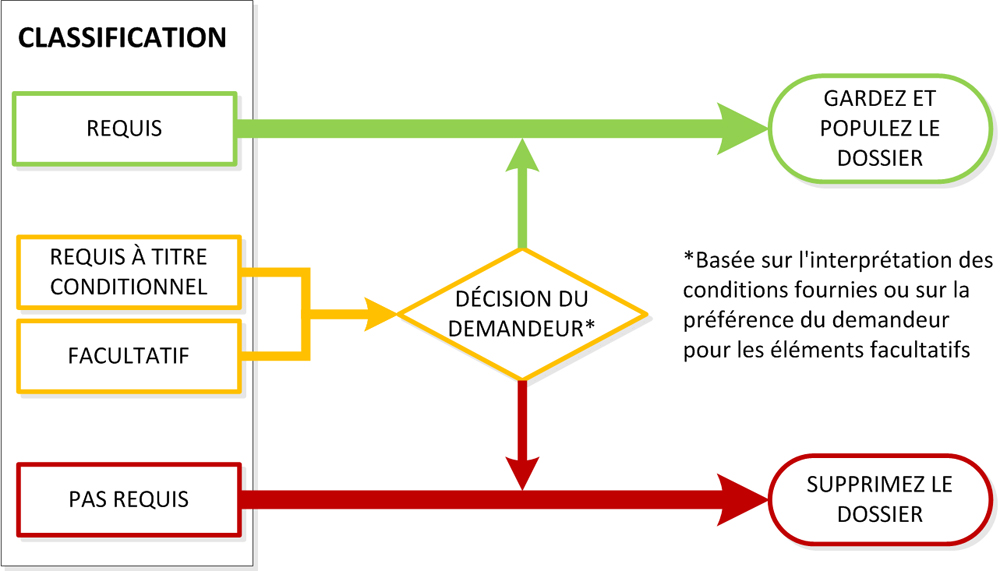
Équivalent textuel
Une figure listant les classifications suivantes dans les cases de gauche: «Obligatoire», «Requis à titre conditionnel», «Facultatif» et «Pas requis». La boîte de décision en forme de losange au centre de la figure indique « Décision du demandeur ». Le côté droit de la figure montre deux actions énumérées dans les ovales: gardez et populez le dossier et supprimez le dossier. Une flèche connecte la case «Obligatoire» à la case «Gardez et populez le dossierr» indiquant que tout dossier requis doit être conservé. Une flèche relie les cases «Requis à titre conditionnel» et «Facultatif» au losange « Décision du demandeur ». Le losangede la décision du demandeur comporte des flèches menant à l'ovale « Supprimer le dossier » ou « Gardez et populez le dossier ». Il y a une note associée à la «Décision du demandeur» qui indique « soit par interprétation de la condition définie au niveau régional pour le contenu conditionnel, soit par préférence pour le contenu optionnel ».
3.2 Conventions d'appellation de dossier
Les dossiers indiqués dans les modèles fournis seront numérotés et nommés conformément aux exigences de la TdM, à l'exception des titres personnalisés, qui seront numérotés et nommés par l'utilisateur. Plus précisément, dans le modèle de structure de dossiers de l'IMDRF, le nom de ces dossiers comprendra le terme « [Custom] » et il sera adapté pour décrire des particularités de l'étude (p. ex. [Study description, study identifier, date of initiation]). Le nom des dossiers [Custom] ou [Trial Details] ne doit pas dépasser 50 caractères (en incluant le numéro de section). Les abréviations dans les noms de dossiers sont prévisibles et acceptables.
Remarque : Il existe des contraintes en ce qui concerne l'appellation des fichiers et des dossiers pour s'assurer de ne pas dépasser la longueur maximale imposée par le système pour les chemins d'accès aux fichiers. Les demandeurs doivent être conscients que les systèmes d'exploitation des ordinateurs ont des limites et qu'ils doivent donc restreindre au minimum les noms des fichiers et des chemins d'accès dans leurs présentations.
Le dernier chiffre du numéro de titre doit être modifié, le cas échéant, pour garantir une présentation dans le bon ordre des dossiers personnalisés lorsque plus d'une étude est incluse.
Par exemple, pour le titre Physical and Mechanical Characterization, le premier dossier personnalisé de l'étude devrait être nommé « 3.05.01.01 [description de l'étude, identificateur de l'étude, date d'ouverture] » et le second dossier personnalisé sur l'étude devrait être nommé « 3.05.01.02 [description de l'étude, identificateur de l'étude, date d'ouverture] ». La numérotation de séquence devrait comprendre deux chiffres (p. ex. 3.05.01.01… 3.05.01.10).
Des dossiers « Overview » ont été créés dans le modèle de dossiers lorsque les lignes directrices de l'IMDRF comprennent une exigence en ce qui a trait au contenu d'un dossier principal. Cette structure de dossiers a été conçue pour veiller à ce que l'ordre des renseignements présentés soit conservé dans un environnement Windows. Par exemple, dans la section 3.05.06, Biocompatibility & Toxicology Evaluation de la structure nDDIV, un sous-dossier est nommé « 3.05.06.00-Overview » dans le modèle. Le contenu exigé dans les lignes directrices de l'IMDRF pour la section 3.05.06 devrait être placé dans ce dossier.
3.3 Format et appellation des fichiers
Les fichiers en format de document portable (PDF) sont à privilégier, bien que d'autres formats comme ceux de Microsoft Office (.docx, .pptx, .xlsx) soient aussi acceptés dans certaines régions.
Lignes directrices propres à Santé Canada
Conformément aux lignes directrices de l'IMDRF, les fichiers en format de document portable (PDF) (versions 1.7, PDF/A-1 et PDF/A-2) sont à privilégier pour Santé Canada, bien que d'autres formats comme ceux de Microsoft Office 2010 (.docx, .pptx, .xlsx) soient aussi acceptés.
Voici, notamment, des formats de fichiers qui ne sont pas acceptés :
- les documents PDF contenant des pièces jointes;
- les fichiers cache de vignettes (Thumbs.db);
- les fichiers de messages Outlook (.msg);
- les fichiers de sauvegarde (~*.docx);
- les fichiers d'images (.jpeg, .bmp, .tff);
- les documents contenant des macros (p. ex., .docm);
- les fichiers d'archives (p. ex. .zip) sauf dans les cas où la soumission sera transmise par courriel et que le fichier d'archive contient la totalité de la soumission. Voir les directives sur la transmission par courriel à la section 3.1.2 – Courriel dans les Lignes directrices de Santé Canada pour les demandes d'homologation d'instruments médicaux fondées sur la TdM de l'IMDRF.
Le demandeur devrait créer tous les fichiers PDF directement à partir des documents d'origine lorsque c'est possible, plutôt qu'en les numérisant. Les documents PDF conçus en numérisant des documents sur papier ont une qualité bien inférieure aux documents d'origine, comme un document Word, et ils devraient donc être évités dans la mesure du possible. Les documents numérisés, en particulier les tableaux et les graphiques, sont plus difficiles à lire et ne permettent pas aux examinateurs de copier et de coller le texte.
Pour tous les documents numérisés, vous devriez les soumettre au processus de reconnaissance optique des caractères (ROC) pour que le texte soit interrogeable. Vérifiez que le contenu a été correctement converti : (1) en surlignant une zone de texte et (2) en cherchant un mot ou une phrase. Si la recherche du mot ou de la phrase ne donne aucun résultat, la ROC n'a pas reconnu le texte. Nous reconnaissons que la ROC pourrait ne pas être possible dans certains cas pour les documents contenant des figures et des images.
La plupart des noms de fichiers sont définis par l'utilisateur et limités à 50 caractères (incluant l'extension et le numéro de section). Les noms de fichier devraient être significatifs et devraient donner des détails sur leur contenu. Lorsque de multiples fichiers sont jugés nécessaires dans un dossier donné, les méthodes d'affectation des noms pour les fichiers devraient veiller à ce que les fichiers soient présentés dans l'ordre convenu. Par exemple, dans le dossier nommé « 2.04.01-Comprehensive Device Description & Principle of Operation » les fichiers doivent apparaître dans l'ordre suivant :
- 2.04.01.00-Comprehensive Device Description and Principle of Operation.pdf;
- 2.04.01.01– Engineering drawings.pdf.
Les titres utilisés par l'IMDRF sont saisis un à un avec les dossiers indiqués dans les modèles de dossiers, à l'exception de ce qui suit :
- les titres Summary ou Synopsis;
- le titre Full Report;
- le titre Statistical Data.
Ces titres doivent être inclus sous forme de fichiers directement dans les dossiers [Custom] ou [Trial Details]. Ces fichiers doivent être nommés afin de s'assurer de conserver l'ordre utilisé dans la table des matières de l'IMDRF (c.-à-d. d'abord Summary/Synopsis, puis Full Report et en troisième Statistical Data).
Remarque : Il existe des contraintes en ce qui concerne l'appellation des fichiers et des dossiers pour s'assurer de ne pas dépasser la longueur maximale imposée par le système pour les chemins d'accès aux fichiers. Les demandeurs doivent être conscients que les systèmes d'exploitation des ordinateurs ont des limites et qu'ils doivent donc restreindre au minimum les noms des fichiers et des chemins d'accès dans leurs présentations.
Les modèles de dossiers de l'IMDRF et les caractéristiques liées à l'appellation ont été conçus dans le but de s'assurer que les organismes de réglementation peuvent recevoir et stocker les présentations sans atteindre les limites du système d'exploitation. Il est recommandé que les demandeurs examinent la longueur du chemin d'accès (c.-à-d. les noms de tous les dossiers et fichiers imbriqués et des extensions de fichier) avant la transmission pour vérifier que le chemin d'accès compte au plus 200 caractères.
3.4 Limites quant à la taille des fichiers et des présentations
Aucun fichier PDF de la présentation ne doit dépasser 100 Mo. De nombreux documents fournis dans un seul fichier PDF ne sont pas acceptés.
La présentation totale ne devrait pas dépasser 4 Go pour s'assurer qu'elle sera acceptée par toutes les régions participantes.
3.5 Sécurité des documents
Les fichiers ne doivent pas avoir de paramètres de sécurité. Plus précisément :
- les fichiers ne doivent pas être protégés par un mot de passe empêchant de les ouvrir;
- les fichiers doivent permettre l'impression, la sélection du texte et des graphiques ainsi que l'ajout ou la modification de notes et de champs de formulaire.
Les demandeurs doivent utiliser des systèmes de téléchargement sécuritaires ou des services de messagerie de bonne réputation pour protéger la transmission aux organismes de réglementation.
3.6 Mise en signets des fichiers PDF
Il est aussi important que les fichiers PDF soient correctement structurés, au moyen d'une table des matières interne adéquatement balisée à l'aide de signets. Voici les bonnes pratiques de structuration recommandées :
- les documents de dix pages ou plus devraient avoir leur propre table des matières interne;
- lors de la création des signets, la fonction d'agrandissement devrait être réglée à « héritier du zoom » de façon à ce que la page de destination affiche l'information au même niveau d'agrandissement que celui utilisé par l'examinateur pour le reste du document;
- les sections, sous-sections, tableaux, figures et annexes doivent être mis en signet;
- il faut éviter les pièces jointes dans les fichiers PDF;
- une hiérarchisation des signets trop élevée est peu efficace. Dans la plupart des cas, une hiérarchisation comportant trois niveaux est suffisante. P. ex. :
- 1. Titre
- 1.1 Sous-titre
- 1.1.1 Sous-sous-titre
- 1.1 Sous-titre
- 1. Titre
On reconnaît que les signets sont générés automatiquement à partir des titres du document; néanmoins, il est recommandé d'en limiter le nombre.
Définissez l'onglet de navigation pour afficher le « Panneau Signets et page ». Il est ainsi possible d'établir la vue initiale du document lorsque le fichier est ouvert. S'il n'y a pas de signets, sélectionnez « Page seule ». La disposition des pages et le zoom devraient être réglés à « Par défaut ».
3.7 Liens hypertextes dans les fichiers PDF
Les hyperliens sont utilisés pour améliorer la navigation dans l'ensemble des documents PDF et sont encouragés. Les hyperliens peuvent être désignés par des encadrés en utilisant de minces lignes ou un texte en bleu, ou vous pouvez utiliser des encadrés invisibles pour les liens hypertextes dans une table des matières afin d'éviter d'occulter le texte. Les hyperliens dans le corps du document, visant à étayer les annotations, les sections connexes, les références, les annexes, les tableaux ou les figures qui ne sont pas situés sur la même page, sont utiles et renforcent l'efficacité de la navigation.
Les hyperliens entre les documents sont acceptés, mais il faut veiller à ce que les liens créés entre les différents documents fonctionnent au moment de la réception de la demande par l'organisme de réglementation (l'utilisation de liens internes est recommandée). Il appartient au demandeur de s'assurer que les hyperliens fonctionnent. Les liens doivent aussi comprendre des renvois à des sections ou des pages précises au cas où le lien serait rompu.
3.8 Règles concernant le niveau de détail
Il n'y a pas de limite au nombre de fichiers autorisés par titre dans une présentation, toutefois, les lignes directrices suivantes doivent être prises en compte.
- Des efforts doivent être déployés pour rédiger des documents communiquant clairement le contenu décrit dans les documents de l'IMDRF In Vitro Diagnostic Medical Device Market Authorization Table of Contents (IVD MA ToC) ou Non-In Vitro Diagnostic Medical Device Market Authorization Table of Contents (nlVD MA ToC), plutôt que de simplement inclure des documents existants contenant des renseignements superflus qui ne sont pas nécessaires sous un titre en particulier. Par exemple, il est moins utile d'inclure un certain nombre de fiches de données de sécurité sous le titre « 2.4.1 –Comprehensive Device Description and Principle of Operation » que de résumer les détails précis concernant ce titre.
- Lorsque de multiples fichiers sont jugés nécessaires, les méthodes d'affectation des noms pour les fichiers devraient veiller à ce que les fichiers soient présentés dans l'ordre convenu. Par exemple, dans le dossier nommé « 2.04.01-Comprehensive Device Description & Principle of Operation » les fichiers doivent apparaître dans l'ordre suivant :
- 2.04.01.00-Comprehensive Device Description and Principle of Operation.pdf
- 2.04.01.01– Engineering drawings.pdf
3.9 Pagination
Les pages de la présentation devraient être numérotées de façon à ce qu'il soit facile de se référer aux renseignements grâce au numéro de page. La pagination devrait être appliquée à chaque document (c.-à-d. le fichier physique). Cela peut être fait en numérotant les pages par section ou par chapitre (p. ex. 2.04.01-1, 2.04.01-2).
Lignes directrices propres à Santé Canada
Santé Canada a défini une série de règles de validation décrites dans le document Avis : Règles de validation des transactions réglementaires envoyées à Santé Canada en format « électronique autre que le format eCTD ». Des outils de validation sont offerts gratuitement par certains fournisseurs de logiciels sur les présentations réglementaires. Les demandeurs sont encouragés à valider leur présentation au moyen de ces règles avant de la soumettre à Santé Canada.
Remarque importante :
Il convient d'ignorer les erreurs concernant la règle de validation « C05‑Convention d'appellation » lorsqu'elles résultent de l'utilisation des caractères requis pour une présentation fondée sur la TdM qui ne correspond pas aux exigences de l'International Council for Harmonisation (ICH) (p. ex. lettres majuscules, « . »).
La longueur maximale de 200 caractères pour les chemins de fichiers définie dans cette règle de validation demeure une exigence pour les présentations fondées sur la TdM.
Annexe 1 : Conseils utiles
A. Matrices de classification et classe de titre
Comme les documents sur la TdM sont exhaustifs, ce ne sont pas tous les titres qui sont requis pour tous les types de présentations et toutes les administrations. Les documents fondés sur la TdM devraient donc être utilisés de concert avec un document distinct créé pour chaque administration participante – une matrice de classification.
Que sont les matrices de classification?
Les matrices de classification sont des tableaux qui définissent la classe de chaque titre dans la TdM (p. ex. requis [R], non requis [NR], requis conditionnellement [RC], facultatif [F], facultatif, mais recommandé [FR]).
Chaque administration possède sa propre matrice de classification.
Les types de présentations pris en charge sont indiqués séparément dans la matrice. Par exemple, le tableau 3 montre les quatre premiers titres du chapitre 1 pour les nouvelles demandes d'homologation d'instruments de classe III de Santé Canada. Il convient de souligner que si le titre est requis conditionnellement (RC), les conditions seront décrites dans la colonne « Condition ».
| Code (Niveau TdM) |
Titre | Nouvelles demandes C3 | |
|---|---|---|---|
| Classification | Condition | ||
| Chapitre 1 - Renseignements administratifs régionaux | |||
| CH1.01 | Lettre d'accompagnement | R | Aucune condition |
| CH1.02 | Table des matières de la présentation | NR | Aucune condition |
| CH1.03 | Liste de termes et des acronymes | R | Aucune condition |
| CH1.04 | Formulaire de demande / Renseignements administratifs | R | Aucune condition |
Où peut-on trouver les matrices de classification?
Vous pouvez obtenir les matrices de classification auprès des organismes de réglementation régionaux.
Comment utilise-t-on la matrice de classification avec la TdM?
Voici les grandes étapes de l'utilisation des matrices de classification :
- obtenir la matrice de classification auprès de l'administration concernée;
- établir le type de présentation et vérifier qu'il s'inscrit dans la portée de la matrice de classification en vigueur de l'administration;
- élaborer la structure de votre présentation selon les directives fournies pour ce type de présentation. Tous les titres classés « non requis (NR) » ne devraient pas être inclus dans la présentation. Les titres classés « requis conditionnellement (RC) » doivent être examinés en tenant compte du type de matériel ou des conditions stipulées dans la matrice de classification. Le demandeur doit aborder TOUS les titres requis (R) dans la présentation.
Par exemple, beaucoup de types de présentations n'exigent que quelques éléments du chapitre 6B. Un exemple précis est une nouvelle demande d'homologation d'instrument de classe IV de Santé Canada. Dans ce cas, la matrice de classification est indiquée dans la figure 2 ci-dessous.
Dans ce cas, le chapitre 6B ne comprendrait que trois ou quatre titres (surlignés en vert), selon si oui ou non la condition indiquée pour le titre classé RC établit que le titre est jugé pertinent pour cette présentation.
| Code (Niveau TdM) |
Titre | Nouvelles demandes C4 | |
|---|---|---|---|
| Classification | Condition | ||
| Chapter 6b – Renseignements sur le système de gestion de la qualité | |||
| CH6B.1 | Table des matières du chapitre | NR | Aucune condition |
| CH6B.2 | Renseignements sur le système de gestion de la qualité | NR | Aucune condition |
| CH6B.3 | Renseignements sur les responsabilités de gestion | NR | Aucune condition |
| CH6B.4 | Renseignements sur la gestion des ressources | NR | Aucune condition |
| CH6B.5 | Plan sur la qualité des dispositifs | R | Aucune condition |
| CH6B.6 | Renseignements sur la réalisation de produits | R | Aucune condition |
| CH6B.6.1 | Renseignements sur la conception et le développement | NR | Aucune condition |
| CH6B.6.2 | Renseignements sur les achats | NR | Aucune condition |
| CH6B.6.3 | Renseignements sur les contrôles de production et de service | R | Aucune condition |
| CH6B.6.4 | Renseignements sur le contrôle de la surveillance et de mesure des dispositifs | NR | Aucune condition |
| CH6B.7 | Renseignements sur la mesure, l'analyse et l'amélioration du système de gestion de la qualité | NR | Aucune condition |
| CH6B.8 | Autres renseignements sur le système de gestion de la qualité propre au dispositif | RC | Lorsque l'organisme de réglementation demande des renseignements (par l'entremise de documents d'orientation ou d'autres communications), mais qu'ils ne correspondent à aucun autre titre de ce chapitre. |
Remarque importante
Chaque matrice de classification est conçue en fonction d'un éventail de documents sources, notamment les lois, les directives, les règlements ou les documents d'orientation de chaque organisme de réglementation. Lorsqu'il y a contradiction entre des exigences de la matrice de classification et ces documents sources, ces derniers ont préséance.
De quelle façon les matrices de classification seront-elles utilisées dans le futur?
Il est à noter que la TdM et les matrices ont été conçues pour être interprétées par un système électronique de présentation des demandes, comme un système conforme à la norme sur la présentation réglementée des produits.
La vision à long terme consiste à intégrer ces matrices aux systèmes de présentation électronique des demandes, comme un système conforme à la norme sur la PRP, pour concevoir automatiquement des trousses de demande électronique respectant les exigences définies de l'organisme de réglementation recevant la demande.
Qu'en est-il des classes de titres définies dans les documents fondés sur la TdM?
Les titres sont aussi classés dans les documents fondés sur la TdM dans les catégories IMDRF; IMDRF, FR; ou Régional. Ces termes sont définis dans les documents fondés sur la TdM.
La classification des titres est présentée dans les documents fondés sur la TdM pour donner une indication quant à la pertinence de tout titre donné pour une administration en particulier, et plus important encore, donner une idée des moments où un demandeur doit examiner le contenu commun dans le contexte d'une administration en particulier. Les matrices de classification fournissent avec plus de précision la classification requise par l'administration ainsi que le type de présentation et elles devraient être utilisées à titre de dernière référence pour ce type de renseignements.
B. Dossiers « Overview » et titres personnalisés
La TdM a été conçue avec une souplesse suffisante pour permettre l'utilisation d'une même structure pour différentes classes de risque. Une sous-structure en particulier est reproduite tout au long du document. Cette structure comporte un titre principal, un titre secondaire personnalisé pour chaque étude ou donnée probante particulière et un sous-sous-titre comprenant un résumé et un rapport complet. Par exemple, le titre « Physical and Mechanical Characterization » est structuré comme suit.
| Titre du chapitre | Description du contenu |
|---|---|
| Caractérisation physique et mécanique | Le titre principal résume toutes les études qui entrent dans cette catégorie (c.-à-d. essais physiques et mécaniques). Chacun des titres principaux est légèrement différent alors consultez le document fondé sur la TdM pour connaître le contenu associé à ces titres. |
| [Description, identificateur et date de début de l'étude]] | Il s'agit d'un titre personnalisé fondé sur l'étude précise décrite ci-dessous – aucun contenu à ce niveau. |
| Résumé | Un résumé de l'étude précise décrite dans le dossier personnalisé susmentionné. |
| Rapport complet | Le rapport d'essai pour l'essai décrit dans le titre personnalisé susmentionné. |
Remarques importantes :
- Dans l'application de la structure de dossiers, le contenu du titre principal est compris dans un dossier « Overview » sous le titre (3.5.01.00 dans l'exemple ci-dessous).
- Lorsque de nombreuses études se retrouvent sous un titre en particulier, les études doivent être présentées de manière séquentielle sous le titre principal, comme montré ci-dessous.
Exemple de structure de dossiers à appliquer
Figure 3 – Capture d'écran démontrant la hiérarchie des dossiers dans le chapitre 3 avec des entêtes sur-mesure ainsi que des résumés et des rapports complets.

Équivalent textuel
La hiérarchie des dossiers avec un dossier racine nommé 3.05.01-Physique mécanique et les sous-répertoires 3.05.01.00– Sommaire qui contient un fichier PDF 3.05.01-Sommaire.pdf, 3.05.01.01– Component A Test de fatigue, MT4203, 2010-10-10 qui contient deux fichiers 1-Résumé.pdf et 2-Rapport.pdf, et 3.05.01.02-Test d'usure de l'ensemble B, MT4584, 2011-01-23 qui contient deux fichiers 1-Résumé.pdf et 2-Rapport.pdf.
Le contenu au niveau du titre principal inclus dans le dossier « Overview » vise à expliquer le contexte de toutes les études présentées plus bas. Le sommaire doit être une description de haut niveau, par exemple :
Caractérisation physique et mécanique (exemple pour un insert utilisé dans la hanche)
Selon les risques associés à l'insert, les évaluations suivantes ont été examinées :
- tests d'usure;
- tests d'extraction;
- …
Toutefois, puisque le mécanisme de verrouillage et la géométrie générale étaient identiques à ceux des versions précédentes, nous n'avons pas jugé nécessaire de répéter les tests d'extraction pour le nouveau concept. Les tests d'usure ont été jugés nécessaires en raison de la modification du processus de fabrication pour utiliser l'UHMWPE. Une copie des tests déjà réalisés a été ajoutée aux fins de référence et ils avaient déjà été examinés dans le cadre de la demande XYZ.
Tests d'usure – Ces tests ont été réalisés sur le plus gros élément indiqué dans la présente demande : taille 36, déviation de +4mm selon l'ASTM F1714 pour 10 MC. Les résultats des tests d'usure compte tenu du volume et de la morphologie se sont avérés comparables à ceux des instruments testés en clinique dans des conditions identiques.
L'usure est une des principales causes d'échec clinique pour les prothèses de la hanche. Cette caractérisation montre que les propriétés de cet instrument en matière d'usure sont semblables sur le plan du volume et de la morphologie à celles des instruments dont l'efficacité a été démontrée en clinique.
C. Énoncés pour les titres sans objet
De nombreux titres dans la présentation nécessitent un énoncé expliquant pourquoi une catégorie en particulier ne s'applique pas à une présentation donnée. Le niveau de soutien pour de tels énoncés varie et ils peuvent prendre la forme des catégories décrites ci-dessous.
Remarque importante :
Les matrices de classification régionale sont la source officielle en ce qui a trait au contenu soumis. Lorsque l'interprétation de la classification régionale indique qu'un titre est NON REQUIS, conformément à la section 4.1 du document Assembly and Technical Guide for IMDRF Table of Contents Submissions, le titre doit être exclu en totalité de la présentation.
| Catégorie | Description | Mesures recommandées |
|---|---|---|
Catégorie 1 – Sans objet pour la présentation |
Dans ce cas, il est évident que l'information ne s'applique pas à l'instrument. Par exemple, des preuves de la sûreté du matériel biologique ne sont pas nécessaires si aucun matériel biologique n'est utilisé dans l'instrument. |
La recommandation varie selon l'administration et/ou le type de présentation et en fonction de la classification du titre. Si la classification régionale pour le type de présentation indique que le titre est REQUIS, aucune explication n'est nécessaire et l'énoncé « Sans objet pour la présentation » suffit. Si la classification régionale pour le type de présentation indique que le titre est NON REQUIS (que ce soit explicitement ou en interprétant la condition), le titre doit être exclu en totalité de la présentation. |
Catégorie 2 – Pertinence possible pour la présentation, mais manifestement sans objet |
Dans ce cas, l'information peut être pertinente dans certaines situations, mais dans ce contexte précis, il est évident qu'elle ne s'applique pas. Par exemple, dans le cas où un fabricant modifie la méthode de stérilisation, mais que l'instrument demeure inchangé. Aucun renseignement sur la biosécurité n'est donc fourni. |
La recommandation varie selon l'administration et/ou le type de présentation et en fonction de la classification du titre. Si la classification régionale pour le type de présentation indique que le titre est REQUIS, il faut expliquer plus en détail le contexte précis, mais l'explication peut se limiter à quelques phrases. Par exemple, « la modification de la méthode de stérilisation n'a aucune incidence sur la sûreté de la source des matières biologiques qui a déjà été examinée ». Si la classification régionale pour le type de présentation indique que le titre est NON REQUIS (que ce soit explicitement ou en interprétant la condition), le titre doit être exclu en totalité de la présentation. |
Catégorie 3 – Pertinence pour la présentation, mais exclu |
Dans ce cas, l'information devrait être fournie dans la présentation, mais elle a été omise après un examen approfondi. Par exemple, des tests de démontage pour un nouveau système de prothèse de la hanche modulaire sont habituellement prévus pour ce type d'instrument, mais ils ont été omis. |
Dans ces cas, l'interprétation de la matrice de classification régionale devrait donner lieu à la classification REQUIS. Des documents scientifiques détaillés à l'appui de la décision de ne pas effectuer ces essais doivent être présentés et toutes les références pertinentes doivent être fournies dans la présentation afin d'appuyer la justification. |
D. Système de gestion de la qualité (chapitres 6A vs 6B)
La TdM comprend deux chapitres sur le système de gestion de la qualité. Les chapitres 6A et 6B de la TdM ont été rédigés en utilisant les termes concernant le système de gestion de la qualité employés dans la norme ISO 13485-2003. C'est dans le chapitre 6A que l'entreprise place les procédures opératoires normalisées (PON) qu'elle utilise pour mettre en œuvre son système de gestion de la qualité général de haut niveau. Dans le chapitre 6B, l'entreprise place les documents et les dossiers que l'entreprise utilise pour mettre en œuvre les PON liées au système de gestion de la qualité décrites au chapitre 6A.
Notes de bas de page
- Note de bas de page 1
-
Voir le fichier IMDRF Standard ToC Folder Structure.
- Note de bas de page 2
-
Pour une description complète des exigences habituelles et régionales en ce qui a trait au contenu de chaque titre, veuillez consulter les adresses suivantes : IMDRF In Vitro Diagnostic Medical Device Market Authorization Table of Contents (IVD MA ToC) (http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-140630-rps-ivd-toc.pdf) ou IMDRF Non-In Vitro Diagnostic Device Market Authorization Table of Contents (nIVD MA ToC). (http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-140630-rps-nivd-toc.pdf)
- Note de bas de page 3
-
Veuillez noter que l'utilisation de documents PDF et de dossiers est un format intermédiaire conçu pour mettre à l'essai une table des matières normalisée et que les futures mises en œuvre pourraient être plus conviviales. Il faut du temps pour que de multiples examinateurs ouvrent chaque fois une chaîne de dossiers pour se rendre compte qu'ils sont vides ou pour s'assurer qu'ils le sont. Il n'est pas habituel pour les organismes de réglementation de modifier des présentations, p. ex. en supprimant des documents ou des dossiers inutiles, puisque ces organismes doivent préserver l'intégrité de la présentation d'origine. Le groupe de travail sur la TdM de l'IMDRF apprécie vos efforts pour supprimer les dossiers vides et fournir un contenu correspondant aux lignes directrices régionales avant la présentation.
- Note de bas de page 4
-
Les conditions pour les titres « requis conditionnellement » sont décrites dans les matrices de classification.
Détails de la page
- Date de modification :