
Impuretés de nitrosamine dans les médicaments : Lignes directrices

Télécharger au format PDF
(1.54 Mo, 69 pages)
Organisation : Santé Canada
Date publiée : 2022-04-04
Mises à jour : 2024-03-15
Sur cette page
- Contexte
- Généralités
- Innocuité
- Qualité
- Annexes
- Annexe 1 : Limites acceptables (LA) établies pour les impuretées de N-nitrosamines
- Annexe 2 : Lignes directrices sur les impuretés de nitrosamine et l'évaluation des risques pour les changements survenus après l'avis de conformité (AC) dans le cas des nouveaux produits médicamenteux contenant des IPA synthétiques et semi-synthétiques (mis à jour)
- Annexe 3 : Conditions du test d'Ames amélioré
- Annexe 4 : Approche de catégorisation de la puissance cancérogène (ACPC) dans le cas des N-nitrosamines
Contexte
Ces lignes directrices présentent la réflexion et les recommandations actuelles de Santé Canada sur les problèmes liés aux impuretés de N-nitrosamine (ci-après appelées de manière interchangeable « impuretés de nitrosamine ou nitrosamines »). Ces lignes directrices peuvent faire l'objet de modification à mesure que de nouveaux renseignements deviennent disponibles et si d'autres lignes directrices sont nécessaires pour les demandeurs et les détenteurs d'une autorisation de mise sur le marché (DAMM).
Le document original de questions et réponses (Q et R) sur les nitrosamines a été distribué aux détenteurs d'autorisation de mise sur le marché (DAMM) le 26 novembre 2019. Ce document a fait l'objet d'un certain nombre de révisions et a été mis à jour en tant que lignes directrices pour fournir plus de précisions et d'informations aux fabricants d'ingrédients pharmaceutiques actifs (IPA), aux fabricants de produits médicamenteux, aux détenteurs d'autorisation de mise sur le marché (DAMM) et aux importateurs d'IPA et de produits médicamenteux.
Dans les présentes lignes directrices, les changements par rapport à la version précédente sont identifiés ci-dessous par les descriptifs « nouveau » ou « mis à jour » (selon le cas). Les renseignements sur un thème similaire ont été rassemblés sous des titres généraux (c'est-à-dire sous les titres Généralités, Innocuité et Qualité).
Toutes les demandes de renseignements concernant les lettres de Santé Canada indiquées ci-dessous peuvent être transmises comme suit :
- « Information aux DAMM des produits pharmaceutiques destinés à l'usage humain concernant des impuretés de nitrosamine : Demande d'évaluation du risque lié à la présence d'impuretés de nitrosamine dans les produits pharmaceutiques destinés à l'usage humain contenant des ingrédients pharmaceutiques actifs obtenus par synthèse chimique » (2 octobre 2019)
- Envoyer par courriel à l'adresse bpsenquiries@hc-sc.gc.ca
- « Information aux DAMM des produits pharmaceutiques destinés à l'usage humain concernant des impuretés de nitrosamine : Demande d'évaluation du risque lié à la présence d'impuretés de nitrosamine dans les produits biologiques et radiopharmaceutiques » (15 décembre 2020)
- Envoyer par courriel à l'adresse brdd.nitrosamines.dmbr@hc-sc.gc.ca
Si vous avez des questions au sujet des présentes lignes directrices, vous pouvez envoyer un courriel à brdd.nitrosamines.dmbr@hc-sc.gc.ca.
Généralités
Portée et responsabilités
1. Produits médicamenteux visés par la demande d'examen de Santé Canada
La demande d'évaluation du risque lié à la présence d'impuretés de nitrosamine de Santé Canada décrite dans la lettre du 2 octobre 2019 s'applique aux produits pharmaceutiques destinés à l'usage humain portant un numéro d'identification du médicament (DIN) et qui contiennent des IPA obtenus par synthèse chimique et semi-synthétiques. Cela comprend :
- les produits médicamenteux sur ordonnance et en vente libre
- les excipients et matières premières obtenus par synthèse chimique utilisés dans la fabrication de produits médicamenteux
On considère également que les produits suivants s'inscrivent dans la portée de la demande d'examen de Santé Canada :
- les produits médicamenteux qui ont été approuvés, mais qui ne sont pas encore commercialisés
- les produits médicamenteux approuvés dont la demande de DIN est en suspens
La demande d'évaluation du risque lié à la présence d'impuretés de nitrosamine a été étendue à tous les produits biologiques et radiopharmaceutiques destinés à l'usage humain, comme l'indiquait la lettre de Santé Canada du 15 décembre 2020.
Toutes les protéines du plasma humain, les vaccins et les produits issus de la fermentation cellulaire sont classés comme des produits biologiques. Ils font donc partie de la portée de la demande d'évaluation des risques.
Pour en savoir plus, veuillez consulter la lettre de Santé Canada du 15 décembre 2020.
Tous les produits en vente libre portant un DIN, comme les produits antiseptiques topiques, les produits de toilette et d'hygiène personnelle et les écrans solaires, sont visés par l'évaluation s'ils contiennent un IPA synthétisé chimiquement ou semi-synthétique. Cela s'applique, quelles que soient la voie d'administration ou les propriétés esthétiques.
Les produits, qui ne font pas partie de la portée des lettres du 2 octobre 2019 et du 15 décembre 2020, comprennent les cosmétiques (qui n'ont pas de DIN). Les catégories de produits médicamenteux suivantes sont également exclues pour le moment : les agents antimicrobiens, les produits vétérinaires (y compris les produits de santé vétérinaires) et les produits de santé naturels. Les produits désinfectants utilisés sur les surfaces dures ne font pas non plus partie de la portée des produits à évaluer pour le moment.
2. Délais pour effectuer les évaluations des risques (étape 1), les tests de confirmation (étape 2) et les changements à l'autorisation de mise en marché (étape 3)
Pour les produits médicamenteux contenant des IPA synthétisés chimiquement et semi-synthétiques, les mesures concernant les nitrosamines doivent se conformer aux étapes suivantes :
- Étape 1 : évaluations des risques d'ici le 31 mars 2021
- Étape 2 : tests de confirmation d'ici le 1er octobre 2022
- Étape 3 : changements à apporter à l'autorisation de mise en marché d'ici le 1er août 2025
Pour les produits biologiques et radiopharmaceutiques, les mesures concernant les nitrosamines doivent se conformer aux étapes suivantes :
- Étape 1 : évaluations des risques d'ici le 30 novembre 2021
- Étape 2 : tests de confirmation d'ici le 30 novembre 2023
- Étape 3 : changements à apporter à l'autorisation de mise en marché d'ici le 1er août 2025
3. Résultats des évaluations des risques (étape 1) et documents devant être fournis à Santé Canada (mis à jour)
La documentation relative à l'évaluation des risques doit être conservée par le DAMM, à moins que des impuretés de nitrosamine soient détectées dans l'IPA, le produit médicamenteux ou les deux pendant les tests de confirmation. À la suite des essais de confirmation du produit médicamenteux, Santé Canada doit être informé si une impureté de nitrosamine est détectée à un niveau supérieur à la limite acceptable (LA) établie (voir l'annexe 1 pour une liste des LA établies) pour l'impureté de nitrosamine en question, ou à un niveau supérieur à la limite acceptable établie en utilisant l'approche de catégorisation de la puissance cancérogène (ACPC, consulter la section 24 et l'annexe 4) si une limite acceptable n'a pas été établie par Santé Canada. Les résultats des tests de confirmation doivent être présentés au moment d'informer Santé Canada de la détection et renseignements détaillés sur l'évaluation des risques doivent être disponibles sur demande. Consulter la section 15.
Pour les impuretés de nitrosamine répertoriées à l'annexe 1 qui sont classées comme non mutagènes, la soumission de l'évaluation des risques et des résultats des tests de confirmation n'est pas requise, et ces impuretés doivent être contrôlées conformément aux lignes directrices Q3A et Q3B de l'ICH.
Veuillez noter que Santé Canada peut demander d'examiner l'évaluation des risques du DAMM pour tous les produits et demandera ces renseignements directement au DAMM, au besoin.
Les importateurs canadiens dont la licence d'établissement de produits pharmaceutiques (LEPP) est assortie de conditions pour les tests de nitrosamine dans les antagonistes des récepteurs de l'angiotensine II (aussi appelés « sartans ») peuvent fournir des renseignements à l'appui pour modifier ou supprimer les conditions. Ils doivent soumettre les évaluations des risques liés à l'IPA et aux produits médicamenteux ainsi que les résultats des tests effectués aux étapes 1 et 2 à des fins d'examen.
Envoyer par courriel à foreign.site-etranger@hc-sc.gc.ca.
Les importateurs canadiens peuvent demander une copie de l'évaluation des risques et des résultats des tests du DAMM pour faciliter cette demande. Les DAMM peuvent également fournir l'évaluation des risques demandée et les renseignements connexes à Santé Canada au nom de l'importateur canadien. Dans ce cas, le DAMM doit préciser le parti au nom duquel l'évaluation des risques et les renseignements connexes sont présentés.
4. Détermination des priorités et de l'ordre dans lequel les produits doivent être examinés
Les DAMM devraient utiliser une approche fondée sur le risque pour déterminer l'ordre dans lequel ils doivent examiner leurs produits médicamenteux. Afin d'établir l'ordre dans lequel les produits doivent être examinés, les DAMM devraient tenir compte d'un certain nombre de facteurs, notamment :
- les principes énoncés dans la directive Q9 de l'International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) : Gestion des risques liés à la qualité
- la dose quotidienne maximale du produit médicamenteux
- la voie d'administration
- la durée d'utilisation
- les indications et les considérations liées aux populations spéciales, comme les femmes enceintes et les enfants
- le profil toxicologique de l'IPA
- Par exemple, l'évaluation du risque de présence d'impuretés de nitrosamine dans les traitements contre le cancer, dont l'IPA est un mutagène puissant, pourrait être considérée comme étant moins prioritaire et faire l'objet d'une analyse à la suite des IPA de priorité plus élevée
- les considérations relatives au marché, comme la disponibilité du produit sur le marché canadien et le nombre de patients traités avec le produit médicamenteux
- de nouveaux renseignements intérieurs et internationaux selon lesquels une ou plusieurs impuretés de nitrosamine ont été décelées dans un IPA (ou un IPA structurellement semblable) ou un produit médicamenteux sont communiqués
- la présence d'éléments structurels dans l'IPA ou de conditions dans les processus de fabrication et d'emballage de l'IPA ou du produit médicamenteux, qui sont propices à la formation de nitrosamines (par exemple, la présence d'amines secondaires ou tertiaires dans l'IPA)
L’annexe 1 doit être consultée pour connaître IPA et les produits pharmaceutiques qui peuvent contenir contiennent des impuretés de nitrosamines. La littérature évaluée par les pairs (par exemple, M.K. Parr, J.F. Joseph, Journal of Pharmaceutical and Biomedical Analysis 164 (2019) 536–549) et d'autres sources d'information (par exemple, les communications réglementaires) doivent également être consultées pour les IPA et les produits pharmaceutiques connus pour contenir des impuretés de nitrosamine.
5. Collaboration des DAMM avec les fabricants d'IPA et de produits médicamenteux pour effectuer des évaluations des risques
Après avoir reçu l'autorisation de mise en marché au Canada, les DAMM sont responsables de l'innocuité, de l'efficacité et de la qualité de leurs produits médicamenteux et de la réalisation des évaluations des risques. À ce titre, ils doivent :
- collaborer avec les fabricants d'IPA et de produits médicamenteux pour examiner leurs processus de fabrication d'IPA et de produits médicamenteux afin d'effectuer des évaluations des risques
- tenir compte des connaissances des fabricants d'IPA et de produits médicamenteux sur les processus de fabrication, les sources potentielles de contamination et les autres causes fondamentales de la formation et de la présence d'impuretés de nitrosamines
Les fabricants d'IPA et de produits médicamenteux devraient mettre à la disposition des DAMM les renseignements nécessaires à la réalisation des évaluations des risques.
Si le risque de formation d'impuretés de nitrosamine a été évalué au cours de la phase de mise au point des processus de fabrication de l'IPA ou du produit médicamenteux, les renseignements tirés de l'évaluation peuvent être utilisés pour appuyer l'évaluation.
6. Responsabilités des fabricants d'IPA, des fabricants d'excipients, des fabricants de produits médicamenteux, des DAMM et des importateurs
Après avoir reçu l'autorisation, les DAMM sont responsables d'assurer l'innocuité, l'efficacité et la qualité continues des produits médicamenteux sur le marché canadien. Cela comprendrait la mise en œuvre d'un programme de surveillance continue pour détecter les tendances en matière de qualité. Un tel programme devrait reposer sur des contrôles appropriés pour les matières premières, toutes les étapes de transformation, les paramètres de processus critiques et les attributs de qualité critiques.
Au moment des évaluations des risques liés à la présence potentielle d'impuretés de nitrosamines, les DAMM devraient effectuer des évaluations de risques robustes au moyen d'une approche holistique. Une évaluation détaillée de toutes les étapes du cycle de vie du produit devrait être effectuée et comprendre une évaluation des facteurs de risque et des causes fondamentales potentielles de la présence de nitrosamines, y compris ceux identifiés à la section 29.
Il incombe aux DAMM de s'assurer que les évaluations des risques sont faites par du personnel possédant des qualifications et des compétences acceptables (par exemple, une formation, des connaissances et une expérience pratique pertinentes). Les fabricants d'IPA, d'excipients et de produits médicamenteux doivent mettre les renseignements à la disposition du DAMM.
Dans le contexte du contrôle des impuretés de nitrosamine, les fabricants et les importateurs doivent respecter toutes les conditions spécifiées sur leur LEPP. Cela pourrait comprendre des restrictions ou des exigences supplémentaires précises en ce qui a trait aux essais et aux enquêtes sur les impuretés de nitrosamines.
7. Incapacité de respecter les délais prescrits pour effectuer des évaluations des risques (étape 1), des tests de confirmation (étape 2) et des changements à l'autorisation de mise en marché (étape 3)
Compte tenu des risques associés à la présence de nitrosamine dans les produits médicamenteux, les DAMM devraient prendre toutes les mesures nécessaires pour effectuer les 3 étapes le plus tôt possible et dans les délais prescrits.
Le 14 avril 2021, Santé Canada a envoyé une lettre à tous les DAMM de produits médicamenteux contenant des ingrédients pharmaceutiques actifs synthétisés chimiquement pour demander des renseignements sur l'état d'avancement de l'étape 1 des évaluations des risques. Les DAMM devraient fournir une annexe 1, une annexe 2 ou une annexe 3 dûment remplie, selon le cas, conformément aux instructions de la lettre de suivi du 14 avril 2021. Si des évaluations des risques n'ont pas été effectuées pour tous les produits commercialisés, approuvés et inactifs ou si elles ne sont pas terminées, l'annexe 3 doit être remplie.
Les DAMM qui ne sont pas en mesure de respecter les délais pour les étapes 2 et 3 en raison de circonstances exceptionnelles peuvent présenter une demande de prolongation à Santé Canada. Cela devrait être fait le plus tôt possible. La demande doit contenir des renseignements pertinents, y compris les progrès réalisés à ce jour, les raisons pour lesquelles les échéances n'ont pas été respectées, les travaux restants et les échéanciers prévus pour l'achèvement.
Afin d'établir l'ordre dans lequel les IPA et les produits médicamenteux doivent être examinés (évaluation des risques, tests de confirmation et changements à l'autorisation de mise en marché), les DAMM devraient utiliser les principes de gestion des risques liés à la qualité. Consulter la directive Q9 de l'ICH, les Lignes directrices sur les Bonnes pratiques de fabrication (BPF) de Santé Canada 0001 (pour les produits médicamenteux) et 0104 (pour les IPA). Consulter également l'information de la section 4.
Les demandes de prolongation seront évaluées au cas par cas. Transmettre ces demandes comme suit :
- pour les produits médicamenteux contenant des IPA synthétisés chimiquement ou semi-synthétiques : bpsenquiries@hc-sc.gc.ca
- pour les produits biologiques et radiopharmaceutiques : brdd.nitrosamines.dmbr@hc-sc.gc.ca
8. Déclarations des fabricants et des fournisseurs au lieu d'effectuer des évaluations des risques
Les déclarations fournies par les fabricants et/ou les fournisseurs ne peuvent pas remplacer une évaluation globale solide des risques par le DAMM. Bien que les connaissances et les compétences offertes par les fabricants soient valables et encouragées pour soutenir le processus d'évaluation des risques, les déclarations des fabricants ou des fournisseurs ne remplacent pas une évaluation de risques documentée par le DAMM.
9. Réalisation des tests de confirmation (étape 2) sans évaluation des risques préalable (étape 1)
L'étape de l'évaluation des risques (étape 1) est nécessaire pour déterminer les facteurs de risque, les causes fondamentales possibles et l'étendue des impuretés de nitrosamine qui ont le potentiel d'être formées ou introduites dans l'IPA ou le produit médicamenteux. Si le risque d'une ou plusieurs impuretés de nitrosamine est détecté, cette connaissance est utilisée pour guider le développement et la validation des méthodes de tests appropriées requises pour l'étape du test de confirmation (étape 2).
Cette connaissance permet également d'établir une stratégie de contrôle appropriée et de mettre en œuvre des changements pour prévenir la présence de nitrosamines.
Par conséquent, il n'est pas approprié de passer directement aux tests de confirmation (étape 2) sans avoir terminé l'étape d'évaluation des risques (étape 1).
10. Application des résultats d'une évaluation des risques et d'un test de confirmation pour un produit médicamenteux qui est commercialisé à l'extérieur du Canada à un médicament dont la vente est autorisée au Canada
Les DAMM sont responsables de s'assurer que les évaluations des risques et, s'il y a lieu, les tests de confirmation sont pertinents pour le produit médicamenteux dont la vente a été autorisée au Canada.
Si une évaluation des risques et des tests de confirmation ont été effectués pour un produit médicamenteux dont l'utilisation est autorisée à l'extérieur du Canada, il peut être possible d'utiliser cette information aux fins de l'évaluation des risques et des tests de confirmation du produit pharmaceutique dont la vente est autorisée au Canada. Dans ce scénario, les 2 produits médicamenteux doivent être identiques (par exemple, composition, concentration, processus de fabrication, sources d'IPA et d'excipients, sites de fabrication).
Les DAMM devraient préparer une justification écrite lorsque l'évaluation des risques et les résultats des tests de confirmation d'un produit dans un autre pays seront pris en compte. Ils devraient être prêts à fournir cette justification à Santé Canada sur demande. Cette justification doit être incluse dans les communications à Santé Canada si des impuretés de nitrosamines sont détectées dans le produit médicamenteux à la suite des essais de confirmation si une impureté de nitrosamine est détectée à un niveau supérieur à la limite acceptable établie pour l'impureté de nitrosamine en question, ou à un niveau supérieur à la limite acceptable établie (voir l'annexe 1) en utilisant l'approche de catégorisation de la puissance cancérogène (ACPC, consulter la section 24 et l'annexe 4) si une limite acceptable n'a pas été établie par Santé Canada. Consulter la section 15.
11. Évaluation des risques liés à la présence de nitrosamine applicable à un produit médicamenteux importé au Canada dans le cadre du Programme d'accès spécial (PAS)
Les entreprises pourraient devoir effectuer des évaluations des risques liés à la présence de nitrosamine pour les produits médicamenteux dont la vente n'est pas autorisée au Canada, mais qui sont offerts au Canada dans le cadre du PAS. Reportez-vous aux approches décrites dans les lettres du 2 octobre 2019 et du 15 décembre 2020 de Santé Canada et dans le présent document.
Si l'évaluation des risques liés à la présence de nitrosamine ou les résultats des tests de confirmation (le cas échéant) indiquent un risque de présence d'une impureté de nitrosamine, aviser le PAS par courriel à sapd-pasm@hc-sc.gc.ca.
Pour protéger la santé et la sécurité des patients qui ont accès à des produits médicamenteux non autorisés, tout nouveau renseignement important sur l'innocuité, l'efficacité et la qualité des produits médicamenteux commercialisés dans le cadre du PAS devrait être transmis aux praticiens et au PAS.
12. Tests de confirmation lorsqu'une évaluation des risques conclut à l'absence de risque de contamination à la nitrosamine
Les DAMM doivent effectuer une évaluation approfondie et rigoureuse des risques. Dans les lettres de Santé Canada du 2 octobre 2019 et du 15 décembre 2020, Santé Canada partageait certaines sources potentielles d'impuretés de nitrosamines. Consulter la section 29 pour en savoir plus sur les facteurs de risque et les causes fondamentales possibles de la présence d'impuretés de nitrosamines.
Les DAMM devraient préparer un rapport comprenant les considérations, les étapes et les conclusions avec une justification. Le rapport devrait indiquer clairement les nitrosamines qui pourraient être formées, le cas échéant. Si l'on conclut qu'il n'existe pas de risque de présence de nitrosamines, on ne s'attend pas à ce que des tests de confirmation soient effectués.
Si un risque de formation ou de présence de nitrosamines est détecté, des tests de confirmation devraient être effectués en utilisant des méthodes adéquatement validées et suffisamment sensibles (voir le numéro 36). À la suite des essais de confirmation du produit médicamenteux, Santé Canada doit être informé si une impureté de nitrosamine est détectée à un niveau supérieur à la limite acceptable établie pour l'impureté de nitrosamine en question, ou à un niveau supérieur à la limite acceptable établie en utilisant l'approche de catégorisation de la puissance cancérogène (ACPC, consulter la section 24 et l'annexe 4) si une limite acceptable n'a pas été établie par Santé Canada. Les adresses de déclaration sont indiquées à la section 15.
13. Gestion et soumission des modifications à apporter à l'autorisation de mise sur le marché relativement aux mesures d'atténuation des risques à l'étape 3 (mis à jour)
Les modifications à apporter à l'autorisation de mise sur le marché relativement aux mesures d'atténuation des risques à l'étape 3 devraient être soumises à Santé Canada en temps opportun en format eCTD ou dans un autre format électronique que eCTD dans le Portail commun de demandes électroniques (PCDE).
Puisque les modifications liées à l'atténuation des risques de l'étape 3 découlent de préoccupations de sécurité potentielles, les données scientifiques doivent faire l'objet d'un examen critique par Santé Canada, et l'autorisation subséquente doit être accordée avant la mise en œuvre par le fabricant. Par conséquent, à l'exception de certains changements mineurs (voir plus de détails décrits ci-dessous dans cette section) les modifications liées à l'atténuation des risques de l'étape 3 pour les produits pharmaceutiques contenant des IPA synthétisés chimiquement et semi-synthétiques doivent être présentées en tant que Niveau I – Suppléments ou de demande de modification après l'émission d'une identification numérique de drogue (DIN), selon le cas. Concernant les produits biologiques et radiopharmaceutiques, les modifications doivent être présentées en tant que Niveau I – Suppléments, Niveau II – Préavis de modification, ou de demande de modification après l'émission d'une DIN, selon le cas. Pour des exemples de changements liés à l'atténuation des risques, voir le numéro 20 et l'annexe 2.
Lors du dépôt d'un supplément, d'un préavis de modification ou d'une demande de modification après l'émission d'une DIN pour l'autorisation de mise en marché, les demandeurs doivent indiquer clairement dans la lettre d'accompagnement que les changements proposés visent à atténuer les risques liés aux nitrosamines (étape 3 de la demande d'évaluation du risque lié à la présence d'impuretés de nitrosamine de Santé Canada en ce qui concerne les produits médicamenteux approuvés).
Un résumé des investigations sur les causes fondamentales et la conclusion concernant la(les) cause(s) fondamentale(s) confirmée(s) de la présence de nitrosamines dans le produit pharmaceutique doivent être inclus dans la section 3.2.P.2.
Lorsqu'il est proposé d'ajouter des limites individuelles ou cumulatives d'LA pour les nitrosamines à la spécification approuvée d'une substance médicamenteuse ou d'un produit médicamenteux (sur la base de l'utilisation de la ACPC ou d'autres approches), les DAMM doivent gérer ces changements en tant que niveau I – Suppléments.
En ce qui concerne les changements proposés qui ne sont pas liés à l'étape 3 de la demande d'examen des produits médicamenteux approuvés (c'est-à-dire les changements qui ne sont pas liés à l'atténuation des risques pour les produits médicamenteux approuvés), les changements devraient être gérées et, le cas échéant, soumises conformément à la ligne directrice Changements survenus après l'avis de conformité (AC) : Document sur la qualité et Ligne directrice - Changements effectués après l'émission d'une identification numérique de drogue (DIN). Voir la section 20 et l'annexe 2.
Pour les produits pharmaceutiques contenant des API chimiquement synthétisés ou semi-synthétiques, lorsque des limites d'LA pour les impuretés de nitrosamine individuelles et, le cas échéant, des limites pour les niveaux cumulatifs d'impuretés de nitrosamine, sont déjà incluses dans une spécification approuvée de substance médicamenteuse ou de produit médicamenteux les changements suivants peuvent être gérés conformément à la Ligne directrice : Changements survenus après l'avis de conformité (AC) : Document sur la qualité de Santé Canada en tant que changements de niveau III (avis annuels) :
- le resserrement de ces limites individuelles ou cumulatives d'LA de nitrosamines dans les spécifications approuvées de la substance médicamenteuse ou du produit pharmaceutique
- l'assouplissement de ces limites individuelles ou cumulatives d'LA dans les spécifications approuvées de la substance médicamenteuse ou du produit pharmaceutique afin d'adopter les limites d'LA énumérées à l'annexe 1 de la présente ligne directrice
- suppression d'un essai de détection d'une impureté de type nitrosamine dans la spécification d'une substance médicamenteuse ou d'un produit pharmaceutique, avec une justification scientifique appropriée.
De plus, les changements suivants peuvent être gérés conformément à la Ligne directrice : Changements survenus après l'avis de conformité (AC) : Document sur la qualité de Santé Canada en tant que changements de niveau III (avis annuels):
- l'ajout d'un essai et de critères d'acceptation à la spécification d'une substance médicamenteuse pour une impureté de type nitrosamine, qui est basé sur un certificat de conformité (CEP) valide délivré par la Direction européenne de la qualité du médicament & soins de santé (DEQM).
- l'ajout d'un essai et de critères d'acceptation à la spécification d'une substance médicamenteuse ou d'un produit médicamenteux pour une impureté de type nitrosamine classée comme non mutagène dans l'annexe 1
Lorsqu'une proposition est faite pour assouplir les limites individuelles ou cumulatives d'LA pour les nitrosamines déjà incluses dans les spécifications d'une substance médicamenteuse ou d'un produit médicamenteux approuvé (sur la base de l'utilisation de la ACPC ou d'autres approches) lorsqu'une limite d'LA n'est pas répertoriée à l'annexe 1, les DAMM doivent gérer ces changements en tant que niveau I – Suppléments.
14. Vente d'un produit médicamenteux si des changements (spécifications, contrôles) à l'autorisation de mise en marché (étape 3) soumis à titre de supplément, de préavis de modification ou d'une demande de modification effectuée après l'émission d'une DIN sont toujours à l'étude
La commercialisation continue d'un produit médicamenteux dépend des niveaux d'impuretés de nitrosamine qui sont détectés et du risque de présence d'impuretés au moment de la notification à Santé Canada. Certains des résultats de l'évaluation de Santé Canada peuvent comprendre des rappels ou des demandes d'arrêt de vente jusqu'à ce que les risques soient atténués et que des mesures correctives et préventives appropriées soient mises en place pour veiller à ce que tous les lots mis sur le marché répondent aux critères d'acceptation pour chaque impureté de nitrosamine (et les nitrosamines multiples, le cas échéant). Les deux résultats auraient une incidence sur la commercialisation continue du produit pendant l'examen du supplément, du préavis de modification ou de la demande de modification après l'émission d'une DIN. Consulter la section 13.
15. Communication avec Santé Canada à la suite des essais de confirmation si des impuretés de nitrosamine sont détectées
Suite des analyses de confirmation du produit pharmaceutique, les DAMM doivent informer Santé Canada si des impuretés de nitrosamines sont détectées à un niveau supérieur à la limite acceptable établie (voir l'annexe 1) pour l'impureté de nitrosamine en question, ou à un niveau supérieur à la limite acceptable établie en utilisant l'approche de catégorisation de la puissance cancérogène (ACPC, consulter la section 24 et l'annexe 4) si une limite acceptable n'a pas été établie par Santé Canada, dans le produit médicamenteux à la suite des essais de confirmation. Les résultats des analyses de confirmation doivent accompagner la notification à Santé Canada par les DAMM.
Santé Canada reconnaît les défis auxquels sont confrontés les DAMM pour ce qui est de diminuer les niveaux d'impuretés de nitrosamine dans leurs produits médicamenteux tout en maintenant l'approvisionnement pour les Canadiens. Afin de minimiser les impacts sur le marché canadien des médicaments, les DAMM sont priés de consulter Santé Canada avant de prendre quelque mesure que ce soit relativement à la mise sur le marché d'un produit médicamenteux présentant des impuretés de nitrosamine.
Ces informations devraient être envoyées selon les directives suivantes :
| Emplacement de l'entreprise | Adresse d'envoi du rapport |
|---|---|
| Nouveau-Brunswick, Terre-Neuve-et-Labrador, Nouvelle-Écosse, Île-du-Prince-Édouard, Québec | Unité Est de la conformité des produits de santé |
| Ontario | Unité Centre de la conformité des produits de santé |
| Manitoba, Saskatchewan, Alberta, Colombie-Britannique, Yukon, Territoires du Nord-Ouest, Nunavut | Unité Ouest de la conformité des produits de santé |
Si les nitrosamines ne sont pas détectées pendant les tests de confirmation (c'est-à-dire qu'elles sont inférieures à la limite de détection appropriée de la méthode d'essai validée), ou si elles sont détectées à un niveau inférieur à la limite acceptable établie pour l’impureté de nitrosamine en question ou à la limite acceptable établie en utilisant l'approche de catégorisation de la puissance cancérogène (ACPC, consulter la section 24 et l'annexe 4) si une limite acceptable n'a pas été établie par Santé Canada, les DAMM ne sont pas tenus d’en informer Santé Canada. Toutefois, le rapport d'évaluation des risques, les résultats des tests analytiques et la documentation de validation de la méthode d'analyse devraient être conservés par le DAMM et mis à la disposition de Santé Canada sur demande. Voir la section 13 pour obtenir des renseignements sur la façon d’informer Santé Canada des modifications apportées à l’autorisation de mise en marché.
16. L'information nécessaire à l'évaluation des risques n'est pas fournie par le fabricant de l'IPA ou du produit médicamenteux
Les DAMM sont responsables de veiller à l'innocuité, à l'efficacité et à la qualité continues des médicaments sur le marché canadien. Lorsque des renseignements jugés essentiels à la réalisation de l'évaluation des risques n'ont pas été fournis au DAMM pour des raisons de confidentialité ou pour d'autres raisons, les DAMM peuvent solliciter les services d'un tiers (comme un consultant) pour travailler directement avec les fabricants pour achever l'évaluation des risques.
Le recours à un tiers peut également constituer une approche appropriée lorsque le DAMM :
- dispose de tous les renseignements nécessaires pour effectuer l'évaluation des risques du fabricant, mais
- n'a pas de personnel possédant les qualifications nécessaires (par exemple, la formation et l'expérience pratique pertinentes) pour effectuer l'évaluation des risques
Pour de plus amples renseignements sur les activités externalisées, veuillez consulter le document suivant :
Autrement, le DAMM devrait envisager de déléguer l'évaluation des risques aux fabricants d'IPA et de produits médicamenteux. Dans ce scénario, le DAMM continue de devoir garantir l'innocuité, l'efficacité et la qualité des produits médicamenteux.
Le DAMM devrait s'assurer, au moyen d'une vérification interne ou par un tiers, que :
- des évaluations des risques ont été effectuées par du personnel possédant des qualifications acceptables (une formation et une expérience pratique pertinentes)
- les fabricants ont tenu compte de tous les facteurs de risques et de toutes les origines possibles des impuretés de nitrosamines (y compris ceux mentionnés dans la lettre du 15 décembre 2020 concernant les produits biologiques et radiopharmaceutiques et ceux indiqués à la section 29)
17. Autres attentes relatives aux DAMM si des impuretés de nitrosamines sont présentes dans le produit médicamenteux (mis à jour)
Lorsqu'une ou plusieurs impuretés de nitrosamine sont détectées à la suite des essais de confirmation (pour les nitrosamines multiples, consulter la section 27), en plus d'aviser Santé Canada, on s'attend à ce que les DAMM aient fait ou soient en train de faire ce qui suit, le cas échéant :
- une évaluation des risques pour la santé posés par la présence de nitrosamines, en indiquant les mesures à prendre, au besoin, pour les lots en circulation sur le marché canadien
- Dans les cas où des mesures de rappel de produits seraient justifiées, le guide GUI0039 (Guide pour le retrait de drogues et de produits de santé naturels) de Santé Canada devrait être consulté pour obtenir les procédures à suivre
- une évaluation permettant de déterminer si le produit est considéré comme médicalement nécessaire ou médicalement important et si une perturbation de l'approvisionnement du produit est à prévoir advenant la prise d'une mesure liée à la mise sur le marché du produit
- un rapport d'enquête détaillé évaluant toutes les causes fondamentales possibles de la présence de l'impureté (ou impuretés) de nitrosamine détectée et décrivant les mesures correctives et préventives
- des enquêtes conformes aux procédures écrites
- une évaluation de tous les changements éventuels aux installations, matériaux, équipements et/ou processus, visant à réduire les niveaux d'impuretés de nitrosamine, par un système formel de contrôle des changements
- un plan d'atténuation des risques comprenant la mise en place d'une stratégie de contrôle appropriée pour la ou les nitrosamine(s) détectée(s) pour veiller à ce que, lors des prochaines étapes, les concentrations d'impuretés des nitrosamines soient constamment inférieures aux limites acceptables à la fin de la période de contre-essais pour l'IPA ou de la durée de vie du produit médicamenteux (se reporter à l'annexe 1 pour obtenir la liste des limites acceptables établies).
Les DAMM ne doivent pas oublier de soumettre les modifications à l'autorisation de mise en marché conformément à l'étape 3 de la lettre du 2 octobre 2019. Consulter la section 13 sur la façon de soumettre les modifications.
Santé Canada peut également utiliser ces avis pour demander des mesures supplémentaires. Par exemple, l'origine des impuretés de nitrosamines peut être attribuée au type de procédé chimique utilisé, et le plan d'atténuation des risques peut nécessiter l'établissement d'une stratégie de contrôle par les fabricants pour chaque impureté de nitrosamine détectée conformément à la directive M7 de l'ICH.
Nous pouvons également demander des mesures supplémentaires à d'autres DAMM du même produit pour atténuer les risques cernés et protéger la santé et la sécurité de la population canadienne, au besoin.
Pour en savoir plus sur l'établissement des spécifications et des contrôles, consulter les sections 34 et 35, respectivement.
18. Évaluation des progrès réalisés relativement à cette demande d'examen du risque lié à la présence d'impuretés de nitrosamines
Le 14 avril 2021, Santé Canada a envoyé une lettre faisant suite à celle du 2 octobre 2019. Elle demandait à tous les DAMM de produits médicamenteux contenant des ingrédients pharmaceutiques actifs synthétisés chimiquement et semi-synthétiques des renseignements sur l'état d'avancement de l'étape 1 (évaluations des risques).
De plus, Santé Canada pourrait faire l'évaluation au moyen de :
- vérifier l'état d'avancement, l'adéquation et la robustesse des évaluations de risques, des résultats des essais de confirmation et de toute autre document justificatif lié à cette demande pendant les inspections de BPF, les projets de gestion proactive des risques pour mesurer les progrès et la vérification de la conformité lors de la réception d'une plainte ou d'une notification
- la demande d'information au moment où des changements sont apportés à l'autorisation de mise en marché existante d'un produit ou à la licence d'établissement d'un médicament
Nous sommes conscients de l'importance de cette demande. Nous continuerons de mobiliser des intervenants afin d'examiner toutes les options pour gérer les risques associés aux nitrosamines.
19. Approche pour les produits médicamenteux dont la présentation est prévue ou qui sont déjà soumis à Santé Canada
Dans le cas des IPA et des produits médicamenteux en cours de développement, la formation ou l'introduction d'impuretés de nitrosamines doit être évitée dès le départ, dans la mesure du possible.
Si la formation ou l'introduction de nitrosamines est inévitable, les processus de fabrication devraient avoir démontré leur capacité à réduire systématiquement les niveaux d'impuretés de nitrosamines à des niveaux inférieurs à la limite acceptable. Une stratégie de contrôle, basée sur la compréhension du produit et du processus, devrait être établie pour chaque impureté de nitrosamine présente dans l'IPA et/ou le produit médicamenteux.
Dans le cas des produits médicamenteux dont la présentation est prévue ou qui ont déjà été soumis, les DAMM devraient procéder de façon proactive à une évaluation des risques pour déceler la présence éventuelle d'impuretés de nitrosamines dans le produit médicamenteux (si ce n'est pas déjà fait) en tenant compte des considérations et des étapes prévues pour les produits approuvés dans les communications de Santé Canada. Dans le cas des présentations prévues, les sections pertinentes du format de dossier technique commun (Common Technical Document [CTD]) de la présentation de drogue devraient inclure de l'information sur les évaluations des risques.
Un résumé des évaluations des risques liés aux impuretés de nitrosamines dans le produit médicamenteux doit être placé dans la section 3.2.P.2 du CTD. Ce résumé doit comprendre suffisamment de détails pour permettre à Santé Canada d'évaluer la pertinence et la rigueur de l'évaluation des risques. Les attentes relatives au contenu du résumé et à la discussion concernant les évaluations des risques se trouvent à la section 20.
Les résultats des tests de confirmation et la stratégie de contrôle mise à jour (le cas échéant) devraient également être inclus dans la présentation de drogue (par exemple, aux sections 3.2.S.2, 3.2.S.4, 3.2.S.7, 3.2.P.3, 3.2.P.4, 3.2.P.5, 3.2.P.8, etc.).
Pour les demandes soumises qui sont présentement en examen, on pourrait demander au DAMM et au promoteur de fournir l'évaluation des risques et les résultats des tests de confirmation dans le cadre du processus d'évaluation. Veuillez consulter la section 20 pour en savoir plus.
20. Évaluation des risques liés à la présence possible d'impuretés de nitrosamines dans le cadre du contenu attendu des nouvelles présentations
Les évaluations des risques liés à la présence potentielle d'impuretés de nitrosamines devraient être effectuées systématiquement pendant le développement del'IPA et du produit médicamenteux. Les résultats de l'évaluation des risques liés aux impuretés de nitrosamines dans le produit médicamenteux et la justification de la stratégie de contrôle proposée à l'égard des impuretés de nitrosamine devraient être présentés aux fins d'évaluation dans les présentations de drogue nouvelle (PDN), les présentations abrégées de drogue nouvelle (PADN), les demandes d'identification numérique de drogues (DDIN) pour un produit pharmaceutique (accompagnées de données sur la chimie et la fabrication [C et F]), les demandes d'identification numérique de drogues pour un produit biologique (DDINB), les suppléments, les préavis de modification et les demandes de modifications après l'émission d'une DIN (consulter la section 13). Pour de plus amples renseignements sur les considérations relatives aux impuretés mutagènes et les principes de gestion des risques liés à la qualité, veuillez consulter les documents suivants :
- Lignes directrices sur les bonnes pratiques de fabrication des drogues (GUI-0001)
- Bonnes pratiques de fabrication des ingrédients pharmaceutiques actifs (GUI-0104)
- Directive M7 de l'ICH (Format PDF)
- Directive Q9 de l'ICH (Format PDF)
Tous les PDN, PADN, DDIN (avec les données C et F), DDINB et tous les suppléments, les préavis de modification et les demandes de modifications après l'émission d'une DIN (pour les changements de qualité qui peuvent influer sur la présence possible d'impuretés de nitrosamines dans l'IPA ou le produit médicamenteux) devraient inclure un résumé et une discussion sur les évaluations des risques pour la formation ou la présence potentielle d'impuretés de nitrosamines dans le produit médicamenteux. Cette exigence est la suivante :
- à compter du 1er avril 2021, pour les PDN, PADN et les suppléments de produits pharmaceutiques contenant des IPA synthétisés chimiquement et semi-synthétiques
- à compter du 1er avril 2021 pour les DDIN (avec les données C et F), y compris les demandes de modifications après l'émission d'une DIN pour des changements de qualité liés à des produits pharmaceutiques contenant des IPA synthétisés chimiquement et semi-synthétiques;
- à compter du 30 novembre 2021, pour les PDN, les suppléments et les préavis de modification pour des produits biologiques et radiopharmaceutiques;
- à compter du 30 novembre 2021 pour les DDINB (avec les données C et F) et les demandes de modifications après l'émission d'une DIN pour des produits biologiques et radiopharmaceutiques.
Le résumé et la discussion sur l'évaluation des risques du produit médicamenteux doivent comprendre suffisamment de détails pour permettre à Santé Canada d'évaluer la pertinence et la rigueur de l'évaluation des risques. Ils doivent comporter une discussion sur les facteurs de risque et les causes fondamentales potentielles pris en compte en fonction des connaissances spécifiques du produit médicamenteux et de ses composants (y compris l'IPA). Les listes de contrôle qui ne sont pas suffisamment détaillées doivent être évitées. Le résumé et la discussion doivent comprendre les éléments suivants :
- identification de toute tierce partie (par exemple, fournisseurs, fabricants, consultants) qui a été autorisée à effectuer l'évaluation des risques au nom du demandeur;
- détermination des facteurs de risque intrinsèques et extrinsèques liés à la formation ou à l'introduction d'impuretés de nitrosamines provenant de tous les composants de produits médicamenteux, de même que des considérations de qualité/conformité;
- identification des nitrosamines potentiellement formées ou introduites;
- information sur le processus établi ou les contrôles analytiques et la manière dont ils peuvent atténuer le risque;
- données scientifiques à l'appui (par exemple, résultats de tests de confirmation) et calculs;
- une conclusion générale sur le risque de présence de nitrosamines dans le produit médicamenteux, accompagnée d'une justification scientifique appropriée.
Pour les suppléments, les préavis de modification et les demandes de modifications après l'émission d'une DIN (pour les changements de qualité qui peuvent influer sur la présence potentielle d'impuretés de nitrosamines dans l'IPA ou le produit pharmaceutique), le résumé et la discussion de l'évaluation des risques doivent porter uniquement sur l'impact des changements proposés sur les impuretés de nitrosamines par rapport au produit pharmaceutique approuvé. Citons, à titre d'exemples de changements pouvant influer sur la présence potentielle d'impuretés de nitrosamine dans le cas d'un produit médicamenteux approuvé, les modifications apportées aux processus de fabrication d'une substance médicamenteuse ou d'un produit médicamenteux, les modifications apportées à la composition d'un produit médicamenteux (IPA, excipients), l'introduction d'une nouvelle forme posologique, ainsi que les modifications apportées au système de fermeture du contenant.
Voir l'annexe 2 pour plus de détails sur les impuretés de nitrosamine et les évaluations des risques dans les cas de changements survenant après l'AC concernant de nouveaux produits médicamenteux qui contiennent des IPA synthétisés par voie chimique ou des IPA semi-synthétiques.
Pour les demandes d'essais cliniques (DEC) (comme décrit à la section 9.1 de la directive M7 de l'ICH) :
- Dans le cas des essais cliniques de phase 1 d'une durée de 14 jours ou moins, inclure une description des efforts déployés pour atténuer les risques de présence d'impuretés mutagènes axées sur les impuretés de classe 1 et de classe 2, ainsi que celles désignées par l'expression « cohorte préoccupante » (par exemple, les impuretés de nitrosamines)
- Pour les essais cliniques de phase 1 de plus de 14 jours et pour les essais cliniques de phase 2 et 3, inclure également les impuretés de classe 3 qui nécessitent des contrôles analytiques
Le défaut d'inclure ces renseignements pourrait entraîner des demandes de renseignements supplémentaires, des retards dans le processus d'examen et la prise de décisions négatives.
21. Contrôles pour détecter la présence de nitrosamines dans les IPA achetés pour la préparation en pharmacie
La Politique sur la fabrication et la préparation en pharmacie de produits pharmaceutiques au Canada (POL-0051) établit un cadre stratégique pour aider à distinguer les activités relatives à la préparation magistrale des produits médicamenteux des activités relatives à leur fabrication. Au Canada, la préparation des médicaments en pharmacie est une activité exercée surtout par les pharmaciens et fait partie intégrante de leur pratique professionnelle. Cette activité est réglementée par les organismes de réglementation provinciaux et territoriaux.
Cette politique indique que les préparations magistrales doivent être soit :
- produites à partir d'un IPA autorisé utilisé dans un produit médicamenteux homologué pour usage au Canada
- énumérées dans une pharmacopée reconnue (par exemple, USP/NF, Ph., Eur., Ph. Int., BP, Codex – Annexe B de la Loi sur les aliments et drogues)
Santé Canada recommande donc que les principes énoncés dans la lettre du 2 octobre 2019 et dans la présente directive soient pris en compte lors de l'achat d'IPA et de la préparation de produits en pharmacie.
Les professionnels de la santé et les entreprises qui font des préparations magistrales sont invités à consulter la page Web sur les nitrosamines pour se tenir au courant des médicaments et des rappels visés par la présence de nitrosamines. Cette page est mise à jour régulièrement et comprend également des renseignements généraux sur les impuretés de nitrosamine, les mesures prises par Santé Canada pour régler le problème et les résultats de nos tests.
Communications
22. Mobilisation des intervenants en vue d'assurer une communication continue avec l'industrie
Santé Canada est déterminé à partager l'information avec les intervenants et à maintenir la transparence, à mesure que nous continuons d'analyser et de mieux comprendre cette situation mondiale en pleine évolution.
Jusqu'à présent, Santé Canada a partagé ouvertement l'information avec les intervenants, y compris les sources possibles d'impuretés de nitrosamines, les causes fondamentales, les facteurs de risque et les nouvelles conclusions. Nous avons organisé des séances à l'intention des intervenants en janvier 2020, en février 2021 et en octobre 2021. Nous pourrions en organiser d'autres à l'avenir, au besoin.
Nous avons également créé une page Web sur les impuretés de nitrosamine dans les médicaments. La page Web comprend ce qui suit :
- des résumés des produits médicamenteux qui ont été visés ou rappelés en raison de la présence de nitrosamines
- les résultats d'analyse des niveaux d'impuretés de nitrosamines dans plusieurs produits
Des discussions sont en cours pour déterminer les méthodes les plus appropriées et les plus efficaces pour continuer de mobiliser les intervenants à mesure que de nouveaux renseignements deviennent disponibles, afin d'assurer une approche coordonnée et uniforme pour régler cette question complexe.
23. Collaboration entre Santé Canada et les organismes de réglementation mondiaux au sujet des problèmes liés aux impuretés de nitrosamine dans les produits médicamenteux
Santé Canada collabore régulièrement avec des partenaires internationaux en matière de réglementation, y compris ceux d'Europe, des États-Unis, du Japon, de la Suisse, de Singapour, de l'Australie et du Brésil, ainsi qu'avec l'Organisation mondiale de la Santé. Nous espérons mieux comprendre les problèmes liés aux impuretés de nitrosamines, harmoniser les exigences et les actions à prendre, le cas échéant, et partager l'information conformément à nos accords de confidentialité.
Les discussions entre le consortium d'autorités réglementaires se sont poursuivies, avec le Nitrosamines International Strategic Group (NISG), qui a été formé en 2018. Le NISG a commencé à se réunir régulièrement en mettant l'accent sur le partage des connaissances sur le marché et les autres mesures réglementaires. En raison d'un intérêt mutuel des organismes de réglementation participants du NISG à avoir des discussions plus approfondies sur les questions techniques et les développements scientifiques, un sous-groupe de ces mêmes organismes de réglementation, le Nitrosamines International Technical Working Group (NITWG), a été créé fin 2020.
Lorsqu'elles déterminent les mesures réglementaires appropriées pour remédier à la présence d'impuretés de nitrosamine dépassant la limite acceptable dans les produits médicamenteux à usage humain, les juridictions individuelles doivent déterminer des délais et les mesures pour protéger au mieux la sécurité des patients tout en respectant le cadre réglementaire.
Innocuité
24. Limites jugées acceptables par Santé Canada pour les impuretés de nitrosamine
Les limites acceptables ont été établies pour plusieurs nitrosamines (annexe 1). Ces limites acceptables sont jugées appropriées pour toutes les voies d'administration et devraient s'appliquer à la dose quotidienne maximale (DQM) du produit médicamenteux.
Les limites acceptables peuvent être établies de différentes façons.
Lorsque les données d'un composé particulier pour une impureté de nitrosamine sont fiables, les DAMM et demandeurs peuvent :
Établir une limite acceptable en fonction de données fiables sur des composés particuliers
- En procédant à une extrapolation linéaire de la dose permettant d'obtenir une incidence tumorale de 50 % (DT50) pour un excès du risque de cancer de 1 sur 105 en utilisant la valeur DT50 la plus pertinente selon une étude de cancérogénicité rigoureuse (voir l'addenda de la directive M7 de l'ICH en ce qui concerne le choix d'une étude de cancérogénicité appropriée).
- En offrant un résultat négatif du test d'Ames amélioré, conforme aux bonnes pratiques de laboratoire (BPL), ainsi qu'aux conditions améliorées décrites à l'annexe 3 afin de justifier une limite de 1,5 µg/jour.
- En fournissant des données négatives sur la mutagénicité in vivo (notamment un résultat négatif d'analyse du potentiel mutagène selon l'Essai 488 de l'OCDE, intitulé Essais de mutations génétiques des cellules somatiques et germinales de rongeurs transgéniques) pour justifier le fait de contrôler une impureté de nitrosamine conformément aux recommandations des directives Q3A et Q3B de l'ICH.
Lorsque les données d'un composé particulier pour une impureté de nitrosamine ne sont pas fiables, les DAMM et demandeurs peuvent :
Établir une limite acceptable en fonction d'une évaluation des relations structure-activité (RSA) et utiliser un substitut de lecture croisée pour lequel il y a des données suffisantes sur des composés particuliers
Pour justifier un substitut approprié de lecture croisée, l'évaluation des RSA devrait tenir compte de la similitude structurelle (de façon globale et au site local de l'activation), de la similitude des caractéristiques physicochimiques, des facteurs stériques et électroniques ayant une incidence sur la réactivité, et de la similitude métabolique (par exemple, la voie métabolique et la stabilité/réactivité des métabolites).
En cas de substitut approprié de lecture croisée pour déterminer la limite acceptable, la DT50 devrait provenir d'une étude de cancérogénicité suffisamment rigoureuse. Les paramètres dont on doit tenir compte incluent une description adéquate de la conception de l'étude et une analyse histopathologique appropriée, le nombre de groupes de doses (ainsi, les études portant sur une seule dose ne sont pas jugées appropriées), le nombre d'animaux pour chaque groupe de doses, la durée de l'exposition, la voie d'administration et la relation entre la dose et la réponse. Voir l'addenda de la directive M7 de l'ICH pour obtenir des instructions quant à la façon de choisir une étude appropriée de cancérogénicité.
Conformément aux pratiques internationales en matière de réglementation, Santé Canada continuera d'utiliser des calculs fondés sur la masse (plutôt que des calculs fondés sur la molarité) pour calculer les limites acceptables pour les impuretés de nitrosamine lorsqu'un analogue est sélectionné pour la lecture croisée, et s'attend à ce que les demandeurs et les DAMM fassent de même.
Établir une limite acceptable au moyen de l'approche de catégorisation de la puissance cancérogène (ACPC)
L'approche de catégorisation de la puissance cancérogène consiste à classer une nitrosamine dans une catégorie de puissance cancérogène prédite.
Il existe cinq catégories de puissance cancérogène et chacune d'entre elles prévoit une limite acceptable de 18 ng/jour à 1 500 ng/jour.
Une nitrosamine est classée dans une catégorie de puissance cancérogène prédite en fonction de l'évaluation des atomes d'hydrogène alpha et des caractéristiques structurelles d'activation et de désactivation présentes dans la nitrosamine. Voir l'annexe 4 pour obtenir une description de l'approche, ainsi que des exemples de cas pour illustrer l'application de l'approche de catégorisation de la puissance cancérogène.
Les DAMM et les demandeurs devraient également consulter les sections suivantes pour obtenir de plus amples renseignements :
- section 27 sur la présence de nitrosamines multiples
- section 28 sur l'application d'une limite inférieure à la durée de vie
- section 31 sur les nitrosamines qui devraient être incluses dans les évaluations des risques et les essais de confirmation
25. Limites acceptables pour les impuretés de nitrosamine présentes dans les produits médicamenteux qui sont visés par la directive S9 de l'ICH ou lorsque l'IPA est génotoxique
Si une impureté de nitrosamine est détectée dans un produit pharmaceutique, biologique ou radiopharmaceutique destiné à des indications pour les cancers avancés (définies dans la portée de la directive S9 de l'ICH), l'impureté peut être contrôlée conformément aux recommandations du document de questions et réponses de la directive S9 de l'ICH.
Si une impureté de nitrosamine est identifiée dans un produit médicamenteux dont l'IPA est génotoxique à des concentrations thérapeutiques, l'impureté peut être contrôlée selon les limites pour les impuretés non mutagènes. Reportez-vous aux directives Q3A et Q3B de l'ICH.
26. Communication de l'information si les limites acceptables sont révisées à l'avenir
Santé Canada continue de collaborer avec les organismes de réglementation internationaux pour déterminer les limites acceptables pour les impuretés de nitrosamines. Santé Canada a l'intention de communiquer en temps opportun aux DAMM tout changement apporté aux limites acceptables des impuretés de nitrosamines.
Les limites provisoires acceptables avaient été originellement communiquées aux DAMM pour cinq impuretés de nitrosamine dans les antagonistes des récepteurs de l'angiotensine II (aussi appelés « sartans »). Elles étaient en place jusqu'au 30 septembre 2020 et ne seront pas réduites à un niveau inférieur.
27. Limite acceptable si plusieurs nitrosamines sont détectées dans un IPA ou un produit médicamenteux
Si un IPA ou un produit médicamenteux risque de contenir plus d'une impureté de nitrosamine actuelle ou potentielle, l'exposition quotidienne totale (cumulée) devrait être limitée à celle de la nitrosamine ayant la limite acceptable la plus prudente selon la dose quotidienne maximale du produit médicamenteux.
Exemples :
- Si un produit médicamenteux contient à la fois de la NDMA et du NMBA, l'exposition quotidienne totale ou cumulative des deux nitrosamines devrait être limitée à 96,0 ng/jour.
- Si un produit médicamenteux contient à la fois de la NDMA et de la NDEA, l'exposition quotidienne totale ou cumulative des deux nitrosamines devrait être limitée à 26,5 ng/jour.
Si un demandeur ou un DAMM propose de vérifier la présence de multiples impuretés de nitrosamine dans un IPA ou un produit médicamenteux à l'aide d'une autre méthode, Santé Canada évaluera l'acceptabilité de l'approche au cas par cas. Toute autre méthode proposée devrait garantir que l'excès de risque de cancer ne dépasse pas 1 sur 100 000.
En ce qui concerne les impuretés de nitrosamine jugées non mutagènes, les recommandations contenues dans les directives Q3A et Q3B de l'ICH s'appliquent, et il n'est pas nécessaire d'inclure ces impuretés dans une limite pour les nitrosamines totales.
28. Détermination d'une limite d'une durée moindre que la durée de vie (LTL) en tenant compte des principes énoncés dans la directive M7 de l'ICH si une impureté de nitrosamine est présente dans un produit médicamenteux qui est administré pendant moins qu'une durée de vie
En tenant compte des profils de risque des nitrosamines et de la possibilité d'un effet biologique additif, les limites acceptables indiquées à l'annexe 1 sont considérées comme appropriées pour l'administration d'un produit médicamenteux pendant une durée moindre que la durée de vie ou pendant toute la durée de vie.
Si une impureté de nitrosamine ne peut être contrôlée selon la limite acceptable, Santé Canada pourrait considérer une limite provisoire acceptable de nitrosamine supérieure à la limite acceptable. Nous le ferons au cas par cas et seulement dans des circonstances exceptionnelles (par exemple, pour éviter une pénurie de produits médicamenteux jugés médicalement nécessaires ou médicalement importants).
Lorsqu'un demandeur ou un DAMM propose une limite provisoire supérieure à la limite acceptable pour une impureté de nitrosamine, Santé Canada tiendra compte de ce qui suit :
- la nécessité médicale ou l'importance médicale du produit médicamenteux
- les niveaux d'impuretés observés dans des lots représentatifs
- les autres facteurs de gestion des risques (par exemple, la disponibilité des produits médicamenteux de remplacement sur le marché canadien)
Nous considérerons une limite provisoire supérieure à la limite acceptable pour une impureté de nitrosamine comme mesure transitoire seulement, jusqu'à ce que des changements appropriés visant à réduire le niveau de l'impureté de nitrosamine à une valeur égale ou inférieure à la limite acceptable ait été mis en œuvre mis en place.
En ce qui concerne les impuretés de nitrosamine jugées non mutagènes, les recommandations contenues dans les directives Q3A et Q3B de l'ICH s'appliquent.
Qualité
29. Facteurs de risque et causes fondamentales possibles à prendre en considération pour la présence d'impuretés de nitrosamine dans les produits pharmaceutiques destinés à l'usage humain lors de l'évaluation des risques
La connaissance des facteurs de risque et des causes fondamentales possibles de la présence d'impuretés de nitrosamines continue d'évoluer. Les demandeurs et les DAMM devraient se tenir au courant des facteurs de risque et des causes fondamentales possibles que Santé Canada et d'autres organismes de réglementation ont cernés dans leurs guides et publications évaluées par les pairs. La collaboration des experts techniques de qualité des partenaires réglementaires internationaux du NITWG a mené à l'élaboration et à la publication de l'article « Regulatory Experiences with Root Causes and Risk Factors for Nitrosamine Impurities in Pharmaceuticals » (Horne et al. J. Pharm. Sci. 2023, 112, 1166-1182). Cette publication a pour but de communiquer les expériences et les renseignements actuels concernant la qualité sur les causes fondamentales, les facteurs de risque et les mesures d'atténuation des risques liés aux impuretés de nitrosamines dans les produits pharmaceutiques destinés aux humains.
La formation ou l'introduction de nitrosamines peut se produire au cours du processus de fabrication de l'IPA ou du produit médicamenteux. Elle peut également se produire par des mécanismes de dégradation au cours de la période de nouvelle analyse de l'IPA et de la durée de conservation du produit médicamenteux.
Une conception et/ou des contrôles inadéquats des processus, ainsi que des lacunes dans la surveillance de la qualité et de la conformité, peuvent contribuer à la présence d'impuretés de nitrosamine dans les IPA et les produits médicamenteux au-delà des limites acceptables. Les demandeurs et les DAMM devraient tenir compte à la fois des facteurs intrinsèques et extrinsèques lorsqu'ils évaluent les risques liés à la présence d'impuretés de nitrosamines.
Les causes fondamentales possibles et confirmées et les facteurs de risque liés à la présence de nitrosamines dans les produits médicamenteux comprennent les suivantes :
- Nitrosation d'une amine secondaire ou tertiaire pendant la fabrication d'IPA ou de produits médicamenteux, avec élimination en aval insuffisante de la nitrosamine formée, et/ou pendant le stockage d'IPA ou de produits médicamenteux (les conditions courantes de nitrosation impliquent la combinaison d'amines et d'ions nitrite dans des conditions acides)
- Les sources d'amines comprennent les IPA, les intermédiaires d'IPA, les matières de départ, les réactifs, les solvants, les catalyseurs, les produits secondaires de réaction et les produits de dégradation. Certains ingrédients non médicinaux peuvent également contenir des amines dans leur structure. Les amines responsables de la formation de nitrosamines stables comprennent les amines secondaires et les amines tertiaires. Les sels d'ammonium quaternaire sont également des précurseurs potentiels des nitrosamines. Les amines primaires et tertiaires peuvent contenir des amines secondaires sous forme d'impuretés. Les amines peuvent être présentes sous forme d'impuretés dans les amides ou se former par la dégradation des amides (par exemple, par l'hydrolyse). La nitrosation d'amines tertiaires suivie de la désalkylation peut produire une ou plusieurs amines secondaires, qui peuvent ensuite subir une nitrosation pour produire de multiples nitrosamines.
- Les précurseurs et les sources des agents de nitrosation comprennent :
- l'ion nitrite utilisé intentionnellement dans un procédé de fabrication (par exemple, comme utilisé dans la chimie de la diazotation ou comme agent réducteur de l'ion azoture)
- le nitrite présent sous forme d'impureté dans les réactifs (par exemple, l'azoture de sodium), les ingrédients non médicinaux courants (par exemple, la cellulose microcristalline, le stéarate de magnésium)
- les oxydes d'azote (par exemple, NO, N2O3)
- l'acide nitrique
- les halogénures de nitrosyle
- les nitrites d'alkyle et les composés nitrés (par exemple, le nitrométhane)
- l'eau potable et/ou l'eau purifiée contenant du nitrite
- Utilisation d'une nitrosamine comme matière de base ou intermédiaire synthétique, avec conversion incomplète de la nitrosamine et/ou élimination en aval insuffisante
- Réaction de l'ion nitrite et d'une amine dans des conditions de procédé avec pH > 7 sous catalyse par un composé carbonylé, avec élimination en aval insuffisante
- Oxydation d'un groupe fonctionnel hydrazine dans un IPA, matière de base, intermédiaire ou réactif pour produire une nitrosamine, avec élimination en aval insuffisante
- Utilisation de certains matériaux dans les composants de fermeture des conteneurs, tels que :
- nitrocellulose, que l'on trouve dans certaines feuilles de couverture contenant un apprêt d'impression à base de nitrocellulose utilisées pour l'emballage alvéolé
- certains types d'accélérateurs de vulcanisation (par exemple, diéthyldithiocarbamate de sodium thiourée, thiruam) utilisés dans la fabrication du caoutchouc
- Utilisation de matériaux recyclés (par exemple, solvants, réactifs, catalyseurs) contaminés par des nitrosamines et/ou des précurseurs de nitrosamine
- Contamination croisée de matériaux contenant des nitrosamines ou leurs précurseurs dans des installations où sont fabriqués plusieurs produits (par exemple, par l'utilisation d'équipement partagé)
- Mauvais fonctionnement d'une étape du processus (par exemple, pendant les séparations de phase liquide-liquide), destinée à éliminer les nitrosamines
- Utilisation de certaines opérations de fabrication qui pourraient faciliter le contact entre les précurseurs de nitrosamine (par exemple, granulation humide) ou introduction d'agents ou de précurseurs de nitrosation (par exemple, des oxydes d'azote pendant le séchage sur lit fluidisé)
30. Composants des produits médicamenteux à prendre en compte dans les évaluations des risques
Tous les composants du produit médicamenteux devraient être considérés comme des sources potentielles d'impuretés de nitrosamine, ou de leurs agents de nitrosation et amines précurseurs, dans le contexte du processus désigné et des conditions d'entreposage. Par exemple, certains excipients peuvent contenir des niveaux résiduels de nitrite (voir Wu, Y. et al. AAPS PharmSciTech 2011, 12(4), 1246-1263 et Boetzel et al. J. Pharm. Sci. 2023, 112, 1615-1624) ou d'amines réactives dans leur structure moléculaire, ce qui, dans certaines procédures de fabrication ou conditions d'entreposage, peut mener à la formation d'impuretés de nitrosamine.
Consulter la section 29 pour en savoir plus sur les facteurs de risque et les causes fondamentales possibles à prendre en considération.
31. Impuretés de nitrosamines à prendre en compte dans l'évaluation des risques (étape 1) et les essais de confirmation (étape 2) (mis à jour)
Compte tenu du caractère unique de chaque processus de fabrication d'IPA et de produit médicamenteux, il convient de noter que la liste des nitrosamines figurant à l'annexe 1 des présentes lignes directrices n'est pas exhaustive et ne représente pas toutes les nitrosamines potentiellement présentes dans les IPA et les produits médicamenteux. À l'inverse, les nitrosamines figurant à l'annexe 1 peuvent ne pas être des impuretés potentielles dans tous les IPA et les produits médicamenteux.
Les DAMM et les demandeurs devraient s'assurer que les évaluations des risques envisagent et déterminent les possibilités de toutes les impuretés de nitrosamines qui pourraient être formées ou introduites. Toutes les nitrosamines susceptibles d'être formées ou introduites doivent être incluses dans le programme de tests de confirmation (étape 2).
Dans le cas des nitrosamines qui ne figurent pas à l'annexe 1, les DAMM et les demandeurs devraient suivre la section 24 pour les directives sur la façon d'établir une limite acceptable.
32. Méthodes d'essai fournies par Santé Canada
Plusieurs organismes de réglementation, dont Santé Canada, le Réseau européen des laboratoires officiels de contrôle des médicaments et la Food and Drug Administration (FDA) des États-Unis, ont publié et partagé des méthodes d'essai. Ces méthodes peuvent être utilisées, bien qu'il n'y ait aucune obligation de le faire.
Dans tous les cas, les entreprises devraient utiliser des méthodes analytiques adéquatement validées et suffisamment sensibles et effectuer des essais dans une installation conforme aux bonnes pratiques de fabrication. Si d'autres méthodes sont utilisées, il n'est pas nécessaire de vérifier la méthode auprès de Santé Canada avant de l'utiliser.
Les méthodes d'analyse devraient être de nature quantitative (au lieu des tests basés sur les limites) et devraient être entièrement validées avant le début des tests de confirmation. S'ils sont utilisés, les tests basés sur les limites devraient être accompagnés d'une justification scientifique appropriée dans la documentation sur l'évaluation des risques. Par exemple :
- une démonstration que le test de limite est valide à la limite acceptable ou en dessous de celle-ci.
- des preuves à l'appui qui indiquent qu'il n'y a pas d'augmentation de la concentration d'impuretés de nitrosamines au fil du temps
Sauf justification contraire, la validation de la méthode devrait être faite en utilisant le produit médicamenteux dont l'utilisation est autorisée au Canada.
Lorsqu'il existe plusieurs concentrations d'un produit médicamenteux et que la validation est susceptible de couvrir de nombreuses concentrations, la justification du choix de la concentration du produit utilisée pour la validation devrait être décrite dans le protocole de validation.
33. Validation de la limite de quantification (LQ) pour les procédures d'analyse des impuretés de nitrosamine (mis à jour)
La LQ pour les procédures analytiques destinées à l'analyse quantitative des impuretés individuelles de nitrosamine dans les IPA et les produits pharmaceutiques doit être égale ou inférieure à la LA établie (annexe 1), la LA établie à l'aide de la ACPC (voir le numéro 24 et l'annexe 4) si une LA n'a pas été établie par Santé Canada, ou la limite ICH Q3A ou Q3B pertinente pour une impureté nitrosamine qui a été classée comme non mutagène dans l'annexe 1.
Les procédures analytiques doivent être validées avec une LQ inférieure ou égale à 10 % de la limite acceptable pour une nitrosamine individuelle, si une proposition visant à ne pas tester systématiquement la nitrosamine dans les spécifications du produit médicamenteux est prévue (voir le numéro 34).
Les procédures analytiques doivent être validées avec une LQ inférieure ou égale à 30 % de la limite acceptable pour une nitrosamine individuelle, si une proposition d'essai périodiques (skip) est prévue.
34. Inclusion de tests de routine pour les impuretés de nitrosamine dans les spécifications de l'IPA et/ou du produit médicamenteux
La spécification de l'IPA devrait inclure un test et un critère d'acceptation pour chaque impureté de nitrosamine dans les cas suivants :
- lorsque le risque de présence de nitrosamine est considéré comme élevé
- lorsque la concentration de toute nitrosamine se trouve à des niveaux significatifs (par exemple, plus de 30 % de la limite acceptable) durant des tests de confirmation
Exemples où le risque de nitrosamines est considéré comme élevé :
- potentiel de formation de nitrosamines lors du stockage
- présence de groupes fonctionnels précurseurs de nitrosamine dans l'IPA
- formation ou introduction tardive d'une impureté de nitrosamine dans le processus de fabrication
Lorsque plusieurs nitrosamines sont détectées dans un IPA, une limite cumulative pour les nitrosamines devrait également être incluse dans la spécification en utilisant l'une des approches décrites dans la section 27.
Des tests de routine pour les impuretés de nitrosamine devraient être inclus dans la spécification dans les cas suivants :
- le potentiel d'introduction de nitrosamine durant la fabrication, le conditionnement et le stockage du produit médicamenteux est déterminé
- une impureté de nitrosamine est détectée dans le produit médicamenteux durant les tests de confirmation et la cause fondamentale est inconnue
Lorsqu'un tel risque est détecté, il convient d'inclure un critère de test et d'acceptation pour les spécifications de libération et de durée de conservation. Lorsque plusieurs nitrosamines sont détectées, le contrôle des nitrosamines totales en utilisant l'une des approches décrites dans la section 27 doit être inclus dans la spécification. Par ailleurs, des limites de contrôle exprimées sur la base d'une impureté individuelle (par exemple, une limite pour chaque nitrosamine fixée à un pourcentage de sa limite acceptable de sorte que la somme des limites acceptables en % pour chaque nitrosamine spécifiée ne dépasse pas 100 %) peuvent être proposées avec une justification appropriée. D'autres approches pour établir une spécification appropriée lorsque plusieurs nitrosamines sont concernées peuvent être acceptables si elles sont dûment justifiées.
La présence d'une ou de plusieurs nitrosamines à une concentration inférieure à 10 % de la limite acceptable dans un produit médicamenteux constitue un risque toxicologique négligeable; si la cause première de la présence de ces impuretés de nitrosamine est comprise et que des mesures de contrôle appropriées ont été mises en place pour garantir que ces impuretés seront toujours inférieures à 10 % de la limite acceptable, il n'est pas nécessaire de mentionner ces impuretés dans les spécifications des produits médicamenteux. Les nitrosamines présentes à moins de 10 % de leur limite acceptable respective ne doivent pas être prises en compte dans le calcul des limites pour les nitrosamines totales.
Les DAMM devraient tester tous les nouveaux lots de produits médicamenteux pour les nitrosamines et ne mettre sur le marché que les lots qui répondent aux critères d'acceptation des nitrosamines individuelles (et multiple, le cas échéant). Les tests de routine de tous les lots de produits médicamenteux devraient se poursuivre jusqu'à ce que la cause fondamentale soit pleinement comprise et que d'autres contrôles ou mesures d'atténuation des risques (par exemple, contrôles de processus, spécifications des matières premières, etc.) aient été mis en œuvre. Il faut veiller à ce que les impuretés de nitrosamine soient systématiquement en dessous de la limite acceptable à l'avenir.
35. Options de contrôle possibles des impuretés de nitrosamine dans l'IPA
Les options de contrôle des impuretés de nitrosamines incluent :
- des tests de routine sur l'IPA (option 1 de la directive M7 de l'ICH)
- un contrôle dans les spécifications d'un produit intermédiaire avec un critère d'acceptation à la limite acceptable (option 2 de la directive M7 de l'ICH) (lorsque la ou les causes fondamentales de la présence de nitrosamine ont été établies sans équivoque)
- un contrôle dans les spécifications d'un produit intermédiaire avec un critère d'acceptation qui dépasse la limite acceptable (option 3 de la directive M7 de l'ICH) (lorsque la ou les causes fondamentales ont été établies sans équivoque et que la justification de la limite proposée est appuyée par une compréhension des contrôles, du devenir, de l'élimination et des processus connexes [par exemple, études d'ajout volontaire et d'élimination])
Les propositions relatives à une stratégie de contrôle à l'égard des impuretés de nitrosamines dans une nouvelle demande d'autorisation de mise sur le marché (option 4 de la directive M7 de l'ICH) seront évaluées au cas par cas. Une proposition de stratégie de contrôle (option 4 de la directive M7 de l'ICH) peut ne pas convenir lorsque la concentration de toute impureté de nitrosamine dans un IPA est supérieure à 30 % de la limite acceptable. Toutefois, une telle stratégie peut être acceptable lorsque la compréhension du procédé a été démontrée par des études « sur le devenir et d'élimination », par l'identification des paramètres du procédé qui ont une incidence sur les niveaux d'impuretés de nitrosamine et lorsque la stratégie est appuyée par des données analytiques appropriées. Les calculs de prévision du facteur d'élimination devraient être appuyés par des données analytiques appropriées.
Cette information devrait être fournie dans la nouvelle demande d'autorisation de mise en marché, avec des copies des procédures analytiques et des rapports de validation de la méthode.
Consulter également la section 34 pour obtenir de l'information sur les tests de routine pour les impuretés de nitrosamine dans l'IPA et/ou la spécification du produit médicamenteux.
36. Attentes en matière de tests de confirmation
Pendant les tests de confirmation, les DAMM devraient tester le produit médicamenteux pour déterminer les niveaux d'impuretés de nitrosamines.
La mise à l'essai de l'IPA est également recommandée si l'évaluation des risques indique que l'IPA est une source potentielle d'impuretés de nitrosamines dans le médicament. Dans de tels cas, les résultats des tests de l'IPA peuvent être utilisés pour appuyer les études des causes fondamentales et l'élaboration d'une stratégie de contrôle justifiée des impuretés de nitrosamines dans l'IPA.
Si un produit médicamenteux est offert en plusieurs concentrations de la même forme pharmaceutique et que les mêmes facteurs de risque s'appliquent à chacune d'elles, il est possible de rationaliser les essais en ne testant que la concentration correspondant au scénario le plus défavorable. L'approche du scénario le plus défavorable doit être justifiée par le DAMM au cas par cas. La justification doit être documentée dans l'évaluation des risques du système de qualité pharmaceutique du DAMM.
Si, malgré des efforts considérables, il apparaît qu'une impureté de nitrosamine ne peut être synthétisée ou isolée puis purifiée, cela peut indiquer que la nitrosamine n'existe pas, qu'elle est instable ou qu'il n'y a pas de risque qu'elle se forme. Dans de tels cas, il pourrait ne pas être nécessaire d'effectuer des essais de confirmation. Une justification détaillée doit être fournie au cas par cas, conformément à des principes scientifiques appropriés. Une justification scientifique comprenant des données expérimentales qui résument les efforts réalisés pour synthétiser et/ou isoler puis purifier l'impureté doit être incluse dans le résumé et dans la discussion de l'évaluation des risques, dans la présentation réglementaire, et être documentée dans le système de qualité pharmaceutique des DAMM.
37. Laboratoires d'analyse effectuant des essais de nitrosamine et inscription sur la LEPP
Le laboratoire d'analyse utilisé pour les tests de confirmation de la présence de nitrosamines (étape 2) n'a pas à être inscrit sur la LEPP pour le moment. Toutefois, dans tous les cas, les tests de confirmation doivent être effectués dans une installation conforme aux BPF. Un laboratoire d'analyse étranger, s'il est utilisé pour effectuer les essais de confirmation de la nitrosamine, doit :
- avoir été jugé conforme aux BPF par Santé Canada
- faire l'objet d'une inspection valide des BPF par un organisme de réglementation compétent ou qualifié qui démontre une conformité aux normes actuelles des BPF
Les médicaments de spécialité médicale sont ceux qui ne nécessitent pas d'ordonnance, mais qui sont généralement prescrits par un médecin praticien à titre de produits d'usage professionnel (par exemple, solutions d'hémodialyse, nitroglycérine). En ce qui concerne les tests de médicaments de spécialité médicale et en vente libre, un rapport d'audit d'entreprise ou de consultant est acceptable pour démontrer la conformité aux BPF si aucun rapport d'inspection par des organismes de réglementation ou des organismes qualifiés n'est disponible.
Pour en savoir plus sur les éléments de preuve acceptables concernant les BPF et les exigences réglementaires, consulter les directives suivantes :
Toutefois, les laboratoires d'analyse doivent être mentionnés dans l'annexe pertinente de la LEPP s'ils effectuent les tests suivants :
- tests de nitrosamine utilisés pour libérer des IPA et des produits médicamenteux destinés au marché canadien
- tests faisant partie de la spécification de l'IPA ou du produit médicamenteux, notamment
- les tests imposés au titre des modalités relatives aux sartans
Pour obtenir des conseils sur les attentes de Santé Canada à l'égard des installations d'analyse ou pour toute autre question connexe, veuillez envoyer un courriel à foreign.site-etranger@hc-sc.gc.ca.
38. Nombre et types de lots de produits médicamenteux devant être testés dans le cadre des tests de confirmation des produits commercialisés et des présentations de nouveaux produits
Pour les produits commercialisés, le nombre de lots à tester doit être proportionnel au risque. Voici des exemples de risques élevés :
- formation ou introduction tardive d'une impureté de nitrosamine dans un procédé de fabrication
- présence de groupes fonctionnels précurseurs de nitrosamine dans l'IPA
- potentiel de formation de nitrosamines lors du stockage
Les DAMM et les fabricants devraient soumettre à des tests un nombre représentatif de lots du produit médicamenteux, le cas échéant, en fonction de l'évaluation des risques (par exemple, des lots qui sont représentatifs des sources des composants, des procédés et des sites de fabrication, des dates de fabrication).
Si la cause fondamentale de la présence de nitrosamine a été déterminée et démontrée scientifiquement, et qu'on s'attend à ce que les niveaux d'impuretés soient uniformes d'un lot à l'autre (par exemple, comme le démontrent les études d'ajout volontaire et d'élimination), des tests devraient être effectués sur 10 % des lots annuels, ou 3 par année, selon la valeur la plus élevée. Les tests doivent comprendre à la fois les lots nouvellement produits et les échantillons conservés des lots qui n'ont pas atteint leur date d'expiration. Si moins de 3 lots sont fabriqués chaque année, tous les lots qui n'ont pas atteint leur date d'expiration doivent être testés.
Il n'est pas nécessaire de soumettre les plans ou protocoles d'essai (par exemple, un protocole pour le nombre et le type de lots à tester) à Santé Canada aux fins de l'évaluation et de l'approbation avant d'entreprendre des tests de confirmation.
Si des impuretés de nitrosamine sont détectées à des concentrations importantes (approchant, atteignant ou dépassant les limites acceptables), des lots supplémentaires du produit médicamenteux sur le marché canadien et qui n'ont pas atteint leur date d'expiration devraient faire l'objet de tests de confirmation. Dans de tels cas, les DAMM pourraient être tenus de tester tous les lots sur le marché canadien qui n'ont pas atteint leur date d'expiration.
Pour les PDN, les PADN, les DDIN (avec données C et F), les DDINB, les suppléments, les préavis de modification et les demandes de modifications après l'émission d'une DIN (pour les changements de qualité qui peuvent avoir un impact lié à la présence potentielle de nitrosamines dans la substance médicamenteuse ou le produit médicamenteux, veuillez consulter la section 13), au moins 6 lots pilotes ou 3 lots à l'échelle commerciale devraient être soumis à des tests de confirmation lorsqu'un risque de nitrosamines a été détecté. Un plus grand nombre de résultats de lots doivent être soumis à une évaluation lorsque le risque de présence de nitrosamine est élevé. En voici quelques exemples :
- la formation ou l'introduction tardive d'une impureté de nitrosamine
- la présence de groupes fonctionnels précurseurs de nitrosamine dans l'IPA
- les problèmes de stabilité liés à la formation de nitrosamines pendant la période de contre-essai ou de durée de conservation
Des tests de stabilité des lots pour une impureté de nitrosamine devraient être effectués dans les cas suivants :
- il existe un risque que les concentrations de nitrosamine augmentent dans l'IPA ou le produit médicamenteux au fil du temps
- le potentiel d'augmentation au fil du temps n'est pas clair
Le cas échéant, la présentation du médicament doit contenir au moins 6 mois de données sur la stabilité recueillies sous des conditions accélérées et à long terme dans le ou les systèmes de fermeture de contenant proposés dans le cadre des essais de détection d'impuretés de nitrosamine.
Annexes
Annexe 1 : Limites acceptables (LA) établies pour les impuretées de N-nitrosamines
- Voir l'annexe 1 séparée.
Annexe 2 : Lignes directrices sur les impuretés de nitrosamine et l'évaluation des risques pour les changements survenus après l'avis de conformité (AC) dans le cas des nouveaux produits médicamenteux contenant des IPA synthétiques et semi-synthétiques (mis à jour)
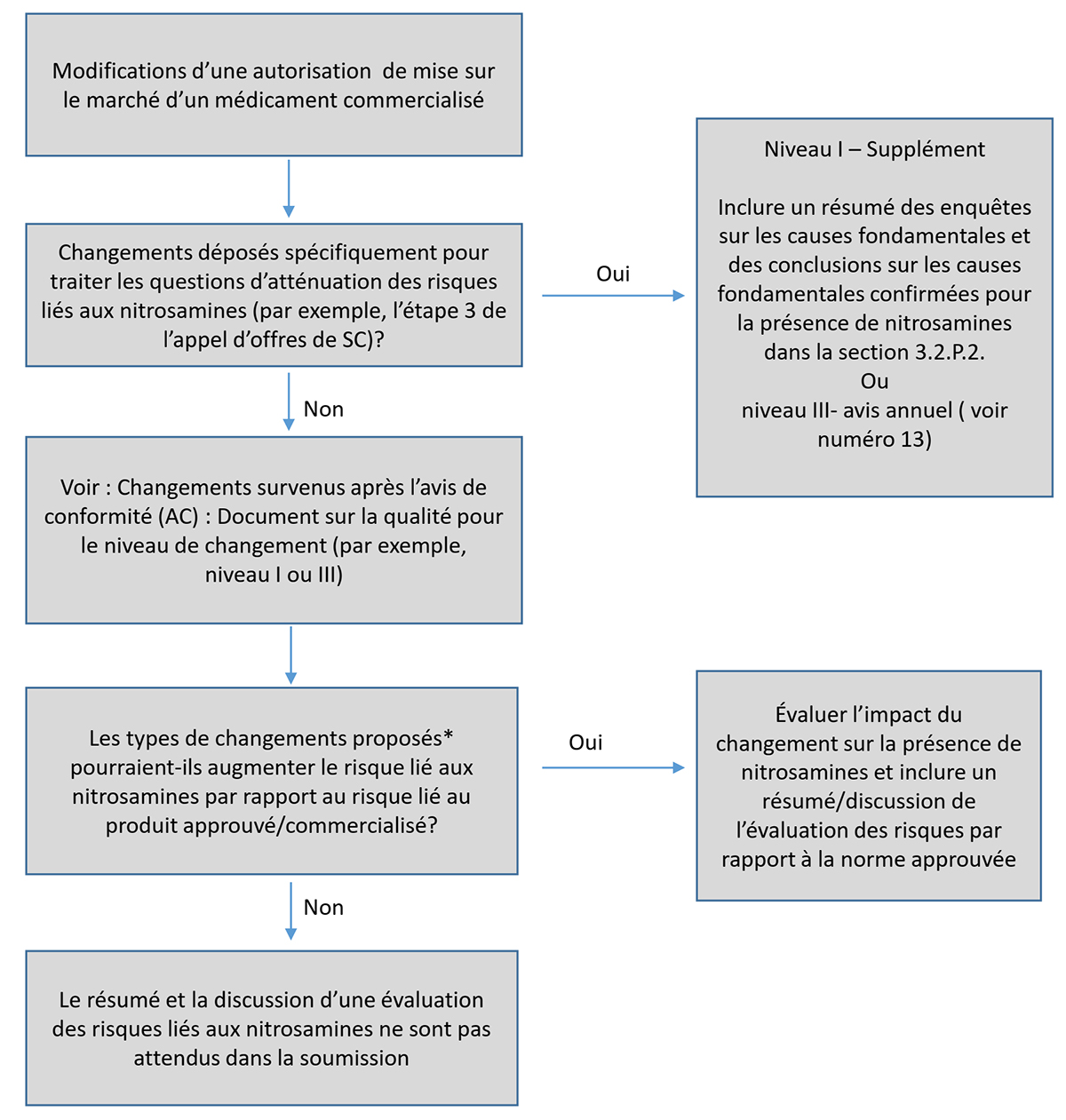
* Par exemple, des changements dans les processus de fabrication de la substance médicamenteuse ou du produit médicamenteux, des changements dans la composition du produit médicamenteux (IPA, excipients), des changements dans la forme posologique, des changements dans le système de fermeture du contenant.
Annexe 2: Description textuel
L'annexe 2 contient un arbre décisionnel fournissant des conseils aux détenteurs d’une autorisation de mise en marché sur les exigences réglementaires en matière de soumission liées aux nitrosamines. Cette annexe décrit également certains changements qui peuvent avoir un impact sur la possibilité de formation de nitrosamine lorsqu'un produit subit une modification.
Annexe 3 : Conditions du test d'Ames amélioré
La ligne directrice no 471 de l'Organisation de coopération et de développement économiques (OCDE) intitulée « Essai de mutation réverse sur les bactéries » fournit des recommandations normalisées pour la réalisation de l'essai de mutation réverse sur les bactéries (aussi appelé test d'Ames) afin d'évaluer le potentiel mutagène d'un composé d'essai. Pour les nitrosamines, des conditions améliorées pour le test d'Ames sont recommandées en raison de la sensibilité réduite signalée du test dans des conditions normalisées pour certaines nitrosamines (p. ex., NDMA). De plus, on sait très peu de choses sur la sensibilité du test d'Ames aux impuretés de nitrosamine dans les substances médicamenteuses (INSM), une classe récemment reconnue d'impuretés de nitrosamine structurellement liées à la substance médicamenteuse. Les INSM ont généralement une plus grande variété de groupes fonctionnels présents que les nitrosamines de faible poids moléculaire (comme la NDMA) déjà étudiées.
Si un test d'Ames standard produit un résultat positif, il n'est pas nécessaire d'effectuer un test supplémentaire en utilisant des conditions d'essai améliorées.
Les conditions améliorées du test d'Ames présentées ci-dessous sont éclairées par les travaux menés par le National Center for Toxicological Research (NCTR) de la FDA (Li et. coll., 2023), ainsi que par d'autres groupes, et ont été évaluées en fonction d'une variété de nitrosamines, y compris les INSM. L'évaluation des conditions du test d'Ames pour les nitrosamines se poursuit dans le but de déterminer les conditions du test d'Ames les plus robustes. Les conditions améliorées du test d'Ames décrites ci-dessous seront mises à jour au besoin. Les écarts par rapport aux conditions recommandées devraient être justifiés.
Souches d'essai : Les souches d'essai S. typhimurium TA98, TA100, TA1535, TA1537 et E. coli WP2 uvrA (pKM101) devraient être incluses.
Type d'essai et temps de préincubation : La méthode de préincubation, et non d'incorporation sur plaque, devrait être utilisée. Le temps de préincubation recommandé est de 30 minutes.
Espèces et concentration de S9 : Les tests d'Ames doivent être effectués en l'absence d'une fraction post-mitochondriale (S9), ainsi qu'en présence d'une fraction S9 de 30 % du foie du rat, ainsi que d'une fraction S9 de 30 % du foie du hamster. Les fractions post-mitochondriales (S9s) du rat et du hamster doivent être préparées à partir de rongeurs traités avec des inducteurs d'enzymes du cytochrome P450 (par exemple, une combinaison de phénobarbital et de β-naphtoflavone).
Contrôle négatif (solvant/véhicule) : Les solvants doivent être compatibles avec le test d'Ames conformément à la ligne directrice OCDE 471. Les solvants peuvent comprendre, sans s'y limiter :
- Eau
- Solvants organiques tels que l'acétone, le méthanol et le DMSO.
Lorsqu'un solvant organique est utilisé, le volume minimal devrait être utilisé dans le mélange de préincubation avec justification qui indique que le volume de solvant n'interfère pas avec l'activation métabolique de la nitrosamine.
Témoins positifs : Les témoins positifs simultanés spécifiques à chaque souche devraient être inclus conformément à la ligne directrice OCDE 471.
Deux nitrosamines dont on sait qu'elles sont mutagènes en présence de la fraction S9 devraient également être incluses comme témoins positifs.
Le choix des témoins positifs de nitrosamine doit être justifié en fonction du métabolisme anticipé de la nitrosamine et des enzymes du cytochrome P450 les plus susceptibles d'être impliqués. De plus, si un solvant organique est utilisé pour dissoudre le composé d'essai, il est recommandé, dans la mesure du possible, que le volume de solvant organique utilisé pour dissoudre les témoins positifs à la nitrosamine donne une concentration semblable à celle du composé d'essai dans le mélange de préincubation.
Voici des témoins positifs de nitrosamine à considérer :
- NDMA (CAS 62-75-9)
- 1-cyclopentyl-4-nitrosopipérazine (numéro CAS 61379-66-6)
- Une INSM
Toutes les autres recommandations pour le test d'Ames devraient suivre la ligne directrice 471 de l'OCDE.
Références :
Ligne directrice 471 d'essai de l'OCDE « Essai de mutation réverse sur les bactéries », 2020
Li et coll., « Revisiting the mutagenicity and genotoxicity of N-nitroso propranolol in bacterial and human in vitro assays », Regulatory Pharmacology and Toxicology, 2023
Annexe 4 : Approche de catégorisation de la puissance cancérogène (ACPC) dans le cas des N-nitrosamines
Ce document décrit une approche permettant d'affecter une impureté de type N-nitrosamine (y compris les impuretés liées à une substance médicamenteuse de type nitrosamine) à une catégorie de puissance cancérogène prédite, avec une limite acceptable correspondante, sur la base d'une évaluation des caractéristiques structurelles activatrices ou désactivatrices présentes dans la molécule. Dans le contexte du présent document, les caractéristiques activatrices ou désactivatrices sont définies comme des sous-structures moléculaires associées à une augmentation ou à une diminution, respectivement, de la puissance cancérogène.
L'approche de catégorisation de la puissance cancérogène est basée sur les concepts de relations structure-activité (SAR) décrits dans des publications scientifiques récentes pour les composés de N-nitrosamineNote de bas de page 1 et utilise également un ensemble d’environ 80 N-nitrosamines avec des valeurs DT50 chez le rat provenant de la base de données sur la puissance cancérogène (CPDB) et/ou de la base de données sur la cancérogénicité de Lhassa (LCDB)Note de bas de page 2, des classifications de la puissance relative telles que définies par Rao et al. (1979)Note de bas de page 3, et/ou des limites acceptables basées sur des analyses de substitution réalisées antérieurement.Note de bas de page 4 Cette approche suppose que le mécanisme d'α-hydroxylation de l'activation métaboliqueNote de bas de page 5 est responsable de la réponse mutagène et cancérogène très puissante observée avec de nombreuses N-nitrosamines. Les caractéristiques structurelles qui augmentent ou diminuent directement la favorabilité du mécanisme d'activation, ou qui augmentent la clairance de la nitrosamine par d'autres voies biologiques, devraient avoir un effet correspondant sur la puissance cancérogène. Il est donc possible de prédire le potentiel mutagène et cancérogène d'une N-nitrosamine sur la base de ses caractéristiques structurelles.
Il est reconnu que la science évolue dans la prédiction du potentiel mutagène et de la puissance cancérogène sur la base des concepts de SAR. Par conséquent, la méthode de catégorisation de la puissance cancérogène décrite dans le présent document est une approche prudente qui représente les meilleures données scientifiques disponibles à l'heure actuelle et qui devrait être affinée et élargie au fur et à mesure que de nouvelles données seront disponibles. Il peut s'agir d'affiner les limites acceptables associées aux catégories de puissance cancérogène prédites et de modifier les caractéristiques structurelles et les scores d'activation et de désactivation qui leur sont associés.
L'approche de catégorisation de la puissance cancérogène s'applique aux N-nitrosamines comportant un atome de carbone de part et d'autre du groupe N-nitroso, et où le carbone n'est pas directement lié par une double liaison à un hétéroatome (les N-nitrosamides, les N-nitrosourées, les N-nitrosoguanidines et les autres structures apparentées sont donc exclues). En outre, l'approche de catégorisation de la puissance cancérogène ne s'applique pas aux N-nitrosamines dont le groupe N-nitroso se trouve à l'intérieur d'un cycle aromatique (par exemple, l'indole nitrosé). Pour les N-nitrosamines contenant deux groupes N-nitroso, le groupe dont la puissance cancérogène prédite est la plus élevée (c'est-à-dire le groupe dont la catégorie de puissance numérique est la plus basse) définit la limite acceptable pour l'ensemble de la moléculeNote de bas de page 6. Les carbones α et β sont définis par rapport au groupe N-nitroso, comme l'indique la figure 1.
Figure 1. Représentation structurale des carbones α et β sur une N-nitrosamine
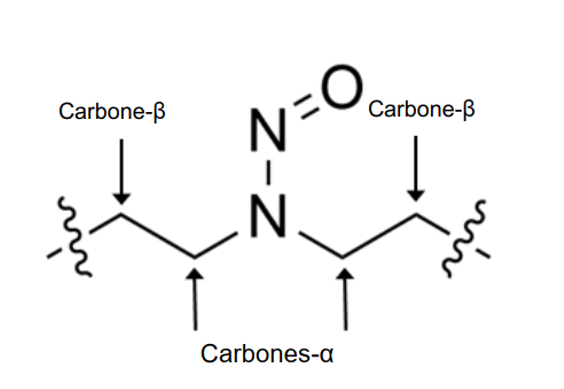
Figure 1: Description textuel
Représentation structurale des carbones α et β sur une N-nitrosamine
Le processus de prédiction de la catégorie de puissance cancérogène appropriée est décrit dans la figure 2. Le tableau 1 résume les cinq catégories de puissance cancérogène prédites et les limites acceptables (LA) qui leur sont associées. Les tableaux permettant de calculer le niveau de puissance mentionné à la figure 2 sont présentés à l'annexe A, et des exemples de calculs sont présentés à l'annexe B.
Figure 2. Diagramme de flux permettant de prédire la catégorie de puissance d'une N-nitrosamine
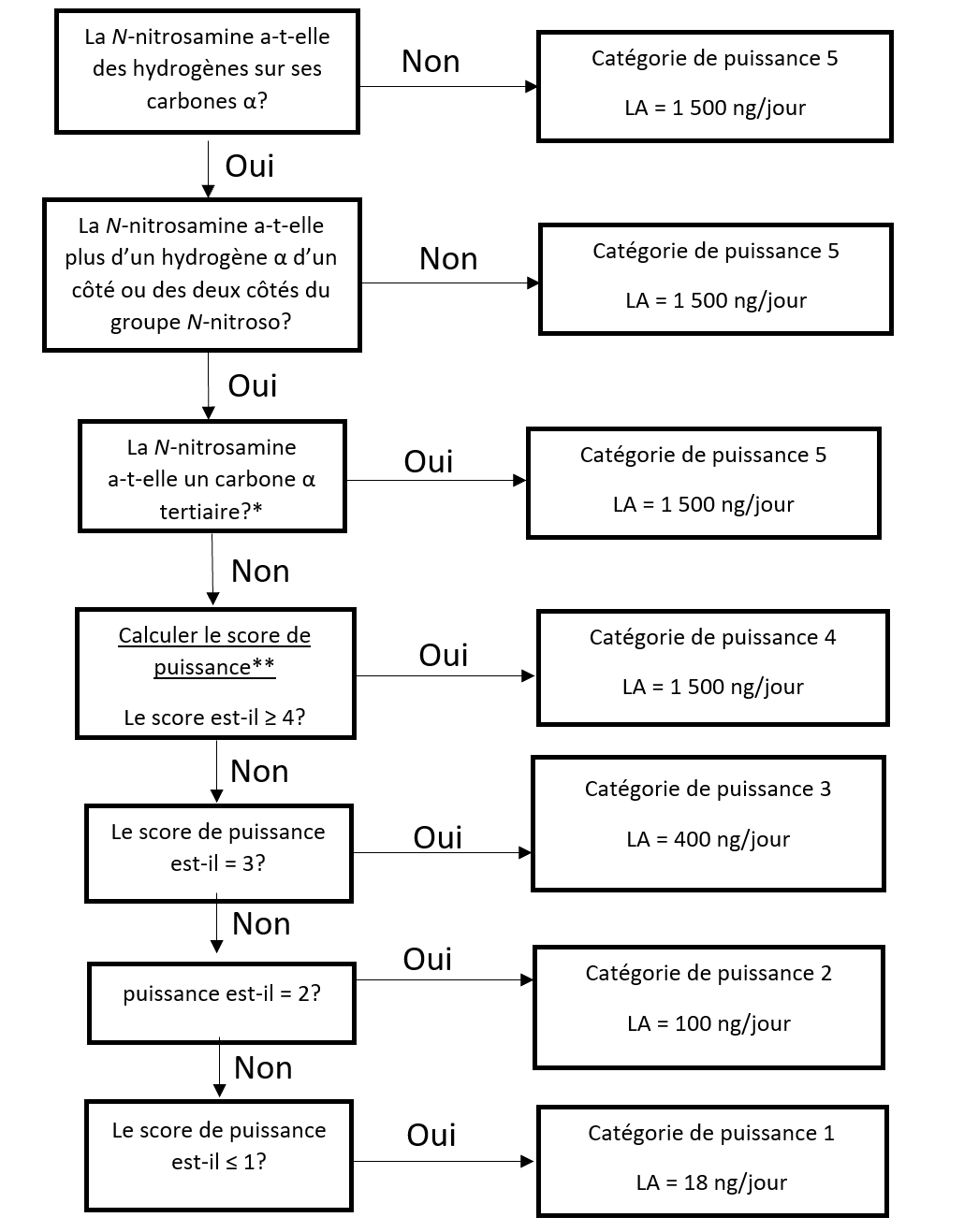
* Un carbone α tertiaire est défini comme un atome de carbone α dans un état d'hybridation sp3, lié à trois autres atomes de carbone.
** Pour calculer le score de puissance, voir l'annexe A.
Figure 2: Description textuel
La figure 2 montre ,étape par étape, un processus permettant d'attribuer une catégorie de puissance à une N-nitrosamine. Chaque étape propose deux options, et les étapes sont suivies jusqu'à ce qu'une catégorie de puissance de 1, 2, 3, 4 ou 5 soit atteinte. La catégorie de puissance détermine la limite d'absorption acceptable à laquelle l'impureté N-nitrosamine peut être contrôlée.
| Catégorie de puissance | Limite acceptable recommandée (ng/jour) | Commentaires |
|---|---|---|
| 1 | 18 | La limite acceptable recommandée de 18 ng/jour est égale au seuil de risque toxicologique (SRT) propre à la classe pour les impuretés de type N-nitrosamineNote de bas de page *. Les N-nitrosamines classées dans la catégorie 1 sont censées avoir une puissance cancérogène élevée; cependant, le SRT propre à la classe dans le cas des impuretés de N-nitrosamine est réputé offrir une protection suffisante pour les patients. |
| 2 | 100 | La limite acceptable recommandée de 100 ng/jour est représentative de deux N-nitrosamines puissantes ayant fait l'objet de tests rigoureux, la N-nitrosodiméthylamine (NDMA) et la 4-(méthylnitrosamino)-1-(3-pyridyl)-1-(butanone) (NNK), pour lesquelles les limites acceptables recommandées sont de 96 ng/jour et de 100 ng/jour respectivement. Les N-nitrosamines classées dans la catégorie 2 sont censées avoir une puissance cancérogène qui n'est pas supérieure à celle de la NDMA et du NNK. |
| 3 | 400 | Comparativement à la catégorie de puissance 2, les N-nitrosamines de cette catégorie ont une puissance cancérogène plus faible en raison, notamment, de la présence d'une caractéristique structurelle faiblement désactivatrice. La limite acceptable recommandée a été fixée de manière à réduire de quatre fois la puissance cancérogène par rapport à la catégorie 2. |
| 4 | 1 500 | Les N-nitrosamines classées dans la catégorie 4 peuvent être activées métaboliquement par une voie d'α-hydroxylation, mais leur puissance cancérogène devrait être faible, notamment parce que la voie n'est pas favorisée en raison d'influences stériques ou électroniques, ou parce que les voies de clairance sont favorisées. La limite acceptable recommandée de 1 500 ng/jour est conforme au SRT selon la Directive M7 de l'ICHNote de bas de page **. |
| 5 | 1 500 | Les N-nitrosamines classées dans la catégorie 5 ne devraient pas être activées métaboliquement par une voie d'α-hydroxylation en raison d'un encombrement stérique ou de l'absence d'hydrogènes α, ou devraient former des espèces instables qui ne réagissent pas avec l'ADN. La limite acceptable recommandée de 1 500 ng/jour est conforme au SRT selon la Directive M7 de l'ICHNote de bas de page **. |
|
||
Annexe A. Calcul du score de puissance
Dans le cas des N-nitrosamines non incluses dans la catégorie de puissance 5, le score de puissance correspond à la somme du score des hydrogènes α (tableau 2), du score des caractéristiques désactivatrices (tableau 3) et du score des caractéristiques activatrices (tableau 4), d'après les caractéristiques structurelles sélectionnées présentes dans la N-nitrosamine. La structure de la N-nitrosamine devrait correspondre exactement à l'une des définitions des hydrogènes α du tableau 2, mais elle peut contenir plusieurs ou aucune des caractéristiques structurelles identifiées dans les tableaux 3 et 4. Lorsqu'une ou plusieurs caractéristiques des tableaux 3 et 4 sont présentes dans la N-nitrosamine, le score de puissance doit être calculé de la façon indiquée dans l'encadré ci-dessous. Si la N-nitrosamine ne présente aucune des caractéristiques des tableaux 3 et 4, le score de puissance sera égal au score des hydrogènes α.
Score de puissance = score des hydrogènes α + score des caractéristiques désactivatrices (somme de tous les scores pour les caractéristiques présentes dans la N-nitrosamine) + score des caractéristiques activatrices (somme de tous les scores pour les caractéristiques présentes dans la N-nitrosamine)
| Nombre d'atomes d'hydrogène sur chaque carbone α, en ordre croissant | Exemple | Score des hydrogènes α |
|---|---|---|
| 0,2 |  |
3Note de bas de page * |
| 0,3 | 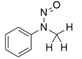 |
2 |
| 1,2 | 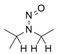 |
3 |
| 1,3 | 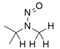 |
3 |
| 2,2 | 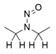 |
1 |
| 2,3 |  |
1 |
|
||
Caractéristique désactivatrice |
Exemple | Score des caractéristiques désactivatrices individuelles |
|---|---|---|
| Groupement acide carboxylique n'importe où sur la molécule |  |
+3 |
| Groupe N-nitroso dans un cycle pyrrolidine |  |
+3 |
| Groupe N-nitroso dans un cycle à 6 chaînons contenant au moins un atome de soufre |  |
+3 |
| Groupe N-nitroso dans un cycle à 5 ou 6 chaînonsNote de bas de page * | 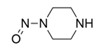 |
+2 |
| Groupe N-nitroso dans un cycle morpholine | 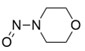 |
+1 |
| Groupe N-nitroso dans un cycle à 7 chaînons | 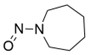 |
+1 |
| Chaînes de ≥ 5 atomes d'hydrogène consécutifs (cycliques ou acycliques) de part et d'autre d'un groupe N-nitroso acyclique. Dans chaque chaîne, il ne peut y avoir plus de 4 atomes dans le même cycle. | 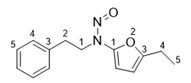 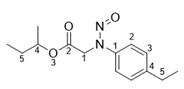 |
+1 |
| Groupe électroattracteurNote de bas de page ** lié au carbone α d'un seul côté du groupe N-nitroso (cyclique ou acyclique) |  |
+1 |
| Groupes électroattracteursNote de bas de page ** liés à des carbones α de part et d'autre du groupe N-nitroso (cyclique ou acyclique) |  |
+2 |
| Groupe hydroxyleNote de bas de page *** lié au carbone β d'un seul côté du groupe N-nitroso (cyclique ou acyclique) |  |
+1 |
| Groupe hydroxyle lié au carbone β de part et d'autre du groupe N-nitroso (cyclique ou acyclique) |  |
+2 |
|
||
Tableau 4. Liste des caractéristiques activatrices et des scores connexes. Pour calculer le score des caractéristiques activatrices, on doit additionner les scores individuels de toutes les caractéristiques énumérées présentes dans la structure de la N-nitrosamine. Chaque ligne de caractéristique activatrice dans le tableau ne peut être comptée qu'une seule fois. Ces exemples ne se veulent pas exhaustifs.
Caractéristique activatrice |
Exemple | Score des caractéristiques activatrices individuelles |
|---|---|---|
| Groupe aryle lié au carbone α (c'est-à-dire substituant benzylique ou pseudo-benzylique sur le groupe N-nitroso) |  |
-1 |
| Groupe méthyle lié à un carbone β (cyclique ou acyclique) |  |
-1 |
Annexe B. Exemples de calculs pour la catégorisation de la puissance cancérogène d'après un diagramme de flux
Exemple 1 – N-nitroso-félodipine
L'exemple 1 montre comment le diagramme de flux de l'approche de catégorisation de la puissance cancérogène (figure 2) peut être appliqué à la N-nitrosamine « N-nitroso-félodipine ». La N-nitroso-félodipine est classée dans la catégorie de puissance 5 avec une limite acceptable recommandée de 1 500 ng/jour.
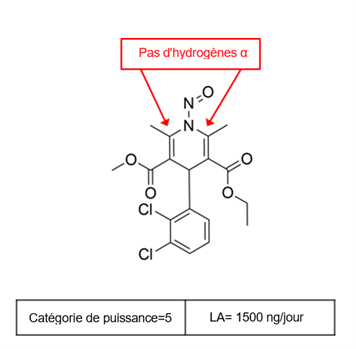
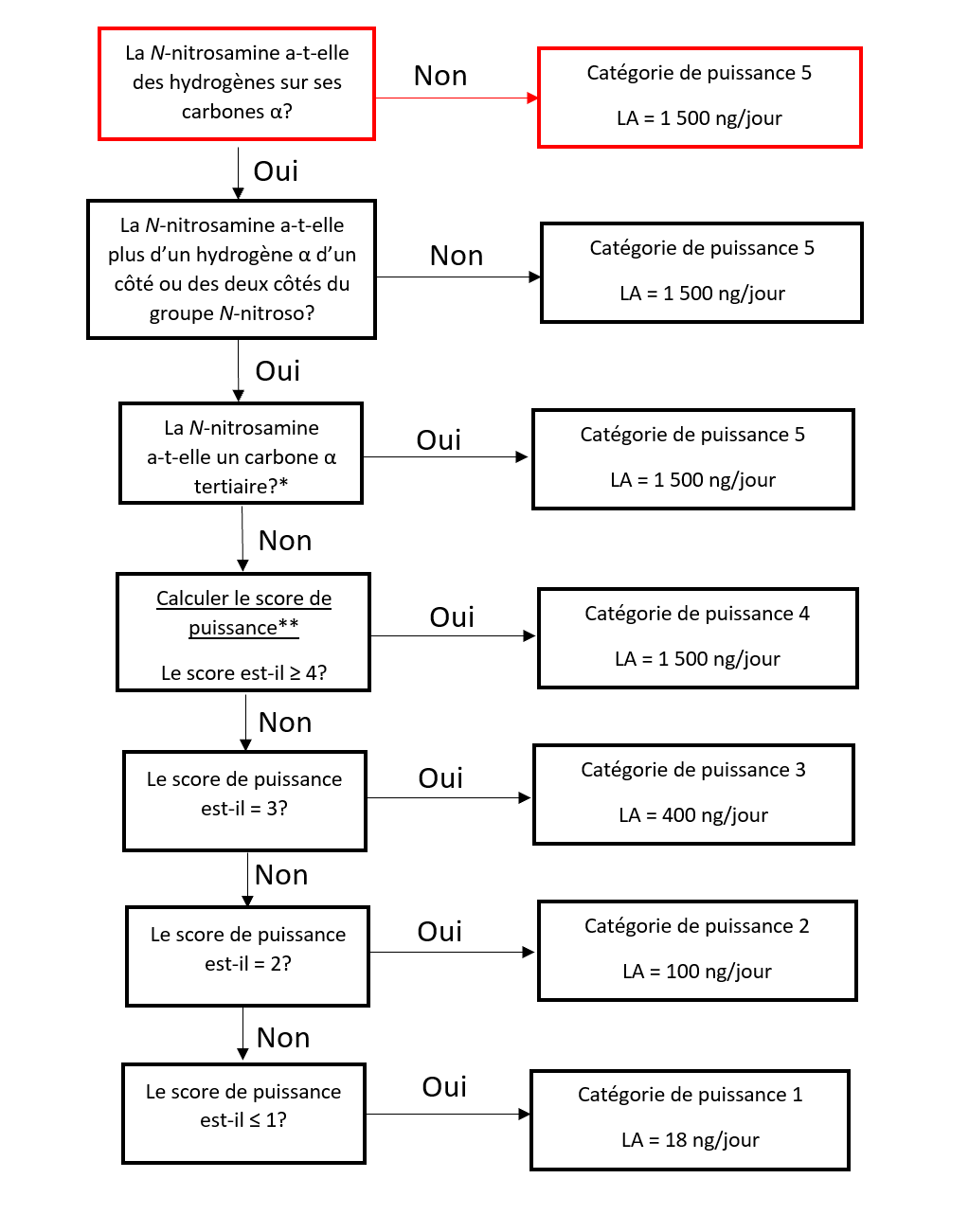
Exemple 1 - Équivalent textuel
La figure de l'exemple 1 montre un processus étape par étape d'attribution d'une catégorie de puissance pour la N-nitroso-félodipine. L'exemple montre comment les étapes ont mené à la catégorie de puissance 5.
Exemple 2 – N-nitroso-énalapril
L'exemple 2 montre comment le diagramme de flux de l'approche de catégorisation de la puissance cancérogène (figure 2) peut être appliqué à la N-nitrosamine « N-nitroso-énalapril ». Le N-nitroso-énalapril est classé dans la catégorie de puissance 5 avec une limite acceptable recommandée de 1 500 ng/jour.
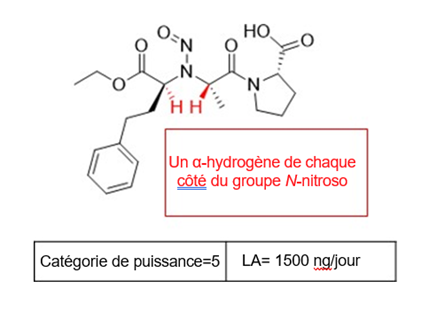
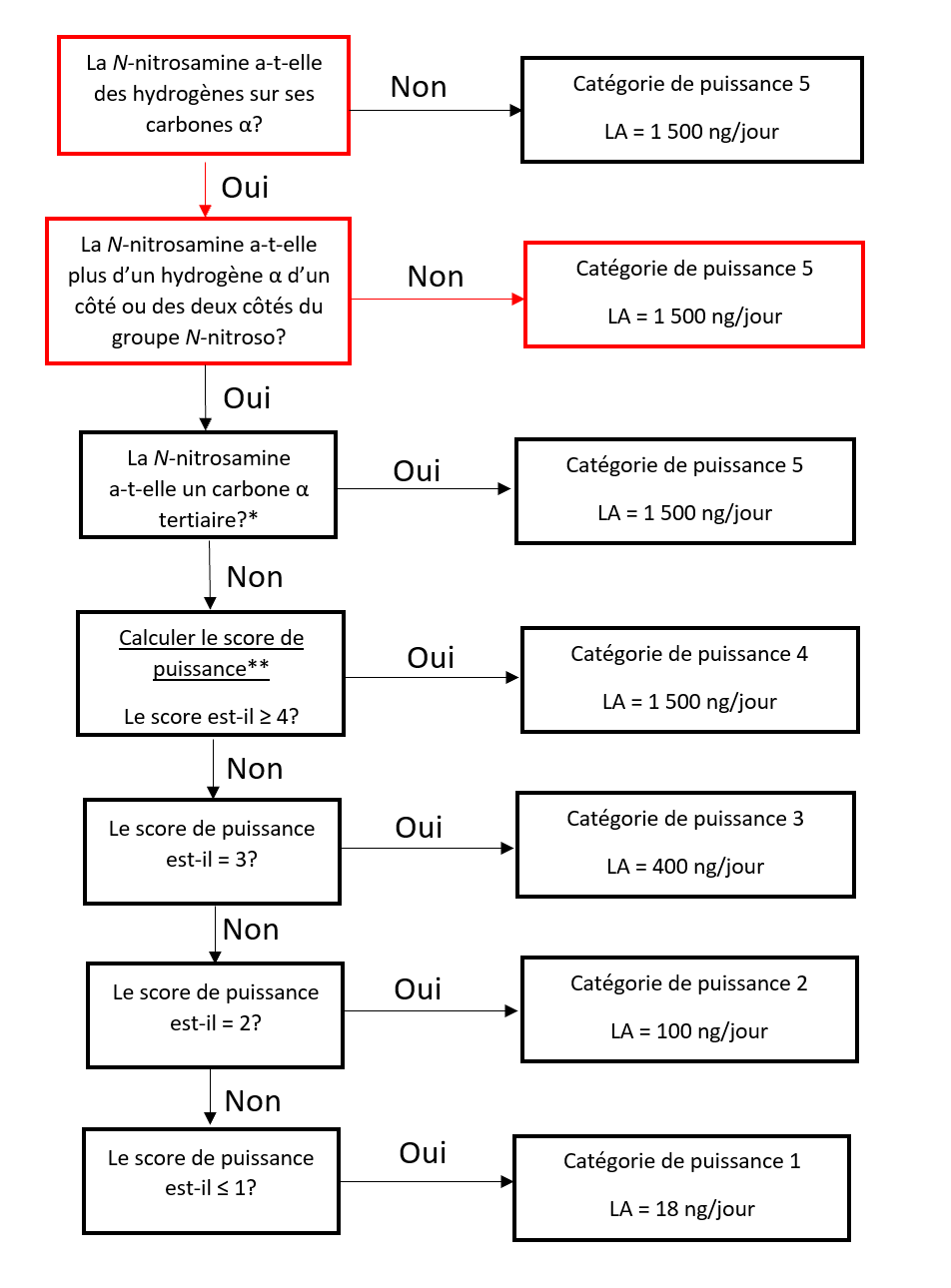
Exemple 2 - Équivalent textuel
La figure de l'exemple 2 montre un processus étape par étape d'attribution d'une catégorie de puissance pour la N-nitroso-énalapril. L'exemple montre comment les étapes ont mené à la catégorie de puissance 5.
Exemple 3 – N-nitroso-kétamine
L'exemple 3 montre comment le diagramme de flux de l'approche de catégorisation de la puissance cancérogène (figure 2) peut être appliqué à la N-nitrosamine « N-nitroso-kétamine ». La N-nitroso-kétamine est classée dans la catégorie de puissance 5 avec une limite acceptable recommandée de 1 500 ng/jour.
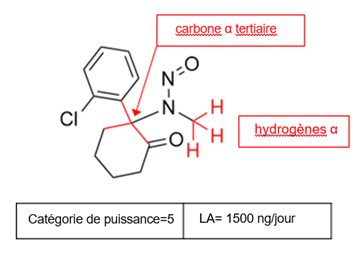
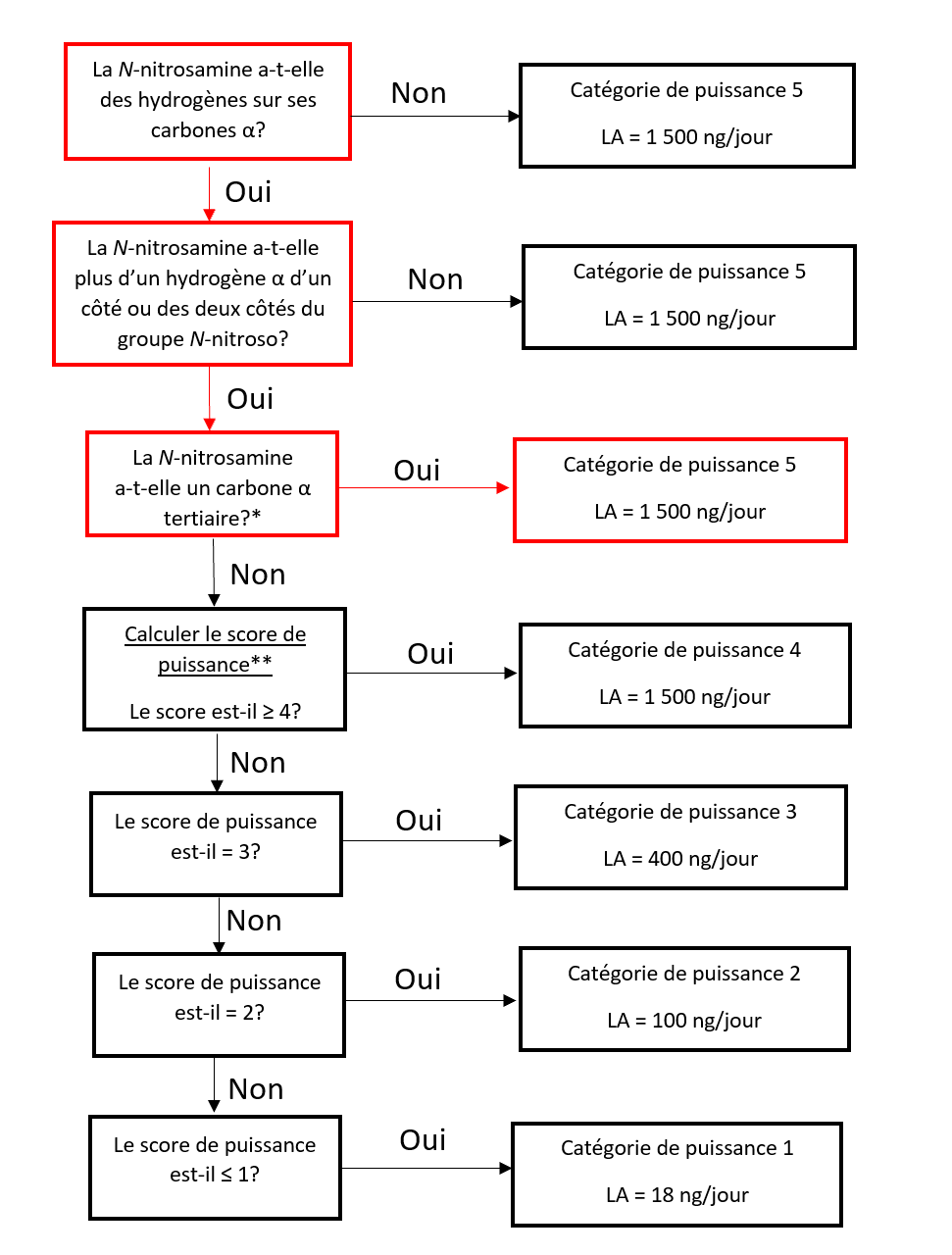
Exemple 3 - Équivalent textuel
La figure de l'exemple 3 montre un processus étape par étape d'attribution d'une catégorie de puissance pour la N-nitroso-kétamine. L'exemple montre comment les étapes ont mené à la catégorie de puissance 5.
Exemple 4 – N-nitroso-nébivolol
L'exemple 4 montre comment le diagramme de flux de l'approche de catégorisation de la puissance cancérogène (figure 2) peut être appliqué à la N-nitrosamine « N-nitroso-nébivolol ». Le N-nitroso-nébivolol est classé dans la catégorie de puissance 4 avec une limite acceptable recommandée de 1 500 ng/jour.
Nombre d'Hydrogènes α |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| 2,2 | 1 |  |
Caractéristiques de Désactivation |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| Groupe hydroxyle lié à des β-carbones*** de part et d'autre du groupe N-nitroso (cyclique ou acyclique) | +2 |  |
| Chaîne de ≥ 5 atomes d'hydrogène consécutifs (cyclique ou acyclique) de part et d'autre d'un groupe N-nitrosé acyclique. Pas plus de 4 atomes dans chaque chaîne ne peuvent se trouver dans le même anneau. | +1 |  |
Aucune Caractéristique d'Activation Présente |
Score de Puissance= 1+2+1=4 |
Catégorie de puissance 4 |
LA= 1 500 ng/jour |
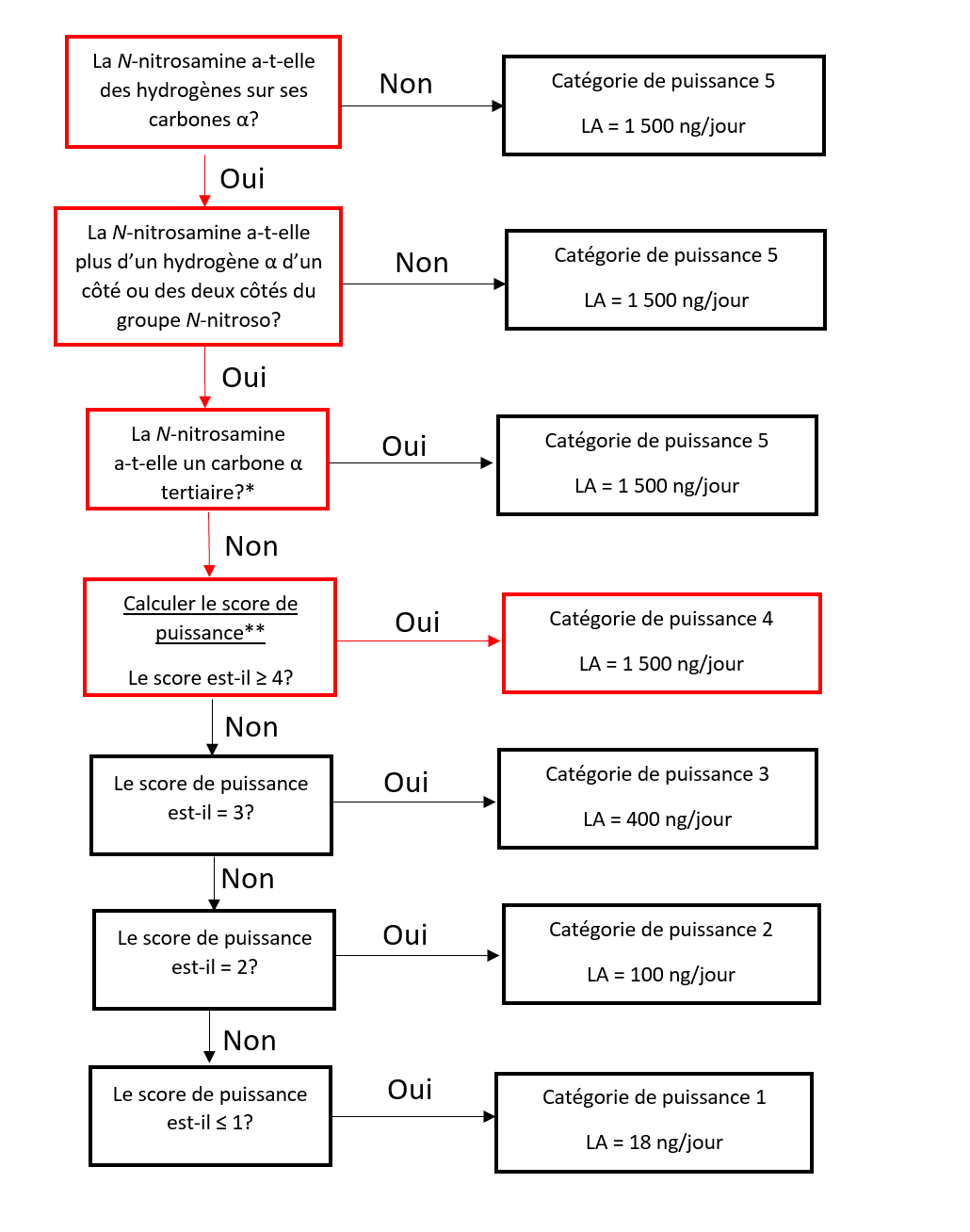
Exemple 4 - Équivalent textuel
La figure de l'exemple 4 montre un processus étape par étape d'attribution d'une catégorie de puissance pour la N-nitroso-nébivolol. L'exemple montre comment les étapes ont mené à la catégorie de puissance 4.
Exemple 5 – N-nitroso-méropénème
L'exemple 5 montre comment le diagramme de flux de l'approche de catégorisation de la puissance cancérogène (figure 2) peut être appliqué à la N-nitrosamine « N-nitroso-méropénème ». Le N-nitroso-méropénème est classé dans la catégorie de puissance 4 avec une limite acceptable recommandée de 1 500 ng/jour.
| Nombre d'Hydrogènes α | Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| 1,2 | 3 |  |
Caractéristiques de Désactivation |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| Groupe acide carboxylique à n'importe quel endroit de la molécule | +3 |  |
| Groupe N-nitroso dans un cycle pyrrolydine | +3 |  |
| Groupe électroattracteur lié à un carbone a d'un seul côté du groupe N-nitroso (cyclique ou acyclique) | +1 | 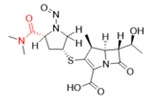 |
Aucune Caractéristique d'Activation Présente |
Score de Puissance= 3+3+3+1=10 |
Catégorie de puissance 4 |
LA= 1500 ng/jour |
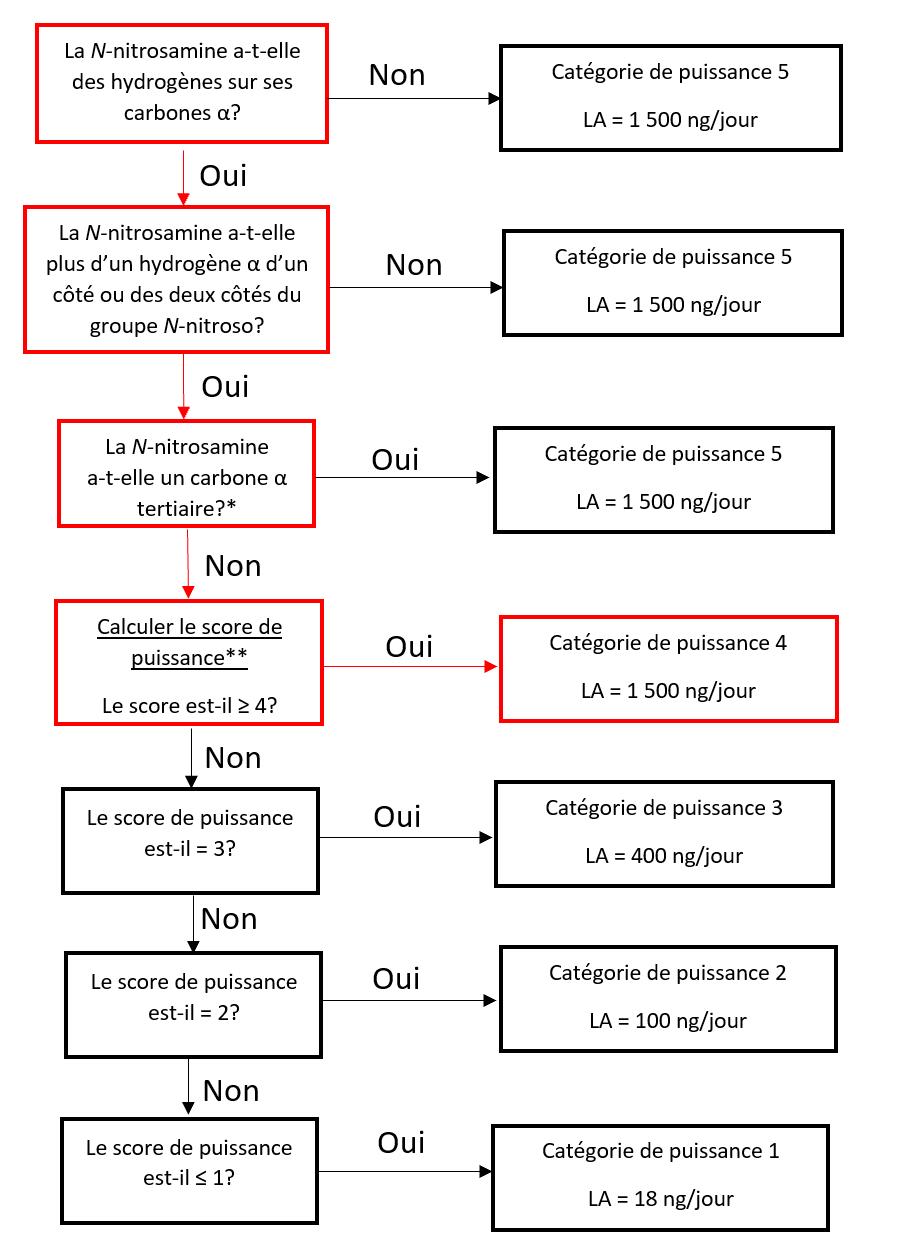
Exemple 5 - Équivalent textuel
La figure de l'exemple 5 montre un processus étape par étape d'attribution d'une catégorie de puissance pour la N-nitroso-méropénème. L'exemple montre comment les étapes ont mené à la catégorie de puissance 4.
Exemple 6 – N-nitroso-desloratadine
L'exemple 6 montre comment le diagramme de flux de l'approche de catégorisation de la puissance cancérogène (figure 2) peut être appliqué à la N-nitrosamine « N-nitroso-desloratadine ». La N-nitroso-desloratadine est classée dans la catégorie de puissance 3 avec une limite acceptable recommandée de 400 ng/jour.
Nombre d'Hydrogènes α |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| 2,2 | 1 |  |
Caractéristiques de Désactivation |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| Groupe N-nitroso dans un cycle à 5 ou 6 chaînons | +2 |  |
Aucune Caractéristique d'Activation Présente |
Score de Puissance= 1+2=3 |
Catégorie de puissance 3 |
LA= 400 ng/jour |
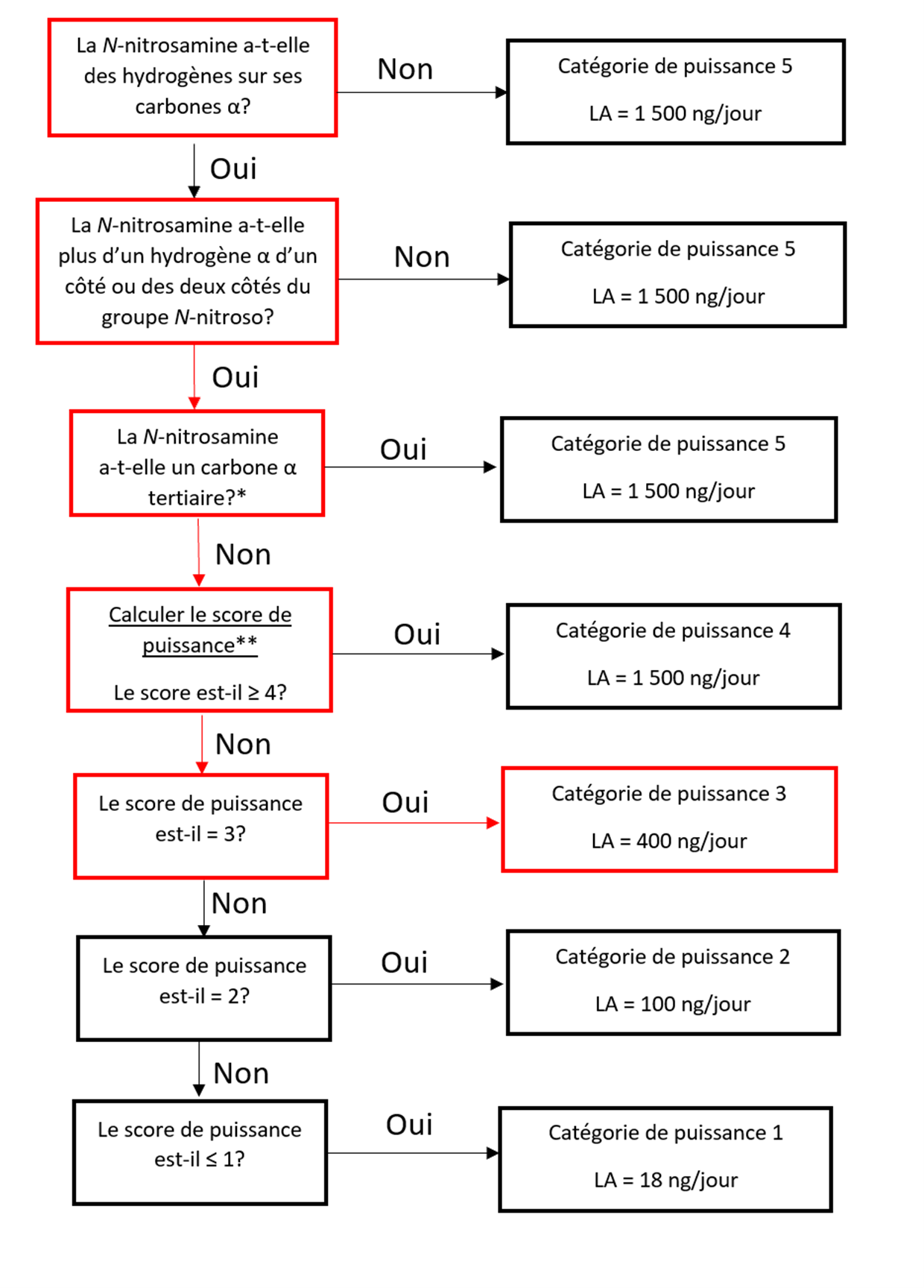
Exemple 6 - Équivalent textuel
La figure de l'exemple 6 montre un processus étape par étape d'attribution d'une catégorie de puissance pour la N-nitroso-desloratadine. L'exemple montre comment les étapes ont mené à la catégorie de puissance 3.
Exemple 7 – N-nitroso-sertraline
L'exemple 7 montre comment le diagramme de flux de l'approche de catégorisation de la puissance cancérogène (figure 2) peut être appliqué à la N-nitrosamine « N-nitroso-sertraline ». La N-nitroso-sertraline est classée dans la catégorie de puissance 2 avec une limite acceptable recommandée de 100 ng/jour.
Nombre d'Hydrogènes α |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| 1,3 | 3 |  |
Aucune Caractéristiques de Désactivation |
Caractéristique d'Activation Présente |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| Groupe N-nitroso dans un cycle à 5 ou 6 chaînons | -1 |  |
Score de Puissance= 3-1=2 |
Catégorie de puissance 2 |
LA= 100 ng/jour |
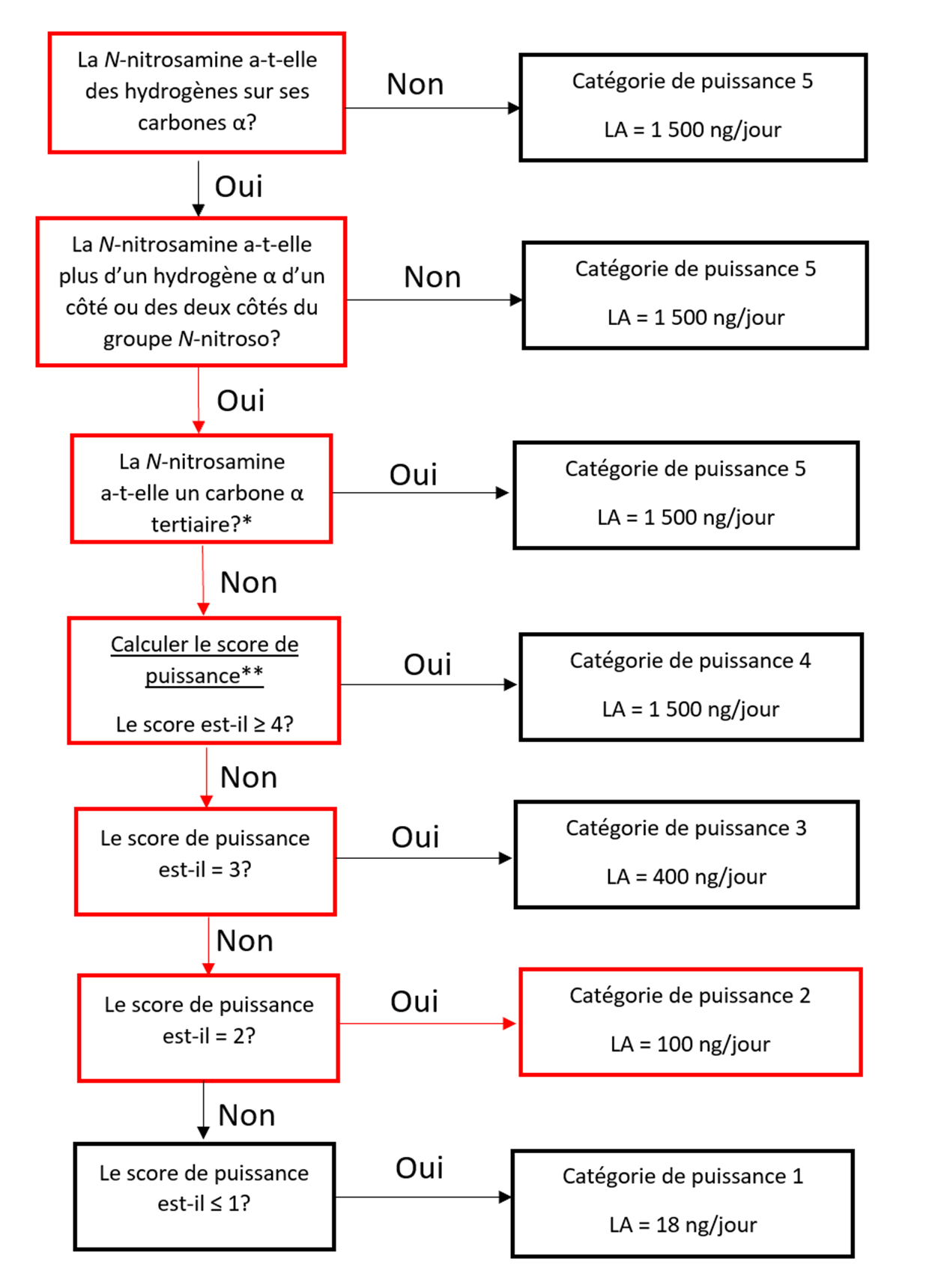
Exemple 7 - Équivalent textuel
La figure de l'exemple 7 montre un processus étape par étape d'attribution d'une catégorie de puissance pour la N-nitroso-sertraline. L'exemple montre comment les étapes ont mené à la catégorie de puissance 2.
Exemple 8 – N-nitroso-lorcasérine
L'exemple 8 montre comment le diagramme de flux de l'approche de catégorisation de la puissance cancérogène (figure 2) peut être appliqué à la N-nitrosamine « N-nitroso-lorcasérine ». La N-nitroso-lorcasérine est classée dans la catégorie de puissance 1 avec une limite acceptable recommandée de 18 ng/jour.
Nombre d'Hydrogènes α |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| 2,2 | 1 | 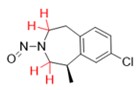 |
Caractéristiques de Désactivation |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| Groupe N-nitroso dans un cycle à 7 chaînons | +1 | 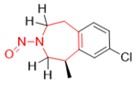 |
Aucune Caractéristique d'Activation Présente |
Score |
Caractéristique Surlignée en Rouge |
|---|---|---|
| Groupe méthyle lié au carbone β (cyclique ou acyclique) | -1 | 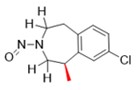 |
| Score de Puissance= 1+1-1=1 | Catégorie de puissance 1 | LA= 18 ng/jour |
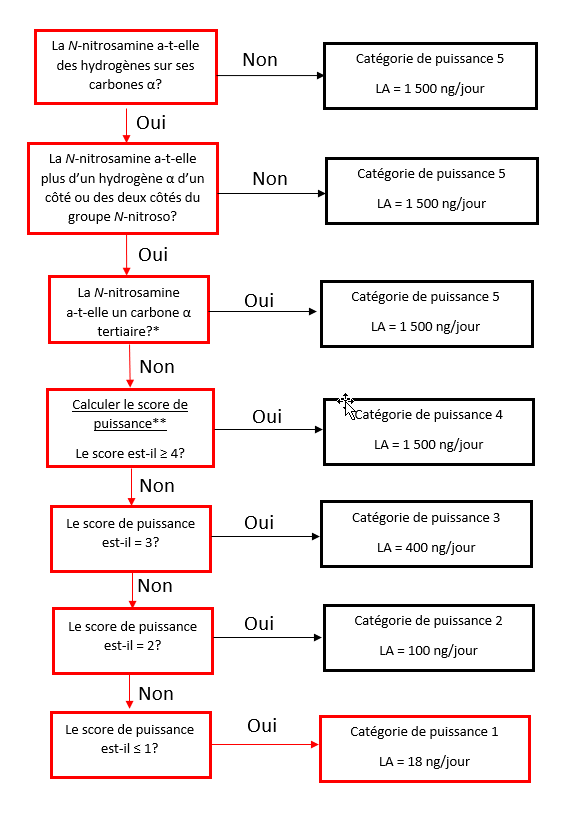
Exemple 8 - Équivalent textuel
La figure de l'exemple 8 montre un processus étape par étape d'attribution d'une catégorie de puissance pour la N-nitroso-lorcasérine. L'exemple montre comment les étapes ont mené à la catégorie de puissance 1.
Notes de bas de page
- Note de bas de page 1
-
Voir, par exemple, Cross KP et Ponting DJ, 2021. Developing Structure-Activity Relationships for N-NitrosamineActivity, Comput Toxicol, 20:100186; Thomas R, Tennant RE, Oliveira AAF, et Ponting DJ, 2022. What makes a Potent Nitrosamine? Statistical Validation of Expert-Derived Structure-Activity Relationships, Chem Res Toxicol, 35:1997-2013; et Ponting DJ, Dobo KL, Kenyon MO, et Kalgutkar AS, 2022. Strategies for Assessing Acceptable Intakes for Novel N-NitrosaminesDerived From Active Pharmaceutical Ingredients, J Med Chem, 65:15584-15607.
- Note de bas de page 2
-
Voir la Base de données sur la cancérogénicité de Lhassa https://carcdb.lhasalimited.org/
- Note de bas de page 3
-
Rao TK, Young JA, Lijinsky W et Epler JL, 1979. Mutagenicity of Aliphatic Nitrosamines in Salmonella typhimurium, Mutat Res, 66:1-7.
- Note de bas de page 4
-
Questions et réponses pour les détenteurs/demandeurs d'autorisation de mise sur le marché concernant l'avis du CHMP sur la saisine au titre de l'article 5, paragraphe 3.
- Note de bas de page 5
-
Li Y, Hecht SS, 2022. Metabolic Activation and DNA Interactions of Carcinogenic N-Nitrosamines to Which Humans Are Commonly Exposed, Int J Mol Sci, 23:4559.
- Note de bas de page 6
-
Pour les N-nitrosaminescontenant plus de deux groupes N-nitroso, le demandeur ou le fabricant doit communiquer avec l'organisme de réglementation des médicaments concerné pour obtenir des conseils supplémentaires.
Détails de la page
- Date de modification :