
Guidance Document: Conduct and Analysis of Comparative Bioavailability Studies
Date Adopted: 2012/02/08
Revised Date: 2023/01/30
Effective Date: 2018/09/01 (for submissions filed on or after September 1, 2018)
Document Change Log
Date: June 8, 2018
Change: Addition of a sentence at the end of the third paragraph.
“The choice of a variance balanced design (Williams’ Design) or separate incomplete block design should be justified.”
Location (section, paragraph): Section 2.3.1
Nature of and/or reason for change: Clarification of statistical requirements.
Change: Addition of section 2.3.3 Pharmacodynamic Studies
Location (section, paragraph): Section 2.3.3
Nature of and/or reason for change: Some information transferred from the Standards guidance since the information was more relevant to study design than to standards.
Change: Modification of the second paragraph
Location (section, paragraph): Section 2.3.5
Nature of and/or reason for change: Editorial clarification of the language.
Change: Removal of the sentences regarding sampling of urine.
Location (section, paragraph): Section 2.4.6
Nature of and/or reason for change: Urine data is no longer used for assessment of comparative bioavailability and the information is therefore no longer relevant.
Change: Removal of two sentences in the first paragraph regarding urine as a biological sample.
Location (section, paragraph): Section 2.4.7
Nature of and/or reason for change: Urine data is no longer used for assessment of comparative bioavailability and the information is therefore no longer relevant.
Change: Removal of a paragraph regarding urine collection and reporting.
Location (section, paragraph): Section 2.4.7
Nature of and/or reason for change: Urine data is no longer used for assessment of comparative bioavailability and the information is therefore no longer relevant.
Change: Modification of the following sentence:
“Sometimes the concentration of drug in a fluid other than blood or urine may correlate better with effect.”
Location (section, paragraph): Section 2.4.7
Nature of and/or reason for change: Urine data is no longer used for assessment of comparative bioavailability and the information is therefore no longer relevant.
Change: Removal of a paragraph regarding metabolite concentrations in the urine not considered an acceptable assessment of bioequivalence.
Location (section, paragraph): Section 2.6.1
Nature of and/or reason for change: Urine data is no longer used for assessment of comparative bioavailability and the information is therefore no longer relevant.
Change: Modification of the following sentence.
“The bioanalytical methods used to measure the drug, or metabolite, in plasma, blood or serum, or urine should be suitable for their intended purpose.”
Location (section, paragraph): Section 2.6.2
Nature of and/or reason for change: Urine data is no longer used for assessment of comparative bioavailability and the information is therefore no longer relevant.
Change: Addition of the following paragraph:
“For more detailed expectations on stability experiments to be conducted during bioanalytical method validation, please refer to the Notice: Clarification of bioanalytical method validation procedures (October 8, 2015: Addendum, March 9, 2016). This document is available on the Health Canada website.”
Location (section, paragraph): Section 2.6.2
Nature of and/or reason for change: Updating reference to the European Medicines Agency guidelines.
Change: Removal of pharmacokinetic parameters l) and m) as well as the following paragraph:
Where comparative bioavailability is based upon urine data, the following parameters should be reported:
Location (section, paragraph): Section 2.7.2
Nature of and/or reason for change: Urine data is no longer used for assessment of comparative bioavailability and the information is therefore no longer relevant.
Change: Addition of the following after point k)
Where the multiphasic plasma concentration profile of a modified-release product has been demonstrated to be integral to the therapeutic effect, the following parameter should be reported:
l) Area under the concentration versus time curve over a defined time interval after drug administration (pAUC).
Location (section, paragraph): Section 2.7.2
Nature of and/or reason for change: Introduction of partial AUC parameter for multiphasic modified-release dosage forms.
Change: Addition of a line in Table A1-D after AUCReftmax with the following definition:
pAUC: Partial area under the curve over a defined time interval after drug administration.
Location (section, paragraph): Section A1.3
Nature of and/or reason for change: Definition needed for new criterion (partial AUC) introduced for multiphasic modified-release products.
Change: Removal of the following:
Ae0-T - Cumulative amount of drug excreted in the urine, measured to the last sampling time.
Rmax - Maximum rate of urinary drug excretion.
Location (section, paragraph): Appendix 2
Nature of and/or reason for change: Urine data is no longer used for assessment of comparative bioavailability and the information is therefore no longer relevant.
Change: Minor addition and revision to the definition of “Modified-release dosage form” to include multiphasic drug product formulations.
From:
To provide, after single administration, multiple peaks and troughs in the serum concentration-time curves similar to those achieved after repeated dosing with the conventional formulation (i.e., multiphasic modified-release dosage forms).
To:
To provide, after single administration, multiple peaks and troughs concentration-time curves (i.e., multiphasic modified-release dosage forms).
Location (section, paragraph): Appendix 2
Nature of and/or reason for change: Multiphasic modified-release products now addressed in this document.
Change: Addition of the term pAUC
Location (section, paragraph): Appendix 2
Nature of and/or reason for change: Criterion related to multiphasic modified-release products now addressed in this document.
Date: January 30, 2023
Change: Update reference to guidance on bioanalytical method validation
Location (section, paragraph): Section 2.6, 3rd paragraph and section 2.6.2 second paragraph
Nature of and/or reason for change: EMA guidance on bioanalytical method validation and related Health Canada notice superseded by ICH M10 guidance
Foreword
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable guidance documents.
Table of Contents
- 1 Introduction
- 1.1 Policy Objectives
- 1.2 Policy Statements
- 1.3 Scope and Application
- 1.4 Background
- 2 Guidance for Implementation
- 2.1 Planning a Comparative Bioavailability Study
- 2.2 Selection of Subjects for a Study
- 2.2.1 Choice of Subjects
- 2.2.2 Inclusion / Exclusion Criteria
- 2.3 Study Design
- 2.3.1 Parallel versus Cross-over
- 2.3.1.1 Number of Subjects
- 2.3.2 Alternate Study Designs
- 2.3.2.1 Group Sequential Designs
- 2.3.2.2 Adaptive Designs
- 2.3.3 Pharmacodynamic studies
- 2.3.4 Accounting for Drop-outs and Withdrawals
- 2.3.5 Outlier Consideration
- 2.3.1 Parallel versus Cross-over
- 2.4 Study Conduct
- 2.4.1 Standardization
- 2.4.2 Blinding
- 2.4.3 Administration of Food and Fluid
- 2.4.3.1 Fasted Study
- 2.4.3.2 Fed Study
- 2.4.4 Posture and Physical Activity
- 2.4.5 Interval Between Doses
- 2.4.6 Sampling Times
- 2.4.7 Sample Collection
- 2.4.8 Handling of Samples
- 2.4.9 Identification of Adverse Events
- 2.5 Test and Reference Drug Products
- 2.5.1 Chemistry
- 2.5.2 Dosage and Strength
- 2.5.3 Selection of Reference Product
- 2.6 Bioanalytical Methodology
- 2.7 Analysis of Data
- 2.7.1 Presentation of Data
- 2.7.2 Pharmacokinetic Parameters
- 2.7.3 Data Collection
- 2.7.4 Statistical Analysis
- 2.7.4.1 Outlier analysis
- 2.7.4.2 Model Fitting
- 2.7.4.3 Testing of Fixed Effects
- 2.7.4.4 Estimation of Random Effects
- 2.7.4.5 Analysis of Data
- Appendix 1: Sample Analysis for a Comparative Bioavailability Study
- Appendix 2: Glossary of Terms
1 Introduction
1.1 Policy Objectives
To provide sponsors of new drug submissions with the information necessary to comply with Sections C.08.002(2)(h), C.08.002.1(2)(c)(ii) and C.08.003(3) of the Food and Drug Regulations (Regulations) with respect to comparative bioavailability studies used in support of the safety and efficacy of a drug.
1.2 Policy Statements
Comparative bioavailability studies should be conducted in accordance with generally accepted clinical practices that are designed to ensure the protection of the rights, safety and well-being of subjects and the good clinical practices referred to in Division 5 of the Regulations and described in the International Conference on Harmonisation (ICH) Guidance (Topic E6) on Good Clinical Practice. The principles of Good Manufacturing Practice should be adhered to wherever applicable, as indicated in Part C, Division 2 of the Regulations.
The recommendations included in this guidance respecting study design and conduct, validation of bioanalytical methodology and statistical analysis of data should be followed in order to ensure compliance with the Regulations.
1.3 Scope and Application
This guidance is intended to be applied to all comparative bioavailability studies which provide pivotal evidence of the safety and efficacy of a product, with the exception of subsequent-entry biologic products. Examples of cases where this guidance applies are:
- comparative bioavailability studies in support of the bioequivalence of subsequent-entry products to the Canadian Reference Product;
- bridging studies where the formulation to be marketed is different from the formulation used in the pivotal clinical trials;
- studies in support of significant post-approval changes and line extensions;
- safety studies for non-systemic drugs, where systemic drug concentrations may be measured for safety assessment of products with drugs that are intended to act locally, for example, drugs administered by metered-dose inhaler.
- comparative bioavailability studies in support of Drug Identification Number (DIN) Applications.
While this guidance is oriented toward solid oral dosage formulations, both immediate- and modified-release, the principles and standards described may also be applied, as appropriate, to other oral dosage forms and non-injectable formulations such as transdermal patches, suppositories, etc. that are intended to deliver medication to the systemic circulation.
This guidance document should be read in conjunction with the associated Health Canada document entitled: Comparative Bioavailability Standards: Formulations Used for Systemic Effects.
1.4 Background
Bioavailability is an important attribute of formulations of drugs used for systemic effects. It is defined as the rate and extent of drug entry into the systemic circulation.
Bioavailability is most frequently assessed by serial measurements of the drug in the systemic circulation. These serial measurements provide a plasma, serum or whole blood concentrationtime profile from which a number of important pharmacokinetic parameters can be calculated, including the area under the curve (AUC), the maximum observed concentration (Cmax) and the time when Cmax is reached (tmax). The AUC represents a surrogate of the amount of drug absorbed into the systemic circulation. Both tmax and Cmax are complex functions that position the point in time when the rate of input and loss are the same. Despite a lack of robustness for these parameters, it is commonly viewed that Cmax is a reasonable metric to define absorption rate. For many drugs, AUC and Cmax together can characterize the concentration-time profile for comparative purposes.
Comparison of the AUC values following oral versus intravenous administration of an equivalent dose of the same active ingredient provides an estimate of absolute bioavailability for most drugs. Comparison of the plasma concentration-time profiles of the drug between the test and reference products containing the same amount of the same active ingredient(s) provides an estimate of relative bioavailability.
If the test and reference products are comparable dosage forms and contain identical amounts of the identical medicinal ingredient(s), they are said to be bioequivalent when the profiles of the drug are similar. The degree of similarity between the profiles needed to establish bioequivalence is determined by the appropriate statistical assessment and by meeting standards established for the particular drug and formulations being compared (refer to Health Canada guidance document: Comparative Bioavailability Standards: Formulations Used for Systemic Effects).
Bioequivalence implies that the test product can be expected to have the same therapeutic effects and safety profile as the reference product when administered to patients under the conditions specified in the labelling.
In the absence of an adequate methodology for bioavailability testing, alternative approaches such as pharmacodynamic studies can be used. In some instances, equivalence may have to be determined by clinical trials with therapeutic end-points.
2 Guidance for Implementation
The acceptability of data from comparative bioavailability studies will be assessed in accordance with the principles enunciated in Division 5 of the Regulations and the ICH Guidance (Topic E6) on Good Clinical Practice. These documents will help sponsors to understand the requirements for submissions to Health Canada, pursuant to the Regulations, even if the studies or portions thereof are conducted in other countries.
2.1 Planning a Comparative Bioavailability Study
The objectives of the study should be clearly defined in the protocol.
A rationale should be provided to justify which bioequivalence standards will be applied. Scientific justification should be provided for any deviation from the guidance set out in this document (for example (e.g.), analyte upon which bioequivalence will be assessed, or deviation from a high fat/high calorie meal in studies conducted under fed conditions). Sponsors are encouraged to consult with Health Canada, in advance of the study, if deviations are substantial.
Among the topics covered by the Regulations and the ICH guidance on Good Clinical Practice, and therefore not repeated in detail here are: Institutional review boards, investigators, and clinical, laboratory and bioanalytical facilities.
2.2 Selection of Subjects for a Study
In general, subjects should be selected so as to reduce (1) risk to study subjects and (2) inter- and intra-subject variability that is not attributable to the drug itself.
2.2.1 Choice of Subjects
To minimize variability, comparative bioavailability studies are usually conducted with normal, healthy volunteers (male and/or female). It is generally accepted that conclusions regarding comparative bioavailability, drawn from studies with healthy volunteers, can be expected to hold in the patient population. It is more difficult to conduct cross-over comparative bioavailability studies in patients, in part due to potential disease progression. In some cases, it may be necessary to conduct studies in patients who are already receiving the drug (e.g., when the drug safety profile precludes administration to healthy volunteers). The variability of the disease states in study patients will be an important consideration in deciding the size of the cohort needed to satisfy the standards.
2.2.2 Inclusion / Exclusion Criteria
The following attributes should be addressed to reduce pharmacokinetic variability not related to differences between products and to prevent undue harm to study subjects.
- Age
Subjects should be between the age of legal majority and the age of onset of ageassociated changes in organic function. This description typically coincides with an age range of 18 to 55 years, inclusive. - Height and Weight
Subjects should preferably have a Body Mass Index within 18.5 and 30 kg/m². - Health
The health of the volunteers should be determined by the supervising physician through a medical examination including a review of medical history and the results of routine tests of liver, kidney, and hematological functions. Aberrant laboratory values should be rechecked and a summary should be presented along with the physician's opinion as to their potential impact on the study's conclusions.
Testing for alcohol and drugs of abuse should be conducted prior to drug administration in each period. - Safety
An electrocardiogram (ECG) should be included in the study documentation if the drug is known to cause ECG changes.
Subjects who have any significant systemic illness or unstable medical condition which could lead to difficulty complying with the protocol, should be excluded.
The investigators should ensure that female volunteers are not pregnant, lactating, or likely to become pregnant during the study. Confirmation regarding pregnancy should be obtained by urine or serum tests prior to drug administration in each period.
2.3 Study Design
2.3.1 Parallel versus Cross-over
The standard study design used is a two-period cross-over, in which each subject is given the test and reference formulations. The advantage of the cross-over design is that the intra-subject error is used in the construction of the confidence intervals for comparing mean differences; the intra-subject error is always lower than the inter-subject error used in a parallel design.
Replicated cross-over designs may also be used, where the formulations are tested more than once in the same subjects. The main advantage of these designs is that fewer subjects are required; however, they must appear for more periods.
In cases where more than two formulations are under study, or are studied under different conditions, a higher order design (that is (i.e.), more periods and sequences) should be considered. Since the intra-subject error term of these designs has more degrees of freedom, smaller sample sizes are often adequate. The choice of a variance balanced design (Williams’ Design) or separate incomplete block design should be justified.
A cross-over design without a drug-free period between formulations may be employed for studies conducted in patients in whom it would be unethical to discontinue treatment during a washout period. Instead of a drug-free washout period, the study drugs are administered long enough, prior to sampling, to allow elimination of the previously administered formulation.
Parallel designs may be useful when studying drugs with very long elimination half-lives or some depot formulations. The error term used is the inter-subject variance.
2.3.1.1 Number of Subjects
The number of subjects to be used in a comparative bioavailability study should be estimated by considering the objectives of the study, the study design, the drug products being compared and the conditions under which the study is carried out. The drug and drug product determine the particular standard which needs to be met. A complete literature search should be conducted in order to understand the drug and drug product. The standard, the expected mean difference between the test and reference formulations and the anticipated intra-subject variance for the parameters stated in the standard, as well as the power, determine the number of subjects. All calculations are to be based on maintaining the overall Type I error rate at 5%. The minimum number of subjects is 12, but a larger number is usually required.
2.3.2 Alternate Study Designs
When the proposed estimate of the intra-subject variance from the literature has large uncertainty, it is possible to collect the data in stages based on the observed intra-subject variance from the first stage. Two strategies for collecting data in stages are Group Sequential Designs and Adaptive Designs. For both types of designs the overall Type I error rate should be maintained at 5% and the algorithm should be defined a priori in the protocol. These approaches can be used for both cross-over and parallel designs.
2.3.2.1 Group Sequential Designs
Collection of data under a group sequential design is based on fixed sample sizes (Ni) at each ith stage. It is recommended that only two stages be used since these trials are very small compared to clinical outcome studies. The first stage N1 is generally based on the most likely intra-subject variance estimate with some added subjects to protect against drop-outs. The additional subjects required for the second stage N2 is usually based on a worst-case scenario using a larger intrasubject variance estimate, such that N1 plus N2 is equal to the estimated sample size for the larger intra-subject variance. Usually the strategy with this design is to accept bioequivalence at the first stage and only go to the second stage when the intra-subject variance from the first stage is very large. It is recommended to use the same alpha for both stages based on the method by Pocock (SJ Pocock. Group sequential methods in the design and analysis of clinical trials Biometrika 1977; 64(2): 191-199), which gives an alpha of 0.0294 for this case. This method precludes the need for a stage effect in the model.
2.3.2.2 Adaptive Designs
When there is very little information on the intra-subject variance, another approach similar to the sequential design is the adaptive design where the second stage sample size is based on the estimated intra-subject variance from the first stage. Method C in Potvin et al. (D. Potvin et al. Sequential design approaches for bioequivalence studies with crossover designs Pharmaceut. Statist. 2008; 7: 245-262) is recommended.
2.3.3 Pharmacodynamic studies
In cases where pharmacokinetic endpoints cannot be reliably measured, it may be acceptable to establish in vivo comparability using pharmacodynamic studies. The use and design of pharmacodynamic studies should be justified.
The design of pharmacodynamic studies should take into consideration the underlying pathology and natural history of the condition being treated. If baseline conditions are not reproducible, it may be necessary to use a parallel-group design rather than a cross-over design as normally preferred. Patients who are non-responders should be excluded from the study by prior screening, based on identifying criteria stated in the protocol. Any important placebo effects should also be considered, as comparisons between drug products can be made only after a priori consideration of such effects in the study design. A placebo cross-over phase may be necessary to evaluate placebo effects.
The doses used in pharmacodynamic studies should be selected to produce a range of response values that support a thorough characterization of the response over the time post-dose, as well as the evaluation of differences between the test and reference products. Neither the test nor the reference product should produce a maximal response in the course of the study, since it may be impossible to distinguish differences between formulations given in doses producing maximum or near-maximum effects. The investigation of dose-response relationships may be a necessary part of the design.
The pharmacodynamic endpoint measured should be a pharmacological or therapeutic effect that is relevant to the claims of efficacy. The response should be measurable using quantitative methods that have been validated for precision, accuracy, reproducibility, and specificity. Repeated measurements of the response over time should be made under double-blind conditions in an instrument-produced or instrument-recorded fashion to provide a record of the pharmacodynamic events which are substitutes for plasma concentrations. In instances where such measurements are not possible, recordings on visual analogue scales may be used. In other instances where the data are limited to qualitative (categorized) measurements, special statistical analysis will be required.
2.3.4 Accounting for drop-outs and withdrawals
A fixed number of subjects, in addition to the number estimated by the sample size calculation, should be recruited into the study. This strategy allows for possible drop-outs. All subjects who provide evaluable data for both test and reference products in a cross-over study, or for one treatment in a parallel study, should be included in the statistical analysis.
Reasons for withdrawal of subjects administered at least one dose of drug (e.g., adverse drug reaction) should be reported, and the subject's plasma (or blood or serum) concentration data should be provided. The results of the bioanalysis of all samples from subjects who were withdrawn from the study should be reported. If a subject withdraws from the study for personal reasons or because of non-compliance with the protocol (e.g., positive drug test) before completing at least two periods of the study, the subject’s blood samples do not have to be analysed.
Subjects who vomit should be evaluated for continued participation in the study based on the potential impact of the vomiting on the integrity of the study results. The evaluation should take place as soon as possible after the episode(s) of vomiting and before analysis of the study samples is initiated.
The concentration-time profiles of subjects who exhibit pre-dose concentrations higher than 5% of the corresponding Cmax should be excluded from the statistical analysis, provided the wash-out period between doses was appropriate. The concentration-time profiles of subjects who exhibit pre-dose concentrations equal to or less than 5% of the corresponding Cmax should be included in the statistical analysis without correction.
2.3.5 Outlier Consideration
Comparative bioavailability studies are small studies compared to other clinical trials. One or two extreme values could have a large effect on the inference to be made from these small studies. The usual parametric assumptions and estimation are not robust against extreme values.
Specific procedures to identify and account for outliers should be pre-specified in the protocol. No more than 5% of the subjects may be considered to be outliers, unless there are 20 or fewer subjects, in which case only 1 subject may be removed. Any protocol for handling outliers should be followed before the results of the analysis are summarised into confidence intervals (i.e., regardless of whether results meet the standard, the outlier protocol should be followed).
The protocol for handling outliers should include the following.
- The observation(s) should be identified by an outlier test. It is recommended that a simple outlier test such as a studentised residual being greater than 3 be used.
- The observation(s) should be outside the range of all the other observations regardless of formulation. In other words, the procedure should only identify observations which are very different from all others collected.
- The subject in question should be identified as an outlier for all parameters, for either the test or reference product, upon which the bioequivalence decision is to be based. Parameters of interest are usually an AUC and Cmax measure, but in some instances other parameters are required.
Re-testing of subjects identified as outliers is not recommended.
2.4 Study Conduct
2.4.1 Standardization
Every effort should be made to standardize the study conditions in every phase of the study; for example, study drug administration should occur at approximately the same time on each study day. Subject's posture, exercise, diet, smoking, and alcohol use should also be standardized. It is preferable to use non-smokers; where smokers are included, they should be identified.
Volunteers should not take any other drug, including non-prescription drugs, natural health products, alcoholic beverages or dietary items that have an effect on P450 enzymes and the PGP efflux pump (e.g., grapefruit juice and St. John's wort, respectively). These restrictions should be in place for an appropriate interval before and during the study. Protocol violations with respect to the use of restricted foods and health products should be reported (dose and time of administration). The decision on whether to include or exclude the results from a subject who has violated the protocol should be made before the statistical analysis starts.
2.4.2 Blinding
To avoid study bias, comparative bioavailability studies should be conducted in such a way that the subjects are not aware of which product (test or reference) is being administered. Furthermore, the persons checking for adverse reactions and those conducting the bioanalysis of samples should not know the treatment sequence.
2.4.3 Administration of Food and Fluid
If there is a documented serious safety risk to subjects from single-dose administration of the drug or drug product in either the absence or presence of food, then an appropriately designed study conducted in the indicated condition of use (fed or fasted state) may be acceptable for purposes of bioequivalence assessment. This approach should be scientifically justified a priori by the sponsor. If steady-state studies are conducted, the food and fluid conditions and restrictions noted below should apply on the preceding evening and on the day the plasma profiles are to be obtained.
2.4.3.1 Fasted Study
The administration of food and fluid should be controlled carefully. Normally, subjects should fast for 8 hours before drug administration. A fast means that no food or solids are to be consumed, although alcohol-free, xanthine-free and flavonoid-free clear fluids are permissible the night prior to the study. Water may be permitted up to one hour before drug administration. The dose should be taken with water of a standard volume (150 to 250 milliliters) and at a standard temperature. One hour after drug administration xanthine- and flavonoid-free fluids are permitted. Four hours after drug administration, a standard meal may be taken. All meals should be standardized and repeated on each day of drug administration.
When comparing the performance of two orally disintegrating dosage forms that are intended to be taken without water, the comparative bioavailability study should be designed to challenge the formulation under the most discriminatory conditions. For such dosage formulations, water should not be administered from one hour prior to dosing, concurrent with dosing and up to one hour post dosing.
For solid oral dosage forms with labelling that allows for alternate modes of administration (e.g., sprinkling on a soft food or dispersion in water) it is recommended that sponsors contact Health Canada prior to commencement of the study to verify the most appropriate mode of drug administration for the biostudy. To support the alternative administration options, additional data may also be required. For example, data should be provided to demonstrate that the technology used in the formulation is robust and that controlled-release properties, if any, are not altered during the proposed period of time by exposure to the foods or liquids specified in the labelling. The products should remain stable during the time of exposure. In addition, if the product is used in conjunction with an administration device, testing with the relevant device (e.g., various syringes and nasogastric tubes) may also be required to assess factors such as settling and clumping of drug granules, clogging of the device, or residual drug in the device.
2.4.3.2 Fed Study
The meal used in a comparative bioavailability study conducted under fed conditions should allow maximal perturbation of systemic bioavailability of the drug from the drug product. This is generally a high-fat, high-calorie meal. Thus, the default meal, for comparative bioavailability studies under fed conditions, should be a high-fat, high-calorie meal.
A high-fat (approximately 50% of total caloric content of the meal) and high-calorie (approximately 800 to 1000 kilocalories) meal should derive approximately 150, 250, and 500-600 kilocalories from protein, carbohydrate, and fat, respectively. One example of a high-fat, high-calorie test meal is the following breakfast: 2 eggs fried in butter, 2 strips of bacon, 2 slices of toast with butter, 120 grams of hash browns and 240 milliliters of whole milk.
Use of a meal other than a high-fat, high-calorie meal should only occur under exceptional circumstances and should be scientifically justified, a priori, by the submission sponsor. A possible justification for use of a meal other than a high fat, high calorie meal would be a documented serious safety risk to subjects from single-dose administration of the drug or drug product in the presence of such a meal.
The test meal should be consumed within a 30-minute interval prior to administration of the drug product.
2.4.4 Posture and Physical Activity
For most drugs, subjects should not be allowed to recline until at least two hours after drug ingestion. Physical activity and posture should be standardized as much as possible to limit effects on gastrointestinal blood flow and motility. The same pattern of posture and physical activity should be maintained for each study period.
2.4.5 Interval between Doses
The interval between study days should be long enough to permit elimination of essentially all of the previous dose from the body. The minimum time between treatments should be the same for all subjects and, to account for variability in elimination rate between subjects, normally should be not less than 10 times the me terminal half-life of the drug. Normally, the interval between study days should not exceed three to four weeks.
2.4.6 Sampling Times
The duration of sampling in a study should be sufficient to account for at least 80% of the known AUC to infinity (AUCI). This period is usually at least three times the terminal half-life of the drug.
To permit calculation of the relevant pharmacokinetic parameters, a minimum of 12 samples should be collected per subject per dose. Inter-subject variability, as well as such factors as the potential for erratic behaviour of some formulations under some conditions (for example, food may affect release from an enteric-coated product), should be taken into consideration in both the total number of samples collected and the sampling schedule. The exact times at which the samples are taken should be recorded and spaced such that the following information can be estimated accurately:
- Cmax
- the area under the concentration-time curve to the time of the last quantifiable concentration (AUCT) is at least 80% of AUCI, and
- the terminal disposition rate constant of the drug (λ)
There may be considerable inaccuracies in the estimates of the terminal disposition rate constant if the constant is estimated from linear regression using only a few points. To reduce these inaccuracies it is preferable that three or more points be determined during the terminal log-linear phase of the curve.
2.4.7 Sample Collection
Blood should be the biological fluid sampled to measure the concentrations of the drug. In most cases the drug may be measured in plasma; however, in some cases, whole blood or serum may be more appropriate for bioanalysis.
Sometimes the concentration of drug in a fluid other than blood may correlate better with effect. Nevertheless, the drug must first be absorbed prior to distribution to the other fluids such as the cerebrospinal fluid or bronchial secretions. Thus, for bioavailability estimations, blood is still to be sampled and assayed.
2.4.8 Handling of Samples
Samples should be collected, processed and stored under conditions that have been shown not to cause significant degradation or inter-conversion of the analytes.
2.4.9 Identification of Adverse Events
Section C.05.001 of the Regulations defines an Adverse Event as "any adverse occurrence in the health of a clinical trial subject who is administered a drug, that may or may not be caused by the administration of the drug, and includes an adverse drug reaction". Consequently, all unfavourable and unintended signs (including an abnormal laboratory finding, for example), symptoms, or disease temporally associated with the use of a drug are to be reported, whether or not they are considered to be related to the drug. (See also ICH guidance Topic E2A; Clinical Safety Data Management: Definitions and Standards for Expedited Reporting).
In some cases, adverse events are due to factors other than the active ingredient in a formulation. The rate of absorption and excipients within formulations may affect the frequency, onset, and severity of adverse events. The incidence, severity, and duration of all adverse events observed during the study should be reported. The probability that an adverse event is drug-induced is to be judged by the investigator.
The same observer and format for eliciting and recording information on adverse events should be used for both test and reference products. Questions concerning adverse events should be asked during each sampling period by the "blinded" observer. For drugs with known adverse events (e.g., metallic taste, postural hypotension, or cardiac dysrhythmia) the specific questions should be raised. In asking the questions, the interviewer should avoid leading the subject to believe that the events are expected or unexpected. Furthermore, the subject should be questioned in private. Observations, such as blood pressure measurement and electrocardiogram, should be performed and recorded at the time the events are known to occur with respect to the time of administration.
2.5 Test and Reference Drug Products
The required characteristics of the test and reference drug products that should be documented include quality, dosage, strength, lot numbers and the identity of the reference product used in the study.
2.5.1 Chemistry
The test and reference products should meet a Schedule B or other applicable standard acceptable to Health Canada. The chemistry and manufacturing guidances for preclinical and new drug submissions should be consulted for an interpretation of the general technical requirements listed in sections C.08.005(1) and C.08.002(2) respectively.
2.5.2 Dosage and Strength
In comparative bioavailability studies, the same dose of each product should be used, preferably as single dosage form units. The lots for comparative bioavailability testing should be representative of proposed market production batches. The lots for comparative bioavailability testing should be taken from a batch that is a minimum of 10% of the commercial batch size or 100,000 units, whichever is greater, unless otherwise justified. The lots should be produced using the same type of equipment and procedures, and for modified-release formulations, the same site, as proposed for market production. The validity of the biolot used in the comparative bioavailability study could be undermined by evidence of inadequate development of the product or the manufacturing process which could result in an inconsistent and/or poor quality product. A comparative bioavailability study using such a biolot would not be considered adequate to support the safety and efficacy of the proposed commercial product.
For products in which the proportions of excipients and the dissolution characteristics are similar, comparative bioavailability studies may not be required for all strengths. Whether all strengths should be tested will depend on the extent to which the formulation differs among strengths and the results of the comparative dissolution studies. Further guidance may be found in the Therapeutic Products Directorate Policy: Bioequivalence of Proportional Formulations - Solid Oral Dosage Forms.
When a modified-release product in the form of a scored tablet possesses the claim that a portion of the tablet may be administered to provide a proportional dose, evidence should be presented to justify the claim. Split tablets from scored tablets are considered independent dosage units and the uniformity of content should be established in the split tablets of all types, irrespective of the type of drug release. For modified release tablets the evidence submitted should include information on product design and development indicating that splitting of the tablet would not adversely affect safety and performance, and that use of in vitro drug release (dissolution) data is justified.
2.5.3 Selection of Reference Product
For a new drug substance (i.e., the first market entry), the reference product should be the formulation used in the pivotal clinical trials.
For other pivotal bioequivalence studies, the reference product should be the Canadian reference product as defined in Section C.08.001.1 of the Regulations:
- a drug in respect of which a notice of compliance is issued pursuant to section C.08.004 [of the Regulations] and which is marketed in Canada by the innovator of the drug;
- a drug, acceptable to the Minister, that can be used for the purpose of demonstrating bioequivalence on the basis of pharmaceutical and, where applicable, bioavailability characteristics, where a drug in respect of which a notice of compliance has been issued pursuant to section C.08.004 [of the Regulations] cannot be used for that purpose because it is no longer marketed in Canada; or
- a drug, acceptable to the Minister, that can be used for the purpose of demonstrating bioequivalence on the basis of pharmaceutical and, where applicable, bioavailability characteristics, in comparison to a drug referred to in paragraph (a).
For further guidance see the Therapeutic Products Directorate Guidance document entitled Use of a Foreign-Sourced Reference Product as a Canadian Reference Product.
2.6 Bioanalytical Methodology
Comparative bioavailability determinations rely on well-characterized and validated bioanalytical methods that are able to generate reliable estimates of analyte concentrations.
The bioanalytical laboratory should maintain a complete written set of standard operating procedures to cover all aspects of method validation and subject sample analysis. In addition, the method validation and subject sample analyses should be fully documented.
The principles and procedures for bioanalytical method validation and analysis of study samples described in the ICH M10 Guideline: Bioanalytical Method Validation and Study Sample Analysis should be followed.
2.6.1 Bioanalysis of Subject Samples
Determination of comparative bioavailability should be based on data for the parent drug.
Waiver of the measurement of the parent drug will not be considered, unless the pharmacokinetics of the formulated drug cannot be reliably estimated, e.g., the parent drug is not detectable due to rapid biotransformation. In such instances, the use of metabolite data may be acceptable. The measured metabolite should be a primary (first step) and major one, and appropriate scientific justification for a waiver of the measurement of the parent drug and the use of metabolite data should be provided. The choice of using the metabolite instead of the parent drug is to be clearly stated, a priori, in the objective of the study in the study protocol.
For the purpose of this guidance, a pro-drug is to be treated as a 'parent drug'. That is, if the substance released from the dosage form is absorbed intact and is reliably measurable in the systemic circulation, it should be used in the assessment of comparative bioavailability.
It is generally not necessary to measure both parent drug and metabolite levels for the purpose of comparative bioavailability assessment. However, quantitation of metabolite levels may sometimes be helpful, e.g., to explain extreme values caused by metabolic changes within a subject.
In the case of chiral drugs, instances where the in vivo disposition of each enantiomer is pertinent to the comparative bioavailability assessment of two oral solid dosage forms of similar type containing a defined ratio of enantiomers would be rare. For further guidance on this issue see the Therapeutic Products Directorate guidance document entitled Stereochemical Issues in Chiral Drug Development.
2.6.2 Assay Method Validation
The bioanalytical methods used to measure the drug, or metabolite, in plasma, blood or serum should be suitable for their intended purpose. They should be reproducible, selective, and sufficiently sensitive, precise, and accurate. When these attributes have been shown to be adequate in the hands of the test laboratory, the investigators can then undertake the bioavailability study.
2.7 Analysis of Data
Bioanalysis of all samples should be completed prior to the initiation of the pharmacokinetic and statistical analyses.
2.7.1 Presentation of Data
The concentrations of the drug in plasma for each subject, the sampling time, and the formulation should be tabulated. Unadjusted, measured concentrations should be provided.
Deviations from the protocol (e.g., missed samples or late collection of samples) should be clearly identified in the tables.
Two graphs should be drawn for each subject and two for the mean values of all subjects, one linear and the other semilogarithmic. On these graphs, the drug concentrations from the reference and the test formulations should be plotted against the sampling times. Usually, the individual semilogarithmic graphs should display the regression lines that are employed to estimate the terminal disposition rate constant (λ) for the two formulations. For drugs with a long half-life where AUC0-72h is measured; λ, the terminal elimination half-life (t½) and AUCI may not be required to be estimated and the regression lines may not need to be presented.
2.7.2 Pharmacokinetic Parameters
Estimates of the following pharmacokinetic parameters should be tabulated for each subject-formulation combination:
- AUCT
- AUCI
- AUCT/AUCI
- Cmax
- tmax
- λ
- t½
Where the time to onset of action is important, the following parameter should also be reported:
- The area under the curve to tmax of the reference product, calculated for each study subject (AUCReftmax).
Where multiple dose studies are conducted, the following parameters should also be reported:
- Minimum observed concentration (Cmin).
- Pre-dose concentrations determined immediately before a dose at steady state (Cpd).
- Area under the concentration versus time curve, over the dosing interval (AUCtau).
Where multiple plasma concentration profile of a modified-release product has been demonstrated to be integral to the therapeutic effect, the following parameter should be reported:
- Area under the concentration versus time curve over a restricted time interval after drug administration (pAUC).
- Additional pharmacokinetic parameters may also be presented, but the methods used to estimate them should be fully described. The means and coefficients of variation should be given for each parameter and for each formulation.
2.7.3 Data Collection
If sequential or adaptive design is used, a description of how changes were made to collection of data should be provided.
2.7.4 Statistical Analysis
2.7.4.1 Outlier analysis
Identification of extreme values should be presented. Results of the proposed outlier test from the study protocol should be listed for each parameter. For instance if the studentised residual is used, only values greater than 3 may be considered extreme values. The minimum and maximum values for each parameter should also be identified. Only those subjects who are identified as outliers for all parameters may be removed.
2.7.4.2 Model Fitting
By definition the cross-over design is a mixed effects model with fixed and random effects. The basic two period cross-over can be analysed according to a simple fixed effects model and least squares means estimation. Identical results will be obtained from a mixed effects analysis such as Proc Mixed in SAS®. If the mixed model approach is used, parameter constraints should be defined in the protocol. Higher order models, such as replicated cross-over designed, should be analysed with the mixed model approach in order to estimate random effects properly. It is recommended to use PROC MIXED rather than PROC GLM in such cases when using SAS®.
2.7.4.3 Testing of Fixed Effects
A summary of the testing of sequence, period and formulation effects should be presented. Explanations for significant effects should be given.
2.7.4.4 Estimation of Random Effects
A summary of the estimates of inter-subject and intra-subject variances should be presented. For replicated cross-over designs, estimates of interaction between subject and formulation and within formulation variance estimates should be given.
2.7.4.5 Analysis of Data
The analyses should include all evaluable data for all subjects (see Section 2.3.3, "Accounting for Drop-outs and Withdrawals") on measured data. Analysis based on less data should be justified.
Analysis should be carried out on the logarithmically transformed AUCT and Cmax data. The analysis and results for each parameter should be reported on a separate page as detailed in Appendix 1, "Sample Analysis for a Comparative Bioavailability Study". The reported results should include:
- arithmetic means and CVs (across subjects) for each product;
- testing and estimates for fixed and random effects;
- AUCT and Cmax ratios of geometric means for test versus reference products;
- the appropriate confidence interval about the parameter being analysed.
Appendix 1: Sample Analysis for a Comparative Bioavailability Study
The following tables and figures illustrate data collected and used in a sample bioavailability study. An analysis of this data is also shown.
Although a comparative bioavailability study may include many formulations, the basic analysis is the same - each test formulation is compared to a reference formulation.
The analysis of any comparative bioavailability study should have the following sections:
- A randomization scheme for the design, where all subjects randomized into the study are included and identified by code, sequence, and dates of the dosing periods for both test and reference formulations (see Section A1.1.).
- A summary of drug concentrations (graphic and quantitative) at each sampling time for each subject for both test and reference formulations (see Section A1.2.).
- A summary of the estimates of the parameters as defined in Section A1.3 for both test and reference formulations, including the means, standard deviations, and CVs (see Section A1.4.).
- A formal statistical analysis of the relevant parameters with comparisons of the test formulations to the reference formulations (see Sections A1.5 through A1.9.).
All the sample statistical analyses that follow have the minimum two formulations (test and reference) given on two dosing days or periods.
A1.1 Randomization Scheme of the Design
Shown in Table A1-A is the randomization scheme for the cross-over design used in the study. In any study, all subjects who were randomized into the study should be included. Even those subjects that did not complete the study should be included and identified accordingly. Subject numbers that appear on informed consent forms and reporting forms should be given. Also, if any other subject identification code was used, it should be given here. The sequence to which the subject was randomized should be given. Finally, all dosing periods and dates should be given.
A1.2 Summary of Drug Concentrations
Tables A1-B and A1-C show a list of the concentrations at each sampling time for each subject for the test and reference formulations, respectively. If any concentration is missing, it should be identified, and the reason it is missing given (e.g., lost sample; sample not collected).
Although no formal statistical analysis is required at each sampling time, it is recommended that summary statistics be given at each sampling time for each formulation. It is also helpful if the lower limit of quantitation of the bioanalytical method is given in this table.
|
Subject
|
Period
|
|||
|---|---|---|---|---|
|
Number
|
ID
|
Sequence
|
May 14, 2008
|
May 21, 2008
|
| 001 | A | TR | T | R |
| 002 | B | RT | R | T |
| 003 | C | RT | R | T |
| 004 |
D | TR | T | No data |
| 005 | E | TR | T | R |
| 006 | F | RT | R | T |
| 007 | G | TR | T | R |
| 008 | H | RT | R | T |
| 009 | I | TR | T | R |
| 010 |
J | RT | No data |
No data |
| 011 | K | RT | R | T |
| 012 | L | TR | T | R |
| 013 | M | TR | T | R |
| 014 | N | RT | R | T |
| 015 | O | RT | R | T |
| 016 | P | TR | T | R |
| 017 | Q | RT | R | T |
| 018 | R | TR | T | R |
|
||||
|
ID
|
Seq
|
Period
|
Sampling Times (hours)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
0.0
|
0.33
|
0.66
|
1.0
|
1.5
|
2.0
|
3.0
|
4.0
|
6.0
|
8.0
|
12.0
|
16.0
|
|||
| A | TR | 14 May | 0.00 | BLQ |
52.01 | 95.03 | 122.20 | 77.88 | 65.15 | 46.24 | 19.20 | 14.99 | BLQ |
BLQ |
| B | RT | 21 May | 0.00 | BLQ |
56.66 | 80.85 | 102.00 | 86.41 | 63.81 | 49.20 | 24.00 | 11.37 | 8.24 | BLQ |
| C | RT | 21 May | 0.00 | 28.63 | 201.50 | 189.80 | 188.70 | 136.20 | 97.64 | 64.53 | 32.08 | 20.63 | 14.59 | BLQ |
| E | TR | 14 May | 0.00 | BLQ |
9.04 | 34.32 | 47.70 | 52.79 | 59.47 | 32.61 | 17.61 | 8.76 | BLQ |
BLQ |
| F | RT | 21 May | 0.00 | BLQ |
55.33 | 66.40 | 58.97 | 48.29 | 43.19 | 34.23 | 17.30 | 6.15 | BLQ |
BLQ |
| G | TR | 14 May | 0.00 | BLQ |
33.15 | 45.64 | 54.19 | 34.13 | 32.78 | 21.55 | 10.75 | 8.35 | BLQ |
BLQ |
| H | RT | 21 May | 0.00 | 35.38 | 79.14 | 100.90 | 70.71 | 48.43 | 30.73 | 26.19 | 8.65 | 6.83 | BLQ |
BLQ |
| I | TR | 14 May | 0.00 | BLQ |
64.57 | 76.52 | 89.51 | 86.21 | 69.04 | 50.96 | 21.55 | 13.71 | 7.55 | BLQ |
| K | RT | 21 May | 0.00 | BLQ |
79.34 | 99.41 | 154.80 | 58.60 | 57.12 | 32.57 | 19.82 | BLQ |
BLQ |
BLQ |
| L | TR | 14 May | 0.00 | 14.78 | 55.54 | 56.88 | 46.87 | 37.29 | 28.75 | 25.20 | BLQ |
BLQ |
BLQ |
BLQ |
| M | TR | 14 May | 0.00 | BLQ |
BLQ |
BLQ |
BLQ |
BLQ |
8.37 | 23.15 | 19.74 | 16.49 | 5.74 | 5.18 |
| N | RT | 21 May | 0.00 | BLQ |
37.76 | 28.58 | 21.56 | 19.02 | 13.25 | 12.44 | 6.38 | BLQ |
BLQ |
BLQ |
| O | RT | 21 May | 0.00 | BLQ |
27.85 | 43.30 | 43.30 | 32.57 | 29.59 | 25.42 | 16.89 | 7.68 | BLQ |
BLQ |
| P | TR | 14 May | 0.00 | BLQ |
68.25 | 52.57 | 51.97 | 28.64 | 23.70 | 12.74 | BLQ |
BLQ |
BLQ |
BLQ |
| Q | RT | 21 May | 0.00 | BLQ |
5.90 | 13.00 | 27.54 | 13.32 | 12.34 | 9.81 | 9.73 | BLQ |
BLQ |
BLQ |
| R | TR | 14 May | 0.00 | BLQ |
18.92 | 35.77 | 53.93 | 60.43 | 47.44 | 41.72 | 16.66 | 8.87 | 5.49 | BLQ |
| If there are more samples, please continue to list as described above until all samples have been recorded. | ||||||||||||||
| MEAN | No data | No data | 0.00 | 4.92 | 52.81 | 63.69 | 70.87 | 51.26 | 42.62 | 31.80 | 15.04 | 7.73 | 2.60 | 0.32 |
| STD | No data | No data | 0.00 | 11.26 | 47.05 | 45.04 | 49.76 | 33.66 | 24.64 | 15.42 | 8.60 | 6.57 | 4.42 | 1.29 |
| CV | No data | No data | No data | 228.66 | 89.09 | 70.22 | 70.22 | 65.66 | 57.79 | 48.51 | 57.18 | 84.94 | 169.84 | 400 |
|
||||||||||||||
|
ID
|
Seq
|
Period
|
Sampling Times (hours)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
0.0
|
0.33
|
0.66
|
1.0
|
1.5
|
2.0
|
3.0
|
4.0
|
6.0
|
8.0
|
12.0
|
16.0
|
|||
| A | TR | 14 May | 0.00 | BLQ |
116.40 | 124.60 | 126.20 | 107.60 | 45.65 | 33.22 | 16.11 | 12.60 | BLQ |
BLQ |
| B | RT | 21 May | 0.00 | BLQ |
88.45 | 121.40 | 206.90 | 179.00 | 84.53 | 40.02 | 38.01 | 15.12 | 5.39 | BLQ |
| C | RT | 14 May | 0.00 | BLQ |
BLQ |
95.57 | 122.80 | 103.20 | 101.70 | 57.65 | 23.85 | 14.59 | 6.29 | BLQ |
| E | TR | 21 May | 0.00 | BLQ |
3723 | 37.26 | 35.90 | 28.87 | 28.48 | 25.10 | 24.91 | 6.72 | BLQ |
BLQ |
| F | RT | 14 May | 0.00 | BLQ |
29.25 | 62.88 | 64.26 | 84.67 | 45.21 | 25.05 | 17.18 | 8.47 | BLQ |
BLQ |
| G | TR | 21 May | 0.00 | BLQ |
6.89 | 50.04 | 55.27 | 51.68 | 38.58 | 26.19 | 7.79 | BLQ |
BLQ |
BLQ |
| H | RT | 14 May | 0.00 | BLQ |
113.50 | 218.70 | 125.80 | 69.77 | 45.03 | 32.78 | 18.55 | 5.42 | BLQ |
BLQ |
| I | TR | 21 May | 0.00 | BLQ |
181.90 | 135.80 | 96.51 | 90.50 | 62.58 | 30.43 | 18.50 | BLQ |
BLQ |
BLQ |
| K | RT | 14 May | 0.00 | BLQ |
42.71 | 58.75 | 59.68 | 54.37 | 44.35 | 22.94 | 11.58 | 6.95 | BLQ |
BLQ |
| L | TR | 21 May | 0.00 | BLQ |
14.29 | 21.32 | 24.32 | 25.56 | 25.51 | 10.49 | 5.49 | BLQ |
BLQ |
BLQ |
| M | TR | 21 May | 0.00 | BLQ |
8.21 | 48.87 | 57.05 | 56.32 | 42.08 | 24.79 | 16.54 | 15.81 | 7.60 | BLQ |
| N | RT | 14 May | 0.00 | BLQ |
47.20 | 34.90 | 34.90 | 24.19 | 20.11 | 8.08 | 7.27 | BLQ |
BLQ |
BLQ |
| O | RT | 14 May | 0.00 | BLQ |
BLQ |
20.35 | 70.88 | 70.60 | 70.38 | 40.51 | 26.93 | 8.20 | BLQ |
BLQ |
| P | TR | 21 May | 0.00 | BLQ |
39.23 | 86.29 | 97.46 | 52.26 | 40.53 | 26.74 | 12.54 | BLQ |
BLQ |
BLQ |
| Q | RT | 14 May | 0.00 | BLQ |
BLQ |
30.86 | 88.38 | 37.67 | 29.28 | 14.99 | 6.38 | BLQ |
BLQ |
BLQ |
| R | TR | 21 May | 0.00 | BLQ |
BLQ |
24.84 | 59.27 | 98.82 | 69.98 | 46.50 | 23.46 | 9.91 | 6.96 | BLQ |
| If there are more samples, please continue to list as described above until all samples have been recorded. | ||||||||||||||
| MEAN | No data | No data | 0.00 | No data | 45.33 | 73.28 | 82.85 | 70.94 | 49.62 | 29.09 | 17.19 | 6.49 | 1.64 | No data |
| STD | No data | No data | 0.00 | No data | 53.30 | 54.49 | 46.24 | 39.78 | 22.51 | 12.88 | 8.83 | 5.98 | 2.96 | No data |
| CV | No data | No data | No data | No data | 117.59 | 74.37 | 55.82 | 56.08 | 45.37 | 44.28 | 51.38 | 92.23 | 180.73 | No data |
|
||||||||||||||
A1.3 List of Parameters and Definitions
Table A1-D shows a list of the parameters used in the analysis and their definitions. If any other parameters are used, they should also be clearly defined.
Table A1-D: Parameter Definitions
|
Parameter |
Definition
|
|---|---|
| Cmax | Maximum observed concentration. |
| tmax | Sampling time at which Cmax occurred. |
| AUCT | The area under the curve to the last quantifiable concentration calculated from observed data at specific time points. |
| AUCI | Area to infinity = AUCT + CT/λ, where CT is the estimated concentration at LQCT. |
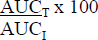 |
Percent of the area measured by AUCT relative to the extrapolated AUCI. |
| AUCRefTmax | Area under the curve, for a test product, to the tmax of the reference product, calculated for each study subject. |
| pAUC | Partial area under the curve over a defined time interval after drug administration. |
| λ | Terminal disposition rate constant calculated from the points on the log-linear end of the concentration versus time curve. |
| TLIN | Time point where log-linear elimination begins. |
| LQCT | Lowest Quantifiable Concentration Time. Time at which the last concentration occurred that is above the lower limit of quantitation. |
| t½ | Drug half-life = ln(2)/λ = 0.693/λ. |
A1.4 Summaries of Parameter Estimates
Tables A1-E and A1-F list, for each subject, the estimates of the parameters defined in Table A1-D for the test and reference formulations respectively. Summary statistics (arithmetic means, standard deviations, and CVs, or medians and ranges) should be given for each formulation.
|
ID
|
Seq
|
Period
|
Test Formulation
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Cmax
(ng/mL) |
tmax
(h) |
AUC
T
(ng·h/mL) |
AUC
I
(ng·h/mL) |
AUC
T
(%) |
λ
(h -1) |
TLIN
(h) |
LQCT
(h) |
t½
(h) |
|||
| A | TR | 14 May | 122 | 1.50 | 365 | 409 | 89 | 0.3002 | 2.0 | 8.0 | 2.3 |
| B | RT | 21 May | 102 | 1.50 | 405 | 432 | 94 | 0.2384 | 3.0 | 12.0 | 2.9 |
| C | RT | 21 May | 202 | 0.66 | 703 | 774 | 91 | 0.1776 | 4.0 | 12.0 | 3.9 |
| E | TR | 14 May | 59 | 3.00 | 233 | 256 | 91 | 0.3680 | 3.0 | 8.0 | 1.9 |
| F | RT | 21 May | 66 | 1.00 | 247 | 265 | 93 | 0.3902 | 3.0 | 8.0 | 1.8 |
| G | TR | 14 May | 54 | 1.50 | 178 | 205 | 87 | 0.2768 | 3.0 | 8.0 | 2.5 |
| H | RT | 21 May | 101 | 1.00 | 246 | 263 | 94 | 0.3437 | 3.0 | 8.0 | 2.0 |
| I | TR | 14 May | 90 | 1.50 | 408 | 433 | 94 | 0.2486 | 3.0 | 12.0 | 2.8 |
| K | RT | 21 May | 155 | 1.50 | 315 | 372 | 85 | 0.3379 | 3.0 | 6.0 | 2.1 |
| L | TR | 14 May | 57 | 1.00 | 140 | 331 | 42 | 0.1318 | 3.0 | 4.0 | 5.3 |
| M | TR | 14 May | 23 | 4.00 | 165 | 195 | 85 | 0.1485 | 6.0 | 16.0 | 4.7 |
| N | RT | 21 May | 38 | 0.66 | 88 | 113 | 78 | 0.2620 | 2.0 | 6.0 | 2.6 |
| O | RT | 21 May | 43 | 1.00 | 183 | 215 | 85 | 0.2671 | 3.0 | 8.0 | 2.6 |
| P | TR | 14 May | 68 | 0.66 | 122 | 148 | 83 | 0.5031 | 1.5 | 4.0 | 1.4 |
| Q | RT | 21 May | 28 | 1.50 | 68 | 113 | 60 | 0.1833 | 1.5 | 6.0 | 3.8 |
| R | TR | 14 May | 60 | 2.00 | 275 | 292 | 94 | 0.2546 | 3.0 | 12.0 | 2.7 |
| If there are more samples, please continue to list as described above until all samples have been recorded. | |||||||||||
| MEAN |
No data | No data | 79 | 1.50 | 259 | 301 | 84 | 0.2770 | 3.0 | 8.0 | 2.8 |
| STD | No data | No data | 48 | 0.89 | 158 | 164 | 14 | 0.0967 | 1.1 | 3.3 | 1.1 |
| CV | No data | No data | 61 | 59.35 | 61 | 54 | 17 | 34.92 | 37.3 | 38.5 | 37.9 |
|
|||||||||||
|
ID
|
Seq
|
Period
|
REFERENCE FORMULATION
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Cmax
(ng/mL) |
tmax
(h) |
AUC
T
(ng·h/mL) |
AUC
I
(ng·h/mL) |
AUC
T
(%) |
λ
(h -1) |
TLIN
(h) |
LQCT
(h) |
t½
(h) |
|||
| A | TR | 21 May | 126 | 1.50 | 375 | 418 | 90 | 0.2660 | 3.0 | 8.0 | 2.6 |
| B | RT | 14 May | 207 | 1.50 | 595 | 613 | 97 | 0.2900 | 3.0 | 12.0 | 2.4 |
| C | RT | 14 May | 123 | 1.50 | 471 | 492 | 96 | 0.2666 | 4.0 | 12.0 | 2.6 |
| E | TR | 21 May | 37 | 1.00 | 190 | 224 | 85 | 0.2653 | 3.0 | 8.0 | 2.6 |
| F | RT | 14 May | 85 | 2.00 | 257 | 285 | 90 | 0.3114 | 3.0 | 8.0 | 2.2 |
| G | TR | 21 May | 55 | 1.50 | 175 | 190 | 92 | 0.5437 | 3.0 | 6.0 | 1.3 |
| H | RT | 14 May | 219 | 1.00 | 382 | 398 | 96 | 0.4047 | 2.0 | 8.0 | 1.7 |
| I | TR | 21 May | 182 | 0.66 | 361 | 406 | 89 | 0.3837 | 3.0 | 6.0 | 1.8 |
| K | RT | 14 May | 60 | 1.50 | 218 | 236 | 83 | 0.3580 | 3.0 | 8.0 | 1.9 |
| L | TR | 21 May | 26 | 2.00 | 92 | 105 | 88 | 0.4208 | 2.0 | 6.0 | 1.6 |
| M | TR | 21 May | 57 | 1.50 | 269 | 327 | 82 | 0.1373 | 6.0 | 12.0 | 5.1 |
| N | RT | 14 May | 47 | 0.66 | 106 | 125 | 85 | 0.3246 | 2.0 | 6.0 | 2.1 |
| O | RT | 14 May | 71 | 1.50 | 290 | 313 | 93 | 0.4028 | 3.0 | 8.0 | 1.7 |
| P | TR | 21 May | 97 | 1.50 | 230 | 266 | 87 | 0.3644 | 2.0 | 6.0 | 1.9 |
| Q | RT | 14 May | 88 | 1.50 | 144 | 156 | 92 | 0.4964 | 3.0 | 6.0 | 1.4 |
| R | TR | 21 May | 99 | 2.00 | 344 | 369 | 93 | 0.2370 | 4.0 | 12.0 | 2.9 |
| If there are more samples, please continue to list as described above until all samples have been recorded. | |||||||||||
| MEAN |
No data | No data | 99 | 1.50 | 281 | 308 | 90 | 0.3420 | 3.0 | 8.0 | 2.2 |
| STD | No data | No data | 59 | 0.41 | 136 | 138 | 4 | 0.1017 | 1.0 | 2.0 | 0.9 |
| CV | No data | No data | 60 | 29.05 | 48 | 45 | 5 | 29.7262 | 32.6 | 29.2 | 39.4 |
|
|||||||||||
A1.5 AUCT Analysis
Tables A1-G, A1-H, and A1-I provide the complete analysis required for AUCT. Table A1-G lists the AUCT estimates on the raw scale and the log scale. Also given is the test AUCT as a percentage of the reference AUCT. Summary statistics are calculated for each variable.
Table A1-H gives the analysis of variance (ANOVA) for the cross-over design model for ln(AUCT). This analysis gives the appropriate intra-subject variance estimate, MS (Residual), for the calculation of the 90% confidence interval. Any significant effects in the model, other than Subject(Seq), should be investigated. The intra-subject and inter-subject CVs should also be calculated.
|
Effects
|
Num df
|
Den df
|
F Value
|
Prob > F
|
|---|---|---|---|---|
|
Seq
|
1 | 14 | 0 | 0.7699 |
|
Period
|
1 | 14 | 0 | 0.5751 |
|
Form
|
1 | 14 | 1 | 0.1916 |
|
||||
Equation 2: Guidance Document: Conduct and Analysis of Comparative Bioavailability Studies
- Intra-subject coefficient of variation equals 100 times open first bracket e to the power of open second bracket the mean square residual close second bracket minus 1 close first bracket to the power of 0.5 equals 100 times open first bracket e to the power of open second bracket 0.0729 close second bracket minus 1 close first bracket to the power of 0.5 equals 27.49 percent
- Inter-subject coefficient of variation equals 100 times open first bracket e to the power of open second bracket the mean square subject nested within sequence close second bracket minus 1 close first bracket to the power of 0.5 equals 100 times open first bracket e to the power of open second bracket 0.2648 close second bracket minus 1 close first bracket to the power of 0.5 equals 55.06 percent
- where the mean square residual equals the mean square residual and the mean square subject nested within sequence equals the mean square subject nested within sequence

The AUC ratio estimate and its 90% confidence interval are derived in the calculations shown in Table A1-J. Because this study had a balanced design (i.e., an equal number of subjects per sequence) the difference is simply the difference in the arithmetic means of the ln(AUC)s. If the study was not balanced, then the least-squares mean estimate for each formulation should be used to form this difference, together with the appropriate standard error.
Table A1-J: AUCT (ng·h/mL) Analysis - Calculations
Equation 3: Guidance Document: Conduct and Analysis of Comparative Bioavailability Studies
- The difference is equal to the test mean minus the reference mean which is equal to 5.3903 minus 5.5217 which equals -0.1314
- The standard error of the difference equals the square root of open bracket 2 times the mean square residual divided by the sample size close bracket which equals the square root of open bracket 2 times 0.0729 divided by 16 close bracket which equals 0.0955
- The area under the curve ratio equals 100 times e to the power of the difference which equals 100 times e to the power of open bracket 5.3908 minus 5.5217 close bracket which equals 87.68 percent
- 90% confidence limits
- Lower, Upper equals 100 times e to the power of open bracket the difference plus or minus t-statistic, where alpha is equal to 0.05 and degrees of freedom is equal to 14, times the standard error of the difference close bracket
- Lower equals 100 times e to the power of open bracket -0.1314 minus 1.761 times 0.0955 close bracket which equals 75.41 percent
- Upper equals 100 times e to the power of open bracket -0.1314 plus 1.761 times 0.0955 close bracket which equals 103.74 percent

A1.6 Cmax Analysis
The necessary information and summary for the analyses of Cmax are shown in Tables A1-K - A1-N.
|
Effects
|
Num df
|
Den df
|
F Value
|
Prob > F
|
|---|---|---|---|---|
|
Seq
|
1 | 14 | 1.02 | 0.3306 |
|
Period
|
1 | 14 | 0.13 | 0.7264 |
|
Forme
|
1 | 14 | 1.77 | 0.2052 |
|
||||
Equation 4: Guidance Document: Conduct and Analysis of Comparative Bioavailability Studies
- Intra-subject coefficient of variation equals 100 times open first bracket e to the power of open second bracket the mean square residual close second bracket minus 1 close first bracket to the power of 0.5 equals 100 times open first bracket e to the power of open second bracket 0.2048 close second bracket minus 1 close first bracket to the power of 0.5 equals 45.25 percent
- Inter-subject coefficient of variation equals 100 times open first bracket e to the power of open second bracket the mean square subject nested within sequence close second bracket minus 1 close first bracket to the power of 0.5 equals 100 times open first bracket e to the power of open second bracket 0.1610 close second bracket minus 1 close first bracket to the power of 0.5 equals 40.12 percent

Table A1-N : Cmax Analysis - Calculations
Equation 5: Guidance Document: Conduct and Analysis of Comparative Bioavailability Studies
- The difference is equal to the test mean minus the reference mean which is equal to 4.2109 minus 4.4244 which equals -0.2135
- The standard error of the difference equals the square root of open bracket 2 times the mean square residual divided by the sample size close bracket which equals 0.1600
- The maximum observed concentration ratio equals 100 times e to the power of the difference which equals 100 times e to the power of open bracket 4.2109 minus 4.4244 close bracket which equals 80.77 percent
- 90% confidence limits
- Lower, Upper equals 100 times e to the power of open bracket the difference plus or minus t-statistic, where alpha is equal to 0.05 and degrees of freedom is equal to 14, times the standard error of the difference close bracket
- Lower equals 100 times e to the power of open bracket -0.2135 minus 1.761 times 0.1600 close bracket which equals 61.94 percent
- Upper equals 100 times e to the power of open bracket -0.2135 plus 1.761 times 0.1600 close bracket which equals 107.06 percent

A1.7 Concentration versus Time Profiles (Subject A)
Figure 1 shows a plot of the concentration versus time profile for subject A. Each plot should include profiles for all formulations given to that subject. Similar profiles should be given for each subject.
Figure 1: Concentration-Time Profile for Subject A
Figure 1 is a line graph showing concentration (in ng/mL) versus time (in hours) profiles, from 0 to 16 hours, for Test and Reference formulations in subject A.
- At 0.0 hours the concentration for Test is at 0 ng/mL and Reference is also at 0 ng/mL
- At 0.33 hours the concentration for the Test is at 0 ng/mL and Reference is also at 0 ng/mL
- At 0.66 hours the concentration for Test is at 52.01 ng/mL and Reference is at 116.40 ng/mL
- At 1.0 hours the concentration for Test is at 95.03 ng/mL and Reference is at 124.60 ng/mL
- At 1.5 hours the concentration for Test is at 122.20 ng/mL and Reference is at 126.20 ng/mL
- At 2.0 hours the concentration for Test is at 77.88 ng/mL and Reference is at 107.60 ng/mL
- At 3.0 hours the concentration for Test is at 65.15 ng/mL and Reference is at 45.65 ng/mL
- At 4.0 hours the concentration for Test is at 46.24 ng/mL and Reference is at 33.22 ng/mL
- At 6.0 hours the concentration for Test is at 19.20 ng/mL and Reference is at 16.11 ng/mL
- At 8.0 hours the concentration for Test is at 14.99 ng/mL and Reference is at 12.60 ng/mL.
At 12.0 and 16.0 hours, the concentrations for test and reference formulations were below the limit of quantitation and no points appear on the graph.

Figure 2 gives a plot of the ln (concentration) versus time profile for subject A. This plot should contain the regression lines from which the terminal disposition rate constants (λ) were estimated. This line should start and end at the time points considered to be in the log-linear elimination phase. Any point that was not used to estimate the regression line should be identified.
Figure 2: Ln (concentration) - Time Profile for Subject A
Figure 2 is a line graph showing the natural log (ln) of concentration (in ng/mL) versus time (in hours) profiles, from 0 to 16 hours, for Test and Reference formulations in subject A. Regression lines from which the terminal disposition rate constants (λ) were estimated, are also shown.
- At 0.66 hours the ln(concentration) for Test is ln(52.01) and Reference is ln(116.40)
- At 1.0 hours the ln(concentration) for Test is ln(95.03) and Reference is ln(124.60)
- At 1.5 hours the ln(concentration) for Test is ln(122.20) and Reference is ln(126.20)
- At 2.0 hours the ln(concentration) for Test is ln(77.88) and Reference is ln(107.60)
- At 3.0 hours the ln(concentration) for Test is ln(65.15) and Reference is ln(45.65)
- At 4.0 hours the ln(concentration) for Test is ln(46.24) and Reference is ln(33.22)
- At 6.0 hours the ln(concentration) for Test is ln(19.20) and Reference is ln(16.11)
- At 8.0 hours the ln(concentration) for Test is ln(14.99) and Reference is ln(12.60)
At 12.0 and 16.0 hours, the concentrations for test and reference formulations were below the limit of quantitation and no points appear on the graph.

Figure 3 shows a profile of the arithmetic means over all subjects for each formulation and sampling time.
Figure 3: Average Concentration-Time Profile for All Subjects
Figure 3 is a line graph showing the profiles of arithmetic mean concentration (in ng/mL) over all subjects for each formulation and sampling time (in hours).
- At 0.0 hours the mean concentration for Test is 0 ng/mL and Reference is also 0 ng/mL
- At 0.33 hours the mean concentration for the Test is 4.92 ng/mL and Reference is 0 ng/mL
- At 0.66 hours the mean concentration for Test is 52.81 ng/mL and Reference is 45.33 ng/mL
- At 1.0 hours the mean concentration for Test is 63.69 ng/mL and Reference is 73.28 ng/mL
- At 1.5 hours the mean concentration for Test is 70.87 ng/mL and Reference is 82.85 ng/mL
- At 2.0 hours the mean concentration for Test is 51.26 ng/mL and Reference is 70.94 ng/mL
- At 3.0 hours the mean concentration for Test is 42.65 ng/mL and Reference is 49.62 ng/mL
- At 4.0 hours the mean concentration for Test is 31.80 ng/mL and Reference is at 29.09 ng/mL
- At 6.0 hours the mean concentration for Test is 15.04 ng/mL and Reference is at 17.19 ng/mL
- At 8.0 hours the mean concentration for Test is 7.73 ng/mL and Reference is 6.49 ng/mL
- At 12.0 hours the mean concentration for Test is 2.60 ng/mL and Reference is 1.64 ng/mL
- At 16.0 hours the mean concentration for Test is 0.32 ng/mL and Reference is 0.0 ng/mL

Figure 4 shows a profile of the ln (arithmetic means) over all subjects for each formulation and sampling time.
Figure 4: Ln (average concentration) - Time Profile for All Subjects
Figure 4 is a line graph showing the natural log (ln) of arithmetic mean concentration (in ng/mL) over all subjects for each formulation and sampling time (in hours).
- At 0.33 hours the (ln) mean concentration for the Test is (ln) 4.92. (Reference concentration was 0 ng/mL)
- At 0.66 hours the (ln) mean concentration for Test is (ln) 52.81 and Reference is (ln) 45.33
- At 1.0 hours the (ln) mean concentration for Test is (ln) 63.69 and Reference is (ln) 73.28
- At 1.5 hours the (ln) mean concentration for Test is (ln) 70.87 and Reference is (ln) 82.85
- At 2.0 hours the (ln) mean concentration for Test is (ln) 51.26 and Reference is (ln) 70.94
- At 3.0 hours the (ln) mean concentration for Test is (ln) 42.65 and Reference is (ln) 49.62
- At 4.0 hours the (ln) mean concentration for Test is (ln) 31.80 and Reference is (ln) 29.09
- At 6.0 hours the (ln) mean concentration for Test is (ln) 15.04 and Reference is (ln) 17.19
- At 8.0 hours the (ln) mean concentration for Test is (ln) 7.73 and Reference is (ln) 6.49
- At 12.0 hours the (ln) mean concentration for Test is (ln) 2.60 and Reference is (ln) 1.64
- At 16.0 hours the (ln) mean concentration for Test is (ln) 0.32 (Reference concentration was 0 ng/mL)

Appendix 2: Glossary of Terms
- Adverse event
- Any adverse occurrence in the health of a clinical trial subject who is administered a drug, that may or may not be caused by the administration of the drug, and includes an adverse drug reaction
- AUC (area under the curve)
- The area under the concentration versus time curve.
- AUC I (AUC to infinity)
- The area obtained by extrapolating to infinity the AUC T. This can be calculated by adding C T/λ to AUC T where C T is the estimated last quantifiable concentration and λ is the terminal disposition rate constant.
- AUC ratio
- The ratio of geometric means of the test and reference AUCs. It is calculated as the antilogarithm of the difference between the means of the logarithms (ln) of the test and reference AUCs.
- AUC Reftmax
- The area under the curve, for a test product, to the time of the maximum concentration of the reference product, calculated for each study subject.
- AUC T (AUC to the last quantifiable concentration)
- The area under the curve versus time curve to the time of the AUC to the time of the last quantifiable concentration. AUCT is calculated from observed data at specific time points.
- AUC tau (AUC over a dosing interval)
- Area under the concentration versus time curve at steady state, over the dosing interval in a multiple-dose study.
- AUC0-72h (AUC 0-72 hours)
- The area under the concentration versus time curve from time 0 to 72 hours.
- Bioavailability
- The rate and extent of absorption of a drug into the systemic circulation.
- Bioequivalence
- A high degree of similarity in the bioavailabilities of two pharmaceutical products (of the same galenic form) from the same molar dose, that are unlikely to produce clinically relevant differences in therapeutic effects, or adverse effects, or both.
- Bioequivalent
- Test and reference products are bioequivalent when they contain an identical drug or drugs and, after comparison in an appropriate bioavailability study, are found to meet the standards for rate and extent of absorption specified in the Guidance Document Comparative Bioavailability Standards: Formulations Used for Systemic Effects.
- C max (maximum observed concentration)
- The observed maximum or peak concentration.
- C max ratio
- The ratio of geometric means of the test and reference C max. It is calculated as the antilogarithm of the difference between the means of the logarithms (ln) of the test and reference C max.
- C min (minimum concentration)
- Minimum observed concentration during steady state.
- C pd (pre-dose concentration)
- Pre-dose concentrations determined immediately before a dose at steady state.
- Excipient
- Any ingredient, excluding the drug substances, incorporated in a formulation for the purpose of enhancing stability, usefulness or elegance, or facilitating preparation; for example, base, carrier, coating, colour, flavour, preservative, stabilizer, and vehicle.
- Formulation
- An ingredient or mixture of specific ingredients; that is, drug substances and excipients in specific amounts, defining a given product.
- Label
- Includes any legend, word, or mark attached to, included in, belonging to, or accompanying any drug or package. (Section 2 of the Food and Drugs Act.)
- Maximum observed concentration (C max)
- See C max.
- Modified-release dosage form
- A dosage form for which the drug-release characteristics of time-course or drug-release location are chosen to accomplish therapeutic or convenience objectives not offered by conventional dosage forms.
Modified-release dosage forms are drug formulations that differ from conventional formulations in the rate at which the drug is released. For the purpose of this guidance, modified-release forms include formulations designed to meet one or more of the following objectives:- To delay disintegration, de-aggregation, or dissolution so that the drug's rate of degradation is altered.
- To delay or decrease the rate of absorption so that the likelihood of gastrointestinal or other adverse effects is diminished (e.g., enteric-coated forms).
- To provide effective drug concentrations for a longer period of time after a single dose.
- To deliver the drug initially at a rate similar to that obtained with the conventional form, and to provide effective drug concentrations for a longer period of time. To minimize fluctuations in drug concentrations during the dosing interval.
- To provide, after single administration, multiple peaks and troughs in the concentration-time curves (i.e. multiphasic modified-release dosage forms).
- 90% Confidence interval
- An interval about the estimated value that provides 90% assurance that it contains the true value.
- pAUC
- Partial area under the concentration versus time curve defined over a restricted time interval after drug administration.
- Pro-drug
- An inactive (or much less active) precursor that is bio-transformed to the active drug.
- Rate of absorption
- The rate at which a drug reaches the systemic circulation after oral administration.
- Standard meal
- A meal of known carbohydrate, protein, fat, and fluid composition.
- Terminal disposition rate constant (λ)
- The rate constant estimated from the slope of the terminal portion of the ln (drug concentration) versus time curve. The terminal half-life (t ½) is calculated from this constant (t ½=ln 2/λ).
- Time of maximum observed concentration (t max)
- The time after administration of the drug at which C max is observed.
Page details
- Date modified: