
Ecological State of the Science Report on Decabromodiphenyl Ether (decaBDE): chapter 3
3. Evidence of Transformation
- 3.1 In Vivo Transformation
- 3.1.1 Information Evaluated in the Screening Assessment for PBDEs
- 3.1.2 New Evidence of Debromination in Vivo
- 3.1.3 Conceptual Models of Metabolism in Vivo
- 3.1.4 Implications for Bioaccumulation Assessment
- 3.2 Debromination in the Environment
This section examines the transformation of decaBDE to determine whether there is concern respecting its transformation to bioaccumulative products in organisms (i.e., in vivo by metabolism) and in the environment (i.e., by abiotic and biodegradation processes). The evidence provided by studies quantifying transformation may be confounded by the following limitations:
- Unknown or uncharacterized purity of the parent compound, and the presence (often unquantified) of lower brominated PBDEs in the materials used (e.g., studies typically used decaBDE with a purity of approximately =98%). There is a potential that lower brominated PBDE congeners may be present as impurities in the food used in in vivo transformation studies. Some of the lower congeners may have higher accumulation factors than decaBDE. Thus, even if these impurities were present in the food (or the decaBDE test substance itself) at concentrations below the detection limit, they could still accumulate to measurable concentrations in the organism during the course of the experiment.
- Analytical challenges, which could influence the accuracy of the results (see Section 2).
- Use of laboratory studies to make inferences respecting processes and rates of transformation in the environment.
These issues are discussed in more detail later in this section.
3.1.1 Information Evaluated in the Screening Assessment for PBDEs
The following summarizes the evidence regarding debromination of decaBDE which was available during and prior to 2004 and considered in the screening assessment for PBDEs (Environment Canada 2006b).
In a decaBDE feeding study with juvenile carp, Stapleton and Baker (2003) and Stapleton et al. (2004) found that seven transformed products of decaBDE accumulated in the whole fish and liver tissues over the exposure period. These were identified as penta- to octaBDEs with only BDE154 and -155 (i.e., hexaBDEs) being positively identified (personal communications from HM Stapleton to Environment Canada, July 2009; unreferenced). The unidentified hepta- and octaBDE congeners have recently been determined to be BDE188, -202 and -197 (Stapleton et al. 2006). It also appeared that higher brominated PBDEs, such as octaBDE, may be further debrominating to lower brominated PBDE homologues. By comparing the structure of decaBDE with those of its identified degradation products, Stapleton and Baker (2003) suggested that this debromination may be due to the action of deiodinase enzymes which normally act on thyroid hormone. The bioavailability of decaBDE (parent chemical plus metabolites) was estimated at 0.44%. This value could be higher if other metabolites were present (not determined in these studies). It should be noted that the alternative explanation provided by Stapleton and Baker (2003) for the transformation of decaBDE, i.e., that it could have also been due to the action of intestinal microflora, has recently been refuted by Benedict et al. (2007), whose study confirmed that carp intestinal flora cannot debrominate PBDEs.
In their feeding study with juvenile rainbow trout, Kierkegaard et al. (1999) detected several hexa-, hepta-, octa- and nonaBDEs in liver and muscle tissues of the treated fish and some of these congeners were not measured in the food. It was not possible to determine in this study whether their presence was a result of decaBDE metabolism or the efficient adsorption of congeners initially present in trace amounts in the food.
Metabolism studies using rats (Norris et al. 1973, 1974; El Dareer et al. 1987) suggest that decaBDE has low bioaccumulation potential in mammalian species. For instance, Norris et al. (1973, 1974) dosed male and female rats with 1.0 mg of 14C-labelled decaBDE as a suspension in corn oil and found that approximately 90.6% of the administered 14C-labelled decaBDE was excreted in the feces within 24 h, and by 48 h, all of the administered chemical had been excreted. Tissue accumulation studies in which rats were fed diets of decaBDE at a rate of 0.1 mg/kg body weight (bw)/d showed that bromine contents in various tissues were not significantly greater than those of the controls. The bromine content of the adipose tissue of decaBDE-dosed rats was found to be significantly increased at the p<0.03 level but not at the p<0.01 level when compared with the controls (Norris et al. 1973, 1974). Further discussion (beyond that presented in Canada 2006 or Environment Canada 2006b) respecting the El Dareer et al. (1987) study is provided below (also cited as NTP 1986).
The National Toxicology Program (NTP 1986) and El Dareer et al. (1987) evaluated the adsorption of 14C-labelled decaBDE (97.9 to 99.2% pure) using F344/N rats. DecaBDE (BDE209) was administered through diet (total food consumption was 50 000 mg/d, on days 1-7 and 9-11 amended with unradiolabelled decaBDE and on day 8 with radiolabelled decaBDE). The total dose of decaBDE ranged from 3718 to 3826 mg/kg/d. The results showed that after 72 hours of exposure, from 91.3 ± 4% to 101 ± 4% of the radioactivity was recovered in the excreted feces. In a further study, the researchers fed both unlabelled and radiolabelled decaBDE (~22-25 mg/kg bw) in food (total food consumption 48 000 mg/d). The rats were fed with unradiolabelled decaBDE in food on days 1-7 and 9, or 9-10, or 9-11, and radiolabelled decaBDE in food on day 8. They determined that 82.5 to 86.4% of the radiolabelled decaBDE was recovered from the feces, and total recovery ranged from 83.2 to 89.3%. Excretion in urine accounted for about 0.01% of the administered dose of decaBDE. They detected trace levels of radioactivity in most tissues, with the highest levels found in liver, kidney, lung, skin and adipose tissue. The authors continued with an intravenous study in which rats were injected with 1.07 mg/kg 14C-decaBDE and were sacrificed 72 hours after dosing. The results revealed that 74% of the dose was found in the feces and gut contents, suggesting that biliary excretion was important. The authors determined that the excreted material was mainly unchanged decaBDE (up to 40%) and three metabolites (unidentified) (up to 49.8%). In a final study, biliary excretion over 4 hours was evaluated after intravenous administration of 0.9 mg/kg of 14C-decaBBDE to rats. The authors found that 7.17% of the dose appeared in the bile after 4 hours. The researchers indicated that one unidentified metabolite was observed in the bile.
Mörck et al. (2003) conducted a feeding study to investigate possible distribution pathways and metabolites of decaBDE in rats. This study involved orally dosing rats with radiolabelled decaBDE. The decaBDE substance used in the study was synthesized by a multi-step process involving the bromination of 14C-labelled phenol ultimately giving 14C-decaBDE (purity determined at > 98%). Before administration, the radiolabelled decaBDE was diluted in unlabelled decaBDE synthesized using the same process (also with a purity of > 98%). This study used the radioactivity associated with non-extractable residues, lipid-bound residues, water-soluble residues, phenolic metabolites and neutral metabolites/parent compound to make inferences about the uptake and metabolic transformation of decaBDE. A total of 8 “conventional” rats and 2 bile-duct-cannulated rats were dosed orally by gavage with 3 mmol/kg, 15 Ci/mol radiolabelled decaBDE in a dosing vehicle (lutrol/soya phospholipone) designed to maximize decaBDE solubility and absorption. Feces were collected for analysis from the conventional and cannulated rats at 24-hour intervals, while bile was collected and analyzed from the cannulated rats at 4, 12, 24, 48 and 72 hours post-dosing. The conventional rats were sacrificed at day 3 post-dose (n=4) and day 7 post-dose (n=3) for analysis of tissues.
Radioactivity related to decaBDE was present in the feces, bile and tissues of exposed rats as a combination of non-extractable residues, lipid-bound residues, water-soluble residues, phenolic metabolites and neutral metabolites/parent compound, indicating the potential presence of parent decaBDE and debrominated metabolites, as well as conjugated metabolites and metabolites bound to lipid or other tissues. In conventional rats, phenolic metabolites increased from ~8% to ~14% in the three days post-dose while neutral metabolites/parent compound decreased from ~29% to ~17% in the three days post-dose. Bile contained predominantly lipid-bound residues (decreasing from ~60% to ~11% in the three days post-dose) and water soluble residues (increasing from ~9% to ~62% in the three days post-dose).
The study found that approximately 90% of the single oral dose was excreted via feces. By totalling the amount of 14C collected in urine and feces and subtracting that from 100%, the authors estimated that 9% remained in the body (i.e., as parent compound and metabolites). The highest concentrations of radioactivity (on a fresh weight basis) were found in adrenals, kidney, heart and liver after both 3 and 7 d. On a lipid weight basis, plasma and liver had the highest levels; levels in other tissues were low. The high absorption (relative to previous studies) of decaBDE was likely due to the use of the lutrol/soya phospholipone carrier. Ten percent of the dose excreted in feces came from bile. Of the total radioactivity excreted in feces, it was inferred that approximately 65% of the dose was eliminated as metabolites. In this study, metabolites were those characterized as non-extractable, water-soluble, lipid-bound, phenolic metabolites and parent compound/neutral metabolites. Thus, there is some uncertainty that the 65% included some proportion of bound parent compound or metabolites.
To characterize decaBDE metabolites, the authors further manipulated and analyzed the phenolic and neutral metabolites / parent compound fractions. Detection was conducted using GC/MS (ECNI). Based on their findings, the authors inferred, although they did not positively determine, the presence of debrominated, phenolic and methoxylated metabolites. Non-transformed decaBDE dominated the neutral metabolites / parent compound fraction of both feces and tissues, but small amounts (< 0.5%) of three nona-brominated metabolites were also present. Analysis of the phenolic fraction of feces and tissues also inferred the presence of hydroxylated and hydroxyl-methoxylated (i.e., guiacol-type) substances with five to seven bromine atoms which the researchers inferred were penta- to heptaBDEs.
From the metabolite characterization, the authors speculated that the potential sequence of events in metabolic transformation of decaBDE would involve debromination followed by hydroxylation of the exposed benzene ring to form an ortho catechol either directly via an arene oxide or by monohydroxylation followed by secondary oxidation of the resulting phenol. The resulting ortho catechol would then be methylated, potentially by the action of catechol-O-methyltransferase to form the observed guaiacol (hydroxymethoxylated) metabolites. The initial debromination step would have to remove least 2 bromines from the hydroxylated/methoxylated ring but appeared to also remove bromines from the other ring.
The authors also speculated that cytochrome P450 enzymes might be responsible for the large proportion bound to residues present in the small intestine. The action of cytochrome P450 following debromination is consistent with the formation of phenols and catechols, as well as the formation of reactive intermediates that bind covalently with macromolecules, which would explain the high level of bound residues in the small intestine.
3.1.2 New Evidence of Debromination in Vivo
Fish
Stapleton et al. (2006) exposed 45 juvenile rainbow trout to spiked food containing decaBDE (Cambridge Isotopes Laboratories, > 98% pure) for a period of 5 months. The debrominated products accounted for approximately 73% of the total PBDE burden in the carcasses. These products were identified as primarily BDE208 (nonaBDE) and BDE202 and -201 (octaBDEs) with a small fraction of BDE188 (heptaBDE). NonaBDEs (primarily BDE207 and -208) accounted for 26% of the burden in serum with only minor amounts of octaBDEs present, and untransformed decaBDE accounting for the remainder (approximately 68%). In the liver, the burden was primarily decaBDE with only a small fraction of lower brominated PBDEs (primarily nonaBDEs). The predominance of BDE202 as a product of decaBDE debromination was similar between rainbow trout (observed here) and carp from a previous study, and has been viewed as a potential marker for transformation as this congener has apparently not been measured in the commercial DecaBDE or OctaBDE products. The researchers also found that concentrations of the hepta- and octaBDE congeners increased in concentration over the duration of the exposure. Based on the total body burden of hepta- to decaBDE, uptake of decaBDE was estimated at 3.2% of total dose. Since no measurement was made of other metabolites, the researchers suggest that the estimate of uptake could be higher.
Stapleton et al. (2006) also conducted an in vitro study using rainbow trout liver microsome fractions to confirm the metabolic capacity of rainbow trout tissues and potential metabolites. This was partnered with an in vitro study using carp liver microsome fractions that was also undertaken to confirm the metabolic capacity of carp demonstrated by Stapleton et al. (2004). In this study, liver microsomes were prepared from three fish and incubated for 1 and 24 h at 25 °C. Analysis was conducted for PBDE congeners; however, recovery was low for the 1 h incubation (< 60%) in comparison to the 24 h incubation (> 80%). The study also had a control consisting of a boiled microsome fraction. Both carp and rainbow trout liver microsomes were observed to transform decaBDE even after the 1-h incubation. The carp liver microsomes debrominated decaBDE to hexa-, hepta-, octa- and nonaBDEs, although very little production of the nonaBDE congeners was observed (mirroring the in vivo experiment) in both 1- and 24-h incubations. After 24 h, the carp microsomes transformed 65% of the decaBDE mass (30% was transformed to hexaBDE congeners). The liver microsomes of rainbow trout transformed as much as 22% of decaBDE to octa- and nonaBDEs in the 24-h incubation. The controls showed no transformation of decaBDE.
The authors concluded that their results supported the hypothesis that deiodinase enzymes were catalyzing debromination of decaBDE; however, they also cautioned that it was not possible to rule out the concurrent or alternative action of P450 enzymes. Stapleton’s work shows that removal of bromine atoms occurs preferentially from the meta- or para-substituted positions (personal communication from HM Stapleton to Environment Canada, January 2008; unreferenced).
Tomy et al. (2004) studied the uptake, by juvenile lake trout,of 12 tetra- to heptaBDEs plus decaBDE from spiked commercial fish food. Three lower brominated PBDE congeners (unknown penta- and hexaBDE, and BDE140) appeared to be bioformed in the exposed fish. The authors hypothesized that debromination of decaBDE was a potential explanation since they were not specifically dosed to the organisms. However, it should be noted that each of the PBDE congeners dosed in this study had purities of > 96%, and thus there is uncertainty regarding whether these congeners were in the dosed material as impurities. They suggested that the structural similarity of BDEs to thyroxine (T4) could mean that deiodinase enzymes were debrominating higher brominated PBDEs to lower brominated PBDEs. The authors concluded that the degree biotransformation, especially for decaBDE, was likely to vary considerably between species, leading to high potential interspecies variability in bioaccumulation.
La Guardia et al. (2007) examined the potential for in vivo debromination of decaBDE in aquatic organisms inhabiting the receiving environment of a WWTP located inRoxboro, North Carolina, which, based on releases reported by industry to the U.S. EPA’s Toxics Release Inventory, was determined to receive wastewater from a large plastics manufacturing facility. The PBDE congener profile was tracked from the WWTP effluent to the receiving environment sediments and to biota in order to evaluate whether significant debromination was occurring. In 2002, samples of wastewater sludge, sediments and biota (sunfish, creek chub and crayfish) were collected. Then in fall 2005, samples of wastewater sludge, sediments and biota (sunfish only) were collected. Aquatic biota sampling involved the use of minnow traps at a location 15 m downstream of the WWTP outfall. All samples were extracted and purified using size-exclusion chromatography analyzed for PBDEs GC/MS in ECNI mode and EI mode.
A total of 23 PBDE congeners were detected in the biota samples. Of these, decaBDE was only detected in 2002 samples of sunfish (2880 µg/kg lipid) and crayfish (21 600 µg/kg lipid). The much higher concentration in crayfish was attributed to the sediment-association of this species and the authors speculated that crayfish could form a link from sediments to pelagic organisms. The authors also speculated that the lack of detected decaBDE concentrations in chub could be due to an enhanced ability of this species to metabolize decaBDE. Chub are closely related to carp, which have previously been demonstrated to have an enhanced capability to debrominate decaBDE (Stapleton et al. 2004). Two octa- and three heptaBDE congeners which were not detected in sludge and sediments were found in chub tissues, suggesting bioformation of these homologues. Based on these findings, the authors concluded that decaBDE is bioavailable in natural environments and could undergo metabolic debromination in the field, resulting in bioformation of lower brominated PBDEs.
Lebeuf et al. (2006) examined the effects of decaBDE and PCB126 on hepatic concentrations of PBDEs and methoxy-PBDEs in Atlantic tomcod. The decaBDE used in their experiment (DE-83R, Great Lakes Chemical Corp.) consisted of greater than 96% BDE209, with BDE153, -183 and -203 detected in amounts of between 0.00024 and 0.034% on a mass basis. Further characterization revealed that as many as seven nonspecified PBDEs were qualitatively detected (including four heptaBDEs and three octaBDEs), in addition to the three nonaBDEs. The fish used in the study were captured in the St. Lawrence estuary in 2001 and were acclimated to laboratory conditions for about 6 months. The fish were fed twice a week with capelin until the beginning of April, and then with rainbow smelts until the end of the study. After the acclimation period, 200 fish were randomly distributed in groups of 25 fish and placed into eight 500-L fibreglass tanks. On day 0, fish from half the tanks were anaesthetized in water. Half of these fish (i.e., composing 4 tanks) were injected interaperitoneally with a dose of PCB126, and the other half were injected with corn oil alone. The PCB126 was injected to evaluate the impact of cytochrome P4501A (CYP1A) induction on the biotransformation of injected PBDEs contained in the decaBDE commercial product administered to the fish. The fish from two of the four tanks that had received PCB126 and the fish from two of the four tanks that had received corn oil alone were injected with decaBDE (dose 400 ng/g fish; fish from the remaining tanks received a dose of corn oil alone) after 21 d.
Fish were sampled and analyzed for decaBDE and potential transformation products after seven weeks following decaBDE administration. The study found BDE209, -208, -207, -206, -203 and three unidentified octaBDEs in the liver of the fish. All these congeners were essentially absent in the control fish. These were also measured in the decaBDE product administered in the experiment, and thus, their presence cannot be attributed exclusively to biotransformation. Also, the presence of these substances could have been due to thermal degradation of the decaBDE product during analysis, or due to transformation in the tank due to fecal egestion of decaBDE. Despite an increase in EROD activity in the liver of tomcod dosed with PCB126 and decaBDE compared to decaBDE alone, no further increases of PBDE hepatic concentrations were observed. However, depleted concentrations of BDE17 and 6-methoxy-BDE47 (both were measured in the control fish) were found in the fish injected with decaBDE compared to control fish. The researchers attributed this to activated hepatic metabolic enzymes other than CYP1A. Fish with the injected PCB126 showed an even more significant depletion of BDE17 than those with decaBDE treatment and significantly lower concentrations of BDE203. While the fish in this study only exhibited limited capacity to metabolize decaBDE, the study demonstrates the importance of methodologies to evaluate the potential sources of substances detected in organisms following administration of decaBDE products.
The finding of Lebeuf et al. (2006) that CYP1A appears to play a limited role in the transformation of PBDEs is consistent with that of Valters et al. (2005), who found limited evidence of cytochrome P450 metabolism in common carp plasma, as hydroxylated PBDEs represented only 0.8% of the fraction of the total PBDE compound. Benedict et al. (2007) also concluded (at least based on their study using BDE99) that it is doubtful that CYPs are responsible for debromination of BDE99. In their evaluation of BDE99 debromination in carp microflora and microsomes, they concluded that an endogenous process that occurs with approximately equal activities in the intestine and liver microsomes appears responsible for debromination of BDE99 to BDE47. They also showed that this process can be inhibited by the presence of reverse thyronine. The exact metabolic mechanism could not be fully elucidated from their study, but they noted that their work suggests that debromination may involve thyroid hormone deiodinases.
Nyholm et al. (2009) used zebrafish (Danio rerio) as a vertebrate model organism to study the uptake, metabolite formation and elimination of 11 structurally diverse BFRs, including BDE28, BDE183 and BDE209 (purity not reported). Solutions containing a mixture of BFRs were made from stock solutions of individual BFRs in iso-octane or tetrahydrofuran, and evaporated almost to dryness. The residues were taken up in ethanol. Resulting mixed BFR solutions were added to a chironomid feed, giving nominal concentrations of 1 and 100 nmol/g of each molecular species on a dry weight basis. A control feed was made with ethanol only. Zebrafish were fed with BFR-treated or control chironomid feed for a 42-d exposure period and then with untreated feed for a 14-d elimination period. Samples were extracted and analyzed by GC/MS.
Zebrafish exposed to high-dose feed (100 nmol/g) for 42 d had detectable BDE209 concentrations after the 2-week elimination period, but concentrations were tenfold lower than those of BDE 28 (which was determined to have the highest concentration among BFRs analyzed). Uptake efficiency was lowest for BDE209 (< 1%) compared to other BFRs studied, and elimination half life was estimated at 6.5 d. No relationship was found between the log Kow values of the BFRs and the levels determined in fish. The study detected several compounds in fish tissues that may have been metabolites of the dosed BFRs. Notably, several hexaBDEs were tentatively identified by full-scan GC/MS and these compounds may have been debromination products of BDE183 or BDE209; however, one cannot discount the possibility that they were impurities in the material dosed to the organisms. No further discussion of potential debromination of BDE209 was mentioned in this study.
Terrestrial Organisms
Sandholm et al. (2003) conducted a study of the bioavailability, absorption and metabolic transformation of decaBDE in Sprague-Dawley rats. A total of 36 male rats weighing 200-220 g each were dosed with decaBDE at a dosage rate of 2 µmol/kg and the bioavailability, elimination and metabolite formation in plasma were monitored over a 6-d period. One subset of 18 rats was dosed orally by gavage while a second subset of 18 rats was dosed intravenously. Blood plasma was monitored at regular intervals (at 1, 3, 6, 24, 48, 72, 96, 120 and 144 h) over the 6-d monitoring period. In addition, plasma samples collected from a separate 7-d single-dose study with radiolabelled decaBDE (Mörck et al. 2003) were analyzed to determine the total radioactivity associated with neutral and phenolic fractions in plasma of rats exposed to decaBDE.
The oral bioavailability was estimated to be approximately 26%, although the sampling methodology may have resulted in an artificially high determination of bioavailability. Oral bioavailability was defined as the fraction of administered parent compound reaching systemic circulation. The authors indicated that the 26% level was much higher than that reported in early studies with decaBDE (i.e., by Norris et al. 1975; NTP 1986; El Dareer et al. 1987). Based on the analysis of plasma samples from the 7-d study with radiolabelled decaBDE, the phenolic radioactivity in plasma at days 3 and 7 was 4 times that of the neutral fraction, suggesting significant oxidative metabolism of neutral decaBDE, debrominated metabolites, and higher exposure to phenolic metabolites than to neutral parent/metabolite compounds. The presence of phenolic metabolites compared with neutral compounds indicated that the adsorption of BDE209 would have been greater than their calculated level of 26%. The elimination of decaBDE from plasma was multi-phased with terminal (i.e., longest) half-lives of 51 h (oral exposure) and 58 h (intravenous exposure).
The neutral compounds in plasma were identified as decaBDE (> 99.5%) as well as three nonaBDEs (< 0.5%). In addition to the neutral compounds, 13 phenolic metabolites were determined in plasma with the major metabolites inferred to consist of a hydroxyl-octaBDE, a hydroxyl-nonaBDE and a guaiacol-type hydroxymethoxy-hexaBDE. Based on the observed parent and metabolite composition, the following general sequence for metabolic transformation of decaBDE was proposed:
- Reductive debrominationof decaBDE
- Subsequent oxidation tophenolic metabolites
- Formation ofguaiacol-type metabolites via an arene oxide and dihydrodiol, or via twosequential oxidation steps followed by methylation
The authors also described how hydroxyl-octaBDE and hydroxyl-nonaBDE could have affinity for transthyretin (TTR), which normally functions as a transport protein for thyroxine in plasma. The binding to TTR could explain the high fraction of phenolic metabolites observed in plasma and may cause the phenolic metabolites to persist as bound residues in plasma. The binding of phenolic metabolites could disrupt thyroxine transport in blood, resulting in adverse hormonal effects. This behaviour was reported by the authors to be similar to that reported for PCB metabolites.
Hakk and Letcher (2003) summarized the findings of Orn and Klasson-Wehler (1998), Hakk et al. (2002) and Mörck and Klasson-Wehler (2001) with respect to the fecal and biliary metabolites formed by rats dosed with decaBDE. They concluded that decaBDE underwent oxidative debromination to guiacols (hydroxyl-methoxylated BDEs), hydroxylated BDEs and nonaBDEs. Additional discussions and conclusions by Hakk and Letcher (2003) included the following points:
- A likely metabolic pathway between decaBDE and hydroxylated or hydroxymethoxylated BDEs is debromination as a first step, followed by the formation of an arene oxide, and subsequent hydroxylation and/or methoxylation.
- PBDE mixtures are inducers of Phase I and Phase II metabolism, suggesting that these metabolic pathways are also important for PBDEs.
- Hydroxylated BDEs are likely to compete with thyroxine for binding to TTR.
- Guiacol metabolites could further oxidize to quinones, which are highly reactive and would bind to cellular macromolecules. Thus, bound residues may make up a significant proportion of metabolite body burdens.
Huwe and Smith (2007a, 2007b) examined the dietary accumulation, debromination and elimination of decaBDE in rats. The burden in body tissues and feces of one nonaBDE congener (BDE207) and two octaBDE congeners (BDE201 and BDE197) were higher than could be explained by the total dose of each congener associated with their background concentrations in rat feed. The authors concluded that the elevated burdens were the result of reductive debromination of decaBDE. The formation of BDE197 and -207 from BDE209 results from meta debromination(s), while para and meta debrominations are responsible for BDE201 formation. The authors proposed that the action of deiodinase enzymes may be involved in the meta debromination of decaBDE. These deiodinase enzymes are normally involved with the meta dehalogenation of the thyroid hormone thyroxine and are present in many tissues of the body.
However, the total burden of BDE207, -201 and -197 accounted for only 1% of the total decaBDE dose, suggesting that either debromination was not the primary metabolic pathway or that the debrominated products rapidly underwent further metabolism. Overall, 45% of the dosed decaBDE was unaccounted for in rat tissues and feces and the authors speculated that the formation of bound and/or hydroxylated metabolites that were not included in their analysis was a likely explanation for the incomplete mass balance of decaBDE.
Kierkegaard et al. (2007) reported the findings of a 3-month feeding study with two dairy cows. The congener profile in adipose and organ tissue differed from the profile in silage and it appeared that BDE207, -197, -196 and -182 accounted for a much higher proportion of the total burden in tissue than in silage. Tracing of potential degradation of decaBDE during extraction and cleanup with radiolabelled standards ruled out photolytic debromination as a source of these hepta- to nonaBDEs during extraction/cleanup. The authors also discounted higher dietary absorption of these congeners as an explanation for the observed differences between feed and tissues since enhanced absorption was not apparent for similar congeners (i.e., BDE183 compared to BDE182). Therefore, it was concluded that the observed increase of BDE207, -197, -196 and -182 was likely due to reductive debromination of decaBDE. The authors proposed the following two potential transformation pathways for decaBDE:
- Ortho debromination to BDE206, followed bymeta debromination to BDE196, followed by meta debromination to BDE182
- Meta debromination to BDE207, followed by meta debromination to BDE197
In a study by Riu et al. (2008), Wistar rats were dosed orally with 99.8% pure [14C]-radiolabelled decaBDE dispersed in peanut oil, gestational days 16 to 19. Urine and feces were collected daily over this 4-d period. The animals were sacrificed on day 20 of gestation (24 h after the last dose of 14C-decaBDE) and the amounts of radioactivity, corresponding decaBDE levels, and metabolic profiles were determined in various organs and tissues. Extraction and analytical methods were developed to achieve the radio-chromatographic separation of decaBDE metabolites formed in vivo.
More than 19% of the 4-d administered dose was recovered in the body (tissues + carcass) of the rats, demonstrating relatively high absorption of decaBDE as well as efficient distribution to tissues. The fecal route was the main excretion pathway, accounting for approximately 66.3% of the administered dose, whereas urinary excretion was very low (accounting for approximately 0.11% of the total dose). In feces extracts, approximately 97% of the radiolabel was found to be unchanged decaBDE. Characterization of the metabolites in feces and tissues, conducted using liquid chromatography/mass spectrometry (LC/MS) nuclear magnetic resonance (NMR), identified all 3 nonaBDEs, an unidentified octaBDE and a hydroxylated octaBDE derivative. Together (excreta + tissues + carcass), metabolites accounted for approximately 7% of the total radioactivity administered to rats. In addition, a very limited amount of an additional metabolite was also detected (not enough for structural identification); however, it could not be ruled out that the compound corresponded to a hydroxylated heptaBDE. Additional unidentified polar metabolites were detected in urine, with similar compounds found in the contents of the intestine, but not the feces.
Riu et al. (2008) found decaBDE residues to be highest in the adrenals, ovaries, liver, kidney and heart, and their calculated values were significantly higher than those of plasma. Liver was reported as the target tissue for decaBDE (11 mg/kg, corresponding to 6.5% of the administered dose). However, the highest residual levels were found in endocrine glands: the adrenals (33 mg/kg) and ovaries (16 mg/kg). Both decaBDE and its metabolites were reported to cross the placental barrier in rats following oral administration (approximately 0.5% of the total dose was found in the fetuses).
Van den Steen et al. (2007) investigated the accumulation in tissues and debromination of BDE209 in European starlings (Sturnus vulgaris), using silastic tube implants as modes of exposure. The implants were inserted under the skin by a small incision along the ribs. The implants contained BDE209 (purity not given, but sourced from Wellington Laboratories, Guelph, Canada ) dissolved in iso-octane and mixed in peanut oil. The iso-octane was evaporated off by heating the oil mixture to a constant weight. After preparation, there were no detectable levels of other PBDE congeners in the oil solution. In total, seven adult male starlings were used in the study, with four receiving an implanted dose of 46.8 ± 2.2 µg BDE209, and with three (the control group) receiving an implant with only peanut oil. During the 76-d exposure period, blood samples were taken every 3-7 d. The birds were then were euthanized and pectoral muscle and liver were excised. Blanks consisting of water instead of blood were included in each sample batch and their PBDE concentrations were subtracted from values determined for biotic samples.
Before implantation, BDE209 concentrations in blood were below the limit of quantitation (i.e., 0.8 and 0.5 µg/L for BDE209 and other BDEs, respectively). Accumulated BDE209 peaked on day 10 (16 ± 4.1 µg/L) in the blood of the exposed starlings. After this peak, there was a decline to 3.3 ± 0.4 µg/L in blood at the end of the 76-d exposure period, which suggested elimination. Tissue concentrations were below the limit of quantitation in the control group (5.6 and 2.9 µg/kg lipid in muscle and liver, respectively). In the exposed group, the muscle concentrations were about two times those in liver (i.e., 461 and 340 µg/kg lipid in muscle, compared with 269 and 337 µg/kg lipid in liver). Their study found that various PBDEs were present in both the control and exposed group muscle and liver tissues, with the differences being most pronounced for the nona- (BDE206, -207, and -208) and octaBDEs (BDE196 and -197). The octaBDEs BDE203 and -205 did not differ much between the groups. The authors concluded that this study provided evidence for the transformation of BDE209 to lower brominated PBDEs in songbirds. The significance of these findings is difficult to interpret since the composition of the test material was not provided, and thus the accumulation of nona- and octaBDEs from the test material cannot be ruled out.
Chen et al. (2008) measured BDE209 in all 114 eggs in a peregrine falcon (Falco peregrinus) egg study in the northeastern United States. Eggs were collected between 1996 and 2006 (excluding 1997 and 1998), and concentrations ranged from 1.4 to 420 ng/g ww. The authors noted that in addition to BDE209, eight nona- and octaBDE congeners frequently detected in their peregrine falcon study have rarely been reported in wildlife. Together with BDE209, these congeners composed 16-57% of total PBDEs in urban eggs and 4.9-53% in rural eggs. The researchers noted that the concentration of BDE201 was not in proportion with this congener’s presence in the OctaBDE commercial mixture and this was suggestive that it was formed in vivo from higher brominated PBDEs. In addition, they detected BDE202 in 107 of 114 eggs. Since BDE202 is not known to be found in OctaBDE or DecaBDE mixtures, they also speculated that its presence could be attributed to metabolic transformation of higher brominated PBDEs.
3.1.3 Conceptual Models of Metabolism in Vivo
This section summarizes pathways of metabolism based on those proposed in reviewed studies. This analysis respects that some studies contain uncertainties (e.g., potential impurities in the test substance administered to organisms, lack of chemical specificity in analyses of metabolites). Based on the reviewed literature, the amount of decaBDE which apparently may have been transformed to lesser-brominated PBDEs like nona-, octa- and heptaBDEs in vivo appears to be generally low, on the order of a small percent of the total decaBDE dosed. However, some studies have made inferences respecting higher levels of metabolism (potentially up to 45 to 65% of the total dose of decaBDE), which could be a result of metabolic transformation products including hydroxylated and hydroxymethoxylated PBDEs and/or the formation of bound residues.
Fish
Based on currently available studies and information reviewed in this report by Stapleton et al. (2004, 2006), personal communications from HM Stapleton to Environment Canada (January 2008 and July 2009) (unreferenced), Kierkegaard et al. (1999) and Tomy et al. (2004) (respecting the uncertainties in these latter two studies), the following pathways for metabolic transformation of decaBDE are deduced:
- DecaBDE (i.e., BDE209) isdebrominated in fish as a first step in metabolism, producing at least debrominated hepta- to nonaBDEs, but also potentially penta- and hexaBDEs.
- Thyroid hormone deiodinaseenzymes which normally remove iodine from thyroxine appear to be involvedin this debromination pathway.
- Bromine has been observed to be preferentially removed from the meta and para positions.
Mammals
According to the discussion and findings of Sandholm et al. (2003), Mörck et al. (2003), Hakk and Letcher (2003), Huwe and Smith (2007a, 2007b) and Kierkegaard et al. (2007) the following pathways of transformation are deduced:
- Reductive debromination to nona-, octa-and heptaBDEs is the likely first step in the metabolism of decaBDE.
- Similar to fish, debromination may be theresult of action by deiodinase enzymes.
- The debrominated neutral metabolites thenappear to undergo hydroxylation to potentially form phenols or catechols, potentially via an arene oxide. This could involve the action of cytochrome P450 enzymes.
- The potentially formed hydroxylated BDEs may compete with thyroxine for binding to TTR, athyroxine transport protein present in blood serum.
- The catechols may then be methylated, potentially by the action of catechol-O-methyltransferase, to form theobserved guaicols.
- The guiacol metabolites could furtheroxidize to quinones, which are highly reactive and would bind to cellularmacromolecules.
- The reactive intermediates would also be subject to rapid conjugation via Phase II metabolic processes, leading towater-soluble metabolites which would be excreted via bile and feces, aswas observed in conventional and cannulated rats.
3.1.4 Implications for Bioaccumulation Assessment
The existing evidence for the debromination and metabolism of decaBDE suggests metabolic pathways which lead to the formation of the following:
- Lower brominated PBDEs (down to heptaBDE congeners in mammals and potentially down to pentaBDE congeners in fish)
- Possibly hydroxylated BDEs (only shown inmammals)
- Possibly hydroxymethoxylated BDEs (onlyshown in mammals)
- Unknown products (potential formation inboth mammals and fish)
In order to evaluate whether these metabolites have the potential to bioaccumulate, or biomagnify in food webs, model predictions were made using the BAF-QSAR model and the terrestrial biomagnifications model of Gobas et al. (2003). It is acknowledged that in some cases some of the lower brominated PBDEs may have field or laboratory measurements of BAFs or BMFs. However, field and laboratory bioaccumulation data for the bioformed hydroxylated and hydroxymethoxylated BDE metabolites are generally lacking, and application of the models for all types of metabolites was seen as a consistent way to inform our knowledge regarding the bioaccumulation and biomagnification potential of these metabolites. The PBDE screening assessment supporting working document (Environment Canada 2006b) provides a summary of the measured aquatic BCFs and BAFs for the PentaBDE and OctaBDE commercial products. These studies showed that BCFs and BAFs exceed 5000 for tetra-, penta- and hexaBDEs (this is consistent with the findings of the modelling exercise conducted for this review).
For making BAF predictions with the BAF-QSAR model, the log Kow of each metabolite was based on reported measurements wherever possible. In the absence of reported measurements, log Kow was estimated using the Experimental Value Adjustment (EVA) method in the KOWWIN estimation program. The EVA uses a reference log Kow from another structurally close substance (in this case decaBDE) and corrects predictions for metabolites based on structural differences with decaBDE. For each metabolite, two prediction scenarios were conducted: the first with no correction for metabolic transformation, and the second corrected for metabolism based on the laboratory observations of Stapleton et al. (2004) and Tomy et al. (2004). Where it was not possible to estimate kM for lower brominated PBDEs from laboratory data, reasonable approximations were made that took into consideration rates of metabolism for similar congeners. For hydroxylated and hydroxymethoxylated BDEs, there was no information that would allow for estimation of kM, and a value of 0.026/day (i.e., equal to that estimated for decaBDE) normalized to the body weight of the model’s middle trophic level fish at 15oC (~0.02/day), was chosen for illustrative purposes. These metabolites are expected to undergo further metabolism; however, the actual rates of metabolism are unknown. The chosen value and resulting BAFs represent reasonable hypothetical predictions. Further details of log Kow and kM estimates are provided in Appendix D.
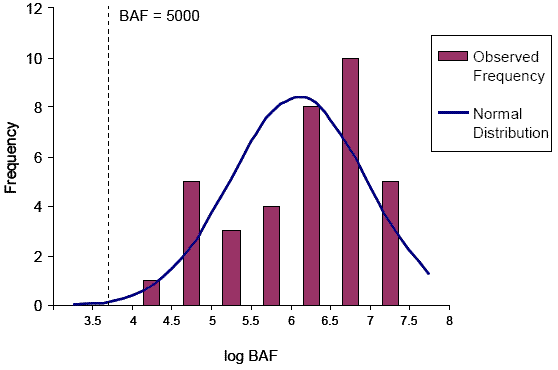
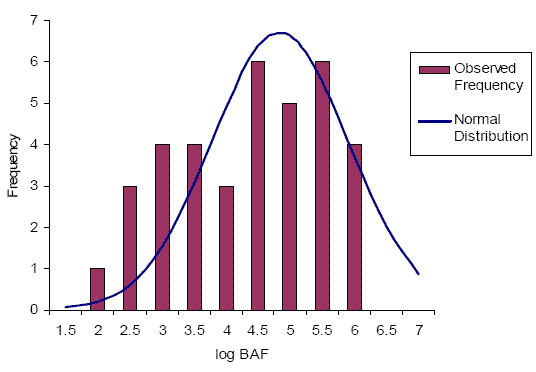
Figure 3-1 provides frequency histograms of the pooled BAF predictions for all potential metabolites and for the middle trophic level. In the absence of metabolism, the BAFs of all metabolites are predicted to exceed 5000. However, with consideration given to metabolic transformation (a more realistic scenario), the predicted BAFs range from 295 to approximately 6 000 000. Based on the fit of a normal distribution to the corrected BAF histogram, it is estimated that approximately 74% of the metabolite BAF predictions exceed 5000. The metabolite groups illustrated to exceed the BAF of 5000 include hydroxymethoxy-penta- to nonaBDEs, hydroxy-hexaBDEs, and penta- to octaBDEs. The remaining 26% of BAF predictions that are below 5000 consist of hydroxyl-octa- to nonaBDEs and nonaBDEs. The large proportion of high BAFs, even when metabolic transformation is considered, suggests that many decaBDE metabolites could potentially be bioaccumulative.
For the hydroxylated and hydroxymethoxy metabolites the actual rate of metabolism is unknown and therefore the BAF predictions are uncertain. If the secondary transformation of these metabolites occurred much faster than approximately 0.02/d then it is possible that the BAFs would be much lower. At the same time, it is possible that in certain species the enzymes responsible for Phase II metabolism might be less developed or lacking, resulting in persistence of these metabolites in tissues and much higher BAFs that approach those of the non-corrected values.
For the lower brominated PBDE metabolites, the metabolism-corrected BAF predictions are considered to have higher certainty than the predictions for the substituted forms since the kM values chosen for the model were based on laboratory observations in most cases (the one exception was for nonaBDEs, which were assumed to be metabolized at a rate based on that determined for decaBDE). It should be noted that the half-lives chosen for model inputs were often relatively short (metabolism is relatively fast) compared to the maximum half-lives observed in lab studies (refer to Appendix D). Therefore the predicted BAFs are less conservative (i.e., lower) than if the longest observed half-lives were chosen. Despite the use of less conservative half-lives, the predicted BAFs for penta- to octaBDEs exceeded 5000.
It is acknowledged that decaBDE metabolites are formed in tissues and accumulation in tissue from water-phase exposure (as quantified by the BAF) may be of limited relevance for decaBDE. However, these BAF predictions are still useful for providing a perspective on the bioaccumulation potential of the metabolite chemicals. The model results raise concerns that many of the metabolites formed in fish as a result of the accumulation and metabolism of decaBDE are potentially bioaccumulative and may be prone to biomagnification in food webs, potentially resulting in increased exposure and risk to upper trophic level organisms.
Figure 3-1: Combined Frequency Distributions of Predicted BAFs for Metabolites of DecaBDE (Predictions for Middle Trophic Level)
(a) No Correction for Metabolism
BMF predictions for decaBDE metabolites in wolves were made using a spreadsheet version of the Gobas et al. (2003) model. Log Kow values were chosen based on the rationale described in Appendix D, whereas log koa for each metabolite was estimated using KOAWIN and by inputting corrected log Kow values first. Two prediction scenarios were modelled: the first did not consider metabolic transformation, while the second considered metabolism based on the laboratory observations of Huwe and Smith (2007a, 2007b). Where it was not possible to estimate kM for lower brominated PBDEs from laboratory data, reasonable approximations were made by considering rates of metabolism for similar congeners. For hydroxylated and hydroxymethoxylated BDEs, there was no information which would allow for the estimation of kM and thus a value of 0.004/d was selected for illustrative purposes. This value corresponds to the longest observed half-life for decaBDE, nonaBDEs and octaBDEs derived by Huwe and Smith (2007a, 2007b), normalized to the body weight of wolf used in the Gobas model. The hydroxylated and hydroxymethoxylated BDE metabolites are expected to undergo further metabolism; however, the actual rates are unknown.
Calculation of BMFs for the BDEs were corrected for dietary assimilation efficiency according to the ED vs. log Kow relationship described in Kelly et al. (2004) for humans as a representative homeotherm. In the absence of metabolism, the BMFs of all metabolites are predicted to be very high, with BMFs ranging from ~4 to ~89 for chemicals within the ranges of log Kow and log koa estimated for decaBDE metabolites. However, with consideration given to metabolic transformation (a more realistic scenario), the predicted BMFs are lower, ranging from 5 to 6. All metabolites’ BMF values were > 1, primarily due to a relatively high dietary assimilation efficiency assumed for these substances.
In mammals, the actual rate of metabolism is unknown for the hydroxylated and hydroxymethoxy metabolites and therefore the BMF predictions are uncertain and may be high. If the secondary transformation of these metabolites occurs much faster than 0.012/d then it is possible that the BMF would be lower and possibly not exceed 1. At the same time, it is possible that in certain species, the enzymes responsible for Phase II metabolism might be less developed or lacking, resulting in persistence of these metabolites and much higher BMFs that approach the BMF values estimated without consideration given to metabolic transformation.
Because these decaBDE metabolites are formed in tissues, the BMF, which quantifies the potential for chemical transfer from prey tissues to predator tissues, is seen as an accurate indicator of bioaccumulation and biomagnification potential. The predicted BMFs, which exceed 1 for all metabolites, raise concerns that the metabolites formed in mammals as a result of the accumulation and metabolism of decaBDE have the potential to biomagnify in food webs, potentially resulting in increased exposure and risk to upper trophic level organisms.
Abiotic Degradation
The following bullets summarize the evidence regarding abiotic debromination of decaBDE which was considered in the screening assessment for PBDEs (Environment Canada 2006a, Environment Canada 2006b).
- Norris et al. (1973, 1974) exposed solid decaBDE (98% decaBDE and 2% nonaBDE) to sunlight in water. The bromine concentration in water increased during the experiment, suggesting that photodegradation was occurring. However, it was not possible to determine the relevance of this study to photolysis in the environment.
- Watanabe and Tatsukawa (1987) carried out photolysis experiments with decaBDE (97% decaBDE and 3% nonaBDE) in a mixture of hexane, benzene and acetone (8:1:1). After 16 h of exposure to ultraviolet (UV) light, decaBDE was found to debrominate primarily to tri- to octaBDEs, but brominated furans with 1 to 6 bromine atoms were also formed. Under sunlight, similar patterns of transformation were determined.
- Söderström et al. (2004) undertook photodegradation studies in which decaBDE (Dow FR-300 BA; exact composition was not provided, but contained traces of octa- and nonaBDEs) was dissolved in toluene and applied as a thin layer to silica gel, sand, soil or sediment. The toluene was evaporated off in the dark. Exposure was to natural sunlight and to UV light generated by three mercury lamps. Commercial DecaBDE applied to solid matrices was observed to transform following a pathway of reductive debromination with products including primarily hexa- to nona-BDEs with some occurrence of penta and tetra products (i.e., BDEs 47, -100, -99, -119). Only trace amounts of lower brominated PBDEs were formed. Photodegradation occurred fastest in toluene and silica gel (estimated half-life of less than 15 min), followed by sand (half-life of 12-13 h), sediment (half-life of 30-60 h) and soil (half-life of 150-200 h). In addition to lower brominated PBDEs, tetra- and pentabrominated dibenzofurans (tetra- and pentaBDFs) were detected as phototransformation products of decaBDE adsorbed to sand, sediment and soil. Based on the relative amounts of debromination products in the samples, the authors concluded that the origin of BDE47, -99, -100, -153 and -183, which are common in environmental samples, was “probably primarily from emissions of technical PentaBDE products and possibly from other degradation pathways of decaBDE.”
- Jafvert and Hua (2001) conducted multiple photodegradation studies of decaBDE (98% purity) on hydrated surfaces of glass and silica sand particles, humic acid-coated silica particles, and glass surfaces in contact with aqueous solutions. The studies were conducted under simulated and natural sunlight. The amount of decaBDE remaining after light exposure for 60-72 h ranged from 29 to 88% depending on the type of surface, test conditions and light used. The presence of unidentified compounds was inferred in some of the treatments. Further analysis of the unidentified compounds was largely inconclusive, although there was some evidence of the formation of hexa- to nonaBDEs.
- Hua et al. (2003) precipitated decaBDE onto surfaces (quartz glass, silica particles and humic acid-coated silica particles), hydrated them with reagent-grade water, and irradiated the system with artificial sunlight or with natural sunlight. After 60-72 h of irradiation, 29-56% of the initial decaBDE remained on the quartz surface, depending on the type of light and wavelength used. Addition of humic acid slowed the rate of decaBDE decay. Small amounts of nona- and octaBDEs were likely formed as degradation by-products. BDE47 and -99 were not formed at detectable levels of the analytical method used (high-performance liquid chromatography, HPLC).
- Palm et al. (2003) investigated the photodegradation of decaBDE dispersed in toluene, dichloromethane or a solvent mixture of hexane:benzene:acetone (8:1:1) and then irradiated with filtered (300 nm) artificial xenon lamps. A half-life of approximately 30 minutes was determined for all the test systems used. Reductive debromination was found to occur, first with three isomers of nonaBDE formed, then six isomers of octaBDE, then to several isomers of heptaBDE, and finally to trace amounts of hexaBDE. Seventy-five percent of decaBDE degradation followed a pathway of debromination, while the products of the remaining 25% could not be determined. In a follow-up study conducted under natural sunlight over 2 d, the researchers found complete disappearance of decaBDE, with nona- to pentaBDEs formed. A similar pattern was observed when decaBDE was dispersed in tetrahydrofuran (THF) and exposed to artificial sunlight for 84 h; however, PBDEs with fewer than 5 bromine atoms per molecule were also formed.
- Palm et al. (2003) also investigated the photochemical transformation of decaBDE in an aerosol smog chamber in which decaBDE was adsorbed to silicon dioxide at a concentration of about 1% by weight. The particles were suspended in water, atomized, dried and dispersed as an aerosol in a smog chamber. The conditions used were assumed to maximize the degradation potential of decaBDE. Although the conditions may not replicate atmospheric transport, the particle size used (diameter of approximately 1µm in the aerosol) can be considered environmentally relevant. When the aerosol was exposed to simulated sunlight (fluorescent lamps) and/or hydroxyl radicals, the rate of degradation of decaBDE was just barely measurable, at a rate of < 6 x 1013 cm3molecule-1s-1 for reaction with hydroxyl radicals. The products of reaction were not determined.
- Palm et al. (2004) provided early results of photolysis experiments with decaBDE adsorbed on pyrogenic silicon dioxide as a carrier material in aqueous suspension. The half-life of decaBDE exposed to UV light in aqueous suspension was reported as 8.8 h, which was much slower than in non-aqueous solvents. Polybrominated dibenzofurans (PBDFs) were confirmed as short-lived intermediates in this study. There was no information available on the other transformation products that were formed.
- Keum and Li (2005) investigated the debromination of decaBDE in aqueous solution when in contact with the reducing agents zerovalent iron, iron sulphide and sodium sulphide. In the experiments with zerovalent iron, decaBDE was rapidly transformed to lower brominated PBDEs. Approximately 90% of the parent was converted to mono- to hexaBDE congeners after 40 d. During the initial reaction period (up to 5 d), decaBDE was predominantly transformed into hexa- and heptaBDEs, but tetra- and pentaBDEs were predominant after 14 d. The results demonstrated reductive debromination. The experiments with iron sulphide and sodium sulphide also showed transformation of decaBDE to lower brominated PBDEs, but the rate was slower than that found in the presence of the zerovalent iron. A similar profile of transformation products was found to that determined in the experiment using zerovalent iron. The Government of the United Kingdom (United Kingdom 2005) points out that the actual conditions used in the Keum and Li (2005) experiment are not directly related to the environment and that concentrations of sulfides present in the environment are a few orders of magnitude lower than those used in this study. Thus, in the environment, other chemical parameters could influence reaction rate. Nevertheless, similar reactants to those used in this study (e.g., iron-bearing minerals and sulphide ions) are present in sediments and soils under anaerobic conditions, although it is uncertain whether environmental conditions are appropriate to activate similar reactions under natural conditions.
Biodegradation
The following bullets summarize the evidence regarding biodegradation of decaBDE that was considered in the screening assessment for PBDEs (Canada 2006; Environment Canada 2006b):
- The biodegradation of decaBDE was reported by MITI (1992). Under aerobic conditions using activated sludge inoculum, no degradation was detected after two weeks of exposure as measured by biological oxygen demand.
- The Chemical Manufacturers Association Brominated Flame Retardant Industry Panel (CMABFRIP 2001) investigated the anaerobic biodegradation of 14C-labelled decaBDE in a sediment-water system over 32 weeks. The decaBDE tested was a mixture of unlabelled substance (97.4% decaBDE, 2.5% nonaBDE and 0.04% octaBDE) with 14C-labelled decaBDE (radiolabelled decaBDE with a purity of 96%). Overall, decaBDE was found to be stable under the conditions tested. The study determined that less than 1% of the total radioactivity was found as 14CO2 and 14CH4, indicating that essentially no mineralization had occurred.
- Gerecke et al. (2005) undertook experiments under anaerobic conditions using sewage sludge as inoculum in glass bottles (100 mL) filled with a 1-cm layer of glass beads. These containers were then spiked with technical DecaBDE (98% purity, 10.0 nmol/sample) and incubated at 37 ±1 °C in the dark for up to 238 d. The DecaBDE contained trace amounts of nonaBDEs (i.e., BDE 206 at approximately 2% on a molar basis, and BDEs 207 and 208 at approximately 0.04% each on a molar basis). No octaBDEs were detected in the sample (detection limit was 0.005 nmol/sample). To stimulate degradation, primers (i.e., 4-bromobenzoic acid, 2,6-dibromobiphenyl, tetrabromobisphenol A, hexabromocyclododecane and decabromobiphenyl) were added in the amount of 9-11 nmol/sample. Two other experiments were carried out with two nonaBDE congeners to determine the fate of these potential decaBDE degradation products. Starch and yeast (50 g) were added immediately before the bottle was filled with 20 mL of freshly collected digested sewage sludge. The bottles were tightly capped and incubated at 37 ±1 °C for up to 238 d in the dark. Analysis by GC/HRMS indicated that decaBDE decreased by 30% within 238 d in the experiments with the primers (i.e., from 11.2 to 7.9 nmol/bottle) and this corresponded to a pseudo-first-order degradation rate constant of 1 x 10-3d-1. Without the primers, the rate constant was 50% lower. DecaBDE (BDE209) was observed to degrade to 2 nonaBDEs and 6 octaBDEs. This was indicative of reductive debromination. All three nonaBDE congeners in the separate bottles were also found to undergo reductive debromination. The mass balance between the loss of decaBDE and the formation of transformation products illustrated that 3 nmol disappeared, while only 0.5 mol of transformation products were identified. The researchers noted that this may have resulted from the formation of unidentified transformation products or bound (non-extractable) decaBDE residues, or from imprecisions in the analytical procedure used. The study demonstrated that the debromination of decaBDE proceeded most readily by the loss of bromine from the para and meta positions, as shown by the formation of BDE208 or -207.
3.2.1 New Evidence of Debromination in the Environment
Abiotic Degradation
Bezares-Cruz et al. (2004) investigated the photodegradation of decaBDE (98% pure obtained from Great Lakes Chemical Company) dissolved in hexane and exposed to natural sunlight. DecaBDE (i.e., BDE209) at a concentration of 2-5 mM in hexane degraded rapidly to lower brominated PBDE congeners in the presence of sunlight, with up to 99% reduction in the decaBDE concentration in 30 minutes (exposure to summer sunlight). First-order degradation rate constants were estimated at 1.86´10-3/s. in July sunlight (half-life = 6.2 minutes) and 1.11´10-3/s in October sunlight (half-life = 10.4 minutes). Unfortunately, the use of hexane as a solvent in this study makes it difficult to extrapolate these results to a natural setting. The authors concluded that photodegradation in nature would be limited by sorption of decaBDE to particulates, light attenuation by humic materials and lower concentrations of hydrogen donor chemicals--which may also be less favourable hydrogen donors in the aquatic environment. Stapleton (2006a) also concluded that “degradation of decaBDE dissolved in water (or organic solvents) is not expected to be of environmental relevance” and this is supported by the fact that decaBDE has extremely low water solubility. Since this study is not applicable to natural settings, the results were not considered further in the assessment of whether decaBDE is undergoing significant debromination to lower brominated PBDE congeners in the environment.
The photodegradation of decaBDE in an 80:20 methanol/water mixture, in pure methanol, in tetrahydrofuran and in water/humic acids mixtures exposed to artificial ultra-violet (UV) light was investigated by Eriksson et al. (2004). The decaBDE used in the experiments had a purity of approximately 98%. When exposed to UV light, decaBDE dissolved in the organic solvents or solvent:water mixture degraded rapidly, with degradation rate constants of approximately 4´10-4/s in the methanol/water mixture (half-life ~ 0.5 h), 6.5´10-4/s in methanol (half-life ~ 0.3 h) and 8.3´10-4/s in pure tetrahydrofuran (half-life ~ 0.23 h). The use of organic solvents and artificial conditions not representative of natural settings makes it difficult to extrapolate these results to the environment. The results of this study were therefore not considered relevant to environmental conditions.
The water / humic acid mixture tested by Eriksson et al. (2004) can potentially provide a better simulation of potential photodegradation in a natural setting since humic acids are often present in aquatic systems and could play a role in photodegradation. To prepare the solution for this experiment, 20 mL of a saturated solution of decaBDE in ethanol was combined with 10 mL of ethanol containing 50 mg of humic acid. Approximately 10 mL of ethanol was evaporated using a nitrogen stream, and the remaining solution was combined with 2 L of water. The solution was then heated and maintained at 80 °C for 1 hour under a constant flow of nitrogen. The solution was cooled to room temperature and then transferred to cylindrical reaction vessels for the irradiation experiments. The Government of the United Kingdom ( United Kingdom 2007a) estimated a final humic substances concentration for this experiment of approximately 25 mg/L and concluded that traces of ethanol may have been present since it was unclear how much ethanol would have been lost by heating at 80 °C. The experiments were replicated between 2 and 5 times but it is not clear how many replicates were conducted specifically for the water / humic acid treatment.
The rate of degradation of decaBDE in water with humic acid was 3´10-5/s (half-life ~ 6.4 h). Although the products were not presented in detail in the article, they were described as being almost identical to those determined for the methanol/water experiment which produced a range of lower brominated PBDEs (including 3 nonaBDEs, at least 7 octaBDEs, 8 heptaBDEs and small amounts of hexaBDEs), mono- to pentabromodibenzofurans (mono- to pentaBDFs), and possibly brominated-methoxylated-dibenzofurans. One key difference with the water / humic acid experiment was the formation of a higher proportion of pentaBDFs. Because both water and humic acid are naturally occurring substances, it is possible that similar reactions could occur in the natural environment. However, the actual environmental rates and degree of debromination are uncertain since this experiment used artificial light and it is uncertain what fraction of decaBDE in the environment might be associated with humic acids, as opposed to particulates. Although the authors also reported results for the water-only treatment, there was a high level of difficulty with this experiment due to the extremely low water solubility of decaBDE. As a consequence, the findings of the water-only experiment were highly uncertain.
In their study respecting the effect of sewage sludge application on concentrations of PBDEs in soils and earthworms, Sellström et al. (2005) provide a brief discussion of an investigation of photolytic debromination of BDE209 amended in soil. The BDE209 was amended with soil from Björketorp, placed in glass test tubes and exposed to artifical UV light on a “rocking/rolling” apparatus for 0, 7, 14 and 21 d. Controls were also exposed to the same agitation, but were protected from the light. Ultimately, soils showed no evidence of photolytic breakdown. The authors concluded that soil appears to encapsulate and shield contaminants so that they are less likely to break down when exposed to sunlight.
Hagberg et al. (2006) observed the formation of PBDF congeners as a result of the photolytic decomposition of decaBDE in toluene. DecaBDE (i.e., BDE209) was dissolved in toluene to a concentration of 2x106 µg/L and then irradiated with UV radiation from a fluorescent tube with or without filtering to generate UV-A (320-400 nm), UV-AB (280-400 nm) or UV-ABC (250-400 nm) light. The light exposures were conducted in petri dishes for 2, 4, 8 and 16 h (parallel exposures) and a dark control was also conducted. All samples were subjected to a serial clean-up technique to separate PBDFs from PBDEs, and identification and quantification of PBDFs were made using HRGC/HRMS. The analysis technique was able to identify PBDFs with 6 or fewer bromines; hepta- and octaBDFs were not included in the analysis scheme. After decaBDE:toluene solution was irradiated with UV-A, UV-AB or UV-ABC, 27 mono- to hexaBDFs were detected, with the majority of products being tetra- to hexaBDFs. The PBDFs formed accounted for 0.31% (UV-A), 0.35% (UV-AB) and 1.2% (UV-ABC) of the initial amount of decaBDE on a molar basis and the authors suggested a trend toward greater transformation (but similar transformation products) at lower wavelengths. The authors also concluded that the observed mono- to hexaBDFs were likely the result of stepwise debromination of higher PBDFs since mono-to hexaBDEs were not detected in any of the irradiated solutions. While these findings demonstrate the possibility of decaBDE transformation to PBDFs, they are considered to have low applicability to the natural environment.
Barcellos da Rosa et al. (2003) studied the photolysis of decaBDE dissolved in toluene. DecaBDE (BDE209, Sigma-Aldrich, 98% purity) was dissolved in toluene at a concentration of 0.31 mM and exposed to light from a 500-watt (W) high-pressure xenon lamp. Following light exposure, samples of the decaBDE:toluene solution were analyzed by GC - flame ionization detector (FID) to identify and quantify decaBDE and debrominated congeners down to hexaBDEs. The decaBDE congener was observed to undergo exponential decay with a photolysis rate constant of 3x10-4/s. Several debrominated congeners were observed in the decaBDE:toluene solutions and the degradation of decaBDE was inferred to proceed via sequential debromination to nona-, octa- and heptaBDEs. The authors concluded that while their results demonstrated the potential for and pathway of photolytic debromination, further work to confirm the environmental relevance of photolysis in toluene was needed. Overall, the findings of this study are considered to have limited applicability to the environment.
Rahm et al. (2005) conducted a study to determine the relative susceptibility of a variety of compounds, including decaBDE, to hydrolysis via nucleophilic aromatic substitution. When decaBDE was reacted with sodium methoxide dissolved in methanol, the estimated half-life for the hydrolysis reaction was 0.028 h, indicating a rapid hydrolysis reaction. For lower brominated PBDEs, the rate of reaction decreased by roughly a factor of 10 for each bromine removed, relative to decaBDE. The authors concluded that decaBDE would be susceptible to hydrolysis by nucleophilic compounds in the environment. However, given that sodium methoxide is not normally present at significant concentrations in the environment and that reaction catalyzed by mineral surfaces, enzymes, etc., would be the main reactant for hydrolysis, there is a high level of uncertainty in extrapolating these results to the natural environment.
Geller et al. (2006) report the findings of a photolysis experiment on decaBDE. DecaBDE (BDE 209 congener; 98% purity) was dissolved in 3 mL of tetrahydrofuran (THF) at saturation (10 g/L, also containing additional particulates of solid-phase decaBDE), and irradiated with four lamps for a period of up to 48 h. Following irradiation, samples were analyzed by HPLC and GC-MS using electron impact ionization with identification of degradation products based on retention times. The photolysis products included hepta- to nonaBDEs as well as tri- to hexabromodibenzofurans. Comparison with the chromatogram for 2,3,7,8-substituted dibenzofurans indicated that these were not major degradation products. These findings have similar shortcomings to other studies using organic solvents and are not considered environmentally relevant.
Kuivikko et al. (2007) investigated the photodegradation of decaBDE dissolved in isooctane and combined the findings with a model to predict the photodegradation half-life in two marine systems--the Baltic Sea and the Atlantic Ocean. DecaBDE (BDE 209; purity > 98.3%) was dissolved in isooctane (250 ng/mL), the solution was placed in quartz GC-autosampler vials and the vials were exposed to natural light in Helsinki, Finland, in a shallow pool for 60 minutes on October 5. The concentration of parent decaBDE was analyzed four to five times during the irradiation; each time there were three replicates, a dark control and an isooctane blank. The quantity of decaBDE was analyzed using GC-MS. The concentration of decaBDE decreased according to first-order kinetics, with a half-life of approximately 0.03 d.
Kuivikko et al. (2007) reported a quantum yield for decaBDE of 0.28 ±0.04. The quantum yield was used in model simulations to predict the photodegradation half-life in the Baltic Sea (both surface and 10 m mixing layer) and the Atlantic Ocean (40 m mixing layer only). The model simulations assumed environmentally relevant concentrations (3-4 pg/L and 30-40 pg/L) in the water phase and typical summer solar radiation in the Baltic Sea, and also accounted for light attenuation by dissolved and particulate matter. Kuivikko et al. (2007) predicted mixing zone half-lives of 1.8 d (Baltic Sea) and 0.4 d ( Atlantic Ocean), which were the same for both decaBDE concentrations.
While the model appears to simulate natural conditions, it is important to consider that decaBDE in the water column would be primarily sorbed to suspended particulates rather than dissolved. Thus, although the degradation of the dissolved phase is predicted to be relatively rapid, this degradation would represent only a very small amount of the total decaBDE present in the water column. Therefore, there is uncertainty as to whether the predicted half-lives would be a representative portrayal of the persistence and degradation of decaBDE in the water column.
Ahn et al. (2006a) investigated the photochemical debromination of decaBDE sorbed to major components of soil/sediment/mineral aerosols including clay minerals and noncrystalline metal oxides which are known to have electron transferring capacities. The decaBDE used in the study had a purity of 98%. Test matrices included montmorillonite, kaolinite, organic-carbon-rich natural sediment (16.4% OC content), aluminum hydroxide, iron oxide and manganese dioxide. Each of the decaBDE-amended test matrices (250 mg) was combined with 500 mL water and the mixtures were then irradiated with artificial light or natural sunlight. Irradiation with artificial light used four 24-W lamps with peak output at 350 nm for a period of 14 d. Natural sunlight exposure was conducted between July and November 2004, in West Lafayette, Indiana, for a period of up to 101 d (further exposure in November/December did not result in additional degradation of decaBDE). Samples of decaBDE were quantified using HPLC, while the debrominated products were quantified using GC - electron capture detector (GC-ECD).
The dark and light controls showed no signs of degradation although the study reported minor peaks of nona- and octaBDEs in the dark control which were likely impurities in the decaBDE formulation. For the artificial light exposures, half-lives for montmorillonite, kaolinite, sediment and aluminum hydroxide were 36, 44, 150 and 178 d, respectively. In natural light, half-lives for decaBDE using montmorillonite, kaolinite and sediment were 261, 408 and 990 d, respectively, with negligible degradation on aluminum hydroxide (note that these half-lives represent days of continuous exposure to sunlight, as opposed to actual days including light and dark exposure). Degradation of decaBDE was negligible for iron oxide and manganese dioxide under either lighting scheme. It is important to note that all of these half-lives are longer than the light exposure times, suggesting the potential for uncertainty in these estimates. The half-lives for the natural light exposures suggest that decaBDE photodegradation occurs slowly in the natural setting.
The fastest degradation was observed on montmorillonite and kaolinite and these matrices were the focus for identification of debromination products. Identified products for kaolinite and montmorillonite exposed to sunlight included nonaBDEs (i.e., BDE208, -207, -206) and octaBDEs (BDE197 and -196), as well as trace amounts of tri- to heptaBDEs over longer sunlight exposure times. Higher fractions of tri-to heptaBDEs were also observed for the artificial light exposures. Several unidentified octaBDE products were also observed in the chromatograms. The formation of the identified products was consistent with stepwise debromination, initially forming nona-, then octa- and then heptaBDEs.
Ahn et al. (2006b) investigated metal oxide mediated debromination of decaBDE using birnessite (a naturally occurring manganese oxide mineral) in THF:water and water:catechol reactor systems. The purity of the decaBDE used for the experiments was 98%. The amended birnessite was prepared by combining 0.1 mL of a stock solution containing 1 mg/mL decaBDE dissolved in THF with 50 mg of birnessite in 15-mL test tubes and then air-drying for one day to remove the THF. The first set of experiments examined the degradation of decaBDE sorbed to birnessite in THF:water systems. For these experiments, the decaBDE-amended birnessite was mixed with 5 mL of THF:water solutions at ratios ranging from 0:10 to 10:0. Follow-up experiments examined (i) the reactivity of dissolved decaBDE in THF:water at a ratio of 7:3 by varying the amount of treated birnessite from 0 to 50 mg/mL, and (ii) the role of THF as the hydrogen donor for debromination of decaBDE, determined by measuring the production of succinic acid in the system. All treatments were conducted in triplicate and shaken in the dark for a period of 24 h with subsampling for analysis at various times. A separate set of experiments investigated the degradation of decaBDE sorbed to birnessite in water systems, in the presence of the naturally occurring hydrogen donor, catechol. Catechol was combined (0.003, 0.045 and 46.135 mmol) with 5 mL of water and mixed with 50 mg of decaBDE-amended birnessite. All experimental treatments were shaken in the dark for 23 d with subsampling at various times to track the degradation of decaBDE. DecaBDE (i.e., BDE209) was extracted in THF and analyzed using HPLC while the potential products were quantified using GC-ECD and identified by matching peak retention times with known retention times for lower brominated PBDEs.
The experiments using combined THF:water reactor systems produced rapid degradation of decaBDE with > 75% transformation of decaBDE over the 24-hour period for some THF:water ratios. However, the follow-up experiments determined that THF was reacting with the birnessite to form succinnic acid, and in the process acted as a hydrogen donor for the debromination of decaBDE sorbed to the birnessite. Thus, the relatively rapid observed degradation rates appeared to depend on the presence of THF. Because THF is not normally present in the environment, the rate of decaBDE degradation is not considered realistic with respect to conditions present in the natural environment. The rapid reaction rates in the THF:water experiments did allow for identification of debrominated products. The lower brominated products produced after 24 h included tetra- to nonaBDEs. The reaction appeared to proceed in a stepwise manner with decaBDE being initially degraded rapidly to nonaBDEs, followed by further stepwise debromination to lower brominated PBDEs.
The second set of experiments using water:catechol reaction systems were used to examine potential for decaBDE degradation in the presence of the naturally occurring substance catechol. No significant degradation was observed over the 23-d period in the experiments with 0.003-0.045 mmol catechol. However, slow degradation was observed with 46 mmol catechol, with the mass of decaBDE in the reaction vessels decreasing from approximately 0.1 mmol to 0.085 mmol during the 23-d experiment. Although the products were not quantified for the water:catechol systems, it is possible that they would follow a similar pattern to the THF:water systems. Although the rate of reaction was slow under the simulated natural conditions, it is possible that decaBDE may eventually debrominate over time in the presence of naturally occurring minerals and hydrogen donors, such as catechol.
Stapleton and Dodder (2006) reported the findings of a study of photolytic debromination of decaBDE in house dust. The dust used for the study was a National Institute of Standards and Technology standard reference material (SRM) prepared from vacuum cleaner bag contents from homes, motels and hotels in the U.S. The dust is known to contain PBDE (including decaBDE), and certified PBDE concentrations were available for 15 congeners. Photolysis studies were conducted with both the SRM dust in its existing form and with SRM dust without PBDEs (these were removed by Soxhlet extraction) but then spiked with a known quantity of decaBDE to yield a concentration of 2180 µg/kg dw. Analysis of the “cleaned” dust prior to spiking confirmed that decaBDE was not detected (< 0.2 mg/kg). Samples (0.5 g) of each dust material were placed in UV cuvettes and exposed to natural outdoor sunlight in Gaithersburg, Maryland, between 9 a.m. and 5 p.m. on days when no precipitation was forecast, for a total of 200 h. Three replicates were included for each experiment along with three control samples for each dust material, which were covered in aluminum foil.
The concentrations of decaBDE were found to decrease in both dust materials, with first-order degradation rates of 2.3x10-3/h in spiked dust and 1.7x10-3/h in natural dust, corresponding to half lives of 301 and 408 h in sunlight, respectively. The authors speculated that the longer half-life in natural dust might be due to matrix factors which attenuated the amount of light reaching the decaBDE molecules, or that sorbing materials present in the natural dust were removed by the extraction process, making the spiked decaBDE more available for photodegradation. An additional explanation was that solid decaBDE with a limited surface area for irradiation might be present in the natural house dust. The spiked dust samples were also analyzed for lower brominated degradation products. At the end of the exposure, approximately 38% of the initial decaBDE concentration had been lost or degraded. Part of the loss (i.e., equivalent to approximately 13%) was due to debromination to predominantly the three nonaBDE congeners, but also due to lesser amounts of octa- and heptaBDE congeners. The remaining 25% of the original decaBDE concentration could not be accounted for and was lost to unknown pathways and/or products. The Stapleton and Dodder (2006) study suggested that the presence of BDE201 and -202 may provide a marker for debromination of BDE209. In the OctaBDE commercial mixture, BDE201 makes up a very small component (i.e., from below detection to 0.8%), and BDE203 is not detected.
This study provides reasonably strong evidence that photodegradation of decaBDE on dust can occur under natural conditions in the environment and that lower brominated PBDEs can be formed. The authors noted that while the actual degree of sunlight exposure to household dust might be limited by windows and shading, dust in cars would be subjected to much higher levels of sunlight, making debromination of decaBDE on dust in cars potentially significant.
The Government of the United Kingdom ( United Kingdom 2007a) describes the findings of an additional study by Stapleton (2006b) on the debromination of decaBDE in house dust. The methods were similar to those of Stapleton and Dodder (2006) except that the exposure was carried out during the hours of 9 a.m. to 4 p.m. for up to a total of 90 h. Sunlight exposures were conducted between July and August 2004 at a mean temperature of 27.4 °C. Over the 90-hour exposure period, decaBDE concentration in the house dust decreased from 2180 µg/kg dw to 1570 µg/kg dw, indicating that approximately 28% of decaBDE degraded. The start and end concentrations of decaBDE in the dark control were not statistically different. The half-life for decaBDE was estimated at 216 h (continuous sunlight exposure, or 27 d assuming 8 h sunlight per day). Lower brominated PBDEs were detected as degradation products and these included three nonaBDEs, six octaBDEs and one heptaBDE. The mass balance of decaBDE and lower brominated PBDE congeners indicated that approximately 17% of the original decaBDE was unaccounted for, suggesting the formation of alternative (unidentified) products or volatilization of lower brominated PBDEs.
Gerecke (2006) determined the reaction quantum yield of decaBDE on kaolinite and measured light penetration in this mineral in order to calculate the photodegradation rate of decaBDE. The decaBDE used in the study had a purity of 98%. Light penetration into kaolinite was estimated using the Kubelka-Munk theory, which relies on the measurement of light absorption (k) and scattering (s). Thin layers of kaolinite were prepared on quartz glass with 10 different thicknesses prepared for estimating k and s. The kaolinite was spiked with decaBDE in an isooctane/toluene mixture (95/5, v/v). Although the spiking method is not provided in detail, it is presumed that after spiking the solvent was evaporated off, leaving a solid layer of decaBDE-spiked kaolinite on quartz glass, since the light exposures were conducted for either dry kaolinite or wetted kaolinite. The spiked kaolinite layers were irradiated with sunlight at noon on clear summer days in Dubendorf, Switzerland . The experiments were conducted in a water bath to maintain constant temperature and the study included dark controls as well as precautions to avoid light exposure outside of the specified times. The concentrations of decaBDE and potential products were analyzed using GC/MS.
Sunlight had only very limited penetration in the kaolinite below 50 mm and the authors concluded that only decaBDE sorbed to particles at the very surface of soils would have the potential to undergo photolysis. In the experiments with decaBDE-spiked kaolinite, half-lives of 76 and 73 minutes were determined for dry and wet conditions. However, the observed degradation was non-exponential, likely due to the large difference in degradation rate between the upper and lower sides of the kaolinite layer. Under dry conditions, degradation products were identified as lower brominated PBDEs, whereas for wet conditions, the products were not identified, suggesting that the products were not lower brominated PBDEs. These findings demonstrate that while photodegradation of decaBDE sorbed to mineral solids is possible in the environment, the rate and amount degraded is highly dependent on the penetration of light into soil and mineral layers.
Nose et al. (2007) studied the degradation pathways of standard decaBDE (purity not noted, but purchased from Wako Pure Chemical Industries, Ltd.) during hydrothermal treatment. Their evaluation was carried out in a micro autoclave made of stainless steel filled with 40 mL of distilled water. The decaBDE material was initially dissolved in toluene, and then spiked into the autoclave chamber, which was sealed. Temperature was controlled, with the first heating of 25 min to 300 ºC and a pressure of 8 MPa. The experiment was repeated at different time intervals (0, 10, 30, 60, 120, 240 and 360 min), defined as the processing time. The reaction time was defined as the sum of the heating time and the processing time. The chamber was then cooled down to 100 ºC using a fan, and then soaked in ice water for 20 min.
The study found some decomposition (~45%) after approximately 12 min at 200 °C, and almost complete breakdown (i.e. > 99%) after 10 min at 300 °C. Debromination to nonaBDE was determined; however, debromination of meta and para positions to BDE208 and -207 occurred at a faster pace than debromination of the ortho position to BDE206. The experiment was also conducted with other lower brominated PBDEs with similar findings. Thus, the authors concluded that the reactivities of bromine on the para and meta positions were relatively high, while the reactivity of the ortho bromine was extremely low in hydrothermal treatment. The study also confirmed the formation of PBDD/DFs during the study. While having limited applicability to the natural environment, the study appears to confirm the findings of other studies respecting preferential meta- and para-position debromination (e.g., Stapleton et al. 2006; Gerecke et al 2005; Huwe and Smith 2007a, 2007b). As noted in the Canadian screening assessment on PBDEs (Canada 2006; Environment Canada 2006), under certain combustion/pyrolysis and photolysis conditions, all PBDEs (including DecaBDE) can form brominated dibenzofurans and dibenzo-p-dioxins. These transformation products are brominated analogues of the Toxic Substances Management Policy Track 1 polychlorinated dibenzofurans and dibenzo-p-dioxins. Complete destruction of DecaBDE and any possible breakdown products appears to occur with exposure to temperatures of 800 °C and above for 2 seconds (European Communities 2002).
Li et al. (2007) examined the debromination of decaBDE (DE-83R, > 97% purity obtained from Great Lakes Chemical Company) by resin-bound zerovalent iron nanoparticles. The study involved the use of about 50 test tubes prepared with decaBDE in acetone, mixed with an equal volume of distilled water to form an 8-mL solution. Then 2 g of resin containing zerovalent iron was added. The test tubes were capped and shaken for 1 h to 10 d in a water bath (25 ±0.5 °C). Analyses were performed using GC with an ECD detector, and HRGC/HRMS. The results demonstrated rapid debromination of BDE209 after about 8 h, which the authors determined to follow first-order transformation kinetics resulting in a rate constant of 0.28 ±0.04/h and half-life of 2.5 h. Disappearance of BDE209 occurred, with the subsequent formation of nona- to triBDEs in a sequential manner. All three nonaBDEs appeared in significant amounts within 1 h, steadily increased for 8 h, then decreased below detection after 24 h. The congener pattern dominance shifted to heptaBDEs after 2 d, and then hexaBDEs were most abundant. After 10 d, pentaBDEs were present in significant amounts.
The authors noted that the identification of reaction products was challenging, mainly due to the lack of appropriate PBDE standards at the time of their experiments, with many peaks not matching any of those for 43 available chemical standards. Issues of co-elution were identified. However, all three nonaBDEs were identified with confidence and two of five octaBDE peaks were identified (i.e., BDE197 and -196). The hepta- to tetraBDEs that were confidently identified included BDE183, -153, -154, -99 and -47 (all contain both para-position bromines). Overall, the presence of unidentified peaks makes it difficult to conclusively confirm the positional preference of debromination pathways. Li et al. (2007) also conducted an identical study using PCB209, which dechlorinated at a much slower rate than the debromination of BDE209. After 10 d, only 21% of PCB209 was lost, with only the formation of nona- and octaPCBs identified. Overall, the use of zerovalent iron nanoparticles means that this study has uncertain relevance to the environment.
Kajiwara et al. (2008) examined DecaBDE photolysis in plastics under natural sunlight. High-impact polystyrene (HIPS) was added to toluene containing 100 µg/mL of DecaBDE and shaken overnight to achieve complete dissolution; toluene subsequently evaporated in the dark. Crushed television casing and solidified HIPS samples compounded with the technical mixtures were further pulverized in a liquid nitrogen chamber. Resultant materials were screened with a vibrating stainless-steel sieve apparatus. The fine sieved powder (106-300 µm) was used in the irradiation experiment (samples of HIPS+DecaBDE and TV casing). Aliquots of the powdered plastic were transferred to quartz tubes, sealed with Teflon plugs. A subset of HIPS+DecaBDE samples hydrated with 0.5 mL of hexane-washed water was also prepared to examine the effect of water on PBDE photolysis. Tubes were kept inside the laboratory in the dark at room temperature until the sunlight irradiation experiment was performed.
The HIPS+DecaBDE samples showed a gradual disappearance of BDE209 upon exposure to sunlight. No degradation was found in the dark controls throughout the experiment. After one week of exposure to sunlight, the concentration of BDE209 declined to approximately 50% of the initial level, indicating prompt photodegradation in the plastic matrix. Hydrated HIPS+DecaBDE samples showed more rapid degradation of BDE209 than non-hydrated samples. The authors suggested that this accelerated degradation might be explained by the fact that water played a role as a hydrogen donor, promoting debromination.
Kajiwara et al. (2008) state that previous studies have focused on the photodegradation of PBDE dissolved in solvents or absorbed to particles, and that their study is the first to show the photolysis of BDE209 compounded into a polymer matrix. Assuming a first-order reaction, they report a calculated half-life for BDE209 transformation in HIPS of 51 d. The authors highlight that this half-life for plastic is longer than those measured in sand, sediments and soils but equivalent to the half-lives measured in house dust.
During the study, the HIPS+DecaBDE samples showed that concentrations of hexa- to nonaBDE congeners increased several-fold after one week of exposure, and the authors interpreted that debromination of BDE209 resulted to some extent in the formation of lower brominated PBDE congeners. After this first week, the concentrations of hexa- to nonaBDE congeners remained constant or decreased slightly, despite the continuous decrease in BDE209. Congeners BDE47, -99 and -100 were not detected and stepwise debromination of PBDEs was not clearly observed in this study. At the end of the exposure period, the total PBDE concentration was less than 20% of the initial level, and the proportion of BDE209 to total PBDEs had decreased from 90% to 44%. Kajiwara et al. (2008) reported the photolytic formation of tri- to octaBDFs in their HIPS+DecaBDE samples. The concentration of total PBDFs showed an increase (greater than 40 times) on day 7 of exposure, while BDE209 concentration decreased.
With respect to the TV casing samples, the study showed no disappearance of BDE209 and formation of lower brominated PBDE congeners throughout the 224 d of sunlight irradiation, in contrast to HIPS+DecaBDE samples. Two possible explanations for the differences in degradation profiles were suggested in the study: 1) the difference in initial concentrations of BDE209 (HIPS+DecaBDE and the TV casing samples contained 0.1 and 10% BDE209, respectively) influenced degradation rates; and/or 2) the effects of the other plastic additives in TV casings, such as pigments, UV absorbers, and stabilizers, potentially affected light penetration depth in each plastic sample.
Raff and Hites (2007) examined the importance of photolysis in the removal of PBDEs from the atmosphere by comparing the photolysis frequencies of PBDE congeners to the first-order removal rate constants for oxidant reactions and dry and wet deposition. The study compared atmospheric removal processes affecting PBDE (i.e., focused on BDE47, -99 and -209) fate using a box model simulating the atmosphere above Lake Superior. The model results indicated that photolysis results in the highest loss (90% of the total losses) of BDE-47 and -99 from the atmosphere above Lake Superior. However, for BDE209, more than 90% of the removal is the result of precipitation events. The authors suggested that this explains why the PBDE congener profile found in sediments from Siskiwit Lake (located on a remote island in Lake Superior) and the Great Lakes appear to be enriched in BDE209 compared to other congeners. These results are consistent with congener profiles obtained from air-particle samples collected in the Arctic that showed significant depletion of BDE209 relative to tetra- and pentabrominated PBDE congeners. The main conclusion of the study is that deposition processes control the loss of BDE209 from the atmosphere and are responsible for the enhancement of BDE209 found in sediment samples from the Great Lakes.
Biodegradation
Gerecke et al. (2006) report the findings of follow-up experiments to their 2005 study. The anaerobic biodegradation of technical DecaBDE (98% purity) in digested sewage sludge was investigated using the same test system as Gerecke et al. (2005) but with only single primers, either 2,6-dibromophenol or 4-bromobenzoic acid. The use of each primer resulted in debromination of decaBDE via loss of para-position bromine to BDE208 but the reaction was slow, with a half-life exceeding 700 d. In the absence of a primer, the half-life was longer, up to 1400 d. The authors also conducted field monitoring of decaBDE in a sewage treatment plant to determine whether biodegradation occurred in a full-scale anaerobic digester. Grab samples of influent, reactor and outlet sewage sludge were collected from a WWTP at Dubendorf, Switzerland . The concentration of decaBDE in sludge was observed to decrease between the influent and outlet streams, suggesting that transformation occurred in the full-scale digester. However, the authors cautioned that the relatively short residence time in the reactor (28 d) and low level of replication (i.e., only one set of grab samples) meant that these results should be considered preliminary and not necessarily unequivocal.
The biodegradation of decaBDE (> 98% purity) by cultures of anaerobic bacteria was investigated by He et al. (2006). The cultures of anaerobic bacteria included Dehalococcoides ethenogenes 195, Sulfurospirillum multivorans and Dehalococcoides sp. strain BAV1 (each of which have been shown to dechlorinate organochlorine compounds), as well as an enriched autotrophic culture containing D. ethenogenes 195, and an enrichment containing a number of Dehalococcoides spp. All cultures were grown on ~500 mM trichlorethane (TCE) with the exception of Dehalococcoides sp. strain BAV1, which was grown on vinyl chloride.
The experiments were conducted in 160-mL serum bottles containing a culture medium and carbon source appropriate for each organism. For D. ethenogenes (an autotrophic culture), no carbon source was used. Prior to inoculation the test vessels were sealed to ensure anaerobic conditions and then autoclaved for 25 minutes at 121 °C. To initiate each experiment, 5 mL of a decaBDE stock solution in trichloroethane (TCE, ~1 mM decaBDE) was added to the test vessel for a final concentration of 1 mM TCE and 0.1 mM decaBDE. The bottles were inoculated with one of the active cultures described above at 5% or 10% v/v. The test samples were incubated in the dark at 30 °C for a period of up to 12 months, with weekly sampling of 1 mL of culture for the analysis of decaBDE and degradation products. Each experiment was conducted with two replicates per treatment and repeated at least once to confirm the results. Abiotic control samples were also included in the experimental design. PBDE congeners were detected using GC-ECD.
The experiment with S. multivorans exhibited rapid dechlorination of TCE, but no degradation of decaBDE within the first weeks of incubation. After an additional two months of incubation without replacement of TCE, decaBDE was observed to degrade to non-detectable levels, while octa- and heptaBDEs became detectable in both replicates. No degradation was evident in the abiotic controls, indicating that the disappearance of decaBDE was due to anaerobic degradation. These findings suggest that under appropriate conditions, certain bacteria may debrominate decaBDE. No degradation of decaBDE was seen in the experiments using any of the other cultures.
Parsons et al. (2004) investigated the potential for reductive debromination of decaBDE in anaerobic sediment suspensions. The experiments were conducted using sediment collected at Hansweert in the Western Scheldt, which is known to contain high concentrations of decaBDE. A total of 20 g of the sediment were suspended in 20 mL of anaerobic medium and the suspensions were spiked with decaBDE (14 mg/g sediment) and incubated anaerobically at room temperature in the dark (details of the anaerobic test system were not provided). Sterilized controls were also conducted (details of the sterilization procedure was not provided). At varying intervals over an approximately 205-day test period (read from graph), experimental and control samples were extracted in hexane/acetone, subjected to a clean-up process and analyzed using GC-low resolution mass spectrometry (LRMS).
Concentrations of decaBDE in the spiked samples decreased significantly over the first two months of incubation and the decrease coincided with the appearance of new chromatogram peaks which were tentatively identified from their retention times and mass spectra as nonaBDEs. The concentrations of nonaBDEs were generally too low to be quantifiable and the authors speculated that nonaBDEs were further debrominating to lower brominated PBDE congeners. There is significant uncertainty in these findings with respect to anaerobic sediment debromination since a similar decrease in decaBDE was also found in the sterile controls. The authors suggested that this was due to incomplete control sterilization as confirmed by the production of methane upon addition of lactic, pyruvic and acetic acids to the control preparations. However, it is also possible that photodegradation of decaBDE occurred during sample handling, extraction and analysis. Therefore, although Parsons et al. (2004) found a decrease in decaBDE in anaerobic sediment suspensions, this decrease cannot be attributed unequivocally to anaerobic degradation.
Parsons et al. (2007) report the findings of an additional investigation of reductive debromination of decaBDE in anaerobic sediment microcosms. The experiments were conducted using sediment collected at Hansweert in the Western Scheldt, which is known to contain high concentrations of decaBDE. A total of 10 g of the sediment were suspended in 50 mL of anaerobic medium (containing acetate, lactate and pyruvate). The suspensions were then spiked with decaBDE or individual nonaBDE congeners and incubated at room temperature in the dark. At varying intervals over an approximate 260-day test period (read from graph), experimental and control samples were extracted in hexane/acetone, subjected to a clean-up process and analyzed using GC/MS in selected ion monitoring mode.
There was no measurable decrease in decaBDE in the active and control microcosms, although nonaBDEs were detected in the decaBDE-spiked samples at much higher concentrations than known background levels in the study sediments
Knoth et al. (2007) conducted a monitoring study of PBDEs, including decaBDE, in sewage sludge from 11 municipal WWTPs in Germany . A total of 39 sludge samples from different stages of the treatment process--primary sludge, secondary excess sludge and (dewatered) digested sludge--were collected from March 2002 to June 2003. The samples were subjected to a process of sterilization, freeze-drying, and spiking with stable isotope standards, followed by Soxhlet extraction in toluene, four-column clean-up of the toluene extraction, and reduction to 100 mL. The extracted and cleaned-up sample was analyzed by GC- select ion monitoring (electron ionization) (SIM(EI+))LRMS to quantify decaBDE and lower brominated PBDE congeners.
The congener profiles in the various treatment plants and treatment stages were dominated by decaBDE, which ranged from 97.1 to 2217 ng/g dw. Debromination of decaBDE to lower brominated PBDEs during treatment was either not occurring or too slow to be detected during the total retention time of sludge (11-13 d) in German WWTPs.
La Guardia et al. (2007) examined potential for in vivo and environmental debromination of decaBDE in a WWTP and its receiving environment. Concentrations of decaBDE and lower brominated PBDEs were monitored in sludge, sediments, and fish in the receiving environment of a WWTP located in Roxboro, North Carolina (further study details in Section 2.2). The PBDE congener profile was tracked from WWTP to receiving environment sediments and to biota in order to evaluate whether significant debromination was occurring. For the sludge samples, 17 PBDE congeners were identified in 2002 while 18 PBDE congeners were identified in 2005. The major congener in sludge was BDE209, accounting for 60% (58 800 µg/kg dw) of the total PBDE burden in 2002 and 87% (37 400 µg/kg dw) of the total PBDE burden in 2005. The sludge congener profiles were similar to the technical formulations (PentaBDE and DecaBDE), suggesting minimal debromination in WWTP sludge. In receiving environment sediments the major PBDE congeners included BDE209, -206, -99 and -47, with BDE209 making up > 89% of the total BDE burden in each of the sediment samples. The decaBDE concentration in sediments was highest between 1 and 6 km downstream of the outfall, with maximum levels ranging from 3240 mg/kg OC to 2450 mg/kg OC. The lower brominated PBDE congeners were attributed to the PentaBDE technical formulation, suggesting minimal debromination of decaBDE in surficial sediments.
Tokarz et al. (2008) conducted concurrent PBDE (i.e., BDE209, -99 and -47) experiments using anaerobic sediment microcosms and a cosolvent-enhanced biomimetic system. The sediment microcosms contained natural sediments with no detectable PBDEs collected from Celery Bog Park, West Lafayette, Indiana. The PBDEs were dissolved in a toluene solution and added to sediments, and then the solvent was evaporated off. This mixture was then blended with wet sediments to form a concentration of approximately 5.0 µg/g and 3 µg/g for BDE99 and -47, and BDE29, respectively. The microcosms were fed methanol and dextrose to ensure the formation of anaerobic conditions, and to provide electron donors. Autoclaved control systems were implemented. The biomimetic experiment involved the use of Teflon-capped glass vials with 0.03 mM of BDE209, -99 or -47 mixed with 5.0 mM titanium citrate and 0.2 mM vitamin B12 in 0.33 M TRIZMA buffer solution containing tetrahydrofuran. Controls were used without titanium citrate. Debromination products were identified and quantified using GC-ECD and GC-MS.
Tokarz et al. (2008) notes that cobalamins, such as coenzyme vitamin B12, have the ability to mediate reductive dehogenation of halorganics. Cobalamins from decaying cells have been iscolated from environmental samples. In addition, they indicate that vitamin B12 has ubiquitous presence in the environment in anaerobic micro-organisms.
The biomimetic system demonstrated reductive debromination at decreasing rates with decreasing bromination (e.g., half-life of 18 seconds for BDE209 and almost 60 d for BDE47). In natural sediment microcosms, the half-life for BDE209 was estimated to range from 6 to 50 years, with an average of 14 years, based on observations over 3.5 years. After 8 months, BDE47 decreased approximately 30% without a consistent concurrent increase in daughter debromination products. While complete debromination to diphenyl ether was not ruled out, it appeared unlikely since no intermediate products were identified. The researchers speculated that there could have been formation of hydroxylated and methoxylated derivatives of tetraBDE. The researchers synthesized their data from both systems and proposed major debromination pathways for sediment and biomimetic systems as follows: BDE209 > nonaBDEs (BDE206, -207 -208) > octaBDEs (BDE196, -197) > heptaBDEs (BDE191, -184, 2 unknown heptaBDEs) > hexaBDEs (BDE138, -128, -154, -153) > pentaBDEs (BDE119, -99) > tetraBDEs (BDE66, -47, -49) > triBDEs (BDE28, -17). Specifically, at the end of the 3.5-year study, their analysis of BDE209 degradation in sediments identified BDE208, -197, -196, -191, -128, -184, -138, and -128, as well as 3 unidentified octaBDEs and 2 unidentified heptaBDEs.
The findings of Tokarz et al. (2008) showing prolonged persistence of decaBDE in sediment microcosms are supported by a field study of Eljarrat et al. (2008) and Sellström et al. (2005). Eljarrat et al. (2008) examined the fate of PBDEs in sewage sludge from five municipal WWTPs after agricultural application in Spain in 2005. PBDE concentration in sewage sludge ranged from 197 to 1185 ng/g dw, with BDE209 being the predominant congener, ranging in concentrations from 80.6 to 1083 ng/g dw. Concentrations of BDE209 in soils ranged from 14.6 to 1082 ng/g dw at seven agricultural sites. High concentrations (i.e., 71.7 ng/g dw) were even found at one site that had not received sludge application for four years, illustrating the persistence of BDE209 in soils. Sellström et al. (2005) collected PBDE samples from sites that received past sewage sludge amendments. They sampled soil from three research stations (with reference plots and sewage-sludge-amended plots) and two farms (reference and amended/flooded soils) in Sweden . They determined that decaBDE concentrations in soil ranged from 0.015 to 22 000 ng/g dw. Highest levels were noted at the farm site that had not received amendments for 20 years.
Kohler et al. (2008) examined sediment concentrations and temporal trends of deca-, nona- and octaBDEs utilizing a sediment core from Greifensee, a small lake located near Zürich, Switzerland . In their study, they note that PBDEs first appeared in sediment layers corresponding to the mid-1970s. The authors identified all three nonaBDEs and at least seven octaBDEs (BDE202, -201, -197/204, -198/203, -196/200, -205 and -194) in the lake’s surface sediments using reference materials. While deca- and nonaBDE concentrations were found to increase rapidly with time, the increase of the octaBDE concentrations was slower. Congener patterns of the octa- and nonaBDE congeners in the sediments did not change with time, and thus transformation of PBDEs was not said to be observed in the sediments over the 30-year span. However, the authors noted different relative proportions of nona- and octaBDEs in the sediments in comparison to the commercial PBDE mixtures. Notably, they detected BDE202 which is apparently not reported in any of the PBDE mixture and they speculated that its presence could have been due to transformation of decaBDE. Also, they noted that congener patterns of octaBDE were dissimilar to those of the commercial PBDE mixtures, but similar to patterns in house dust and profiles of photodegradation products of decaBDE. Consequently, they suggested biotic and/or abiotic transformation processes are involved between the release of the technical products into the environment and their final residues in sediments that are responsible for the changes of congener patterns that they observed in the sediments of the Swiss lake.
Zhou et al. (2007) evaluated the ability of white rot fungi to degrade BDE209 in a liquid culture medium, and the effects of Tween 80 and ß -cyclodextrin on the degradation of BDE209 by white rot fungi. White rot fungi have been shown to rapidly oxidize and mineralize many aromatic compounds, including PCBs. In order to improve BDE bioavailability, “solubilizing” agents such as the surfactant Tween 80 or cyclodextrin were added to their test systems. The authors note that the application of this technology is most suited for bioremediation applications. Overall, this study has unknown relevance to environmental conditions.
Zhou et al. (2007) utilized 1 mL of decaBDE (98% pure) in dichloromethane added to 250-mL flasks. The solvent was then allowed to evaporate, resulting in a total BDE209 mass of 16 µg coating the bottom of the flask systems. A 100-mL aliquot of aqueous medium (distilled water, malt extract, glucose, peptone and yeast extract mixture) was added to each flask. White rot fungi were inoculated to the liquid culture and the test system was shaken for 10 d in the dark. Identical test systems were also implemented utilizing additions of Tween 80 and cyclodextrin concentrations ranging from 0 to 900 mg/L. Analyses were conducted using HPLC with a UV detector. The test systems with only white rot fungi added showed a decrease of 42.2% over 10 d in the amount of BDE209 in the test system. The sterile controls showed no significant degradation over time. Tween 80 was found to enhance BDE209 degradation at an appropriate concentration (maximum degradation 96.5% over 10 d). Cyclodextrin was also shown to enhance BDE209 degradation (maximum degradation of 78.4% over 10 d). Transformation products were not identified in this study.
Orihel et al. (2009) presented an ongoing study that may provide some key information on the environmental transformation and aquatic bioaccumulation of decaBDE. Their field mesocosm study compares sediment and periphyton BDE209 concentrations from low (dose=0.039 g of DecaBDE), medium (dose=0.28 g of DecaBDE) and high (dose=2.3 g of DecaBDE) treatments against a control mesocosm. Based on preliminary results, the authors suggest that DecaBDE breakdown had occurred since lower brominated PBDE products were observed in surface sediments as soon as 1 month after DecaBDE addition to mesocosm water. In their study, penta- to hepta-BDEs were observed in the medium- and high-mesocosm treatments, but not in the low-treatment or control mesocosms. DecaBDE was only observed in periphyton in the medium- and high-treatment mesocosms. nonaBDE was observed in periphyton in the high mesocosm after 1 month, but not after 4 months. Hexa-, hepta- and octaBDEs were not detected in periphyton after 1 month, but were after 4 months in the medium- and high-treatment mesocosms. In the coming years, the results will be finalized.
3.2.2 Conceptual Model of Transformation in the Environment
The likelihood, rates and potential products of decaBDE debromination will depend upon the medium in/on which it is present, and the rate of various degradation processes (e.g., photodegradation, abiotic degradation, biodegradation). To illustrate the partitioning properties of decaBDE under various possible release scenarios, Level III fugacity modelling using the CHEMCAN model (Webster et al. 2004) was conducted and parameterized for the “Ontario Mixed Wooded Plain” region. The primary model parameter inputs included
- log Kow = 8.7 (Wania and Dugani 2003; see Appendix C).
- log koa = 12 (maximum value allowed in model, indicating that decaBDE is essentially non-volatile; estimated values for decaBDE exceed 15).
- degradation rates = negligible in air, water, soil and sediment.Footnote 7
Based on its chemical properties, decaBDE is expected to be primarily sorbed to organic fractions. Table 3-1 summarizes the predicted fractions in each medium for releases to air, water or soil. From Table 3-1 it is apparent that
- A large fraction (> 96%) of decaBDE inthe environment is expected to be associated with either soils orsediments (depending on whether the release is to soil or the aquaticenvironment) and, within these bulk compartments, decaBDE is associated almostentirely with the solid phase.
- Although < 3.4% of decaBDE in theenvironment is expected to be associated with bulk air or bulk waterphases, it is expected that of the mass of decaBDE present in bulk air orbulk water > 95% will be sorbed to either aerosol or suspendedsediment.
- Regardless of the release scenario, lessthan 0.1% of decaBDE in the environment is predicted to be found dissolvedin water or in the vapour phase.
Thus, studies that examine the debromination of decaBDE present in soils and sediments or sorbed to particulate and/or biota phases in air or water likely give the best indication of the potential significance of a transformation process in the environment. For studies that examine the debromination of decaBDE in solution or in vapour phase, the significance of transformation is less certain and is likely low since the dissolved/vapour fraction of decaBDE in the environment is predicted to be extremely low. According to Stapleton (2006a),
In the environment, it is expected that BDE 209 will be found primarily bound to solids in the water column, and bound to particles in the atmosphere. Therefore, degradation of BDE 209 dissolved in water (or organic solvents) is not expected to be of environmental relevance.
A further consideration for evaluating the available information on debromination in the environment is how well the experimental conditions represent those that would be expected under natural settings. For example, in experiments that use organic solvents to disperse or dissolve decaBDE, extrapolation to normal environmental conditions is difficult since artificial organic solvents used in laboratory studies are not commonly found in the environment and the solubility of decaBDE in water is much lower than that of organic solvents. Furthermore, organic solvents are often much stronger hydrogen donors than water or other naturally occurring substances, resulting in faster rates and potentially non-representative patterns of decaBDE transformation. In the environment, many different donor molecules would be present, and thus substitution of bromine by hydrogen is unlikely to be the sole route of transformation. This creates uncertainty as to how well degradation studies that use organic solvents would represent expected degradation of decaBDE in the environment. Other experimental conditions also require scrutiny to ensure relevance to those that may be seen in the environment. For instance, experiments using natural sunlight or simulated natural sunlight are likely to better represent natural conditions than those using artificial light. It is also not likely that materials such as zerovalent iron nanoparticles, specialized microbial cultures and fungal cultures will be found in great abundance in the environment.
In general, laboratory studies examining decaBDE transformation were frequently conducted under conditions that were favourable for decaBDE debromination and are not necessarily representative of typical conditions in the environment. Thus, while some transformation of decaBDE in the environment is plausible, data showing accumulations of decaBDE in the environment (e.g., see Canada 2006; Environment Canada 2006b) also imply recalcitrance.
Table 3-1: Predictions of the CHEMCAN Model for DecaBDE in the "Ontario Mixed Wooded Plain" Region
| 1 kg/year to waterFootnote a |
1 kg/year to soilFootnote a |
1 kg/year to airFootnote a |
|
|---|---|---|---|
| Mass in bulk air (kg) | 1.27E-07 | 2.17E-07 | 2.23E-03 |
| Fraction of total mass in bulk air | 1.46E-06% | 7.64E-08% | 3.70E-03% |
| In bulk air,fraction as vapour | 0.21% | 0.21% | 0.21% |
| In bulk air,fraction sorbed to aerosol | 99.79% | 99.79% | 99.80% |
| Mass in bulk water (kg) | 0.31 | 0.31 | 9.49E-02 |
| Fraction of total mass in bulk water | 3.38% | 0.11% | 0.16% |
| In bulk water,fraction dissolved in water | 0.12% | 0.01% | 0.12% |
| In bulk water,fraction sorbed to suspended particles | 95.25% | 95.35% | 95.25% |
| In bulk water,fraction accumulated in fish | 4.63% | 4.63% | 4.64% |
| Mass in bulk soil (kg) | 3.32E-03 | 274.36 | 57.59 |
| Fraction of total mass in bulk soil | 0.00% | 96.80% | 95.35% |
| In bulk soil,fraction in gas phase in air spaces | 0.00% | 0.00% | 0.00% |
| In bulk soil,fraction dissolved in soil water | 0.00% | 0.00% | 0.00% |
| In bulk soil,fraction sorbed to soil solids | 100.00% | 100.00% | 100.00% |
| Mass in bulk sediment (kg) | 8.78E+00 | 8.78 | 2.71 |
| Fraction of total mass in bulk sediment | 96.60% | 3.10% | 4.49% |
| In bulk sediments,fraction dissolved in porewater | 0.00% | 0.00% | 0.00% |
| In bulk sediments,fraction sorbed to sediment solids | 100.00% | 100.00% | 100.00% |
Figure 3-1 integrates the available information regarding possible environmental debromination pathways for decaBDE in a conceptual model while Table 3-2 provides a list of the products which are potentially formed from each debromination process. A tabulated summary of the environmental debromination information is provided in Appendix E.
Studies focusing on decaBDE sorbed to particulates or solid phases conducted in the absence of organic solvents, with natural sunlight, may provide a realistic indication of decaBDE transformation in the environment. Figure 3-2a summarizes the photodegradation pathways for decaBDE sorbed to particulate or solid phases. DecaBDE (i.e., BDE209) sorbed to dust or other dry minerals and particulates appears susceptible to relatively rapid transformation, with half-lives ranging from 76 min (decaBDE sorbed to a thin film of kaolinite; Gerecke 2006) to 408 h (decaBDE sorbed to house dust; Stapleton and Dodder 2006). Transformation appears to follow stepwise reductive debromination to form hexa- to nonaBDEs. A few studies also indicated alternative reaction pathways to either tetra- and pentaBDFs or unidentified products.
DecaBDE (i.e., BDE209) sorbed to particulates in aqueous systems appears to photodegrade at a generally slower rate, with half-lives ranging from 73 minutes (decaBDE sorbed to a thin film of kaolinite in presence of water; Gerecke et al. 2006) to 990 d (decaBDE sorbed to sediment organic carbon and exposed to sunlight; Ahn et al. 2006a). In addition, some studies have also demonstrated significant degradation (up to 71%) over 60-72 h. The identified products included hexa- to nonaBDEs and some lower brominated PBDEs. The potential for photolytic transformation of the sorbed decaBDE appears to be modulated by the ability of sunlight to penetrate water and the sorbing matrix. Gerecke et al. (2006) determined that light penetrated only 50 mm into kaolinite, suggesting that the photodegradation of decaBDE would be mainly limited to that amount of the chemical present on exposed surfaces. In surface waters, sunlight reaching suspended and bottom sediments would also experience some level of attenuation. Thus, it can be concluded that while photodegradation on solid phases can occur at significant rates, only a very small fraction of the total decaBDE in the environment (i.e., adsorbed to particulates or on solid surfaces) which has contact with sunlight would be susceptible to photodegradation. Also, depending on matrix characteristics (e.g., shielding capacity) and level of exposure to sunlight, photodegradation will be limited.
In addition to photodegradation, decaBDE sorbed to solids may also be subject to biodegradation processes in aquatic sediments, soils or WWTPs. Figure 3-2b summarizes biodegradation pathways for decaBDE. The results of biodegradation studies are somewhat mixed. Early studies (MITI 1992; CMABFRIP 2001) focused on decaBDE mineralization and these indicated very little, if any, degradation; however, these studies did not specifically determine the production of potential transformation products. Tokarz et al. (2008) showed that decaBDE in natural sediment microcosms had a half-life that ranged from approximately 6 to 50 years, with an average of 14 years. In laboratory studies using activated sludge, Gerecke et al. (2005, 2006) determined half-lives ranging from 693 to 1400 d depending on the presence/absence of primer, and identified debromination products as octa- and nona-BDEs. In a separate study, He et al. (2006) observed complete transformation of decaBDE to hepta- and octaBDEs over 2 months with one anaerobic culture (Sulfurospirillum multivorans) but negligible degradation with other anaerobic cultures. The monitoring studies of WWTP sludge (Knoth et al. 2007; La Guardia et al. 2007) provide little evidence of decaBDE debromination in WWTPs, possibly because the residence time in WWTPs is too short for significant debromination to be observed. In addition, sediment microcosm studies by Parsons et al. (2004, 2007) fail to provide evidence of significant debromination, although Parsons et al. (2007) did identify small amounts of nonaBDEs. Thus, while the experimental conditions of the activated sludge studies are environmentally relevant, it is possible that the rates of degradation are too slow, or the cultures used are too specific, for the observed debromination to be significant in the environment. Overall, it appears that photodegradation may be more significant than biodegradation for decaBDE sorbed to solids.
Figure 3-2: Conceptual Model of Possible Environmental Transformation Pathways for DecaBDE
A) Photodegradation on Particulates (Dust, Soil, Aerosol and Sediment) in Dry and Aqueous Systems
- Photodegradation of DecaBDE sorbed to dust, aerosol, suspended sediment and other solids occurs faster than biodegradation but slower than photodegradation in solution, likely due to limited light penetration into solid surfaces.
- > 95% of DecaBDE in bulk air and bulk water will be sorbed to either aerosol or suspended sediment.
- However, < 3.4% of DecaBDE in the environment is expected to be associated with bulk air or bulk water, reducing the significance of photodegradation on solid phases within these comparmtents.
- DecaBDE sorbed to dust and other surfaces indoors, outdoors and in automobiles could have significant light exposure and potentially significant photodegradation.
- DecaBDE at soil surfaces could also be subject to light exposure and photodegradation.
- Photodegradation of DecaBDE sorbed to solid phases may be the most important degradation pathway in the environment.
- Other pathways leading to unidentified products could also occur, including further debromination/transformation of PBDEs or PBDFs and ring cleavage.

- Biodegradation occurs slowly in activated sludge experiments and may be limited in sediments and soils with less ideal biodegradation conditions.
- However, a large fraction (>96%) of decaBDE in the environment is expected to be associated with either soils or sediments, depending on the compartment of release.
- In soil and sediments, biodegradation will be the primary transformation pathway since light penetration would be limited, reducing photodegradation.
- Given the low rates of biodegradation, the significance of these pathways in the environment as a whole is uncertain.
- Although not shown on the figure, it is also possible that other, unidentified products may be formed including substances formed via pathway of ring cleavage.

- Photodegradation occurs rapidly for decaBDE in solution which is irradiated by sunlight or artificial light.
- However, less than 0.1% of decaBDE in the environment is predicted to be dissolved in solution.
- Therefore, although the reaction is rapid, photodegradation in solution is not expected to result in significant debromination of decaBDE in the environment as a whole.
- Other pathways leading to unidentified products could also occur, including further debromination/transformation of PBDEs or PBDFs and ring cleavage.

- Abiotic degradation may occur for decaBDE in the presence of reducing agents or sorbed to minerals in the presence of natural hydrogen donors such as catechol (although the observed degradation in the presence of catechol is slow). Most laboratory studies demonstrating abiotic degradatin (other than photolysis) appear to have limited relevance to the environment.
- Hydrolysis has also been demonstrated but it is unknown whether this would occur under normal environmental conditions.
- Because there are relatively few studies of abiotic degradation, the significance of abiotic processes is highly uncertain.
- It is possible that abiotic degradation could be a significant, albeit slow, process in soils and sedements where light does not penetrate and biodegradation occurs slowly.
- Other pathways leading to unidentified products could also occur, including ring cleavage.

Table 3-2: Summary of DecaBDE Transformation Products Observed in Laboratory Studies with some Relevance to Environmental Settings
| Congener Group Formed |
Tar- geted for Virtual Elimi- nation under CEPA 1999 |
DecaBDE Transformation Process | ||||
|---|---|---|---|---|---|---|
| Photodegradation Sorbed to Solids in Dry and Aqueous Systems (references in brackets) |
Photode- gradation Dissolved in Natural Solvents (references in brackets) |
Photode- gradation Dissolved in Artificial Organic SolventsFootnote b (references in brackets) |
Biodegradation (references in brackets) |
Abiotic Degradation (references in brackets) |
||
| nonaBDEs | X (Söderstrom et al. 2004, Jafvert and Hua 2001Footnote c, Hua et al. 2003, Ahn et al. 2006a, Stapleton and Dodder 2006, Stapleton 2006b, Kajiwara et al. 2008) | X (Geller et al. 2006) | X (Watanabe and Tatsukawa 1987, Palm et al. 2003, Bezares-Cruz et al. 2004, Eriksson et al. 2004, Geller et al. 2006, Barcellos da Rosa et al. 2003) | X (Gerecke et al. 2005, He et al. 2006, Gerecke et al. 2006, Kajiwara et al. 2008Footnote d&Footnote e) | X (Keum and Li 2005, Ahn et al. 2006bFootnote b, Tokarz et al. 2008Footnote b) | |
| octaBDEs | X (Söderstrom et al. 2004, Jafvert and Hua 2001Footnote c, Hua et al. 2003, Ahn et al. 2006a, Stapleton and Dodder 2006, Stapleton 2006b, Kajiwara et al. 2008) | X (Geller et al. 2006) | X (Watanabe and Tatsukawa 1987, Palm et al. 2003, Bezares-Cruz et al. 2004, Eriksson et al. 2004, Geller et al. 2006, Barcellos da Rosa et al. 2003) | X (Gerecke et al. 2005, He et al. 2006, Kajiwara et al. 2008Footnote d&Footnote e) | X (Keum and Li 2005, Ahn et al. 2006bFootnote b, Tokarz et al. 2008Footnote b) | |
| heptaBDEs | X (Söderstrom et al. 2004, Jafvert and Hua 2001Footnote c, Ahn et al. 2006a, Stapleton and Dodder 2006, Stapleton 2006b, Kajiwara et al. 2008) | X (Geller et al. 2006) | X (Watanabe and Tatsukawa 1987, Palm et al. 2003, Bezares-Cruz et al. 2004, Eriksson et al. 2004, Geller et al. 2006, Barcellos da Rosa et al. 2003) | X (He et al. 2006, Kajiwara et al. 2008Footnote d&Footnote e) | X (Keum and Li 2005, Ahn et al. 2006bFootnote b) | |
| hexaBDEs | X | X (Söderstrom et al. 2004, Jafvert and Hua 2001Footnote c, Ahn et al. 2006a, Kajiwara et al. 2008) | X (Geller et al. 2006) | X (Watanabe and Tatsukawa 1987, Palm et al. 2003Footnote a.1, Bezares-Cruz et al. 2004, Eriksson et al. 2004Footnote a.1) | X (Kajiwara et al. 2008Footnote d&Footnote e) | X (Keum and Li 2005, Ahn et al. 2006bFootnote b, Tokarz et al. 2008Footnote b) |
| pentaBDEs | X | X (Ahn et al. 2006aFootnote a.1) | X (Watanabe and Tatsukawa 1987, Bezares-Cruz et al. 2004) | X (Kajiwara et al. 2008Footnote e) | X (Keum and Li 2005, Ahn et al. 2006bFootnote b, Tokarz et al. 2008Footnote b) | |
| tetraBDEs | X | X (Ahn et al. 2006aFootnote a.1) | X (Watanabe and Tatsukawa 1987, Bezares-Cruz et al. 2004) | X (Kajiwara et al. 2008Footnote e) | X (Keum and Li 2005, Ahn et al. 2006bFootnote b, Tokarz et al. 2008Footnote b) | |
| triBDEs | X (Ahn et al. 2006aFootnote a.1) | X (Watanabe and Tatsukawa 1987, Bezares-Cruz et al. 2004) | X (Kajiwara et al. 2008Footnote e) | X (Tokarz et al. 2008Footnote b) | ||
| pentaBDFs | X (Kajiwara et al. 2008) | |||||
| heptaBDFs | X (Kajiwara et al. 2008) | |||||
| hexaBDFs | X (Kajiwara et al. 2008) | X (Hagberg et al. 2006) | ||||
| triBDFs | (Söderstrom et al. 2004, Kajiwara et al. 2008) | X (Geller et al. 2006, Hagberg et al. 2006) | ||||
| tetraBDFs | X (Söderstrom et al. 2004, Kajiwara et al. 2008) | X (Geller et al. 2006, Hagberg et al. 2006) | ||||
| triBDFs | (Kajiwara et al. 2008) | X (Geller et al. 2006, Hagberg et al. 2006) | ||||
| diBDFs | X (Hagberg et al. 2006Footnote a.1) | |||||
| monoBDFs | X (Hagberg et al. 2006Footnote a.1) | |||||
| Unidentified products | X ( Stapleton and Dodder 2006, Stapleton 2006b, Gerecke 2006, Kajiwara et al. 2008) | X (Palm et al. 2003) | ||||
BDEs - brominated diphenyl ethers; BDFs - brominated dibenzofurans
The results of Gerecke (2006) have not been included because the decaBDE degradation products were described as lower brominated PBDEs, with no indication of which PBDEs were formed.
The results of Parsons et al. (2004) have not been included because degradation to nona- and lower brominated PBDEs was observed in both the control and treatment groups.
Although photodegradation experiments with decaBDE dissolved in water potentially represent environmentally relevant conditions, the significance of any observed debromination is uncertain since only a very low fraction of decaBDE would be present in the dissolved phase. Figure 3-2c provides a conceptual model of photodebromination pathways for decaBDE dissolved in water/solvent systems. Two of the associated studies are notable because the results are potentially relevant to the natural environment. Eriksson et al. (2004) observed relatively rapid transformation (half-life = 6.4 h) of decaBDE in water/humic acid mixtures with hexa- to nonaBDEs formed. Because artificial light was used, there is some uncertainty whether the rate would be as fast in natural sunlight. Kuivikko et al. (2006, 2007) observed a photodegradation half-life of 0.3 d for decaBDE in isooctane but conducted modelling to estimate half-lives ranging from 0.2 to 1.8 d in Baltic Sea and Atlantic Ocean surface waters.
Laboratory-based studies on the transformation of decaBDE provide support for a conclusion that transformation to lower BDEs and BDFs should be occurring in the environment. However, there is a dichotomy between the findings of these studies and the results of monitoring studies in terms of realistic support for significant transformation of decaBDE in the environment. If these laboratory-based studies provide accurate depictions of transformation in the environment, one would expect to find similarities in the patterns observed in the environment and those described as transformation products of decaBDE in laboratory studies. One would expect to see widespread occurrence of the decaBDE transformation products in various environmental media, such as sediments. The Canadian screening assessment on PBDEs (Canada 2006) showed how monitoring studies are dominated by congener profiles that are consistent with the PentaBDE and OctaBDE commercial mixtures. The United Kingdom (2007a) notes that the available monitoring data provide limited circumstantial evidence for a correlation between concentrations of decaBDE and lower brominated PBDEs in some environmental matrices (e.g., citing the findings of Voorspels et al. (2006a, 2007b). The United Kingdom (2007a) also notes that, in the study by Gerecke et al. (2005), biodegradation appeared to preferentially remove the para-position bromines from the BDE molecule, but that these PBDEs are not routinely observed in the environment.
Besides photodegradation and biodegradation, decaBDE is susceptible to abiotic debromination in the absence of light exposure as shown under laboratory conditions. Figure 3-2d summarizes the transformation pathways for these other abiotic degradation processes. Keum and Li (2005) observed up to 90% transformation of decaBDE dissolved in deionized water over 40 d in the presence of reducing agents such as zerovalent iron, iron sulphide and sodium sulphide. Li et al. (2007) showed complete disappearance after 8 h in a water-acetone system containing nanoscale zerovalent iron, while Rahm et al. (2005) observed a half-life of 0.028 h for decaBDE dissolved in methanol and reacted with sodium methoxide. Similar to photodegradation studies using organic solvents, there is uncertainty as to how well these studies represent natural conditions and whether such processes may take place under natural conditions.
3.2.3 Implications for Bioaccumulation Assessment
The existing laboratory evidence for the transformation and debromination of decaBDE through environmental processes such as photodegradation, abiotic degradation and biodegradation indicates potential transformation pathways which lead to the formation of
- lower brominated PBDEs;
- brominated dibenzofurans; and
- unidentified degradation products.
However, the laboratory studies also indicate that both the lower brominated PBDEs and PBDFs would, themselves, be susceptible to further degradation and the actual significance of the formation of these products is unknown. Palm et al. (2003) and Eriksson et al. (2004) have shown that the rate of transformation of PBDEs decreases as the number of bromine atoms/molecule decreases. With decreasing bromination, diphenyl ethers have a lessened overlap with absorption spectra of wavelengths >290 nm, which is the range of the solar spectrum at ground level, and thus it is expected that they would be less susceptible to decay than decaBDE. Since lower brominated PBDEs demonstrate slower rates of phototransformation, it could be speculated that in a system with a single input of decaBDE, a steady-state build-up of lower brominated PBDEs could occur. However, some level of continued photodegradation down to the biphenyl ether could also likely occur. The magnitude of the build-up would be difficult to predict since the formation and subsequent degradation of PBDEs depends on their relative rates of formation and degradation in the environment, both of which are not known. In addition, the magnitude of accumulation would depend on decaBDE loading rates to the environment, which are also difficult to predict (United Kingdom 2004).
In order to evaluate whether these decaBDE transformation products have the potential to bioaccumulate or biomagnify in food webs, the BAF and BMF models from Section 3.1 will be used (see Appendix D for model input values). This analysis assumes that the transformations of decaBDE observed in the laboratory studies discussed in this report are also occurring in the natural environment; however, it is acknowledged that this is a subject of uncertainty. As noted earlier, in the absence of metabolism, the BAFs of hexa- to nonaBDEs are predicted to exceed 5000. When metabolic transformation is considered (a more realistic scenario), BAF estimates for hexaBDEs to nonaBDEs still exceed 5000, but are orders of magnitude lower.
BAF estimates for brominated dibenzofuran products were also undertaken. For these transformation products, there was no information that would allow for estimation of kM, and a value equal to that estimated for decaBDE was chosen for illustrative purposes. It is likely that PBDFs would undergo further metabolism; however, the actual rates of metabolism are unknown. Although the chosen value and resulting BAFs represent reasonable hypothetical predictions, it is important to acknowledge that these predictions are uncertain. Further detail of log Kow and kM estimates are provided in Appendix D. In the absence of metabolism, the BAFs for penta- and hexaBDFs are predicted to exceed 5000 for the middle trophic level fish, while BAF predictions for tri- and tetraBDFs are below 5000. With consideration given to metabolic transformation (a more realistic scenario), the status of the BDFs with respect to the BAF criterion of 5000 remains unchanged, but values are somewhat lower overall.
In the absence of metabolism, the BMFs of hexa- to nonaBDEs are predicted to be very high, ranging from ~130 to ~150. However, when metabolic transformation (a more realistic scenario) is considered, the predicted BMFs for hexa- to nonaBDEs ranged from 5 to 6. The metabolism-corrected BMFs exceeded 1 for all metabolites, primarily due to a relatively high dietary assimilation efficiency calculated for these substances. For the BDF products, a similar BMF of 120 to 150 for penta- and hexaBDFs was predicted, while BMFs predictions for tri- and tetraBDFs were 8 and 45, respectively. When corrected for metabolism, the BMF values ranged from 4 to 6 (a similar kM value was used for all BDFs).
The modelling analysis of potential environmental transformation products of decaBDE predicted BAFs that predominantly exceed 5000 and BMFs exceeding 1 for practically all of the metabolites, because of relatively high dietary assimilation efficiency and optimal log Kow and log koa values for bioaccumulation via passive diffusion. This suggests concerns that the transformation products formed in the environment as a result of the photodegradation, biodegradation, and possibly abiotic degradation, of decaBDE, are potentially bioaccumulative and biomagnifying in food webs, and may result in increased exposure and risk to upper trophic level organisms.