
Determination of ammonia in whole tobacco: T-302
1 Scope of application
1.1
Applicable to the isolation and quantification of ammonia in whole tobacco by High Performance Liquid Chromatograpy (HPLC).
2 Normative references
2.1
Health Canada Official Method T-115. Determination of Tar, Water, Nicotine and Carbon Monoxide in Mainstream Tobacco Smoke, 2016.
2.2
Health Canada Official Method T-402. Preparation of Sample for Testing of Cigarettes, Tobacco Sticks, Cigarette Tobacco, Cigars, Little Cigars, Kreteks, Bidis, Leaf, Pipe and Smokeless Tobacco, 2016.
2.3
International Organization for Standardization, ISO 8243 Cigarettes - Sampling. 2013.
2.4
International Organization for Standardization, ISO 15592-1 Fine-Cut Tobacco and smoking articles made from it - Methods of sampling, conditioning and analysis - Part 1: Sampling. 2001.
3 Definitions
3.1
Refer to T-301 for definitions of terms used in this document.
4 Method summary
4.1
Tobacco is placed into a vial and lyophilized for 48 hours. The dried tobacco is then ground using a screen on a bench-top grinder.
4.2
The freeze-dried ground tobacco is weighed into a culture tube with cap. The sample is extracted into diluted H2SO4 on a wrist-action shaker. This mixture is then filtered into a scintillation vial for storage from which an aliquot is taken for analysis by cation exchange chromatography.
4.3
The sample is injected onto a cation exchange analytical column that uses a carboxylic acid/phosphonic acid functional group to achieve separation of ammonium and monovalent cations. In order to adequately resolve sodium from the ammonium cation for quantification, a methane sulphonic acid solution is used as the mobile phase. After the ammonium ion has eluted, a gradient using H2SO4 is used to remove any divalent cations and quaternary amines that may be present in the sample.
4.4
Quantification is obtained using the peak area response of ammonium sulphate using a conductivity detector.
Warning: The testing and evaluation of certain products against this test method may require the use of materials and/or equipment that are potentially hazardous and this document does not purport to address all the safety aspects associated with its use. Anyone using this test method has the responsibility to consult with the appropriate authorities and to establish health and safety practices in conjunction with all existing applicable regulatory requirements prior to its use.
5 Apparatus and equipment
5.1
Freeze dryer, Lyophilizer or equivalent.
5.2
Wrist-action shaker.
5.3
Analytical balance measuring to at least 4 decimal places.
5.4
Bench top grinder with #40 (40 sections /square inch) screen.
5.5
Glass fibre syringe filter, 25 mm × 0.45 µm or equivalent.
5.6
Volumetric flasks, 25, 50, and 100 mL.
5.7
Disposable syringe, 5 mL or equivalent.
5.8
Screw top vials with aluminum lined caps, 7 mL or equivalent.
5.9
Septa, 8 mm, blue TFE/SIL, 60 MIL or equivalent.
5.10
Autosampler vials and caps, 2 mL or equivalent.
5.11
High Performance Liquid Chromatograph (HPLC) consisting of:
5.11.1
Refrigerated autosampler.
5.11.2
Conductivity detector, Dionex or equivalent.
5.11.3
Cation trap, Dionex or equivalent.
5.11.4
Conductivity suppressor, Dionex or equivalent.
5.12
Cation exchange analytical column, 250 mm × 4 mm, Dionex IonPac or equivalent.
5.13
Cation exchange guard column, Dionex IonPac or equivalent.
5.14
Polymethylpentene (PMP) Erlenmeyer flasks with screw top caps, 125 ml, and/or 16 mL culture tubes with High Density Polyethylene (HDPE) screw top caps (depending on extraction weights and volumes used) or equivalent.
6 Reagents and supplies
6.1
All reagents shall be at least analytical reagent grade.
Note: Wherever possible, reagents are identified by their Chemical Abstract Service [CAS] registry numbers in square brackets.
6.2
Ammonium Sulphate - [7783-20-2] > 99 % purity.
6.3
Methanesulphonic Acid (MSA) - [75-75-2] 100 % purity.
6.4
Sulphuric Acid - [7664-93-9] > 96 % purity.
6.5
Water, Type I (as outlined in ASTM D1193, Table 1: Processes for Reagent Water Production, Note A).
7 Preparation of glassware
7.1
Clean and dry glassware in a manner to ensure that contamination from residues on glassware does not occur.
7.2
Immediately prior to use, rinse all extraction tubes with 0.1N H2SO4, then 3 times with Type I water.
8 Preparation of solutions
8.1
Sulphuric Acid, 0.10N - Stock Standards Solution
8.1.1
Carefully add 5.108 g of H2SO4 (96 % w/w) to 900 mL of Type I water.
8.1.2
Mix and dilute to 1 L with Type I water.
8.2
Sulphuric Acid, 0.025N - Extraction Solution
8.2.1
Carefully add 1.277 g of H2SO4 (96 % w/w) to 900 mL of Type I water.
8.2.2
Mix and dilute to 1 L with Type I water.
8.3
Sulphuric Acid, 0.20N - Solution C (Ion Chromatography)
8.3.1
Carefully add 10.216 g of H2SO4 (96 % w/w) to 900 mL of Type I water.
8.3.2
Mix and dilute to 1 L with Type I water.
8.4
MSA 0.003N - Solution A (Ion Chromatography)
8.4.1
Carefully add 0.2883 g of methanesulphonic acid (MSA) to 900 mL of Type I water.
8.4.2
Mix and dilute to 1 L with Type I water.
9 Preparation of standards
9.1
Primary Ammonium Stock
9.1.1
Weigh 0.20 g of ammonium sulphate into a 50 mL volumetric flask.
9.1.2
Dissolve in 0.1N H2SO4.
9.1.3
Make up to volume with 0.1N H2SO4.
9.1.4
Prepare a stock solution fresh every 10 days.
Note: This corresponds to a 1.0898 mg/mL NH4+ ion stock solution.
9.1.5
Working Standards
| Standards No. |
Volume of Standard (µL) |
Final Volume (mL) |
Concentration [µg/mL] |
|---|---|---|---|
| 0 | 0 | 25 | 0.000 |
| 1 | 250 | 25 | 10.898 |
| 2 | 175 | 25 | 7.6283 |
| 3 | 75 | 25 | 3.2693 |
| 4 | 75 | 50 | 1.6346 |
| 5 | 50 | 100 | 0.5449 |
| 6 | 20 | 100 | 0.2180 |
Note: All run standards are made to volume to have a 0.025N H2SO4 concentration.
Note: Prepare fresh solutions every 5 days.
Note: All weights, volumes, and purity must be recorded and used to accurately calculate the standard concentrations.
Note: Additional standards may have to be prepared to cover the range of anticipated responses for test samples.
10 Sampling
10.1
The sampling of tobacco products for the purpose of testing shall be in accord with ISO 8243 as specified.
10.2
The sampling of kreteks, little cigars, bidis, tobacco sticks for the purpose of testing shall be in accord with ISO 8243, but modified such that the term "cigarette" is substituted with "kreteks", "little cigars", "bidis" or "tobacco sticks", whereby the term "carton" is equivalent to 200 units.
10.3
The sampling of cigars for the purpose of testing shall be in accord ISO 8243, but modified such that the term "cigarette" is substituted with "cigar", whereby 200 units of cigarette is equivalent to 200 grams of cigar.
10.4
The sampling of cigarette tobacco for the purpose of testing shall be in accord with ISO 15592-1.
10.5
The sampling of leaf tobacco, pipe tobacco or smokeless tobacco shall be in accord with ISO 15592-1 but modified such that the term "fine-cut" is substituted with "leaf tobacco", "pipe tobacco" or "smokeless tobacco".
11 Tobacco product preparation
11.1
The initial preparation of tobacco products for the purpose of testing shall be as specified in T-402.
12 Sample preparation
12.1
Freeze dry the whole tobacco in a lyophilizer for a minimum of 48 hours.
12.2
Mill the whole tobacco in a bench-top grinder to a 40 mesh size.
12.3
Weigh 100 mg of ground tobacco into a 16 mL culture tube.
Note: Depending on the amount of sample available, the amount of sample used for analysis may be modified as long as the sample appears homogeneous and the ratio of sample weight to extraction solution remains the same (i.e. 0.1 g to 10 mL solution; 1:100).
12.4
Add 10 mL of extraction solution and shake for 60 minutes on a wrist action shaker.
12.5
Allow the mixture to settle (approximately one hour) and filter the solution through a syringe filter into an 8 mL storage vial noting to rinse the vial initially with approximately 1 mL of sample.
12.6
Pipette 250 µL of the filtrate directly into a 2 mL autosampler vial then add 1000 µL of extraction solution (1:5 dilution).
Note: The actual dilution is dependent upon the anticipated level in the test sample. The filtrate may require different dilutions (or none at all) to fall within the prepared calibration range. All dilutions are made with the extraction solution.
Note: Dilutions are not required to be performed in this manner. Dilutions using volumetric flasks may be used (but are more time consuming and susceptible to contamination).
13 Sample analysis
13.1
Dionex ED-40 Conditions
- Suppressor Conductivity (SRS):
- 100 mA
- Scale:
- 20 µS
- Output:
- Offset
- Offset:
- 1% of Full Scale
Note: These settings are detector-dependent and may have to be modified in order to achieve a linear response over the range of concentrations for the analyte of interest.
Note: Suppression may or may not be required, dependent on anticipated levels.
13.2
Autosampler: Injection Volume
13.2.1
Analyze using a 20 µL injection.
13.3
Column Temperature: 30 °C
13.4
Mobile Phase / Gradient Conditions (Tertiary Gradient System)
- Solvent A:
- 0.003N MSA
- Solvent B:
- Type I water
- Solvent C:
- 0.2N H2SO4
- Flow:
- 1.0 mL/minute
| Time (min) |
Composition | ||
|---|---|---|---|
| A (%) |
B (%) |
C (%) |
|
| 0.00 | 100 | 0 | 0 |
| 24.00 | 100 | 0 | 0 |
| 24.01 | 50 | 39 | 11 |
| 40.00 | 50 | 39 | 11 |
| 42.00 | 99 | 1 | 0 |
| 45.00 | 99 | 1 | 0 |
| 45.00 | Method End Action: | Equilibrate | |
Equilibration Time: 9.00 minutes
Note: Adjustments to the gradient may be required, depending on instrument and column conditions as well as the resolution of the analyte peak.
14 Calculations
14.1
Determination of Response Factor (RF)
14.1.1
Prepare a calibration curve by plotting the concentration of NH4+ ion in the standard versus the peak area response from the conductivity detector.
Note: Inject the first standard a minimum of 2 times until the response and retention time are constant.
14.1.2
The RF is the slope of the line as determined by linear regression (area counts/unit concentration).
Note: A quadratic calibration curve may be required at higher concentrations.
14.2
Determination of Ammonium Ion
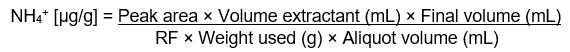
Determination of Ammonium Ion: Text description
NH4+ [µg/g] = Peak area × Volume extractant (mL) × Final volume (mL)
divided by
RF × Weight used (g) × Aliquot volume (mL)
where the aliquot volume (mL) is the volume transferred to the autosampler vial.
Note: Results are calculated on a 'dry matter' basis since a dried sample is used for the weight.
14.3
Determination of Ammonia
NH3 [µg/g] = NH4+ [µg/g] × 17/18
where 17/18 corrects for molecular weight
15 Quality control
15.1
For a typical chromatogram, see Appendix 1.
15.2
Typical Control Parameters
Note: If the control measurements are outside the tolerance limits of the expected values, appropriate investigation and action must be taken.
15.2.1
Laboratory Reagent Blank (LRB)
To detect potential contamination during the sample preparation and analysis processes, include a laboratory reagent blank (LRB). The LRB consists of all reagents and materials used in performing the analysis on test samples and is analyzed as a test sample.
15.2.2
Laboratory Fortified Blank (LFB)
To detect potential loss of analyte during the sample preparation and analysis processes, include a laboratory fortified blank (LFB). The LFB consists of all reagents and materials used in performing the analysis on test samples plus fortification with a known concentration of at least one of the analytes of interest. The level of fortification should reflect the range of typical results for that sample. The LFB is then analyzed as a test sample.
15.2.3
Laboratory Fortified Matrix (LFM)
To detect potential matrix interferences, include a laboratory fortified matrix (LFM). During the sample preparation and/or analysis processes, divide a test sample and fortify an aliquot with at least one of the analytes of interest in known concentration. The level of fortification should reflect the range of typical results for that sample. The LFM is then analyzed as a test sample.
15.2.4
Laboratory Control Sample
To assess the overall performance of an analysis, a control sample is analyzed. The results of the control sample should be compared, using appropriate statistical techniques, to 'expected values' generated by the laboratory or, if none exist, to values found in literature. This provides information to the laboratory, on test accuracy and precision.
15.2.5
Standard as Sample
To assess the stability of the analytical system, a standard is analyzed as a sample. The results of this standard should be compared, using appropriate statistical techniques, to expected concentrations.
15.3
Recoveries and Levels of Contamination
15.3.1
Typical recoveries of Laboratory Fortified Blanks (LFB) are 60-100 % and of Laboratory Fortified Matrix (LFM) are 95-120 % when a spiked solution (or sample) is carried out through the entire extraction process.
15.3.2
Typical Laboratory Reagent Blanks (LRB) have a value of 2 ± 8 µg/g. Contamination is usually associated with inadequate cleaning of glassware.
15.4
Limit of Detection (LOD) and Limit of Quantification (LOQ)
15.4.1
The LOD can be determined as 3 times the standard deviation of results obtained by analyzing the lowest standard level a minimum of 10 times over several days.
15.4.1.1
A typical value for LOD determined in this manner is 18 µg/g.
15.4.2
The LOQ can be determined as 10 times the standard deviation of results obtained by analyzing the lowest standard level a minimum of 10 times over several days.
15.4.2.1
A typical value for LOQ determined in this manner is 60 µg/g.
15.5
Stability of Reagents and Samples
15.5.1
Primary standards should be prepared fresh every 10 days and stored at 4 °C.
15.5.2
Run standards should be prepared fresh from the stock solution weekly and stored at 4 °C.
15.5.3
Samples are stable for approximately one week if kept in a sealed vial at 4 °C.
16 References
16.1
Dionex Corporation. IonPac CS12A Analytical column, installation instructions and troubleshooting guide. Document No. 031132: Revision 01. 1995.
16.2
Nanni, E. J. et al. 1990. Separation and quantitation of monovalent anionic and cationic species in mainstream cigarette smoke aerosols by high-performance ion chromatography. Journal of Chromatographic Science. 28: 432-436.
16.3
Risner, C. H. and Conner, J. M. 1991. Collection of ammonia in indoor air by means of a weak cation exchange cartridge. Environmental Toxicology and Chemistry. 10: 1417-1423.
16.4
ASTM International, ASTM Standard D1193-06(2011). Standard Specifications for Reagent Water.
Appendix 1
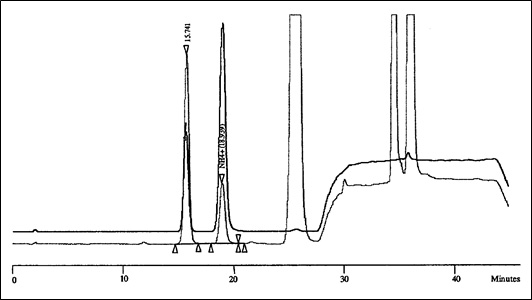
A Typical Chromatogram - An overlay of a standard (dark line) and a reference tobacco sample (faint line) with a 5% offset.: Text description
This figure demonstrates a typical chromatogram for the overlay of a standard and a tobacco reference sample with a 5% offset.