
Determination of nitrate in whole tobacco: T-308
1 Scope of application
1.1
This method is applicable to determine nitrate in whole tobacco by an automated continuous flow colorimetric analyzer.
1.2
This method does not distinguish between nitrate and nitrite found in tobacco since all nitrate is converted to nitrite in the analysis.
2 Normative references
2.1
Health Canada Official Method T-402. Preparation of Sample for Testing of Cigarettes, Tobacco Sticks, Cigarette Tobacco, Cigars, Little Cigars, Kreteks, Bidis, Leaf, Pipe and Smokeless Tobacco, 2016.
2.2
Health Canada Official Method T-115. Determination of Tar, Water, Nicotine and Carbon Monoxide in Mainstream Tobacco Smoke, 2016.
2.3
International Organization for Standardization, ISO 8243 Cigarettes - Sampling. 2013.
2.4
International Organization for Standardization, ISO 15592-1 Fine-Cut tobacco and smoking articles made from it - Methods of sampling, conditioning and analysis - Part 1: Sampling. 2001.
2.5
AOAC INTERNATIONAL, AOAC Official Method 968.07 Nitrogen (Nitrate and Nitrite) in Animal Feed, Colorimetric Method. Official Methods of Analysis of AOAC INTERNATIONAL 19th Ed., 2012.
3 Definitions
3.1
Refer to T-301 for definitions of terms used in this document.
4 Method summary
4.1
Ground sample is extracted with acetic acid and then filtered.
4.2
The extract is then analyzed by an automated continuous flow colorimetric analyzer, where each sample undergoes on-line dialysis to remove interferences.
4.3
The nitrate ion is reduced upon reaction with hydrazine in the presence of copper.
4.4
The nitrite formed is reacted with sulfanilamide under acidic conditions to yield a diazo compound which when mixed with N -(1-napthyl)-ethylenediamine forms a red coloured complex.
4.5
A single channel colorimeter monitors the eluant. Nitrate found in whole tobacco is quantified by comparison to an external standard calibration.
4.6
The analysis should be completed within 24 hours of extraction.
Warning: The testing and evaluation of certain products against this test method may require the use of materials and/or equipment that are potentially hazardous and this document does not purport to address all the safety aspects associated with its use. Anyone using this test method has the responsibility to consult with the appropriate authorities and to establish health and safety practices in conjunction with all existing applicable regulatory requirements prior to its use.
5 Apparatus and equipment
5.1
Robot Coupe Model RSI 2V scientific batch processor or equivalent.
5.2
Analytical balance measuring to at least 4 decimal places.
5.3
Polymethylpentene (PMP) Erlenmeyer flasks with screw-caps, 125 mL or equivalent.
5.4
Wrist-action shaker or equivalent.
5.5
Disposable syringe, 5 mL.
5.6
Syringe filter, 0.45 µm PVDF or equivalent.
5.7
Continuous flow auto-analyzer (or equivalent) with:
5.7.1
Autosampler.
5.7.2
Peristaltic pump.
5.7.3
Nitrate manifold designed for auto-analyzer used (See figure 1).
5.7.4
Single channel colorimeter equipped with 15 mm flow cell and 550 nm filter.
5.8
Volumetric flasks with ground glass joints, 100 mL.
5.9
Graduated cylinders, 50 mL.
5.10
Glass filter funnel.
5.11
Magnetic stirrer.
5.12
Variable volume pipettor or equivalent, 1000 µL (for standards preparation).
5.13
Sample cups for autoanalyzer.
5.14
Volumetric flask, 1 L.
6 Reagents and supplies
6.1
All reagents shall be at least analytical reagent grade.
Note: Wherever possible, reagents are identified by their Chemical Abstract Service [CAS] registry numbers in square brackets.
6.2
Acetic Acid - [64-19-7] glacial, HPLC grade.
6.3
Brij-35 - [9002-92-0] 30 % w/v in water.
6.4
Cupric Sulphate Pentahydrate - [7758-99-8].
6.5
Hydrazine Sulphate - [10034-93-2].
6.6
Hydrochloric Acid - [7647-01-0].
6.7
Methanol - [67-56-1].
6.8
N-1-Napthylethylenediamine Dihydrochloride - [1465-25-4].
6.9
Ortho Phosphoric Acid - [7664-38-2] 98 %.
6.10
Potassium Nitrate - [7757-79-1].
6.11
Sodium Acetate, anhydrous - [127-09-3].
6.12
Sodium Hydroxide - [1310-73-2].
6.13
Sodium Nitrite - [7632-00-0].
6.14
Sulfanilamide - [63-74-1].
6.15
Triton X-100 - [92046-34-9].
6.16
Water, Type I (as outlined in ASTM D1193, Table 1: Processes for Reagent Water Production, Note A).
7 Preparation of glassware
7.1
Clean and dry glassware in a manner to ensure that contamination from residues on glassware does not occur.
8 Preparation of solutions
Note: The large volumes of solution described in these preparations may not be necessary if only small volumes of sample are to be analyzed. However, it is important to maintain the proper concentrations of solutions prepared.
8.1
50 % Triton X-100 Solution
8.1.1
Mix 50 mL Triton X-100 with 50 mL methanol. Transfer to a storage bottle.
8.2
Colour Reagent
8.2.1
Add 700 mL of Type I water to a 1 litre volumetric flask.
8.2.2
Add 100 mL of concentrated phosphoric acid.
8.2.3
Dissolve 40 g of sulfanilamide in the above solution. Heat if necessary to dissolve completely.
8.2.4
Add 2.0 g of N-1-napthylethylenediamine dihydrochloride.
8.2.5
Allow the mixture to cool to room temperature, add 0.5 mL Brij-35 and make to volume with Type I water.
8.2.6
Store at 4 °C in an amber bottle. The solution is stable for one month.
8.3
Copper Sulphate Solution
8.3.1
Cupric Sulphate Stock Solution
8.3.1.1
Dissolve 1.25 g of cupric sulphate in Type I water and make to 500 mL.
8.3.2
Add 4.0 mL of the cupric sulphate stock solution to Type I water in a 1 litre volumetric flask.
8.3.3
Add 0.5 mL Brij-35 and make to volume with Type I water.
8.4
Hydrazine Sulphate Solution
8.4.1
Hydrazine sulphate stock solution:
8.4.1.1
Dissolve 25.0 g of hydrazine sulphate in 800 mL of Type I water. Dilute to 1 litre. The stock solution is stable for 6 months if kept in a tightly stoppered amber bottle.
8.4.2
Add 36 mL of stock hydrazine sulphate to 900 mL of Type I water in a one litre volumetric flask.
8.4.3
Add 0.5 mL Brij-35 and make up to volume with Type I water.
8.4.4
Store in an amber bottle. This is stable for one month.
8.5
0.2N Sodium Hydroxide
8.5.1
Dissolve 8.0 g of sodium hydroxide in about 800 mL of Type I water.
8.5.2
Add 0.5 mL Brij-35, allow the solution to cool and make to one litre with Type I water.
8.6
Acetate Buffer (pH 3.7)
8.6.1
Dissolve 16.4 g of anhydrous sodium acetate in about 300 mL of Type I water.
8.6.2
Add 100 mL of acetic acid, 1 mL of 50 % Triton X-100 solution and dilute to one litre.
8.7
Dialyzer Solution
8.7.1
Dilute 1 mL of Brij-35 to 1000 mL with Type I water.
8.8
Tobacco Extraction Solution (5% Acetic Acid)
8.8.1
Dilute 50 mL of glacial acetic acid to 1000 mL with Type I water.
8.9
1.0N Hydrochloric Acid
8.9.1
Carefully add 83 mL of hydrochloric acid to about 800 mL of Type I water in a one litre volumetric flask. Mix well.
8.9.2
Cool and make to volume with Type I water.
8.9.3
This solution is used for the cleaning of the copper sulphate lines of the autoanalyzer to prevent the build-up of copper between projects or groupings of analyses.
9 Preparation of standards
9.1
Primary Standard Potassium Nitrate (2000 ppm Nitrogen)
9.1.1
Dissolve 14.442 g of desiccated potassium nitrate in about 800 mL of 5 % acetic acid extraction solution (in a one litre volumetric flask).
9.1.2
Dilute to volume with 5 % acetic acid extraction solution.
9.2
Potassium Nitrate Calibration Standards
9.2.1
Take appropriate volumes (0.20-3.0 mL) of the nitrate primary stock solution and dilute to 100 mL with the extraction solution to give calibration standards with approximate nitrogen (from nitrate) concentrations in the ranges detailed below:
| Standard No. |
Volume Primary KNO3 Standard (mL) |
Final Volume (mL) |
Nitrogen [ppm] |
|---|---|---|---|
| Blank 0 | 0 | 100 | 0.0 |
| 1 | 0.2 | 100 | 4 |
| 2 | 0.5 | 100 | 10 |
| 3 | 1.0 | 100 | 20 |
| 4 | 2.0 | 100 | 40 |
| 5 | 3.0 | 100 | 60 |
Note: Additional standards may have to be prepared to cover the range of anticipated responses for test samples.
9.2.2
Prepare nitrate calibration standards fresh every 5 days.
9.3
Primary Stock Sodium Nitrite (2000 ppm Nitrogen)
9.3.1
Dissolve 9.852 g of desiccated sodium nitrite in about 800 mL of 5 % acetic acid extraction solution (in a one litre volumetric flask).
9.3.2
Dilute to volume with 5 % acetic acid extraction solution.
9.4
Sodium Nitrite Calibration Check Standard
9.4.1
Take 2.0 mL of the nitrite primary stock solution and dilute to 100 mL with the extraction solution to give a 40 ppm nitrogen (from nitrite) calibration standard.
9.4.2
Use immediately to check the reduction efficiency.
9.4.3
Prepare nitrite calibration standards fresh as required.
10 Sampling
10.1
The sampling of cigarettes for the purpose of testing shall be in accord with ISO 8243.
10.2
The sampling of kreteks, little cigars bidis, tobacco sticks for the purpose of testing shall be in accord with ISO 8243, but modified such that the term "cigarette" is substituted with "kreteks", "little cigars", "bidis" or "tobacco sticks", whereby the term "carton" is equivalent to 200 units.
10.3
The sampling of cigars for the purpose of testing shall be in accord ISO 8243, but modified such that the term "cigarette" is substituted with "cigar", whereby 200 units of cigarette is equivalent to 200 grams of cigar.
10.4
The sampling of cigarette tobacco for the purpose of testing shall be in accord with ISO 15592-1.
10.5
The sampling of leaf tobacco, pipe tobacco or smokeless tobacco shall be in accord with ISO 15592-1 but modified such that the term "fine-cut" is substituted with "leaf tobacco", "pipe tobacco" or "smokeless tobacco".
11 Tobacco product preparation
11.1
Preparation of Test Sample
11.1.1
The preparation of tobacco products for the purpose of testing shall be as specified in T-402.
11.1.2
Analyze the test samples on an 'as received' basis and correct the results to a 'dry matter' basis, if required, by determining the moisture in the homogenized product.
12 Sample preparation
12.1
Extraction of Whole Tobacco
12.1.1
Weigh 0.5 g of the test sample into a 125 mL PMP Erlenmeyer flask with cap.
12.1.2
Add 50 mL of 5 % acetic acid to the flask and cap.
12.1.3
Clamp the flasks onto the armature of the wrist-action shaker and agitate for 15 minutes.
12.1.4
Filter the whole tobacco extract directly into appropriately labelled autoanalyzer sample cups using a syringe filter attached to a 5 mL disposable syringe.
13 Sample analysis
13.1
Instrument Analysis
13.1.1
Continuous Flow Analyzer Setup
Note: This setup is specific to a Technicon AutoAnalyzer system. The use of a different manufacturer's system may require different flows and/or components. (See figure 1.)
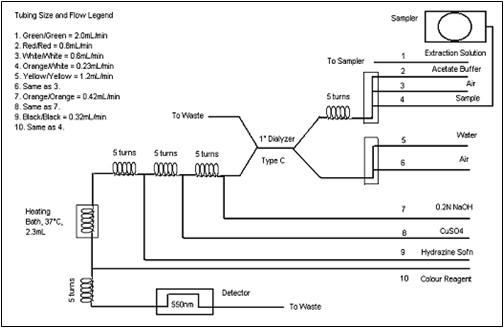
Figure 1: Typical Flow Diagram for a Technicon AutoAnalyzer: Text description
The following figure is a schematic flow diagram of a Technicon AutoAnalyzer, including the detector and heating bath connected on the left of the primary dialyzer, and the sampler on the right of the dialyzer.
Tubing Size and Flow Legend
- Green/Green = 2.0 mL/min
- Red/Red = 0.6mL/min
- White/White = 0.6mL/min
- Orange/White = 0.23mL/min
- Yellow/Yellow = 1.2mL/min
- Same as 3.
- Orange/Orange = 0.42mL/min
- Same as 7.
- Black/Black = 0.32mL/min
- Same as 4.
- Extraction Solution
- Acetate Buffer
- Air
- Sample
- Water
- Air
- 0.2N NaOH
- CuSO4
- Hydrazine Solution
- Colour Reagant
13.1.2
Reduction Efficiency Check
Check each working hydrazine solution for efficient reduction capacity. Run both the 40 ppm nitrate and the nitrite standards. If the apparent nitrogen from nitrate concentration (response) is much lower than the nitrogen from nitrite concentration, adjust the working hydrazine solution by adding 1 mL aliquots of the hydrazine stock solution until the 2 standards show equal responses.
13.1.3
Continuous Flow Analysis
13.1.3.1
Operate the autosampler in the normal manner.
Example: a sampling rate of 20 per hour with a 2:1 sample to wash ratio.
Note: Operating conditions may vary depending on the autoanalyzer being used.
13.1.4
Allow sufficient time for the system to become stable with the reagents being pumped.
13.1.5
Load the sample vials onto the autosampler so that calibration standards are first in the queue, followed by a baseline correction.
13.1.6
Rerun the samples only if the analyte response is out of range or there was a particular problem with the analysis.
13.1.7
Carry out on-line dialysis on the samples.
13.1.8
Place sampling cups containing only 5 % acetic acid (extraction solution) at regular intervals to allow for baseline correction.
13.1.9
The 5 % extraction solution is also used as the sample wash.
14 Calculations
14.1
Construct a Calibration Curve
Prepare a calibration curve for nitrogen by plotting the concentration of the standards versus their respective peak heights in order to determine a response factor (RF).
14.2
Sample Quantification
Quantify the amount of nitrate nitrogen in the extracted solutions by an external standard method, by multiplying the height response of the solution by the response factor (RF).
Nitrate Nitrogen [µg/mL] = Sample Height × RF
14.3
Determination of Nitrate Nitrogen (μ g/g)
Calculate the concentration of nitrate nitrogen automatically by entering the correct multiplier (overall volume the original sample is diluted to (50 mL)) and divisor (the original sample weight (0.5 g)) to find the µg/g:

Figure 14.3: Text description
Nitrate Nitrogen [µg/g] = ppm nitrate nitrogen (µg/mL) × 50 (mL)
divided by
0.5 g sample
14.4
Calculate the nitrate nitrogen on a mg/g basis by using the following:

Figure 14.4: Text description
Nitrate Nitrogen (mg/g) = ppm nitrate nitrogen (µg/g)
divided by
1000 µg/g
14.5
All the results are expressed on an 'as received' basis. These may be expressed on a 'dry matter' basis using the appropriate moisture result.
14.6
All results may be reported as nitrate (mg/g) or nitrate (µg/g) by multiplying the nitrate nitrogen results by 4.43 (the relative weight of nitrate to nitrogen). The results may be expressed in percent (Nitrate (%)).
Nitrate (µg/g) = Nitrate Nitrogen (µg/g) × 4.43
Nitrate (mg/g) = Nitrate Nitrogen (mg/g) × 4.43
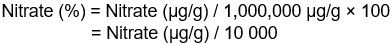
Figure 14.6: Text description
Nitrate (%) = Nitrate (µg/g) / 1,000,000 µg/g × 100
Nitrate (%) = Nitrate (µg/g) / 10 000
15 Quality control
15.1
Typical Control Parameters
Note: If the control measurements are outside the tolerance limits of the expected values, appropriate investigation and action must be taken.
15.1.1
Laboratory Reagent Blank (LRB)
To detect potential contamination during the sample preparation and analysis processes, include a laboratory reagent blank (LRB). The LRB consists of all reagents and materials used in performing the analysis on test samples and is analyzed as a test sample.
15.1.2
Laboratory Fortified Blank (LFB)
To detect potential loss of analyte during the sample preparation and analysis processes, include a laboratory fortified blank (LFB). The LFB consists of all reagents and materials used in performing the analysis on test samples plus fortification with a known concentration of at least one of the analytes of interest. The level of fortification should reflect the range of typical results for that sample. The LFB is then analyzed as a test sample.
15.1.3
Laboratory Fortified Matrix (LFM)
To detect potential matrix interferences, include a laboratory fortified matrix (LFM). During the sample preparation and/or analysis processes, divide a test sample and fortify an aliquot with at least one of the analytes of interest in known concentration. The level of fortification should reflect the range of typical results for that sample. The LFM is then analyzed as a test sample.
15.1.4
Laboratory Control Sample
To assess the overall performance of an analysis, a control sample is analyzed. The results of the control sample should be compared, using appropriate statistical techniques, to 'expected values' generated by the laboratory or, if none exist, to values found in literature. This provides information to the laboratory, on test accuracy and precision.
15.1.5
Standard as Sample
To assess the stability of the analytical system, a standard is analyzed as a sample. The results of this standard should be compared, using appropriate statistical techniques, to expected concentrations.
15.2
Recoveries and Levels of Contamination
15.2.1
A typical LRB is 0.01 ± 0.02 mg/g.
15.2.2
A typical LFB recovery falls in the range 85-115 % recovery.
15.2.3
A typical LFM recovery falls in the range 90-120 % recovery.
15.3
Limit of Detection (LOD) and Limit of Quantification (LOQ)
15.3.1
The LOD can be determined as 3 times the standard deviation of results obtained by analyzing the lowest standard level a minimum of 10 times over several days.
15.3.1.1
A typical value for LOD is 0.15 mg/g nitrate as analyzed.
15.3.2
The LOQ can be determined as 10 times the standard deviation of results obtained by analyzing the lowest standard level a minimum of 10 times over several days.
15.3.2.1
A typical value for LOQ is 0.51 mg/g nitrate as analyzed.
15.4
Stability of Reagents and Supplies
15.4.1
Stock hydrazine sulphate is stable for up to 6 months when stored at room temperature in a tightly stoppered amber bottle.
15.4.2
Hydrazine sulphate working solution is stable for up to one month when stored at room temperature.
15.4.3
Prepare sodium nitrite stock and working standard daily due to the instability of the solutions.
15.4.4
The sulfanilamide colour reagent is stable for up to one month when stored at 4 °C in an amber bottle.
15.4.5
Prepare all primary stock nitrate standards fresh weekly.
15.4.6
Prepare all nitrate working standards and extraction solvents fresh weekly.
15.4.7
Analyze all samples within 48 hours of extraction if the extract is stored in the dark at 4 °C.
16 References
16.1
Bran & Leubbe Inc. Technicon Industrial Systems Corp. Nitrate in Tobacco Extracts. Industrial Method No. 838–87T. 1025 Busch Parkway, Buffalo Grove, Illinois, USA 60090–4516. August 1987.
16.2
ASTM International, ASTM Standard D1193-06(2011). Standard Specifications for Reagent Water.