
Reference method for PCDDs and PCDFs in pulp and paper mill effluents: section 6
Section 6: Gas Chromatographic/Mass Spectrometric Analysis
6.1 Instrumentation
Requirements for the gas chromatograph (GC) are: that it exhibits isothermal temperature stability of ± 0.2°C, or better, over its specified range of operation; that it have capability to accomodate a minimum of three temperature ramps; and that it be configured for use with a capillary column. The capillary column is directly coupled to a high-resolution, double-focussing mass spectrometer (MS) of any geometry. The MS is operator-programmed for time-sequenced acquisition of selected MS data for each congener group. The MS is operated in the electron impact (EI) and selected ion monitoring (SIM) modes.
6.2 Gas Chromatograph (GC) Parameters
Optimum operating conditions must be achieved for both the gas chromatographic separation and the subsequent mass spectrometric analysis of the separated constituents.
To achieve acceptable gas chromatographic separation on a 60 metre DB-5 (polymethyl [5% phenyl] silicone), or equivalent column (see Subsection 6.3), the following set of parameters can be used as a starting point: Injector temperature: 300°C for split-splitless, or ambient for on-column; Interface temperature: 290°C; Temperature program: l) Initial temperature of 100°C for split-splitless or 70°C for on-column and hold for 1 minute; 2) 100 (or 70) to 200°C at 40 °C min-1; 3) 200 to 235°C at 3 °C min-1; 4) hold at 235°C for 10 min.; 5) 235 to 310°C at 8 °C min-1; and 6) hold at 310°C for 15 min.
Optimum settings for GC parameters and appropriate retention time windows for time-sequenced SIM mode analysis of PCDDs/PCDFs on a 60 metre DB-5 column are established from the analysis of Window Defining Mixtures containing the first and last eluting congeners within each homologue group of analytes. The order of elution, as shown in Table 2, is such that five retention windows can be defined, corresponding to the five levels of chlorine substitution (4 Cl to 8 Cl), without any overlap. Under optimum conditions, the interval between the latest eluting TCDD and TCDF congeners and the earliest eluting P5CDF congeners is no more than 0.1 minute. Parameter settings that produce a 2,3,7,8-TCDD retention time of 25 minutes or more will normally represent conditions under which the 4 Cl/5 Cl gap is optimized and the chromatographic performance criterion described in the following subsection can be satisfied.
| Homologue | First Eluting Isomer | Last Eluting Isomer | Retention Time Window* (min) |
|---|---|---|---|
| * On the basis of temperature program given in Subsection 6.2. | |||
| TCDD | 1,3,6,8- | 1,2,8,9- | 25.0 - 29.7 |
| TCDF | 1,3,6,8- | 1,2,8,9- | 23.4 - 29.7 |
| P5CDD | 1,2,4,6,8-/1,2,4,7,9- | 1,2,3,8,9- | 31.5 - 34.0 |
| P5CDF | 1,2,4,6,8-/1,3,4,6,8- | 1,2,3,8,9- | 30.0 - 34.2 |
| H6CDD | 1,2,4,6,7,9-/1,2,4,6,8,9- | 1,2,3,4,6,7- | 35.6 - 37.3 |
| H6CDF | 1,2,3,4,6,8- |
1,2,3,4,8,9- | 35.1 - 37.8 |
| H7CDD | 1,2,3,4,6,7,9- |
1,2,3,4,6,7,8- | 39.9 - 40.7 |
| H7CDF | 1,2,3,4,6,7,8- |
1,2,3,4,7,8,9- | 39.5 - 41.3 |
| OCDD | 47.0 | ||
| OCDF | 47.3 | ||
6.3 Isomer-Specific Separation
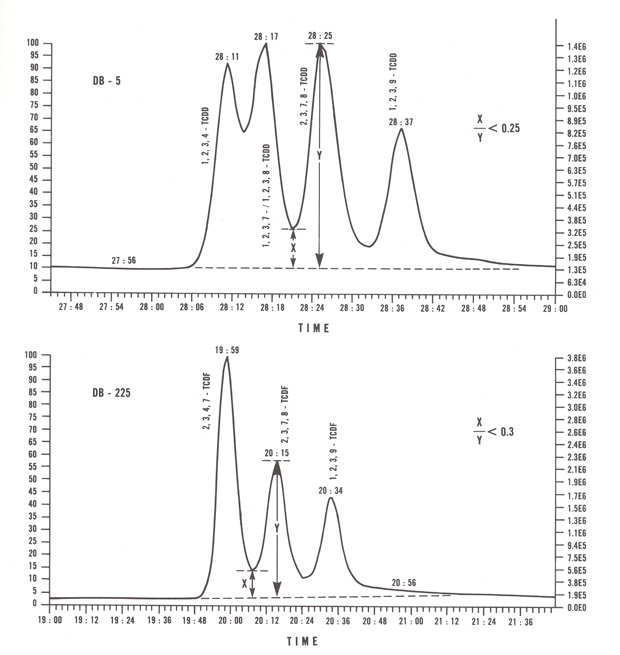
Using helium as carrier gas at an appropriate linear velocity, and with an appropriate oven temperature program, a 60 metre DB-5 column (0.25 mm ID, 0.25 μm film thickness) can adequately separate 2,3,7,8-TCDD from neighbouring isomers 1,2,3,7-/1,2,3,8- and 1,2,3,9-TCDD. In addition, this column easily separates to baseline both hepta-CDD isomers and the four hepta-CDF isomers. However; on a DB-5 column, 2,3,7,8-TCDF cannot be resolved from its neighbouring isomers (i.e., 1,2,4,9-, 2,3,4,8- and 2,3,4,6-). In order to accurately quantify any 2,3,7,8-TCDF that may be present in a sample, analysis on a second column is required. Use of a second column is only mandatory when results from the DB-5 run indicate that the concentration of 2,3,7,8-TCDF may equal or exceed the regulatory level. On a 30 metre DB-225 column, 2,3,7,8-TCDF can be resolved from its neighbouring 2,3,4,7- and 1,2,3,9-isomers.
A 60 metre DB-Dioxin column can also be used as an alternative for 2,3,7,8-TCDD and 2,3,7,8-TCDF analysis because this column is capable of separating 2,3,7,8-TCDD from neighbouring isomers 1,2,4,6-/1,2,4,9- and 1,2,3,7-/1,2,6,8-TCDD, and 2,3,7,8-TCDF from neighbouring isomers 2,3,4,7- and 2,3,4,8-TCDF. However, it cannot be used effectively for homologue analysis.
It is important to recognize that most of the 2,3,7,8-substituted congeners cannot be uniquely identified on a DB-5 column, even under optimum conditions. For this (and any other) column, all of the congeners that would be required to verify the chromatographic separation of individual 2,3,7,8-substituted penta- and hexa-CDD/CDF congeners from their nearest neighbours are not commercially available at the present time. Therefore, amounts reported for target penta- and hexa-CDD/CDF congeners represent maximum possible concentrations. Results for these target analytes must be properly flagged to warn about the possibility of other co-eluting isomers.
Acceptable chromatographic performance, illustrated in Figure 5, is determined by analyzing a Column Performance Mixture containing either 2,3,7,8-TCDD or 2,3,7,8-TCDF, and its nearest neighbours, at equal concentrations. On a 60 metre DB-5 column, the height of the valley between the most closely eluting peaks must not exceed 25% of the 2,3,7,8-TCDD peak height. On a 30 metre DB-225 column, neither valley must exceed 30% of the 2,3,7,8-TCDF peak height. Daily verification of acceptable chromatographic resolution is required.
Figure 5: Acceptable Chromatographic Separation for 2,3,7,8-TCDD and 2,3,7,8-TCDF
Click to enlarge
{kind=link}

If the appropriate valley criterion is not met, it should be evident whether or not minor adjustment of chromatographic parameters alone will be sufficient to bring performance into specification. If that is not the case, it is usually possible to restore performance without replacing the column. Whenever the column is disconnected at either end while trying to restore chromatographic performance, or for any other reason, all other GC and MS performance specifications (see Subsection 6.2 and Subsection 6.4) must be re-established or re-verified before proceeding with sample analysis, and recalibration is required (see Subsection 6.6).
Other well-documented and commercially available GC columns, which are not described in this method, may also be used if the user can demonstrate that the appropriate valley criterion has been met.
Window settings and column performance can be assessed in a single run by combining the appropriate mixtures. By comparing the native or labelled 2,3,7,8-TCDD retention time in subsequent standard and sample runs to that obtained in the combined window/performance run, the fine cutoff point between the tetra and penta windows can be adjusted accurately to compensate for any retention time drift that might occur over the course of a day.
6.4 Mass Spectrometer (MS) Parameters
The mass spectrometer is operated under electron impact (EI) conditions. It is recommended that the electron energy be optimized for detection of PCDD/PCDF molecular cluster ions. This value is expected to be in the range of 28 to 40 eV.
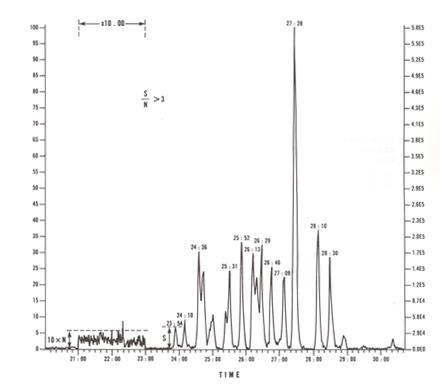
Accurate mass measurements must be made to the third decimal place. In the present method, the mass spectrometer is operated at a static resolution of 10 000 (10% valley). An example of 10 000 resolution is given in Figure 6. Reference perflurokerosene (PFK) is leaked via the reference inlet system and a few scans are acquired to establish a normal calibration file against a PFK reference table for the spectra to be recorded subsequently. Tune the instrument at a minimum of 10 000 resolving power (10% valley) at m/z 304.9824 and, using peak matching, verify that m/z 380.9760 of PFK falls within 5 ppm of the exact value. Acceptable resolution must be confirmed immediately following each series of injections related to the application of this Reference Method. Verification at appropriate intermediate points of lengthy injection series is highly recommended. Hard copies of these measurements (peak shape and peak width) must be available for auditing purposes. The PFK leak is continued throughout the sample data acquisition cycle to ensure that the MS remains locked onto the exact masses of all analytically significant ions.
Figure 6: Peak Display for PFK m/z 304.9824 at 10 000 Resolution

The level of reference compound (PFK) that is allowed to enter the ion source should be monitored and adjusted to ensure that the amplitude of the most abundant lock-mass ion does not exceed 10% of full-scale deflection for the detector parameters used for the analysis. This will allow for more effective monitoring of any sensitivity changes that may occur during analysis.
Each lock-mass must be monitored and its intensity must not vary by more than 10% throughout its respective window. Variations of more than 10% may indicate the presence of co-eluting interferences that may substantially reduce the sensitivity of the instrument (U.S. EPA, 1990). At this stage, re-injection of the sample will not correct the problem; the only viable alternative is further cleanup of the extract, until lock-mass remains constant within 10% throughout the entire duration of its window. Lock-mass traces must be available for possible audit.
At 10 000 or better resolution, and with a total scan time of approximately one second for each group of ions specified in Table 3, inject 1 to 2 μL of PCDD/PCDF calibration standard CS3 (Table 4) into the GC/MS system. The isotope abundance ratio for each and every native and surrogate analyte must agree with the corresponding theoretical ratio within the control limits given in Table 3. If any ratio is outside its control limits, a problem clearly exists which must be identified and corrected before proceeding further. If the problem is found to be related to MS parameter settings, any changes at this stage are likely to alter MS resolution, which must therefore be measured again once acceptable ion abundance ratios have been achieved.
| Window Number | Compound | Quantification Ions (m/z) | Ion Type | Control Limits for Isotope Ratio | |
|---|---|---|---|---|---|
| 1st | 2nd | ||||
| 1 | TCDF | 303.9016 | 305.8987 | M/M+2 | 0.65-0.89 |
| 13C12-TCDF | 315.9419 | 317.9389 | M/M+2 | 0.65-0.89 | |
| TCDD | 319.8965 | 321.8936 | M/M+2 | 0.65-0.89 | |
| 13C12-TCDD | 331.9368 | 333.9339 | M/M+2 | 0.65-0.89 | |
| H6CDPE* | 375.8364 | M+2 | |||
| PFK | 316.9824 | Lock | |||
| 2 | P5CDF | 339.8597 | 341.8567 | M+2/M+4 | 1.32-1.78 |
| 13C12-P5CDF | 351.9000 | 353.8970 | M+2/M+4 | 1.32-1.78 | |
| P5CDD | 355.8546 | 357.8516 | M+2/M+4 | 1.32-1.78 | |
| 13C12-P5CDD | 367.8949 | 369.8919 | M+2/M+4 | 1.32-1.78 | |
| H7CDPE* | 409.7974 | M+2 | |||
| PFK | 366.9792 | Lock | |||
| 3 | H6CDF | 373.8208 | 375.8178 | M+2/M+4 | 1.05-1.43 |
| 13C12-H6CDF | 383.8639 | 385.8610 | M/M+2 | 0.43-0.59 | |
| H6CDD | 389.8157 | 391.8127 | M+2/M+4 | 1.05-1.43 | |
| 13C12-H6CDD | 401.8559 | 403.8529 | M+2/M+4 | 1.05-1.43 | |
| O8CDPE* | 445.7555 | M+4 | |||
| PFK | 380.9760 | Lock | |||
| 4 | H7CDF | 407.7818 | 409.7789 | M+2/M+4 | 0.88-1.20 |
| 13C12-H7CDF | 419.8220 | 421.8191 | M+2/M+4 | 0.88-1.20 | |
| H7CDD | 423.7766 | 425.7737 | M+2/M+4 | 0.88-1.20 | |
| 13C12-H7CDD | 435.8169 | 437.8140 | M+2/M+4 | 0.88-1.20 | |
| N9CDPE* | 479.7165 | M+4 | |||
| PFK | 430.9728 | Lock | |||
| 5 | OCDF | 441.7428 | 443.7398 | M+2/M+4 | 0.76-1.02 |
| OCDD | 457.7378 | 459.7348 | M+2/M+4 | 0.76-1.02 | |
| 13C12-OCDD | 469.7780 | 471.7750 | M+2/M+4 | 0.76-1.02 | |
| D10CDPE* | 513.6775 | M+4 | 0.76-1.02 | ||
| PFK | 454.9728 | Lock | |||
| * Response of the chlorinated diphenyl ether must be absent for PCDF determination as it has a similar isotope ratio. | |||||
| PCDD/PCDFs Standard | pg/μL (in toluene) | |||
|---|---|---|---|---|
| CS1a | CS2 | CS3b | CS4 | |
| Native standardsc,d | ||||
| 2,3,7,8-TCDD | 0.25 | 1 | 5 | 25 |
| 2,3,7,8-TCDF | 0.25 | 1 | 5 | 25 |
| 1,2,3,7,8-P5CDD | 0.50 | 2 | 10 | 50 |
| 1,2,3,7,8-P5CDF | 0.50 | 2 | 10 | 50 |
| 2,3,4,7,8-P5CDF | 0.50 | 2 | 10 | 50 |
| 1,2,3,4,7,8-H6CDD | 0.50 | 2 | 10 | 50 |
| 1,2,3,6,7,8-H6CDD | 0.50 | 2 | 10 | 50 |
| 1,2,3,7,8,9-H6CDD | 0.50 | 2 | 10 | 50 |
| 1,2,3,4,7,8-H6CDF | 0.50 | 2 | 10 | 50 |
| 1,2,3,6,7,8-H6CDF | 0.50 | 2 | 10 | 50 |
| 2,3,4,6,7,8-H6CDF | 0.50 | 2 | 10 | 50 |
| 1,2,3,7,8,9-H6CDF | 0.50 | 2 | 10 | 50 |
| 1,2,3,4,6,7,8-H7CDD | 0.50 | 2 | 10 | 50 |
| 1,2,3,4,6,7,8-H7CDF | 0.50 | 2 | 10 | 50 |
| 1,2,3,4,7,8,9-H7CDF | 0.50 | 2 | 10 | 50 |
| OCDD | 1 | 4 | 20 | 100 |
| OCDF | 1 | 4 | 20 | 100 |
| Surrogates | ||||
| 13C12-2,3,7,8-TCDD | 50 | 50 | 50 | 50 |
| 13C12-2,3,7,8-TCDF | 50 | 50 | 50 | 50 |
| 13C12-1,2,3,7,8-P5CDD | 50 | 50 | 50 | 50 |
| 13C12-1,2,3,7,8-P5CDF | 50 | 50 | 50 | 50 |
| 13C12-1,2,3,6,7,8-H6CDD | 50 | 50 | 50 | 50 |
| 13C12-1,2,3,4,7,8-H6CDF | 50 | 50 | 50 | 50 |
| 13C12-1,2,3,4,6,7,8-H7CDD | 50 | 50 | 50 | 50 |
| 13C12-1,2,3,4,6,7,8-H7CDF | 50 | 50 | 50 | 50 |
| 13C12-OCDD | 100 | 100 | 100 | 100 |
| Recovery Standards | ||||
| 13C12-1,2,3,4-TCDDe | 50 | 50 | 50 | 50 |
| 13C12-1,2,3,7,8,9-H6CDDf | 50 | 50 | 50 | 50 |
| a also used to assess detection limits; b used daily to verify calibration and abundance ratios; c penta- and helpta-CDR, hexa-CDD/CDF isomers listed in order of elution on a DB-5 column; d a solution of native congeners at CS2 concentrations is used as a spiking solution for method performance test (see Subsection 5.2); e retention time marker and recovery standard for tetra- and penta-CDD/CDF; f retention time marker and recovery standard for hexa- and hepta-CDD/CDF and OCDD; |
||||
Calibration standard CSl is used to assess instrumental detection limits. At 10 000 resolution, injection of 0.25 pg of TCDD and TCDF must result in a peak response which is at least five times the background noise level (i.e., S/N ≥ 5) for each of the four monitored ions. If these levels cannot be detected, then instrument parameters are to be adjusted accordingly, while maintaining acceptable resolution. In other words, the instrument is not in acceptable operating condition until all detection and measurement criteria are satisfied simultaneously.
6.5 Criteria for Analyte Identification
Sample components are identified as PCDDs/PCDFs if their GC/MS data satisfy the following criteria:
- Peak responses for each of the two selected molecular cluster ions must be at least three times the background noise level (i.e., S/N ≥ 3, see Subsection 6.7 for a definition of noise).
- Chlorine isotope ratio for the two molecular cluster ions must be within 15 % of the correct ratio (see Table 3).
- Peak maxima for both quantification ions must coincide within two seconds.
- Reported sample concentrations of PCDF must be flagged with an "E" if any peak recorded in the chlorinated Diphenyl ether channel maximizes within two seconds of the retention time of an apparent PCDF congener, and response for the ether ion exceeds 5% of the sum of the peak areas for the two monitored furan ions.
- Sample components are identified as 2,3,7,8-substituted congeners if the following applicable criterion is also satisfied:
- In the case of a congener for which a labelled analogue is present in the surrogate spiking mixture, all native and surrogate ion peak maxima must be coincident within three seconds.
- In the case of a congener for which a labelled analogue is not present in the surrogate spiking mixture, peak maxima for both quantification ions must be coincident with each other within two seconds, and both peaks must be within three seconds of the expected retention time for that congener. Expected retention time is determined on the basis of known relative retention times for these congeners.
- In the case of a congener for which a labelled analogue is present in the surrogate spiking mixture, all native and surrogate ion peak maxima must be coincident within three seconds.
6.6 Quantification
An internal standard method is used to quantify PCDDs/PCDFs. It relies upon consistent linearity of MS response over time and over the calibration range represented by the standard solutions defined in Table 4. This internal standard method is easily integrated into an automated routine for data quantification.
Internal standard quantification is based on the use of Relative Response Factors (RRF). For native standards, the RRF is the ratio of analyte response factor to the response factor of the corresponding labelled surrogate (internal standard). These RRFs remain unchanged over the range of concentration for which MS response is linear. Using these RRFs, along with native and surrogate responses from the sample run, recovery-corrected concentrations of PCDDs/PCDFs are calculated directly. Surrogate recoveries are calculated separately and reported, as these values reflect the overall quality of the concentration data being reported.
Relative response factors for the native standard (RRFn) and for the surrogate standard (RRFs) are calculated using the following equations:


- RRFn
- relative response factor, native standard to surrogate standard;
- RRFs
- relative response factor, surrogate standard to recovery standard;
- Ac
- quantification ion (single or both ions) peak area for native standard;
- Asc
- quantification ion (single or both ions) peak area for the appropriate surrogate standard;
- Arc
- quantification ion (single or both ions) peak area for the 13C 12-1,2,3,4- TCDD or 13C 12-1,2,3,7,8,9- H6CDD
- Cc
- concentration of the native standard (pg/μL);
- Csc
- concentration of the appropriate surrogate standard (pg/μL);
- Crc
- concentration of 13C 12-1,2,3,4- TCDD or 13C 12-1,2,3,7,8,9- H6CDD. (pg/μL)
Using the RRFs, sample concentrations of PCDDs/PCDFs (C) and surrogate standard recoveries (%R) can be calculated as follows:


- C(X)
- recovery-corrected concentration of analyte X (pg/L);
- Ak
- quantification ion (single or both ions) peak area for the "k"th homologous isomer of analyte X (n=1 for isomer-specific analysis);
- Qss
- amount of surrogate standard X added to the sample (pg);
- Ass
- quantification ion (single or both ions) peak area for surrogate standard X in sample extract;
- V
- sample size (L);
- %R(X)
- percent recovery of surrogate standard X;
- Qrs
- amount of 13C 12-1,2,3,4- TCDD (recovery standard for tetra- and penta -CDD/CDF) or 13C 12-1,2,3,7,8,9- H6CDD (recovery standard for hexa- and hepta -CDD/CDF and OCDD) in sample extract (pg);
- Ars
- quantification ion (single or both ions) peak area for 13C 12-1,2,3,4- TCDD or 13C 12-1,2,3,7,8,9- H6CDD in sample extract.
For homologues represented by more than one isomer in the calibration standard solutions, the "homologue-average" RRF is used to quantify all target analytes that are not 2,3,7,8-substituted congeners.
The calibration data from which RRFs are calculated must be of good and definable quality. For initial calibration, RRFs are calculated by analyzing the series of four calibration standard solutions, the compositions of which are given in Table 4. The Relative Standard Deviation (RSD) of the four RRFs must be less than 15% for both 2,3,7,8-TCDD and 2,3,7,8-TCDF, and less than 20% for all other native analytes. This value effectively corresponds to a response linearity criterion. If this criterion is met, then the calibration is successful, and the mean RRF values are used for quantification of subsequent target analyte data. The established calibration curve must be verified by analyzing the calibration verification standard (CS3) at least once during every 12-hour period in which sample analysis occurs (see Section 8).
6.7 Limit of Detection
The Method Detection Limit (MDL) for PCDD/PCDF analysis is defined as the minimum concentration of analyte in the sample that will produce clearly defined peaks with an acceptable chlorine isotope ratio, and witha signal-to-noise ratio equal to three for the quantification ion exhibiting the poorer signal-to-noise ratio. Variables, such as sample matrix, sample size, final extract volume, injection volume used in analysis, surrogate recovery, GC column performance, chromatographic parameters, electronic noise, and instrument sensitivity may directly influence the MDL.
Reported MDL must be corrected for surrogate recovery and is calculated as follows:

- N
- estimated sum of electronic and chemical (matrix) noise expressed as peak height (single or both ions);
- A / H
- area/height ratio for the surrogate standard peak;
- Qss
- amount of surrogate standard added (pg);
- Ass
- surrogate peak area (single or both ions);
- RRFn
- relative response factor, as defined in preceding subsection;
- V
- sample size (L).
Average noise amplitude can be determined manually, as shown in Figure 7. The area/height ratio observed for the corresponding surrogate ion peak(s) in the sample data is used to convert noise amplitude to an area value.
Figure 7: Noise Determination
Click to enlarge
{kind=link}

The noise for each homologue group must be determined from the actual sample chromatograms. If a quantification ion channel contains one or more large peaks that prevent observation of the noise, a portion of the chromatogram should be re-scaled so that the noise level can be assessed.
When detection of any of the seventeen 2,3,7,8-substituted congeners is rejected solely due to an incorrect isotope ratio, reporting requirements are as follows:
- 1) in Figure 8, Part I, report the result as NDR in the pg/L column;
- 2) calculate a concentration value for the congener in the same manner as if the ratio had been within specification and report this value, in brackets, in the Detection Limit column;
- 3) in Figure 8, Part II, bracket the ion(s) used for quantification; and
- 4) report the actual isotope ratio, in brackets, in the adjacent column. The bracketed concentration value reported in Part I is not to be included in the corresponding homologue total.
Figure 8: Effluent Sample Data Sheet - Part I
Click to enlarge
{kind=link}

Figure 8: Effluent Sample Data Sheet - Part II
Click to enlarge
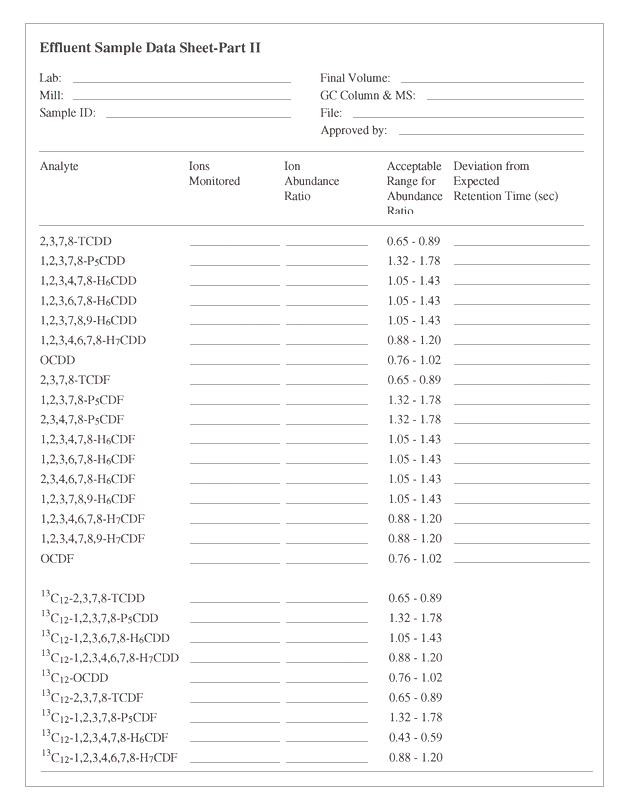{kind=link}

On the basis of these procedures, assuming adequate surrogate recoveries, MDL target ranges for pulp mill effluents are listed in Table 5.
| Analytes | MDL (pg/L)* |
|---|---|
| * For individual analyte peaks including 2,3,7,8-substituted congeners. | |
| TCDD/TCDF | 2 - 5 |
| P5CDD/P5CDF | 4 - 10 |
| H6CDD/H6CDF | 4 - 10 |
| H7CDD/H7CDF | 6 - 15 |
| OCDD/OCDF | 8 - 20 |
6.8 Level of Quantification (LOQ)
The LOQ is defined as the lowest level above which quantitative results may be obtained with a specified degree of confidence. A single LOQ concentration value for the determination of 2,3,7,8-TCDD and 2,3,7,8-TCDF in pulp mill effluents by this method was established from statistical evaluation of data generated in an interlaboratory study. Ten commercial, industrial, and governmental laboratories participated in the study, which involved analysis of eight replicate portions of two pulp mill effluent samples containing low and near-detection-limit levels of 2,3,7,8-TCDD and 2,3,7,8-TCDF. Following screening of data for statistical validity, pooled standard deviations of the measured concentrations were calculated. By defining the LOQ as ten times the standard deviation of measured near-detection-limit levels (Keith et al.,1983), and using a pooled estimate of standard deviation from the interlaboratory study, the LOQ concentration value for 2,3,7,8-TCDD and 2,3,7,8-TCDF was established as 15 ppq. Method precision at or near the LOQ concentration is estimated to be ± 5 ppq at concentrations at the 99% confidence level.