
Module 1: Overview of Vanessa’s Law and reporting requirements

Download the alternative format
(PowerPoint format, 1.8 MB, 31 pages)
Organization: Health Canada
Published: 2019-12-13
Mandatory Reporting of Serious Adverse Drug Reactions and Medical Device Incidents by Hospitals
Educational Support for Mandatory Reporting
- The educational materials provide core content about serious adverse drug reaction (serious ADR) and medical device incident (MDI) reporting that can be used by hospitals, health care leadership, health care providers, patients and families, and educators.
- There are 4 PowerPoint modules:
- Module 1 – Overview of Vanessa’s Law and Reporting Requirements
- Module 2 – Reporting Processes to Health Canada
- Module 3 – Strategies to Promote and Support Mandatory Reporting
- Module 4 – Health Canada’s Review and Communication of Safety Findings
- The educational materials (as entire modules or individual slides or selected content) can be used to explain, describe, or promote serious ADR and MDI reporting. Source of this educational material may be acknowledged as: Educational Support for Mandatory Reporting. Health Canada; 2019.
Goals of the Education Approach
- Support the implementation of a key provision of the Protecting Canadians from Unsafe Drugs Act (Vanessa’s Law) by providing stakeholders with information on Health Canada's new regulatory requirements for serious ADR and MDI reporting
- Describe the reporting processes for hospitals to meet the mandatory reporting requirements
- Provide strategies for health care leadership and health care providers to promote and support reporting of serious ADRs and MDIs documented within hospitals
- Describe Health Canada’s review and communication of safety findings
Module 1 - Learning Outcomes
Completion of Module 1 will enable you to:
- Explain the purpose of Vanessa's Law
- Describe the regulations for mandatory reporting by hospitals of serious ADRs and MDIs
- Recognize the required data elements for mandatory reporting of serious ADRs and MDIs
Module 1 - Outline
- Purpose of Vanessa’s Law
- Regulations for Mandatory Reporting
- Who is required to report?
- What are the definitions of a serious ADR and MDI?
- What products are in scope of these regulations?
- When must hospitals report?
- Required Data Elements
- Serious ADR report
- MDI report
- Key Points to Remember
- Abbreviations
- Resources
Conceptual Model of Serious ADR and MDI Reporting by Hospitals
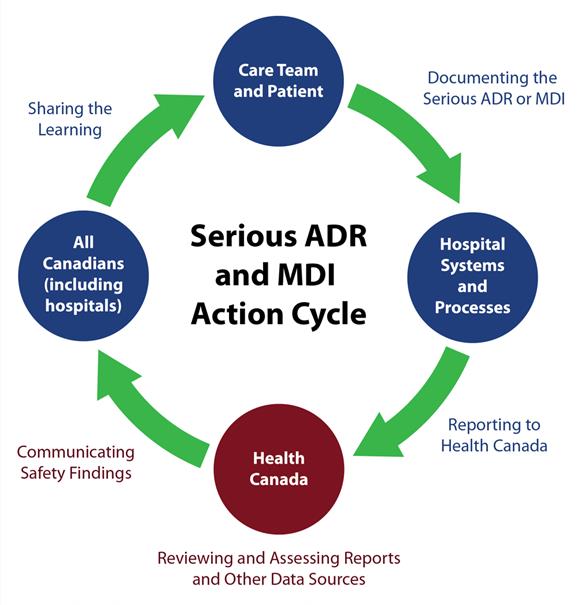
Source: Serious ADR and MDI Action Cycle. ISMP Canada, HSO, CPSI; 2019.
Text description
A circle diagram depicts the action cycle for serious ADR and MDI reporting by hospitals. In summary, the figure illustrates that the care team and patients document the serious ADR or MDI. Reporting a serious ADR or MDI to Health Canada is supported by hospital systems and processes. Health Canada reviews and assesses reports and other data sources. Health Canada communicates safety findings to all Canadians, including hospitals, who are shown to share the learning. This concludes the action cycle.
Purpose of Vanessa’s Law
Protecting Canadians from Unsafe Drugs Act (Vanessa's Law)
The Protecting Canadians from Unsafe Drugs Act (Vanessa's Law) introduces amendments to the Food and Drugs Act to improve Health Canada's ability to:
- collect post-market safety information;
- take appropriate action when a serious risk to health is identified; and
- promote greater confidence in the oversight of therapeutic products by increasing transparency.
Protecting Canadians from Unsafe Drugs Act (Vanessa's Law) Amendments to the Food and Drugs Act include:
- Power to require information, tests or studies
- Power to require a label change/package modification
- Power to recall unsafe therapeutic products
- Ability to disclose information in certain circumstances
- Tougher measures for those that do not comply
- Mandatory reporting of serious adverse drug reactions and medical device incidents by health care institutions*
*Regulatory amendments give effect to this authority and further define health care institutions as hospitals under section C.01.020.1 of the Food and Drug Regulations and section 62 of the Medical Device Regulations.
Who was Vanessa?
- Vanessa Young died in 2000, at the age of 15, of a cardiac arrhythmia after taking cisapride (Prepulsid®) as prescribed.
- A campaign for increased regulation of therapeutic products subsequently led to greater powers for Health Canada to request safety data from hospitals and industry about drugs and medical devices.
- Vanessa’s Law was enacted in 2014 and the mandatory reporting requirements come into effect December 16th, 2019.
Why Is Mandatory Reporting of Serious ADRs and MDIs Important?
- Health Canada is continuously looking for ways to strengthen its knowledge base on product safety in the interest of improving patient outcomes and public health.
- Serious ADR and MDI reports are important sources of information for identifying emerging safety issues.
- Under-reporting and poor quality of reports is an issue in all countries. An international systematic review estimated that only 2-18% (median of approximately 6%) of ADRs are reported.Footnote 1
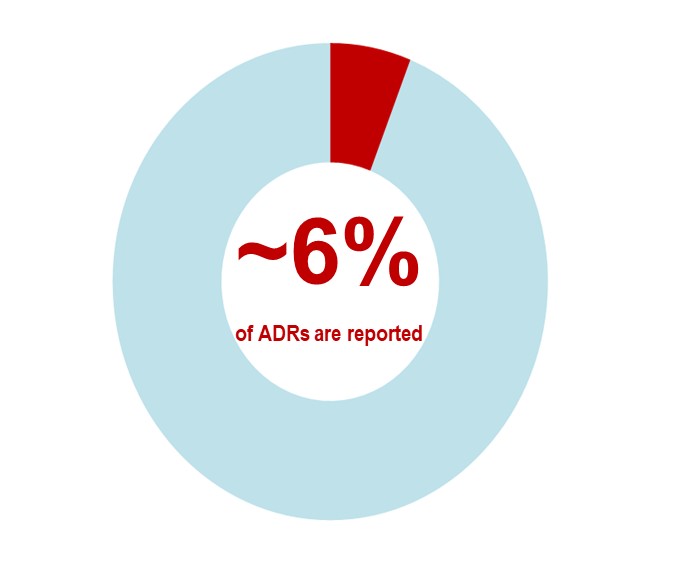
- Footnote 1
-
Hazell L, Shakir SAW. Under-reporting of adverse drug reactions: a systematic review. Drug Saf. 2006;29(5):385-396.
Text description
An international systematic review estimates that approximately 6% of adverse drug reactions are reported.
What Are the Benefits of Serious ADR and MDI Reporting?
Serious ADR and MDI Reporting Contributes to:
- Identification of emerging safety issues related to drugs and medical devices
- Assessment of harm vs. benefit of drugs and medical devices
- Sharing of learning, including warnings and advisories for health care providers, patients, and stakeholders
- Improvement of safety of products through risk mitigation such as a labelling change, a product information update, or a recall
Serious ADR and MDI Reporting and Learning
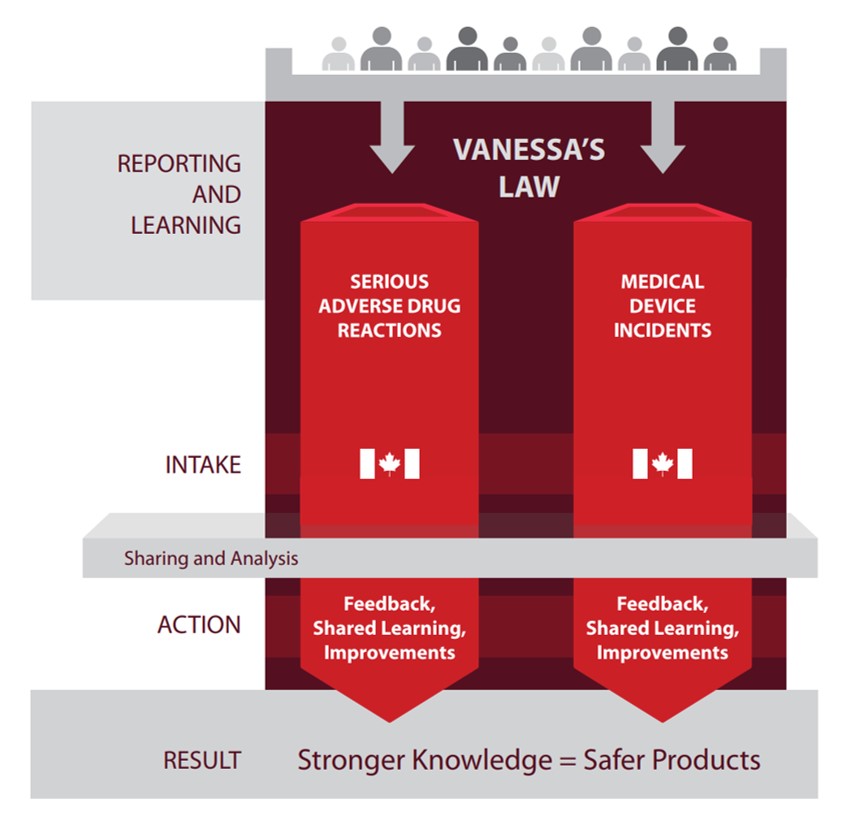
Text description
This infographic shows mandatory reporting inputs and outputs under Vanessa's Law. The infographic illustrates that reporting results in stronger knowledge which leads to safer products. The left column lists the stages of reporting and learning, intake, sharing and analysis, action, and result. The middle and right columns show the flow for serious adverse drug reactions and medical device incidents. The top row shows a group of silhouettes with arrows pointing down to the middle and right columns. The flow is the same for both types of reporting.
Reporting and Learning:
- An individual submits a serious adverse drug reaction or medical device incident report.
Intake:
- Health Canada receives the adverse drug reaction or medical device incident report.
- Health Canada shares and analyses the information.
Action:
- Health Canada provides feedback to those who report, shares learning and improvements.
Result:
- Reporting results in stronger knowledge which lead to safer products
- Under Vanessa’s Law, the reporting of serious ADRs and MDIs to Health Canada is mandatory by hospitals.
- Analysis and shared learning from these reports contribute to achieving the goal of providing safer health care and health products.
Regulations for Mandatory Reporting
Who Is Required to Report?
The regulations apply to all hospitals.
The regulations define a hospital as a facility that:
- is licensed, approved or designated as a hospital by a province or territory, in accordance with the laws of the province or territory, to provide care or treatment to persons suffering from any form of disease or illness; or
- is operated by the Government of Canada and provides health services to in-patients.
Notes:
- Outpatient clinics are subject to the regulations if they are legally part of the hospital, even if they are physically separate from the hospital. On the other hand, clinics that may be physically located within a hospital, but that are not legally part of the hospital, will not be subject to the regulations.
- Health care institutions that are outside the scope of the definition of hospitals, such as private clinics or long-term care facilities (e.g., nursing homes), continue to be encouraged to report on a voluntary basis.
What are the Definitions of a Serious ADR and MDI?
A serious adverse drug reaction (serious ADR) is a noxious and unintended response to a drug that occurs at any dose and that
- requires in-patient hospitalization or prolongation of existing hospitalization,
- causes congenital malformation,
- results in persistent or significant disability or incapacity,
- is life-threatening, or
- results in death.
A medical device incident (MDI) is an incident related to a failure of a medical device or a deterioration in its effectiveness, or any inadequacy in its labelling or in its directions for use that has led to the death or a serious deterioration in the state of health of a patient, user, or other person, or could do so were it to recur.
Note: Hospitals are not required to establish causality; the information to be submitted by the hospital to Health Canada only needs to represent the suspicions of a health care professional that a serious ADR or MDI has been observed.
What Products Are In Scope of these Regulations?
The mandatory reporting requirements for hospitals apply to therapeutic products, including:
- Pharmaceuticals (prescription and non-prescription drugs)
- Biologic drugs (biotechnology products, fractionated blood products, plasma proteins, and vaccines [excluding vaccines administered under a routine immunization program of a province or territory])
- Radiopharmaceutical drugs
- Disinfectants
- Medical devices
- Drugs for an urgent public health need
When in doubt, Health Canada encourages hospitals to report.
Types of Medical Devices Included
The term medical device covers a wide range of health and/or medical instruments used in the treatment, mitigation, diagnosis or prevention of a disease or abnormal physical condition.
Medical devices are classified into Class I (lowest risk) to Class IV (highest risk). Examples are:
- Class I – hospital beds, wheelchairs, leg prostheses
- Class II – infusion sets, syringes, tracheostomy tubes, urethral catheters
- Class III – infusion pumps, anesthesia gas machines, intrauterine devices
- Class IV – pacemakers, defibrillators, breast implants, bone grafts
All classes of medical devices are included in mandatory reporting by hospitals.
When Must Hospitals Report?
The regulations require hospitals to report serious ADRs or MDIs in writing to Health Canada within 30 calendar days of first documentation of the serious ADR or MDI within the hospital.
Information “Within the Control of the Hospital”
The regulations require hospitals to report all documented serious ADRs and all documented MDIs, where the required information is within the control of the hospital.
- Information that is within the control of the hospital is information that would be reasonably accessible within the hospital.
- While it is encouraged for hospitals to take all reasonable steps to retrieve the required information to complete as thorough a report as possible, there is no requirement to do further investigation in order to obtain the pieces of information.
Examples of Documentation Within the Hospital
Examples of serious ADR or MDI documentation within the hospital include:
- a serious ADR or MDI that is identified in a patient’s clinical/medical record;
- a serious ADR or MDI that is identified in a separate report form (electronic or hard copy) that has been completed by a health care professional; and
- a serious ADR or MDI that has been documented in an ADR form or a product complaint form (e.g., an MDI form) as per internal hospital policy, a pathology report, an incident/patient safety learning database, or a computerized prescription recording system.
Health Canada’s Compliance Approach
- Health Canada has implemented an oversight mechanism to verify that reports are being received and that they are complete and provide information of sufficient quality to meet the regulatory requirements.
- When a situation of non-compliance is identified, Health Canada will work with hospitals to help them meet the mandatory reporting requirements under Vanessa’s Law, building on guidance, outreach and education efforts.
- In the event that Health Canada identifies instances of more persistent non-compliance, additional compliance and enforcement measures could be taken by the Regulatory Operations and Enforcement Branch.
Required Data Elements
Required Data Elements for Hospital Mandatory Reporting
- The data elements on the following two slides (for serious ADR and MDI reporting, respectively) are required to be included in reports, if the hospital has this information within its control.
- The data elements marked with a double asterisk (**) are essential in order for the report to be useful for Health Canada and represent the minimal information required when submitting a report.
- If a hospital does not have within its control all of the information for the essential data elements (i.e., those with a double asterisk [**]), it is exempt from having to submit a report.
Serious ADR Report - Required Data Elements
Reporter:
- Contact information: The name of the hospital and the contact information of a representative of that hospital
Suspect product:
- **Name: The drug’s brand name, proper name or common name
- **Drug Number/Code: In the case of a drug imported under Part C, Division 10 of the Food and Drug Regulations (subsection C.10.001(2)), the identifying number or code of the drug, if any, assigned in the country in which the drug was authorized for sale
- DIN: The drug identification number (DIN) assigned for the drug, if applicable
- Concomitants: Any concomitant therapeutic products used by the patient
Patient information:
- **Age/Sex: The patient’s age and sex
- Medical history: Any medical condition of the patient that directly relates to the serious adverse drug reaction
ADR information:
- **ADR description: A description of the serious adverse drug reaction
- Documentation date: The date on which the serious adverse drug reaction was first documented
- Start/End therapy date: The date on which the patient first used the drug and, if applicable, the date on which the patient stopped using the drug
- Start/End ADR date: The date on which the serious adverse drug reaction first occurred and, if applicable, the date on which the patient’s health was restored to its state prior to the reaction
- Outcome: The effect of the serious adverse drug reaction on the patient’s health
**Essential data elements representing minimal information required to submit a report
MDI Report - Required Data Elements
Reporter:
- Contact information: The name of the hospital and the contact information of a representative of that hospital
Suspect Product:
- **Name or identifier: The name or identifier of the medical device, so that it is uniquely identifiable
- Manufacturer Name: The name of the manufacturer of the medical device
- Lot/Serial Number: The lot number of the device or its serial number
MDI information:
- **MDI description: A description of the medical device incident
- Documentation date: The date on which the medical device incident was first documented
- Contributing factors: Any contributing factors to the medical device incident including any medical condition of the patient that directly relates to the medical device incident
- Outcome: The effect of the medical device incident on the patient’s health
**Essential data elements representing minimal information required to submit a report
Methods for Submitting Serious ADR and MDI Reports to Health Canada
From a Hospital System Database
If you are interested in submitting reports electronically (e.g., secure File Transfer Protocol - sFTP, system-to-system exchanges) to Health Canada, please email the Canada Vigilance Program at hc.canada.vigilance.sc@canada.ca
Fax or Mail
The new reporting forms for serious ADR and MDI, together with instructions, are available on the Health Canada website. Download, print, and complete the applicable form and send by fax or by mail to the Canada Vigilance Office.
Fax to: 1-866-678-6789
Mail to:
Canada Vigilance Program
Marketed Health Products Directorate
Health Products and Food Branch
Health Canada
Address Locator 1908C
Ottawa, Ontario K1A 0K9
Directly Online
Complete and submit a report using the online reporting application available at: MedEffect Canada.
Key Points to Remember
- The Protecting Canadians from Unsafe Drugs Act (Vanessa's Law) introduces amendments to the Food and Drugs Act, including mandatory reporting of serious adverse drug reactions (serious ADRs) and medical device incidents (MDIs) by health care institutions.
- The Act aims to improve the quality and quantity of serious ADR and MDI reports to strengthen the safety oversight of therapeutic products.
- The reporting of serious ADRs and MDIs contributes to identification of emerging safety issues, assessment of harm vs. benefit, sharing of learning, and improvement of product safety.
- The mandatory reporting regulations require hospitals to report in writing serious ADRs and MDIs to Health Canada within 30 calendar days of first documentation of the reaction or incident within the hospital.
- These regulations apply to therapeutic products, defined as: pharmaceuticals (prescription and non-prescription), biologic drugs, radiopharmaceutical drugs, disinfectants and medical devices.
- There are required data elements for mandatory reporting of serious ADRs and MDIs.
Abbreviations
- ADR:
- Adverse Drug Reaction
- DIN:
- Drug Identification Number
- MDI:
- Medical Device Incident
- sFTP:
- Secure File Transfer Protocol
Resources
- Guidance Document - Guidance on the Risk-Based Classification System for Non-In Vitro Diagnostic Devices (non-IVDDs)
- Mandatory reporting of serious adverse drug reactions and medical device incidents by hospitals - Guidance document
- MedEffect Canada
- Protecting Canadians from Unsafe Drugs Act (Vanessa’s Law) Amendments to the Food and Drugs Act (Bill C-17)
- Regulations Amending the Food and Drug Regulations (Serious Adverse Drug Reaction Reporting - Hospitals): SOR/2019-190
- Regulations Amending the Medical Devices Regulations (Medical Device Incident Reporting - Hospitals): SOR/2019-191
For additional information, please contact the Canada Vigilance Program at:
Email: hc.canada.vigilance.sc@canada.ca
Telephone: 1-866-234-2345
Acknowledgments
- All materials were developed by the collaborating parties: Health Canada, Institute for Safe Medication Practices Canada (ISMP Canada), Health Standards Organization (HSO), and the Canadian Patient Safety Institute (CPSI).
- Any stakeholder interested in using the materials should acknowledge Health Canada as the owner and source: Educational Support for Mandatory Reporting. Health Canada; 2019.
Page details
- Date modified: