
Page 4: Guidelines for Canadian Drinking Water Quality: Guideline Technical Document – Haloacetic Acids
10.0 Health effects in laboratory animals and in vitro test systems
10.1 Acute toxicity
| Compound | Oral LD50 (mg/kg bw)Table 7 Footnote a | Clinical observations | |||
|---|---|---|---|---|---|
| Rats | Mice | Rabbits | Guinea pigs | ||
| MCA acid | 90.4 for a 1% solutionTable 7 Footnote 1) ~200 for a 6% solutionTable 7 Footnote 1) | 260Table 7 Footnote 2) | n/aTable 7 Footnote b | n/a | Apathy, hypoactivity, disorders of balance, lacrimation, dyspnoea, cyanosisTable 7 Footnote 3) |
| MCA salt | 76Table 7 Footnote 3) | 165-255Table 7 Footnote 3),Table 7 Footnote 4) | n/a | 80Table 7 Footnote 3) | Apathy, weight lossTable 7 Footnote 3) |
| DCA salt | 4480Table 7 Footnote 3) | 4845-5500Table 7 Footnote 3),Table 7 Footnote 5) | n/a | n/a | Semi-narcosis, narcosisTable 7 Footnote 3) |
| TCA acid | 400Table 7 Footnote 6) | n/a | n/a | n/a | n/a |
| TCA salt | 3320-5000Table 7 Footnote 3),Table 7 Footnote 7) | 3640-4870Table 7 Footnote 3),Table 7 Footnote 7),Table 7 Footnote 8) | 4000Table 7 Footnote 7) | n/a | Semi-narcosis, narcosisTable 7 Footnote 3) |
| MBA salt | 177Table 7 Footnote 9) | n/a | n/a | n/a | Excessive drinking, hypomobility, laboured breathing, mild diarrhoeaTable 7 Footnote 9) |
| DBA salt | 1737Table 7 Footnote 9) | n/a | n/a | n/a | Excessive drinking, hypomobility, laboured breathing, mild diarrhoeaTable 7 Footnote 9) |
| Compound | Dermal LD50 (mg/kg bw)Table 8 Footnote a | Clinical observations | |||
|---|---|---|---|---|---|
| Rats | Rabbits | ||||
| MCA acid | >400 (5% solution)Table 8 Footnote 1) 305-800 (40-50% solution)Table 8 Footnote 1) 145 (acid)Table 8 Footnote 2) | 250 (50% solution)Table 8 Footnote 1) | Apathy, hypoactivity, lacrimation, piloerection, panting, prone positionTable 8 Footnote 1) | ||
| MCA salt | >2000 (no deaths)Table 8 Footnote 1) | n/aTable 8 Footnote b | Decreased activity, squatting posture, stilted and staggering gait, irregular breathing, moist rales, diarrhoeaTable 8 Footnote 1) | ||
| TCA salt | >2000Table 8 Footnote 3),Table 8 Footnote 4) | n/a | |||
10.2 Short-term exposure
10.2.1 Monochloroacetic acid
In a 13-week gavage study, B6C3F1 mice (20 per sex per dose) received MCA (as an acid in deionized water) at doses of 0, 25, 50, 100, 150 or 200 mg/kg bw per day, 5 days per week. Interim evaluations were done on five mice per dose after 4 and 8 weeks of treatment. Mortality was increased only at the highest dose and mostly in male mice. The final mean body weight and the mean weight gain were significantly less in the 200 mg/kg bw per day females compared with controls. In female mice only, absolute and relative liver weights were significantly increased at the highest dose. Cholinesterase levels were decreased at the two highest dose groups in female mice. Hepatocellular cytoplasmic vacuolations were found following histopathological examination and were related to metabolic derangement occurring in moribund animals. No evidence of peroxisome proliferation in the liver was reported. The authors set the no-observed-adverse-effect level (NOAEL) at 100 mg/kg bw per day (Bryant et al., 1992; NTP, 1992).
In the same study as above, six groups of F344 rats (20 per sex per dose) received MCA (as an acid in deionized water) by gavage at 0, 30, 60, 90, 120 or 150 mg/kg bw per day, 5 days per week, for 13 weeks. Interim evaluations were done on five rats per dose group after 4 and 8 weeks of treatment. Toxic effects were observed at every dose level. Almost all animals died at 90 mg/kg bw per day and above. Relative heart weights were significantly decreased in both sexes at 60 mg/kg bw per day and only in females at 30 mg/kg bw per day. A dose-related increase in the incidence and severity of cardiomyopathy was observed in both sexes at 60 mg/kg bw per day and above. In male rats, relative liver and kidney weights were increased at 30 and 60 mg/kg bw per day; in female rats, relative liver weight was increased at 60 mg/kg bw per day only. During the treatment, blood urea nitrogen levels were increased in male rats at 90 mg/kg bw per day and up and in female rats at 60 mg/kg bw per day and up; however, there was no microscopic evidence of kidney or liver damage. Significant dose-related increases in serum levels of alanine transaminase (ALT) and aspartate transaminase (AST) were seen in both sexes at 60 mg/kg bw per day and above. Lymphocyte counts were decreased at 30 mg/kg bw per day and above, but were related to stress. The authors set the NOAEL at 30 mg/kg bw per day based on cardiac effects in both sexes (Bryant et al., 1992; NTP, 1992).
10.2.2 Dichloroacetic acid
In a 7-week drinking water study, male Sprague-Dawley rats received DCA (sodium salt) at dose levels of 50 or 1100 mg/kg bw per day (Stacpoole et al., 1990). The high-dose rats had severe hind limb weakness; however, light microscopic examination of the peripheral nerves did not detect any changes. Thiamine deficiency was also detected at the high dose, as measured by transketolase activity in red cells. No clinical signs or effects on transketolase activity were seen at the low dose. The authors stated that the toxicity seen in rats in this study is associated with signs typical of thiamine deficiency and that hind limb weakness and other neuropathic manifestations of chronic thiamine deficiency in animals are considered to be due to changes in central, rather than peripheral, nervous system structure and function.
In a 12-week dietary study, hind limb weakness and abnormal gait also appeared in male Wistar rats (n = 6) exposed to DCA (neutralized). Approximate doses varied from 4 mmol/kg bw per day (516 mg/kg bw per day) at the beginning of the study to about 2.5 mmol/kg bw per day (323 mg/kg bw per day) at the end of the study (Yount et al., 1982). Decreased nerve conduction velocity was also detected in several nerves (sural and motor), as well as a decrease in the diameter of the tibial nerves. Decreased weight gain as a result of decreased food consumption and the presence of hepatomegaly were among the other toxic effects observed.
In a 3-month oral gavage study, DCA (sodium salt) was administered to Sprague-Dawley rats (10 per sex per dose) at dose levels of 0, 125, 500 or 2000 mg/kg bw per day, with an additional five rats per sex dosed with 0 or 2000 mg/kg bw per day allowed a 4-week recovery period (Katz et al., 1981). Death was seen in two rats per sex at the high dose. Hind limb paralysis occurred in 27% of rats of both sexes at the high dose. In the 4-week recovery group, one rat per sex afflicted by the paralysis seemed to have recovered completely. Histopathological examination showed the brain and testes as the main target organs. Oedematous brain lesions, characterized by vacuolation of the myelinated white tracts, were seen in the cerebrum and, to a lesser extent, the cerebellum. Combined incidence rates for these were 60% at the low dose and 100% at the middle and high doses. In the high-dose recovery group, brain lesions persisted in three of eight rats. Body weight was decreased in all treated rats and was associated with reduced food consumption. A significant increase was observed in relative liver weight (both sexes, all doses), relative kidney weight (females, all doses) and relative adrenal weight (males, 500 mg/kg bw per day and above; females, 2000 mg/kg bw per day). Clinical chemistry showed a mild depression of the erythroid parameters at the middle and high doses and a decrease in blood glucose and lactate levels at all levels. Testicular effects were also seen and are discussed in Section 10.5.2.
In a 90-day drinking water study, male Sprague-Dawley rats (n = 10) were treated with DCA (neutralized) at doses of 0, 50, 500 or 5000 mg/L (0, 4, 35 or 350 mg/kg bw per day) (Mather et al., 1990). Decreased body weight and water consumption were observed at 35 mg/kg bw per day and above. Relative liver and kidney weights were decreased at doses of 35 mg/kg bw per day and above. However, histological and biochemical signs of liver and kidney damage, as well as an increase in the hepatic peroxisomal beta-oxidation activity, were seen only in the highest dose group. Increases in relative spleen weight were seen at the top dose in the absence of histopathological effects. No effects on immunological functions were seen.
In a 13-week drinking water study (NTP, 2000), B6C3F1 mice (10 per sex per dose) were exposed to DCA (neutralized to pH 5) doses of 0, 67, 125, 250, 500 or 1000 mg/L (0, 9, 16, 32, 61 and 124 mg/kg bw per day for males and 0, 10, 18, 38, 72 and 132 mg/kg bw per day for females). Dose-related increases were seen in liver weight and incidence of cytoplasmic vacuolation change in hepatocytes (females at 10 mg/kg bw per day and above; males at 32 mg/kg bw per day and above). No clinical signs or deaths were observed during the study. In the high-dose group, mild leukopenia, neutropenia and monocytopenia were observed in some male mice, which were potentially chemical related. The authors set NOAELs of <32 mg/kg bw for males and <10 mg/kg bw for females, both based on microscopic liver lesions.
NTP (2000) also dosed Fischer-344 rats (10 per sex per dose) with the same protocol and doses as above (calculated doses: males: 0, 5, 9.3, 18.8, 39.2 and 81.4 mg/kg bw per day; females: 0, 5.9, 10.0, 20.9, 43.8 and 94.7 mg/kg bw per day). A significant decrease in body weight gain was noted in high-dose males during weeks 4-13. No other significant effects were observed.
In a 13-week subchronic study (Katz et al., 1981), 10- to 12-month-old beagle dogs (3-4 per sex per dose) received DCA (sodium salt) in gelatin capsules at doses of 0, 50, 75 or 100 mg/kg bw per day, with one additional dog per sex dosed with 0 or 100 mg/kg bw per day and allowed a 4-week recovery period. In female dogs only, food consumption was reduced at all doses; however, dose-dependent weight losses at all dose levels were observed in both sexes during the treatment but reversed upon cessation. One female and one male died at the 75 and 100 mg/kg bw per day dose, respectively, and signs of adverse effects observed prior to their deaths included anorexia, ataxia, hind limb weakness and reduced activity. Other adverse effects related to the treatment include emesis (75 and 100 mg/kg bw per day), bloody stools (100 mg/kg bw per day) and paralysis (100 mg/kg bw per day). Also reported in all dose groups (both sexes) were a high incidence of ocular anomalies: bilateral lenticular opacities, injected bulbar conjunctivae and superficial corneal vascularization with a tendency for keratoconjunctivitis sicca. Haematological parameters were depressed at all dose levels in both sexes. Liver and kidney parameters were not affected by the DCA treatment in dogs. An increase in the incidence of lung consolidation was observed in all doses (both sexes). Histopathological examination of the brain revealed that dogs (in all treated groups) suffered slight to moderate vacuolization of white myelinated tracts in the cerebrum and, to a lesser extent, in the cerebellum. Increases in the incidence of haemosiderin-laden Kupffer cells in the liver and cystic mucosal hyperplasia in the gall bladder were observed at all dose levels, even 5 weeks after cessation of the treatment. Prostate and testicular changes were also seen at 50 mg/kg bw per day and above and are discussed in Section 10.5.2. The authors noted that beagle dogs are more susceptible to cataract formation than any other species (Katz et al., 1981).
In another subchronic dog study (Cicmanec et al., 1991), 4-month-old male and female beagle dogs (five per sex per dose) received DCA (neutralized to pH 7.4) in gelatin capsules at 0, 12.5, 39.5 or 72 mg/kg bw per day for 90 days. The controls received encapsulated distilled water. Three high-dose animals died as a result of dehydration and pneumonia. Overt clinical signs such as dyspnoea (middle and high doses) and partial paralysis (high dose only) were observed in both sexes. Diarrhoea was present in the mid- and high-dose animals; some dogs that were highly dehydrated required fluid therapy. Inflammation of the ocular membranes was accompanied by swelling; discharge was clear at the low and middle doses and became purulent in the high-dose group. A dose-related decrease in body weight gain was observed in all treated animals. Relative liver weights were increased in all female dose groups; however, kidney and lung weights were increased only in the high-dose group. In male dogs, the effects in these organs were less consistent. Apparent increases in ALT, AST and lactate dehydrogenase were observed in both sexes in the high-dose group. Erythrocytes and haemoglobin concentrations were significantly reduced in both sexes in the high-dose group from day 30 and beyond. Upon microscopic examination, lesions were apparent in the liver, brain, lung, pancreas and testes. Hepatic vacuolization was observed in most of the treated dogs (both sexes) as well as in a few control animals. In the brain, mild vacuolization of the white myelinated tracts of the cerebrum, cerebellum and/or spinal cord was present in all exposed groups. Pneumonia and bronchopneumonia was observed in most treated dogs; more severe effects were seen in the mid- and high-dose groups. Pancreatic acinar associated with chronic inflammation was observed in many mid-and high-dose animals (both sexes). Testicular (all doses) and prostatic effects (middle and high doses) were seen and are discussed in Section 10.5.2.
10.2.3 Trichloroacetic acid
In a 90-day study (Mather et al., 1990), male Sprague-Dawley rats (10 per dose) were treated with TCA (neutralized) in their drinking water at dose levels of 0, 50, 500 or 5000 mg/L (0, 4.1, 36.5 or 355 mg/kg bw per day). Water consumption was decreased (statistically significant) at the middle and high doses compared with controls. A decrease in body weight was observed for all dose groups, but was not statistically significant. The high-dose group also had an increase in relative liver and kidney weights and a significant increase in hepatic peroxisomal activity.
10.2.4 Monobromoacetic acid
No subchronic studies on MBA were located in the literature.
10.2.5 Dibromoacetic acid
To determine the effects on the liver, male B6C3F1 mice (five per dose) were administered DBA (neutralized) at 0, 300, 1000 or 2000 mg/L in drinking water for up to 12 weeks (Kato-Weinstein et al., 2001). All animals were sacrificed at 20 weeks of age. Decreased water consumption and body weight were seen at the highest dose. A dose-related increase in relative liver weights was seen at 1000 mg/L and above at 12 weeks. Increased absolute and relative liver weights were seen at all time points (4, 8 and 12 weeks) at the highest dose. A significant increase in the total liver glycogen content was seen at the highest dose at 12 weeks. Dose-related decreases in serum glucose and serum insulin concentrations were seen at 1000 mg/L and above at 12 weeks (Kato-Weinstein et al., 2001). DBA displayed effects similar to those of DCA; both caused an increase in glycogen content and a depression of serum insulin concentrations. This may suggest that common mechanisms are involved with dihaloacetates.
A 13-week drinking water study in both B6C3F1 mice and F344 rats was reported by Melnick et al. (2007) and in greater detail in NTP (2007). Animals (10 per dose per sex per species) were exposed to DBA (neutralized to pH 5) in drinking water at doses of 0, 125, 250, 500, 1000 or 2000 mg/L (equivalent to 0, 10, 20, 40, 90 and 166 mg/kg bw per day in male rats and 0, 12, 23, 48, 93 and 181 mg/kg bw per day in female rats; 0, 16, 30, 56, 115 and 230 mg/kg bw per day in male mice and 0, 17, 34, 67, 132 and 260 mg/kg bw per day in female mice). No clinical signs or deaths were observed. A significant decrease in final mean body weights were seen in both species and sexes at the highest dose, however only a slight decrease in water consumption was seen at weeks 1 and/or 13. Dose-related increases in liver weights were noted in both sexes of mice at 500 mg/L and above and in rats at 125 mg/L and above. Dose-related increases in the severity of cytoplasmic vacuolation in hepatocytes were seen in mice (both sexes at 1000 mg/L and above), although these lesions were present in both controls and treated mice. It was noted that the increase in severity corresponded to increased liver weights. In rats, an increased incidence of cytoplasmic vacuolation in hepatocytes was noted in males at 500 mg/L and above and in females at 2000 mg/L. Haematopoietic cell proliferation (mild to minimal) was observed in the spleen of high-dose female rats. A significant increase in the incidence of cellular hypertrophy of the pituary gland was observed in high-dose male rats; however, these effects were considered secondary to testicular atrophy. Testicular effects were seen in rats and mice and are outlined in Section 10.5.5.
Moser et al. (2004) and Phillips et al. (2002) examined the neurotoxic potential of DBA. Adolescent male and female F344 rats (12 per sex per group) were administered DBA (acid) at 0, 200, 600 and 1500 mg/L (estimated as 0, 20, 72 and 161 mg/kg bw per day) for 6 months in drinking water. No treatment-related deaths were noted. A decrease in weight gain was noted in the high-dose rats (both sexes). Functional observational battery test results revealed dose-related neuromuscular toxicity at the middle and high doses, such as limb weakness, mild changes in gait and hypotonia. Sensorimotor responses were depressed at all treatment levels. Decreased activity and chest clasping were also noted at the highest dose. Neuropathological evaluation revealed degeneration of spinal cord nerve fibres in the mid- and high-dose groups. Neuronal vacuolization of the spinal cord (mostly in grey matter and occasionally in white matter) increased in severity and incidence at the middle dose and above. The authors set a noobserved-effect level (NOEL) of 20 mg/kg bw per day based on neuropathological effects but were unable to establish one for neurobehavioural effects due to sensorimotor changes at that level.
Clinical signs of neurotoxic effects were also seen in a male reproductive study (Linder et al., 1995). Male rats (n = 10) were dosed with 250 mg DBA/kg bw daily for up to 42 days, after which dosing was discontinued due to severe toxicity; clinical signs consisted of abnormal posturing, light tremor, atypical movement of limbs and difficulty in moving hind limbs. Rats were allowed to recover for a 6-month interval; some of the effects either diminished (abnormal gait) or disappeared (tremors). No other overt signs of toxicity were seen at the lower doses in male rats (Linder et al., 1995).
NTP (1999a) examined the immunotoxicity of DBA in female mice in four separate studies investigating different end-points. Groups of female B6C3F1 mice (eight per dose) were exposed to DBA in drinking water at concentrations of 0, 125, 250, 500, 1000 or 2000 mg/L per day (calculated as 0, 14-20, 33-39, 68-73, 132-150 or 236-285 mg/kg bw per day) for a period of 28 days. No signs of overt toxicity were seen, and water consumption was unaffected. Only high-dose females had a 40% decrease in body weight gain compared with controls. In one experiment, an increase in spleen weight was observed, while none was seen in a second experiment using similar doses. Several indicators of immunological response were affected. A statistically significant dose-related increase in the number of spleen macrophages was seen at 500 mg/L and above, indicating an immunotoxic response in the spleen. Humoral immunity was also affected, as seen by a decrease of spleen immunoglobulin M antibody-forming cell response to sheep erythrocytes at doses of 500 mg DBA/L and above. A lowest-observed-adverse-effect level (LOAEL) of 500 mg/L (68-73 mg/kg bw per day) and a NOAEL of 250 mg/L (33-39 mg/kg bw per day) were established for immunotoxicity by U.S. EPA (2005a).
McCay et al. (2000) reported in an abstract that no effects on the immune system were seen in a 28-day study in which female B6C3F1 mice were dosed daily via drinking water with DBA concentrations of 0, 250, 500 or 1000 mg/L. Changes in relative and/or absolute liver and thymus weights were the only effects seen.
10.3 Long-term exposure and carcinogenicity
Long-term studies with MCA failed to produce tumours in rodents. In contrast, DCA, TCA and DBA produced liver tumours in laboratory animals. However, these liver tumours were seen only in mice, and not in rats, exposed to TCA and DBA, whereas both rats and mice were affected when exposed to DCA. However in rats treated with DBA, tumours in other organs were detected. No adequate long-term/carcinogenicity studies on MBA were identified. The results of these carcinogenicity studies relating to liver tumours are summarized in Table 9, while the details of these studies are described in the sections below.
| HAA | Dosing route | Doses (mg/kg bw per day) | Duration | Acid or salt | Strain/ species | Sex | Liver tumours found | Study author |
|---|---|---|---|---|---|---|---|---|
| MCA | Gav | 0, 15, 30 | 2 years | A | F344 rats | M & F | None | NTP, 1992 |
| Gav | 0, 50, 100 | 2 years | A | B6C3F1 mice | M & F | None | NTP, 1992 | |
| DW | 0, 3.5, 26, 59.9 | 2 years | S | F344 rats | M | None | DeAngelo et al., 1997 | |
| DCA | DW | 0, 140, 280 | 52 weeks | S | B6C3F1 mice | M | HA, HC | Bull et al., 1990 |
| DW | 0, 7.6, 77, 410, 486 | 60-75 weeks | ?Table 9 Footnote b | B6C3F1 mice | M | HA, HC | DeAngelo et al., 1991; U.S. EPA, 1991 | |
| DW | 0, 93 | 104 weeks | S | B6C3F1 mice | M | HA, HC | Daniel et al., 1992 | |
| DW | 0, 8, 84, 168, 315, 429 | 90-100 weeks | S | B6C3F1 mice | M | HA, HC | DeAngelo et al., 1999 | |
| DW | 0, 40, 120, 330 | 360 or 576 days | S | B6C3F1 mice | F | HA, HC | Pereira, 1996 | |
| DW | 0, 3.6, 40.2, 139.1 | 100 weeks | S | F344 rats | M | HA, HC | DeAngelo et al., 1996 | |
| TCA | DW | 0, 178, 319 | Up to 52 weeks | S | B6C3F1 mice | M | HA, HC | Bull et al., 1990 |
| DW | 0, 7, 71, 595 | 60 weeks | possible S | B6C3F1 mice | M | HA, HC | DeAngelo and Daniel, 1990; U.S. EPA, 1991 | |
| DW | 0, 64, 212, 640 | 360 or 576 days | S | B6C3F1 mice | F | HA, HC | Pereira, 1995, 1996; Pereira and Phelps, 1996 | |
| DW | 0, 71, 583 | 104 weeks | ?Table 9 Footnote b | B6C3F1 mice | F | HA, HC | U.S. EPA, 1991 | |
| DW | 0, 3.6, 32.5, 364 | 104 weeks | S | F344 rats | M | None | DeAngelo and Daniel, 1992; DeAngelo et al., 1997 | |
| DBA | DW | 0, 4, 35, 65 | 104 weeks | S | B6C3F1 mice | F | HA, HC | Melnick et al. 2007; NTP, 2007 |
| DW | 0, 4, 45, 87 | 104 weeks | S | B6C3F1 mice | M | HA, HC | Melnick et al. 2007; NTP, 2007 | |
| DW | 0, 2, 25 45 | 104 weeks | S | F344 rats | F | NoneTable 9 Footnote b | Melnick et al. 2007; NTP, 2007 | |
| DW | 0, 2, 20, 40 | 104 weeks | S | F344 rats | M | NoneTable 9 Footnote b | Melnick et al. 2007; NTP, 2007 | |
10.3.1 Monochloroacetic acid
No tumours were observed in two different strains of mice (18 per sex per strain) gavaged with MCA (acid) in distilled water at 46 mg/kg bw per day from the age of 7 days to 4 weeks, after which MCA was mixed directly in the diet at 149 mg/kg for a total of 18 months (Innes et al., 1969).
In a 2-year carcinogenicity bioassay, F344/N rats (70 per sex per dose) were dosed with MCA (acid) in deionized water at 0, 15 or 30 mg/kg bw per day by gavage, 5 days per week (NTP, 1992). The mean body weight of high-dose male rats was reduced after 30 weeks. Survival rates were significantly reduced for the high-dose male and low- and high-dose female rats, but were not treatment related. Overall, there were no treatment-related increases for nonneoplastic lesions or neoplasia reported. The authors concluded that "there was no evidence of carcinogenic activity"
for MCA in male and female F344/N rats at the dose levels used in the study.
In the same 2-year bioassay, B6C3F1 mice (60 per sex per dose) were dosed with MCA (acid) in deionized water by gavage at 0, 50 or 100 mg/kg bw per day, 5 days per week (NTP, 1992). A significant reduction was noted for mean body weight in high-dose females (after 52 weeks) and in survival rates for high-dose males. The incidences of acute nasal inflammation ranged from mild to minimal in severity but were not treatment related. The incidence of metaplasia of the olfactory epithelium was significantly greater in high-dose females. No other significant increases in non-neoplastic lesions were observed in either sex. No treatment-related increases for any neoplasia were reported. The authors stated that "there was no evidence of carcinogenic activity"
for MCA in B6C3F1 mice (both sexes) at the dose levels used in the study.
In a 104-week drinking water study, male F344/N rats (50 per dose) were exposed to MCA (neutralized) at 0, 50, 500 or 1100 mg/LFootnote 1 (0, 3.5, 26 or 59.9 mg/kg bw per day) (DeAngelo et al., 1997). MCA treatment had no significant effect on survival, but drinking water consumption and final mean body weights were significantly reduced in the two highest dose groups. Absolute and relative spleen weights were significantly increased (74% and 80% over controls, respectively) for rats consuming 3.5 mg MCA/kg bw per day; however, this increase was seen in the absence of gross or microscopic lesions. Although decreases in absolute and relative spleen weights were observed at the two highest doses, only the absolute weight was statistically lower at the highest dose (59.9 mg/kg bw per day). Exposure to 26 mg/kg bw per day significantly decreased absolute liver and kidney weights and relative liver weights and increased relative testes weights. At the 59.9 mg/kg bw per day dose, absolute kidney and absolute and relative liver weights were reduced, while relative testes weights were increased. Serum enzyme analysis revealed no treatment-related effects on either AST or ALT, and MCA treatment did not enhance peroxisome proliferation or hepatocyte proliferation. No MCA-induced liver pathology was observed during the study, nor were there any significant lesions found in any non-hepatic tissues. Most common age-related spontaneous changes present in rat tissues were within historical controls except for myocardial degeneration and chronic/active inflammation of the nasal cavities, which were seen at an increased incidence at 104 weeks but not at earlier sacrifice periods or in the lower dose groups. There were no significant increases in either the prevalence or multiplicity of hepatocellular adenomas, carcinomas or hyperplastic nodules in any of the MCA-treated animals compared with controls. Similarly, neoplastic changes in non-hepatic tissues were consistent with those reported in historical controls. The authors concluded that the decreased body weights and other pathology observed in the mid-dose groups were not significant and set the NOEL for carcinogenicity at 500 mg/L (26 mg/kg bw per day) for MCA (DeAngelo and Daniel, 1992; DeAngelo et al., 1997).
However, Health Canada did not agree with the decision to ignore statistically significant changes in body weight and liver, kidney and testes weights, particularly since these changes are consistent with a dose-related trend. At the next lower dose of 50 mg/L (3.5 mg/kg bw per day), the only significant change was an increase in absolute and relative spleen weights. This change can be considered idiosyncratic (a physiological peculiarity), since there is no dose-related trend towards an increase in spleen weight. At the next dose, 500 mg/L (26 mg/kg bw per day), the absolute and relative spleen weights were lower than control, while the absolute weight was significantly lower at the highest dose. Based on these considerations, Health Canada derived a NOAEL of 3.5 mg/kg bw per day.
10.3.2 Dichloroacetic acid
Several studies on the potential carcinogenicity of DCA have been conducted and have been summarized in Table 9. Increased incidences of either liver tumours (adenomas and carcinomas) or preneoplastic lesions (e.g., hyperplastic nodules and altered hepatic foci) were reported when mice and rats were exposed to drinking water containing 0.5-5 g DCA/L for periods ranging from 52 to 104 weeks. The details of the studies are reported below.
In a 52-week drinking water study (Bull et al., 1990), groups of male B6C3F1 mice (n = 61, 11, 50) were exposed to DCA (neutralized) at 0, 1000 or 2000 mg/L (0, 140 or 280 mg/kg bw per day; WHO, 2005), and a group of 10 females was exposed to 2000 mg DCA/L for up to 52 weeks. Interim sacrifices of five male mice from the 2000 mg/L group were carried out at 15, 24 and 37 weeks. All other animals were sacrificed on week 52, including 11 males that had their exposure terminated at 37 weeks, followed by 15 weeks of recovery before sacrifice. A significant dose-related increase in the absolute and relative liver weights was noted in treated males at 37 or 52 weeks. Livers from all DCA-treated animals were enlarged (hepatomegaly), with a uniform distribution of marked cytomegaly and extensive accumulation of glycogen in hepatocytes as well as multi-focal areas of necrosis with lymphocyte infiltration throughout the liver. Glycogen-poor basophilic foci of cellular alteration were observed at 24 and 37 weeks in the central lobes of livers from males treated with 2000 mg DCA/L. After 52 weeks of treatment, hyperplastic nodules, adenomas and carcinomas were observed in the livers of males in the 2000 mg/L group. In contrast, the only hepatoproliferative lesions observed in the other groups were one hyperplastic nodule in a male mouse from each of the control and 1000 mg/L dose groups and hyperplastic nodules in three females. No hepatocellular carcinomas were reported in males where DCA treatment was terminated at 37 weeks; by 52 weeks, two animals had adenomas, and six had hyperplastic nodules. The relationship between mean total dose of DCA and total number of hepatic lesions (nodules + adenomas + carcinomas) per mouse was nonlinear, with the number of lesions increasing sharply as the dose increased from 1000 to 2000 mg/L (Bull et al., 1990).
Male B6C3F1 mice (30 per dose) were administered DCA in their drinking water at 50, 500 or 5000 mg/L for 60 and/or 75 weeks (DeAngelo et al., 1991; U.S. EPA, 1991). The control group received 2 g sodium chloride/L. In a concurrent study, mice were exposed to 3500 mg DCA/L and killed after 60 weeks. In this experiment, the control group received acetic acid. Time-weighted mean daily doses of 7.6, 77, 410 and 486 mg/kg bw per day were calculated for the 50, 500, 3500 and 5000 mg/L concentrations, respectively. Mice exposed to 3500 and 5000 mg/L had their final body weights reduced compared with the control group: 87% and 83% of the control value, respectively. The relative liver weights were increased at the three highest dose groups compared with the control value: 118%, 230% and 351% for 500, 3500 and 5000 mg/L, respectively.
Hyperplastic liver nodules (a non-neoplastic nodular lesion) were observed primarily at the two highest doses: 58% and 83% incidence at 3500 and 5000 mg/L, respectively. Mice receiving 5000 mg/L (after 60 weeks) had a 90% prevalence of liver neoplasia (carcinomas and adenomas), with a mean multiplicity of 4.50 tumours per animal; those receiving 3500 mg/L had a 100% prevalence, with a tumour multiplicity of 4.0 tumours per animal. The tumour prevalence and multiplicity in the two lowest dose groups at 75 weeks did not significantly differ from the control value. The control group had no liver tumours. The authors concluded that there was a significant positive dose-related trend in age-adjusted prevalence of liver tumours. The authors concluded that DCA exhibited a threshold of at least 500 mg/L (77 mg/kg bw per day) for tumour response in mice. A steep increase in tumour incidence occurred at 3500 mg/L (410 mg/kg bw per day), which also represents the maximum incidence attained (DeAngelo et al., 1991; U.S. EPA, 1991).
In a 104-week drinking water study (Daniel et al., 1992), DCA (neutralized) was administered to a group of 33 male B6C3F1 mice at a concentration of 500 mg/L (93 mg/kg bw per day), with one interim sacrifice (n = 5) at 30 weeks. There were no significant treatment-related effects on drinking water consumption, relative body weight gain or relative or absolute spleen, kidney or testes weight. Absolute and relative liver weights were increased at both 30 and 104 weeks. The most notable treatment-related non-neoplastic hepatic effects observed were cytomegaly, necrosis and chronic active inflammation. After 104 weeks, there was a significant increase in the incidence of hepatocellular carcinomas and hepatocellular adenomas. The combined incidences of carcinoma + adenoma and carcinoma + adenoma + nodule were significantly increased in treated animals compared with controls. Mice killed at the end of the study had hepatocellular carcinomas (15/24, compared with 2/20 in the control groups); hepatocellular adenomas (10/24, compared with 1/20 in the controls); and carcinomas or adenomas (18/24, compared with 3/20 in the controls). Other effects observed were hyperplastic nodules in 2/24 treated mice, hepatocellular necrosis in 8/24 treated mice (compared with 1/20 in the controls) and cytomegaly in 22/24 treated mice (compared with 1/20 in the controls) (Daniel et al., 1992). (Data were not provided for interim necropsies at week 30.)
In a 90- to 100-week drinking water study, male B6C3F1 mice (n = 35-71) were exposed to DCA (neutralized) at 50, 500, 1000, 2000 or 3500 mg/L (8, 84, 168, 315 and 429 mg/kg bw per day) (DeAngelo et al., 1999). The control group consisted of 88 mice. Interim sacrifices were performed throughout the study, except at the lowest dose. At final sacrifice, the body weights were decreased at the two highest doses. A dose-dependent increase in liver weight at 84 mg/kg bw per day and above was seen at 26 and 52 weeks and at 315 mg/kg bw per day and above at 100 weeks. Liver toxicity was demonstrated at 168 mg/kg bw per day and above, as indicated by an increase in serum liver enzymes and histopathology. Mortality at the two highest doses was statistically significant when compared with the control group. There was a significant increase in the incidence of hepatocellular carcinoma in male mice. At 26 weeks, no tumours were seen in the livers of any of the mice, but at 52 and 78 weeks, the incidence of hepatocellular carcinoma was significantly (P < 0.05) elevated in the highest dose group (Table 10). At the terminal sacrifice, the incidence of hepatocellular carcinoma was significantly elevated (P < 0.05) at the three highest dose groups. Hepatocellular adenomas were first observed at the high dose at 26 weeks; however, only at 100 weeks was the incidence of hepatocellular adenomas significantly (P < 0.05) increased (no dose-response) in the three highest dose groups (Table 10).
| Dose (mg/kg bw per day) |
Prevalence of male mice with hepatocellular carcinomas (%) | Prevalence of male mice with hepatocellular adenomas (%) | ||||
|---|---|---|---|---|---|---|
| Week 52 | Week 78 | Week 100 | Week 52 | Week 78 | Week 100 | |
| 0 (water control) | 0 | 10 | 26 | 0 | 10 | 10 |
| 8 | n/rFootnote a | n/r | 33 | n/r | n/r | n/r |
| 84 | 0 | 0 | 48 | 10 | 10 | 20 |
| 168 | 0 | 20 | 71Footnote b | 10 | 20 | 51.4Footnote b |
| 315 | 20 | 50 | 95Footnote b | 0 | 50 | 42.9Footnote b |
| 429 | 50 | 70Footnote b | 100Footnote b | 50 | 50 | 45Footnote b |
Liver peroxisome proliferation was significantly elevated only in the high-dose group after 26 weeks, but not at 52 weeks. In contrast, hepatocyte proliferation was not significantly different from the control rates at any of the doses that produced tumours. The authors could not determine a NOEL for hepatocarcinogenicity based on the significant increase in hepatocellular carcinoma multiplicity at the lowest dose (0.58 compared with 0.28 in the control), but concluded that DCA produced a dose-related increase in the incidence of hepatocellular carcinoma that was not associated with either liver peroxisome or hepatocyte proliferation. Endocrine disruption and liver cell necrosis were proposed as playing important roles in the carcinogenesis (DeAngelo et al., 1999).
Groups of 7- to 8-week-old B6C3F1 female mice (n = 40-90 and n = 134 in the control) were administered DCA (neutralized) in drinking water for either 360 or 576 days at concentrations of 0, 2.0, 6.67 or 20.0 mmol/L (0, 40, 120 or 330 mg/kg bw per day) (Pereira, 1996; IPCS, 2000). An additional group of animals (n = 50) was exposed intermittently to 20.0 mmol DCA/L in drinking water in a 72-day cycle, 24 days with exposure and 48 days without. The 72-day cycle was repeated until the mice were sacrificed, so that the total dose was the same as for those continuously exposed to 6.67 mmol DCA/L. Body weights were reduced in the high-dose group after 35 weeks. There was a dose-related increase in relative liver weight and vacuolated hepatocytes. For the 576-day exposure groups, there was a significant increase in hepatic foci, hepatocellular adenomas and hepatocellular carcinomas at the high dose and an increased incidence of foci and adenomas (but not carcinomas) at the middle dose, compared with controls. The intermittent DCA group also displayed a significantly increased incidence of altered hepatic foci at 576 days, but no significant increases in neoplastic response (Pereira, 1996).
In a modified carcinogenesis bioassay (DeAngelo et al., 1996), 28-day-old male F344 rats (n = 50-78) were exposed to DCA (neutralized) at 0, 50, 500 or 1600Footnote 2 mg/L (estimated time-weighted mean daily doses: 0, 3.6, 40.2 and 139.1 mg/kg bw) in drinking water for 100 weeks. There were no significant differences in water consumption or survival for any of the treatment groups when compared with controls. Terminal body weights and relative liver and kidney weights were reduced only in the 1600 mg/L group. Testicular effects are reported in Section 10.5.2. The only non-neoplastic treatment-related hepatic change was hepatocellular cytoplasmic vacuolization, attributed to DCA-induced increases in glycogen deposition. The combined hepatocellular adenoma and hepatocellular carcinoma prevalence was significantly increased (P < 0.05) in the 500 mg/L group compared with controls, as was the total hepatoproliferative lesion prevalence (hyperplastic nodules, adenomas and carcinomas). Significant dose-related trends were observed for hepatocellular adenoma and hepatocellular carcinoma (P < 0.05 for each) prevalence, combined hepatocellular adenoma and hepatocellular carcinoma prevalence and total hepatoproliferative lesions. In the 1600 mg/L group, increased prevalences of hepatocellular carcinoma, combined hepatocellular carcinoma and hepatocellular adenoma and total hepatoproliferative lesions were observed. At the high dose, DCA induced hepatocyte peroxisome proliferation. DCA treatment depressed hepatocyte proliferation at 14 weeks; at the other time periods, it remained depressed, but did not differ significantly from the control group. The authors concluded that DCA is a hepatocarcinogen in male F344 rats. Male F344 rats were found to be more sensitive to DCA exposure than male B6C3F1 mice based on a previous study by DeAngelo et al. (1991). The authors set a NOEL of 3.6 mg/kg bw per day (DeAngelo et al., 1996).
The ability of DCA to act as a promoter of carcinogenesis in the liver was observed in several studies. According to Pereira (1995), "promotion is defined as the enhancement of the progression of initiated cells to precancerous lesions and tumors."
A tumour promotion study was conducted in male B6C3F1 mice with DCA (neutralized) (Herren-Freund and Pereira, 1986; Herren-Freund et al., 1987). Tumour incidences (hepatocellular adenomas and carcinomas) were significantly elevated for mice with and without initiation compared with the controls. The authors concluded that DCA is a complete hepatocarcinogen in B6C3F1 mice (Herren-Freund et al., 1987).
The potential of DCA (neutralized) to promote tumours was also investigated in female B6C3F1 mice (Pereira, 1995; Pereira and Phelps, 1996). An increased incidence of hepatic foci and adenomas was seen in initiated mice exposed to the highest dose of DCA for either 31 or 52 weeks compared with the corresponding animals receiving initiation or DCA alone. A second-order relationship (i.e., exponential relationship) was observed between the concentration of DCA and the yield of total lesions (foci + adenomas + carcinomas) at both 31 and 52 weeks. Upon termination of treatment, the foci of altered liver hepatocytes and adenomas regressed.
Two separate initiation-promotion studies, conducted by Bull et al. (2004) and Pereira et al. (1997), looked at the interaction between DCA and TCA (both tumour promoters involving different mechanisms) in B6C3F1 mice. Different results between the two studies may be related to doses used.
Bull et al. (2004) demonstrated that the interactions between TCA and DCA were limited by additivity: the lowest effective doses with TCA and DCA showed additivity, whereas the effects tended to be inhibitory (suppression of overall growth rate) as the doses increased. DCA and TCA selectively promote the growth of different types of initiated cells, by stimulating the clonal expansion in one cell type while inhibiting it in another cell type. Such a process would explain the presence of a mixed phenotype of tumours as well as the suppression of the overall tumour growth rate in the present study (Bull et al., 2004).
In contrast, Pereira et al. (1997) observed synergistic effects with a mixture of TCA and DCA at high doses; the yields of foci of altered hepatocytes and total lesions produced were greater than the sums of the yields produced by the two HAAs administered alone, whereas additive or inhibitory effects were seen with the mixtures at lower doses. The mixtures of DCA-TCA produced more foci of altered hepatocytes than adenomas, similar to the effects seen when DCA was administered alone. In addition, the phenotype of these lesions resembled those of DCA-induced tumours, suggesting that DCA may predominate in determining the characteristics of the proliferative lesions.
10.3.2.1 Mechanisms of carcinogenicity
Based on the results of the studies described above and summarized in Table 9, DCA has been shown to be a liver carcinogen in two species of rodents, mice and rats. DCA is thought to be a "complete carcinogen,"
since it has induced tumours at both low doses in long-term assays and high doses in shorter-term assays when administered alone; doses from these studies ranged from 50 to 5000 mg/L. More than one mode of action may explain DCA-induced carcinogenicity, and several hypotheses have been put forward, as described below. In all likelihood, a number of events would be significant to tumour development in the rodent under bioassay conditions. Uncertainty exists, however, as to which events may be relevant to human exposure to DCA at environmental levels.
10.3.2.1.1 Genotoxicity
Mixed reviews were seen with regard to DCA-induced carcinogenicity mediated through a genotoxic mechanism. Previous reviews by IARC (1995) and ILSI (1997) reported that DCA was not genotoxic, whereas a more recent review by IPCS (2000) reported that DCA had some ability to induce genotoxic effects but only at high concentrations; it concluded that at low doses, DCA was either not acting or acting minimally through a genotoxic mechanism. The U.S. National Center for Environmental Assessment (U.S. EPA, 2003c) reported that more recent literature indicated that DCA is a direct-acting genotoxic agent. IARC (2004) recently reported that DCA is genotoxic (in vivo and in vitro) but may be acting indirectly via an epigenetic mechanisn. Owing to the lack of causal data, U.S. EPA (2003c) took the cautious position that DCA might be genotoxic at high doses while uncertainty remained for the lower doses.
10.3.2.1.2 Peroxisome proliferation
Some hepatocarcinogens have been shown to increase the number and/or size of liver peroxisomes (peroxisome proliferation) in rodents (U.S. EPA, 2003c). Peroxisome proliferatoractivated receptors (PPARs) are a class of nuclear receptors that regulate this proliferation and are believed to be responsible for the initiation of certain cellular events leading to transformation (as well as other reported effects) for liver carcinogens. Although peroxisome proliferation has been correlated with carcinogenesis, the actual mechanism of carcinogenesis as it is related to peroxisome proliferation is unknown (Bull, 2000). Species differences are seen with relation to the expression of various PPARs; humans seem less responsive to a variety of peroxisome proliferators than rodents, and, as a result, controversy exists as to whether these compounds are even carcinogenic to humans (Bull, 2000; U.S. EPA, 2003c).
Studies conducted by DeAngelo et al. (1989, 1999), Daniel et al. (1992), Mather et al. (1990) and Pereira (1995, 1996) showed that DCA was a weak peroxisome proliferator in rodents.
Seemingly, much lower doses of DCA are needed to induce liver tumours compared with those that cause a significant increase in peroxisome proliferation (U.S. EPA, 2003c). In conclusion, U.S. EPA, (2003c), Thai et al. (2003) and Bull (2000) do not consider this mechanism an important route in DCA tumorigenesis.
10.3.2.1.3 Down-regulation of insulin
The glycogen content of liver cells of DCA-treated mice can vary depending on the state of the cell; altered hepatic foci and tumours are glycogen poor, whereas normal liver cells accumulate large amounts of glycogen (Lingohr et al., 2001), thus suggesting a possible link between glycogen and hepatic tumours. Glycogen levels are mediated primarily by insulin via metabolism in the liver (Lingohr et al., 2001). Insulin may have other roles, such as acting as an agent that triggers mitosis (mitogen) for normal and malignant liver cells and suppressing apoptosis (Lingohr et al., 2001).
Lingohr et al. (2001) investigated the effects of DCA on insulin levels and expression of insulin-controlled signalling proteins in normal liver tissue and DCA-induced liver tumour tissues of male B6C3F1 mice treated with DCA (neutralized) at 100-2000 mg/L in drinking water for 2-10 weeks. Decreases were seen in insulin receptor proteins, serum insulin levels and protein kinase B expression after 2 weeks in DCA-treated mice. In contrast, increases in glycogen (which preceded these effects) were seen as early as 1 week, suggesting that DCA-induced alterations in insulin, insulin receptors and possibly protein kinase B resulted from a compensatory down-regulation of the insulin pathway triggered by high glycogen levels in the liver. An in vitro study by Lingohr et al. (2002) using isolated hepatocytes showed similar results: increased glycogen levels (independent of insulin) and effects on insulin signalling proteins.
These studies show an apparent down-regulation of insulin and insulin receptor activity after relatively short durations of treatment.
10.3.2.1.4 Tumour promotion, alterations in cell replication and death
Stauber and Bull (1997) studied the differences in phenotype and cell replicative behaviour of liver tumours induced by DCA and TCA in male B6C3F1 mice. Clear differences in phenotype were seen between DCA- and TCA-induced tumours. The study also showed the extent to which changes in cell replication within tumours and normal hepatocytes were influenced by DCA to increase tumour formation. Stauber et al. (1998) also conducted in vitro studies, which duplicated these results.
Several mechanisms of action are proposed for tumour promoters. Bull et al. (2004) suggested that promoters should influence mainly tumour size rather than tumour numbers. This behaviour can be observed with DCA. Miller et al. (2000) investigated, by means of magnetic resonance imaging, the growth rates of liver tumours in male B6C3F1 mice given DCA (neutralized) at 2000 mg/L in their drinking water for 48 weeks - a treatment period ensuring induction of small liver tumours. Results showed that continued growth of the tumours was entirely dependent upon DCA treatment. The authors suggested that DCA's main effect in tumour induction is mediated through accelerated growth of spontaneously initiated cells and that it may be due to the suppression of apoptosis and modification of cell replication rates.
Another hypothesis for promoters is the suppression of apoptosis. Snyder et al. (1995) studied the frequency of spontaneous apoptosis in liver hepatocytes of male B6C3F1 mice when given DCA at dose levels of 0, 500 or 5000 mg/L in their drinking water for 5-30 days. DCA significantly reduced apoptosis in treated mice in a dose-dependent manner relative to the untreated controls. The authors suggest that disrupting the apoptosis process can result in the outgrowth of initiated cells (by suppressing the ability of the liver to remove initiated cells, preneoplastic cells) and thus leading to the formation of tumours, rather than by induction of selective proliferation of initiated cells.
In a review of TCE and its metabolites (Bull, 2000), the data suggest that the modification of cell signalling pathways resulting in cell replication, selection and apoptosis may be an important contributor to the hepatocarcinogenicity of DCA.
In a hepatic cell proliferation study (Pereira, 1995, 1996), groups of 10 female B6C3F1 mice were exposed to DCA (neutralized) in drinking water at concentrations of 0.26, 0.86 or 2.6 g/L (52, 172 or 520 mg/kg bw per day) until sacrifice at day 5, 12 or 33. A concentration-related increase in cell proliferation was seen after 5 days of treatment with DCA but not at 12 and 33 days, except for an apparent increase at the high dose at 12 days. Therefore, DCA treatment caused a transient increase in hepatic cell proliferation (Pereira, 1995, 1996).
Increases in hepatocyte proliferation were observed in other short-term studies (Carter et al., 1995; Stauber and Bull (1997). Decreased cell proliferation was seen at higher doses and with chronic dosing periods (Bull, 2000; U.S. EPA, 2003c).
Carter et al. (2003) conducted a histopathological analysis of hepatic lesions from male mice from the carcinogencity study by DeAngelo et al. (1999) in order to identify and quantify the different phenotypes of hepatocellular lesions, such as altered hepatic foci, large foci of cellular alteration, adenomas and carcinomas. As a result of the analysis, three different lesion sequences were proposed during mouse liver carcinogenesis - altered hepatic foci, large foci of cellular alteration and adenomas - which demonstrated neoplastic progression with time. The analysis also demonstrated that some toxic adaptive changes in non-involved liver were related to this neoplastic progression, regardless of the dose and length of exposure. According to Carter et al. (2003), the homeostasis of liver cells is altered by DCA, which leads to negative selection of cells (by suppressing apoptosis, a natural process for eliminating initiated cells), with a new state of differentiation resistant to DCA toxicity, thereby allowing the growth and/or survival of these initiated cells (i.e., preneoplastic cells and lesions). These dose-related effects occur at less than 1 g/L, which the authors considered as the inflection point of the dose-response curve for carcinogenesis.
10.3.2.1.5 Other mechanisms: hypomethylation
Another hypothesis for non-genotoxic carcinogens may be through an epigenetic mechanism involving DNA methylation (Pereira et al., 2004). The reduction in the level of DNA methylation (hypomethylation) is a common event in most cancers, including liver cancer (Pereira et al., 2004).
DNA methylation, a DNA modification that occurs naturally, takes place when a methyl group is added to the 5-position carbon of the cytosine ring to form 5-methylcytosine; the methyl group is supplied by S-adenosylmethionione, while the reaction is catalysed by DNA methyl-transferase (Ge et al., 2001; Pereira et al., 2004). Disruption of these different processes of DNA methylation may lead to hypomethylation (Tao et al., 2000).
DCA (neutralized) reduced the level of DNA methylation (hypomethylation) in the liver and as a result induced foci of altered hepatocytes and hepatocellular adenomas when given to female B6C3F1 mice in drinking water (Pereira et al., 2004). The authors suggested that there may be a correlation between the prevention of carcinogen-induced DNA methylation and prevention of liver tumours and that DNA hypomethylation may be critical for the carcinogenic effects of DCA.
Tao et al. (1998) showed in a promotion study that DCA reduced the levels of 5-methylcytosine in DNA of liver tumours and that the neoplastic progression of the liver lesions, from adenomas to carcinomas, seemed associated with a decrease in the level of 5-methylcytosine in DNA. In another study, Tao et al. (2000) reported that DCA decreased the methylation in the promoter regions for 2 proto-oncogenes, c-jun and c-myc, and increased the expression of their mRNA and proteins, while the addition of methionine prevented these changes. Both these proto-oncogenes are known to participate in the control of cell proliferation (U.S. EPA, 2003c).
Stauber and Bull (1997) identified that DCA-induced lesions had a phenotype that was cjun immunoreactive.
Uncertainty exists regarding the actual importance that decreased methylation associated with an increase in the expression of the mRNA bears on the mediation of tumorigenicity of DCA (U.S. EPA, 2003c).
10.3.3 Trichloroacetic acid
Several studies on the potential carcinogenicity of TCA have been conducted and have been summarized in Table 9. Increased incidences of liver tumours (adenomas and carcinomas) were reported when mice were exposed to drinking water containing 500-5000 mg TCA/L for periods ranging from 52 to 104 weeks. No liver tumours were found in rats. The details of the studies are reported below.
In a 1-year chronic drinking water study (Bull et al., 1990), groups of male B6C3F1 mice (n = 61, 11 and 50) were exposed to TCA (neutralized) at 0, 1000 or 2000 mg/L (0, 178 and 319 mg/kg bw per day, respectively; WHO, 2004b), and a group of 10 females was exposed to 2000 mg/L in drinking water for up to 52 weeks. Interim sacrifices were made throughout the study, but all animals were sacrificed by week 52. Dose-related accumulation of lipofuscin (indicative of intracellular lipid peroxidation) in the liver was seen in mice at 52 weeks but not in males terminated at 37 weeks. Dose-related increases in the incidence of hepatoproliferative lesions - namely, hyperplastic nodules, adenomas and hepatocellular carcinomas - were seen in mice at 1000 mg/L and above at 52 weeks. Large concentrations of lipofuscin were found in areas surrounding the hepatoproliferative lesions but were absent from the lesions themselves. There was a linear relationship between the mean total dose of TCA consumed over 52 weeks and the number of hepatic lesions (nodules + adenomas + carcinomas) per mouse, although the incidence of lesions per mouse for animals in the 37-week exposure group was less than what would have been predicted on the basis of the total dose administered. No hyperplastic or neoplastic lesions were observed in any of the female mice treated with TCA. In rats treated with TCA, the liver showed only small increases in hepatic cell size, modest accumulation of glycogen, absence of focal necrosis and only marginal induction of cell proliferation and organ hypertrophy (Bull et al., 1990).
In a 60-week drinking water study (DeAngelo and Daniel, 1990), groups of male B6C3F1 mice (number not specified) were administered TCA (possibly neutralized based on a similar study with DCA by DeAngelo et al., 1991) at 0, 50, 500 or 5000 mg/L (0, 7, 71 and 595 mg/kg bw per day). A dose-related increase in the incidences of hyperplastic nodules, adenomas and carcinomas was seen. The hepatocellular tumour incidence was 55.2% at the high dose, 37.9% at the middle dose and 13.37% in the untreated control group. At the lowest dose (50 mg/L), no significant difference in tumour prevalence or multiplicity was observed compared with the control group. Hyperplastic nodules of the liver were seen in the highest dose only (prevalence: 24.1%), while none was seen at the lower dose or in the control group. Chronic inflammation and liver necrosis were observed in the two highest dose groups near the end of the study.
In another chronic study, female B6C3F1 mice (n = 10-40) received TCA (neutralized) at 2.0, 6.67 or 20.0 mmol/L (64, 212 and 640 mg/kg bw per day) in their drinking water for 360 or 576 days (Pereira, 1996; Pereira and Phelps, 1996). The control group received 640 mg sodium chloride/kg bw per day. At the high dose, after 360 days of exposure, a significant increase in hepatocellular carcinoma incidence and multiplicity was seen; after 576 days of exposure, significant increases in altered hepatic foci, hepatocellular adenoma and hepatocellular carcinoma incidences and yields were seen. After 576 days, the middle dose also had increased incidences and yields of foci and carcinomas. The combined numbers of total lesions (foci + adenomas + carcinomas) and total tumours (adenomas + carcinomas) appeared to be linearly related to TCA dose. The authors concluded that TCA is a peroxisome proliferator and that the basophilic staining of the tumours is consistent with other peroxisome proliferators (Pereira, 1995, 1996; Pereira and Phelps, 1996).
In a review of the carcinogenicity of TCA in rodents by the U.S. EPA (1991), the results from a carcinogenicity study of TCA (form unknown) in female B6C3F1 mice (number unknown) were reported. Groups of female mice were administered 0, 500 or 4500 mg/L (0, 71 or 583 mg TCA/kg bw per day; WHO, 2004b) in drinking water for 104 weeks. The combined liver tumour incidence (adenomas + carcinomas) was significantly increased in the high-dose group. The high dose had a 64% hepatocellular tumour incidence, and the lowest dose had a tumour incidence of 16.7%; the incidence was 7.7% in the untreated control group. The LOAEL as derived by U.S. EPA (1991) in female mice is 71 mg/kg bw per day based on the presence of tumours.
There was no evidence of increased liver tumours in male F344 rats in a 2-year carcinogenicity study in which groups (50 per dose) were administered TCA (neutralized) at 0, 50, 500 or 5000 mg/L (0, 3.6, 32.5 or 364 mg/kg bw per day) in drinking water (DeAngelo and Daniel, 1992; DeAngelo et al., 1997). In the high-dose group, significant decreases were seen in body weights and in absolute liver weights, while increases in serum levels of ALT and palmitoyl conenzyme A activity (a marker of hepatic peroxisome proliferation) were seen. The authors set a NOEL of 364 mg/kg bw per day. A NOAEL based on non-neoplastic effects is derived at 32.5 mg/kg bw per day.
The ability of TCA to act as a promoter of carcinogenesis in the liver was also examined in several studies. Parnell et al. (1986, 1988) studied the initiation and promotion abilities of TCA (neutralized) using rat hepatic enzyme-altered foci bioassays. The authors concluded that TCA was a weak peroxisome proliferator and a liver tumour promoter, but not an initiator of liver tumours in Sprague-Dawley rats (Parnell et al., 1986, 1988).
The potential of TCA (neutralized) to promote tumours was also investigated in male B6C3F1 mice (Herren-Freund and Pereira, 1986; Herren-Freund et al., 1987). The incidence of hepatocellular adenomas and carcinomas was significantly increased for both types of treatment: those pretreated with an initiator or not. The authors concluded that TCA was hepatocarcinogenic, regardless of pretreatment with an initiator (Herren-Freund and Pereira, 1986; Herren-Freund et al., 1987).
In a second promotion study, Pereira (1995); Pereira and Phelps (1996) used groups of female B6C3F1 mice. A small increase in the incidence of neoplastic changes was noted at the highest dose in TCA groups without prior initiation. In contrast, when mice were pretreated with an initiator and then exposed to TCA, a significant increase in the incidence and multiplicity of adenomas and carcinomas was seen at the higher doses. The total number of neoplastic lesions per mouse was linearly related to the TCA dose with time.
10.3.4 Monobromoacetic acid
No long-term studies on MBA were identified.
10.3.5 Dibromoacetic acid
Although an abstract published by Bull (1995) referred to a 2-year limited drinking water study on the carcinogenic potential of DBA and other brominated haloacetates in B6C3F1 mice and F344 rats, the final version has yet to be published.
However, a more recent 2-year carcinogenicity drinking water study on DBA was published by Melnick et al. (2007) and with greater detail by NTP (2007). Groups of F344/N rats (50 per sex per dose) and B6C3F1 mice (50 per sex per dose) were administered DBA (neutralized to pH 5) in drinking water at 0, 50, 500 or 1000 mg/L (equivalent to 0, 2, 20 and 40 mg/kg bw per day for male rats and 0, 2, 25 and 45 mg/kg bw per day for female rats; 0, 4, 45 and 87 mg/kg bw per day for male mice and 0, 4, 35 and 65 mg/kg bw per day for female mice). No effect on survival was seen in either species, nor were there any changes in body weight in mice. However, in rats (both sexes), mean body weight was decreased at the two highest doses compared with controls, whereas water consumption was decreased at the highest dose (both sexes). Neoplastic lesions were observed at multiple sites in both rats and mice.
In male rats, a significant increase in malignant mesotheliomas of the abdominal cavity was observed at the highest dose. In female rats, a positive increasing trend in the incidence of monocellular cell leukaemia, a haematopoietic (involved in the formation of blood cells) neoplasm, was noted that was significant at the highest dose. In contrast, male rats showed a significant increase in monocellular cell leukaemia at the lowest dose and no increase at the highest dose (which was similar to concurrent controls and historical controls), but incidences seen at the low and middle doses exceeded historical controls. Other non-cancer effects included significant increased incidence of lesions in the liver (cystic degeneration, minimal to mild) of all exposed groups of males, in the lung (alveolar epithelial hyperplasia) of the two highest dose groups in females, and in the kidney (nephropathy) of all exposed groups of females.
In mice, neoplasms were observed in both the liver and lung. A significant increase in the incidence of multiple hepatocellular adenoma and hepatocellular adenoma or carcinoma (combined) was observed in all treated males and in females at 500 mg/L and above, whereas a significant increase in the incidence of hepatocellular carcinoma was observed only in high-dose males and in mid-dose females. A significant increase in the incidence of hepatoblastoma was observed in male mice at 500 mg/L and above. The incidence of alveolar/bronchiolar adenoma exceeded the historical controls in males at 500 mg/L and 1000 mg/L, although the increase was significant only in males at 500 mg/L. In females, a non-significant increase in lung neoplasms was observed. Non-cancer effects, non-significant increases in the incidence of alveolar epithelial hyperplasia, were observed at all doses in male mice. An increased incidence of splenic haematopoieisis was also observed in high-dose male mice (NTP, 2007).
NTP (2007) indicates that there was clear evidence of carcinogenic activity of DBA in mice, based on increased incidences of hepatocellular neoplasm (both sexes) and hepatoblastomas (male only). It also finds some evidence of carcinogenic activity in rats based on an increased incidence of malignant mesothelioma in males and an increased incidence and positive trend of mononuclear cell leukaemia in females.
10.4 Mutagenicity and genotoxicity
10.4.1 Monochloroacetic acid
There was no evidence of genotoxic potential in mutagenicity studies in bacteria using Salmonella typhimurium (Rannug et al., 1976; NTP, 1992; BG Chemie, 1993; Giller et al., 1997; ECETOC, 1999). Mostly positive results were seen in assays using mammalian cells (mouse lymphoma assay) (Amacher and Turner, 1982; McGregor et al., 1987) , but these may be due to changes in pH or cytotoxicity (ECETOC, 1999). In vitro DNA repair/damage assays involving Escherichia coli, S. typhimurium and cultured mammalian cells were largely negative (Gross et al., 1982; Ono et al., 1991; Chang et al., 1992; NTP, 1992; BG Chemie, 1993; Giller et al., 1997; ECETOC, 1999), except for one study looking at DNA strand breaks in Chinese hamster ovary cells, which was positive (Plewa et al., 2002). Clastogenic studies were mostly negative with MCA (Galloway et al., 1987; Sawada et al., 1987; Giller et al., 1997), except for one study in which MCA (acid) induced sister chromatid exchange in Chinese hamster ovary cells without S9 only (Galloway et al., 1987). In an in vivo bone marrow assay, positive results were seen via the intraperitoneal route, but not by the oral or subcutaneous route (Bhunya and Das, 1987). Details were lacking in this study. Sperm shape abnormalities were seen in an intraperitoneal injection study in mice only at the top two doses, but this study was poorly reported (Bhunya and Das, 1987).
10.4.2 Dichloroacetic acid
There was mostly negative evidence of genotoxic potential in mutagenicity studies in bacteria using S. typhimurium (Herbert et al., 1980; Matsuda et al., 1991; DeMarini et al., 1994; Fox et al., 1996; Giller et al., 1997; Meier et al., 1997), and equivocal results were seen with mammalian cells (Fox et al., 1996; Harrington-Brock et al., 1998). Clastogenic studies demonstrated largely negative results (Fox et al., 1996; Giller et al., 1997; Meier et al., 1997). DNA repair assays with bacteria were generally positive, but involved DCA as a free acid. DNA damage assays (strand breaks) involving mammalian cells (in vitro and in vivo) were largely negative (Chang et al., 1992; Plewa et al., 2002), except for one oral study involving mice and rats where the acid was used (Nelson and Bull, 1988; Nelson et al., 1989). Dose-related sperm head abnormalities were seen in mice following oral gavage with DCA (sodium salt) at doses of 1125-4500 mg/kg bw per day (Meier et al., 1997).
10.4.3 Trichloroacetic acid
There was no evidence of genotoxic potential in mutagenicity studies in bacteria (IARC, 1995; Kargalioglu et al., 2002; NTP, 2003a), except in one modified Ames study, which was weakly positive (Giller et al., 1997). Weakly positive results were also seen in the one gene mutation study with mammalian cells (Harrington-Brock et al., 1998). In vitro DNA damage assays with mammalian cells were negative (Chang et al., 1992; Plewa et al., 2002), but mixed results were seen when TCA was administered in vivo (Nelson and Bull, 1988; Nelson et al., 1989; Chang et al., 1992). TCA gave mostly negative results for DNA damage/repair with bacterial systems (Ono et al., 1991; Giller et al., 1997).
Clastogenic studies (in vitro and in vivo) also demonstrated equivocal results (Bhunya and Behera, 1987; MacKay et al., 1995; Giller et al., 1997; Meier et al., 1997). Harrington-Brock et al. (1998) reported that positive results for clastogenicity and mutagenicity may be seen in in vitro mammalian assays as a result of low pH, especially in the presence of metabolic activation.
Sperm shape abnormalities in mice were equivocal with intraperitoneal administration but negative with oral dosing (Bhunya and Behera, 1987). Gap-junctional intercellular communication was seen in liver cells of mice (in vitro) (IARC, 1995).
10.4.4 Monobromoacetic acid
There is some evidence to suggest that MBA is weakly mutagenic. Mixed results were seen in the Ames assay with MBA (Saito et al., 1995; Kohan et al., 1998; Kargalioglu et al., 2002; NTP, 2003b). Kargalioglu et al., (2002) reported in their Ames assay that MBA was more mutagenic than DBA or MCA. There was also slight evidence of genotoxic potential in in vitro DNA damage assays. MBA (acid) failed to induce primary DNA damage in an SOS chromotest (E. coli) with and without S9 and failed to increase the frequency of micronuclei in a newt micronucleus test (Giller et al., 1997). In contrast, MBA induced DNA strand breaks in the absence of S9, following a 1-hour treatment, using L-1220 mouse leukaemia cells (Stratton et al., 1981). The number of strand breaks increased further when the chemical was removed (Stratton et al., 1981). The authors suggested that MBA may act as an alkylating agent due to the presence of strand breaks (indicator of direct DNA damage).
10.4.5 Dibromoacetic acid
There is some evidence to suggest that DBA is weakly mutagenic. Mixed results were seen with the Ames test with S. typhimurium (Saito et al., 1995; Giller et al., 1997; Morita et al., 1997; Kohan et al., 1998; Kargalioglu et al., 2002; NTP, 2007). Positive results were seen for genotoxic potential in in vitro DNA damage assays. DBA induced primary DNA damage in an SOS chromotest using E. coli with and without metabolic activation (Giller et al., 1997), and induced DNA strand breaks in Chinese hamster ovary cells as measured in a comet assay (Plewa et al., 2002). There was also some evidence of clastogenicity (increased frequencies of micronucleated normochromatic erythrocytes using peripheral blood) in male mice, but not in female mice, when DBA was administered orally via drinking water as part of a 13-week study (NTP, 2007). No clastogenic activity was seen in the newt micronucleus test (Giller et al., 1997).
10.5 Reproductive and developmental toxicity
10.5.1 Monochloroacetic acid
No reproductive studies were identified. No adequate developmental studies were conducted with MCA. Two studies are reported below. However, they lack important information; one study originates from an abstract, and the other one uses only one dose.
One developmental assay (oral gavage with MCA acid in distilled water at 0, 17, 35, 70 or 140 mg/kg bw per day from gestation days 6 to 15) using Long-Evans rats was published in an abstract by Smith et al. (1990). This study suggested developmental effects (laevocardia in the high-dose group) in the presence of maternal toxicity. No statistical data were provided in the abstract, and no final study was published to confirm these results.
In a second developmental study (using only one dose), a group of 10 pregnant Sprague-Dawley rats was given MCA (neutralized) in drinking water at a concentration of 1570 mg/L (193 mg/kg bw per day) on gestation days 1-22 (Johnson et al., 1998). The control group consisted of 55 females. A significant decrease in body weight gain was observed in exposed dams relative to controls. The average amount of drinking water consumed on a daily basis per maternal rat was lower in treated dams than in the control group. No adverse reproductive, developmental or teratogenic effects were reported; however, a complete fetal examination for internal or skeletal abnormalities was not conducted.
10.5.2 Dichloroacetic acid
10.5.2.1 Developmental studies
Several studies on the potential developmental toxicity of DCA in female rats have been conducted and are detailed below and summarized in Table 11.
| Doses (mg/kg bw per day) | Species | Developmental effects | LOAEL / NOAEL | Reference |
|---|---|---|---|---|
| 0, 14, 140, 400,900, 1400, 1900 or 2400 (GDTable 11 Footnote a 6-15) | Long-Evanshooded rats (19-21/dose) |
|
NOAEL for developmental toxicity: 14 mg/kg bw per day | Smith et al., 1992 |
Four studies:
|
Long-Evans rats (7-11/dose) |
|
Epstein et al., 1992 | |
| 0 or 300 (GD 6-15) | Sprague-Dawley rats (19-20/dose) |
|
Fisher et al., 2001 | |
In a developmental study (Smith et al., 1992), pregnant Long-Evans hooded rats (19-21 per dose) were administered DCA (neutralized) by gavage in two separate studies at dose levels of 0, 14, 140 or 400 or of 0, 900, 1400, 1900 or 2400 mg/kg bw per day on days 6-15 of gestation, inclusively. There were dose-related reductions in adjusted maternal weight gain (140 mg/kg bw per day and above), dose-related increases in relative liver weights (all doses) and dose-related increases in relative kidney and spleen weights (400 mg/kg bw per day and above). Treatment-related maternal lethality was seen at 1400 mg/kg bw per day and above (1/19 [5.3%], 2/19 [10.5%] and 5/21 [24%], respectively). The pregnancy rates, total number of implants per litter and preimplantation losses were not affected by treatment. There was a dose-related increase in post-implantation loss rate (900 mg/kg bw per day and above), and the number of live fetuses per litter was reduced at the highest dose (at which significant maternal toxicity [lethality] was observed). A dose-related decrease in fetal body weight and crown-rump length was observed at 400 mg/kg bw per day and above. Dose-related increases in external (1400 mg/kg bw per day and above), total soft tissue (140 mg/kg bw per day and above), cardiovascular (400 mg/kg bw per day and above), urogenital (1400 mg/kg bw per day and above) and orbital (900 mg/kg bw per day and above) malformations were observed. Lower frequencies of urogenital (bilateral hydronephrosis, renal papilla, stage one) and orbital anomalies were seen compared with other malformations. The main fetal target for DCA was the heart and major vessels. The most common heart defect in fetuses occurred between the ascending aorta and the right ventricle and was identified as high interventricular septal defect. The second most common heart malformation was overt interventricular septal defect. The authors set the NOAEL for developmental toxicity at 14 mg/kg bw per day.
Four studies were performed with pregnant Long-Evans rats (7-11 per dose) exposed to DCA (neutralized) to determine the most sensitive developmental period and also to characterize heart defects (Epstein et al., 1992). In the first study, rats were treated by oral intubation with 1900 mg DCA/kg bw per day on gestation days 6-8, 9-11 or 12-15; in the second study, with 2400 mg/kg bw per day on gestation day 10, 11, 12 or 13; and in the third study, with 3500 mg/kg bw per day on gestation day 9, 10, 11, 12 or 13. No significant maternal toxicity was observed in the first three studies. The mean percentages of heart malformations were significantly higher (statistically) in the group treated with 1900 mg DCA/kg bw per day on gestation days 9-11 (7.2% per litter) and 12-15 (15.1% per litter) compared with the combined controls. The cardiac defects seen were either high interventricular septal defect or a conventional interventricular septal defect. Lower incidences of cardiac malformations were observed with the 2400 mg DCA/kg bw per day dose (gestation day 10: 2.5% per litter; gestation day 12: 3.3% per litter; gestation days 11 and 13: 0%). The incidences on gestation days 10 and 12 were significantly different from the combined controls. At the higher dose, 3500 mg/kg bw per day, the incidences of heart defects were not increased, but this study showed that dosing on gestation days 9, 10 and 12 (3.6%, 2.9% and 2.9% per litter, respectively) would produce these defects, and the incidences were significantly different from combined control (0.5% per litter). No heart defects were seen on gestation day 11 or 13 (Epstein et al., 1992).
In a fourth study (Epstein et al., 1992) to characterize heart defects, six dams were orally intubated with 1900 mg DCA/kg bw per day on days 6-15 of pregnancy, and 56 fetuses from the six litters as well as eight control fetuses from four litters were harvested for light microscopic examination of the heart. Examination revealed that 25 of 56 fetuses (45%) had cardiovascular defects; 24 had hearts characterized by high interventricular septal defects (five of these hearts also presented a membranous-type interventricular septal defect), and one had an isolated occurrence of an interventricular septal defect, membranous type.
In a more recent study, Fisher et al. (2001) gavaged a group of 20 pregnant female Sprague-Dawley rats with DCA (neutralized) at 300 mg/kg bw per day during gestation days 6-15. A control group (n = 19) was dosed with water. No malformations of the heart were seen, contrary to those reported in previous studies by Smith et al. (1992) and Epstein et al. (1992). It is possible that this study was not sensitive enough, given the high background of heart malformations (on a per litter basis) seen in the water controls (31.6%; higher than in the treated animals: 30%), which might have masked the effects in the DCA treatment group. The Smith et al. (1992) study showed only one or two cardiovascular defects at doses below 300 mg/kg bw per day. It is also possible that the strain differences in the rats and differences in the purity of the test agents used may account for the incongruent findings between the two set of studies.
10.5.2.2 Reproductive studies
Adult Sprague-Dawley male rats (n = 24 per dose) were given single oral doses of DCA (neutralized) at 0, 1500 or 3000 mg/kg bw to study testicular toxicity and then sacrificed (n = 8 per time period) on day 2, 14 or 28 (Linder et al., 1997a). No clinical signs of toxicity were seen in treated rats. Body weights were not statistically different from controls at any of the three time points. Mild effects on spermiation (i.e., delays) and changes in the degree of resorption of residual bodies were seen at both doses; these effects persisted to varying degrees throughout the observation period (Linder et al., 1997a).
In a parallel study by the same author, another group of male rats (n = 8-26) was given multiple oral doses of DCA (neutralized) at 0, 18, 54, 160, 480 or 1440 mg/kg bw per day for up to 14 days (Linder et al., 1997a). No clinical signs of toxicity were observed in the treated animals, except for reduced weight gain, which was seen at the three top doses by day 14. Delayed spermiation and formation of atypical residual bodies were observed in all treated males, except at the lowest dose. An increased number of fused epididymal sperm occurred after day 5 at doses of 160 mg/kg bw per day and above. A decrease in percentage of motile sperm was seen on day 9 at 480 mg/kg bw per day and above and on day 14 at 160 mg/kg bw per day and above. On day 14, epididymal sperm count was decreased at 160 mg/kg bw per day and above, while a decrease in epididymal sperm weight was seen at 480 mg/kg bw per day and above. Distorted sperm heads and acrosomes were seen in step 15 spermatids after 14 days at 480 mg/kg bw per day and above. Testicular lesions developed with greater severity as the duration of the dosing and dose levels increased.
Long-Evans male rats (n = 8-19) were gavaged daily with DCA (sodium salt) at 0, 31.3, 62.5 or 125 mg/kg bw for 10 weeks (Toth et al., 1992). On day 70, each male was mated overnight with one untreated, pro-estrous female to assess fertility. Males were then sacrificed on day 75 and females on day 14 of gestation. Pregnancy and implantation rates were not significantly different from the female controls. However, in high-dose females, a reduction in the number of live implants was observed. In males, decreased weights were seen in the epididymis and preputial glands at 31.3 mg/kg bw per day and above and in the accessory organs (prostate and seminal vesicles) at the highest dose (125 mg/kg bw per day). At the two highest doses, sperm morphology was affected and epididymal sperm counts were decreased. Effects on the testis and spermiation (a reduction in late-step spermatid head counts) were seen only in the highest dose group. The authors set a LOAEL of 31.3 mg/kg bw per day for DCA (sodium salt) (equivalent to 26.7 mg DCA/kg bw per day), based on adverse male reproductive effects on the accessory organs and sperm (Toth et al., 1992).
In contrast, in a shorter 7-week subchronic drinking water study, Sprague-Dawley rats had normal testicular histopathology and sperm production when exposed to DCA (sodium salt) at 1100 mg/kg bw per day (Stacpoole et al., 1990). Normal histopathology and testis weights were also seen in male Sprague-Dawley rats (10 per dose) when treated with DCA (neutralized) in their drinking water at lower doses of 0, 50, 500 or 5000 mg/L (0, 4, 35 or 350 mg/kg bw per day) as part of a 90-day drinking water study (Mather et al., 1990).
In a 3-month gavage study (Katz et al., 1981), DCA (sodium salt) was administered to groups of male and female Sprague-Dawley rats (10 per sex per dose) at dose levels of 0, 125, 500 or 2000 mg/kg bw per day. An additional five rats per sex dosed with 0 or 2000 mg/kg bw per day were allowed a recovery period of 4 weeks. No adverse reproductive effects were observed in female rats, whereas adverse effects were observed in males only at the two highest doses. Testicular germinal epithelial degeneration was observed in 40% of mid-dose males and 100% of high-dose males. High-dose males showed aspermatogenesis and formation of syncytial giant cells in the germinal epithelium, whereas no spermatozoa were seen in the epididymis ducts. Syncytial giant cells were also seen in 20% of males from the mid-dose group. In the high-dose recovery group (n = 5), half of the males showed germinal epithelium regeneration, 75% were aspermatogenic and all showed loss of germinal epithelium.
As part of another 90-day study (Cicmanec et al., 1991), 4-month-old male and female beagle dogs (five per sex per dose) received DCA (neutralized) in gelatin capsules at 0, 12.5, 39.5 or 72 mg/kg bw daily. No significant weight changes were seen in the ovaries or the testes compared with controls. No effects of DCA on uterine histopathology in treated females were seen. Testicular changes (syncytial giant cell formation and degeneration of testicular germinal epithelium) were observed in all treated males, and prostatic glandular atrophy was observed in the mid- and high-dose males. A dose-related increase in severity of testicular lesions were seen at the mid- and high-dose groups.
In a 13-week subchronic study (Katz et al., 1981), similar testicular effects were observed in 10- to 12-month-old male beagle dogs (3-4 per dose) administered DCA (sodium salt) in capsules at doses of 0, 50, 75 or 100 mg/kg bw per day. Testicular changes in the germinal epithelium (degeneration of germinal epithelium, vacuolation of Leydig cells and formation of syncytial giant cells) as well as prostate gland atrophy were seen in all treated males. One male from the high-dose group was allowed to recover for 4 weeks post-treatment; the prostate appeared normal, and there was evidence of germinal epithelium regeneration with spermatogenesis.
DeAngelo et al. (1996) reported, as part of a modified carcinogenesis bioassay with male F344 rats, that testicular and relative testicular weights were slightly increased in the mid-dose group (500 mg/L), but absolute testes weights were decreased in the high-dose group (1600 mg/L).
10.5.3 Trichloroacetic acid
No reproductive studies were identified
In a developmental study, pregnant Long-Evans rats (n = 20-26) were treated with TCA (neutralized) at 0, 330, 800, 1200 or 1800 mg/kg bw per day (as sodium salt) by gavage on gestation days 6-15. No treatment-related maternal deaths were observed during the study. A dose-dependent increase in the frequency of resorptions per litter (34%, 62% and 90% of implants resorbed at dose levels of 800, 1200 and 1800 mg/kg bw per day, respectively) was observed at maternally toxic doses (decreased weight gain and dose-related increases in spleen and kidney weights). At all dose levels, a dose-related reduction in fetal weight and length as well as a dose-related increase in the frequency of soft tissue anomalies (from 9% at 330 mg/kg bw per day to 97% at 1800 mg/kg bw per day) were seen. Soft tissue anomalies were found mostly in the cardiovascular system and consisted of interventricular septal defects and laevocardia; however, the authors reported that this strain of rats is somewhat susceptible to laevocardia. At the two highest doses, an increase in skeletal malformations (principally in the orbit) was also observed. The maternal LOAEL is considered to be 330 mg/kg bw per day based on weight changes in the spleen and kidney. The developmental LOAEL is considered to be 330 mg/kg bw per day based on an increase in the frequency of soft tissue anomalies (Smith et al., 1989). No NOAEL was determined; however, a benchmark dose of 218 mg/kg bw per day was calculated for developmental toxicity (Health Canada, 2004b) for purposes of comparison with other end-points seen with TCA.
Previous developmental studies with TCE have shown an increase in congenital cardiac lesions in rats; however, it was suggested that metabolites of TCE were possibly responsible for these effects (Johnson et al., 1998). As a result, researchers conducted developmental studies with several metabolites of TCE, including TCA, to identify the responsible metabolites. The two studies that included TCA are described below but are inconclusive for the observed heart defects, since they used different methodologies and obtained conflicting results.
In a study by Johnson et al. (1998), pregnant Sprague-Dawley rats were given TCA (neutralized) in drinking water at concentrations of 0 mg/L (n = 55) or 2730 mg/L (n = 11) (0 or 290 mg/kg bw per day) on gestation days 1-22. A significant decrease in body weight gain was observed in treated dams, relative to controls. Developmental effects included a statistically significant increase in the number of resorptions, in the number of implantation sites and in cardiac soft tissue malformations (10.53% versus 2.15% for controls).
Fisher et al. (2001) gavaged a group of 19 pregnant female Sprague-Dawley rats with TCA (neutralized) at 300 mg/kg bw per day during gestation days 6-15. A control group (n = 19) was dosed with water. Mean maternal body weight gain was significantly less than controls on gestation days 7-15 and 18-21. A statistically significant reduction in fetal body weight (on a per fetus basis and on a per litter basis) was seen in the treated group. No malformations of the heart were seen at 300 mg/kg bw per day, contrary to those reported in the previous study by Smith et al. (1989).
10.5.4 Monobromoacetic acid
No well-conducted developmental studies were done with MBA. Randall et al. (1991) published an abstract on a study with MBA (acid) in Long-Evans rats suggesting possible soft tissue malformations in pups in the presence of maternal toxicity; however, the final study was never published.
No adverse reproductive effects were observed in male Sprague-Dawley rats when administered MBA (neutralized) at 0 or 100 mg/kg bw in a single dose or at 0 or 25 mg/kg bw per day for 14 days (Linder et al., 1994a).
10.5.5 Dibromoacetic acid
No effect on early pregnancy was seen in groups of mature female Holtzman rats (eight per dose per experiment) when gavaged with DBA (neutralized) at 0, 62.5, 125 or 250 mg/kg bw per day during gestation days 1-8. A group of dams was sacrificed on gestation day 9 or 20. The only effect, an increase in serum 17ß -estradiol at the highest dose, was seen in dams sacrificed on gestation day 9 (Cummings and Hedge, 1998).
No adequate developmental studies were conducted. However, two developmental screening assays (oral gavage with neutralized DBA) using CD-1 mice were published in abstracts by Narotsky et al. (1996, 1997). Although these studies suggested developmental effects in the presence/absence of maternal toxicity, no statistical data were provided in either abstract, and no final study was published to confirm these results.
Dose-related alterations of estrous cyclicity in female Sprague-Dawley rats were observed at doses of 90 and 270 mg/kg bw per day when rats were dosed for 14 consecutive days in drinking water with DBA (neutralized) (Balchak et al., 2000), but no alterations were seen at lower doses (10 or 30 mg/kg bw per day) in the same study or during a 20-week exposure to a dose of 5, 16 or 33 mg/kg bw per day (Murr and Goldman, 2005).
In a two-generation drinking water study, groups of Sprague-Dawley rats (30 per sex per dose) were treated with DBA (97% purity in deionized water) continuously via the drinking water at concentrations of 0, 50, 250 or 650 mg/L (equivalent to 0, 4.4-11.6, 22.4-55.6 and 52.4-132.0 mg/kg bw per day, respectively) (Christian et al., 2002). Doses were determined based on a range-finding reproductive/developmental study by Christian et al. (2001). Reduced water consumption was seen at all dose levels of the parental (P) and F1 generations. In the top dose group of the P and F1 generations, clinical signs associated with reduced water consumption, reduced body weights and weight gains and reduced food consumption were observed. Food consumption was also decreased in the F1 mid-dose group. Body weight decreases were also seen with all doses of the F1 generation during lactation; as a result, weaning was delayed until day 29. Small delays in sexual maturation (preputial separation, vaginal patency) at the top dose in F1 rats and a significant decrease in anogenital distance on lactation day 22 in F2 male pups at the middle and high doses were also attributed to a general retardation of growth associated with a significant reduction in body weight. Reproductive performance and development of female rats were unaffected at all doses in both P and F1 generations. In males, sperm motility, count and density and abnormal sperm were also unaffected by treatment. However, histopathology of the reproductive organs of P and F1 males in the mid- and high-dose groups revealed altered sperm production (a dose-related increase in retained step 19 spermatids in Stage IX and X tubules, and the presence of abnormal residual bodies in affected seminiferous tubules) and some epididymal tubule changes. Reproductive effects were seen in high-dose F1 males: a significant increase in the unilateral malformation of the reproductive tract (small or absent epididymides, and small testis) was seen. The authors identified a parental NOAEL for general toxicity of 50 mg/L (4.4-11.6 mg/kg bw per day) based on an increase in absolute and relative kidney and liver weights in the absence of histopathology. A U.S. EPA draft report (U.S. EPA, 2005a) has proposed a reproductive/developmental LOAEL of 250 mg/L (or 22.4-55.6 mg/kg bw per day) based on abnormal spermatogenesis in P and F1 males and a reproductive/developmental NOAEL at 50 mg/L (4.4-11.6 mg/kg bw per day).
Testicular effects were observed in a 13-week drinking water study (Melnick et al., 2007; NTP, 2007) in which male B6C3F1 mice and F344 rats were exposed to DBA (neutralized to pH 5) at doses of 0, 125, 250, 500, 1000 or 2000 mg/L (equivalent to 0, 16, 30, 56, 115 and 230 mg/kg bw per day for mice, and 0, 10, 20, 40, 90 and 166 mg/kg bw per day for rats). Testicular atrophy of the germinal epithelium as well as significant reductions in testicular weight, sperm motility and sperm concentrations were observed in high-dose male rats, along with a significant increase in hypospermia. Delayed spermiation with atypical residual bodies was observed at the two highest doses in male mice and only in the middle doses (500 and 1000 mg/L) in rats. The lowest dose showing testicular effects in rats is 500 mg/L. NTP, (2007) reported a NOEL for testicular lesions of 250 mg/L in rats. Similar effects (such as decreased testis weights, delayed spermiation or atypical residual bodies) were observed when rats (1000 mg/L and above) and mice (500 mg/L and above) were dosed for two weeks (Melnick et al., 2007; NTP, 2007).
Spermatotoxic/testicular effects consisting of testicular atrophy and sperm alteration (motility, morphology, count, fused or abnormal sperm, increased retention of step 19 spermatids, atypical residual bodies) were also observed in Sprague-Dawley rats given single or repeated oral doses of TCA (neutralized or acid) (Linder et al., 1994a, b, 1995; Vetter et al., 1998; Tsuchiya et al., 2000; Holmes et al., 2001).
Reproductive ability was compromised in male rats when gavaged with DBA (neutralized) (Linder et al., 1995). Abnormal sperm morphology, decreased sperm counts and motility and behaviour changes (during mating) in the males were noted.
Preliminary results of recently published abstracts (Klinefelter et al., 2000; Veeramachaneni et al., 2000; Bodensteiner et al., 2001; Veeramacheneni, 2002) suggest disruption in pubertal development, reproductive function, spermatogenesis and fertility in male rats and/or rabbits and a reduction of the population of primordial follicles in female rabbits when DBA (neutralized) is administered with the first exposure in utero from gestation day 15 throughout life. No final study was published to confirm these results.
11.0 Classification and assessment
11.1 Monochloroacetic acid
There was no evidence of carcinogenicity of MCA in mice or rats in a lifetime study (NTP, 1992). Tests for mutagenicity and genotoxicity of MCA were also largely negative. Based on the lack of evidence for carcinogenicity, MCA has therefore been classified in Group IV.D in this assessment, unlikely to be carcinogenic to humans (Health Canada, 1994).
MCA has not been evaluated by the International Agency for Research on Cancer (IARC). The U.S. EPA (2003b) reported that, under the 1999 Draft Revised Guidelines for Carcinogen Risk Assessment (U.S. EPA, 1999b), the data for MCA were considered "inadequate for an assessment of human carcinogenic potential."
Several long-term studies conducted with MCA failed to show carcinogenicity. The lowest NOAEL of all the studies was from the 104-week drinking water study with rats by DeAngelo et al. (1997), for which a NOAEL of 3.5 mg/kg bw per day was derived by Health Canada for treatment-related changes in body, liver, kidney and testes weights. This study was chosen for the risk assessment, based on the appropriateness of the vehicle used (drinking water), the absence of significant effects at the low dose, the length of the study as well as the form of MCA administered (i.e., neutralized solution).
The tolerable daily intake (TDI) for MCA is calculated as follows:
Figure 6: The equation used for calculating the tolerable daily intake for Monochloracetic acid (MCA)
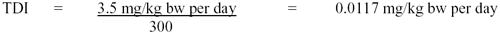
Figure 6: The equation used for calculating the tolerable daily intake for Monochloracetic acid (MCA) - Text Description
The tolerable daily intake (TDI) for Monochloracetic acid (MCA) is 0.0117 milligrams per kilogram body weight (kg bw) per day. This value is calculated by dividing 3.5 milligrams per kilogram body weight per day by 300.where:
- 3.5 mg/kg bw per day is the NOAEL in the DeAngelo et al. (1997) chronic rat study, as derived by Health Canada,
- 300 is the uncertainty factor (x10 for interspecies variation, x10 for intraspecies variation and x3 for database deficiencies, including lack of reproductive/developmental studies).
Using the TDI derived from the NOAEL, a health-based target can be calculated as follows:
Figure 7: The equation used for calculating a health-based value for Monochloracetic acid (MCA)
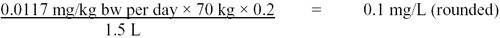
Figure 7: The equation used for calculating a health-based value for Monochloracetic acid (MCA) - Text Description
The health-based target value for Monochloracetic acid (MCA) is 0.1 milligrams per litre (rounded). This value is calculated by taking the tolerable daily intake (previously calculated) 0.0117 milligrams per kilogram body weight per day and multiplying it by 70 kilograms and then multiplying by 0.2. This value is then divided by 1.5 litres to achieve the health-based target value.where:
- 0.0117 mg/kg bw per day is the TDI, as derived above,
- 70 kg is the average body weight of an adult,
- 0.2 is the proportion of the daily intake allocated to drinking water; this is a default value, since there are insufficient data to calculate the actual value,
- 1.5 L is the average daily consumption of drinking water by an adult.
11.2 Dichloroacetic acid
Liver tumours in both mice and rats have been reported in several carcinogenicity bioassays (Herren-Freund et al., 1987; Bull et al., 1990; Daniel et al., 1992; Richmond et al., 1995; DeAngelo et al., 1996, 1999; Pereira and Phelps, 1996) and are considered sufficient evidence to classify DCA as an animal carcinogen. The exact mechanism for tumorigenicity has not been identified, and tests for mutagenicity and genotoxicity have been mostly negative or equivocal in bacterial and mammalian test systems. It is unknown at this time if the carcinogenicity of DCA is mediated by a non-genotoxic mechanism. DCA did not induce peroxisome proliferation (Tong et al., 1998b).
DCA has been classified in Group II, probably carcinogenic to humans, with sufficient evidence in animals and inadequate evidence in humans (Health Canada, 1994). IARC (2004) recently classified DCA as Group 2B, possibly carcinogenic to humans on the basis of sufficient evidence of its carcinogenicity in experimental animals and inadequate evidence of its carcinogenicity in humans. In the 2006 edition of the U.S. drinking water standards and health advisories, U.S. EPA (2006) considers DCA as "likely to be carcinogenic to humans"
based on the 2005 U.S. EPA Guidelines for Carcinogen Risk Assessment. This is "based on current data and the lack of conclusive data regarding the mode of action of DCA at environmentally relevant doses"
(U.S. EPA, 2003c). There is no information available on the mutagenic effects of DCA in humans (CHEMINFO, 2003c).
Liver tumours in both mice and rats have been reported in several carcinogenicity bioassays with DCA. Liver tumours (hepatocellular carcinomas) in mice were chosen for the cancer risk assessment, as the only rat study available used a high dose that was not well tolerated by the rats and had to be decreased several times. Therefore, cancer risks have been estimated on the basis of results of an adequate long-term drinking water study (90-100 weeks) in male B6C3F1 mice, which was conducted by DeAngelo et al. (1999). Liver tumours (hepatocellular carcinomas) seen in male mice showed an appropriate dose-response relationship. Other considerations for choosing the study for the risk assessment were the appropriateness of the vehicle used (drinking water), the form of DCA administered (i.e., neutralized solution), the length of the study as well as the use of numerous dose groups (five doses and a control group). A NOEL could not be determined for hepatocarcinogenicity because of the significant increase in hepatocellular carcinoma multiplicity observed at the lowest dose, 8 mg/kg bw per day (0.58 compared with 0.28 in the control) (DeAngelo et al., 1999).
Based on the classification of probable carcinogen and the uncertainty surrounding whether or not the carcinogenicity of DCA is mediated by a non-genotoxic mechanism, Health Canada has chosen to use a low-dose linear risk extrapolation for calculating the cancer risk. This is consistent with the most recent (2005) U.S. EPA Guidelines for Carcinogen Risk Assessment, which state that "when the weight of evidence evaluation of all available data are insufficient to establish the mode of action for a tumour site and when scientifically plausible based on the available data, linear extrapolation is used as a default approach, because linear extrapolation generally is considered to be a health-protective approach. Nonlinear approaches generally should not be used in cases where the mode of action has not been ascertained."
Therefore the unit risk can be assessed by the linearized multistage (LMS) method (Health Canada, 2004a), using the number of male B6C3F1 mice in each dose group having hepatocellular carcinomas following exposure to DCA in drinking water for 90-100 weeks (DeAngelo et al., 1999).
In this method, the multistage model is fit to the dose-response data, and the upper 95% confidence limit on the linear term is taken to be the unit risk. The multistage model is given by:
Figure 1: The equation which provides the multistage model
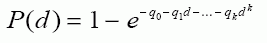
Figure 1: The equation which provides the multistage model - Text Description
The probability of an animal developing a tumour at a certain dose P(d) is found by calculating 1 minus the constant e (2.718) to a range of exponents determined by the known dose, the appropriate dose response curve, and the number of dose groups in the study data.where d is dose, k is the number of dose groups in the study (excluding control), P(d) is the probability of the animal developing a tumour at dose d and qi > 0, i = 0, ..., k are parameters to be estimated. The unit risk is defined as the increase in excess risk per unit dose, where excess risk is given by
Figure 2: The equation used for calculating the amount of excess risk
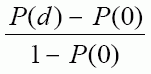
Figure 2: The equation used for calculating the amount of excess risk - Text Description
The excess risk is calculated by taking the probablilty of an animal developing a tumor at a certain dose P(d) minus the probabilty of an animal developing a tumor at background exposure only P(0) divided by 1 minus P(0).The unit risk is applicable at very low doses, presumably in the range where humans would be exposed. For a small dose, d, the excess risk can be shown to be approximately equal to q1d.
Thus, when the background P(0) is small, q1 represents the slope (i.e., change in risk per increase of unit dose) of the dose-response curve in the low-dose region. In practice, the upper 95% confidence limit on q1 is used and is denoted by q1*. This is the unit risk for the LMS method.
The multistage model was fit using THRESH (Howe, 1995), and the unit risk was calculated by the LMS method without prior application of the kinetic adjustment factor. The P-value for the chi-square lack of fit test was 0.87, indicating that the model fit the data adequately. The units of the unit risks were converted to concentrations in drinking water by multiplying by the drinking rate of a human (1.5 L/day) and dividing by the standard body weight of a human (70 kg).
An animal-to-human kinetic adjustment factor (KA) (also known as allometric scaling factor, which corrrects for body weight differences between animals and humans) can also be applied. The animal-to-human kinetic adjustment factor (KA) is given by:
Figure 3. The equation used to adjust for body weight differences between animals and humans

Figure 3. The equation used to adjust for body weight differences between animals and humans - Text Description
The kinetic adjustment factor is calculated by taking the animal weight in kilograms and dividing it by the standard body weight of a human (70 kg), and the resulting value is placed to the exponent of 1/4.where 70 kg is the standard body weight of a human. Since the allometric scaling factor is applied to the unit risk after modelling, a mouse body weight of 43.9 g was used in the above formula. This was the average body weight in the control group.
The estimated calculated unit lifetime human cancer risk associated with the ingestion of DCA at 1 µg/L in drinking water is 1.02 × 10-6 (based on liver tumours, hepatocellular carcinomas).
In the context of drinking water guidelines, Health Canada has defined the term "essentially negligible"
as a range from one new cancer above background per 100 000 people to one new cancer above background per 1 million people (i.e., 10-5 to 10-6) over a lifetime.
| Lifetime risk | Concentration in drinking water (µg/L) |
|---|---|
| 10-4 | 98.1 (rounded to 100) |
| 10-5 | 9.81 (rounded to 10) |
| 10-6 | 0.98 (rounded to 1) |
Using the most conservative concentration in drinking water estimated for a 10-5 lifetime human cancer risk, a health-based target of 0.01 mg/L (10 µg/L) is derived for DCA in drinking water.
An additional analysis was undertaken in which the kinetic adjustment factor was applied to the experimental doses before modelling the data. This was done in order to facilitate comparisons with the U.S. EPA's (2003c) methodology. The multistage model was fit using THRESH (Howe, 1995), and unit risks were calculated by LMS, with prior application of the kinetic adjustment factor. The results demonstrated that applying the animal-to-human kinetic adjustment factor before or after fitting the model had no impact on the resulting unit risks or concentrations.
11.3 Trichloroacetic acid
TCA administered in drinking water has consistently been shown to produce liver tumours in mice but not in rats. The mechanism underlying the mouse liver tumours was determined to be based on peroxisome proliferation, which may or may not be relevant to humans (Cattley et al., 1998). TCA was seen as weakly genotoxic.
Based on evidence of carcinogenicity being limited to one species of rodents, the mouse, and inadequate evidence in humans, TCA has been classified in Group III in this assessment, possibly carcinogenic to humans (Health Canada, 1994). According to IARC (1995), there is inadequate evidence for its carcinogenicity in humans and limited evidence for its carcinogenicity in experimental animals. U.S. EPA (1986) classified TCA as a possible human carcinogen, whereas U.S. EPA (2003b), under the 1999 Draft Revised Guidelines for Carcinogen Risk Assessment (U.S. EPA, 1999b), stated that there is suggestive evidence for TCA carcinogenicity, but the data are not sufficient to assess human carcinogenicity.
Long-term drinking water studies produced liver tumours in mice, but not in rats. Since it is not known whether the underlying mechanism is relevant to humans, a long-term rat study with a non-cancer end-point was chosen for the risk assessment. The 104-week drinking water study by DeAngelo et al. (1997) also showed the lowest NOAEL of 32.5 mg/kg bw per day based on treatment-related changes (decreased body weight, increased liver serum enzyme activity and liver histopathology compared with control animals). Other considerations for choosing the study included the appropriateness of the vehicle used (drinking water), the absence of significant effects at the low dose, the length of the study and the form of TCA administered (i.e., neutralized solution).
The TDI for TCA can be calculated as follows:
Figure 4: The equation used to calculate the tolerable daily intake (TDI) for Trichloroacetic acid (TCA)
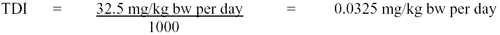
Figure 4: The equation used to calculate the tolerable daily intake (TDI) for Trichloroacetic acid (TCA) - Text Description
The tolerable daily intake (TDI) for Trichloroacetic acid (TCA) is 0.0325 milligrams (mg) per kilogram of body weight (kg bw) per day. This value is calculated by dividing 32.5 milligrams per kilogram of body weight per day by 1000.where:
- 32.5 mg/kg bw per day is the NOAEL in the DeAngelo et al. (1997) chronic rat study,
- 1000 is the uncertainty factor (x10 for interspecies variation, x10 for intraspecies variation and x10 for database deficiencies, including lack of a multigenerational reproductive study, and possible carcinogenicity).
Using the TDI derived from the NOAEL, a health-based target can be calculated as follows:
Figure 5: The equation used for calculating the health-based value for Trichloroacetic acid (TCA)
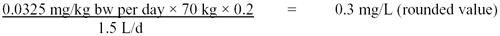
Figure 5: The equation used for calculating the health-based value for Trichloroacetic acid (TCA) - Text Description
The health based value for Trichloroacetic acid (TCA) is 0.3 milligrams per litre (rounded). This value is calculated by taking the tolerable daily intake (previously calculated) 0.0325 milligrams per kilogram of body weight and multiplying it by 70 kilograms and then multiplying by 0 .2. This value is then divided by 1.5 litres per day to achieve the health based value.where:
- 0.0325 mg/kg bw per day is the TDI, as derived above,
- 70 kg is the average body weight of an adult,
- 0.2 is the proportion of the daily intake allocated to drinking water; this is a default value, since there are insufficient data to calculate the actual value,
- 1.5 L/d is the average daily consumption of drinking water by an adult.
11.4 Monobromoacetic acid
There are insufficient data on the toxicity of MBA identified in this document to establish a health-based target. Acute oral studies have shown MBA to be acutely toxic. However, no subchronic, chronic or carcinogenic studies were conducted or published with MBA, nor was a standard developmental or multigeneration study conducted. Mixed results were seen with regards to mutagenicity/genotoxicity studies.
MBA has been classified in Group VI in this assessment, unclassifiable with respect to carcinogenicity in humans, based on inadequate data from animal studies (Health Canada, 1994).
U.S. EPA (2005a) reported that, under the 1999 Draft Revised Guidelines for Carcinogen Risk Assessment, the data for MBA were considered "inadequate for an assessment of human carcinogenic potential."
MBA has not been evaluated or classified by IARC.
11.5 Dibromoacetic acid
Based on the only oral acute toxicity study, DBA has been shown to be moderately toxic. Available subchronic and chronic studies suggest that the liver is the target organ. Reproductive studies suggest male-mediated effects, whereas the reporting of developmental studies is limited to abstracts (limited data) but may suggest fetotoxicity. Additional studies need to be conducted in order to adequately assess the developmental toxicity of DBA. Available mutagenicity studies are limited and show mixed results.
In a recent 2-year drinking water study in mice and rats conducted by NTP (2007) (also published as a summary by Melnick et al., 2007), DBA was shown to be a multiple-organ carcinogen in laboratory animals, with tumour induction seen in the liver and lung of mice, in the abdominal cavity (mesotheliomas) of male rats and in the haematopoietic system (monocellular cell leukaemia) in female rats. As a result, Health Canada has classified DBA as Group II, probably carcinogenic to humans, based on sufficient evidence in animals and inadequate evidence in humans (Health Canada, 1994). DBA has not been evaluated or classified by IARC. In 2005, before the publication of these new studies, the U.S. EPA (2005a) had reported that, based on the 1999 Draft Revised Guidelines for Carcinogen Risk Assessment, the data for DBA were considered "inadequate for an assessment of human carcinogenic potential."
Based on DBA's new classification as a probable carcinogen, the linearized multistage method was chosen to calculate unit risks. The application of the multistage model is described in Section 11.2.
The multistage models were fit using THRESH (Howe, 1995) and the unit risks were calculated (Health Canada, 2007a). An animal-to-human kinetic adjustment factor (KA) was applied to the final unit risks assuming a rat weighs 0.35 kg, a mouse weighs 0.03 kg and a human weighs 70 kg. The KA formula is also described in Section 11.2. A chi-square lack of fit test was performed for the model fits. The degrees of freedom for this test are equal to k minus the number of qi's whose estimates are non-zero. A p-value less than 0.05 indicates a significant lack of fit. Based on this criterion, a few models exhibited a significant lack of fit due to an uneven dose-response. Although no simple model will adequately describe these data, these models do provide a reasonable visual fit. This is in contrast to DCA, where the model fit the data adequately. The calculations for the unit risks (raw and converted using the allometic scaling factor) for DBA and lack of fit p-values are displayed in Health Canada (2007b).
The estimated unit lifetime risks associated with ingestion of 1 µg/L of DBA in drinking water were estimated to range from 0.14 × 10-6 to 4.26 × 10-6. The unit risk range was derived from mesotheliomas observed in male rats (0.14 × 10-6) as its lower bound (least sensitive) and hepatocellular adenoma/carcinoma in male mice (4.26 × 10-6) as its upper bound (most sensitive).
| Lifetime risk | Concentration in drinking water (µg/L) |
|---|---|
| 10-4 | 23.5 - 701.6 |
| 10-5 | 2.3 - 70.2 |
| 10-6 | 0.23 - 7.0 |
Using the most conservative concentration in drinking water estimated for a 10-5 lifetime human cancer risk, a health-based target of 0.002 mg/L (2 µg/L) (rounded) is derived for DBA in drinking water.
11.6 International considerations
Several agencies have reviewed HAAs and have established guidelines or standards as a function of various factors, such as best available technology and/or health effects.
WHO (2004a, b, 2005) has established separate guideline values for three of the HAAs: MCA (20 µg/L), DCA (provisional guideline value of 50 µg/L) and TCA (200 µg/L). Although the World Health Organization (WHO) calculated a health-based guideline value of 40 µg/L for DCA based on a 10-5 upper-bound excess lifetime cancer risk, the guideline is provisional because "the data on treatment are insufficient to ensure that the 40 µg/litre value is technically achievable under a wide range of circumstances"
(WHO, 2005). In regards to MBA and DBA, WHO (2004c) considered the databases inadequate for the derivation of guideline values.
The U.S. EPA (2006) has used a different approach and established a single maximum contaminant level (MCL) for all five HAAs (HAA5) of 0.06 mg/L based on best available technology, as well as individual non-enforceable maximum contaminant level goals of 0.03 mg/L for MCA, 0 for DCA (based on its carcinogenicity) and 0.02 mg/L for TCA.
12.0 Rationale / Errata*
Because haloacetic acids are formed in drinking water primarily as a result of chlorination of organic matter present in raw water supplies, it is important to recognize the substantial benefits to health associated with disinfection by chlorination. The use of chlorine has virtually eliminated waterborne microbial diseases, because of its ability to kill or inactivate essentially all enteric pathogenic microorganisms, including viruses and bacteria from the human intestinal tract. Chlorine is the most convenient and easily controlled disinfectant; it is a strong oxidant for which a residual can be maintained in the distribution system to prevent bacterial regrowth. Although the use of chlorine can lead to the formation of DBPs such as HAAs, efforts to manage HAA levels in drinking water must not compromise the effectiveness of disinfection.
HAAs and THMs are the two major groups of DBPs found in drinking water and generally at the highest levels. The concentrations of these contaminants can be used as indicators of the total loading of all DBPs that may be found in drinking water supplies. In the absence of information on other DBPs, control and management of HAAs and THMs should reduce exposure to and risk from other by-products. When appropriate drinking water treatment strategies are implemented to reduce HAAs and THMs, the levels of other halogenated DBPs may also be reduced in the process.
There are sufficient scientific data available to derive health-based targets for four HAAs: MCA, DCA, TCA and DBA. MCA is classified as Group IV (unlikely to be carcinogenic to humans). A health-based target concentration of 0.1 mg/L can be calculated for MCA in drinking water, based on changes in body, liver, kidney and testes weights observed in rats. DCA is classified in Group II (probably carcinogenic to humans), based on sufficient evidence in animals and inadequate evidence in humans. A health-based target concentration of 0.01 mg/L can be calculated for DCA in drinking water, based on liver tumours observed in both mice and rats. TCA is classified in Group III (possibly carcinogenic to humans), based on limited evidence of carcinogenicity in experimental animals and inadequate evidence in humans. A health-based target concentration of 0.3 mg/L can be calculated for TCA in drinking water. Although animal studies have shown a link between exposure to TCA and liver tumours in mice only, it is still uncertain whether the mechanism causing these tumours is relevant to humans. MBA is classified in Group VI (unclassifiable with respect to carcinogenicity in humans), based on inadequate data from animal studies. No health-based target concentration can be established for MBA at this time. DBA is classified in Group II (probably carcinogenic to humans), based on sufficient evidence in animals and inadequate evidence in humans. A health-based target concentration of 0.002 mg/L can be calculated for DBA in drinking water, based on tumours in several organs observed in both mice and rats.
Recent Canadian exposure data for surface water sources show that the HAAs consistently found at the highest concentrations in Canadian distribution systems were DCA and TCA.
Although the proportion of each HAA will vary according to conditions, DCA can commonly represent 40-60% of the total HAA concentration. DBA makes up only a small fraction (generally less than 6%)Footnote 3 of the total HAA concentration.
The removal of HAAs after their formation in drinking water supplies is not considered to be the best approach to reduce exposure to HAAs. The most efficient and practical way to reduce HAA concentrations in finished waters is to prevent their formation, primarily through the removal of organic precursors. Although pH adjustments may help reduce HAA formation, they may cause a corresponding increase in the formation of other DBPs, including THMs.
Although health-based targets can be established for four of the five HAAs, and considering the technological limitations associated with reducing individual HAA levels in drinking water while maintaining effective disinfection, the Federal-Provincial-Territorial Committee on Drinking Water is establishing a MAC of 0.08 mg/L (80 µg/L) for Total HAA5 in drinking water based on a running annual average rather than individual guidelines. This is consistent with the approach taken by the U.S. EPA, which established a maximum contaminant level based on best available technology for these same HAAs.
Errata
Section 12, page 61
Although health-based targets can be established for four of the five HAAs, and considering the technological limitations associated with reducing individual HAA levels in drinking water while maintaining effective disinfection, the Federal-Provincial-Territorial Committee on Drinking Water is establishing a MAC of 0.08 mg/L (80 µg/L) for Total HAA5 in drinking water based on a running annual average rather than individual guidelines. This is consistent with the approach taken by the U.S. EPA, which established a maximum contaminant level based on best available technology for these same HAAs.
The MAC of 0.08 mg/L for total HAAs in drinking water is established, using a risk management approach based on the following considerations:
- 1a. Although the health-based target for DBA is the lowest value calculated, it is also found as a very small proportion of the HAA mixture and is not considered to be the appropriate driver for a health-based guideline. Mean levels of DBA observed are well below the health-based target of 0.002 mg/L for DBA.
- 1b. The health-based target for DCA is the lowest value that should be considered. The concentration of DCA in drinking water representing an
"essentially negligible"
risk is 0.01 mg/L, but this level cannot be achieved in distribution systems without compromising the effectiveness of disinfection. - 2. The PQLs, based on the capability of laboratories to routinely measure HAAs within reasonable limits of precision and accuracy, vary according to the individual HAAs and the method used. However, all the PQL values are well below the MAC.
- 3. The MAC must be achievable at reasonable cost. Based on the limited available Canadian data, it is estimated that 88% of existing treatment plants serving populations over 5000 and 56% of existing treatment plants serving populations less than 5000 can be expected to achieve HAA concentrations of 0.08 mg/L. By optimization of treatment processes and the use of advance treatment technologies to remove organic compounds prior to disinfection, it is possible to reduce HAA5 below 0.08 mg/L.
The estimated lifetime cancer risk associated with the ingestion of drinking water containing HAAs at 0.08 mg/L is greater than the range that is considered generally to be "essentially negligible"
(i.e., between 10-5 and 10-6). Based on the incidence of liver cancer in animal studies for DCA, the estimated lifetime risk associated with ingestion of water containing HAAs at 0.08 mg/L is 3.2 x 10-5 to 4.8 x 10-5 for DCA proportions of 40% to 60%, respectively, of total HAAs. Although exposure to HAAs at the guideline level may carry a lifetime risk from DCA that is higher than would normally be considered negligible, this risk is calculated using a very conservative approach that typically overestimates potential risk.
It is recommended that utilities strive to maintain HAA levels as low as reasonably achievable without compromising the effectiveness of disinfection. As part of its ongoing guideline review process, Health Canada will continue to monitor new research in this area and recommend any change(s) to the guideline that it deems necessary.
13.0 References
ACGIH (American Conference of Governmental Industrial Hygienists). 2001. Trichloroethylene BEI. CAS # 79-01-6. In: Documentation of the threshold limit values and biological exposure indices. 7th edition. ACGIH, Cincinnati, OH.
Allen, B.C. and Fisher, J.W. 1993. Pharmacokinetic modeling of trichloroethylene and trichloroacetic acid in humans. Risk Anal., 13(1): 71-86.
Amacher, D.E. and Turner, G.N. 1982. Mutagenic evaluation of carcinogens and non-carcinogens in the L5178Y/TK assay utilizing postmitochondrial fractions (S9) from normal rat liver. Mutat. Res., 97: 49-65.
Anderson, W.B., Board, P.G., Gargano, B. and Anders, M.W. 1999. Inactivation of glutathione transferase zeta by dichloroacetic acid and other fluorine-lacking alpha-haloalkanoic acids. Chem. Res. Toxicol., 12(12): 1144-1149.
APHA (American Public Health Association), AWWA (American Water Works Association) and WEF (Water Environment Federation). 2005. Standard methods for the examination of water and wastewater. 21th edition. APHA, Washington, DC.
Aranda-Rodriguez, R., Benoit, F.M., Lebel, G.L. and Jay, B. 2002. Chlorinated disinfection by-products (CDBPs) in Canadian drinking water - a survey of 27 small systems. In: CWWA Tenth National Conference on Drinking Water, Halifax, Nova Scotia. Canadian Water and Wastewater Association, Ottawa, Ontario. pp. 140-150.
Ashford, R.D. 1994. Ashford's dictionary of industrial chemicals. Wavelength Publications Ltd., London, UK.
Bader, E.L., Hrudey, S.E. and Froese, K.L. 2005. Urinary excretion half life of trichloroacetic acid as a biomarker of exposure to chlorinated drinking water disinfection by-products. Occup. Environ. Med., 61: 715-716.
Bakeas, E.B., Economou, A.G., Siskos, P.A. and Frank, H. 2003. Determination of chloroacetates in atmospheric particulate matter. Environ. Sci. Technol., 37(11): 2336-2339.
Balchak, S.K., Hedge, J.M., Murr, A.S., Mole, M.L. and Goldman, J.M. 2000. Influence of the drinking water disinfection by-product dibromoacetic acid on rat estrous cyclicity and ovarian follicular steroid release in vitro. Reprod. Toxicol., 14(6): 533-539.
Berardi, M.R., Snyder, R., Waritz, R.S. and Cooper, K.R. 1987. Monochloroacetic acid toxicity in the mouse associated with blood-brain barrier damage. Fundam. Appl. Toxicol., 9(3): 469-479.
BG Chemie. 1993. Monochloroacetic acid. In: Toxicological evaluations. Vol. 6. Potential health hazards of existing chemicals. Springer-Verlag, Berlin. pp. 1-32.
Bhat, H.K. and Ansari, G.A.S. 1989. Covalent interactions of chloroacetic and acetic acids with cholesterol. J. Biochem. Toxicol., 4(3): 189-193.
Bhat, H.K., Ahmed, A.E. and Ansari, G.A.S. 1990. Toxicokinetics of monochloroacetic acid: A whole-body autoradiography study. Toxicology, 63: 35-43.
Bhunya, S.P. and Behera, B.C. 1987. Relative genotoxicity of trichloroacetic acid (TCA) as revealed by different cytogenetic assays: bone marrow chromosome aberration, micronucleus and sperm-head abnormality in the mouse. Mutat. Res., 188: 215-221.
Bhunya, S.P. and Das, P. 1987. Bone marrow chromosome aberration and sperm-head abnormality in mice in vivo induced by monochloroacetic acid (MCA). Chromosome Inf. Serv., 42: 28-30.
Bodensteiner, K.J., Veeramachaneni, R., Klinefelter, G.R., Kane, C.M., Higuchi, T.T., Moeller, C.L. and Sawyer, H.R. 2001. Chronic exposure to dibromoacetic acid (DBA), a commonly occurring disinfection by-product in drinking water, diminishes primordial follicle populations in the rabbit. Biol. Reprod., 64: 122 (abstract).
Boethling, R.S. and Alexander, M. 1979. Effect of concentration of organic chemicals on their biodegradation by natural microbial communities. Appl. Environ. Microbiol., 37: 1211-1216.
Bove, F.J., Fulcomer, M.C., Klot, J.B., Esmart, J., Dufficy, E.M. and Savrin, J.E. 1995. Public drinking water contamination and birth outcomes. Am. J. Epidemiol., 141: 850-862.
Bryant, B.J., Jokinen, M.P., Eustis, S.L., Thompson, M.B. and Abdo, K.M. 1992. Toxicity of monochloroacetic acid administered by gavage to F344 rats and B6C3F1 mice for up to 13 weeks. Toxicology, 72(1): 77-87.
Budavari, S., O'Neil, M.J. and Smith, A. (eds.). 1996. The Merck index: an encyclopedia of chemicals, drugs and biologicals. Merck and Co., Inc., Whitehorse, NJ. pp. 2158, 3095, 9757-9758.
Bull, R.J. 1995. Carcinogenic properties of brominated haloacetates. Disinfection by-products in drinking water; critical issues in health effects research. ILSI Workshop Report. Health and Environmental Science Institute, International Life Sciences Institute, Washington, DC. p. 29.
Bull, R.J. 2000. Mode of action of liver tumor induction by trichloroethylene and its metabolites, trichloroacetate and dichloroacetate. Environ. Health Perspect., 108(Suppl. 2): 241-259.
Bull, R.J., Sanchez, I.M., Nelson, M.A., Larson, J.L. and Lansing, A.J. 1990. Liver tumour induction in B6C3F1 mice by dichloroacetate and trichloroacetate. Toxicology, 63: 341-359.
Bull, R.J., Sasser, L.B. and Lei, X.C. 2004. Interaction in the tumor-promoting activity of carbon tetrachloride, trichloroacetate, and dichloroacetate in the liver of male B6C3F1 mice. Toxicology, 199: 169-183.
Carter, J.H., Carter, H.W. and DeAngelo, A.B. 1995. Biochemical, pathologic and morphometric alterations induced in male B6C3F1 mouse liver by short-term exposure to dichloroacetic acid. Toxicol. Lett., 81: 55-71.
Carter, J.H., Carter, H.W., Deddens, J.A., Hurst, B.M., George, M.H. and DeAngelo, A.B. 2003. A 2-year dose- response study of lesion sequences during hepatocellular carcinogenesis in the male B6C3F1 mouse given the drinking water chemical dichloroacetic acid. Environ. Health Perspect., 111(1): 53-64.
Cattley, R.C., Deluca, J., Elcombe, C., Fenner-Crisp, P., Lake, B.G., Marsman, D.S., Pastoor, T.A., Popp, J.A., Robinson, D.E., Schwetz, B., Tugwood, J. and Wahli, W. 1998. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regul. Toxicol. Pharmacol., 27: 47-60.
CDBP (Chlorinated Disinfection By-Products) Task Group. 2000. Chlorinated disinfection by-products. CDBP Task Group, Health Canada, March.
Chang, L.W., Daniel, F.B. and DeAngelo, A.B. 1992. Analysis of DNA strand breaks induced in rodent liver in vivo, hepatocytes in primary culture, and a human cell line by chlorinated acetic acids and chlorinated acetaldehydes. Environ. Mol. Mutagen., 20: 277-288.
Chemada Fine Chemicals. 2002. Bromoacetic acid material safety data sheet.
CHEMINFO. 2003a. Monochloroacetic acid solutions. CAS # 79-11-8. Record No. 516. Chemical profiles created by Canadian Centre for Occupational Health and Safety, Hamilton, Ontario.
CHEMINFO. 2003b. Monochloroacetic acid. CAS # 79-11-8. Record No. 767. Chemical profiles created by Canadian Centre for Occupational Health and Safety, Hamilton, Ontario.
CHEMINFO. 2003c. Dichloroacetic acid. CAS # 79-43-6. Record No. 540. Chemical profiles created by Canadian Centre for Occupational Health and Safety, Hamilton, Ontario.
CHEMINFO. 2003d. Trichloroacetic acid solutions. CAS # 76-03-9. Record No. 766. Chemical profiles created by Canadian Centre for Occupational Health and Safety, Hamilton, Ontario.
CHEMINFO. 2003e. Trichloroacetic acid solid. CAS # 76-03-9. Record No. 539. Chemical profiles created by Canadian Centre for Occupational Health and Safety, Hamilton, Ontario.
Christian, M.S., York, R.G., Hoberman, A.M., Diener, R.M., Fisher, L.C. and Gates, G.A. 2001. Biodisposition of dibromoacetic acid (DBA) and bromodichloromethane (BDCM) administered to rats and rabbits in drinking water during range-finding reproduction and developmental toxicity studies. Int. J. Toxicol., 20(4): 239-253.
Christian, M.S., York, R.G., Hoberman, A.M., Frazee, J., Fisher, L.C., Brown, W.R. and Creasy, D.M. 2002. Oral (drinking water) two-generation reproductive toxicity study of dibromoacetic acid (DBA) in rats. Int. J. Toxicol., 21(4): 237-276.
Cicmanec, J.L., Condie, L.W., Olson, G.R. and Wang, S.-R. 1991. 90-day toxicity study of dichloroacetate in dogs. Fundam. Appl. Toxicol., 17: 376-389.
Cornett, R., James, M., Henderson, G., Cheung, J., Shroads, A. and Stacpoole, P. 1999. Inhibition of glutathione S-transferase and tyrosine metabolism by dichloroacetate: A potential unifying mechanism for its altered biotransformation and toxicity. Biochem. Biophys. Res. Commun., 262: 752-756.
Cummings, A.M. and Hedge, J.M. 1998. Dibromoacetic acid does not adversely affect early pregnancy in rats. Reprod. Toxicol., 12(4): 445-448.
Curry, S.H., Chu, P.I., Baumgartner, T.G. and Stacpoole, P.W. (1985) Plasma concentrations and metabolic effects of intravenous sodium dichloroacetate. Clin. Pharmacol. Ther., 37: 89-93.
Daniel, F.B., DeAngelo, A.B., Stober, J.A., Olson, G.R. and Page, N.P. 1992. Hepatocarcinogenicity of chloral hydrate, 2-chloroacetaldehyde and dichloroacetic acid in B6C3F1 mouse. Fundam. Appl. Toxicol., 19: 159-168.
Daubert, T.E. and Danner, R.P. 1989. Physical and thermodynamic properties of pure chemical data compilation. Taylor and Francis, Washington, DC.
DeAngelo, A.B. and Daniel, F.B. 1990. The comparative carcinogenicity of dichloroacetic (DCA) and trichloroacetic acid (TCA) in the male B6C3F1 mouse. Toxicologist, 10: 148 (abstract).
DeAngelo, A.B. and Daniel, F.B. 1992. An evaluation of the carcinogenicity of the chloroacetic acids in the male F344 rat. Toxicologist, 12: 206 (abstract).
DeAngelo, A.B., Daniel, F.B., McMillan, L., Wernsing, P. and Savage, R.E., Jr. 1989. Species and strain sensitivity to the induction of peroxisome proliferation by chloroacetic acids. Toxicol. Appl. Pharmacol., 101: 285-298.
DeAngelo, A.B., Daniel, F.B., Stober, J.A. and Olson, G.R. 1991. The carcinogenicity of dichloroacetic acid in the male B6C3F1 mouse. Fundam. Appl. Toxicol., 16: 337-347.
DeAngelo, A.B., Daniel, F.B., Most, B.M. and Olson, G. 1996. The carcinogenicity of dichloroacetic acid in the male Fischer 344 rat. Toxicology, 114: 207-221.
DeAngelo, A.B., Daniel, F.B., Most, B.M. and Olson, G.R. 1997. Failure of monochloroacetic acid and trichloroacetic acid administered in the drinking water to produce liver cancer in male F344/N rats. J. Toxicol. Environ. Health, 52: 425-445.
DeAngelo, A.B., George, M.H. and House, D.E. 1999. Hepatocarcinogenicity in the male B6C3F1 mouse following a lifetime exposure to dichloroacetic acid in the drinking water: dose response determination and modes of action. J. Toxicol. Environ. Health, 58: 485-507.
DeMarini, D.M., Perry, E. and Sheldon, M.L. 1994. Dichloroacetic acid and related compounds: induction of prophage in E. coli and mutagenicity and mutation spectra in Salmonella TA100. Mutagenesis, 9(5): 429-437.
Demint, R.J., Pringle, J.C., Hattrup, A., Bruns, V.F. and Frank, P.A. 1975. Residues in crops irrigated with water containing trichloroacetic acid. J. Agric. Food Chem., 23(1): 81-84.
EC (European Commission). 2003. Monochloroacetic acid (MCAA) environmental part. Scientific Committee on Toxicity, Ecotoxicity and the Environment, Health & Consumer Protection Directorate-General, EC, Brussels.
ECETOC (European Centre for Ecotoxicology and Toxicology of Chemicals). 1999. Monochloroacetic acid (CAS No. 79-11-8) and its sodium salt (CAS No. 3926-62-3). Joint Assessment of Commodity Chemicals No. 38. ECETOC, Brussels, June.
ECETOC (European Centre for Ecotoxicology and Toxicology of Chemicals). 2001. Monochloroacetic acid (CAS No. 79-11-8). Human acute intoxication from monochloroacetic acid: proposals for therapy. Technical Report No. 81. ECETOC, Brussels, November.
Edwards, M. and Dudi, A. (2004) Role of chlorine and chloramine in corrosion of lead-bearing plumbing materials. J. Am. Water Works Assoc., 96(10): 69-81.
Epstein, D.L., Nolen, G.A., Randall, J.L., Christ, S.A., Read, E.J., Stober, J.A. and Smith, M.K 1992. Cardiopathic effects of dichloroacetate in the fetal Long-Evans rat. Teratology, 46(3): 225-235.
Evans, O.B. 1982. Dichloroacetate tissue concentrations and its relationship to hypolactatemia and pyruvate dehydrogenase activation. Biochem. Pharmacol., 31(19): 3124-3126.
Feldhaus, K., Hudson, D., Rogers, D., Horowitz, R.S., Brent, J., Dart, R.C. and Gomez, H. 1993. Pediatric fatality associated with accidental oral administration of monochloroacetic acid (MCA). Vet. Hum. Toxicol., 35: 344 (abstract).
Fisher, J.W., Channel, S.R., Eggers, J.S., Johnson, P.D., MacMahon, K.L., Goodyear, C.D., Sudberry, G.L., Warren, D.A., Latendresse, J.R. and Graeter, L.J. 2001. Trichloroethylene, trichloroacetic acid, and dichloroacetic acid: do they affect fetal rat heart development? Int. J. Toxicol., 20: 257-267.
Forkert, P.G., Lash, L., Tardif, R., Tanphaichitr, N., Vandevoort, C. and Moussa, M. 2003. Identification of trichloroethylene and its metabolites in human seminal fluid of workers exposed to trichloroethylene. Drug Metab. Dispos., 31(3): 306-311.
Fox, A.W., Yang, X., Murli, H., Lawlor, T.E., Cifone, M.A. and Reno, F.E. 1996. Absence of mutagenic effects of sodium dichloroacetate. Fundam. Appl. Toxicol., 32: 87-95.
Galloway, S.M., Armstrong, M.J., Reuben, C., Colman, S., Brown, B., Cannon, C., Bloom, A.D., Nakamura, F., Ahmed, M., Duk, S., Rimpo, J., Margolin, B.H., Resnick, M.A., Anderson, B. and Zeiger, E. 1987. Chromosome aberrations and sister chromatid exchanges in Chinese hamster ovary cells: evaluations of 108 chemicals. Environ. Mutagen., 10: 1-175.
Ge, R., Yang, S., Kramer, P.M., Tao, L. and Pereira, M.A. 2001. The effect of dichloroacetic acid on DNA methylation and cell proliferation in B6C3F1 mice. J. Biochem. Mol. Toxicol., 15: 100-106.
Giller, S., Le Curieux, F., Erb, F. and Marzin, D. 1997. Comparative genotoxicity of halogenated acetic acids found in drinking water. Mutagenesis, 12(5): 321-328.
Gonzalez-Leon, A., Schultz, I.R., Xu, G. and Bull, R.J. 1997. Pharmacokinetics and metabolism of dichloroacetate in the F344 rat after prior administration in drinking water. Toxicol. Appl. Pharmacol., 146: 189-195.
Gosselin, R.E., Smith, R.P. and Hodge, H.C. 1984. Clinical toxicology of commercial products. 5th edition. Williams and Wilkins, Baltimore, MD.
Grant, W.M. and Schuman, J.S. 1993. Toxicology of the eye: Effects on the eyes and visual system from chemicals, drugs, metals and minerals, plants, toxins and venoms. 4th edition. C.C. Thomas, Springfield, IL. p. 993.
Gross, B.J., Branchflower, R.V., Burke, T.R., Lees, D.E. and Pohl, L. 1982. Bone marrow toxicity in vitro of chloramphenicol and its metabolites. Toxicol. Appl. Pharmacol., 64: 557-565.
Hansch, C. , Leo, A., Hoekman, D.H. and Heller, S. 1995. Exploring QSAR. Hydrophobic, electronic, and steric constants. ACS Professional Reference Book. American Chemical Society, Washington, DC.
Harrington-Brock, K., Doerr, C.L. and Moore, M.M. 1998. Mutagenicity of three disinfection by-products: di- and trichloroacetic acid and chloral hydrate in L5178Y/TK+/!!3.72 mouse lymphoma cells. Mutat. Res., 413: 265-276.
Hashimoto, H., Azumar, T. and Otsuki, A. 1998. Distribution, sources, and stability of haloacetic acids in Tokyo Bay, Japan. Environ. Toxicol. Chem., 17 (5): 798-805.
Heal, M.R., Reeves, N.M. and Cape, J.N. 2003. Atmospheric concentrations and deposition of trichloroacetic acid in Scotland: results from a 2 year sampling program. Environ. Sci. Technol., 37(12): 2627-2633.
Health Canada. 1994. Canadian Environmental Protection Act. Human health risk assessment for priority substances. Catalogue No. En40-215/41E. Minister of Supply and Services Canada, Ottawa, Ontario.
Health Canada. 1995. A national survey of chlorinated disinfection by-products in Canadian drinking water. Report No. 95-EHD-197. Environmental Health Directorate, Health Protection Branch, Health Canada, Ottawa, Ontario.
Health Canada. 2003. Personal communication from R. Aranda-Rodriguez, Chemistry Research Division, Environmental Health Science Bureau, Safe Environments Programme, Healthy Environments and Consumer Safety Branch, Health Canada, Ottawa, Ontario.
Health Canada. 2004a. Unit risk for dichloroacetic acid (DCA) in drinking water. Report prepared by M. Walker, Biostatistics Unit, Healthy Environments and Consumer Safety Branch, Health Canada, Ottawa, Ontario.
Health Canada. 2004b. Benchmark dose for TCA in drinking water. Report prepared by M. Walker, Biostatistics Unit, Healthy Environments and Consumer Safety Branch, Health Canada, Ottawa, Ontario.
Health Canada. 2007a. Unit risk for dibromoacetic acid (DBA) in drinking water. Report prepared by M. Walker, Biostatistics Unit, Healthy Environments and Consumer Safety Branch, Health Canada, Ottawa, Ontario, February 15.
Health Canada. 2007b. Calculations for the unit risk (raw and converted) for dibromoacetic acid (DBA) in drinking water. Report prepared by M. Walker, Biostatistics Unit, Healthy Environments and Consumer Safety Branch, Health Canada, Ottawa, Ontario, February 16.
Heithersay, G.S. and Wilson, D.F. 1988. Tissue responses in the rat to trichloroacetic acid - an agent used in the treatment of invasive cervical resorption. Aust. Dental J., 33(6): 451-461.
Herbert, V., Gardner, A. and Colman, N. 1980. Mutagenicity of dichloroacetate, an ingredient of some formulation of pangamic acid (trade-named "vitamin B15"). Am. J. Clin. Nutr., 33: 1179-1182.
Herren-Freund, S.L. and Pereira, M.A. 1986. Carcinogenicity of by-products of disinfection in mouse and rat liver. Environ. Health Perspect., 69: 59-65.
Herren-Freund, S.L., Pereira, M.A., Khoury M.D. and Olson, G. 1987. The carcinogenicity of trichloroethylene and its metabolites, trichloroacetic acid and dichloroacetic acid, in mouse liver. Toxicol. Appl. Pharmacol., 90: 183-189.
Hodgeson, J.W. and Becker, D. 1992. Method 552.1: Determination of haloacetic acids and Dalapon in drinking water by ion exchange liquid-solid extraction and gas chromatography with an electron capture detector. In: Methods for the determination of organic compounds in drinking water - Supplement II. EPA/600/R-92/129. U.S. Environmental Protection Agency, Cincinnati, OH. Table 2, p. 552.1-22.
Hoechst AG. 1974. Unpublished results (74.0414) [cited in OECD, 2000].
Hoekstra, E.J. 2003. Review of concentrations and chemistry of trichloroacetate in the environment. Chemosphere, 52(2): 355-369.
Holmes, M., Suarez, J.D., Roberts, N.L., Mole, M.L., Murr, A.S. and Klinefelter, G.R. 2001. Dibromoacetic acid, a prevalent by-product of drinking water disinfection, compromises the synthesis of specific seminiferous tubule proteins following both in vivo and in vitro exposures. J. Androl., 22(5): 878-890.
Howe, R.B. 1995. THRESH: A computer program to compute a reference dose from quantal animal toxicity data using the benchmark dose method. ICF Kaiser Engineers, Inc., Ruston, LA.
HSDB (Hazardous Substances Data Bank). 2003. Trichloroacetic acid. Hazardous Substances Databank No. 1779. U.S. National Library of Medicine, Bethesda, MD.
IARC (International Agency for Research on Cancer). 1995. Dichloroacetic acid and trichloroacetic acid. In: Dry cleaning, some chlorinated solvents and other industrial chemicals. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 63. IARC, Lyon.
IARC (International Agency for Research on Cancer). 2004. Some drinking-water disinfectants and contaminants, including arsenic. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 84. IARC, Lyon.
ILSI (International Life Sciences Institute). 1997. An evaluation of EPA's proposed guidelines for carcinogenic risk assessment using chloroform and dichloroacetate as case studies: report of an expert panel. Health and Environmental Sciences Institute, ILSI, Washington, DC, November.
Innes, J.M.R., Ulland, B.M., Valerio, M.G., Petrucelli, L., Fishbein, L., Hart, E.R., Pallotta, A.J., Bates, R.R., Falk, H.L., Gart, J.J., Klein, M., Mitchell, I. and Peters, J. 1969. Bioassay of pesticides and industrial chemicals for tumorigenicity in mice: a preliminary note. J. Natl. Cancer Inst., 42(6): 1101-1114.
IPCS (International Programme on Chemical Safety). 2000. Disinfectants and disinfectant by-products. Environmental Health Criteria 216. IPCS, World Health Organization, Geneva.
Izumi, M., Hirayama, Y., Sugai, K., Fukumizu, M., Hanaoka, S., Sasaki, M., Kaga, M. and Murayama, K. 2003. [Adverse effects of dichloroacetate in a girl with mitochondrial disorder.] No To Hattatsu, 35(1): 54-58 (in Japanese).
James, M.O., Yan, Z., Cornett, R., Jayanti, V.M., Henderson, G.N., Davydova, N., Katovich, M.J., Pollock, B. and Stacpoole, P.W. 1998. Pharmacokinetics and metabolism of [14C]dichloroacetate in male Sprague-Dawley rats. Identification of glycine conjugates, including hippurate, as urinary metabolites of dichloroacetate. Drug Metab. Dispos., 26(11): 1134-1143 [cited in U.S EPA, 2003c].
Johnson, P.D., Dawson, B.V. and Goldberg, S.J. 1998. Cardiac teratogenicity of trichloroethylene metabolites. J. Am. Coll. Cardiol., 32(2): 540-545.
Jones, A.R. and Wells, G. (1981) The comparative metabolism of 2-bromoethanol and ethylene oxide in the rat. Xenobiotica, 11(11): 763-770.
Juuti, S. and Hoekstra, E. 1998. New directions: the origins and occurrence of trichloroacetic acid. Atmos. Environ., 32(17): 3059-3060.
Kaphalia, B.S., Bhat, H.K., Khan, M.F. and Ansari, G.A.S. 1992. Tissue distribution of monochloroacetic acid and its binding to albumin in rats. Toxicol. Ind. Health, 8(1-2): 53-61.
Kargalioglu, Y., McMillan, B.J., Minear, R.A.and Plewa, M.J. 2002. Analysis of the cytotoxicity and mutagenicity of drinking water disinfection by-products in Salmonella typhimurium. Teratogen. Carcinogen. Mutagen., 22(2): 113-128.
Kato-Weinstein, J., Stauber, A.J., Orner, G.A., Thrall, B.D. and Bull, R.J. 2001. Differential effects of dihalogenated and trihalogenated acetates in the liver of B6C3F1 mice. J. Appl. Toxicol., 21: 81-89.
Katz, R., Tai, C.N., Diener, R.M., McConnell, R.F. and Semonick, D.E. 1981. Dichloroacetate, sodium: 3-month oral toxicity study in rats and dogs. Toxicol. Appl. Pharmacol., 57: 273-287.
Ketcha, M.M., Stevens, D.K., Warren, D.A., Bishop, C.T. and Brashear, W.T. 1996. Conversion of trichloroacetic acid to dichloroacetic acid in biological samples. J. Anal. Toxicol., 20: 236-241.
Keys, D.A., Schultz, I.R., Mahle, D.A. and Fisher, J.W. 2004. A quantitative description of suicide inhibition of dichloroacetic acid in rats and mice. Toxicol. Sci., 82: 381-393.
Kim, H. and Weisel, C.P. 1998. Dermal absorption of dichloro- and trichloroacetic acids from chlorinated water. J. Expos. Anal. Environ. Epidemiol., 8(4): 555-575.
King, W.D., Dodds, L., Allen, A.C., Armson, B.A., Fell, D. and Nimrod, C. 2005. Haloacetic acids in drinking water and risk for stillbirth. Occup. Environ. Med., 62(2): 124-127.
Klinefelter, G.R., Strader, L., Suarez, J., Roberts, N., Holmes, M. and Mole, L. 2000. Dibromoacetic acid, a drinking water disinfection-by-product, alters male reproductive development and fertility. Biol. Reprod., 62(Suppl. 1): 285 (abstract).
Koenig, G., Lohmar, E. and Rupprich, N. 2002 Chloroacetic acids. In: Ullmann's encyclopedia of industrial chemistry. John Wiley & Sons, Inc.
Kohan, M.J., Huggins-Clark, G. and George, S.E. 1998. Mutagenicity of chlorinated and brominated acetic acids. 29th Annual Meeting of the Environmental Mutagen Society, Anaheim, CA, March 21-26. Environ. Mol. Mutagen., 31(Suppl. 29): 36 (abstract).
Kondo, M., Nishihara, T., Shimamoto, T., Koshikawa, T., Itio, T., Sawamura, R. and Tanaka, T. 1988. Biodegradation test of chemicals by cultivation methods. Eisei Kagaku, 34(2): 188-195 [cited in HSDB, 2003].
Krasner, S.W., McGuire, M.J., Jacangelo, J.G., Patania, N.L., Reagan, K.M. and Aieta., E.M. 1989. The occurrence of disinfection by-products in US drinking water. J. Am. Water Works Assoc., 81: 41-53.
Larson, J.L. and Bull, R.J. 1992. Metabolism and lipoperoxidative activity of trichloroacetate and dichloroacetate in rats and mice. Toxicol. Appl. Pharmacol., 115: 268-277.
Lash, L.H., Fisher, J.W., Lipscomb, J.C. and Parker, J.C. 2000. Metabolism of trichloroethylene. Environ. Health Perspect., 108(Suppl. 2): 177-200.
Lewinstein, I. and Rotstein, I. 1992. Effect of trichloroacetic acid on the microhardness and surface morphology of human dentin and enamel. Endodont. Dent. Traumatol., 8: 16-20.
Lewis, R.J., Sr. (ed.). 2001. Hawley's condensed chemical dictionary. 14th edition. John Wiley & Sons, Inc., New York, NY. pp. 251, 361, 1124.
Lide, D.R. (ed.). 2003-2004. CRC handbook of chemistry and physics. 84th edition. CRC Press Inc., Boca Raton, FL.
Lin, E.L., Mattox J.K. and Daniel, F.B. 1993. Tissue distribution, excretion and urinary metabolites of dichloroacetic acid in the male Fischer 344 rat. J. Toxicol. Environ. Health, 38(1): 19-32.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Suarez, J.D. and Dyer, C.J. 1994a. Acute spermatogenic effects of bromoacetic acids. Fundam. Appl. Toxicol., 22(3): 422-430.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Suarez, J.D., Roberts, N.L. and Dyer, C.J. 1994b. Spermatotoxicity of dibromoacetic acid in rats after 14 daily exposures. Reprod. Toxicol., 8(3): 251-259.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Narotsky, M.G., Suarez, J.D., Roberts, N.L. and Perreault, S.D. 1995. Dibromoacetic acid affects reproductive competence and sperm quality in the male rat. Fundam. Appl. Toxicol., 28(1): 9-17.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Suarez, J.D. and Roberts, N. 1997a. Spermatotoxicity of dichloroacetic acid. Reprod. Toxicol., 11(5): 681-688.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Veeramachaneni, D.N.R., Roberts, N.L. and Suarez, J.D. 1997b. Histopathologic changes in the testes of rats exposed to dibromoacetic acid. Reprod. Toxicol., 11(1): 47-56.
Lingohr, M.K., Thrall, B.D. and Bull, R.J. 2001. Effects of dichloroacetate (DCA) on serum insulin levels and insulin-controlled signaling proteins in livers of male B6C3F1 mice. Toxicol. Sci., 59: 178-184.
Lingohr, M.K., Bull, R.J., Kato-Weinstein, J. and Thrall, B.D. 2002. Dichloroacetate stimulates glycogen accumulation in primary hepatocytes through insulin-independent mechanism. Toxicol. Sci., 68: 508-515.
Lukas, G., Vyas, K.H., Brindle, S.D., LeSher, A.R. and Wagner, W.E., Jr. 1980. Biological disposition of sodium dichloroacetate in animals and humans after intravenous administration. J. Pharmacol. Sci., 69(4): 419-421.
Lumpkin, M.H., Bruckner, J.V., Campbell, J.L., Dallas, C.E., White, C.A. and Fisher, J.W. 2003. Plasma binding of trichloroacetic acid in mice, rats, and humans under cancer bioassay and environmental exposure conditions. Drug Metab. Dispos., 31(10): 1203-1207.
Lytle, D.A. and Schock, M.R. (2005) Formation of Pb(IV) oxides in chlorinated water. J. Am. Water Works Assoc., 97(11): 102-114.
MacKay, J.M., Griffiths, K., Fox, D.A., Howeard, C.A., Coutts, C., Wyatt, I. and Styles, J.A. 1995. Trichloroacetic acid: investigation into the mechanism of chromosomal damage in the in vitro human lymphocyte cytogenetic assay and the bone marrow micronucleus test. Carcinogenesis, 16: 1127-1133.
Manitoba Department of Conservation. 2004. Personal communication from D. Rocan, Manitoba Department of Conservation, Winnipeg, Manitoba, to N. Edmonds, Health Canada, Ottawa, Ontario.
Maruthamuthu, P. and Huie, R.E. 1995. Ferric ion assisted photooxidation of haloacetates. Chemosphere, 30(11): 2199-2207.
Mather, G.G., Exon, J.H. and Koller, L.D. 1990. Subchronic 90 day toxicity of dichloroacetic and trichloroacetic acid in rats. Toxicology, 64: 71-80.
Matsuda, H., Ose, Y., Nagase, H., Sato, T., Kito, H. and Sumida, K. 1991. Mutagenicity of ozonation and chlorination products from p-hydroxybenzaldehyde. Sci. Total Environ., 103: 141-149.
McCay, J.A., Musgrove, D.L., Brown, R.D., Johnson, N.A., Karrow, N.A., Guo, T.L., Germolec, D.R. and White, K.L., Jr. 2000. Exposure to disinfection by-product dibromoacetic acid does not alter immune function of host resistance. Toxicologist, 54: 157 (abstract).
McGregor, D.B., Martin, R., Cattanach, P., Edwards, I., McBride, D. and Caspary, W.J. 1987. Responses of the L5178Y tk+/tk- mouse lymphoma cell forward mutation assay to coded chemicals. I: Results for nine compounds. Environ. Mutagen., 9(2): 143-160.
Meier, J.R., Stewart, B.E. and Blazak, W.F. 1997. Genotoxicity studies of sodium dichloroacetate and sodium trichloroacetate. Environ. Sci., 5(2): 95-108.
Meister, R.T. (ed.). 2002. Farm chemicals handbook. Vol. 88. Meister Publishing Co., Willoughby, OH. p. C-380.
Melnick, R.L., Nyska, A., Foster, P.M., Roycroft, J.H. and Kissling, G.E. 2007. Toxicity and carcinogenicity of the water disinfection byproduct, dibromoacetic acid, in rats and mice. Toxicology, 230(2-3): 126-136.
Meusel, M. and Rehm, H.J. 1993. Biodegradation of dichloroacetic acid by freely suspended and adsorptive immobilized Xanthobacter autotrophicus GJ10 in soil. Appl. Microbiol. Biotechnol., 40: 165-171 [cited in Williams et al., 1998].
Miller, J.H., Minard, K., Wind, R.A., Orner, G.A., Sasser, L.B. and Bull, R.J. 2000. In vivo MRI measurements of tumor growth induced by dichloroacetate: implications for mode of action. Toxicology, 145: 115-125.
Miller, J.W. and Uden, P.C. 1983. Characterization of nonvolatile chlorination products of humic substances. Environ. Sci. Technol., 17(3): 150-157.
Mills, C.J., Bull, R.J., Cantor, K.P., Reif, J., Hrudey, S.E., Huston, P. and an Expert Working Group. 1998. Health risks of drinking water chlorination by-products: Report of an expert working group. Chronic Dis. Can., 19(3)..
Monster, A.C., Boersma, G. and Duba, W.C. 1979. Kinetics of trichloroethylene in repeated exposure of volunteers. Int. Arch. Occup. Environ. Health, 42: 283-292.
Moore, G.W., Swift, L.L., Rabinowitz, D., Crofford, O.B., Oates, J.A. and Stacpoole, P.W. 1979. Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Atherosclerosis, 33: 285-293.
Morita, H., Tadaki, S., Nozaka, T., Omura, T., Haga, M. and Tanaka, A. 1997. Mutagenicity and concentration of chlorination by-products in tap water in Saitama. Environ. Mutagen. Res., 19: 127-134.
Morris, E.D. and Bost, J.C. 2002. Acetic acid, halogenated derivatives. In: Kirk-Othmer encyclopedia of chemical technology. 5th edition. John Wiley & Sons, Inc.
Morrison, J. 1946. Toxicity of certain halogen substituted aliphatic acids for white mice. J. Pharmacol. Exp. Ther., 86: 336-338.
Morrison, J.L. and Leake, C.D. 1941. Monochloroacetic acid as a food beverage stabilizer. Univ. Calif. Publ. Pharmacol. J., 1: 397-421.
Moser, V.C., Phillips, P.M., Levine, A.B., McDaniel, K.L., Sills, R.C., Jortner, B.S. and Butt, M.T. 2004. Neurotoxicity produced by dibromoacetic acid in drinking water of rats. Toxicol. Sci., 79: 112-122.
Mowrer, J. and Nordin, J. 1987. Characterization of halogenated organic acids in flue gases from municipal waste incinerators. Chemosphere, 16(6): 1181-1192.
Muller, G., Spassovski, M. and Henschler, D. 1974. Metabolism of trichloroethylene in man. II. Pharmacokinetics of metabolites. Arch. Toxicol., 32: 283-295.
Munch, D.J., Munch, J.W. and Pawlecki, A.M. 1995. Method 552: Determination of haloacetic acids and Dalapon in drinking water by liquid-liquid extraction, derivatization, and gas chromatography with electron capture detection. In: Methods for the determination of organic compounds - Supplement III. EPA/600/R-95/131. U.S. Environmental Protection Agency, Cincinnati, OH.
Murr, A.S. and Goldman, J.M. 2005. Twenty-week exposures to the drinking water disinfection by-product dibromoacetic acid: reproductive cyclicity and steroid concentrations in the female Sprague-Dawley rat. Reprod. Toxicol., 20: 73-80.
Narotsky, M.G., Hamby, B.T., Best, D.S. and Hunter, E.S. 1996. In vivo developmental effects of dibromoacetic acid (DBA) and dichloroacetic acid (DCA) in mice. Teratology, 53: 96-97 (abstract).
Narotsky, M.G., Hamby, B.T. and Best, D.S. 1997. Developmental effects of dibromoacetic acid (DBA) in a segment II study in mice. Teratology, 55(1): 67 (abstract).
Nelson, M.A. and Bull, R.J. 1988. Induction of strand breaks in DNA by trichloroethylene and metabolites in rat and mouse liver in vivo. Toxicol. Appl. Pharmacol., 94: 45-54.
Nelson, M.A., Lansing, A.J., Sanchez, I.M., Bull, R.J. and Springer, D.L. 1989. Dichloroacetic acid and trichloroacetic acid-induced DNA strand breaks are independent of peroxisome proliferation. Toxicology, 58: 239-248.
Newfoundland and Labrador Department of Environment. 2003. Personal communication from M. Goebel, Newfoundland and Labrador Department of Environment, St. John's, Newfoundland and Labrador, to N. Edmonds, Health Canada, Ottawa, Ontario.
Nikolaou, A.D., Kostopoulou, M.N. and Lekkas, T.D. 1999. Organic by-products of drinking water chlorination. Global Nest: Int. J., 1(3): 143-156.
NIOSH (National Institute for Occupational Safety and Health). 1990. Unpublished provisional data as of 7/1/90. National Occupational Exposure Survey (1981-83). NIOSH, Cincinnati, OH [cited in WHO, 2004c].
NTP (National Toxicology Program). 1992. Toxicology and carcinogenesis studies of monochloroacetic acid (CAS No. 79-11-8) in F344/N rats and B6C3F1 mice (gavage studies). NIH Publication No. 92-2851. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
NTP (National Toxicology Program). 1999a. The immunotoxicity of dibromoacetic acid (CAS No. 631-64-1). Dose range-finding study in female B6C3F1 mice. NTP Study No. IMM98001. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
NTP (National Toxicology Program). 1999b. Final range-finding report. Immunotoxicity of dibromoacetic acid in female B6C3F1 mice. Final report submitted to NTP by K.L. White and J.A. Munson, Medical College of Virginia [cited in U.S. EPA, 2005a].
NTP (National Toxicology Program). 2000. Final report on the subchronic toxicity study of dichloroacetic acid to Fischer-344 rats and B6C3F1 mice. Report submitted to the NTP under Contract No. N01-ES-85420, September 1999. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
NTP (Natort ional Toxicology Program). 2003a. Dichloroacetic acid (Executive summary). Clinical Evaluation Committee draft rep submitted by Arthur D. Little, Inc. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
NTP (National Toxicology Program). 2003b. Testing status for bromoacetic acid. M920034. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
NTP (National Toxicology Program). 2007. NTP technical report on the toxicology and carcinogenesis studies of dibromoacetic acid (CAS No. 631-64-1) in F344/N rats and B6C3F1 mice (drinking water studies). NTP Technical Report Series No. 537. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
OECD (Organisation for Economic Co-operation and Development). 2000. SIDS initial assessment profile. Trichloroacetic acid. CAS No. 76-03-9. Vol. 6, Part II, June. United Nations Environment Programme publication.
OEHHA (Office of Environmental Health Hazard Assessment). 1999. Evidence on the carcinogenicity of trichloroacetic acid and its salts (PDF Version - 137 K). July draft. Reproductive and Cancer Hazard Assessment Section, OEHHA, California Environmental Protection Agency.
Ono, Y., Somiya, I. and Kawamura, M. 1991. The evaluation of genotoxicity using DNA repairing test for chemicals produced in chlorination and ozonation processes. Water Sci. Technol., 23(1-3): 329-338.
Ontario Ministry of Environment and Energy. 2003. Personal communication from A. Socha, Ontario Ministry of Environment and Energy, Toronto, Ontario.
Parnell, M.J., Koller, L.D., Exon, J.H. and Arnzen, J.M. 1986. Trichloroacetic acid effects on rat liver peroxisomes and enzyme-altered foci. Environ. Health Perspect., 69: 73-79.
Parnell, M.J., Exon, J.H. and Koller, L.D. 1988. Assessment of hepatic initiation-promotion properties of trichloroacetic acid. Arch. Environ. Contam. Toxicol., 17: 429-436.
Parrish, J.M., Austin, E.W., Stevens, D.K., Kinder, D.H. and Bull, R.J. 1996. Haloacetate-induced oxidative damage to DNA in the liver of male B6C3F1 mice. Toxicology, 110: 103-111.
Pereira, M.A. 1995. Effect of dichloroacetic acid and trichloroacetic acid on cell proliferation in B6C3F1 mice. American Water Works Association Research Foundation and American Water Works Association, Denver, CO.
Pereira, M.A. 1996. Carcinogenic activity of dichloroacetic acid and trichloroacetic acid in the liver of female B6C3F1 mice. Fundam. Appl. Toxicol., 31: 192-199.
Pereira, M.A. and Phelps, J.B. 1996. Promotion by dichloroacetic acid and trichloroacetic acid of N-methyl-Nnitrosourea-initiated cancer in the liver of female B6C3F1 mice. Cancer Lett., 102: 133-141.
Pereira, M.A., Li, K. and Kramer, P.M. 1997. Promotion by mixtures of dichloroacetic acid and trichloroacetic acid of N-methyl-N-nitrosourea-initiated cancer in the liver of female B6C3F1 mice. Cancer Lett., 115: 15-23.
Pereira, M.A., Wang, W., Kramer, P.M. and Tao, L. 2004. Prevention by methionine of dichloroacetic acid-induced liver cancer and DNA hypomethylation in mice. Toxicol. Sci., 77: 243-248.
Peters, R.J. 2003. Chloroacetic acids in European soils and vegetation. J. Environ. Monit., 5(2): 275-280.
Phillips, M., Levine, A., McDaniel, K.L., Sills, R.C. and Moser, V.C. 2002. Neurotoxicity produced by dibromoacetic acid in drinking water of rats. Toxicologist, 66(1-S): 251 (abstract).
Plewa, M.J., Kargalioglu,Y., Vankerk, D., Minear, R.A. and Wagner, E.D. 2002. Mammalian cell cytotoxicity and genotoxicity analysis of drinking water disinfection by-products. Environ. Mol. Mutagen., 40: 134-142.
Ploeg, J., Hall, G. and Janssen, D. 1991. Characterization of haloacid dehalogenases from Xanthobacter autotrophicus GJ10 and sequencing of dhIB gene. J. Bacteriol., 173: 7925-7933 [cited in Williams et al., 1998].
Pourmoghaddas, H. and Stevens, P.C. 1995. Relationship between trihalomethanes and haloacetic acids with total organic halogen during chlorination. Water Res., 29: 2059-2062.
Randall, J.L., Christ, S.A., Horton Perez, P., Nolen, G.A., Read, E.J. and Smith, M.K. 1991. Developmental effects of 2-bromoacetic acid in the Long Evans rat. Teratology, 43(5): 454 (abstract).
Rannug, U., Gothe, R. and Wachtmeister, C.A. 1976. The mutagenicity of chloroethylene oxide, chloroacetaldehyde, 2-chloroethanol and chloroacetic acid, conceivable metabolites of vinyl chloride. Chem. Biol. Interact., 12(3-4): 251-263.
Raymer, J.H., Pellizzari, E.D. and Hu, Y. 2001. Exposures to water disinfection byproducts via food. National Center for Environmental Research STAR Drinking Water Progress Review Meeting, February 22-23, U.S. Environmental Protection Agency [cited in WHO, 2004a,b, 2005].
Reckhow, D.A., Singer, P.C. and Malcolm, R.L. 1990. Chlorination of humic materials: by-product formation and chemical interpretations. Environ. Sci. Technol., 24: 1655-1664.
Reid Crowther & Partners Ltd. 2000. Canadian water treatment study: water treatment and disinfection byproducts. Winnipeg, Manitoba, September.
Reimann, S., Grob, K. and Frank, H. 1996. Environmental chloroacetic acids in foods analyzed by GC-ECD. Mitt. Geb. Lebensmittelunters. Hyg., 87(2): 212-222 [cited in WHO, 2004a,b, 2005].
Richardson, S.D., Thurston, A.D., Caughran, T.V., Chen, P.H., Collette, T.W. and Floyd, T.L. 1999. Identification of new drinking water disinfection byproducts formed in the presence of bromide. Environ. Sci. Technol., 33: 3378-3383.
Richmond, R.E., Carter, J.H., Carter, H.W., Daniel, F.B. and DeAngelo, A.B. 1995. Immunohistochemical analysis of dichloroacetic acid (DCA)-induced hepatocarcinogenesis in male Fischer (F344) rats. Cancer Lett., 92: 67-76.
Rogers, D.R. 1995. Accidental fatal monochloroacetic acid poisoning. Am. J. Forensic Med. Pathol., 16(2): 115-116.
Saghir, S.A. and Rozman, K.K. 2003. Kinetics of monochloroacetic acid at subtoxic doses in rats after single oral and dermal administrations. Toxicol. Sci., 76: 51-64.
Saito, H., Isoda, S., Kato, M. and Nagaoka, N. 1995. Mutagenic activity of indoor swimming pool water. Environ. Mutat. Res. Commun., 17: 169-177.
Sawada, M., Sofuni, T. and Ishidate, M., Jr. 1987. Cytogenetic studies on 1,1-dichloroethylene and its two isomers in mammalian cells in vitro and in vivo. Mutat. Res., 187: 157-163.
Schock, M.R. and Giani, R. (2004) Oxidant/disinfectant chemistry and impacts on lead corrosion. In: Proceedings of the 2004 American Water Works Association Water Quality Technology Conference, San Antonio, TX.
Schroll, R., Bierling, B., Cao, G., Dorfler, U., Lahaniati, M., Langenbach, T., Scheunert, I. and Winkler, R. 1994. Uptake pathways of organic chemicals from soil by agricultural plants. Chemosphere, 28(2): 297-303 [cited in WHO, 2004b].
Schultz, I.R., Merdink, J.L., Gonzalez-Leon, A. and Bull, R.J. 1999. Comparative toxicokinetics of chlorinated and brominated haloacetates in F344 rats. Toxicol. Appl. Pharmacol., 158(2): 103-114.
Schultz, I.R., Merdink, J.L., Gonzalez-Leon, A. and Bull, R.J. 2002. Dichloroacetate toxicokinetics and disruption of tyrosine catabolism in B6C3F1 mice: dose-response relationships and age as a modifying factor. Toxicology, 173: 229-247.
Scott, B.F., Spencer, C., Marvin, C.H., Mactavish, D.C. and Muir, D.C.G. 2002. Distribution of haloacetic acids in water columns of the Laurentian Great Lakes and Lake Malawi. Environ. Sci. Technol., 36: 1893-1898.
Serjeant, E.P. and Dempsey, B. 1979. Ionisation constants of organic acids in aqueous solution. International Union of Pure and Applied Chemistry (IUPAC) Chemical Data Series No. 23. Pergamon Press, New York, NY. p. 989.
Sidebottom, H. and Franklin, J. 1996. The atmospheric fate and impact of hydrochlorofluorocarbons and chlorinated solvents. Pure Appl. Chem., 68(9): 1757-1769.
Singer, P.C. 1993. Formation and characterization of DBPs. In: Safety of water disinfection: balancing chemical and microbial risks. G.F. Craun (ed.). ILSI Press, Washington, DC.
Smith, M.K., Randall, J.L., Read, E.J. and Stober, J.A. 1989. Teratogenic activity of trichloroacetic acid in the rat. Teratology, 40: 445-451.
Smith, M.K., Randall, J.L., Read, E.J. and Stober, J.A. 1990. Developmental effects of chloroacetic acid in the Long-Evans rat. Teratology, 41(5): 593.
Smith, M.K., Randall, J.L., Read, E.J. and Stober, J.A. 1992. Developmental toxicity of dichloroacetate in rat. Teratology, 46(3): 217-223.
Snyder, R.D., Pullman, J., Carter, J.H., Carter, H.W. and DeAngelo, A.B. 1995. In vivo administration of dichloroacetic acid suppresses spontaneous apoptosis in murine hepatocytes. Cancer Res., 55(17): 3702-3705.
Spruijt, L., Naviaux, R.K., McGowan, K.A., Nyhan, W.L., Sheean, G., Haas, R.H. and Barshop, B.A. 2001. Nerve conduction changes in patients with mitochondrial diseases treated with dichloroacetate. Muscle-Nerve, 24(7): 916-924.
Stacpoole, P.W. 1989. The pharmacology of dichloroacetate. Metabolism, 38(11): 1124-1144.
Stacpoole, P.W., Gonzalez, M.G., Vlasak, J., Oshiro, Y. and Bodor, N. 1987. Dichloroacetate derivatives. Metabolic effects and pharmacodynamics in normal rats. Life Sci., 41: 2167-2176.
Stacpoole, P.W., Hardwood, H.J., Jr., Cameron, D.F., Curry, S.H., Samuelson, D.A., Cornwell, P.E. and Sauberlich, H.E. 1990. Chronic toxicity of dichloroacetate: possible relation to thiamine deficiency in rats. Fundam. Appl. Toxicol., 14: 327-337.
Stacpoole, P.W., Henderson, G.N., Yan, Z. and James, M.O. 1998a. Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect., 106(Suppl. 4): 989-994.
Stacpoole, P.W., Henderson, G.N., Yan, Z., Cornett, R. and James, M.O. 1998b. Pharmacokinetics, metabolism, and toxicology of dichloroacetate. Drug Metab. Rev., 30(30): 499-539.
Stauber, A.J. and Bull, R.J. 1997. Differences in phenotype and cell replicative behavior of hepatic tumors by dichloroacetate (DCA) and trichloroacetate (TCA). Toxicol. Appl. Pharmacol., 144: 235-246.
Stauber, A.J., Bull, R.J. and Thrall, B.D. 1998. Dichloroacetate and trichloroacetate promote clonal expansion of anchorage-independent hepatocytes in vivo and in vitro. Toxicol. Appl. Pharmacol., 150: 287-294.
Stratton, C.E., Ross, W.E. and Chapman, S. 1981. Cytotoxicity and deoxyribonucleic acid damage associated with bromoacetate. Biochem. Pharmacol., 30: 1497-1500.
Sutinen, S., Juuti, S., Koivisto, L., Turunen, M. and Ruuskanen, J. 1995. The uptake of and structural changes induced by trichloroacetic acid in the needles of Scot pine seedlings. J. Exp. Bot., 46(290): 1223-1231 [cited in WHO, 2004b].
Tao, L., Kramer, P.M., Ge, R. and Pereira, M.A. 1998. Effect of dichloroacetic acid and trichloroacetic acid on DNA methylation in liver tumors of female B6C3F1 mice. Toxicol. Sci., 43: 139-144.
Tao, L., Yan, S., Xie, M., Kramer, P.M. and Pereira, M.A. 2000. Effect of trichloroethylene and its metabolites, dichloroacetic acid and trichloroacetic acid, on the methylation and expression of c-jun and c-myc protooncogenes in mouse liver: Prevention by methionine. Toxicol. Sci., 54: 399-407.
Templin, M.V., Parker, J.C. and Bull, R.J. 1993. Relative formation of dichloroacetate and trichloroacetate from trichloroethylene in male B6C3F1 mice. Toxicol. Appl. Pharmacol., 123: 1-8.
Thai, S.F., Allen, J.W., DeAngelo, A.B., George, M.H. and Fuscoe, J.C. 2003. Altered gene expression in mouse livers after dichloroacetic acid exposure. Mutat. Res., 543(2): 167-180.
Tomlin, C. (ed.). 1994. The pesticide manual: world compendium. 10th edition. British Crop Protection Council, Surrey, and the Royal Society of Chemistry, Cambridge. pp. 940-994.
Tong, Z., Board, P.G. and Anders, M.W. 1998a. Glutathione transferase zeta-catalyzed biotransformation of dichloroacetic acid and other alpha-haloacids. Chem. Res. Toxicol., 11: 1332-1338.
Tong, Z., Board, P.G. and Anders, M.W. 1998b. Glutathione transferase zeta catalyzes the oxygenation of the carcinogen DCA to glyoxylic acid. Biochem. J., 331(2): 371-374.
Toth, G.P., Kelty, K.C., George, E.L., Read, E.J. and Smith, M.K. 1992. Adverse male reproductive effects following subchronic exposure of rats to sodium dichloroacetate. Fundam. Appl. Toxicol., 19: 57-63.
Tsuchiya, T., Ooyama, N., Murakami, T., Sano, F., Sugimoto, J. and Mutai, M. 2000. Collaborative work to evaluate toxicity on male reproductive organs by repeated dose studies in rats. 25). Effects of 2- and 4-week repeated-dosing of dibromoacetic acid. J. Toxicol. Sci., 25(Special Issue): 241-249.
Uchiyama, H., Nakajima, T., Yagi, O. and Nakahara T. 1992. Role of heterotrophic bacteria in complete mineralization of trichloroethylene by Methylocystis sp. strain M. Appl. Environ. Microbiol., 58 (9): 3067-3071 [cited in Williams et al., 1998].
U.S. EPA (United States Environmental Protection Agency). 1986. Guidelines for carcinogen risk assessment, 1986. Fed. Regist., 51(185): 33992-34003.
U.S. EPA (United States Environmental Protection Agency). 1991. Toxicology of the chloroacetic acids, by-products of the drinking water disinfection process. II. The comparative carcinogenicity of dichloroacetic acid and trichloroacetic acid: implication for risk assessment. Document No. HERL-0820. Deliverable No. 3101. Health Effects Research Laboratory, U.S. EPA, Research Triangle Park, NC.
U.S. EPA (United States Environmental Protection Agency). 1992. Methods for the determination of organic compounds in drinking water - Supplement II. EPA-600/R-92/129. U.S. EPA, Cincinnati, OH.
U.S. EPA (United States Environmental Protection Agency). 1994. Final draft for the criteria document on chlorinated acids/aldehydes/ketones/alcohols. EPA 68-C2-0139. Prepared for Health and Ecological Criteria Division, Office of Science and Technology, Office of Water, U.S. EPA, Washington, DC.
U.S. EPA (United States Environmental Protection Agency). 1995. Methods for the determination of organic compounds in drinking water - Supplement III. EPA-600/R-95/131. U.S. EPA, Cincinnati, OH.
U.S. EPA (United States Environmental Protection Agency). 1999a. EPA guidance manual - Alternative disinfectants and oxidants. EPA-815-R-99-014. U.S. EPA, Washington, DC, April.
U.S. EPA (United States Environmental Protection Agency). 1999b. Draft revised guidelines for carcinogen risk assessment. U.S. EPA, Washington, DC, July.
U.S. EPA (United States Environmental Protection Agency). 1999c. Microbial and Disinfection Byproduct Rules Simultaneous Compliance Guidance Manual. EPA-815-R-99-015. U.S. EPA, Washington, DC, August.
U.S. EPA (United States Environmental Protection Agency). 2003a. Approved methods for microorganisms, disinfectants and disinfection byproducts. U.S. EPA, Washington, DC, June.
U.S. EPA (United States Environmental Protection Agency). 2003b. Addendum to drinking water criteria document for monochloracetic acid and trichloroacetic acid (final). Office of Science and Technology, Office of Water, U.S. EPA, Washington, DC, July.
U.S. EPA (United States Environmental Protection Agency). 2003c. Toxicological review of dichloroacetic acid (PDF Version - 1706 K) (CAS No. 79-43-6). In support of summary information on the Integrated Risk Information System (IRIS). EPA 635/R-03/007. National Center for Environmental Assessment, U.S. EPA, Washington, DC, August.
U.S. EPA (United States Environmental Protection Agency). 2003d. National Primary Drinking Water Regulations: Stage 2 Disinfectants and Disinfection Byproducts Rule; National Primary and Secondary Drinking Water Regulations: Approval of Analytical Methods for Chemical Contaminants, U.S. EPA, Washington, DC, August 18.
U.S. EPA (United States Environmental Protection Agency). 2003e. Determination of haloacetic acids and Dalapon in drinking water by liquid-liquid microextraction, derivatization, and gas chromatography with electron capture detection. EPA 815-B-03-002. Office of Ground Water and Drinking Water, U.S. EPA, Cincinnati, OH, July.
U.S. EPA (United States Environmental Protection Agency). 2005a. Drinking water criteria document: Brominated acetic acids. Document No. EPA-822-R-05-007. Office of Science and Technology, Office of Water, U.S. EPA, Washington, DC, November.
U.S. EPA (United States Environmental Protection Agency). 2005b. Technologies and costs document for the Final Long Term 2 Enhanced Surface Water Treatment Rule and Final Stage 2 Disinfectants and Disinfection Byproducts Rule. EPA-815-R-05-013. U.S. EPA, Washington, DC, December.
U.S. EPA (United States Environmental Protection Agency). 2006. 2006 edition of the drinking water standards and health advisories. EPA 822-R-06-013. Office of Water, U.S. EPA, Washington, DC, August.
Veeramachaneni, D.N.R. 2002. The rabbit may represent a better animal model for determining reproductive risk associated with DBPs. Toxicologist, 66(1-S): 331 (abstract).
Veeramachaneni, D.N.R., Higuchi, T.T., Palmer, J.S. and Kane, C.M. 2000. Dibromoacetic acid, a disinfection by-product in drinking water, impairs sexual function and fertility in male rabbits. Biol. Reprod., 62(Suppl. 1): 246 (abstract).
Verschueren, K. 2001. Handbook of environmental data on organic chemicals. 4th edition. John Wiley & Sons, Inc., New York, NY.
Vetter, C.M., Miller, J.E., Crawford, L.M., Armstrong, M.J., Clair, J.H., Conner, M.W., Wise, L.D. and Skopek, T.R. 1998. Comparison of motility and membrane integrity to assess rat sperm viability. Reprod. Toxicol., 12(2): 105-114.
Volkel, W., Friedewald, M., Lederer, E., Pahler, A., Parker, J. and Dekant, W. 1998. Biotransformation of perchloroethene: Dose-dependent excretion of trichloroacetic acid, dichloroacetic acid, and N-acetyl-S(trichlorovinyl)-L-cysteine in rats and humans after inhalation. Toxicol. Appl. Pharmacol., 153: 20-27.
Weast, R.C. (ed.). 1973. Handbook of chemistry and physics. 54th edition. CRC Press Inc., Cleveland, OH.
WHO (World Health Organization). 2004a. Monochloroacetic acid in drinking water (PDF Version - 74 K). WHO/SDE/WSH/03.04/85. WHO, Geneva.
WHO (World Health Organization). 2004b. Trichloroacetic acid in drinking water (PDF Version - 153 K). WHO/SDE/WSH/03.04/120. WHO, Geneva.
WHO (World Health Organization). 2004c. Brominated acetic acids in drinking water (PDF Version - 144 K). WHO/SDE/WSH/03.04/79. WHO, Geneva.
WHO (World Health Organization). 2005. Dichloroacetic acid in drinking water (PDF Version - 184 K). WHO/SDE/WSH/05.08/121. WHO, Geneva.
Williams, S.L., Williams, R.L. and Yuan, J. 1998. Bacterial degradation of haloacetic acids in the distribution system. Proceedings of the Water Quality Technical Conference, San Diego, CA, November 1-4. American Water Works Association, Denver, CO.
Woodard, G., Lange, S.W., Nelson, K.W. and Calvery, H.O. 1941. The acute oral toxicity of acetic, chloracetic, dichloroacetic and trichloroacetic acids. J. Ind. Hyg. Toxicol., 23: 78-82.
WSSA (Weed Science Society of America). 1983. Trichloroacetic acid. In: Herbicide handbook of the Weed Science Society of America. 5th edition. WSSA, Champaign, IL. 447 pp.
Xie, Y.F. 2004. Disinfection byproducts in drinking water: Formation, analysis, and control. Lewis Publishers, CRC Press, Boca Raton, FL.
Xu, G., Stevens, D.K. and Bull, R.J. 1995. Metabolism of bromodichloroacetate in B6C3F1 mice. Drug Metab. Dispos., 23(12): 1412-1416.
Xu, X. and Weisel, C.P. 2003. Inhalation exposure to haloacetic acids and haloketones during showering. Environ. Sci. Technol., 37(3): 569-576.
Xu, X., Mariano, T.M., Laskin, J.D. and Weisel, C.P. 2002. Percutaneous absorption of trihalomethanes, haloacetic acids, and haloketones. Toxicol. Appl. Pharmacol., 184(1): 19-26.
Yllner, S. 1971. Metabolism of chloroacetate-14C in the mouse. Acta Pharmacol. Toxicol., 30(1-2): 69.
Yount, E.A., Felten, S.Y., O'Connor, B.L., Peterson, R.G., Powell, R.S., Yum, M.N. and Harris, R.A. 1982. Comparison of metabolic and toxic effects of 2-chloroproprionate and dichloroacetate. J. Pharmacol. Exp. Ther., 222(2): 501-508.
Yu, K.O., Barton, H.A., Mahle, D.A. and Frazier, J.M. 2000. In vivo kinetics of trichloroacetate in male Fischer 344 rats. Toxicol. Sci., 54: 302-311.
ZirChrom Separations, Inc. Undated. Dissociation constants of organic acids and bases.
Appendix A: List of acronyms
- ALARA
- as low as reasonably achievable
- ALT
- alanine transaminase
- ANSI
- American National Standards Institute
- APHA
- American Public Health Association
- AST
- aspartate transaminase
- CAS
- Chemical Abstracts Service
- CDBP
- chlorinated disinfection by-product
- DBA
- dibromoacetic acid
- DBP
- disinfection by-product
- DCA
- dichloroacetic acid
- DNA
- deoxyribonucleic acid
- ECD
- electron capture detection
- EPA
- Environmental Protection Agency ( USA)
- GAC
- granular activated carbon
- GC
- gas chromatography
- GST-zeta
- glutathione-S-transferase-zeta
- HAA
- haloacetic acid
- HAA5
- total haloacetic acids; refers to the total of monochloroacetic acid, dichloroacetic acid, trichloroacetic acid, monobromoacetic acid and dibromoacetic acid
- IARC
- International Agency for Research on Cancer
- KA
- kinetic adjustment factor
- kg bw
- kilogram body weight
- LD50
- median lethal dose
- LMS
- linearized multistage
- LOAEL
- lowest-observed-adverse-effect level
- MAC
- maximum acceptable concentration
- MBA
- monobromoacetic acid
- MCA
- monochloroacetic acid
- MDL
- method detection limit
- mRNA
- messenger ribonucleic acid
- MTBE
- methyl tert-butyl ether
- NOAEL
- no-observed-adverse-effect level
- NOEL
- no-observed-effect level
- NOM
- natural organic matter
- NSF
- NSF International
- NTP
- National Toxicology Program ( USA)
- NTU
- nephelometric turbidity unit
- PCE
- perchloroethylene (tetrachloroethene)
- PPAR
- peroxisome proliferator activated receptor
- ppm
- parts per million
- PQL
- practical quantitation limit
- SCC
- Standards Council of Canada
- TCA
- trichloroacetic acid
- TCE
- trichloroethylene
- TDI
- tolerable daily intake
- THM
- trihalomethane
- UV
- ultraviolet
- WHO
- World Health Organization