
Page 4 : Recommandations pour la qualité de l'eau potable au Canada : document technique – les acides haloacétiques
10.0 Effets sur la santé chez les animaux de laboratoire et sur les systèmes d'essai in vitro
10.1 Toxicité aiguë
Les tableaux 7 et 8 présentent des détails concernant les études disponibles sur la toxicité aiguë par voie orale et cutanée.
| Composé | DL50 cutanée (mg/kg p.c.)Tableau 8 note de bas de page a | Observations cliniques | |
|---|---|---|---|
| Rats | Lapins | ||
| MCA, acide | >400 (solution à 5 %)Tableau 8 note de bas de page 1) 305-800 (solution à 40-50 %)Tableau 8 note de bas de page 1) 145 (acide)Tableau 8 note de bas de page 2) | 250 (solution à 50 %)Tableau 8 note de bas de page 1) | Apathie, hypoactivité, larmoiement horripilation, halètement, position couchée sur le ventreTableau 8 note de bas de page 1) |
| MCA, sel | >2000 (aucun décès)Tableau 8 note de bas de page 1) | n.d.Tableau 8 note de bas de page b | Activité réduite, accroupissement, démarche guindée et titubante, respiration irrégulière, râles humides, diarrhéeTableau 8 note de bas de page 1) |
| TCA, sel | >2000Tableau 8 note de bas de page 3),Tableau 8 note de bas de page 4) | n.d. | |
Notes de bas de page
|
|||
10.2 Exposition de courte durée
10.2.1 Acide monochloroacétique
Au cours d'une étude par gavage d'une durée de 13 semaines, des souris B6C3F1 (20 par sexe et par dose) ont reçu des doses de 0, 25, 50, 100, 150 ou 200 mg/kg p.c. par jour de MCA (sous forme acide dans de l'eau désionisée) cinq jours par semaine. On a soumis cinq souris par dose à des évaluations intermédiaires après quatre et huit semaines de traitement. La mortalité a augmenté uniquement à la dose la plus forte et surtout chez les souris mâles. Le poids corporel moyen final et le gain de poids moyen étaient beaucoup moins élevés chez les femelles qui avaient reçu 200 mg/kg p.c. par jour que chez les sujets témoins. Chez les souris femelles seulement, le poids absolu et relatif du foie a augmenté considérablement à la dose la plus élevée. Les concentrations de cholinestérase ont diminué chez les souris femelles des deux groupes qui avaient reçu les doses les plus élevées. On a constaté à la suite d'un examen histopathologique la présence de vacuolisations cytoplasmiques hépatocellulaires qu'on a associée à un trouble du métabolisme survenant chez les animaux moribonds. Aucun signe de prolifération des peroxysomes dans le foie n'a été relevé. Les auteurs ont fixé la dose sans effet nocif observé (NOAEL) à 100 mg/kg p.c. par jour (Bryant et coll., 1992; NTP, 1992).
Au cours de la même étude, six groupes de rats F344 (20 par sexe et par dose) ont reçu par gavage des doses de 0, 30, 60, 90, 120 ou 150 mg/kg p.c. par jour de MCA (sous forme acide dans de l'eau désionisée) cinq jours par semaine pendant 13 semaines. On a soumis cinq rats par groupe de dose à des évaluations intermédiaires après quatre et huit semaines de traitement. Des effets toxiques ont été observés à toutes les doses. Presque tous les animaux sont morts à une dose de 90 mg/kg p.c. par jour et plus. Le poids relatif du cour des sujets avait diminué considérablement chez les deux sexes à une dose de 60 mg/kg p.c. par jour et chez les femelles seulement à une dose de 30 mg/kg p.c. par jour. On a observé une augmentation liée à la dose de l'incidence et de la gravité de la cardiomyopathie chez les deux sexes à des doses de 60 mg/kg p.c. par jour et plus. Chez les rats mâles, le poids relatif du foie et des reins a augmenté à des doses de 30 et 60 mg/kg p.c. par jour. Chez les rats femelles, le poids relatif du foie a augmenté à une dose de 60 mg/kg p.c. par jour seulement. Au cours du traitement, les concentrations d'azote uréique du sang ont grimpé chez les rats mâles ayant reçu des doses de 90 mg/kg p.c. par jour et plus et chez les rats femelles ayant reçu des doses de 60 mg/kg p.c. par jour et plus. Il n'y avait toutefois aucun signe microscopique de lésions des reins ou du foie. On a constaté d'importantes hausses des concentrations sériques d'alanine aminotransférase (ALT) et d'aspartate aminotransférase (AST) liées à la dose chez les sujets des deux sexes exposés à des doses de 60 mg/kg p.c. par jour et plus. Le nombre de lymphocytes a diminué à des doses de 30 mg/kg p.c. par jour et plus, mais cette diminution était liée au stress. Les auteurs ont fixé la NOAEL à 30 mg/kg p.c. par jour en se basant sur les effets cardiaques chez les deux sexes (Bryant et coll., 1992; NTP, 1992).
10.2.2 Acide dichloroacétique
Dans une étude sur l'eau potable d'une durée de sept semaines, des rats Sprague-Dawley mâles ont reçu des doses de 50 ou 1 100 mg/kg p.c. par jour de DCA (sel de sodium) (Stacpoole et coll., 1990). Les rats qui avaient reçu une dose élevée présentaient une grande faiblesse des membres postérieurs, mais l'examen au microscope optique des nerfs périphériques n'a pas révélé de changements. On a aussi détecté un déficit de la thiamine à la dose élevée, mesuré par l'activité transcétolasique érythrocytaire. On n'a constaté aucun signe clinique ni effet sur l'activité transcétolasique à la dose faible. Les auteurs ont déclaré qu'il y avait un lien entre la toxicité constatée chez les rats au cours de cette étude et des signes typiques de déficits en thiamine; ils ont également considéré que la faiblesse des membres postérieurs et autres manifestations neuropathiques d'un déficit chronique en thiamine chez les animaux étaient attribuables à des changements de la structure et du fonctionnement du système nerveux central plutôt que du système nerveux périphérique.
Au cours d'une étude sur l'alimentation d'une durée de 12 semaines, une faiblesse des membres postérieurs et une démarche anormale ont été observées chez des rats Wistar mâles (n = 6) exposés au DCA (neutralisé). Les doses approximatives ont varié de 4 mmol/kg p.c. par jour (516 mg/kg p.c. par jour) au début de l'étude jusqu'à 2,5 mmol/kg p.c. par jour (323 mg/kg p.c. par jour) à la fin de l'étude (Yount et coll., 1982). On a aussi détecté un ralentissement de la conduction nerveuse dans plusieurs nerfs (saphène et moteur), ainsi qu'une diminution du diamètre des nerfs tibiaux. La diminution du gain de poids à la suite d'une réduction de la consommation d'aliments et la présence d'une hépatomégalie sont au nombre des autres effets toxiques enregistrés.
Au cours d'une étude par gavage oral d'une durée de trois mois, on a administré à des rats Sprague-Dawley (10 par sexe et par dose) des doses de 0, 125, 500 ou 2 000 mg/kg p.c. par jour de DCA (sel de sodium) et on a laissé une période de récupération de quatre semaines à cinq rats par sexe traités aux doses de 0 ou 2 000 mg/kg p.c. par jour (Katz et coll., 1981). Deux rats de chaque sexe ayant reçu la dose élevée sont morts. Il y a eu paralysie des membres postérieurs chez 27 % des rats des deux sexes qui avaient reçu la dose élevée. Chez les sujets du groupe ayant bénéficié de quatre semaines pour se rétablir, un rat par sexe atteint de paralysie a semblé se rétablir complètement. L'examen histopathologique a révélé que le cerveau et les testicules constituaient les principaux organes cibles. On a décelé la présence de lésions cérébrales oedémateuses, caractérisées par la vacuolisation des faisceaux blancs myélinisés dans le cerveau et, à un degré moindre, dans le cervelet. Les taux combinés d'incidence de ces lésions s'élevaient à 60 % chez les sujets qui avaient reçu de faibles doses et à 100 % chez ceux qui avaient reçu des doses moyennes et élevées. Chez les sujets traités à la dose élevée ayant bénéficié de la période de récupération, les lésions cérébrales ont persisté chez trois rats sur huit. Le poids corporel a diminué chez tous les rats traités, et cette diminution a été associée à une baisse de la consommation d'aliments. On a observé une augmentation importante du poids relatif du foie (chez les deux sexes, à toutes les doses), du poids relatif des reins, (chez les femelles, à toutes les doses) et du poids relatif des surrénales (chez les mâles à 500 mg/kg p.c. par jour et plus; chez les femelles à 2 000 mg/kg p.c. par jour). Les tests de chimie clinique ont révélé une légère baisse des paramètres érythroïdes aux doses moyennes et élevées et une diminution de la glycémie et des concentrations de lactate à toutes les doses. Des effets sur les testicules ont aussi été relevés, et seront abordés à la section 10.5.2.
Dans une étude sur l'eau potable d'une durée de 90 jours, on a administré à des rats Sprague-Dawley mâles (n = 10) des doses de 0, 50, 500 ou 5 000 mg/L (0, 4, 35 ou 350 mg/kg p.c. par jour) de DCA (neutralisé) (Mather et coll., 1990). On a observé une baisse du poids corporel et une diminution de la consommation d'eau à des doses de 35 mg/kg p.c. par jour et plus. Le poids relatif du foie et des reins a diminué à des doses de 35 mg/kg p.c. par jour et plus. On a toutefois constaté des signes histologiques et biochimiques de dommages au foie et aux reins, ainsi qu'une augmentation de la bêta-oxydation des peroxysomes hépatiques seulement chez les sujets qui avaient reçu la dose la plus élevée. Des augmentations du poids relatif de la rate ont également été enregistrées à la dose la plus élevée en l'absence d'effets histopathologiques. Aucun effet sur la fonction immunologique n'a été relevé.
Au cours d'une étude sur l'eau potable d'une durée de 13 semaines (NTP, 2000), on a exposé des souris B6C3F1 (10 par sexe et par dose) à des doses de 0, 67, 125, 250, 500 ou 1 000 mg/L (0, 9, 16, 32, 61 et 124 mg/kg p.c. par jour chez les mâles et de 0, 10, 18, 38, 72 et 132 mg/kg p.c. par jour chez les femelles) de DCA (neutralisé à un pH de 5). On a constaté des augmentations liées à la dose du poids du foie et de l'incidence de changements de la vacuolisation cytoplasmique dans les hépatocytes (chez les femelles à 10 mg/kg p.c. par jour et plus; chez les mâles à 32 mg/kg p.c. par jour et plus). Aucun signe clinique ni décès n'ont été observés au cours de l'étude. Dans le groupe ayant reçu la dose élevée, certaines souris mâles présentaient une leucopénie légère, une neutropénie et une monocytopénie qui pourraient être liées à des effets chimiques. Les auteurs ont fixé des NOAEL de <32 mg/kg p.c. pour les mâles et de <10 mg/kg p.c. pour les femelles, les deux étant fondées sur les lésions microscopiques du foie.
Le NTP (2000) a aussi administré à des rats Fischer-344 (10 par sexe et par dose) le même protocole et les mêmes doses que ci-dessus (doses calculées : mâles : 0, 5, 9,3, 18,8, 39,2 et 81,4 mg/kg p.c. par jour; femelles : 0, 5,9, 10,0, 20,9, 43,8 et 94,7 mg/kg p.c. par jour). On a constaté une diminution importante du gain de poids corporel chez les mâles qui avaient reçu la dose élevée au cours des semaines 4 à 13. Aucun autre effet significatif n'a été observé.
Dans le cadre d'une étude sur l'exposition subchronique d'une durée de 13 semaines (Katz et coll., 1981), des chiens beagle âgés de 10 à 12 mois (3 à 4 par sexe et par dose) ont reçu des doses de 0, 50, 75 ou 100 mg/kg p.c. par jour de DCA (sel de sodium) en capsules de gélatine; un chien de plus par sexe a reçu une dose de 0 ou 100 mg/kg p.c. par jour et on lui a accordé une période de récupération de quatre semaines. Chez les chiennes seulement, on a réduit la consommation d'aliments pour toutes les doses; on a toutefois observé des pertes de poids liées à la dose à toutes les doses chez les deux sexes au cours du traitement, et la perte de poids a cessé après l'arrêt du traitement. Une femelle et un mâle sont morts après avoir été exposés à une dose de 75 et 100 mg/kg p.c. par jour, respectivement, et l'on a relevé des signes d'effets indésirables avant leur mort, dont l'anorexie, l'ataxie, la faiblesse des membres postérieurs et une diminution de l'activité. Les autres effets indésirables liés au traitement comprenaient les vomissements (75 et 100 mg/kg p.c. par jour), les selles sanguinolentes (100 mg/kg p.c. par jour) et la paralysie (100 mg/kg p.c. par jour). On a aussi signalé à toutes les doses (chez les deux sexes) une incidence élevée d'anomalies oculaires : opacités lenticulaires bilatérales, conjonctives bulbaires injectées et vascularisation superficielle de la cornée avec tendance à la kératoconjonctivite sèche. Les paramètres hématologiques avaient diminué à toutes les doses chez les sujets des deux sexes. Le traitement au DCA n'a pas eu d'effet sur les paramètres hépatiques et rénaux chez les chiens. On a observé une augmentation de l'incidence de la consolidation pulmonaire à toutes les doses (chez les deux sexes). L'étude histopathologique du cerveau a révélé que les chiens (de tous les groupes traités) présentaient une vacuolisation variant de légère à moyenne des faisceaux myélinisés blancs du cerveau et, à un degré moindre, au niveau du cervelet. On a observé à toutes les doses, même cinq semaines après la fin du traitement, des augmentations du nombre de cellules de Kupffer chargées d'hémosidérine dans le foie et de l'incidence de l'hyperplasie de la muqueuse kystique dans la vésicule biliaire. On a aussi remarqué des changements au niveau de la prostate et des testicules à des doses de 50 mg/kg p.c. par jour et plus (voir la section 10.5.2). Les auteurs ont signalé que les beagles étaient plus susceptibles de développer des cataractes que les autres espèces (Katz et coll., 1981).
Dans une autre étude portant sur l'exposition subchronique de chiens (Cicmanec et coll., 1991), des beagles mâles et femelles âgés de quatre mois (5 par sexe et par dose) ont reçu des doses de 0, 12,5, 39,5 ou 72 mg/kg p.c. par jour de TCA (neutralisé à un pH de 7,4) en capsules de gélatine pendant 90 jours. Les témoins ont reçu de l'eau distillée en capsules. Les animaux exposés à une dose élevée sont morts de déshydratation et de pneumonie. On a observé chez les sujets des deux sexes des signes cliniques évidents comme la dyspnée (doses moyennes et élevées) et la paralysie partielle (dose élevée seulement). Les animaux exposés à la dose moyenne et à la dose élevée ont eu la diarrhée et certains chiens très déshydratés ont eu besoin d'une thérapie liquidienne. L'inflammation des membranes oculaires était accompagnée d'odème; les sécrétions étaient claires aux doses faibles et moyennes et sont devenues purulentes chez les sujets exposés à la dose élevée. Une diminution du gain de poids corporel liée à la dose a été observée chez tous les animaux exposés. Le poids relatif du foie a augmenté chez toutes les femelles, mais le poids des reins et des poumons n'a augmenté que chez les sujets exposés à la dose élevée. Les effets sur ces organes étaient moins uniformes chez les chiens mâles. On a observé des augmentations apparentes de l'ALT, de l'AST et de la lactate déshydrogénase chez les sujets des deux sexes exposés à la dose élevée. Les concentrations d'érythrocytes et d'hémoglobine ont diminué considérablement chez les sujets des deux sexes exposés à la dose élevée à compter du jour 30. L'examen microscopique a révélé des lésions apparentes dans le foie, le cerveau, les poumons, le pancréas et les testicules. Une vacuolisation hépatique a été relevée chez la plupart des chiens traités (des deux sexes), ainsi que chez quelques animaux témoins. Dans le cerveau, on a remarqué une vacuolisation légère des faisceaux myélinisés blancs du cerveau, du cervelet et de la moelle épinière chez tous les groupes exposés. On a également observé une pneumonie et une bronchopneumonie chez la plupart des chiens traités; les effets les plus graves sont surtout apparus chez les sujets exposés aux doses moyennes et élevées. Chez beaucoup d'animaux (des deux sexes) exposés aux doses moyennes et élevées, on a décelé la présence dans le pancréas de cellules acineuses associées à une inflammation chronique. Des effets sur les testicules (toutes les doses) et la prostate (doses moyennes et élevées) ont aussi été constatés et seront examinés dans la section 10.5.2.
10.2.3 Acide trichloroacétique
Dans une étude d'une durée de 90 jours (Mather et coll., 1990), on a administré à des rats Sprague-Dawley mâles (10 par dose) des doses de 0, 50, 500 ou 5 000 mg/L (0, 4,1, 36,5 ou 355 mg/kg p.c. par jour) de TCA (neutralisé) dans leur eau potable. La consommation d'eau a diminué (de façon statistiquement significative) aux doses moyennes et élevées comparativement aux sujets témoins. Une diminution du poids corporel a été observée chez les sujets de tous les groupes, mais elle n'était pas statistiquement significative. On a aussi constaté une augmentation du poids relatif du foie et des reins des sujets exposés à la dose élevée ainsi qu'un accroissement considérable de l'activité des peroxysomes hépatiques.
10.2.4 Acide monobromoacétique
On n'a trouvé dans les publications aucune étude sur l'exposition subchronique au MBA.
10.2.5 Acide dibromoacétique
Afin de déterminer ses effets sur le foie, on a administré à des souris B6C3F1 mâles (cinq par dose) du DBA (neutralisé) à des doses de 0, 300, 1 000 ou 2 000 mg/L dans l'eau potable pendant une période maximale de 12 semaines (Kato-Weinstein et coll., 2001). Tous les animaux ont été sacrifiés à l'âge de 20 semaines. On a constaté à la dose la plus élevée une diminution de la consommation d'eau et du poids corporel, ainsi qu'une augmentation liée à la dose du poids relatif du foie à la dose de 1 000 mg/L et plus après 12 semaines. Le poids absolu et relatif du foie a augmenté à tous les points dans le temps (4, 8 et 12 semaines) à la dose la plus élevée, et la concentration totale de glycogène dans le foie était beaucoup plus grande à la dose la plus élevée après 12 semaines. Une diminution des concentrations de glucose et d'insuline sériques liée à la dose a été enregistrée à 1 000 mg/L et plus après 12 semaines (Kato-Weinstein et coll., 2001). Le DBA a produit des effets semblables à ceux du DCA : les deux ont causé une augmentation de la concentration de glycogène et une chute des concentrations d'insuline sérique, ce qui peut indiquer que les dihaloacétates font intervenir des mécanismes communs.
Les résultats d'une étude sur l'eau potable d'une durée de 13 semaines qui a porté sur des souris B6C3F1 et des rats F344 ont été signalés par Melnick et coll. (2007) et décrits plus en détail par le NTP (2007) : les animaux (10 par dose, par sexe et par espèce) ont été exposés à des doses de 0, 125, 250, 500, 1 000 ou 2 000 mg/L de DBA (neutralisé à un pH de 5) dans l'eau potable (équivalant à 0, 10, 20, 40, 90 et 166 mg/kg p.c. par jour chez les rats mâles et 0, 12, 23, 48, 93 et 181 mg/kg p.c. par jour chez les rats femelles; ainsi qu'à 0, 16, 30, 56, 115 et 230 mg/kg bw per day chez les souris mâles et 0, 17, 34, 67, 132 et 260 mg/kg chez les souris femelles). Aucun signe clinique ni décès n'ont été observés. On a constaté une diminution significative du poids corporel moyen final chez les deux espèces et les deux sexes exposés à la dose la plus forte, mais seulement une légère baisse de la consommation d'eau aux semaines 1 et/ou 13. Le poids du foie a augmenté en fonction de la dose chez les souris des deux sexes exposées à 500 mg/L et plus et chez les rats exposés à 125 mg/L et plus. Des augmentations de la gravité de la vacuolisation cytoplasmique liées à la dose ont aussi été observées dans les hépatocytes des souris (des deux sexes exposées à 1 000 mg/L et plus), bien que ces lésions fussent présentes chez les souris témoins et traitées. On a constaté que l'augmentation de la gravité correspondait à une hausse du poids du foie. Chez les rats, une augmentation de l'incidence de la vacuolisation cytoplasmique dans les hépatocytes a été relevée chez les mâles exposés à 500 mg/L et plus et chez les femelles exposées à 2 000 mg/L. Une prolifération des cellules hématopoïétiques (légère à minime) a été observée dans la rate des rats femelles exposés à la dose élevée. Une augmentation significative de l'incidence de l'hypertrophie cellulaire au niveau de l'hypophyse était visible chez les rats mâles exposés à la dose élevée, mais ces effets étaient considérés comme secondaires à une atrophie testiculaire. Des effets testiculaires ont été observés chez les rats et les souris et sont décrits à la section 10.5.5.
Moser et coll. (2004) et Phillips et coll. (2002) ont étudié le potentiel neurotoxique du DBA. Ils ont administré à des rats F344 mâles et femelles adolescents (12 par sexe par groupe) des doses de 0, 200, 600 et 1 500 mg/L (estimées à 0, 20, 72 et 161 mg/kg p.c. par jour) de DBA (acide) pendant six mois dans leur eau potable. Aucune mort liée au traitement n'a été constatée. Les chercheurs ont observé une diminution du gain de poids chez les rats (des deux sexes) exposés à la dose élevée. Les résultats d'une batterie de tests d'observation fonctionnelle ont révélé une toxicité neuromusculaire liée à la dose chez les sujets exposés aux doses moyennes et élevées, notamment une faiblesse des membres, de légers changements de la démarche et une hypotonie. Les réponses sensorimotrices étaient réduites à tous les niveaux de traitement. On a aussi observé, à la dose la plus élevée, une diminution de l'activité et un resserrement des pattes au niveau de la cage thoracique. L'évaluation neuropathologique a révélé une dégénérescence des fibres nerveuses de la moelle épinière chez les sujets exposés aux doses moyennes et élevées. La gravité et l'incidence de la vacuolisation des neurones de la moelle épinière (principalement dans la substance grise et, à l'occasion, dans la substance blanche) ont augmenté à la dose moyenne et aux doses plus fortes. Les auteurs ont fixé une dose sans effet observé (NOEL) de 20 mg/kg p.c. par jour en se fondant sur les effets neuropathologiques, mais ils n'ont pu en calculer une pour les effets neurocomportementaux en raison de changements sensorimoteurs à ce niveau.
Des signes cliniques d'effets neurotoxiques ont aussi été relevés au cours d'une étude sur la reproduction chez les mâles (Linder et coll., 1995). On a administré à des rats mâles (n = 10) des doses de 250 mg/kg p.c. par jour de DBA pendant une période maximale de 42 jours, et l'on a ensuite cessé de leur administrer la dose à cause d'une toxicité importante. Les signes cliniques observés étaient les suivants : postures anormales, tremblement léger, mouvement atypique des membres et difficulté à bouger les membres postérieurs. On a accordé aux rats une période de récupération de six mois. Certains des effets ont diminué (démarche anormale) ou ont disparu (tremblements). Aucun signe évident de toxicité n'a été constaté aux doses inférieures chez les rats mâles (Linder et coll., 1995).
Le NTP (1999a) a étudié l'immunotoxicité du DBA chez les souris femelles dans quatre études distinctes portant sur différents effets immunotoxiques. Des groupes de souris B6C3F1 femelles (huit par dose) ont été exposés au DBA dans l'eau potable à des concentrations de 0, 125, 250, 500, 1 000 ou 2 000 mg/L par jour (0, 14-20, 33-39, 68-73, 132-150 ou 236-285 mg/kg p.c. par jour) pendant 28 jours. Aucun signe de toxicité évidente n'est apparu et la consommation d'eau n'a pas changé. On a noté une diminution de 40 % du gain de poids corporel comparativement aux témoins uniquement chez les femelles exposées à la dose élevée. Au cours d'une expérience, le poids de la rate a augmenté, mais pas dans une seconde expérience portant sur des doses semblables. Plusieurs indicateurs de réponse immunologique ont été touchés. On a constaté une augmentation statistiquement significative et liée à la dose du nombre de macrophages dans la rate à 500 mg/L et plus, ce qui indique une réponse immunotoxique dans cet organe. L'immunité humorale a aussi été touchée, comme l'indique une diminution de la réponse des cellules productrices d'immunoglobulines M à des érythrocytes de mouton dans la rate des souris exposées à des doses de 500 mg/L et plus de DBA. L'U.S. EPA (2005a) a établi une dose minimale entraînant un effet nocif observé (LOAEL) de 500 mg/L (68-73 mg/kg p.c. par jour) et une NOAEL de 250 mg/L (33-39 mg/kg p.c. par jour) pour l'immunotoxicité.
McCay et coll. (2000) ont signalé dans un abrégé n'avoir constaté aucun effet sur le système immunitaire au cours d'une étude d'une durée de 28 jours pendant laquelle des souris B6C3F1 femelles ont reçu des doses quotidiennes de 0, 250, 500 ou 1 000 mg/L de DBA dans l'eau potable. Les changements du poids relatif ou absolu du foie et du thymus étaient les seuls effets observés.
10.3 Exposition de longue durée et cancérogénicité
Des études de longue durée sur le MCA n'ont pas produit de tumeurs chez les rongeurs. Par contre, le DCA, le TCA et le DBA ont causé chez des animaux de laboratoire des tumeurs au foie. Toutefois, après une exposition au TCA et au DBA, seules les souris présentaient des tumeurs au foie, et non les rats, alors qu'après une exposition au DCA, les rats aussi bien que les souris étaient atteints. Cependant, chez les rats exposés au DBA, on a détecté des tumeurs dans d'autres organes. Aucune étude de longue durée adéquate sur la cancérogénicité du MBA ou du DBA n'a été retracée. Les résultats de ces études de cancérogénicité relatives aux tumeurs du foie sont résumés dans le tableau 9 et décrits en détail dans les sections qui suivent.
| AHA | Voie d'adminis-tration | Doses (mg/ kg p.c. ou sel/ foie par jour) | Durée | Acide ou sel | Lignée Sexe/ espèce | Sexe | Tumeur du foie décou- verte |
Auteur de l'étude |
|---|---|---|---|---|---|---|---|---|
| MCA | G | 0, 15, 30 | 2 ans | A | rats F344 | M et F | Aucune | NTP, 1992 |
| G | 0, 50, 100 | 2 ans | A | souris B6C3F1 | M et F | Aucune | NTP, 1992 | |
| EP | 0, 3,5, 26, 59,9 | 2 ans | S | rats F344 | M | Aucune | DeAngelo et coll., 1997 | |
| DCA | EP | 0, 140, 280 | 52 semaines | S | souris B6C3F1 | M | AH, CH | Bull et coll., 1990 |
| EP | 0, 7,6, 77, 410, 486 | 60-75 semaines | ?Tableau 9 note de bas de page b | souris B6C3F1 | M | AH, CH | DeAngelo et coll., 1991; U.S. EPA, 1991 | |
| EP | 0, 93 | 104 semaines | S | souris B6C3F1 | M | AH, CH | Daniel et coll., 1992 | |
| EP | 0, 8, 84, 168, 315, 429 | 90-100 semaines | S | souris B6C3F1 | M | AH, CH | DeAngelo et coll., 1999 | |
| EP | 0, 40, 120, 330 | 360 or 576 jours | S | souris B6C3F1 | F | AH, CH | Pereira, 1996 | |
| EP | 0, 3,6, 40,2, 139,1 | 100 semaines | S | rats F344 | M | AH, CH | DeAngelo et coll., 1996 | |
| TCA | EP | 0, 178, 319 | Up to 52 semaines | S | souris B6C3F1 | M | AH, CH | Bull et coll., 1990 |
| EP | 0, 7, 71, 595 | 60 semaines | possible S | souris B6C3F1 | M | AH, CH | DeAngelo et Daniel, 1990; U.S. EPA, 1991 | |
| EP | 0, 64, 212, 640 | 360 ou 576 jours | S | souris B6C3F1 | F | AH, CH | Pereira, 1995, 1996; Pereira et Phelps, 1996 | |
| EP | 0, 71, 583 | 104 semaines | ?Tableau 9 note de bas de page b | souris B6C3F1 | F | AH, CH | U.S. EPA, 1991 | |
| EP | 0, 3.6, 32.5, 364 | 104 semaines | S | rats F344 | M | Aucune | DeAngelo et Daniel, 1992; DeAngelo et coll., 1997 | |
| DBA | EP | 0, 4, 35, 65 | 104 semaines | S | souris B6C3F1 | F | AH, CH | Melnick et coll. 2007; NTP, 2007 |
| EP | 0, 4, 45, 87 | 104 semaines | S | souris B6C3F1 | M | AH, CH | Melnick et coll. 2007; NTP, 2007 | |
| EP | 0, 2, 25 45 | 104 semaines | S | rats F344 | F | AucuneTableau 9 note de bas de page b | Melnick et coll. 2007; NTP, 2007 | |
| EP | 0, 2, 20, 40 | 104 semaines | S | rats F344 | M | AucuneTableau 9 note de bas de page b | Melnick et coll. 2007; NTP, 2007 | |
Notes de bas de page
|
||||||||
10.3.1 Acide monochloroacétique
On n'a observé aucune tumeur chez des souris de deux lignées différentes (18 par sexe par lignée) gavées au MCA (acide) dans de l'eau distillée à une dose de 46 mg/kg p.c. par jour dès l'âge de sept jours et jusqu'à quatre semaines. On a ensuite mélangé le MCA directement dans l'alimentation, à une dose de 149 mg/kg p.c. pendant 18 mois au total (Innes et coll., 1969).
Au cours d'un essai biologique sur la cancérogénicité d'une durée de deux ans, on a administré par gavage à des rats F344/N (70 par sexe et par dose), dans de l'eau désionisée, des doses de 0, 15 ou 30 mg/kg p.c. de MCA (acide) par jour, cinq jours par semaine (NTP, 1992). Le poids corporel moyen des rats mâles exposés à la dose élevée avait diminué après 30 semaines. Les taux de survie ont chuté considérablement chez les mâles exposés à la dose élevée et chez les femelles exposées à la dose faible et à la dose élevée, mais ils n'étaient pas liés au traitement. Dans l'ensemble, on n'a constaté aucune augmentation des lésions non néoplasiques ni des néoplasies signalées liée au traitement. Les auteurs ont conclu qu'il n'y avait « aucun signe d'activité cancérogène »
dans le cas du MCA chez les rats F344/N mâles et femelles exposés aux doses utilisées au cours de l'étude.
Au cours du même essai biologique d'une durée de deux ans, on a administré par gavage à des souris B6C3F1 (60 par sexe et par dose), dans de l'eau désionisée, des doses de 0, 50 ou 100 mg/kg p.c. par jour de MCA (acide) cinq jours par semaine (NTP, 1992). On a constaté une diminution importante du poids corporel moyen chez les femelles exposées à la dose élevée (après 52 semaines) et des taux de survie chez les mâles exposés à la dose élevée. La gravité des cas d'inflammation nasale aiguë variait de légère à minime, mais ceux-ci n'étaient pas liés au traitement. L'incidence de la métaplasie de l'épithélium olfactif était beaucoup plus élevée chez les femelles exposées à la dose élevée. On n'a observé aucune autre augmentation significative des lésions non néoplasiques chez les sujets de l'un ou l'autre sexe. Aucune augmentation des néoplasies non liée au traitement n'a été signalée. Les auteurs ont déclaré qu'il « n'y avait aucun signe d'activité cancérogène »
du MCA chez les souris B6C3F1 (des deux sexes) exposées aux doses utilisées au cours de l'étude.
Dans une étude sur l'eau potable d'une durée de 104 semaines, on a exposé des rats F344/N (50 par dose) à des doses de 0, 50, 500 ou 1 100 mg/LFootnote 1 (0, 3,5, 26 ou 59,9 mg/kg p.c. par jour) de MCA (neutralisé) (DeAngelo et coll., 1997). Le traitement au MCA n'a eu aucun effet significatif sur la survie, mais la consommation d'eau potable et le poids corporel moyen final ont diminué considérablement chez les sujets des groupes exposés aux deux doses les plus fortes. Le poids absolu et relatif de la rate a augmenté considérablement (74 % et 80 % par rapport aux témoins respectivement) chez les rats qui avaient consommé 3,5 mg de MCA/kg p.c. par jour, mais cette augmentation ne s'accompagnait pas de lésions macroscopiques ou microscopiques. Même si l'on a observé des diminutions du poids absolu et relatif de la rate aux deux doses les plus fortes, seul le poids absolu était statistiquement plus faible à la dose la plus élevée (59,9 mg/kg p.c. par jour). L'exposition à une dose de 26 mg/kg p.c.par jour a entraîné une baisse significative du poids absolu du foie et des reins et du poids relatif du foie, ainsi qu'unehausse du poids relatif des testicules. À la dose de 59,9 mg/kg p.c. par jour, le poids absolu des reins et le poids absolu et relatif du foie ont diminué, tandis que le poids relatif des testicules a augmenté. L'analyse des enzymes sériques a révélé que le traitement n'avait aucun effet sur l'AST ou l'ALT et que le traitement au MCA n'avait pas favorisé la prolifération des peroxysomes ni celle des hépatocytes. On n'a observé aucune pathologie hépatique provoquée par le MCA au cours de l'étude, et l'on n'a pas trouvé non plus de lésions importantes dans les tissus non hépatiques. La plupart des changements spontanés les plus courants liés à l'âge qu'on a décelés dans des tissus de rats se situaient à l'intérieur des plages pour les témoins historiques, sauf dans le cas de la dégénérescence du myocarde et de l'inflammation chronique/active des cavités nasales, dont l'incidence a augmenté après 104 semaines, mais pas au cours de périodes antérieures de sacrifice, ni chez les sujets exposés à la dose la plus faible. Il n'y a pas eu d'augmentations importantes de la prévalence ni de la multiplicité des adénomes hépatocellulaires, des carcinomes ou des nodules hyperplasiques chez aucun des animaux exposés au MCA comparativement aux animaux témoins. De même, les changements néoplasiques des tissus non hépatiques étaient conformes à ceux signalés chez les témoins historiques. Les auteurs ont conclu que la diminution du poids corporel et les autres phénomènes pathologiques observés chez les sujets des groupes exposés à la dose moyenne n'étaient pas significatifs et ont fixé à 500 mg/L (26 mg/kg p.c. par jour) la NOEL pour la cancérogénicité du MCA (DeAngelo et Daniel, 1992; DeAngelo et coll., 1997).
Santé Canada n'était toutefois pas d'accord avec la décision de ne pas tenir compte de changements statistiquement significatifs du poids du corps, du foie, des reins et des testicules,étant donné notamment que ces changements sont conformes à une tendance liée à la dose. À la dose inférieure suivante de 50 mg/L (3,5 mg/kg p.c. par jour), le seul changement significatif était une augmentation du poids absolu et relatif de la rate. On peut considérer que ce changement est idiosyncrasique (particularité physiologique) puisqu'il n'y a pas de tendance àune augmentation du poids de la rate qui est liée à la dose. À la dose suivante, soit 500 mg/L (26 mg/kg p.c. par jour), le poids absolu et le poids relatif de la rate étaient inférieurs à ceux des sujets témoins, tandis que le poids absolu était beaucoup plus faible à la dose la plus élevée. Compte tenu de ces facteurs, Santé Canada a calculé une NOAEL de 3,5 mg/kg p.c. par jour.
10.3.2 Acide dichloroacétique
Plusieurs études sur la cancérogénicité possible du DCA ont été effectuées et sont répertoriées dans le tableau 9. Des incidences accrues de tumeurs du foie (adénomes et carcinomes) ou de lésions prénéoplasiques (p. ex. nodules hyperplasiques et foyers hépatiques altérés) ont été signalées à la suite de l'exposition de souris et de rats à de l'eau potable contenant des concentrations de 0,5-5 g/L de DCA pendant des périodes variant de 52 à 104 semaines. Les détails des études suivent.
Au cours d'une étude sur l'eau potable d'une durée de 52 semaines (Bull et coll., 1990), on a exposé des groupes de souris B6C3F1 mâles (n = 61, 11, 50) à des doses de 0, 1 000 ou 2 000 mg/L (0, 140 ou 280 mg/kg p.c. par jour; OMS, 2005) de DCA (neutralisé), et un groupe de 10 femelles à 2 000 mg de DCA/L pendant une période maximale de 52 semaines. Entre temps, cinq souris mâles du groupe exposé à 2 000 mg/L ont été sacrifiées après 15, 24 et 37 semaines. Tous les autres animaux ont été sacrifiés à la 52e semaine, y compris 11 mâles que l'on avait cessé d'exposer après 37 semaines et qui avaient eu 15 semaines pour se rétablir avant leur sacrifice. Une augmentation importante, liée à la dose, du poids absolu et relatif du foie a été enregistrée après 37 ou 52 semaines chez les mâles traités. Tous les animaux traités au DCA avaient le foie hypertrophié (hépatomégalie) et l'on a constaté une distribution uniforme d'une cytomégalie marquée, ainsi qu'une accumulation importante de glycogène dans les hépatocytes, de même que de multiples foyers de nécrose avec infiltration de lymphocytes dans tout le foie. Des foyers basophiles d'altération cellulaire pauvres en glycogène ont été observés après 24 et 37 semaines dans le lobe central du foie des mâles exposés à 2 000 mg de DCA/L. Après 52 semaines de traitement, des nodules hyperplasiques, des adénomes et des carcinomes étaient visibles dans le foie des mâles exposés à 2 000 mg/L. Par contre, les seules lésions hépatoprolifératives observées chez les sujets des autres groupes étaient un nodule hyperplasique chez une souris mâle du groupe témoin et une autre souris mâle du groupe exposé à 1 000 mg/L, et des nodules hyperplasiques chez trois femelles. Aucun carcinome hépatocellulaire n'a été signalé chez les mâles qui avaient cessé d'être exposés au DCA après 37 semaines. Après 52 semaines, deux animaux avaient des adénomes et six, des nodules hyperplasiques. Le lien entre la dose totale moyenne de DCA et le nombre total de lésions hépatiques (nodules + adénomes + carcinomes) par souris était non linéaire et le nombre de lésions grimpait en flèche lorsque la dose passait de 1 000 à 2 000 mg/L (Bull et coll., 1990).
On a administré à des souris B6C3F1 mâles (30 par dose) 50, 500 ou 5 000 mg/L de DCA dans l'eau potable pendant 60 et 75 semaines (DeAngelo et coll., 1991; U.S. EPA, 1991). Les sujets du groupe témoin ont reçu 2 000 mg de chlorure de sodium/L. Dans une étude parallèle, on a exposé des souris à 3 500 mg de DCA/L et on les a sacrifiées après 60 semaines. Au cours de cette expérience, les sujets du groupe témoin ont reçu de l'acide acétique. On a calculé des doses quotidiennes moyennes pondérées en fonction du temps de 7,6, 77, 410 et 486 mg/kg par jour pour les concentrations de 50, 500, 3 500 et 5 000 mg/L respectivement. Le poids corporel final des souris exposées à 3 500 et 5 000 mg/L a diminué comparativement à celui des sujets du groupe témoin : 87 % et 83 % de la valeur témoin respectivement. Le poids relatif du foie a augmenté aux trois doses les plus élevées comparativement à celui des sujets témoins : 118 %, 230 % et 351 % pour les concentrations de 500, 3 500 et 5 000 mg/L respectivement.
On a observé l'apparition de nodules hépatiques hyperplasiques (lésion nodulaire non néoplasique) principalement aux deux doses les plus élevées : incidence de 58 % et 83 % à 3 500 et 5 000 mg/L respectivement. Les souris qui avaient reçu 5 000 mg/L (après 60 semaines) présentaient une prévalence de néoplasie du foie (carcinomes et adénomes) de 90 % et une multiplicité moyenne de 4,50 tumeurs par animal. Les sujets qui avaient reçu 3 500 mg/L présentaient une prévalence de 100 % et une multiplicité de 4,0 tumeurs par animal. La prévalence et la multiplicité des tumeurs chez les sujets exposés aux deux doses les plus faibles au bout de 75 semaines ne différaient pas de façon significative par rapport à la valeur témoin. Les sujets du groupe témoin n'avaient pas de tumeurs du foie. Les auteurs ont conclu qu'il existait une tendance positive significative liée à la dose en ce qui concerne la prévalence des tumeurs du foie corrigée en fonction de l'âge. Ils ont également conclu que la valeur-seuil du DCA était d'au moins 500 mg/L (77 mg/kg p.c. par jour) pour la réponse tumorale chez les souris. L'incidence des tumeurs a grimpé en flèche à une concentration de 3 500 mg/L (410 mg/kg p.c. par jour), ce qui représente aussi l'incidence maximale atteinte (DeAngelo et coll., 1991; U.S. EPA, 1991).
Au cours d'une étude sur l'eau potable d'une durée de 104 semaines (Daniel et coll., 1992), on a administré à un groupe de 33 souris B6C3F1 mâles 500 mg/L (93 mg/kg p.c. par jour) de DCA (neutralisé) et procédé à un sacrifice en cours d'étude (n = 5) après 30 semaines. On n'a pas constaté d'effets significatifs liés au traitement sur la consommation d'eau potable, le gain relatif de poids corporel ou le poids relatif ou absolu de la rate, des reins ou des testicules. Les poids absolu et relatif du foie avaient augmenté après 30 et 104 semaines. La cytomégalie, la nécrose et l'inflammation active chronique ont constitué les effets hépatiques non néoplasiques les plus notables liés au traitement. Après 104 semaines, l'incidence des carcinomes et des adénomes hépatocellulaires avait augmenté de façon significative. Les incidences combinées de carcinome + adénome et de carcinome + adénome + nodule avaient progressé considérablement chez les animaux traités comparativement aux animaux témoins. Les souris sacrifiées à la fin de l'étude étaient porteuses de carcinomes hépatocellulaires (15/24, comparativement à 2/20 chez les témoins), d'adénomes hépatocellulaires (10/24, comparativement à 1/20 chez les témoins) et de carcinomes ou d'adénomes (18/24, comparativement à 3/20 chez les témoins). Parmi les autres effets observés, citons les nodules hyperplasiques chez 2 souris traitées sur 24, la nécrose hépatocellulaire chez 8 souris traitées sur 24 (par rapport à 1 sur 20 chez les témoins) et une cytomégalie chez 22 souris traitées sur 24 (par rapport à 1 sur 20 chez les témoins) (Daniel et coll., 1992). (On n'a pas fourni de données sur les nécropsies intermédiaires effectuées à la semaine 30.)
Au cours d'une étude sur l'eau potable d'une durée de 90 à 100 semaines, on a exposé des souris B6C3F1 mâles (n = 35-71) à 50, 500, 1 000, 2 000 ou 3500 mg/L (8, 84, 168, 315 et 429 mg/kg p.c. par jour) de DCA (neutralisé) (DeAngelo et coll., 1999). Le groupe témoin comportait 88 souris. On a procédé à des sacrifices intermédiaires pendant toute l'étude, sauf chez les sujets exposés à la dose la plus faible. Lors du sacrifice final, les poids corporels avaient diminué aux deux doses les plus élevées. On a constaté une augmentation du poids du foie liée à la dose chez les sujets ayant reçu 84 mg/kg p.c. par jour et plus après 26 et 52 semaines, ainsi que chez ceux ayant reçu 315 mg/kg p.c. par jour et plus après 100 semaines. Une hépatotoxicité a été démontrée à une dose de 168 mg/kg p.c. par jour et plus, comme l'indiquent l'augmentation des enzymes hépatiques dans le sérum et l'histopathologie. La mortalité aux deux doses les plus élevées était statistiquement significative comparativement au groupe témoin. L'incidence du carcinome hépatocellulaire chez les souris mâles a augmenté considérablement. Après 26 semaines, on n'a détecté aucune tumeur dans le foie d'aucune des souris, mais après 52 et 78 semaines, l'incidence du carcinome hépatocellulaire a augmenté de façon significative (P < 0,05) chez les sujets exposés à la dose la plus élevée (tableau 10). Au moment du sacrifice final, l'incidence du carcinome hépatocellulaire avait augmenté de façon significative (P < 0,05) chez les sujets exposés aux trois doses les plus élevées. On a observé pour la première fois après 26 semaines l'apparition d'adénomes hépatocellulaires chez les sujets exposés à la dose élevée; toutefois, l'incidence des adénomes hépatocellulaires n'a augmenté de façon significative (P < 0,05) (sans dose-réponse) chez les sujets exposés aux trois doses les plus fortes (tableau 10) après 100 semaines.
| Dose (mg/ kg p.c. par jour) | Prévalence des carcinomes hépatocellulaires chez les souris mâles (%) | Prévalence des adénomes hépatocellulaires chez les souris mâles (%) | ||||
|---|---|---|---|---|---|---|
| Semaine 52 | Semaine 78 | Semaine 100 | Semaine 52 | Semaine 78 | Semaine 100 | |
| 0 (témoin eau) | 0 | 10 | 26 | 0 | 10 | 10 |
| 8 | n.d.Tableau 10 note de bas de page a | n.d. | 33 | n.d. | n.d. | n.d. |
| 84 | 0 | 0 | 48 | 10 | 10 | 20 |
| 168 | 0 | 20 | 71Tableau 10 note de bas de page b | 10 | 20 | 51,4Tableau 10 note de bas de page b |
| 315 | 20 | 50 | 95Tableau 10 note de bas de page b | 0 | 50 | 42,9Tableau 10 note de bas de page b |
| 429 | 50 | 70Tableau 10 note de bas de page b | 100Tableau 10 note de bas de page b | 50 | 50 | 45Tableau 10 note de bas de page b |
Notes de bas de page
|
||||||
La prolifération des peroxysomes hépatiques a augmenté considérablement uniquement chez les sujets exposés à la dose élevée après 26 semaines, mais pas après 52 semaines. Par ailleurs, la prolifération des hépatocytes ne présentait pas de différence significative par rapport aux taux chez les témoins à n'importe laquelle des doses qui ont produit des tumeurs. Les auteurs n'ont pu déterminer de NOEL pour l'hépatocancérogénicité en se fondant sur l'augmentation significative de la multiplicité des carcinomes hépatocellulaires à la dose la plus faible (0,58 comparativement à 0,28 chez les témoins), mais ils ont conclu que le DCA produisait une augmentation, liée à la dose, de l'incidence des carcinomes hépatocellulaires qui n'avait aucun lien avec la prolifération des peroxysomes hépatiques ou des hépatocytes. Ils ont avancé que la perturbation du système endocrinien et la nécrose des hépatocytes jouaient un rôle important dans la cancérogenèse (DeAngelo et coll., 1999).
On a administré à des groupes de souris femelles B6C3F1 âgées de sept à huit semaines (n = 40-90 et n = 134 pour les groupes témoins) du DCA (neutralisé) dans l'eau potable, pendant 360 ou 576 jours, à des concentrations de 0, 2,0, 6,67 ou 20,0 mmol/L (0, 40, 120 ou 330 mg/kg p.c. par jour) (Pereira, 1996; PISC, 2000). Un autre groupe d'animaux (n = 50) a été exposé de façon intermittente à 20,0 mmol de DCA/L dans l'eau potable pendant un cycle de 72 jours, soit 24 jours d'exposition et 48 jours sans exposition. On a répété le cycle de 72 jours jusqu'à ce que les souris soient sacrifiées. La dose totale reçue par les souris était donc la même que celle qu'avaient reçue les sujets exposés sans interruption à 6,67 mmol de DCA/L. Les poids corporels avaient diminué chez les sujets exposés à la dose élevée après 35 semaines. On a constaté une augmentation, liée à la dose, du poids relatif du foie et des hépatocytes vacuolisés. Chez les sujets des groupes exposés pendant 576 jours, on a observé une augmentation importante des foyers hépatiques, des adénomes hépatocellulaires et des carcinomes hépatocellulaires à la dose élevée, ainsi qu'une incidence accrue des foyers hépatiques et des adénomes (mais non des carcinomes) à la dose moyenne, comparativement aux sujets témoins. Les sujets exposés par intermittence au DCA présentaient aussi une incidence significativement accrue d'altération des foyers hépatiques après 576 jours, mais aucune augmentation importante de la réponse néoplasique (Pereira, 1996).
Au cours d'un essai biologique modifié sur la cancérogenèse (DeAngelo et coll., 1996), des rats F344 mâles âgés de 28 jours (n = 50-78) ont été exposés à des concentrations de 0, 50, 500 ou 1 600Footnote 2 mg/L de DCA (neutralisé) (doses quotidiennes moyennes estimatives pondérées en fonction du temps : 0, 3,6, 40,2 et 139,1 mg/kg p.c.) dans l'eau potable pendant 100 semaines. Il n'y avait pas de différences significatives au niveau de la consommation d'eau ou de la survie chez aucun des groupes traités comparativement aux groupes témoins. Le poids corporel et les poids relatifs du foie et des reins à la fin de l'essai ont diminué seulement chez les sujets exposés à 1 600 mg/L. Les effets sur les testicules sont décrits à la section 10.5.2. La vacuolisation du cytoplasme hépatocellulaire, attribuée à des augmentations du dépôt de glycogène causées par le DCA, a constitué le seul changement hépatique non néoplasique lié au traitement. La prévalence combinée d'adénomes hépatocellulaires et de carcinomes hépatocellulaires a augmenté de façon significative (P < 0,05) chez les sujets exposés à 500 mg/L comparativement aux témoins, tout comme la prévalence totale de lésions hépatoprolifératives (nodules hyperplasiques, adénomes et carcinomes). On a observé des tendances significatives liées à la dose, pour ce qui est de la prévalence de l'adénome et du carcinome hépatocellulaires (P < 0,05 dans chaque cas), de la prévalence de l'adénome et du carcinome hépatocellulaires combinés et des lésions hépatoprolifératives totales. Les sujets exposés à 1 600 mg/L présentaient des taux accrus de carcinome hépatocellulaire, de carcinome et d'adénome hépatocellulaires combinés, et de lésionshépatoprolifératives totales. À la dose élevée, le DCA a provoqué une prolifération des peroxysomes des hépatocytes. Le traitement au DCA a réduit la prolifération d'hépatocytes après 14 semaines. Aux autres intervalles, la prolifération est demeurée réduite, mais ne différait pas de façon significative par rapport au groupe témoin. Les auteurs ont conclu que le DCA était un agent hépatocancérogène chez les rats F344 mâles, et que ceux-ci étaient plus sensibles à l'exposition au DCA que les souris B6C3F1 mâles, si l'on se fonde sur une étude antérieure réalisée par DeAngelo et coll. (1991). Les auteurs ont fixé la NOEL à 3,6 mg/kg p.c. par jour (DeAngelo et coll., 1996).
Plusieurs études ont révélé que le DCA pouvait agir comme promoteur de la cancérogenèse dans le foie. Selon Pereira (1995), [TRADUCTION] « on entend par promotion le fait de favoriser l'évolution des cellules initiées en lésions et tumeurs précancéreuses »
.
On a procédé à une étude sur la promotion des tumeurs par le DCA (neutralisé) chez des souris B6C3F1 mâles (Herren-Freund et Pereira, 1986; Herren-Freund et coll., 1987). Les incidences des tumeurs (adénomes et carcinomes hépatocellulaires) étaient considérablement élevées chez les souris initiées ou non, comparativement aux sujets témoins. Les auteurs ont conclu que le DCA était un hépatocancérogène complet chez les souris B6C3F1 (Herren-Freund et coll., 1987).
Le potentiel de promotion de tumeurs du DCA (neutralisé) a été étudié chez des souris B6C3F1 femelles (Pereira, 1995; Pereira et Phelps, 1996). On a constaté une incidence accrue d'adénomes et de foyers hépatiques chez les souris initiées exposées à la dose la plus élevée de DCA pendant 31 ou 52 semaines comparativement aux animaux correspondants initiés ou exposés au DCA seulement. On a observé un lien de deuxième ordre (c.-à-d. un rapport exponentiel) entre la concentration de DCA et le nombre total de lésions produites (foyers + adénomes + carcinomes) après 31 et 52 semaines. Lorsqu'on a mis fin au traitement, les foyers d'hépatocytes altérés et d'adénomes ont régressé.
Deux études distinctes sur l'initiation et la promotion réalisées par Bull et coll. (2004) et Pereira et coll. (1997) ont porté sur l'interaction entre le DCA et le TCA (deux promoteurs tumoraux faisant intervenir des mécanismes différents) chez la souris B6C3F1. Les différences dans les résultats des deux études peuvent être attribuables aux doses utilisées.
Bull et coll. (2004) ont démontré que les interactions entre le TCA et le DCA étaient limitées par l'additivité : les doses efficaces les plus faibles de DCA et de TCA sont additives, tandis que leurs effets ont tendance à être inhibiteurs (suppression du taux de croissance global) à mesure que les doses augmentent. Le DCA et le TCA ont favorisé de façon sélective la multiplication de différents types de cellules initiées en stimulant l'expansion clonale dans un type de cellules tout en l'inhibant dans un autre type. Un tel phénomène expliquerait la présence d'un phénotype mixte de tumeurs, ainsi que la suppression du taux global de croissance tumorale dans l'étude (Bull et coll., 2004).
Pereira et coll. (1997) ont observé par contre des effets synergiques provoqués par un mélange de TCA et de DCA à fortes doses. Les foyers d'hépatocytes altérés et le nombre total de lésions produites étaient plus importants que la somme des effets produits par les deux AHA administrés seuls; mais à des doses plus faibles, les mélanges avaient des effets additifs ou inhibiteurs. Les mélanges de DCA et de TCA ont produit plus de foyers d'hépatocytes altérés que d'adénomes, ce qui ressemble aux effets relevés pour le DCA administré seul. Le phénotype de ces lésions ressemblait en outre aux phénotypes des tumeurs provoquées par le DCA, ce qui indique que ce dernier peut être prédominant dans la détermination des caractéristiques des lésions prolifératives.
10.3.2.1 Mécanismes de la cancérogénicité
Compte tenu des résultats des études décrits ci-dessus et résumés au tableau 9, il appert que le DCA est un agent hépatocancérogène chez deux espèces de rongeurs, les souris et les rats. On croit que le DCA est un « cancérogène complet »
puisqu'il a provoqué l'apparition de tumeurs à la fois à faible dose au cours d'essais de longue durée et à forte dose dans le cadre d'essais de plus courte durée lorsqu'il est administré seul. Les doses utilisées au cours de ces études ont varié de 50 à 5 000 mg/L. Plus d'un mode d'action peut expliquer la cancérogénicité induite par le DCA et l'on a avancé à cet égard plusieurs hypothèses, décrites ci-dessous. Selon toute probabilité, de nombreux événements contribueraient à l'apparition de tumeurs chez les rongeurs dans les conditions d'un essai biologique. L'incertitude règne toutefois quant aux événements qui peuvent jouer un rôle dans l'exposition humaine au DCA à des concentrations présentes dans l'environnement.
10.3.2.1.1 Génotoxicité
Les avis sont partagés pour ce qui est de la cancérogénicité induite par le DCA par l'intermédiaire d'un mécanisme génotoxique. Des études antérieures du CIRC (1995) et de l'ILSI (1997) ont indiqué que le DCA n'était pas génotoxique, tandis qu'une étude plus récente du PISC (2000) a signalé que le DCA pouvait produire des effets génotoxiques, mais seulement à des concentrations élevées, et a conclu qu'à faible dose, l'effet du DCA médié par un mécanisme génotoxique était nul ou minime. Selon le National Center for Environmental Assessment (U.S. EPA, 2003c), des études plus récentes indiquent que le DCA est un agent génotoxique à action directe. Le CIRC (2004) a signalé récemment que le DCA était génotoxique (in vivo et in vitro), mais qu'il pouvait agir indirectement par un mécanisme épigénétique. En raison du manque de données sur les liens de cause à effet, l'U.S. EPA (2003c) a adopté une position prudente, à savoir que le DCA pourrait être génotoxique à fortes doses, tandis que l'incertitude persiste quant aux doses plus faibles.
10.3.2.1.2 Prolifération des peroxysomes
On a démontré que certains agents hépatocancérogènes augmentaient le nombre ou la taille des peroxysomes hépatiques (prolifération des peroxysomes) chez les rongeurs (U.S. EPA, 2003c). Les récepteurs activés de la prolifération des peroxysomes (PPAR) sont une catégorie de récepteurs nucléaires qui régulent cette prolifération et que l'on croit à l'origine de l'initiation de certains événements cellulaires aboutissant à une transformation (ainsi qu'à d'autres effets signalés) en agents hépatocancérogènes. Même si l'on a établi un lien entre la prolifération des peroxysomes et la cancérogenèse, le mécanisme réel de la cancérogenèse par rapport à la prolifération des peroxysomes demeure inconnu (Bull, 2000). Il existe des différences entre espèces sur le plan de l'expression de divers PPAR : les êtres humains semblent en effet réagir moins que les rongeurs à tout un éventail de proliférateurs des peroxysomes. C'est pourquoi la question de la cancérogénicité de ces composés pour les êtres humains est controversée (Bull, 2000; U.S. EPA, 2003c).
Des études réalisées par DeAngelo et coll. (1989, 1999), Daniel et coll. (1992), Mather et coll. (1990) et Pereira (1995, 1996) ont montré que le DCA était un faible proliférateur de peroxysomes chez les rongeurs.
Il faut apparemment des doses beaucoup plus faibles de DCA pour provoquer l'apparition de tumeurs hépatiques que pour causer une augmentation importante de la prolifération des peroxysomes (U.S. EPA, 2003c). En conclusion, l'U.S. EPA, (2003c), Thai et coll. (2003) et Bull (2000) ne considèrent pas ce mécanisme comme une voie importante de tumorigenèse pour le DCA.
10.3.2.1.3 Régulation négative de l'insuline
Le contenu en glycogène des hépatocytes de souris traitées au DCA peut varier en fonction de l'état de la cellule. Les tumeurs et foyers hépatiques altérés sont pauvres en glycogène, tandis que les hépatocytes normaux accumulent d'importantes quantités de glycogène (Lingohr et coll., 2001); il pourrait ainsi y avoir un lien entre le glycogène et les tumeurs hépatiques. Les concentrations de glycogène sont contrôlées principalement par l'insuline via le métabolisme dans le foie (Lingohr et coll., 2001). L'insuline peut avoir d'autres rôles, comme celui de déclencher la mitose (mitogène) des hépatocytes normaux et malins et de supprimer l'apoptose cellulaire (Lingohr et coll., 2001).
Lingohr et coll. (2001) ont étudié les effets du DCA sur les concentrations d'insuline et l'expression des protéines de signalisation contrôlées par l'insuline dans le tissu hépatique normal et dans le tissu de tumeurs du foie provoquées par le DCA chez des souris B6C3F1 mâles auxquelles on a administré de 100 à 2 000 mg de DCA (neutralisé)/L dans l'eau potable pendant deux à 10 semaines. On a constaté des diminutions des protéines réceptrices d'insuline, des concentrations d'insuline sérique et de l'expression de la protéine kinase B après deux semaines chez les souris traitées au DCA. Par contre, le glycogène avait augmenté (avant ces effets) dès la semaine 1, ce qui indique que les altérations de l'insuline, des récepteurs de l'insuline et, peutêtre, de la protéine kinase B provoquées par le DCA résultaient d'une régulation négative compensatoire de la voie de l'insuline déclenchée par des concentrations élevées de glycogène dans le foie. Une étude in vitro réalisée par Lingohr et coll. (2002) sur des hépatocytes isolés a produit des résultats semblables : hausse des concentrations de glycogène (indépendante de l'insuline) et effets sur les protéines de signalisation de l'insuline.
Ces études mettent en évidence une régulation négative de l'insuline et de l'activité des récepteurs de l'insuline après des traitements d'une durée relativement brève.
10.3.2.1.4 Promotion tumorale, altération de la réplication et de la mort cellulaires
Stauber et Bull (1997) ont étudié les différences au niveau du phénotype et du mode de réplication cellulaire des tumeurs du foie provoquées par le DCA et le TCA chez des souris B6C3F1 mâles. Ils ont relevé des différences claires au niveau du phénotype entre les tumeurs provoquées par le DCA et celles induites par le TCA. L'étude a aussi démontré dans quelle mesure le DCA influait sur les changements de la réplication cellulaire à l'intérieur de tumeurs et d'hépatocytes normaux pour accroître la formation de tumeurs. Stauber et coll. (1998) ont aussi réalisé des études in vitro qui ont donné les mêmes résultats.
Plusieurs mécanismes d'action ont été suggérés pour les promoteurs tumoraux. Bull et coll. (2004) ont avancé que les promoteurs devraient agir principalement sur la taille des tumeurs plutôt que sur leur nombre. Il est possible d'observer ce comportement avec le DCA. Miller et coll. (2000) ont étudié, au moyen de l'imagerie par résonnance magnétique (IRM), les taux de croissance des tumeurs du foie chez des souris B6C3F1 mâles auxquelles on avait administré 2 000 mg/L de DCA (neutralisé) dans l'eau potable pendant 48 semaines - période de traitement qui garantit l'induction de petites tumeurs du foie. Les résultats ont démontré que la croissance continue des tumeurs dépendait totalement du traitement au DCA. D'après les auteurs, le principal effet du DCA sur l'induction de tumeurs est médié par la croissance accélérée de cellules spontanément initiées et peut être attribuable à la suppression de l'apoptose et à la modification des taux de réplication cellulaire.
La suppression de l'apoptose constitue une autre hypothèse dans le cas des promoteurs. Snyder et coll. (1995) ont étudié la fréquence de l'apoptose spontanée dans les hépatocytes de souris B6C3F1 mâles qui avaient reçu des doses de 0, 500 ou 5 000 mg/L de DCA dans leur eau potable pendant 5 à 30 jours. Le DCA a réduit considérablement l'apoptose chez les souris traitées par rapport aux sujets témoins non traités; la réduction était liée à la dose. Les auteurs estiment que la perturbation du phénomène de l'apoptose peut provoquer la croissance excessive des cellules initiées et, partant, l'apparition de tumeurs, en supprimant la capacité du foie d'éliminer les cellules initiées (cellules prénéoplasiques) plutôt qu'en induisant la prolifération sélective des cellules initiées.
Les données produites par une étude sur le TCE et ses métabolites (Bull, 2000) indiquent que la modification des voies de signalisation des cellules qui entraîne la réplication, la sélection et l'apoptose cellulaires peut contribuer considérablement à l'hépatocancérogénicité du DCA.
Au cours d'une étude sur la prolifération des hépatocytes (Pereira, 1995, 1996), on a exposé des groupes de 10 souris B6C3F1 femelles à des concentrations de 260, 860 ou 2 600 mg/L (52, 172 ou 520 mg/kg p.c. par jour) de DCA (neutralisé) dans l'eau potable jusqu'à leur sacrifice aux jours 5, 12 ou 33. On a constaté une augmentation de la prolifération cellulaire liée à la concentration après cinq jours de traitement au DCA, mais non après 12 et 33 jours, à l'exception d'une augmentation apparente à la dose élevée après 12 jours. Le traitement au DCA a ainsi causé une augmentation transitoire de la prolifération des hépatocytes (Pereira, 1995, 1996).
Des augmentations de la prolifération des hépatocytes ont également été observées au cours d'autres études de courte durée (Carter et coll., 1995; Stauber et Bull, 1997). On a constaté une diminution de la prolifération cellulaire à des doses plus élevées pendant des périodes d'exposition chronique (Bull, 2000; U.S. EPA, 2003c).
Carter et coll. (2003) ont procédé à une analyse histopathologique des lésions hépatiques chez des souris mâles utilisées au cours de l'étude de DeAngelo et coll. (1999) sur la cancérogénicité afin de définir et de quantifier les différents phénotypes des lésions hépatocellulaires, comme les foyers hépatiques altérés, les gros foyers d'altération cellulaire, les adénomes et les carcinomes. À la suite de l'analyse, ils ont proposé trois séquences différentes de formation de lésions pendant la cancérogenèse dans le foie de souris - foyers hépatiques altérés, gros foyers d'altération cellulaire et adénomes - qui évoluent en néoplasie avec le temps. L'analyse a aussi démontré l'existence d'un lien entre certains changements adaptatifs toxiques dans le foie non atteint et cette évolution vers la néoplasie, sans égard à la dose et à la durée de l'exposition. Selon Carter et coll. (2003), le DCA altère l'homéostase des hépatocytes, ce qui entraîne une sélection négative de cellules (par la suppression de l'apoptose, phénomène naturel d'élimination des cellules initiées) et un nouvel état de différenciation qui résiste à la toxicité du DCA, permettant ainsi aux cellules initiées (c.-à-d. cellules et lésions prénéoplasiques) de se multiplier ou de survivre. Ces effets liés à la dose se produisent à des concentrations de moins de 1 000 mg/L, que les auteurs ont considérées comme le point d'inflexion de la courbe doseréponse dans le processus de cancérogenèse.
10.3.2.1.5 Autres mécanismes : hypométhylation
Une autre hypothèse concernant les agents cancérogènes non génotoxiques se fonde sur un mécanisme épigénétique mettant en cause la méthylation de l'ADN (Pereira et coll., 2004). La baisse du niveau de méthylation (hypométhylation) de l'ADN est un phénomène courant dans la plupart des cancers, y compris celui du foie (Pereira et coll., 2004).
Modification naturelle de l'ADN, la méthylation de l'ADN se produit lorsqu'un groupe méthyle s'ajoute au carbone en position 5 de l'anneau de cytosine pour former de la 5-méthylcytosine. Le groupe méthyle est fourni par la S-adénosylméthionine, tandis que la réaction est catalysée par l'ADN méthyltransférase (Ge et coll., 2001; Pereira et coll., 2004). Une perturbation de ces différents phénomènes de méthylation de l'ADN peut entraîner une hypométhylation (Tao et coll., 2000).
Le DCA (neutralisé) a réduit la méthylation de l'ADN (hypométhylation) dans le foie et provoqué l'apparition de foyers d'hépatocytes altérés et d'adénomes hépatocellulaires lorsqu'on l'a administré dans l'eau potable à des souris B6C3F1 femelles (Pereira et coll., 2004). Les auteurs ont indiqué qu'il pourrait y avoir un lien entre la prévention de la méthylation de l'ADN provoquée par un agent cancérogène et la prévention de tumeurs du foie, et que l'hypométhylation de l'ADN pouvait jouer un rôle crucial dans les effets cancérogènes du DCA.
Tao et coll. (1998) ont montré au cours d'une étude de promotion que le DCA réduisait les concentrations de 5-méthylcytosine dans l'ADN des tumeurs du foie et qu'il semblait y avoir un lien entre l'évolution néoplasique des lésions hépatiques, d'adénomes en carcinomes, et une baisse de la concentration de 5-méthylcytosine dans l'ADN. Dans une autre étude, Tao et coll. (2000) ont signalé que le DCA réduisait la méthylation dans les régions promotrices de deux proto-oncogènes, c-jun et c-myc, et augmentait l'expression de leur ARNm et de leurs protéines, tandis que l'ajout de méthionine prévenait ces changements. On sait que ces deux proto-oncogènes participent au contrôle de la prolifération cellulaire (U.S. EPA, 2003c).
Stauber et Bull (1997) ont déterminé que les lésions provoquées par le DCA avaient un phénotype immunoréactif au c-jun.
L'incertitude règne quant à l'importance réelle qu'une réduction de la méthylation associée à une augmentation de l'expression de l'ARNm peut avoir pour la médiation de la tumorigénicité du DCA (U.S. EPA, 2003c).
10.3.3 Acide trichloroacétique
Plusieurs études ont été effectuées sur la cancérogénicité possible du TCA et sont répertoriées dans le tableau 9. On a signalé des incidences accrues de tumeurs du foie (adénomes et carcinomes) chez des souris exposées à de l'eau potable contenant de 500 à 5 000 mg de TCA/L pendant des périodes variant de 52 à 104 semaines. Aucune tumeur du foie n'a été détectée chez les rats. Les détails des études suivent.
Dans le cadre d'une étude sur l'exposition chronique à l'eau potable d'une durée d'un an (Bull et coll., 1990), on a exposé des groupes de souris B6C3F1 mâles (n = 61, 11 et 50) au TCA (neutralisé) à des concentrations de 0, 1 000 ou 2 000 mg/L (0, 178 et 319 mg/kg p.c. par jour respectivement; OMS, 2004b), et un groupe de 10 femelles à 2 000 mg/L dans l'eau potable pendant une période maximale de 52 semaines. On a procédé à des sacrifices tout au long de l'étude, et tous les animaux restants ont été sacrifiés à la 52e semaine. On a constaté une accumulation, liée à la dose, de la lipofuscine (indicateur d'une peroxydation des lipides intracellulaires) dans le foie de souris après 52 semaines, mais non chez les mâles sacrifiés après 37 semaines. Des augmentations, liées à la dose, de l'incidence des lésions hépatoprolifératives - c.-à-d. de nodules hyperplasiques, d'adénomes et de carcinomes hépatocellulaires - ont été observées après 52 semaines chez les souris exposées à 1 000 mg/L et plus. On a également constaté la présence d'importantes concentrations de lipofuscine dans les régions voisines des lésions hépatoprolifératives, et non dans les lésions mêmes. Il y avait un rapport linéaire entre la dose totale moyenne de TCA consommée en 52 semaines et le nombre de lésions hépatiques (nodules + adénomes + carcinomes) par souris, même si l'incidence des lésions par souris dans le groupe exposé pendant 37 semaines a été inférieure à ce que l'on aurait prédit, compte tenu de la dose totale administrée. Aucune des souris femelles exposées au TCA ne présentait de lésions hyperplasiques ou néoplasiques. Chez les rats traités au TCA, on a relevé uniquement de faibles augmentations de la taille des hépatocytes, une modeste accumulation de glycogène, l'absence de nécrose focale et une induction marginale seulement de la prolifération cellulaire et de l'hypertrophie du foie (Bull et coll., 1990).
Au cours d'une étude sur l'eau potable d'une durée de 60 semaines (DeAngelo et Daniel, 1990), on a administré à des groupes de souris B6C3F1 mâles (dont le nombre n'était pas précisé) du TCA (peut-être neutralisé compte tenu d'une étude semblable sur le DCA réalisée par DeAngelo et coll. en 1991) à des concentrations de 0, 50, 500 ou 5 000 mg/L (0, 7, 71 et 595 mg/kg p.c. par jour). On a constaté une augmentation liée à la dose des incidences de nodules hyperplasiques, d'adénomes et de carcinomes. L'incidence de tumeurs hépatocellulaires a atteint 55,2 % à la dose élevée, 37,9 % à la dose moyenne et 13,37 % chez les sujets du groupetémoin non traité. À la dose la plus faible (50 mg/L), aucune différence importante dans la prévalence ou la multiplicité des tumeurs n'a été observée comparativement au groupe témoin. Des nodules hyperplasiques du foie ont été détectés chez les sujets exposés à la dose la plus élevée seulement (prévalence : 24,1 %), mais aucun chez les sujets exposés aux doses faibles ou chez les témoins. Vers la fin de l'étude, on a observé une inflammation chronique et une nécrose du foie chez les sujets des groupes exposés aux deux doses les plus élevées.
Dans une autre étude sur l'exposition chronique, des souris B6C3F1 femelles (n = 10-40) ont reçu du TCA (neutralisé) à des concentrations de 2,0, 6,67 ou 20,0 mmol/L (64, 212 et 640 mg/kg p.c. par jour) dans leur eau potable pendant 360 ou 576 jours (Pereira, 1996; Pereira et Phelps, 1996). Les sujets du groupe témoin ont reçu 640 mg de chlorure de sodium/kg p.c. par jour. Après 360 jours d'exposition à la dose élevée, on a constaté une augmentation importante de l'incidence du carcinome hépatocellulaire et de la multiciplicité des tumeurs. Après 576 jours d'exposition, on a noté des augmentations importantes de l'incidence et de la production de foyers hépatiques altérés, d'adénomes et de carcinomes hépatocellulaires. Après 576 jours, la dose moyenne avait aussi entraîné des augmentations de l'incidence et de la production de foyers et de carcinomes. Il semblait y avoir un lien linéaire entre la dose de TCA et le nombre total combiné de lésions (foyers + adénomes + carcinomes) et de tumeurs (adénomes + carcinomes). Les auteurs ont conclu que le TCA était un proliférateur de peroxysomes et que la coloration basophile des tumeurs correspondait à celle que produisent d'autres proliférateurs de peroxysomes (Pereira, 1995, 1996; Pereira et Phelps, 1996).
Une étude réalisée par l'U.S. EPA (1991) sur la cancérogénicité du TCA chez les rongeurs a fait état des résultats d'une étude sur la cancérogénicité du TCA (forme inconnue) chez des souris B6C3F1 femelles (nombre inconnu). Des groupes de souris femelles ont reçu 0, 500 ou 4 500 mg/L de TCA (0, 71 ou 583 mg/kg p.c. par jour; OMS, 2004b) dans l'eau potable pendant 104 semaines. L'incidence des tumeurs du foie combinées (adénomes + carcinomes) a augmenté considérablement chez le groupe exposé à la dose élevée. L'incidence des tumeurs hépatocellulaires a atteint 64 % chez les sujets exposés à la dose élevée et 16,7 % chez ceux qui ont été exposés à la dose la plus faible, comparativement à 7,7 % chez le groupe témoin non traité. La LOAEL, calculée par l'U.S. EPA (1991) chez les souris femelles sur la base de la présence de tumeurs, est de 71 mg/kg p.c. par jour.
Une étude sur la cancérogénicité d'une durée de deux ans au cours de laquelle on a administré à des groupes de rats F344 mâles (50 par dose) du TCA (neutralisé) à des concentrations de 0, 50, 500 ou 5 000 mg/L (0, 3,6, 32,5 ou 364 mg/kg p.c. par jour) dans l'eau potable n'a pas révélé de signes d'augmentation du nombre de tumeurs du foie (DeAngelo et Daniel, 1992; DeAngelo et coll., 1997). Chez les sujets exposés à la dose élevée, on a constaté des diminutions importantes du poids corporel et du poids absolu du foie, tandis qu'on a relevé des augmentations des concentrations sériques d'ALT et de l'activité de la palmitoyl-coenzyme A (marqueur de la prolifération des peroxysomes hépatiques). Les auteurs ont fixé une NOEL de 364 mg/kg p.c. par jour. Une NOAEL de 32,5 mg/kg p.c. par jour a été établie sur la base des effets non néoplasiques.
Plusieurs études ont aussi porté sur la capacité du TCA d'agir comme promoteur de la cancérogenèse. Parnell et coll. (1986, 1988) ont étudié les capacités d'initiation et de promotion du TCA (neutralisé) au moyen d'essais biologiques sur des foyers hépatiques altérés par des enzymes chez le rat. Les auteurs ont conclu que le TCA était un faible proliférateur de peroxysomes et un promoteur des tumeurs du foie, mais non un initiateur de tumeurs du foie chez les rats Sprague-Dawley (Parnell et coll., 1986, 1988).
On a aussi étudié, chez des souris B6C3F1 mâles (Herren-Freund et Pereira, 1986; Heren-Freund et coll., 1987), le potentiel du TCA (neutralisé) comme promoteur tumoral. L'incidence des adénomes et des carcinomes hépatocellulaires a augmenté considérablement dans le cas des deux types de traitement, à savoir avec ou sans prétraitement au moyen d'un initiateur. Les auteurs ont conclu que le TCA était hépatocancérogène, qu'il y ait eu ou non prétraitement au moyen d'un initiateur (Herren-Freund et Pereira, 1986; Heren-Freund et coll., 1987).
Au cours d'une deuxième étude sur la promotion, Pereira (1995) et Pereira et Phelps (1996) ont utilisé des groupes de souris B6C3F1 femelles. Ils ont constaté une faible augmentation de l'incidence des changements néoplasiques à la dose la plus élevée chez les sujets exposés au TCA sans initiation préalable. Par contre, lorsque des souris ont subi un traitement préalable au moyen d'un initiateur et ont été exposées ensuite au TCA, une augmentation importante de l'incidence et de la multiplicité des adénomes et des carcinomes a été observée aux doses élevées. Il y avait un rapport linéaire entre le nombre total de lésions néoplasiques par souris et la dose de TCA au fil du temps.
10.3.4 Acide monobromoacétique
On n'a pas trouvé d'études de longue durée sur le MBA.
10.3.5 Acide dibromoacétique
Un abrégé publié par Bull (1995) mentionne une étude sur l'eau potable d'une durée de deux ans qui a porté sur le potentiel cancérogène du DBA et d'autres haloacétates bromés chez les souris B6C3F1 et les rats F344, mais aucune version finale n'a été publiée.
Toutefois, une étude plus récente d'une durée de 2 ans sur la cancérogénicité du DBA dans l'eau potable a été publiée par Melnick et coll. (2007) et analysée plus en détail par le NTP (2007). Des groupes de rats F344/N (50 par sexe et par dose) et de souris B6C3F1 (50 par sexe et par dose) ont reçu du DBA (neutralisé à un pH de 5) dans l'eau potable à des concentrations de 0, 50, 500 ou 1 000 mg/L (équivalant à 0, 2, 20 et 40 mg/kg p.c. par jour dans le cas des rats mâles et à 0, 2, 25 et 45 mg/kg p.c. par jour dans le cas des rats femelles; ainsi qu'à 0, 4, 45 et 87 mg/kg p.c. par jour pour les souris mâles et 0, 4, 35 et 65 mg/kg p.c. par jour pour les souris femelles). Aucun effet sur la survie des deux espèces ni changement dans le poids corporel des souris n'ont été observés. Chez les rats (des deux sexes), cependant, le poids corporel moyen a diminué chez les sujets exposés aux deux doses les plus élevées comparativement aux témoins, alors que la consommation d'eau a diminué chez ceux exposés à la plus forte dose (les deux sexes). Des lésions néoplasiques ont été détectées dans plusieurs sièges chez les rats comme chez les souris.
Chez les rats mâles, une augmentation importante du nombre de mésothéliomes malins de la cavité abdominale a été enregistrée dans le groupe exposé à la plus forte dose. Chez les rats femelles, une tendance positive à la hausse de l'incidence de la leucémie à cellules mononucléées, un type de tumeur hématopoïétique (participant à la formation des globules sanguins), a été relevée et était significative à la dose la plus forte.Par contre, l'incidence de la leucémie à cellules mononucléées a augmenté de façon significative chez les rats mâles exposés à la plus faible dose mais pas chez ceux exposés à la dose la plus forte (phénomène qu'on retrouvait également chez les témoins concomitants et les témoins historiques); les incidences enregistrées aux doses faibles et moyennes dépassaient toutefois celles relevées chez les témoins historiques. Au nombre des autres effets non cancéreux observés figuraient une augmentation significative de l'incidence des lésions du foie (dégénérescence kystique, de minime à légère) chez tous les groupes de mâles exposés, des lésions du poumon (hyperplasie épithéliale alvéolaire) dans les groupes exposés aux deux doses les plus élevées chez les femelles, et des lésions du rein (néphropathie) dans tous les groupes de femelles exposés.
Chez les souris, des tumeurs ont été observées tant au niveau du foie que du poumon. Une augmentation significative de l'incidence de l'adénome hépatocellulaire multiple et de l'adénome ou du carcinome hépatocellulaire (combinés) a été relevée chez tous les mâles traités et chez les femelles exposées à 500 mg/L et plus, alors que l'augmentation de l'incidence du carcinome hépatocellulaire n'était significative que chez les mâles exposés à une dose élevée et chez les femelles exposées à une dose moyenne. Une hausse significative de l'incidence de l'hépatoblastome a été observée chez les souris mâles exposées à 500 mg/L et plus. L'incidence de l'adénome alvéolaire/bronchiolaire chez les mâles dépassait celle relevée chez les témoins historiques aux concentrations de 500 mg/L et 1 000 mg/L, mais la hausse était significative uniquement chez les mâles exposés à 500 mg/L. Chez les femelles, une augmentation non significative des tumeurs pulmonaires a été observée. Des effets non cancéreux ainsi que des augmentations non significatives de l'incidence de l'hyperplasie épithéliale alvéolaire ont été observés à toutes les doses chez les souris mâles. Une augmentation de l'incidence de l'hématopoïèse splénique a également été constatée chez les souris mâles exposées à une forte dose (NTP, 2007).
Selon le NTP, (2007), les données montrent clairement l'activité cancérogène du DBA chez les souris, compte tenu de l'augmentation de l'incidence de tumeurs hépatocellulaires (chez les deux sexes) et d'hépatoblastomes (chez les mâles seulement). Le NTP a également recueilli certaines preuves de l'activité cancérogène chez les rats, fondées sur l'augmentation de l'incidence du mésothéliome malin chez les mâles et sur une hausse et une tendance positive de l'incidence de la leucémie à cellules mononucléées chez les femelles.
10.4 Mutagénicité et génotoxicité
10.4.1 Acide monochloroacétique
Des études de mutagénicité effectuées sur des bactéries au moyen de Salmonella typhimurium (Rannug et coll, 1976; NTP, 1992; BG Chemie, 1993; Giller et coll., 1997; ECETOC, 1999) n'ont révélé aucun signe de potentiel génotoxique. On a obtenu des résultats essentiellement positifs au cours de tests portant sur des cellules de mammifères (test du lymphome de souris) (Amacher et Turner, 1982; McGregor et coll., 1987), mais ils peuvent être attribuables à des changements du pH ou de la cytotoxicité (ECETOC, 1999). Des tests in vitro de détection de lésions ou de réparations de l'ADN utilisant Escherichia coli, S. typhimurium et des cultures de cellules de mammifères ont donné des résultats en grande partie négatifs (Gross et coll., 1982; Ono et coll., 1991; Chang et coll. 1992; NTP, 1992; BG Chemie, 1993; Giller et coll., 1997; ECETOC, 1999), sauf dans le cas d'une étude portant sur les cassures de brins d'ADN dans des cellules d'ovaire de hamsters chinois, qui a donné un résultat positif (Plewa et coll., 2002). Les études portant sur les effets clastogènes ont donné des résultats surtout négatifs avec le MCA (Galloway et coll., 1987; Sawada et coll., 1987; Giller et coll., 1997), sauf dans un cas où le MCA (acide) a provoqué des échanges de chromatides soeurs seulement dans des cellules d'ovaire de hamster chinois sans S9 (Galloway et coll., 1987). Au cours d'un test sur cellules de moelle osseuse in vivo, on a obtenu des résultats positifs par la voie intrapéritonéale, mais non par la voie orale ou sous-cutanée (Bhunya et Das, 1987). Cette étude était cependant peu détaillée. On a constaté des anomalies de la forme des spermatozoïdes au cours d'une étude par injection intrapéritonéale chez les souris seulement aux deux doses les plus fortes, mais le rapport d'étude était médiocre (Bhunya et Das, 1987).
10.4.2 Acide dichloroacétique
Les études de mutagénicité portant sur des bactéries et effectuées au moyen de S. typhimurium (Herbert et coll., 1980; Matsuda et coll., 1991; DeMarini et coll., 1994; Fox et coll., 1996; Giller et coll., 1997; Meier et coll., 1997) ont donné des résultats surtout négatifs, et celles portant sur des cellules de mammifères ont donné des résultats équivoques (Fox et coll., 1996; Harrington-Brock et coll., 1998). Des études sur les effets clastogènes ont produit des résultats en grande partie négatifs (Fox et coll., 1996; Giller et coll., 1997; Meier et coll., 1997). Les tests de réparation de l'ADN sur bactéries ont donné des résultats en général positifs, mais utilisaient le DCA sous forme d'acide libre. Les tests de détection des lésions de l'ADN (cassure de brins) dans des cellules de mammifères (in vitro et in vivo) ont donné des résultats en grande partie négatifs (Chang et coll., 1992; Plewa et coll., 2002), sauf dans le cas d'une étude sur l'exposition par voie orale de souris et de rats au cours de laquelle on a utilisé l'acide (Nelson et Bull, 1988; Nelson et coll., 1989). Des anomalies liées à la dose ont été relevées au niveau de la tête des spermatozoïdes chez les souris auxquelles on avait administré oralement par gavage des doses journalières de 1 125-4 500 mg/kg p.c. de DCA (sel de sodium) (Meier et coll., 1997).
10.4.3 Acide trichloroacétique
Des études de mutagénicité chez les bactéries n'ont fourni aucune preuve du potentiel génotoxique (CIRC, 1995; Kargalioglu et coll., 2002; NTP, 2003a), à l'exception d'une étude au moyen du test d'Ames modifié qui a donné un résultat faiblement positif (Giller et coll., 1997). On a aussi obtenu des résultats faiblement positifs au cours de la seule étude de mutation génétique portant sur des cellules de mammifères (Harrington-Brock et coll., 1998). Des études in vitro des lésions de l'ADN effectuées sur des cellules de mammifères ont donné des résultats négatifs (Chang et coll., 1992; Plewa et coll., 2002), mais les résultats étaient contradictoires lorsque le TCA était administré in vivo (Nelson et Bull, 1988; Nelson et coll., 1989; Chang et coll., 1992). Le TCA a produit surtout des résultats négatifs pour ce qui est des lésions de l'ADN et des réparations de l'ADN avec des systèmes bactériens (Ono et coll., 1991; Giller et coll., 1997).
Des études des effets clastogènes (in vitro et in vivo) ont aussi produit des résultats équivoques (Bhunya et Behera, 1987; MacKay et coll., 1995; Giller et coll., 1997; Meier et coll., 1997). Harrington-Brock et coll. (1998) ont signalé que l'on pouvait obtenir des résultats positifs pour la clastogénicité et la mutagénicité dans des tests in vitro sur des cellules de mammifères lorsque le pH est faible, surtout en présence d'une activation métabolique.
Les anomalies morphologiques des spermatozoïdes de souris étaient équivoques à la suite d'une administration intrapéritonéale, mais négatives à la suite d'une ingestion par voie orale (Bhunya et Behera, 1987). Une communication intercellulaire par jonction lacunaire a été observée dans des hépatocytes de souris (in vitro) (CIRC, 1995).
10.4.4 Acide monobromoacétique
Certaines données indiquent que le MBA possède un faible pouvoir mutagène. On a obtenu des résultats contradictoires pour le MBA au moyen du test d'Ames (Saito et coll., 1995; Kohan et coll., 1998; Kargalioglu et coll., 2002; NTP, 2003b). Kargalioglu et coll., (2002) ont signalé dans leur rapport de test d'Ames que le MBA était plus mutagène que le DBA ou le MCA. Selon les études in vitro des lésions de l'ADN, il y avait aussi quelques signes de potentiel génotoxique. Le MBA (acide) n'a pas causé de lésions primaires de l'ADN au cours d'un test SOS Chromotest (E. coli) avec et sans S9 et n'a pas accru la fréquence de micronoyaux dans un test des micronoyaux sur tritons (Giller et coll., 1997). Par contre, le MBA a provoqué des cassures de brins d'ADN en l'absence de S9 à la suite d'un traitement d'une heure au moyen de cellules de leucémie de souris L-1220 (Stratton et coll., 1981). Le nombre de cassures de brins a augmenté encore davantage lorsqu'on a enlevé le produit chimique (Stratton et coll., 1981). Les auteurs ont avancé que le MBA pouvait faire fonction d'agent alkylant à cause de la présence de cassures de brins (indicateur de lésions directes de l'ADN).
10.4.5 Acide dibromoacétique
Des données indiquent que le DBA a un faible pouvoir mutagène. Le test d'Ames avec S. typhimurium a produit des résultats contradictoires (Saito et coll., 1995; Giller et coll., 1997; Morita et coll., 1997; Kohan et coll., 1998; Kargalioglu et coll., 2002; NTP, 2007). Seules les études des lésions de l'ADN effectuées in vitro ont produit des résultats positifs pour le potentiel génotoxique. Le DBA a causé des lésions primaires de l'ADN au cours d'un SOS Chromotest pour lequel on a utilisé E. coli avec et sans activation métabolique (Giller et coll., 1997) et a induit des cassures de brins d'ADN dans des cellules d'ovaire de hamster chinois, selon un test des comètes (Plewa et coll., 2002). Il y avait aussi des signes de clastogénicité (fréquences accrues d'érythrocytes normochromatiques dans le sang périphérique chez les souris mâles, mais non chez les souris femelles, après l'administration de DBA par voie orale dans l'eau potable au cours d'une étude d'une durée de 13 semaines (NTP, 2007). On n'a constaté aucune activité clastogène au cours du test des micronoyaux sur tritons (Giller et coll., 1997).
10.5 Toxicité pour la reproduction et le développement
10.5.1 Acide monochloroacétique
On n'a pas trouvé d'études sur la reproduction. Il n'existe pas d'études adéquates au sujet de l'effet du MCA sur le développement. Dans les deux études mentionnées ci-dessous, il manque des renseignements importants : l'une provient d'un abrégé et dans l'autre, on utilise une dose seulement.
Une étude sur le développement (administration orale par gavage dans de l'eau distillée de 0, 17, 35, 70 ou 140 mg/kg p.c. par jour de MCA acide pendant les jours 6 à 15 de la gestation) portant sur des rats Long-Evans a été publiée sous forme abrégée par Smith et coll. (1990). Les auteurs indiquent qu'il y a des effets sur le développement (lévocardie congénitale chez les sujets exposés à la dose élevée) en présence de toxicité maternelle. L'abrégé ne fournit pas de données statistiques et aucune étude finale n'a été publiée pour confirmer ces résultats.
Au cours d'une deuxième étude sur le développement (où l'on a utilisé une dose seulement), on a administré à un groupe de 10 rats Sprague-Dawley femelles gravides du MCA
(neutralisé) dans l'eau potable à une concentration de 1 570 mg/L (193 mg/kg p.c. par jour) pendant les jours 1 à 22 de la gestation (Johnson et coll., 1998). Le groupe témoin comptait 55 femelles. Une diminution importante du gain de poids corporel a été observée chez les femelles exposées par rapport aux femelles témoins. Le volume moyen d'eau potable consommé quotidiennement par rat femelle gravide était moins élevé chez les femelles traitées que dans le groupe témoin. Les auteurs n'ont pas signalé d'effets sur la reproduction ou le développement ni d'effets tératogènes, mais ils n'avaient pas procédé à un examen complet du fotus pour y repérer d'éventuelles anomalies internes ou squelettiques.
10.5.2 Acide dichloroacétique
10.5.2.1 Études sur le développement
On a réalisé plusieurs études sur la toxicité possible du DCA pour le développement chez les rats femelles. Ces études sont décrites en détail ci-dessous et résumées au tableau 11.
| Doses (mg/kg p.c.par jour) | Espèce | Effets sur le développement | LOAEL/ NOAEL | Référence |
|---|---|---|---|---|
| 0, 14, 140, 400, 900, 1 400, 1 900 ou 2 400 (JGTableau 11 note de bas de page a 6-15) | Rats à capuchon Long-Evans (19-21/dose) |
|
NOAEL pour les effets toxiques sur le développement : 14 mg/kg p.c. par jour | Smith et coll., 1992 |
Quatre études :
|
Rats Long-Evans (711/dose) |
|
Epstein et coll., 1992 | |
| 0 ou 300 (JG 6-15) | Rats Sprague-Dawley (19-20/dose) |
|
Fisher et coll., 2001 | |
Notes de bas de page
|
||||
Dans le cadre d'une étude sur le développement (Smith et coll., 1992), des rats à capuchon Long-Evans gravides (19-21 par dose) ont reçu par gavage, dans deux études distinctes, du DCA (neutralisé) à des doses de 0, 14, 140 ou 400, ou de 0, 900, 1 400, 1 900 ou 2 400 mg/kg p.c. par jour au cours des jours 6 à 15 de la gestation inclusivement. On a constaté des réductions liées à la dose du gain rajusté de poids chez la mère (140 mg/kg p.c. par jour ou plus), des augmentations du poids relatif du foie liées à la dose (toutes les doses) et des augmentations du poids relatif des reins et de la rate liées à la dose (400 mg/kg p.c. par jour et plus). La létalité maternelle liée au traitement est survenue à 1 400 mg/kg p.c. par jour et plus (1/19 [5,3 %], 2/19 [10,5 %] et 5/21 [24 %], respectivement). Le traitement n'a pas eu d'effet sur les taux de gestation, le nombre total d'implantations par portée et les pertes préimplantation. On a enregistré une augmentation du taux de pertes postimplantation liées à la dose (900 mg/kg p.c. par jour et plus), et le nombre de fotus vivants par portée a diminué à la dose la plus élevée (à laquelle on a observé une toxicité [létalité] maternelle importante). On a relevé également une diminution, liée à la dose, du poids corporel et de la longueur totale du fotus à 400 mg/kg p.c. par jour et plus. On a observé des augmentations liées à la dose des malformations externes (1 400 mg/kg p.c. par jour et plus), des tissus mous totaux (140 mg/kg p.c. par jour et plus), cardiovasculaires (400 mg/kg p.c. par jour et plus), urogénitales (1 400 mg/kg p.c. par jour et plus) et orbitaires (900 mg/kg p.c. par jour et plus). Comparativement à d'autres malformations, les anomalies urogénitales (hydronéphrose bilatérale, papilles rénales, stade un) et orbitaires étaient moins fréquentes. Le DCA avait comme cible principale le cour et les gros vaisseaux du fotus. La malformation cardiaque la plus courante chez les fotus était située entre l'aorte ascendante et le ventricule droit et constituait une malformation de la partie supérieure du septum interventriculaire. La deuxième des malformations cardiaques les plus courantes était la malformation apparente du septum interventriculaire. Dans le cas de la toxicité pour le développement, les auteurs ont fixé la NOAEL à 14 mg/kg p.c. par jour.
Quatre études ont été menées sur des rats femelles Long-Evans gravides (7-11 par dose) exposés au DCA (neutralisé) afin de déterminer la période de développement la plus sensible et de caractériser les malformations cardiaques (Epstein et coll., 1992). Dans la première étude, on a administré aux rats, par intubation orale, 1 900 mg de DCA/kg p.c. par jour au cours des jours 6 à 8, 9 à 11 ou 12 à 15 de la gestation. Dans la deuxième, on leur a administré 2 400 mg/kg p.c. par jour, les jours 10, 11, 12 ou 13 de la gestation. Pendant la troisième, les sujets ont reçu 3 500 mg/kg p.c. par jour les jours 9, 10, 11, 12 ou 13 de la gestation. On n'a relevé aucune toxicité maternelle importante au cours des trois premières études. Les pourcentages moyens de malformations cardiaques étaient beaucoup plus élevés (statistiquement) chez les sujets qui avaient reçu 1 900 mg de DCA/kg p.c. par jour les jours 9 à 11 (7,2 % par portée) et 12 à 15 de la gestation (15,1 % par portée) comparativement aux totaux combinés chez les témoins. Les malformations cardiaques observées étaient soit une malformation de la partie supérieure du septum interventriculaire ou une malformation classique du septum interventriculaire. On a obtenu des incidences plus faibles de malformations cardiaques à la dose de 2 400 mg de DCA/kg p.c. par jour (jour 10 de la gestation : 2,5 % par portée; jour 12 : 3,3 % par portée; jours 11 et 13 : 0 %). Les incidences de l'exposition les jours 10 et 12 de la gestation étaient trèsdifférentes de celles des témoins combinés. À la dose la plus élevée, soit 3 500 mg/kg p.c. par jour, les incidences de malformations cardiaques n'ont pas augmenté, mais l'étude a démontré que l'administration de doses les jours 9, 10 et 12 de la gestation (3,6 %, 2,9 % et 2,9 % par portée respectivement) produirait ces malformations et que les incidences étaient très différentes de celles des témoins combinés (0,5 % par portée). On n'a constaté aucune malformation cardiaque à la suite d'une exposition les jours 11 ou 13 de la gestation (Epstein et coll., 1992).
Dans une quatrième étude (Epstein et coll., 1992) visant à caractériser les malformations cardiaques, on a intubé oralement six femelles pour leur administrer 1 900 mg de DCA/kg p.c. par jour les jours 6 à 15 de la gestation et on a examiné le cour de 56 fotus des six portées au microscope optique, de même que huit fotus témoins provenant de quatre portées. L'examen a révélé que 25 fotus sur 56 (45 %) présentaient des malformations cardiovasculaires, soit une malformation de la partie supérieure du septum interventriculaire chez 24 fotus (cinq des cours en question présentaient aussi une malformation du septum interventriculaire de type membraneux) et un cas isolé de malformation du septum interventriculaire de type membraneux.
Au cours d'une étude plus récente, Fisher et coll. (2001) ont administré par gavage à un groupe de 20 rats Sprague-Dawley femelles gravides du DCA (neutralisé) à raison de 300 mg/kg p.c. par jour pendant les jours 6 à 15 de la gestation. Un groupe témoin a reçu de l'eau (n = 19). Les chercheurs n'ont constaté aucune malformation cardiaque, contrairement à ce qu'avaient signalé Smith et coll. (1992) et Epstein et coll. (1992) dans des études antérieures. Il se peut cependant que cette étude n'ait pas été assez sensible, vu le niveau de fond élevé de malformations cardiaques (par portée) présentes chez les témoins qui ont reçu de l'eau (31,6 %; niveau plus élevé que chez les animaux traités, où il était égal à 30 %), ce qui a pu masquer les effets chez les sujets exposés au DCA. L'étude de Smith et coll. (1992) a révélé une à deux malformations cardiovasculaires seulement à des doses de moins de 300 mg/kg p.c. par jour. Des différences au niveau des lignées chez les rats et de la pureté des agents utilisés dans les tests pourraient aussi expliquer les divergences dans les observations des deux séries d'études.
10.5.2.2 Études sur la reproduction
On a administré à des rats mâles Sprague-Dawley adultes (n = 24 par dose) des doses orales uniques de 0, 1 500 ou 3 000 mg/kg p.c. de DCA (neutralisé) afin d'étudier la toxicité pour les testicules, puis on les a sacrifiés (n = 8 par période) les jours 2, 14 ou 28 (Linder et coll., 1997a). Aucun signe clinique de toxicité n'a été relevé chez les rats exposés. Les poids corporels ne différaient pas, sur le plan statistique, de ceux des sujets témoins à aucun des trois points dans le temps. On a constaté des effets bénins sur la spermiation (c.-à-d. retards) et des changements du degré de résorption des corps résiduels aux deux doses; ces effets ont persisté à des degrés variables durant toute la période d'observation (Linder et coll., 1997a).
Dans une étude parallèle réalisée par le même auteur, on a administré à un autre groupe de rats mâles (n = 8-26) de multiples doses orales de 0, 18, 54, 160, 480 ou 1 440 mg/kg p.c. de DCA (neutralisé) par jour pendant une période maximale de 14 jours (Linder et coll., 1997a). On n'a observé aucun signe clinique de toxicité chez les animaux traités, à l'exception d'une diminution du gain de poids constatée aux trois doses les plus fortes le jour 14. On a observé un retard de la spermiation et la formation de corps résiduels atypiques chez tous les mâles exposés, sauf à la dose la plus faible. Les doses de 160 mg/kg p.c. par jour et plus ont entraîné une augmentation du nombre de spermatozoïdes épididymaires soudés après le jour 5. On a constaté une diminution du pourcentage de spermatozoïdes motiles le jour 9, à des doses de 480 mg/kg p.c. par jour et plus, et le jour 14 à des doses de 160 mg/kg p.c. et plus. Le jour 14, le nombre de spermatozoïdes épididymaires a diminué aux doses de 160 mg/kg p.c. et plus alors que leur poids a diminué à des doses de 480 mg/kg p.c. par jour et plus. On a relevé la présence de difformités de la tête de spermatozoïdes et des acrosomes chez les spermatides au stade 15 après 14 jours d'exposition à une dose de 480 mg/kg p.c. par jour et plus. La gravité des lésions testiculaires subies a augmenté en fonction de la durée de l'exposition et de l'importance des doses.
Des rats mâles Long-Evans (n = 8-19) ont reçu par gavage quotidien 0, 31,3, 62,5 ou 125 mg/kg p.c. de DCA (sel de sodium) pendant 10 semaines (Toth et coll., 1992). Le jour 70, on a accouplé chaque mâle pendant la nuit à une femelle non traitée pendant la phase de proestrus afin d'évaluer la fertilité. On a ensuite sacrifié les mâles le jour 75, et les femelles le jour 14 de la gestation. Les taux de gestation et d'implantation n'étaient pas très différents de ceux des témoins femelles. Chez les femelles exposées à des doses élevées, on a toutefois observé une réduction du nombre d'implants vivants. Chez les mâles, le poids de l'épididyme et des glandes préputiales a diminué aux doses de 31,3 mg/kg p.c. et plus par jour de même que celui des organes annexes (prostate et vésicules séminales) à la dose la plus élevée (125 mg/kg p.c par jour). Les deux doses les plus élevées ont eu un effet sur la morphologie des spermatozoïdes, et le nombre de spermatozoïdes épididymaires a baissé. Les auteurs ont constaté des effets sur les testicules et la spermiation (réduction du nombre de spermatides en fin de stade) seulement chez les sujets exposés à la dose la plus élevée. Ils ont fixé à 31,3 mg/kg p.c. par jour la LOAEL pour le DCA (sel de sodium) (ce qui équivaut à 26,7 mg de DCA/kg p.c. par jour) en se fondant sur les effets indésirables sur la reproduction touchant les organes annexes et les spermatozoïdes chez les mâles (Toth et coll., 1992).
Par contre, une étude de toxicité subchronique de plus courte durée (sept semaines) sur l'eau potable a révélé que l'histopathologie des testicules et la production de spermatozoïdes étaient normales chez des rats Sprague-Dawley exposés à 1 100 mg/kg p.c. de DCA (sel de sodium) par jour (Stacpoole et coll., 1990). On a aussi constaté que l'histopathologie et le poids des testicules étaient normaux chez des rats Sprague-Dawley mâles (10 par dose) exposés à des doses plus faibles de 0, 50, 500 ou 5 000 mg/L (0, 4, 35 ou 350 mg/kg p.c. par jour) de DCA (neutralisé) dans l'eau potable au cours d'une étude sur l'eau potable d'une durée de 90 jours (Mather et coll., 1990).
Dans une étude par gavage d'une durée de trois mois (Katz et coll., 1981), des groupes de rats Sprague-Dawley mâles et femelles (10 par sexe et par dose) ont reçu des doses de 0, 125, 500 ou 2 000 mg/kg p.c. par jour de DCA (sel de sodium). On a accordé une période de récupération de quatre semaines à cinq autres rats par sexe exposés à 0 ou 2 000 mg/kg p.c. par jour. On n'a relevé aucun effet indésirable sur la reproduction chez les rats femelles, tandis qu'on en a observé chez les rats mâles aux deux doses les plus élevées seulement. Une dégénérescence de l'épithélium des cellules germinales du testicule était visible chez 40 % des mâles exposés à la dose moyenne et chez 100 % des mâles exposés à la dose élevée. On a constaté une aspermatogenèse et la formation de cellules géantes syncytiales dans l'épithélium germinal chez les mâles exposés à la dose élevée, ainsi que l'absence de spermatozoïdes dans les canaux épididymaires. Des cellules géantes syncytiales étaient aussi présentes chez 20 % des mâles exposés à la dose moyenne. Dans le groupe de dose élevée ayant bénéficié d'une période de récupération (n = 5), la moitié des mâles présentaient une régénération de l'épithélium germinal, 75 % une aspermatogénie et 100 % une perte d'épithélium germinal.
Dans le cadre d'une autre étude d'une durée de 90 jours (Cicmanec et coll., 1991), des beagles mâles et femelles âgés de quatre mois (cinq par sexe et par dose) ont reçu 0, 12,5, 39,5 ou 72 mg/kg p.c. par jour de DCA (neutralisé) dans des capsules de gélatine. On n'a constaté aucun changement important dans le poids des ovaires ou des testicules comparativement aux sujets témoins, ni aucun effet du DCA sur l'histopathologie de l'utérus chez les femelles exposées. Des changements au niveau des testicules (formation de cellules géantes syncytiales et dégénérescence de l'épithélium germinal du testicule) ont été enregistrés chez tous les mâles exposés, de même qu'une atrophie de la prostate chez les mâles exposés à la dose moyenne et à la dose élevée. La gravité des lésions des testicules a augmenté en fonction de la dose chez les sujets exposés à la dose moyenne et à la dose élevée.
Au cours d'une étude sur l'exposition subchronique d'une durée de 13 semaines (Katz et coll., 1981), des chercheurs ont observé des effets semblables sur les testicules chez des beagles mâles âgés de 10 à 12 mois (3-4 par dose) auxquels ils avaient administré des doses de 0, 50, 75 ou 100 mg/kg p.c. par jour de DCA (sel de sodium) dans des capsules. Tous les mâles exposés présentaient des changements testiculaires au niveau de l'épithélium germinal (dégénérescence de l'épithélium germinal, vacuolisation des cellules de Leydig et formation de cellules géantes syncytiales), ainsi qu'une atrophie de la prostate. On a laissé un mâle du groupe exposé à la dose élevée se rétablir pendant quatre semaines après le traitement. La prostate semblait normale et il y avait des signes de régénération de l'épithélium germinal avec la spermatogenèse.
Dans le cadre d'un essai biologique modifié de cancérogenèse chez des rats F344 mâles, DeAngelo et coll. (1996) ont signalé que le poids absolu et le poids relatif des testicules augmentaient légèrement chez les sujets exposés à la dose moyenne (500 mg/L), mais que le poids absolu diminuait chez ceux exposés à la dose élevée (1 600 mg/L).
10.5.3 Acide trichloroacétique
On n'a pas trouvé d'études sur la reproduction.
Dans une étude sur le développement, des rats Long-Evans femelles gravides (n = 20-26) ont reçu par gavage 0, 330, 800, 1 200 ou 1 800 mg/kg p.c. par jour de TCA (neutralisé) (sous forme de sel de sodium) les jours 6 à 15 de la gestation. Les chercheurs n'ont observé aucune mort liée au traitement chez les mères au cours de l'étude. Ils ont relevé une augmentation, liée à la dose, de la fréquence des résorptions par portée (34 %, 62 % et 90 % des implants se sont résorbés à des doses de 800, 1 200 et 1 800 mg/kg p.c. par jour respectivement) à des doses toxiques pour la mère (diminution de la prise de poids et augmentation, liée à la dose, du poids de la rate et des reins). Ils ont constaté à toutes les doses une réduction, liée à la dose, du poids et de la longueur du fotus, ainsi qu'une augmentation, également liée à la dose, de la fréquence des anomalies des tissus mous (de 9 % à 330 mg/kg p.c. par jour à 97 % à 1 800 mg/kg p.c. par jour). Les anomalies des tissus mous ont touché principalement l'appareil cardiovasculaire et il s'agissait de malformations du septum interventriculaire et de lévocardie congénitale. Les auteurs ont toutefois signalé que cette lignée de rats était un peu sensible à la lévocardie. Les deux doses les plus élevées ont aussi provoqué une augmentation des malformations squelettiques (principalement au niveau de l'orbite). On considère que la LOAEL chez la mère est de 330 mg/kg p.c. par jour, compte tenu des changements de poids au niveau de la rate et du rein. La LOAEL pour le développement est de 330 mg/kg p.c. par jour, compte tenu des augmentations de la fréquence des anomalies des tissus mous (Smith et coll., 1989). On n'a pas fixé de NOAEL, mais on a calculé une dose repère de 218 mg/kg p.c. par jour pour les effets toxiques sur le développement (Santé Canada, 2004b) à des fins de comparaison avec d'autres effets constatés dans le cas du TCA.
Des études antérieures sur le développement portant sur le TCE ont révélé une augmentation des lésions cardiaques congénitales chez les rats, mais on a avancé que ces effets pouvaient être attribuables à des métabolites du TCE (Johnson et coll., 1998). C'est pourquoi des chercheurs ont réalisé des études sur le développement qui ont porté sur plusieurs métabolites du TCE, y compris le TCA, afin d'identifier les métabolites responsables. Les deux études qui ont englobé le TCA sont décrites ci-dessous, mais elles ne sont pas concluantes en ce qui concerne les malformations cardiaques observées, puisqu'elles ont utilisé des méthodologies différentes et produit des résultats contradictoires.
Dans une étude, Johnson et coll. (1998) ont administré à des rats Sprague-Dawley femelles gravides des concentrations de 0 mg/L (n = 55) ou 2 730 mg/L (n = 11) (0 ou 290 mg/kg p.c. par jour) de TCA (neutralisé) dans l'eau potable au cours des jours de gestation 1 à 22. Ils ont observé une diminution importante de la prise de poids corporel chez les femelles traitées par rapport aux témoins. Parmi les effets sur le développement figurait une augmentation statistiquement significative du nombre de résorptions, de sites d'implantation et de malformations des tissus mous cardiaques (10,53 % contre 2,15 % chez les témoins).
Fisher et coll. (2001) ont administré par gavage à un groupe de 19 rats Sprague-Dawley femelles gravides une dose de 300 mg/kg p.c. par jour de TCA (neutralisé) pendant les jours 6 à 15 de la gestation. Ils ont administré de l'eau à un groupe témoin (n = 19). La prise de poids moyenne chez la mère a été beaucoup moins importante que chez les témoins les jours 7 à 15 et 18 à 21 de la gestation. Les chercheurs ont constaté une réduction statistiquement significative du poids corporel du fotus (selon le fotus et selon la portée) chez les sujets exposés. Ils n'ont pas observé de malformation cardiaque à 300 mg/kg p.c. par jour, contrairement à Smith et coll. qui en avaient signalé dans leur étude précédente (1989).
10.5.4 Acide monobromoacétique
Le MBA n'a fait l'objet d'aucune étude rigoureuse quant à ses effets sur le développement. MBA. Randall et coll. (1991) ont publié un abrégé d'une étude sur le MBA (acide) chez des rats Long-Evans, qui indique une possibilité de malformation des tissus mous chez les petits en présence d'effets toxiques chez la mère, mais l'étude finale n'a jamais été publiée.
On n'a pas observé d'effets indésirables sur la reproduction chez des rats Sprague-Dawley mâles auxquels on avait administré 0 ou 100 mg/kg p.c. de MBA (neutralisé) en une seule dose ou 0 ou 25 mg/kg p.c. par jour pendant 14 jours (Linder et coll., 1994a).
10.5.5 Acide dibromoacétique
Des chercheurs n'ont constaté aucun effet sur le début de la gestation chez des groupes de rats Holtzman femelles à maturité (huit par dose et par expérience) auxquels on avait administré par gavage 0, 62,5, 125 ou 250 mg/kg p.c. de DBA (neutralisé) par jour au cours des jours 1 à 8 de la gestation. Un groupe de femelles a été sacrifié le jour 9 ou le jour 20 de la gestation. Le seul effet observé était une élévation de la concentration de 17ß -oestradiol sérique à la dose la plus élevée, chez les femelles sacrifiées le jour 9 de la gestation (Cummings et Hedge, 1998).
On n'a réalisé aucune étude adéquate sur le développement. Deux essais de dépistage d'effets sur le développement (gavage oral avec du DBA neutralisé) portant sur des souris CD-1 ont été publiés sous forme d'abrégés par Narotsky et coll. (1996, 1997). Même si les études ont laissé entendre qu'il y avait des effets sur le développement en présence ou en l'absence d'effets toxiques chez la mère, on n'a fourni aucune donnée statistique dans l'un ou l'autre des abrégés, ni publié d'étude finale pour confirmer ces résultats.
Des modifications, liées à la dose, du cycle ostral chez des rats Sprague-Dawley femelles ont été observées à des doses de 90 et 270 mg/kg p.c. par jour lorsqu'on a administré aux animaux pendant 14 jours consécutifs du DBA (neutralisé) dans l'eau potable (Balchak et coll., 2000), mais on n'a constaté aucune altération aux doses les plus faibles (10 ou 30 mg/kg p.c. par jour) au cours de la même étude ou d'une exposition d'une durée de 20 semaines à une dose de 5, 16 ou 33 mg/kg p.c. par jour (Murr et Goldman, 2005).
Dans une étude sur l'eau potable portant sur deux générations, des groupes de rats Sprague-Dawley (30 par sexe et par dose) ont reçu du DBA (à une pureté de 97 % dans de l'eau désionisée) en continu dans leur eau potable à des concentrations de 0, 50, 250 ou 650 mg/L (équivalant à 0, 4,4-11,6, 22,4-55,6 et 52,4-132,0 mg/kg p.c. par jour respectivement) (Christian et coll., 2002). Les doses ont été calculées à partir d'une étude de Christian et coll. (2001) sur la reproduction et le développement visant à délimiter la gamme de doses à utiliser. Les chercheurs ont constaté une réduction de la consommation d'eau à toutes les doses chez les parents (P) et la génération F1. Chez les sujets du groupe exposés à la dose la plus forte des générations P et F1, ils ont observé des signes cliniques associés à la réduction de la consommation d'eau, une baisse du poids corporel, une diminution de la prise de poids et une réduction de la consommation d'aliments. La consommation d'aliments a aussi diminué chez les sujets du groupe F1 exposés à la dose moyenne. Toutes les doses administrées à la génération F1 pendant la lactation ont entraîné une diminution du poids corporel, ce qui a nécessité le report du sevrage au jour 29. De légers retards de la maturation sexuelle (séparation du prépuce, ouverture du vagin) chez les rats F1 qui avaient reçu la dose la plus forte et une diminution importante de la distance anogénitale le jour 22 de la lactation chez les petits mâles F2 des groupes exposés aux doses moyennes et fortes ont aussi été attribués à un retard général de croissance associé à une réduction importante du poids corporel. La performance reproductrice et le développement des rats femelles n'ont pas changé à toutes les doses pour les générations P et F1. Chez les mâles, le traitement n'a pas eu d'effet non plus sur la motilité des spermatozoïdes, leur nombre et leur densité, ni sur les spermatozoïdes anormaux. L'histopathologie des organes reproducteurs des mâles P et F1 exposés aux doses moyenne et élevée a toutefois révélé une altération de la production de spermatozoïdes (augmentation liée à la dose des spermatides retenus au stade 19 dans les tubules aux stades IX et X, et présence de corps résiduels anormaux dans les tubules séminifères touchés), ainsi que des changements au niveau des tubules épididymaires. On a constaté des effets sur la reproduction chez les mâles F1 exposés à la dose élevée, soit une augmentation importante de la malformation unilatérale du tractus reproducteur (petitesse ou absence des épididymes et petits testicules). Les auteurs ont établi, pour la toxicité générale, une NOAEL parentale de 50 mg/L (4,4-11,6 mg/kg p.c. par jour) en se fondant sur une augmentation du poids absolu et relatif des reins et du foie en l'absence d'histopathologie. Dans un projet de rapport, l'U.S. EPA, 2005a a proposé pour la reproduction et le développement une LOAEL de 250 mg/L (ou 22,4-55,6 mg/kg p.c. par jour) basée sur une spermatogenèse anormale chez les mâles P et F1 et une NOAEL de 50 mg/L (4,4-11,6 mg/kg p.c. par jour).
Des effets au niveau des testicules ont été observés dans une étude d'une durée de 13 semaines portant sur l'eau potable (Melnick et coll., 2007; NTP, 2007) au cours de laquelle des souris B6C3F1 mâles et des rats F344 mâles ont été exposés à des doses de 0, 125, 250, 500, 1 000 ou 2 000 mg/L de DBA (neutralisé à un pH de 5) (équivalant à 0, 16, 30, 56, 115 et 230 mg/kg p.c. par jour chez les souris et à 0, 10, 20, 40, 90 et 166 mg/kg p.c. par jour chez les rats). Une atrophie testiculaire de l'épithélium germinal de même que des réductions importantes du poids des testicules, de la motilité et des concentrations des spermatozoïdes ont été relevées chez les rats mâles exposés à une forte dose, de même qu'une augmentation importante de l'hypospermie. Un retard de la spermiation accompagné de la formation de corps résiduels atypiques a été constaté chez les souris mâles exposées aux deux doses les plus fortes et chez les rats exposés uniquement aux doses moyennes (500 et 1 000 mg/L). La dose la plus faible associée à des effets testiculaires chez les rats s'élevait à 500 mg/L. Le NTP, (2007) a fait état d'une NOEL de 250 mg/L pour les lésions testiculaires chez le rat. Des effets similaires (tels qu'une diminution du poids des testicules, un retard de la spermiation ou la présence de corps résiduels atypiques) ont été relevés chez les rats (1 000 mg/L et plus) et les souris (500 mg/L et plus) exposés pendant plus de deux semaines (Melnick et coll., 2007; NTP, 2007).
Des effets spermatoxiques et testiculaires, à savoir une atrophie des testicules et une altération des spermatozoïdes (motilité, morphologie, nombre, spermatozoïdes soudés ou anormaux, rétention accrue des spermatides au stade 19, corps résiduels atypiques), ont aussi été observés chez des rats Sprague-Dawley auxquels on a administré une dose unique ou des doses répétées de TCA (neutralisé ou acide) par voie orale (Linder et coll., 1994a, b, 1995; Vetter et coll., 1998; Tsuchiya et coll., 2000; Holmes et coll., 2001).
La capacité de reproduction a été compromise chez les rats mâles gavés au DBA (neutralisé) (Linder et coll., 1995). On a constaté une morphologie anormale des spermatozoïdes, une réduction de leur nombre et de leur motilité, ainsi que des changements de comportement (au cours de l'accouplement) chez les mâles.
Les résultats préliminaires d'études publiées récemment sous forme d'abrégés (Klinefelter et coll., 2000; Veeramachaneni et coll., 2000, 2002; Bodensteiner et coll., 2001) indiquent une perturbation du développement à la puberté, de la fonction reproductrice, de la spermatogenèse et de la fertilité chez les rats et/ou les lapins mâles, ainsi qu'une réduction de la population de follicules primordiaux chez les lapins femelles lorsqu'on administre du DBA (neutralisé) lors de la première exposition in utero à compter du jour 15 de la gestation et durant toute la vie. On n'a pas publié d'études finales pour confirmer ces résultats.
11.0 Classification et évaluation
11.1 Acide monochloroacétique
Une étude longitudinale n'a révélé aucun signe de cancérogénicité du MCA chez les souris ou les rats (NTP, 1992). Les tests portant sur la mutagénicité et la génotoxicité du MCA ont aussi donné des résultats en grande partie négatifs. Compte tenu du manque de preuves relatives à la cancérogénicité, on a donc classé le MCA au cours de cette évaluation dans le groupe IV.D des substances peu susceptibles d'être cancérogènes pour l'être humain (Santé Canada, 1994).
Le Centre international de recherche sur le cancer (CIRC) n'a pas évalué le MCA. L'U.S. EPA (2003) a signalé que, selon les Draft Revised Guidelines for Carcinogen Risk Assessment (U.S. EPA, 1999b), les données sur le MCA étaient « inadéquates pour permettre d'évaluer le potentiel cancérogène pour l'être humain »
.
Plusieurs études de longue durée réalisées sur le MCA n'ont pas mis en évidence d'effets cancérogènes. La NOAEL la plus basse de toutes les études provient de l'étude sur l'eau potable réalisée sur des rats pendant 104 semaines par DeAngelo et coll. (1997); à partir de cette étude, Santé Canada a calculé une NOAEL de 3,5 mg/kg p.c. par jour pour les changements dans le poids du corps, du foie, des reins et des testicules liés à l'exposition. On a choisi cette étude pour évaluer le risque, compte tenu de la pertinence du véhicule utilisé (eau potable), de l'absence d'effets importants à faible dose, de la durée de l'étude, ainsi que de la forme de MCA administrée (c.-à-d. en solution neutralisée).
La dose journalière tolérable (DJT) de MCA se calcule ainsi :
Figure 6: l'équation utilisée pour le calcul de la dose journalière tolérable (DJT) de l'acide monochloroacétique (MCA)
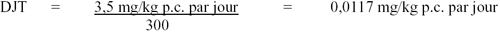
Figure 6: l'équation utilisée pour le calcul de la dose journalière tolérable (DJT) de l'acide monochloroacétique (MCA) - Description textuel
La dose journalière tolérable (DJT) de l'acide monochloroacétique (MCA) est égale à 0,0117 milligrammes par kilogramme de poids corporel (kg pc) par jour. Cette valeur a été calculée en divisant 3,5 milligrammes par kilogramme de poids corporel par jour par 300.où :
- 3,5 mg/kg p.c. par jour représente la NOAEL (déterminée par Santé Canada) dans l'étude sur l'exposition chronique de rats réalisée par DeAngelo et coll. (1997);
- 300 représente le facteur d'incertitude (× 10 pour les variations entre espèces, × 10 pour la variation intraspécifique et × 3 pour les lacunes de la base de données, y compris le manque d'études portant sur la reproduction ou le développement).
À partir de la DJT dérivée de la NOAEL, il est possible de calculer un objectif basé sur la santé de la façon suivante :
Figure 7 : l'équation utilisée pour le calcul d'un objectif basé sur la santé pour l'acide monochloroacétique (MCA)
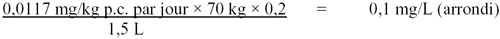
Figure 7 : l'équation utilisée pour le calcul d'un objectif basé sur la santé pour l'acide monochloroacétique (MCA) - Description textuel
L'objectif basé sur la santé pour l'acide monochloroacétique (MCA) est égale à 0,1 milligramme par litre (valeur arrondie). Cette valeur a été calculée en multipliant la dose journalière tolérable (précédemment calculée) 0,0117 milligrammes par kilogramme de poids corporel par jour par 70 kilogrammes, puis par 0,2. La valeur obtenue a par la suite été divisée par 1,5 litres pour avoir l'objectif basé sur la santé.où :
- 0,0117 mg/kg p.c. par jour représente la DJT calculée ci-dessus;
- 70 kg représente le poids moyen d'un adulte;
- 0,2 représente la proportion de la dose journalière attribuée à l'eau potable. Il s'agit d'une valeur par défaut puisqu'il n'y a pas suffisamment de données pour calculer la valeur réelle;
- 1,5 L représente la consommation quotidienne moyenne d'eau potable chez l'adulte.
11.2 Acide dichloroacétique
Des tumeurs du foie ont été signalées tant chez les souris que chez les rats au cours de plusieurs essais biologiques de cancérogénicité (Herren-Freund et coll., 1987; Bull et coll., 1990; Daniel et coll., 1992; Richmond et coll., 1995; DeAngelo et coll., 1996, 1999; Pereira et Phelps, 1996) et sont considérées comme des preuves suffisantes pour classer le DCA comme agent cancérogène pour les animaux. Le mécanisme exact de la tumorigénicité n'a pas été déterminé, et des tests portant sur le pouvoir mutagène et génotoxique ont donné des résultats surtout négatifs ou équivoques dans des systèmes de tests sur des bactéries et des mammifères. On ne sait pas pour le moment si la cancérogénicité du DCA est médiée par un mécanisme non génotoxique. Le DCA n'a pas provoqué de prolifération des peroxysomes (Tong et coll., 1998b).
Le DCA a été classé dans le Groupe II des substances probablement cancérogènes pour l'être humain, compte tenu de l'existence de preuves suffisantes chez les animaux mais non chez les êtres humains (Santé Canada, 1994). Le CIRC (2004) a classé récemment le DCA dans le Groupe 2B des substances possiblement cancérogènes pour l'être humain, en se fondant sur des preuves suffisantes de sa cancérogénicité chez les animaux de laboratoire et inadéquates chez l'être humain. Dans son édition de 2006 des normes et avis sanitaires relatifs à l'eau potable, l'U.S. EPA (2006) considère que le DCA est probablement cancérogène pour l'être humain, d'après ses Guidelines for Carcinogen Risk Assessment de 2005. Elle se base sur « les données courantes et le manque de données concluantes sur le mode d'action du DCA à des doses pertinentes sur le plan environnemental »
(U.S. EPA, 2003c). Il n'y a pas de renseignements disponibles sur les effets mutagènes du DCA chez l'être humain (CHEMINFO, 2003c).
Plusieurs essais biologiques portant sur la cancérogénicité du DCA ont signalé des tumeurs au foie, chez les souris comme chez les rats. On a choisi les tumeurs du foie (carcinomes hépatocellulaires) chez les souris pour évaluer le risque de cancer, car au cours de la seule étude disponible sur les rats, on a utilisé une dose élevée que les rats ne toléraient pas bien et qu'il a fallu réduire à plusieurs reprises. On a donc calculé les risques de cancer en fonction des résultats d'une étude adéquate de longue durée (90-100 semaines) sur l'eau potable chez des souris B6C3F1 mâles, réalisée par DeAngelo et coll. (1999). Dans le cas des tumeurs du foie (carcinomes hépatocellulaires) constatées chez les souris mâles, un rapport dose-réponse approprié a été noté. La pertinence du véhicule utilisé (eau potable), la forme du DCA administré (c.-à-d. solution neutralisée), la durée de l'étude, ainsi que l'utilisation de nombreux groupes de doses (cinq doses et un groupe témoin), sont au nombre des autres facteurs ayant motivé le choix de cette étude pour l'évaluation du risque. On n'a pas pu calculer de NOEL dans le cas de l'hépatocancérogénicité à cause de l'augmentation importante de la multiplicité des carcinomes hépatocellulaires observés à la dose la plus faible, soit 8 mg/kg p.c. par jour (0,58 comparativement à 0,28 chez les sujets témoins) (DeAngelo et coll., 1999).
Comme le DCA est classé parmi les substances probablement cancérogènes et qu'on ignore si sa cancérogénicité est médiée par un mécanisme non génotoxique, Santé Canada a décidé d'utiliser une extrapolation linéaire du risque associé à une faible dose pour calculer le risque de cancer. Cette façon de procéder concorde avec les dernières Guidelines for Carcinogen Risk Assessment de l'U.S. EPA (2005), selon lesquelles [TRADUCTION] « lorsque l'évaluation de toutes les données dont on dispose ne permet pas d'établir le mode d'action pour un siège de tumeur et lorsqu'une telle approche est scientifiquement plausible d'après les données disponibles, on a recours à une extrapolation linéaire à défaut de mieux, parce que l'extrapolation linéaire est généralement considérée comme une approche qui protège la santé. Les approches non linéaires ne devraient pas en règle générale être utilisées dans des cas où le mode d'action n'a pas été établi. »
Le risque unitaire peut donc être évalué par la méthode linéarisée à degrés multiples (Santé Canada, 2004a) en utilisant le nombre de souris B6C3F1 mâles dans chaque groupe de dose qui ont développé des carcinomes hépatocellulaires après avoir été exposées au DCA dans l'eau potable pendant 90-100 semaines (DeAngelo et coll., 1999).
Dans cette méthode, on adapte le modèle à degrés multiples aux données sur la doseréponse et on applique au risque unitaire la limite supérieure de l'intervalle de confiance à 95 % pour la composante linéaire. On calcule le modèle à degrés multiples au moyen de l'équation suivante :
Figure 1 : l'équation traduisant le modèle à degrés multiples
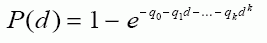
Figure 1 : l'équation traduisant le modèle à degrés multiples - Description textuel
P(d), la probabilité qu'un animal développe une tumeur à une dose donnée d, est estimée en soustrayant de 1 la constante e (2,718) exponentielle un facteur qui est fonction d'un intervalle de doses déterminées, de la courbe dose réponse appropriée et du nombre de groupes de doses utilisées dans l'étude.
où d représente la dose; k, le nombre de groupes de dose dans l'étude (à l'exclusion du groupe témoin); P(d), la probabilité que l'animal ait une tumeur à la dose d; et où qi > 0, i = 0, ..., k sont les paramètres à calculer. On entend par risque unitaire l'augmentation du risque excédentaire par dose unitaire; le risque excédentaire se calcule par l'équation
Figure 2 : L'équation utilisée pour le calcul du risque excédentaire
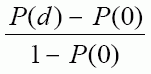
Figure 2 : L'équation utilisée pour le calcul du risque excédentaire - Description textuel
Le risque excédentaire se calcule en soustrayant de P(d) (la probabilité qu'un animal développe une tumeur à une dose donnée d) P(0) (la probabilité correspondant à l'exposition de fond), le tout divisé par 1 moins P(0).
Le risque unitaire est applicable à de très faibles doses, vraisemblablement dans la plage à laquelle les êtres humains seraient exposés. Pour une faible dose, d, on peut démontrer que le risque excédentaire équivaut à peu près à q1d. Ainsi, lorsque l'exposition de fond P(0) est faible, q1 représente la pente (c.-à-d. le changement du risque en fonction de l'augmentation de la dose unitaire) de la courbe dose-réponse dans la région à faible dose. En pratique, on utilise la limite supérieure de l'intervalle de confiance à 95 % sur q1 et on la représente par q1*. Il s'agit du risque unitaire calculé par la méthode linéarisée à degrés multiples. On a adapté le modèle à degrés multiples en utilisant la méthode THRESH (Howe, 1995) et calculé le risque unitaire par la méthode linéarisée à degrés multiples sans appliquer auparavant le facteur de rajustement cinétique. La valeur P utilisée pour les tests de manque d'ajustement (chi-carré) était de 0,87, ce qui indique que le modèle était bien adapté aux données. On a converti les unités des risques unitaires en concentrations dans l'eau potable en multipliant par le taux de consommation d'un être humain (1,5 L/jour) et en divisant par le poids corporel type d'un humain (70 kg).
Il est aussi possible d'appliquer un facteur de rajustement cinétique d'animal à humain (KA, aussi appelé facteur d'échelle allométrique, qui corrige les différences dans le poids du corps entre les animaux et les humains), que l'on calcule au moyen de l'équation suivante :
Figure 3 : l'équation utilisée pour l'ajustement allométrique de la différence de poids entre l'animal et l'humain

Figure 3 : l'équation utilisée pour l'ajustement allométrique de la différence de poids entre l'animal et l'humain - Description textuel
Le facteur de rajustement cinétique d'animal à humain est calculé en divisant le poids corporel de l'animal en kilogrammes, par le poids corporel standard de l'homme qui est de 70 kilogrammes. La valeur obtenue est élevée à l'exposant un quart (1/4).où 70 kg représente le poids corporel type d'un humain. Comme le facteur d'échelle allométrique est appliqué au risque unitaire après la modélisation, on a utilisé un poids de souris de 43,9 g dans la formule ci-dessus. Il s'agissait du poids moyen des sujets du groupe témoin.
Le risque unitaire à vie de cancer pour l'être humain associé à l'ingestion de 1 µg de DCA/L dans l'eau potable a été estimé à 1,02 × 10-6 (fondé sur la présence de tumeurs du foie et de carcinomes hépatocellulaires).
Dans le cadre des recommandations pour la qualité de l'eau potable, Santé Canada entend par « essentiellement négligeable »
une plage allant d'un nouveau cas de cancer de plus que le niveau de fond pour 100 000 personnes à un nouveau cas de cancer de plus que le niveau de fond pour 1 million de personnes (c.-à-d. 10-5 à 10-6) au cours de la durée d'une vie.
| Risque à vie | Concentration dans l'eau potable (µg/L) |
|---|---|
| 10-4 | 98,1 (arrondi à 100) |
| 10-5 | 9,81 (arrondi à 10) |
| 10-6 | 0,98 (arrondi à 1) |
À partir de la concentration la plus prudente dans l'eau potable estimée pour un risque à vie de cancer chez l'être humain de 10-5, on dérive un objectif basé sur la santé de 0,01 mg/L (10 µg/L) pour le DCA dans l'eau potable.
On a entrepris une autre analyse au cours de laquelle on a appliqué le facteur de rajustement cinétique aux doses expérimentales avant de modéliser les données. Cette opération visait à faciliter les comparaisons avec la méthodologie de l'U.S. U.S. EPA (2003c). On a adapté le modèle à degrés multiples en utilisant l'outil THRESH (Howe, 1995) et calculé les risques unitaires au moyen de la méthode linéarisée à degrés multiples après avoir appliqué au préalable le facteur de rajustement cinétique. Les résultats ont démontré que l'application du rajustement cinétique de l'animal à l'humain avant ou après l'adaptation du modèle n'avait aucun effet sur les risques unitaires calculés ni sur les concentrations.
11.3 Acide trichloroacétique
On a démontré de façon constante que le TCA administré dans l'eau potable causait l'apparition de tumeurs du foie chez la souris mais non chez le rat. Il a été établi que le mécanisme à l'origine de tumeurs du foie chez la souris repose sur la prolifération des peroxysomes, ce qui peut ou non être pertinent pour l'être humain (Cattley et coll., 1998). Le TCA a été considéré comme faiblement génotoxique.
Comme il existe des données probantes indiquant que la cancérogénicité du DBA est limitée à une seule espèce de rongeurs, la souris, et vu que les données concernant l'être humain sont inadéquates, le TCA a été classé au cours de cette évaluation dans le Groupe III des substances possiblement cancérogènes pour l'être humain (Santé Canada, 1994). Selon le CIRC (1995), il n'y a pas suffisamment de preuves de sa cancérogénicité chez l'être humain et les données sur sa cancérogénicité chez les animaux de laboratoire sont limitées. L'U.S. EPA (1986) avait classé le TCA comme agent possiblement cancérogène pour l'être humain, mais en vertu des Draft Revised Guidelines for Carcinogen Risk Assessment de 1999 (U.S. EPA, 1999b), elle a déclaré ensuite (2003b) qu'il existait des données indiquant que le TCA était cancérogène, mais qu'elles ne suffisaient pas pour évaluer sa cancérogénicité pour l'être humain.
Des études de longue durée sur l'eau potable ont mis au jour des tumeurs du foie chez la souris, mais non chez le rat. Comme on ne sait pas si le mécanisme sous-jacent est pertinent pour l'être humain, on a choisi pour évaluer le risque une étude de longue durée sur le rat avec un paramètre final autre que le cancer. L'étude sur l'eau potable d'une durée de 104 semaines réalisée par DeAngelo et coll. (1997) a aussi montré que la NOAEL la plus faible, soit 32,5 mg/kg p.c. par jour, était fondée sur des changements liés au traitement (baisse du poids corporel, augmentation de l'activité des enzymes sériques dans le foie et histopathologie hépatique comparativement aux animaux témoins). La pertinence du véhicule utilisé (eau potable), l'absence d'effets importants à faible dose, la durée de l'étude et la forme sous laquelle le TCA a été administré (c.-à-d. solution neutralisée) sont au nombre des autres raisons pour lesquelles on a choisi cette étude.
Il est possible de calculer la DJT dans le cas du TCA de la façon suivante :
Figure 4 : l'équation utilisée pour le calcul de la dose journalière tolérable (DJT) de l'acide trichloroacétique (TCA)
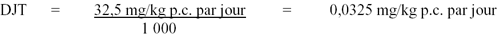
Figure 4 : l'équation utilisée pour le calcul de la dose journalière tolérable (DJT) de l'acide trichloroacétique (TCA) - Description textuel
La dose journalière tolérable (DJT) de l'acide trichloroacétique (TCA) est égale à 0,0325 milligrammes par kilogramme de poids corporel (kg pc) par jour. Cette valeur a été calculée en divisant 32,5 milligrammes par kilogramme de poids corporel par jour par 1000.où :
- 32,5 mg/kg p.c. par jour représente la NOAEL établie dans l'étude sur l'exposition chronique de rats publiée par DeAngelo et coll. (1997);
- 1 000 représente le facteur d'incertitude (× 10 pour la variation entre espèces, × 10 pour la variation intraspécifique et × 10 pour les lacunes des bases de données, y compris le manque d'études multigénérationnelles sur la reproduction ainsi que la cancérogénicité possible).
En utilisant la DJT dérivée de la NOAEL, il est possible de calculer un objectif basé sur la santé de la façon suivante :
Figure 5 : l'équation utilisée pour le calcul d'un objectif basé sur la santé pour l'acide trichloroacétique (TCA)
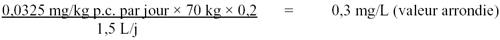
Figure 5 : l'équation utilisée pour le calcul d'un objectif basé sur la santé pour l'acide trichloroacétique (TCA) - Description textuel
La dose journalière tolérable (DJT) de l'acide trichloroacétique (TCA) est égale à 0,0325 milligrammes par kilogramme de poids corporel (kg pc) par jour. Cette valeur a été calculée en divisant 32,5 milligrammes par kilogramme de poids corporel par jour par 1000.où :
- 0,0325 mg/kg p.c. par jour représente la DJT calculée ci-dessus;
- 70 kg représente le poids moyen d'un adulte;
- 0,2 représente la proportion de la dose journalière attribuée à l'eau potable; il s'agit d'une valeur par défaut, puisqu'il n'y a pas suffisamment de données pour calculer la valeur réelle;
- 1,5 L/j représente la consommation quotidienne moyenne d'eau potable chez l'adulte.
11.4 Acide monobromoacétique
Ce document ne contient pas suffisamment de données sur la toxicité du MBA pour établir un objectif basé sur la santé. Des études d'exposition aiguë par voie orale ont démontré que le MBA présentait une toxicité aiguë. On n'a toutefois pas réalisé ni publié d'études sur l'exposition subchronique ou chronique au MBA, ni sur la cancérogénicité du MBA; on n'a pas effectué non plus d'études normalisées sur le développement ou sur de multiples générations. Les études sur la mutagénicité et la génotoxicité ont produit des résultats contradictoires.
Le MBA a été classé au cours de cette évaluation dans le Groupe IV des substances inclassables quant à leur cancérogénicité pour l'être humain, à cause de l'inadéquation des données tirées des études animales (Santé Canada, 1994). En vertu des Draft Revised Guidelines for Carcinogen Risk Assessment de 1999, l'U.S. EPA (2005a) a aussi classé les données sur le MBA comme [TRADUCTION] « inadéquates pour évaluer le potentiel cancérogène chez l'être humain »
. Le CIRC n'a pas évalué ni classé le MBA.
11.5 Acide dibromoacétique
Selon la seule étude de toxicité aiguë orale qui a été effectuée, le DBA exerce des effets modérément toxiques. Des études de toxicité subchronique et chronique semblent indiquer que le foie est l'organe cible. Des études sur la reproduction font état d'effets médiés par le sexe masculin, mais on ne dispose comme rapports d'études sur le développement que d'abrégés (données limitées), qui semblent néanmoins indiquer une fototoxicité. D'autres études doivent être effectuées afin d'évaluer convenablement les effets toxiques du DBA sur le développement. Les études de mutagénicité disponibles sont limitées et les résultats contradictoires.
Une étude récente de 2 ans sur l'eau potable chez la souris et le rat réalisée par le NTP (2007) (également publiée sous forme de résumé par Melnick et coll., 2007) a montré que le DBA avait un effet cancérogène sur plusieurs organes chez les animaux de laboratoire, des tumeurs étant induites dans le foie et le poumon des souris, dans la cavité abdominale (mésothéliomes) des rats mâles et dans le système hématopoïétique (leucémie à cellules mononucléées) des rats femelles. Santé Canada a donc classé le DBA dans le Groupe II des substances probablement cancérogènes pour l'être humain, compte tenu de l'existence de preuves suffisantes chez les animaux mais non chez les humains (Santé Canada, 1994). Le DBA n'a pas été évalué ni classé par le CIRC. En 2005, avant la publication de ces nouvelles études,l'EPA des États-Unis (2005a) a indiqué que, d'après les Draft Revised Guidelines for Carcinogen Risk Assessment de 1999, les données concernant le DBA étaient considérées « inadéquates pour une évaluation du potentiel cancérogène chez l'être humain »
.
Compte tenu de la nouvelle classification du DBA parmi les substances probablement cancérogènes, on a choisi la méthode linéarisée à degrés multiples pour calculer les risques unitaires. L'application du modèle à degrés multiples est décrite à la section 11.2.
On a adapté les modèles à degrés multiples en utilisant la méthode THRESH (Howe, 1995) et calculé les risques unitaires (Santé Canada (2007a). Un facteur de rajustement cinétique d'animal à humain (KA) a été appliqué aux risques unitaires finaux, en présumant qu'un rat pèse 0,35 kg, une souris 0,03 kg et un humain 70 kg. La formule pour le KA est également décrite à la section 11.2. Un test de manque d'ajustement chi-carré a été effectué pour les ajustements du modèle. Les degrés de liberté pour ce test équivalent à k moins le nombre de qi dont les valeurs estimatives sont autres que zéro. Une valeur P inférieure à 0,05 indique un manque d'ajustement significatif. Lorsqu'on utilisait ce critère, certains modèles présentaient un manque d'ajustement significatif en raison d'une courbe dose-réponse inégale. Bien qu'aucun modèle simple ne puisse décrire adéquatement ces données, ce type de modèles offre un ajustement visuel raisonnable. En revanche, dans le cas du DCA, le modèle s'ajuste bien aux données. Les calculs pour les risques unitaires (bruts et convertis au moyen d'un facteur d'échelle allométrique) associés au DBA et les valeurs P pour le manque d'ajustement sont présentés dans le document de Santé Canada (2007b).
Les risques unitaires estimés à vie associés à l'ingestion de 1 µg/L de DBA dans l'eau potable variaient entre 0,14 × 10-6 et 4,26 × 10-6. La plage de risque unitaire a été calculée sur la base de l'incidence des mésothéliomes observée chez les rats mâles (0,14 x 10-6), utilisée comme limite inférieure (la moins sensible), et l'incidence des adénomes/carcinomes hépatocellulaires chez les souris mâles (4,26 × 10-6), utilisée comme limite supérieure (la plus sensible).
| Risque à vie | Concentration dans l'eau potable (µg/L) |
|---|---|
| 10-4 | 23,5 - 701,6 |
| 10-5 | 2,3 - 70,2 |
| 10-6 | 0,23 - 7,0 |
À partir de la concentration la plus prudente dans l'eau potable estimée pour un risque à vie de cancer chez l'être humain de 10-5, on dérive un objectif basé sur la santé de 0,002 mg/L (2 µg/L) (arrondi) pour le DBA dans l'eau potable.
11.6 Considérations internationales
Plusieurs agences ont passé en revue les AHA et établi des lignes directrices ou des normes en fonction de divers facteurs, comme la meilleure technologie disponible ou les effets sur la santé.
L'OMS (2004a, b, 2005) a formulé des recommandations distinctes pour trois des AHA : le MCA (20 µg/L), le DCA (valeur provisoire recommandée de 50 µg/L) et le TCA (200 µg/L). Bien que l'Organisation mondiale de la santé (OMS) ait recommandé une valeur basée sur la santé de 40 µg/L pour le DCA en se fondant sur une limite supérieure de 10-5 pour le risque excédentaire à vie de cancer, la recommandation est provisoire parce que [TRADUCTION] « les données sur le traitement sont insuffisantes pour qu'on puisse garantir que la valeur de 40 µg/litre est techniquement réalisable dans une multitude de circonstances »
(OMS, 2005). Pour ce qui est du MBA et du DBA, l'OMS (2004c) a jugé que les bases de données étaient inadéquates pour qu'on puisse formuler des recommandations.
La U.S. EPA (2006) a opté pour une approche différente et établi une concentration maximale unique de contaminants de 0,06 mg/L pour les cinq AHA (AHA5) en se fondant sur la meilleure technologie disponible, de même que des concentrations maximales de contaminants cibles non exigibles de 0,03 mg/L pour le MCA, de 0 pour le DCA (en raison de sa cancérogénicité) et de 0,02 mg/L pour le TCA.
12.0 Justification / Errata
Comme les AHA présents dans l'eau potable proviennent principalement de la chloration des matières organiques qui se trouvent dans l'eau brute, il importe de reconnaître les avantages importants pour la santé qui découlent de la désinfection par chloration. Le chlore a à peu près éliminé les maladies microbiennes d'origine hydrique parce qu'il peut détruire ou inactiver presque tous les micro-organismes entériques pathogènes, y compris les bactéries et les virus intestinaux d'origine humaine. Le chlore est le désinfectant le plus facile à utiliser et à surveiller. Oxydant puissant, il peut, à l'état résiduel dans le réseau de distribution d'eau, empêcher la recroissance bactérienne. Même si l'utilisation du chlore peut entraîner la formation de SPD comme les AHA, les efforts de gestion des concentrations d'AHA dans l'eau potable ne doivent pas compromettre l'efficacité de la désinfection.
Les AHA et les THM constituent les deux principaux groupes de SPD présents dans l'eau potable et s'y trouvent en général aux concentrations les plus élevées. Il est possible d'utiliser les concentrations de ces contaminants comme indicateurs de la charge totale de tous les SPD qui peuvent être présents dans les approvisionnements d'eau potable. En l'absence d'information sur d'autres SPD, le contrôle et la gestion des AHA et des THM devraient réduire l'exposition aux autres sous-produits et le risque qui en découle. Lorsqu'on met en oeuvre des stratégies appropriées de traitement de l'eau potable pour réduire les concentrations d'AHA et de THM, le procédé peut aussi réduire les concentrations d'autres SPD halogénés.
On dispose de suffisamment de données scientifiques pour établir des objectifs basés sur la santé pour quatre AHA : le MCA, le DCA, le TCA et le DBA. Le MCA est classé parmi les substances du Groupe IV (peu susceptibles d'être cancérogènes pour l'être humain). Une valeur cible basée sur la santé de 0,1 mg/L peut être calculée pour le MCA dans l'eau potable, à partir des changements observés dans le poids du corps, du foie, du rein et des testicules chez le rat. Le DCA est classé comme une substance du groupe II (probablement cancérogène pour l'être humain), compte tenu de l'existence de preuves suffisantes chez les animaux mais non chez les humains. Une valeur cible basée sur la santé de 0,01 mg/L peut être établie pour le DCA dans l'eau potable sur la base des tumeurs du foie retrouvées chez les souris et les rats. On a classé le TCA dans le Groupe III (possiblement cancérogène pour l'être humain), compte tenu de preuves limitées de sa cancérogénicité chez les animaux de laboratoire et de preuves inadéquates chez l'être humain. Une valeur cible basée sur la santé de 0,3 mg/L peut être établie pour le TCA dans l'eau potable. Bien que des études sur des animaux aient mis en évidence un lien entre l'exposition au TCA et les tumeurs hépatiques chez les souris uniquement, on ne sait pas encore si le mécanisme à l'origine de ces tumeurs joue un rôle chez les humains. Le MBA est classé dans le Groupe VI (inclassable en ce qui concerne la cancérogénicité chez l'être humain), car les données provenant d'études sur des animaux sont inadéquates. Aucune valeur cible basée sur la santé ne peut être établie pour le MBA pour le moment. Le DBA est classé dans le Groupe II (probablement cancérogène pour l'être humain), car on dispose de preuves suffisantes chez les animaux mais inadéquates chez les humains. Une valeur cible basée sur la santé de 0,002 mg/L peut être établie pour le DBA dans l'eau potable, compte tenu des tumeurs observées au niveau de plusieurs organes chez les souris et les rats.
Des données récentes sur l'exposition au Canada à des sources d'eau de surface montrent que le DCA et le TCA sont les AHA qui présentent toujours les concentrations les plus élevées dans les réseaux de distribution canadiens. Bien que la proportion de chaque AHA varie selon les conditions, le DCA peut fréquemment représenter de 40 à 60 % de la concentration totale d'AHA. Le DBA ne constitue qu'une petite fraction (en général de moins de 6 %)Footnote 3 de la concentration totale d'AHA.
L'élimination des AHA après leur formation dans les approvisionnements d'eau potable n'est pas considérée comme la meilleure solution pour réduire l'exposition aux AHA. Le moyen le plus efficient et le plus pratique d'abaisser les concentrations d'AHA dans les eaux prêtes au débit consiste à en prévenir la formation, principalement en éliminant les précurseurs organiques. Même si des ajustements du pH peuvent aider à réduire la formation d'AHA, ils peuvent entraîner une augmentation correspondante de la formation d'autres SPD, dont les THM.
Bien qu'il soit possible d'établir des valeurs cibles basées sur la santé pour quatre des cinq AHA et compte tenu des limites techniques relatives à la réduction des concentrations de chaque AHA dans l'eau potable tout en maintenant une désinfection efficace, le Comité fédéralprovincial-territorial sur l'eau potable a fixé à 0,08 mg/L (80 µg/L) la CMA pour les AHA5 totaux dans l'eau potable sur la base d'une moyenne courante annuelle, plutôt que de formuler des recommandations individuelles. Cette approche concorde avec celle de l'U.S. EPA, qui a établi une concentration maximale de contaminants en fonction des meilleures techniques disponibles pour ces mêmes AHA.
Errata
Section 12, page 71
Bien qu'il soit possible d'établir des valeurs cibles basées sur la santé pour quatre des cinq AHA et compte tenu des limites techniques relatives à la réduction des concentrations de chaque AHA dans l'eau potable tout en maintenant une désinfection efficace, le Comité fédéral-provincial-territorial sur l'eau potable a fixé à 0,08 mg/L (80 µg/L) la CMA pour les AHA5totaux dans l'eau potable sur la base d'une moyenne courante annuelle, plutôt que de formuler des recommandations individuelles. Cette approche concorde avec celle de l'U.S. EPA, qui a établi une concentration maximale de contaminants en fonction des meilleures techniques disponibles pour ces mêmes AHA.
On établit à 0,08 mg/L la CMA pour les AHA totaux dans l'eau potable, suivant une stratégie de gestion du risque basée sur les considérations suivantes :
- 1a) Bien que la valeur cible basée sur la santé pour le DBA soit la plus faible valeur calculée, cette substance est également présente en très faibles proportions dans le mélange d'AHA et n'est pas considérée comme le facteur approprié à prendre en compte pour formuler une recommandation basée sur la santé. Les concentrations moyennes de DBA relevées sont bien en-deçà de la valeur cible basée sur la santé de 0,002 mg/L pour cette substance.
- 1b) La valeur cible basée sur la santé pour le DCA est la plus faible valeur dont il faut tenir compte. La concentration de DCA dans l'eau potable qui représente un risque « essentiellement négligeable » est de 0,01mg/L, mais elle est impossible à atteindre dans les réseaux de distribution sans compromettre l'efficacité de la désinfection.
- 2. Les LPAQ, fondées sur la capacité des laboratoires de mesurer systématiquement les AHA dans des limites raisonnables de précision et d'exactitude, varient selon chaque AHA et selon la méthode utilisée. Toutefois, toutes les valeurs pour les LPAQ sont bien en-deçà de la CMA.
- 3. Il doit être possible d'atteindre la CMA à un coût raisonnable. Les données limitées qui existent au Canada indiquent que l'on peut s'attendre à ce que 88 % des usines de traitement qui desservent des populations de plus de 5 000 habitants et 56 % de celles qui desservent des populations de moins de 5 000 habitants puissent atteindre des concentrations d'AHA de 0,08 mg/L. En optimisant les procédés de traitement et en ayant recours à des technologies avancées de traitement pour éliminer les composés organiques avant la désinfection, il sera possible d'obtenir des concentrations d'AHA5 de moins de 0,08mg/L.
Le risque estimé à vie de cancer associé à l'ingestion d'eau potable contenant des concentrations de 0,08 mg/L d'AHA est plus élevé que la plage considérée en général comme « essentiellement négligeable » (c.-à-d. entre 10-5 et 10-6). En se fondant sur l'incidence du cancer du foie dans les études animales sur le DCA, on a calculé que le risque estimé à vie de cancer associé à l'ingestion d'eau potable contenant des concentrations de 0,08 mg/L d'AHA s'élevait entre 3,2 x 10-5 et 4,8 x 10-5 pour des proportions de 40 à 60 % de DCA dans les AHA totaux, respectivement. Bien que l'exposition aux AHA à la concentration recommandée puisse présenter un risque à vie associé au DCA plus élevé que ce qui serait normalement considéré comme négligeable, ce risque est calculé en utilisant une approche très prudente qui surestime en général le risque potentiel.
Il est recommandé que les services de distribution d'eau fassent tout ce qui est en leur pouvoir pour maintenir les concentrations d'AHA au niveau le plus bas qu'il soit raisonnablement possible d'atteindre sans compromettre l'efficacité de la désinfection. Dans le cadre de son processus permanent de révision des recommandations, Santé Canada continuera de suivre les nouvelles recherches à ce sujet et recommandera au besoin toute modification jugée appropriée.
13.0 Bibliographie
Certains hyperliens donnent accès à des sites d'organismes qui ne sont pas assujettis à la Loi sur les langues officielles. L'information qui s'y trouve est donc dans la langue du site.
ACGIH (American Conference of Governmental Industrial Hygienists). 2001. Trichloroethylene BEI. CAS # 79-01-6. Dans : Documentation of the threshold limit values and biological exposure indices. 7e éd. ACGIH, Cincinnati, OH.
Allen, B.C. et Fisher, J.W. 1993. Pharmacokinetic modeling of trichloroethylene and trichloroacetic acid in humans. Risk Anal., 13(1) : 71-86.
Amacher, D.E. et Turner, G.N. 1982. Mutagenic evaluation of carcinogens and non-carcinogens in the L5178Y/TK assay utilizing postmitochondrial fractions (S9) from normal rat liver. Mutat. Res., 97 : 49-65.
Anderson, W.B., Board, P.G., Gargano, B. et Anders, M.W. 1999. Inactivation of glutathione transferase zeta by dichloroacetic acid and other fluorine-lacking alpha-haloalkanoic acids. Chem. Res. Toxicol., 12(12) : 1144-1149.
APHA (American Public Health Association), AWWA (American Water Works Association) et WEF (Water Environment Federation). 2005. Standard methods for the examination of water and wastewater. 21e éd. APHA, Washington, DC.
Aranda-Rodriguez, R., Benoit, F.M., Lebel, G.L. et Jay, B. 2002. Chlorinated disinfection by-products (CDBPs) in Canadian drinking water - a survey of 27 small systems. Dans : Dixième conférence nationale sur l'eau potable de l'ACEPU, Halifax, Nouvelle-Écosse. Association canadienne des eaux potables et usées, Ottawa, Ontario. p. 140-150.
Ashford, R.D. 1994. Ashford's dictionary of industrial chemicals. Wavelength Publications Ltd., Londres, Royaume-Uni.
Bader, E.L., Hrudey, S.E. et Froese, K.L. 2005. Urinary excretion half life of trichloroacetic acid as a biomarker of exposure to chlorinated drinking water disinfection by-products. Occup. Environ. Med., 61 : 715-716.
Bakeas, E.B., Economou, A.G., Siskos, P.A. et Frank, H. 2003. Determination of chloroacetates in atmospheric particulate matter. Environ. Sci. Technol., 37(11) : 2336-2339.
Balchak, S.K., Hedge, J.M., Murr, A.S., Mole, M.L. et Goldman, J.M. 2000. Influence of the drinking water disinfection by-product dibromoacetic acid on rat estrous cyclicity and ovarian follicular steroid release in vitro. Reprod. Toxicol., 14(6) : 533-539.
Berardi, M.R., Snyder, R., Waritz, R.S. et Cooper, K.R. 1987. Monochloroacetic acid toxicity in the mouse associated with blood-brain barrier damage. Fundam. Appl. Toxicol., 9(3) : 469-479.
BG Chemie. 1993. Monochloroacetic acid. Dans : Toxicological evaluations. Vol. 6. Potential health hazards of existing chemicals. Springer-Verlag, Berlin. p. 1-32.
Bhat, H.K et Ansari, G.A.S. 1989. Covalent interactions of chloroacetic and acetic acids with cholesterol. J. Biochem. Toxicol., 4(3) : 189-193.
Bhat, H.K., Ahmed, A.E. et Ansari, G.A.S. 1990. Toxicokinetics of monochloroacetic acid: A whole-body autoradiography study. Toxicology, 63 : 35-43.
Bhunya, S.P. et Behera, B.C. 1987. Relative genotoxicity of trichloroacetic acid (TCA) as revealed by different cytogenetic assays: bone marrow chromosome aberration, micronucleus and sperm-head abnormality in the mouse. Mutat. Res., 188 : 215-221.
Bhunya, S.P. et Das, P. 1987. Bone marrow chromosome aberration and sperm-head abnormality in mice in vivo induced by monochloroacetic acid (MCA). Chromosome Information Service, 42 : 28-30.
Bodensteiner, K.J., Veeramachaneni, R., Klinefelter, G.R., Kane, C.M., Higuchi, T.T., Moeller, C.L. et Sawyer, H.R. 2001. Chronic exposure to dibromoacetic acid (DBA), a commonly occurring disinfection by-product in drinking water, diminishes primordial follicle populations in the rabbit. Biol. Reprod., 64 : 122 (abrégé).
Boethling, R.S. et Alexander, M. 1979. Effect of concentration of organic chemicals on their biodegradation by natural microbial communities. Appl. Environ. Microbiol., 37 : 1211-1216.
Bove, F.J., Fulcomer, M.C., Klot, J.B., Esmart, J., Dufficy, E.M. et Savrin, J.E. 1995. Public drinking water contamination and birth outcomes. Am. J. Epidemiol., 141 : 850-862.
Bryant, B.J., Jokinen, M.P., Eustis, S.L., Thompson, M.B. et Abdo, K.M. 1992. Toxicity of monochloroacetic acid administered by gavage to F344 >rats and B6C3F1 mice for up to 13 weeks. Toxicology, 72(1) : 77-87.
Budavari, S., O'Neil, M.J. et Smith, A. (dir.). 1996. The Merck index: an encyclopedia of chemicals, drugs and biologicals. Merck et Co., Inc., Whitehorse, NJ. p. 2158, 3095, 9757-9758.
Bull, R.J. 1995. Carcinogenic properties of brominated haloacetates. Disinfection by-products in drinking water; critical issues in health effects research. ILSI Workshop Report. Health and Environmental Science Institute, International Life Sciences Institute, Washington, DC. p. 29.
Bull, R.J. 2000. Mode of action of liver tumor induction by trichloroethylene and its metabolites, trichloroacetate and dichloroacetate. Environ. Health Perspect., 108 (Suppl. 2) : 241-259.
Bull, R.J., Sanchez, I.M., Nelson, M.A., Larson, J.L. et Lansing, A.J. 1990. Liver tumour induction in B6C3F1 mice by dichloroacetate and trichloroacetate. Toxicology, 63 : 341-359.
Bull, R.J., Sasser, L.B. et Lei, X.C. 2004. Interaction in the tumor-promoting activity of carbon tetrachloride, trichloroacetate, and dichloroacetate in the liver of male B6C3F1 mice. Toxicology, 199 : 169-183.
Carter, J.H., Carter, H.W. et DeAngelo, A.B. 1995. Biochemical, pathologic and morphometric alterations induced in male B6C3F1 mouse liver by short-term exposure to dichloroacetic acid. Toxicol. Lett., 81 : 55-71.
Carter, J.H., Carter, H.W., Deddens, J.A., Hurst, B.M., George, M.H. et DeAngelo, A.B. 2003. A 2-year dose-response study of lesion sequences during hepatocellular carcinogenesis in the male B6C3F1 mouse given the drinking water chemical dichloroacetic acid. Environ. Health Perspect., 111(1) : 53-64.
Cattley, R.C., Deluca, J., Elcombe, C., Fenner-Crisp, P., Lake, B.G., Marsman, D.S., Pastoor, T.A., Popp, J.A., Robinson, D.E., Schwetz, B., Tugwood, J. et Wahli, W. 1998. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regul. Toxicol. Pharmacol., 27 : 47-60.
CE (Commission européenne). 2003. Monochloroacetic acid (MCAA) environmental part. Comité scientifique sur la toxicité, l'écotoxicité et l'environnement, Direction générale Santé et protection des consommateurs, CE, Bruxelles.
Chang, L.W., Daniel, F.B. et DeAngelo, A.B. 1992. Analysis of DNA strand breaks induced in rodent liver in vivo, hepatocytes in primary culture, and a human cell line by chlorinated acetic acids and chlorinated acetaldehydes. Environ. Mol. Mutagen., 20 : 277-288.
Chemada Fine Chemicals. 2002. Bromoacetic acid material safety data sheet.
CHEMINFO. 2003a. Monochloroacetic acid solutions. No CAS 79-11-8. Registre no 516. Profils chimiques établis par le Centre canadien d'hygiène et de sécurité au travail, Hamilton, Ontario.
CHEMINFO. 2003b. Monochloroacetic acid. No CAS 79-11-8. Registre no 767. Profils chimiques établis par le Centre canadien d'hygiène et de sécurité au travail, Hamilton, Ontario.
CHEMINFO. 2003c. Dichloroacetic acid. No CAS 79-43-6. Registre no 540. Profils chimiques établis par le Centre canadien d'hygiène et de sécurité au travail, Hamilton, Ontario.
CHEMINFO. 2003d. Trichloroacetic acid solutions. No CAS 76-03-9. Registre no 766. Profils chimiques établis par le Centre canadien d'hygiène et de sécurité au travail, Hamilton, Ontario.
CHEMINFO. 2003e. Trichloroacetic acid solid. No CAS 76-03-9. Registre no 539. Profils chimiques établis par le Centre canadien d'hygiène et de sécurité au travail, Hamilton, Ontario.
Christian, M.S., York, R.G., Hoberman, A.M., Diener, R.M., Fisher, L.C. et Gates, G.A. 2001. Biodisposition of dibromoacetic acid (DBA) and bromodichloromethane (BDCM) administered to rats and rabbits in drinking water during range-finding reproduction and developmental toxicity studies. Int. J. Toxicol., 20(4) : 239-253.
Christian, M.S., York, R.G., Hoberman, A.M., Frazee, J., Fisher, L.C., Brown, W.R. et Creasy, D.M. 2002. Oral (drinking water) two-generation reproductive toxicity study of dibromoacetic acid (DBA) in rats. Int. J. Toxicol., 21(4) : 237-276.
Cicmanec, J.L., Condie, L.W., Olson, G.R. et Wang, S.-R. 1991. 90-day toxicity study of dichloroacetate in dogs. Fundam. Appl. Toxicol., 17 : 376-389.
CIRC (Centre international de recherche sur le cancer). 1995. Dichloroacetic acid and trichloroacetic acid. Dans : Dry cleaning, some chlorinated solvents and other industrial chemicals. Monographies du CIRC sur l'évaluation des risques de cancérogénicité pour l'homme, Vol. 63. CIRC, Lyon.
CIRC (Centre international de recherche sur le cancer). 2004. Some drinking-water disinfectants and contaminants, including arsenic. Monographies du CIRC sur l'évaluation des risques de cancérogénicité pour l'homme, Vol. 84. CIRC, Lyon.
Cornett, R., James, M., Henderson, G., Cheung, J., Shroads, A. et Stacpoole, P. 1999. Inhibition of glutathione S-transferase and tyrosine metabolism by dichloroacetate: A potential unifying mechanism for its altered biotransformation and toxicity. Biochem. Biophys. Res. Commun., 262 : 752-756.
Cummings, A.M. et Hedge, J.M. 1998. Dibromoacetic acid does not adversely affect early pregnancy in rats. Reprod. Toxicol., 12(4) : 445-448.
Curry, S.H., Chu, P.I., Baumgartner, T.G. et Stacpoole, P.W. (1985) Plasma concentrations and metabolic effects of intravenous sodium dichloroacetate. Clin. Pharmacol. Ther., 37: 89-93.
Daniel, F.B., DeAngelo, A.B., Stober, J.A., Olson, G.R. et Page, N.P. 1992. Hepatocarcinogenicity of chloral hydrate, 2-chloroacetaldehyde and dichloroacetic acid in B6C3F1 mouse. Fundam. Appl. Toxicol., 19 : 159-168.
Daubert, T.E. et Danner, R.P. 1989. Physical and thermodynamic properties of pure chemical data compilation. Taylor et Francis, Washington, DC.
DeAngelo, A.B. et Daniel, F.B. 1990. The comparative carcinogenicity of dichloroacetic (DCA) and trichloroacetic acid (TCA) in the male B6C3F1 mouse. Toxicologist, 10 : 148 (abrégé).
DeAngelo, A.B. et Daniel, F.B. 1992. An evaluation of the carcinogenicity of the chloroacetic acids in the male F344 rat. Toxicologist, 12 : 206 (abrégé).
DeAngelo, A.B., Daniel, F.B., McMillan, L., Wernsing, P. et Savage, R.E. Jr. 1989. Species and strain sensitivity to the induction of peroxisome proliferation by chloroacetic acids. Toxicol Appl. Pharmacol., 101 : 285-298.
DeAngelo, A.B., Daniel, F.B., Stober, J.A. et Olson, G.R. 1991. The carcinogenicity of dichloroacetic acid in the male B6C3F1 mouse. Fundam. Appl. Toxicol., 16 : 337-347.
DeAngelo, A.B., Daniel, F.B., Most, B.M. et Olson, G. 1996. The carcinogenicity of dichloroacetic acid in the male Fischer 344rat. Toxicology, 114 : 207-221.
DeAngelo, A.B., Daniel, F.B., Most, B.M. et Olson, G.R. 1997. Failure of monochloroacetic acid and trichloroacetic acid administered in the drinking water to produce liver cancer in male F344/N rats. J. Toxicol. Environ. Health, 52 : 425-445.
DeAngelo, A.B., George, M.H. et House, D.E. 1999. Hepatocarcinogenicity in the male B6C3F1 mouse following a lifetime exposure to dichloroacetic acid in the drinking water: dose response determination and modes of action. J. Toxicol. Environ. Health, 58 : 485-507.
DeMarini, D.M., Perry, E. et Sheldon, M.L. 1994. Dichloroacetic acid and related compounds: induction of prophage in E. coli and mutagenicity and mutation spectra in Salmonella TA100. Mutagenesis, 9(5) : 429-437.
Demint, R.J., Pringle, J.C., Hattrup, A., Bruns, V.F. et Frank, P.A. 1975. Residues in crops irrigated with water containing trichloroacetic acid. J. Agric. Food Chem., 23(1) : 81-84.
ECETOC (Centre d'écologie et de toxicologie de l'industrie chimique européenne). 1999. Monochloroacetic acid (CAS No. 79-11-8) and its sodium salt (CAS No. 3926-62-3). Joint Assessment of Commodity Chemicals No 38. ECETOC, Bruxelles, juin.
ECETOC (Centre d'écologie et de toxicologie de l'industrie chimique européenne). 2001. Monochloroacetic acid (CAS No. 79-11-8). Human acute intoxication from monochloroacetic acid: proposals for therapy. Rapport technique no 81. ECETOC, Bruxelles, novembre.
Edwards, M. et Dudi, A. (2004) Role of chlorine and chloramine in corrosion of lead-bearing plumbing materials. J. Am. Water Works Assoc., 96(10) : 69-81.
Epstein, D.L., Nolen, G.A., Randall, J.L., Christ, S.A., Read, E.J., Stober, J.A. et Smith, M.K 1992. Cardiopathic effects of dichloroacetate in the fetal Long-Evans rat. Teratology, 46(3) : 225-235.
Evans, O.B. 1982. Dichloroacetate tissue concentrations and its relationship to hypolactatemia and pyruvate dehydrogenase activation. Biochem. Pharmacol., 31(19) : 3124-3126.
Feldhaus, K., Hudson, D., Rogers, D., Horowitz, R.S., Brent, J., Dart, R.C. et Gomez, H. 1993. Pediatric fatality associated with accidental oral administration of monochloroacetic acid (MCA). Vet. Hum. Toxicol., 35 : 344 (abrégé).
Fisher, J.W., Channel, S.R., Eggers, J.S., Johnson, P.D., MacMahon, K.L., Goodyear, C.D., Sudberry, G.L., Warren, D.A., Latendresse, J.R. et Graeter, L.J. 2001. Trichloroethylene, trichloroacetic acid, and dichloroacetic acid: do they affect fetal rat heart development? Int. J. Toxicol., 20 : 257-267.
Forkert, P.G., Lash, L., Tardif, R., Tanphaichitr, N., Vandevoort, C. et Moussa, M. 2003. Identification of trichloroethylene and its metabolites in human seminal fluid of workers exposed to trichloroethylene. Drug Metab. Dispos., 31(3) : 306-311.
Fox, A.W., Yang, X., Murli, H., Lawlor, T.E., Cifone, M.A. et Reno, F.E. 1996. Absence of mutagenic effects of sodium dichloroacetate. Fundam. Appl. Toxicol., 32 : 87-95.
Galloway, S.M., Armstrong, M.J., Reuben, C., Colman, S., Brown, B., Cannon, C., Bloom, A.D., Nakamura, F., Ahmed, M., Duk, S., Rimpo, J., Margolin, B.H., Resnick, M.A., Anderson, B. et Zeiger, E. 1987. Chromosome aberrations and sister chromatid exchanges in Chinese hamster ovary cells: evaluations of 108chemicals. Environ. Mutagen., 10 : 1-175.
Ge, R., Yang, S., Kramer, P.M., Tao, L. et Pereira, M.A. 2001. The effect of dichloroacetic acid on DNA methylation and cell proliferation in B6C3F1 mice. J. Biochem. Mol. Toxicol., 15 : 100-106.
Giller, S., Le Curieux, F., Erb, F. et Marzin, D. 1997. Comparative genotoxicity of halogenated acetic acids found in drinking water. Mutagenesis, 12(5) : 321-328.
Gonzalez-Leon, A., Schultz, I.R., Xu, G. et Bull, R.J. 1997. Pharmacokinetics and metabolism of dichloroacetate in the F344 rat after prior administration in drinking water. Toxicol. Appl. Pharmacol., 146 : 189-195.
Gosselin, R.E., Smith, R.P. et Hodge, H.C. 1984. Clinical toxicology of commercial products. 5e éd. Williams and Wilkins, Baltimore, MD.
Grant, W.M. et Schuman, J.S. 1993. Toxicology of the eye: Effects on the eyes and visual system from chemicals, drugs, metals and minerals, plants, toxins and venoms. 4e éd. C.C. Thomas, Springfield, IL. p. 993.
Gross, B.J., Branchflower, R.V., Burke, T.R., Lees, D.E. et Pohl, L. 1982. Bone marrow toxicity in vitro of chloramphenicol and its metabolites. Toxicol. Appl. Pharmacol., 64 : 557-565.
Groupe de travail sur les SPCD (sous-produits chlorés de désinfection). 2000. Chlorinated disinfection by-products. Groupe de travail sur les SPCD, Santé Canada, mars.
Hansch, C., Leo, A., Hoekman, D.H. et Heller, S. 1995. Exploring QSAR. Hydrophobic, electronic, and steric constants. ACS Professional Reference Book. American Chemical Society, Washington, DC.
Harrington-Brock, K., Doerr, C.L. et Moore, M.M. 1998. Mutagenicity of three disinfection by-products: di- and trichloroacetic acid and chloral hydrate in L5178Y/TK+/- -3.72 mouse lymphoma cells. Mutat. Res., 413 : 265-276.
Hashimoto, H., Azumar, T. et Otsuki, A. 1998. Distribution, sources, and stability of haloacetic acids in Tokyo Bay, Japan. Environ. Toxicol. Chem., 17 (5) : 798-805.
Heal, M.R., Reeves, N.M. et Cape, J.N. 2003. Atmospheric concentrations and deposition of trichloroacetic acid in Scotland: results from a 2year sampling program. Environ. Sci. Technol., 37(12) : 2627-2633.
Heithersay, G.S. et Wilson, D.F. 1988. Tissue responses in the rat to trichloroacetic acid - an agent used in the treatment of invasive cervical resorption. Aust. Dental J., 33(6) : 451-461.
Herbert, V., Gardner, A. et Colman, N. 1980. Mutagenicity of dichloroacetate, an ingredient of some formulation of pangamic acid (trade-named "vitamin B15"
). Am. J. Clin. Nutr., 33 : 1179-1182.
Herren-Freund, S.L. et Pereira, M.A. 1986. Carcinogenicity of by-products of disinfection in mouse and rat liver. Environ. Health Perspect., 69 : 59-65.
Herren-Freund, S.I., Pereira, M.A., Khoury M.D. et Olson, G. 1987. The carcinogenicity of trichloroethylene and its metabolites, trichloroacetic acid and dichloroacetic acid, in mouse liver. Toxicol. Appl. Pharmacol., 90 : 183-189.
Hodgeson, J.W. et Becker, D. 1992. Method 552.1: Determination of haloacetic acids and Dalapon in drinking water by ion exchange liquid-solid extraction and gas chromatography with an electron capture detector. Dans : Methods for the determination of organic compounds in drinking water - Supplement II. EPA/600/R-92/129. Environmental Protection Agency des États-Unis, Cincinnati, OH. Tableau 2, p. 552.1-22.
Hoechst AG. 1974. Résultats non publiés (74.0414) [cité dans OCDE, 2000].
Hoekstra, E.J. 2003. Review of concentrations and chemistry of trichloroacetate in the environment. Chemosphere, 52(2) : 355-369.
Holmes, M., Suarez, J.D., Roberts, N.L., Mole, M.L., Murr, A.S. et Klinefelter, G.R. 2001. Dibromoacetic acid, a prevalent by-product of drinking water disinfection, compromises the synthesis of specific seminiferous tubule proteins following both in vivo and in vitro exposures. J. Androl., 22(5) : 878-890.
Howe, R.B. 1995. THRESH: A computer program to compute a reference dose from quantal animal toxicity data using the benchmark dose method. ICF Kaiser Engineers, Inc., Ruston, LA.
HSDB (Hazardous Substances Data Bank). 2003. Trichloroacetic acid. Hazardous Substances Databank no 1779. U.S. National Library of Medicine, Bethesda, MD.
ILSI (International Life Sciences Institute). 1997. An evaluation of EPA's proposed guidelines for carcinogenic risk assessment using chloroform and dichloroacetate as case studies: report of an expert panel. Health and Environmental Sciences Institute, ILSI, Washington, DC, novembre.
Innes, J.M.R., Ulland, B.M., Valerio, M.G., Petrucelli, L., Fishbein, L., Hart, E.R., Pallotta, A.J., Bates, R.R., Falk, H.L., Gart, J.J., Klein, M., Mitchell, I. et Peters, J. 1969. Bioassay of pesticides and industrial chemicals for tumorigenicity in mice: a preliminary note. J. Natl. Cancer Inst., 42(6) : 1101-1114.
Izumi, M., Hirayama, Y., Sugai, K., Fukumizu, M., Hanaoka, S., Sasaki, M., Kaga, M. et Murayama, K. 2003. [Effets secondaires du dichloroacétate sur une fillette souffrant de troubles mitochondriaux.] No To Hattatsu, 35(1) : 54-58 (en japonais).
James, M.O., Yan, Z., Cornett, R., Jayanti, V.M., Henderson, G.N., Davydova, N., Katovich, M.J., Pollock, B. et Stacpoole, P.W. 1998. Pharmacokinetics and metabolism of [14C]dichloroacetate in male Sprague-Dawley rats. Identification of glycine conjugates, including hippurate, as urinary metabolites of dichloroacetate. Drug Metab. Dispos., 26(11) : 1134-1143 [cité dans U.S. EPA, 2003c].
Johnson, P.D., Dawson, B.V. et Goldberg, S.J. 1998. Cardiac teratogenicity of trichloroethylene metabolites. J. Am. Coll. Cardiol., 32(2) : 540-545.
Jones, A.R. et Wells, G. 1981. The comparative metabolism of 2-bromoethanol and ethylene oxide in the rat. Xenobiotica, 11(11) : 763-770.
Juuti, S. et Hoekstra, E. 1998. New directions: the origins and occurrence of trichloroacetic acid. Atmos. Environ., 32(17) : 3059-3060.
Kaphalia, B.S., Bhat, H.K., Khan, M.F. et Ansari, G.A.S. 1992. Tissue distribution of monochloroacetic acid and its binding to albumin in rats. Toxicol. Ind. Health, 8(1-2) : 53-61.
Kargalioglu, Y., McMillan, B.J., Minear, R.A. et Plewa, M.J. 2002. Analysis of the cytotoxicity and mutagenicity of drinking water disinfection by-products in Salmonella typhimurium. Teratogen. Carcinogen. Mutagen., 22(2) : 113-128.
Kato-Weinstein, J., Stauber, A.J., Orner, G.A., Thrall, B.D. et Bull, R.J. 2001. Differential effects of dihalogenated and trihalogenated acetates in the liver of B6C3F1 mice. J. Appl. Toxicol., 21 : 81-89.
Katz, R., Tai, C.N., Diener, R.M., McConnell, R.F. et Semonick, D.E. 1981. Dichloroacetate, sodium: 3-month oral toxicity study in rats and dogs. Toxicol. Appl. Pharmacol., 57 : 273-287.
Ketcha, M.M., Stevens, D.K., Warren, D.A., Bishop, C.T. et Brashear, W.T. 1996. Conversion of trichloroacetic acid to dichloroacetic acid in biological samples. J. Anal. Toxicol., 20 : 236-241.
Keys, D.A., Schultz, I.R., Mahle, D.A. et Fisher, J.W. 2004. A quantitative description of suicide inhibition of dichloroacetic acid in rats and mice. Toxicol. Sci., 82 : 381-393.
Kim, H. et Weisel, C.P. 1998. Dermal absorption of dichloro- and trichloroacetic acids from chlorinated water. J. Expos. Anal. Environ. Epidemiol., 8(4) : 555-575.
King, W.D., Dodds, L., Allen, A.C., Armson, B.A., Fell, D. et Nimrod, C. 2005. Haloacetic acids in drinking water and risk for stillbirth. Occup. Environ. Med., 62(2) : 124-127.
Klinefelter, G.R., Strader, L., Suarez, J., Roberts, N., Holmes, M. et Mole, L. 2000. Dibromoacetic acid, a drinking water disinfection-by-product, alters male reproductive development and fertility. Biol. Reprod., 62 (Suppl. 1) : 285 (abrégé).
Koenig, G., Lohmar, E. et Rupprich, N. 2002. Chloroacetic acids. Dans : Ullmann's encyclopedia of industrial chemistry. John Wiley & Sons, Inc.
Kohan, M.J., Huggins-Clark, G. et George, S.E. 1998. Mutagenicity of chlorinated and brominated acetic acids. 29th Annual Meeting of the Environmental Mutagen Society, Anaheim, CA, 21-26 mars. Environ. Mol. Mutagen., 31 (Suppl. 29) : 36 (abrégé).
Kondo, M., Nishihara, T., Shimamoto, T., Koshikawa, T., Itio, T., Sawamura, R. et Tanaka, T. 1988. Biodegradation test of chemicals by cultivation methods. Eisei Kagaku, 34(2) : 188-195 [cité dans HSDB, 2003].
Krasner, S.W., McGuire, M.J., Jacangelo, J.G., Patania, N.L., Reagan, K.M. et Aieta., E.M. 1989. The occurrence of disinfection by-products in US drinking water. J. Am. Water Works Assoc., 81 : 41-53.
Larson, J.L. et Bull, R.J. 1992. Metabolism and lipoperoxidative activity of trichloroacetate and dichloroacetate in rats and mice. Toxicol. Appl. Pharmacol., 115 : 268-277.
Lash, L.H., Fisher, J.W., Lipscomb, J.C. et Parker, J.C. 2000. Metabolism of trichloroethylene. Environ. Health Perspect., 108 (Suppl. 2) : 177-200.
Lewinstein, I. et Rotstein, I. 1992. Effect of trichloroacetic acid on the microhardness and surface morphology of human dentin and enamel. Endodont. Dent. Traumatol., 8 : 16-20.
Lewis, R.J., Sr. (dir.). 2001. Hawley's condensed chemical dictionary. 14e éd. John Wiley & Sons, Inc., New York, NY. p. 251, 361, 1124.
Lide, D.R. (dir.). 2003-2004. CRC handbook of chemistry and physics. 84e éd. CRC Press Inc., Boca Raton, FL.
Lin, E.L., Mattox J.K. et Daniel, F.B. 1993. Tissue distribution, excretion and urinary metabolites of dichloroacetic acid in the male Fischer 344 rat. J. Toxicol. Environ. Health, 38(1) : 19-32.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Suarez, J.D. et Dyer, C.J. 1994a. Acute spermatogenic effects of bromoacetic acids. Fundam. Appl. Toxicol., 22(3) : 422-430.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Suarez, J.D., Roberts, N.L. et Dyer, C.J. 1994b. Spermatotoxicity of dibromoacetic acid in rats after 14daily exposures. Reprod. Toxicol., 8(3) : 251-259.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Narotsky, M.G., Suarez, J.D., Roberts, N.L. et Perreault, S.D. 1995. Dibromoacetic acid affects reproductive competence and sperm quality in the male rat. Fundam. Appl. Toxicol., 28(1) : 9-17.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Suarez, J.D. et Roberts, N. 1997a. Spermatotoxicity of dichloroacetic acid. Reprod. Toxicol., 11(5) : 681-688.
Linder, R.E., Klinefelter, G.R., Strader, L.F., Veeramachaneni, D.N.R., Roberts, N.L. et Suarez, J.D. 1997b. Histopathologic changes in the testes of rats exposed to dibromoacetic acid. Reprod. Toxicol., 11(1) : 47-56.
Lingohr, M.K., Thrall, B.D. et Bull, R.J. 2001. Effects of dichloroacetate (DCA) on serum insulin levels and insulin-controlled signaling proteins in livers of male B6C3F1 mice. Toxicol. Sci., 59 : 178-184.
Lingohr, M.K., Bull, R.J., Kato-Weinstein, J. et Thrall, B.D. 2002. Dichloroacetate stimulates glycogen accumulation in primary hepatocytes through insulin-independent mechanism. Toxicol. Sci., 68 : 508-515.
Lukas, G., Vyas, K.H., Brindle, S.D., LeSher, A.R. et Wagner, W.E., Jr. 1980. Biological disposition of sodium dichloroacetate in animals and humans after intravenous administration. J. Pharmacol. Sci., 69(4) : 419-421.
Lumpkin, M.H., Bruckner, J.V., Campbell, J.L., Dallas, C.E., White, C.A. et Fisher, J.W. 2003. Plasma binding of trichloroacetic acid in mice, rats, and humans under cancer bioassay and environmental exposure conditions. Drug Metab. Dispos., 31(10) : 1203-1207.
Lytle, D.A. et Schock, M.R. 2005. Formation of Pb(IV) oxides in chlorinated water. J. Am. Water Works Assoc., 97(11) : 102-114.
MacKay, J.M., Griffiths, K., Fox, D.A., Howard, C.A., Coutts, C., Wyatt, I. et Styles, J.A. 1995. Trichloroacetic acid: investigation into the mechanism of chromosomal damage in the in vitro human lymphocyte cytogenetic assay and the bone marrow micronucleus test. Carcinogenesis, 16 : 1127-1133.
Maruthamuthu, P. et Huie, R.E. 1995. Ferric ion assisted photooxidation of haloacetates. Chemosphere, 30(11) : 2199-2207.
Mather, G.G., Exon, J.H. et Koller, L.D. 1990. Subchronic 90day toxicity of dichloroacetic and trichloroacetic acid in rats. Toxicology, 64 : 71-80.
Matsuda, H., Ose, Y., Nagase, H., Sato, T., Kito, H. et Sumida, K. 1991. Mutagenicity of ozonation and chlorination products from p-hydroxybenzaldehyde. Sci. Total Environ., 103 : 141-149.
McCay, J.A., Musgrove, D.L., Brown, R.D., Johnson, N.A., Karrow, N.A., Guo, T.L., Germolec, D.R. et White, K.L., Jr. 2000. Exposure to disinfection by-product dibromoacetic acid does not alter immune function of host resistance. Toxicologist, 54 : 157 (abrégé).
McGregor, D.B., Martin, R., Cattanach, P., Edwards, I., McBride, D. et Caspary, W.J. 1987. Responses of the L5178Y tk+/tk- mouse lymphoma cell forward mutation assay to coded chemicals. Dans : Results for nine compounds. Environ. Mutagen., 9(2) : 143-160.
Meier, J.R., Stewart, B.E. et Blazak, W.F. 1997. Genotoxicity studies of sodium dichloroacetate and sodium trichloroacetate. Environ. Sci., 5(2) : 95-108.
Meister, R.T. (dir.). 2002. Farm chemicals handbook. Vol. 88. Meister Publishing Co., Willoughby, OH. p. C-380.
Melnick, R.L., Nyska, A., Foster, P.M., Roycroft, J.H. et Kissling, G.E. 2007. Toxicity and carcinogenicity of the water disinfection byproduct, dibromoacetic acid, in rats and mice. Toxicology, 230(2-3) : 126-136.
Meusel, M. et Rehm, H.J. 1993. Biodegradation of dichloroacetic acid by freely suspended and adsorptive immobilized Xanthobacter autotrophicus GJ10 in soil. Appl. Microbiol. Biotechnol., 40 : 165-171 [cité dans Williams et coll., 1998].
Miller, J.H., Minard, K., Wind, R.A., Orner, G.A., Sasser, L.B. et Bull, R.J. 2000. In vivo MRI measurements of tumor growth induced by dichloroacetate: implications for mode of action. Toxicology, 145 : 115-125.
Miller, J.W. et Uden, P.C. 1983. Characterization of nonvolatile chlorination products of humic substances. Environ. Sci. Technol., 17(3) : 150-157.
Mills, C.J., Bull, R.J., Cantor, K.P., Reif, J., Hrudey, S.E., Huston, P. et un groupe d'experts. 1998. Risques pour la santé liés à la consommation de sous-produits de la chloration de l'eau potable : rapport d'un groupe d'experts. Mal. Chron. Can., 19(3).
Ministère de la Conservation du Manitoba. 2004. Communication personnelle de D. Rocan, ministère de la Conservation du Manitoba, Winnipeg, Manitoba, à N. Edmonds, Santé Canada, Ottawa, Ontario.
Ministère de l'Environnement et de l'Énergie de l'Ontario. 2003. Communication personnelle de A. Socha, ministère de l'Environnement et de l'Énergie de l'Ontario, Toronto, Ontario.
Ministère de l'Environnement de Terre-Neuve-et-Labrador. 2003. Communication personnelle de M. Goebel, ministère de l'Environnement de Terre-Neuve-et-Labrador, St. John's, Terre-Neuve-et-Labrador, à N. Edmonds, Santé Canada, Ottawa, Ontario.
Monster, A.C., Boersma, G. et Duba, W.C. 1979. Kinetics of trichloroethylene in repeated exposure of volunteers. Int. Arch. Occup. Environ. Health, 42 : 283-292.
Moore, G.W., Swift, L.L., Rabinowitz, D., Crofford, O.B., Oates, J.A. et Stacpoole, P.W. 1979. Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Atherosclerosis, 33 : 285-293.
Morita, H., Tadaki, S., Nozaka, T., Omura, T., Haga, M. et Tanaka, A. 1997. Mutagenicity and concentration of chlorination by-products in tap water in Saitama. Environ. Mutagen. Res., 19 : 127-134.
Morris, E.D. et Bost, J.C. 2002. Acetic acid, halogenated derivatives. Dans : Kirk-Othmer encyclopedia of chemical technology. 5e éd. John Wiley & Sons, Inc.
Morrison, J. 1946. Toxicity of certain halogen substituted aliphatic acids for white mice. J. Pharmacol. Exp. Ther., 86 : 336-338.
Morrison, J.L. et Leake, C.D. 1941. Monochloroacetic acid as a food beverage stabilizer. Univ. Calif. Publ. Pharmacol. J., 1 : 397-421.
Moser, V.C., Phillips, P.M., Levine, A.B., McDaniel, K.L., Sills, R.C., Jortner, B.S. et Butt, M.T. 2004. Neurotoxicity produced by dibromoacetic acid in drinking water of rats. Toxicol. Sci., 79 : 112-122.
Mowrer, J. et Nordin, J. 1987. Characterization of halogenated organic acids in flue gases from municipal waste incinerators. Chemosphere, 16(6) : 1181-1192.
Muller, G., Spassovski, M. et Henschler, D. 1974. Metabolism of trichloroethylene in man.II. Pharmacokinetics of metabolites. Arch. Toxicol., 32 : 283-295.
Munch, D.J., Munch, J.W. et Pawlecki, A.M. 1995. Method 552: Determination of haloacetic acids and Dalapon in drinking water by liquid-liquid extraction, derivatization, and gas chromatography with electron capture detection. Dans : Methods for the determination of organic compounds - Supplement III. EPA/600/R-95/131. Environmental Protection Agency des États-Unis, Cincinnati, OH.
Murr, A.S. et Goldman, J.M. 2005. Twenty-week exposures to the drinking water disinfection by-product dibromoacetic acid: reproductive cyclicity and steroid concentrations in the female Sprague-Dawley rat. Reprod. Toxicol., 20 : 73-80.
Narotsky, M.G., Hamby, B.T., Best, D.S. et Hunter, E.S. 1996. In vivo developmental effects of dibromoacetic acid (DBA) and dichloroacetic acid (DCA) in mice. Teratology, 53 : 96-97 (abrégé).
Narotsky, M.G., Hamby, B.T. et Best, D.S. 1997. Developmental effects of dibromoacetic acid (DBA) in a segment II study in mice. Teratology, 55(1) : 67 (abrégé).
Nelson, M.A. et Bull, R.J. 1988. Induction of strand breaks in DNA by trichloroethylene and metabolites in rat and mouse liver in vivo. Toxicol. Appl. Pharmacol., 94 : 45-54.
Nelson, M.A., Lansing, A.J., Sanchez, I.M., Bull, R.J. et Springer, D.L. 1989. Dichloroacetic acid and trichloroacetic acid-induced DNA strand breaks are independent of peroxisome proliferation. Toxicology, 58 : 239-248.
Nikolaou, A.D., Kostopoulou, M.N. et Lekkas, T.D. 1999. Organic by-products of drinking water chlorination. Global Nest: Int. J., 1(3) : 143-156.
NIOSH (National Institute for Occupational Safety and Health). 1990. Données provisoires non publiées, à partir du 7/1/90. National Occupational Exposure Survey (1981-83). NIOSH. Cincinnati, OH [cité dans OMS, 2004c].
NTP (National Toxicology Program). 1992. Toxicology and carcinogenesis studies of monochloroacetic acid (CAS No. 79-11-8) in F344/N rats and B6C3F1 mice (gavage studies). Publication NIH no 92-2851. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
NTP (National Toxicology Program). 1999a. The immunotoxicity of dibromoacetic acid (CAS No. 631-64-1). Doserange-finding study in female B6C3F1 mice. Étude NTP no IMM98001. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services. Résumé disponible sur : http://ntp.niehs.nih.gov/index.cfm?objectid=03E8C5E4-A831-D3E7-BCD72E17943C93CA.
NTP (National Toxicology Program). 1999b. Final range-finding report. Immunotoxicity of dibromoacetic acid in female B6C3F1 mice. Rapport final soumis au NTP par K.L. White et J.A. Munson, Medical College of Virginia [cité dans U.S. EPA, 2005a].
NTP (National Toxicology Program). 2000. Final report on the subchronic toxicity study of dichloroacetic acid to Fischer-344 rats and B6C3F1 mice. Rapport soumis au NTP sous le no de contrat N01-ES-85420, septembre 1999. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
NTP (National Toxicology Program). 2003a. Dichloroacetic acid (sommaire). Clinical Evaluation Committee Rapport préliminaire soumis par Arthur D. Little, Inc. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services.
NTP (National Toxicology Program). 2003b. Testing status for bromoacetic acid. M920034. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services. Disponible sur : http://ntp.niehs.nih.gov/index.cfm?objectid=BDA478F3-123F-7908-7BF175759DDA357A.
NTP (National Toxicology Program ). 2007. NTP technical report on the toxicology and carcinogenesis studies of dibromoacetic acid (CAS No. 631-64-1) in F344/N rats and B6C3F1 mice (drinking water studies). NTP Rapport technique Series No. 537. NTP, National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services. Disponible sur : http://ntp.niehs.nih.gov/index.cfm?objectid=8831333E-F1F6-975E-71D4F287C2229308
OCDE (Organisation de coopération et de développement économiques). 2000. SIDS initial assessment profile. Trichloroacetic acid. CAS No. 76-03-9. Vol. 6, Partie II, juin. Publication du Programme des Nations-Unies pour l'environnement.
OEHHA (Office of Environmental Health Hazard Assessment). 1999. Evidence on the carcinogenicity of trichloroacetic acid and its salts (Version PDF - 137 Ko) . Juillet. Reproductive and Cancer Hazard Assessment Section, OEHHA, California Environmental Protection Agency.
OMS (Organisation mondiale de la santé). 2004a. Monochloroacetic acid in drinking water (Version PDF - 74 Ko). WHO/SDE/WSH/03.04/85. OMS, Genève. Disponible sur : http://www.who.int/water_sanitation_health/dwq/chemicals/monochloroaceticacid.pdf.
OMS (Organisation mondiale de la santé). 2004b. Trichloroacetic acid in drinking water (Version PDF - 153 Ko). WHO/SDE/WSH/03.04/120. OMS, Genève.
OMS (Organisation mondiale de la santé). 2004c. Brominated acetic acids in drinking water (Version PDF - 144 Ko). WHO/SDE/WSH/03.04/79. OMS, Genève.
OMS (Organisation mondiale de la santé). 2005. Dichloroacetic acid in drinking water (Version PDF - 184 Ko). WHO/SDE/WSH/05.08/121. OMS, Genève.
Ono, Y., Somiya, I. et Kawamura, M. 1991. The evaluation of genotoxicity using DNA repairing test for chemicals produced in chlorination and ozonation processes. Water Sci. Technol., 23(1-3) : 329-338.
Parnell, M.J., Koller, L.D., Exon, J.H. et Arnzen, J.M. 1986. Trichloroacetic acid effects on rat liver peroxisomes and enzyme-altered foci. Environ. Health Perspect., 69 : 73-79.
Parnell, M.J., Exon, J.H. et Koller, L.D. 1988. Assessment of hepatic initiation-promotion properties of trichloroacetic acid. Arch. Environ. Contam. Toxicol., 17 : 429-436.
Parrish, J.M., Austin, E.W., Stevens, D.K., Kinder, D.H. et Bull, R.J. 1996. Haloacetate-induced oxidative damage to DNA in the liver of male B6C3F1 mice. Toxicology, 110 : 103-111.
Pereira, M.A. 1995. Effect of dichloroacetic acid and trichloroacetic acid on cell proliferation in B6C3F1 mice. American Water Works Association Research Foundation et American Water Works Association, Denver, CO.
Pereira, M.A. 1996. Carcinogenic activity of dichloroacetic acid and trichloroacetic acid in the liver of female B6C3F1 mice. Fundam. Appl. Toxicol., 31 : 192-199.
Pereira, M.A. et Phelps, J.B. 1996. Promotion by dichloroacetic acid and trichloroacetic acid of N-methyl-Nnitrosourea-initiated cancer in the liver of female B6C3F1 mice. Cancer Lett., 102 : 133-141.
Pereira, M.A., Li, K. et Kramer, P.M. 1997. Promotion by mixtures of dichloroacetic acid and trichloroacetic acid of N-methyl-N-nitrosourea-initiated cancer in the liver of female B6C3F1 mice. Cancer Lett., 115 : 15-23.
Pereira, M.A., Wang, W., Kramer, P.M. et Tao, L. 2004. Prevention by methionine of dichloroacetic acid-induced liver cancer and DNA hypomethylation in mice. Toxicol. Sci., 77 : 243-248.
Peters, R.J. 2003. Chloroacetic acids in European soils and vegetation. J. Environ. Monit., 5(2) : 275-280.
Phillips, M., Levine, A., McDaniel, K.L., Sills, R.C. et Moser, V.C. 2002. Neurotoxicity produced by dibromoacetic acid in drinking water of rats. Toxicologist, 66 (1-S) : 251 (abrégé).
PISC (Programme international sur la sécurité des substances chimiques). 2000. Disinfectants and disinfectant by-products. Critère d'hygiène de l'environnement 216. Organisation mondiale de la santé, Genève.
Plewa, M.J., Kargalioglu,Y., Vankerk, D., Minear, R.A. et Wagner, E.D. 2002. Mammalian cell cytotoxicity and genotoxicity analysis of drinking water disinfection by-products. Environ. Mol. Mutagen., 40 : 134-142.
Ploeg, J., Hall, G. et Janssen, D. 1991. Characterization of haloacid dehalogenases from Xanthobacter autotrophicus GJ10 and sequencing of dhIB gene. J. Bacteriol., 173 : 7925-7933 [cité dans Williams et coll., 1998].
Pourmoghaddas, H. et Stevens, P.C. 1995. Relationship between trihalomethanes and haloacetic acids with total organic halogen during chlorination. Water Res., 29 : 2059-2062.
Randall, J.L., Christ, S.A., Horton Perez, P., Nolen, G.A., Read, E.J. et Smith, M.K. 1991. Developmental effects of 2-bromoacetic acid in the Long Evans rat. Teratology, 43(5) : 454 (abrégé).
Rannug, U., Gothe, R. et Wachtmeister, C.A. 1976. The mutagenicity of chloroethylene oxide, chloroacetaldehyde, 2-chloroethanol and chloroacetic acid, conceivable metabolites of vinyl chloride. Chem. Biol. Interact., 12(3-4) : 251-263.
Raymer, J.H., Pellizzari, E.D. et Hu, Y. 2001. Exposures to water disinfection byproducts via food. National Center for Environmental Research STAR Drinking Water Progress Review Meeting, 22-23 février. Environmental Protection Agency des États-Unis [cité dans OMS, 2004a,b, 2005].
Reckhow, D.A., Singer, P.C. et Malcolm, R.L. 1990. Chlorination of humic materials: by-product formation and chemical interpretations. Environ. Sci. Technol., 24 : 1655-1664.
Reid Crowther & Partners Ltd. 2000. Canadian water treatment study: water treatment and disinfection byproducts. Winnipeg, Manitoba, septembre.
Reimann, S., Grob, K. et Frank, H. 1996. Environmental chloroacetic acids in foods analyzed by GC-ECD. Mitt. Geb. Lebensmittelunters. Hyg., 87(2) : 212-222 [cité dans OMS, 2004a,b, 2005].
Richardson, S.D., Thurston, A.D., Caughran, T.V., Chen, P.H., Collette, T.W. et Floyd, T.L. 1999. Identification of new drinking water disinfection byproducts formed in the presence of bromide. Environ. Sci. Technol., 33 : 33783383.
Richmond, R.E., Carter, J.H., Carter, H.W., Daniel, F.B. et DeAngelo, A.B. 1995. Immunohistochemical analysis of dichloroacetic acid (DCA)-induced hepatocarcinogenesis in male Fischer (F344) rats. Cancer Lett., 92 : 67-76.
Rogers, D.R. 1995. Accidental fatal monochloroacetic acid poisoning. Am. J. Forensic Med. Pathol., 16(2): 115-116.
Saghir, S.A. et Rozman, K.K. 2003. Kinetics of monochloroacetic acid at subtoxic doses in rats after single oral and dermal administrations. Toxicol. Sci., 76 : 51-64.
Saito, H., Isoda, S., Kato, M. et Nagaoka, N. 1995. Mutagenic activity of indoor swimming pool water. Environ. Mutat. Res. Commun., 17 : 169-177.
Santé Canada. 1994. Loi canadienne sur la protection de l'environnement. L'évaluation du risque à la santé humaine des substances d'intérêt prioritaire. No catalogue En40-215/41E. Ministère de l'Approvisionnement et des Services Canada, Ottawa, Ontario.
Santé Canada. 1995. Étude nationale sur les sous-produits de désinfection chlorés dans l'eau potable au Canada. Rapport no 95-DHM-197. Direction de l'hygiène du milieu, Direction générale de la protection de la santé, Santé Canada, Ottawa, Ontario.
Santé Canada. 2003. Communication personnelle de R. Aranda-Rodriguez, Division de la recherche en chimie, Bureau de la science de la santé environnementale, Programme de la sécurité des milieux, Direction générale de la santé environnementale et de la sécurité des consommateurs, Santé Canada, Ottawa, Ontario.
Santé Canada. 2004a. Unit risk for dichloroacetic acid (DCA) in drinking water. Rapport préparé par M. Walker, Unité biostatistique, Direction générale de la santé environnementale et de la sécurité des consommateurs, Santé Canada, Ottawa, Ontario.
Santé Canada. 2004b. Benchmark dose for TCA in drinking water. Rapport préparé par M. Walker, Unité biostatistique, Direction générale de la santé environnementale et de la sécurité des consommateurs, Santé Canada, Ottawa, Ontario.
Santé Canada. 2007a. Unit risk for dibromoacetic acid (DBA) in drinking water. Rapport préparé par M. Walker, Unité biostatistique, Direction générale de la santé environnementale et de la sécurité des consommateurs, Santé Canada, Ottawa, Ontario, 15 février.
Santé Canada. 2007b. Calculations for the unit risk (raw and converted) for dibromoacetic acid (DBA) in drinking water. Rapport préparé par M. Walker, Unité biostatistique, Direction générale de la santé environnementale et de la sécurité des consommateurs, Santé Canada, Ottawa, Ontario, 16 février.
Sawada, M., Sofuni, T. et Ishidate, M., Jr. 1987. Cytogenetic studies on 1,1-dichloroethylene and its two isomers in mammalian cells in vitro and in vivo. Mutat. Res., 187 : 157-163.
Schock, M.R. et Giani, R. 2004. Oxidant/disinfectant chemistry and impacts on lead corrosion. Dans : Proceedings of the 2004American Water Works Association Water Quality Technology Conference, San Antonio, TX.
Schroll, R., Bierling, B., Cao, G., Dorfler, U., Lahaniati, M., Langenbach, T., Scheunert, I. et Winkler, R. 1994. Uptake pathways of organic chemicals from soil by agricultural plants. Chemosphere, 28(2) : 297-303 [cité dans OMS, 2004b].
Schultz, I.R., Merdink, J.L., Gonzalez-Leon, A. et Bull, R.J. 1999. Comparative toxicokinetics of chlorinated and brominated haloacetates in F344 rats. Toxicol. Appl. Pharmacol., 158(2) : 103-114.
Schultz, I.R., Merdink, J.L., Gonzalez-Leon, A. et Bull, R.J. 2002. Dichloroacetate toxicokinetics and disruption of tyrosine catabolism in B6C3F1 mice: dose-response relationships and age as a modifying factor. Toxicology, 173 : 229-247.
Scott, B.F., Spencer, C., Marvin, C.H., Mactavish, D.C. et Muir, D.C.G. 2002. Distribution of haloacetic acids in water columns of the Laurentian Great Lakes and Lake Malawi. Environ. Sci. Technol., 36 : 1893-1898.
Serjeant, E.P. et Dempsey, B. 1979. Ionisation constants of organic acids in aqueous solution. International Union of Pure and Applied Chemistry (IUPAC) Chemical Data Series No. 23. Pergamon Press, New York, NY. p. 989.
Sidebottom, H. et Franklin, J. 1996. The atmospheric fate and impact of hydrochlorofluorocarbons and chlorinated solvents. Pure Appl. Chem., 68(9) : 1757-1769.
Singer, P.C. 1993. Formation and characterization of DBPs. Dans : Safety of water disinfection: balancing chemical and microbial risks. G.F. Craun (dir.). ILSI Press, Washington, DC.
Smith, M.K., Randall, J.L., Read, E.J. et Stober, J.A. 1989. Teratogenic activity of trichloroacetic acid in the rat. Teratology, 40 : 445-451.
Smith, M.K., Randall, J.L., Read, E.J. et Stober, J.A. 1990. Developmental effects of chloroacetic acid in the Long-Evans rat. Teratology, 41(5) : 593.
Smith, M.K., Randall, J.L., Read, E.J. et Stober, J.A. 1992. Developmental toxicity of dichloroacetate in rat. Teratology, 46(3) : 217-223.
Snyder, R.D., Pullman, J., Carter, J.H., Carter, H.W. et DeAngelo, A.B. 1995. In vivo administration of dichloroacetic acid suppresses spontaneous apoptosis in murine hepatocytes. Cancer Res., 55(17) : 3702-3705.
Spruijt, L., Naviaux, R.K., McGowan, K.A., Nyhan, W.L., Sheean, G., Haas, R.H. et Barshop, B.A. 2001. Nerve conduction changes in patients with mitochondrial diseases treated with dichloroacetate. Muscle-Nerve, 24(7) : 916-924.
Stacpoole, P.W. 1989. The pharmacology of dichloroacetate. Metabolism, 38(11) : 1124-1144.
Stacpoole, P.W., Gonzalez, M.G., Vlasak, J., Oshiro, Y. et Bodor, N. 1987. Dichloroacetate derivatives. Metabolic effects and pharmacodynamics in normal rats. Life Sci., 41 : 2167-2176.
Stacpoole, P.W., Hardwood, H.J., Jr., Cameron, D.F., Curry, S.H., Samuelson, D.A., Cornwell, P.E. et Sauberlich, H.E. 1990. Chronic toxicity of dichloroacetate: possible relation to thiamine deficiency in rats. Fundam. Appl. Toxicol., 14 : 327-337.
Stacpoole, P.W., Henderson, G.N., Yan, Z. et James, M.O. 1998a. Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect., 106 (Suppl. 4) : 989-994.
Stacpoole, P.W., Henderson, G.N., Yan, Z., Cornett, R. et James, M.O. 1998b. Pharmacokinetics, metabolism, and toxicology of dichloroacetate. Drug Metab. Rev., 30(30) : 499-539.
Stauber, A.J. et Bull, R.J. 1997. Differences in phenotype and cell replicative behavior of hepatic tumors by dichloroacetate (DCA) and trichloroacetate (TCA). Toxicol. Appl. Pharmacol., 144 : 235-246.
Stauber, A.J., Bull, R.J. et Thrall, B.D. 1998. Dichloroacetate and trichloroacetate promote clonal expansion of anchorage-independent hepatocytes in vivo and in vitro. Toxicol. Appl. Pharmacol., 150 : 287-294.
Stratton, C.E., Ross, W.E. et Chapman, S. 1981. Cytotoxicity and deoxyribonucleic acid damage associated with bromoacetate. Biochem. Pharmacol., 30 : 1497-1500.
Sutinen, S., Juuti, S., Koivisto, L., Turunen, M. et Ruuskanen, J. 1995. The uptake of and structural changes induced by trichloroacetic acid in the needles of Scot pine seedlings. J. Exp. Bot., 46(290) : 1223-1231 [cité dans OMS, 2004b].
Tao, L., Kramer, P.M., Ge, R. et Pereira, M.A. 1998. Effect of dichloroacetic acid and trichloroacetic acid on DNA methylation in liver tumors of female B6C3F1 mice. Toxicol. Sci., 43 : 139-144.
Tao, L., Yan, S., Xie, M., Kramer, P.M. et Pereira, M.A. 2000. Effect of trichloroethylene and its metabolites, dichloroacetic acid and trichloroacetic acid, on the methylation and expression of c-jun and c-myc protooncogenes in mouse liver: Prevention by methionine. Toxicol. Sci., 54 : 399-407.
Templin, M.V., Parker, J.C. et Bull, R.J. 1993. Relative formation of dichloroacetate and trichloroacetate from trichloroethylene in male B6C3F1 mice. Toxicol. Appl. Pharmacol., 123 : 1-8.
Thai, S.F., Allen, J.W., DeAngelo, A.B., George, M.H. et Fuscoe, J.C. 2003. Altered gene expression in mouse livers after dichloroacetic acid exposure. Mutat. Res., 543(2) : 167-180.
Tomlin, C. (dir.). 1994. The pesticide manual: world compendium. 10e éd. British Crop Protection Council, Surrey, and the Royal Society of Chemistry, Cambridge. p. 940-994.
Tong, Z., Board, P.G. et Anders, M.W. 1998a. Glutathione transferase zeta-catalyzed biotransformation of dichloroacetic acid and other alpha-haloacids. Chem. Res. Toxicol., 11 : 1332-1338.
Tong, Z., Board, P.G. et Anders, M.W. 1998b. Glutathione transferase zeta catalyzes the oxygenation of the carcinogen DCA to glyoxylic acid. Biochem. J., 331(2) : 371-374.
Toth, G.P., Kelty, K.C., George, E.L., Read, E.J. et Smith, M.K. 1992. Adverse male reproductive effects following subchronic exposure of rats to sodium dichloroacetate. Fundam. Appl. Toxicol., 19 : 57-63.
Tsuchiya, T., Ooyama, N., Murakami, T., Sano, F., Sugimoto, J. et Mutai, M. 2000. Collaborative work to evaluate toxicity on male reproductive organs by repeated dose studies in rats. Effects of 2- and 4-week repeated-dosing of dibromoacetic acid. J. Toxicol. Sci., 25 (numéro spécial) : 241-249.
Uchiyama, H., Nakajima, T., Yagi, O. et Nakahara T. 1992. Role of heterotrophic bacteria in complete mineralization of trichloroethylene by Methylocystis sp. strain M. Appl. Environ. Microbiol., 58(9) : 3067-3071 [cité dans Williams et coll., 1998].
U.S. EPA (Environmental Protection Agency des États-Unis). 1986. Guidelines for carcinogen risk assessment, 1986. Fed. Regist., 51(185) : 33992-34003. Disponible sur : http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?deid=54933.
U.S. EPA (Environmental Protection Agency des États-Unis). 1991. Toxicology of the chloroacetic acids, by-products of the drinking water disinfection process. II. The comparative carcinogenicity of dichloroacetic acid and trichloroacetic acid: implication for risk assessment. Document no HERL-0820. No 3101. Health Effects Research Laboratory, U.S. EPA, Research Triangle Park, NC.
U.S. EPA (Environmental Protection Agency des États-Unis). 1992. Methods for the determination of organic compounds in drinking water - Supplement II. EPA-600/R-92/129. U.S. EPA, Cincinnati, OH.
U.S. EPA (Environmental Protection Agency des États-Unis). 1994. Final draft for the criteria document on chlorinated acids/aldehydes/ketones/alcohols. EPA 68-C2-0139. Préparé pour la Health and Ecological Criteria Division, Office of Science and Technology, Office of Water, U.S. EPA, Washington, DC.
U.S. EPA (Environmental Protection Agency des États-Unis). 1995. Methods for the determination of organic compounds in drinking water - Supplement III. EPA-600/R-95/131. U.S. EPA, Cincinnati, OH.
U.S. EPA (Environmental Protection Agency des États-Unis). 1999a. EPA guidance manual - Alternative disinfectants and oxidants. EPA-815-R-99-014. U.S. EPA, Washington, DC, avril.
U.S. EPA (Environmental Protection Agency des États-Unis). 1999b. Draft revised guidelines for carcinogen risk assessment. U.S. EPA, Washington, DC, juillet. Disponible sur : http://cfpub.epa.gov/ncea/raf/cancer.cfm.
U.S. EPA (Environmental Protection Agency des États-Unis). 1999c. Microbial and Disinfection Byproduct Rules Simultaneous Compliance Guidance Manual. EPA-815-R-99-015. U.S. EPA, Washington, DC, août.
U.S. EPA (Environmental Protection Agency des États-Unis). 2003a. Approved methods for microorganisms, disinfectants and disinfection byproducts). U.S. EPA, Washington, DC, juin.
U.S. EPA (Environmental Protection Agency des États-Unis). 2003b. Addendum to drinking water criteria document for monochloracetic acid and trichloroacetic acid (final). Office of Science and Technology, Office of Water, U.S. EPA, Washington, DC, juillet.
U.S. EPA (Environmental Protection Agency des États-Unis). 2003c. Toxicological review of dichloroacetic acid (Version PDF - 1706 Ko) (CAS No. 79-43-6). In support of summary information on the Integrated Risk Information System (IRIS). EPA 635/R-03/007. National Center for Environmental Assessment, U.S. EPA, Washington, DC. Août.
U.S. EPA (Environmental Protection Agency des États-Unis). 2003d. National Primary Drinking Water Regulations: Stage 2 Disinfectants and Disinfection Byproducts Rule; National Primary and Secondary Drinking Water Regulations: Approval of Analytical Methods for Chemical Contaminants, U.S. EPA, Washington, DC, 18 août.
U.S. EPA (Environmental Protection Agency des États-Unis). 2003e. Determination of haloacetic acids and Dalapon in drinking water by liquid-liquid microextraction, derivatization, and gas chromatography with electron capture detection. EPA 815-B-03-002. Office of Ground Water and Drinking Water, U.S. EPA, Cincinnati, OH, juillet.
U.S. EPA (Environmental Protection Agency des États-Unis). 2005a. Drinking water criteria document: Brominated acetic acids. No de document EPA-822-R-05-007. Office of Science and Technology, Office of Water, U.S. EPA, Washington, DC, novembre.
U.S. EPA (Environmental Protection Agency des États-Unis). 2005b. Technologies and costs document for the Final Long Term 2Enhanced Surface Water Treatment Rule and Final Stage 2Disinfectants and Disinfection Byproducts Rule. EPA-815-R-05-013. U.S. EPA, Washington, DC, décembre.
U.S. EPA (Environmental Protection Agency des États-Unis). 2006. 2006 edition of the drinking water standards and health advisories. EPA 822-R-06-013. Office of Water, U.S. EPA, Washington, DC, août.
Veeramachaneni, D.N.R. 2002. The rabbit may represent a better animal model for determining reproductive risk associated with DBPs. Toxicologist, 66(1-S) : 331 (abrégé).
Veeramachaneni, D.N.R., Higuchi, T.T., Palmer, J.S. et Kane, C.M. 2000. Dibromoacetic acid, a disinfection by-product in drinking water, impairs sexual function and fertility in male rabbits. Biol. Reprod., 62 (Suppl. 1) : 246 (abrégé).
Verschueren, K. 2001. Handbook of environmental data on organic chemicals. 4e éd. John Wiley & Sons, Inc., New York, NY.
Vetter, C.M., Miller, J.E., Crawford, L.M., Armstrong, M.J., Clair, J.H., Conner, M.W., Wise, L.D. et Skopek, T.R. 1998. Comparison of motility and membrane integrity to assess rat sperm viability. Reprod. Toxicol., 12(2) : 105-114.
Volkel, W., Friedewald, M., Lederer, E., Pahler, A., Parker, J. et Dekant, W. 1998. Biotransformation of perchloroethene: Dose-dependent excretion of trichloroacetic acid, dichloroacetic acid, and N-acetyl-S(trichlorovinyl)-L-cysteine in rats and humans after inhalation. Toxicol. Appl. Pharmacol., 153 : 20-27.
Weast, R.C. (dir.). 1973. Handbook of chemistry and physics. 54e éd. CRC Press Inc., Cleveland, OH.
Williams, S.L., Williams, R.L. et Yuan, J. 1998. Bacterial degradation of haloacetic acids in the distribution system. Proceedings of the Water Quality Technical Conference, San Diego, CA, 1-4 novembre. American Water Works Association, Denver, CO.
Woodard, G., Lange, S.W., Nelson, K.W. et Calvery, H.O. 1941. The acute oral toxicity of acetic, chloracetic, dichloroacetic and trichloroacetic acids. J. Ind. Hyg. Toxicol., 23 : 78-82.
WSSA (Weed Science Society of America). 1983. Trichloroacetic acid. Dans : Herbicide handbook of the Weed Science Society of America. 5e éd. WSSA, Champaign, IL. 447 p.
Xie, Y.F. 2004. Disinfection byproducts in drinking water: Formation, analysis, and control. Lewis Publishers, CRC Press, Boca Raton, FL.
Xu, G., Stevens, D.K. et Bull, R.J. 1995. Metabolism of bromodichloroacetate 4 in B6C3F1 mice. Drug Metab. Dispos., 23(12) : 1412-1416.
Xu, X. et Weisel, C.P. 2003. Inhalation exposure to haloacetic acids and haloketones during showering. Environ. Sci. Technol., 37(3) : 569-576.
Xu, X., Mariano, T.M., Laskin, J.D. et Weisel, C.P. 2002. Percutaneous absorption of trihalomethanes, haloacetic acids, and haloketones. Toxicol. Appl. Pharmacol., 184(1) : 19-26.
Yllner, S. 1971. Metabolism of chloroacetate-14C in the mouse. Acta Pharmacol. Toxicol., 30(1-2) : 69.
Yount, E.A., Felten, S.Y., O'Connor, B.L., Peterson, R.G., Powell, R.S., Yum, M.N. et Harris, R.A. 1982. Comparison of metabolic and toxic effects of 2-chloroproprionate and dichloroacetate. J. Pharmacol. Exp. Ther., 222(2) : 501-508.
Yu, K.O., Barton, H.A., Mahle, D.A. et Frazier, J.M. 2000. In vivo kinetics of trichloroacetate in male Fischer 344rats. Toxicol. Sci., 54 : 302-311.
ZirChrom Separations, Inc. Sans date. Dissociation constants of organic acids and bases.
Annexe A : Liste des sigles
- ADN
- acide désoxyribonucléique
- AHA
- acide haloacétique
- AHA5
- acides haloacétiques totaux : désigne le total des acides monochloroacétique, dichloroacétique, trichloroacétique, monobromoacétique et dibromoacétique
- ALARA
- as low as reasonably achievable (niveau le plus bas qu'il soit raisonnablement possible d'atteindre)
- ALT
- alanine transaminase
- ANSI
- American National Standards Institute
- APHA
- American Public Health Association
- ARNm
- acide ribonucléique messager
- AST
- aspartate transaminase
- CAG
- charbon actif en grains
- CAS
- Chemical Abstracts Service
- CCN
- Conseil canadien des normes
- CG
- chromatographie en phase gazeuse
- CIRC
- Centre international de recherche sur le cancer
- CMA
- concentration maximale acceptable
- DBA
- acide dibromoacétique
- DCA
- acide dichloroacétique
- DCE
- détecteur à capture d'électrons
- DJT
- dose journalière tolérable
- DL50
- dose létale 50
- EPA
- Environmental Protection Agency ( É.-U.)
- GST-zêta
- glutathion S-transférase-zêta
- KA
- facteur de rajustement cinétique
- kg p.c.
- kilogramme de poids corporel
- LDM
- limite de détection de la méthode
- LOAEL
- dose minimale entraînant un effet nocif observé
- LPAQ
- limite pratique de l'analyse quantitative
- MBA
- acide monobromoacétique
- MCA
- acide monochloroacétique
- MON
- matière organique naturelle
- MTBE
- éther méthyltertiobutylique
- NOAEL
- dose sans effet nocif observé
- NOEL
- dose sans effet observé
- NSF
- NSF International
- NTP
- National Toxicology Program ( É.-U.)
- OMS
- Organisation mondiale de la santé
- PCE
- perchloroéthylène (tétrachloroéthylène)
- PPAR
- récepteur activé de la prolifération des peroxysomes
- ppm
- parties par million
- SPCD
- sous-produit chloré de désinfection
- SPD
- sous-produit de désinfection
- TCA
- acide trichloroacétique
- TCE
- trichloroéthylène
- THM
- trihalométhane
- UTN
- unité de turbidité néphélométrique
- UV
- ultraviolet