
2022 Proposed updates to the PMPRB Guidelines
New Guidelines will not be implemented on January 1, 2023. The Interim Guidance issued by the Board on August 18, 2022, will remain in place until further notice.
Draft Guidelines 2022
Draft Guidelines 2022

Table of Contents
- I. Introduction
- II. Interpretation
- III. Legal Framework and Jurisdiction
- IV. Rights Holder Filings
- V. Criteria That May Trigger an Investigation
- VI. Special Provisions and Timing
- VII. Investigations
- VIII. Undertakings
- IX. Excessive Price Hearing Process and Remedies
- X. Failure to File Hearing
- XI. Complaints
- XIII. Appendices
I. Introduction
1. The Patented Medicine Prices Review Board (PMPRB) was created in 1987 to protect consumers from excessively priced patented medicines. Its creation arose out of concern that stronger patent protection for pharmaceuticals might cause their prices to rise to unacceptable levels.Footnote1 The PMPRB is a quasi-judicial body composed of up to five Board Members,Footnote2 assisted by such civil servantsFootnote3 (“Staff”) as are necessary for the proper conduct of the Board.
2. These Guidelines, which are issued by the Board pursuant to subsection 96(4) of the Patent Act (“Act”), are intended to provide transparency to stakeholders regarding the procedures typically used by Staff when monitoring the prices of patented medicines. In particular, they explain the criteria Staff will consider in determining whether the price of a patented medicine warrants a more in-depth review in the form of an investigation.Footnote4 This is significant because the opening of an investigation may lead to a recommendation from Staff to the Chairperson to hold a hearing into whether the price of the patented medicine is excessive under s. 85 of the Act. If such a hearing is deemed to be in the public interest by the Chairperson, and a panel composed of Board Members (“Hearing Panel”) finds that the patented medicine was priced excessively in any market, it may order the rights holder to reduce the price. Furthermore, it may take measures to offset any excess revenues that may have been earned through sales of the patented medicine at an excessive price.
3. The opening of an investigation into the price of a patented medicine by Staff does not mean that it is excessive. Conversely, the closing of an investigation does not mean that the price of a patented medicine is not excessive. Investigations are simply administrative triage procedures designed to enable the most efficient prioritization of the Board’s resources. A legal determination as to whether the price of a patented medicine is excessive can only be done by a Hearing Panel following a public hearing on the matter.Footnote5
II. Interpretation
4. These Guidelines supersede all previous guidance documents, policy communiqués and written or verbal statements of any kind by the PMPRB regarding the administration of the price review process, including all previous versions of the PMPRB’s Compendium of Guidelines, Policies and Procedures. The Guidelines should be read in conjunction with the Act, the Patented Medicines Regulations (“Regulations”), the appendices to these Guidelines and other related guidance documents published by the PMPRB from time to time, including the Help section of the online filing tool, which takes the place of the Patentee’s Guide to Reporting.
5. In accordance with s. 96(4) of the Act, the Guidelines are not binding on Staff, the Chairperson, Hearing Panels, or rights holders, and are not intended to create any legal rights or presumptions, to restate the law, or to constitute a definitive statement on the interpretation of the legislation related to the PMPRB. In any given case, the approach taken by Staff and the ultimate resolution of issues will depend on the particular circumstances of the matter in question. Final interpretation of the law is the responsibility of the Board (sitting as a Hearing Panel) and is subject to review by the courts.
6. While the Board has the power to order that the price of a medicine be reduced to a non-excessive level following a public hearing, the Board does not set or mandate prices for patented medicine and these Guidelines are not intended to be read as pricing guidelines. Accordingly, rights holders do not require permission or approval from the PMPRB to sell their products in Canada.
7. Certain aspects of these Guidelines may be revisited by the PMPRB in light of experience and changing circumstances. Guidelines and policies issued by the PMPRB are developed in an open manner with opportunities for full consultation with interested parties. When any changes to the Guidelines are considered, stakeholders will be consulted by the PMPRB in accordance with the commentary process established under s. 96(5) of the Act.
8. The Guidelines do not provide an exhaustive description of all steps that may be taken or all issues that may arise in the context of a price review. In exceptional circumstances or in the event of a hearing, any methods or tests deemed appropriate and consistent with the Act and Regulations may be used by the PMPRB, regardless of whether they are addressed in the Guidelines or otherwise differ from the approach set out therein.
9. For additional information on these Guidelines or Staff’s general approach to monitoring and reviewing prices, please see the PMPRB’s website or contact Staff at the following:
Patented Medicine Prices Review Board
Box L40
Standard Life Centre
333 Laurier Avenue West
Suite 1400
Ottawa, Ontario
K1P 1C1
Attention: Secretary of the Board
III. Legal Framework and Jurisdiction
10. The PMPRB has a dual mandate: in its regulatory role, it monitors and reviews the prices of patented medicines to ensure they are not excessive; in its reporting role, it provides information on pricing trends in the pharmaceutical industry via its Annual Reports.Footnote6 Further to a directive from the Minister of Health under s. 90 of the Act, the PMPRB also supports informed and evidence-based health policy by reporting on medicine price, utilization and cost trends under the National Prescription Drug Utilization Information System (NPDUIS) initiative.
11. The PMPRB Board consists of five members appointed by the Governor–in-Council under s. 91 of the Act for terms of up to five (5) years, renewable once. The Chairperson of the Board acts as the Chief Executive Officer (CEO) of the PMPRB and has supervision over and direction of its work. The PMPRB employs public servants (i.e., Staff) pursuant to s. 94 of the Act to carry out its day-to-day work. The PMPRB’s Executive Director is its senior public servant, Chief Operating Officer (COO) and Chief Financial Officer (CFO) and is responsible for the management of Staff.
12. The PMPRB is established under the Act as an independent, quasi-judicial body.To ensure this independence and autonomy, no express or implicit power is provided under the Act to Health Canada or any other government entity to direct the PMPRB in the exercise of its regulatory function. The PMPRB maintains an arm’s length relationship from the Minister of Health (who is responsible for the sections of the Act pertaining to the PMPRB), the Minister of Innovation, Science and Economic Development (who is responsible for the Act as a whole) and its various stakeholders. Similarly, the PMPRB is structured in a manner that separates the work and functions of Staff, the Chairperson and Board Members: investigation, litigation and reporting functions reside with Staff and are separate from the adjudication functions that are reserved for Board Members only.
13. Under the Act, the PMPRB has jurisdiction to determine whether a patented medicine is or has been sold by a rights holder at an excessive price in any market in Canada.Footnote7 The term “rights holder” is definedFootnote8 in the Act as, in respect of an invention pertaining to a medicine, a patentee and the person for the time being entitled to the benefit of a certificate of supplementary protection for that invention, and includes, if any other person is entitled to exercise rights in relation to the certificate, that other person in respect of those rights. The term “patentee” is definedFootnote9 in the Act as a person who is entitled to the benefit of a patent for an invention for a period, including any other person entitled to exercise rights in relation to the patent, such as a holder of an express or implied license.Footnote10
14. An invention pertains to a medicine if the invention is intended or capable of being used for medicine or for the preparation or production of medicine.Footnote11 The phrase “pertain to a medicine” has a broad meaning. The Federal Court of Appeal has determined that the nature of that connection may be “tenuous”.Footnote12 It may be satisfied, for example, where there may “only be a slender thread of a connection between a patented invention and the medicine sold in Canada”Footnote13.Footnote14
15. The term “medicine” is defined in the Act as including a drug (i.e., a substance or a mixture of substances manufactured, sold or represented for use in (i) the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state, or its symptoms, in human beings or animals; or (ii) restoring, correcting or modifying organic functions in human beings or animals)Footnote15 and a medicinal ingredient.Footnote16 Unless otherwise specified, in these Guidelines, a reference to a “medicine” includes all dosage forms and strengths (e.g. all Drug Identification Numbers or “DINs” for medicines that have been assigned a DIN) of the medicine.
16. The PMPRB recognizes that the term “medicinal ingredient” is generally understood to mean the “active pharmaceutical ingredient” (API) used as raw materials during the manufacture of the finished dosage form. Patented medicines under the PMPRB’s jurisdiction include vaccines, topical preparations, anaesthetics and diagnostic products used in vivo, regardless of delivery mechanism (i.e., trans-dermal patch, capsule, injectable, inhaler). However, the PMPRB does not consider medical devices, in vitro diagnostic products and disinfectants that are not used in vivo to be patented medicines for the purpose of price review provisions in the Act.
17. The PMPRB has jurisdiction during the life of an eligible and issued patent including the pre-grant period (from the patent application date). The PMPRB also has jurisdiction for the extended period of protection granted via a certificate of supplementary protection.Footnote17
18. The PMPRB’s jurisdiction over the price at which a patented medicine is sold in any market in Canada persists after the patent has been dedicated and until the cancellation or surrender of the patent pursuant to the express provisions of the Act or the expiry of the term of the patent. Patent dedication is not expressly recognized in the Act as a mechanism by which patent rights may be terminated before the normal expiry of the patent term.
19. The PMPRB reviews the prices of patented medicines sold at arm’s-length by rights holders. Sales in Canada may include, but are not limited to, patented medicines subject to a Notice of Compliance (NOC), the Special Access Programme (“SAP”), the List of Drugs for an Urgent Public Health Need, or Clinical Trial Applications. The PMPRB has no authority over prices charged by parties other than rights holders, such as prices charged by wholesalers or retailers, or over pharmacists’ professional fees.
20. Orders issued by the PMPRB are enforceable in the same manner as orders of the Federal Court or any superior court in Canada and may be enforced by the PMPRB or by the Federal Court. Decisions embodied in orders issued by the PMPRB may be subject to judicial review by the Federal Court in accordance with administrative law principles and the Federal Courts Act.
21. In addition to ss. 79–103 of the Act, the PMPRB is responsible for the administration of the Regulations and of the PMPRB Rules of Practice and Procedure.
IV. Rights Holder Filings
22. Rights holders and former rights holders are required by law to submit information to the PMPRB. The information that must be submitted is set out in s. 82 of the Act and in the Regulations. Rights holders are responsible for compliance with filing obligations. These statutory obligations cannot be waived or amended by Staff.
23. As per s. 7 of the Regulations, rights holders shall submit required filings using the electronic forms on the PMPRB’s website. The forms must bear the electronic signature of an authorized individual who certifies that the information is true and complete. These forms are available through the PMPRB’s online filing tool.
24. It is the responsibility of each rights holder to independently ensure that the information filed with the PMPRB (including domestic and foreign prices) is accurate. Ad hoc audits of rights holder filings, including pricing, revenue and patent information, may be conducted from time to time by Staff. In the event of such an audit, rights holders may be asked to provide additional supporting materials and/or corrections or confirmation of the information filed.
25. Failure to file required information within the specified period or the filing of erroneous or false information may have significant consequences for rights holders or former rights holders. An order for certain remedies may be sought by Staff from a Hearing Panel, including an ex parte order requiring that the missing information be submitted. Alternatively, the matter may lead to summary conviction proceedings under s. 76.1(1) of the Act. Further, the filing of false information is an indictable offence under s 76 of the Act that, on conviction, can lead to monetary fines or terms of imprisonment.
26. The Act provides for the confidentiality of information submitted to the PMPRB in certain circumstances. Specifically, information or documents provided to the PMPRB in accordance with the provisions dealing with pricing information in s. 80, 81 and 82 of the Act, or in any proceeding relating to excessive prices under s. 83, are privileged and cannot be disclosed to the public without authorization of the disclosing party, unless such information has been disclosed at a public hearing under s. 83 of the Act or is subject to the exceptions outlined in s. 87(2) of the Act.
27. Information provided to the PMPRB may be subject to certain provisions in the Access to Information Act and the Privacy Act.
28. Information that rights holders or former rights holders may be required to file under the Regulations includes, but is not limited to:
- A notification describing a rights holder’s intention to offer a patented medicine for sale in a market in Canada in which such medicine has not previously been sold (i.e., the first sale of the medicine), along with related information (Notification of Intention to Sell a Patented Medicine);
- Prescribed information relating to the identity and characteristics of a patented medicine, such as the product monograph for the medicine or equivalent information, and any DINs assigned for each dosage form and strength of the medicine;
- Prescribed information relating to the price of a patented medicine, such as information concerning the price at which each dosage form and strength of the medicine is or has been sold in any market in Canada or in any of the eleven countries set out in the Regulations (the “PMPRB11”).
29. For prices filed by rights holders for the PMPRB11, Staff convert local currency prices filed for the PMPRB11 into Canadian dollars using exchange rates calculated as the simple average of the thirty-six (36) monthly average noon spot exchange rates for each country as published by the Bank of Canada. During a medicine’s introductory period, the thirty-six (36) months ending in the second month of the previous reporting period (i.e., February or August) is generally used. Subsequently, the thirty-six (36) months ending in the second month of the reporting period under review is generally used to look at prices filed by the rights holders (for Canada and for the PMPRB11). Where there are multiple list prices filed for the same country, the lowest such price is generally used.
V. Criteria That May Trigger an Investigation
30. Investigations are reviews conducted by Staff to identify and prioritize matters that may be brought to the attention of the Chairperson and could potentially lead to a hearing. When conducting an investigation, Staff review information provided by the rights holder and potentially any relevant information obtained from other sources.
31. Investigations are purely administrative in nature. Board Members are not involved in the investigation process. Investigations cannot, in and of themselves, lead to a determination that the price of a patented medicine is excessive. If an investigation results in a recommendation to the Chairperson that a public hearing be held, and the Chairperson agrees, the Hearing Panel seized with the matter must undertake an independent de novo review of the price of the patented medicine to determine whether it is excessive under s. 83 of the Act. As such, it is open to Staff and the rights holder(s) to advance arguments and positions in a hearing that are different from what was discussed during the investigation.
32. Staff may open an investigation into the price of a patented medicine when certain criteria are present. These criteria are based on the factors in s. 85 of the Act and have been developed with the intention of making the most efficient use of the PMPRB’s human and financial resources. The criteria Staff considers vary depending on whether a medicine is an “existing” medicine or a “new” medicine as defined below.
- Existing medicines: (i) all dosage forms and strengths of medicines for which an NOC was issued prior to July 1, 2022 regardless of whether those dosage forms and strengths have been approved for new indications (without a DIN change) after July 1, 2022, (ii) new dosage forms and strengths of these medicines to which an NOC was issued on or after July 1, 2022; and (iii) all dosage forms and strengths of medicines for an authorized sale under the “Special Access Programme” for the Sale of New Drugs for Emergency Treatment under Part C Division 8 of the Food and Drug Regulations was made prior to July 1, 2022.
- New medicines: are all other dosage forms and strengths of medicines that are not Existing medicines.
33. The following criteria applies to all medicines (i.e., existing medicines and new medicines):
- A complaint is received in respect of the pricing of the medicine; or
- The list priceFootnote18 increased by more than the changes in the Consumer Price Index (CPI); or
- No international prices were filed by the rights holder.
34. The following additional criteria applies to existing medicines:
- The list price of any dosage form or strength of the medicine exceeds the highest international price for the PMPRB11 based on pricing information provided by the rights holder.
35. The following additional criteria applies to new medicines in the specific circumstances described:
- The list price exceeds the median international price for the PMPRB11; or
- The list price falls between the median and the lowest international price for the PMPRB11, but exceeds the top of the domestic therapeutic class comparator prices (“dTCC”); or
- The list price exceeds the midpoint between the top of the dTCC and lowest international price for the PMPRB11, and the top of the dTCC is more than 50% lower than the lowest international price.
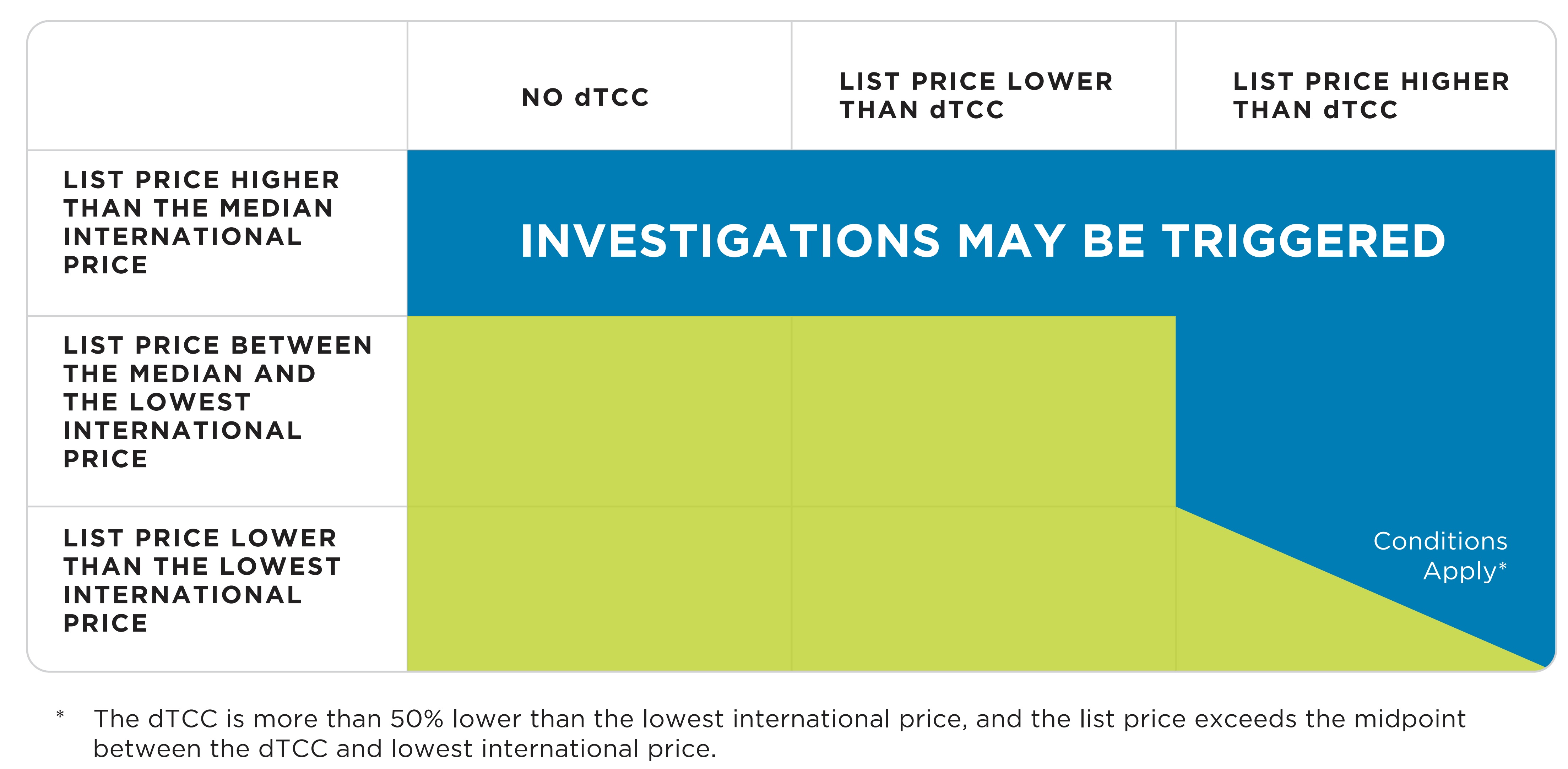
Figure description
This figure illustrates the additional circumstances that may trigger an investigation into the
prices of new patented medicines, as follows:
The list price exceeds the median international price; or
The list falls between the median and the lowest international price, but exceeds the top of the
domestic therapeutic class comparator prices (“dTCC”); or
The list price exceeds the midpoint between the top of the dTCC and lowest international price,
and the top of the dTCC is more than 50% lower than the lowest international price.
VI. Special Provisions and Timing
36. For Biosimilars,Footnote19 medicines for veterinary use, over-the-counter (OTC) medicines, and vaccines,Footnote20 an investigation may be opened only when a complaint is received.
37. For GenericFootnote21 medicines, an investigation may only be opened when a complaint is received, and:
- The rights holder of the medicine is the only company in Canada which is selling a generic version of the medicine in Canada; and
- The medicine is not the subject of a pricing agreement with the pan-Canadian Pharmaceutical Alliance (pCPA) to which it is compliant. The onus of proving to Staff that a medicine is subject to, and compliant with, a pricing agreement with pCPA will rest with the rights holder.
38. For all medicines, Staff will wait until the rights holder has filed sales for the patented medicine at prices that are above zero ($0) dollars to determine whether the investigation criteria for a patented medicine are met. For all medicines, Staff will wait until two reporting periods have passed, starting on the period during which these Guidelines come into effect, to determine whether the investigation criteria for a patented medicine are met.
VII. Investigations
39. The rights holder will be notified of the criteria that led to the opening of an investigation and the medicine will be identified in the PMPRB’s Annual Report as “Under Investigation” until such time as the investigation is closed.
40. An investigation will remain open until one of the following occurs:
- Staff closes the investigation for administrative reasons; or
- The Chairperson closes the investigation following the receipt of an acceptable Undertaking from the rights holder; or
- The investigation is moot because the matter has proceeded to a hearing following the issuance of a Notice of Hearing.
41. The outcome of an investigation will depend on a number of factors. Medicines which are perceived by Staff as being at high risk of excessive pricing are most likely to proceed to a hearing or result in an undertaking. Staff’s analysis will focus on the strength of the evidence, the degree to which the price of the medicine deviates from the investigation criteria levels, the extent to which the facts are aligned with the jurisprudence interpreting the s. 85 factors and whether the investigation raises new or unique questions that warrant judicial elucidation. As always, for reasons of administrative efficiency and resource optimization, Staff will focus its efforts on achieving an outcome that avoids the need for litigation.
42. The closing of an investigation by Staff is an administrative act and does not constitute a legal determination or an admission by the PMPRB that the price of the patented medicine is not excessive. As such, it does not preclude the possibility of the opening or re-opening of further investigation(s) in respect of the same medicine or the commencement of a hearing in the future.
43. When an investigation into the price of a patented medicine is completed and the matter is not resolved to the satisfaction of Staff, the Executive Director may submit a report to the Chairperson recommending that a hearing take place. The Chairperson may decide to issue a Notice of Hearing if he or she is of the opinion that doing so is in the public interest. A decision to issue a Notice of Hearing is not adjudicative and no analysis is undertaken by the Chairperson as to whether the facts alleged by Staff are, or will be, proven. Until a matter is brought before a Hearing Panel at a public hearing, no other Board member is informed of the results of Staff’s review or investigation into the price of a patented medicine.
44. The decision of whether the price of a patented medicine is excessive is made by the Hearing Panel alone on the basis of evidence and argument advanced by the parties in a public hearing.
VIII. Undertakings
45. At any time prior to the issuance of a Notice of Hearing, a rights holder may choose to submit a proposed undertakingFootnote22 to Staff. An undertaking is a formal promise by the rights holder to reduce its price(s) and/or offset any potential excess revenues from the sale of a patented medicine that is subject to an investigation. Offers to offset potential excess revenues may be based on netFootnote23 as opposed to list prices but offers to reduce list prices are also relevant in this context. A proposed undertaking does not constitute an admission by the rights holder that the price of the patented medicine is excessive. Undertakings are purely voluntary on the part of the rights holder.
46. Discussions of undertakings with rights holders are conducted by Staff and do not involve the Chairperson. If discussions result in an undertaking proposal that Staff believes would be acceptable to the Chairperson, it will be referred to them by the Executive Director for consideration. The decision to accept an undertaking is within the sole purview of the Chairperson.
47. The consideration of an undertaking is an administrative procedure and does not constitute an admission or determination by the PMPRB that the price submitted by the rights holder, or used to calculate a potential revenue offset, is not excessive. However, the acceptance of an undertaking by the Chairperson will result in the closing of an investigation.
48. Where a rights holder offers to offset potential excess revenues as part of an undertaking, any amounts collected are directed by the Receiver General to the Consolidated Revenue Fund, and do not form part of the PMPRB’s Budget. Subject to s. 103 of the Act, the Minister may enter into agreements with any province respecting the distribution of, and may pay to that province out of the Consolidated Revenue Fund, amounts received or collected by the Receiver General under s. 83 or s. 84 or in respect of an undertaking given by a rights holder or former rights holder that is accepted by the Board in lieu of holding a hearing or making an order under s. 83, less any costs incurred in relation to the collection and distribution of those amounts.
49. The PMPRB reports publicly on all undertakings that the Chairperson has accepted as warranting the closing of an investigation. In submitting a signed undertaking, a rights holder must consent to its publication either in full or redacted form. The reported information can include disclosure of a copy of the undertaking or terms included in the undertaking. The reported information may appear in the PMPRB’s Annual Report, on the PMPRB’s website, in the PMPRB’s publications such as the NEWSletter, and on social media platforms.
50. Requests for discussions or proposals for undertakings “without prejudice” cannot be considered by Staff. Undertakings are not settlement agreements as they take place before the issuance of a Notice of Hearing. However, parts of the discussions between rights holders and Staff that relate to the content of the rights holder’s filings may be subject to the protections set out in s. 87 and s. 88 of the Act. In addition, provisions of the Access to Information Act may apply.
51 Once a Notice of Hearing has been issued, Staff can no longer engage in discussions with the rights holder on a potential undertaking. In the alternative, the rights holder may pursue the negotiation of a settlement agreement with Staff, but its terms must ultimately be approved by the Hearing Panel. Unlike undertakings, requests for settlement agreements or proposals are considered by Staff on a “without prejudice” basis.
IX. Excessive Price Hearing Process and Remedies
52. PMPRB hearings are public proceedings. During a hearing, submissions and evidence from the parties are heard by a Hearing Panel consisting of at least two Board members. The Hearing Panel determines whether a patented medicine is being or has been sold at an excessive price in any market in Canada by taking into consideration the available information relating to the factors set out in s. 85 of the Act.
53. For more information about hearings, please consult the PMPRB Rules of Practice and Procedure, the published standard set of procedures to be followed by all participants in hearings before the PMPRB. The Rules set out the PMPRB’s procedures in accordance with the requirement under the Act to resolve matters as informally and expeditiously as the circumstances and considerations of fairness permit. Practice directions and further information about previous and ongoing hearings are also publicly available on the PMPRB’s website
54. Under the Act, the PMPRB is empowered to make remedial orders when it finds, following a hearing, that a rights holder (or former rights holder) is selling, or has sold, a patented medicine in any market in Canada at an excessive price.Footnote24
55. In broad terms, the PMPRB has the power to impose two main forms of remedy after a hearing: (i) orders directing the rights holder to cause the maximum price at which the rights holder sells the patented medicine in a market to be reduced to such level as the PMPRB considers not to be excessive; and (ii) orders directing the rights holder to offset the amount of the excess revenues estimated by it to have been derived by the rights holder from the sale of the patented medicine at an excessive price by either (a) reducing the price at which the rights holder sells the patented medicine; (b) reducing the price at which the rights holder sells one other medicine to which a patented invention of the rights holder pertains; or (c) paying to His Majesty in right of Canada an amount specified in the order.
56. If a Hearing Panel finds that the rights holder or former rights holder has engaged in a policy of selling the patented medicine at an excessive price, it may order the rights holder to offset up to twice the amount of excess revenues estimated by it to have been derived from the sale of the patented medicine at an excessive price.Footnote25 The extent and duration of sales of the patented medicine at an excessive price may be considered by the PMPRB in making this finding and order.
X. Failure to File Hearing
57. When it is the opinion of Staff that a rights holder has failed or refused to provide the PMPRB with the pricing, sales, or revenues and like information required by law, the Executive Director may submit a report to the Chairperson. The Chairperson may decide to issue a Notice of Hearing if he or she is of the opinion that it is in the public interest to hold a hearing to determine whether the rights holder has, in fact, breached the reporting requirements of the Act and Regulations.
58. As with excessive price hearings, a decision to issue a Notice of Hearing is not adjudicative and no analysis is undertaken by the Chairperson as to whether the facts alleged by Staff are, or will be, proven. Until a matter is brought before a Hearing Panel at a public hearing, no other Board member is informed of the results of Staff’s review or investigation into the matter. The decision of whether a rights holder has failed to file required information is made by the Hearing Panel alone after the public hearing is held. If, as the result of such a hearing, the Hearing Panel finds that the rights holder is in breach of its reporting requirements, it may order the rights holder to provide the PMPRB with the required information and documents as per s. 81 and/or s. 88 of the Act.
59. In addition, as per s. 76.1(1) of the Act, every person who contravenes or fails to comply with the filing requirements set out in s. 80, 81, 82 or 88, or any order made thereunder, is guilty of an offence punishable on summary conviction and liable to a fine or to imprisonment.
XI. Complaints
60. Any individual or group who believes that the price of a patented medicine in any market in Canada is excessive may submit a complaint to the PMPRB. A complaint may be submitted by telephone, in writing, or electronically using the contact information available on the PMPRB “How to Make a Complaint” page.
61. Generally, whenever a complaint is received, Staff will open an investigation. The complainant is not part of that investigation or of any resulting hearing (unless the complainant applies to become an intervener in the hearing). The complainant is not required to provide any documents or evidence to the PMPRB. Any investigation is based on materials provided by the rights holder or otherwise obtained by Staff.
62. Due to limitations on disclosure set out in s. 87 and s. 88 of the Act and in the Access to Information Act, the complainant is only informed that the complaint has been received, and of the outcome of an investigation if the process results in an undertaking or a Notice of Hearing.
XIII. Appendices
The information detailed in these Appendices is for illustrative purposes only. It is not intended to be directive nor exhaustive, and it should not be relied upon as such.
As with the rest of the Guidelines, the Appendices should be understood to be examples of the sort of approaches that may be considered by Staff during an investigation. However, an investigation may or may not use the approaches outlined in the Appendices, as circumstances and applicable legislation warrant. Similarly, whether or not an investigation uses the approaches described in the Appendices, an investigation does not make any determination that prices are excessive or non-excessive.
A. Scientific Review Process: identifying TCC, dosage regimens and reference strength
The PMPRB’s scientific review is an evidence-based process that considers clinical, and other relevant information about the patented medicine for the purpose of determining its therapeutic class comparators (“TCC”), dosage regimens and reference strength.
If a patented medicine that has previously been subject to scientific review is approved for a new indication, a new scientific review producing a new therapeutic class comparison may be conducted, and the rights holder will be advised accordingly.
The scientific review of a patented medicine is based on information from a variety of sources such as those set out below:
Rights holder Submission – Rights holders may provide Staff with a brief submission document which clearly explains the rationale for the rights holder’s proposals relative to the medicines identified for comparison purposes, and comparable dosage regimens. Information on submission procedures is available on the PMPRB online filing tool.
Research by a Drug Information Centre (DIC) – Staff may use the services of various drug information centres to obtain scientific information, such as clinical trial information, clinical practice guidelines, etc. The basis of the review by the DIC is the product monograph or information similar to that contained in a product monograph if a NOC has not been granted.
Research by Staff – Staff may also update research and supplement data and evidence from the rights holder and DIC using other sources.
PMPRB Human Drug Advisory Panel (HDAP) – On an ad hoc basis, Staff may consult with the HDAP to provide clinical context pertaining to the scientific information that is being considered by Staff.
B. Domestic Therapeutic Class Comparison (dTCC)
The dTCC compares a patented medicine’s list price with the list prices of other medicines identified by scientific review for comparison purposes.
Identification of Medicines for Comparison Purposes
The World Health Organization (WHO) Collaborating Centre for Drug Statistics Methodology’s Anatomical Therapeutic Chemical (ATC) Classification System is often used by Staff in the selection of medicines to be used for comparison purposes.
The medicines used for comparison purposes will typically be those identified under the ATC classification system at the sub-class level above the single chemical substance. This will normally be the fourth sub-class level but could include the next higher subclass or another sub-class. In some instances, it may be appropriate to select from the fifth or single chemical substance level.
A medicine of the same ATC therapeutic class as the patented medicine under review may be omitted if it is unsuitable for comparison.
Typically, where there are multiple sellers of a medicine identified as a comparator, the lowest price is used for that comparator. The final therapeutic class comparison price will typically be the highest price across all comparable medicines.
For a patented medicine that is a new dosage form or strength of the same medicinal ingredient as one or more existing medicines, its comparators will usually be those existing medicines that are available in the same or comparable dosage form and have the same indication or use, regardless of whether the dosage regimens are the same.
For a product that is a combination of medicines, where each of the medicines of the combination are sold in Canada and have the same indication or use, its comparators are generally limited to the component medicines.
Comparable Dosage Regimens
The comparable dosage regimen used for comparison purposes will normally be the maximum of the usual recommended dosage in the Product Monograph (or similar information), taking into account relevant clinical variables. The most appropriate strength of the medicine will be chosen for a particular dosage regimen. Generally, a dosage regimen based on a course of treatment will be applicable to acute indications, while a per-day regimen (based on maintenance dose) will be applicable to chronic indications.
DTCC Price Sources
Rights holders are not specifically required by law to file prices for the patented medicine’s comparators. Public sources will typically be used for the prices of the comparators in order to conduct a dTCC. Provincial formularies will generally be the starting point in Staff’s identification of prices. The lowest price for each of the medicines identified for comparison purposes will typically be used.
Any medicine (patented or non-patented) identified for comparison purposes may be excluded from a dTCC test as appropriate, for instance, if Staff has reason to believe it is being sold at an excessive price.
DTCC Calculation
Following the identification of medicines for comparison purposes and of the lowest price for each medicine, the cost of comparable courses of treatment for each medicine will generally be calculated by identifying the most expensive, and dividing this cost of treatment by the constituent units to establish a per-unit price.
Generic Prices
With respect to domestic and international price comparisons for Generic medicines that are subject to an investigation, Staff may sometimes consider using the domestic and international prices of the brand reference product as comparators. If the reference brand product is no longer being sold in the Canadian or the international market, Staff may sometimes consider using the former brand reference product price as a comparator.
C. Reasonable Relationship Comparison and Comparable Dosage Forms
Reasonable Relationship Comparison
The Reasonable Relationship (RR) comparison may be conducted for a new additional strength of a patented medicine with other existing strengths, where the new additional strength has the same medicinal ingredient, indication, dosage regimen, and same or comparable dosage form as the existing strength(s).
When there are multiple strengths of a patented medicine at introduction, a reference strength will generally be selected based on scientific considerations, and the rights holder will be notified.
Once the reference strength is identified, the additional investigation criteria for the other strengths will typically be applied based on the proportional relationship between the strengths.
This means that, usually, an investigation may be opened into the new higher additional strength where its price is greater than the price per standard unit of the reference strength.
Staff may reassess the reference strength of a medicine if additional strengths become available.
Comparable Dosage Forms
The following are generally considered comparable dosage forms for the purpose of the RR test. Formulations within each group are usually considered comparable, but dosage forms in a different group are typically not.
Topical (T)
- Aerosol
- Aerosol (foam)
- Cream
- Disc (extended release)
- Disc
- Dressings
- Gel
- Gel (controlled release)
- Liposomes
- Liquid
- Lotion
- Ointment
- Pad
- Paint
- Paste
- Patch
- Patch (extended release)
- Pencil
- Plaster
- Powder
- Shampoo
- Soap Bar
- Solution
- Sponge
- Spray
- Spray (bag-on-valve)
- Spray (metered dose)
- Stick
- Strip
- Swab
- Tincture
Nasal (N) / Pulmonary (P)
- Aerosol
- Aerosol-metered dose
- Drops
- Gas
- Metered dose preparation
- Powder
- Powder (metered dose)
- Solution
- Solution (extended release)
- Spray
- Spray (metered dose)
- Stick
Oral Solid (S)
- Bar (chewable)
- Caplet
- Capsule
- Effervescent granules
- Effervescent powder
- Effervescent tablet
- Film (soluble)
- Globules
- Granules
- Gum
- Lozenge
- Modified release caplet
- Modified release capsule
- Modified release tablet
- Pellet
- Piece (chewable)
- Powder (extended release)
- Strip
- Tablet
- Tablet (chewable)
- Tablet (oral disintegrating)
- Tablet for suspension
- Wafer
Oral Liquid (L)
- Drops
- Elixir
- Emulsion
- Gel
- Granules for solution
- Granules for suspension
- Granules for suspension (delayed release)
- Granules for suspension (extended release)
- Liquid
- Modified release liquid
- Powder (extended release)
- Powder for solution
- Powder for suspension
- Solution
- Solution (extended release)
- Spray
- Suspension
- Suspension (extended release)
- Syrup
- Syrup (extended release)
- Tea (herbal)
- Tincture
Vaginal (V)
- Cone
- Cream
- Douche
- Foam
- Gel
- Gel (controlled release)
- Implant
- Insert
- Insert (extended release)
- Ovule
- Pellet
- Ring (slow release)
- Sponge
- Suppository
- Suppository (sustained release)
- Tampon
- Vaginal tablet
- Vaginal tablet (effervescent)
Parenteral (J)
- Bolus
- Implant
- Kit
- Liposomes
- Modified release injection
- Pellet (implantable)
- Powder for solution
- Powder for suspension (sustained-release)
- Solution
- Solution (extended release)
- Suspension for emulsion
- Suspension (extended release)
Otic (E) / Ophthalmic (Y)
- Drops
- Gel
- Gel (controlled release)
- Implant
- Insert
- Insert (extended release)
- Liquid
- Modified release ocular device
- Ointment
- Powder for solution
- Solution
- Solution (extended release)
- Suspension
Rectal (R)
- Cream
- Enema
- Foam
- Insert
- Ointment
- Ovule
- Stick
- Suppository
- Suppository (sustained release)
- Suspension
- Suspension (extended release)
Dental / Sublingual Buccal (M)
- Emulsion
- Film (soluble)
- Floss
- Gel
- Gel (controlled release)
- Gum
- Lozenge
- Metered-dose pump
- Modified release buccal tablet
- Mouthwash (gargle)
- Paste
- Powder (effervescent)
- Powder for suspension
- Solution
- Solution (extended release)
- Spray – buccal
- Spray – sublingual
- Stick
- Strip
- Sublingual tablet
- Suspension
- Suspension (extended release)
- Swab
- Tablet (orally disintegrating)
- Tablet
- Tooth paste
- Tooth powder
- Wafer
Backgrounder 2022
Backgrounder 2022

Table of Contents
Introduction
This document is intended to be read as a companion piece to the proposed Draft Guidelines. It supports readers’ understanding of the Draft Guidelines by providing a plain language contextual explanation of key changes from previous iterations of the Guidelines. This document offers a high-level summary of these changes and is not intended to serve as an exhaustive treatment of the whole of the Draft Guidelines. It is being shared with the aim of fostering a more informed and productive consultation process. As is true of the Guidelines themselves, this Backgrounder is not in any way binding on the Board, Board Staff or rights holders.
In developing the Draft Guidelines, the Board took into account the feedback received from stakeholders and the public as part of recent consultations on the 2020 Guidelines and in response to the July 2022 Notice and Comment on the Interim Guidance. The Board was also mindful of the evolving nature of its operating environment and its continuing duty to use its consumer protection powers in a responsible and efficient manner.
Background
On April 14, 2022, the Minister of Health announced that the Government would proceed with amendments to the Patented Medicine Regulations (“Regulations”) resulting in a newly constituted Schedule of comparator countries and reduced reporting requirements for medicines believed to be at the lowest risk of excessive pricing. The amended Regulations came into force on July 1, 2022.
In order to give effect to the Regulations and in furtherance of the Board’s longstanding commitment to modernize and simplify its administrative framework, the PMPRB is proposing the new Draft Guidelines and is inviting its stakeholders and interested members of the public to provide feedback during a 60-day consultation period. In addition to the present document which explains certain key changes in the proposed Draft Guidelines, the PMPRB will be hosting a series of webinars for industry and the public. Requests from individual stakeholders to meet with the PMPRB to discuss the Draft Guidelines will be considered on a case-by-case basis during the consultation period. The deadline for providing written submissions is December 5, 2022.
The PMPRB remains committed to hearing from all Canadians who have an interest in and opinion on how it carries out its regulatory mandate and looks forward to receiving constructive and meaningful feedback from this consultation process with a view to issuing final Guidelines by end of year.
What’s different about the Draft Guidelines?
Section 85 of the Patent Act (“Act”) contemplates intervention by the Board only where the price of a patented medicine is found to be “excessive”, which is determined based on a set of broadly expressed factors. Given the open-ended nature of the exercise contemplated under the Act, many of the core administrative concepts which give effect to the PMPRB’s regulatory authority have been developed through the Guidelines, which the Board is authorized to make under subsection 96(4) of the Act, subject to consulting first with relevant stakeholders. While the Guidelines, by definition, do not have force of law, they have been held by the courts to be useful both for the PMPRB and the public and may legitimately inform the Board’s reasoning in an excessive price hearing. Throughout the PMPRB’s 35-year history, as the pharmaceutical market and regulatory landscape have evolved, so too have the Guidelines, with multiple substantive changes having taken place over time. The changes described below are a continuation of that decades-long effort.
Aside from referencing the new Schedule of 11 comparator countries (“PMPRB11”), these Draft Guidelines contain a number of substantive changes that are intended to streamline how Staff monitors and reviews patented medicine prices and reduce their administrative burden on rights holders. On the whole, the changes described below make for a more pragmatic, less prescriptive set of Guidelines that better resemble modern guidance documents issued by other federal regulators. In conceiving the changes explained below, the Board was cognizant of the fact that there is no one right way to carry out its regulatory responsibilities under the Act and Regulations. As with previous iterations of the Guidelines, the approach taken by Staff and the ultimate resolution of issues will depend on the particular circumstances of the matter in question. Final interpretation of the law is the responsibility of the Board (sitting as a Hearing Panel) and is subject to review by the courts.
1. Investigation criteria
Unlike current and previous iterations of the Guidelines, the Draft Guidelines do not separate patented medicines into different therapeutic categories and assign highly particularized excessive price tests that change from year to year. The Board believes that such an approach is not mandated by law and that it is possible to craft the bright-line guidance sought by rights holders while clearly delineating the respective roles and responsibilities of the Board and Staff. The adoption of the concept of “investigation criteria” in the Draft Guidelines is an alternative approach that serves to achieve both these objectives.
The proposed criteria that can give rise to an investigation are simple, straightforward and anchored in the section 85 factors at all times. For “new medicines”, a concerted effort has been made in the Draft Guidelines to devise criteria that will only trigger an investigation when at least two of the factors are engaged whenever possible. This should significantly reduce the number of overall investigations and enable staff to focus resources on cases where the potential risk of excessive pricing is highest.
Under the Draft Guidelines, when the criteria are met and an investigation is opened, Staff will not presume the price of the medicine to be excessive. It is only after considering the totality of the circumstances surrounding the price of the medicine, through the lens of the section 85 factors, that Staff will close the investigation or recommend to the Chairperson that a hearing be commenced. As always, for reasons of administrative efficiency and resource optimization, where Staff is of the view that such a recommendation is warranted, it will work with the rights holder to achieve an outcome that avoids the need for litigation.
2. List Prices
Under the Draft Guidelines, the proposed investigation criteria apply to list prices only and will not fluctuate annually based on average transaction prices (ATP) the year before and a formula derived from CPI. This should make the potential triggering of an investigation more stable and predictable for rights holders. It also allows for more consistent and robust “apples-to-apples” price comparisons with the pricing information that is either filed by rights holders or is otherwise available to Staff from public sources, which mainly consists of domestic and international list prices for a medicine and/or its therapeutic comparators. Moreover, because list prices are by definition the highest prices that rights holders charge in the market, they are the most relevant prices for Staff to consider in the context of opening an investigation into potential excessive pricing. Under the Regulations, rights holders will continue to be required to file their ATP with the PMPRB, but Staff will only look at this information in the context of proposed undertakings by rights holders to offset potential excess revenues from the sale of a patented medicine that is subject to an investigation.
3. New and existing medicines
The Draft Guidelines distinguish between “existing” and “new” medicines and propose to apply less probing investigation criteria to the former. Existing medicines are defined as having received an NOC prior to the coming into force of the amended Regulations on July 1, 2022, and include line extensions of these same medicines. All other medicines are considered new medicines. This is a policy choice by the Board and a concession to rights holders whose expectations may have been raised by the Cost Benefit Analysis (CBA) that accompanied the publication of the original amendments to the Regulations. The Board is of the view that the misalignment of Canadian and international prices is best addressed by applying more probing investigation criteria to new medicines on a go forward basis.
It is important to note that this distinction is solely for administrative purposes and does not arise from a transitional provision in the Regulations, as was the case under the original amendments. As a result, from a legal standpoint, all patented medicines are subject to the same section 85 factors and have the same reporting obligations (with certain exceptions described below). Accordingly, once an investigation is opened, Staff will apply the same level of regulatory scrutiny to all medicines regardless of NOC date in determining whether to recommend the commencement of a hearing to the Chairperson.
4. Waiver of filing requirements for medicines believed to be at lower risk of excessive pricing
Under the amended Regulations, patented over-the-counter (OTC) medicines, certain non-prescription controlled substances, generic and veterinary medicines are not required to file pricing and other information with the PMPRB unless specifically requested to do so by the PMPRB. The Draft Guidelines propose to operationalize this by only opening an investigation in the event that a complaint is received in respect of one of these types of products. In response to feedback from its consultation on the 2020 Guidelines, the Board has opted to expand this list of products for administrative purposes to include biosimilars and vaccines, as these are also thought to be at lower risk of excessive pricing. Biosimilars are, in many ways, subject to similar market conditions as generic medicines. Regulating biosimilars on a complaints-basis is also consistent with the Canadian Agency for Drugs and Technologies in Health’s (CADTH’s) recent decision to no longer review biosimilars for cost effectiveness in order to simplify and streamline market access for these products. As for vaccines, most are subject to a public tendering process designed to award the contract to the bidder with the best value proposition. It should be noted that all the foregoing products remain subject to the Board’s jurisdiction provided they are patented and a complaint in respect of any one of them will automatically result in the opening of an investigation, which may or may not result in a recommendation to commence an excessive price hearing.
Webinars
Webinars
The PMPRB will host public consultations online on the draft Guidelines.
Public Webinars: The PMPRB will host two webinars in English and in French for all interested stakeholders on the following dates:
English webinar
Date: Thursday, November 3, 2022
Time: 11:00 to 12:00 EST
Presentation (PDF 1.4 MB)
French webinar
Date: Tuesday, November 8, 2022
Time: 11:00 to 12:00 EST
Industry Webinars: The PMPRB will also host webinars for the representatives of the pharmaceutical industry, by invitation only:
English webinar
Date: Thursday, November 3, 2022
Time: 13:00 to 14:00 EST
French webinar
Date: Tuesday, November 8, 2022
Time: 13:00 to 14:00 EST
Submissions
Below are the submissions the PMPRB received during the 2022 Guideline Consultation Period.