
PMPRB Draft Guidelines Consultation
The coming-into-force of the amended Patented Medicines Regulations (“Regulations”) has been further delayed until July 1, 2021.
Consultations for the June 2020 Draft Guidelines are now closed. Written feedback submitted to the PMPRB is available under the Feedback tab.
The written submissions for the November 2019 Draft Guidelines are also available on the PMPRB website.
Draft Guidelines 2020
PMPRB Draft Guidelines 2020
Table of Contents
- I. Preface
- II. Interpretation
- III. Legal Framework
- IV. Filing Requirements Pertaining to Price Reviews
- V. Price Review Process
- VI. Reassessment
- VII. Investigations
- VIII. Voluntary Compliance Undertaking (VCU)
- IX. Hearing Recommendation
- X. Excessive Price Hearing Process and Remedies
- XI. Failure to File Hearing
- XII. Complaints
- XIII. Appendices
- A. Domestic Therapeutic Class Comparison (dTCC) and International Therapeutic Class Comparison (iTCC)
- B. Reasonable Relationship Test and Comparable Dosage Forms
- C. Pharmacoeconomic Value Assessment
- D. Market Size Adjustment Methodology
- E. Scientific Review Process
- F. Summary of Compliance Timelines
I. Preface
1. The Patented Medicine Prices Review Board (PMPRB) was created in 1987 as the consumer protection “pillar” of a major set of reforms to the Patent Act (“Act”). The PMPRB is a quasi-judicial body with a regulatory mandate to prevent pharmaceutical patentees from charging consumers excessive prices during the statutory monopoly period. Its creation arose out of concern that stronger patent protection for medicines might cause their prices to rise unacceptably and become unaffordable to consumers. As a member of the Health Portfolio, the PMPRB contributes to a modern and sustain- able health system by ensuring that Canadians continue to have access to patented medicines at non-excessive prices.
2. These Guidelines, which are issued pursuant to subsection 96(4) of the Act, are intended to provide transparency and predictability to patentees regarding the process typically engaged in by public servant employees of the PMPRB (“Staff”) in assessing whether a patented medicine appears to be priced excessively in any market in Canada. The Guidelines also provide an overview of the processes that patentees should be aware of regarding their filing obligations under the Patented Medicines Regulations (“Regulations”).
3. If a patented medicine appears to be priced excessively based on these Guidelines and an acceptable Voluntary Compliance Undertaking (“VCU”) has not been sub- mitted by the patentee, the Chairperson may receive a recommendation from Staff that a hearing be held on the matter. If such a hearing is deemed to be in the public interest by the Chairperson, and it is confirmed at a hearing by a panel composed of Board members (“Hearing Panel”) that the patented medicine was priced excessively in any market, an order may be issued to the patentee that the price be reduced and that measures be taken to offset any excess revenues that may have been earned through sales of the patented medicine at an excessive price.
II. Interpretation
4. The Guidelines provide information on the PMPRB’s general approach to the price review process and investigations. They supersede all previous guidance documents, policy communiqués and written or verbal statements of any kind by the PMPRB regarding the administration of the price review process and investigations, including all previous versions of the PMPRB’s Compendium of Guidelines, Policies and Procedures. The Guidelines should be read in conjunction with the Act, the Regulations, the appendices to these Guidelines and other related guidance documents published in the future by the PMPRB from time to time, including the Help section of the online filing tool, which takes the place of the previous Patentee’s Guide to Reporting.
5. In accordance with subsection 96(4) of the Act, these Guidelines are not binding on Staff, the Chairperson, Hearing Panels, or patentees, and are not intended to create any legal rights or presumptions, to restate the law, or to constitute a definitive statement on the interpretation of the legislation related to the PMPRB. In any given case, the enforcement decisions of Staff and the ultimate resolution of issues will depend on the particular circumstances of the matter in question. Final interpretation of the law is the responsibility of the Board (sitting as a Hearing Panel) and is subject to review by the courts.
6. Certain aspects of these Guidelines may be revisited by the PMPRB in light of experience and changing circumstances. Guidelines and policies issued by the PMPRB are developed in an open manner with opportunities for full consultation with interested parties. When any changes to the Guidelines are considered, stakeholders will be consulted by the PMPRB in accordance with the commentary process established under subsection 96(5) of the Act.
7. Every reasonable effort will be made by the PMPRB to assist patentees in understanding the Guidelines and their application. For example, upon request, patentees will be advised by Staff on the appropriate methodologies to be applied in the price review of patented medicines sold in Canada. In addition, upon request and if there is sufficient information satisfying the Board that the price at which a patentee is selling or proposes to sell a patented medicine would not be found to be excessive, a non-binding certificate to that effect may be issued under subsection 98(4) of the Act.
8. The Guidelines do not provide an exhaustive description of all steps that may be taken or all issues that may arise in the context of a price review. In exceptional circumstances or in the event of a hearing, any methods or tests deemed appropriate and consistent with the Act and Regulations may be used by the PMPRB, regardless of whether they are addressed in the Guidelines or otherwise differ from the approach set out therein. In no case will Staff or Board members be bound or limited by these Guidelines.
9. For additional information on these Guidelines or Staff’s general approach to price reviews and investigations, please see the PMPRB’s website or contact Staff at the following:
Patented Medicine Prices Review Board
Box L40
Standard Life Centre
333 Laurier Avenue West
Suite 1400
Ottawa, Ontario
K1P 1C1
Attention: Secretary of the Board
III. Legal Framework
10. The PMPRB was established in 1987 as part of a sweeping set of amendments to the Patent Act brought into effect by Bill C-22Footnote 1. These amendments strengthened patent protection for medicines, both in terms of the scope of patentable subject matter and length of the patent-derived exclusivity period, thereby creating what policy makers believed to be a more favourable investment climate in Canada for pharmaceutical research and development (R&D). As a concession to opponents of these changes who were concerned that stronger patent protection for medicines might cause prices to rise to unacceptable levels and become unaffordable to Canadians, Bill C-22 also created the PMPRB. The PMPRB has a regulatory mandate to ensure that the prices of patented medicines are not excessive. In essence, Bill C-22 sought to strike a balance between the need to recognize and reward pharmaceutical innovation by providing patentees with a period of market exclusivity and ensuring that prices charged during that exclusivity period remain reasonable. The PMPRB’s existence as the only sector-specific price ceiling regulator under the Act reflects a recognition by policy makers that the unfettered ability to set prices for patented medicines is not in the public interest given the unique harm that can ensue if consumers are made to pay excessive prices for them.
11. The PMPRB has a dual mandate: in its regulatory role, it protects consumers by ensuring that the prices of patented medicines are not excessive; in its reporting role, it provides information on pricing trends in the pharmaceutical industry via its Annual Reports. Further to a directive from the Minister of Health under section 90 of the Act, the PMPRB also supports informed and evidence-based health policy by reporting on medicine price, utilization and cost trends under the National Prescription Drug Utilization Information System (NPDUIS) initiative.
12. The PMPRB Board consists of five members appointed by the Governor–in-Council under section 91 of the Act for terms of up to five (5) years, renewable once. The Chairperson of the Board acts as the Chief Executive Officer (CEO) of the PMPRB and has supervision over and direction of its work. The PMPRB employs public servants (i.e., Staff) pursuant to section 94 of the Act to carry out its day-to-day work. The PMPRB’s Executive Director is its senior public servant, Chief Operating Officer (COO) and Chief Financial Officer (CFO), and is responsible for the management of Staff.
13. The PMPRB is established under the Act as an independent, quasi-judicial body. To ensure this independence and autonomy, no express or implicit power is provided under the Act to Health Canada or any other government entity to direct the PMPRB in the exercise of its regulatory function. The PMPRB maintains an arm’s length relationship from the Minister of Health (who is responsible for the sections of the Act pertaining to the PMPRB), the Minister of Innovation, Science and Economic Development (who is responsible for the Act as a whole) and its various stakeholders. Similarly, the PMPRB is structured in a manner that separates the work and functions of Staff, the Chairperson and Board members. Investigation, litigation and reporting functions reside with Staff and are separate from the adjudication functions that are reserved for Board members only.
14. The monitoring of patentees’ compliance with regulatory filing requirements and the administrative price reviews of patented medicines are the responsibility of Staff. When a patented medicine appears to be priced excessively and the issue cannot be resolved through voluntary price reduction and/or measures to offset revenues from sales at that price by the patentee, the matter may be brought by the Executive Director to the Chairperson who will determine whether a hearing is in the public interest. If the decision to hold a hearing is made, a Notice of Hearing is issued and a Hearing Panel is appointed by the Chairperson to adjudicate the matter. The Chairperson’s decision to issue a Notice of Hearing is a purely administrative act and does not express his or her view of the merits of the underlying matter.
15. The PMPRB reviews the prices of patented medicines sold at arm’s-length by patentees. Sales in Canada may include, but are not limited to, patented medicines subject to a Notice of Compliance (NOC), the Special Access Programme, the List of Drugs for an Urgent Public Health Need, Clinical Trial Applications, or Investigational New Drugs. The PMPRB has no authority over prices charged by parties other than patentees, such as prices charged by wholesalers or retailers, or over pharmacists’ professional fees.
16. Under the Act, the PMPRB is given jurisdiction to determine whether a patented medicine is or has been sold by a rights holder (a patentee, former patentee or the person for the time being entitled to the benefit of a certificate of supplementary protection for an invention pertaining to a medicine) at an excessive price in any market in Canada.Footnote 2 The term “patentee” is defined in the Act as a person who is entitled to the benefit of a patent for an invention for a period, including any other person entitled to exercise rights in relation to the patent, such as a holder of an express or implied license.Footnote 3
17. An invention pertains to a medicine if the invention is intended or capable of being used for medicine or for the preparation or production of medicine. The phrase “pertain to a medicine” has a broad meaning. The Federal Court of Appeal has determined that the nature of that connection may be “tenuous”Footnote 4. It is satisfied, for example, where there may “only be a slender thread of a connection between a patented invention and the medicine sold in Canada”Footnote 5.Footnote 6
18. The term “medicine” is defined in the Act as including a drug (i.e., a substance or a mixture of substances manufactured, sold or represented for use in (i) the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state, or its symptoms, in human beings or animals; or (ii) restoring, correcting or modifying organic functions in human beings or animals) and a medicinal ingredient.Footnote 7 Unless otherwise specified, in these Guidelines, a reference to a “medicine” includes all dosage forms and strengths (i.e., all Drug Identification Numbers or “DINs”) of the medicine.
19. The PMPRB recognizes that the term “medicinal ingredient” is generally understood to mean the “active pharmaceutical ingredient” (API) used as raw materials during the manufacture of the finished dosage form. Patented medicines under the PMPRB’s juris- diction include vaccines, topical preparations, anaesthetics and diagnostic products used in vivo, regardless of delivery mechanism (i.e., trans-dermal patch, capsule, injectable, inhaler). However, the PMPRB does not consider medical devices, in vitro diagnostic products and disinfectants that are not used in vivo to be patented medicines for the purpose of price review provisions in the Act.
20. The PMPRB has jurisdiction during the life of an eligible and issued patent including the pre-grant period (from the patent application date). The PMPRB also has jurisdiction for the extended period of protection granted via a certificate of supplementary protection.Footnote 8
21. The PMPRB’s jurisdiction over the price at which a patented medicine is sold in any market in Canada persists after the patent has been dedicated and until the cancellation or surrender of the patent pursuant to the express provisions of the Act or the expiry of the term of the patent. Patent dedication is not expressly recognized in the Act as a mechanism by which patent rights may be terminated before the normal expiry of the patent term.
22. Orders issued by the Board are enforceable in the same manner as orders of the Federal Court or any superior court in Canada and may be enforced by the PMPRB or by the Federal Court. Decisions embodied in orders issued by the PMPRB may be subject to judicial review by the Federal Court in accordance with administrative law principles and the Federal Courts Act.
IV. Filing Requirements Pertaining to Price Reviews
23. Access to timely and accurate information regarding the sale of patented medicines is necessary for the PMPRB to fulfil its regulatory mandate. Therefore, patentees and former patentees are required to submit this information to the PMPRB.
24. The information that must be supplied is set out in section 82 of the Act and in the Regulations. Further information to assist patentees in identifying the content and form of information to be supplied and the strict deadlines set out in the legislation may be found in the Help section of the online filing tool, which takes the place of the previous Patentee’s Guide to Reporting. Patentees are responsible for compliance with filing obligations. These statutory obligations cannot be waived or amended by Staff.
25. Information that patentees or former patentees may be required to file under the Regulations includes, but is not limited to:
- A notification describing a patentee’s intention to offer a patented medicine for sale in a market in Canada in which such patented medicine has not previously been sold (i.e., the first sale of the patented medicine), along with related information (Notification of Intention to Sell a Patented Medicine);
- Prescribed information relating to the identity and characteristics of a patented medicine, such as the product monograph for the patented medicine or equivalent information, and the DINs assigned for each dosage form and strength of the patented medicine;
- Prescribed information relating to the price of a patented medicine, such as information concerning the price at which each dosage form and strength of the patented medicine is or has been sold in any market in Canada or in any of the eleven countries set out in the Regulations (the “PMPRB11”); or
- Prescribed information relating to cost-utility analyses prepared by publicly funded Canadian health technology assessment (HTA) agencies, for which the outcomes are expressed as the cost per quality-adjusted life year (QALY) for each indication that is the subject of the analysis; and
- Prescribed information relating to the estimated maximum use of the patented medicine in Canada for a given time period, by quantity of the patented medicine in final dosage form.
26. As per s. 7 of the Regulations, patentees shall submit required filings by email using the electronic forms on the PMPRB’s website. The forms should bear the electronic signature of an authorized individual who certifies that the information is true and complete.
27. It is the responsibility of each patentee to independently ensure that the information filed with the PMPRB (including domestic and foreign prices) is accurate. Ad hoc audits of patentee filings, including pricing, revenue and patent information, may be conducted by Staff. In the event of such an audit, patentees may be asked to provide additional supporting materials and/or corrections or confirmation of the information filed.
28. The failure to file required information within the specified period or the filing of erroneous or false information may have significant consequences for patentees or former patentees. An order granting certain remedies may be issued by the Board, including an ex parte order requiring that the missing information be submitted. Alternatively, the matter may lead to summary conviction proceedings under subsection 76.1(1) of the Act. Further, the filing of false information is an indictable offence under section 76 of the Act that, on conviction, can lead to monetary fines or terms of imprisonment.
29. The Act provides for the confidentiality of information supplied to the PMPRB in certain circumstances. Specifically, information or documents provided to the PMPRB in accordance with the provisions dealing with pricing information in sections 80, 81 and 82 of the Act, or in any proceeding relating to excessive prices under section 83, is privileged and cannot be disclosed to the public without authorization of the disclosing party, unless such information has been disclosed at a public hearing under section 83 of the Act or is subject to the exceptions outlined in section 87(2) of the Act.
30. Information provided to the PMPRB may be subject to certain provisions in the Access to Information Act and the Privacy Act.
V. Price Review Process
31. The price review process consists of a series of steps whereby (i) patented medicines are divided into categories depending on their date of introduction and on criteria related to their market characteristics and (ii) ceiling prices are identified and used to assess the patented medicines’ prices. Price reviews are normally conducted by Staff using the methods and tests set out in these Guidelines based on the information filed by the patentees and/or obtained by Staff from relevant outside sources such as public formularies.
32. As an initial review step, patented medicines are divided into four broad categories: (1) Grandfathered patented medicines; (2) Line Extensions of Grandfathered patented medicines; (3) Gap medicines; and (4) New patented medicines.
33. Grandfathered patented medicines include the dosage strengths and forms for patented medicines for which the patentee was assigned a DIN prior to August 21, 2019 regardless of whether those dosage strengths and forms have been approved for new indications (without a DIN change) after August 21, 2019.
34. Line Extensions of Grandfathered patented medicines are new dosage forms and strengths of Grandfathered patented medicines to which a DIN was assigned on or after August 21, 2019.
35. Gap Medicines are patented medicines for which a DIN was assigned on or after August 21, 2019 and the first sale in Canada took place prior to January 1, 2021.
36. New patented medicines include all other dosage strengths and forms of patented medicines
37. The price review factors under s. 85(1) of the Act for the four categories of patented medicines are as follows.
- Grandfathered patented medicines:
- The prices at which the medicine has been sold in the relevant market
- The prices at which other medicines in the same therapeutic class have been sold in the relevant market
- The prices at which the medicine and other medicines in the same therapeutic class have been sold in countries other than Canada
- Changes in the Consumer Price Index (CPI)
- New patented medicines, Line Extensions and Gap medicines:
- The prices at which the medicine has been sold in the relevant market
- The prices at which other medicines in the same therapeutic class have been sold in the relevant market
- The prices at which the medicine and other medicines in the same therapeutic class have been sold in countries other than Canada
- Changes in the Consumer Price Index (CPI)
- The pharmacoeconomic value in Canada of the medicine
- The size of the market for the medicine in Canada
- The gross domestic product in Canada and the gross domestic product per capita in Canada
38. The specific review processes and tests applicable to New patented medicines, Grandfathered patented medicines, Line Extension patented medicines and Gap patented medicines are explained in greater detail hereafter.
A. Price Review Process for New Patented Medicines
39. Information filed by patentees and information obtained from other sources is reviewed by the PMPRB to assess whether any patented medicine introduced in Canada appears to be priced excessively. The following diagram illustrates the review process for New patented medicines:
Schematic of Draft Guidelines
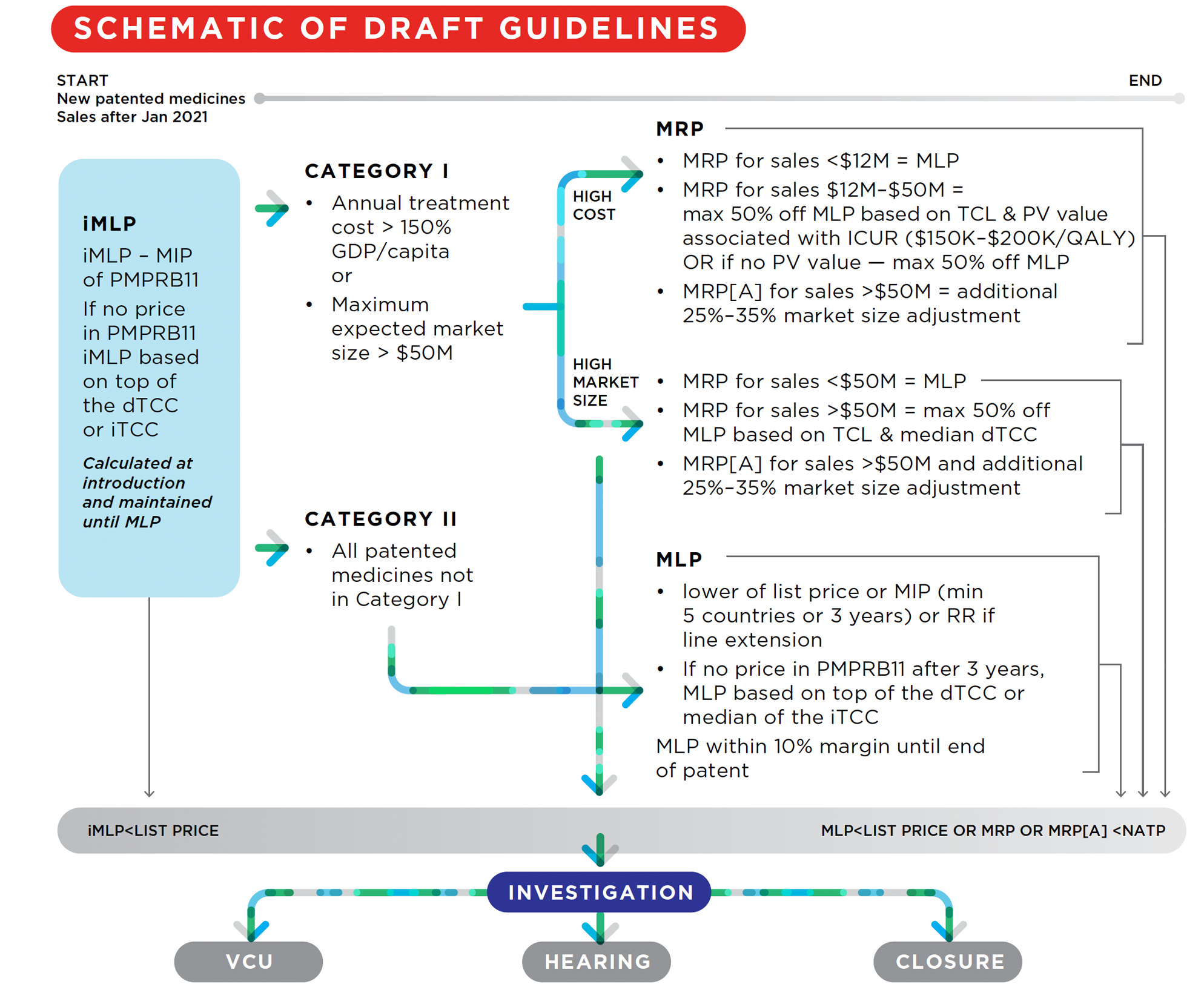
Figure description
This is a flowchart describing generally illustrating the review process for New patented medicines (patented medicines with sales after January 2021):
First, a iMLP is determined based on the MIP of the available PMPRB11 prices. If there are not prices in the PMPRB11 countries, the iMLP is based on the top of the dTCC or the iTCC. The iMLP is calculated at introduction and maintained until the MLP is set. If the patented medicine’s list price is above the iMLP, an investigation can be opened, which may lead to either a VCU, a hearing, or the closure of the investigation.
Then, the patented medicine is classified as either category I (medicines whose annual treatment costs are greater than 150% of GDP/Capita and/or medicines with a maximum expected market size greater than $50) or Catetgory II (all patented medicines not in Category I).
Category I medicines are subject to a MRP and a MLP. The test used for the MRP for category I medicines that have annual costs greater than 150% of GDP/capita are different for different sales. In either case, if the patented medicine’s average transaction price is greater than the MRP, an investigation can be opened, which may lead to either a VCU, a hearing, or the closure of the investigation.
For sales made where the annual market size is less than $12 million, the MRP is equal to the MLP.
For sales made after the annual market size rises to between $12 million and $50 million, the MRP is equal to a discount percentage based on the TCL and PV value associated with the ICUR (for ICURs between $150K-$200K per QALY) which can be up to a maximum 50% discount off the MLP. If there is to PV value, the maximum discount of 50% off the MLP is applied.
For sales made after the annual market size rises above $50 million, the MRP is subject to an additional market size adjustment which can range between 25% to 35%.
The test used for the MRP for category I medicines that have a maximum expected market size that is greater than $50 million are also different for different sales.
For sales made where the annual market size is less than $50 million, the MRP is equal to the MLP.
For sales made after the annual market size rises above $50 million, the MRP is based on a discount off the MLP which itself is based on the TCL and median dTCC and can be up to a maximum 50% discount, an additional 25%-35% market size adjustment may also apply.
Category II medicines are only subject to a MLP. The MLP is equal to either the lower of the list price or the MIP (once there is data for a minimum of 5 countries or 3 years have elapsed since introduction) or the RR if the patented medicine is a line extension. If, after 3 years post-introduction, there are no prices available for the PMPRB11 countries, the MLP is based on the top of the dTCC or the median of the iTCC. The MLP is maintained within a 10% margin of the MIP until the end of PMPRB jurisdiction. If the patented medicine’s list price is greater than the MLP, an investigation can be opened, which may lead to either a VCU, a hearing, or the closure of the investigation.
Each of the above steps is described in greater detail hereafter.
Step 1: iMLP
40. At introduction (i.e upon the first sale in any market in Canada), an Interim Maximum List Price ceiling is set (the “iMLP”) for the sale of the patented medicine. The iMLP is set at the median international ex-factory list price (“MIP”) for the PMPRB11 countries for which the patentee has provided information during the Interim Period (defined below). Patentees must ensure that the patented medicine’s grossFootnote 9 publicly available Canadian ex-factory price list price is no higher than the iMLP for the interim period, failing which the price may be subject to additional review or investigation by Staff.
41. If the patentee has not filed international price information for the PMPRB11 countries, the iMLP is set by the top of the domestic Therapeutic Class Comparison (“dTCC”). The dTCC will be calculated based on the highest cost of treatment across the comparator medicines, derived by taking into account the lowest public price of each comparator (see Appendix A for further details).
42. The iMLP is only calculated once and applies during the Interim Period, which lasts until the earlier of:
- three (3) years from the date of the introduction of the patented medicine in Canada; or
- the date when the patentee has filed international price information for at least five (5) of the PMPRB11 countries.
At the end of the Interim Period, the MLP will be set (see Step 2), and the iMLP will cease to apply.
43. If, at introduction, the patentee files international price information for at least five (5) of the PMPRB11 countries, the iMLP does not apply and the MLP is set immediately (see section below).
Step 2: MLP
44. Subject to the procedure described above, the iMLP will be replaced by a Maximum List Price (“MLP”).
45. If the patentee has filed international prices by the end of the Interim Period, the MLP is set by the MIP. Otherwise, the MLP is set by the top of the dTCC. The dTCC will be calculated based on the highest cost of treatment across the comparator medicines, derived by taking into account the lowest public price of each comparator (see Appendix A for further details).
46. If the patentee has not filed international prices by the end of the Interim Period, and there are no domestic therapeutic class comparators, the MLP may be set at the median of the international Therapeutic Class Comparison (“iTCC”).
47. There may be circumstances where the MLP is lower than the iMLP set during the Interim Period. In these circumstances, patentees will be granted until the next reporting period after the MLP is set to ensure the list price of the patented medicine is lowered to a level that is no higher than the MLP.
48. As explained in greater detail in section VI (“Reassessment”), if in subsequent periods, the prevailing MIP exceeds the MLP by more than 10%, the MLP may be adjusted based on the actual lagged change in the consumer price index (“CPI”)Footnote 10. For patented medicines with multiple DINs, comparison of the MLP against the MIP is only conducted for the DIN in respect of which the MLP was initially set by the MIP, and not for DINs in respect of which the MLPs were set by the Reasonable Relationship (“RR”) test (described in detail in Appendix B). However, if a DIN’s MLP is adjusted for CPI, additional MLPs set by the RR test in reference to this MLP will be adjusted in accordance with the RR methodology.
49. The MLP of the patented medicine may also be subject to reassessment and adjustment due to a 10% or greater variance from the MIP as described above (see section VI “Reassessment”).
50. As with the iMLP, patentees must ensure that the patented medicine’s list price is no higher than the MLP for the period during which it is applicable, failing which the price may be subject to additional review or investigation by Staff.
a) List Price of the patented medicine in Canada
51. As per the Regulations, patentees must file information about the list price for each dosage form, strength and package size (i.e., for each DIN) of the patented medicine in each province and territory.
52. List prices may vary within Canadian geographic markets (i.e., between provinces or territories). Where there are multiple list prices in different markets, the highest list price will be used for the purpose of the iMLP and MLP comparison.
b) List Prices of the patented medicine in the PMPRB11
53. International price tests are based on the information filed by the patentee. Where there are multiple list prices in the same country, the lowest price will generally be used. The prices for all countries in the PMPRB11 provided by the patentee will be used.
54. Guidance on potential sources of ex-factory prices is available through the Help section of the online filing tool.
55. When comparing prices in the PMPRB11, the local currency is converted into Canadian dollars using exchange rates calculated as the simple average of the thirty-six (36) monthly average noon spot exchange rates for each country (taken to eight (8) decimal places) as published by the Bank of Canada. For a patented medicine’s introductory period, the thirty-six (36) months ending in the second month of the previous reporting period (i.e., February or August) will be used. Subsequently, the thirty-six (36) months ending in the second month of the reporting period under review will be used.
Step 3: MRP/MRP[A] (Category I New Patented Medicines Only)
56. New patented medicines will be classified as either Category I or Category II based on certain market characteristics. In addition to the iMLP/MLP, Category I patented medicines are also subject to a “Maximum Rebated Price” ceiling (“MRP”) that may be further adjusted based on the size of the market under certain circumstances, thus becoming a MRP[Adjusted] (“MRP[A]”). Both the calculation of the MRP and of the MRP[A] are described below and in Appendix C and Appendix D.
57. The MRP takes into account Therapeutic Criteria Level (TCL) (levels I-IV and the scientific review process are described in Appendix E; the levels are generally based on scientific information including therapeutic effect, clinical impact and QALY gain), pharmacoeconomic value and market size for the patented medicine (see Appendix C). Patentees must ensure that the patented medicine’s net price in Canada, (i.e., its average transaction price or “ATP”Footnote 11) is no higher than the MRP, failing which the price may be subject to additional review or investigation by Staff. Guidance on calculating the net price of a patented medicine, including the treatment of free goods and rebates is available through the Help section of the online filing tool.
a) Classifying a patented medicine as Category I
58. A New patented medicine will be classified as Category I if it meets either of the following criteria:
- 12-month treatment cost greater than 150% of GDP per capita: based on the introductory period pricing information filed by the patentee, the patented medicine’s 12-month treatment cost will be calculated by Staff based on the maximum dosage per course of treatment listed in the product monograph; the maximum number of courses of treatment per 12 months, based on the nature of the condition, clinical practices, and other relevant criteria; and the highest Canadian list price. If a list price is not available, the national net price will be used.
- Estimated or actual market size (revenue) exceeds annual market size threshold: The market size for categorization purposes can be based on (i) an estimate filed under the Regulations or (ii) actual revenue filed under the Regulations. The annual market size threshold pursuant to which a New patented medicine will be classified as Category 1 is $50 million.Footnote 12
59. All other New patented medicines will be classified as Category II.
60. In addition, even if they would otherwise meet the Category I criteria, all new patented BiosimilarsFootnote 13 and new patented GenericFootnote 14 medicines will be classified as Category II.
b) MRP and MRP[A] calculation
61. The MRP is calculated depending on the relevant criteria that led to the patented medicine’s categorization as Category I and the availability and content of a cost-utility analysis.
62. For patented medicines that have (1) a 12-month treatment cost greater than 150% of GDP per capita and estimated or actual maximum revenue greater than $12 million per year; and (2) an available cost-utility analysis, the MRP is calculated as follows:
- The Incremental Cost-Utility Ratio (“ICUR”) measured in cost per quality-adjusted life years (“QALYs”) for each indication of the patented medicine will be identified from the cost-utility analyses filed by the patentee.
- The price at which the patented medicine’s ICUR would be equivalent to the Pharmacoeconomic Value Threshold (“PVT”) will be identified (the “Pharmacoeconomic Price” or “PEP”).
- The ICUR will be compared against the applicable PVT and reduction floor, based on its Therapeutic Criteria Level (see Appendix C, “Pharmacoeconomic Value Assessment” and chart reproduced below).
Price adjustment based on Therapeutic Criteria Level for MRP calculation Therapeutic Criteria Level
(See Appendix E – The Scientific Review Process)PVT Reduction Floor off MLP Level I $200K/ QALY 20% Level II $150K/ QALY 30% Level III $150K/ QALY 40% Level IV $150K/ QALY 50% Pharmacoeconomic analysis is a cost minimization Median of dTCC subject to 50% floor No pharmacoeconomic assessment 50% of MLP - The MRP will be determined assuming that the first $12 million are realized for quantities sold at the MLP, while the remaining sales up to $50 million are realized for quantities sold at the PEP. The MRP may be further adjusted for market size – thus becoming a MRP[A]- if the patented medicine realizes actual (as opposed to estimated) annual sales such that, if priced at the MRP set by the PEP subject to the reduction floor, actual revenues would be in excess of $50 million (see Appendix D, “Market Size Adjustment Methodology”). Because this adjustment takes place at predictable sales levels, the patentee will be informed of what both its current MRP and expected future MRP[A] once the sales of the patented medicine reach certain levels.
63. If the procedure above results in a MRP or MRP[A] that exceeds the MLP, the MRP will be set at the same level as the MLP.
64. For patented medicines that have a 12-month treatment cost greater than 150% of GDP per capita and estimated maximum revenue greater than $12 million per year; but do not have an available cost-utility analysis or if the analysis submitted does not allow for the determination of the MRP as described above, the MRP is set at 50% of the MLP. A MRP[A] based on this MRP may be calculated if applicable (see Appendix C).
65. For patented medicines that have been categorized as Category I based solely on market size, the MRP is calculated as follows:
- If the actual market size is less than $50 million, the MRP is set at the MLP.
- If the actual market size is more than $50 million, the MRP is set as the median of the dTCC taking into account the applicable floor as per the table in Appendix A and the application of market size adjustments as per Appendix D, and a MRP[A] applies.
66. The MRP/MRP[A] is recalculated annually.
67. The following different MRP/MRP[A] calculation pathways described above are illustrated visually below.
Category 1 - MRP
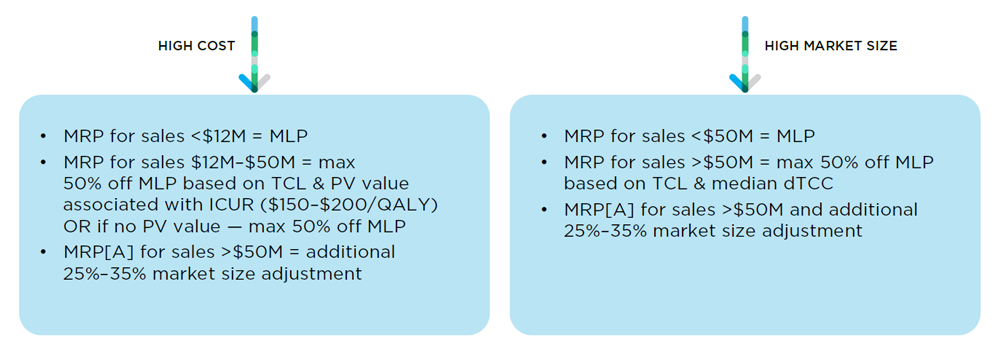
Figure description
This is a schema illustrating the calculation of the MRP for Category I patented medicines.
The test used for the MRP for Category I medicines that have high annual costs are different for different sales.
For sales made where the annual market size is less than $12 million, the MRP is equal to the MLP.
For sales made after the annual market size rises to between $12 million and $50 million, the MRP is equal to a discount percentage based on the TCL and VP associated with the ICUR (for ICURs between $150K-$200K per QALY) which can be up to a maximum 50% discount off the MLP. If there is to VP value, the maximum discount of 50% off the MLP is applied.
For sales made after the annual market size rises above $50 million, the MRP is subject to an additional market size adjustment which can range between 25% to 35%
The test used for the MRP for Category I medicines that have a high market size are also different for different sales.
For sales made where the annual market size is less than $50 million, the MRP is equal to the MLP
For sales made after the annual market size rises above $50 million, the MRP is based on a discount off the MLP which itself is based on the TCL and median dTCC and can be up to a maximum 50% discount, an additional 25%-35% market size adjustment may also apply.
Step 4: Identification of Relevant Indication
68. For patented medicines with more than one approved indication, the indication for which the MRP (and MLP, if applicable) will be assessed will be determined by Staff (the “Relevant Indication”). This could occur at introduction, or as part of a reassessment if additional indications are approved during the life cycle of the patented medicine (see section VI).
69. For Category I patented medicines, the Relevant Indication will be the indication triggering the Category I classification criteria for annual treatment cost in these Guidelines. For Category I patented medicines where more than one, or no, indications meet this threshold, and for Category II patented medicines, the Relevant Indication will be the indication treating the condition with the highest prevalence (i.e., the largest patient population) or estimated use.
Step 5: Compliance Review Timeline
70. The timing for compliance with the MRP/MLP ceilings described in this section is as follows:
- iMLP: patentees must comply with the iMLP at market entry if the iMLP is known at that time, or within one (1) reporting period when the iMLP is known.
- MLP: patentees must comply with the MLP at market entry if the MLP is known at that time, or within one (1) reporting period when the MLP is set subsequent to an iMLP.
- MRP/MRP[A]: patentees must comply with the MRP/MRP[A] within two (2) reporting periods of the MRP/MRP[A] being known.
B. Price Review Process for Grandfathered, Line Extension and Gap and Patented Medicines
71. Grandfathered, Line Extension and Gap patented medicines will be subject to a MLP but not to a MRP unless there is not enough available data for the MLP calculation, in which case a iMLP will be identified once as per the procedures described in paras. 40 and 41 until there is enough data to set the MLP.
72. The MLP for Grandfathered and Line Extension medicines is set at the lower of
- the highest international price (“HIP”) for the PMPRB11 countries for which the patentee has provided information; or
- the patented medicine’s ceiling (e.g. the “NEAP”) under the Guidelines applicable prior to the issuance of these Guidelines.
73. The HIP and list prices for Grandfathered and Line Extension patented medicines will be calculated in the same manner as for New patented medicines under these Guidelines (see section V(A), “Price Review Process for New Patented Medicines”).
74. The MLP for Gap patented medicines is set at the lower of
- the MIP for the PMPRB11 countries for which the patentee has provided information; or
- the patented medicine’s ceiling (e.g. the “NEAP”) under the Guidelines applicable prior to the issuance of these Guidelines.
75. Patentees must ensure that the patented medicine’s list price is no higher than the MLP for the period during which it is applicable, failing which the price may be subject to additional review or investigation by Staff.
76. For Grandfathered, Line Extension and Gap patented medicines, if the MLP is set by the patented medicine’s NEAP and if there is evidence that its calculation was uncharacteristically low due to the reporting of benefits, the patentee may submit a request for a higher MLP. Any such request must include a proposed MLP, details and supported documentation of the reported benefits and historic list price changes, as well as any other relevant information specified by Staff. If the requisite information supports a reassessment, the MLP will be adjusted to the lower of (i) the applicable international price test for the PMPRB11 countries for which the patentee has provided information, or (ii) the highest compliant list price of the patented medicine under the Guidelines applicable prior to the issuance of these Guidelines.
77. Patentees are expected to comply with the MLP within one reporting period once the MLP is set for Line Extension patented medicines and within two (2) reporting periods for Grandfathered or Gap patented medicines.
VI. Reassessment
78. The categories or price ceilings of patented medicines may be reassessed to ensure that they remain relevant in light of material changes in market conditions.
Reassessment of New Patented Medicines
79. In the case of New patented medicines, a reassessment may be conducted if any of the following situations arise:
- A patented medicine (Category I or Category II) is approved for a new indication;
- A Category II patented medicine has sales exceeding the market size threshold (see Appendix D), contrary to the initial estimate filed by the patentee; or
- A Category I patented medicine’s cost-utility analysis is updated;
- If in two consecutive subsequent periods, the prevailing MIP is lower than the MLP by more than 10%.
80. A patented medicine that is approved for a new indication may have its Relevant Indication changed in accordance with the procedures described in section V. A new indication may alter the patented medicine’s market size, therapeutic class comparators, and cost-effectiveness. As a result, there may be an increase or decrease in the MRP.
81. A Category II patented medicine that is approved for a new indication (except for Biosimilars and Generic medicines) may be reassessed as Category I if it triggers the relevant screening criteria. For example, if the patented medicine’s actual revenues increase above the annual Market Size Threshold, contrary to the initial market size estimate filed by the patentee.
82. A reassessment from Category II to Category I will result in the patented medicine being given a MRP.
83. If in subsequent periods, the prevailing MIP is lower than the MLP by more than 10%, the MLP will be reset by the prevailing MIP. If the patented medicine has additional DINs with MLPs set by the RR test (see Appendix B), those MLPs will be adjusted accordingly.
Reassessment of Grandfathered, Line Extension and Gap Patented Medicines
84. In the case of Grandfathered, Line Extension and Gap patented medicines, if in two consecutive subsequent periods, the prevailing HIP is lower than the MLP, the MLP will be reset by the prevailing HIP. If in two consecutive subsequent periods, the prevailing MIP is lower than the MLP by more than 10%, the MLP will be reset by the prevailing MIP.
Compliance with Reassessed Ceilings
85. The patentee will be notified in the event that reassessment criteria have been triggered and will be subsequently notified of the adjusted MLP/MRP. The price of the patented medicine must come into compliance with the new MLP/MRP as follows:
- MLP: within one (1) reporting period of being notified of the new MLP.
- MRP: within two (2) reporting periods of being notified of the new MRP.
VII. Investigations
86. An investigation is an in-depth review of the price of a patented medicine conducted by Staff. As part of an investigation, information provided by the patentee and information that may be obtained from other sources is reviewed by Staff. Investigations are purely administrative in nature and no Board members are involved in this process. If an investigation results in a hearing, the Hearing Panel must undertake an independent de novo review of the price of the patented medicine to determine whether it is excessive under section 83 of the Act. Accordingly, the positions taken by Staff or the patentee(s) during the investigation may differ from those taken during a hearing.
87. The criteria for commencing an investigation have been developed with the intention of making the most efficient use of the PMPRB’s human and financial resources. The fact that the price of a patented medicine is not subject to an investigation does not necessarily mean that its price is not excessive and vice-versa. It only means that the investigation criteria under the Guidelines have not been met in the particular circumstances.
A. Investigation Criteria
88. In general, an investigation will be commenced by Staff when any of the following situations occur:
- the price of any dosage form or strength of a patented medicine appears to be above the corresponding applicable price ceiling by more than 5%; or
- the cumulative potential revenues earned as a result of pricing above applicable ceiling(s) (“potential excess revenues”) appear to exceed $50,000 for the patented medicine (i.e., across all dosage forms and strengths of the patented medicine) in a calendar year; or
- A complaint from any source is received at any time (including prior to the applicable date for the assessment of compliance with a particular ceiling).
89. Notwithstanding the above, in the case of patented Biosimilars, patented Generic medicines, patented medicines for veterinary use and over the counter (OTC) patented medicines, an investigation will only be commenced by Staff if a complaint is received.
90. Notwithstanding para. 88, the price of any patented medicine which appears on the List of Drugs for Exceptional Importation and Sale set out in accordance with s. 3 of the March 30, 2020 Interim Order Respecting Drugs, Medical Devices and Foods for a Special Dietary Purpose in Relation to COVID-19 will not be subject to an investigation unless a complaint is received from either the federal Minister of Health or any of her provincial or territorial counterparts.
91. When the price of a patented medicine is higher than the applicable ceilings under the Guidelines but does not meet the criteria for commencing an investigation, the patentee will be notified and the patented medicine will be reported in the PMPRB’s Annual Report as “Does Not Trigger Investigation.” In such an instance, no immediate action will be taken by Staff; however, in order to avoid an investigation at a later date, the patentee should ensure that its price is reduced to a level no higher than the applicable ceiling price and offset any revenues that may have accrued, failing which the price may be subject to eventual investigation by Staff.
B. Investigation Process
92. When an investigation is commenced by Staff, the patentee will be notified and the patented medicine will be reported in the PMPRB’s Annual Report as “Under Investigation.” Price reviews and investigations cannot result in a legal determination that the price of a patented medicine is excessive under the Act. Such a determination can only be made by a Hearing Panel after the patentee or former patentee has been provided with a reasonable opportunity to be heard, as required by section 83 of the Act.
1. Additional Review of Filings
93. When a patented medicine comes under investigation, its pricing history from introduction will be reviewed by Staff. All information filed by the patentee is analyzed and further clarification may be sought. For example, if a price seems to be in error or unexpected, or if there are discrepancies in the patentee’s filings, the patentee may be asked for an explanation or to provide additional supporting materials. Any relevant materials not filed by the patentee, such as dTCC and iTCC list prices, may also be considered by Staff. In addition, an analysis of the appropriateness of the applicable category and tests is conducted by Staff to take into account the facts of the case and the particularities of the relevant markets in which the patented medicine is sold. For example, Staff may consider whether the actual market size is materially lower than the estimated market size, or whether the patented medicine is a vaccine, blood product or other product subject to a tendering process.
94. Staff may utilize any of the tests described in the Guidelines and modifications or variations of those tests (e.g., MIP instead of HIP or median as opposed to the top of the dTCC) depending what it believes most appropriate to the factual circumstances surrounding the price of the patented medicine under investigation.
2. Calculation of Potential Excess Revenues
95. In the event that the price of a patented medicine exceeds the ceilings established under the Guidelines, the patentee will be notified that its price(s) are “outside the thresholds set out in the Guidelines” and the applicable price ceilings will be identified. Cumulative potential excess revenues will also begin to be calculated by Staff based on net prices filed by the patentee regardless of the type of price ceiling or test being used (iMLP, MLP, or MRP/MRP[A]). Because the PMPRB has jurisdiction over the patented medicine in the pre-grant patent period, potential excess revenues related to its sales during that period will be included in the calculations.
96. In some cases, the tests and ceilings used during the investigation may differ from the initial thresholds that led to the triggering of the investigation. In such cases, the investigation ceilings (as opposed to the triggering ceilings) will be used to calculate potential excess revenues.
97. Finally, in the event of a hearing, Staff may seek a remedy in the form of excess revenues that differs from the cumulative potential excess revenues calculated during an investigation. In addition, where Staff believes the patentee or former patentee has engaged in a policy of selling the patented medicine at an excessive price, it may seek an order that the patentee offset up to twice the amount of excess revenues.
3. Investigation Outcomes
98. Possible outcomes of an investigation include:
- A Voluntary Compliance Undertaking (“VCU”), as described in section VIII;
- The issuance of a Notice of Hearing if, upon the recommendation of Staff, the Chairperson considers it to be in the public interest; and/or
- The closure of the investigation.
99. The closure of an investigation may follow from a VCU or a determination that further examination of the price of a patented medicine is not warranted at that point in time in view of facts and considerations that come to light during the investigation.
100. The closure of an investigation is an administrative act and does not constitute a legal determination or an admission by the PMPRB that the price of the patented medicine is not excessive. The closure of an investigation does not preclude the possibility of the opening or re-opening of further investigation(s) or the commencement of a hearing in the future.
VIII. Voluntary Compliance Undertaking (VCU)
101. At any time prior to the issuance of a Notice of Hearing, a patentee may submit a VCU to Staff. A VCU is a promise by the patentee to reduce its price and/or offset any potential excess revenues from the sale of a patented medicine that is subject to an investigation. A proposed VCU does not constitute an admission by the patentee that the price of the patented medicine is excessive.
102. VCU negotiations with patentees are conducted by Staff, and it is the PMPRB’s policy that the Chairperson not be involved in those discussions. If negotiations result in a VCU proposal that Staff believes would be acceptable to the Chairperson, it will be referred to the Chairperson for his or her consideration. Staff cannot independently determine whether a VCU proposal is acceptable and cannot make any assurances to the patentee regarding the likelihood that the Chairperson will consider it acceptable.
103. The consideration of a VCU is an administrative procedure and does not constitute an admission or determination by the PMPRB that the price submitted by the patentee, or used to calculate a revenue offset, is not excessive. However, the acceptance of a VCU by the Chairperson will result in the closure of an investigation.
104. The PMPRB reports publicly on all VCUs that the Chairperson has accepted. In submitting a signed VCU a patentee must consent to its publication either in full or redacted form. The reported information can include disclosure of a copy of the VCU or terms included in the VCU. The reported information may appear in the PMPRB’s Annual Report, on the PMPRB’s website, in the PMPRB’s publications such as the NEWSletter, and on social media platforms.
105. Requests for VCU negotiations or proposals “without prejudice” cannot be considered by Staff. VCUs are unilateral promises by patentees and not settlement agreements. However, parts of any discussions between patentees and Staff that relate to the content of the patentee’s filings may be subject to the protections set out in sections 87 and 88 of the Patent Act. In addition, provisions of the Access to Information Act may apply.
106. If a Notice of Hearing has been issued, patentees may still pursue settlement through the negotiation of a settlement agreement, which must be approved by the Hearing Panel. Requests for settlement agreements or proposals are considered by Staff on a “without prejudice” basis.
IX. Hearing Recommendation
107. When an investigation into the price of a patented medicine is completed and the matter is not resolved with the patentee, the Executive Director may submit a report to the Chairperson. The Chairperson, in his or her capacity as the CEO of the PMPRB, may decide to issue a Notice of Hearing if he or she is of the opinion that it is in the public interest. A decision to issue a Notice of Hearing is not adjudicative and no analysis is undertaken by the Chairperson as to whether the facts alleged by Staff are, or will be, proven. Until a matter is brought before a Hearing Panel at a public hearing, no other Board member is informed of the results of Staff’s review or investigation into the price of a patented medicine.
108. The decision of whether the price of a patented medicine is excessive is made by the Hearing Panel alone after the public hearing is held.
X. Excessive Price Hearing Process and Remedies
109. PMPRB hearings are public proceedings. During a hearing, submissions and evidence from the parties are heard by a Hearing Panel consisting of at least two Board members. The Hearing Panel determines whether a patented medicine is being or has been sold at an excessive price in any market in Canada by taking into consideration the available information relating to the factors set out in section 85 of the Act.
110. For more information about hearings, please consult the PMPRB Rules of Practice and Procedure, the published standard set of procedures to be followed by all participants in hearings before the Board. The Rules set out the Board’s procedures in accordance with the requirement under the Act to resolve matters as informally and expeditiously as the circumstances and considerations of fairness permit. Practice directions and further information about previous and ongoing hearings are also publicly available on the PMPRB’s website.
111. Under the Act, the PMPRB is empowered to make remedial orders when it is found, following a hearing, that a patentee (or former patentee) is selling, or has sold, a patented medicine in any market in Canada at an excessive price.Footnote 15
112. In broad terms, the PMPRB has the power to impose two main forms of remedy after a hearing: (i) orders directing the patentee to cause the maximum price at which the patentee sells the patented medicine in that market to be reduced to such level as the Board considers not to be excessive; and (ii) orders directing the patentee to offset the amount of the excess revenues estimated by it to have been derived by the patentee from the sale of the patented medicine at an excessive price by either (a) reducing the price at which the patentee sells the patented medicine; (b) reducing the price at which the patentee sells one other medicine to which a patented invention of the patentee pertains; or (c) paying to Her Majesty in right of Canada an amount specified in the order.
113. If a Hearing Panel finds that the patentee or former patentee has engaged in a policy of selling the patented medicine at an excessive price, it may order the patentee to offset up to twice the amount of excess revenues estimated by it to have been derived from the sale of the patented medicine at an excessive price. The extent and duration of sales of the patented medicine at an excessive price will be considered by the Board in making this finding and order.
XI. Failure to File Hearing
114. When it is the opinion of Staff that a patentee has failed or refused to provide the PMPRB with the pricing, sales, or revenues and like information required by law, the Executive Director may recommend to the Chairperson to hold a public hearing to determine whether the patentee has, in fact, breached the reporting requirements of the Act and Regulations. If, as the result of such a hearing, the Hearing Panel finds that the patentee is in breach of its reporting requirements, it may order the patentee to provide the PMPRB with the required information and documents as per section 81 and/or section 88 of the Act.
115. In addition, as per subsection 76.1(1) of the Act, every person who contravenes or fails to comply with the filing requirements set out in section 80, 81, 82 or 88, or any order made thereunder, is guilty of an offence punishable on summary conviction and liable to a fine or to imprisonment.
XII. Complaints
116. Any individual or group who believes that the price of a patented medicine is excessive may submit a complaint to the PMPRB. A complaint may be submitted by telephone, in writing, or electronically using the contact information available on the PMRPB’s “How to Make a Complaint” page.
117. Complaints are a trigger for an investigation by Staff into the price of a patented medicine. The complainant is not part of that investigation or of any resulting hearing (unless the complainant applies to become an intervener in the hearing). The complainant is not required to provide any documents or evidence to the PMPRB. Any investigation will be based on materials provided by the patentee or otherwise obtained by Staff.
118. Due to limitations on disclosure set out in sections 87 and 88 of the Patent Act and in the Access to Information Act, the complainant will be only be informed of the outcome of the investigation if the process results in a VCU or a Notice of Hearing.
XIII. Appendices
A. Domestic Therapeutic Class Comparison (dTCC) and International Therapeutic Class Comparison (iTCC)
dTCC TEST
As described in section V of these Guidelines, the domestic Therapeutic Class Comparison (“dTCC”) test is used as part of the price ceiling calculation in certain circumstances. The dTCC test compares a patented medicine’s Price with the list prices of other medicines identified by scientific review for comparison purposes.
For Category I medicines with an actual market size exceeding $50 million, the MRP based on dTCC are set as the floors set out in the table below.
| Therapeutic Criteria Level (See Appendix E – The Scientific Review Process) |
dTCC Reduction Floor |
|---|---|
| Level I | 20% off MLP (dTCC not applicable) |
| Level II | 30% |
| Level III | 40% |
| Level IV | 50% |
Identification of medicines for comparison purposes
The World Health Organization (WHO) Collaborating Centre for Drug Statistics Methodology’s Anatomical Therapeutic Chemical (ATC) Classification System is used in the selection of medicines to be used for comparison purposes.
The medicines used for comparison purposes will typically be those identified under the ATC classification system at the sub-class level above the single chemical substance. This will normally be the fourth sub-class level but could include the next higher sub-class or another sub-class. In some instances, it may be appropriate to select from the fifth or single chemical substance level.
A medicine of the same ATC therapeutic class as the medicine under review may be omitted if it is unsuitable for comparison. For example, a medicine with a primary indication other than the primary indication of the patented medicine under review may be omitted from the comparison.
All medicines identified for comparison that have the same approved indication or use as the Relevant Indication of the patented medicine under review will be included in the review. This review is based on Staff research and may include additional research by a Drug Information Centre (DIC) or consideration of evidence submitted by patentees.
Where there are multiple sellers of a medecine identified as a comparable medicine, the lowest price of the comparable medicine will be identified. The final therapeutic class comparison price is the highest price across all comparable medicines for the MLP and the median for the MRP.
For a patented medicine that is a new dosage form or strength of the same medicinal ingredient as one or more existing medicines, its comparators will be those existing medicines that are available in the same or comparable dosage form and have the same indication or use. This will apply regardless of whether the dosage regimens are the same or differ materially.
For a product that is a combination of medicines, where each of the medicines of the combination are sold in Canada and have the same indication or use, its comparators will be limited to the component medicines.
Comparable dosage regimens
The comparable dosage regimen used for comparison purposes will normally be the maximum of the usual recommended dosage in the Product Monograph (or similar information) taking into account relevant clinical variables. The most appropriate strength of the medicine will be chosen for a particular dosage regimen. Generally, a dosage regimen based on a course of treatment will be applicable to acute indications, while a per-day regimen (based on maintenance dose) will be applicable to chronic indications.
Price sources
Patentees do not file prices for the patented medicine’s comparators. Public sources will be used for the prices of the medicines used for comparison purposes in order to conduct a dTCC test. Provincial formularies will be the starting point in Staff’s identification of public prices. The lowest public price for each of the medicines identified for comparison purposes will be used. Any medicine (patented or non-patented) identified for comparison purposes may be excluded from a dTCC test if Staff has reason to believe it is being sold at an excessive price.
dTCC test
Following the identification of medicines for comparison purposes and of their lowest public price for each medicine, the cost of comparable courses of treatment for each medicine will be calculated. These costs of treatment will be ordered and the top or the median identified. In the event of an even number of medicines used for comparison purposes, the median will be the simple average of the middle two costs of treatment. This median cost of treatment will then be divided by the constituent units of the comparable course of treatment for the medicine under review to establish a per-unit price.
iTCC test
As described in section V of these Guidelines, the international Therapeutic Class Comparison (“iTCC”) test may be used in certain circumstances. The iTCC test compares a patented medicine’s list price with the list prices of other medicines identified by scientific review for comparison purposes in the eleven (11) comparator countries listed in the Regulations.
Identification of medicines for comparison purposes and comparable dosage regimens
The iTCC test uses the same medicines identified for comparison purposes and comparable dosage regimens as the dTCC test, following the procedures set out above. In cases where no domestic comparators have been identified, Staff may assess whether there are additional medicines approved for the same approved indication or use as the medicine under review in any of the comparator countries.
Price sources
Patentees do not file international prices for the international comparators to the patented medicine. National formularies will be the starting point in Staff’s identification of public prices. Publicly available ex-factory prices for the comparator medicines will be used in order to conduct an iTCC test. The lowest public price for each of the medicines identified for comparison purposes will be used.
Any medicine (patented or non-patented) identified for comparison purposes may be excluded from an iTCC test if Staff has reason to believe it is being sold at an excessive price.
iTCC test
Following the identification of medicines for comparison purposes and of the lowest public price for each medicine, the cost of comparable courses of treatment for each comparator medicine in each comparator country will be calculated. These costs of treatment will be ordered and the median identified for each country. In the event of an even number of comparator medicines used for comparison purposes, the median will be the simple average of the middle two costs of treatment. These medians will be ordered in a “median of the medians” will be identified. This median cost of treatment will then be divided by the constituent units of the comparable course of treatment for the medicine under review to establish a per-unit price. Local currencies will be converted to Canadian dollars using the methodology described in section V of these Guidelines.
B. Reasonable Relationship Test and Comparable Dosage Forms
Reasonable Relationship Test
The Reasonable Relationship (RR) test may be conducted to determine the MLP of a new additional strength of a patented medicine with other existing strengths, where the new additional strength has the same medicinal ingredient, indication, dosage regimen, and same or comparable dosage form as the existing strength(s). There is no RR test until a MLP is set for the reference strength.
MLP
Where a patented medicine is not or is no longer in the Interim period, the first strength that met the criteria for the transition from iMLP to MLP will become the reference strength. If there are multiple strengths that meet this criterion within a reporting period, the reference strength will be selected among these based on scientific considerations. Once the reference strength is identified, the ceiling for the new additional higher strengths will be established based on the proportional relationship between the strengths, subject to a HIP cap for the new additional strength. Specifically, the MLP of the new higher additional strength will be set to be equivalent to the price per standard unit of the reference strength. The MLP for the new additional lower strength will not be set higher than the price of the reference strength provided that this does not result in a price for the new additional strength that is above the HIP. If the list price for the strength that is getting the MLP set by the RR is lower than the calculation, the MLP should be set by the lower of the list price and the RR test.
MRP
The MRP is calculated as a reduction from the MLP. Therefore, the RR test application for a new additional strength of a patented medicine subject to the MRP will be done at the MLP level for the reference strength and the corresponding price reduction percentage will be carried over to the new additional strengths of the patented medicine.
MRP[A]
The MRP[A] is an adjustment based on the MRP as a function of market size and will thus change accordingly.
Comparable dosage forms
The following are considered comparable dosage forms for the purpose of the RR test. Formulations within each group are considered comparable, but dosage forms in a different group are not.
Topical (T)
- Aerosol
- Aerosol (foam)
- Cream
- Disc (extended release)
- Disc
- Dressings
- Gel
- Gel (controlled release)
- Liposomes
- Liquid
- Lotion
- Ointment
- Pad
- Paint
- Paste
- Patch
- Patch (extended release)
- Pencil
- Plaster
- Powder
- Shampoo
- Soap Bar
- Solution
- Sponge
- Spray
- Spray (bag-on-valve)
- Spray (metered dose)
- Stick
- Strip
- Swab
- Tincture
Nasal (N) / Pulmonary (P)
- Aerosol
- Aerosol-metered dose
- Drops
- Gas
- Metered dose preparation
- Powder
- Powder (metered dose)
- Solution
- Solution (extended release)
- Spray
- Spray (metered dose)
- Stick
Oral Solid (S)
- Bar (chewable)
- Caplet
- Capsule
- Effervescent granules
- Effervescent powder
- Effervescent tablet
- Film (soluble)
- Globules
- Granules
- Gum
- Lozenge
- Modified release caplet
- Modified release capsule
- Modified release tablet
- Pellet
- Piece (chewable)
- Powder (extended release)
- Strip
- Tablet
- Tablet (chewable)
- Tablet (oral disintegrating)
- Tablet for suspension
- Wafer
Oral Liquid (L)
- Drops
- Elixir
- Emulsion
- Gel
- Granules for solution
- Granules for suspension
- Granules for suspension (delayed release)
- Granules for suspension (extended release)
- Liquid
- Modified release liquid
- Powder (extended release)
- Powder for solution
- Powder for suspension
- Solution
- Solution (extended release)
- Spray
- Suspension
- Suspension (extended release)
- Syrup
- Syrup (extended release)
- Tea (herbal)
- Tincture
Vaginal (V)
- Cone
- Cream
- Douche
- Foam
- Gel
- Gel (controlled release)
- Implant
- Insert
- Insert (extended release)
- Ovule
- Pellet
- Ring (slow release)
- Sponge
- Suppository
- Suppository (sustained release)
- Tampon
- Vaginal tablet
- Vaginal tablet (effervescent)
Parenteral (J)
- Bolus
- Implant
- Kit
- Liposomes
- Modified release injection
- Pellet (implantable)
- Powder for solution
- Powder for suspension (sustained-release)
- Solution
- Solution (extended release)
- Suspension for emulsion
- Suspension (extended release)
Otic (E) / Opthalmic (Y)
- Drops
- Gel
- Gel (controlled release)
- Implant
- Insert
- Insert (extended release)
- Liquid
- Modified release ocular device
- Ointment
- Powder for solution
- Solution
- Solution (extended release)
- Suspension
Rectal (R)
- Cream
- Enema
- Foam
- Insert
- Ointment
- Ovule
- Stick
- Suppository
- Suppository (sustained release)
- Suspension
- Suspension (extended release)
Dental/Sublingual Buccal (M)
- Emulsion
- Film (soluble)
- Floss
- Gel
- Gel (controlled release)
- Gum
- Lozenge
- Metered-dose pump
- Modified release buccal tablet
- Mouthwash (gargle)
- Paste
- Powder (effervescent)
- Powder for suspension
- Solution
- Solution (extended release)
- Spray – buccal
- Spray – sublingual
- Stick
- Strip
- Sublingual tablet
- Suspension
- Suspension (extended release)
- Swab
- Tablet (orally disintegrating)
- Tablet
- Tooth paste
- Tooth powder
- Wafer
C. Pharmacoeconomic Value Assessment
As described in section V of these Guidelines, a pharmacoeconomic value assessment is used as part of the calculation of the MRP. Under the Regulations, patentees are required to file prescribed information relating to all pharmacoeconomic analyses of a patented medicine prepared by publicly funded Canadian organizations if the pro-rated cost of the treatment for that patented medicine over a 12-month period is greater than or equal to 150% of GDP per capita at the time of the publication of the analysis.
The typical source of pharmacoeconomic analysis used for this assessment will be the Common Drug Review (CDR) Pharmacoeconomic Reports and pan-Canadian Oncology Drug Review (pCODR) Final Economic Guidance Reports of the Canadian Agency for Drugs and Technologies in Health (CADTH). Analyses developed by the Institut national d’excellence en santé et services sociaux (INESSS) in its Évaluations aux fins d’inscription or the National Advisory Committee on Immunization (NACI) will also be considered.
As part of this assessment, Staff calculations will rely on the base case reanalysis conducted by the public agency, as opposed to the analysis conducted with the base case model submitted by the patentee. This will be a cost-utility analysis model in which health outcomes are expressed as QALYs. The Incremental Cost-Utility Ratio (“ICUR”) measured in cost per quality-adjusted life years (“QALYs”) for each indication of the patented medicine will be identified from the cost-utility analyses filed by the patentee.
If the agency’s report does not include a cost-utility analysis, but a cost-minimization modelFootnote 16, a dTCC will be conducted. In such cases, the MRP will be based on the median of the dTCC, subject to a 50% reduction floor off the MLP and subject to market size adjustments as discussed in Appendix D. For patented medicines with multiple indications, the pharmacoeconomic assessment for the Relevant Indication, as determined by the procedure in section V of these Guidelines, will be used.
The ICUR will be compared against the applicable Pharmacoeconomic Value ThresholdFootnote 17 (“PVT”), based on the Therapeutic Criteria Levels provided in the table below. The MRP is based on the PEP (the price at which the patented medicine’s ICUR would be equivalent to the PVT) with the proviso that if the PEP results in a reduction below the floor indicated in the table, the MRP is set so that the price reduction is no greater than the applicable floor.
The PMPRB relies on agencies to publish estimates enabling the calculation of the PEP in their reporting.
| Therapeutic Criteria Level (See Appendix E – The Scientific Review Process) |
PVT | Reduction Floor off MLP |
|---|---|---|
| Level I | $200K/ QALY | 20% |
| Level II | $150K/ QALY | 30% |
| Level III | $150K/ QALY | 40% |
| Level IV | $150K/ QALY | 50% |
| Pharmacoeconomic analysis is a cost minimization | Median of dTCC subject to 50% floor | |
| No pharmacoeconomic assessment | 50% of MLP | |
D. Market Size Adjustment Methodology
As described in section V of the Guidelines, a market size adjustment is applied to Category I patented medicines when actual revenues exceed $50 million across all dosage forms and strengths of the patented medicine (i.e., all DINs combined). This adjustment will be applied annually in accordance with the following table:
| Annual revenues | MRP | Incremental MLP adjustment factor | Price‡ adjustment factor used to calculate MLP adjustment factor |
|---|---|---|---|
| <$12M | MLP | 0% | 0% |
| $12M-50M | Greater of PEP and Floor | ($12M - $12M * (MRP/MLP)) / Revenues + (MRP/MLP) | |
| $50M-$100M | -25% | ($21.5M - $9M * (MRP/MLP)) / Revenues + (1 - 25%) * (MRP/MLP) | |
| >$100M | -35% | ($32M - $7.8M * (MRP/MLP)) / Revenues + (1 - 35%) * (MRP/MLP) |
‡ Lower of the MLP and List Price
| Annual revenues | MRP | Incremental MLP adjustment factor | Price‡ adjustment factor used to calculate MLP adjustment factor |
|---|---|---|---|
| <50M | MLP | 0% | 0% |
| $50M-$100M | Lowest of the MLP and the media of the dTCC | -25% | ($50M - $37.5M * (MRP/MLP)) / Revenues + (1 - 25%) * (MRP/MLP) |
| >$100M | -35% | ($56.7M - $32.5M * (MRP/MLP)) / Revenues + (1 - 35%) * (MRP/MLP) |
‡ Lower of the MLP and List Price
For example, a patented medicine requiring an ICUR and realizing between $50 million and $100 million in revenues based on units sold at the MRP set by the PEP will have its MRP[A] set as follows:
MRP[A] = MLP * ($21.5M - $9M * (MRP/MLP)) / Revenue + (1 - 25%) * (MRP/MLP)
After the initial market size adjustment, a patented medicine’s MRP will only be readjusted following an increase in annual units sold. A patented medicine’s MRP will not be readjusted following a decrease in annual units sold, or if its actual realized revenues fall into a lower tier.
E. Scientific Review Process
The PMPRB’s scientific review is an evidence-based process that identifies the Therapeutic Criteria, appropriate therapeutic comparators, and comparable dosage regimen of a patented medicine for the purpose of informing the price review process.
The scientific review of a new patented medicine is based on information from a variety of sources such as those set out below:
- Patentee Submission – Patentees may provide Staff with a brief submission document which clearly explains the rationale for the patentee’s proposals relative to the therapeutic criteria, medicines identified for comparison purposes, and comparable dosage regimens. Information on submission procedures is available on the PMPRB online filing tool.
- Research by a Drug Information Centre (DIC) – Staff may use the services of various drug information centres to obtain scientific information, such as clinical trial information, clinical practice guidelines, etc. The basis of the review by the DIC is the product monograph or information similar to that contained in a product monograph if a Notice Of Compliance (NOC) has not been granted.
- Research by Staff – Staff may also update research and supplement data and evidence from the patentee and DIC using other sources.
- PMPRB Human Drug Advisory Panel (HDAP) - On an ad hoc basis, Staff may consult with the HDAP to provide clinical context pertaining to the scientific information that is being considered by Staff.
Therapeutic Criteria
The Therapeutic Criteria considered by Staff for New patented medicines are set out below. For patented medicines with multiple approved indications or multiple uses, the assessment of Therapeutic Criteria will be based on the Relevant Indication:
Therapeutic Criteria Level I
The patented medicine is the first medicine to be sold in Canada that effectively treats a particular illness or effectively addresses a particular indication in a clinically impactful manner. Clinically impactful improvement includes improvements in quality of life, mortality or significant reductions in disease severity or healthcare utilization, preferably based on high quality evidence using hard clinical endpoints rather than surrogate outcomes. A high QALY gain is normally associated with medicines at this level.
Therapeutic Criteria Level II
The patented medicine provides a considerable improvement in therapeutic effect, relative to other medicines sold in Canada, in a clinically impactful manner. Clinically impactful improvement includes improvements in quality of life, mortality or significant reductions in disease severity or healthcare utilization, preferably based on high quality, active-comparator controlled evidence using hard clinical endpoints rather than surrogate outcomes. High quality network-meta-analysis may also be considered. A high QALY gain is normally associated with medicines at this level.
Therapeutic Criteria Level III
The patented medicine provides moderate absolute improvement in therapeutic effect, relative to other medicines sold in Canada. The medicine may have (a) an increase in clinically relevant efficacy; (b) be associated with a reduction in the incidence or grade of important adverse reactions; or (c) be associated with clinically relevant increased ease of use characteristics (e.g. route of administration, convenience, increased compliance, etc.), however, these improvements may provide limited meaningful clinical impact or may be based on lower quality clinical evidence. Patented medicines at this level are normally associated with moderate incremental QALY gains with a relatively high degree of certainty or a high QALY gains with a relatively low degree of certainty.
Therapeutic Criteria Level IV
The patented medicine provides no or slight improvement relative to other medicines sold in Canada. Alternatively, or in addition, the patented medicine has very limited (or no) robust clinical evidence available to clearly identify a medicine’s degree of clinically relevant therapeutic improvement relative to other medicines sold in Canada. Limited or no additional QALY gains or significant uncertainty due to poor quality evidence are associated with medicines at this level.
Identification of Medicines for Comparison Purposes
The World Health Organization (WHO) Collaborating Centre for Drug Statistics Methodology’s Anatomical Therapeutic Chemical (ATC) Classification System may be used in the selection of medicines for comparison purposes. The medicines used for comparison purposes will typically be those identified under the ATC classification system at the sub-class level above the single chemical substance. This will normally be the fourth sub-class level but could include the next higher sub-class or another sub-class. In some instances, it may be appropriate to select from the fifth or single chemical substance level. A medicine of the same ATC therapeutic class as the medicine under review may be omitted if it is unsuitable for comparison. For example, a medicine with a primary indication/use other than the primary indication/use of the patented medicine under review may be omitted from the comparison.
All medicines used for comparison purposes will typically have the same approved indication or use as the patented medicine under review.
If the iTCC includes medicines with the same approved indication or use as the patented medicine under review, that information may be considered.
For a patented medicine that is a new dosage form or strength of the same medicinal ingredient as one or more existing medicines, its comparators will be those existing medicines that are available in the same or comparable dosage form and have the same indication or use. This will apply regardless of whether the dosage regimens are the same or differ materially.
For a product that is a combination of medicines, where each of the medicines of the ombination are sold in Canada and have the same indication or use, its comparators will be limited to the component medicines.
Identification of Comparable Dosage Regimens
The comparable dosage regimen used for comparison purposes will normally be the maximum of the usual recommended dose in the product monograph (or similar information) taking into account relevant clinical variables. The most appropriate strength of the medicine will be chosen for a particular dosage regimen. Generally, a dosage regimen based on a course of treatment will be applicable to acute indications, while a per-day regimen (based on maintenance dose) will be applicable to chronic indications.
F. Summary of Compliance Timelines
| Patented Medicine Category | iMLP | MLP (3 years or 5 countries min.) | MRP | Reassessment | Compliance Assessment |
|---|---|---|---|---|---|
| Grandfathered | N/A | Lower of NEAP and HIP | N/A | If MLP>HIP | 2 reporting periods (Dec. 2021) |
| Line extension of grandfathered | Lower of list and HIP | Lower of list and HIP | N/A | If MLP>HIP | 1 reporting period |
| Gap | Lower of list and MIP | Lower of list, NEAP/MAPP and MIP | N/A | If MLP>MIP±10% | 2 reporting periods (Dec. 2021) |
| New Category I | MIP | Lower of list and MIP (or dTCC or iTCC if no IP*) | Lower of MLP and MRP (X% off MLP based on PEP, dTCC subject to TCL floor and market size adjustment if applicable) | Market size trigger Updated therapeutic/HTA evidence New relevant indication | MLP – 1 reporting period MRP – 2 reporting periods |
| Line extension of new Category I | MIP | RR or level pricing subject to HIP | Same X% off MLP as reference strength | As above for all DINs | MLP – 1 reporting period MRP – 2 reporting periods |
| New Category II | MIP | Lower of list and MIP (or dTCC or iTCC if no IP) | N/A | N/A | 1 reporting period |
| Line extension of new Category II | MIP | RR or level pricing subject to HIP | N/A | N/A | 1 reporting period |
*IP: international price
Backgrounder 2020
Backgrounder 2020
On June 2020 Draft Guidelines: Explanation of Changes from November 2019 Draft Guidelines

Table of Contents
- The PMPRB Guidelines
- The PMPRB Guidelines Consultation
- Reaching out to Canadians: Overview of the PMPRB Consultation
- Explanation of Changes to the Draft Guidelines
- 1. Domestic Therapeutic Class Comparison (dTCC)
- 2. Maximum List Price (MLP) Test - International Price Comparison
- 3. Classifying a patented medicine as Category I
- 4. Pharmacoeconomic Value (PV) Threshold & Accounting for Therapeutic Comparators – High Cost Medicines
- 5. Market Size Adjustments
- 6. Confidentiality of Maximum Rebated Price (MRP)
- 7. Regulatory review of patented biosimilars and generics
- 8. Other issues raised by stakeholders that have been addressed by these changes
The PMPRB Guidelines
The Patented Medicine Prices Review Board (PMPRB) is a quasi-judicial body with a regulatory mandate to prevent pharmaceutical patentees from charging consumers excessive prices during the statutory monopoly period. Its creation arose out of concern that stronger patent protection for medicines might cause their prices to rise unacceptably to the detriment of consumers.
Pursuant to subsection 96(4) of the Patent Act, the PMPRB issues guidelines (“Guidelines”) which are intended to provide transparency and predictability to patentees regarding the process typically engaged in by public servant employees of the PMPRB (“Staff”) in seeking to determine whether a patented medicine appears to be priced excessively in any market in Canada.
Before making or amending its Guidelines, the PMPRB has an obligation to consult under subsection 96(5) of the Patent Act. The PMPRB takes its obligations in this regard very seriously and has pursued all necessary steps to ensure a meaningful consultation process predicated on an open and transparent dialogue with Canadians.
The PMPRB Guidelines Consultation
On January 1, 2021, the amended Patented Medicines Regulations (“Amended Regulations”)Footnote 1 will come into force. Changes to the PMPRB’s pricing Guidelines are necessary for the Amended Regulations to be implemented and to enable the PMPRB’s move to a more risk-based approach to regulating the ceiling prices of patented medicines. On November 21, 2019, the PMPRB published a draft set of new Guidelines (“the November 2019 Draft”) for consultation with stakeholders and the public. Extensive feedback followed and the written submissions we received are available on the PMPRB website.
In response to the feedback received during the consultation period, the PMPRB has made a number of substantive changes to the November 2019 Draft. These changes are reflected in a second draft set of Guidelines published on June 19, 2020 (“the June 2020 Draft”) which the PMPRB is now consulting on for a period of 30-days.Footnote 2 The purpose of the present document is to explain in general terms the nature of the changes and why they were made. It is intended to be read as a companion piece to the June 2020 Draft to support the reader’s understanding of the changes. A similar document accompanied the publication of the November 2019 Draft Guidelines and is available on the PMPRB website. As is the case for the Guidelines, the Backgrounder is not binding on the PMPRB or patentees.
The publication of the June 2020 Draft Guidelines and the ensuing 30-day consultation process is the last and final step in a process that dates back to the release of the PMPRB’s Discussion Paper on Guidelines Modernization in June 2016, and follows the roadmap for reform laid out in its 2015-2018 Strategic Plan. From the beginning, the PMPRB’s consultation process has been consistent with government best practices to ensure maximum inclusion, clarity, and productive discussion.
The deadline for providing written submissions to the PMPRB on the June 2020 Draft Guidelines is July 20, 2020. The PMPRB remains committed to hearing from all Canadians who have an interest in and opinion on how it carries out its regulatory mandate and looks forward to receiving constructive and meaningful feedback from this consultation process with a view to issuing final Guidelines in the fall of 2020.
Reaching out to Canadians: Overview of the PMPRB Consultation
The publication of the November 2019 Draft Guidelines was followed by an 85-day consultation period. During that time, the PMPRB sought to engage with as many stakeholders as possible in various ways, including face-to-face meetings across the country, day-long outreach sessions in Ottawa with industry and civil society, webinars and working groups.
The following is a summary of the PMPRB’s consultation efforts following the release of the November 2019 Draft Guidelines:
- Health Partners Working Groups: two one-day outreach sessions in November 2019 and January 2020 with health partner representatives including public drug plan representatives, the Canadian Agency for Drugs and Technologies in Health (CADTH), Institut national d'excellence en santé et services sociaux (INESSS), pan-Canadian Pharmaceutical Alliance (pCPA), Health Canada, and Cancer Agencies.
- An Industry Forum: a one-day outreach session in December 2019 with representatives from Innovative Medicines Canada (IMC), BIOTECanada and some of their member companies.
- A Civil Society Forum: a one-day outreach session in December 2019 with patient groups and other non-institutional stakeholders.
- An Industry Webinar: in January 2020 with 187 participants from across the pharmaceutical industry.
- Cross country consultation meetings: over 60 meetings with more than 260 participants from stakeholder groups across the country. These included ministries of health, public and private payers, patient and patient groups, clinicians, industry and trade associations, pharmacists and distributors, health care organizations, etc.
- Bilateral meetings with pharmaceutical companies, trade associations and consultants: over 40 meetings, mainly in Ottawa.
Specific information on dates, locations and stakeholder groups the PMPRB met with, as well as electronic versions of the documents that were discussed at those meetings are available on the PMPRB website.
The PMPRB’s Board had planned to host a public policy forum in Ottawa on March 18, 2020 but was unable to do so because of the emerging COVID-19 pandemic. To provide stakeholders with an opportunity to convey any new or different information they may have intended to present to the Board at that event, the PMPRB extended the timeline for written submissions from interested parties until March 18, 2020.
The PMPRB received 123 written submissions during the consultation period from a diverse array of stakeholders. One-third (33%) of the submissions received were from patentees and their industry associations, and another third (33%) came from consumer and patient advocacy groups. The PMPRB also received almost 900 letters from individuals or patients, the majority of which were from Cystic Fibrosis patients and their caregivers as part of an advocacy initiative spearheaded by Cystic Fibrosis Canada. The 123 written submissions are available on the PMPRB website.
| Category | Submissions (#) | Submissions (%) |
|---|---|---|
| Consumer/patient advocacy total | 41 | 33% |
| Patentee | 34 | 28% |
| Patentee association | 4 | 3% |
| Generics/biosimilars | 2 | 2% |
| Patentee/patentee association total | 40 | 33% |
| Distributor/consultant/pharmacist | 11 | 9% |
| Industry associations (e.g. life sciences) | 6 | 5% |
| Consultant | 2 | 2% |
| Industry total | 19 | 15% |
| Union | 7 | 6% |
| Clinician | 4 | 3% |
| Academic | 3 | 2% |
| Think tank | 1 | 1% |
| International | 1 | 1% |
| Civil academic/clinician/think tank total | 16 | 13% |
| Public (e.g. agency, health authority, government) | 5 | 4% |
| Private insurance | 2 | 2% |
| Public entity or private insurance total | 7 | 6% |
| Grand Total | 123 | 100% |
The efforts outlined above are part of a process that will continue even after the Guidelines are implemented. The PMPRB will work with stakeholders to ensure that the impact of the Guidelines is well understood and to minimize any operational or compliance issues arising from their early application. Adjustments to the Guidelines will be made if it becomes clear that certain aspects of the new regime are not operating as intended, subject as always to further consultation with stakeholders and the public.
Explanation of Changes to the Draft Guidelines
In issuing Guidelines, the PMPRB must reconcile seemingly conflicting public policy objectives, namely, facilitating access to patented medicines at non-excessive prices while recognizing the legitimate interest of pharmaceutical patentees in maximizing the value of their intellectual property. Not surprisingly, the PMPRB’s diverse stakeholder community holds divergent and even diametrically opposing points of view on the policy rationale for the Guidelines and the Amended Regulations upon which they are based. Accordingly, the PMPRB recognizes that consensus is not achievable when consulting on measures to modernize its regulatory framework. However, the PMPRB has made every effort throughout the consultation process to foster a productive, fair and transparent dialogue with our stakeholders and to listen carefully to their concerns. The changes explained below are the result of that effort and represent our best attempt to be responsive to the competing feedback we have received from stakeholders and to craft a modern-day regime that continues to encourage voluntary compliance.
What follows is a concise description of the key changes between the November 2019 and June 2020 Draft Guidelines, the stakeholder feedback upon which they are based, and an explanation of the Board’s reasoning in making them. This information is presented to provide context and is not intended to constitute an exhaustive treatment of all the relevant feedback the PMPRB received in respect of each issue, or of the analysis that prompted the change.
1. Domestic Therapeutic Class Comparison (dTCC)
Proposed Approach in November 2019 Draft Guidelines
One of the factors for assessing whether a price is excessive as set out in s. 85 of the Patent Act is “the prices at which other medicines in the same therapeutic class have been sold in the relevant market” (s. 85(1)(b)).
The November 2019 Draft Guidelines proposed to address this factor in two main ways. First, it provided that the Maximum List Price (MLP) for New patented medicines would be set by the lower of the Median International List Price (MIP) for the PMPRB11 comparator countries and the domestic Therapeutic Class Comparison (“dTCC”), subject to a floor set by the Lowest International Price (LIP) for the PMPRB11 countries. Second, it provided that if a cost-utility analysis prepared by a publicly funded Canadian organization was not available for a Category I patented medicine, then the Maximum Rebated Price (MRP) would be set by the lower of the LIP, the dTCC or the international Therapeutic Class Comparison (“iTCC”), with further adjustments based on the Market Size Adjustment Methodology.
Both the dTCC and the iTCC would be calculated based on the median cost of treatment across the comparator medicines, derived by taking into account the lowest public price and price source.
Summary of Stakeholder Feedback
Patentees object to the proposed approach, asserting that it would push Canadian list prices towards the LIP and fails to recognize that medicines within a class can have differing levels of therapeutic benefit. Patentees also recommend that the dTCC and the iTCC be calculated based on the highest rather than the median cost of treatment across the comparator medicines.
Public and private payers and other stakeholders involved in health care delivery did not express any concerns with applying the TCC factor in the manner described above.
Analysis
While few patented medicinesFootnote 3 are launched in therapeutic areas dominated by older, genericized medicines, the Board recognizes that the proposed approach would likely have the effect of pushing the MLP for these medicines to the LIP. As a result, the Board has decided to forego this approach where the patentee has filed prices for the medicine in PMPRB11 countries. It is believed that the international prices of the medicine may already reflect, to some extent, the availability and pricing of comparator medicines in those markets.
The Board did elect to retain the domestic dTCC whenever PMPRB11 prices for a medicine are not available. In such cases, the dTCC test will be calculated using the top of the class instead of the median. The dTCC and iTCC will also be retained in the context of setting the MRP for large market size Category 1 medicinesFootnote 4. The Board views the median as the more appropriate bar in this instance given that the prices used for conducting the TCC are gross (list) prices instead of net prices.
Description of the Change in the June 2020 Draft Guidelines
For New patented medicines, the MLP will be set by the MIP for the PMPRB11 comparator countries if the patentee has filed prices for at least one country. If there are no available prices in the PMPRB11 countries, then the MLPFootnote 5 will be set by the dTCC, calculated based on the highest instead of the median cost of treatment across the comparator medicines, derived by taking into account the lowest public price in Canada. If the patentee has not filed international prices by the end of the interim period (maximum 3 years), and there are no domestic therapeutic class comparators, the MLP may be set at the median of the iTCC.
If during the lifecycle of the medicine, it is launched in other countries, the MLP will be subject to the reassessment criteria, which contemplates an increase or a decrease depending on where the Canadian list price sits vis-à-vis international list prices.
The dTCC will be considered in the establishment of the MRP for Category I medicines with an actual market size exceeding $50 million, as described in Section 5. In such a case, the dTCC will be based on the median cost of treatment across the comparator medicines, derived by taking into account the lowest public price in Canada and subject to the applicable Therapeutic Criteria Level (TCL) floor.
2. Maximum List Price (MLP) Test - International Price Comparison
Proposed Approach in November 2019 Draft Guidelines
One of the excessive pricing factors set out in s. 85 of the Patent Act is “the prices at which the medicine and other medicines in the same therapeutic class have been sold in countries other than Canada” (S. 85 (1) (c)).
The November 2019 Draft Guidelines proposed that the Maximum List Price (MLP) be set by the lower of the Median International Price (MIP) for the PMPRB11 comparator countries and the domestic Therapeutic Class Comparison (“dTCC”), subject to a floor set by the Lowest International Price (LIP) in the PMPRB11. If the patentee does not file international prices by the end of the three years (“Interim Period”), and there are no domestic therapeutic class comparators, it further proposed that the MLP be set using the international Therapeutic Class Comparison (“iTCC”).
Summary of Stakeholder Feedback
Patentees, the biosimilar industry, distributors, industry consultants and some patients and patient groups argue that the MIP should be replaced by the Highest International Price (HIP) for Grandfathered medicines, and make a number of points in support of that position. First, they claim that the MIP approach incorrectly assumes that all prices above the median of the comparator countries are excessive. Second, they view the application of the MIP for Grandfathered medicines as inconsistent with the Cost Benefit Analysis (CBA) conducted by Health Canada as part of the Regulatory Impact Analysis Statement (RIAS) that accompanied the Amended Regulations. Third, they maintain that the application of the MIP to the prices of Grandfathered medicines will have a significant impact on industry revenues and result in drug shortages and a decline in support services currently available to patients.
“Recognizing that the application of the new basket is mandated in the amended Regulations, it would be more appropriate to apply the current pricing rules (highest international price comparison or HIPC) to establish the list price ceiling (MLP) for existing medicines. Even with the HIPC, list price reductions will be achieved for existing products, as a result of the change in the international reference basket (PMPRB11)”
Janssen
Conversely, some public and private payers and other patient groups take the position that Canadians have been paying excessive prices for patented medicines for years and view the MIP of the new PMPRB11 basket of comparator countries as a reasonable way to ensure gross (list) prices in Canada align with international norms. In addition, these stakeholders stress that the MIP of the PMPRB11 is the most appropriate price test to adequately protect cash-paying customers from excessive list prices. This is very important given that a significant portion of sales are borne directly by Canadians through cash payments and as contributions to co-payments and deductibles.
Health researchers and provincial cancer agencies support the proposed approach for calculating the MLP and urged the PMPRB not to use the HIP test. One consumer advocacy group applauded the updated schedule of comparator countries and noted that it will “make price-comparisons much more equitable for the Canadian pharmaceutical consumers”.
“...nous ne pouvons qu’accueillir favorablement tout nouveau processus de fixation et de plafonnement des prix s’appuyant sur les données probantes et les pratiques exemplaires, de même que l’utilisation d’une nouvelle annexe de pays de comparaison (le CEPMB11) visant à établir les nouvelles règles de comparaison des prix...”
Centrale des syndicats du Québec
Analysis
The Patent Act does not require the Board to adopt any specific threshold based on the PMPRB11 prices, only that these be considered.
It is acknowledged that, in the main, the Cost Benefit Analysis (CBA) assumes MLP ceilings that are generally closer to the highest and median price in the PMPRB11 countries for Grandfathered and New patented medicines respectively. At the same time, the purpose of the CBA was to assess the impact of the Amended Regulations in isolation. It should not be read as precluding changes to the current Guidelines, as significant such changes are necessary to implement the Amended Regulations in the first place.
The Board’s analysis of the impact of the application of the MIP to Grandfathered medicines indicates that it may be less significant than claimed by certain stakeholders who oppose it. List prices are not reflective of true net prices paid by a large segment of the Canadian market and the true impact on the net revenue will thus be less than the nominal impact on the list prices. That being said, the Board has decided to apply the HIP test to Grandfathered medicines as a concession to patentees whose expectations may have been raised by the CBA and in recognition of the impact of changing the schedule of comparator countries. The Board is of the view that the misalignment of Canadian prices vis-à-vis international prices is best addressed by applying the MIP test for New patented medicines moving forward. As a general rule, the Board feels that Canadian list prices higher than international norms smacks of excessiveness and the MIP is an appropriate litmus test for ensuring that Canadian list prices do not become excessive in the future.
Description of Change in the June 2020 Draft Guidelines
Grandfathered Patented Medicines and their Line Extensions
The gross (list) price ceiling of Grandfathered patented medicines will be set as the lower of the HIP for the PMPRB11 countries, and the applicable ceiling under the previous Guidelines. The MLP of Line Extensions of Grandfathered medicines (i.e., new strengths and dosage forms) will also be set by the HIP.
Gap and New Patented Medicines
The MIP test will be retained for setting the MLP for Gap medicines, (i.e., medicines for which a DIN was assigned on or after August 21, 2019 and the first sale in Canada took place prior to January 1, 2021) and New patented medicines (i.e., all other patented medicines for which a DIN was assigned on or after August 21, 2019).
3. Classifying a patented medicine as Category I
Proposed Approach in November 2019 Draft Guidelines
The November 2019 Draft Guidelines proposed that a patented medicine be classified as Category I if it meets either of the following criteria:
- 12-month treatment cost greater than 50% of Gross Domestic Product (GDP) per capita, and/or
- an estimated or actual market size (revenue) exceeds annual Market Size Threshold, initially set at $25 million.
Summary of Stakeholder Feedback
Pharmaceutical patentees oppose the criteria and associated thresholds. As regards the latter, they contend that the thresholds are too low and would result in a majority of New patented medicines falling into Category I, contrary to the PMPRB’s stated intent in adopting a risk-based regulatory approach. Other stakeholders, notably some patient groups and pharmacy associations, echoed concerns about the proportion of patented medicines likely to be screened in as Category I.
“These guidelines are inherently biased against drugs for small, difficult-to-diagnose and previously untreated patient populations with poorly documented natural history of disease and little clinical evidence of direct links between biomarkers and other outcome measures. This description would apply to almost all rare diseases. …. It is ridiculous to introduce a cost-effectiveness assessment (CEA) at the time of launch…”
Canadian Organization for Rare Disorders (CORD)
Patient groups concerned with access to rare disease medicines are particularly troubled by the fact that virtually all such medicines would fall into Category I due to their high annual treatment costs and, as such, be subject to the new Pharmacoeconomic Value (PV) factor. These products rarely have favorable pharmacoeconomic profiles because of their extreme high prices, which patentees claim are necessary to recoup research and development costs from a relatively small patient population.
Conversely, some representatives of civil society and healthcare organizations are of the view that a lower threshold for annual treatment cost should apply if the PMPRB is to properly scrutinize all patented medicines that are beyond the means of many consumers.
For their part, union groups express strong support for the inclusion of market size as a criterion for Category I medicines, citing the significant impact some medicines have on health insurance plans despite falling under the PV threshold. They note the large year-over-year increases their members’ drug plans have struggled with recently due to medicines that would be considered low cost under the November 2019 Draft Guidelines.
Finally, patentees assert that there should be a mechanism to move medicines out of Category I and into Category II if their revenues fall below the specified thresholds.
Analysis
Further analysis of the data suggests that a significant percentage of medicines would realize over $25 million in annual revenue at some point over their patent life. Considering that this data reflects historical trends, it is likely an underestimate of expected future revenues for patented medicines launched more recently.
In addition, with high cost medicines increasingly dominating the market, the observation that the proposed Category 1 criteria would capture a significant number of new medicines is not without merit. As a result, the Board has concluded that in order for its risk-based approach to be administratively feasible for PMPRB Staff and patentees, higher thresholds are warranted, both in terms of treatment cost and market size. To address concerns related to rare disease medicines in particular, the Board has decided to carve out from Category I high cost medicines that are expected to realize below a certain minimum amount in annual revenue.
Description of Changes in June 2020 Draft Guidelines
The new market size threshold will be $50M in annual revenues and the new treatment cost threshold will be 150% of GDP per capita. It is estimated these revised thresholds will result in approximately 25% of new medicines triggering the Category I criteria, which is more in keeping with the PMPRB’s original intent in moving to a risk-based approach. These medicines are expected to account for a majority of patented medicine sales (68%), which will in essence ensure that the PMPRB exercises greater regulatory scrutiny over a minority of medicines that account for the majority of sales.
Medicines that realize less than $12M in annual revenue will not be subject to an MRP even when they exceed the new annual treatment cost threshold. This will ensure that medicines that treat rare diseases are not discouraged from coming to Canada out of concern that their net price will be substantially reduced by regulation. Recent data suggests that a sizable portion of patented medicines (42.5%) do not realize $12M in annual revenues in any of the first 10 years on the market (Figure 1). These medicines only accounted for 3.7% of patented sales, suggesting that even cumulatively these medicines do not give rise to affordability concerns.
Figure 1. Patented medicines in Canada, share of medicines and share of sales, by maximum annual sales
Figure description
This bar graph depicts the share of medicines and their corresponding share of total sales by various market size: $12M+, $25M+, $50M+ and $100M+. The results on the left are based on the maximum annual sales by the third year and the results on the right are based on the maximum annual sales by the tenth year since medicines’ introduction.
*Included patented drugs launched after 1998 in Canada; Sample Size: N=639 for the 3-year analysis; N=338 for the 10-year analysis
Data source: PMPRB, 2018; CPI applied to bring historical sales into 2018 value
4. Pharmacoeconomic Value (PV) Threshold & Accounting for Therapeutic Comparators – High Cost Medicines
Proposed Approach in Draft Guidelines
Under the November 2019 Draft Guidelines, Category I medicines that are required to report cost-utility analysis would have a maximum rebated price (MRP) ceiling based on the level at which the patented medicine’s Incremental Cost-Effectiveness Ratio (“ICER”) would equate to the Pharmacoeconomic Value (PV) threshold of $60,000 per Quality-Adjusted-Life-Year (QALY).
For patented medicines for rare diseases with an estimated total prevalence no greater than 1 in 2,000 across all approved indications, the MRP would be set at 50% above the level at which the ICER would equate to the PV threshold of $60,000 QALY.
“Breast cancer is the poster child for some of the worst practices of pharmaceutical companies, for example, trying to get extremely highly priced drugs on the provincial drug formularies by mobilizing women with breast cancer through “patient advocacy” groups to directly pressure those governments. This tactic has been used for stage 4 breast cancer drugs whose actual benefits do not live up to the claims of the drug companies and whose side effects greatly diminish the quality of life of the women taking them”
~ Breast Cancer Action Quebec
Summary of Stakeholder Feedback
Patentees are fundamentally opposed to the introduction of PV as a factor for the PMPRB to consider in determining what constitutes an excessive price and, not surprisingly, take issue with the approach proposed for its application in the November 2019 Draft Guidelines. In addition to citing operational and technical challenges arising from its application, patentees and some patient groups objected to the proposed PV threshold of $60,000/QALY as unreasonably low, especially for medicines for rare diseases, notwithstanding the 50% higher ceiling afforded to this class of products. Whereas some patient groups believe that the PV factor should never apply to medicines for rare diseases, others are extremely concerned with the very high opportunity cost associated with reimbursing these products, especially given that evidence of their clinical benefits at introduction is often ambiguous at best.
Patentees also take issue with the proposed $60,000/QALY PV threshold as an arbitrary one-size-fits-all approach that runs the risk of exposing what public drug plans are paying under confidential product listing agreements (PLAs). They argue for a more flexible approach that recognizes and rewards therapeutic benefit, as is the case under the current Guidelines, but did not volunteer an alternative which would satisfy them. Specific suggestions on this point did come from some public insurers, who recommend PV thresholds of $100,000/QALY and $120,000/QALY for medicines that are therapeutically superior, or even higher for medicines that are potentially curative.
Lastly, in addition or in the alternative to advocating for a more flexible approach predicated on therapeutic benefit, some patentees suggest that the application of the PV threshold be subject to some form of stop-loss mechanism which would blunt its impact and provide much needed certainty to industry about the worst case scenario under the new regime.
Analysis
Pharmacoeconomic value is now a s.85(1) factor and the Board has a statutory obligation to consider it. However, until such time as there is more developed empirical evidence in Canada on opportunity cost in the public health system, an argument exists for erring in favour of more generous thresholds that are aligned with the higher end of what is seen internationally and that provide greater certainty and predictability for patentees.
By way of comparison, in the United Kingdom, the National Institute for Health and Care Excellence (NICE) has an explicit cost effectiveness threshold of €30,000/QALY. However, there are certain cases where NICE will allow a higher threshold of £50,000/QALY for end of life treatments and €100,000 to €300,000 for “Highly Specialized Technologies” (HSTs) depending on the absolute QALY gain associated with the medicine. In other countries such as the Netherlands and Norway, the thresholds depend on the severity of the disease, among other factors. In the Netherlands, an informal cost-effectiveness threshold of €20K/QALY (low burden) to €80K/QALY (high burden) is used. Japan has recently implemented a tiered cost-effectiveness assessment scheme that requires a downward price adjustment, the amount of which depends on the drug’s cost-effectiveness. The price reduction is limited to 10-15% of the pre-adjustment National Health Insurance reimbursement price
“…. the MRP concept has no price floor whatsoever and will result in prices below the lowest of the PMPRB 11 schedule. This is contrary to the government’s policy intent….”
Innovative Medicines Canada (IMC)
In addition to the above considerations, the Cost Benefit Analysis (CBA) for the Amended Regulations also assumed a maximum price reduction of 50% for high priority New patented medicinesFootnote 6.
Description of Change in June 2020 Draft Guidelines
The following PV thresholds and capped price reduction floors will be applied to set the MRP for high cost Category I medicines based on an evaluation of their therapeutic criteria (for revenues in excess of $12 million annually:
Table 1. Pharmacoeconomic Value Thresholds
| Therapeutic Criteria Level (See Appendix E – The Scientific Review Process) |
PV Threshold | Reduction Floor off MLP |
|---|---|---|
| Level I | $200K/ QALY | 20% |
| Level II | $150K/ QALY | 30% |
| Level III | $150K/ QALY | 40% |
| Level IV | $150K/ QALY | 50% |
| Pharmacoeconomic analysis is a cost minimization | Median of dTCC subject to 50% floor | |
| No pharmacoeconomic assessment | 50% of MLP | |
It should be noted that for Category I high cost medicines, in the absence of a cost-utility analysis being provided by the patentee, a maximum reduction of 50% will automatically be applied.
5. Market Size Adjustments
Proposed Approach in November 2019 Draft Guidelines
For high cost Category I medicines, the November 2019 Draft Guidelines proposed that the Maximum Rebated Price (MRP) be further adjusted for market size if annual quantities of the medicine are sold such that, after the application of the Pharmacoeconomic Value (PV) Threshold, annual revenues would be in excess of $25 million. These further adjustments would result in additional 10% reductions in revenue increments of $25 million to a maximum reduction of 50% for annual revenue in excess of $125 million. They would apply equally to other Category I medicines that are not high cost but are so categorized because their estimated or actual annual revenues exceed the $25 million market size threshold.
Summary of Stakeholder Feedback
The intensity of patentee opposition to the Market Size factor is second only to the sentiment reserved for the PV factor. In general, patentees feel that the effect of this factor is to import into the PMPRB regime considerations that should lie within the exclusive purview of public and private payers, not a federal ceiling price regulator. In addition, many patentees argue that the $25 million increment is arbitrary and that not allowing the MRP to increase if revenue falls below the specified thresholds is inconsistent and unfair.
Stakeholders who are supportive of the Market Size factor recognize that demand and prevalence need to be taken into account in setting ceiling prices that contribute meaningfully to the sustainability of Canada’s health care system. They note that a small number of medicines can account for significant expenditures and pose immediate affordability challenges in the system without necessarily being ‘high cost’.
Analysis
Like PV, Market Size is now a s.85(1) factor and the Board has a statutory obligation to consider it. However, a case can be made that the initial $25 million threshold is too low and the number of incremental adjustments proposed in the November 2019 Draft Guidelines is unduly onerous on patentees. A retrospective analysis indicates that approximately one-quarter of patented medicines realize over $50 million in annual revenues over the first 10 years of market exclusivity, while only 14% realize over $100 million.
Many of the other relevant points raised by stakeholders in this context have already been addressed in section 3 of above.
Description of Change in June 2020 Draft Guidelines
The revised thresholds for the Market Size adjustment and the corresponding reductions required at these thresholds are set out in the tables below.
Table 2. Market Size Adjustments
| Annual revenues | MRP | Incremental MLP adjustment factor | Price‡ adjustment factor used to calculate MLP adjustment factor |
|---|---|---|---|
| <$12M | MLP | 0% | 0% |
| $12M-50M | Greater of PEP and Floor | ($12M - $12M * (MRP/MLP)) / Revenues + (MRP/MLP) | |
| $50M-$100M | -25% | ($21.5M - $9M * (MRP/MLP)) / Revenues + (1 - 25%) * (MRP/MLP) | |
| >$100M | -35% | ($32M - $7.8M * (MRP/MLP)) / Revenues + (1 - 35%) * (MRP/MLP) |
‡ Lower of the MLP and List Price
| Annual revenues | MRP | Incremental MLP adjustment factor | Price‡ adjustment factor used to calculate MLP adjustment factor |
|---|---|---|---|
| <50M | MLP | 0% | 0% |
| $50M-$100M | Lowest of the MLP and the media of the dTCC | -25% | ($50M - $37.5M * (MRP/MLP)) / Revenues + (1 - 25%) * (MRP/MLP) |
| >$100M | -35% | ($56.7M - $32.5M * (MRP/MLP)) / Revenues + (1 - 35%) * (MRP/MLP) |
‡ Lower of the MLP and List Price
For Category I medicines that do not meet the annual treatment cost threshold of 150% of GDP per capita but with an actual market size exceeding $50 million, the MRP will be based on the lower of the Maximum List Price (MLP) and the domestic Therapeutic Class Comparison (dTCC) adjusted by the applicable factor. The dTCC will be derived based on the median cost of treatment across the comparator medicines, using the lowest public price. The impact of the dTCC will be subject to a price reduction floor that varies based on the therapeutic criteria level.
| Therapeutic Criteria Level | dTCC Floor |
|---|---|
| Level I | 20% off MLP (dTCC not applicable) |
| Level II | 30% |
| Level III | 40% |
| Level IV | 50% |
6. Confidentiality of Maximum Rebated Price (MRP)
Proposed Approach in Draft Guidelines
Upon the coming-into-force of the Amended Regulations, all patentees are to report price and revenue information that is net of all adjustments including discounts, rebates and free goods and services, to any party that pays for, or reimburses, the patented medicine. This will ensure that the PMPRB has a complete and accurate picture of what patentees are truly charging for their medicines in Canada. Any information filed by patentees with the PMPRB in this respect enjoys the full protection of the confidentiality provisions of the Patent Act.
As already explained, the November 2019 Draft Guidelines proposed that for high cost Category I patented medicines the MRP would be calculated by applying a $60,000 per Quality-Adjusted- Life-Year (QALY) Pharmacoeconomic Value (PV) threshold to the Incremental Cost-Effectiveness Ratio (“ICER”). For patented medicines for rare diseases, the MRP would be set at 50% above the level at which the ICER would equate to the PV threshold of $60,000/QALY. Finally, the MRP for all Category I patented medicines would be subject to 10% reductions in revenue increments of $25 million to a maximum reduction of 50% for annual revenue in excess of $125 million.
Summary of Stakeholder Feedback
Patentees are very concerned with the impact of the new regime on their ability to continue to negotiate confidential discounts and rebates with payers in Canada. They do not appear to dispute that their filings with the PMPRB will remain confidential, but are worried that the approach taken in the November 2019 Draft Guidelines would enable third parties (including payers in other countries) to back calculate a rough estimate of the MRP and extrapolate the levels of price reductions from the list price that would be required in order for the patentee to comply with it. Patentees believe that this would be possible for three reasons. First, the cost-utility reports they will be required to file with the PMPRB are made publicly available by the organizations that issue them (e.g. Canadian Agency for Drugs and Technologies in Health (CADTH) and Institut national d'excellence en santé et services sociaux (INESSS)). Second, the MRP tests are transparently set out in the PMPRB’s Guidelines. Third, list price and estimated revenue data are also publicly available from various public sources other than the PMPRB. Patentees believe if other countries and competitors are able to estimate the magnitude of the difference between gross (list) and net price in Canada, this would put their global business model at risk and imperil the introduction of New patented medicines in Canada.
“The new maximum rebated price (MRP) calculation methodology, when combined with publicly available data, may allow third parties to reverse engineer or estimate net prices.”
Innovative Medicines Canada (IMC)
Stakeholders who express the opposite view believe just as strongly that confidential pricing is a key contributing factor to skyrocketing pharmaceutical prices, both domestically and internationally in recent years and that it is imperative that this phenomenon be brought into the light, not only in Canada but around the world.
“The fact that the information underlying the calculation of the … MRP is confidential will introduce additional obscurity into pharmaceutical prices which is unfortunate.”
~ Expensive Drugs for Rare Diseases Advisory Committee
Analysis
The Board recognizes the difficulty inherent in crafting ‘brightline’ price tests that are predictable to patentees but do not enable competitors to back calculate rough estimates of their confidential discounts to third parties. At the same time, it notes that a number of the issues raised by patentees, such as the public availability of pharmacoeconomic reports issued by CADTH and INESSS and the public availability of list prices and revenue estimates, are well established features of the status quo in Canada and cannot be unwound by the Guidelines. Formal and informal cost-utility thresholds are standard in a number of countries and yet patentees continue to sell their products in those markets. That being said, the Board is not unsympathetic to the concerns raised by patentees and believes that the revised approach to calculating the MRP in the June 2020 Draft Guidelines has the dual benefit of being responsive to both these concerns and the desire expressed by both patentees and many patient groups to retain an assessment of therapeutic criteria as a key input in setting ceiling prices.
Description of Change in June 2020 Draft Guidelines
As explained in section 4, the MRP calculation will be a function of the Therapeutic Criteria Level (TCL) assigned to the patented medicine and the applicable PV threshold and price floor that corresponds to it. The TCL of a patented medicine will be known only to the PMPRB and the patentee.
7. Regulatory review of patented biosimilars and generics
Proposed Approach in November 2019 Draft Guidelines
The Amended Regulations removed the requirement for patentees of certain types of patented medicines to file identity, price and sale information with the PMPRB, unless so requested in response to a complaint. These include patented veterinary medicines, an expanded subset of medicines that do not require a prescription (i.e., over-the-counter – “OTC” medicines) and generic medicines that meet the regulatory definition. Where a complaint is received in respect of one of these types of patented medicines, they would be automatically considered Category II for investigation purposes.
The November 2019 Draft Guidelines made no special provision for ‘biosimilar’ medicines (i.e., subsequent entry biologics approved on the basis of a comparison to an innovator’s reference biologic) which do not fall within the definition of “generic” medicines in the Amended Regulations. Consequently, patentees of these medicines would be required to file price information with the PMPRB and could fall into either Category I or Category II.
Summary of Stakeholder Feedback
Makers of biosimilar medicines, the generic pharmaceutical industry and some patentees argue that there are other types of patented medicines that should be considered at low risk of excessive pricing from an administrative standpoint and thus exempt from the reporting obligations under the Amended Regulations. In their view, this exemption should apply to biosimilars, the brand version of medicines that face generic competition and patented generic medicines that are authorized for sale by regulatory pathways under the Food and Drug Regulations other than just an Abbreviated New Drug Submission (ANDS).
Biosimilar makers in particular stress that their market in Canada is still in its infancy and should be shielded to the greatest extent possible from needless regulatory burden. They contend that having biosimilars subject to the same degree of regulatory scrutiny from the PMPRB as the reference biologic product flies in the face of efforts elsewhere in the health system to reap the potential savings associated with this fledgling market. Regulating biosimilars on a complaints-basis is also said to be more aligned with Canadian Agency for Drugs and Technologies in Health’s (CADTH’s) recent decision to no longer conduct Health Technology Assessment (HTA) reviews of biosimilars in order to simplify and streamline market access for these products. Alternatively, if biosimilars are not exempt from PMPRB oversight, biosimilar makers argue that the domestic price of the reference biologic should be the only comparator because international price comparisons to other patented biosimilars would be inappropriate.
Analysis
Although the Board cannot exempt patentees of medicines from the prescribed filing requirements under the Amended Regulations, from a day to day administrative standpoint, it does have some flexibility in the degree to which certain non-exempt medicines are proactively scrutinized. The Board is of the view that a strong case can be made that expanding the scope of exempt patented medicines beyond the strict regulatory definition for administrative purposes is consistent with a risk-based approach to regulating ceiling prices.
Description of Change in the June 2020 Draft Regulations
Patented biosimilars, generic medicines, medicines for veterinary use and OTC patented medicines will only be subject to investigation if a complaint is received by the PMPRB. In such an event, medicines of this kind will be deemed to be Category II and treated as such notwithstanding the existence of properties that might otherwise meet the criteria for Category I screening. Unlike patented medicines which have reduced reporting requirements under the Amended Regulations, patented biosimilars and generics approved through non-ANDS pathways have a legal requirement to report to the PMPRB.
8. Other issues raised by stakeholders that have been addressed by the above described changes
Medicines for rare diseases
As already explained, the November 2019 Draft Guidelines proposed a 50% higher Maximum Rebated Price (MRP) for medicines with an estimated total prevalence no greater than 1 in 2,000 across all approved indications. The revisions to the MRP approach in the June 2020 Draft Guidelines no longer distinguish between medicines for rare and non-rare conditions. Instead, a full exemption from the MRP has been made for all medicines which would otherwise qualify as Category I because of their annual treatment costs if their annual revenues is below $12M. This measure, coupled with the higher Pharmacoeconomic Value (PV) thresholds and capped price reductions for medicines based on their Therapeutic Criteria Level (TCL), go a long way to address concerns raised by certain stakeholders about unfair treatment of medicines for rare diseases under the new regime.
Reasonable Relationship (RR) Test
The November 2019 Draft Guidelines proposed that the Reasonable Relationship (RR) test may be conducted to determine the Maximum List Price (MLP) or Maximum Rebated Price (MRP) of a new or additional strength of a patented medicine with other existing strengths, where the new or additional strength has the same medicinal ingredient, indication, dosage regimen, and same or comparable dosage from as the existing strength(s). The MLP or MRP of the new strength would have been set to be equivalent to the price per standard unit of the existing strength(s). The approach was also intended to be applied when multiple strengths of new medicine are first sold simultaneously, and some strengths are identified specifically as loading, titration, or reduction doses.
Patentees have concerns that this approach would discourage the launch of new strengths that are potentially more convenient for patients. This would particularly be the case for medicines that would otherwise be priced at level with other existing strengths.
In view of these concerns, the Board has decided to revert to a simplified version of the RR test used under the current Guidelines. The test will benchmark one strength as the reference strength and set the ceiling for the other strengths (new or existing) based on their proportional relationship to it, subject to a Highest International Price (HIP) cap. In order to accommodate the practice of level pricing, patentees will be allowed to price lower strengths up to the MLP of the reference strength subject to the HIP. This approach addresses the issue of level pricing within the confines of international price limits.
The RR test will be applied to New patented medicines (i.e., medicines for which a DIN was assigned on or after August 21, 2019 and the first sale took place on or after January 1, 2021).
Consultation News
September 17, 2020
Update - PMPRB Price Regulation of Drugs Authorized for Use in COVID-19
In advance of the upcoming publication of the final PMPRB Guidelines, the PMPRB would like to provide stakeholders with early notice of the special consideration that document affords to patented medicines appearing on the List of Drugs for Exceptional Importation and Sale and on the list(s) published by Health Canada pursuant to the September 16, 2020 Interim Order Respecting the Importation, Sale and Advertising of Drugs for Use in Relation to COVID-19. If a patented medicine appears on any of these lists, it will only be subject to review or investigation by PMPRB Staff if a pricing complaint is received from the federal Minister of Health or any of her provincial or territorial counterparts. This will be case for as long as the Interim Orders are in effect and the patented medicine remains listed. In the event that the requisite complaint is received, an investigation will take place and Staff will review the price of the patented medicine in accordance with the usual process and procedure under the Guidelines.
Upon the expiry or repeal of the Interim Orders or the removal of a patented medicine from the applicable list, absent a pre-existing complaint, price reviews for these medicines will be based on prevailing international and domestic list prices, and not on introductory discounted prices in Canada.
This policy has been adopted as part of a government wide effort to provisionally ease the regulatory pathway for drugs and medical devices urgently needed for COVID-19 diagnosis, treatment, mitigation or prevention. The PMPRB will continue to monitor the evolving situation regarding the COVID-19 virus in Canada and the appropriateness and feasibility of this policy.
July 8, 2020
Impact of the recent Federal Court decision and deadline extension for written submissions
On June 29, 2020, Justice Manson of the Federal Court issued his decision in respect of Innovative Medicines Canada’s (“IMC”) application for judicial review of the recent amendments to the Patented Medicines Regulations (the “Amended Regulations”) which will come into force on January 1, 2021. In his decision, Justice Manson upheld the new section 85(1) excessive pricing factors and the new Schedule of comparator countries but found that subsection 3(4) of the Amended Regulations, which relates to the calculation of net prices, is outside the scope of the Patent Act and thus ultra vires the Governor-in-Council’s regulation making authority. The effect of this decision is that, upon coming into force of the Amended Regulations in January 2021, subsection 4(4) of the Patented Medicines Regulations in their current form will remain in effect. The PMPRB is reviewing the decision to evaluate its impact but, at this time, does not believe any substantive changes to the June 2020 Draft Guidelines are required as a result. However, we invite stakeholders to share any views they may have regarding the import of Justice Manson’s decision as part of their written submission to the PMPRB in the context of the current consultation on the June 2020 Draft Guidelines. The parties to these judicial review proceedings, the Attorney General of Canada and IMC, currently have until September of this year to appeal the decision to the Federal Court of Appeal.
In order to provide parties with more time to address this recent development in their written submissions, and in response to multiple requests for an extension to the 30-day consultation period, the deadline to submit written feedback has been changed from July 20, 2020 to August 4, 2020. For more information on how to submit feedback, please visit the Consultation Portal.
June 19, 2020
June 2020 Draft Guidelines now available
The June 2020 Draft Guidelines are available for public comment until July 20, 2020. To submit your feedback, please visit the Submit Feedback section. All written submissions will be made available to the public on this website.
The PMPRB will host two public consultations online on the draft Guidelines:
- Industry Webinar – June 29 from 1:30 to 4:30 (EST)
- This webinar is limited to representatives of the pharmaceutical industry
- Public Webinar – July 8 from 1:30 to 4:30 (EST)
- This webinar is open to all interested stakeholders
The PMPRB will also host two dedicated sessions with its health partners (June 25) and private insurers (July 7) by invitation.
In addition, the PMPRB will be hosting three Research Webinars on topical analyses expected to inform the consultation.
Research Webinar 1: June 23 from 1:30 to 3:00 (EST)
- Insight into the spending on expensive drugs for rare diseases
- Insights into the market size of patented medicines in Canada
Research Webinar 2: July 6 from 1:30 to 3:00 (EST)
- Drug pricing and its impact on R&D investments, clinical trials and availability of medicines in Canada
Research Webinar 3: July 16 from 1:30 to 3:00 (EST)
- Drug shortages in Canada
To participate, please visit the Webinar Registration section.
June 1, 2020
Message from the Chairperson – New coming into force date for the Patented Medicines Regulations
The amended Patented Medicines Regulations (“Regulations”) will now come into force on January 1, 2021.
As previously communicated, the PMPRB will move forward with the issuance of a revised set of draft Guidelines, followed by a written consultation period. The new draft will be published on the week of June 15, 2020, and the consultation period will be for 30 days. More details on the next and final phase of the PMPRB’s consultations will be announced closer to the date of publication of the revised draft Guidelines.
The PMPRB will continue to work with its stakeholders and interested members of the public to ensure that a fully operational set of Guidelines is ready in time for the coming into force of the regulatory amendments in January 2021. The final Guidelines will include transitional measures which will provide patentees sufficient time to take the necessary steps to come into voluntary compliance with the relevant price ceilings for both new and existing patented medicines.
April 15, 2020
Update to the Submissions section
It has recently come to our attention that a number of stakeholder submissions the PMPRB received on the Draft Guidelines were not made publicly available on the PMPRB’s consultation portal. Further to an internal audit of its consultation inbox, the PMPRB has identified six such submissions originating from the following individuals or organizations:
- Bill Swan
- Canadian Generic Pharmaceutical Association
- Canadian Health Policy Institute
- Dr. Martha L. McKinney
- Life Saving Therapies Network
- Lorna Kosteniuk, RN
We apologize to our stakeholder community for this inadvertent omission and encourage anyone who has filed a submission with the PMPRB on the Draft Guidelines that still does not appear on our consultation portal to contact us directly. Thank you.
April 2, 2020
Message from the Chairperson
On July 1, 2020, the amended Patented Medicines Regulations will come into force. Changes to the PMPRB’s pricing Guidelines are necessary in order for the regulatory amendments to be implemented. On November 21, 2019, the PMPRB published a draft set of new Guidelines for consultation with stakeholders and the public. Extensive feedback was received and all written submissions are now available on the PMPRB website.
The PMPRB will be making significant changes to the draft Guidelines in response to the feedback it has received. At this time, the PMPRB intends to publish a revised draft set of Guidelines later this spring and will provide stakeholders and the public with a limited period of time in which to submit their views in writing prior to the Guidelines being finalized.
The PMPRB is monitoring the evolving situation regarding the Covid-19 virus in Canada very closely and will reassess whether the next steps it currently intends to take with respect to the revised set of draft Guidelines are appropriate and feasible closer to the date of their publication.
The PMPRB will continue to work with its stakeholders to ensure that a fully operational set of Guidelines is ready in time for the coming into force of the regulatory amendments. In the meantime, patentees can rest assured that the final Guidelines will include transitional measures which will provide them ample time to take the necessary steps to come into voluntary compliance with the relevant price ceilings for both new and existing patented medicines.
March 12, 2020
News
In view of the evolving situation with COVID-19 and the Public Health Agency of Canada’s advice on gatherings at this time, the PMPRB is no longer holding a public policy forum. To provide stakeholders with an opportunity to convey any new or different information that may have been presented to the Board at the forum, the PMPRB is accepting further written submissions from interested parties until March 18, 2020. Any submissions received by March 18, 2020 will be carefully considered by the Board, and stakeholders will be contacted by the PMPRB for clarifications on their materials as necessary.
February 11, 2020
Highlights and News
- Last chance! The deadline to provide written submissions is February 14, 2020. All written submissions will be made available to the public on this website.
- During this last week in the consultation phase, the PMPRB will continue to meet with stakeholders in Ottawa.
Meetings and discussions
- The PMPRB met with Innomar Strategies Inc. on February 3 in Ottawa.
February 3, 2020
Highlights and News
- Less than two weeks left! The deadline to provide written submissions is February 14, 2020. All written submissions will be made available to the public on this website.
Meetings and discussions
- The PMPRB traveled to Nova Scotia on January 30 to consult with:
- Drug Evaluation Unit, Nova Scotia Health Authority
- Advocates for the Care of the Elderly
- Association of Dalhousie Retirees and Pensioners
- College and University Retiree Associations of Canada
- Independent Voices for Safe and Effective Drugs
- Nova Scotia Health Coalition
- Nova Scotia Department of Health
- Faces of Pharmacare
- The PMPRB met with the New Brunswick Medical Society on January 31 in New Brunswick.
January 27, 2020
Highlights and News
- The PMPRB will visit New Brunswick and Nova Scotia on January 30 and 31 to consult with stakeholders, including:
- Nova Scotia Health Coalition
- Faces of Pharmacare
- Nova Scotia Department of Health and Wellness; and
- New Brunswick Medical Society.
- Reminder: February 14, 2020 is the new deadline to provide written submissions. All written submissions will be made available to the public on this website.
Meetings and discussions
- The PMPRB met with the Canadian Association of Pharmacy Distribution Management (CAPDM) and IQVIA on January 22 in Ottawa.
- The PMPRB hosted Health Partners for a discussion on the Draft Guidelines on January 23.
- The presentation from the January 17 webinar with patentees (PDF 1.9 MB) is now available.
January 20, 2020
Highlights and News
- February 14, 2020 is the new deadline to provide written submissions. All written submissions will be made available to the public on this website.
Meetings and discussions
- On January 14, the PMPRB consulted with members of Medicago and the Institut national d’excellence en santé et en services sociaux (INESS) in Québec City, Québec.
- On January 17, the PMPRB hosted a webinar with patentees to provide information on the Guidelines and answer questions.
- Meetings originally planned for the week of January 13 in New Brunswick and Nova Scotia are being rescheduled.
January 17, 2020
February 14, 2020 is the new deadline to provide written submissions. All written submissions will be made available to the public on this website.
January 13, 2020
Highlights and News
- During the week of January 13, 2020, the PMPRB will be touring the east coast of Canada, visiting:
- Quebec City, Quebec
- Fredericton, New Brunswick
- Halifax, Nova Scotia
- January 31, 2020 is the new deadline to provide written submissions. All written submissions will be made available to the public on this website.
Meetings and discussions
- On January 6 and 7, the PMPRB traveled to Toronto, Ontario to consult with members of the pharmaceutical industry, civil society and government, including:
- the Ministry of Health and Long Term Care
- Canadian Association of Provincial Cancer Agencies
- Cancer Care Ontario (now a part of Ontario Health)
- McKesson Canada
- GlaxoSmithKline Inc. (GSK)
- From January 8 to 10, the PMPRB held bilateral meetings in Toronto with members of the pharmaceutical industry, including:
- Lundbeck Canada Inc.
- Paladin Labs Inc.
- Merck Canada Inc.
- Boehringer Ingelheim (Canada) Ltd
December 23, 2019
Highlights and News
- Next planned meetings with members of the pharmaceutical industry and civil society are scheduled for January 6 and January 7, 2020, in Toronto, Ontario.
- During the week of January 13, 2020, the PMPRB will be touring the east coast of Canada, visiting:
- Halifax, Nova Scotia
- Fredericton, New Brunswick
- Montreal, Quebec
- The content presented to industry members and civil society from the December 9 and December 10 forums are now available online.
- January 31, 2020 is the new deadline to provide written submissions. All written submissions will be made available to the public on this website.
Meetings and discussions
- On December 16 and 17, the PMPRB traveled to Toronto, Ontario to consult with health partners, members of industry and patient advocacy groups, including members of Neighbourhood Pharmacy Association of Canada
The PMPRB still encourages all its stakeholders and the interested public to submit written feedback prior to January 31, 2020.
December 16, 2019
Highlights and News
- The PMPRB will be in Toronto, Ontario this week to participate in bilateral meetings with stakeholders.
- January 31, 2020 is the new deadline to provide written submission. All written submissions will be made available to the public on this website.
Meetings and discussions
The PMPRB held a one-day outreach session with a number of industry representatives on December 9. During this forum, the focus was on the proposed amendments to the guidelines, how to operationalize the draft guidelines, going through the next steps for how the PMPRB will implement the guidelines, and gathering productive feedback.
On December 10, the PMPRB hosted a one-day forum for civil society. The objective was to bring together informed non-industry and non-institutional stakeholders who represent the diverse voices of the very consumer community the PMPRB intends to protect with its new drug pricing framework. During the session, the topics covered were: the changes within the pharmaceutical market, the proposed amendments to the Guidelines, and the next steps for how the PMPRB will implement the guidelines.
Meetings with stakeholders will continue into the new year as the PMPRB travels across eastern Canada.
The PMPRB is also encouraging all its stakeholders and the interested public to submit written feedback prior to January 31, 2020.
December 9, 2019
Highlights and News
- January 31, 2020 is the new deadline to provide written submission. All written submissions will be made available to the public on this website.
- The PMPRB is in Ottawa, Ontario this week. A one-day outreach session focused on compliance and operational issues will be held with number of industry representatives on December 9. On December 10, the PMPRB will host a one-day forum for civil society.
Meetings and discussions
The PMPRB was in British Columbia, Alberta, Saskatchewan and Manitoba on the week of December 2 and consulted with various stakeholders and partners including representatives of the following:
- British Columbia Ministry of Health
- Therapeutics Initiative
- Arthritis Consumer Experts
- HepCBC
- Alberta Blue Cross
- Health Coalition of Alberta
- Institute of Health Economics
- GlaxoSmithKline Inc.
- Alberta Ministry of Health
The PMPRB also had the chance to meet with clinicians and researchers from the University of British Columbia, the Vancouver General Hospital and the BC Children's Hospital.
Next week
The PMPRB will continue to participate in bilateral meetings that have been requested by members of the industry.
Additional opportunities to participate in our consultation process and provide feedback are offered more broadly to industry stakeholders. This includes the possibility for individual companies to request bilateral meetings with the PMPRB at any time during the consultation period.
The PMPRB is also encouraging all its stakeholders and the interested public to submit written feedback prior to January 31, 2020.
December 2, 2019
Highlights and News
- The PMPRB is looking forward to stakeholder feedback on the Draft Guidelines, which were launched on November 21, 2019
Meetings and discussions
This week, the PMRPB will be meeting with provincial and territorial health ministries, private insurers and other important participants in the health care system in British Columbia, Alberta, Saskatchewan and Manitoba.
Next week
The PMPRB will be in Ottawa, Ontario during the week of December 9. A one-day outreach session focused on compliance and operational issues will be held with number of industry representatives. On December 10, the PMPRB will host a one-day forum for civil society.
November 21, 2019
Highlights and News
- On November 21, The PMPRB released its new draft Guidelines and launched a 60-day consultation period with stakeholders and interested members of the public.
- The PMPRB also published the Consultation Backgrounder with information about how you can participate and answers to some of the questions you may have.
Meetings and discussions
The PMPRB will be travelling during the week of December 2 to meet with various stakeholders in:
- Victoria and Vancouver, British Columbia
- Edmonton, Alberta
- Regina, Saskatchewan
- Winnipeg, Manitoba
Feedback
Below are the submissions the PMPRB received during the June 2020 Guideline Consultation Period before the August 4, 2020 deadline. Individual submissions have not been posted unless the individual is writing in their capacity as a medical professional, an academic; or a representative of a stakeholder organization.
The PMPRB acknowledges and thanks the thousands of Canadians who shared their thoughts on the Guidelines during the consultation process. Every submission was carefully reviewed and considered by the PMPRB.
Webinars
The PMPRB hosted two public consultations online on the draft Guidelines.
- Industry Webinar – June 29 (PDF - 2.4 MB)
- This webinar was limited to representatives of the pharmaceutical industry
- Public Webinar – July 8 (PDF - 2.4 MB)
- This webinar was open to all interested stakeholders
The PMPRB also hosted two dedicated sessions with its health partners (June 25) and private insurers (July 7).
Research Webinars
In addition, the PMPRB hosted three Research Webinars on topical analyses to inform the consultation.
Research Webinar 1: June 23 (PDF - 1.5 MB)
- Insight into the spending on expensive drugs for rare diseases
- Insights into the market size of patented medicines in Canada
Research Webinar 2: July 6 (PDF - 2.6 MB)
- Drug pricing and its impact on R&D investments, clinical trials and availability of medicines in Canada
Research Webinar 3: July 16 (PDF - 2.9 MB)
- Drug shortages in Canada