
Consultation sur les Lignes directrices provisoires du CEPMB
L’entrée en vigueur du Règlement sur les médicaments brevetés modifié (le « Règlement ») a de nouveau été reportée au 1er juillet 2021.
La consultation sur les Lignes directrices provisoires de juin 2020 est maintenant terminée. Les observations écrites présentées au CEPMB sont accessibles sous l’onglet Commentaires.
Les soumissions écrites sur la version de novembre 2019 des Lignes directrices provisoires sont également accessibles sur le site Web du CEPMB.
Lignes directrices provisoires 2020
Lignes directrices du CEPMB 2020
Table des matières
- I. Préface
- II. Interprétation
- III. Cadre juridique
- IV. Exigences en matière de présentation de renseignements dans le cadre de l’examen des prix
- V. Processus d’examen des prix
- VI. Réévaluation
- VII. Enquêtes
- VIII. Engagement de conformité volontaire (ECV)
- IX. Recommandations relatives à l’audience
- X. Processus d’audience relativement aux prix excessifs et recours
- XI. Audience pour défaut de présenter les renseignements requis
- XII. Plaintes
- XIII. Annexes
- A. Comparaison selon la catégorie thérapeutique des prix nationaux (CCTn) et comparaison selon la catégorie thérapeutique des prix internationaux (CCTi)
- B. Test de la relation raisonnable et formes posologiques comparables
- C. Évaluation de la valeur pharmacoéconomique
- D. Méthode de rajustement en fonction de la taille du marché
- E. Processus d’examen scientifique
- F. Sommaire des calendriers de mise en conformité
I. Préface
1. Le Conseil d’examen du prix des médicaments brevetés (CEPMB), créé en 1987 en tant que pilier de la protection des consommateurs dans le cadre d’importantes réformes de la Loi sur les brevets (la « Loi »), est un organisme quasi judiciaire doté d’un mandat de réglementation dont l’objectif est de veiller à ce que les titulaires de brevets (ou brevetés) pharmaceutiques ne facturent pas des prix excessifs aux consommateurs pendant la période de monopole de droit. La mise sur pied du CEPMB découle de la crainte qu’une protection accrue des brevets des médicaments puisse entraîner une hausse inacceptable de leur prix et les rendre inabordables pour les consommateurs. À titre de membre du portefeuille de la Santé, le CEPMB contribue à un système de santé moderne et viable en veillant à ce que les Canadiens continuent d’avoir accès aux médicaments brevetés à des prix non excessifs.
2. Les présentes Lignes directrices, établies conformément au paragraphe 96(4) de la Loi, visent à assurer aux brevetés la transparence et la prévisibilité du processus habituellement suivi par les fonctionnaires du CEPMB (le « personnel ») pour déterminer si le prix d’un médicament breveté semble excessif sur un des différents marchés au Canada. Les Lignes directrices présentent également un aperçu des processus que les brevetés devraient connaître en ce qui a trait à leurs obligations en matière de présentation de renseignements en vertu du Règlement sur les médicaments brevetés (le « Règlement »).
3. Si le prix d’un médicament breveté semble excessif au regard des Lignes directrices et que le breveté n’a pas soumis d’engagement de conformité volontaire (ECV) acceptable, le personnel peut recommander au président de tenir une audience sur la question. Si une telle audience est jugée dans l’intérêt public par le président et qu’un panel d’audience composé de membres du Conseil (le « panel d’audience ») confirme que le prix du médicament breveté était excessif sur tout marché, le CEPMB peut rendre une ordonnance obligeant le breveté à baisser le prix du médicament et à prendre des mesures pour compenser l’excédent des recettes que lui aurait procuré la vente du médicament au prix excessif.
II. Interprétation
4. Les Lignes directrices fournissent des renseignements sur l’approche générale du CEPMB à l’égard du processus d’examen des prix et des enquêtes. Elles remplacent l’ensemble des documents d’orientation, des communiqués de politique et des énoncés écrits ou verbaux antérieurs du CEPMB concernant l’administration du processus d’examen des prix et des enquêtes, y compris les versions antérieures du Compendium des politiques, des Lignes directrices et des procédures du CEPMB. Les Lignes directrices doivent être lues en parallèle avec la Loi, le Règlement, les annexes desdites Lignes directrices et les autres documents d’orientation connexes que le CEPMB publiera à l’avenir, dont la section d’aide de l’outil de dépôt en ligne, qui remplace l’ancien Guide du breveté.
5. Conformément au paragraphe 96(4) de la Loi, les Lignes directrices ne lient ni le personnel, ni le président, ni les panels d’audience, ni les brevetés, et elles ne visent pas à créer des droits ou des présomptions juridiques, à réaffirmer la loi ou à faire une déclaration définitive concernant l’interprétation des dispositions législatives se rapportant au CEPMB. Dans chaque cas, les décisions du personnel en matière d’application de la loi et la résolution définitive des questions dépendront des circonstances particulières de l’affaire en cause. L’interprétation définitive de la loi relève du Conseil (qui siège en tant que panel d’audience) et est soumis à un contrôle judiciaire par les tribunaux.
6. Certains aspects des Lignes directrices peuvent être revus par le CEPMB à la lumière de l’expérience et de l’évolution des circonstances. Les Lignes directrices et les politiques émises par le CEPMB sont élaborées de façon transparente et permettent une consultation complète des parties intéressées. Lorsqu’il envisagera d’apporter des modifications aux Lignes directrices, le CEPMB consultera les intervenants conformément au processus de consultation établi en vertu du paragraphe 96(5) de la Loi.
7. Le CEPMB fera tous les efforts raisonnables pour aider les brevetés à comprendre les Lignes directrices et leur application. Par exemple, le personnel informera sur demande les brevetés des méthodes à appliquer dans le cadre de l’examen du prix des médicaments brevetés vendus au Canada. En outre, si on lui en fait la demande et s’il dispose de suffisamment de renseignements pour le convaincre que le prix auquel le breveté vend ou a l’intention de vendre un médicament breveté ne serait pas jugé excessif, le Conseil peut délivrer un certificat non contraignant en ce sens en vertu du paragraphe 98(4) de la Loi.
8. Les Lignes directrices ne décrivent pas de manière exhaustive toutes les mesures qui peuvent être prises ou tous les problèmes qui peuvent survenir dans le contexte d’un examen du prix. Dans des circonstances exceptionnelles ou lors d’une audience, le CEPMB peut utiliser des méthodes ou des tests jugés appropriés et conformes à la Loi et au Règlement, même s’il n’en est pas question dans les Lignes directrices ou s’ils diffèrent de l’approche qui y est énoncée. En aucun cas le personnel ou les membres du Conseil ne seront liés ou limités par les Lignes directrices.
9. Pour obtenir de plus amples renseignements au sujet de ces Lignes directrices ou de l’approche générale du personnel à l’égard de l’examen des prix et des enquêtes, veuillez consulter le site Web du CEPMB ou communiquer avec le personnel à l’adresse suivante :
Conseil d’examen du prix des médicaments brevetés
C.P. L40
Centre Standard Life
333, avenue Laurier Ouest
Bureau 1400
Ottawa (Ontario)
K1P 1C1
À l’attention de : Secrétaire du Conseil
III. Cadre juridique
10. Le CEPMB a été créé en 1987 après que des modifications d’envergure apportées à la Loi sur les brevets sont entrées en vigueur par l’adoption du projet de loi C-22Note de bas de page 1. Ces modifications ont permis de renforcer la protection des brevets des médicaments, tant du point de vue de la portée de l’objet brevetable que de la période d’exclusivité fondée sur les brevets, instaurant ainsi au Canada ce que les décideurs croyaient être un climat d’investissement plus propice à la recherche et au développement (R-D) pharmaceutique. En guise de concession faite aux opposants à ces modifications, qui craignaient que la protection renforcée des brevets des médicaments entraîne une hausse inacceptable des prix et rende ces produits inabordables pour les Canadiens, on a également créé le CEPMB dans la foulée du projet de loi C-22. Le mandat du CEPMB consiste à s’assurer que le prix des médicaments brevetés n’est pas excessif. Essentiellement, le projet de loi C-22 cherchait à établir un équilibre entre la nécessité de reconnaître et de récompenser l’innovation pharmaceutique par l’octroi d’une période d’exclusivité de marché aux brevetés et la nécessité de veiller à ce que les prix demandés au cours de cette période d’exclusivité restent raisonnables. Comme en témoigne le statut du CEPMB, qui est le seul organisme sectoriel de réglementation des prix plafonds en vertu de la Loi, les décideurs reconnaissent que la capacité absolue d’établir le prix des médicaments brevetés n’est pas dans l’intérêt public, étant donné le préjudice unique qui peut découler des prix excessifs de ces médicaments pour les consommateurs.
11. Le CEPMB est investi d’un double rôle : dans son rôle de réglementation, il protège les consommateurs en veillant à ce que le prix des médicaments brevetés ne soit pas excessif; dans son rôle de rapport, il fournit des renseignements sur les tendances des prix dans l’industrie pharmaceutique au moyen de son rapport annuel. Conformément à une directive du ministre de la Santé prise en application de l’article 90 de la Loi, le CEPMB appuie également la politique éclairée et fondée sur des données probantes en matière de santé en rendant compte des tendances relatives aux prix, à l’utilisation et aux coûts des médicaments dans le cadre de l’initiative du Système national d’information sur l’utilisation des médicaments prescrits (SNIUMP).
12. Le CEPMB est composé de cinq membres nommés par le gouverneur en conseil en vertu de l’article 91 de la Loi. La durée maximale de leur mandat est de cinq ans, renouvelable une seule fois. Le président du CEPMB agit comme premier dirigeant du Conseil et, à ce titre, il en assure la direction. Aux termes de l’article 94 de la Loi, le CEPMB emploie des fonctionnaires (le « personnel ») pour l’exercice de ses activités quotidiennes. Le directeur exécutif du CEPMB est son haut fonctionnaire, son chef de l’exploitation et son dirigeant principal des finances, et il est responsable de la gestion du personnel.
13. Le CEPMB a été créé en vertu de la Loi en tant qu’organisme indépendant quasi judiciaire. Afin d’assurer l’indépendance et l’autonomie du Conseil, la Loi ne confère aucun pouvoir exprès ou implicite à Santé Canada ou à toute autre entité gouvernementale qui permettrait de diriger le CEPMB dans l’exercice de ses fonctions de réglementation. Le CEPMB n’a aucun lien de dépendance avec le ministre de la Santé (qui est responsable des articles de la Loi applicables au CEPMB), le ministre de l’Innovation, des Sciences et du Développement économique (qui est responsable de la Loi dans son ensemble) et ses différents intervenants. De même, le CEPMB est structuré de manière à ce que les activités et les fonctions du personnel, du président et des membres du Conseil soient distinctes. En effet, le personnel s’acquitte des fonctions liées aux enquêtes, aux litiges et aux rapports, qui sont distinctes des fonctions d’arbitrage, réservées aux membres du Conseil.
14. La surveillance de la conformité des brevetés aux exigences réglementaires en matière de présentation de renseignements et l’examen administratif du prix des médicaments brevetés relèvent du personnel. Lorsque le prix d’un médicament breveté semble excessif et que la question ne peut pas être résolue au moyen d’une baisse volontaire du prix ou de mesures visant à compenser les recettes procurées au breveté par la vente du médicament à ce prix, le directeur exécutif peut porter le dossier à l’attention du président, qui déterminera si la tenue d’une audience est dans l’intérêt public. S’il décide de tenir une audience, le président émettra un avis d’audience et constituera un panel d’audience chargé de trancher la question. La décision du président d’émettre un avis d’audience est de nature purement administrative et n’exprime pas son point de vue sur le bien-fondé du dossier sous-jacent.
15. Le CEPMB examine le prix des médicaments brevetés vendus sans lien de dépendance par les brevetés. Les ventes au Canada peuvent comprendre notamment les médicaments brevetés assujettis à un avis de conformité (AC), les médicaments obtenus par le Programme d’accès spécial, les médicaments figurant sur la Liste des drogues utilisées pour des besoins urgents en matière de santé publique, les médicaments visés par des demandes d’essais cliniques et les nouveaux médicaments expérimentaux. Le CEPMB n’a aucun pouvoir sur les prix demandés par des parties autres que les brevetés, comme les prix pratiqués par les grossistes ou les détaillants, ou sur les honoraires des pharmaciens.
16. En vertu de la Loi, le CEPMB a compétence pour déterminer si un médicament breveté est ou a été vendu par un titulaire de droits (le titulaire d’un brevet, l’ancien titulaire d’un brevet ou la personne ayant pour le moment droit à l’avantage d’un certificat de protection supplémentaire délivré à l’égard d’une invention liée à un médicament) à un prix excessif sur un marché au CanadaNote de bas de page 2. Le terme « titulaire de brevet » (ou breveté) est défini dans la Loi comme une personne ayant droit à l’avantage d’un brevet pour une invention pendant une période, ainsi que quiconque était titulaire d’un brevet pour une telle invention ou exerce ou a exercé les droits d’un titulaire, par exemple le titulaire d’une licence implicite ou explicite.Note de bas de page 3
17. Une invention est liée à un médicament si elle est destinée à des médicaments ou à la préparation ou la production de médicaments, ou susceptible d’être utilisée à de telles fins. L’expression « liée à un médicament » a un sens large. La Cour d’appel fédérale a déterminé que la nature de ce lien peut être « ténue »Note de bas de page 4 [traduction]. Par exemple, il ne faut parfois qu’« un lien, aussi ténu soit-il, entre l’invention brevetée et le médicament vendu au Canada »Note de bas de page 5 [traduction].Note de bas de page 6
18. Au sens de la Loi, le terme « médicament » s’entend notamment d’une drogue (c.-à-d. une substance ou un mélange de substances qui est fabriqué, vendu ou présenté comme pouvant servir à l’une des fins suivantes : (i) le diagnostic, le traitement, l’atténuation, la prévention d’une maladie, d’un désordre, d’un état physique anormal ou de leurs symptômes, chez l’être humain ou les animaux; (ii) la restauration, la correction ou la modification des fonctions organiques chez l’être humain ou les animaux) et d’un ingrédient médicinalNote de bas de page 7. Sauf indication contraire, toute mention d’un « médicament » dans les présentes Lignes directrices inclut toutes les formes posologiques et toutes les concentrations (c.-à-d. tous les numéros d’identification de médicament ou « DIN ») du médicament.
19. Le CEPMB reconnaît que le terme « ingrédient médicinal » signifie généralement « ingrédient pharmaceutique actif » (IPA) utilisé comme matière première au cours de la fabrication de la forme posologique finale. Les médicaments brevetés relevant de la compétence du CEPMB comprennent les vaccins, les préparations topiques, les anesthésiques et les produits diagnostiques utilisés in vivo, peu importe leur mode d’administration (c.-à-d. timbre transcutané, capsule, produit injectable, inhalateur). Toutefois, le CEPMB ne considère pas les instruments médicaux, les produits diagnostiques in vitro et les désinfectants qui ne sont pas utilisés in vivo comme étant des médicaments brevetés aux fins de l’application des dispositions de la Loi relatives à l’examen des prix.
20. Le CEPMB a compétence pendant toute la durée d’un brevet admissible et délivré, ce qui comprend la période précédant la délivrance du brevet (à partir de la date de la demande de brevet). Le CEPMB a également compétence pendant la période de protection prolongée accordée par un certificat de protection supplémentaire.Note de bas de page 8
21. Le CEPMB continue d’avoir compétence sur le prix auquel un médicament breveté est vendu sur un marché canadien après la cession du brevet et jusqu’à l’annulation ou à l’abandon du brevet conformément aux dispositions expresses de la Loi ou à la péremption du brevet. La cession de brevets n’est pas expressément reconnue dans la Loi comme un mécanisme d’extinction des droits que confère un brevet avant la péremption normale du brevet.
22. Les ordonnances rendues par le CEPMB sont exécutoires de la même manière que les ordonnances de la Cour fédérale ou de toute cour supérieure au Canada, et elles peuvent être exécutées par le Conseil ou par la Cour fédérale. Les décisions contenues dans les ordonnances rendues par le CEPMB peuvent faire l’objet d’un contrôle judiciaire par la Cour fédérale, conformément aux principes de droit administratif et à la Loi sur les Cours fédérales.
IV. Exigences en matière de présentation de renseignements dans le cadre de l’examen des prix
23. Le CEPMB doit avoir accès à des renseignements exacts et opportuns au sujet de la vente des médicaments brevetés pour pouvoir s’acquitter de son mandat de réglementation. Par conséquent, les brevetés et les anciens brevetés sont tenus de présenter ces renseignements au CEPMB.
24. Les renseignements à fournir sont énoncés à l’article 82 de la Loi et dans le Règlement. Les brevetés peuvent trouver d’autres détails qui les aideront à déterminer le contenu et la forme des renseignements à fournir et les échéances strictes à respecter qui sont établies par la loi dans la section d’aide de l’outil de dépôt en ligne, qui remplace le Guide du breveté. Les brevetés sont responsables de la conformité avec leurs obligations en matière de présentation de renseignements. Ces obligations statutaires ne peuvent être levées ou modifiées par le personnel.
25. Les renseignements que les brevetés ou les anciens brevetés peuvent être tenus de fournir comprennent ce qui suit (liste non exhaustive) :
- Un avis décrivant l’intention du breveté de mettre en vente sur un marché canadien un médicament breveté qui n’y a jamais été vendu (c.-à-d. la première vente du médicament breveté) et les renseignements connexes (Notification de l’intention de vendre un médicament breveté);
- Les renseignements réglementaires sur l’identification et les caractéristiques d’un médicament breveté, tels que la monographie du médicament breveté ou les renseignements équivalents, et les DIN octroyés à chaque forme posologique et à chaque concentration du médicament breveté;
- Les renseignements réglementaires sur le prix d’un médicament breveté, comme le prix auquel chaque forme posologique et chaque concentration du médicament breveté est ou a été vendu sur tout marché canadien ou dans n’importe lequel des onze pays indiqués dans le Règlement (le « CEPMB11 »);
- Les renseignements réglementaires sur les analyses coût-utilité préparées par un organisme canadien d’évaluation des technologies de la santé (ÉTS) financé par l’État, dont les résultats sont exprimés en fonction du coût par année de vie ajustée en fonction de la qualité (AVAQ), pour chaque indication faisant l’objet de l’analyse;
- Les renseignements réglementaires sur l’utilisation maximale approximative d’un médicament breveté au Canada, en fonction de la quantité prévue des ventes du médicament breveté sous sa forme posologique finale.
26. En vertu du paragraphe 7 du Règlement, les brevetés doivent présenter les renseignements exigés par courriel en utilisant les formulaires électroniques disponibles sur le site Web du CEPMB. Les formulaires doivent porter la signature électronique d’une personne autorisée qui atteste que les renseignements sont exacts et complets.
27. Il incombe à chaque breveté de s’assurer de manière indépendante que les renseignements présentés au CEPMB (dont les prix nationaux et internationaux) sont exacts. Le personnel peut effectuer une vérification ponctuelle des renseignements fournis par les brevetés, ce qui comprend les renseignements sur les prix, les recettes et les brevets. Dans l’éventualité d’une telle vérification, les brevetés peuvent être appelés à fournir d’autres documents à l’appui, ou encore à corriger ou à confirmer des renseignements fournis.
28. Le défaut de soumettre les renseignements requis dans le délai imparti ou le fait de présenter des renseignements erronés ou faux peuvent avoir des conséquences graves pour les brevetés ou les anciens brevetés. Si les circonstances le justifient, le Conseil peut rendre une ordonnance accordant certains recours, dont une ordonnance ex parte qui exige que les renseignements manquants soient présentés. Le dossier peut également mener à des poursuites par procédure sommaire en vertu du paragraphe 76.1(1) de la Loi. En outre, la présentation de faux renseignements constitue un acte criminel en vertu de l’article 76 de la Loi, lequel peut entraîner une amende ou un emprisonnement sur déclaration de culpabilité.
29. La Loi prévoit la confidentialité des renseignements fournis au CEPMB dans certaines circonstances. En effet, les renseignements ou les documents fournis au CEPMB en application des dispositions des articles 80, 81 et 82 de la Loi qui concernent les renseignements sur les prix ou dans le cadre d’une poursuite relative aux prix excessifs intentée en vertu de l’article 83 sont protégés et ne peuvent être divulgués au public sans l’autorisation de la partie qui les a fournis, à moins que ces renseignements n’aient été divulgués lors d’une audience publique tenue aux termes de l’article 83 de la Loi ou ne soient visés par les exceptions énoncées au paragraphe 87(2) de la Loi.
30. Les renseignements fournis au CEPMB peuvent être assujettis à certaines dispositions de la Loi sur l’accès à l’information et de la Loi sur la protection des renseignements personnels.
V. Processus d’examen des prix
31. Le processus d’examen des prix comprend une série d’étapes au cours desquelles (i) les médicaments brevetés sont regroupés par catégorie selon la date de leur lancement et des critères liés à leurs caractéristiques commerciales et (ii) les prix plafonds sont établis et utilisés pour évaluer le prix des médicaments brevetés. En général, c’est le personnel qui effectue l’examen des prix en utilisant les méthodes et les tests énoncés dans les présentes Lignes directrices, d’après les renseignements fournis par le breveté ou obtenus par le personnel auprès de sources externes pertinentes, comme les listes de médicaments des régimes publics.
32. Dans une première étape d’examen, les médicaments brevetés sont divisés en quatre grandes catégories : (1) médicaments brevetés bénéficiant de droits acquis; (2) élargissements de gammes de médicaments brevetés bénéficiant de droits acquis; (3) médicaments de transition; (4) nouveaux médicaments brevetés.
33. Les médicaments brevetés bénéficiant de droits acquis comprennent les concentrations et les formes posologiques de médicaments brevetés pour lesquels un DIN a été attribué au breveté avant le 21 août 2019, même si ces concentrations et ces formes ont été approuvées pour de nouvelles indications (sans changement de DIN) après le 21 août 2019.
34. Les élargissements de gammes de médicaments brevetés bénéficiant de droits acquis sont de nouvelles formes posologiques et concentrations de médicaments brevetés bénéficiant de droits acquis auxquels un DIN a été attribué à partir du 21 août 2019.
35. Les médicaments de transition sont des médicaments brevetés pour lesquels un DIN a été attribué à partir du 21 août 2019 et dont la première vente au Canada a eu lieu avant le 1er janvier 2021.
36. Les nouveaux médicaments brevetés comprennent toutes les autres concentrations et formes posologiques de médicaments brevetés
37. Les facteurs d’examen des prix en vertu du paragraphe 85(1) de la Loi pour les quatre catégories de médicaments brevetés sont les suivants :
- Médicaments brevetés bénéficiant de droits acquis :
- Le prix de vente du médicament sur un marché canadien
- Le prix de vente de médicaments de la même catégorie thérapeutique sur un marché canadien
- Le prix de vente du médicament et d’autres médicaments de la même catégorie thérapeutique à l’étranger;
- Les variations de l’indice des prix à la consommation (IPC)
- Nouveaux médicaments brevetés, élargissements de gammes et médicaments de transition :
- Le prix de vente du médicament sur un marché canadien
- Le prix de vente de médicaments de la même catégorie thérapeutique sur un marché canadien
- Le prix de vente du médicament et d’autres médicaments de la même catégorie thérapeutique à l’étranger
- Les variations de l’indice des prix à la consommation (IPC)
- La valeur pharmacoéconomique du médicament au Canada
- La taille du marché du médicament au Canada
- Le produit intérieur brut au Canada et le produit intérieur brut par habitant au Canada
38. Les processus d’examen et les tests qui s’appliquent aux nouveaux médicaments brevetés, aux médicaments brevetés bénéficiant de droits acquis, aux élargissements de gammes de médicaments brevetés et aux médicaments brevetés de transition sont expliqués plus en détail ciaprès.
A. Processus d’examen du prix des nouveaux médicaments brevetés
39. Le CEPMB examine les renseignements fournis par le breveté et les renseignements obtenus d’autres sources pour déterminer si le prix d’un médicament breveté lancé sur un marché canadien semble excessif. Le diagramme ci-après illustre le processus d’examen du prix des nouveaux médicaments brevetés :
Schéma des lignes directrices provisoires
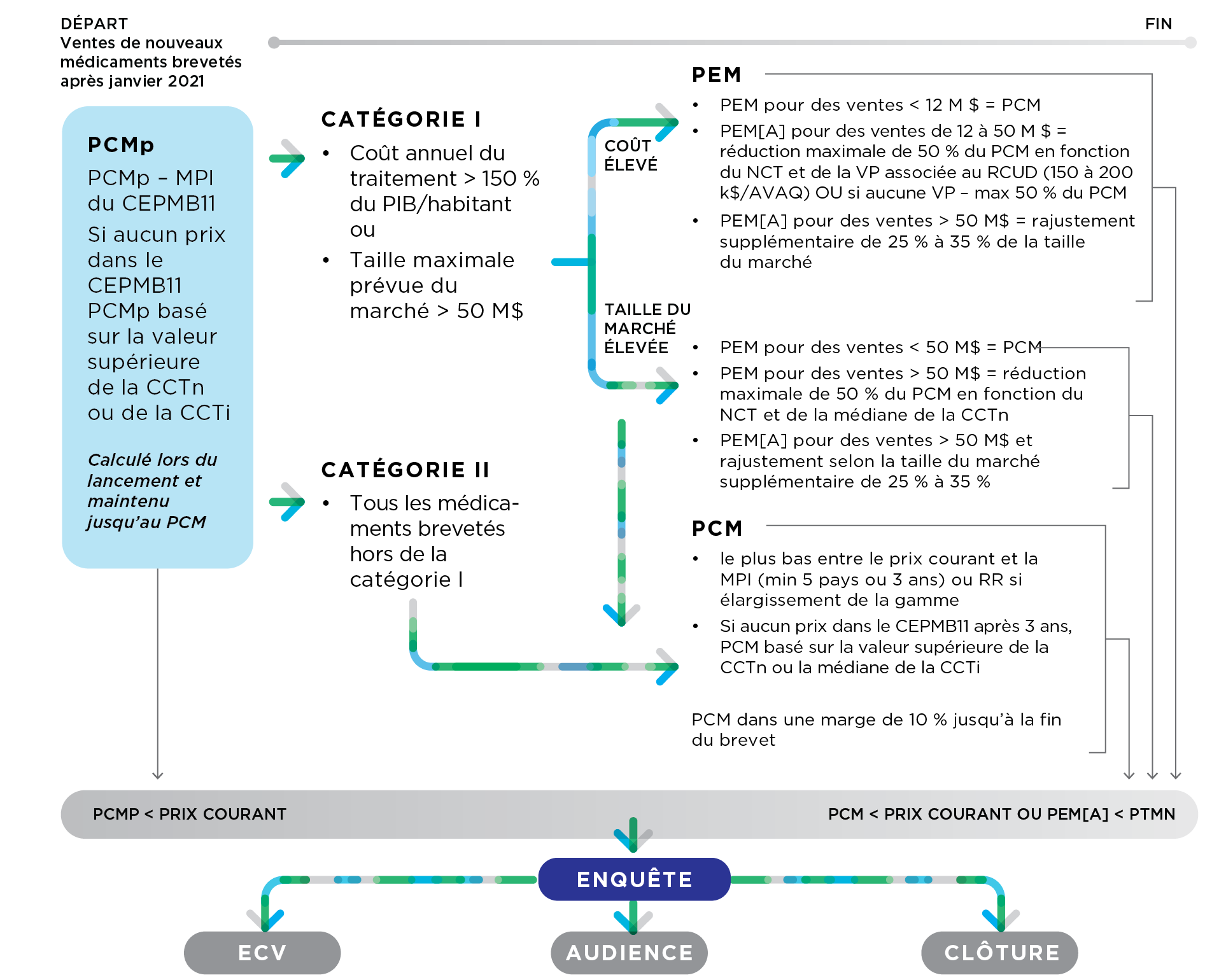
Description de la figure
Il s'agit d'un schéma décrivant de manière générale le processus d'examen des nouveaux médicaments brevetés (médicaments brevetés dont les ventes sont postérieures à janvier 2021) :
Premièrement, un PCMp est déterminé sur la base de la MPI des prix du CEPMB11 disponibles. S'il n'y a pas de prix dans les pays du CEPMB11, le PCMp est basé sur le maximum de la CCTn ou de la CCTi. Le PCMp est calculé lors du lancement et maintenu jusqu'à ce que le PCM soit fixé. Si le prix courant du médicament breveté est supérieur au PCMp, une enquête peut être ouverte, qui peut conduire soit à un ECV, soit à une audience, soit à la clôture de l'enquête.
Ensuite, le médicament breveté est classé soit dans la catégorie I (médicaments dont le coût annuel du traitement est supérieur à 150 % du PIB/habitant et (ou) médicaments dont la taille maximale prévue du marché est supérieure à 50 millions de dollars), soit dans la catégorie II (tous les médicaments brevetés qui ne font pas partie de la catégorie I).
Les médicaments de la catégorie I sont soumis à un PEM et à un PCM. Les tests utilisés pour le PEM pour les médicaments de catégorie I dont le coût annuel est supérieur à 150 % du PIB/habitant sont différents selon les ventes. Dans les deux cas, si le prix de transaction moyen du médicament breveté est supérieur au PEM, une enquête peut être ouverte, qui peut déboucher soit sur un ECV, soit sur une audience, soit sur la clôture de l'enquête.
Pour les ventes effectuées sur un marché dont la taille annuelle est inférieure à 12 millions de dollars, le PEM est égal au PCM.
Pour les ventes effectuées après que la taille annuelle du marché ait atteint entre 12 et 50 millions de dollars, le PEM est égal à un pourcentage de réduction basé sur le NCT et le VP associés au RCUD (pour les RCUD entre 150 000 et 200 000 dollars par AVAQ) qui peut aller jusqu'à une réduction maximale de 50 % sur le PCM. S'il n'y a pas de valeur VP, la réduction maximale de 50 % sur le PCM est appliquée.
Pour les ventes effectuées après que la taille annuelle du marché ait dépassé 50 millions de dollars, le PEM est soumis à un ajustement supplémentaire de la taille du marché qui peut varier entre 25 % et 35 %.
Les tests utilisés pour le PEM pour les médicaments de la catégorie I dont la taille maximale prévue du marché est supérieure à 50 millions de dollars sont également différents selon les ventes.
Pour les ventes effectuées sur un marché dont la taille annuelle est inférieure à 50 millions de dollars, le PEM est égal au PCM.
Pour les ventes réalisées après que la taille annuelle du marché ait dépassé 50 millions de dollars, le PEM est basée sur une réduction du PCM qui lui-même est basée sur le NCT et la CCTn médiane et peut aller jusqu'à une réduction maximale de 50 %. Un ajustement supplémentaire de 25 à 35 % de la taille du marché peut également s'appliquer.
Les médicaments de catégorie II ne sont soumis qu'à un seul PCM. Le PCM est égal soit au plus bas entre le prix courant et la MPI (une fois qu'il y a des données pour un minimum de 5 pays ou que 3 ans se sont écoulés depuis le lancement) ou au RR si le médicament breveté est un élargissement de la gamme. Si, trois ans après le lancement, aucun prix n'est disponible pour les pays du CEPMB11, le PCM est basé sur le sommet de la CCTn ou la médiane de la CCTi. Le PCM est maintenu dans une marge de 10 % de la MPI jusqu'à la fin de la compétence du CEPMB. Si le prix courant du médicament breveté est supérieur au PCM, une enquête peut être ouverte, qui peut conduire soit à un ECV, soit à une audience, soit à la clôture de l'enquête.
Chacune des étapes est décrite plus en détail ci-dessous.
Étape 1 : PCMp
40. Au moment du lancement d’un médicament (c’est-à-dire lors de la première vente sur un marché canadien), un prix courant maximum provisoire (le « PCMp ») est fixé pour la vente du médicament breveté. Le PCMp correspond à la médiane des prix courants internationaux départ usine (« MPI ») des pays du CEPMB11 pour lesquels le breveté a fourni des renseignements pendant la période provisoire (définie ci-dessous). Le breveté doit s’assurer que le prix canadien départ usine brutNote de bas de page 9 accessible au public du médicament breveté (« prix courant ») ne dépasse pas le PCMp pour la période provisoire au cours de laquelle il s’applique, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
41. Si le breveté n’a pas soumis de renseignements sur les prix internationaux pour les pays du CEPMB11, le PCMp est fixé par la valeur supérieure de la comparaison selon la catégorie thérapeutique des prix nationaux (« CCTn »). La CCTn sera calculée sur la base du coût de traitement le plus élevé parmi les médicaments de comparaison, obtenu en tenant compte du prix public le plus bas de chaque médicament de comparaison (voir l’annexe A pour plus de détails).
42. Le PCMp n’est calculé qu’une seule fois et s’applique pendant la période provisoire, qui dure jusqu’à la première des deux dates suivantes :
- trois (3) ans à compter de la date de lancement du médicament breveté au Canada;
- la date à laquelle le breveté fournit des renseignements sur les prix internationaux d’au moins cinq pays du CEPMB11.
À la fin de la période provisoire, on établit le PCM (voir l’étape 2), et le PCMp cesse de s’appliquer.
43. Si, au moment du lancement du médicament, le breveté fournit de l’information sur les prix internationaux pour au moins cinq des pays du CEPMB11, le PCMp ne s’applique pas et le PCM est fixé immédiatement (voir la section ci-dessous).
Étape 2 : PCM
44. Sous réserve de la procédure décrite ci-dessus, le PCMp est remplacé par un prix courant maximum ou « PCM ».
45. Si le breveté a déposé des prix internationaux avant la fin de la période provisoire, le PCM est fixé par la MPI. Sinon, le PCM est fixé par la valeur supérieure de la CCTn. La CCTn sera calculée sur la base du coût de traitement le plus élevé parmi les médicaments de comparaison, obtenu en tenant compte du prix public le plus bas de chaque médicament de comparaison (voir l’annexe A pour plus de détails).
46. Si le breveté n’a pas soumis de prix international à la fin de la période provisoire, et qu’il n’existe pas de comparateur de classe thérapeutique au niveau national, le PCM peut être fixé selon la médiane de la comparaison selon la catégorie thérapeutique des prix internationaux (« CCTi »).
47. Il peut arriver que le PCM soit inférieur au PCMp établi au cours de la période provisoire. Le cas échéant, le breveté dispose jusqu’à la prochaine période de référence après l’établissement du PCM pour s’assurer que le prix courant du médicament breveté est réduit à un niveau qui ne dépasse pas le PCM.
48. Comme il est expliqué plus en détail dans la section VI (« Réévaluation »), si le PCM a été fixé par la MPI et que, dans les périodes ultérieures, la MPI en vigueur dépasse le PCM de plus de 10 %, le PCM peut être ajusté sur la base de la variation réelle retardée de l’indice des prix à la consommation (« IPC »)Note de bas de page 10. Pour les médicaments brevetés ayant plusieurs DIN, la comparaison du PCM avec la MPI n’est effectuée que pour le DIN pour lequel le PCM a initialement été fixé par la MPI, et non pour les DIN pour lesquels les PCM ont été fixées par le test de la relation raisonnable (« RR ») (décrit en détail à l’annexe B). Toutefois, si le PCM d’un DIN est ajusté en fonction de l’IPC, les PCM supplémentaires fixés par le test de la RR en référence à ce PCM seront ajustés conformément à la méthodologie de la RR.
49. Le PCM du médicament breveté peut également faire l’objet d’une réévaluation et d’un rajustement en raison d’un écart de 10 % ou plus par rapport à la MPI comme décrit ci-dessus (voir section VI « Réévaluation »).
50. Comme c’est le cas pour le PCMp, le breveté doit s’assurer que le prix courant du médicament breveté ne dépasse pas le PCM pour la période au cours de laquelle il s’applique, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
a) Prix courant du médicament breveté au Canada
51. Conformément au Règlement, le breveté doit présenter des renseignements sur le prix courant de chaque forme posologique, de chaque concentration et de chaque format d’emballage (c. à d. pour chaque DIN) du médicament breveté dans chaque province et territoire.
52. Les prix courants peuvent varier à l’intérieur des marchés géographiques canadiens (c. à d. entre les provinces ou les territoires). Lorsqu’il y a plusieurs prix courants sur différents marchés, le prix courant le plus élevé est utilisé aux fins de la comparaison du PCMp et du PCM.
b) Prix courants du médicament dans les pays du CEPMB11
53. Les tests des prix internationaux sont fondés sur les renseignements fournis par le breveté. Lorsqu’il y a plusieurs prix courants dans le même pays, le prix le plus bas est généralement utilisé. Les prix de tous les pays du CEPMB11 pour lesquels le breveté a fourni des renseignements sont utilisés.
54. Des conseils sur les sources potentielles de prix départ usine sont présentés dans la section d’aide de l’outil de soumission en ligne.
55. Pour comparer les prix dans les pays du CEPMB11, on convertit la monnaie locale en dollars canadiens en utilisant les taux de change calculés comme la moyenne simple des trente six (36) taux de change mensuels moyens au comptant à midi pour chaque pays (ramenés à huit [8] décimales) comme publiés par la Banque du Canada. Pour la période de lancement d’un médicament breveté, les trente six (36) mois se terminant au deuxième mois de la période de référence précédente (c’est-à-dire février ou août) seront utilisés. Par la suite, les trente six (36) mois se terminant au cours du deuxième mois de la période de référence qui est examinée seront utilisés.
Étape 3 : PEM/PEM[A] (Nouveaux médicaments brevetés de la catégorie I seulement)
56. Les nouveaux médicaments brevetés sont classés comme étant des médicaments brevetés de catégorie I ou de catégorie II en fonction de certaines caractéristiques commerciales. En plus du PCMp/PCM, les médicaments brevetés de la catégorie I sont également soumis à un plafond de « prix escompté maximum » (« PEM ») qui peut être ajusté en fonction de la taille du marché dans certaines circonstances, devenant ainsi un PEM [ajusté] (« PEM[A] »). Le calcul du PEM et du PEM[A] est décrit ci-dessous et dans les annexes C et D.
57. Le PEM prend en compte le niveau de critères thérapeutiques (NCT) (les niveaux I à IV et le processus d’examen scientifique sont décrits à l’annexe E; les niveaux sont généralement fondés sur des renseignements scientifiques, notamment l’effet thérapeutique, l’incidence clinique et le gain d’AVAQ), la valeur pharmacoéconomique et la taille du marché pour le médicament breveté (voir l’annexe C). Le breveté doit s’assurer que le prix net du médicament breveté au Canada (c. à d. le « prix de transaction moyen » ou « PTM »Note de bas de page 11) ne dépasse pas le PEM, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel. Des conseils sur le calcul du prix net d’un médicament breveté, y compris le traitement des produits gratuits et des rabais, sont présentés dans la section d’aide de l’outil de soumission en ligne.
a) Classification d’un médicament breveté de catégorie I
58. Les nouveaux médicaments brevetés de catégorie I sont les médicaments brevetés qui satisfont l’un ou l’autre des critères suivants :
- Le coût de traitement de 12 mois est supérieur à 150 % du PIB par habitant : sur la base des informations sur les prix de la période de lancement fournies par le breveté, le personnel calcule le coût de traitement sur 12 mois du médicament selon la dose maximale par traitement indiquée dans la monographie du produit; le nombre maximal de traitements par période de 12 mois en fonction de la nature du problème de santé, des pratiques cliniques et d’autres critères pertinents; le prix courant canadien le plus élevé. Si aucun prix courant n’est disponible, le prix net national est utilisé.
- La taille estimée ou réelle du marché (recettes) dépasse le seuil annuel de la taille du marché : La taille du marché à des fins de classification peut être basée sur (i) une estimation déposée en vertu du Règlement ou (ii) les recettes réelles déposées en vertu du Règlement. Le seuil annuel de la taille du marché à partir duquel un nouveau médicament breveté sera classé dans la catégorie I est de 50 millions de dollars.Note de bas de page 12
59. Tous les autres nouveaux médicaments brevetés sont des médicaments brevetés de catégorie II.
60. En outre, même s’ils répondent aux critères de la catégorie I, tous les nouveaux médicaments biosimilaires brevetésNote de bas de page 13 et les nouveaux médicaments génériques brevetésNote de bas de page 14 seront classés dans la catégorie II.
b) Calcul du PEM et du PEM[A]
61. Le PEM est calculé en fonction des critères pertinents qui ont conduit à la classification du médicament breveté dans la catégorie I et de la disponibilité et du contenu d’une analyse coût-utilité.
62. Pour les médicaments brevetés qui ont (1) un coût de traitement sur 12 mois supérieur à 150 % du PIB par habitant et un revenu maximal estimé ou réel supérieur à 12 millions de dollars par an et (2) une analyse coût-utilité disponible, le PEM est calculé comme suit :
- Le rapport coût-utilité différentiel (« RCUD ») mesuré en années de vie ajustées en fonction de la qualité (« AVAQ ») pour chaque indication du médicament breveté est calculé à partir des analyses coût-utilité présentées par le breveté.
- Le prix auquel le RCUD du médicament breveté serait équivalent au seuil de la valeur pharmacoéconomique (« SVP ») est déterminé (le « prix pharmacoéconomique » ou « PP »).
- Le RCUD est comparé au SVP et au plancher de réduction applicables, en fonction de son niveau de critères thérapeutiques (voir l’annexe C, « Évaluation de la valeur pharmacoéconomique » et le tableau reproduit ci-dessous).
Rajustement du prix basé sur le niveau de critères thérapeutiques pour le calcul du PEM Niveau de critères thérapeutiques
(voir annexe E – Processus d’examen scientifique)SVP Plancher de réduction du PCM Niveau I 200 k$/ AVAQ 20 % Niveau II 150 k$/ AVAQ 30 % Niveau III 150 k$/ AVAQ 40 % Niveau IV 150 k$/ AVAQ 50 % L’analyse pharmacoéconomique est une minimisation des coûts Médiane de la CCTn soumise à un plancher de 50 % Aucune évaluation pharmacoéconomique 50 % du PCM - Le PEM sera déterminé en supposant que les 12 premiers millions de dollars sont réalisés pour les quantités vendues au PCM, tandis que les ventes restantes jusqu’à 50 millions de dollars sont réalisées pour les quantités vendues au PP. Le PEM peut être ajusté davantage en fonction de la taille du marché – devenant ainsi un PEM[A] – si le médicament breveté réalise des ventes annuelles réelles (par opposition à une estimation) telles que, si son prix est fixé au PEM établi par le PP soumis au plancher de réduction, les recettes réelles seraient supérieures à 50 millions de dollars (voir l’annexe D « Méthode de rajustement en fonction de la taille du marché »). Comme ce rajustement a lieu à des niveaux de vente prévisibles, le breveté sera informé de son PEM actuel et de son PEM[A] futur prévu une fois que les ventes du médicament breveté auront atteint certains niveaux.
63. Si, au terme de la procédure décrite ci-dessus, le PEM ou PEM[A] dépasse le PCM, le PEM est établi au même niveau que le PCM.
64. Pour les médicaments brevetés dont le coût du traitement sur 12 mois est supérieur à 150 % du PIB par habitant et dont le revenu maximal estimé est supérieur à 12 millions de dollars par an, mais qui ne disposent pas d’une analyse coût-utilité ou si l’analyse présentée ne permet pas de déterminer le PEM comme décrit ci-dessus, le PEM est fixé à 50 % du PCM. Un PEM[A] basé sur ce PEM peut être calculé, le cas échéant (voir l’annexe C).
65. Pour les médicaments brevetés qui ont été classés dans la catégorie I sur la seule base de la taille du marché, le PEM est calculé comme suit :
- Si la taille réelle du marché est inférieure à 50 millions de dollars, le PEM est fixé au PCM.
- Si la taille réelle du marché est supérieure à 50 millions de dollars, le PEM est fixé à la médiane de la CCTn en tenant compte du plancher applicable selon le tableau de l’annexe A et de l’application des rajustements de la taille du marché selon l’annexe D, et un PEM[A] s’applique.
66. Le PEM ou PEM[A] est recalculé annuellement.
67. Les différentes méthodes de calcul des PEM/PEM[A] décrites ci-dessus sont illustrées visuellement ci-après.
Categorie 1 – PEM

Description de la figure
Il s'agit d'un schéma illustrant les différentes méthodes de calcul des PEM pour les médicaments brevetés de catégorie I.
Les tests utilisés pour le PEM pour les médicaments de catégorie I dont le coût annuel est élevé sont différents selon les ventes.
Pour les ventes effectuées sur un marché dont la taille annuelle est inférieure à 12 millions de dollars, le PEM est égal au PCM. Pour les ventes effectuées après que la taille annuelle du marché ait atteint entre 12 et 50 millions de dollars, le PEM est égal à un pourcentage de réduction basé sur le NCT et le VP associés au RCUD (pour les RCUD entre 150 000 et 200 000 dollars par AVAQ) qui peut aller jusqu'à une réduction maximale de 50 % sur le PCM. S'il n'y a pas de valeur VP, la réduction maximale de 50 % sur le PCM est appliquée.
Pour les ventes effectuées après que la taille annuelle du marché ait dépassé 50 millions de dollars, le PEM est soumis à un ajustement supplémentaire de la taille du marché qui peut varier entre 25 % et 35 %.
Les tests utilisés pour le PEM pour les médicaments de la catégorie I dont la taille du marché est élevée sont également différents selon les ventes.
Pour les ventes effectuées sur un marché dont la taille annuelle est inférieure à 50 millions de dollars, le PEM est égal au PCM. Pour les ventes réalisées après que la taille annuelle du marché ait dépassé 50 millions de dollars, le PEM est basée sur une réduction du PCM qui elle-même est basée sur le NCT et la médiane de la CCTn et peut aller jusqu'à une réduction maximale de 50 %. Un ajustement supplémentaire de 25 à 35 % de la taille du marché peut également s'appliquer.
Étape 4 : Détermination de l’indication pertinente
68. Pour les médicaments brevetés ayant plus d’une indication approuvée, le personnel déterminera l’indication pour laquelle le PEM (et le PCM, le cas échéant) sera évalué (l’« indication pertinente »). Cela pourrait se produire lors du lancement, ou dans le cadre d’une réévaluation si des indications supplémentaires sont approuvées pendant le cycle de vie du médicament breveté (voir la section VI).
69. S’il s’agit d’un médicament breveté de catégorie I, l’indication pertinente sera l’indication qui remplit le critère relatif au coût annuel de traitement aux fins de la classification des médicaments brevetés de catégorie I, tel qu’il est énoncé dans les présentes Lignes directrices. Si le médicament breveté de catégorie I possède plus d’une indication qui remplit ce critère ou n’en possède aucune, ou s’il s’agit d’un médicament de catégorie II, l’indication pertinente sera l’indication associée au problème de santé ayant la prévalence la plus élevée (c.-à-d. la plus grande population de patients) ou l’utilisation estimée.
Étape 5 : Calendrier de l’examen de la conformité
70. Le calendrier de mise en conformité avec les plafonds de PEM/PCM décrits dans cette section est le suivant :
- PCMp : Le breveté doit se conformer au PCMp lors de l’entrée sur le marché si celui-ci est connu à ce moment-là, ou au cours d’une (1) période de référence lorsque le PCMp est connu.
- PCM : Le breveté doit se conformer au PCM lors de l’entrée sur le marché si celui-ci est connu à ce moment-là, ou au cours d’une (1) période de référence lorsque le PCM est fixé après un PCMp.
- PEM/PEM[A] : Le breveté doit se conformer PEM/PEM[A] dans les deux (2) périodes de référence du PEM/PEM[A] connues.
B. Processus d’examen du prix pour les médicaments brevetés bénéficiant de droits acquis, les élargissements de gammes et les médicaments de transition
71. Les médicaments brevetés bénéficiant de droits acquis, les élargissements de gammes et les médicaments de transition sont assujettis à un PCM, mais pas à un PEM.
72. Le PCM pour les médicaments bénéficiant de droits acquis et les élargissements de gammes est fixé à la plus faible des deux valeurs suivantes
- le plus élevé des prix internationaux (« PEPI ») pour les pays du CEPMB11 pour lesquels le breveté a fourni des renseignements;
- le plafond du médicament breveté (p. ex. le « PMNE ») en vertu des Lignes directrices applicables avant la publication des présentes Lignes directrices.
73. La méthode de calcul du PEPI et du prix courant des médicaments brevetés bénéficiant de droits acquis et des élargissements de gammes est la même que la méthode prévue dans les présentes Lignes directrices pour les nouveaux médicaments brevetés (voir la section V[A], « Processus d’examen du prix des nouveaux médicaments brevetés »).
74. Le PCM pour les médicaments brevetés de transition est fixé à la plus faible des deux valeurs suivantes
- la MPI pour les pays du CEPMB11 pour lesquels le breveté a fourni des informations;
- le plafond du médicament breveté (p. ex. le « PMNE ») en vertu des Lignes directrices applicables avant la publication des présentes Lignes directrices.
75. Le breveté doit s’assurer que le prix courant du médicament breveté ne dépasse pas le PCM pour la période au cours de laquelle il s’applique, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
76. Pour les médicaments brevetés bénéficiant de droits acquis, les élargissements de gammes et les médicaments brevetés de transition, si le PCM est fixé par le PMNE du médicament breveté et s’il est prouvé que son calcul était anormalement bas en raison de la déclaration d’avantages, le breveté peut soumettre une demande de PCM plus élevée. Toute demande de ce type doit inclure une proposition de PCM, des détails et des documents justificatifs des avantages déclarés et des changements historiques de prix courants, ainsi que toute autre information pertinente indiquée par le personnel. Si les renseignements requis justifient une réévaluation, le PCM sera ajusté au prix le plus bas entre (i) les tests des prix internationaux applicables pour les pays du CEPMB11 pour lesquels le breveté a fourni des renseignements et (ii) le prix courant conforme le plus élevé du médicament breveté selon les Lignes directrices applicables avant la publication des présentes Lignes directrices.
77. Le breveté est tenu de se conformer au PCM dans un délai d’une période de référence une fois que le PCM a été établi pour un élargissement de gamme d’un médicament breveté et dans un délai de deux (2) périodes de référence pour un médicament breveté bénéficiant de droits acquis ou un médicament breveté de transition.
VI. Réévaluation
78. Il est possible que l’on réévalue la catégorie ou le prix plafond d’un médicament breveté pour s’assurer qu’ils restent pertinents à la suite de changements importants dans les conditions du marché.
Réévaluation des nouveaux médicaments brevetés
79. En ce qui concerne les nouveaux médicaments brevetés, il peut y avoir une réévaluation dans l’une ou l’autre des situations suivantes :
- Une nouvelle indication d’un médicament breveté (de catégorie I ou II) est approuvée;
- Les ventes d’un médicament breveté de catégorie II ont dépassé le seuil de taille du marché (voir l’annexe D), contrairement à l’estimation initiale présentée par le breveté;
- L’analyse coût-utilité d’un médicament breveté de catégorie I est mise à jour;
- Si le PCM est fixé par la MPI et que, dans deux périodes consécutives ultérieures, la MPI en vigueur est inférieure au PCM de plus de 10 %.
80. Un médicament breveté qui est approuvé pour une nouvelle indication peut voir son indication pertinente modifiée conformément aux procédures décrites dans la section V. L’attribution d’une nouvelle indication peut avoir une incidence sur la taille du marché du médicament breveté, les médicaments de comparaison appartenant à la même catégorie thérapeutique et le coût-efficacité. Il peut donc y avoir une augmentation ou une diminution du PEM.
81. Un médicament breveté de catégorie II qui est approuvé pour une nouvelle indication (à l’exception des biosimilaires et des médicaments génériques) peut être réévalué en catégorie I s’il répond aux critères de sélection pertinents. Par exemple, si les recettes réelles du médicament breveté augmentent au-delà du seuil annuel de la taille du marché, contrairement à l’estimation initiale de la taille du marché déposée par le breveté.
82. Une réévaluation de la catégorie II à la catégorie I entraînera l’attribution d’un PEM au médicament breveté.
83. Si le PCM est fixé par la MPI et que, dans des périodes ultérieures, la MPI en vigueur est inférieure au PCM de plus de 10 %, le PCM sera redéfini par la MPI en vigueur. Si le médicament breveté a des DIN supplémentaires avec des PCM fixés par le test de la RR (voir l’annexe B), ces PCM seront ajustés en conséquence.
Réévaluation des médicaments brevetés bénéficiant de droits acquis, des élargissements de gammes et des médicaments brevetés de transition
84. Dans le cas des médicaments brevetés bénéficiant de droits acquis, des élargissements de gammes et des médicaments brevetés de transition, si le PCM est fixé par le PEPI et que, dans deux périodes subséquentes ultérieures, le PEPI en vigueur est inférieur au PCM, le PCM sera redéfini par le PEPI en vigueur. Si le PCM est fixé par la MPI et que, dans deux périodes subséquentes ultérieures, la MPI en vigueur est inférieure au PCM de plus de 10 %, le PCM sera redéfini par la MPI en vigueur.
Respect des plafonds réévalués
85. Le breveté sera informé en cas de réévaluation des critères et sera ensuite informé du PCM/PEM ajusté. Le prix du médicament breveté doit être conforme au nouveau PCM/PEM comme suit :
- PCM : dans un délai d’une (1) période de référence à compter de la notification du nouveau PCM.
- PEM : dans un délai de deux (2) périodes de référence à compter de la notification du nouveau PEM.
VII. Enquêtes
86. Une enquête est un examen approfondi du prix d’un médicament breveté réalisé par le personnel. Dans le cadre d’une enquête, le personnel examine les renseignements fournis par le breveté et les renseignements pouvant être obtenus d’autres sources. Les enquêtes sont de nature purement administrative, et aucun membre du Conseil ne participe au processus. Si une enquête entraîne la tenue d’une audience, le panel d’audience doit effectuer un examen de novo indépendant du prix du médicament breveté afin de déterminer s’il est excessif au sens de l’article 83 de la Loi. Par conséquent, les positions prises par le personnel ou par le breveté au cours de l’enquête peuvent différer de celles prises au cours d’une audience.
87. Les critères justifiant la tenue d’une enquête ont été élaborés dans le but d’assurer l’utilisation la plus efficace des ressources humaines et financières du CEPMB. Le fait que le prix d’un médicament breveté ne soit pas visé par une enquête ne signifie pas nécessairement que son prix ne soit pas excessif, et inversement. Cela signifie uniquement que les critères d’enquête prévus dans les Lignes directrices n’ont pas été remplis dans les circonstances particulières.
A. Critères d’enquête
88. En règle générale, le personnel lancera une enquête si aux moins une des conditions suivantes est présente :
- Lorsque le prix de toute forme posologique ou concentration d’un médicament breveté semble être supérieur de plus de 5 p. 100 au prix plafond applicable correspondant;
- Lorsque les recettes cumulatives potentielles tirées de la vente à un prix supérieur au prix plafond applicable (« excédent des recettes potentielles ») semblent dépasser 50 000 dollars pour l’ensemble des produits du breveté qui contiennent le même médicament breveté (c.-à-d. tous les DIN contenant le même ingrédient médicinal) au cours d’une année civile;
- Lorsqu’une plainte est déposée par quiconque a à tout moment (y compris avant la date applicable pour l’évaluation du respect d’un plafond particulier).
89. Nonobstant ce qui précède, dans le cas des biosimilaires brevetés, des médicaments génériques brevetés, des médicaments brevetés à usage vétérinaire et des médicaments brevetés en vente libre, une enquête ne sera ouverte par le personnel que si une plainte est reçue.
90. Nonobstant le paragraphe 88, le prix de tout médicament breveté figurant sur la Liste des drogues destinées aux importations et aux ventes exceptionnelles établie conformément à l’article 3 de l’Arrêté d’urgence concernant les drogues, les instruments médicaux et les aliments à des fins diététiques spéciales dans le cadre de la COVID-19 du 30 mars 2020 ne fera l’objet d’une enquête que si un plainte est reçue de la part de la ministre fédérale de la Santé ou de tout homologue provincial ou territorial.
91. Lorsque le prix d’un médicament breveté est supérieur aux prix plafonds applicables prévus dans les Lignes directrices, mais que les critères justifiant la tenue d’une enquête ne sont pas remplis, le breveté en est avisé, et le médicament breveté se voit attribuer la mention « Ne justifie pas une enquête » dans le rapport annuel du CEPMB. Le cas échant, le personnel ne prend aucune mesure immédiate; toutefois, afin d’éviter une enquête à une date ultérieure, le breveté doit s’assurer que le prix du médicament breveté est réduit à un niveau ne dépassant pas le prix plafond applicable et compenser les recettes qui peuvent avoir été tirées, sans quoi il pourrait faire l’objet d’une enquête par le personnel.
B. Processus d’enquête
92. Lorsque le personnel lance une enquête, le breveté en est avisé, et le médicament breveté se voit attribuer la mention « Sous enquête » dans le rapport annuel du CEPMB. Le processus d’examen des prix et le processus d’enquête ne peuvent pas donner lieu à une décision judiciaire établissant que le prix d’un médicament breveté est excessif au sens de la Loi. Une telle décision ne peut être prise qu’une fois que le breveté ou l’ancien breveté a eu la possibilité raisonnable de présenter ses observations, conformément à l’article 83 de la Loi.
1. Examen supplémentaire des renseignements présentés
93. Lorsqu’un médicament breveté fait l’objet d’une enquête, le personnel examine l’historique des prix du médicament depuis son lancement. Tous les renseignements fournis par le breveté sont analysés, et d’autres éclaircissements peuvent être demandés. Par exemple, si un prix semble erroné ou inattendu, ou s’il y a des divergences dans les renseignements présentés par le breveté, celui-ci pourrait devoir fournir une explication ou d’autres documents à l’appui. Le personnel pourrait également prendre en considération tous les renseignements pertinents qui n’ont pas été soumis par le breveté, comme les prix courants obtenus avec la CCTn et la CCTi. En outre, le personnel analyse la pertinence de la catégorie et des tests applicables en fonction des faits du dossier et des particularités des marchés pertinents sur lesquels le médicament breveté est vendu. Par exemple, le personnel peut examiner si la taille réelle du marché est sensiblement inférieure à la taille estimée du marché, ou si le médicament breveté est un vaccin, un produit sanguin ou un autre produit soumis à un processus d’appel d’offres.
94. Le personnel peut utiliser tout test décrit dans les Lignes directrices et toute modification ou variation de ces tests (par exemple, la MPI au lieu du PEPI ou la médiane par opposition au sommet de la CCTn) selon ce qu’il juge le plus approprié au vu des circonstances factuelles entourant le prix du médicament breveté faisant l’objet de l’enquête.
2. Calcul de l’excédent des recettes potentielles
95. Lorsque le prix d’un médicament breveté est supérieur aux prix plafonds prévus dans les Lignes directrices, le breveté est informé que le prix du médicament breveté « dépasse les seuils établis dans les Lignes directrices », et les prix plafonds ou tests applicables sont identifiés. Le personnel commence également à calculer l’excédent cumulatif des recettes potentielles en fonction des prix nets présentés par le breveté, quel que soit le prix plafond ou test utilisé (PCMp, PCM ou PEM/PEM[A]). Comme le CEPMB a compétence sur le médicament breveté au cours de la période précédant la délivrance du brevet, l’excédent des recettes qu’aurait procuré la vente du médicament pendant cette période est inclus dans les calculs.
96. Dans certains cas, les tests et les plafonds utilisés au cours de l’enquête peuvent différer des seuils initiaux qui ont mené au déclenchement de l’enquête. Dans de tels cas, les plafonds de l’enquête (par opposition aux plafonds de déclenchement) seront utilisés pour calculer les recettes excédentaires potentielles.
97. Enfin, si une audience a lieu, le personnel peut exercer un recours dans le cadre duquel l’excédent des recettes diffère de l’excédent cumulatif des recettes potentielles calculé lors de l’enquête. De plus, lorsque le personnel estime que le breveté ou l’ancien breveté s’est livré à une politique de vente d’un médicament breveté à un prix excessif, il peut, par ordonnance, lui enjoindre de compenser jusqu’au double de l’excédent des recettes.
3. Résultats de l’enquête
98. Les résultats possibles d’une enquête sont les suivants :
- Un engagement de conformité volontaire (« ECV »), tel qu’il est décrit dans la section VIII;
- L’émission d’un avis d’audience si, sur recommandation du personnel, le président estime que la tenue d’une audience est dans l’intérêt public;
- La clôture de l’enquête.
99. La clôture d’une enquête peut découler d’un ECV ou d’une décision selon laquelle un examen approfondi du prix d’un médicament breveté n’est pas justifié à ce moment-là, compte tenu des faits et des considérations qui ont été mis en lumière au cours de l’enquête.
100.La clôture d’une enquête est une mesure administrative; il ne s’agit pas d’une décision judiciaire établissant que le prix du médicament breveté n’est pas excessif ni d’un aveu du CEPMB en ce sens. La clôture d’une enquête n’exclut pas la possibilité que l’on rouvre une enquête, que l’on en lance une nouvelle ou que l’on tienne une audience dans l’avenir.
VIII. Engagement de conformité volontaire (ECV)
101. Le breveté peut présenter un ECV au personnel en tout temps avant l’émission d’un avis d’audience. Un ECV est une promesse par laquelle le breveté s’engage à baisser le prix du médicament visé par l’enquête ou à compenser l’excédent des recettes que lui aurait procuré la vente de ce médicament à ce prix. Une proposition d’ECV ne constitue pas un aveu du breveté quant au caractère excessif du prix du médicament.
102. Le personnel est chargé de mener les négociations relatives à l’ECV avec le breveté et, selon la politique du CEPMB, le président ne peut pas participer aux discussions. Si les négociations aboutissent à une proposition d’ECV qui, de l’avis du personnel, serait acceptable pour le président, la proposition sera soumise au président aux fins d’examen. Le personnel ne peut pas déterminer de façon indépendante si une proposition d’ECV est acceptable, et il ne peut pas donner de garantie au breveté quant à la probabilité que le président accepte la proposition.
103. L’examen d’un ECV est une procédure administrative; il ne s’agit pas d’une décision du CEPMB établissant que le prix du médicament présenté par le breveté ou utilisé pour le calcul du montant de la compensation n’est pas excessif ni d’un aveu du CEPMB en ce sens. Toutefois, l’acceptation d’un ECV par le président donnera lieu à la clôture de l’enquête.
104. Le CEPMB rend compte publiquement de tous les ECV acceptés par le président. Lorsqu’il présente un ECV signé, le breveté doit consentir à sa publication en version entière ou expurgée. Les renseignements publiés peuvent comprendre le contenu de l’ECV, dont les conditions qui y sont énoncées. Ces renseignements peuvent figurer dans le rapport annuel du CEPMB, sur le site Web du CEPMB, dans les publications du CEPMB, comme La Nouvelle, et sur les plateformes de médias sociaux.
105. Le personnel ne peut pas se pencher sur les demandes de négociations ou de propositions d’ECV « sans préjudice ». Les ECV sont des promesses unilatérales faites par les brevetés et ne sont pas des ententes entre le CEPMB et les brevetés. Néanmoins, certains aspects des discussions entre les brevetés et le personnel qui ont trait au contenu des présentations du breveté peuvent être soumis aux protections énoncées aux articles 87 et 88 de la Loi. En outre, les dispositions dans la Loi sur l’accès à l’information peuvent s’appliquer.
106. Lorsqu’un avis d’audience est émis, le breveté peut quand même négocier un règlement sous forme d’entente, laquelle doit être approuvée par le panel d’audience. Le personnel examine les demandes d’accord ou de proposition de règlement « sans préjudice ».
IX. Recommandations relatives à l’audience
107. Lorsqu’une enquête sur le prix d’un médicament breveté est terminée et que l’affaire n’est pas réglée avec le breveté, le directeur exécutif peut présenter un rapport au président. Le président, en sa qualité de premier dirigeant du CEPMB, peut décider d’émettre un avis d’audience s’il estime que la tenue d’une audience est dans l’intérêt public. La décision d’émettre un avis d’audience n’est pas judiciaire ou quasi judiciaire, et le président n’effectue pas d’analyse pour déterminer si les faits allégués par le personnel sont ou seront prouvés. Aucun autre membre du Conseil n’est informé de l’enquête ni des résultats de l’examen du personnel sur le prix d’un médicament breveté tant que l’affaire n’est pas soumise à un panel d’audience au cours d’une audience publique.
108. La décision quant au caractère excessif du prix d’un médicament breveté est prise par le panel d’audience seulement, après la tenue de l’audience publique.
X. Processus d’audience relativement aux prix excessifs et recours
109. Les audiences du CEPMB sont publiques. Lors d’une audience, un panel d’audience, composé d’au moins deux membres du Conseil, entend les observations et les éléments de preuve des parties. Le panel détermine si un médicament breveté est ou a été vendu à un prix excessif sur n’importe quel marché canadien en tenant compte des renseignements disponibles concernant les facteurs énoncés à l’article 85 de la Loi.
110. Pour obtenir de plus amples renseignements sur les audiences, veuillez consulter les Règles de pratique et de procédure du CEPMB, l’ensemble normalisé et publié de procédures que tous les participants à des audiences devant le Conseil doivent suivre. Les Règles établissent les procédures du CEPMB conformément à l’exigence prévue dans la Loi selon laquelle le Conseil doit résoudre les questions soumises à son attention sans formalisme, en procédure expéditive, dans la mesure où les circonstances et l’équité le permettent. Les instructions relatives à la pratique et d’autres renseignements au sujet des audiences en cours ou passées sont à la disposition du public sur le site Web du CEPMB.
111. En vertu de la Loi, le CEPMB est habilité à rendre des ordonnances correctives lorsqu’il est déterminé, à la suite d’une audience, qu’un breveté (ou un ancien breveté) vend ou a vendu un médicament breveté à un prix excessif sur un marché canadienNote de bas de page 15
112. En termes généraux, le CEPMB a le pouvoir d’exercer des deux grandes voies de recours après une audience: i) des ordonnances enjoignant au breveté de faire baisser le prix maximum auquel il vend le médicament breveté sur ce marché à un niveau que le Conseil ne juge pas excessif ; et ii) des ordonnances enjoignant au breveté de compenser le montant des recettes excessives qu’il estime avoir tirées de la vente du médicament breveté à un prix excessif soit a) en réduisant le prix auquel il vend le médicament breveté ; (b) en réduisant le prix auquel le breveté vend un autre médicament auquel se rattache une de ses inventions brevetées ; ou (c) en versant à Sa Majesté du chef du Canada un montant spécifié dans l’ordonnance.
113. Si le panel d’audience conclut que le breveté ou l’ancien breveté s’est livré à une politique de vente du médicament breveté à un prix excessif, il peut, par ordonnance, lui enjoindre de compenser, selon lui, jusqu’au double de l’excédent des recettes procuré par la vente du médicament breveté au prix excessif. Le Conseil examinera l’étendue et la durée des ventes du médicament breveté au prix excessif lorsqu’il arrivera à cette conclusion et rendra l’ordonnance.
XI. Audience pour défaut de présenter les renseignements requis
114. Lorsque le personnel estime qu’un breveté a omis ou a refusé de fournir au CEPMB les renseignements sur les prix, les ventes ou les recettes, ou d’autres renseignements semblables exigés par la loi, le directeur exécutif peut recommander au président de tenir une audience publique pour déterminer si le breveté a bel et bien manqué aux exigences en matière de présentation de renseignements prévues par la Loi et le Règlement. Si, à la suite d’une audience, le panel d’audience conclut que le breveté a manqué à ces exigences, il peut, par ordonnance, lui enjoindre de fournir au CEPMB les renseignements et les documents requis en vertu de l’article 81 ou 88 de la Loi.
115. En outre, aux termes du paragraphe 76.1(1) de la Loi, quiconque contrevient aux exigences en matière de présentation de renseignements énoncées aux articles 80, 81, 82 ou 88 ou à une ordonnance prise sous le régime de l’un ou l’autre de ces articles commet une infraction et encourt, sur déclaration de culpabilité par procédure sommaire, une amende ou un emprisonnement.
XII. Plaintes
116. Une personne ou un groupe qui estime que le prix d’un médicament breveté est excessif peut déposer une plainte au CEPMB par téléphone, par écrit ou par voie électronique. Les coordonnées à utiliser se trouvent sur la page « Comment déposer une plainte » du site Web du CEPMB.
117. Toute plainte déclenche une enquête sur le prix du médicament breveté visé par la plainte. Le plaignant ne participe pas à l’enquête ni aux audiences qui en résultent (à moins qu’il ne présente une demande pour participer aux audiences à titre d’intervenant). Le plaignant n’est pas tenu de fournir des documents ou des éléments de preuve au CEPMB. Les enquêtes sont fondées sur les documents fournis par le breveté ou obtenus autrement par le personnel.
118. En raison des limites en matière de communication énoncées aux articles 87 et 88 de la Loi sur les brevets et dans la Loi sur l’accès à l’information, le plaignant est informé du résultat de l’enquête uniquement si le processus mène à un ECV ou à un avis d’audience.
XIII. Annexes
A. Comparaison selon la catégorie thérapeutique des prix nationaux (CCTn) et comparaison selon la catégorie thérapeutique des prix internationaux (CCTi)
Test de la CCTn
Tel qu’il est décrit dans la section V des présentes Lignes directrices, le test de la comparaison selon la catégorie thérapeutique des prix nationaux (« CCTn ») sert à calculer le seuil maximum du prix d’un médicament breveté dans certains cas. Le test de la CCTn permet de comparer le prix d’un médicament breveté avec le prix courant d’autres médicaments sélectionnés au terme d’un examen scientifique.
Pour les médicaments de catégorie I dont la taille réelle du marché dépasse 50 millions de dollars, le PEM basé sur la CCTn est fixé selon les planchers indiqués dans le tableau ci-dessous.
| Niveau de critères thérapeutiques (Voir annexe E – Processus d’examen scientifique) |
Plancher de réduction de la CCTn |
|---|---|
| Niveau I | 20 % du PCM (la CCTn ne s’applique pas) |
| Niveau II | 30 % |
| Niveau III | 40 % |
| Niveau IV | 50 % |
Sélection des médicaments aux fins de comparaison
La sélection des médicaments à utiliser aux fins de comparaison est fondée sur le Système de classification anatomique thérapeutique chimique (ATC) du Centre collaborateur de l’Organisation mondiale de la Santé (OMS) pour la méthodologie sur l’établissement des statistiques concernant les produits médicamenteux.
Les médicaments utilisés aux fins de comparaison appartiennent généralement à la sous-catégorie du système ATC située immédiatement au-dessus de la substance chimique simple. Il s’agit habituellement du quatrième niveau de sous-catégorie, mais il peut également s’agir de la sous-catégorie supérieure suivante ou d’une autre sous-catégorie. Dans certains cas, la sélection peut devoir être faite au cinquième niveau, soit au niveau de la substance chimique simple.
Un médicament de la même catégorie thérapeutique du système ATC que le médicament à l’étude peut être omis s’il ne se prête pas à la comparaison. Par exemple, un médicament comportant une indication ou un usage principal autre que l’indication ou l’usage principal du médicament breveté à l’étude peut être omis de la comparaison.
Tous les médicaments sélectionnés aux fins de comparaison qui comportent la même indication approuvée ou le même usage que l’indication pertinente du médicament breveté à l’étude sont inclus dans l’examen. Cet examen repose sur les recherches effectuées par le personnel et peut comprendre d’autres recherches menées par un centre d’information sur les médicaments ou une analyse des données probantes présentées par les brevetés.
Lorsqu’il y a plusieurs vendeurs d’un médicament identifié en tant que médicament comparable, le prix le plus bas des médicaments sera identifié. Le prix final de la comparaison selon la catégorie thérapeutique est le prix le plus élevé de l’ensemble des médicaments comparables pour le PCM et la médiane pour le PEM.
Lorsque le médicament breveté est une nouvelle forme posologique ou concentration du même ingrédient médicinal contenu dans un ou plusieurs médicaments existants, les médicaments existants qui sont offerts dans la même forme posologique ou dans une forme posologique comparable et qui comportent la même indication ou le même usage servent de médicaments de comparaison, et ce, que les régimes posologiques soient identiques ou diffèrent sensiblement.
Lorsque le produit est une combinaison de médicaments qui sont chacun vendus au Canada et qui comportent tous la même indication ou le même usage, les médicaments de comparaison sont limités aux médicaments composants.
Régimes posologiques comparables
Le régime posologique comparable utilisé aux fins de comparaison correspond normalement au régime posologique maximal habituellement recommandé dans la monographie de produit (ou dans des renseignements semblables), et il tient compte des variables cliniques pertinentes. La concentration est déterminée en fonction du régime posologique du médicament à l’étude. En règle générale, un régime posologique fondé sur un traitement s’appliquera aux indications aiguës, tandis qu’un régime quotidien (selon la dose d’entretien) sera utilisé pour les indications chroniques.
Sources des prix
Les brevetés ne présentent pas de renseignements sur le prix des médicaments sélectionnés aux fins de comparaison avec un médicament breveté. On consulte d’abord les Listes de médicaments des régimes provinciaux pour obtenir le prix d’un médicament de comparaison dans le cadre du test de la CCTn. Pour chaque médicament de comparaison sélectionné, on utilise le prix public le plus bas. Le personnel peut décider d’exclure du test de la CCTn un médicament (breveté ou non) sélectionné aux fins de comparaison s’il a des raisons de croire que le médicament est vendu à un prix excessif.
Test de la CCTn
Après avoir sélectionné les médicaments qui se prêtent à la comparaison et déterminé le prix public le plus bas de chaque médicament, on calcule le coût d’un traitement comparable pour chaque médicament. Les différents coûts de traitement sont triés, et le plafond ou la médiane, établi(e). Lorsqu’un nombre pair de médicaments est utilisé aux fins de comparaison, la médiane correspond à la moyenne simple des deux coûts de traitement du milieu. Le coût médian de traitement est ensuite divisé par le nombre d’unités constituantes du traitement comparable pour le médicament à l’étude, ce qui permet d’établir un prix unitaire.
Test de la CCTI
Tel qu’il est décrit dans la section V des présentes Lignes directrices, le test de la comparaison selon la catégorie thérapeutique des prix internationaux (« CCTi ») peut être utilisé dans certains cas. Le test de la CCTi permet de comparer le prix d’un médicament breveté avec le prix courant d’autres médicaments sélectionnés au terme d’un examen scientifique aux fins de comparaison dans les onze pays de comparaison énumérés dans le Règlement.
Sélection des médicaments aux fins de comparaison et régimes posologiques comparables
Le test de la CCTi se fonde sur les mêmes médicaments sélectionnés aux fins de comparaison et les mêmes régimes posologiques comparables que ceux utilisés pour le test de la CCTn, conformément aux procédures établies précédemment. Lorsqu’aucun médicament de comparaison n’a été trouvé au pays, le personnel peut déterminer si d’autres médicaments comportent la même indication approuvée ou le même usage que le médicament à l’étude dans l’un des pays de comparaison.
Sources des prix
Les brevetés ne présentent pas de renseignements sur les prix internationaux des médicaments sélectionnés à l’étranger aux fins de comparaison avec un médicament breveté. On emploie le prix départ usine accessible au public des médicaments sélectionnés aux fins de comparaison dans le cadre du test de la CCTi. Pour chaque médicament de comparaison sélectionné, on utilise le prix public le plus bas.
Le personnel peut décider d’exclure du test de la CCTi un médicament (breveté ou non) sélectionné aux fins de comparaison s’il a des raisons de croire que le médicament est vendu à un prix excessif.
Test de la CCTi
Après avoir sélectionné les médicaments qui se prêtent à la comparaison et déterminé le prix public le plus bas de chaque médicament, on calcule le coût d’un traitement comparable pour chaque médicament dans chaque pays de comparaison. Les différents coûts de traitement sont triés, et la médiane de chaque pays est établie. Lorsqu’un nombre pair de médicaments est utilisé aux fins de comparaison, la médiane correspond à la moyenne simple des deux coûts de traitement du milieu. Ces médianes sont triées, et une « médiane des médianes » est définie. Le coût médian de traitement est ensuite divisé par le nombre d’unités constituantes du traitement comparable pour le médicament à l’étude, ce qui permet d’établir un prix unitaire. Les devises étrangères sont converties en dollars canadiens selon la méthode décrite à la section V des présentes Lignes directrices.
B. Test de la relation raisonnable et formes posologiques comparables
Test de la relation raisonnable
Le test de la relation raisonnable (RR) peut servir à déterminer le PCM d’une nouvelle concentration supplémentaire d’un médicament breveté qui est déjà offert dans une ou plusieurs autres concentrations, pourvu que la nouvelle concentration comporte le même ingrédient médicinal, la même indication, le même régime posologique et la même forme posologique (ou une forme posologique comparable) que la ou les concentrations existantes. Il n’y a pas de test de la RR tant qu’un PCM n’est pas fixé pour la concentration de référence.
PCM
Lorsqu’un médicament breveté n’est pas ou n’est plus dans la période provisoire, la première concentration qui a satisfait aux critères de transition du PCMp au PCM deviendra la concentration de référence. S’il existe plusieurs concentrations qui répondent à ce critère au cours d’une période de référence, la concentration de référence sera choisie parmi celles-ci sur la base de considérations scientifiques. Une fois la concentration de référence définie, le plafond pour les nouvelles concentrations supplémentaires sera établi sur la base de la relation proportionnelle entre les concentrations, sous réserve d’un plafond du PEPI pour la nouvelle concentration supplémentaire. Plus précisément, le PCM de la nouvelle concentration supplémentaire plus élevée sera fixé de manière à être équivalent au prix par unité standard de la concentration de référence. Toutefois, le PCM pour la nouvelle concentration supplémentaire inférieure ne sera pas fixé à un niveau supérieur au prix de la concentration de référence, à condition que cela n’entraîne pas un prix pour la nouvelle concentration supplémentaire supérieur au PEPI.
PEM
Le PEM est calculé comme une réduction par rapport au PCM. Par conséquent, la demande de test de la RR pour une nouvelle concentration supplémentaire d’un médicament breveté soumis au PEM sera faite au niveau du PCM pour la concentration de référence et le pourcentage de réduction de prix correspondant sera reporté sur les nouvelles concentrations supplémentaires du médicament breveté.
PEM[A]
Le PEM[A] est un ajustement basé sur le PEM en fonction de la taille du marché et changera donc en conséquence.
Formes posologiques comparables
Les formes posologiques ci-après sont considérées comme étant des formes posologiques comparables pour les besoins du test de la RR. Les formulations d’un même groupe sont considérées comme étant comparables, mais les formes posologiques de groupes différents ne le sont pas.
Topique (T)
- Aérosol
- Aérosol (mousse)
- Crème
- Disque (à libération prolongée)
- Disque
- Pansement
- Gel
- Gel (à libération contrôlée)
- Liposomes
- Liquid
- Lotion
- Onguent
- Tampon
- Peinture
- Pâte
- Timbre
- Timbre (à libération prolongée)
- Crayon
- Plâtre
- Poudre
- Shampoing
- Savon en pain
- Solution
- Éponge
- Vaporisateur
- Vaporisateur (valve à poche)
- Vaporisateur (à dose mesurée)
- Bâtonnet
- Bandelette
- Écouvillon
- Teinture
Nasal (N) ou pulmonaire (P)
- Aérosol
- Aérosol à dose mesurée
- Gouttes
- Gaz
- Préparation à dose mesurée
- Poudre
- Poudre (à dose mesurée)
- Solution
- Solution (à libération prolongée)
- Vaporisateur
- Vaporisateur (à dose mesurée)
- Bâtonnet
Solide administré par voie orale (S)
- Barre à croquer
- Caplet
- Capsule
- Granules effervescents
- Poudre effervescente
- Comprimé effervescent
- Pellicule (soluble)
- Globules
- Granules
- Gomme à mâcher
- Pastille
- Caplet à libération modifiée
- Capsule à libération modifiée
- Comprimé à libération modifiée
- Granulés
- Morceau à croquer
- Poudre (à libération prolongée)
- Bandelette
- Comprimé
- Comprimé (à croquer)
- Comprimé (à dissolution orale)
- Comprimé pour suspension
- Cachet
Liquide administré par voie orale (L)
- Gouttes
- Solution buvable
- Émulsion
- Gel
- Granules pour solution
- Granules pour suspension
- Granules pour suspension (à libération retardée)
- Granules pour suspension (à libération prolongée)
- Liquide
- Liquide à libération modifiée
- Poudre (à libération prolongée)
- Poudre pour solution
- Poudre pour suspension
- Solution
- Solution (à libération prolongée)
- Vaporisateur
- Suspension
- Suspension (à libération prolongée)
- Sirop
- Sirop (à libération prolongée)
- Tisane
- Teinture
Vaginal (V)
- Cône
- Crème
- Douche
- Mousse
- Gel
- Gel (à libération contrôlée)
- Implant
- Insert
- Insert (à libération prolongée)
- Suppositoire
- Granulés
- Anneau (à libération lente)
- Éponge
- Suppositoire
- Suppositoire (à libération soutenue)
- Tampon
- Comprimé vaginal
- Comprimé vaginal (effervescent)
Parentéral (J)
- Bolus
- Implant
- Trousse
- Liposomes
- Injection à libération modifiée
- Pellet (implantable)
- Poudre pour solution
- Poudre pour suspension (à libération soutenue)
- Solution
- Solution (à libération prolongée)
- Suspension pour émulsion
- Suspension (à libération prolongée)
Otique (E) ou ophtalmique (Y)
- Gouttes
- Gel
- Gel (à libération contrôlée)
- Implant
- Insert
- Insert (à libération prolongée)
- Liquide
- Dispositif oculaire à libération modifiée
- Onguent
- Poudre pour solution
- Solution
- Solution (à libération prolongée)
- Suspension
Rectal (R)
- Crème
- Lavement
- Mousse
- Insert
- Onguent
- Suppositoire
- Bâtonnet
- Suppositoire
- Suppositoire (à libération soutenue)
- Suspension
- Suspension (à libération prolongée)
Dentaire, sublingual ou buccal (M)
- Émulsion
- Pellicule (soluble)
- Soie dentaire
- Gel
- Gel (à libération contrôlée)
- Gomme à mâcher
- Pastille
- Pompe à dose mesurée
- Comprimé buccal à libération modifiée
- Rince-bouche (gargarisme)
- Pâte
- Poudre effervescente
- Poudre pour suspension
- Solution
- Solution (à libération prolongée)
- Vaporisateur – buccal
- Vaporisateur – sublingual
- Bâtonnet
- Bandelette
- Comprimé sublingual
- Suspension
- Suspension (à libération prolongée)
- Écouvillon
- Comprimé (dissolution orale)
- Comprimé
- Dentifrice
- Poudre dentifrice
- Cachet
C. Évaluation de la valeur pharmacoéconomique
Comme il est décrit dans la section V des présentes Lignes directrices, l’évaluation de la valeur pharmacoéconomique sert à calculer le PEM d’un médicament. Le Règlement exige que les brevetés présentent des renseignements relatifs à toutes les analyses pharmacoéconomiques d’un médicament breveté préparées par des organismes canadiens financés par l’État si le coût de traitement calculé au prorata du médicament breveté au cours d’une période de 12 mois est supérieur ou égal à 50 % du PIB par habitant au moment de la publication de l’analyse.
Les rapports pharmacoéconomiques du Programme commun d’évaluation des médicaments (PCEM) et les rapports finaux d’orientation économique du Programme pancanadien d’évaluation des anticancéreux (PPEA) de l’Agence canadienne des médicaments et des technologies de la santé (ACMTS) sont les sources habituelles d’analyses pharmacoéconomiques utilisées pour les besoins de l’évaluation de la valeur pharmacoéconomique. Les analyses préparées par l’Institut national d’excellence en santé et services sociaux (INESSS) dans ses Évaluations aux fins d’inscription ou par le Comité consultatif national de l’immunisation (CCNI) sont également étudiées.
Dans le cadre de l’évaluation de la valeur pharmacoéconomique, les calculs du personnel sont fondés sur la nouvelle analyse du scénario de référence menée par l’organisme public, par opposition à l’analyse effectuée à l’aide du modèle de scénario de référence présenté par le breveté. Il s’agit d’un modèle d’analyse coût-utilité dans lequel les résultats en matière de santé sont exprimés sous forme d’AVAQ. Le rapport coût-utilité différentiel (« RCUD ») mesuré en années de vie ajustées en fonction de la qualité (« AVAQ ») pour chaque indication du médicament breveté sera calculé à partir des analyses coût-utilité présentées par le breveté.
Si le rapport de l’organisme ne contient pas d’analyse coût-utilité, mais un modèle de minimisation des coûtsNote de bas de page 16, une CCTn pourra être effectuée. Dans ce cas, le PEM sera basé sur la médiane de la CCTn, sous réserve d’un plancher de réduction du PCM de 50 % et d’ajustements en fonction de la taille du marché, comme indiqué à l’annexe D. Pour les médicaments brevetés à indications multiples, l’évaluation pharmacoéconomique pour l’indication pertinente, comme déterminée par la procédure de la section V des présentes Lignes directrices, sera utilisée.
Le RCUD sera comparé au seuil de la valeur pharmacoéconomiqueNote de bas de page 17 (« SVP ») applicable, sur la base des niveaux de critères thérapeutiques indiqués dans le tableau ci-dessous. La PEM est basée sur le PP (le prix auquel le RCUD du médicament breveté serait équivalent au SVP), à condition que si le PP entraîne une réduction en dessous du plancher indiqué dans le tableau, le PEM soit fixé de manière à ce que la réduction de prix ne soit pas supérieure au plancher applicable.
Le CEPMB compte sur les organismes pour publier dans leurs rapports des estimations qui lui permettent de calculer le PP.
| Niveau de critères thérapeutiques (voir annexe E – Processus d’examen scientifique) |
SVP | Plancher de réduction du PCM |
|---|---|---|
| Niveau I | 200 k$/ AVAQ | 20 % |
| Niveau II | 150 k$/ AVAQ | 30 % |
| Niveau III | 150 k$/ AVAQ | 40 % |
| Niveau IV | 150 k$/ AVAQ | 50 % |
| L’analyse pharmacoéconomique est une minimisation des coûts | Médiane de la CCTn soumise à un plancher de 50 % | |
| Aucune évaluation pharmacoéconomique | 50 % du PCM | |
D. Méthode de rajustement en fonction de la taille du marché
Comme il est décrit dans la section V des Lignes directrices, un rajustement de la taille du marché est appliqué aux médicaments brevetés de la catégorie I lorsque les recettes réelles dépassent 50 millions de dollars pour toutes les formes posologiques et toutes les concentrations du médicament breveté (c.-à-d. tous les DIN combinés). On applique ce rajustement chaque année conformément au tableau suivant :
| Recettes annuelles | PEM | Facteur de rajustement du PCM supplémentaire |
Facteur de rajustement des prix‡ utilisé pour le calcul du facteur de rajustement du PCM |
|---|---|---|---|
| < 12 M$ | PCM | 0 % | 0 % |
| 12 M$ à 50 M$ | Le plus grand entre le PP et le plancher | (12 M$ - 12 M$ * (PEM/PCM)) / Recettes + (PEM/PCM) | |
| 50 M$ à 100 M$ | -25 % | (21,5 M$ - 9 M$ * (PEM/PCM)) / Recettes + (1 - 25 %) * (PEM/PCM) | |
| >100 M$ | -35 % | (32 M$ - 7,8 M$ * (PEM/PCM)) / Recettes + (1 - 35 %) * (PEM/PCM) |
‡ Plus bas entre le PCM et le prix courant
| Recettes annuelles | PEM | Facteur de rajustement du PCM supplémentaire | Facteur de rajustement des prix‡ utilisé pour le calcul du facteur de rajustement du PCM |
|---|---|---|---|
| < 50 M$ | PCM | 0 % | 0 % |
| $50M-$100M | Le plus bas entre le PCM et la médiane de la CCTn | -25 % | (50 M$ - 37,5 M$ * (PEM/PCM)) / Recettes + (1 - 25 %) * (PEM/PCM) |
| > 100 M$ | -35 % | (56,7 M$ - 32,5 M$ * (PEM/PCM)) / Recettes + (1 - 35 %) * (PEM/PCM) |
‡ Plus bas entre le PCM et le prix courant
Par exemple, un médicament breveté nécessitant un RCUD et réalisant entre 50 et 100 millions de dollars de recettes sur la base des unités vendues au PEM fixé par le PP aura son PEM[A] fixé comme suit :
PEM[A] = PCM * (21,5 M$ - 9 M$ * (PEM/PCM)) / Recettes + (1 - 25 %) * (PEM/PCM)
Après le rajustement initial de la taille du marché, le PEM d’un médicament breveté ne sera réajusté qu’à la suite d’une augmentation des unités annuelles vendues. Le PEM d’un médicament breveté ne sera pas réajusté à la suite d’une diminution des unités annuelles vendues, ou si ses recettes réelles réalisées tombent dans un niveau inférieur.
E. Processus d’examen scientifique
L’examen scientifique du CEPMB est un processus fondé sur des données probantes qui détermine les critères thérapeutiques, les comparateurs thérapeutiques appropriés et le régime posologique comparable d’un médicament breveté afin d’éclairer le processus d’examen du prix.
L’examen scientifique d’un nouveau médicament breveté est basé sur des renseignements provenant de diverses sources telles que celles qui sont présentées ci-dessous.
- Présentation du breveté – Le breveté peut fournir au personnel un bref document de présentation qui explique clairement la raison d’être de sa proposition quant aux critères thérapeutiques, aux médicaments retenus pour la comparaison et aux régimes posologiques comparables. Des renseignements sur les procédures de présentation sont disponibles sur l’outil de soumission en ligne du CEPMB.
- Recherches menées par un centre d’information sur les médicaments (CIM) – Le personnel peut faire appel aux services de divers centres d’information sur les médicaments pour obtenir des renseignements scientifiques, tels que des renseignements sur les essais cliniques, les directives de pratique clinique, etc. La base de l’examen par le CIM est la monographie du produit ou des renseignements similaires à ceux contenus dans une monographie de produit si un avis de conformité (AC) n’a pas été accordé.
- Recherches menées par le personnel – Le personnel peut également mettre à jour les recherches et compléter les données et les données probantes du breveté et du CIM en utilisant d’autres sources.
- Groupe consultatif sur les médicaments pour usage humain (GCMUH) – Le personnel peut, de façon ponctuelle, consulter le GCMUH quant aux aspects cliniques des renseignements scientifiques à l’étude.
Critères thérapeutiques
Les critères thérapeutiques pris en compte par le personnel pour les nouveaux médicaments brevetés sont indiqués ci-dessous. Pour les médicaments brevetés ayant plusieurs indications approuvées ou plusieurs usages, l’évaluation des critères thérapeutiques sera basée sur l’indication pertinente :
Critères thérapeutiques de niveau I
Le médicament breveté est le premier médicament de sa catégorie disponible sur le marché canadien qui est efficace sur le plan clinique pour le traitement d’une maladie ou pour une indication donnée. Les améliorations qui ont une incidence sur le plan clinique sont définies comme étant celles qui contribuent à augmenter la qualité de vie, à diminuer la mortalité ou à réduire de façon marquée la gravité de la maladie ou l’utilisation des soins de santé, de préférence sur la base de données probantes de haute qualité utilisant des paramètres cliniques concrets plutôt que des résultats dévaluation de substitution. Un gain élevé des AVAQ est normalement associé aux médicaments à ce niveau.
Critères thérapeutiques de niveau II
Le médicament breveté apporte une amélioration considérable des bienfaits thérapeutiques, par rapport à d’autres médicaments vendus au Canada, d’une manière efficace sur le plan clinique. Les améliorations qui ont une incidence sur le plan clinique sont définies comme étant celles qui contribuent à augmenter la qualité de vie, à diminuer la mortalité ou à réduire de façon marquée la gravité de la maladie ou l’utilisation des soins de santé, de préférence sur la base de données probantes de haute qualité fondées sur un comparateur actif utilisant des paramètres cliniques concrets plutôt que des résultats dévaluation de substitution. Une méta-analyse de réseau de haute qualité peut également être envisagée. Un gain élevé des AVAQ est normalement associé aux médicaments à ce niveau.
Critères thérapeutiques de niveau III
Le médicament breveté apporte une amélioration absolue modérée des bienfaits thérapeutiques, par rapport à d’autres médicaments vendus au Canada. Le médicament peut a) entraîner une augmentation de l’efficacité cliniquement pertinente; b) être associé à une réduction de l’incidence ou du degré des effets indésirables importants; c) être associé à des caractéristiques de facilité d’utilisation accrue pertinentes sur le plan clinique (p. ex. voie d’administration, commodité, conformité accrue, etc.), cependant, ces améliorations peuvent avoir une incidence clinique valable limitée ou être basées sur des données probantes cliniques de moindre qualité. Les médicaments brevetés à ce niveau sont normalement associés à des gains d’AVAQ graduels modérés avec un degré de certitude relativement élevé ou à des gains d’AVAQ élevés avec un degré de certitude relativement faible.
Critères thérapeutiques de niveau IV
Le médicament breveté n’apporte aucune amélioration ou une légère amélioration par rapport aux autres médicaments vendus au Canada. Par ailleurs, ou en plus, le médicament breveté dispose de très peu (ou pas) de données probantes cliniques solides pour établir clairement le degré d’amélioration thérapeutique pertinente sur le plan clinique par rapport à d’autres médicaments vendus au Canada. À ce niveau, les médicaments sont associés à des gains d’AVAQ limités ou inexistants, ou à une incertitude importante due à des données probantes de mauvaise qualité.
Sélection des médicaments aux fins de comparaison
Le Système de classification anatomique thérapeutique chimique (ATC) du Centre collaborateur de l’Organisation mondiale de la Santé (OMS) pour la méthodologie sur l’établissement des statistiques concernant les produits médicamenteux peut être utilisé pour la sélection des médicaments à utiliser aux fins de comparaison. Les médicaments utilisés aux fins de comparaison appartiennent généralement à la sous-catégorie du système ATC située immédiatement au-dessus de la substance chimique simple. Il s’agit habituellement du quatrième niveau de sous-catégorie, mais il peut également s’agir de la sous-catégorie supérieure suivante ou d’une autre sous-catégorie. Dans certains cas, la sélection peut devoir être faite au cinquième niveau, soit au niveau de la substance chimique simple. Un médicament de la même catégorie thérapeutique du système ATC que le médicament à l’étude peut être omis s’il ne se prête pas à la comparaison. Par exemple, un médicament comportant une indication ou un usage principal autre que l’indication ou l’usage principal du médicament breveté à l’étude peut être omis de la comparaison.
Tous les médicaments utilisés à des fins de comparaison auront généralement la même indication ou utilisation approuvée que le médicament breveté qui est examiné.
Si la CCTi inclut des médicaments ayant la même indication ou utilisation approuvée que le médicament breveté examiné, cette information peut être prise en compte.
Lorsque le médicament breveté est une nouvelle forme posologique ou concentration du même ingrédient médicinal contenu dans un ou plusieurs médicaments existants, les médicaments existants qui sont offerts dans la même forme posologique ou dans une forme posologique comparable et qui comportent la même indication ou le même usage servent de médicaments de comparaison. Cela s’appliquera indépendamment du fait que le régime posologique soit le même ou diffère sensiblement.
Lorsque le produit est une combinaison de médicaments qui sont chacun vendus au Canada et qui comportent tous la même indication ou le même usage, les produits de comparaison sont limités aux médicaments de la composition.
Détermination des régimes posologiques comparables
Le régime posologique comparable utilisé aux fins de comparaison correspond normalement au régime posologique maximum habituellement recommandé dans la monographie de produit (ou dans des renseignements semblables), et il tient compte des variables cliniques pertinentes. La concentration est déterminée en fonction du régime posologique du médicament à l’étude. En règle générale, un régime posologique fondé sur un traitement s’appliquera aux indications aiguës, tandis qu’un régime quotidien (selon la dose d’entretien) sera utilisé pour les indications chroniques.
F. Sommaire des calendriers de mise en conformité
| Catégorie du médicament breveté | PCMp | PCM (au moins 3 ans ou 5 pays) | PEM | Réévaluation | Examen de la conformité |
|---|---|---|---|---|---|
| Médicament bénéficiant de droits acquis | S. O. | Plus bas entre le PMNE et le PEPI | S. O. | Si PCM > PEPI | 2 périodes de référence (déc. 2021) |
| Élargissement de gamme d’un médicament bénéficiant de droits acquis | Plus bas entre le prix courant et le PEPI | Plus bas entre le prix courant et le PEPI | S. O. | Si PCM > PEPI | 1 période de référence |
| Médicament de transition | Plus bas entre le prix courant et la MPI | Plus bas entre le prix courant, le PMNE/ PMMP et la MPI | S. O. | Si PCM > MPI ± 10 % | 2 périodes de référence (déc. 2021) |
| Nouveau médicament de catégorie I | MPI | Plus bas entre le prix courant et la MPI (ou CCTn/CCTi si aucun PI*) | Plus bas entre le PCM et le PEM (X % déduit du PCM selon le PP, CCTn sous réserve du plancher du NCT et du rajustement en fonction de la taille du marché, le cas échéant) | Déclenchement dû à la taille du marché Preuves thérapeutique/ÉTS mises à jour Nouvelle indication pertinente |
PCM - 1 période de référence PEM - 2 périodes de référence |
| PCM - 1 période de référence PEM - 2 périodes de référence | MPI | RR ou prix echelonné à concurrence du PEPI | Même X % déduit du PCM que la concentration de référence | Comme ci-dessus pour tous les DIN | PCM - 1 période de référence PEM - 2 périodes de référence |
| Nouveau médicament de catégorie II | MPI | Plus bas entre le prix courant et la MPI (ou CCTn/CCTi si aucun PI) | S. O. | S. O. | 1 période de référence |
| Élargissement de gamme d’un nouveau médicament de catégorie II | MPI | RR ou prix echelonné à concurrence du PEPI | S. O. | S. O. | 1 période de référence |
*PI : prix international
Document d’information 2020
Document d’information 2020
Sur les Lignes directrices provisoires de juin 2020 : Explication des changements par rapport aux Lignes directrices provisoires de novembre 2019

PDF version (738 Ko)
Table des matières
- Les lignes directrices du CEPMB
- La consultation relative aux Lignes directrices du CEPMB
- Main tendue aux Canadiens : Aperçu du processus de consultation du CEPMB
- Explication des changements apportés aux Lignes directrices provisoires
- 1. Comparaison selon la catégorie thérapeutique des prix nationaux (CCTn)
- 2. Test du prix courant maximum (PCM) – Comparaison des prix internationaux
- 3. Classification d’un médicament breveté de catégorie I
- 4. Seuil de la valeur pharmacoéconomique (VP) et prise en compte des comparateurs thérapeutiques – Médicaments à coût élevé
- 5. Rajustements en fonction de la taille du marché
- 6. Confidentialité du prix escompté maximum (PEM)
- 7. Examen réglementaire des médicaments biosimilaires et génériques brevetés
- 8. Autres questions soulevées par les intervenants et abordées par les changements décrits précédemment
Les lignes directrices du CEPMB
Le Conseil d’examen du prix des médicaments brevetés (CEPMB) est un organe quasi judiciaire dont le mandat réglementaire est d’empêcher les titulaires de brevets pharmaceutiques de faire payer aux consommateurs des prix excessifs durant la période de monopole prévue par la loi. Cet organe est né de la crainte que le renforcement de la protection des médicaments par les brevets n’entraîne une hausse inacceptable des prix au détriment des consommateurs.
Aux termes du paragraphe 96(4) de la Loi sur les brevets, le CEPMB formule des lignes directrices (« Lignes directrices ») destinées à assurer, pour les titulaires de brevets, la transparence et la prévisibilité du processus habituellement suivi par les fonctionnaires du CEPMB (« personnel ») pour déterminer si le prix d’un médicament breveté semble excessif sur un marché canadien.
Avant d’élaborer ou de modifier ses Lignes directrices, le CEPMB est tenu de lancer des consultations aux termes du paragraphe 96(5) de la Loi sur les brevets. Le CEPMB prend très au sérieux ses obligations à cet égard et a adopté toutes les mesures nécessaires pour assurer un processus de consultation valable reposant sur un dialogue ouvert et transparent avec les Canadiens.
La consultation relative aux Lignes directrices du CEPMB
Le 1er janvier 2021, le Règlement sur les médicaments brevetés modifié (« Règlement modifié »)Note de bas de page 1 entrera en vigueur. Les modifications apportées aux Lignes directrices du CEPMB concernant les prix sont nécessaires pour que le Règlement modifié soit mis en œuvre et que le CEPMB puisse réglementer les prix plafonds des médicaments brevetés en passant à une approche davantage axée sur les risques. Le 21 novembre 2019, le CEPMB a publié une série provisoire de nouvelles Lignes directrices (« la version provisoire de novembre 2019 ») aux fins de consultation avec les intervenants et le public. Un grand nombre de commentaires ont été formulés et les observations écrites reçues peuvent être consultées sur le site web du CEPMB.
Le CEPMB a apporté un certain nombre de changements de fond à la version provisoire de novembre 2019 en réponse aux commentaires reçus durant la période de consultation. Ces changements se retrouvent dans une deuxième série provisoire de Lignes directrices publiées le 19 juin 2020 (la version provisoire de juin 2020), à l’égard de laquelle le CEPMB lance à présent des consultations pour une période de 30 joursNote de bas de page 2. Le présent document a pour objet d’expliquer en termes généraux la nature des changements et leur justification. Il est destiné à être lu comme un document d’accompagnement de la version provisoire de juin 2020 et à aider le lecteur à comprendre les changements. Un document semblable avait accompagné la publication des Lignes directrices provisoires de novembre 2019 et il peut être consulté sur le site web du CEPMB. Comme c’est le cas des Lignes directrices, ce document d’information n’est contraignant ni pour le CEPMB ni pour les titulaires de brevets.
La publication des Lignes directrices provisoires de juin 2020 et la consultation de 30 jours qui lui succédera sont la dernière étape d’un processus qui a débuté par la publication du document de discussion sur la modernisation des Lignes directrices du CEPMB en juin 2016, et suit la feuille de route de réformes exposées dans son Plan stratégique 2015-2018. Dès le départ, le processus de consultation du CEPMB était conforme aux meilleures pratiques gouvernementales visant à assurer un maximum d’inclusion, de clarté et de discussions productives.
La date limite de présentation au CEPMB d’observations écrites concernant les Lignes directrices provisoires de juin 2020 est le 20 juillet 2020. Le CEPMB reste à l’écoute de tous les Canadiens intéressés par la manière dont il exerce son mandat réglementaire et qui ont une opinion sur le sujet. Le CEPMB attend avec intérêt de recevoir des commentaires valables et constructifs dans le cadre de ce processus de consultation en vue de publier des Lignes directrices définitives à l’automne 2020.
Main tendue aux Canadiens : Aperçu du processus de consultation du CEPMB
La publication des Lignes directrices provisoires de novembre 2019 a été suivie par une consultation de 85 jours durant laquelle le CEPMB s’est efforcé d’interagir avec autant d’intervenants que possible selon différentes modalités, y compris des rencontres dans tout le pays, des séances d’information d’une journée à Ottawa avec des représentants de l’industrie et de la société civile, des webinaires et des groupes de travail.
Voici un résumé des efforts de consultation déployés par le CEPMB après la publication des Lignes directrices provisoires de novembre 2019 :
- Groupes de travail avec des partenaires de la santé : Deux séances d’information d’une journée en novembre 2019 et en janvier 2020 avec des représentants de partenaires de la santé, y compris les régimes publics d’assurance-médicaments, l’Agence canadienne des médicaments et des technologies de la santé (ACMTS), l’Institut national d’excellence en santé et services sociaux (INESSS), l’Alliance pancanadienne pharmaceutique (APP), Santé Canada et des agences de lutte contre le cancer.
- Forum de l’industrie : Une séance d’information d’un jour en décembre 2019 avec des représentants de Médicaments novateurs Canada (MNC), BIOTECanada et certaines de leurs sociétés membres.
- Forum de la société civile : Une séance d’information d’un jour en décembre 2019 avec des groupes de patients et d’autres intervenants non affiliés à des institutions.
- Webinaire de l’industrie : en janvier 2020 avec 187 participants de toute l’industrie pharmaceutique.
- Rencontres de consultation à travers le pays : plus de 60 rencontres avec plus de 260 participants issus de groupes d’intervenants de tout le pays. Il s’agissait notamment de ministères de la Santé, de payeurs publics et privés, de patients et de groupes de patients, de cliniciens, de représentants de l’industrie et d’associations commerciales, de pharmaciens et de distributeurs, d’organisations de soins de santé, etc.
- Rencontres bilatérales avec des compagnies pharmaceutiques, des associations commerciales et des consultants : plus de 40 rencontres, principalement à Ottawa.
Des renseignements précis sur les dates, les lieux et les groupes d’intervenants que le CEPMB a rencontrés, ainsi que la version électronique des documents ayant été évoqués durant ces rencontres peuvent être consultés sur le site web du CEPMB.
Le CEPMB avait l’intention d’organiser un forum de politique publique à Ottawa le 18 mars 2020, mais il n’a pu le faire en raison de la pandémie émergente de la COVID 19. Pour permettre aux intervenants de transmettre au Conseil toute information, nouvelle ou différente, qu’ils souhaitaient peut-être présenter à cette occasion, le CEPMB a prolongé au 18 mars 2020 le délai de soumission des observations écrites par les parties intéressées.
Le CEPMB a reçu, durant la période de consultation, 123 observations écrites provenant d’un large éventail d’intervenants. Un tiers (33 %) des observations reçues émanait de titulaires de brevets et d’associations regroupant des représentants de leur industrie, un autre tiers (33 %) provenait de groupes de consommateurs et de défense des intérêts des patients. Le CEPMB a également reçu presque 900 lettres d’individus ou de patients; la majorité d’entre elles provenait de patients atteints de fibrose kystique et de leurs soignants, mobilisés dans le cadre d’une initiative de sensibilisation lancée par Fibrose kystique Canada. Les 123 observations écrites peuvent être consultées sur le site web du CEPMB.
| Catégorie | Observations (nbre) | Observations (%) |
|---|---|---|
| Consommateurs/Groupes de défense des intérêts des patients (Total) | 41 | 33 % |
| Titulaires de brevets | 34 | 28 % |
| Associations de titulaires de brevets | 4 | 3 % |
| Médicaments génériques/biosimilaires | 2 | 2 % |
| Titulaires de brevets/associations de titulaires de brevets (Total) | 40 | 33 % |
| Distributeurs/consultants/pharmaciens | 11 | 9 % |
| Associations de l’industrie (p. ex., sciences de la vie) | 6 | 5 % |
| Consultants | 2 | 2 % |
| Industrie (Total) | 19 | 15 % |
| Syndicats | 7 | 6 % |
| Cliniciens | 4 | 3 % |
| Universitaires | 3 | 2 % |
| Groupes de réflexion | 1 | 1 % |
| International | 1 | 1 % |
| Universitaires civils/cliniciens/groupes de réflexion (Total) | 16 | 13 % |
| Entités publiques (p. ex. organismes, autorités sanitaires, gouvernements) | 5 | 4 % |
| Assurances privées | 2 | 2 % |
| Entités publiques ou assurances privées (Total) | 7 | 6 % |
| Total général | 123 | 100 % |
Les efforts décrits précédemment s’inscrivent dans un processus qui se poursuivra même après la mise en œuvre des Lignes directrices. Le CEPMB travaillera de concert avec les intervenants pour que l’impact des Lignes directrices soit bien compris et que tout problème d’observance ou opérationnel découlant de leur application précoce réduit au minimum. Des ajustements seront apportés aux Lignes directrices s’il devient apparent que certains aspects du nouveau régime ne fonctionnent pas comme prévu, sous réserve, comme toujours, de consultations additionnelles avec les intervenants et le public.
Explication des changements apportés aux Lignes directrices provisoires
Lorsqu’il formule des lignes directrices, le CEPMB doit concilier des objectifs de politique publique apparemment contradictoires, à savoir faciliter l’accès aux médicaments brevetés à des prix non excessifs tout en reconnaissant l’intérêt légitime à ce que les titulaires de brevets pharmaceutiques maximisent la valeur de leur propriété intellectuelle. Il n’est pas surprenant que les divers intervenants aient des points de vue divergents, voire diamétralement opposés, quant au fondement politique des Lignes directrices et du Règlement modifié sur lequel elles reposent. Par conséquent, le CEPMB reconnaît que les consultations concernant des mesures destinées à moderniser son cadre réglementaire ne peuvent aboutir à un consensus. Le CEPMB s’est pourtant efforcé tout au long du processus de consultation de favoriser un dialogue productif, juste et transparent avec nos intervenants et d’écouter attentivement leurs préoccupations. Les modifications expliquées ci-après sont le résultat de cet effort et constituent notre tentative la plus aboutie de prise en compte des commentaires contradictoires que nous ont adressés les intervenants, et d’élaboration d’un régime moderne qui continue de favoriser le respect volontaire des directives.
Les pages qui suivent donnent une description concise des principaux changements intervenus entre les Lignes directrices provisoires de novembre 2019 et celles de juin 2020, des commentaires des intervenants ayant inspiré ces changements et du raisonnement suivi par le Conseil lorsqu’il les a introduits. Cette information est présentée à titre contextuel et ne doit pas passer pour une description exhaustive de tous les commentaires pertinents reçus par le CEPMB à l’égard de chaque question, ou de l’analyse ayant motivé les changements.
1. Comparaison selon la catégorie thérapeutique des prix nationaux (CCTn)
Approche proposée dans les Lignes directrices provisoires de novembre 2019
L’un des facteurs énoncés à l’article 85 de la Loi sur les brevets et permettant d’évaluer si un prix est excessif est « le prix de vente de médicaments de la même catégorie thérapeutique sur un tel marché » (al. 85(1)b)).
Dans les Lignes directrices provisoires de novembre 2019, ce facteur est abordé de deux manières principales. Premièrement, elles prévoyaient fixer le prix courant maximum (PCM) des nouveaux médicaments brevetés à la plus faible des deux valeurs suivantes : la médiane des prix courants internationaux (MPI) en vigueur dans les pays de comparaison du CEPMB11 et la comparaison selon la catégorie thérapeutique des prix nationaux (CCTn), sous réserve d’un plancher fixé par le prix international le plus bas (PIPB) pour les pays du CEPMB11. Deuxièmement, elles précisaient qu’en l’absence d’une analyse coût-utilité préparée par un organisme canadien financé par l’État pour les médicaments brevetés de catégorie I, le prix escompté maximum (PEM) correspondrait à la plus faible des valeurs suivantes : le PIPB, la CCTn ou la comparaison selon la catégorie thérapeutique des prix internationaux (CCTi), les ajustements additionnels étant fondés sur la méthode de rajustement en fonction de la taille du marché.
La CCTn et la CCTi étaient alors calculées en se basant sur le coût médian de traitement par tous les médicaments de comparaison, obtenu en tenant compte du prix public le plus bas et de la source des prix.
Résumé des commentaires des intervenants
Les titulaires de brevets qui s’opposent à l’approche proposée affirment qu’elle entraînera une réduction des prix courants canadiens vers le PIPB et qu’elle ne tient pas compte des avantages thérapeutiques variables associés aux médicaments d’une catégorie donnée. Les titulaires de brevets recommandent également que les CCTn et CCTi soient calculées en se fondant sur le coût le plus élevé plutôt que sur le coût médian de traitement par tous les médicaments de comparaison.
Aucune préoccupation liée à l’application de la CCTn de la manière décrite précédemment n’a été soulevée par les payeurs publics et privés ou par d’autres intervenants travaillant dans l’administration des soins de santé.
Analyse
Malgré le faible nombre de médicaments brevetésNote de bas de page 3 lancés dans les catégories thérapeutiques dominées par des médicaments plus anciens et génériques, le Conseil reconnaît que l’approche proposée aura probablement pour effet de réduire le PCM de ces médicaments jusqu’au PIPB. Le Conseil a donc décidé de renoncer à cette approche lorsque le titulaire de brevet dépose les prix du médicament en vigueur dans les pays du CEPMB11. Nous pensons que les prix internationaux du médicament pourraient déjà refléter, dans une certaine mesure, la disponibilité et le prix des médicaments de comparaison sur ces marchés.
Le Conseil a décidé de conserver le test de CCTn lorsque les prix du médicament dans les pays du CEPMB11 ne sont pas disponibles. Dans un tel cas, le test de CCTn sera calculé en fonction du coût le plus élevé, et non médian, dans la catégorie en cause. La CCTn et la CCTi continueront également d’être utilisées pour fixer le PEM de médicaments de catégorie I dont la taille de marché est élevéeNote de bas de page 4. Pour le Conseil, la valeur médiane est la plus appropriée dans ce cas, étant donné que les prix utilisés pour effectuer la CCT sont des prix bruts (courants) et non des prix nets.
Description des changements dans les Lignes directrices provisoires de juin 2020
Le prix courant maximum (PCM) des nouveaux médicaments brevetés sera fixé par la médiane des prix internationaux (MPI) disponibles dans les pays de comparaison du CEPMB11, pour autant que le titulaire de brevet ait déclaré les prix en vigueur dans au moins un pays. En l’absence de telles données, le PCMNote de bas de page 5 sera alors établi par la CCTn calculée en fonction du coût du traitement le plus élevé, et non médian, parmi tous les médicaments de comparaison, obtenu en tenant compte du prix public le plus bas au Canada. Si le titulaire de brevet n’a pas déclaré les prix internationaux avant la fin de la période provisoire (maximum de trois ans), et qu’il n’existe aucun traitement de comparaison dans la catégorie thérapeutique nationale, le PCM pourra être fixé à la médiane de la CCTi.
Si pendant sa durée de vie, le médicament est lancé dans d’autres pays, le PCM sera soumis au critère de réévaluation qui envisage une augmentation ou une réduction selon le positionnement du prix courant canadien par rapport aux prix courants internationaux.
La CCTn sera considérée dans l’établissement du PEM de médicaments de catégorie I dont la taille de marché réelle dépasse 50 millions de dollars, comme nous l’expliquons à la section 5. La CCTn sera alors fondée sur le coût médian du traitement parmi tous les médicaments de comparaison obtenu en tenant compte du prix public le plus bas au Canada et sous réserve du plancher lié au niveau de critères thérapeutiques (NCT) applicable.
2. Test du prix courant maximum (PCM) – Comparaison des prix internationaux
Approche proposée dans les Lignes directrices provisoires de novembre 2019
L’un des facteurs énoncés à l’article 85 de la Loi sur les brevets et permettant d’évaluer le caractère excessif d’un prix est « le prix de vente du médicament et d’autres médicaments de la même catégorie thérapeutique à l’étranger » (al. 85(1)c)).
Selon les Lignes directrices provisoires de novembre 2019, le prix courant maximum (PCM) est fixé en fonction de la plus faible des deux valeurs suivantes : la médiane des prix internationaux (MPI) en vigueur dans les pays de comparaison du CEPMB11 et la comparaison selon la catégorie thérapeutique des prix nationaux (CCTn), sous réserve d’un prix plancher fixé au prix international le plus bas (PIPB) dans les pays du CEPMB11. Les Lignes directrices proposaient en outre, au cas où le titulaire de brevet ne déclarait pas les prix internationaux avant la fin des trois ans (« période provisoire ») et qu’il n’existait aucun médicament de comparaison dans la catégorie thérapeutique nationale, que le PCM soit fixé en fonction de la comparaison selon la catégorie thérapeutique des prix internationaux (CCTi).
Résumé des commentaires des intervenants
Les titulaires de brevets, l’industrie des biosimilaires, les distributeurs, les consultants de l’industrie et certains patients et groupes de patients soutiennent que la MPI devrait être remplacée par le plus élevé des prix internationaux (PEPI) pour les médicaments bénéficiant de droits acquis, et avancent un certain nombre d’arguments à l’appui de cette position. Premièrement, ils affirment que l’approche fondée sur la MPI présume à tort que tous les prix supérieurs à la médiane dans les pays de comparaison sont excessifs. Deuxièmement, ils estiment que l’application de la MPI aux médicaments bénéficiant de droits acquis ne concorde pas avec l’analyse coûts-avantages (ACA) menée par Santé Canada dans le cadre du résumé de l’étude d’impact de la réglementation (le REIR) accompagnant le Règlement modifié. Troisièmement, ils maintiennent que l’application de la MPI aux prix des médicaments bénéficiant de droits acquis aura d’importantes répercussions sur les recettes de l’industrie et aboutira à des pénuries de médicaments et à une réduction des services de soutien actuellement offerts aux patients.
« Étant donné que l’application du nouveau panier est dictée par le Règlement modifié, il serait plus opportun d’appliquer les règles actuelles de fixation des prix (comparaison avec le plus élevé des prix internationaux ou PEPI) pour établir le prix courant maximal (PCM) des médicaments existants. Même au regard du PEPI, les prix courants des produits existants seront réduits du fait du passage au nouveau panier de référence international (CEPMB11) »
Janssen
Inversement, selon certains payeurs publics et privés et d’autres groupes de patients, cela fait des années que les Canadiens paient des prix excessifs pour les médicaments brevetés, et la MPI en vigueur dans le nouveau panier de pays de comparaison du CEPMB11 constitue un moyen raisonnable de s’assurer que les prix bruts (courants) au Canada sont conformes aux normes internationales. De plus, ces intervenants soulignent que la MPI dans les pays du CEPMB11 représente le critère de prix le plus approprié pour offrir aux personnes qui paient en espèces une protection contre les prix courants excessifs. Ceci est d’autant plus important qu’une grande partie des ventes sont directement attribuables aux paiements en espèces et aux contributions aux quotes-parts et aux franchises des Canadiens.
Des chercheurs dans le domaine de la santé et des organismes provinciaux de lutte contre le cancer appuient l’approche proposée aux fins du calcul du PCM et demandent instamment au CEPMB de ne pas utiliser le test du PEPI. Un groupe de défense de consommateurs s’est félicité de la mise à jour de l’annexe des pays comparateurs et fait remarquer qu’elle [TRADUCTION] « rendra bien plus équitables les comparaisons de prix pour les consommateurs canadiens de produits pharmaceutiques ».
“...nous ne pouvons qu’accueillir favorablement tout nouveau processus de fixation et de plafonnement des prix s’appuyant sur les données probantes et les pratiques exemplaires, de même que l’utilisation d’une nouvelle annexe de pays de comparaison (le CEPMB11) visant à établir les nouvelles règles de comparaison des prix...”
Centrale des syndicats du Québec
Analyse
Selon la Loi sur les brevets, le Conseil n’est pas tenu d’adopter un seuil particulier fondé sur les prix en vigueur dans les pays du CEPMB11, mais doit uniquement les considérer.
Nous reconnaissons que pour l’essentiel, l’analyse coûts-avantages (ACA) se base sur des plafonds de PCM qui se rapprochent généralement davantage du prix le plus élevé et du prix médian dans les pays du CEPMB11 pour les médicaments bénéficiant de droits acquis et les nouveaux médicaments brevetés, respectivement. En même temps, l’ACA avait pour objet d’évaluer l’impact du Règlement modifié de manière isolée. Évidemment, elle ne doit pas être interprétée comme un obstacle à la modification des Lignes directrices actuelles, dont le remaniement en profondeur est nécessaire en premier lieu à la mise en œuvre du Règlement modifié.
D’après l’analyse du Conseil, l’impact de l’application de la MPI sur les médicaments bénéficiant de droits acquis pourrait être moins important que ne le prétendent certains intervenants qui s’y opposent. Les prix courants ne reflètent pas les prix nets réels versés par un segment important du marché canadien et l’impact réel sur les recettes nettes sera donc inférieur à l’impact nominal sur les prix courants. Cela dit, le Conseil a décidé d’appliquer le critère du PEPI aux médicaments bénéficiant de droits acquis, à titre de concession aux titulaires de brevets dont les attentes pourraient avoir été avivées par l’ACA et pour reconnaître l’impact associé à la modification de l’annexe des pays de comparaison. Le Conseil estime que l’application à venir du test de la MPI aux nouveaux médicaments brevetés est la plus à même de corriger le décalage des prix canadiens par rapport aux prix internationaux. Le Conseil estime en règle générale que les prix courants canadiens qui dépassent les normes internationales paraissent excessifs et que la MPI est le critère déterminant qui permettra de s’assurer que ces prix courants ne deviendront pas excessifs.
Description des changements dans les Lignes directrices provisoires de juin 2020
Médicaments brevetés bénéficiant de droits acquis et élargissements de leurs gammes
Le prix brut (courant) plafond des médicaments brevetés bénéficiant de droits acquis sera fixé à la plus faible des deux valeurs suivantes : le PEPI pour les pays du CEPMB11, et le plafond applicable au titre des Lignes directrices antérieures. Le PCM associé aux élargissements de gammes de médicaments bénéficiant de droits acquis (c. à d. nouvelles concentrations et formes posologiques) sera également fixé en fonction du PEPI.
Nouveaux médicaments brevetés et médicaments brevetés de transition
Le test de la MPI sera maintenu pour ce qui est de fixer le PCM des médicaments de transition (c. à d. médicaments auxquels un numéro DIN a été attribué à compter du 21 août 2019 et dont la première vente au Canada est survenue avant le 1er janvier 2021) et des nouveaux médicaments brevetés (c. à d. tout autre médicament breveté auquel un numéro DIN a été attribué depuis le 21 août 2019).
3. Classification d’un médicament breveté de catégorie I
Approche proposée dans les Lignes directrices provisoires de novembre 2019
Les Lignes directrices provisoires de novembre 2019 proposaient la classification de catégorie I pour les médicaments brevetés remplissant l’un des critères suivants :
- Coût du traitement de 12 mois supérieur à 50 % du produit intérieur brut (PIB) par habitant, et (ou)
- Taille réelle ou estimée du marché (recettes) supérieure au seuil annuel de la taille du marché, initialement fixé à 25 millions de dollars.
Résumé des commentaires des intervenants
Les titulaires de brevets pharmaceutiques contestent les critères et seuils afférents; ils estiment que ces seuils sont trop bas et entraîneraient la classification de la majorité des nouveaux médicaments brevetés dans la catégorie I, ce qui va à l’encontre de l’intention déclarée du CEPMB d’adopter une approche réglementaire fondée sur les risques. D’autres intervenants, notablement des groupes de patients et d’associations de pharmacies ont repris à leur compte les préoccupations concernant la proportion de médicaments brevetés susceptibles d’être sélectionnés dans la catégorie I.
« Ces lignes directrices sont intrinsèquement défavorables aux médicaments destinés à de petites populations de patients non encore traités et atteints de maladies difficiles à diagnostiquer, dont l’histoire naturelle n’est pas bien établie et à l’égard desquelles il existe peu de données cliniques démontrant des liens directs entre les biomarqueurs et d’autres indicateurs de résultats. Cette description pourrait s’appliquer à presque toutes les maladies rares. [...] Il est ridicule de présenter une évaluation coût-efficacité (ECA) à l’étape du lancement [...] »
Canadian Organization for Rare Disorders (CORD)
Les groupes de patients préoccupés par l’accès aux médicaments pour maladies rares sont particulièrement troublés par le fait que ces médicaments relèveraient pratiquement tous de la catégorie I, en raison des coûts annuels élevés de traitement, et qu’ils seraient donc soumis au nouveau facteur de la valeur pharmacoéconomique (VP). Ces médicaments présentent rarement des profils pharmacoéconomiques favorables en raison de leurs prix extrêmement élevés, que les titulaires de brevets croient nécessaires pour récupérer les dépenses en recherche et développement consacrées à une population de patients relativement petite.
Inversement, certains représentants de la société civile et organismes de soins de santé estiment qu’un seuil plus faible devrait s’appliquer aux coûts annuels de traitement si le CEPMB doit correctement examiner de manière minutieuse tous les médicaments brevetés que de nombreux consommateurs n’ont pas les moyens de se procurer.
Pour leur part, les groupes syndicaux expriment un soutien vigoureux à l’inclusion de la taille du marché comme critère pour les médicaments de catégorie I et citent à cet égard certains médicaments qui, même s’ils se situent en dessous du seuil de VP, ont un impact sur les régimes d’assurance-maladie. Ils signalent que récemment le coût des régimes d’assurance-médicaments de leurs membres a considérablement augmenté d’une année à l’autre à cause de médicaments qui seraient considérés comme peu coûteux aux termes des Lignes directrices provisoires de novembre 2019.
Enfin, les titulaires de brevets affirment qu’un mécanisme devrait être prévu pour faire passer des médicaments de la catégorie I à la catégorie II si les recettes qu’ils génèrent s’avèrent inférieures aux seuils prévus.
Analyse
Une analyse approfondie des données laisse penser qu’un pourcentage significatif de médicaments générerait des recettes annuelles de plus de 25 millions de dollars pendant la durée de vie du brevet. Attendu que ces données reflètent des tendances historiques, il s’agit probablement d’une sous-estimation des recettes futures escomptées pour les médicaments brevetés lancés plus récemment.
En outre, étant donné que les médicaments à coût élevé dominent de plus en plus le marché, l’observation selon laquelle les critères envisagés aux fins de la catégorie 1 concerneraient un grand nombre de nouveaux médicaments n’est pas sans fondement. Par conséquent, le Conseil a conclu qu’il est justifié d’imposer des seuils plus élevés, à la fois en termes de coût de traitement et de taille de marché, et ce, afin que son approche fondée sur les risques soit plus réalisable sur le plan administratif pour le personnel du CEPMB et les titulaires de brevets. Pour dissiper les préoccupations concernant les médicaments pour maladies rares en particulier, le Conseil a décidé de retirer de la catégorie I les médicaments à coût élevé qui devraient générer des recettes annuelles inférieures à un seuil minimal.
Description des changements dans les Lignes directrices provisoires de juin 2020
Le nouveau seuil de taille du marché sera établi à 50 millions de dollars de recettes annuelles tandis que le nouveau seuil de coût du traitement sera fixé à 150 % du PIB par habitant. Certaines estimations indiquent que près de 25 % des nouveaux médicaments entraîneront, du fait de ces seuils révisés, la mise en jeu des critères de catégorie I, ce qui concorde davantage avec l’intention initiale du CEPMB de passer à une approche fondée sur les risques. Ces médicaments devraient représenter la majorité des ventes de médicaments brevetés (68 %), ce qui permettra essentiellement au CEPMB d’exercer une surveillance plus étroite sur une minorité de médicaments qui comptent pour la majorité des ventes.
Les médicaments qui génèrent moins de 12 millions de dollars en recettes annuelles ne seront pas soumis au PEM même s’ils dépassent le nouveau seuil de coût annuel du traitement. Ainsi, l’arrivée sur le marché canadien de médicaments pour maladies rares ne pourra pas être découragée par crainte que leur prix net ne connaisse une réduction substantielle en raison de normes réglementaires. Des données récentes donnent à penser qu’une proportion assez importante de médicaments brevetés (42,5 %) ne génèrent 12 millions de dollars de recettes annuelles pendant aucune des dix premières années de leur commercialisation (figure 1). Ces médicaments ne représentent que 3,7 % des ventes de médicaments brevetés, ce qui laisse penser que même pris cumulativement, ils ne soulèvent pas de préoccupation en matière de capacité financière.
Figure 1. Médicaments brevetés au Canada, part des médicaments et part des ventes, selon les ventes annuelles maximales
Description de la figure
Ce graphique à barres illustre la part des médicaments et leurs parts correspondantes des ventes totales selon différentes tailles de marché : plus de 12 millions de dollars, plus de 25 millions de dollars, plus de 50 millions de dollars et plus de 100 millions de dollars. Les résultats présentés à la gauche reposent sur le seuil des ventes annuelles dans les 3 premières années et les résultats présentés à la droite reposent sur le seuil des ventes annuelles dans les 10 premières années après le lancement du médicament.
*Y compris les médicaments brevetés lancés après 1998 au Canada; taille de l’échantillon : n = 639 pour l’analyse à trois ans; n = 338 pour l’analyse à dix ans.
Source des données : CEPMB 2018; application de l’IPC pour actualiser les données historiques sur les ventes en 2018
4. Seuil de la valeur pharmacoéconomique (VP) et prise en compte des comparateurs thérapeutiques – Médicaments à coût élevé
Approche proposée dans les Lignes directrices provisoires
Suivant les lignes directrices provisoires de novembre 2019, le prix escompté maximum (PEM) des médicaments de catégorie I devant donner lieu à une analyse coût-utilité serait soumis à un plafond basé sur le niveau auquel le rapport coût-efficacité différentiel (RCED) du médicament breveté correspondrait au seuil de la valeur pharmacoéconomique (VP) de 60 000 $ par année de vie ajustée en fonction de la qualité (AVAQ).
En ce qui concerne les médicaments brevetés pour maladies rares dont la prévalence totale estimée est inférieure à 1 sur 2 000 pour toutes les indications approuvées, le PEM fixé serait 50 % plus élevé que le niveau auquel le RCED correspondrait au SVP de 60 000 $ par AVAQ.
« Le cancer du sein illustre parfaitement certaines des pires pratiques des sociétés pharmaceutiques, comme celle qui consiste à essayer d’inscrire des médicaments extrêmement coûteux sur les listes de médicaments provinciales en mobilisant les femmes atteintes d’un cancer du sein par l’entremise de groupes de "défense des intérêts des patients" pour faire directement pression sur ces gouvernements. Cette tactique a été utilisée dans le cas de certains médicaments contre le cancer du sein de stade 4 dont les avantages réels ne sont pas à la hauteur des prétentions des compagnies pharmaceutiques et dont les effets secondaires diminuent considérablement la qualité de vie des femmes qui les prennent »
~ Action cancer du sein du Québec
Résumé des commentaires des intervenants
Les titulaires de brevets s’opposent fondamentalement à ce que le CEPMB prenne en compte la VP pour déterminer ce qui constitue un prix excessif, et donc, contestent l’approche que les Lignes directrices provisoires de novembre 2019 proposent pour l’appliquer. En plus de citer des difficultés opérationnelles et techniques découlant de son application, les titulaires de brevets et certains groupes de patients s’opposent au seuil proposé de VP de 60 000 $/AVAQ qu’ils jugent déraisonnablement faible, surtout en ce qui touche les médicaments pour maladies rares et malgré un prix plafond 50 % plus élevé réservé à cette catégorie de médicaments. Alors que certains groupes de patients estiment que le facteur de la VP ne devrait jamais s’appliquer aux médicaments pour maladies rares, d’autres sont très préoccupés par les coûts de renonciation extrêmement élevés associés au remboursement de ces produits, d’autant plus que les données attestant leurs bienfaits cliniques au moment de leur introduction sur le marché sont, dans le meilleur des cas, souvent ambiguës.
Les titulaires de brevets contestent également le seuil de VP proposé de 60 000 $/AVAQ qu’ils assimilent à une approche uniforme et arbitraire susceptible d’exposer les montants remboursés par les régimes publics d’assurance-médicaments au titre des ententes confidentielles relatives à l’inscription des médicaments (ERIM). Ils préconisent une approche plus souple qui reconnaît et récompense les avantages thérapeutiques, comme le font les Lignes directrices actuelles, mais ne proposent aucune solution de rechange qu’ils jugeraient satisfaisante. Des suggestions précises ont été faites à cet égard par certains assureurs publics, qui ont recommandé des seuils de VP de 100 000 $/AVAQ et de 120 000 $/AVAQ pour les médicaments supérieurs d’un point de vue thérapeutique ou même davantage pour les médicaments possiblement curatifs.
Enfin, en plus de préconiser une approche plus souple fondée sur les bienfaits thérapeutiques, certains titulaires de brevets suggèrent à titre subsidiaire que l’application du seuil de VP soit soumise à une sorte de mécanisme d’arrêt des pertes qui atténuerait son impact et donnerait aux acteurs de l’industrie la certitude dont ils ont bien besoin pour envisager le scénario le plus défavorable au titre du nouveau régime.
Analyse
La valeur pharmacoéconomique est à présent un facteur à prendre en compte aux termes du paragraphe 85(1) et le Conseil est tenu par la loi de l’examiner. Cependant, tant que nous ne disposerons pas de données plus empiriques sur le coût de renonciation dans le système de santé publique au Canada, un argument peut être avancé en faveur de seuils plus généreux qui concordent avec les montants supérieurs pratiqués à l’échelle internationale et qui offrent aux titulaires de brevets davantage de certitude et de prévisibilité.
À titre de comparaison, le National Institute for Health and Care Excellence (NICE) au Royaume-Uni prévoit un seuil explicite coût-efficacité de 30 000 €/AVAQ. Cependant, NICE autorisera dans certains cas des seuils plus élevés, 50 000 £/AVAQ pour les traitements de fin de vie et 100 000 à 300 000 € pour les technologies hautement spécialisées (Highly Specialized Technologies – [HST]), dépendamment du gain absolu en AVAQ associé au médicament. Dans d’autres pays, comme les Pays-Bas et la Norvège, le seuil dépend de la gravité de la maladie, entre autres facteurs. Aux Pays-Bas, un seuil coût-efficacité informel de 20 000 €/AVAQ (fardeau léger) à 80 000 €/AVAQ (fardeau élevé) est utilisé. Le Japon a récemment mis en œuvre un régime d’évaluation coût-efficacité comportant plusieurs niveaux et qui impose un ajustement des prix à la baisse, dont le montant dépend du rapport coût-efficacité associé au médicament. La réduction de prix est limitée à 10 15 % du prix remboursé par le régime national d’assurance maladie avant l’ajustement.
« [...] le concept de PEM ne comporte pas le moindre plancher et aboutira à des prix inférieurs au plus bas prix en vigueur dans les pays du CEPMB11. Cela est contraire à l’intention politique du gouvernement [...] »
Médicaments novateurs Canada
En plus des considérations précédentes, l’analyse coûts-avantages (ACA) aux fins du Règlement modifié présumait également une réduction maximale de 50 % du prix pour les nouveaux médicaments brevetés hautement prioritairesNote de bas de page 6.
Description des changements dans les Lignes directrices provisoires de juin 2020
Les seuils de VP et planchers de réduction des prix plafonnés suivants seront appliqués pour établir le PEM des médicaments à coût élevé de catégorie I en fonction d’une évaluation de leurs critères thérapeutiques (pour les recettes annuelles qui s’élèvent à plus de 12 millions de dollars) :
Tableau 1. Seuils de valeurs pharmacoéconomiques (SVP)
| Niveau de critères thérapeutiques (voir annexe E – Processus d’examen scientifique) |
SVP | Plancher de réduction du PCM |
|---|---|---|
| Niveau I | 200 k$/AVAQ | 20 % |
| Niveau II | 150 k$/AVAQ | 30 % |
| Niveau III | 150 k$/AVAQ | 40 % |
| Niveau IV | 150 k$/AVAQ | 50 % |
| L’analyse pharmacoéconomique est une minimisation des coûts | Médiane de la CCTn soumise à un plancher de 50 % | |
| Aucune évaluation pharmacoéconomique | 50 % du PCM | |
Il convient de noter que pour les médicaments à coût élevé de catégorie I, une réduction maximale de 50 % sera automatiquement appliquée si le titulaire de brevet ne fournit aucune analyse coût-utilité.
5. Rajustements en fonction de la taille du marché
Approche proposée dans les Lignes directrices provisoires de novembre 2019
En ce qui concerne les médicaments à coût élevé de catégorie I, les Lignes directrices provisoires de novembre 2019 proposaient que le prix escompté maximum (PEM) soit encore ajusté en fonction de la taille du marché si les ventes annuelles de médicaments génèrent des recettes annuelles dépassant 25 millions de dollars après application du seuil de valeur pharmacoéconomique (VP). Ces nouveaux ajustements se traduiraient par des réductions graduelles de 10 % sur les tranches de recettes de 25 millions de dollars, pouvant aller jusqu’à une réduction maximale de 50 % pour les recettes annuelles supérieures à 125 millions de dollars. Ils s’appliqueraient également aux autres médicaments de catégorie I dont le coût n’est pas élevé, mais qui relèvent de cette catégorie parce que les recettes annuelles réelles ou estimées qu’ils engendrent dépassent le seuil de 25 millions de dollars de taille du marché.
Résumé des commentaires des intervenants
L’intensité de l’opposition des titulaires de brevets au facteur de la taille du marché ne le cède qu’au sentiment qu’inspire le facteur de la VP. En général, les titulaires de brevets estiment que ce facteur a pour effet d’introduire au sein du régime du CEPMB des considérations qui devraient relever exclusivement de la compétence des assureurs publics et privés, et non d’un organisme fédéral. De plus, de nombreux titulaires de brevets soutiennent que l’augmentation de 25 millions de dollars est arbitraire et qu’il est incohérent et injuste de ne pas envisager une hausse du PEM si les recettes chutent en dessous des seuils prévus.
Les intervenants favorables au facteur relatif à la taille du marché reconnaissent que la demande et la prévalence doivent être prises en compte dans la fixation des prix plafonds qui influent de manière prépondérante sur la viabilité du système de soins de santé canadien. Ils font remarquer qu’une partie significative des dépenses peut concerner un petit nombre de médicaments, lesquels posent des problèmes immédiats de capacité financière au sein du système, sans nécessairement être qualifiés de médicaments à coût élevé.
Analyse
Comme la VP, la taille de marché est désormais un facteur à prendre en compte aux termes du paragraphe 85(1) et le Conseil est tenu par la loi de l’examiner. Cependant, d’aucuns pourraient faire valoir que le seuil initial de 25 millions de dollars est trop bas et que le nombre d’ajustements graduels proposés dans les Lignes directrices provisoires de novembre 2019 est indûment onéreux pour les titulaires de brevets. Selon une analyse rétrospective, près d’un quart des médicaments brevetés génèrent plus de 50 millions de dollars en recettes annuelles durant les 10 premières années d’exclusivité de marché, tandis que seuls 14 % d’entre eux génèrent plus de 100 millions de dollars.
Un grand nombre d’autres arguments pertinents soulevés par les intervenants dans ce contexte ont déjà été abordés dans la section 3 précédente.
Description des changements dans les Lignes directrices provisoires de juin 2020
Les seuils révisés aux fins de l’ajustement en fonction de la taille du marché et les réductions correspondantes qu’ils nécessitent sont exposés dans les tableaux ci après.
Tableau 2. Rajustements en fonction de la taille du marché
| Recettes annuelles | PEM | Facteur de rajustement du PCM supplémentaire | Facteur de rajustement des prix‡ utilisé pour le calcul du facteur de rajustement du PCM |
|---|---|---|---|
| < 12 M$ | PCM | 0 % | 0 % |
| 12 M $-50 M$ | Le plus grand entre le PP et le prix plancher | (12 M$ - 12 M$ * (PEM/PCM)) / Recettes + (PEM/PCM) | |
| 50 M$ - 100 M$ | -25 % | (21,5 M$ - 9 M$ * (PEM/PCM)) / Recettes + (1 - 25 %) * (PEM/PCM) | |
| >100 M$ | -35 % | (32 M$ - 7,8 M$ * (PEM/PCM)) / Recettes + (1 - 35 %) * (PEM/PCM) |
‡ Plus bas entre le PCM et le prix courant
| Recettes annuelles | PEM | Facteur de rajustement du PCM supplémentaire | Facteur de rajustement des prix‡ utilisé pour le calcul du facteur de rajustement du PCM |
|---|---|---|---|
| < 50 M$ | PCM | 0 % | 0 % |
| 50 M$ - 100 M$ | Le plus bas entre le PCM et la médiane de la CCTn | -25 % | (50 M$ - 37,5 M$ * (PEM/PCM)) / Recettes + (1 - 25 %) * (PEM/PCM) |
| >100 M$ | -35 % | (56,7 M$ - 32,5 M$ * (PEM/PCM)) / Recettes + (1 - 35 %) * (PEM/PCM) |
‡ Plus bas entre le PCM et le prix courant
S’agissant des médicaments de catégorie I n’atteignant pas le seuil de coût du traitement annuel de 150 % du PIB par habitant, mais dont la taille réelle de marché dépasse 50 millions de dollars, le PEM sera fondé sur la valeur la plus faible entre le prix courant maximum (PCM) et la comparaison selon la catégorie thérapeutique des prix nationaux (CCTn) ajustée en fonction du facteur applicable. La CCTn sera obtenue en fonction du coût médian du traitement dans tous les pays de comparaison, en utilisant le prix public le plus bas. L’impact de la CCTn sera soumis à un plancher de réduction des prix qui varie en fonction du niveau de critères thérapeutiques.
| Niveau de critères thérapeutiques | Plancher de la CCTn |
|---|---|
| Niveau I | 20 % du PCM (CCTn non applicable) |
| Niveau II | 30 % |
| Niveau III | 40 % |
| Niveau IV | 50 % |
6. Confidentialité du prix escompté maximum (PEM)
Approche proposée dans les Lignes directrices provisoires
Dès l’entrée en vigueur du Règlement modifié, tous les titulaires de brevets devront communiquer les renseignements concernant les prix et les recettes, déduction faite de tous les ajustements, et notamment des ristournes, à toute partie qui paie ou rembourse les médicaments brevetés, des rabais et des biens et services offerts gratuitement. Le CEPMB aura ainsi une idée précise et complète du prix réel net que les titulaires de brevets facturent pour leurs médicaments au Canada. Tout renseignement qu’ils déposeront auprès du CEPMB à cet égard bénéficiera de la protection totale des dispositions de la Loi sur les brevets concernant la confidentialité.
Comme nous l’avons déjà expliqué, les Lignes directrices provisoires de novembre 2019 proposaient que le PEM des médicaments brevetés de catégorie I à coût élevé soit calculé en appliquant au rapport coût-efficacité différentiel (RCED) le seuil de valeur phamacoéconomique (VP) de 60 000 $ par année de vie ajustée en fonction de la qualité (AVAQ). En ce qui concerne des médicaments brevetés pour maladies rares, le PEM serait fixé 50 % au dessus du niveau auquel le RCED est égal au seuil de VP de 60 000 $/AVAQ. Enfin, le PEM pour tous les médicaments brevetés de catégorie I sera réduit de 10 % par recettes supplémentaires de 25 millions de dollars jusqu’à une réduction maximale de 50 % pour les recettes annuelles dépassant 125 millions de dollars.
Résumé des commentaires des intervenants
Les titulaires de brevets craignent que le nouveau régime mine leur capacité à continuer de négocier des ristournes et des rabais confidentiels avec les payeurs au Canada. Ils ne semblent pas contester que les rapports qu’ils déposent auprès du CEPMB demeureront confidentiels, mais ils craignent que l’approche adoptée dans les Lignes directrices provisoires de novembre 2019 permette à des tiers (y compris des payeurs dans d’autres pays) de rétrocalculer une estimation sommaire du PEM et d’extrapoler les niveaux de réductions du prix courant qui seraient requises pour s’y conformer. Toujours selon les brevetés, cela est possible pour trois raisons. Premièrement, les rapports coût-utilité qu’ils devront déposer auprès du CEPMB sont rendus publics par les organismes qui les délivrent (p. ex. l’Agence canadienne des médicaments et des technologies de la santé [ACMTS] et l’Institut national d’excellence en santé et services sociaux [INESSS]). Deuxièmement, les critères de PEM sont énoncés en toute transparence dans les Lignes directrices du CEPMB. Troisièmement, le prix courant et les données sur les recettes estimées sont également accessibles auprès de diverses sources publiques autres que le CEPMB. D’après les titulaires de brevets, si d’autres pays et concurrents arrivent à estimer l’ampleur de la différence entre les prix bruts (courants) et les prix nets au Canada, leur modèle d’entreprise global serait compromis et l’introduction de nouveaux médicaments brevetés au Canada mise en péril.
« La nouvelle méthode de calcul du prix escompté maximum (PEM), combinée aux données publiques, pourrait permettre à des tiers de rétrocalculer ou d’estimer les prix nets ».
Médicaments novateurs Canada (MNC)
Les intervenants qui pensent le contraire croient tout aussi fermement que la confidentialité des prix a fondamentalement contribué à l’explosion des prix pharmaceutiques au cours des dernières années, aussi bien à l’échelle nationale qu’internationale et qu’il est impératif que ce phénomène soit mis en lumière, non seulement au Canada, mais dans le monde entier.
« Le fait que les renseignements qui sous-tendent le calcul du […] PEM soient confidentiels rendra encore plus opaques les prix pharmaceutiques, ce qui est malencontreux ».
Innovative Medicines Canada (IMC)
Analyse
Le Conseil reconnaît qu’il est difficile en soi d’établir des seuils de démarcation des prix qui soient prévisibles pour les titulaires de brevets mais qui ne permettent pas aux concurrents d'estimer approximativement les ristournes confidentielles accordées à des tierces parties. En même temps, il fait observer qu’un certain nombre des problèmes soulevés par les titulaires de brevets, tels que la mise à la disposition du public des rapports pharmacoéconomiques publiés par l’ACMTS et l’INESSS ou celle des prix courants et des estimations de recettes sont des éléments bien établis du statu quo qui prévaut au Canada et que les Lignes directrices ne peuvent pas défaire. Des seuils coût-utilité formels et informels sont les standards retenus dans un certain nombre de pays où les titulaires de brevets continuent pourtant de vendre leurs produits. Cela dit, le Conseil n’est pas insensible aux préoccupations qu’ils soulèvent et estime que l’approche révisée de calcul du PEM décrite dans les Lignes directrices provisoires de juin 2020 présente le double avantage de tenir compte de ces deux préoccupations et du désir exprimé et par les titulaires de brevets et par de nombreux groupes de patients pour qu’une évaluation des critères thérapeutiques demeure un facteur clé dans l’établissement des prix plafonds.
Description des changements dans les Lignes directrices provisoires de juin 2020
Comme nous l’expliquions à la section 4, le calcul du PEM dépendra du niveau de critères thérapeutiques (NCT) attribué au médicament breveté ainsi que du seuil de VP applicable et du prix plancher qui lui correspond. Le NCT d’un médicament breveté ne sera connu que du CEPMB et du titulaire de brevet.
7. Examen réglementaire des médicaments biosimilaires et génériques brevetés
Approche proposée dans les Lignes directrices provisoires de novembre 2019
Le Règlement modifié a retiré, sauf en cas de plainte, l’exigence selon laquelle les titulaires de brevets de certains médicaments brevetés doivent déposer auprès du CEPMB des renseignements concernant l’identification, les prix et les ventes. C’est le cas notamment des médicaments vétérinaires brevetés, d’un vaste sous-groupe de médicaments ne nécessitant pas d’ordonnance (c. à d. les médicaments en vente libre) et de médicaments génériques qui satisfont à la définition réglementaire. Si une plainte est reçue à l’égard de l’un de ces types de médicaments brevetés, ils seront alors automatiquement considérés comme relevant de la catégorie II aux fins de l’enquête.
Les Lignes directrices provisoires de novembre 2019 ne faisaient aucune mention particulière des biosimilaires (c. à d. des médicaments biologiques biosimilaires approuvés en fonction d’une comparaison avec le produit de référence d’une société novatrice) qui ne tombent pas sous le coup de la définition des médicaments génériques dans le Règlement modifié. Par conséquent, les titulaires des brevets en question seraient tenus de déposer auprès du CEPMB des renseignements sur les prix et ces médicaments pourraient relever de la catégorie I ou de la catégorie II.
Résumé des commentaires des intervenants
Les fabricants de biosimilaires, l’industrie pharmaceutique de produits génériques et certains titulaires de brevets soutiennent que d’autres types de médicaments brevetés doivent être considérés comme présentant un faible risque de prix excessif sur le plan administratif et ainsi être soustraits aux exigences prévues dans le Règlement modifié en matière de rapports. D’après eux, cette exemption devrait s’appliquer aux biosimilaires, aux médicaments de marque faisant face à une concurrence par des compagnies génériques et aux médicaments génériques brevetés dont la vente est autorisée par des voies réglementaires autres que la présentation abrégée de drogue nouvelle (PADN) au titre du Règlement sur les aliments et drogues.
Les fabricants de biosimilaires soulignent en particulier que leur marché au Canada en est encore à ses balbutiements et qu’ils devraient être soustraits le plus possible aux exigences réglementaires inutiles. Ils soutiennent que le fait de soumettre les produits biosimilaires au même niveau de contrôle réglementaire de la part du CEPMB que les produits biologiques de référence va à l’encontre des efforts consentis dans d’autres champs du système de santé pour bénéficier des économies que ce marché naissant permet de réaliser. D’aucuns prétendent que la réglementation des biosimilaires fondée sur les plaintes est plus conforme à la décision récente de l’Agence canadienne des médicaments et des technologies de la santé (ACMTS) de ne plus soumettre ce type de médicaments à une évaluation des technologies de la santé (ETS), et ce, afin de simplifier et de rationaliser l’accès au marché de ces produits. Par conséquent, si les biosimilaires ne sont pas soustraits à la surveillance du CEPMB, leurs fabricants soutiennent que le prix national des médicaments biologiques de référence doit constituer le seul comparateur, car il serait inapproprié d’effectuer des comparaisons avec les prix internationaux d’autres biosimilaires brevetés.
Analyse
Bien que le Conseil ne puisse pas exempter les fabricants de médicaments brevetés des exigences de présentation prévues par le Règlement modifié, du point de vue de l’administration courante, il a une certaine latitude quant à la mesure dans laquelle certains médicaments non exemptés sont soumis à un examen proactif. Le Conseil estime que de solides arguments peuvent étayer l’idée selon laquelle l’élargissement du champ des médicaments brevetés exemptés au-delà de la stricte définition réglementaire, et à des fins administratives, est compatible avec une approche fondée sur les risques de la réglementation des prix plafonds.
Description des changements dans la version provisoire du règlement de juin 2020
Les biosimilaires brevetés, les médicaments génériques, les médicaments destinés à un usage vétérinaire et les médicaments brevetés en vente libre ne donneront lieu à une enquête que si une plainte est reçue par le CEPMB. Ces médicaments seront alors considérés comme relevant de la catégorie II et traités comme tels nonobstant l’existence de propriétés qui pourraient autrement satisfaire aux critères de sélection de la catégorie I. Contrairement aux médicaments brevetés à l’égard desquels les exigences prévues par le Règlement modifié en matière d’établissement de rapports ont été allégées, les fabricants de biosimilaires et de génériques brevetés ayant été approuvés autrement que par l’entremise de PADN sont tenus en vertu de la Loi de présenter des rapports au CEPMB.
8. Autres questions soulevées par les intervenants et abordées par les changements décrits précédemment
Médicaments pour maladies rares
Comme nous l’avons déjà expliqué, les Lignes directrices provisoires de novembre 2019 proposaient un prix escompté maximum (PEM) 50 % plus élevé pour les médicaments destinés au traitement d’une affection dont la prévalence totale estimée est inférieure à 1 sur 2 000 pour toutes les indications approuvées. Les révisions de l’approche en matière de PEM décrites dans les Lignes directrices provisoires de juin 2020 ne font plus de distinction entre les médicaments destinés au traitement des maladies rares et non rares. Au lieu de cela, une exemption totale de l’exigence du PEM est prévue pour tous les médicaments qui génèrent des recettes annuelles inférieures à 12 millions de dollars et qui relèveraient autrement de la catégorie I en raison des coûts annuels de traitement. Cette mesure, associée à la hausse du seuil de valeur pharmacoéconomique (VP) et aux réductions de prix plafonnées en fonction du niveau de critères thérapeutiques (NCT) permet de dissiper dans une large mesure les préoccupations liées au traitement qualifié d'injuste par certains intervenants réservé aux médicaments pour maladies rares au titre du nouveau régime.
Test de la relation raisonnable (RR)
Les Lignes directrices provisoires de novembre 2019 proposaient que le test de la relation raisonnable (RR) puisse servir à déterminer le prix courant maximum (PCM) ou le prix escompté maximum (PEM) d’une concentration supplémentaire ou nouvelle d’un médicament breveté qui est déjà offert à d’autres concentrations, pourvu que la concentration nouvelle ou supplémentaire porte sur le même ingrédient médicinal, la même indication, le même régime posologique et la même forme posologique ou une forme posologique comparable que la ou les concentrations existantes. Le PCM ou le PEM de la nouvelle concentration était alors fixé de manière à équivaloir au prix par unité standard de la ou des concentrations existantes. L’approche était également censée s’appliquer lorsque plusieurs concentrations d’un nouveau médicament étaient initialement vendues simultanément, et que certaines concentrations étaient précisément associées à des doses d’attaque, des doses d’ajustement posologique ou des doses de réduction.
Les titulaires de brevets craignent que cette approche décourage le lancement de nouvelles concentrations potentiellement plus commodes pour les patients. Ce serait le cas en particulier de médicaments dont le prix serait autrement fixé au même niveau que celui des autres concentrations existantes.
Compte tenu de ces préoccupations, le Conseil a décidé de revenir à une version simplifiée du test de la RR utilisé dans la version actuelle des Lignes directrices. Le critère servira à établir une concentration de référence et à fixer le plafond des autres concentrations (nouvelles ou existantes) selon leur rapport proportionnel à celle-ci, sous réserve du plafonnement du plus élevé des prix internationaux (PEPI). Pour qu’ils puissent s’adapter à la pratique des niveaux de tarification, les titulaires de brevets seront autorisés à fixer le prix des concentrations inférieures jusqu’au niveau du PCM de la concentration de référence, sous réserve du PEPI. Cette approche permet de traiter la question du niveau des prix en restant dans les limites du prix international.
Le test de la RR sera appliqué aux nouveaux médicaments brevetés (c. à d. les médicaments pour lesquels un numéro DIN a été attribué depuis le 21 août 2019 et dont la première vente est survenue le 1er janvier 2021 ou après cette date).
Nouvelles sur la consultation
Le 17 septembre 2020
Mise à jour – Réglementation, par le CEPMB, du prix des médicaments autorisés à être utilisés dans la lutte contre la COVID-19
En prévision de la publication prochaine de la version définitive de ses Lignes directrices, le CEPMB souhaite informer les intervenants de l’attention particulière que ce document accorde aux médicaments brevetés figurant dans la Liste des drogues destinées aux importations et aux ventes exceptionnelles et dans toute liste publiée par Santé Canda conformément à l'Arrêté d'urgence concernant l'importation, la vente et la publicité de drogues à utiliser relativement à la COVID-19 du 16 septembre 2020. Si un médicament breveté figure sur l’une de ces listes ou sur les deux, il ne fera l’objet d’un examen ou d’une enquête par le personnel du CEPMB que si de la ministre fédérale de la Santé ou l’un de ses homologues provinciaux ou territoriaux formule une plainte concernant son prix. Ce sera le cas tant que les décrets provisoires seront en vigueur et que le médicament breveté restera inscrit sur la liste. Si la plainte est jugée recevable, une enquête aura lieu et le personnel examinera le prix du médicament breveté conformément à la procédure habituelle prévue par les Lignes directrices.
Lorsque les décrets provisoires seront expirés ou abrogés, ou qu’un médicament breveté sera retiré de la liste applicable, et en l’absence d’une plainte, les prix des médicaments brevetés seront réexaminés en fonction des prix de liste internationaux et nationaux en vigueur, et non en fonction des prix de lancement réduits au Canada.
Cette politique a été adoptée dans le cadre d’un effort gouvernemental visant à assouplir provisoirement le processus de réglementation des médicaments et des instruments médicaux nécessaires au diagnostic, au traitement, à l’atténuation ou à la prévention de la COVID-19. Le CEPMB continuera de suivre l’évolution de la situation relative au virus responsable de la COVID‑19 au Canada, et de vérifier la pertinence et la faisabilité de cette politique.
Le 8 juillet 2020
Répercussions de la décision de la Cour fédérale et report de la date d'échéance pour les observations écrites
Le 29 juin 2020, le juge Manson de la Cour fédérale a publié sa décision concernant la demande de contrôle judiciaire de Médicaments novateurs Canada (MNC) des récentes modifications apportées au Règlement sur les médicaments brevetés (le « Règlement modifié ») qui entrera en vigueur le 1er janvier 2021. Dans sa décision, le juge Manson a maintenu les changements concernant les facteurs relatifs aux prix excessifs et la nouvelle annexe sur les pays de comparaison, mais a statué que le paragraphe 3(4) du règlement modifié, ayant trait au calcul des prix nets, est hors de la portée de la Loi sur les brevets et, par conséquent, au-delà du pouvoir de réglementation du gouverneur en conseil. En raison de cette décision, lorsque le règlement modifié entrera en vigueur en janvier 2021, le paragraphe 4(4) du Règlement sur les médicaments brevetés demeurera dans sa forme actuelle. Le CEPMB examine la décision afin d'en évaluer l'effet, mais, pour le moment, ne croit pas que des modifications importantes devront être apportées aux Lignes directrices provisoires de juin 2020. Toutefois, nous invitons les intervenants à partager leurs opinions concernant l'application de la décision du juge Manson dans le cadre de leur présentation écrite au CEPMB dans le contexte de la consultation en cours sur les Lignes directrices provisoires de juin 2020. Les parties à cette procédure de contrôle judiciaire, le procureur général du Canada et MNC, ont jusqu'au mois de septembre 2020 pour faire appel de la décision auprès de la Cour d'appel fédérale.
Afin de donner aux parties plus de temps pour aborder cette évolution récente dans leurs observations écrites, et suite à plusieurs demandes de prolongation de la période de consultation de trente jours, la date limite pour présenter des commentaires par écrit est reportée du 20 juillet 2020 au 4 août 2020. Pour de plus amples renseignements concernant la présentation de commentaires, veuillez accéder au portail de consultation.
Le 19 juin 2020
Publication de Lignes directrices provisoires de juin 2020
On invite les commentaires du public sur les Lignes directrices provisoires de juin 2020 jusqu’au 20 juillet 2020. Pour présenter une réponse, veuillez consulter la section Soumettre commentaires. Toutes les soumissions écrites seront publiées sur ce site Web.
Le CEPMB tiendra deux consultations en ligne sur les Lignes directrices provisoires. Pour vous inscrire, veuillez cliquer sur le lien ci-dessous :
- Webinaire à l'intention de l’industrie – le 29 juin de 13 h 30 à 16 h 30 (HNE)
- Ce webinaire est réservé aux représentants de l’industrie pharmaceutique
- Webinaire à l'intention du public – le 8 juillet de 13 h 30 à 16 h 30 (HNE)
- Ce webinaire est ouvert à toutes les parties intéressées
Le CEPMB organisera également deux séances réservées aux partenaires de la santé (le 25 juin) et aux assureurs privés (le 7 juillet), accessibles via invitation.
De plus, le CEPMB offrira trois webinaires à l’intention des chercheurs sur des analyses thématiques susceptibles d’éclairer la consultation.
1er webinaire à l’intention des chercheurs : le 23 juin de 13 h 30 à 15 h (HNE)
- Aperçu des dépenses en médicaments onéreux pour les maladies rares
- Aperçu de la taille du marché des médicaments brevetés au Canada
2e webinaire à l’intention des chercheurs : le 6 juillet de 13 h 30 à 15 h (HNE)
- Le prix des médicaments et les répercussions sur les investissements en R-D, les essais cliniques et l'accès aux médicaments au Canada
3e webinaire à l’intention des chercheurs : le 16 juillet de 13 h 30 à 15 h (HNE)
- Les pénuries de médicaments au Canada
Pour y participer, veuillez consulter la page Inscription aux webinaires
Le 1er juin 2020
Message du président - nouvelle date d'entrée en vigueur du Règlement sur les médicaments brevetés
Le Règlement sur les médicaments brevetés (« Règlement ») modifié entrera désormais en vigueur le 1er janvier 2021.
Comme indiqué précédemment, le CEPMB procédera à la publication d’un ensemble révisé de Lignes directrices provisoires, ce qui sera suivi d’une période de commentaires écrits. Les nouvelles Lignes directrices provisoires seront publiées la semaine du 15 juin 2020, et la période de consultation durera 30 jours. De plus amples détails sur la prochaine et dernière phase de consultation du CEPMB seront communiqués à l’approche de la date de publication des Lignes directrices provisoires révisées.
Le CEPMB poursuivra sa collaboration avec ses intervenants afin de veiller à la mise au point d’un ensemble de Lignes directrices pleinement opérationnel à temps pour l’entrée en vigueur du règlement modifié en janvier 2021. Les Lignes directrices définitives comprendront des dispositions transitoires accordant aux brevetés amplement le temps de prendre les mesures nécessaires pour se conformer volontairement aux prix plafonds applicables pour les médicaments brevetés nouveaux et existants.
Le 15 avril 2020
Mise à jour de la section « Soumissions »
Nous avons appris récemment que certaines soumissions des intervenants sur les Lignes directrices provisoires reçues par le CEPMB n’avaient pas été publiées sur le portail de consultation du CEPMB. Suite à une vérification interne de sa boîte de réception propre à la consultation, le CEPMB a cerné six soumissions manquantes de la part des personnes et organisations suivantes :
- Bill Swan
- Canadian Generic Pharmaceutical Association
- Canadian Health Policy Institute
- Dr Martha L. McKinney
- Life Saving Therapies Network
- Lorna Kosteniuk, RN
Nous regrettons cette omission involontaire et nous invitons tout intervenant qui a présenté une soumission au CEPMB sur les Lignes directrices provisoires et dont le nom ne figure toujours pas sur notre portail de consultation à communiquer avec nous directement. Merci.
Le 2 avril 2020
Message du président
Le 1er juillet 2020, le Règlement sur les médicaments brevetés modifié entrera en vigueur. Pour mettre en œuvre les modifications réglementaires, des changements aux Lignes directrices sur les prix du CEPMB sont nécessaires. Le 21 novembre 2019, le CEPMB a publié de nouvelles Lignes directrices provisoires aux fins de consultation auprès des intervenants et des membres du public. De vastes commentaires ont été reçus et toutes les soumissions écrites sont maintenant accessibles sur le site Web du CEPMB.
Le CEPMB apportera des modifications importantes aux Lignes directrices provisoires en réponse à la rétroaction reçue. À l’heure actuelle, le CEPMB prévoit publier un ensemble révisé de Lignes directrices provisoires plus tard ce printemps, et il accordera aux intervenants et aux membres du public une certaine période pour présenter leurs opinions par écrit avant la mise au point des Lignes directrices définitives.
Le CEPMB surveille de près la situation relativement à la COVID-19 au Canada et il réévaluera s’il est approprié et réaliste d’entreprendre les prochaines étapes actuellement envisagées à l’approche de la date de publication prévue des Lignes directrices provisoires révisées.
Le CEPMB poursuivra sa collaboration avec ses intervenants afin de veiller à la mise au point d’un ensemble de Lignes directrices pleinement opérationnel à temps pour l’entrée en vigueur du règlement modifié. Dans l’intervalle, les brevetés peuvent être assurés que les Lignes directrices définitives comprendront des dispositions transitoires leur accordant amplement le temps de prendre les mesures nécessaires pour se conformer volontairement aux prix plafonds applicables pour les médicaments brevetés nouveaux et existants.
Le 12 mars 2020
Nouvelles
Après avoir examiné attentivement l’évolution de la situation concernant le COVID-19, et à la lumière des recommandations de l’Agence de la santé publique du Canada sur les rassemblements durant cette période, le CEPMB a décidé de ne pas tenir un Forum des politiques publiques. Afin de donner aux parties intéressées l'occasion de communiquer toute information nouvelle ou différente qui aurait pu être présentée au Conseil lors du forum, le CEPMB accepte d'autres soumissions écrites des parties intéressées jusqu’au 18 mars 2020. Toute soumission reçue d’ici le 18 mars 2020 sera soigneusement examinée par le Conseil et les parties prenantes seront contactées par le CEPMB pour obtenir des éclaircissements sur leurs documents, le cas échéant.
Le 11 février 2020
Faits saillants et nouvelles
- Dernière chance! La date limite pour présenter une soumission écrite est le 14 février 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
- Durant la dernière semaine du processus de consultation, le CEPMB poursuivra ses rencontres avec ses intervenants à Ottawa.
Réunions et discussions
- Le 3 février, des représentants du CEPMB ont rencontré Innomar Strategies Inc. à Ottawa.
Le 3 février 2020
Faits saillants et nouvelles
- Il reste moins de deux semaines! La date limite pour présenter une soumission écrite est le 14 février 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
Réunions et discussions
- Le 30 janvier, des représentants du CEPMB se sont déplacés en Nouvelle-Écosse pour consulter les intervenants suivants :
- Nova Scotia Health Coalition
- le ministère de la Santé et du Mieux-être de la Nouvelle-Écosse
- Faces of Pharmacare
- Unité de l’évaluation des médicaments, Régie de la santé de la Nouvelle-Écosse
- Advocates for the Care of the Elderly
- Association of Dalhousie Retirees and Pensioners
- Associations des retraités des universités et collèges du Canada
- Independent Voices for Safe and Effective Drugs
- Le 31 janvier, des représentants du CEPMB ont rencontré la Société médicale du Nouveau-Brunswick au Nouveau-Brunswick.
Le 27 janvier 2020
Faits saillants et nouvelles
- Les 30 et 31 janvier, des représentants du CEPMB se déplaceront Nouveau-Brunswick et en Nouvelle-Écosse pour consulter des intervenants, notamment :
- Nova Scotia Health Coalition;
- Faces of Pharmacare;
- le ministère de la Santé et du Mieux-être de la Nouvelle-Écosse;
- la Société médicale du Nouveau-Brunswick.
- Rappel : La nouvelle date limite pour présenter une soumission écrite est le 14 février 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
Réunions et discussions
- Le 22 janvier, le CEPMB a rencontré l'Association canadienne de la gestion de l'approvisionnement pharmaceutique (ACGAP) et IQVIA à Ottawa.
- Le 23 janvier, le CEPMB a accueilli ses partenaires de la santé afin de discuter des Lignes directrices provisoires.
- Le webinaire à l’intention des brevetés (PDF 1,8 Mo) présenté le 17 janvier est maintenant accessible.
Le 20 janvier 2020
Faits saillants et nouvelles
- La nouvelle date limite pour présenter une soumission écrite est le 14 février 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
Réunions et discussions
- Le 14 janvier, le CEPMB a consulté des membres de Medicago et de l’Institut national d’excellence en santé et en services sociaux (INESS) à Québec (Québec).
- Le 17 janvier, le CEPMB a tenu un webinaire à l’intention des brevetés afin de leur fournir des renseignements sur les Lignes directrices et de répondre aux questions.
- Les rencontres qui étaient prévues la semaine du 13 janvier au Nouveau-Brunswick et en Nouvelle-Écosse sont reportées à une date ultérieure.
Le 17 janvier 2020
La nouvelle date limite pour présenter une soumission écrite est le 14 février 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
Le 13 janvier 2020
Faits saillants et nouvelles
- La semaine du 13 janvier 2020, le CEPMB fera le tour de l’Est canadien, s’arrêtant aux villes suivantes :
- Québec (Québec)
- Fredericton (Nouveau-Brunswick)
- Halifax (Nouvelle-Écosse)
- La nouvelle date limite pour présenter une soumission écrite est le 31 janvier 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
Réunions et discussions
- Les 6 et 7 janvier, des représentants du CEPMB se sont déplacés à Toronto (Ontario) afin de consulter des membres de l’industrie pharmaceutique, de la société civile et du gouvernement, notamment :
- le ministère de la Santé et des soins de longue durée
- l’Association canadienne des organismes provinciaux de lutte contre le cancer
- Action Cancer Ontario (qui fait maintenant partie de Santé Ontario)
- McKesson Canada
- GlaxoSmithKline Inc. (GSK)
- Entre les 8 et 10 janvier, des représentants du CEPMB ont tenu de réunions bilatérales à Toronto avec des membres de l’industrie pharmaceutique, notamment :
- Lundbeck Canada Inc.
- Laboratoires Paladin Inc.
- Merck Canada Inc.
- Boehringer Ingelheim (Canada) Ltée
Le 23 décembre 2019
Faits saillants et nouvelles
- Les prochaines rencontres auprès des membres de l’industrie pharmaceutique et de la société civile sont prévues les 6 et 7 janvier 2020, à Toronto (Ontario).
- La semaine du 13 janvier 2020, le CEPMB fera le tour de l’Est canadien, s’arrêtant aux villes suivantes :
- Halifax (Nouvelle-Écosse)
- Fredericton (Nouveau-Brunswick)
- Montréal (Québec)
- Les renseignements présentés aux membres de l’industrie et de la société civile dans le cadre des forums du 9 décembre et du 10 décembre sont maintenant accessibles en ligne.
- La nouvelle date limite pour présenter une soumission écrite est le 31 janvier 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
Réunions et discussions
- Les 16 et 17 décembre, des représentants du CEPMB se sont déplacés à Toronto (Ontario) afin de consulter des partenaires de la santé, des membres de l’industrie et des groupes de patients, notamment des membres de l’Association canadienne des pharmacies de quartier.
Le CEPMB invite toujours tous les intervenants et les membres intéressés du public à présenter leurs soumissions écrites avant le 31 janvier 2020.
Le 16 décembre 2019
Faits saillants et nouvelles
- Cette semaine, des représentants du CEPMB seront à Toronto (Ontario) afin de prendre part à des réunions bilatérales avec des intervenants.
- La nouvelle date limite pour présenter une soumission écrite est le 31 janvier 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
Réunions et discussions
Le 9 décembre, une séance de sensibilisation d’un jour axée sur la conformité et les questions opérationnelles a été tenue auprès de certains représentants de l’industrie. Le forum a mis l’accent sur les modifications proposées aux Lignes directrices, l’opérationnalisation des Lignes directrices provisoires, les prochaines étapes de la mise en œuvre des Lignes directrices par le CEPMB, et la rétroaction positive.
Le 10 décembre, le CEPMB a tenu un forum de la société civile d’un jour. L’objectif de la journée était de rassembler des intervenants renseignés qui ne font pas partie de l’industrie ni de l’institution et qui représentent la diversité des voix des consommateurs que le nouveau cadre de prix des médicaments du CEPMB a l’intention de protéger. Dans le cadre de la séance, on a traité de l’évolution du marché pharmaceutique, des modifications proposées aux Lignes directrices, et des prochaines étapes concernant la mises en œuvre de ces dernières par le CEPMB.
Au début de l’année prochaine, les représentants du CEPMB continueront de rencontrer des intervenants à travers l'Est du Canada.
Le CEPMB invite également tous les intervenants et les membres intéressés du public à présenter leurs soumissions écrites avant le 31 janvier 2020.
Le 9 décembre 2019
Faits saillants et nouvelles
- La nouvelle date limite pour présenter une soumission écrite est le 31 janvier 2020. Toutes les soumissions écrites seront accessibles au public sur ce site Web.
- Cette semaine, des représentants du CEPMB sont à Ottawa (Ontario). Le 9 décembre, une séance de sensibilisation d’un jour axée sur la conformité et les questions opérationnelles sera tenue auprès de certains représentants de l’industrie. Le 10 décembre, le CEPMB tiendra un forum de la société civile d’un jour.
Réunions et discussions
La semaine du 2 décembre, des représentants du CEPMB se sont déplacés en Colombie-Britannique, en Alberta, en Saskatchewan et au Manitoba. Ils ont tenu des dialogues ouverts et constructifs avec divers intervenants et partenaires, notamment de représentants des organismes suivants :
- Ministère de la Santé de la Colombie-Britannique
- Therapeutics Initiative
- Arthritis Consumer Experts
- HepCBC
- Alberta Blue Cross
- Health Coalition of Alberta
- Institute of Health Economics
- GlaxoSmithKline
- Ministère de la Santé de l’Alberta
Les représentants du CEPMB ont également eu l’occasion de rencontrer des cliniciens et des chercheurs de l'Université de la Colombie-Britannique, de l'Hôpital général de Vancouver et de l’Hôpital pour enfants de la Colombie-Britannique
La semaine prochaine
Le CEPMB poursuivra sa consultation auprès de différents membres de l’industrie lors de réunions bilatérales.
D’autres opportunités sont offertes aux membres de l’industrie afin de participer à la consultation et de fournir vos commentaires. Ceci inclut la possibilité aux compagnies de solliciter une rencontre bilatérale avec le CEPMB pendant la période de consultation.
Le CEPMB encourage aussi tous ses intervenants et membres du public à soumettre leurs commentaires par écrit avant le 31 janvier, 2020.
Le 2 décembre 2019
Faits saillants et nouvelles
- Le CEPMB attend avec intérêt les commentaires des intervenants sur les Lignes directrices provisoires, qui ont été publiées le 21 novembre 2019.
Réunions et discussions
Le 21 novembre 2019, le CEPMB a tenu une discussion par téléphone avec des représentants des services de santé de l’Alberta (Alberta Health Services).
Cette semaine, le CEPMB rencontrera des ministères provinciaux et territoriaux de la santé, des assureurs privés et d’autres acteurs importants au sein du système des soins de santé de la Colombie-Britannique, de l’Alberta, de la Saskatchewan et du Manitoba.
La semaine prochaine
La semaine du 9 décembre, des représentants du CEPMB seront à Ottawa (Ontario). Une séance de sensibilisation d’un jour axée sur la conformité et les questions opérationnelles sera tenue auprès de certains représentants de l’industrie. Le 10 décembre, le CEPMB tiendra un forum de la société civile d’un jour.
Le 21 novembre 2019
Faits saillants et nouvelles
- Le 21 novembre, le CEPMB a publié ses nouvelles Lignes directrices provisoires et a entamé une période de consultation de 60 jours auprès des intervenants et des membres intéressés du public.
- Le CEPMB a aussi publié un document d’information sur la consultation, lequel explique comment participer à la consultation et répond à certaines questions.
Réunions et discussions
La semaine du 2 décembre, des représentants du CEPMB se déplaceront aux villes suivantes afin de rencontrer divers intervenants :
- Victoria et Vancouver (Colombie-Britannique)
- Edmonton (Alberta)
- Regina (Saskatchewan)
- Winnipeg (Manitoba)
Commentaires
Vous trouverez ci-dessous les soumissions que le CEPMB a reçues pour la période de consultation des lignes directrices de juin 2020 avant la date limite du 4 août 2020.Les observations individuelles ne sont pas affichées, sauf si le soumissionnaire présente une observation à titre de professionnel de la santé, d’universitaire ou de représentant d’une organisation intéressée.
Le CEPMB reconnait et remercie les milliers de Canadiens qui ont exprimé leurs vues sur les Lignes directrices au cours de ce processus de consultation. Toutes les soumissions ont été examinées avec soin par le personnel du CEPMB.
Webinaires
Le CEPMB a tenu deux consultations publiques en ligne sur les Lignes directrices provisoires.
- Webinaire à l'intention de l’industrie – le 29 juin (PDF - 1,4 Mo)
- Ce webinaire était réservé aux représentants de l’industrie pharmaceutique
- Webinaire à l'intention du public – le 8 juillet (PDF - 2,6 Mo)
- Ce webinaire était ouvert à toutes les parties intéressées
Le CEPMB a également organisé deux séances réservées aux partenaires de la santé (le 25 juin) et aux assureurs privés (le 7 juillet).
Webinaires à l’intention des chercheurs
De plus, le CEPMB a offert trois webinaires à l’intention des chercheurs sur des analyses thématiques susceptibles d’éclairer la consultation.
1er webinaire à l’intention des chercheurs : le 23 juin (PDF - 1,7 Mo)
- Aperçu des dépenses en médicaments onéreux pour les maladies rares
- Aperçu de la taille du marché des médicaments brevetés au Canada
2e webinaire à l’intention des chercheurs : le 6 juillet (PDF - 2,8 Mo)
- Le prix des médicaments et les répercussions sur les investissements en R-D, les essais cliniques et l'accès aux médicaments au Canada
3e webinaire à l’intention des chercheurs : le 16 juillet (PDF - 2,7 Mo)
- Les pénuries de médicaments au Canada