
Lignes directrices du CEPMB 2019
Ces lignes directrices provisoires ont été publiées en novembre 2019. La consultation sur cette version des lignes directrices provisoires a pris fin. Pour obtenir la dernière version des lignes directrices provisoires, veuillez consulter la page Consultation sur les Lignes directrices provisoires du CEPMB.
Lignes directrices provisoires
Table des matières
- I. Préface
- II. Interprétation
- III. Cadre juridique
- IV. Exigences en matière de présentation de renseignements dans le cadre de l’examen des prix
- V. Processus d’examen des prix
- VI. Réévaluation
- VII. Enquêtes
- VIII. Engagement de conformité volontaire (ECV)
- IX. Recommandations relatives à l’audience
- X. Processus d’audience relativement aux prix excessifs et recours
- XI. Audience pour défaut de présenter les renseignements requis
- XII. Plaintes
- XIII. Annexes
- A. Comparaison selon la catégorie thérapeutique des prix nationaux (CCTn) et comparaison selon la catégorie thérapeutique des prix internationaux (CCTi)
- B. Test de la relation raisonnable et formes posologiques comparables
- C. Évaluation de la valeur pharmacoéconomique
- D. Méthode de rajustement en fonction de la taille du marché
I. Préface
1. Le Conseil d’examen du prix des médicaments brevetés (CEPMB), créé en 1987 en tant que pilier de la protection des consommateurs dans le cadre d’importantes réformes de la Loi sur les brevets (la « Loi »), est un organisme quasi judiciaire doté d’un mandat de réglementation dont l’objectif est de veiller à ce que les titulaires de brevets (ou brevetés) pharmaceutiques ne facturent pas des prix excessifs aux consommateurs pendant la période de monopole de droit. La mise sur pied du CEPMB découle de la crainte qu’une protection accrue des brevets des médicaments puisse entraîner une hausse inacceptable de leur prix et les rendre inabordables pour les consommateurs. À titre de membre du portefeuille de la Santé, le CEPMB contribue à un système de santé moderne et viable en veillant à ce que les Canadiens continuent d’avoir accès aux médicaments brevetés à des prix raisonnables.
2. Les présentes Lignes directrices, établies conformément au paragraphe 96(4) de la Loi, visent à assurer aux brevetés la transparence et la prévisibilité du processus habituellement suivi par les fonctionnaires du CEPMB (le « personnel ») pour déterminer si le prix d’un médicament breveté semble excessif sur un des différents marchés au Canada. Les Lignes directrices présentent également un aperçu des processus que les brevetés devraient connaître en ce qui a trait à leurs obligations en matière de présentation de renseignements en vertu du Règlement sur les médicaments brevetés (le « Règlement »).
3. Si le prix d’un médicament breveté semble excessif au regard des Lignes directrices et que le breveté n’a pas soumis d’engagement de conformité volontaire (ECV) acceptable, le personnel peut recommander au président de tenir une audience sur la question. Si une telle audience est jugée dans l’intérêt public par le président et qu’un panel d’audience composé de membres du Conseil (le « panel d’audience ») confirme que le prix du médicament breveté était excessif sur tout marché, le CEPMB peut rendre une ordonnance obligeant le breveté à baisser le prix du médicament et à prendre des mesures pour compenser l’excédent des recettes que lui aurait procuré la vente du médicament au prix excessif.
II. Interprétation
4. Les Lignes directrices fournissent des renseignements sur l’approche générale du CEPMB à l’égard du processus d’examen des prix et des enquêtes. Elles remplacent l’ensemble des documents d’orientation, des communiqués de politique et des énoncés écrits ou verbaux antérieurs du CEPMB concernant l’administration du processus d’examen des prix et des enquêtes, y compris les versions antérieures du Compendium des politiques, des Lignes directrices et des procédures du CEPMB. Les Lignes directrices doivent être lues en parallèle avec la Loi, le Règlement, les annexes desdites Lignes directrices et les autres documents d’orientation connexes que le CEPMB publie de temps à autre, dont le Guide du breveté.
5. Conformément au paragraphe 96(4) de la Loi, les Lignes directrices ne lient ni le personnel, ni le président, ni les panels d’audience, ni les brevetés, et elles ne visent pas à créer des droits ou des présomptions juridiques, à réaffirmer la loi ou à faire une déclaration définitive concernant l’interprétation des dispositions législatives se rapportant au CEPMB. Les décisions du personnel en matière d’application de la loi et la résolution définitive des questions dépendront des circonstances particulières de l’affaire en cause. L’interprétation définitive de la loi relève du Conseil (qui siège en tant que panel d’audience) et des tribunaux.
6. Certains aspects des Lignes directrices peuvent être revus par le CEPMB à la lumière de l’expérience et de l’évolution des circonstances. Les Lignes directrices et les politiques émises par le CEPMB sont élaborées de façon transparente et permettent une consultation complète des parties intéressées. Lorsqu’il envisagera d’apporter des modifications aux Lignes directrices, le CEPMB consultera les intervenants conformément au processus de consultation établi en vertu du paragraphe 96(5) de la Loi.
7. Le CEPMB fera tous les efforts raisonnables pour aider les brevetés à comprendre les Lignes directrices et leur application. Par exemple, le personnel informera sur demande les brevetés des méthodes à appliquer dans le cadre de l’examen du prix d’un nouveau médicament breveté vendu au Canada. En outre, si on lui en fait la demande et s’il dispose de suffisamment de renseignements pour le convaincre que le prix auquel le breveté vend ou a l’intention de vendre un médicament breveté ne serait pas jugé excessif, le Conseil peut délivrer un certificat non contraignant en ce sens en vertu du paragraphe 98(4) de la Loi.
8. Les Lignes directrices ne décrivent pas de manière exhaustive toutes les mesures qui peuvent être prises ou tous les problèmes qui peuvent survenir dans le contexte d’un examen du prix. Dans des circonstances exceptionnelles ou lors d’une audience, le CEPMB peut utiliser des méthodes ou des tests jugés appropriés et conformes à la Loi et au Règlement, même s’il n’en est pas question dans les Lignes directrices ou s’ils diffèrent de l’approche qui y est énoncée. En aucun cas le personnel ou les membres du Conseil ne seront liés ou limités par les Lignes directrices.
9. Pour obtenir de plus amples renseignements sur l’application des dispositions pertinentes de la Loi, du Règlement, des Lignes directrices ou de l’approche générale du personnel à l’égard de l’examen des prix et des enquêtes, veuillez consulter le site Web du CEPMB ou communiquer avec le personnel à l’adresse suivante :
Conseil d’examen du prix des médicaments brevetés
C.P. L40
Centre Standard Life
333, avenue Laurier Ouest
Bureau 1400
Ottawa (Ontario)
K1P 1C1
À l’attention de : Secrétaire du Conseil
III. Cadre juridique
10. Le CEPMB a été créé en 1987 après que des modifications d’envergure apportées à la Loi sur les brevets sont entrées en vigueur par l’adoption du projet de loi C-22Note de bas de page 1. Ces modifications ont permis de renforcer la protection des brevets des médicaments, tant du point de vue de la portée de l’objet brevetable que de la période d’exclusivité fondée sur les brevets, instaurant ainsi au Canada ce que les décideurs croyaient être un climat d’investissement plus propice à la recherche et au développement (R-D) pharmaceutique. En guise de concession faite aux opposants à ces modifications, qui craignaient que la protection renforcée des brevets des produits pharmaceutiques entraîne une hausse inacceptable des prix et rende ces produits inabordables pour les Canadiens, on a également créé le CEPMB dans la foulée du projet de loi C-22. Le CEPMB est un organisme de protection des consommateurs dont le mandat de réglementation consiste à s’assurer que le prix des médicaments brevetés n’est pas excessif. Essentiellement, le projet de loi C-22 cherchait à établir un équilibre entre la nécessité de reconnaître et de récompenser l’innovation pharmaceutique par l’octroi d’une période d’exclusivité de marché aux brevetés et la nécessité de veiller à ce que les prix demandés au cours de cette période d’exclusivité restent raisonnables. Comme en témoigne le statut du CEPMB, qui est le seul organisme sectoriel de réglementation des prix en vertu de la Loi, les décideurs reconnaissent que la capacité absolue d’établir le prix des médicaments brevetés n’est pas dans l’intérêt public, étant donné le préjudice unique qui peut découler du non-accès à ces médicaments par les consommateurs.
11. Le CEPMB est investi d’un double rôle : dans son rôle de réglementation, il protège les consommateurs en veillant à ce que le prix des médicaments brevetés ne soit pas excessif; dans son rôle de rapport, il fournit des renseignements sur les tendances des prix dans l’industrie pharmaceutique au moyen de son rapport annuel. Conformément à une directive du ministre de la Santé prise en application de l’article 90 de la Loi, le CEPMB appuie également la politique éclairée et fondée sur des données probantes en matière de santé en rendant compte des tendances relatives aux prix, à l’utilisation et aux coûts des médicaments dans le cadre de l’initiative du Système national d’information sur l’utilisation des médicaments prescrits (SNIUMP).
12. Le CEPMB est composé de cinq membres nommés par le gouverneur en conseil en vertu de l’article 91 de la Loi. La durée maximale de leur mandat est de cinq ans, renouvelable une seule fois. Le président du CEPMB agit comme premier dirigeant du Conseil et, à ce titre, il en assure la direction. Aux termes de l’article 94 de la Loi, le CEPMB emploie des fonctionnaires (le « personnel ») pour l’exercice de ses activités quotidiennes. Le directeur exécutif du CEPMB est son haut fonctionnaire, son chef de l’exploitation et son dirigeant principal des finances, et il est responsable de la gestion du personnel.
13. Le CEPMB a été créé en vertu de la Loi en tant qu’organisme indépendant quasi judiciaire. Afin d’assurer l’indépendance et l’autonomie du Conseil, la Loi ne confère aucun pouvoir exprès ou implicite à Santé Canada ou à toute autre entité gouvernementale qui permettrait de diriger le CEPMB dans l’exercice de ses fonctions de réglementation. Le CEPMB n’a aucun lien de dépendance avec le ministre de la Santé (qui est responsable des articles de la Loi applicables au CEPMB), le ministre de l’Innovation, des Sciences et du Développement économique (qui est responsable de la Loi dans son ensemble) et ses différents intervenants. De même, le CEPMB est structuré de manière à ce que les activités et les fonctions du personnel, du président et des membres du Conseil soient distinctes. En effet, le personnel s’acquitte des fonctions liées aux enquêtes, aux litiges et aux rapports, qui sont distinctes des fonctions d’arbitrage, réservées aux membres du Conseil.
14. La surveillance de la conformité des brevetés aux exigences réglementaires en matière de présentation de renseignements et l’examen administratif du prix des médicaments brevetés relèvent du personnel. Lorsque le prix d’un médicament breveté semble excessif et que la question ne peut pas être résolue au moyen d’une baisse volontaire du prix ou de mesures visant à compenser les recettes procurées au breveté par la vente du médicament à ce prix, le personnel peut porter le dossier à l’attention du président, qui déterminera si la tenue d’une audience est dans l’intérêt public. S’il décide de tenir une audience, le président émettra un avis d’audience et constituera un panel d’audience chargé de trancher la question. La décision du président d’émettre un avis d’audience est de nature purement administrative et n’exprime pas son point de vue sur le bien-fondé du dossier sous-jacent.
15. Le CEPMB examine le prix des médicaments brevetés vendus sans lien de dépendance par les brevetés (c.-à-d. le prix « départ usine » ou prix courant). Les ventes au Canada peuvent comprendre notamment les médicaments assujettis à un avis de conformité (AC), les médicaments obtenus par le Programme d’accès spécial, les médicaments figurant sur la Liste des drogues utilisées pour des besoins urgents en matière de santé publique, les médicaments visés par des demandes d’essais cliniques et les nouveaux médicaments expérimentaux. Le CEPMB n’a aucun pouvoir sur les prix demandés par des parties autres que les brevetés, comme les prix pratiqués par les grossistes ou les détaillants, ou sur les honoraires des pharmaciens.
16. En vertu de la Loi, le CEPMB a compétence pour déterminer si un médicament breveté est ou a été vendu par un titulaire de droits (le titulaire d’un brevet, l’ancien titulaire d’un brevet ou la personne ayant pour le moment droit à l’avantage d’un certificat de protection supplémentaire délivré à l’égard d’une invention liée à un médicament) à un prix excessif sur un marché au CanadaNote de bas de page 2. Le terme « titulaire de brevet » (ou breveté) est défini dans la Loi comme une personne ayant droit à l’avantage d’un brevet pour une invention pendant une période, ainsi que quiconque était titulaire d’un brevet pour une telle invention ou exerce ou a exercé les droits d’un titulaire, par exemple le titulaire d’une licence implicite ou expliciteNote de bas de page 3.
17. Une invention est liée à un médicament si elle est destinée à des médicaments ou à la préparation ou la production de médicaments, ou susceptible d’être utilisée à de telles fins. L’expression « liée à un médicament » a un sens large. La Cour d’appel fédérale a déterminé que la nature de ce lien peut être « ténue »Note de bas de page 4 [traduction]. Par exemple, il ne faut parfois qu’« un lien, aussi ténu soit-il, entre l’invention brevetée et le médicament vendu au Canada »Note de bas de page 5 [traduction].Note de bas de page 6
18. Au sens de la Loi, le terme « médicament » s’entend notamment d’une drogue (c.-à-d. une substance ou un mélange de substances qui est fabriqué, vendu ou présenté comme pouvant servir à l’une des fins suivantes : (i) le diagnostic, le traitement, l’atténuation, la prévention d’une maladie, d’un désordre, d’un état physique anormal ou de leurs symptômes, chez l’être humain ou les animaux; (ii) la restauration, la correction ou la modification des fonctions organiques chez l’être humain ou les animaux) et d’un ingrédient médicinalNote de bas de page 7. Sauf indication contraire, toute mention d’un « médicament » dans les présentes Lignes directrices inclut toutes les formes posologiques et toutes les concentrations (c.-à-d. tous les DIN) du médicament.
19. Le CEPMB reconnaît que le terme « ingrédient médicinal » signifie généralement « ingrédient pharmaceutique actif » (IPA) utilisé comme matière première au cours de la fabrication de la forme posologique finale. Les médicaments brevetés relevant de la compétence du CEPMB comprennent les vaccins, les préparations topiques, les anesthésiques et les produits diagnostiques utilisés in vivo, peu importe leur mode d’administration (c.-à-d. timbre transcutané, capsule, produit injectable, inhalateur). Toutefois, le CEPMB ne considère pas les instruments médicaux, les produits diagnostiques in vitro et les désinfectants qui ne sont pas utilisés in vivo comme étant des médicaments brevetés aux fins de l’application des dispositions de la Loi relatives à l’examen des prix.
20. Le CEPMB a compétence pendant toute la durée d’un brevet admissible et délivré, ce qui comprend la période précédant la délivrance du brevet. Le CEPMB a également compétence pendant la période de protection prolongée accordée par un certificat de protection supplémentaire.Note de bas de page 8
21. Le CEPMB continue d’avoir compétence sur le prix auquel un médicament breveté est vendu sur un marché canadien après la cession du brevet et jusqu’à l’annulation ou à l’abandon du brevet conformément aux dispositions expresses de la Loi ou à la péremption du brevet. La cession de brevets n’est pas expressément reconnue dans la Loi comme un mécanisme d’extinction des droits que confère un brevet avant la péremption normale du brevet.
22. Les ordonnances rendues par le CEPMB sont exécutoires de la même manière que les ordonnances de la Cour fédérale ou de toute cour supérieure au Canada, et elles peuvent être exécutées par le Conseil ou par la Cour fédérale. Les décisions contenues dans les ordonnances rendues par le CEPMB peuvent faire l’objet d’un contrôle judiciaire par la Cour fédérale, conformément aux principes de droit administratif et à la Loi sur les Cours fédérales.
IV. Exigences en matière de présentation de renseignements dans le cadre de l’examen des prix
23. Le CEPMB doit avoir accès à des renseignements exacts et opportuns au sujet de la vente des médicaments brevetés pour pouvoir s’acquitter de son mandat de réglementation. Par conséquent, les brevetés et les anciens brevetés sont tenus de présenter ces renseignements au CEPMB.
24. Les renseignements à fournir sont énoncés à l’article 82 de la Loi et dans le Règlement. Les brevetés peuvent trouver d’autres détails qui les aideront à déterminer le contenu et la forme des renseignements à fournir et les échéances strictes à respecter qui sont établies par la loi dans le Guide du breveté, un document de référence publié par le personnel. Le respect des obligations en matière de présentation de renseignements est la responsabilité exclusive des brevetés. Ces obligations statutaires ne peuvent être levées ou modifiées par le personnel.
25. Les renseignements que les brevetés ou les anciens brevetés peuvent être tenus de fournir comprennent ce qui suit (liste non exhaustive) :
- Un avis décrivant l’intention du breveté de mettre en vente sur un marché canadien un médicament breveté qui n’y a jamais été vendu (c.-à-d. la première vente du médicament breveté) et les renseignements connexes (Notification de l’intention de vendre un médicament breveté);
- Les renseignements réglementaires sur l’identification et les caractéristiques d’un médicament breveté, tels que la monographie du médicament breveté ou les renseignements équivalents, et les DIN octroyés à chaque forme posologique et à chaque concentration du médicament breveté;
- Les renseignements réglementaires sur le prix d’un médicament breveté, comme le prix auquel chaque forme posologique et chaque concentration du médicament breveté est ou a été vendu sur tout marché canadien ou dans n’importe lequel des onze pays indiqués dans le Règlement (le « CEPMB11 »);
- Les renseignements réglementaires sur les analyses coût-utilité préparées par un organisme canadien d’évaluation des technologies de la santé (ETS) financé par l’État, dont les résultats sont exprimés en fonction du coût par année de vie ajustée en fonction de la qualité (AVAQ), pour chaque indication faisant l’objet de l’analyse;
- Les renseignements réglementaires sur l’utilisation maximale approximative d’un médicament breveté au Canada, en fonction de la quantité prévue des ventes du médicament breveté sous sa forme posologique finale.
26. En vertu du paragraphe 7 du Règlement, les brevetés doivent présenter les renseignements exigés par courriel en utilisant les formulaires électroniques disponibles sur le site Web du CEPMB. Les formulaires doivent porter la signature électronique d’une personne autorisée qui atteste que les renseignements sont exacts et complets.
27. Il incombe à chaque breveté de s’assurer de manière indépendante que les renseignements présentés au CEPMB (dont les prix nationaux et internationaux) sont exacts. Le personnel peut effectuer une vérification ponctuelle des renseignements fournis par les brevetés, ce qui comprend les renseignements sur les prix, les recettes et les brevets. Dans l’éventualité d’une telle vérification, les brevetés peuvent être appelés à fournir d’autres documents à l’appui, ou encore à corriger ou à confirmer des renseignements fournis.
28. Le défaut de soumettre les renseignements requis dans le délai imparti ou le fait de présenter des renseignements erronés ou faux peuvent avoir des conséquences graves pour les brevetés ou les anciens brevetés. Si les circonstances le justifient, le Conseil peut rendre une ordonnance accordant certains recours, dont une ordonnance ex parte qui exige que les renseignements manquants soient présentés. Le dossier peut également mener à des poursuites par procédure sommaire en vertu du paragraphe 76.1(1) de la Loi. En outre, la présentation de faux renseignements constitue un acte criminel en vertu de l’article 76 de la Loi, lequel peut entraîner une amende ou un emprisonnement sur déclaration de culpabilité.
29. La Loi prévoit la confidentialité des renseignements fournis au CEPMB dans certaines circonstances. En effet, les renseignements ou les documents fournis au CEPMB en application des dispositions des articles 80, 81 et 82 de la Loi qui concernent les renseignements sur les prix ou dans le cadre d’une poursuite relative aux prix excessifs intentée en vertu de l’article 83 sont protégés et ne peuvent être divulgués au public sans l’autorisation de la partie qui les a fournis, à moins que ces renseignements n’aient été divulgués lors d’une audience publique tenue aux termes de l’article 83 de la Loi ou ne soient visés par les exceptions énoncées au paragraphe 87(2) de la Loi.
30. Les renseignements fournis au CEPMB peuvent être assujettis à certaines dispositions de la Loi sur l’accès à l’information et de la Loi sur la protection des renseignements personnels.
V. Processus d’examen des prix
31. Le processus d’examen des prix comprend une série d’étapes au cours desquelles (i) les médicaments sont regroupés par catégorie selon la date de leur lancement et des critères liés à leurs caractéristiques commerciales et (ii) les prix plafonds sont établis et évalués en fonction du prix des médicaments. En général, c’est le personnel qui effectue l’examen des prix en utilisant les méthodes et les tests énoncés dans les présentes Lignes directrices, d’après les renseignements fournis par les brevetés ou obtenus par le personnel auprès de sources externes pertinentes, comme les listes de médicaments des régimes publics.
32. La première étape du processus d’examen consiste à regrouper les médicaments brevetés comme suit : (i) les formes posologiques et les concentrations des médicaments brevetés qui bénéficient de droits acquis et qui ont reçu un numéro d’identification de médicament (DIN) avant le 21 août 2019 et (ii) toutes les autres formes posologiques et concentrations des médicaments brevetés qui n’avaient pas reçu de DIN en date du 21 août 2019 (c.-à-d. les médicaments brevetés ne bénéficiant pas de droits acquis).
33. Les facteurs d’examen du prix des médicaments brevetés bénéficiant de droits acquis [anciens facteurs prévus au paragraphe 85(1)] et de tous les autres médicaments brevetés ne bénéficiant pas de droits acquis [facteurs anciens et nouveaux prévus au paragraphe 85(1)] sont les suivants :
- Médicaments brevetés bénéficiant de droits acquis :
- Le prix de vente du médicament sur un marché canadien;
- Le prix de vente de médicaments de la même catégorie thérapeutique sur un marché canadien;
- Le prix de vente du médicament et d’autres médicaments de la même catégorie thérapeutique à l’étranger;
- Les variations de l’indice des prix à la consommation (IPC).
- Tous les autres médicaments brevetés ne bénéficiant pas de droits acquis :
- Le prix de vente du médicament sur un marché canadien;
- Le prix de vente de médicaments de la même catégorie thérapeutique sur un marché canadien;
- Le prix de vente du médicament et d’autres médicaments de la même catégorie thérapeutique à l’étranger;
- Les variations de l’indice des prix à la consommation (IPC);
- La valeur pharmacoéconomique du médicament au Canada;
- La taille du marché du médicament au Canada;
- Le produit intérieur brut du Canada et le produit intérieur brut par habitant au Canada.
34. Les processus et les tests qui s’appliquent aux médicaments brevetés et aux médicaments brevetés bénéficiant de droits acquis dans le cadre du processus d’examen des prix sont présentés en détail ci-après.
A. Processus d’examen du prix des médicaments brevetés ne bénéficiant pas de droits acquis
35. Le CEPMB examine les renseignements fournis par les brevetés et les renseignements obtenus d’autres sources pour déterminer si le prix d’un médicament breveté lancé sur un marché canadien semble excessif. Le diagramme ci-après illustre le processus d’examen du prix des nouveaux médicaments brevetés :
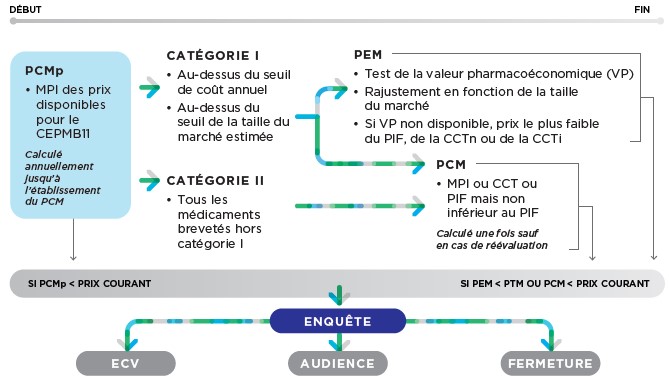
36. Chacune des étapes susmentionnées est décrite en détail ci-après.
Étape 1 : PCMp (tous les médicaments brevetés)
37. Au moment du lancement d’un médicament breveté, on établit un prix plafond provisoire applicable au prix courant (« prix courant maximum provisoire » ou « PCMp ») pour la vente du médicament breveté. Le PCMp correspond à la médiane des prix courants internationaux (« MPI ») des pays du CEPMB11 pour lesquels le breveté a fourni des renseignements pendant la période provisoire. Le breveté doit s’assurer que le prix départ usine brut accessible au publicNote de bas de page 9 du médicament breveté au Canada (« prix courant ») ne dépasse pas le PCMp pour la période au cours de laquelle il s’applique, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
38. Le PCMp est recalculé chaque année et s’applique jusqu’à la première des éventualités suivantes : (i) trois ans à partir de la date de lancement du médicament breveté au Canada ou (ii) la date à laquelle le breveté fournit des renseignements sur les prix internationaux d’au moins cinq pays du CEPMB11. À la fin de la période provisoire, on établit le PCM (voir l’étape 2), et le PCMp cesse de s’appliquer.
Étape 2 : PCM (tous les médicaments brevetés)
39. Sous réserve de la procédure décrite ci-dessus, le PCMp est remplacé par un « prix courant maximum » ou « PCM ». Le PCM est établi en fonction de la valeur la plus faible entre la MPI et la médiane obtenue avec la comparaison selon la catégorie thérapeutique des prix nationaux (« CCTn »), mais il est assujetti à un prix plancher fixé en fonction du prix international le plus faible (« PIF ») parmi les pays du CEPMB11 pour lesquels le breveté a fourni des renseignements à la fin de la période provisoire.
40. Le PCM est établi comme suit :
- La MPI et le PIF sont déterminés.
- Le prix de la CCTn est déterminé (voir l’annexe A, « CCTn et CCTi »).
- Si le prix de la CCTn est supérieur à la MPI, le PCM est établi à la MPI.
- Si le prix de la CCTn est inférieur à la MPI, mais supérieur au PIF, le PCM est établi au prix de la CCTn.
- Si le prix de la CCTn est inférieur au PIF, le PCM est établi au PIF.
41. Il peut arriver que le PCM soit inférieur au PCMp établi au cours de la période provisoire. Le cas échéant, le breveté dispose d’une période de transition jusqu’à la prochaine période de rapport après l’établissement du PCM pour s’assurer que le prix courant du médicament breveté est réduit à un niveau qui ne dépasse pas le PCM.
42. Si le PCM a été établi par la MPI et, lors des périodes ultérieures, la MPI en cours dépasse de plus de 10 p. 100 le PCM, le PCM peut être rajusté par rapport à l’IPC réel retardéNote de bas de page 10, tant que le PCM ne dépasse pas la MPINote de bas de page 11. Le PCM peut aussi être réévalué s’il a été établi par la MPI et, plus tard, la MPI en cours baisse à un niveau plus de 10 p. 100 inférieur au PCM (voir la section VI).
43. Comme c’est le cas pour le PCMp, le breveté doit s’assurer que le prix courant du médicament breveté ne dépasse pas le PCM pour la période au cours de laquelle il s’applique, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
a) Prix courant du médicament breveté au Canada
44. Conformément au Règlement, le breveté doit présenter des renseignements sur le prix courant de chaque forme posologique, de chaque concentration et de chaque format d’emballage (c.-à-d. pour chaque DIN) du médicament breveté dans chaque province et territoire.
45. Le prix courant peut varier entre les marchés canadiens (c.-à-d. entre les provinces et territoires). Lorsqu’il y a plusieurs prix courants sur différents marchés, le prix courant le plus élevé est utilisé aux fins de la comparaison du PCMp et du PCM.
b) Prix courant du médicament breveté dans les pays du CEPMB11
46. La MPI et le PIF sont fondés sur les renseignements fournis par le breveté. Lorsqu’il y a plusieurs prix courants dans le même pays, le prix le plus faible est utilisé. Les prix de tous les pays du CEPMB11 pour lesquels le breveté a fourni des renseignements sont utilisés.
47. Pour comparer les prix dans les pays du CEPMB11, on convertit la monnaie locale en dollars canadiens au moyen des taux de change exprimés sous forme de moyenne simple de la moyenne des taux du cours au comptant à midi pour chaque pays (arrondie à la huitième décimale) publiés par la Banque du Canada. Pour la période d’introduction d’un médicament breveté, la période de trente-six mois se terminant deux mois avant la date de la première période de rapport (c.-à-d. février ou août 2019) sera utilisée. Par la suite, la période de trente-six mois se terminant le deuxième mois après la période de rapport sera utilisée.
Étape 3 : PEM (médicaments brevetés de catégorie 1 seulement)
48. Les médicaments brevetés sont classés comme étant des médicaments brevetés de catégorie I ou de catégorie II en fonction de certaines caractéristiques commerciales. En plus du PCMi et du PCM, les médicaments brevetés de catégorie I sont également assujettis à un plafond pour le « prix escompté maximum » ou « PEM ». Le PEM tient compte de la valeur thérapeutique et des mesures de l’abordabilité (c.-à-d. la valeur pharmacoéconomique et la taille du marché du médicament breveté). Le breveté doit s’assurer que le prix netNote de bas de page 12 du médicament breveté au Canada (c.-à-d. le « prix de transaction moyen » ou « PTM ») ne dépasse pas le PEM, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
a) Classification d’un médicament breveté de catégorie I
49. Les médicaments brevetés de catégorie I sont les médicaments brevetés qui remplissent l’un ou l’autre des critères suivants :
- Le coût de traitement sur 12 mois dépasse 50 p. 100 du PIB par habitant : Une fois que le breveté a présenté les renseignements sur le prix pour la période de lancement, le personnel calcule le coût de traitement sur 12 mois du médicament selon les facteurs suivants : la dose maximale par traitement indiquée dans la monographie du produit; le nombre maximal de traitements par période de 12 mois en fonction de la nature du problème de santé, des pratiques cliniques et d’autres critères pertinents; le prix courant canadien le plus élevé. S’il n’y a pas de prix courant de disponible, le prix net national est utilisé.
- La taille estimée ou réelle du marché (recettes) dépasse le seuil annuel de taille du marché : Le seuil annuel de taille du marché est établi au départ à 25 millions de dollarsNote de bas de page 13.
50. Tous les autres médicaments brevetés sont des médicaments brevetés de catégorie II, y compris les nouveaux DIN octroyés après le 21 août 2019 pour les nouvelles formes posologiques, concentrations ou formats d’emballage de médicaments brevetés dont au moins un DIN date d’avant le 21 août 2019 et dont les nouveaux DIN ne sont pas liés à une nouvelle indication thérapeutique.
a) Calcul du PEM
51. Le PEM est calculé comme suit :
- Le rapport coût-efficacité différentiel (« RCED ») et le coût par année de vie ajustée en fonction de la qualité (« AVAQ ») pour chaque indication du médicament breveté sont déterminés à partir des analyses coût-utilité présentées par le breveté.
- Le RCED est comparé au seuil de la valeur pharmacoéconomique (« SVP ») de 60 000 dollars par AVAQ (voir l’annexe C, « Évaluation de la valeur pharmacoéconomique »).
- Le prix auquel le RCED du médicament breveté serait équivalent au SVP est déterminé (le « prix pharmacoéconomique » ou « PPE »).
- Le PPE peut être rajusté en fonction de la taille du marché si les ventes par unité du médicament breveté au PEM établi par le PPE générerait des recettes annuelles de plus de 25 millions de dollars (voir l’annexe D, « Méthode de rajustement en fonction de la taille du marché »).
- Dans le cas des médicaments brevetés associés à une prévalence totale estimée ne dépassant pas 1 sur 2 000 pour toutes les indications approuvées, le PEM est établi à 50 p. 100 au-dessus du PPE, mais il sera rajusté en fonction de la taille du marché si le médicament breveté génère des recettes annuelles de plus de 12,5 millions de dollars (voir l’annexe D, « Méthode de rajustement en fonction de la taille du marché »).
52. Si, au terme de la procédure décrite ci-dessus, le PEM dépasse le PCM, le PEM est établi au même niveau que le PCM.
b) Circonstances exceptionnelles
53. Si le breveté ne présente pas une analyse coût-utilité préparée par un organisme canadien financé par l’État pour un médicament breveté de catégorie I, ou si l’analyse présentée ne permet pas de déterminer le PEM conformément à la procédure décrite ci-dessus, il est possible d’établir le PEM à l’aide d’autres méthodes, dont la suivante :
- Le PEM est établi en fonction de la valeur la plus faible entre le PIF, la valeur obtenue avec la CCTn et celle obtenue avec la comparaison selon la catégorie thérapeutique des prix internationaux (« CCTi »), puis il est rajusté au moyen de la méthode de rajustement en fonction de la taille du marché.
54. Comme de tels scénarios devraient être rares et dépendront des faits, ils seront traités au cas par cas.
55. Une fois le PEM établi, il ne sera réévalué que si les conditions énumérées dans la section VI sont présentes.
Étape 4 : Détermination de l’indication pertinente
56. Si le médicament breveté a plus d’une indication ou d’un usage approuvé, le personnel déterminera, au moment du lancement du médicament ou dans le cadre d’une réévaluation si d’autres indications sont approuvées au cours du cycle de vie du médicament (voir la section VI), l’indication pertinente pour laquelle le PCM (et le PEM, le cas échéant) sera évalué.
57. S’il s’agit d’un médicament de catégorie I, l’indication pertinente sera l’indication qui remplit le critère relatif au coût annuel de traitement aux fins de la classification des médicaments brevetés de catégorie I, tel qu’il est énoncé dans les présentes Lignes directrices. Si le médicament de catégorie I possède plus d’une indication qui remplit ce critère ou n’en possède aucune, ou s’il s’agit d’un médicament de catégorie II, l’indication pertinente sera l’indication associée au problème de santé ayant la prévalence la plus élevée (c.-à-d. la plus grande population de patients).
B. Processus d’examen du prix des médicaments brevetés bénéficiant de droits acquis
58. Les médicaments brevetés bénéficiant de droit acquis sont assujettis à un PCM, mais pas à un PEM.
59. Le PCM de tous les médicaments brevetés bénéficiant de droits acquis est établi en fonction de la valeur la plus faible entre (i) la MPI des pays du CEPMB11 pour lesquels le breveté a fourni des renseignements et (ii) le prix plafond du médicament breveté fixé conformément aux Lignes directrices applicables avant la publication des présentes Lignes directricesNote de bas de page 14. Si le PCM a été établi par la MPI et, plus tard, la MPI en cours grimpe à un niveau de plus de 10 p. 100 supérieur au PCM, ce PCM peut être rajusté par rapport à l’IPC réel retardéNote de bas de page 15 tant que le PCM ne dépasse pas la MPI. Le PCM peut aussi être réévalué s’il a été établi par la MPI et, plus tard, la MPI en cours baisse à un niveau plus de 10 p. 100 inférieur au PCM (voir la section VI). Le breveté doit s’assurer que le prix courant du médicament breveté ne dépasse pas le PCM pour la période au cours de laquelle il s’applique, sans quoi il pourrait faire l’objet d’un examen supplémentaire ou d’une enquête par le personnel.
60. La méthode de calcul de la MPI et du prix courant des médicaments brevetés bénéficiant de droits acquis est la même que la méthode prévue dans les présentes Lignes directrices pour les nouveaux médicaments brevetés [voir la section V(A), « Processus d’examen du prix des médicaments brevetés ne bénéficiant pas de droits acquis »].
61. Le breveté dispose d’une période de transition jusqu’à la prochaine période de rapport après l’établissement du PCM pour s’assurer que le prix courant du médicament breveté bénéficiant de droits acquis est réduit à un niveau qui ne dépasse pas le PCM, sans quoi il pourrait faire l’objet d’un examen supplémentaire ou d’une enquête par le personnel.
VI. Réévaluation
62. Il est possible que l’on réévalue la catégorie ou le prix plafond d’un médicament breveté pour s’assurer qu’ils restent pertinents à la suite de changements importants dans les conditions du marché ou l’usage du médicament.
63. Pour les médicaments brevetés ne bénéficiant pas de droits acquis, il peut y avoir une réévaluation dans l’une ou l’autre des situations suivantes :
- Une nouvelle indication d’un médicament breveté (de catégorie I ou II) est approuvée;
- Les ventes d’un médicament breveté de catégorie II ont dépassé le seuil de taille du marché (voir l’annexe D), contrairement à l’estimation initiale présentée par le breveté;
- La prévalence totale estimée par le personnel du Conseil associée à un médicament breveté de catégorie I pour toutes les indications approuvées est supérieure à 1 sur 2 000;
- L’analyse coût-utilité associée à un médicament breveté de catégorie I est rajustée.
64. Conformément aux procédures décrites à la section V, il est possible que l’on modifie l’indication pertinente d’un médicament breveté auquel on a attribué une nouvelle indication. L’attribution d’une nouvelle indication peut avoir une incidence sur la taille du marché du médicament breveté, les médicaments de comparaison appartenant à la même catégorie thérapeutique et le coût-efficacité. Il peut donc y avoir une augmentation ou une diminution du PCM ou du PEM.
65. Un médicament breveté de catégorie II auquel on a attribué une nouvelle indication peut passer de la catégorie II à la catégorie I s’il remplit un des critères de classification des médicaments brevetés de catégorie I. Un médicament breveté peut également passer de la catégorie II à la catégorie I si ses recettes sont supérieures au seuil annuel de taille du marché, contrairement à l’estimation initiale de la taille du marché présentée par le breveté. Dans un cas comme dans l’autre, on établira un PEM en raison du changement de catégorie.
66. Si le PEM d’un médicament breveté de catégorie I a été établi en fonction d’une prévalence totale estimée ne dépassant pas 1 sur 2 000 pour toutes les indications et que la prévalence totale réelle dépasse ce seuil, il est possible que l’on réévalue le PEM.
67. Pour tous les médicaments brevetés, qu’ils bénéficient de droits acquis ou non, si le PCM a été établi par la MPI et, plus tard, la MPI en cours baisse à un niveau plus de 10 p. 100 inférieur au PCM, le PCM sera rajusté par rapport à la nouvelle MPI. S’il existe plusieurs DIN pour le médicament breveté dont les PCM ont été établis par le test de la RR (voir l’annexe B) par rapport au PCM ainsi rajusté, les PCM des autres DIN seront également rajustés.
68. Si l’on décide de rajuster le PCM ou le PEM d’un médicament breveté à la suite d’une réévaluation, le breveté en sera avisé. Après réception de l’avis, le breveté disposera d’une période de transition jusqu’à la prochaine période de rapport pour le PEM afin de s’assurer que les prix sont rajustés de manière à ce que le prix du médicament breveté ne dépasse pas le prix plafond rajusté, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
VII. Enquêtes
69. Une enquête est un examen approfondi du prix d’un médicament breveté réalisé par le personnel. Dans le cadre d’une enquête, le personnel examine les renseignements fournis par le breveté et les renseignements pouvant être obtenus d’autres sources. Les enquêtes sont de nature purement administrative, et elles ne peuvent être menées que par le personnel. Aucun membre du Conseil ne participe au processus. Si une enquête entraîne la tenue d’une audience, le panel d’audience doit effectuer un examen de novo indépendant du prix du médicament breveté afin de déterminer s’il est excessif au sens de l’article 83 de la Loi. Par conséquent, les positions prises par le personnel ou par le breveté au cours de l’enquête peuvent différer de celles prises au cours de l’audience.
70. Les critères justifiant la tenue d’une enquête ont été élaborés dans le but d’assurer l’utilisation la plus efficace des ressources humaines et financières du CEPMB. Le fait que le prix d’un médicament breveté n’est pas visé par une enquête ne signifie pas nécessairement que le prix n’est pas excessif; cela signifie uniquement que les critères d’enquête n’ont pas été remplis dans les circonstances particulières.
A. Critères d’enquête
71. En règle générale, le personnel lancera une enquête si aux moins une des conditions suivantes est présente :
- Lorsque le prix de toute forme posologique ou concentration d’un médicament breveté semble être supérieur de plus de 5 p. 100 au prix plafond applicable correspondant;
- Lorsque les recettes cumulatives potentielles tirées de la vente à un prix supérieur au prix plafond applicable (« excédent des recettes potentielles ») semblent dépasser 50 000 dollars pour l’ensemble des produits du breveté qui contiennent le même médicament breveté (c.-à-d. tous les DIN contenant le même ingrédient médicinal) au cours d’une année civile;
- Lorsqu’une plainte est déposée.
72. Lorsque le prix d’un médicament breveté est supérieur aux prix plafonds applicables prévus dans les Lignes directrices, mais que les critères justifiant la tenue d’une enquête ne sont pas remplis, le breveté en est avisé, et le médicament breveté se voit attribuer la mention « Ne justifie pas une enquête » dans le rapport annuel du CEPMB. Le cas échant, le personnel ne prend aucune mesure immédiate; toutefois, le breveté doit s’assurer que le prix du médicament breveté est réduit à un niveau ne dépassant pas le prix plafond applicable et compenser les recettes qui peuvent avoir été tirées conformément à la section B.2, « Calcul de l’excédent des recettes potentielles », sans quoi il pourrait faire l’objet d’une enquête par le personnel.
B. Processus d’enquête
73. Lorsque le personnel lance une enquête, le breveté en est avisé, et le médicament breveté se voit attribuer la mention « Sous enquête » dans le rapport annuel du CEPMB. Le processus d’examen des prix et le processus d’enquête ne peuvent pas donner lieu à une décision judiciaire établissant que le prix d’un médicament breveté est excessif au sens de la Loi. Une telle décision ne peut être prise qu’une fois que le breveté ou l’ancien breveté a eu la possibilité raisonnable de présenter ses observations, conformément à l’article 83 de la Loi.
1. Examen supplémentaire des renseignements présentés
74. Lorsqu’un médicament breveté fait l’objet d’une enquête, le personnel examine l’historique des prix du médicament depuis son lancement. Tous les renseignements fournis par le breveté sont analysés, et d’autres éclaircissements peuvent être demandés. Par exemple, si un prix semble erroné ou inattendu, ou s’il y a des divergences dans les renseignements présentés par le breveté, celui-ci pourrait devoir fournir une explication ou d’autres documents à l’appui. Le personnel pourrait également prendre en considération tous les renseignements pertinents qui n’ont pas été soumis par le breveté, comme les prix courants obtenus avec la CCTn et la CCTi. En outre, le personnel analyse la pertinence des tests applicables en fonction des faits du dossier et des particularités des marchés pertinents sur lesquels le médicament breveté est vendu.
2. Calcul de l’excédent des recettes potentielles
75. Lorsque le prix d’un médicament breveté est supérieur aux prix plafonds prévus dans les Lignes directrices, le breveté est informé que le prix du médicament breveté « dépasse les seuils établis dans les Lignes directrices », et les prix plafonds applicables sont indiqués. Le personnel commence également à calculer l’excédent cumulatif des recettes potentielles en fonction des prix nets présentés par le breveté, quel que soit le prix plafond utilisé (PCMp, PCM ou PEM). Comme le CEPMB a compétence sur le médicament au cours de la période précédant la délivrance du brevet, l’excédent des recettes qu’aurait procuré la vente du médicament pendant cette période est inclus dans les calculs.
76. Au cours d’une audience, le personnel peut exercer un recours dans le cadre duquel l’excédent des recettes diffère de l’excédent cumulatif des recettes potentielles calculé lors de l’enquête. De plus, lorsque le personnel estime que le breveté ou l’ancien breveté s’est livré à une politique de vente d’un médicament breveté à un prix excessif, il peut, par ordonnance, lui enjoindre de compenser jusqu’au double de l’excédent des recettes.
3. Résultats de l’enquête
77. Les résultats possibles d’une enquête sont les suivants :
- Un engagement de conformité volontaire (« ECV »), tel qu’il est décrit dans la section VIII;
- L’émission d’un avis d’audience si, sur recommandation du personnel, le président estime que la tenue d’une audience est dans l’intérêt public;
- La clôture de l’enquête.
78. La clôture d’une enquête peut découler d’un ECV ou d’une décision selon laquelle un examen approfondi du prix d’un médicament breveté n’est pas justifié à ce moment-là, compte tenu des faits et des considérations qui ont été mis en lumière au cours de l’enquête.
79. La clôture d’une enquête est une mesure administrative; il ne s’agit pas d’une décision judiciaire établissant que le prix du médicament breveté n’est pas excessif ni d’un aveu du CEPMB en ce sens. La clôture d’une enquête n’exclut pas la possibilité que l’on rouvre une enquête, que l’on en lance une nouvelle ou que l’on tienne une audience dans l’avenir.
VIII. Engagement de conformité volontaire (ECV)
80. Le breveté peut présenter un ECV au personnel en tout temps avant l’émission d’un avis d’audience. Un ECV est une promesse par laquelle le breveté s’engage à baisser le prix du médicament visé par l’enquête ou à compenser l’excédent des recettes que lui aurait procuré la vente de ce médicament à ce prix. Une proposition d’ECV ne constitue pas un aveu du breveté quant au caractère excessif du prix du médicament.
81. Le personnel est chargé de mener les négociations relatives à l’ECV avec le breveté et, selon la politique du CEPMB, le président ne peut pas participer aux discussions. Si les négociations aboutissent à une proposition d’ECV qui, de l’avis du personnel, serait acceptable pour le président, la proposition sera soumise au président aux fins d’examen. Le personnel ne peut pas déterminer de façon indépendante si une proposition d’ECV est acceptable, et il ne peut pas donner de garantie au breveté quant à la probabilité que le président accepte la proposition.
82. L’examen d’un ECV est une procédure administrative; il ne s’agit pas d’une décision du CEPMB établissant que le prix du médicament présenté par le breveté ou utilisé pour le calcul du montant de la compensation n’est pas excessif ni d’un aveu du CEPMB en ce sens. Toutefois, l’acceptation d’un ECV par le président donnera lieu à la clôture de l’enquête.
83. Le CEPMB rend compte publiquement de tous les ECV acceptés par le président. Lorsqu’il présente un ECV signé, le breveté doit consentir à sa publication en version entière ou expurgée. Les renseignements publiés peuvent comprendre le contenu de l’ECV, dont les conditions qui y sont énoncées. Ces renseignements peuvent figurer dans le rapport annuel du CEPMB, sur le site Web du CEPMB, dans les publications du CEPMB, comme La Nouvelle, et sur les plateformes de médias sociaux.
84. Le personnel ne peut pas se pencher sur les demandes de négociations ou de propositions d’ECV « sans préjudice ». Les ECV sont des promesses unilatérales faites par les brevetés et ne sont pas des ententes entre le CEPMB et les brevetés. Néanmoins, les discussions entre les brevetés et le personnel du Conseil sont sujettes aux normes de protection énoncées aux articles 87 et 88 de la Loi et dans la Loi sur l’accès à l’information.
85. Lorsqu’un avis d’audience est émis, le breveté peut quand même négocier un règlement sous forme d’entente, laquelle doit être approuvée par le panel d’audience. Le personnel examine les demandes d’accord ou de proposition de règlement « sans préjudice ».
IX. Recommandations relatives à l’audience
86. Lorsqu’une enquête sur le prix d’un médicament breveté est terminée et que l’affaire n’est pas réglée avec le breveté, le personnel peut présenter un rapport au président. Le président, en sa qualité de premier dirigeant du CEPMB, peut décider d’émettre un avis d’audience s’il estime que la tenue d’une audience est dans l’intérêt public. La décision d’émettre un avis d’audience n’est d’aucune façon judiciaire ou quasi judiciaire, et le président n’effectue pas d’analyse pour déterminer si les faits allégués par le personnel sont ou seront prouvés. Aucun membre du CEPMB (à l’exception du président du Conseil, conformément à la procédure décrite ci-dessus) n’est informé des résultats de l’examen du personnel sur le prix d’un médicament breveté tant que l’affaire n’est pas soumise à un panel d’audience au cours d’une audience publique.
87. La décision quant au caractère excessif du prix d’un médicament breveté est prise par le panel d’audience seulement, après la tenue de l’audience publique.
X. Processus d’audience relativement aux prix excessifs et recours
88. Les audiences du CEPMB sont publiques. Lors d’une audience, un panel d’audience, composé d’au moins deux membres du Conseil, entend les observations et les éléments de preuve des parties. Le panel détermine si un médicament breveté est ou a été vendu à un prix excessif sur n’importe quel marché canadien en tenant compte des renseignements disponibles concernant les facteurs énoncés à l’article 85 de la Loi.
89. Pour obtenir de plus amples renseignements sur les audiences, veuillez consulter les Règles de pratique et de procédure du CEPMB, l’ensemble normalisé et publié de procédures que tous les participants à des audiences devant le Conseil doivent suivre. Les Règles établissent les procédures du CEPMB conformément à l’exigence prévue dans la Loi selon laquelle le Conseil doit résoudre les questions soumises à son attention sans formalisme, en procédure expéditive, dans la mesure où les circonstances et l’équité le permettent. Les instructions relatives à la pratique et d’autres renseignements au sujet des audiences en cours ou passées sont à la disposition du public sur le site Web du CEPMB.
90. En vertu de la Loi, le CEPMB est habilité à rendre des ordonnances correctives lorsqu’il est déterminé, à la suite d’une audience, qu’un breveté (ou un ancien breveté) vend ou a vendu un médicament breveté à un prix excessif sur un marché canadienNote de bas de page 16.
91. En termes généraux, le CEPMB a le pouvoir d’exercer des deux grandes voies de recours après une audience : (i) enjoindre le breveté de baisser, dans un marché canadien, le prix de vente du médicament dans la mesure et pour la période prévue par l’ordonnance, ou de baisser, dans un marché canadien, le prix de vente de tout autre médicament lié à une invention brevetée du titulaire dans la mesure et pour la période prévue par l’ordonnance; (ii) enjoindre le breveté de payer à Sa Majesté du chef du Canada un montant précisé.
92. Si le panel d’audience conclut que le breveté ou l’ancien breveté s’est livré à une politique de vente du médicament breveté à un prix excessif, il peut, par ordonnance, lui enjoindre de compenser, selon lui, jusqu’au double de l’excédent des recettes procuré par la vente du médicament breveté au prix excessif. Le Conseil examinera l’étendue et la durée des ventes du médicament breveté au prix excessif lorsqu’il arrivera à cette conclusion et rendra l’ordonnance.
XI. Audience pour défaut de présenter les renseignements requis
93. Lorsque le personnel estime qu’un breveté a omis ou a refusé de fournir au CEPMB les renseignements sur les prix, les ventes ou les recettes, ou d’autres renseignements semblables exigés par la loi, il recommande au président de tenir une audience publique pour déterminer si le breveté a bel et bien manqué aux exigences en matière de présentation de renseignements prévues par la Loi et le Règlement. Si, à la suite d’une audience, le panel d’audience conclut que le breveté a manqué à ces exigences, il peut, par ordonnance, lui enjoindre de fournir au CEPMB les renseignements et les documents requis en vertu de l’article 81 ou 88 de la Loi.
94. En outre, aux termes du paragraphe 76.1(1) de la Loi, quiconque contrevient aux exigences en matière de présentation de renseignements énoncées aux articles 80, 81, 82 ou 88 ou à une ordonnance prise sous le régime de l’un ou l’autre de ces articles commet une infraction et encourt, sur déclaration de culpabilité par procédure sommaire, une amende ou un emprisonnement.
XII. Plaintes
95. Une personne ou un groupe qui estime que le prix d’un médicament breveté est excessif peut déposer une plainte au CEPMB par téléphone, par écrit ou par voie électronique. Les coordonnées à utiliser se trouvent sur la page « Comment déposer une plainte » du site Web du CEPMB.
96. Toute plainte déclenche une enquête sur le prix du médicament breveté visé par la plainte. Le plaignant ne participe pas à l’enquête ni aux audiences qui en résultent (à moins qu’il ne présente une demande pour participer aux audiences à titre d’intervenant). Le plaignant n’est pas tenu de fournir des documents ou des éléments de preuve au CEPMB. Les enquêtes sont fondées sur les documents fournis par le breveté ou obtenus autrement par le personnel.
97. En raison des limites en matière de communication énoncées aux articles 87 et 88 de la Loi sur les brevets et dans la Loi sur l’accès à l’information, le plaignant est informé du résultat de l’enquête uniquement si le processus mène à un ECV ou à un avis d’audience.
XIII. Annexes
A. Comparaison selon la catégorie thérapeutique des prix nationaux (CCTn) et comparaison selon la catégorie thérapeutique des prix internationaux (CCTi)
Test de la CCTn
Tel qu’il est décrit dans la section V des présentes Lignes directrices, le test de la comparaison selon la catégorie thérapeutique des prix nationaux (« CCTn ») sert à calculer le PCM d’un médicament breveté et, dans certains cas, son PEM. Le test de la CCTn permet de comparer le prix courant d’un médicament breveté avec le prix courant d’autres médicaments sélectionnés au terme d’un examen scientifique.
Sélection des médicaments aux fins de comparaison
de classification anatomique thérapeutique chimique (ATC) du Centre collaborateur de l’Organisation mondiale de la Santé (OMS) pour la méthodologie sur l’établissement des statistiques concernant les produits médicamenteux.
Les médicaments utilisés aux fins de comparaison appartiennent généralement à la sous-catégorie du système ATC située immédiatement au-dessus de la substance chimique simple. Il s’agit habituellement du quatrième niveau de sous-catégorie, mais il peut également s’agir de la sous-catégorie supérieure suivante ou d’une autre sous-catégorie. Dans certains cas, la sélection peut devoir être faite au cinquième niveau, soit au niveau de la substance chimique simple.
Un médicament de la même catégorie thérapeutique du système ATC que le médicament à l’étude peut être omis s’il ne se prête pas à la comparaison. Par exemple, un médicament comportant une indication ou un usage principal autre que l’indication ou l’usage principal du médicament breveté à l’étude peut être omis de la comparaison.
Tous les médicaments sélectionnés aux fins de comparaison qui comportent la même indication approuvée ou le même usage approuvé que l’indication pertinente du médicament breveté à l’étude sont inclus dans l’examen. Cet examen repose sur les recherches effectuées par le personnel et peut comprendre d’autres recherches menées par un centre d’information sur les médicaments ou une analyse des données probantes présentées par les brevetés. Dans certains cas, le personnel peut également demander des conseils non exécutoires au Groupe consultatif sur les médicaments pour usage humain (GCMUH).
Lorsque le médicament breveté est une nouvelle forme posologique ou concentration du même ingrédient médicinal contenu dans un ou plusieurs médicaments existants, les médicaments existants qui sont offerts dans la même forme posologique ou dans une forme posologique comparable et qui comportent la même indication ou le même usage servent de médicaments de comparaison, et ce, que le régime posologique du médicament nouveau et des médicaments existants soit le même ou diffère sensiblement.
Lorsque le produit est une combinaison de médicaments qui sont chacun vendus au Canada et qui comportent tous la même indication ou le même usage, les médicaments de comparaison sont limités aux médicaments composants.
Régimes posologiques comparables
Le régime posologique comparable utilisé aux fins de comparaison correspond normalement au régime posologique maximal habituellement recommandé dans la monographie de produit (ou dans des renseignements semblables), et il tient compte des variables cliniques pertinentes. La concentration est déterminée en fonction du régime posologique du médicament à l’étude. En règle générale, un régime posologique fondé sur un traitement s’appliquera aux indications aiguës, tandis qu’un régime quotidien (selon la dose d’entretien) sera utilisé pour les indications chroniques.
Sources des prix
Les brevetés ne présentent pas de renseignements sur le prix des médicaments sélectionnés aux fins de comparaison avec un médicament breveté. On consulte d’abord les Listes de médicaments des régimes provinciaux pour obtenir le prix d’un médicament de comparaison dans le cadre du test de la CCTn. Pour chaque médicament de comparaison sélectionné, on utilise le prix public le plus bas. Le personnel peut décider d’exclure du test de la CCTn un médicament (breveté ou non) sélectionné aux fins de comparaison s’il a des raisons de croire que le médicament est vendu à un prix excessif.
Test de la CCTn
Après avoir sélectionné les médicaments qui se prêtent à la comparaison et déterminé le prix public le plus bas de chaque médicament, on calcule le coût d’un traitement comparable pour chaque médicament. Les différents coûts de traitement sont triés, et la médiane, établie. Lorsqu’un nombre pair de médicaments est utilisé aux fins de comparaison, la médiane correspond à la moyenne simple des deux coûts de traitement du milieu. Le coût médian de traitement est ensuite divisé par le nombre d’unités constituantes du traitement comparable pour le médicament à l’étude, ce qui permet d’établir un prix unitaire.
Test de la CCTi
Tel qu’il est décrit dans la section V des présentes Lignes directrices, le test de la comparaison selon la catégorie thérapeutique des prix internationaux (« CCTi ») peut servir à calculer le PEM d’un médicament breveté. Le test de la CCTi permet de comparer le prix courant d’un médicament breveté avec le prix courant d’autres médicaments sélectionnés au terme d’un examen scientifique aux fins de comparaison dans les onze pays de comparaison énumérés dans le Règlement.
Sélection des médicaments aux fins de comparaison et régimes posologiques comparables
Le test de la CCTi se fonde sur les mêmes médicaments sélectionnés aux fins de comparaison et les mêmes régimes posologiques comparables que ceux utilisés pour le test de la CCTn, conformément aux procédures établies précédemment. Lorsqu’aucun médicament de comparaison n’a été trouvé au pays, le personnel peut déterminer si d’autres médicaments comportent la même indication approuvée ou le même usage approuvé que le médicament à l’étude dans l’un des pays de comparaison.
Sources des prix
Les brevetés ne présentent pas de renseignements sur les prix internationaux des médicaments sélectionnés à l’étranger aux fins de comparaison avec un médicament breveté. On emploie le prix départ usine accessible au public des médicaments sélectionnés aux fins de comparaison dans le cadre du test de la CCTi. Pour chaque médicament de comparaison sélectionné, on utilise le prix public le plus bas.
Le personnel peut décider d’exclure du test de la CCTi un médicament (breveté ou non) sélectionné aux fins de comparaison s’il a des raisons de croire que le médicament est vendu à un prix excessif.
Test de la CCTi
Après avoir sélectionné les médicaments qui se prêtent à la comparaison et déterminé le prix public le plus bas de chaque médicament, on calcule le coût d’un traitement comparable pour chaque médicament dans chaque pays de comparaison. Les différents coûts de traitement sont triés, et la médiane de chaque pays est établie. Lorsqu’un nombre pair de médicaments est utilisé aux fins de comparaison, la médiane correspond à la moyenne simple des deux coûts de traitement du milieu. Ces médianes sont triées, et une « médiane des médianes » est définie. Le coût médian de traitement est ensuite divisé par le nombre d’unités constituantes du traitement comparable pour le médicament à l’étude, ce qui permet d’établir un prix unitaire. Les devises étrangères sont converties en dollars canadiens selon la méthode décrite à la section V des présentes Lignes directrices.
B. Test de la relation raisonnable et formes posologiques comparables
Test de la relation raisonnable
Le test de la relation raisonnable (RR) peut servir à déterminer le PCM ou le PEM d’une nouvelle concentration d’un médicament breveté qui est déjà offert dans une ou plusieurs autres concentrations, pourvu que la nouvelle concentration comporte le même ingrédient médicinal, la même indication, le même régime posologique et la même forme posologique (ou une forme posologique comparable) que la ou les concentrations existantes.
Lorsqu’une nouvelle concentration d’un médicament déjà vendu au Canada est lancée et qu’elle satisfait aux exigences susmentionnées du test de la RR, le PCM ou le PEM de la nouvelle concentration est établi comme étant équivalent au prix par unité standard de la ou des concentrations existantes. On procède de la même façon lorsque plusieurs concentrations d’un nouveau médicament sont vendues simultanément pour la première fois et que certaines concentrations sont définies plus précisément comme étant des doses de charge, d’ajustement ou de réduction.
Formes posologiques comparables
Les formes posologiques ci-après sont considérées comme étant des formes posologiques comparables pour les besoins du test de la RR. Les formulations d’un même groupe sont considérées comme étant comparables, mais les formes posologiques de groupes différents ne le sont pas.
Topical (T)
- Aérosol
- Aérosol (mousse)
- Crème
- Disque (à libération prolongée)
- Disque
- Pansement
- Gel
- Gel (à libération contrôlée)
- Liposomes
- Liquide
- Lotion
- Onguent
- Tampon
- Peinture
- Pâte
- Timbre
- Timbre (à libération prolongée)
- Crayon
- Plâtre
- Poudre
- Shampooing
- Savon en pain
- Solution
- Éponge
- Vaporisateur
- Vaporisateur (valve à poche)
- Vaporisateur (à dose mesurée)
- Bâtonnet
- Bandelette
- Écouvillon
- Teinture
Nasale (N) ou pulmonaire (P)
- Aérosol
- Aérosol à dose mesurée
- Gouttes
- Gaz
- Préparation à dose mesurée
- Poudre
- Poudre (à dose mesurée)
- Solution
- Solution (à libération prolongée)
- Vaporisateur
- Vaporisateur (à dose mesurée)
- Bâtonnet
Solide administré par voie orale (S)
- Barre (à croquer)
- Caplet
- Capsule
- Granules effervescents
- Poudre effervescente
- Comprimé effervescent
- Pellicule (soluble)
- Globules
- Granules
- Gomme à mâcher
- Pastille
- Caplet à libération modifiée
- Capsule à libération modifiée
- Comprimé à libération modifiée
- Pellet
- Morceau (à croquer)
- Poudre (à libération prolongée)
- Bandelette
- Comprimé
- Comprimé (à croquer)
- Comprimé (à dissolution orale)
- Comprimé pour suspension
- Cachet
Liquide administré par voie orale (L)
- Gouttes
- Solution buvable
- Émulsion
- Gel
- Granules pour solution
- Granules pour suspension
- Granules pour suspension (à libération retardée)
- Granules pour suspension (à libération prolongée)
- Liquide
- Liquide à libération modifiée
- Poudre (à libération prolongée)
- Poudre pour solution
- Poudre pour suspension
- Solution
- Solution (à libération prolongée)
- Vaporisateur
- Suspension
- Suspension (à libération prolongée)
- Sirop
- Sirop (à libération prolongée)
- Thé (à base d’herbes)
- Teinture
Vaginale (V)
- Cône
- Crème
- Douche
- Mousse
- Gel
- Gel (à libération contrôlée)
- Implant
- Insert
- Insert (à libération prolongée)
- Ovule
- Pellet
- Anneau (à libération lente)
- Éponge
- Suppositoire
- Suppositoire (à libération soutenue)
- Tampon
- Comprimé vaginal
- Comprimé vaginal (effervescent)
Parentérale (J)
- Bolus
- Implant
- Trousse
- Liposomes
- Injection à libération modifiée
- Pellet (implantable)
- Poudre pour solution
- Poudre pour suspension (à libération soutenue)
- Solution
- Solution (à libération prolongée)
- Suspension pour émulsion
- Suspension (à libération prolongée)
Otique (E) ou ophtalmique (Y)
- Gouttes
- Gel
- Gel (à libération contrôlée)
- Implant
- Insert
- Insert (à libération prolongée)
- Liquide
- Dispositif oculaire à libération modifiée
- Onguent
- Poudre pour solution
- Solution
- Solution (à libération prolongée)
- Suspension
Rectale (R)
- Crème
- Lavement
- Mousse
- Insert
- Onguent
- Ovule
- Bâtonnet
- Suppositoire
- Suppositoire (à libération soutenue)
- Suspension
- Suspension (à libération prolongée)
Dentaire, sublinguale ou buccale (M)
- Émulsion
- Pellicule (soluble)
- Soie dentaire
- Gel
- Gel (à libération contrôlée)
- Gomme à mâcher
- Pastille
- Pompe à dose mesurée
- Comprimé buccal à libération modifiée
- Rince-bouche (gargarisme)
- Pâte
- Poudre (effervescente)
- Poudre pour suspension
- Solution
- Solution (à libération prolongée)
- Vaporisateur – buccal
- Vaporisateur – sublingual
- Bâtonnet
- Bandelette
- Comprimé sublingual
- Suspension
- Suspension (à libération prolongée)
- Écouvillon
- Comprimé (à dissolution orale)
- Comprimé
- Dentifrice
- Poudre dentifrice
- Cachet
C. Évaluation de la valeur pharmacoéconomique
Tel qu’il est décrit dans la section V des présentes Lignes directrices, l’évaluation de la valeur pharmacoéconomique sert à calculer le PEM d’un médicament. Le Règlement exige que les brevetés présentent des renseignements relatifs à toutes les analyses pharmacoéconomiques d’un médicament préparées par des organismes canadiens financés par l’État si le coût de traitement calculé au prorata du médicament au cours d’une période de 12 mois est supérieur ou égal à 50 p. 100 du PIB par habitant au moment de la publication de l’analyse.
Sélection du modèle
Les rapports pharmacoéconomiques du Programme commun d’évaluation des médicaments (PCEM) et les rapports finaux d’orientation économique du Programme pancanadien d’évaluation des anticancéreux (PPEA) de l’Agence canadienne des médicaments et des technologies de la santé (ACMTS) sont les sources habituelles d’analyses pharmacoéconomiques utilisées pour les besoins de l’évaluation de la valeur pharmacoéconomique. Les analyses préparées par l’Institut national d’excellence en santé et services sociaux (INESSS) dans ses Évaluations aux fins d’inscription sont également étudiées.
Dans le cadre de l’évaluation de la valeur pharmacoéconomique, les calculs du personnel sont fondés sur la nouvelle analyse du scénario de référence menée par l’organisme public (c.-à-d. l’ACMTS ou l’INESSS), par opposition à l’analyse effectuée à l’aide du modèle de scénario de référence présenté par le breveté. Il s’agit d’un modèle d’analyse coût-utilité dans lequel les résultats en matière de santé sont exprimés sous forme d’AVAQ. Si le rapport de l’organisme ne contient pas d’analyse coût-utilité, on peut avoir recours à un modèle de minimisation des coûts. Si le médicament comporte plusieurs indications, on utilisera l’évaluation pharmacoéconomique de l’indication pertinente, déterminée conformément à la procédure énoncée dans la section V des présentes Lignes directrices.
Calcul du prix pharmacoéconomique (PPE)
Le CEPMB compte sur les organismes pour publier dans leurs rapports des estimations qui lui permettent de calculer le PPE.
Pour les médicaments qui procurent des avantages pour la santé par rapport aux soins actuels, le PPE est calculé comme suit :
PPE = P1(SVP * AVAQ supplémentaires + coût de traitement - coûts supplémentaires) / Coût de traitement
Aux fins du calcul :
- La variable SVP représente le seuil de la valeur pharmacoéconomique de 60 000 dollars par AVAQNote de bas de page 17;
- La variable P1représente le prix courant du médicament utilisé dans les rapports de l’organisme;
- La variable AVAQ supplémentaires représente l’estimation ponctuelle des AVAQ supplémentaires gagnées grâce au médicament par rapport au médicament de comparaison employé dans le modèle d’analyse coût-utilité du scénario de référence de l’organisme, exprimée en valeur actuelle;
- La variable Coûts supplémentaires représente l’estimation ponctuelle des coûts supplémentaires du médicament par rapport au médicament de comparaison utilisé, exprimée en valeur actuelle;
- La variable Coût de traitement représente l’estimation ponctuelle des coûts du médicament par patient au cours de l’horizon temporel étudié dans le rapport de l’organisme, exprimée en valeur actuelle. Cette valeur est limitée au médicament évalué et exclut le coût des autres médicaments utilisés en association avec le médicament évalué ou des médicaments utilisés pour traiter les effets secondaires.
Pour les médicaments qui ne procurent pas d’avantages pour la santé par rapport aux traitements existants, le PPE peut être déterminé de l’une des deux manières suivantes :
-
Si les coûts connexes du médicament fournis par les ressources de soins de santé financées par l’État diffèrent de ceux des soins actuels, le PPE est calculé comme suit :
PPE = P1 (coût de traitement - coûts supplémentaires) / Coût de traitement - Si les coûts connexes du médicament fournis par les ressources de soins de santé financées par l’État sont équivalents à ceux des soins actuels, le PPE est équivalent à la valeur de la CCTn.
Populations de patients multiples
Il peut arriver que l’organisme calcule plusieurs PPE en fonction de sous-populations distinctes. Le cas échéant, le personnel calcule un PPE moyen pondéré en se servant de tous les modèles du scénario de référence qui s’appliquent à une proportion importante (10 p. 100 et plus) de la population cible totale. Si le PPE d’une sous-population particulière tend vers zéro ou l’infini, le CEPMB peut l’exclure de la moyenne pondérée. Comme la population cible est la population qui peut bénéficier du médicament pour l’indication pertinente au Canada, la moyenne pondérée exclut également les groupes de patients pour lesquels les résultats en matière de santé devraient être pires que ceux liés aux soins actuels selon le modèle de l’organisme.
D. Méthode de rajustement en fonction de la taille du marché
Tel qu’il est décrit dans la section V des présentes Lignes directrices, la méthode de rajustement en fonction de la taille du marché est appliquée aux médicaments brevetés de catégorie I dont les ventes par unité généreraient des recettes annuelles supérieures à 25 millions de dollars pour toutes le formes posologiques et concentrations du médicament breveté (tous les DIN) si le médicament breveté était vendu au PEM établi par le PPE. On applique ce rajustement chaque année afin de déterminer le PEM conformément au tableau ci-dessous :
Rajustement en fonction de la taille du marché pour les médicaments de catégorie I
| Recettes annuelles | Facteur de rajustement supplémentaire | PEM | |
|---|---|---|---|
| Médicaments avec un PPE | Médicaments sans PPE | ||
| Moins de 25 M$ | 0 % | PPE | Valeur la plus faible (PIF, CCTn, CCTi) |
| 25 M$ à 50 M$ | -10 % | PPE rajusté en fonction du facteur applicable | Valeur la plus faible (PIF, CCTn, CCTi) rajustée en fonction du facteur applicable |
| 50 M$ à 75 M$ | -20 % | ||
| 75 M$ à 100 M$ | -30 % | ||
| 100 M$ à 125 M$ | -40 % | ||
| 125 M$ et plus | -50 % | ||
Le rajustement du PEM d’un médicament breveté en fonction de la taille du marché est évalué chaque année selon les ventes par unité réelles au cours de l’année civile précédente; toutefois, il n’est pas évalué lorsque les ventes ont été générées sur une période inférieure à la période de rapport. Le rajustement est appliqué progressivement en fonction des unités vendues dans chaque catégorie de recettes de plus de 25 millions de dollarsNote de bas de page 18.
Par exemple, le PEM établi par le PPE pour un médicament avec des recettes annuelles entre 25 millions de dollars et 50 millions de dollars serait rajusté selon la formule suivante :
PEM = [25 M$ + 0,9 * PPE * (Unités - 25 M$/PPE)] / Unités
On calcule la moyenne des tranches supplémentaires pour chaque autre catégorie de recettes procurées par les ventes par unité le médicament breveté. Par exemple, le PEM établi par le PPE pour un médicament avec des recettes annuelles entre 50 millions de dollars et 75 millions de dollars serait rajusté selon la formule suivante :
PEM = [25 M$ + 25 M$ + 0,8 * PPE * (Unités - 25 M$/PPE - 25 M$/0,9 * PPE)] / Unités
Après le rajustement initial en fonction de la taille du marché, le PEM ne peut être rajusté que si le nombre d’unités vendues augmente.Il n’y a pas d’augmentation du PEM d’un médicament breveté si le nombre d’unités vendues baisse ou si les recettes réalisées ultérieurement se situent dans une catégorie inférieure.
Médicaments utilisés dans le traitement des maladies rares
Dans le cas des médicaments brevetés de catégorie I, le personnel évalue également la prévalence du ou des problèmes de santé indiqués. Les médicaments brevetés associés à une prévalence totale estimée ne dépassant pas 1 sur 2 000 pour toutes les indications approuvées sont désignés comme étant des médicaments brevetés « utilisés dans le traitement des maladies rares ». Cette évaluation repose sur les recherches effectuées par le personnel et peut comprendre d’autres recherches menées par un centre d’information sur les médicaments ou un examen des données probantes présentées par les brevetés.
Aux fins du calcul initial du PEM de ces médicaments brevetés, on détermine le PPE et on applique une augmentation de 50 p. 100. Pour les médicaments brevetés dont les ventes par unité généreraient des recettes réelles entre 12,5 millions de dollars et 25 millions de dollars au cours de l’année civile précédente, on calcule ensuite le PEM en appliquant un rajustement supplémentaire fondé sur le PPE conformément à la procédure décrite précédemment pour les autres médicaments de catégorie I. Enfin, le rajustement en fonction de la taille du marché est appliqué aux recettes de chaque catégorie de plus de 25 millions de dollarsNote de bas de page 19.
Rajustement en fonction de la taille du marché pour les médicaments brevetés de catégorie I utilisés dans le traitement des maladies rares
| Recettes annuelles | Facteur de rajustement supplémentaire | PEM | |
|---|---|---|---|
| Médicaments avec un PPE | Médicaments sans PPE | ||
| Moins de 12,5 M$ | +50 % | 1,5 * PPE | Valeur la plus faible (PIF, CCTn, CCTi) |
| 12,5 M$ à 25 M$ | 0 % | PPE | |
| 25 M$ à 50 M$ | -10 % | PPE rajusté en fonction du facteur applicable | Valeur la plus faible (PIF, CCTn, CCTi) rajustée en fonction du facteur applicable |
| 50 M$ à 75 M$ | -20 % | ||
| 75 M$ à 100 M$ | -30 % | ||
| 100 M$ à 125 M$ | -40 % | ||
| 125 M$ et plus | -50 % | ||
Par exemple, le PEM établi par le PPE pour un médicament breveté utilisé dans le traitement d’une maladie rare avec des ventes par unité qui généreraient des recettes annuelles entre 12,5 millions de dollars et 25 millions de dollars serait rajusté selon la formule suivante :
PEM = [12,5 M$ + PPE * (Unités - 12,5 M$/1,5 * PPE)] / Unités
De la même façon, le PEM établi par le PPE pour un médicament breveté utilisé dans le traitement d’une maladie rare avec des ventes par unité qui généreraient des recettes annuelles entre 25 millions de dollars et 50 millions de dollars serait rajusté selon la formule suivante :
PEM = [12,5 M$ + 12,5 M$ + 0,9 * PPE * (Unités - 12,5 M$/1,5 * PPE - 12,5 M$/PPE)] / Unités
Document d’information
Document d’information – Consultation sur les Lignes directrices provisoires du CEPMB 2019
Table des matières
- Consultation sur les Lignes directrices provisoires du CEPMB
- Participation aux consultations
- Questions et réponses
- 1) Pourquoi le CEPMB procède-t-il à des consultations sur les nouvelles Lignes directrices provisoires?
- 2) Les changements proposés dans les Lignes directrices provisoires sont nécessaires pour mettre en oeuvre les modifications au Règlement sur les médicaments brevetés. En quoi consistent ces modifications?
- 3) Qu’y a-t-il de nouveau dans les Lignes directrices provisoires?
- 4) Qu’entendez-vous lorsque vous affirmez que les Lignes directrices provisoires adoptent une approche fondée sur le risque en matière de réglementation des prix?
- 5) Comment les Lignes directrices provisoires simplifient et allègent-elles le processus de conformité des titulaires de brevets?
- 6) En quoi consistent les nouveaux facteurs de l’article 85, et comment le CEPMB les interprète-t-il dans les Lignes directrices provisoires?
- 7) Que signifie la valeur pharmacoéconomique dans le contexte des Lignes directrices provisoires?
- 8) Que signifie la taille du marché dans le contexte des Lignes directrices provisoires?
- 9) Que signifient les termes PIB et PIB par habitant dans le contexte des Lignes directrices provisoires?
- 10) Comment le CEPMB aboutit-il à un seuil de valeur pharmacoénonomique de 60 000 $/AVAQ dans les Lignes directrices provisoires?
- 11) Comment le CEPMB aboutit-il à un seuil de taille de marché de 25 millions $ dans ses Lignes directrices provisoires?
- 12) Pourquoi les Lignes directrices provisoires accordent-elles une hausse de 50 % du PEM des médicaments de catégorie I qui traitent des problèmes de santé ayant une très faible prévalence au Canada?
- 13) Pourquoi les Lignes directrices provisoires n’utilisent-elles pas les mêmes seuils de coût par AVAQ mentionnés à la section de l’analyse coût-avantage du REIR?
- 14) Quelles sont les nouvelles exigences en matière de production de rapports du Règlement modifié?
- 15) Pourquoi le PEM ne s’applique-t-il qu’aux médicaments brevetés de catégorie I?
- 16) Pourquoi le PEM est-il évalué au regard du prix net plutôt que du prix courant?
- 17) Pourquoi le PCM est-il évalué au regard du prix courant plutôt que du prix net?
- 18) Quelles mesures de protection s’appliquent aux renseignements fournis par les brevetés?
- 19) Pourquoi les Lignes directrices provisoires exigent-elles que les brevetés fournissent une analyse coût-utilité uniquement pour les médicaments dont le coût de traitement annuel est de plus de 50 % du PIB par habitant?
- 20) Pourquoi le CEPMB utilise-t-il le rapport de l’ACMTS comme principal modèle source?
- 21) Pourquoi les Lignes directrices provisoires fournissent-elles un seul plafond de prix pour un médicament breveté ayant plusieurs indications?
- 22) Le CEPMB collaborera-t-il avec l’ACMTS et l’INESSS pour s’assurer que les données nécessaires figurent dans leurs rapports?
- 23) Pourquoi les médicaments brevetés bénéficiant de droits acquis sont-ils soumis à la nouvelle annexe des pays de comparaison, et pourquoi la médiane internationale établit-elle leur prix plafond?
- 24) Comment les Lignes directrices provisoires traitent-elles de l’IPC?
- 25) Comment et quand le CEPMB assurera-t-il le suivi et rendra-t-il compte de l’incidence du Règlement modifié et des nouvelles Lignes directrices, notamment s’ils produisent les effets escomptés?
Consultation sur les Lignes directrices provisoires du CEPMB
Seules les Lignes directrices provisoires font l’objet des consultations. Le présent document a un rôle auxiliaire au processus de consultation; il ne fait pas partie des Lignes directrices provisoires, et ne fait pas l’objet de consultations.
Le Conseil d’examen du prix des médicaments brevetés (CEPMB) est un organisme quasi judiciaire doté d’un mandat de réglementation dont l’objectif est d’empêcher les titulaires de brevets pharmaceutiques de facturer des prix excessifs aux consommateurs pendant la période de monopole de droit. Sa création découle de la préoccupation qu’une protection accrue des brevets des médicaments pourrait entraîner une hausse inacceptable des prix de sorte qu’ils deviennent inabordables pour les consommateurs.
En vertu du paragraphe 96(4) de la Loi sur les brevets, le CEPMB doit publier des lignes directrices (les « Lignes directrices ») qui visent à rendre transparent et prévisible le processus entamé par les employés de la fonction publique du CEPMB (le « personnel ») pour leur permettre de déterminer si le prix d’un médicament breveté semble excessif sur le marché canadien. Les Lignes directrices fournissent aussi un aperçu des processus que les titulaires de brevets devraient connaître quant à leurs obligations en matière de déclaration en vertu du Règlement sur les médicaments brevetés (le « Règlement »).
Le 21 novembre 2019, le CEPMB a annoncé une période de consultation de 60 jours concernant les changements proposés aux Lignes directrices (les « Lignes directrices provisoires »). Les consultations visent à recueillir la rétroaction des titulaires de brevets, des intervenants et des membres intéressés du public concernant les Lignes directrices provisoires.
Participation aux consultations
Le CEPMB consultera les Canadiens de la façon suivante :
- Une période initiale de consultation de 60 jours durant laquelle les intervenants et les membres intéressés du public peuvent fournir des propositions écrites concernant les Lignes directrices provisoires;
- Un forum de politiques, où les intervenants seront invités à se présenter en personne devant le Conseil pour faire connaître leurs points de vue;
- La création de groupes de travail pour discuter de questions et d’enjeux particuliers soulevés par les Lignes directrices provisoires.
Les intervenants et les membres intéressés du public ont jusqu’au 14 février 2020 pour fournir leurs propositions écrites. Le calendrier du forum de politiques et des groupes de travail techniques sera communiqué à une date ultérieure. Le CEPMB tient à connaître les opinions et les points de vue des Canadiens et attend impatiemment de recevoir des commentaires constructifs et pertinents sur les nouvelles Lignes directrices provisoires dans le cadre du présent processus de consultation.
Le présent Document d’information sur les consultations, est conçu comme document d’accompagnement des Lignes directrices provisoires et fournit des renseignements contextuels sur les changements clés proposés dans ce document. Il vise à permettre au lecteur de mieux comprendre les Lignes directrices provisoires et à rendre le processus de consultation plus éclairé et productifNote de bas de page 1 en répondant aux questions les plus fréquemment posées présentées ci-après.
Questions et réponses
1) Pourquoi le CEPMB procède-t-il à des consultations sur les nouvelles Lignes directrices provisoires?
Depuis l’établissement du CEPMB il y a trois décennies, le contexte réglementaire dans lequel il évolue a profondément évolué. Plus particulièrement, les activités de recherche et développement menées par l’industrie pharmaceutique, qui portaient auparavant sur les médicaments à petites molécules conventionnels à coût relativement faible pour le traitement de maladies courantes, se concentrent désormais sur les produits biologiques et les thérapies génétiques à coût élevé s’adressant à des populations de patients de taille restreinte. Or, ces nouveaux produits s’exposent souvent à un risque de prix excessif, car ils ont peu (voire pas du tout) de substituts concurrentiels, et parce que la demande pour des traitements innovants et améliorés est très élevée au sein des populations de patients sévèrement touchées. Cela est notamment vrai pour les médicaments inédits, ou pour lesquels les solutions de rechange sont moins efficaces ou encore présentent des effets secondaires moins tolérables.
En outre, il est devenu courant pour les intervenants de l’industrie de négocier des rabais et des réductions par rapport aux prix courants publics pour, en échange, voir leurs produits remboursés par les assureurs publics et privés. Cela permet aux fabricants de faire une discrimination par les prix entre les acheteurs en fonction de leur pouvoir compensateur perçu et de leur capacité à payer. Cela accentue aussi l’écart entre les prix courants publics (p. ex. les prix bruts, ou « départ-usine ») et les prix réels payés sur le marché (p. ex. les prix nets, ou « prix de transaction moyen »).
Les modifications apportées récemment au Règlement sur les médicaments brevetés (le « Règlement ») visent à fournir au CEPMB les outils et les données qu’il lui faut pour exécuter efficacement son mandat de protection des consommateurs à la lumière de ces changements. En vertu du Règlement modifié, le CEPMB peut déterminer, pour les médicaments brevetés, des prix plafonds non-excessifs bruts et nets qui correspondent mieux aux prix des pays aux vues similaires, qui sont plus représentatifs de leur valeur aux yeux des consommateurs canadiens, et qui sont mieux éclairés par les limites d’abordabilité de l’économie canadienne. Les changements apportés aux Lignes directrices provisoires sont nécessaires pour opérationnaliser les modifications réglementaires.
2) Les changements proposés dans les Lignes directrices provisoires sont nécessaires pour mettre en oeuvre les modifications au Règlement sur les médicaments brevetés. En quoi consistent ces modifications?
Le CEPMB détermine si le prix d’un médicament breveté est excessif au regard des facteurs de fixation des prix de l’article 85 de la Loi sur les brevets (la « Loi ») en utilisant les renseignements requis des titulaires de brevets en vertu du Règlement et des données connexes. Le 7 août 2019, le Règlement a été modifié de façon substantielle de trois façons :
1. Autres facteurs réglementaires de fixation des prix de l’article 85 :
- Les modifications offrent un complément aux facteurs dont le CEPMB doit tenir compte au moment de déterminer si le prix d’un médicament breveté est excessif aux termes de l’article 85 de la Loi sur les brevets, notamment la valeur et l’incidence financière d’un médicament sur les consommateurs et le système de santé. Parmi les nouveaux facteurs, notons la valeur phamacoéconomique, la taille du marché, ainsi que le produit intérieur brut (PIB) et le PIB par habitant au Canada.
2. Mise à jour de l’annexe des pays de comparaison (le nouveau « CEPMB11 »)
- Les modifications réexaminent l’annexe des pays de comparaison de sorte qu’elle cadre mieux avec le mandat du CEPMB de protéger les consommateurs des prix excessifs et qu’elle tienne davantage compte des priorités stratégiques du gouvernement du Canada.
3. Modification des exigences en matière de reddition de comptes
- Les modifications atténuent les obligations en matière de reddition de comptes relativement aux médicaments vétérinaires brevetés, aux médicaments en vente libre et aux médicaments génériques, afin que le CEPMB puisse concentrer ses efforts et ses ressources sur les médicaments brevetés dont le prix est plus à risque d’être excessif.
- Les modifications obligent certains titulaires de brevets à signaler l’information relative aux nouveaux facteurs de l’article 85 pour que le CEPMB puisse les administrer efficacement.
- Les modifications obligent les brevetés à signaler l’information sur les prix et les recettes, déduction faite de tous les rajustements de prix, y compris les réductions de prix directes et indirectes, de manière à ce que les activités réglementaires du CEPMB soient fondées sur un portrait réel et complet des prix des médicaments brevetés au Canada.
Les changements corrélatifs aux Lignes directrices sont nécessaires pour donner effet au Règlement modifié et officialiser la transition du CEPMB vers une approche plus efficace et axée sur le risque, le but étant d’offrir aux titulaires de brevets un moyen plus simple et facile de s’y conformer.
La publication des Lignes directrices provisoires est la plus récente et la dernière initiative du processus de consultation qui a commencé avec la publication du document de discussion du CEPMB concernant l’actualisation des Lignes directrices de juin 2016; elle cadre avec la feuille de route en matière de réforme établie dans son Plan stratégique de 2015-2018. Autant les Lignes directrices provisoires et le Règlement modifié qu’elles cherchent à opérationnaliser entreront en vigueur le 1er juillet 2020.
3) Qu’y a-t-il de nouveau dans les Lignes directrices provisoires?
Comme auparavant, les Lignes directrices provisoires fournissent des règles d’application générale qui servent de mécanisme pour déterminer comment les prix plafonds sont habituellement établis par les employés de la fonction publique du CEPMB (le « personnel »). Toutefois, on y propose une nouvelle approche en ce qui a trait à la surveillance et à l’examen des prix des médicaments brevetés, qui prévoit : 1) un système de vérification initiale permettant de déterminer les médicaments dont le prix est plus à risque d’être excessif (p. ex. les médicaments de « catégorie I »); 2) des plafonds distincts pour les prix bruts/courants (le prix courant maximum, ou « PCM ») et les prix nets (le prix escompté maximum, ou « PEM », qui ne s’appliqueront qu’aux médicaments de catégorie I); 3) les tests appliqués aux prix, qui intègrent à la fois les nouveaux et les anciens facteurs de l’article 85, et 4) une procédure de réévaluation pour les médicaments qui peuvent justifier l’établissement d’un plafond plus ou moins élevé en raison des changements des conditions du marché. Les Lignes directrices provisoires fournissent aussi des conseils aux titulaires de brevets sur la façon de se conformer aux nouvelles exigences en matière de déclaration figurant dans le Règlement modifié.
4) Qu’entendez-vous lorsque vous affirmez que les Lignes directrices provisoires adoptent une approche fondée sur le risque en matière de réglementation des prix?
Depuis juin 2018, le CEPMB sollicite des commentaires généraux des intervenants concernant un nouveau cadre proposé pour les Lignes directrices, lequel donnerait effet au Règlement modifié (tel que publié le 2 décembre 2017 dans la Partie I de la Gazette du Canada) et appuierait sa transition vers une approche axée sur le risque de réglementation des prix des médicaments brevetés qui simplifie et allège le respect de la conformité par les titulaires de brevets.
L’adoption d’une approche fondée sur le risque permet au CEPMB d’utiliser plus efficacement ses ressources en accordant la priorité réglementaire à la minorité des médicaments brevetés dont le prix est le plus à risque d’être excessif. Ceci est cohérent avec le but de la politique du gouvernement, soit d’intégrer les nouveaux facteurs de l’article 85 et de limiter l’exigence de consigner les analyses coût-utilité menées par l’Agence canadienne des médicaments et des technologies de la santé (ACMTS) et l’Institut national d’excellence en santé et services sociaux (INESSS) pour les médicaments brevetés dont le coût annuel de traitement dépasse 50 % du PIB par habitant. En vertu de la nouvelle approche, seuls les brevetés des médicaments dont l’analyse a déterminé qu’ils étaient de catégorie I seraient soumis à un examen approfondi au regard des nouveaux facteurs pour établir si leur prix peut être excessif. Les médicaments dont le prix est à plus faible risque d’être excessif seraient classés dans la catégorie II et, ainsi, soumis à une surveillance relativement moindre.
5) Comment les Lignes directrices provisoires simplifient et allègent-elles le processus de conformité des titulaires de brevets?
Les Lignes directrices actuelles et les Lignes directrices provisoires se distinguent par le fait que, une fois établis, les prix plafonds du CEPMB ne fluctuent pas d’année en année selon le prix net ou le prix de transaction moyen (PTM) du médicament breveté l’année précédente. Il s’agit d’une règle très stricte qui exige que les titulaires suivent de très près les PTM annuels et rajustent leurs prix en conséquence pour continuer à se conformer. En vertu des Lignes directrices provisoires, les titulaires de brevets ont l’entière liberté de modifier leurs prix d’année en année, du moment que ceux-ci demeurent en deçà du prix courant maximum (PCM) et du prix escompté maximum (PEM) selon le cas. En outre, la majorité des médicaments brevetés seront classés dans la catégorie II, pour laquelle le respect du PCM sera évalué uniquement au regard des prix courants. Les Lignes directrices provisoires réduisent considérablement le nombre de marchés et de catégories de clients pour lesquels les titulaires de brevets doivent présenter de l’information sur les prix et portent maintenant uniquement sur les prix à l’échelle nationale, provinciale ou territoriale. Par conséquent, les titulaires de brevets n’ont plus besoin de présenter de l’information sur les prix au niveau des ventes aux hôpitaux, pharmacies et grossistes. Par rapport à l’ancien Compendium des politiques, des Lignes directrices et des procédures, cette nouvelle approche est reflétée dans des Lignes directrices provisoires plus générales qui sont plus succinctes, moins techniques et plus faciles à comprendre.
6) En quoi consistent les nouveaux facteurs de l’article 85, et comment le CEPMB les interprète-t-il dans les Lignes directrices provisoires?
Le Règlement modifié intègre trois nouveaux facteurs au paragraphe 85(1), notamment la valeur pharmacoéconomique, la taille du marché, et le produit intérieur brut (PIB)/le PIB par habitant au Canada. Le Résumé de l’étude d’impact de la réglementation (REIR) indique que l’intégration de ces facteurs part du constat que le prix seul ne fournit pas suffisamment de contexte pour évaluer ce qui est jugé excessif dans le climat réglementaire actuel. Plus précisément, un prix qui ne tient pas compte du prix payé par les consommateurs ne tient pas compte d’intrants clés visant à déterminer si le médicament présente un excellent rapport qualité-prix relativement aux technologies qu’il remplacera ou à l’impact plus vaste qu’il pourrait avoir sur la capacité du système de santé à intégrer d’autres technologies de la sorte. Ce sont là d’importants points à prendre en considération à une époque marquée par des enveloppes budgétaires de la santé de plus en plus serrées, une population vieillissante et un nombre croissant de médicaments dont les coûts de traitement annuels moyens se chiffrent à des centaines de milliers de dollars.
7) Que signifie la valeur pharmacoéconomique dans le contexte des Lignes directrices provisoires?
La valeur pharmacoéconomique est la mesure du coût d’un médicament par rapport à son effet bénéfique sur la santé, qui peut être comparé à d’autres médicaments ou technologies de la santé en recourant à une mesure normalisée du bienfait. La mesure normalisée privilégiée par les agences d’évaluation des technologies de la santé (ETS) du monde entier est l’année de vie ajustée en fonction de la qualité (AVAQ). Les coûts et les répercussions sanitaires découlant de la mise en oeuvre d’une nouvelle technologie de la santé auprès d’une population particulière dans un contexte et un système de santé particuliers se traduisent souvent par le rapport coût-efficacité différentiel (RCED), exprimé sous forme de coût par AVAQ gagnée. Les RCED constituent une mesure utile pour quantifier l’ampleur des ressources additionnelles requises pour obtenir une amélioration mesurable de la santé (p. ex. le coût additionnel requis pour gagner une AVAQ).
Dans un système de santé publique, une nouvelle technologie de la santé améliorera les résultats globaux sur la santé seulement si ses effets bénéfiques sur la santé excèdent les coûts de renonciation associés aux ressources additionnelles requises qu’il faut débourser pour cette technologie. Le coût de renonciation se mesure au regard des avantages estimés pour la santé auxquels renoncent les autres patients du système de soins de santé lorsque des ressources fixes et intégralement affectées sont utilisées pour adopter un nouveau médicament. Une telle évaluation du coût de renonciation pour la santé traduit le coût maximal qu’un système de santé peut payer pour les bienfaits sur la santé offerts par une nouvelle technologie sans réduire la santé globale de la population. C’est ce qu’il convient d’appeler « un seuil du côté de l’offre ». Un tel seuil nécessite de connaître le coût marginal d’une AVAQ au sein du système de santé (c.-à-d., le point auquel l’acquisition d’une nouvelle technologie de la santé destinée à un ensemble de patients du système de santé publique entraînera la perte d’une AVAQ pour un autre ensemble de patients du système).
Il est souvent mentionné que le Canada est le seul pays ayant un système de soins de santé financé par l’État qui ne comporte pas d’assurancemédicaments universelle. Il en résulte une mosaïque de payeurs du secteur public et du secteur privé qui ne disposent pas du pouvoir d’achat national pour contrer le monopole de droit des titulaires de brevets. Cette situation de monopole se trouve renforcée lorsqu’une proportion sans cesse grandissante des dépenses publiques et privées est consacrée à des médicaments très coûteux pour lesquels il y a peu, voire pas du tout, de concurrents directs. En exigeant du CEPMB qu’il tienne compte de la valeur pharmacoéconomique de ces médicaments, le Règlement modifié veille à ce que le concept de coût de renonciation soit pris en compte dans l’évaluation du caractère excessif d’un prix. Puisque le marché privé canadien des produits pharmaceutiques est un prolongement du système public et qu’il dépend de celui-ci pour fonctionner, l’intention politique du Règlement modifié est que le CEPMB adopte la perspective du système de santé publique et favorise un « seuil de rentabilité du côté de l’offre » dans l’estimation
du coût de renonciation.8) Que signifie la taille du marché dans le contexte des Lignes directrices provisoires?
L’incidence économique d’un médicament particulier sur les consommateurs dépend à la fois du prix et du volume. Lorsque les assureurs publics et privés sont interpellés pour couvrir le coût d’un médicament pour un nombre important de patients, son prix pourrait rendre ce médicament inaccessible aux consommateurs. Cela peut être vrai même dans le cas des médicaments ayant un profil pharmacoéconomique favorable, puisque la vaste étendue du marché auquel ils s’adressent peut éventuellement entraîner le déplacement de technologies plus rentables et, ainsi, contribuer à la croissance non viable des coûts des soins de santé. Le contraire est aussi vrai pour ce qui est des médicaments s’adressant à des marchés très restreints, puisqu’ils n’ont pas, au cas par cas, tendance à donner lieu à des limites d’abordabilité, même si leur coût de renonciation est très élevé. En exigeant du CEPMB qu’il tienne compte de la taille du marché d’un médicament, le Règlement modifié veille à ce que l’incidence globale de la couverture du médicament pour quiconque en a besoin est prise en compte dans la détermination du caractère excessif du prix du médicament. Il permet aussi au CEPMB de réévaluer les prix des médicaments au fil du temps, au gré de la croissance ou de la décroissance du marché auquel ils s’adressent.
9) Que signifient les termes PIB et PIB par habitant dans le contexte des Lignes directrices provisoires?
Le produit intérieur brut (PIB) sert à mesurer la production économique d’un pays. La croissance du PIB sert à évaluer la mesure dans laquelle la valeur marchande des biens et services produits par une économie, rajustée en fonction de l’inflation, augmente au fil du temps. Le PIB par habitant permet de mesurer la production d’un pays par rapport à sa population. Le premier est un indicateur de richesse sociale, tandis que le second est considéré comme un indicateur du niveau de développement économique au sein de cette société.
Même si l’on sait que le contexte financier varie d’un utilisateur institutionnel à un autre au Canada, la croissance du PIB d’une année à l’autre représente un indice du prix que la population canadienne peut se permettre pour les nouveaux médicaments brevetés qui arrivent sur le marché sur une base annuelle. Le PIB par habitant peut servir à des fins semblables pour fournir une valeur représentative du prix qui serait considéré comme excessif pour les consommateurs individuels. Ces nouveaux facteurs permettent au CEPMB d’évaluer l’incidence économique du prix d’un médicament breveté sur les assureurs et les consommateurs ainsi que d’élaborer des critères d’évaluation et des tests de taille du marché pour les médicaments qui risquent de poser problème sur le plan de l’abordabilité au sein du système de soins de santé canadien.
10) Comment le CEPMB aboutit-il à un seuil de valeur pharmacoénonomique de 60 000 $/AVAQ dans les Lignes directrices provisoires?
Le seuil de 60 000 $ par AVAQ se fonde sur des travaux universitaires préliminaires commandés par le CEPMB pour calculer le coût marginal d’une AVAQ dans le système de santé canadien, sur des estimations empiriques des seuils du côté de l’offre provenant d’autres administrations pertinentes, et sur des indices de tendances historiques relevés dans les évaluations du rapport coût-efficacité par les agences canadiennes d’ETS.
Dans le système de santé canadien, le gouvernement fédéral administre les normes nationales et accorde un soutien financier aux provinces et aux territoires qui sont responsables de la gestion et de la prestation des services de santé. Le seuil de rentabilité du côté de l’offre d’une province ou d’un territoire particulier est fonction, entre autres, du budget accordé à son propre système de soins de santé. Une évaluation des coûts de renonciation associés à la santé pour le système de santé canadien commandée par le CEPMB (Ochalek, Lomas et Claxton, 2018) est arrivée à la conclusion qu’un coût de 30 000 $ par AVAQ serait raisonnable pour le Canada dans son ensemble, de même que, probablement, dans l’ensemble des provinces. Ces chiffres sont conformes, en général, aux estimations empiriques des seuils du côté de l’offre en vigueur dans d’autres administrations présentant des caractéristiques semblables au Canada sur les plans pharmaceutique et de la richesseNote de bas de page 2.
Dans son rapport final, le Groupe de travail du Comité directeur sur les Lignes directrices visant la modernisation du processus d’examen du prix du CEPMB (ci‑apres le « Groupe de travail ») faisait observer qu’aucune agence canadienne d’ETS n’avait, pour l’heure, établi de seuil politique explicite de coût par AVAQ. Cependant, on a rapporté des cas de seuils variant entre 50 000 $ et 100 000 $, les anticancéreux se situant habituellement à l’extrémité supérieure de cette fourchetteNote de bas de page 3.
En donnant effet à ce nouveau facteur dans les Lignes directrices provisoires, le CEPMB doit composer avec le fait que les travaux réalisés au Canada sur les seuils de rentabilité du côté de l’offre en sont encore au stade embryonnaire. Le CEPMB doit aussi tenir compte de l’incertitude relative des valeurs de RCED et des analyses coût-utilité sur lesquelles les RCED se fondent. L’approche préconisée par le CEPMB pour tenir compte de ces considérations contextuelles est orientée par son rôle, à savoir celui d’organisme de réglementation des prix plafonds et non d’organe de fixation des prix, ainsi que par la nécessité de fournir aux patients canadiens l’accès à de nouvelles technologies de la santé. Par conséquent, les Lignes directrices provisoires proposent un seuil de rentabilité initial de 60 000 $ par AVAQ, soit le double de la meilleure estimation actuelle du coût marginal d’une AVAQ dans le système de santé canadien. Le seuil sera recalculé périodiquement pour tenir compte des variations du PIB, de l’ampleur des budgets en santé, de l’évolution de la recherche empirique et de la productivité marginale des services de santé qui font face au déplacement dans la foulée de l’adoption de nouveaux médicaments.
11) Comment le CEPMB aboutit-il à un seuil de taille de marché de 25 millions $ dans ses Lignes directrices provisoires?
En plus d’appliquer un seuil de valeur pharmacoénonomique de 60 000 $ par AVAQ, les Lignes directrices provisoires prévoient que le PEM des médicaments de catégorie I peut être ajusté davantage si les recettes annuelles générées par le produit excèdent le seuil de 25 millions de dollars. Comme mentionné précédemment, l’ajout des facteurs de la taille du marché et du PIB au Règlement reconnaît la tension qui pourrait apparaître entre le rapport qualité-prix à long terme d’un médicament (c.-à-d., sa rentabilité) et son abordabilité à court terme au sein du système de santé. Les hypothèses suivantes ont servi à orienter le calcul du seuil d’abordabilité de 25 millions de dollars, lequel se fonde sur l’application combinée de ces deux facteurs :
- Pour qu’elles soient durables, les dépenses additionnelles pour des médicaments brevetés devraient être proportionnelles à la croissance annuelle anticipée du PIB au Canada.
- Les montants consacrés aux médicaments brevetés dans les dépenses en santé devraient rester les mêmes, car les dépenses en soins de santé augmentent proportionnellement à la croissance du PIB.
- Le nombre de médicaments brevetés prévus chaque année est calculé en tant que moyenne des cinq années précédentes.
- Un impact budgétaire net moyen pour le pays peut être calculé pour chaque nouveau médicament breveté qui absorberait l’enveloppe budgétaire disponible pour toute nouvelle dépense en médicaments brevetés dans l’estimation de la croissance du PIB.
- L’impact budgétaire net moyen pour le pays n’est pas un seuil approprié de taille du marché, car trop de médicaments pourraient avoir un impact budgétaire plus élevé que la moyenne.
- Un seuil correspondant au double de l’impact budgétaire net moyen pour le pays serait plus conforme à une approche fondée sur le risque, car il attire l’attention réglementaire sur les médicaments brevetés dont le coût de renonciation risque d’être le plus élevé en raison de la taille du marché auquel ils s’adressent.
Une croissance des ventes d’un médicament breveté au Canada qui irait dans le sens des hypothèses formulées ci-dessus entraînerait une hausse annuelle de l’ordre de 432 millions de dollars des ventes du médicament breveté. Si l’on tient compte du fait que 36 nouveaux médicaments brevetés sont mis sur le marché chaque année pour absorber cette croissance, cela signifie que chaque médicament générerait, en moyenne, 12 millions $ en ventes de médicaments brevetés, comme indiqué ci-dessous. Le seuil de 25 millions $ représente environ le double de ce montant.
| Ventes annuelles moyennes de médicaments brevetés, de 2014 à 2018 | 14,9 G$ | |
| Croissance annuelle des ventes de médicaments brevetés, en proportion du taux de croissance du PIB |
|
|
| Nombre moyen de nouveaux médicaments brevetés mis sur le marché par année, de 2014 à 2018 | 36 | |
| Seuil d’abordabilité moyen annuel par médicament | 12 M$ |
Selon l’analyse du CEPMB, plus de 75 % des médicaments brevetés mis en marché au cours des 20 dernières années ont réalisé des recettes inférieures à ce seuil durant les trois premières années de vente.
Description longue
Ce diagramme à colonnes présente les percentiles relatifs aux revenus maximums pour les médicaments brevetés sur une période de trois ans suivant leur mise en marché au Canada depuis 1998.
| Rang centile | Percentile relatif aux revenus maximums sur trois ans |
|---|---|
| 25 | 2 millions $ |
| 33,3 | 4 millions $ |
| 50 | 8 millions $ |
| 66,6 | 15 millions $ |
| 75 | 23 millions $ |
| 80 | 32 millions $ |
| 85 | 41 millions $ |
| 90 | 62 millions $ |
| 95 | 108 millions $ |
Remarque : La présente analyse porte sur 639 médicaments brevetés mis en marché au Canada entre 1998 et 2015. L’année de lancement constitue l’année 0.
Source des données: CEPMB
Ces chiffres seront mis à jour périodiquement pour tenir compte de l’évolution de l’IPC et du PIB.
12) Pourquoi les Lignes directrices provisoires accordent-elles une hausse de 50 % du PEM des médicaments de catégorie I qui traitent des problèmes de santé ayant une très faible prévalence au Canada?
En raison de l’acquisition de nouvelles connaissances et des avancées technologiques, de plus en plus de Canadiens reçoivent un diagnostic de maladie ou de trouble rare pour lequel ils doivent obtenir un traitement. Conformément à l’engagement d’aider les Canadiens aux prises avec des maladies rares pris par le Gouvernement du Canada dans le budget de 2019, les Lignes directrices provisoires prévoient un ajustement du PEM des médicaments brevetés servant à traiter des maladies et des troubles rares touchant des populations de patients restreintes (c.-à-d. s’adressant à un petit marché en raison de leur faible prévalence). De fait, l’ajustement signifie que les plafonds des prix nets des médicaments brevetés pour le traitement de maladies et troubles rares et ayant un faible volume de ventes seront plus élevés que les plafonds de ceux servant au traitement de maladies plus répandues. Cela permet aussi une application plus uniforme du facteur de la valeur pharmacoéconomique parmi l’ensemble des médicaments de catégorie I (c.-à-d., l’utilisation du même seuil de 60 000 $ par AVAQ pour tous ces médicaments) tout en produisant des plafonds de PEM substantiellement plus élevés pour les médicaments servant à traiter des maladies et troubles rares.
Le CEPMB a établi qu’une maladie (ou un trouble) est considérée comme rare si elle touche moins de 1 Canadien sur 2000. Cette mesure se fonde sur la définition utilisée par les Instituts de recherche en santé du Canada, par l’Union européenne, dans le rapport de février 2019 du Comité permanent de la santé intitulé « Améliorer l’accès aux traitements pour les Canadiens atteints de maladies et de troubles rares » et par les associations de patients canadiennes.
En vertu des Lignes directrices provisoires, le PEM des médicaments brevetés servant à traiter des maladies et troubles rares sera augmenté de 50 %, du moment que leurs recettes annuelles restent sous le seuil d’abordabilité de 12,5 millions de dollars défini ci-dessus. Cette hausse de 50 % vise à refléter le bénéfice accru que ces médicaments rapportent actuellement par rapport aux médicaments pour les maladies plus répandues. Le PEM sera alors appliqué de façon progressive à toute vente excédant ce seuil.
Le PEM est augmenté de 50 % au-dessus du prix pharmacoeconomique (PPE) pour les revenus (recettes annuelles) jusqu’à 12,5 millions $. Pour les revenus entre 12,5 millions $ et 25 millions $, le PEM est établi au niveau du PPE, tandis que le PEM pour les revenus supérieurs à 25 millions $ est établi par rapport au seuil d’abordabilité utilisé pour les médicaments de catégorie I. Cette approche est basée sur l’analyse des données sur les médicaments introduits entre 2010 et 2015 qui ont un chiffre de patients inférieur à 1 sur 2000 et pour lesquels les PPE étaient disponibles. Ces résultats indiquent qu’en moyenne, le prix de ces médicaments doit être au-dessus de 49 % du PPE atteindre avoir des revenus annuels de 12,5 millions $.
13) Pourquoi les Lignes directrices provisoires n’utilisent-elles pas les mêmes seuils de coût par AVAQ mentionnés à la section de l’analyse coût-avantage du REIR?
Le REIR comporte une section sur l’analyse coût-avantage, qui décrit le calcul de l’impact économique attendu des nouveaux facteurs de fixation des prix prévus à l’article 85. L’analyse coût-avantage se fonde sur un modèle formulant un certain nombre d’hypothèses, notamment que le CEPMB appliquerait des seuils de 50 000 $ par AVAQ pour les médicaments qui servent à traiter des maladies ordinaires, de 150 000 $ par AVAQ pour les médicaments servant à traiter des maladies rares, et de 35 000 $ par AVAQ pour les médicaments qui servent à traiter des maladies à forte prévalence au sein de la population canadienne. Aux fins de la modélisation, ces hypothèses sont présentées comme un scénario possible de mise en oeuvre des nouveaux facteurs prévus à l’article 85, et ne doivent pas servir de seuils obligatoires que le CEPMB doit observer dans ses Lignes directrices. Le CEPMB utilise un seuil de 60 000 $ par AVAQ pour tous les médicaments, et la hausse de 50 % qu’il a accordée au PEM des médicaments servant à traiter les maladies et troubles rares va dans le sens de l’intention politique globale du Règlement modifié.
14) Quelles sont les nouvelles exigences en matière de production de rapports du Règlement modifié?
Le Règlement définit les renseignements que les titulaires de brevets doivent fournir au CEPMB au sujet des prix des médicaments brevetés vendus au Canada et dans d’autres pays, de leurs revenus et de leurs dépenses en R et D. Le Règlement modifié exige que les titulaires de brevets fournissent des renseignements supplémentaires pertinents au regard des nouveaux facteurs prévus à l’article 85, à l’exception des renseignements en lien avec le PIB et le PIB par habitant, ceux-ci étant fournis par Statistique Canada.
En ce qui a trait à la valeur pharmacoéconomique, le Règlement modifié exige des titulaires de brevets qui vendent des médicaments dont le coût annuel est de plus de 50 % du PIB par habitant de fournir au CEPMB toutes les analyses coût-utilité en lien avec ces médicaments publiées par une agence canadienne d’ETS, notamment l’ACMTS ou l’INESSS. Le CEPMB ne répétera pas les analyses pharmacoéconomiques effectuées par l’ACMTS et l’INESSS.
L’obligation pour le titulaire de brevet de fournir l’analyse coût-utilité la plus récemment publiée reçue ne s’applique qu’aux médicaments de catégorie I dont le coût annuel est de plus de 50 % du PIB par habitant. Les analyses coût-utilité ne sont habituellement effectuées qu’à certains moments clés du cycle de vie d’un médicament (p. ex. avant la mise en marché initiale ou suite à l’approbation réglementaire d’une nouvelle indication). Les titulaires de brevets ne sont pas tenus de préparer une analyse coût-utilité si aucune n’a été rendue publique auparavant. Si aucune analyse coût-utilité n’est disponible, le PEM est calculé en recourant à d’autres renseignements, par exemple le prix de médicaments de la même classe thérapeutique, les prix ailleurs dans le monde, et les données sur la taille du marché.
Pour ce qui est de la taille du marché, le Règlement modifié prévoit que les titulaires de brevets soumettent au CEPMB des données sur la consommation maximale estimée du médicament au Canada, par quantité de médicaments vendus, ainsi que la fourchette de dates prévue pour calculer la consommation maximale. Les titulaires rassemblent déjà ce type de données lorsqu’ils élaborent leurs plans d’affaires et leurs prévisions de ventes, ainsi que pour les processus de l’ACMTS. Les titulaires de brevets sont aussi tenus de fournir au CEPMB toute mise à jour de leurs estimations pouvant résulter de l’approbation d’une nouvelle indication ou d’une autre évolution importante du marché pour le médicament en question.
15) Pourquoi le PEM ne s’applique-t-il qu’aux médicaments brevetés de catégorie I?
En vertu des Lignes directrices provisoires, les médicaments de catégorie I sont soumis à la fois à un plafond du prix courant (PCM) et à un plafond du prix net (PEM). Les médicaments brevetés de catégorie II ne sont soumis qu’à un plafond du prix courant (PCM).
L’application du plafond du prix net aux médicaments brevetés de catégorie I se fonde (a) sur la probabilité accrue que ces médicaments aient peu ou pas du tout d’équivalents thérapeutiques, ce qui désavantagerait grandement les consommateurs (les assureurs publics et privés) au moment de négocier un prix acceptable du point de vue social avec le titulaire de brevet; et (b) la probabilité accrue que la taille des marchés auxquels s’adressent ces médicaments soulève des préoccupations sur le plan de l’abordabilité.
16) Pourquoi le PEM est-il évalué au regard du prix net plutôt que du prix courant?
L’évaluation du PEM au regard du prix net vise à traduire la réalité de la tarification confidentielle dans le contexte des médicaments brevetés de catégorie I ainsi qu’à permettre aux titulaires de brevets de respecter plus facilement les plafonds prévus par les Lignes directrices provisoires sans que cela ait une incidence sur le prix courant public.
17) Pourquoi le PCM est-il évalué au regard du prix courant plutôt que du prix net?
L’évaluation du PCM au regard du prix courant plutôt que du prix net permet de comparer des éléments semblables, car les tests effectués pour établir le PCM se fondent tous sur les prix courants nationaux et internationaux, qui ne comportent pas d’escomptes.
18) Quelles mesures de protection s’appliquent aux renseignements fournis par les brevetés?
La Loi contient des mesures de protection qui s’appliquent aux renseignements fournis par les brevetés. En particulier, les renseignements ou documents fournis au Conseil en application des articles 80, 81, 82 ou 83 (p. ex. les recettes, les prix moyens ou les recettes nettes et les déductions et réductions connexes) sont protégés; nul ne peut, après les avoir obtenus en conformité avec la présente loi, sciemment les communiquer ou en permettre la communication sans l’autorisation de la personne qui les a fournis, sauf s’ils ont été divulgués dans le cadre d’une audience publique tenue en vertu de l’article 83 ou s’ils font partie des exceptions énumérées en vertu de l’article 87(2). Les renseignements fournis au CEPMB peuvent aussi être sujets à la Loi sur l’accès à l’information et à la Loi sur la protection des renseignements personnels.
19) Pourquoi les Lignes directrices provisoires exigent-elles que les brevetés fournissent une analyse coût-utilité uniquement pour les médicaments dont le coût de traitement annuel est de plus de 50 % du PIB par habitant?
C’est ce qui est stipulé à l’article 4.1(5) du Règlement modifié.
20) Pourquoi le CEPMB utilise-t-il le rapport de l’ACMTS comme principal modèle source?
Les brevetés qui doivent présenter une analyse coût-utilité pour un médicament de catégorie I peuvent recourir aux analyses de diverses sources, soit : les rapports pharmacoéconomiques du Programme commun d’évaluation des médicaments (PCEM) et les rapports d’orientation économique finaux du Programme pancanadien d’évaluation des anticancéreux (PPEA) de l’ACMTS, ainsi que le modèle élaboré par l’INESSS dans ses Évaluations aux fins d’inscription.
Par défaut, l’approche préconisée par le CEPMB consistera à utiliser les modèles sources de l’ACMTS, ceux-ci étant représentatifs d’une plus grande part de la population canadienne. Si aucun rapport de l’ACMTS n’est disponible, ou si les rapports disponibles ne répondent pas aux critères définis dans les lignes directrices (p. ex. parce qu’ils ne contiennent pas d’analyse coût-utilité ou ne présentent pas une approche de modélisation de la minimisation des coûts), les rapports de l’INESSS seront utilisés.
21) Pourquoi les Lignes directrices provisoires fournissent-elles un seul plafond de prix pour un médicament breveté ayant plusieurs indications?
Dans la foulée des consultations avec les intervenants et les experts techniques, le CEPMB est d’avis que l’établissement de prix pour chaque indication serait extrêmement difficile à mettre en oeuvre au Canada à l’heure actuelle, en raison des contraintes que supposent la saisie de données et la production de rapports sur le volume indication par indication. À la lumière de ces considérations, les Lignes directrices provisoires adoptent l’approche plus simple qui consiste à établir un seul prix plafond pour les médicaments à plusieurs indications. Dans les cas où un médicament breveté présente plus d’une indication, le CEPMB relèvera l’indication la plus susceptible d’avoir l’incidence la plus grande sur les consommateurs (l’« indication pertinente ») et établira ses plafonds à la lumière de cette constatation. Pour les médicaments de catégorie I, il s’agira de l’indication justifiant d’appliquer le critère d’évaluation de catégorie I; pour les médicaments de catégorie I pour lesquels plus d’une indication (ou aucune indication) rencontrent ce seuil, et pour les médicaments de catégorie II, l’indication pertinente sera celle qui traite l’état de santé ayant la plus forte prévalence (c.-à-d. la plus importante population de patients).
22) Le CEPMB collaborera-t-il avec l’ACMTS et l’INESSS pour s’assurer que les données nécessaires figurent dans leurs rapports?
Le CEPMB reconnaît que l’ACMTS et l’INESSS disposent d’une expertise dédiée et d’une réputation d’excellence en matière de production de rapports d’ETS au Canada. Le CEPMB n’a pas l’intention de reprendre le travail effectué par l’ACMTS et l’INESSS à ce titre.
Aussi, le CEPMB collaborera avec l’ACMTS et l’INESSS pour apporter les modifications qui pourraient s’avérer nécessaires aux processus et aux rapports afin de satisfaire aux exigences du nouveau cadre réglementaire.
23) Pourquoi les médicaments brevetés bénéficiant de droits acquis sont-ils soumis à la nouvelle annexe des pays de comparaison, et pourquoi la médiane internationale établit-elle leur prix plafond?
Les dispositions transitoires du Règlement modifié accordent des droits acquis aux médicaments brevetés qui ont reçu un numéro d’identification du médicament (DIN) de Santé Canada avant la publication du Règlement modifié dans la Partie II de la Gazette du Canada le 21 août 2019. Ces droits les exemptent de l’application des nouveaux facteurs de l’article 85. Ils demeurent toutefois soumis à la nouvelle annexe des pays de comparaison que nous appelons maintenant CEPMB11. Les médicaments brevetés vendus au Canada en vertu du Programme d’accès spécial (PAS) avant le 21 août 2019 ne bénéficieront pas de droits acquis, car ces médicaments ne reçoivent pas de DIN. De plus, les produits venant s’ajouter à des gammes de produits existants et qui se voient attribuer un DIN après le 21 août 2019 ne bénéficient pas de droits acquis, même si les produits d’origine connexes qui existaient avant cette date bénéficient de tels droits.
Le PCM des médicaments bénéficiant de droits acquis est établi dans la fourchette inférieure du (i) prix médian dans les pays du CEPMB11 pour lesquels le breveté a fourni de l’information, ou (ii) selon le prix plafond qui était en vigueur avant la publication des présentes Lignes directrices. Le prix plafond médian est considéré comme compatible avec la responsabilité du Canada de payer sa juste part de l’innovation biopharmaceutique mondiale.
Le PCM peut être rajusté selon l’IPC dans certain cas.
24) Comment les Lignes directrices provisoires traitent-elles de l’IPC?
En vertu de l’alinéa 85(1)(d) de la Loi, le CEPMB est tenu de prendre en compte l’indice des prix à la consommation (IPC) au moment de déterminer si le prix d’un médicament breveté est excessif. Dans le contexte des Lignes directrices provisoires, ce facteur est pris en compte de trois façons : 1) le PCM est fixe pendant les trois premières années, ce qui a un effet équivalent à une augmentation du PCM en fonction de l’IPC par rapport au déclin moyen des prix internationaux; 2) quand le PCM est établi par la médiane des prix internationaux, et cette médiane augmente, le PEM peut être rajuste en fonction de l’IPC; et 3) le seuil d’abordabilité sera actualisé périodiquement pour tenir compte des variations de l’IPC. Ainsi, les tests et les plafonds définis dans les Lignes directrices provisoires demeurent pertinents à la lumière des changements au sein de l’économie canadienne tout en veillant à ce que les Canadiens ne paient pas de prix plus élevés que les consommateurs du CEPMB11 pour les médicaments brevetés.
25) Comment et quand le CEPMB assurera-t-il le suivi et rendra-t-il compte de l’incidence du Règlement modifié et des nouvelles Lignes directrices, notamment s’ils produisent les effets escomptés?
Le CEPMB s’est engagé à élaborer ainsi qu’à appliquer le Processus de surveillance et d’évaluation des Lignes directrices (PSELD), une stratégie de surveillance d’envergure, pour évaluer l’incidence des nouvelles Lignes directrices sur les patients, les fournisseurs de soins de santé et d’autres intervenants.
Le PSELD ciblera les domaines clés suivants : 1) l’incidence sur les prix des médicaments, 2) l’incidence sur l’accès aux médicaments, 3) les répercussions sur l’économie et 4) l’incidence sur les processus du CEPMB. Dans tous ces domaines, le CEPMB aura recours à des sources de données administratives, commerciales et internes pour étudier et documenter les changements après la mise en oeuvre des lignes directrices par rapport à la période précédant ces changements. Même si la majorité des indicateurs seront quantitatifs, le PSELD aura également recours à certains indicateurs qualitatifs.
Une proposition détaillée de PSELD, y compris la portée des rapports qui seront rendus publics, sera présentée au Comité du CEPMB aux fins d’approbation au début de l’année 2020. Le plan de PSELD sera rendu public, et un premier rapport du PSELD sera publié après qu’au moins une année complète de données suivant la mise en oeuvre soit disponible aux fins d’analyse. Au cours des années subséquentes, des rapports périodiques seront publiés pour surveiller les répercussions à long terme.
Soumissions
Veuillez noter que les soumissions présentées ci-dessous se rapportent à la version préliminaire des lignes directrices provisoires publiée en novembre 2019.