
Méthode d'évaluation des risques de cancer : Un sondage sur les pratiques actuelles à Santé Canada

Télécharger le format de rechange
(Format PDF, 4,62 Mo, 42 pages)
Organisation : Santé Canada
Date publiée : Novembre 2021
Cat. : 210343
ISBN : 978-0-660-40496-7
Membres du groupe de travail
Jane MacAulay, Division de la qualité de l'eau, Bureau de la qualité de l'air et de l'eau, Direction de la sécurité des milieux, DGSESC
Ivy Moffat, Division de la qualité de l'eau, Bureau de la qualité de l'air et de l'eau, Direction de la sécurité des milieux, DGSESC
Alisa Vespa, Bureau du métabolisme, de l'oncologie et des sciences de la reproduction, Direction des produits thérapeutiques, DGPSA
Catherine Adcock, Division II des effets sur la santé, Direction de l'évaluation sanitaire, ARLA
Ian Aldous, Bureau de l'évaluation, Direction des instruments médicaux, DGPSA
Mohammed Ansari, Bureau des produits pharmaceutiques commercialisés, Direction des produits de santé commercialisés, DGPSA
Alain Beliveau, Bureau du métabolisme, de l'oncologie et des sciences de la reproduction, Direction des produits thérapeutiques, DGPSA
Vinita Chauhan, Bureau de la protection contre les rayonnements des produits cliniques et de consommation, Direction des sciences de la santé environnementale et de la radioprotection, DGSESC
Guosheng Chen, Division de l'innocuité pour les humains, Direction des médicaments vétérinaires, DGPSA
Michelle Deveau, Section de l'évaluation des contaminants de l'air intérieur, Division des sciences de la qualité de l'air, Bureau de la qualité de l'air et de l'eau, Direction de la sécurité des milieux, DGSESC
John Field, Bureau de l'évaluation du risque, Direction de la sécurité des produits de consommation et des produits dangereux, DGSESC
Jason Fine, Division de l'évaluation des médicaments vendus sans ordonnance, Direction des produits de santé naturels et sans ordonnance, DGPSA
Zoe Gillespie, Bureau d'innocuité des produits chimiques, Direction des aliments, DGPSA
Michael Honeyman, Division II des effets sur la santé, Direction de l'évaluation sanitaire, ARLA
Jianli Jiao, Bureau de l'évaluation et du contrôle des substances nouvelles, Direction de la sécurité des milieux, DGSESC
Dustin Johnson, Bureau des essais cliniques, Centre d'excellence réglementaire, statistiques et essais cliniques, Direction des médicaments biologiques et radiopharmaceutiques, DGPSA
Steven R. Jones, Direction des médicaments biologiques et radiopharmaceutiques, DGPSA
Benny Ling, Division de l'évaluation des effets de l'air sur la santé, Bureau de la qualité de l'air et de l'eau, Direction de la sécurité des milieux, DGSESC
Luigi Lorusso, Division de l'évaluation environnementale et des sites contaminés, Bureau de gestion des produits chimiques et de la santé environnementale, Direction de la sécurité des milieux, DGSESC
Jocelyn Moore, Bureau de l'évaluation du risque, Direction de la sécurité des produits de consommation et des produits dangereux, DGSESC
Michel Ntemgwa, Division de l'évaluation des médicaments vendus sans ordonnance, Direction des produits de santé naturels et sans ordonnance, DGPSA
Sanya Petrovic, Division de l'évaluation environnementale et des sites contaminés, Bureau de gestion des produits chimiques et de la santé environnementale, Direction de la sécurité des milieux, DGSESC
Debora Quayle, Bureau de la radioprotection, Direction des sciences de la santé environnementale et de la radioprotection, DGSESC
Jayadev Raju, Section de la toxicologie moléculaire et appliquée, Division de la recherche en toxicologie et en réglementation, Bureau d'innocuité des produits chimiques, Direction des aliments, DGPSA
Tanya Ramsamy, Bureau des essais cliniques, Direction des produits thérapeutiques, DGPSA
Christopher Rowat, Bureau de l'évaluation des risques pour les substances existantes, Direction de la sécurité des milieux, DGSESC
Tim Schrader, Section de la toxicologie moléculaire et appliquée, Division de la recherche en toxicologie et en réglementation, Bureau d'innocuité des produits chimiques, Direction des aliments, DGPSA
Janice Weightman, Section de l'évaluation toxicologique préalable à la mise en marché, Division de l'évaluation du danger des produits chimiques pour la santé, Bureau d'innocuité des produits chimiques, Direction des aliments, DGPSA
Paul White, Bureau de la science et de la recherche en santé environnementale, Direction des sciences de la santé environnementale et de la radioprotection, DGSESC
Ruth Wilkins, Bureau de la protection contre les rayonnements des produits cliniques et de consommation, Direction des sciences de la santé environnementale et de la radioprotection, DGSESC
Deon Williams, Section de l'évaluation toxicologique des contaminants alimentaires, Division de l'évaluation du danger des produits chimiques pour la santé, Bureau d'innocuité des produits chimiques, Direction des aliments, DGPSA
Table des matières
- Sigles et définitions
- 1. Introduction
- 2. Évaluation des risques de cancer à Santé Canada
- 3. Renseignements utilisés pour orienter les évaluations des risques de cancer
- 4. Méthodologie utilisée pour effectuer les évaluations des risques de cancer
- 5. Défis présentés lors de l'évaluation des risques de cancer
- 6. Besoins et directives futurs
- 6.1. Harmonisation
- 6.2. Mise en commun de l'information
- 6.3. Évolution des tendances dans l'évaluation du risque potentiel de cancer chez l'humain
- 6.3.1. Tendance vers une réduction du recours aux études chez les animaux et l'intégration de nouvelles approches pour l'évaluation du risque potentiel de cancer chez l'humain
- 6.3.2. Interprétation des données sur la relation dose-réponse concernant la cancérogénicité
- 6.3.3. Interprétation et intégration potentielle de données quantitatives sur la relation dose-réponse concernant la génotoxicité dans les évaluations des risques de cancer
- 7. Références
Sigles et définitions
- Apica
- Un résultat observable dans un organisme entier, tel qu'un signe clinique ou un état pathologique, qui indique un problème de santé pouvant résulter de l'exposition à un agent toxique
- AQA
- Apport quotidien acceptable; l'estimation de la quantité d'un produit chimique qui peut être ingérée quotidiennement au cours d'une vie sans risque pour la santé appréciable pour le consommateur. L'AQA est exprimé en mg/kg/jour et peut être appliqué dans l'évaluation des substances comme les additifs alimentaires, les résidus de pesticides et les résidus de médicaments vétérinaires dans les aliments.
- AQT
- Apport quotidien tolérable; analogue à AQA. Le terme AQT est utilisé pour les agents qui ne sont pas ajoutés délibérément, comme les contaminants dans l'eau.
- ARLA
- Agence de réglementation de la lutte antiparasitaire
- ATSDR
- Agency for Toxic Substances and Disease Registry
- BCASN
- Bureau de la cardiologie, des allergies et des sciences neurologiques
- BEC
- Bureau des essais cliniques
- BECSN
- Bureau de l'évaluation et du contrôle des substances nouvelles
- BEEP
- Bureau de l'examen et de l'évaluation des produits
- BER
- Bureau de l'évaluation du risque
- BERSE
- Bureau de l'évaluation des risques pour les substances existantes
- BGMIV
- Bureau de la gastroentérologie et des maladies infectieuses et virales
- BGPCSE
- Bureau de gestion des produits chimiques et de la salubrité de l'environnement
- BIPC
- Bureau d'innocuité des produits chimiques
- BMOSR
- Bureau du métabolisme, de l'oncologie et des sciences de la reproduction
- BPPC
- Bureau des produits pharmaceutiques commercialisés
- BPRPCC
- Bureau de la protection contre les rayonnements des produits cliniques et de consommation
- BQAE
- Bureau de la qualité de l'air et de l'eau
- BRP
- Bureau de la radioprotection
- BSRSE
- Bureau de la science et de la recherche en santé environnementale
- CCSN
- Commission canadienne de sûreté nucléaire
- CDR
- Concentration de référence
- CEBS
- Chemical Effects in Biological Systems
- CEPB
- Centre d'évaluation des produits biologiques
- CEPRB
- Centre d'évaluation des produits radiopharmaceutiques et biothérapeutiques
- CERSEC-BEC
- Centre d'excellence réglementaire, statistiques et essais cliniques - Bureau des essais cliniques
- CIRC
- Centre international de recherche sur le cancer
- CMA
- Concentration maximale acceptable
- CPDB
- Carcinogenic Potency Database
- CT
- Concentration tumorigène
- DA
- Direction des aliments
- DDIN
- Demande de numéro d'identification de médicament
- DDR
- Dose de référence
- DEEAS
- Division de l'évaluation des effets de l'air sur la santé
- DEESC
- Division de l'évaluation environnementale et des sites contaminés
- DEMVSO
- Division de l'évaluation des médicaments vendus sans ordonnance
- DES
- Direction de l'évaluation sanitaire
- DGPSA
- Direction générale des produits de santé et des aliments
- DGSESC
- Direction générale de la santé environnementale et de la sécurité des consommateurs
- DIM
- Direction des instruments médicaux
- DMBR
- Direction des médicaments biologiques et radiopharmaceutiques
- DMENO
- Dose minimale entraînant un effet nocif observé
- DMV
- Direction des médicaments vétérinaires
- DPSC
- Direction des produits de santé commercialisés
- DPSNSO
- Direction des produits de santé naturels et sans ordonnance
- DPT
- Direction des produits thérapeutiques
- DQE
- Division de la qualité de l'eau
- DSENO
- Dose sans effet nocif observé
- DSEO
- Dose sans effet observé
- DSM
- Direction de la sécurité des milieux
- DSPCPD
- Direction de la sécurité des produits de consommation et des produits dangereux
- DSSER
- Direction des sciences de la santé environnementale et de la radioprotection
- DT50
- Dose donnant une incidence de tumeurs de 50 % (DT50) à la fin de la durée de vie standard pour les espèces
- EFSA
- Autorité européenne de sécurité des aliments
- EPA des États-Unis
- Environmental Protection Agency des États-Unis
- ÉQDRCHIM
- Évaluation quantitative des risques pour les produits chimiques
- ERSH
- Évaluation du risque pour la santé humaine
- FDA des États-Unis
- Food and Drug Administration des États-Unis
- Génotoxicité
- Terme générique faisant référence à tout changement délétère dans le matériel génétique, quel que soit le mécanisme par lequel le changement est induit (p. ex. mutation ponctuelle, cassure chromosomique)
- GTERS
- Groupe de travail sur l'évaluation des risques scientifiques
- HSDB
- Hazardous Substances Data Bank
- ICH
- Conférence internationale sur l'harmonisation des exigences techniques relatives à l'homologation des produits pharmaceutiques à usage humain (aussi parfois désignée la CIH)
- Impureté mutagène
- Une impureté dans un produit pharmaceutique qui a été démontré mutagène dans un modèle de mutagénicité approprié, p. ex. épreuve de mutagénicité bactérienne
- IPA
- Ingrédient pharmaceutique actif
- IRIS
- Integrated Risk Information System
- IRS
- Incidences du rayonnement sur la santé
- ISA
- Integrated Science Assessment (EPA des États-Unis)
- JECFA
- Comité mixte FAO/OMS d'experts des additifs alimentaires
- JRC
- Centre commun de recherche, service de la Commission européenne
- LCPE
- Loi canadienne sur la protection de l'environnement
- LCSPC
- Loi canadienne sur la sécurité des produits de consommation
- LDQAIR
- Lignes directrices sur la qualité de l'air intérieur résidentiel
- LICDR10
- Limite inférieure de l'intervalle de confiance de la dose de référence; une estimation de la dose inférieure qui est de 95 % certaine pour causer une incidence de cancer d'au plus 10 % chez les rongeurs
- MA
- Mode d'action; séquence biologiquement plausible d'événements clés menant à un effet observé soutenu par des observations expérimentales solides et des données mécanistiques
- ME
- Marge d'exposition
- Mutagène
- Agent chimique, physique ou biologique qui peut introduire des changements permanents et transmissibles au génome d'un organisme exposé
- NRAI
- Niveaux de référence dans l'air intérieur
- NRM
- Niveau de risque minimal; estimation de l'exposition quotidienne à une substance dangereuse qui est susceptible d'être sans risque appréciable d'effets nocifs autres que le cancer sur la santé sur une durée précise de l'exposition
- NTP
- National Toxicology Program
- OCDE
- Organisation de coopération et de développement économiques
- OEBQA
- Outil d'évaluation des bénéfices liés à la qualité de l'air
- OMS
- Organisation mondiale de la Santé
- PDD
- Point de départ; Le point de départ représente une dose provenant de données observées qui est associée à un risque supplémentaire pour un critère d'évaluation spécifique. Cela pourrait être sous forme de DMENO, de DSENO ou de DDR.
- PDN
- Présentation de drogue nouvelle
- PETA
- People for the Ethical Treatment of Animals
- PGPC
- Plan de gestion des produits chimiques
- PISSC
- Programme international sur la sécurité des substances chimiques
- PM2,5
- Fines particules désignant une gamme de particules dans l'air dont le diamètre est inférieur à 2,5 microns (µm).
- PNR
- Programme national sur le radon
- PPAR α
- Récepteur alpha activé de la prolifération des peroxysomes
- R(Q)SA
- Relation (quantitative) structure-activité
- REACH
- Registration, Evaluation, Authorisation and Restriction of Chemicals
- ReCAAP
- Rethinking Carcinogenicity Assessment for Agrochemicals Project
- SECAI
- Section de l'évaluation des contaminants de l'air intérieur
- SOT
- Society of Toxicology
- SPT
- Seuil de préoccupation toxicologique
- TgRasH2
- Modèle de souris transgénique
- ToxCast
- Toxicity Forecaster
- ToxNet
- Toxicology Data Network
- ToxTree
- Toxic Hazard Estimation by decision tree approach
- VBS
- Valeur basée sur la santé
- VICH
- Coopération internationale pour l'harmonisation des exigences techniques pour l'enregistrement des médicaments vétérinaires
- VTR
- Valeur toxicologique de référence
1. Introduction
Santé Canada est le ministère fédéral responsable d'aider les Canadiens à maintenir et à améliorer leur état de santé. Dans le cadre de ce mandat, Santé Canada édicte des règles ou établit des recommandations sur les risques pour la santé d'origine environnementale et la sûreté des produits, y compris les produits de santé (p. ex. médicaments et instruments médicaux), les aliments, l'eau, l'air, les produits de consommation (p. ex. cosmétiques), les médicaments vétérinaires, les pesticides ainsi que les dispositifs et les sources environnementales qui émettent des rayonnements. Un élément essentiel de l'évaluation de la sécurité environnementale et des produits est l'analyse des risques de cancer chez l'humain découlant d'une exposition à des substances présentant un potentiel mutagène, génotoxique ou cancérogène.
Le Groupe de travail sur l'évaluation des risques scientifiques (GTERS) a été établi en 2009 et comprend des évaluateurs de risques ou des gestionnaires de risques de divers secteurs de programme au Ministère. Le GTERS prépare des rapports, des lignes directrices et d'autres livrables pour le Ministère grâce à la création de groupes de travail composés d'experts en la matière. Le GTERS aborde une vaste gamme de problèmes et cherche à améliorer la coordination, l'uniformité et la cohérence des évaluations scientifiques des risques dans l'ensemble de ces programmes.
Le GTERS a créé un sous-groupe de travail de la méthode d'évaluation des risques de cancer afin de mieux comprendre les approches utilisées pour effectuer des évaluations des risques de cancer parmi les divers secteurs de programme de Santé Canada. Compte tenu de l'ampleur du sujet, le consensus a été atteint par le sous-groupe de travail de la méthode d'évaluation des risques de cancer pour élaborer ce document explicatif initial, en visant comme public cible principal les programmes qui mènent ou utilisent des évaluations des risques de cancer, ainsi que ceux qui souhaitent comprendre le contexte actuel d'évaluation des risques de cancer dans l'ensemble de Santé Canada. Le présent rapport présente les approches utilisées pour évaluer les dangers de cancer ou estimer les risques de cancer dans l'ensemble des secteurs de programme, notamment si, et le cas échéant comment, les données sur la génotoxicité, la mutagénicité et la cancérogénicité sont utilisées pour éclairer la méthode d'évaluation des risques de cancer. Enfin, le sous-groupe de travail de la méthode d'évaluation des risques de cancer a été chargé de cerner les occasions de collaboration et d'harmonisation futures, dans la mesure du possible. Pour générer les données requises pour préparer le document explicatif, un sondage a été effectué pour recueillir les renseignements suivants :
- le nom du secteur de programme qui répond au sondage;
- l'initiative législative ou politique appuyant la réalisation des évaluations des risques (ou des dangers) de cancer;
- les objectifs de la réalisation des évaluations des risques (ou des dangers) de cancer;
- la réalisation de nouvelles évaluations « internes » des risques (ou des dangers) de cancer ou l'utilisation d'évaluations des risques (ou des dangers) de cancer d'autres sources;
- les évaluations des risques de cancer consultées ou utilisées pour compléter l'évaluation des risques de cancer dans les secteurs de programme (sources internes et externes);
- les ressources ou les documents d'orientation consultés;
- les sources de données ou les bases de données consultées;
- l'utilisation des résultats d'études de génotoxicité et de mutagénicité pour faire l'évaluation des risques (ou des dangers) de cancer et, le cas échéant, la façon dont ils sont utilisés;
- la description de l'approche générale pour effectuer une évaluation des risques (ou des dangers) de cancer;
- des détails sur les défis rencontrés au cours de l'évaluation des risques (ou des dangers) de cancer;
- tout autre renseignement pertinent pour interpréter la façon dont les évaluations des risques (ou des dangers) de cancer sont effectuées.
Le sondage a été lancé le 27 avril 2018 et a été achevé le 16 janvier 2019. L'information a été obtenue auprès de différents secteurs de Santé Canada (Figure 1).
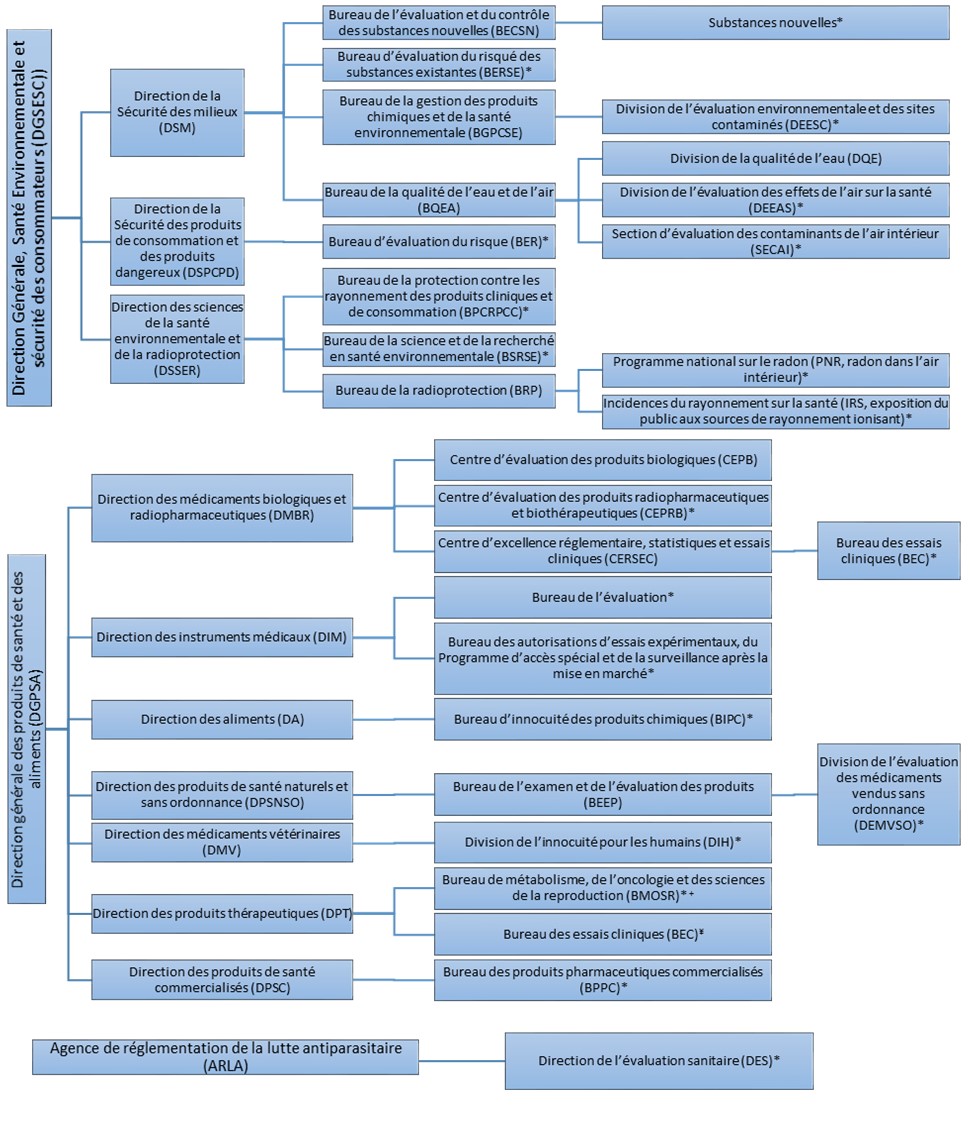
Figure 1 - Organigramme des secteurs ayant répondu au sondage.
Direction Générale, Santé Environnementale et sécurité des consommateurs (DGSESC)
- Direction de la Sécurité des milieux (DSM)
- Bureau de l'évaluation et du contrôle des substances nouvelles (BECSN)
- Substances nouvelles*
- Bureau d'évaluation du risqué des substances existantes (BERSE)*
- Bureau de la gestion des produits chimiques et de la santé environnementale (BGPCSE)
- Division de l'évaluation environnementale et des sites contaminés (DEESC)*
- Bureau de la qualité de l'eau et de l'air (BQEA)
- Division de la qualité de l'eau (DQE)
- Division de l'évaluation des effets de l'air sur la santé (DEEAS)*
- Section d'évaluation des contaminants de l'air intérieur (SECAI)*
- Bureau de l'évaluation et du contrôle des substances nouvelles (BECSN)
- Direction de la Sécurité des produits de consommation et des produits dangereux (DSPCPD)
- Bureau d'évaluation du risque (BER)*
- Direction des sciences de la santé environnementale et de la radioprotection (DSSER)
- Bureau de la protection contre les rayonnement des produits cliniques et de consommation (BPCRPCC)*
- Bureau de la science et de la recherché en santé environnementale (BSRSE)*
- Bureau de la radioprotection (BRP)
- Programme national sur le radon (PNR, radon dans l'air intérieur)*
- Incidences du rayonnement sur la santé (IRS, exposition du public aux sources de rayonnement ionisant)*
Direction générale des produits de santé et des aliments (DGPSA)
- Direction des médicaments biologiques et radiopharmaceutiques (DMBR)
- Centre d'évaluation des produits biologiques (CEPB)
- Centre d'évaluation des produits radiopharmaceutiques et biothérapeutiques (CEPRB)*
- Centre d'excellence réglementaire, statistiques et essais cliniques (CERSEC)
- Bureau des essais cliniques (BEC)*
- Direction des instruments médicaux (DIM)
- Bureau de l'évaluation*
- Bureau des autorisations d'essais expérimentaux, du Programme d'accès spécial et de la surveillance après la mise en marché*
- Direction des aliments (DA)
- Bureau d'innocuité des produits chimiques (BIPC)*
- Direction des produits de santé naturels et sans ordonnance (DPSNSO)
- Bureau de l'examen et de l'évaluation des produits (BEEP)
- Division de l'évaluation des médicaments vendus sans ordonnance (DEMVSO)*
- Bureau de l'examen et de l'évaluation des produits (BEEP)
- Direction des médicaments vétérinaires (DMV)
- Division de l'innocuité pour les humains (DIH)*
- Direction des produits thérapeutiques (DPT)
- Bureau de métabolisme, de l'oncologie et des sciences de la reproduction (BMOSR)* +
- Bureau des essais cliniques (BEC)¥
- Direction des produits de santé commercialisés (DPSC)
- Bureau des produits pharmaceutiques commercialisés (BPPC)*
Agence de réglementation de la lutte antiparasitaire (ARLA)
- Direction de l'évaluation sanitaire (DES)*
Figure 1. Organigramme des secteurs ayant répondu au sondage. *Les secteurs de programme qui ont soumis une réponse au sondage. ¥Bien que le BEC de la DPT n'ait pas été inclus dans le sondage, de l'information a été obtenue de ce secteur de programme pour assurer sa représentation appropriée dans le rapport. +Les réponses au sondage fournies par le BMOSR sont également pertinentes pour le bureau d'examen clinique de la DPT, y compris le Bureau de la gastroentérologie et des maladies infectieuses et virales (BGMIV) et le Bureau de la cardiologie, des allergies et des sciences neurologiques (BCASN).
2. Évaluation des risques de cancer à Santé Canada
Les évaluations des risques de cancer sont menées ou prises en compte dans une certaine mesure par tous les secteurs de programme sondés. Le contenu des évaluations individuelles des risques et la recommandation globale peuvent varier en fonction de plusieurs facteurs, dont les considérations politiques ou législatives et l'objectif dans lequel est menée l'évaluation des risques de cancer. La variation peut également être attribuable aux renseignements disponibles utilisés pour éclairer les évaluations, comme il sera discuté à la section 3.0.
2.1. Considérations politiques ou législatives
La variété d'évaluations des risques de cancer menées à Santé Canada est en grande partie reflétée par les divers mandats des directions et des bureaux qui forment le Ministère. Les membres du sous-groupe de travail ont été interrogés quant au type de facteurs politiques ou législatifs qui oriente les évaluations des risques de cancer dans leur direction ou leur bureau respectif. Les facteurs législatifs comprennent les travaux effectués en vertu de la Loi sur les aliments et drogues pour les représentants de la DGPSA; de laLoi canadienne sur la protection de l'environnement (LCPE), de laLoi sur les dispositifs émettant des radiations, de la Loi canadienne sur la sécurité des produits de consommation (LCSPC), de la Loi sur la santé et la sécurité au travail (LSST) et du Code canadien du travail pour les représentants de la DGSESC, et de la Loi sur les produits antiparasitaires pour ceux de l'ARLA. Il convient de noter que certains travaux de la DGSESC et de l'ARLA relèvent également de la Loi sur les aliments et drogues.
2.2. Objectif des évaluations des risques de cancer
L'objectif de la réalisation et de l'utilisation d'évaluations des risques de cancer dans l'ensemble de Santé Canada varie considérablement (tableau 1). Les répondants de la DGSESC qui effectuent des évaluations des risques de cancer cherchent souvent à quantifier les risques associés à l'exposition à un cancérogène ou à quantifier un niveau d'exposition qui correspond à un risque acceptableNote de bas de page 1. Par exemple, au Bureau de la qualité de l'air et de l'eau, les répondants ont indiqué avoir besoin d'élaborer des évaluations quantitatives des risques pour définir un niveau maximal d'une substance considérée comme acceptable dans l'air intérieur ou dans l'eau potable. De même, au sein du Bureau, la Division des effets de la pollution de l'air sur la santé effectue des évaluations des incidences sur la santé pour déterminer le nombre de résultats sanitaires excédentaires qui sont associés au risque accru attribuable à une variation de la concentration des polluants de l'air. La Division travaille aussi à une estimation permettant de quantifier le risque excédentaire de cancer du poumon attribuable aux polluants atmosphériques que sont les PM2,5 pour une utilisation dans les évaluations des incidences sur la santé. Le processus est semblable à l'ARLA : la Direction de l'évaluation sanitaire effectue des évaluations quantitatives des risques pour préciser les utilisations acceptables d'un pesticide.
Certaines évaluations effectuées à Santé Canada visent à déterminer du point de vue qualitatif si une substance est cancérogène ou non, ou si elle présente un risque de cancérogénicité, sans avoir à établir un niveau sécuritaire d'exposition. Cela représente une approche fondée sur les dangers. Par exemple, dans l'évaluation des médicaments vétérinaires à la DGPSA, lorsqu'il est établi qu'une substance ou l'un de ses métabolites est cause de cancer chez les animaux ou les humains, le produit ne peut généralement pas être approuvé en tant que médicament pour usage auprès des animaux destinés à la consommation. De même, au cours de l'évaluation préalable à la mise en marché des additifs alimentaires au Bureau d'innocuité des produits chimiques de la Direction des aliments de la DGPSA, les composés qui sont des cancérogènes génotoxiques sont rejetés pour toute éventuelle utilisation dans les aliments. Cependant, il convient de noter que dans les évaluations après la mise en marché portant sur des contaminants et des substances toxiques présentes naturellement dans les aliments, les évaluations des risques de cancer sont effectuées pour établir des valeurs toxicologiques de référence ou des valeurs d'orientation fondées sur la santé.
Pour l'évaluation des médicaments à petites molécules, des instruments médicaux et des médicaments biologiques et radiopharmaceutiques au sein de la Direction des produits thérapeutiques, de la Direction des instruments médicaux, de la Direction des médicaments biologiques et radiopharmaceutiques et de la Direction des produits de santé commercialisés, le profil avantages-risques du produit est évalué. Étant donné que les produits sont approuvés pour la mise en marché ou sont maintenus sur le marché uniquement lorsque leur profil avantages-risques est favorable, tant les avantages (p. ex. efficacité du médicament) que les risques sont pris en compte. L'évaluation des risques de cancer comprend un aspect de la compréhension globale du profil avantages-risques du produitNote de bas de page 2.
D'autres utilisations des évaluations des risques de cancer ont aussi été relevées : déterminer les risques pour la santé de la population et les répercussions possibles découlant de l'exposition à une substance, servir de directives dans les scénarios d'exposition du public, évaluer la toxicité et établir des niveaux sécuritaires d'exposition à une substance, vérifier la sûreté d'un produit de consommation ou d'un cosmétique et évaluer la conformité aux normes. Les évaluations des risques de cancer peuvent également éclairer l'établissement d'orientations générales et de santé publique (tableau 1).
| PROGRAMME | OBJECTIF DE L'ÉVALUATION DES RISQUES DE CANCER |
|---|---|
| Direction générale de la santé environnementale et de la sécurité des consommateurs (DGSESC) | |
| Bureau de l'évaluation des risques pour les substances existantes (BERSE) | L'évaluation est effectuée au cours de l'évaluation des substances existantes inscrites sur la Liste intérieure des substances afin de déterminer si elles correspondent à la définition de « toxique » au sens de l'article 64 de la LCPE. |
| Bureau de l'évaluation et du contrôle des substances nouvelles (BECSN) | L'évaluation est effectuée dans le cadre de l'évaluation des risques pour la santé humaine pour les substances nouvelles à déclarer conformément à la version modifiée du Règlement sur les renseignements concernant les substances nouvelles. |
| Bureau de la qualité de l'air et de l'eau (BQAE) - Qualité de l'eau | L'évaluation est effectuée dans le but de calculer les valeurs basées sur la santé (VBS) pour les contaminants dans l'eau potable. Ces VBS orientent les concentrations maximales acceptables (CMA) établies dans les Recommandations pour la qualité de l'eau potable au Canada. |
| Bureau de la qualité de l'air et de l'eau (BQAE) - Air extérieur | Le cancer et le potentiel cancérogène sont pris en compte dans le cadre des évaluations des incidences sur la santé (estimer le nombre de résultats sanitaires excédentaires associés au risque accru attribuable à une variation de la concentration des polluants de l'air) et des évaluations des risques (établir une relation de causalité entre un critère d'effet particulier sur la santé [p. ex. un cancer] et une source de pollution atmosphérique), respectivement. La Division travaille aussi à une estimation permettant de quantifier le risque excédentaire de cancer du poumon attribuable aux polluants atmosphériques que sont les PM2,5 pour une utilisation dans les évaluations des incidences sur la santé. |
| Bureau de la qualité de l'air et de l'eau (BQAE) - Air intérieur | L'évaluation est effectuée pour calculer les limites d'exposition maximales recommandées en fonction de critères sanitaires. Les Lignes directrices sur la qualité de l'air intérieur résidentiel (LDQAIR) contiennent généralement des limites d'exposition à court terme et à long terme. Les niveaux de référence dans l'air intérieur (NRAI) sont des valeurs-guides pour une exposition à vie et reposent sur les évaluations des risques réalisées par d'autres programmes de Santé Canada ou des organismes externes. |
| Bureau de gestion des produits chimiques et de la santé environnementale (BGPCSE) - Division de l'évaluation environnementale et des sites contaminés | L'évaluation est réalisée pour s'assurer que les objectifs d'assainissement en fonction du risque pour les sites contaminés fédéraux sont établis à des niveaux qui favorisent la protection de la santé à l'égard de critères d'effet cancérogène. Des lignes directrices pour la qualité du sol sont établies pour des substances cancérogènes et non cancérogènes dans le but d'établir des recommandations pour la protection de la santé humaine et de l'environnement. |
| Bureau de l'évaluation du risque (BER) | L'évaluation peut être menée en réponse à une préoccupation concernant la sûreté d'un produit de consommation particulier, d'une catégorie de produits de consommation ou d'un ingrédient de cosmétiques. Les facteurs de l'évaluation des risques pour les produits de consommation sont principalement les incidents signalés par le public ou par l'industrie et les informations diffusées par les médias. L'évaluation des risques pour les ingrédients de cosmétiques peut être effectuée pour ces mêmes raisons ou suivant la divulgation de données scientifiques récentes ou la prise de mesures par d'autres organismes de réglementation. |
| Bureau de la protection contre les rayonnements des produits cliniques et de consommation (BPRPCC) | Les rayonnements ionisants (p. ex. rayons X) et les rayons ultraviolets sont des cancérogènes connus. Des évaluations des risques de cancer sont rarement réalisées puisque des évaluations ont déjà bien établi les dangers de cancer associés à diverses formes de rayonnements ionisants et non ionisants. Pour les dispositifs émettant des rayons X et des rayons ultraviolets, le programme évalue leur conformité aux normes nationales et internationales établies pour ce type de dispositifs, lesquelles se fondent à leur tour sur des directives et des principes internationaux de radioprotection et des modèles d'évaluation des risques. De plus, les modèles de risque de cancer acceptés internationalement peuvent être appliqués pour comparer le risque relatif de cancer dans différents scénarios d'exposition aux rayonnements. |
| Bureau de la radioprotection (BRP) - Programme national sur le radon | Ce programme ne mène pas d'évaluations indépendantes de la cancérogénicité du radon parce qu'il s'agit d'un cancérogène du groupe 1 reconnu par le Centre international de recherche sur le cancer (CIRC). Le risque d'un cancer causé par le radon aux fins d'utilisation dans les évaluations a été caractérisé par des organismes internationaux de radioprotection à la suite de l'examen des meilleures données scientifiques disponibles; il est utilisé par le BRP pour établir des orientations et encourager les mesures d'atténuation du radon dans les politiques de santé publique. Le BRP utilise des modèles de risque établis pour le radon afin de calculer les impacts sur la population selon la répartition du radon à l'échelle du Canada. |
| Bureau de la radioprotection (BRP) - Incidences du rayonnement sur la santé | Il est reconnu internationalement que l'exposition à des rayonnements ionisants augmente le risque de cancer. Le risque d'un cancer causé par le rayonnement aux fins d'utilisation dans les évaluations a été caractérisé par des organismes internationaux de radioprotection à la suite de l'examen des meilleures données scientifiques disponibles. Le BRP applique et adapte les orientations et les pratiques exemplaires internationales pour gérer les risques de cancer posés par divers scénarios d'exposition dans le contexte canadien. |
| Bureau de la science et de la recherche en santé environnementale (BSRSE) | Ce bureau effectue des évaluations des risques à des fins de recherche et mène des activités de recherche appliquée pour appuyer la modernisation de l'évaluation des risques. La recherche est menée en collaboration avec des évaluateurs de divers bureaux. Les activités visent souvent l'interprétation des évaluations de la toxicité génétique pour appuyer la détermination du mode d'action et la modernisation de l'évaluation des risques. |
| Direction générale des produits de santé et des aliments (DGPSA) | |
| Bureau du métabolisme, de l'oncologie et des sciences de la reproduction (BMOSR)* | Un nouveau produit pharmaceutique est approuvé pour la mise en marché lorsqu'il est établi que ses avantages dépassent tout méfait possible, avec un degré raisonnable de certitude. L'évaluation du potentiel cancérogène de l'ingrédient pharmaceutique actif (IPA) ainsi que des impuretés pharmaceutiques aide à comprendre le profil de risque du produit. Si une nouvelle impureté ou un niveau supérieur d'une impureté précédemment qualifiée est détecté après la commercialisation, le potentiel cancérogène peut être évalué et le risque potentiel de cancer peut être estimé. |
| Bureau des essais cliniques (BEC) | Des évaluations qualitatives des risques de cancer sont réalisées pour déterminer si le risque pour les participants aux essais cliniques est acceptable selon les lignes directrices acceptées à l'échelle internationale. |
| Direction des instruments médicaux (DIM) | Cette direction n'effectue pas d'évaluations des risques de cancer. |
| Direction des médicaments vétérinaires (DMV) | Le risque cancérogène est considéré comme un élément important dans l'évaluation des médicaments vétérinaires pour protéger la santé humaine et animale, et la salubrité des aliments. |
| Bureau d'innocuité des produits chimiques (BIPC) | Les évaluations des risques de cancer sont utilisées pour orienter l'évaluation d'innocuité préalable à la mise en marché de certains aliments, ingrédients alimentaires, substances utilisées dans le traitement des aliments et matériaux d'emballage alimentaire. Ces évaluations permettent de déterminer si la nourriture ou la substance doit être autorisée pour utilisation et à quel niveau. Les évaluations des risques de cancer sont également utilisées dans les évaluations des risques post-commercialisation de contaminants chimiques et de substances toxiques susceptibles d'être présentes naturellement dans les aliments; les résultats aident à établir si la gestion des risques est nécessaire. |
| Bureau de l'examen et de l'évaluation des produits (BEEP), Division de l'évaluation des médicaments vendus sans ordonnance (DEMVSO) | Des évaluations des risques de cancer ne sont pas effectuées à proprement dit au sein de la Division de l'évaluation des médicaments vendus sans ordonnance. Toutefois, lorsque nécessaire, des données de génotoxicité et de mutagénicité sont évaluées dans les cas suivants : a. au cours de l'examen d'une présentation de drogue nouvelle (PDN) dans le contexte de l'ensemble de données non cliniques (section sur la toxicologie). Dans de rares cas pour des médicaments visés au titre 1 (présentations de DDIN), la génotoxicité et la mutagénicité du produit proposé sont évaluées; b. lorsqu'un contaminant est relevé dans un produit pharmaceutique commercialisé; c. au cours de l'évaluation du profil de risque des impuretés dans un produit pharmaceutique avant sa mise sur le marché, ou pour qualifier des limites d'impureté plus élevées (ensemble de données chimiques et de fabrication de la PDN ou supplément à une PDN). |
| Centre d'évaluation des produits biologiques (CEPB) et Centre d'évaluation des produits radiopharmaceutiques et biothérapeutiques (CEPRB) | Ces centres évaluent le profil avantages-risques des nouveaux médicaments biologiques (produits issus de sources vivantes) avant l'autorisation de la mise sur le marché du médicament. L'évaluation du potentiel cancérogène d'une substance pharmaceutique biologique s'inscrit dans le processus visant à mieux comprendre le profil de risque du produit. Étant donné la nature des produits biologiques, il s'agit d'une approche au cas par cas qui est fondée sur des données scientifiques propres au produit. |
| Centre d'excellence réglementaire, statistiques et essais cliniques - Bureau des essais cliniques (CERSEC-BEC) | L'évaluation des risques de cancer, si nécessaire, contribue à une analyse avantages-risques de l'utilisation proposée d'un médicament biologique expérimental dans le cadre d'un essai clinique. Étant donné la nature des produits biologiques, l'évaluation des risques de cancer suit une approche au cas par cas fondée sur des données scientifiques propres au produit. |
| Bureau des produits pharmaceutiques commercialisés (BPPC) | L'évaluation est réalisée pour veiller à ce que le profil des avantages et des méfaits associés au produit continue de favoriser l'utilisation du médicament ou de l'instrument médical après sa mise en marché. |
| Agence de réglementation de la lutte antiparasitaire | |
| Direction de l'évaluation sanitaire (DES) | L'évaluation est réalisée au cours de l'évaluation de nouvelles matières actives de qualité technique et de la réévaluation de matières actives déjà homologuées afin de s'assurer que les risques sont acceptables. |
| *Bien que cette réponse au sondage ait été fournie par le BMOSR, l'approche décrite est utilisée dans d'autres bureaux d'examen clinique de la DPT, dont le BGMIV et le BCASN. | |
2.3. Élaboration et utilisation des évaluations des risques de cancer
Bien qu'une proportion importante des secteurs de programme effectue régulièrement des évaluations des risques de cancer, nombre d'entre eux comptent sur les évaluations des risques effectuées par d'autres groupes au sein de Santé Canada ou de l'extérieur (p. ex. des évaluations pour le Plan de gestion des produits chimiques [PGPC], d'autres autorités de réglementation ou d'organismes internationaux [p. ex. EPA des États-Unis, EFSA, OMS], ou des évaluations produites par l'industrie [p. ex. sociétés pharmaceutiques]), comme il est indiqué dans le tableau 2. Par conséquent, ce ne sont pas toutes les évaluations des risques de cancer utilisées à Santé Canada qui sont élaborées à l'interne et ce ne sont pas tous les secteurs de programme qui effectuent de nouvelles évaluations « internes » (tableau 2).
| SECTEUR DE PROGRAMME | ÉLABORATION DE NOUVELLES ÉVALUATIONS INTERNES DES RISQUES DE CANCER | UTILISATION D'ÉVALUATIONS DES RISQUES DE CANCER EFFECTUÉES PAR D'AUTRES (P. EX SANTÉ CANADA, ORGANISMES INTERNATIONAUX, INDUSTRIE)* |
|---|---|---|
| DGSESC-DSM-BERSE | ||
| DGSESC-DSM-BECSN | ||
| DGSESC-DSM-BQAE-DQE | ||
| DGSESC-DSM-BQAE-DEEAS | ||
| DGSESC-DSM-BQAE-SECAI | ||
| DGSESC-DSM-DEESC | ||
| DGSESC-DSPCPD-BER | ||
| DGSESC-DSSER-BPRPCC | ||
| DGSESC-DSSER-BRP-PNR | ||
| DGSESC-DSSER-BRP-IRS | ||
| DGPSA-DPT-BMOSR | ||
| DGPSA-DPT-BEC | ||
| DGPSA-DIM | ||
| DGPSA-DPSC-BPPC | ||
| DGPSA-DMV | ||
| DGPSA-DA-BIPC | ||
| DGPSA-DPSNSO-DEMVSO | ||
| DGPSA-DMBR-CEPB/CEPRB/CERSEC-BEC | ||
| ARLA-DES |
3. Renseignements utilisés pour orienter les évaluations des risques de cancer
Le sondage mené par le sous-groupe de travail de la méthode d'évaluation des risques de cancer a révélé de nombreux points communs ainsi que de nombreuses différences quant aux ressources d'orientation consultées et aux sources de données utilisées pour orienter les évaluations des risques de cancer. Ces renseignements sont résumés dans les sections qui suivent.
3.1. Ressources consultées
Les résultats du sondage ont révélé que les programmes consultent plusieurs des mêmes ressources pour effectuer les évaluations des risques de cancer (tableau 3). Le document d'orientation le plus souvent cité s'intitule Guidelines for Carcinogen Risk Assessment (2005a) de l'EPA des États-Unis, avec ses directives supplémentaires intitulées Supplemental Guidance for Assessing Susceptibility from Early-Life Exposure to Carcinogens (2005b). D'autres lignes directrices couramment utilisées dans l'ensemble des programmes sont les Lignes directrices de l'Organisation pour la coopération et le développement économiques (OCDE) pour les essais de produits chimiques, et les notes d'orientation connexes (en anglais seulement) intitulées Guidance Notes for Analysis and Evaluation of Chronic Toxicity and Carcinogenicity Studies (2002), le Conceptual Framework for Evaluating a Mode of Action for Chemical Carcinogenesis (2001) du Programme international sur la sécurité des substances chimiques (PISSC) ainsi que les lignes directrices de la Conférence internationale sur l'harmonisation des exigences techniques relatives à l'homologation des produits pharmaceutiques à usage humain (ICH) portant sur l'évaluation du potentiel génotoxique et cancérogène des produits pharmaceutiques et des impuretés connexes et sur l'évaluation des produits biologiques. D'autres lignes directrices consultées par des programmes particuliers comprennent celles élaborées par l'Organisation mondiale de la Santé (OMS), le Comité mixte FAO/OMS d'experts des additifs alimentaires (JECFA), la Coopération internationale pour l'harmonisation des exigences techniques pour l'enregistrement des médicaments vétérinaires (VICH) et le Centre commun de recherche (JRC [Joint Research Centre]) de la Commission européenne, en plus du Preambule to the Integrated Science Assessments (ISA) (2005) de l'EPA des États-Unis. Ce dernier est consulté pour l'évaluation de la causalité de critères d'effet sur la santé, y compris le cancer. Il convient de noter que certains secteurs du Ministère (p. ex. la Division de l'évaluation environnementale et des sites contaminés et la Division de la qualité de l'eau) ont élaboré leurs propres directives pour un usage externe et interne, respectivement.
| RESSOURCES | TYPE DE RENSEIGNEMENTS |
|---|---|
| EPA des États-Unis – cancer (en anglais seulement) | Évaluation des risques cancérogènes |
| Lignes directrices de l'OCDE pour les essais | Méthodes d'essai acceptées à l'échelle internationale |
| Cadre du PISSC (en anglais seulement) | Cadre d'évaluation du mode d'action cancérogène d'une substance |
| ICH (en anglais seulement) | Lignes directrices scientifiques visant à évaluer l'innocuité, la qualité et l'efficacité des médicaments humains |
| VICH | Exigences techniques pour l'homologation des médicaments vétérinaires |
| JECFA (en anglais seulement) | Innocuité des additifs alimentaires et des contaminants |
| JRC (en anglais seulement) | Conseils scientifiques indépendants à l'appui des politiques de l'UE |
| ISA de l'EPA des États-Unis (en anglais seulement) | Survol des étapes et des critères pour l'élaboration d'évaluations scientifiques intégrées |
3.2. Sources de données
Parmi toutes les sources de données citées pour orienter les évaluations des risques de cancer, les plus courantes sont accessibles au public et comprennent des outils in silico pour effectuer des évaluations en toxicologie computationnelle (tableau 4). Certains secteurs de programme utilisent des bases de données et des outils in silico qui ne sont pas en libre accès. Cela comprend des outils de prévision exclusifs comme Leadscope, des données sur l'exposition aux médicaments de l'IQVIA et des données disponibles par l'entremise de l'Institut canadien d'information sur la santé. De plus, l'ARLA-DES, la DGPSA-DPT-BMOSR/BCASN/BGMIV/BEC, la DGPSA-DMBR-CEPB/CEPRB/CERSEC-BEC, la DGPSA-DPSNSO-DEMVSO, la DGPSA-DPSC, la DGPSA-DA-BIPC, la DGPSA-DMV et la DGSESC-BECSN reçoivent des présentations de l'industrie qui contiennent des données faisant l'objet d'une propriété exclusive pour orienter l'évaluation des risques de cancer.
| SOURCE | TYPE DE RENSEIGNEMENTS |
|---|---|
| Bases de données documentaires | |
| PubMed (en anglais seulement) | Publications |
| Scopus (en anglais seulement) | Publications |
| Rapports nationaux et internationaux | |
| Report on Carcinogens du NTP (en anglais seulement) | Évaluation de la toxicité et de la cancérogénicité de substances |
| CIRC | Publication de monographies traitant des dangers cancérogènes pour les humains |
| ATSDR (en anglais seulement) | Examen de l'exposition environnementale aux substances |
| Évaluations de l'IRIS (en anglais seulement) | Détermination et caractérisation par le programme IRIS de l'EPA du danger que posent pour la santé des produits chimiques présents dans l'environnement |
| Évaluations de l'EFSA | Évaluation des risques que posent des substances chimiques associées à la chaîne alimentaire |
| CCSN | Évaluation des effets environnementaux des installations nucléaires |
| Monographies du JECFA (en anglais seulement) | Évaluation des risques que posent les additifs alimentaires et les contaminants chimiques présents dans les aliments |
| Bases de données : Généralités | |
| CEBS (en anglais seulement) | Compilation de données individuelles et sommaires sur les animaux provenant du programme d'essai du NTP et d'autres déposants dans un seul dépôt électronique |
| Carcinogenic Potency Database (CPDB)* (en anglais seulement) | Compilation de résultats d'études de cancérogénicité à long terme chez les animaux |
| Carcinogenicity Database de Lhasa (en anglais seulement) | Compilation de résultats d'études de cancérogénicité à long terme chez les animaux |
| HSDB** (en anglais seulement) | Données de toxicologie et d'exposition à des substances dangereuses |
| Pubchem (en anglais seulement) | Données sur la composition chimique et la toxicité de substances |
| ToxCast (en anglais seulement) | Données à haut débit pour la toxicité chimique |
| COSMOS** | Évaluation de la toxicité à doses répétées d'ingrédients cosmétiques |
| ToxNet** (en anglais seulement) | Bases de données sur les effets toxiques de substances |
| REACH | Évaluation d'homologations de substances individuelles d'entreprises; évaluation quant à la possibilité de gérer les risques de substances diverses |
| Genomics (en anglais seulement) | Données de toxicogénomique pouvant être utilisées pour cerner les dangers et analyser les mécanismes d'action |
| AOP (en anglais seulement) | Voie associée aux effets indésirables de substances |
| ClinicalTrials (en anglais seulement) | Essais cliniques financés par le secteur privé ou par l'État |
| Bases de données : Pour l'évaluation de la sûreté de produits pharmaceutiques après leur mise en marché | |
| Canada Vigilance | Base de données en ligne des effets indésirables de médicaments |
| Vigilyze É.-U., OMS (en anglais seulement) | Base de données de pharmacovigilance mondiale |
| FDA Adverse Event Reporting System (en anglais seulement) | Base de données des événements indésirables |
| Outils de toxicologie computationnelle | |
| Boîte à outils de l'OCDE | Évaluation des dangers chimiques reproductible et transparente |
| ToxTree (en anglais seulement) | Estimation des dangers toxiques des substances |
| Outil d'évaluation des bénéfices liés à la qualité de l'air (OEBQA) | Estimation des répercussions sur la santé humaine associées aux changements dans la qualité de l'air ambiant au Canada |
| * : N'est plus mis à jour; ** : N'est plus disponible | |
4. Méthodologie utilisée pour effectuer les évaluations des risques de cancer
Les membres du sous-groupe de travail ont été interrogés concernant la méthodologie utilisée pour effectuer les évaluations du risque de cancer. Cela comprenait un compte rendu d'approches utilisées par différents secteurs de programme, en plus de l'utilisation de données de génotoxicité et de cancérogénicité pour orienter l'évaluation du risque de cancer. Comme il a été mentionné précédemment à la section 2.2 de ce rapport, les secteurs de programme individuels mènent des évaluations quantitatives, qualitatives ou les deux types d'évaluations du risque de cancer. De façon générale, lorsque de nouvelles évaluations quantitatives du risque de cancer sont réalisées à l'interne, le risque de cancer lié à l'exposition à une substance peut être calculé (p. ex. développement d'un facteur de pente ou d'un risque unitaire, estimation de l'excès de risque de cancer après l'exposition) ou la marge d'exposition (ME) peut être estimée. De plus amples renseignements sur ces approches sont fournis ci-dessous.
4.1. Approche générale pour évaluer le risque de cancer
Lors d'une évaluation des risques liés au cancer, l'identification des dangers est la première étape et elle est réalisée par tous les secteurs de programme interrogés qui mènent des évaluations du risque de cancer de novo. Dans le contexte des évaluations des risques de cancer, l'identification des dangers demande d'évaluer le poids de la preuve collective et d'établir si un composé présente un potentiel cancérogène. À la suite de cette évaluation, le mécanisme donnant lieu à la formation de tumeur est examiné, et toutes les populations sensibles ou étapes de vie qui nécessitent une considération supplémentaire sont identifiées. Diverses sources de données sont évaluées selon le cas et le secteur de programme, y compris les résultats des études sur la cancérogénicité chez les animaux, les tests de génotoxicité in vitro et in vivo, les études mécanistiques, les matrices épidémiologiques et, plus récemment, les analyses de relation structure-activité (p. ex. R(Q)SA, en lecture avec des composés chimiquement similaires) et les données toxicogénomiques. Pour certains secteurs de programme sondés, la détermination de la causalité de la cancérogénicité est également évaluée en tenant compte des critères de Bradford-Hill modifiés.
La pertinence humaine de la fréquence des constats de tumeurs animales a également été décrite comme étant une considération dans certains secteurs de programme. À titre d'exemple, un certain nombre de composés industriels et thérapeutiques causent des tumeurs hépatiques chez les rats par l'activation du récepteur nucléaire appelé récepteur alpha activé de la prolifération des peroxysomes (PPARα). Un grand nombre d'études mécanistiques ont démontré que les mécanismes menant à la formation de tumeurs chez les rongeurs par l'intermédiaire du PPARα pourraient ne pas être pertinents pour les humains en raison des différences biologiques dans les réponses en aval. Par conséquent, les résultats des tumeurs hépatiques chez les rats ou les souris qui présentent un mode d'action (MA) non génotoxique du PPARα ne sont probablement pas considérés comme étant pertinents pour l'évaluation des risques liés au cancer chez l'humain.
La connaissance du mode d'action d'un composé est utile pour déterminer l'approche d'extrapolation à faible dose d'une substance. Il s'agit d'un élément important du processus d'identification des dangers puisque la réponse à la dose à des niveaux de dose pertinents pour les humains n'est généralement pas examinée dans le cadre d'études sur la cancérogénicité chez les animaux. À cette fin, si le poids de la preuve suggère qu'un composé présente un mode d'action mutagène, on suppose que la courbe dose-réponse linéaire (ou non-seuil) est prise en charge et qu'un composé présente une mode d'action non mutagène, une courbe dose-réponse non linéaire (ou seuil) est prise en charge. Dans les cas où les données probantes concernant le mode d'action ne sont pas claires, une courbe dose-réponse linéaire est considérée comme une approche par défaut conservatrice.
Pour les composés qui présentent un mode d'action sans seuil, le niveau de risque progressif acceptable dans les secteurs de programme interrogés varie de 10-5 à 10-6 (c.-à-d. 1 cas de cancer supplémentaire par 100 000 à 1 000 000 de personnes exposées ou au-dessus de la normale, selon le secteur de programme). À la DPT, alors que le niveau acceptable de risque d'impureté mutagène dans un produit médicamenteux qui présente un mode d'action sans seuil est de 10 -5, un IPA qui présente un potentiel mutagène est considéré comme acceptable pour l'usage humain lorsque l'avantage potentiel est considéré comme supérieur aux risques (p. ex. maladie grave ou menaçant le pronostic vital comme un cancer avancé) (Tableau 5).
Unique aux DPT, DMBR et DPSC de la DGPSA, il s'agit de l'évaluation du risque de cancer dans le cadre d'une évaluation des avantages-risques pour un produit pharmaceutique ou biologique. Une évaluation des avantages-risques d'un médicament est une évaluation globale qui tient compte des avantages cliniques potentiels, des préjudices cliniques potentiels (p. ex. événements indésirables), des risques non cliniques (p. ex. toxicité reproductive, cancérogénicité), des caractéristiques de la population de patients visée (p. ex. gravité de la maladie) et de la disponibilité d'autres options thérapeutiques. Dans le cas de la DMBR et de la DPT, un résultat favorable d'une évaluation des avantages-risques est nécessaire pour accorder l'autorisation du marché pour un nouveau médicament. L'évaluation des essais cliniques dans le cadre de la DPT et de la DMBR se concentre principalement sur le risque du produit en cours d'examen et sur l'acceptabilité de l'atténuation des risques proposée pour l'essai clinique.
De plus, dans le cadre de la pharmacovigilance post-commercialisation à la DPSC, une évaluation du risque de cancer peut être initiée lorsqu'un problème (c.-à-d. un signal d'innocuité) est détecté à partir d'une ou de plusieurs des sources couramment étudiées. Parmi les exemples de sources de signaux d'innocuité figurent, sans s'y limiter, les nouvelles émergentes dans les médias, l'intimation des nouvelles évaluations de l'innocuité menées par d'autres organismes de réglementation, la disproportion observée dans le signalement spontané des réactions indésirables soupçonnées au médicament, une nouvelle étude publiée dans la littérature médicale, les nouvelles découvertes provenant d'études ou de registres de sécurité commandités par l'industrie, etc. Les nouveaux signaux d'innocuité sont ensuite priorisés et évalués en détail afin d'établir un lien de causalité (et la certitude associée) entre une intervention de santé et un cancer. Au sujet des meilleures données probantes actuelles, ces évaluations des signaux d'innocuité conduisent souvent à diverses recommandations fondées sur des données probantes pour gérer et atténuer le risque (p. ex. changements d'étiquetage des produits, communication des risques identifiés aux professionnels de la santé et au public, éducation et formation, distribution contrôlée, retrait de l'autorisation du marché des produits, etc.).
4.2. Approche de la marge d'exposition (ME) à l'évaluation du risque de cancer
La prise en considération de la ME est une autre approche utilisée pour évaluer les préoccupations en matière de sécurité des substances génotoxiques et cancérogènes dans les médicaments, les aliments et l'environnement. Une ME est dérivée en calculant le rapport entre un niveau d'effet indésirable [c.-à-d. le point de départ (PDD) le plus bas pour un critère d'effet pour le cancer] avec une exposition humaine estimée. Une petite ME est associée à une plus grande inquiétude qu'une grande ME. Par exemple, une ME peut être un outil utile pour comprendre s'il existe une préoccupation en matière de sécurité à un niveau particulier d'exposition à un cancérogène (mode d'action génotoxique ou non génotoxique). L'approche de la ME n'offre pas de valeur spécifique au risque de cancer; il s'agit plutôt d'une approche pratique qui fournit de l'information pour déterminer si l'exposition à une substance est préoccupante, sans autre investigation ou débat sur la forme de la courbe dose-réponse dans la plage de faible dose. Bien qu'une estimation quantitative des risques ne soit pas calculée, le mode d'action pour un cancérogène (c.-à-d. génotoxique ou non génotoxique) peut tout de même jouer un rôle important dans l'interprétation d'une ME calculée. Généralement, pour une substance génotoxique et cancérogène, si la ME est ³ 10 000, le niveau de préoccupation en matière de sécurité est considéré comme faible.
Le sondage mené par le sous-groupe de travail de la méthode d'évaluation des risques de cancer a indiqué que plusieurs programmes (p. ex. DA-BIPC, DMV, DSM-BERSE) utilisent l'approche de la ME pour évaluer la préoccupation en matière de sécurité des substances cancérogènes, même si ces programmes peuvent avoir différentes façons de choisir le PDD approprié et d'interpréter l'acceptation de la ME. Pour certains programmes (p. ex. DPT, ARLA-DES), une ME est uniquement calculée pour une substance qui présente un mode d'action non génotoxique.
| PROGRAMME | APPROCHES PRIVILÉGIÉES |
|---|---|
| BERSE |
CT/DT05=concentration tumorigène ou dose tumorigène associée à une augmentation de 5 % de l'incidence ou de la mortalité due au cancer. |
| BECSN |
|
| DQE |
|
| DEEAS |
|
| SECAI |
|
| DEESC |
|
| BER |
|
| BRP/BPRPCC |
|
| PNR |
|
| IRS |
|
| BMOSR |
|
| BEC |
|
| DIM |
|
| DMV |
|
| BIPC |
|
| DEMVSO |
|
| CEPB, CEPRB et CERSEC-BEC |
|
| BPPC |
|
| DES |
|
4.3. Utilisation des données de génotoxicité et de mutagénicité pour éclairer la méthodologie d'évaluation des risques de cancer
Soixante-huit pour cent (68 %) des répondants au sondage utilisent des données de génotoxicité ou de mutagénicité pour éclairer leurs évaluations. Les principales utilisations (Figure 2) de ces données sont d'orienter le mode d'action cancérogène d'une substance (cancérogène génotoxique à action directe ou indirecte) et d'orienter par la suite l'approche d'évaluation des risques (avec seuil ou sans seuil).
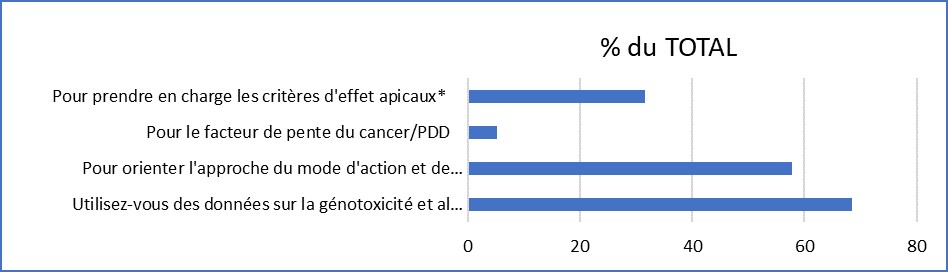
Figure 2. Utilisation des renseignements sur la génotoxicité.
| Information pour l'utilisation des renseignements sur la génotoxicité | Pourcentage (%) du TOTAL |
|---|---|
| Pour prendre en charge les critères d'effet apicaux* | 31,6 |
| Pour le facteur de pente du cancer/PDD | 5,3 |
| Pour orienter l'approche du mode d'action et l'évaluation du risque | 57,9 |
| Utilisez-vous des données sur la génotoxicité et la mutagénicité pour l'évaluation du risque et, si oui, comment? | 68,4 |
Figure 2. Utilisation des renseignements sur la génotoxicité. Selon les réponses fournies par 19 répondants au sondage à Santé Canada. *Les critères d'effet apicaux font référence à des résultats observables dans un organisme entier, comme un signe clinique ou un état pathologique, qui indique un état pathologique qui peut résulter d'une exposition à une substance toxique.
5. Défis présentés lors de l'évaluation des risques de cancer
Plusieurs défis ont été identifiés par le sous-groupe de travail au cours de l'évaluation des risques de cancer et sont décrits ci-dessous. Ces défis se rapportent principalement à la compréhension de la courbe dose-réponse pour les cancérogènes, à l'extrapolation du risque d'une forte dose à l'exposition à faible dose et à l'incertitude dans les données disponibles utilisées pour l'évaluation des risques.
5.1. Détermination de la forme de la courbe dose-réponse dans la portion correspondant aux faibles doses
Après l'identification des dangers (voir la section 4.1), une évaluation de l'exposition et de la dose peut être effectuée pour évaluer le ou les risques chez les humains à des niveaux d'exposition d'intérêt, et peut impliquer l'extrapolation des données de la tumeur animale à partir de doses élevées (c.-à-d. la plage de doses généralement évaluée dans les études de cancérogénicité) à des doses faibles (c.-à-d. la plage posologique généralement pertinente à l'exposition humaine). Comme la forme de la courbe dose-réponse (c.-à-d. linéaire plutôt que non linéaire) détermine si une approche avec seuil ou sans seuil doit être utilisée pour évaluer le risque de cancer, un élément important du processus d'identification des dangers est de déterminer si l'ensemble des données suggère que le composé présente un seuil ou un mode d'action sans seuil. Pour les composés non génotoxiques qui présentent un potentiel cancérogène dans les études sur la cancérogénicité chez les animaux, un mécanisme d'action avec seuil est tenu pour acquis dans les cas où aucune dose sans effet observé (DSEO) ou aucune dose sans effet nocif observé (DSENO) n'est identifiée ou des données sur le mode d'action sont disponibles pour appuyer une approche non linéaire. Certains secteurs de programme ont indiqué qu'il était difficile de déterminer s'il faut utiliser une approche avec ou sans seuil, car les résultats des études réalisées pour évaluer le potentiel génotoxique du composé ne sont pas concluants. Cependant, dans les cas où ce n'est pas clair, un mode d'action sans seuil est souvent considéré comme une approche par défaut.
On considère que les composés mutagènes présentent une courbe de réponse à la dose sans seuil, qui suppose que même une très faible dose d'un mutagène est associée à un effet mutagène et constitue donc un risque cancérogène. Plus récemment, des expériences in vitro et in vivo ont suggéré qu'une courbe sous-linéaire de dose-réponse, ou même un seuil, peut exister à des niveaux faibles en raison de l'influence des mécanismes de réparation de l'ADN et du métabolisme cellulaire. Il a été suggéré qu'une DSEO ou une DSENO peut être établie en fonction des résultats des études de mutagénicité à faible dose, même pour les mutagènes puissants tels que le méthanesulfonate d'éthyle (Muller et Gocke, 2009; ICH M7(R1)). Les répondants ont indiqué qu'il était difficile de déterminer le niveau de preuve nécessaire pour appuyer un seuil de courbe dose-réponse. De plus, certains répondants ont souligné que les données disponibles sont parfois controversées et incohérentes, ce qui rend l'interprétation des données difficile.
5.2. Comment effectuer une extrapolation de dose élevée à faible dose
La forme de la courbe dose-réponse (linéaire et non linéaire) dicte comment une dose élevée à une extrapolation à faible dose est réalisée. Certains répondants ont exprimé de l'incertitude quant à la pertinence des résultats des études à doses élevées à l'évaluation du risque de cancer humain et l'utilisation de ces données pour extrapoler jusqu'au niveau de dose faible généralement associé à l'exposition humaine. De même, les répondants de la DSSER ont exprimé de l'incertitude quant à la relation dose-réponse à des taux d'exposition et à des niveaux conformes à la plupart des scénarios d'exposition au public, ainsi qu'à la pertinence des scénarios d'exposition aux rayonnements à dose élevée/dose élevée en cas de risque de cancer pour les scénarios d'exposition chronique à faible dose. Un manque de données scientifiques de haute qualité a également été cité comme une préoccupation.
Un autre défi associé à l'extrapolation de dose élevée à faible dose, particulièrement pour les composés sans seuil, est déduit si l'estimation du risque de cancer dérivée à l'aide d'approches linéaires est trop conservatrice et ne convient pas à l'estimation du risque de cancer humain. Au BECSN, les études sur la cancérogénicité ne sont généralement pas disponibles, et les résultats des études de génotoxicité sont utilisés en tant que marqueur indirect pour la cancérogénicité dans l'évaluation du risque de cancer. La courbe dose-réponse à partir des tests de génotoxicité n'est pas déterminée, et la linéarité est assumée pour tout génotoxique, y compris les clastogènes. D'après cette évaluation, la substance est identifiée comme une substance hautement prioritaire ou faible priorité pour une considération plus approfondie.
5.3. Incertitude
Le sous-groupe de travail a identifié plusieurs défis liés à l'incertitude en ce qui concerne les données appuyant l'évaluation du risque de cancer ou l'incertitude associée à l'évaluation des risques liés au cancer. Elles comprennent :
- Incertitudes quant aux estimations historiques d'exposition (en particulier pour l'exposition aux rayonnements).
- Incertitude quant à la façon d'estimer la dose quotidienne moyenne pour les expositions chimiques intermittentes ou l'exposition se produisant pour moins d'une vie.
- Incertitude quant à la façon d'estimer le risque de cancer excédentaire à la suite d'une exposition élevée à court terme.
- Manque d'information pour réaliser une évaluation significative du risque de cancer, comme la disponibilité des facteurs de pente du cancer et des données pertinentes sur l'exposition humaine. Il s'agit d'un défi particulier pour les nouvelles substances du BECSN.
- Incertitude quant à l'application des facteurs d'incertitude appropriés lors de l'élimination d'une VBS pour les cancérogènes seuils et l'évaluation de l'adéquation des VBS dérivées, y compris celles dérivées pour diverses sous-populations, le cas échéant.
- Pertinence des données provenant de différentes espèces animales (p. ex. extrapolation d'espèces à espèces, évaluant la pertinence des résultats des tumeurs animales aux humains).
- Incertitude quant à la façon dont les systèmes d'essai in vitro et in vivo reflètent le métabolisme humain ou l'activation chimique.
- Incertitude quant à la façon d'intégrer les données des études R(Q)SA et toxicogénomiques à l'évaluation du risque de cancer humain.
- Vérifier si les résultats des études in vitro et in silico sont suffisants pour conclure qu'un composé ne présente aucun potentiel mutagène, génotoxique ou cancérogène.
- Incertitude quant à l'ensemble des preuves requises pour conclure qu'une substance ne présente pas de mode d'action sans seuil.
- Incertitude associée à l'évaluation de la cancérogénicité dans les groupes d'âge potentiellement sensibles (p. ex. exposition chez les enfants et application des facteurs d'ajustement en fonction de l'âge au facteur de pente du cancer ou au risque unitaire).
- Incertitude des estimations du risque de cancer due au manque de données sur la radiosensibilité. Ce défi a été particulier au Bureau de la protection contre le rayonnement.
- Incertitudes quant à l'établissement d'un lien causal entre un médicament ou un dispositif et le risque de cancer étant donné la preuve existante. Ces incertitudes peuvent être liées à des préoccupations relatives à la plausibilité biologique, à la validité méthodologique des données probantes pour la déduction causale, à la puissance statistique des études, à l'incohérence des conclusions de l'étude, à la généralisabilité des données probantes aux paramètres de santé canadiens, à la production de rapports sur les résultats sélectifs et aux biais publiés.
- Interprétation de la signification des données cancérogènes sur les risques pour la santé par rapport aux expositions environnementales.
Pour gérer l'incertitude associée à une évaluation des risques particulière, un évaluateur des risques peut reconnaître la ou les sources d'incertitude dans les conclusions de l'évaluation des risques et décrire les hypothèses formulées dans l'évaluation des risques avec une justification. Dans certains cas, il peut s'avérer approprié d'appliquer un facteur d'innocuité ou d'incertitude supplémentaire lors de la substitution d'une VBS, d'exiger une plus grande ME, ou d'appliquer le principe de précaution (p. ex. ne pas incorporer les données de R(Q)SA et des études toxicogénomiques dans l'évaluation du risque de cancer jusqu'à ce que les critères d'effet soient mieux validés.
6. Besoins et directives futurs
L'un des objectifs du processus de préparation du présent document explicatif était de préciser les besoins et les occasions de collaboration et d'harmonisation futurs, dans la mesure du possible. À cette fin, les membres du sous-groupe de travail ont discuté des besoins de leurs programmes respectifs et ont relevé d'éventuels secteurs à examiner.
6.1. Harmonisation
Les membres du sous-groupe de travail ont discuté des possibilités d'harmonisation. Il a été généralement convenu que la normalisation de la méthode d'évaluation des risques de cancer à Santé Canada serait difficile puisque les diverses directions générales et directions fondent leurs évaluations des risques de cancer sur des lignes directrices différentes, selon des ensembles de règlements différents. De plus, les évaluations des risques ont des exigences variées et servent des fins différentes. Dans certains cas, l'utilisation de documents d'orientation particuliers est prévue par la loi, ce qui rend leur utilisation obligatoire pour certains secteurs de programme. Dans d'autres cas, l'utilisation de documents d'orientation particuliers est prévue par une convention internationale, rendant leur application plus appropriée dans le contexte international. Cependant, dans le but de promouvoir l'uniformité des approches d'évaluations des risques cancérogènes à l'échelle du Ministère, lorsque cela est possible, la facilitation de la mise en commun de l'information a été définie comme un besoin fondamental. La section suivante porte sur cet aspect.
6.2. Mise en commun de l'information
Étant donné que tous les secteurs de programme recourent, dans une certaine mesure, aux évaluations des risques de cancer, il est nécessaire d'établir un processus efficace de mise en commun de l'information. Loin de se limiter aux évaluations des risques de cancer, ce besoin s'étend plutôt à de nombreux domaines d'évaluation des risques pour les contaminants et autres substances soumis à l'examen de Santé Canada. Les membres du sous-groupe de travail ont exprimé un besoin clair de cerner les secteurs de programme qui élaborent actuellement des évaluations des risques pour une substance donnée ou encore qui ont déjà mené des évaluations des risques sur une substance particulière. La mise en commun de l'information aiderait les évaluateurs des risques à tirer profit du travail effectué par d'autres secteurs de programme du Ministère, en plus d'améliorer l'uniformité des évaluations des risques à Santé Canada.
Certains avantages possibles du fait de faciliter la mise en commun des évaluations des risques à Santé Canada sont notamment une meilleure conformité interne, une efficacité accrue pour la réalisation d'une évaluation des risques sur une substance d'intérêt commun, une caractérisation des risques mieux éclairée (grâce à la compréhension des risques découlant d'autres sources d'exposition à une substance) et des possibilités accrues de collaboration entre les secteurs de programme à l'échelle du Ministère. Il est recommandé d'instaurer un projet de suivi pour explorer les possibilités de mise en commun de l'information et faciliter le processus dans l'ensemble du Ministère. De plus, il serait possible d'envisager d'utiliser dans les évaluations des risques de cancer des données dérivées d'études toxicologiques sur des substances chimiques d'intérêt prioritaire examinées par des chercheurs scientifiques de Santé Canada.
Cela offrirait une occasion de collaboration entre le personnel de recherche et de réglementation, en plus de permettre une évaluation concertée de stratégies de modernisation des méthodes d'évaluation des risques.
6.3. Évolution des tendances dans l'évaluation du risque potentiel de cancer chez l'humain
Le domaine de l'évaluation des risques est en constante évolution. Récemment, l'EPA des États-Unis a pris des engagements pour s'éloigner des essais sur les animaux, ce qui aura une incidence sur la façon dont les évaluations des risques de cancer sont réalisées pour les substances environnementales (et possiblement d'autres). De même, des changements au cadre de travail sur la façon d'évaluer le potentiel cancérogène d'un produit pharmaceutique pointent à l'horizon. De plus, des méthodes d'évaluation des risques de cancer et d'interprétation des données sur la relation dose-réponse concernant la génotoxicité continuent d'être mises au point.
6.3.1. Tendance vers une réduction du recours aux études chez les animaux et l'intégration de nouvelles approches pour l'évaluation du risque potentiel de cancer chez l'humain
Récemment, l'EPA des États-Unis a indiqué qu'elle délaissera les essais sur les animaux et cherchera à réduire de 30 % ses demandes et le financement d'études sur les mammifères d'ici 2025, dans l'objectif d'éliminer toutes les demandes et le financement de telles études d'ici 2035. De plus, elle a indiqué en 2019 que toute étude sur les mammifères demandée ou financée par l'EPA après 2035 devra être approuvée par l'administrateur. Étant donné que les disciplines scientifiques cherchent à réduire, à améliorer et à remplacer les essais sur les animaux, il est raisonnable de supposer que les résultats obtenus de nouvelles méthodes et stratégies d'essais toxicologiques seront de plus en plus intégrés aux évaluations des risques scientifiques, y compris les évaluations des risques de cancer, comme en témoignent les données toxicogénomiques (voir le rapport du GTERS sur la toxicogénomique, 2019) et l'inclusion de données sur la relation dose-réponse concernant la génotoxicité dans l'évaluation des risques de cancer, lorsque cela est approprié et applicable à la substance en cours d'évaluation.
À l'heure actuelle, le potentiel cancérogène des produits pharmaceutiques est évalué dans le cadre d'une étude de deux ans chez la souris et d'une étude de deux ans chez le rat, ou dans le cadre d'une étude de deux ans menée chez une seule espèce de rongeurs (généralement le rat) et d'une étude à court ou moyen terme in vivo sur des rongeurs (p. ex. modèles de cancérogenèse sur des souris transgéniques, comme TgRasH2) (ICH S1A; ICH S1B). Récemment, une évaluation rétrospective d'ensembles de données suggère que la connaissance de la pharmacologie et l'issue de divers essais de toxicité (p. ex. toxicologie génétique, études de toxicité chronique) peuvent fournir suffisamment d'information pour prédire le résultat de l'étude de deux ans chez le rat dans certains cas, ce qui suggère qu'une étude de cancérogénicité chez le rat pourrait ne pas être nécessaire dans ces cas (Sistare et coll., 2011; Van der Laan et coll., 2016). Pour cette raison, il est envisagé de modifier la ligne directrice actuelle S1 de l'ICH sur les essais de cancérogénicité chez les rongeurs afin d'inclure une approche fondée sur le poids de la preuve pour évaluer le risque de cancer chez l'humain, le cas échéant. Afin de définir l'ensemble de critères lorsqu'une étude de deux ans chez le rat offre une valeur ajoutée à l'évaluation des risques de cancer chez l'humain découlant de produits pharmaceutiques à petites molécules, ou pour déterminer si une approche du poids de la preuve pourrait remplacer une étude de deux ans chez le rat, une étude prospective est actuellement menée par le groupe d'experts responsable de la ligne directrice S1 de l'ICH (pour obtenir des détails, consultez le document d'avis réglementaire affiché sur le site Web de l'ICHNote de bas de page 3). L'étude a été lancée en août 2013 et est toujours en cours; le résultat de l'étude orientera les modifications à la version actuelle de la ligne directrice S1 (des rapports d'étape sont publiésNote de bas de page 4). Les modifications devraient introduire une approche plus intégrée à l'évaluation des risques de cancer chez l'humain et faciliter les objectifs de réduction, d'amélioration et de remplacement des essais sur les animaux.
Des études à long terme chez la souris et le rat sont également nécessaires pour évaluer le potentiel cancérogène de pesticides. Étant donné la décision de l'EPA des États-Unis de délaisser les essais sur les animaux, un projet est en cours dans le but de définir des critères pour la levée de l'exigence de la tenue de l'une ou l'autre de ces études pour les pesticides individuels en fonction de données toxicologiques et d'exposition. Le Rethinking Carcinogenicity Assessment for Agrochemicals Project (ReCAAP) est dirigé par des experts du consortium scientifique international People for the Ethical Treatment of Animals (PETA), du gouvernement et de l'industrie qui élaborent une stratégie visant à délaisser l'approche des « cases à cocher », qui comprend des essais biologiques, et à se pencher vers des mécanismes de transmission des maladies pertinents pour l'humain et d'autres sources d'information pertinentes. Les détails de ce projet ont été présentés à la 58e réunion annuelle de la Society of Toxicology (SOT)Note de bas de page 5.
6.3.2. Interprétation des données sur la relation dose-réponse concernant la cancérogénicité
Le sous-groupe de travail a cerné une occasion de mieux caractériser les approches émergentes pour l'évaluation des risques de cancer. Bien que le sondage ait relevé certaines ressources importantes utilisées dans l'interprétation des données de cancérogénicité à Santé Canada, il ne permettait pas un compte rendu détaillé de la façon dont les données sur la relation dose-réponse sont analysées. Un rapport de suivi qui permettrait d'explorer diverses options pour l'utilisation de l'information sur le mode d'action pour l'interprétation des données de cancérogénicité serait utile. Une telle exploration pourrait approfondir diverses options quant à l'utilisation des données de génotoxicité. Plus précisément, un tel rapport pourrait examiner la façon dont différents groupes quantifient le risque; il pourrait inclure un aperçu de l'utilisation des données de génotoxicité, et de l'utilisation connexe d'une approche d'extrapolation linéaire aux faibles doses (sans seuil) par rapport à une approche non linéaire (avec seuil).
6.3.3. Interprétation et intégration potentielle de données quantitatives sur la relation dose-réponse concernant la génotoxicité dans les évaluations des risques de cancer
Dans certains cas, les évaluateurs des risques utilisent des données de génotoxicité pour orienter leurs évaluations des risques de cancer. Ces données sont généralement utilisées de façon qualitative pour déterminer l'approche utilisée pour l'interprétation des données sur la relation dose-réponse concernant la cancérogénicité (c.-à-d. linéaire sans seuil ou non linéaire avec seuil). Cependant, plutôt que le paradigme actuel qui recommande l'utilisation des données de génotoxicité comme éléments à l'appui pour déterminer l'approche utilisée pour évaluer le danger ou le risque de cancer, il existe de plus en plus de travaux qui visent à établir un fondement où la génotoxicité peut être traitée comme un véritable critère d'effet toxicologique (White et coll., 2020). Par conséquent, les évaluateurs des risques sont maintenant confrontés à des questions difficiles. Par exemple, en l'absence de données de cancérogénicité, l'interprétation quantitative de données de mutagénicité in vivo (p. ex. essai de mutation chez des rongeurs transgéniques) peut-elle servir à faire une évaluation fiable des risques de cancer? Autrement, l'extrapolation linéaire aux faibles doses demeure-t-elle une approche valide pour évaluer les risques de cancer? Un rapport qui examine ces problèmes et qui fournit des renseignements généraux aux évaluateurs des risques de Santé Canada pourrait aider l'inclusion de données sur la relation dose-réponse dans une évaluation des risques de cancer pour les groupes pertinents à l'échelle du Ministère, le cas échéant. Le sous-groupe de travail recommande la surveillance continue des ouvrages scientifiques concernant l'utilisation quantitative de données sur la relation dose-réponse concernant la mutagénicité in vivo pour l'évaluation des risques de cancer et la prise de décisions réglementaires.
7. Références
7.1. Documents d'orientation
EFSA. (2019) Guidance on the use of the Threshold of Toxicological Concern approach in food safety assessment. (en anglais seulement)
Santé Canada. (2000) Document de principes : Document technique - Cadre décisionnel pour l'évaluation et la gestion des risques à l'Agence de réglementation de la lutte antiparasitaire.
SPN 2000-01.
Santé Canada. (2008) Document de principes : Utilisation de facteurs d'incertitude et du facteur issu de la Loi sur les produits antiparasitaires dans l'évaluation des risques des pesticides pour la santé humaine. SPN 2008-01.
Santé Canada. (2010a) L'évaluation des risques pour les sites contaminés fédéraux au Canada, Partie V : L'évaluation quantitative détaillée des risques pour la santé humaine associés aux substances chimiques (ÉQDRCHIM)
Santé Canada. (2010b) L'évaluation des risques pour les sites contaminés fédéraux au Canada, Partie II : Valeurs toxicologiques de référence (VTR) de Santé Canada et paramètres de substances chimiques sélectionnées, version 2.0
Santé Canada. (2013) Évaluation des risques pour les sites contaminés fédéraux au Canada : Document d'orientation provisoire sur l'évaluation des risques pour la santé humaine associés à une exposition de courte durée aux substances cancérogènes présentes dans les sites contaminés
ICH M7(R1). (2017) Évaluation et contrôle des impuretés réactives de l'ADN (mutagènes) dans les produits pharmaceutiques pour limiter les risques de cancérogénicité
ICH M3(R2). (2009) Directive sur les études d'innocuité non cliniques requises pour les études cliniques chez l'humain
ICH S1A. (1995) Nécessité des études sur la cancérogénicité des produits pharmaceutiques
ICH S1B. (1997) Évaluation de la cancérogénicité des produits pharmaceutiques
ICH S1C(R2). (2008) Sélection des doses pour les études de cancérogénicité des produits pharmaceutiques et dose limite
ICH S2(R1). (2011) Directive sur l'évaluation de la génotoxicité des produits pharmaceutiques destinés à l'utilisation humaine et interprétation des données
ICH S6(R1). (2011) Évaluation au stade préclinique de la sécurité des produits pharmaceutiques issus de la biotechnologie
ICH Q3A(R2). (2006) Présence d'impuretés dans les nouvelles substances médicamenteuses
ICH Q3B(R2). (2006) Présence d'impuretés dans les nouveaux produits
ICH Q3C(R6). (2016) Impuretés : Directive sur les solvants résiduels
JECFA. (2016) Guidance Document for WHO Monographers and Reviewers Evaluating Veterinary Drug Residues in Food. Comité mixte FAO/OMS d'experts des additifs alimentaires.
OCDE. (2002) Guidance Notes for Analysis and Evaluation of Chronic Toxicity and Carcinogenicity Studies. OECD Environment, Health and Safety Publications. Series on Testing and Assessment No. 35 and Series on Pesticides No. 14. ENV/JM/MONO(2002)19
US EPA. (2015) Preamble to the Integrated Science Assessments (ISA). U.S. Environmental Protection Agency, Washington, DC, EPA/600/R-15/067.
US EPA. (2005a) Guidelines for Carcinogen Risk Assessment.
US EPA. (2005b) Supplemental Guidance for Assessing Susceptibility from Early-Life Exposure to Carcinogens. Risk Assessment Forum.
VICH GL28. (2005) Studies to Evaluate the Safety of Residues of Veterinary Drugs in Human Food: Carcinogenicity testing. International Cooperation on Harmonisation of Technical Requirements for Registration of Veterinary Medicinal Products (VICH).
VICH GL23 (R). (2014) Studies to Evaluate the Safety of Residues of Veterinary Drugs in Human Food: Genotoxicity testing. International Cooperation on Harmonisation of Technical Requirements for Registration of Veterinary Medicinal Products (VICH).
7.2. Références bibliographiques
Annys E., Billington R., Clayton R., Bremm K.D., Graziano M., McKelvie J., Ragan I., Schwarz M., van der Laan J.W., Wood C., Öberg M., Wester P., Woodward K.N. (2014) Advancing the 3Rs in Regulatory Toxicology - Carcinogenicity testing: Scope for Harmonisation and Advancing the 3Rs in Regulated Sectors of the European Union. Regulatory Toxicology and Pharmacology. 69(2): 234-242.
Boobis A., Brown P., Cronin M.T.D., Edwards J., Galli C.L., Goodman J., Jacobs A., Kirkland D., Luijten M., Marsaux C., Martin M., Yang C., Hollnagel H.M. (2017) Origin of the TTC Values for Compounds that are Genotoxic and/or Carcinogenic and an Approach for their Re-Evaluation. Critical Reviews in Toxicology. 47(8): 710-732.
Madia F., Worth A., Corvi R. (2016) Analysis of Carcinogenicity Testing for Regulatory Purposes in the European Union. Review of the Current Demand of in vivo Carcinogenicity Studies Across Sectors. JRC Technical Reports.
Meek, M. E., Boobis, A., Cote, I., Dellarco, V., Fotakis, G., Munn, S., Seed, J., & Vickers, C. (2014). New developments in the evolution and application of the WHO/IPCS framework on mode of action/species concordance analysis. Journal of applied toxicology. 34(1): 1-18.
Muller L. and Gocke E. (2009) Considerations Regarding a Permitted Daily Exposure Calculation for Ethyl Methanesulfonate. Toxicology Letters. 190(3): 330-332.
Sistare F.D., Morton D., Alden C., Christensen J., Keller D., Jonghe S.D., Storer R.D., Reddy M.V., Kraynak A., Trela B., Bienvenu J.G., Bjurström S., Bosmans V., Brewster D., Colman K., Dominick M., Evans J., Hailey J.R., Kinter L., Liu M., Mahrt C., Marien D., Myer J., Perry R., Potenta D., Roth A., Sherratt P., Singer T., Slim R., Soper K., Fransson-Steen R., Stoltz J., Turner O., Turnquist S., van Heerden M., Woicke J., DeGeorge J.J. (2011) An Analysis of Pharmaceutical Experience with Decades of Rat Carcinogenicity Testing: Support for a Proposal to Modify Current Regulatory Guidelines. Toxicologic Pathology. 39(4): 716-744.
Toxicogenomics Working Group (2019). Evaluation of the use of toxicogenomics in risk assessment at Health Canada: An Exploratory Document on Current Health Canada Practices for the Use of Toxicogenomics in Risk Assessment. Prepared for the Task Force on Scientific Risk Assessment.
US EPA (2019). Memorandum on the Directive to Prioritize Efforts to Reduce Animal Testing. From Andrew R. Wheeler. September 10, 2019.
Van der Laan, J.W., Kasper P., Silva Lima B., Jones D.R., Pasanen M. (2016) Critical Analysis of Carcinogenicity Study Outcomes. Relationship with Pharmacological Properties. Critical Reviews in Toxicology. 46(7): 587-614.
White, P.A., Long, A.S., Johnson G.E. (2020) Quantitative Interpretation of Genetic Toxicity Dose‐Response Data for Risk Assessment and Regulatory Decision‐Making: Current Status and Emerging Priorities. Environmental and Molecular Mutagenesis. 61: 66-83.
Notes de bas de page
- Note de bas de page 1
-
Tout au long du présent document, les termes risque « acceptable », dose « acceptable », niveau « acceptable », etc. sont utilisés. Cette terminologie reflète ce qui est généralement utilisé dans les documents d'orientation réglementaire ou dans les règlements.
- Note de bas de page 2
-
À la DPT, l'« évaluation avantages-risques » est désignée « évaluation avantages-méfaits-incertitudes ». Un médicament est approuvé lorsque ses avantages dépassent tout méfait possible, avec un degré raisonnable de certitude.
- Note de bas de page 3
-
https://database.ich.org/sites/default/files/S1%28R1%29_EWG_RND.pdf (en anglais seulement)
- Note de bas de page 4
-
https://database.ich.org/sites/default/files/S1_StatusReport_2019_0802.pdf (en anglais seulement, rapport d'étape le plus récent; consultez le site Web de l'ICH pour des mises à jour)
- Note de bas de page 5
-
https://www.piscltd.org.uk/wp-content/uploads/2019/03/SOT-2019-Poster_Agchem-Bioassay-Waiver_Final_GMH.pdf (en anglais seulement)