
Radioprotection et normes de qualité en mammographie - Procédure de sécurité pour l'installation, l'utilisation et le contrôle des appareils à rayons X mammographique : Code de sécurité 36

Télécharger le format de rechange
(Format PDF, 860 Ko, 82 pages)
Organisation : Santé Canada
Publiée : octobre 2013
Table des matières
- Notes explicatives
- Remerciements
- Introduction
- Objectifs principaux du Code de sécurité
- Section A: Qualifications et responsabilités du personnel
- 1.0 Qualifications et responsabilités du personnel
- 1.1 Propriétaire
- 1.2 Radiologiste qui interprète des mammographies
- 1.3 Technologue en mammographie
- 1.4 Physicien médical
- 1.5 Spécialiste des systèmes informatiques (SSI)
- 1.6 Personnel de réparation et d'entretien
- 1.7 Conservation des registres relatif aux compétences personnelles
- 2.0 Mesures de réduction de l'exposition du personnel aux rayons X
- 3.0 Mesures de réduction de l'exposition du patient aux rayons X
- Section B : Exigences des locaux et des appareils
- 1.0 Exigences des locaux
- 1.1 Critères généraux
- 1.2 Conception et plan des locaux de mammographie
- 1.3 Calculs des blindages
- 2.0 Exigences des appareils de mammographie
- 2.1 Exigences réglementaires des appareils de mammographie
- 2.2 Achat d'appareil
- 2.3 Appareil de mammographie en service
- 2.4 Mise à niveau avec les systèmes d'imagerie numérique
- 2.5 Exigences détaillées concernant les appareils de mammographie
- 3.0 Systèmes de traitement d'images
- 3.1 Systèmes de mammographie film-écran
- 3.2 Systèmes de mammographie numérique
- 3.2.1 Plaques d'imagerie de CR
- 3.2.2 Cassette de CR
- 3.2.3 Tomosynthèse du sein
- 3.2.4 Poste de travail mammographique/Dispositif de visualisation électronique
- 3.2.5 Système d'archivage et de transmission d'images (PACS)
- 3.2.6 Mise en œuvre du PACS
- 3.2.7 Profils d'intégration de l'IHE (Integrating the Healthcare Enterprise)
- 3.2.8 Détection et diagnostic assistée par ordinateur (DAO)
- 3.2.9 Télémammographie
- 3.2.10 Compression d'images numériques
- 4.0 Équipement d'essai
- 5.0 Enquêtes de radioprotection
- 6.0 Élimination des appareils radiographiques
- Section C: Programme d'assurance de la qualité
- 1.0 Introduction
- 2.0 Essais d'acceptation
- 3.0 Procédures et équipement d'essai de contrôle de la qualité
- Annexe I : Organismes fédéraux/provinciaux/territoriaux de sécurité radiologique
- Annexe II : Limites de dose de rayonnement ionisant dans un contexte professionnel
- Annexe III : Calcul de la dose glandulaire moyenne
- Annexe IV : Guides de blindage pour le stockage des films radiographiques
- Annexe V : Unités de mesure du rayonnement
- Annexe VI Lexique
- Référence
Notes explicatives
Le présent document fait partie d'une série de Codes de sécurité préparés par Santé Canada afin d'établir les exigences relatives à l'utilisation sécuritaire des dispositifs émettant des radiations.
L'information contenue dans le présent Code de sécurité a été préparée pour fournir des directives spécifique aux propriétaires d'équipement mammographique, aux radiologistes, aux technologues en mammographie, aux physiciens médicaux et au personnel concerné par les procédures de sécurité, le rendement de l'équipement, la qualité de l'image, la radioprotection et la qualité globale d'un établissement où il se fait de la mammographie.
Le champ d'application de ce Code de sécurité comprend les technologies de mammographie telle que le film /écran, CR (de l'anglais computed radiography), DR (de l'anglais digital radiography) et la tomosynthèse. Les autres modalités d'imagerie mammaire n'utilisant pas les rayons X sont exclues de ce Code.
Le présent Code de sécurité remplace le Code de sécurité 33, Radioprotection dans l'exercice de la mammographie (SC 1995) et le Guide canadien de qualité en mammographie (SC 2002).
Les exigences envers le personnel, les procédures de sécurité, les directives relatives aux appareils et établissements et les mesures d'assurance de la qualité détaillées dans le présent Code de sécurité servent essentiellement à l'instruction et à l'orientation des personnes employées dans les ministères et organismes fédéraux de la fonction publique, de même que celles qui sont régies par le Code canadien du travail. Les établissements de juridiction provinciale ou territoriale peuvent être soumis à des exigences spécifiques associées à leurs statuts. On doit communiquer avec les organismes énumérés à l'annexe I pour obtenir des détails sur les exigences des lois de chaque province et territoire.
Dans ce Code, les termes « doit » ou « doivent » et « devrait » ou « devraient » ont été choisis à dessein. Les termes « doit » ou « doivent» indiquent qu'il s'agit d'une exigence essentielle afin de satisfaire aux normes de protection actuellement acceptées, tandis que les termes « devrait » ou « devraient » indiquent que la recommandation conseillée est fort souhaitable et qu'elle devrait être appliquée lorsqu'il est possible de le faire.
Dans ce domaine où la technologie progresse rapidement et où des problèmes imprévus et uniques surviennent continuellement, ce Code ne peut tenir compte de toutes les situations possibles. L'observation stricte des règles ne peut remplacer un jugement sûr. Les recommandations peuvent être modifiées dans certains cas inhabituels, mais seulement suite à des conseils d'experts en radioprotection. Le présent Code sera examiné et révisé périodiquement et les exigences particulières peuvent être reconsidérées à tout moment s'il devient nécessaire de tenir compte d'une situation imprévue. L'interprétation ou l'élaboration de toute question peut être réalisée en consultant le Bureau de la protection contre les rayonnements des produits cliniques et de consommation, Santé Canada, Ottawa, Ontario K1A 1C1.
Remerciements
Ce document reflète le travail de plusieurs personnes. Il a été préparé et rédigé par Mme Narine Martel de la Division d'imagerie médicale, Bureau de la protection contre les rayonnements des produits cliniques et de consommation. Nous remercions M. Richard Tremblay, M. Charles Steiner et les membres de la Division d'imagerie médicale pour leur assistance au cours de la préparation de ce Code.
Nous aimerions remercier les membres du Groupe de travail sur les normes de qualité en mammographie canadien et l'agrément, créé par le Comité national de l'Initiative canadienne pour le dépistage du cancer du sein:
Elaine Dever, Association canadienne des technologues en radiation médicale
Brenda Mitchell, Action Cancer Ontario (autrefois)
Tom Butterworth, Association canadienne des radiologistes (autrefois)
Andrea Nelson, Association canadienne des radiologistes
Jay Onysko, Agence de la santé publique du Canada
Rasika Rajapakshe, Collège canadien des physiciens en médecine
Rene Shumak, Cancer Care Ontario
Richard Tremblay, Ministère de la Santé et des Services sociaux du Québec (autrefois)
Nancy Wadden, St-Clare's Mercy Hospital, Terre-Neuve
Les contributions des organisations, organismes et associations suivantes, dont les commentaires et suggestions lors de la consultation de ce document ont contribué à l'élaboration de cette version finale du Code, sont remerciés:
Alberta College of Medical Diagnostic and Therapeutic Technologists
APIBQ Association des physiciens et ingénieurs biomédicaux du Québec Comité de radioprotection
Breast Centre Radiology, Edmonton Alberta
British Columbia Centre for Disease Control
British Columbia Interior Health Authority, Diagnostic Imaging Services
Canadian Association of Radiologists
Federal/Provincial/Territorial Radiation Protection Committee
Health Canada, Medical Devices Bureau
Health Prince Edward Island, Provincial Diagnostic Imaging Services
Horizon Health Network New Brunswick, Saint John Regional Hospital
Horizon Health Network New Brunswick, Upper River Valley Hospital
Imaging Research Program, Sunnybrook Research Institute
Ministère de la Santé et des Services sociaux, Laboratoire de Santé Publique du Québec
Northern Alberta Institute of Technology, School of Health Sciences
Ontario Breast Screening Program, Cancer Care Ontario
Ontario Ministry of Health and Long Term Care
Radiology Consultants Associated, Alberta
Saskatchewan Ministry of Labour Relations and Workplace Safety, Radiation Safety Unit
Saskatchewan Cancer Agency, Screening Program for Breast Cancer
Thunder Bay Regional Health Sciences Centre, Linda Buchan Centre for Breast Screening and Assessment
Vancouver Coastal Health Authority, Department of Radiology
Introduction
La mammographie est une méthode d'imagerie efficace pour détecter le cancer du sein. Elle permet de détecter le cancer à un stade précoce et, lorsque qu'un suivi diagnostic et des traitements appropriés sont apportés, de réduire la mortalité due à ce type de cancer. Au cours des vingt dernières années, les développements technologiques ont grandement amélioré la conception de l'équipement mammographique et le rendement de ces appareils, ont amélioré la qualité de l'image et ont permis de réduire les doses de rayonnement. Les procédures mammographiques aux rayons X comptent parmi les procédures radiologiques les mieux gérées. Cela est nécessaire pour assurer l'optimisation de la qualité de l'image en vue de l'interprétation des mammogrammes, et pour réduire au minimum l'exposition au rayonnement des patients.
Le cancer du sein est la forme de cancer la plus courante chez les canadiennes. En 2013, on estime que 23,800 femmes au Canada seront diagnostiquées d'un cancer du sein. Alors que l'incidence du cancer du sein au Canada s'est stabilisée depuis 1999, le taux de mortalité du cancer du sein a diminué de plus de 30 pour cent depuis 1986. L'amélioration significative observée est probablement le résultat des améliorations apportées dans le domaine du dépistage et de l'efficacité du traitement. En 2013, on estime à plus de 5,000 femmes qui mourront du cancer du sein. S'assurer de la qualité de la mammographie, que ce soit pour le dépistage ou pour le diagnostic, est une des composantes importantes dans la gestion du cancer du sein. Pour mettre en place un programme efficace de mammographie, il est essentiel d'effectuer la mammographie de manière à respecter des normes de qualité rigoureuses. La responsabilité de la qualité mammographique est partagée au Canada entre les gouvernements fédéral, provinciaux et territoriaux, et parmi les professionnels du domaine médical qui entretiennent les équipements, accomplissent les procédures et interprètent des mammogrammes.
Le but de ce document est de fournir des conseils, aux établissements de mammographie de dépistage ou de diagnostic, concernant la radioprotection et l'assurance de la qualité. Le contenu du présent document est basé sur, et harmonisé avec des normes canadiennes et internationales de mammographie en vigueur. Ces normes comprennent le Règlement sur les appareils de radiodiagnostic, Partie XII de la Loi sur les dispositifs émettant des radiations, qui réglemente la conception, la construction et le fonctionnement des appareils à mammographie, les exigences du Programme d'agrément en mammographie de l'Association canadienne des radiologistes, les exigences provinciales et les recommandations de la Commission internationale de protection radiologique (CIPR) et l'Agence internationale de l'énergie atomique (AIEA). Il convient de noter que d'autres exigences provinciales ou territoriales en vigueur peuvent remplacer ou compléter les dispositions du présent document.
Actuellement au Canada, la mammographie est réalisée à l'aide de systèmes de mammographie film-écran et de systèmes numériques. Les deux types de systèmes utilisent des rayons X de faible énergie qui pénètrent les tissus du sein, mais ils utilisent un récepteur d'image, ou détecteur, différent pour créer l'image. Dans la mammographie film-écran, un film à rayons X est placé en contact direct avec l'écran. Les photons de rayons X sont capturés par l'écran qui émet une lumière exposant le film. Le film est ensuite développé chimiquement pour produire le mammogramme. Les systèmes de mammographie numérique utilisent des détecteurs avec différentes technologies dans le but de produire des images mammographiques. Ces technologies sont généralement réparties en deux groupes: les détecteurs à conversion directe et les détecteurs à conversion indirecte. Dans les systèmes qui utilisent des détecteurs à conversion directe, les photons de rayon X incidents qui arrivent sur le récepteur d'image interagissent avec un matériau photoconducteur qui convertit directement l'énergie des rayons X en signal électrique transportant l'information sur l'image. Le signal électrique est produit et est affiché presque instantanément. Dans les systèmes qui utilisent un détecteur indirect, un scintillateur est utilisé pour capturer l'énergie des rayons X et pour la convertir en lumière. Les photons de lumière sont ensuite convertis en signal électrique. Les systèmes de plaques photostimulables (ou CR, de l'anglais computed radiography) utilisés en mammographie, font appel à une technologie du type imagerie numérique indirecte. Ces systèmes comprennent une cassette, une plaque d'imagerie qui contient du phosphore photostimulable et un lecteur de plaque d'imagerie. La cassette de CR, chargée avec une plaque d'imagerie, est positionnée dans le système de mammographie, comme on le fait avec les cassettes de film. À l'exposition aux rayons X, la plaque d'imagerie, qui contient un phosphore de stockage photostimulable, stocke l'image latente. La plaque d'imagerie est ensuite lue et une image mammographique est produite.
La dose de rayonnement associée à un examen mammographique bien effectué est très faible et essentiellement et uniquement donnée au tissu du sein. En raison des très faibles énergies de rayons X utilisés en mammographie, il y a très peu de dose à d'autres tissus. Cependant, toute procédure au cours de laquelle il y a exposition au rayonnement ionisant doit être gérée avec soin, car on présume que même de faibles doses de rayonnement risquent d'avoir des effets néfastes sur la santé. Des effets somatiques peuvent se manifester chez les personnes ayant été exposées et sont caractérisés par des changements observables qui se produisent dans les organes de ces personnes. Des effets génétiques peuvent survenir chez les descendants des individus exposés. En mammographie, le risque de défauts génétiques associés à des examens bien effectués est très faible, et dans le cas des femmes ménopausées, il n'y a aucun risque génétique.
Puisqu'il n'est pas possible de mesurer les effets cancérogènes à faibles doses, les estimations des effets des rayonnements peu intenses sont fondées sur une extrapolation linéaire à partir de doses relativement élevées. En raison des incertitudes à l'égard du risque radiologique, un modèle de risque de la protection contre les rayonnements part de l'hypothèse que le risque pour la santé dû à l'exposition aux rayonnements est proportionnel à la dose. On appelle cela l'hypothèse de linéarité sans seuil. Puisque l'effet projeté d'une faible dose augmente la fréquence des effets néfastes que très légèrement au-dessus du niveau « naturel » il est impossible de prouver par l'observation seule, la validité ou l'invalidité de cette hypothèse. Cependant, l'hypothèse de linéarité sans seuil a été largement adoptée en radioprotection et elle a mené à la formulation du principe « ALARA», qui vient de l'expression anglaise « As Low As Reasonably Achievable » aussi faible que raisonnablement réalisable. Le principe « ALARA » est une approche de la protection contre les rayonnements destinée à gérer et maîtriser les expositions aussi faibles que raisonnablement possible des travailleurs exposés au rayonnement et du public en général, en tenant compte des facteurs sociaux et économiques.
En mammographie, il faut considérer quatre aspects principaux dans la radioprotection. Premièrement, les patients ne devraient pas être soumis à des procédures radiographiques non nécessaires. Ceci veut dire que les examens doivent être justifiée, et seulement si l'information nécessaire au diagnostic ne peut pas s'obtenir autrement. Deuxièmement, lorsqu'une procédure est requise, il est essentiel que le patient soit protégé contre une exposition excessive au rayonnement pendant l'examen. Troisièmement, il est nécessaire que les employés au sein de l'établissement soient protégés contre une exposition excessive au rayonnement pendant qu'ils travaillent. Enfin, le personnel et le public en général à proximité de telles installations ont besoin d'une protection adéquate.
Tandis que des limites de doses réglementaires ont été établies pour les travailleurs exposés au rayonnement et le public en général, ces limites ne s'appliquent pas aux doses reçues par un patient soumis à une radiologie médicale. Chez les patients, le risque doit toujours être évalué en fonction de la nécessité clinique d'obtenir un diagnostic ou un traitement précis. On doit toujours s'efforcer de réduire les doses des patients au niveau le plus faible possible compatible avec une qualité optimale d'information nécessaire au diagnostic. Par une coopération étroite entre les professionnels médicaux, les technologues, les physiciens médicaux et autres personnels de soutien, il est possible de suivre un programme de radioprotection efficace et de maintenir un programme de mammographie de haute qualité.
Objectifs principaux du Code de sécurité
Le présent Code de sécurité fournit une orientation à tous les établissements de mammographie, à des fins de dépistage et de diagnostic, dans le but d'offrir et de maintenir des programmes de mammographie efficaces et d'assurer la radioprotection. Le but du présent Code de sécurité est de fournir aux établissements de mammographie l'information nécessaire pour atteindre les principaux objectifs suivants:
- assurer une qualité d'image mammographique optimale tout en réduisant au minimum l'exposition du patient aux rayons X;
- assurer une protection adéquate du personnel utilisant des appareils mammographiques; et
- assurer une protection adéquate des autres membres du personnel et du public en général qui se trouvent à proximité de l'équipement mammographique.
Afin de réaliser ces objectifs, ce Code de sécurité spécifie:
- les qualifications et responsabilités du propriétaire, des radiologistes qui interprètent les mammogrammes, le technologue en mammographie, le physicien médical et le spécialiste des systèmes d'information;
- les pratiques et les procédures pour minimiser les doses de rayonnement aux opérateurs et au public;
- les pratiques et les procédures pour minimiser les doses de rayonnement aux patients tout en maintenant une qualité d'image adéquate;
- les pratiques et les procédures pour s'assurer que les appareils radiographiques sont utilisés de façon sécuritaire;
- les renseignements sur la conception des installations et les exigences en matière de blindage;
- les normes de construction minimales et le rendement des appareils mammographiques;
- l'information nécessaire pour mettre en œuvre et exploiter un programme d'assurance de la qualité pour l'établissement;
- une liste d'essais d'acceptation et de contrôle de la qualité pour les divers types d'appareils mammographique et leurs accessoires; et
- un calendrier de réalisation des essais de contrôle de la qualité.
Ce Code de sécurité comprend trois sections
Section A : Responsabilités et protection
Cette section énonce les responsabilités du propriétaire, des radiologistes, des technologues en mammographie, des physiciens médicaux et des spécialistes des systèmes informatiques pour l'installation, le fonctionnement et le contrôle en toute sécurité des appareils. Elle énonce également les pratiques destinées à minimiser les doses de rayonnement aux patients, au personnel et au public.
Section B : Exigences des locaux et des appareils
Cette section énonce les exigences de conception des locaux et les normes minimales de construction et de rendement des appareils.
Section C : Programme d'assurance de la qualité
Section A: Qualifications et responsabilités du personnel
1.0 Qualifications et responsabilités du personnel
La présente section précise les qualifications et responsabilités de tout le personnel qui travaille en mammographie. Les compétences initiales, l'expérience et l'éducation continue, et le renouvellement des qualifications sont fournis et basés sur un cycle de trois (3) ans. Bien que les responsabilités du personnel décrites plus bas soient énumérées en groupes séparés, pour obtenir le niveau optimal de radioprotection et de qualité d'image, il est impératif qu'une coopération totale existe entre toutes les parties concernées.
1.1 Propriétaire
Le propriétaire est l'ultime responsable de la sécurité radiologique de l'établissement. Par définition, le propriétaire est la personne ou le groupe de personnes qui contrôle la possession et l'utilisation de l'équipement de mammographie. Le propriétaire est soit un individu, une société, un district, une province ou quelque autre entité. ll lui appartient donc de veiller à ce que les appareils utilisés ainsi que les locaux où sont installés les appareils répondent à toutes les normes de radioprotection et qu'un programme de radioprotection soit élaboré, mis en œuvre et tenu par l'établissement. Le propriétaire peut déléguer sa responsabilité aux membres qualifiés. La manière dont cette responsabilité est déléguée dépendra du nombre de membres du personnel, du fonctionnement de l'établissement et de la quantité d'appareils radiographiques possédés. Dans tous les cas, le propriétaire doit s'assurer qu'une ou plusieurs personnes qualifiées sont désignées pour remplir les rôles décrits plus bas.
Le propriétaire est responsable:
- de la mise en place et du déroulement du programme d'assurance de la qualité, pour obtenir la qualité optimale d'imagerie diagnostic, y compris les procédures d'essai de contrôle de la qualité et la tenue de registres;
- de la conformité de l'installation avec toutes les exigences réglementaires applicables;
- de la consultation des agences gouvernementales appropriées;
- au moment de la construction d'une nouvelle installation ou de la modification d'une installation existante, pour s'assurer que les mesures de radioprotection sont appropriées,
- lorsque des appareils à mammographie sont achetés, pour assurer une radioprotection appropriée et pour enregistrer ces appareils auprès de l'organisme approprié,
- fixant des inspections périodiques pour l'établissement. Dans certaines juridictions, l'organisme responsable des inspections a le mandat de fixer le calendrier d'inspection;
- d'établir des conditions de travail sécuritaires;
- de veiller à ce que
- les appareils fonctionnent correctement et soient entretenus de façon appropriée par le personnel compétent et que les appareils datés et non-conformes sont remplacés,
- les appareils détiennent une homologation canadienne des instruments médicaux,
- des normes de fonctionnement sécuritaires soient établies et suivies,
- la vérification aux fins du contrôle de la qualité des appareils à mammographie, de la machine à traiter les films et de l'équipement auxiliaire soit effectuée,
- les technologues aient reçu une formation appropriée dans l'utilisation des appareils,
- les technologues en stage de formation et le personnel inexpérimenté utilisent les appareils à mammographie uniquement lorsqu'ils sont sous la supervision directe d'un technologue autorisé ou certifié,
- s'assurer que les compétences professionnelles du personnel sont maintenues;
- de déclarer quels sont les membres du personnel exposés au rayonnement dans le cadre de leurs fonctions qui sont susceptibles de recevoir une dose de rayonnement dépassant un vingtième de la limite de dose recommandée pour un travailleur sous irradiation, mentionnée à l'annexe II;
- de tenir des registres des doses reçues en milieu de travail par le personnel et enquêter sur toute exposition reçue par le personnel dépassant un vingtième de la limite de dose recommandée;
- de tenir des registres sur les enquêtes de radioprotection, notamment effectuer le résumé des mesures correctives recommandées ou apportées, et inscrire l'établissement, s'il y a lieu, à un service de dosimétrie individuelle; et
- de s'assurer que le personnel comprenne bien le contenu du présent Code.
1.2 Radiologiste qui interprète des mammographies
Tous les radiologistes qui interprètent des mammographies doivent posséder les compétences suivantes :
1.2.1 Compétences initiales
Le radiologiste qui interprète des mammographies doit, avant de pouvoir interpréter seul un mammogramme:
- posséder les compétences requises par les lois ou les règlements fédéraux et provinciaux ou territoriaux pertinents;
- détenir un certificat en Radiologie de diagnostic par le Collège royal des médecins et chirurgiens du Canada ou le Collège des médecins du Québec, OU détenir un certificat en Radiologie de diagnostic par un autre organisme habilité à délivrer des permis et se conformer aux exigences provinciales en matière de permis;
- posséder au moins 40 crédits en Développement professionnel continu (DPC) documenté en imagerie du sein associée à un programme d'apprentissage avec Maintien du certificat (MDC) du Programme du Collège royal des médecins et chirurgiens du Canada. Au moins la moitié de ces crédits doivent être associée à des activités d'apprentissage collectif agréé ou à des activités des programmes d'auto-évaluation et le reste peut être des rencontres, lectures, bandes vidéo, CD-Rom, etc., documentés non agréés. Au moins 15 des crédits de DPC doivent avoir été acquis dans les trois années qui précèdent immédiatement la date à laquelle le médecin a été reconnu comme radiologistes qui interprètent des mammographies. Au moins la moitié de ces 15 crédits doivent être associée à des activités agréés. Le temps de résidence consacré en particulier à la mammographie est admis si le radiologiste produit le document du programme de formation;
- Avoir interprété ou avoir procéder à une seconde lecture d'au moins 480 examens mammographiques au cours de la dernière année immédiatement avant la date à laquelle le radiologiste est reconnu comme un radiologiste qui interprète des mammographies. L'interprétation ou la deuxième lecture doit être effectuée sous la supervision directe d'un radiologiste reconnu comme un radiologiste qui interprète des mammographies. Il est recommandé qu'un minimum de 1000 examens mammographiques soient interprété ou avoir eu une seconde lecture cependant, dans certains cas, il est possible que ce nombre soit inatteignable, il est alors acceptable qu'un nombre minimum de 480 examens mammographiques soit acceptable si les justifications sont transmises expliquant pourquoi le nombre minimum de 1000 mammographies en peut être atteint.
- Avoir au moins 8 heures de formation dans chaque technologie mammographique utilisée (par ex. CR, DR, tomosynthèse) par le radiologiste. Ces 8 heures documentées peuvent être intégrées aux 40 heures prévues dans la section (c) ci-dessus.
1.2.2 Expérience et formation continues
Tous les radiologistes qui interprètent des mammographies doivent maintenir leurs compétences en respectant les exigences suivantes:
- Le radiologiste qui interprète des mammographies devrait interpréter ou procéder à une seconde lecture d'au moins 480 examens mammographiques par année. Il est recommandé qu'un minimum de 1000 examens mammographiques soit interprété ou vu en seconde lecture, cependant dans certaines circonstances ce nombre peut être inatteignable, il est alors acceptable d'interpréter au minimum 480 examen mammographiques. Les justifications qui supportent le fait que le radiologiste ne peut interpréter 1000 examens mammographiques doivent être transmises. Pour les radiologistes qui interprètent des mammographies de dépistage, le nombre recommandé est de 2000 examens mammographiques par an (Coldman et coll., 2006).
- Tous les trois ans, acquérir ou enseigner l'équivalent d'au moins 15 crédits en DPC en imagerie du sein associée à un programme d'apprentissage avec MDC du Programme du Collège royal des médecins et chirurgiens du Canada. Au moins la moitié de cet apprentissage doit être associée à des activités d'apprentissage collectif agréés ou à des activités des programmes d'auto-évaluation et le reste peut être des rencontres, lectures, bandes vidéo, CD Roms, etc., documentés non agrées. Cette formation doit inclure au moins six crédits de formation médicale continue de catégorie I dans chaque technologie mammographique utilisée par le radiologiste dans sa pratique. Les documents doivent être soumis sur demande. Les unités acquises grâce à l'enseignement d'un cours donné ne peuvent être comptées qu'une fois dans les 15 crédits, même si le cours est enseigné plusieurs fois au cours de la période de trois ans; et
- pour qu'un radiologiste puisse commencer à interpréter de manière autonome des mammographies produites à l'aide d'une nouvelle technologie pour laquelle il n'a pas reçu antérieurement de formation, il doit avoir au moins 8 heures de formation dans la nouvelle technologie (par ex. CR, DR).
1.2.3 Renouvellement des compétences
Les radiologistes qui ne remplissent pas les exigences en matière d'expérience ou de formation continues doivent renouveler leur qualification avant de pouvoir interpréter sans supervision des mammographies. Ils procèdent comme suit:
- Les radiologistes qui ne remplissent pas les exigences d'expérience continue de section 1.2.2 doivent, dans les 6 mois qui précèdent immédiatement la reprise de l'interprétation autonome:
- interpréter ou faire une seconde lecture d'au moins 240 examens mammographiques sous la supervision directe d'un radiologiste; ou
- interpréter ou faire une seconde lecture sous la supervision directe d'un radiologiste d'un nombre de mammographie suffisant pour amener le total à au moins 960 examens dans les 24 mois qui précèdent selon la moindre de ces exigences.
- Les radiologistes qui ne remplissent pas les exigences de la section 1.2.2 en matière de formation continue doivent obtenir un nombre suffisant de crédits additionnels de la catégorie I de formation médicale continue en mammographie de façon à atteindre le total requis de 15 crédits au cours des 36 mois avant de pouvoir reprendre l'interprétation autonome.
1.2.4 Responsabilités du radiologiste
Tous les radiologistes qui interprètent des mammographies doivent participer pleinement au programme d'assurance de la qualité, comme suit:
- Communiquer au personnel tout changement dans la qualité de l'image, que ces changements soient dus à un mauvais positionnement, à un paramètre d'exposition inappropriée inappropriée ou à un traitement d'image inadéquat;
- Participer à l'enregistrement et à la conservation des dossiers sur les résultats pour établir la corrélation entre les mammographies positives, les résultats des biopsies effectuées et le nombre de cancers détectés; et
- Comprendre les exigences et recommandations du présent Code de sécurité.
1.3 Technologue en mammographie
1.3.1 Compétences initiales
Le technologue en mammographie doit:
- Posséder les compétences requises par les lois ou les règlements fédéraux et provinciaux ou territoriaux pertinents;
- Détenir un certificat par l'Association canadienne des technologues en radiation médicale (ACTRM) ou par l'Ordre des technologues en imagerie médicale, en radio-oncologie et en électrophysiologie médicale du Québec en technique de radiologie, et être inscrit auprès de l'organisme de réglementation provincial/territorial des technologues en radiologie dans la spécialité/discipline de cette technique radiologique, s'il y a lieu;
- Obtenir les compétences suivantes nécessaires pour appliquer des procédures de mammographie, par le biais de programmes de développement et de cours spécialisés, comme le certificat de l'ACTRM en imagerie du sein ou des cours offerts par d'autres organismes, et en acquérant des connaissances dans les domaines suivants:
- Anatomie du sein et physiologie,
- Techniques de positionnement,
- Prise en charge des patients,
- Fonctionnement d'équipement de mammographie, y compris les systèmes de réseautage et d'archivage,
- Évaluation de l'image, et
- Essais portant sur le contrôle de qualité;
- Avoir au moins 40 heures d'expérience clinique supervisée dans la pratique de la mammographie et avoir réalisé au moins 50 examens mammographiques.
1.3.2 Expériences et formation continues
Tous les technologues en mammographie doivent maintenir leurs compétences dans la pratique de l'imagerie du sein en respectant les exigences suivantes:
- recevoir au moins 15 heures de formation en mammographie chaque 3 ans ou le nombre d'heures précisé dans la réglementation provinciale/territoriale;
- travailler l'équivalent d'au moins 390 heures en mammographie par année pour 3 ans ou le nombre d'heures précisé dans la réglementation provinciale/territoriale; et
- effectuer au moins 480 examens mammographiques chaque année pendant 3 ans. Dans certaines circonstances ce nombre peut ne pas être atteignable, dans ce cas les justifications doivent être transmises pour expliquer pourquoi ce nombre ne peut être atteint.
1.3.3 Renouvellement des compétences
Le technologue en mammographie qui ne respecte pas les exigences de la formation continue doit :
- effectuer au moins 50 examens mammographiques sous la supervision d'un technologue en mammographie qualifié; et
- démontrer sa capacité à faire fonctionner les modalités d'imagerie, particulièrement pour les appareils nouvellement acquis, en assistant à 40 heures de formation pratique, supervisées par un technologue qualifié en mammographie. Cependant dans certaines circonstances, les 40 heures peuvent ne pas être nécessaire, mais dans ce cas, une justification doit être fournie qui explique, les raisons qui motive un nombre d'heures réduit de pratique supervisée et la façon dont le niveau de pratique adéquat supervisé a été évalué.
1.3.4 Responsabilités du technologue en mammographie
Le technologue en mammographie doit participer pleinement au programme d'assurance de la qualité, et:
- s'assurer que le niveau optimal de qualité d'image diagnostique est maintenu;
- communiquer au personnel tout changement dans la qualité d'image;
- signaler au propriétaire tout changement dans le rendement des appareils;
- effectuer quotidiennement et de façon routinière des tests de contrôle de la qualité des appareils de mammographie, du système de traitement d'image (film ou numérique) et de l'équipement auxiliaire et consigner ces tests par écrit;
- reconnaître les dangers des rayonnements liés à leur travail et prendre des mesures pour les réduire;
- connaître les conséquences d'une mauvaise exécution des techniques mammographiques sur la qualité de l'image et l'irradiation des patientes;
- s'efforcer d'éliminer les procédures mammographiques inutiles en réduisant le nombre de reprises, et en réduisant toutes les expositions des patients à leurs valeurs pratiques les plus faibles; et
- comprendre les exigences et les recommandations du présent Code de sécurité.
1.4 Physicien médical
1.4.1 Compétences initiales
Tous les physiciens médicaux qui effectuent des évaluations des centres de mammographie et assurent une surveillance de leur programme d'assurance de la qualité doivent remplir les exigences suivantes:
- Posséder les compétences requises par les lois ou les règlements fédéraux et provinciaux ou territoriaux pertinents;
- Détenir un certificat en Physique de la mammographie par le Collège canadien des physiciens en médecine (CCPM) et être inscrit auprès de l'organisme de réglementation provincial/territorial des physiciens médicaux, comme l'Association des physiciens et ingénieurs biomédicaux du Québec, s'il y a lieu; et
Exigences de la CCPM pour l'accréditation de Physique de la mammographie incluent:
- Avoir un minimum de 15 crédits de formation continue documenté sur les questions pertinentes en matière de mammographie au cours des 3 années précédente, et
- avoir l'expérience de la conduite des évaluations d'au moins 2 installations de mammographie et un total d'au moins 6 unités de mammographie. Pas plus d'une évaluation d'une installation spécifique dans une période de 10 mois ou une unité spécifique dans un délai de 60 jours peut être prise en compte pour cette exigence. L'expérience concernant la conduite des évaluations doit être acquise sous la supervision directe d'un physicien médical certifié en Physique de la mammographie par le CCPM. Dans certaines circonstances, cette exigence peut être inaccessible. Une dérogation peut être accordée par le CCPM, si une justification acceptable est fournie.
1.4.2 Expérience et éducation continues
Tous les physiciens médicaux qui mènent des évaluations des installations de mammographie et s'assurent de la surveillance du programme d'assurance de la qualité de l'installation doivent acquérir l'expérience et la formation continue en conformité avec les exigences de la CCPM.
- Éducation continue
- Le physicien médical doit avoir enseigné ou suivi au moins 15 heures de formation continue sur les questions pertinentes en mammographie pendant les trois années suivant son accréditation ou renouvellement d'accréditation par le CCPM. Cette formation continue doit comprendre des heures de formation se rattachant à chaque technologie mammographique évaluée par le physicien médical durant les évaluations ou de surveillance des programmes d'assurance de la qualité. Les unités acquises en enseignant un cours donné ne peuvent être comptées qu'une seule fois pour l'obtention des 15 heures requises au cours d'une période de trois ans, même si le cours est enseigné plusieurs fois au cours de la période de trois ans.
- Expérience continue
- Le physicien médical doit avoir effectué une évaluation d'au moins 2 centres de mammographie et 6 appareils au total au cours des 3 ans qui suivent l'accréditation ou le renouvellement d'accréditation par le CCPM. Pour satisfaire à cette exigence, on ne peut pas compter plus d'une évaluation d'un centre donné au cours d'une période de 10 mois ou d'un appareil donné au cours d'une période de 60 jours. Dans certaines circonstances, cette exigence peut être inatteignable. Une dérogation peut être accordée par le CCPM, si la justification acceptable est fournie.
- Pour qu'un physicien médical puisse commencer à effectuer de manière autonome des évaluations d'une nouvelle technologie mammographique, c'est-à-dire une autre technologie que celle à laquelle il a été formé en vue de son accréditation initiale, il doit suivre au moins 8 heures de formation dans des unités de la nouvelle technologie. Il est à noter que ce 8 heures peut faire partie du 15 heures requises à la section (a)(i) si une documentation est fournie.
1.4.3 Renouvellement des compétences
Les physiciens médicaux qui ne remplissent pas les exigences en matière d'expérience et de formation continues ne peuvent pas effectuer des évaluations sans la supervision d'un physicien médical qualifié. Pour pouvoir effectuer des évaluations de manière autonome, dans un autre centre, les physiciens médicaux doivent renouveler leur accréditation comme suit:
- Les physiciens médicaux qui ne remplissent pas les exigences en matière de formation continue doivent obtenir un nombre suffisant d'unités de formation continue pour atteindre au total 15 heures dans les trois années précédentes.
- Les physiciens médicaux qui ne remplissent pas les exigences d'expérience continue doivent effectuer un nombre suffisant d'évaluations sous la supervision directe d'un physicien médical qualifié afin d'atteindre le nombre total d'évaluations requis à 2 centres et à 6 appareils au cours des 3 années précédentes. Pour satisfaire aux exigences sur le total requis d'évaluation d'appareils, on ne peut pas utiliser plus d'une évaluation d'un appareil donné au cours d'une période de 60 jours. Dans certaines circonstances, cette exigence peut être inatteignable. Une dérogation peut être accordée par le CCPM, si la justification acceptable est fournie
1.4.4 Responsabilités du physicien médical
Le physicien médical doit:
- vérifier la sécurité d'une installation au moment de la planification et de la construction de celle-ci et s'assurer qu'elle est conforme à tous les règlements applicables;
- examiner régulièrement les mesures de sécurité et recommander au propriétaire les changements nécessaires afin d'assurer la sécurité optimale des patientes et du personnel, et recommander au personnel les méthodes de radioprotection appropriées;
- participer au programme d'assurance de la qualité comme suit:
- s'assurer que le programme d'assurance de la qualité soit correctement appliqué et exécuté,
- vérifier qu'une image de qualité optimale soit obtenue,
- s'assurer que les instruments de contrôle de la qualité appropriés soient disponibles et correctement étalonnés;
- effectuer les essais, tel que requis, des appareils de mammographie, les systèmes d'acquisition et traitement, les écrans et l'équipement auxiliaire, et documenter les résultats conformément aux procédures de tenue de registre appropriées;
- produire dans les 30 jours suivants l'évaluation, un rapport écrit complet qui décrit clairement les résultats (le rapport devrait être disponible dans les 10 jours suivant la vérification.);
- communiquer rapidement les résultats des évaluations à la personne responsable de l'installation (La personne responsable peut différer d'un établissement à un autre. Avant une évaluation, le physicien médical doit demander à l'établissement d'identifier la personne responsable à qui les résultats de l'évaluation doivent être communiqué); et
- comprendre les recommandations du présent Code de sécurité.
1.5 Spécialiste des systèmes informatiques (SSI)
Les établissements qui effectuent le traitement d'images numériques doivent avoir accès à une personne formée et possédant de l'expérience dans la maintenance et le contrôle de la qualité des logiciels et des matériels informatiques. En fonction de l'établissement, cette personne peut-être sur site ou disponible à la demande. La qualification requise pour cette personne dépendra fortement du type d'établissement et du type d'appareil qui y est utilisé. Idéalement, cette personne devra posséder une expertise en technologie de l'information et en technologie du rayonnement.
1.5.1 Compétences initiales
Le SSI:
- doit posséder les compétences requises par les lois ou les règlements fédéraux et provinciaux ou territoriaux pertinents;
- devrait détenir un certificat conforme aux normes reconnues, comme celles de la Society of Imaging Informatics in Medicine ou la PACS Administrators Registry and Certification Association; et
- devrait posséder des connaissances dans les domaines suivants:
- Informatique fondamentale, comme comprendre les composants matériels, le type d'interfaces, les paramètres de configuration, ainsi que les applications de systèmes d'exploitation et de logiciels,
- Notions fondamentales sur les bases de données,
- Notions de réseautage, comme DICOM, HL7, RIS et HIS,
- Systèmes de sécurité et notions visant à assurer la confidentialité des dossiers de patients,
- Terminologie en imagerie médicale,
- Positionnement et caractéristiques de visionnement,
- Caractéristiques d'imagerie des différents modes d'acquisition d'images,
- Déroulement du travail dans l'installation,
- Dispositions législatives fédérales, provinciales, territoriales et institutionnelles, comme la Loi sur la protection des renseignements personnels et les documents électroniques (LPRPDE).
1.5.2 Expérience et formation continues
- Éducation continue
- Le SSI doit avoir suivi au moins 15 heures d'éducation continue concernant des questions pertinentes sur la technologie de l'information sur une période de 3 ans. Cette éducation continue doit inclure des heures de formation appropriées pour les systèmes d'information de l'installation.
- Expérience continue
- Le SSI devrait travailler l'équivalent d'au moins 390 heures en mammographie par année pour 3 ans.
- Avant que le SSI puisse commencer à travailler de manière autonome sur de nouveaux systèmes d'information, c'est-à-dire des systèmes autres que celui pour lequel il a reçu de la formation de compétences initiales, il doit recevoir au moins 8 heures de formation sur le nouveau système d'information.
1.5.3 Renouvellement des compétences
Le SSI qui ne réussit pas à maintenir les compétences continues requises ne peut pas accomplir ses tâches sans la supervision d'un SSI compétent. Avant de réaliser des travaux indépendants, le SSI doit renouveler ses compétences, comme suit:
- Les SSI qui ne remplissent pas les exigences en matière de formation continue doivent obtenir un nombre suffisant d'unités de formation continue pour atteindre au total 15 heures dans les trois années précédentes; et
- Le SSI qui ne réussit pas à respecter les exigences d'expérience continue doit travailler sous la supervision directe d'un SSI qualifié afin de faire passer le nombre total d'heures travaillées à 75 heures pour les 3 dernières années.
1.5.4 Spécialiste des systèmes informatiques
Le SSI doit:
- assurer la confidentialité des dossiers de patients;
- comprendre les politiques et procédures en place au sein de l'établissement;
- comprendre l'importance et les exigences d'un programme d'assurance de la qualité pour les systèmes informatiques; et
- communiquer avec le personnel au sujet de toute modification ou mise à niveau de matériels ou de logiciels des appareils de gestion des données et des conséquences sur les procédures d'exploitation de l'établissement.
1.6 Personnel de réparation et d'entretien
Le personnel de réparation et d'entretien comprend les personnes autorisées à effectuer la réparation et l'entretien des génératrices de rayons X, des systèmes de commande, des systèmes d'imagerie et de leurs logiciels d'opération. En fonction de l'établissement, ces personnes peuvent être sur site ou disponibles à la demande, mais en général, cette fonction est parfois sous-traitée à une organisation extérieure, ou au fabricant des appareils.
Le personnel de réparation et d'entretien doit :
- avoir des compétences et une formation dans
- la réparation et l'entretien des appareils radiologiques d'imagerie, et
- les principes et procédures de radioprotection;
- s'assurer, à la suite d'une procédure de réparation ou d'entretien, que l'appareil est conforme aux normes réglementaires ou aux spécifications du fabricant;
- s'assurer que toutes les procédures de réparation ou d'entretien sont correctement consignées et communiquées au propriétaire et aux autres membres du personnel concernés;
- signaler au propriétaire toute non-conformité par rapport aux procédures de sécurité établies;
- examiner périodiquement les procédures d'entretien et les mettre à jour pour garantir la sécurité optimale des patients et des opérateurs;
- communiquer, si nécessaire, au personnel les besoins d'essais d'acceptation appropriés, de réglage de base et d'essais de contrôle de la qualité; et
- suivre les recommandations du fabricant pour la réparation et l'entretien des appareils.
1.7 Conservation des registres relatif aux compétences personnelles
Les documents relatifs aux compétences personnelles des radiologistes qui interprètent des mammographies, technologues en mammographie, physiciens médicaux et des spécialistes des systèmes informatiques doivent être conservés par l'établissement. La durée de conservation des dossiers doit être conforme aux lois et règlements fédéraux/provinciaux/territoriaux pertinents. Les dossiers des employés, y compris ceux qui ont quitté l'installation, ne doivent pas être éliminés par l'établissement avant qu'il ait été démontré clairement que le personnel possède les compétences nécessaires et qu'il soit accrédité auprès d'un organisme.
2.0 Mesures de réduction de l'exposition du personnel aux rayons X
Les procédures requises et recommandées décrites dans la présente section sont essentiellement orientées vers la protection de la santé au travail. Cependant, le respect de ces procédures assurera également, dans de nombreux cas, la protection des visiteurs et des personnes à proximité d'une installation radiographique. Les pratiques et procédures de travail sécuritaires devraient être considérées comme une base, qu'il conviendrait de renforcer par des exigences supplémentaires, lorsque cela est justifié, pour couvrir des circonstances spéciales dans des installations particulières.
Pour obtenir une sécurité optimale, les opérateurs d'appareils doivent faire tous les efforts raisonnables pour maintenir leur exposition et celle des autres employés raisonnablement en dessous des limites spécifiées à l'annexe II.
2.1 Exigences générales et recommandations
- Une salle de radiologie ne doit pas être utilisée pour plus d'un examen à la fois.
- Seules les personnes dont la présence est indispensable doivent se trouver dans la salle de radiographie lorsqu'un examen radiographique est effectué.
- Le personnel doit, en permanence, se tenir aussi loin que possible du faisceau de rayonnement.
- L'irradiation délibérée d'un individu pour des motifs de formation ou d'évaluation des appareils ne doit jamais se produire.
- Toutes les personnes doivent utiliser au mieux les dispositifs de protection mis à leur disposition.
- Le fonctionnement de l'équipement mammographique doit être contrôlé à partir du panneau de commande situé derrière un écran protecteur ou à l'intérieur d'une cabine de commande. Le technologue doit avoir une protection contre l'exposition au rayonnement au moment des expositions.
- L'opérateur doit avoir une vue nette du patient durant l'examen radiographique et il doit être capable de communiquer avec le patient et/ou les assistants sans quitter la cabine de commande.
- L'équipement mammographique doit être utilisé par des personnes qualifiées seulement qui ont reçu la formation adéquate relativement à l'équipement et aux procédures.
- Tous les opérateurs d'appareils à rayons X ainsi que le personnel (c.-à-d. les infirmières) qui participe couramment à des examens radiographiques, et les autres personnes qui sont susceptibles de recevoir une dose de rayonnement supérieure à un vingtième des limites de dose équivalente admissibles indiquées à l'annexe II, doivent être déclarés travailleurs sous rayonnements et surveiller leurs expositions à l'aide d'un dosimètre individuel. En général le personnel n'opérant que les équipements de mammographie n'est pas déclaré travailleur sous rayonnement puisqu'il ne reçoit pas de dose de rayonnement supérieur à 1mSv. Cependant le personnel considéré comme travailleur sous rayonnement suite aux autres taches à l'extérieur de la salle de mammographie doit porter leurs dosimètres lorsqu'ils réalisent des examens de mammographie.
- Des dosimètres individuels doivent être portés et stockés conformément aux recommandations du fournisseur des services de dosimétrie. Lorsqu'un tablier de protection est porté, le dosimètre individuel doit être porté sous le tablier. Si les yeux ou des extrémités sont susceptibles d'être exposés à des doses bien plus élevées, il est recommandé de porter des dosimètres supplémentaires à ces extrémités. Cependant cela est peu probable dans le contexte des examens de mammographie.
- Lorsque les membres du personnel exposés au rayonnement dans le cadre de leurs fonctions qui sont susceptibles de recevoir une dose de rayonnement supérieure à un vingtième de la limite de dose recommandée mentionnée à l'annexe II, des mesures correctrices doivent être prises pour améliorer les techniques et les mesures de protection.
- Tous les dossiers individuels de dosimétrie doivent être tenus pendant la durée de vie de l'installation.
- Une opératrice doit immédiatement informer son employeur dès qu'elle sait être enceinte, afin que des mesures appropriées soient mise en place pour s'assurer que ses prestations de travail pendant le reste de la grossesse sont compatibles avec la dose maximum recommandée et énoncé à l'annexe II. Selon le type de travail effectué par le salarié, il peut être nécessaire de retirer un membre du personnel enceinte de leurs devoirs de fonctionnement de l'équipement de radiographie. Il est recommandé que la décision de retirer les travailleuses enceintes de leurs fonctions prend en compte les risques d'exposition aux rayonnements associés aux fonctions de l'employé, tel que déterminé par un physicien médical ou un responsable de la radioprotection. En général, pour l'exécution des examens de mammographie, il n'est pas nécessaire de supprimer ou de limiter les devoirs des opérateurs d'équipement de mammographie pendant la grossesse.
- Lorsqu'un accompagnateur ou un membre du personnel doit assister la patiente, il est nécessaire de fournir à ces personnes un vêtement de protection radiologique. Aucune personne ne devrait accomplir ces fonctions de manière régulière.
- Toutes les portes d'entrée d'une salle de rayons X devraient rester fermées lorsqu'un patient est dans la salle et doivent être fermées lorsqu'une exposition radiographique est réalisée.
- Les appareils radiogènes sous tension et prêts à produire des rayons X ne doivent pas être accessibles au public.
3.0 Mesures de réduction de l'exposition du patient aux rayons X
Dans la population en général, la plus grande source d'exposition au rayonnement d'origine humaine est la radiologie diagnostique médicale et dentaire. En effet, l'utilisation de la radiologie à des fins diagnostiques représente dans la population plus de 90 % de la totalité des expositions au rayonnement des sources artificielles de rayonnement.
Pour chaque patient, le risque découlant d'un seul examen radiologique est très faible. Cependant, dans une population donnée, le risque est plus élevé à cause de la multiplication des examens radiographiques et de l'augmentation du nombre des personnes exposées au rayonnement. Par conséquent, il faut diminuer le plus possible le nombre de radiogrammes et le nombre de personnes qui subissent des examens radiographiques et réduire la dose de rayonnement administrée à chaque examen.
Pour atteindre ces objectifs, il est essentiel de limiter les examens radiographiques à ceux qui sont absolument nécessaires et, dans les cas où ils s'imposent, d'éviter toute exposition inutile du patient au rayonnement.
Les procédures requises et recommandées décrites dans cette section et destinées à protéger le patient s'adressent au professionnel de la santé, au radiologiste et au technologue. Elles fournissent des moyens d'éliminer les examens qui ne sont pas absolument nécessaires et de réduire au minimum l'exposition des patients lorsque les examens sont nécessaires.
3.1 Lignes directrices sur la prescription des mammogrammes à des fins diagnostiques
Les expositions non nécessaires des patients au rayonnement peuvent être réduites de façon significative en s'assurant que tous les examens cliniques sont justifiés. Ceci peut se faire en se conformant, autant que possible, à certaines recommandations de base. Ces recommandations sont présentées ci-dessous.
- La prescription d'un examen mammographique devrait s'appuyer sur une évaluation clinique de l'état du patient et avoir pour but d'obtenir une information nécessaire au diagnostic ou au traitement du patient.
- Les examens mammographiques ne devraient pas être effectués s'il n'y a pas eu d'examen clinique préalable du patient.
- Le médecin doit d'abord déterminer s'il y a eu des examens mammographiques antérieurs qui rendraient inutile un nouvel examen ou permettraient à tout le moins de l'abréger. Les clichés ou les rapports des examens antérieurs devraient être étudiés lors de l'évaluation clinique du patient.
- Dans les cas où un patient serait dirigé d'un professionnel de la santé ou d'un hôpital à un autre, les clichés ou les rapports devraient suivre et être étudiés par le professionnel de la santé.
- En ordonnant un examen mammographique, le professionnel de la santé devrait préciser les indications cliniques et l'information qu'il cherche.
- Le nombre de clichés requis doit être réduit au minimum nécessaire à la réalisation des objectifs cliniques de l'examen.
- En prescrivant un examen mammographique à une patiente qui est ou qui croit être enceinte, le médecin doit tenir compte des conséquences pour le foetus d'une exposition au rayonnement. Il est généralement accepté que, bien que la dose de rayonnement reçue par les ovaires et le foetus soit faible en mammographie, les mesures nécessaires devraient être prises durant l'examen pour éviter d'irradier la région abdominale.
- Lorsqu'un mammogramme contient les renseignements que l'on cherche, on devrait éviter d'en ordonner la répétition pour la seule raison que le mammogramme n'est pas de la «meilleure qualité» diagnostique possible.
- Les examens spécialisés ne devraient être effectués que par un radiologiste qualifié ou en collaboration étroite avec lui.
- Le dossier médical du patient devrait inclure les détails de tous les examens radiographiques qu'il a subis.
Des directives plus spécifiques pour la prescription d'examens d'imagerie sont disponibles auprès de l'Association canadienne des radiologistes (CAR) dans leurs lignes directrices pour les examens d'imagerie diagnostique (CAR 2012). Ces lignes directrices fournissent des recommandations sur la pertinence des examens d'imagerie dans un but de diagnostic clinique et de gestion de problèmes spécifiques cliniques ou diagnostiques. L'objectif de ces lignes directrices est d'aider le professionnel de la santé traitant à sélectionner l'examen d'imagerie approprié et par là de réduire l'imagerie inutile en éliminant tout examen d'imagerie qui n'est pas susceptible d'aider à diagnostiquer un patient particulier et en suggérant des procédures alternatives qui n'utilisent pas de rayonnements ionisants, mais qui offrent une précision d'épreuve diagnostique comparable.
3.2 Lignes directrices sur les mammogrammes à des fins de dépistage
Dans le cadre des programmes de dépistage du cancer du sein, les femmes asymptomatiques subissent des examens mammographiques afin que l'on puisse détecter les tumeurs cancéreuses de façon précoce et réduire les décès par le cancer du sein. Dans ces programmes, il est important de réduire au minimum l'exposition au rayonnement des participantes, tout en s'assurant d'avoir des images de qualité diagnostique optimale. Par conséquent, un programme de dépistage du cancer du sein ne doit être établi que si les doses glandulaires moyennes se situent dans les limites acceptées et qu'un programme d'assurance de la qualité est mis en oeuvre.
- Des examens de dépistage mammographique devraient être effectués lorsque les avantages l'emportent sur les risques que comporte une exposition accrue à des doses de rayonnement pour le groupe visé par le programme.
- Le dépistage mammographique ne devrait pas être réalisé chez les femmes enceintes ou qui croient être enceintes, en raison des conséquences pour le foetus d'une exposition au rayonnement. Il est généralement accepté que, bien que la dose de rayonnement reçue par les ovaires et le foetus soit faible en mammographie, l'examen devrait être reporté à une date ultérieure.
- Les participantes qui ont des implants mammaires doivent suivre le même calendrier d'intervalle d'examens mammographiques que celui recommande pour les femmes qui n'en possèdent pas. Cependant, il peut être nécessaire d'utiliser des techniques de compression, de positionnement et de chargements spéciaux. Le personnel de l'installation mammographique doit pouvoir effectuer ces procédures de manière efficace. Les femmes avec des implants mammaires peuvent être référées à un centre de mammographie diagnostic.
- Le nombre des clichés doit être limité au minimum nécessaire à la réalisation des objectifs du programme de dépistage.
- On ne doit pas répéter le mammogramme sous prétexte que la qualité de l'image n'est pas optimale si le cliché fournit les renseignements diagnostiques nécessaires.
- La qualité des mammogrammes doit être vérifiée régulièrement dans le cadre d'un programme d'assurance de la qualité, de sorte qu'elle permette de satisfaire aux exigences diagnostiques tout en limitant le plus possible l'exposition de la patiente.
- Dans le cas des unités de dépistage mammographique mobiles, on recommande de développer les films sur place afin que les techniciens puissent vérifier les clichés, ce qui réduira le rappel des participantes. Cependant, comme il est souvent difficile de stabiliser les machines à développement des films dans des unités de dépistage mammographique mobiles, on doit s'assurer du fonctionnement optimal de ces machines. Si cela n'est pas possible, le traitement des films peut être fait dans un endroit différent.
- On recommande de mettre les mammogrammes précédents à la disposition du radiologiste, y compris les mammogrammes de base, effectués dans le cadre de programmes de dépistage mammographique, afin que celui-ci puisse les examiner.
3.3 Lignes directrices pour effectuer les examens mammographiques
À part l'élimination inutile des examens radiographiques, la méthode la plus efficace de réduire l'exposition du patient est d'utiliser pour ces examens une bonne technique. Ainsi, il est possible d'obtenir une série de radiographies acceptables pour le diagnostic avec des expositions très variées, par le choix des facteurs techniques de l'opération. Il incombe au technologue, au physicien médical et au radiologiste de savoir comment réaliser un examen mammographique en maintenant l'exposition de la patiente ou du participant à la mammographie de dépistage au niveau le plus faible possible.
Les exigences et recommandations qui suivent ont pour but de fournir des directives pour l'opérateur, le physicien médical et le radiologiste dans l'exercice de leur responsabilité envers la réduction de l'exposition des patients.
- Les appareils mammographiques doivent être conçus spécifiquement pour la mammographie et le récepteur d'image doit être compatible avec ces appareils.
- On doit utiliser une combinaison film-écran offrant une bonne qualité d'image sur le plan diagnostique. Il ne faut jamais utiliser de films à exposition directe.
- Lors de la mise à niveau d'un système mammographique en service avec un récepteur d'image numérique (système de CR), le système mammographique doit être calibré pour refléter la sensibilité du récepteur numérique.
- À l'exception des examens mammographiques effectués dans le cadre d'un programme de dépistage, le technologue ne doit jamais effectuer un examen qui n'a pas été prescrit par un professionnel de la santé de la patiente.
- L'exposition de la patiente doit être limitée au strict minimum compatible avec les objectifs de l'examen et la nature de l'information diagnostique recherchée. Pour ce faire, il est nécessaire d'utiliser des techniques compatibles avec l'équipement et d'évaluer occasionnellement leur efficacité.
- Des précautions particulières en vue de la protection des patientes doivent être prises lorsque celles-ci sont enceintes ou croient être enceintes, même si la dose de rayonnement reçue par l'abdomen et le foetus est négligeable durant un examen mammographique normal.
- Avant de faire une mammographie, il faut déterminer si la patiente a des prothèses mammaires. Il peut être nécessaire d'utiliser des techniques spéciales lorsqu'on effectue un examen mammographique chez des patientes ayant des prothèses mammaires, telles que la prise de clichés supplémentaires et l'utilisation de techniques de positionnement, de compression et de charge différentes. Des précautions particulières concernant les techniques de compression doivent être prises étant donné qu'une compression excessive des prothèses durant l'examen mammographique peut entraîner la rupture de celles-ci.
- Les cassettes devraient être chargées au moins 15 minutes à l'avance pour permettre à l'air de s'échapper et pour améliorer ainsi le contact film-écran.
- Il faut prendre soin de bien positionner le sein, en examinant notamment la hauteur et l'angle de la table de support, en appliquant le degré de compression approprié et en évitant les plis de peau.
- En mammographie, il est recommandé que le champ de radiation corresponde à la dimension du récepteur d'image, mais il ne devra pas être plus large que le support du récepteur d'image, excepté au niveau de la paroi thoracique. Le nombre des zones non exposées sur les films devrait être maintenu le plus bas possible pour éviter le masquage. L'exécution de clichés localisés avec collimation est utile dans certains cas, mais elle devrait être considérée comme une technique spéciale.
- L'utilisation d'un col thyroïdien plombé n'est pas recommandée durant les examens mammographiques. La dose de rayonnement reçue à la thyroïde est très faible, car elle est due uniquement au rayonnement diffusé. De plus, l'Association canadienne des radiologistes (CAR) recommande de ne pas utiliser de col thyroïdien, car un tel col pourrait cacher des parties importantes de l'anatomie, nuire au positionnement adéquat du sein durant l'examen et avoir une incidence sur la qualité de l'image mammographique (CAR 2011). D'autres types de protection comme le tablier ou le protège gonade offre peu de protection puisque les appareil de mammographies sont conçus avec un blindage pour assurer la protection du patient contre les rayonnements parasites.
- Le technologue devrait examiner les clichés après leur développement afin de vérifier que les techniques utilisées produisent des images de qualité diagnostique et que les appareils radiographiques fonctionnent correctement.
- Des précautions doivent être prises pour identifier et minimiser la présence d'artefacts. Les artefacts peuvent être dû à la performance des équipements, à la technique d'imagerie, au positionnement, à la propreté et la manipulation, et aux problèmes liés au patient. Par exemple, la compression insuffisante et temps d'exposition trop longue peut entraîner des artefacts de mouvement. Les artefacts de grille peuvent également résulter d'une exposition plus longue. L'antisudorifique que les patients ont appliqué les déodorants et les crèmes avant l'examen mammographique peuvent aussi produire des artefacts. Un artefact peut aussi être dû à inhomogénéité de champ, à des éléments défectueux de détection et de traitement des images numériques.
- Les détails de tout examen mammographique, incluant des reprises, devraient être consignés au dossier du patient.
- Bien que des limites de dose aient été définies pour le personnel exposé au rayonnement et la population en général, aucune limite de dose n'existe pour les patientes soumises aux examens radiographiques. Cependant, on peut établir des niveaux de référence diagnostique en mammographie. Annexe III fourni une description de la méthodologie pour déterminer la dose moyenne glandulaire.
Section B : Exigences des locaux et des appareils
1.0 Exigences des locaux
1.1 Critères généraux
Lors de la planification des locaux de radiographie médicale, il faut s'assurer que les personnes à proximité des locaux ne sont pas exposées à des niveaux de rayonnement qui dépassent les limites d'exposition légales actuelles. On doit prendre des mesures appropriées pour garantir la présence d'équipement de protection adéquat afin de se conformer aux exigences suivantes:
- les niveaux de rayonnement dans les zones contrôlées, occupées de façon courante uniquement par les travailleurs sous irradiation, doivent être tels qu'aucun d'eux ne soit exposé dans l'exercice de ses fonctions à plus de 20 mSv par année; et
- les niveaux de rayonnement dans les zones non contrôlées doivent être tels que personne ne reçoive plus de 1 mSv par année.
L'annexe II fournit une description détaillée des limites de dose réglementaires. Pour les locaux de mammographie, les zones contrôlées sont habituellement dans les zones immédiates où les appareils de mammographie sont utilisés. Les travailleurs dans ces secteurs sont essentiellement des opérateurs d'appareils tels que des technologues en mammographie qui sont formés à l'utilisation adéquate des appareils et à la protection contre les rayonnements. Les zones non contrôlées sont celles occupées par les personnes telles que les patients, les visiteurs de l'établissement et les employés qui ne travaillent pas habituellement avec les sources de rayonnement ou autour de celles-ci (NCRP 2004).
Le rayonnement près du récepteur d'image des appareils mammographiques atteint généralement un niveau tel que les limites décrites plus haut pourraient être dépassées selon la conception de l'équipement, les techniques utilisées et la charge de travail. Cependant, étant donné que les appareils mammographiques nécessitent une faible tension radiogène, on peut réduire l'intensité du rayonnement grâce à un écran de protection approprié entre la patiente et le technologue et en combinant de façon convenable la distance de la source de rayonnement et les écrans de protection. On peut également restreindre l'accès à toutes les zones dans lesquelles la limite de dose recommandée peut être dépassée.
1.2 Conception et plan des locaux de mammographie
Dans les étapes préliminaires de la conception et de la planification d'une installation de mammographie, on devrait mettre en œuvre trois étapes pour garantir qu'une protection adéquate est en place afin de fournir le niveau de radioprotection nécessaire:
- Préparation des plans des locaux;
- Considérations pour la conception et l'aménagement de la salle; et
- Détermination des paramètres qui régissent les exigences en matière de blindage de protection.
1.2.1 Préparation des plans des locaux
Afin de déterminer les exigences en matière de blindage de protection d'une installation radiologique, un plan d'implantation doit être préparé, identifiant clairement les composants suivants:
- Les dimensions et la forme de la salle où les appareils mammographiques sont utilisés et l'orientation physique de la salle (une marque indiquant le nord).
- L'emplacement où il est prévu de placer les appareils mammographiques et la plage de mouvement du tube à rayon X.
- L'emplacement du panneau de commande.
- L'emplacement, l'utilisation, le niveau d'occupation et l'accessibilité des salles adjacentes, ainsi que les salles situées au-dessus et en dessous de l'installation.
- La désignation des salles adjacentes, selon qu'elles doivent être désignées comme zone contrôlée ou non contrôlée. Les zones contrôlées, principalement occupées par des travailleurs sous irradiation, sont soumises à la limite de 20 mSv par an, tandis que les zones non contrôlées, principalement occupées par des travailleurs non exposés au rayonnement, sont soumises à la limite de 1 mSv par an. Dans les zones non contrôlées, où des populations sensibles au rayonnement sont présentes, par exemple les salles pédiatriques, un niveau de contrainte de 0,30 mSv par an devrait être utilisé.
- L'emplacement où le traitement des images est effectué, c.-à-d. l'emplacement des chambres noires, la zone de stockage des films, l'emplacement des cassettes CR, le lecteur de plaque CR et les postes de travail informatique.
- La position de toutes les fenêtres, portes, persiennes, etc., qui peuvent affecter les exigences en matière de radioprotection.
- Les matériaux prévus et en service utilisés pour construire les murs, le plancher, et le plafond, ainsi que leurs épaisseurs comprenant des matériaux supplémentaires actuellement utilisés, ou dont l'utilisation est prévue, comme les barrières de protection contre les rayonnements.
- L'application des barrières de protection. L'application des barrières de protection. En mammographie, l'ensemble image-récepteur agit comme un dispositif d'arrêt de faisceau primaire. Un plan d'implantation doit indiquer que l'écran de séparation entre l'appareil mammographique et la zone occupée agit comme une barrière de protection secondaire pour atténuer le rayonnement diffusé et le rayonnement de fuite.
1.2.2 Considérations sur la conception et l'aménagement des salles
Lors de la conception de l'aménagement des locaux de mammographie, il faut prendre en considération les recommandations générales suivantes.
- Les salles de mammographie, avec des appareils fixes, accessible de différentes zones, devraient être équipées d'une porte coupe-feu à fermeture automatique, et elles doivent être identifiées par des écriteaux de mise en garde comportant le symbole d'avertissement contre les rayons X ainsi que l'inscription « Personnel autorisé seulement ». Des variantes acceptables de symboles de mise en garde contre les rayons X sont données au Règlements relatifs aux dispositifs émettant des rayonnements pour les Appareils de radiodiagnostic.
- Les appareils de mammographie portatifs utilisés couramment en un endroit sont considérés comme des installations fixes et le blindage nécessaire pour les appareils et la salle doivent être déterminés en conséquence.
- Les salles contenant les appareils de mammographie devraient être conçues pour fournir un espace de travail adéquat à l'opérateur d'appareils et permettre le mouvement aisé des patients.
- Les appareils de mammographie devraient être positionnés dans la salle de telle sorte que, lors d'une irradiation, personne ne puisse entrer à l'insu de l'opérateur.
- Le panneau de commande et la fenêtre d'observation doivent posséder des propriétés de protection telles qu'aucun opérateur dans son travail n'est exposé à plus de 0,4 mSv/semaine. Le principe ALARA exige qu'une protection supplémentaire soit spécifiée dans la conception pour réduire davantage l'exposition de l'opérateur, chaque fois que ceci peut raisonnablement être fait.
- Le blindage doit être construit pour constituer une barrière ininterrompue, et en cas d'utilisation du plomb, il devrait être adéquatement soutenu pour éviter tout « affaissement ».
1.2.3 Détermination des paramètres régissant les exigences de blindage structural
L'épaisseur du matériau de blindage, comme le plomb, le béton ou le panneau de placoplâtre (gypse), requise pour réduire les niveaux de rayonnement aux doses limites recommandées peut être déterminée par des calculs. En général, l'exposition des individus au rayonnement dépend principalement de la quantité de rayonnement produite par la source, de la distance entre la personne exposée et la source du rayonnement, du temps qu'un individu passe dans la zone irradiée et de la quantité de blindage de protection entre l'individu et la source de rayonnement.
Étant donné que la mammographie est réalisée à des tensions radiogène faibles, les installations de mammographie permanentes n'ont pas nécessairement besoin d'un blindage additionnel, en plus du niveau de protection assuré par les panneaux de placoplâtre (gypse) de type courant que l'on utilise habituellement dans la construction. Une attention spéciale doit être accordée à la radioprotection lorsque l'on utilise de l'équipement de mammographie mobile ou temporaire. Pour tous les types d'équipements de mammographie, un expert qualifié doit être consulté pour s'assurer que le niveau de radioprotection de l'installation est adéquat.
Les paramètres énumérés ci-dessous doivent être pris en considération pour le calcul de l'épaisseur de la barrière de protection. On devrait tenir compte de modifications futures possibles de chacun ou dans tous ces paramètres, y compris les augmentations des coefficients d'utilisation et d'occupation, la tension et le volume d'utilisation du tube radiogène, ainsi que les modifications des techniques qui peuvent nécessiter des appareils auxiliaires.
1. La charge maximale de travail, (W) ou la répartition de la charge de travail.
La charge de travail est une mesure de la durée opérationnelle ou du volume d'utilisation des appareils radiographiques. Une répartition du volume d'utilisation indique le volume d'utilisation sur la gamme des tensions de fonctionnement. La répartition du volume d'utilisation et le spectre du volume d'utilisation peuvent être déterminés en enregistrant la tension de fonctionnement et le produit courant-durée de chaque irradiation prise dans chaque série de rayons X sur une période de temps fixe (c.-à-d. une semaine). Si les charges de travail réelles ne sont pas disponibles, on peut consulter le tableau 1, qui présente des charges de travail totales estimées pour une installation de mammographie. (NCRP 2004)
| Charge de travail total par patient (mA min / patient) |
Nombre typique de patients (par semaine de 40 heures) | Charge de travail total par semaine (mA min/semaine) | |||
|---|---|---|---|---|---|
| Moyenne | Occupé | Moyenne | Occupé | ||
| Salle de mammographie | 6,7 | 80 | 160 | 550 | 1075 |
2. Le Coefficient d'occupation (T)
Le coefficient d'occupation est la fraction de temps pendant lequel la zone considérée est occupée par la personne (employé ou public) qui passe le plus de temps à cet emplacement pendant le fonctionnement de l'appareil à rayons X. Le tableau suivant présente les coefficients d'occupation recommandés.
| Coefficient d'occupation | Description |
|---|---|
| T = 1 | Les bureaux administratifs et les zones d'accueil, les laboratoires, les pharmacies et autres zones pleinement occupées par un individu, les salles d'attente fréquentées, les aires de jeux intérieures d'enfants, les salles de radiographie adjacentes, les zones d'observation des images, les postes d'infirmières, les salles de commande, les pièces habitées. |
| T=1/2 | Les salles utilisées par les patients pour les examens et les traitements. |
| T=1/5 | Couloirs, chambres de patients, salles des employés, toilettes des employés. |
| T=1/8 | Portes de corridor. |
| T=1/20 | Toilettes publiques, zones non surveillées de distributeurs automatiques, salles de stockage, zones extérieures avec des places assises, salles d'attente non surveillées, zones d'attente des patients. |
| T=1/40 | Zones extérieures avec uniquement des piétons de passage ou une circulation d'automobiles, des aires de stationnement non surveillées, des aires de débarquement (non surveillées), des greniers, des escaliers, des ascenseurs non surveillés, des placards de service. |
3. Le Coefficient d'utilisation (U)
Le coefficient d'utilisation est la fraction du volume d'utilisation pendant lequel le faisceau rayonnant est pointé dans la direction considérée. En mammographie, l'ensemble image-récepteur agit comme un dispositif d'arrêt de faisceau primaire et, par conséquent, U=0. Seul le rayonnement secondaire doit être considéré. Le coefficient d'utilisation pour le calcul des blindages de protection secondaire est toujours 1.
1.3 Calculs des blindages
Pour une salle de mammographie, les calculs des blindages doivent être effectués pour les barrières de protection secondaires. Étant donné que l'ensemble image-récepteur agit comme une barrière de protection primaire, il n'est pas nécessaire d'avoir d'autres barrières primaires. Les blindages de protection secondaires sont nécessaires pour fournir une protection contre le rayonnement diffusé et le rayonnement de fuite.
Les calculs de protection détaillés pour les installations de mammographie ne devraient être effectués que par des personnes ayant des connaissances à jour de la conception du blindage structurel et des méthodes acceptables pour effectuer ces calculs. Il est recommandé d'effectuer ces calculs de blindage à l'aide de la méthodologie présentée dans le rapport n 147 du National Council on Radiation Protection and Measurements (NCRP): Structural Shielding Design for Medical X-Ray Imaging Installations (NCRP 2004). Cependant, il faut souligner que les objectifs de la conception du blindage spécifiés dans le rapport n 147 du NCRP ne sont pas adoptés dans le présent Code de sécurité. Les valeurs des objectifs de la conception du blindage peuvent être plus basses, mais ne doivent pas dépasser les limites énoncées à la section B1.1 pour les zones contrôlées et non contrôlées. En raison de l'envergure de l'information, la méthodologie du NCRP 147, comprenant des équations, des tableaux et des figures, n'est pas fournie dans le présent Code de sécurité.
L'information exposée dans les sections B1.1 et B1.2 ainsi que les plans finaux de l'établissement doivent être examinés par l'organisme gouvernemental responsable approprié. Les établissements radiologiques de juridiction provinciale ou territoriale devraient communiquer avec l'organisme responsable dans leur province ou territoire respectif figurant dans la liste à l'annexe I.
1.3.1 Blindage des films radiographiques et des cassettes CR
Les contenants de stockage des films doivent être convenablement blindés pour éviter une exposition excessive des films par les rayons X. Les films doivent être suffisamment protégés pour réduire le niveau de rayonnement des films stockés à moins de 0.1 mGy pendant la période de stockage. Lorsque les films sont chargés dans des cassettes, les niveaux d'exposition au rayonnement devraient être inférieurs à 0,5 µGy et l'augmentation qui en résulte dans la base en plus du voile devrait être inférieure à 0,05 DO. L'Appendix IV donne une orientation sur les exigences relatives au blindage en vue du stockage des films. Les valeurs présentées à l'Appendix IV sont très conservatives, mais elles permettent la protection des films contre l'exposition au rayonnement dans la plupart des circonstances. Étant donné que les cassettes CR sont utilisées plus fréquemment et donc la période de stockage est plus courte, la limite de 0.5 µGy est aussi considérée comme un niveau de blindage suffisant pour les cassettes CR. (NCRP 147). En général, les cassettes CR sont stockées derrière le panneau plombé transparent des technologues en mammographie. Le panneau transparent doit avoir une étiquette permanente indiquant l'équivalence en plomb du panneau.
2.0 Exigences des appareils de mammographie
2.1 Exigences réglementaires des appareils de mammographie
Tous les appareils de mammographie neufs, d'occasion ou remis à neuf, ainsi que leurs accessoires, qui sont vendus, importés ou distribués au Canada, doivent se conformer aux exigences de la Loi sur les dispositifs émettant des radiations, la Loi sur les aliments et drogues ainsi que leurs règlements. Il s'agit du Règlement sur les dispositifs émettant des radiations et du Règlement sur les instruments médicaux. Le Règlement sur les dispositifs émettant des radiations, Annexe II, Partie XII (Appareils de radiodiagnostic) établit les exigences relatives à l'information et à l'étiquetage, la construction et le rendement de l'équipement mammographique à rayons X. Le Règlement sur les instruments médicaux tient compte de toutes les autres considérations en matière de sûreté et d'efficacité. Il incombe au fabricant ou au distributeur de veiller à ce que l'équipement soit conforme aux exigences de ces règlements avant d'importer ou de vendre au Canada. Les établissements de juridiction provinciale ou territoriale peuvent être soumis à des exigences spécifiées au titre de leurs statuts. L'Association canadienne de normalisation et le service public d'électricité provincial devraient être consultés pour de plus amples renseignements.
La Loi sur les dispositifs émettant des radiations, la Loi sur les aliments et drogues ainsi que leurs règlements sont disponible aux ressources en ligne des lois et règlements codifiés du Canada du Ministère de la Justice (http://laws-lois.justice.gc.ca/fra/lois/R-1/index.html, http://laws-lois.justice.gc.ca/fra/lois/F-27/index.html).
2.2 Achat d'appareil
L'achat d'un appareil d'imagerie médicale est l'un des investissements les plus importants d'un établissement d'imagerie. Il est donc essentiel de s'assurer que l'on obtient la conception souhaitée et le meilleur rendement qualité prix. Une description du processus d'achat d'appareil d'imagerie médicale se trouve ci-dessous
2.2.1 Analyse des besoins
Une analyse des besoins doit être effectuée afin d'identifier le type et les spécifications des appareils requis pour satisfaire les besoins d'imagerie clinique par rayons X. Lors d'une analyse des besoins, les points principaux qu'il convient de considérer sont les types d'examens que l'établissement prévoit d'effectuer avec l'appareil, et le niveau de rendement que doit avoir l'appareil. Il faudrait également savoir si le personnel de l'établissement possède le savoir-faire nécessaire à l'utilisation de l'appareil, s'il y a un espace suffisant pour l'installation du nouvel appareil, et à quelle date l'appareil doit être installé et opérationnel dans l'établissement. À ce stade, tous les membres du personnel qui utiliseront régulièrement l'appareil devraient être consultés afin qu'ils fassent part de leurs commentaires et suggestions.
2.2.2 Caractéristique de l'appareil
Les caractéristiques de l'appareil doivent être préparées en connaissant parfaitement les besoins cliniques et les conditions opérationnelles, ainsi que les spécifications du fabricant et les exigences en matière de réglementation. La liste des caractéristiques de l'appareil fourni au distributeur devrait identifier le type d'appareil radiographique nécessaire et les types de procédures cliniques que l'on prévoit effectuer avec l'appareil. Il faudrait également identifier tous les composants du système et fournir une description complète de la conception, de la fabrication et des caractéristiques de rendement de chaque composant. Leur niveau de rendement devrait permettre à la majorité des fabricants de répondre aux exigences de rendement avec des composants et des gammes de produits rapidement utilisables. Toutes les exigences pertinentes énoncées dans le présent Code de sécurité et toute autre exigence telle que spécifiée par l'organisme responsable de l'établissement devraient être également traitées dans le cahier des charges de l'appareil. Les conditions électriques, mécaniques et environnementales susceptibles d'affecter le rendement de l'appareil devraient également être incluses.
La liste des besoins devrait aussi inclure d'autres renseignements pertinents comme les détails concernant l'installation et le calibrage de l'appareil par le distributeur ainsi que les délais associés, le type de garantie et le plan d'entretien, et si la formation du personnel par le fabricant est requise . En général, le cahier des charges de l'appareil doit identifier tous les critères qui doivent être satisfaits pour l'acceptation de l'appareil.
Les dispositifs d'essai nécessaires pour effectuer les procédures quotidiennes et mensuelles de contrôle de la qualité que l'établissement ne possède pas déjà doivent être achetés en même temps que l'appareil radiographique.
2.2.3 Analyse de la proposition du vendeur et du contrat d'achat
Les propositions du vendeur doivent être examinées à fond pour garantir que les spécifications de l'appareil fourni par le vendeur répondent aux besoins identifiés de l'établissement. La proposition du vendeur devrait comprendre l'installation et le calibrage de l'appareil, les garanties, le délai de livraison, les plans d'entretien, l'outillage d'essai du contrôle de la qualité, la formation du personnel ainsi que tous les autres critères inclus dans la liste des besoins de l'acheteur concernant l'appareil.
Le contrat d'achat devrait faire état des rubriques et conditions de l'achat précisées dans la liste des besoins de l'établissement et dans la proposition du vendeur qui a été acceptée par l'acheteur et le vendeur. Toutes les conditions pour l'acceptation de l'appareil doivent être clairement précisées, ainsi que les recours possibles si les conditions de l'acceptation ne sont pas respectées. Un contrat d'achat détaillé et concis garantira la livraison de l'appareil dans le respect des délais et des prix.
2.2.4 Essais d'acceptation
Les essais d'acceptation doivent être effectués avant toute utilisation clinique de l'appareil. Les essais d'acceptation sont un processus destiné à vérifier la conformité aux spécifications de rendement de l'appareil radiographique tel que rédigé dans le contrat d'achat. Ils doivent également vérifier que le rendement de l'appareil est conforme au cahier des charges du fabricant et aux règlements fédéraux, provinciaux et territoriaux. Les essais d'acceptation doivent être effectués ou supervisés par un physicien médical possédant une connaissance approfondie du type particulier d'appareil radiologique et des règlements pertinents. Cette personne ne doit avoir aucun lien avec le fabricant.
Les essais d'acceptation d'un système médical à rayons X comportent plusieurs étapes importantes. Ce sont:
- la vérification que les composants ou les systèmes livrés correspondent à ce qui a été commandé;
- la vérification de l'intégrité et de la stabilité de la mécanique du système, notamment des mécanismes de sécurité, la libération automatique du patient, les commandes motrices, le verrouillage des commandes;
- la vérification que les inspections appropriées des installations électriques ont été effectuées, y compris la sécurité de l'électricité et la fluctuation de la ligne de transport électrique;
- la vérification du rendement des rayons X; et
- la vérification des performances de l'imagerie ou du diagnostic.
Des renseignements plus détaillés sur les essais d'acceptation des appareils de mammographie sont disponibles dans les publications de la Commission électrotechnique internationale (IEC 2007).
Les essais de rendement des rayons X effectués lors des essais d'acceptation devraient également refléter les exigences décrites au paragraphe B2.5. Les résultats des essais d'acceptation devraient servir à établir des valeurs et des limites de référence d'acceptation relatives au rendement opérationnel de l'appareil radiographique. Ces valeurs et ces limites de référence sont essentielles au programme d'assurance de la qualité.
2.3 Appareil de mammographie en service
Lorsque possible, il faudrait mettre à niveau les appareils de mammographie en service afin d'incorporer autant de caractéristiques de sécurité et de rendement que possible qui sont requises pour des nouveaux appareils de mammographie, telle que spécifié dans le Règlement sur les dispositifs émettant des radiations en vigueur durant cette période. Il est à noter que la Loi sur les dispositifs émettant des radiations exige que les remplacements de tout composant ou sous-ensemble d'un appareil à rayons X, pour laquelle une norme de fabrication ou de rendement a été spécifiée dans les règlements applicables à la classe d'appareil radiographique, soient en conformité avec les normes en vigueur au moment du remplacement. De plus, les composants et/ou logiciels mis à niveau doivent être autorisés par Santé Canada. L'information sur l'homologation d'équipement mammographique, de logiciels, les accessoires sont disponibles auprès de la Direction des produits thérapeutiques, Bureau des matériels médicaux de Santé Canada (information sur le contact disponible à l'Annexe I). Le propriétaire d'une installation mammographique doit s'assurer que toute amélioration ou tout changement à l'équipement ou au logiciel respecte la réglementation fédérale, provinciale ou territoriale applicable. Tout changement ou toute amélioration à l'équipement ayant une incidence sur la qualité de l'image ou sur la dose de rayonnement reçue doit faire l'objet d'essais d'acceptation réalisés par un physicien médical.
2.4 Mise à niveau avec les systèmes d'imagerie numérique
Au moment d'intégrer un récepteur d'image numérique (ex un système CR) à un système mammographique existant ou à un nouveau système, le propriétaire de l'installation doit s'assurer que le récepteur d'images numériques respecte les exigences de la Loi sur les dispositifs émettant des radiations et de ses règlements, ainsi que de la Loi sur les aliments et drogues et du Règlement sur les instruments médicaux. De plus, le système mammographique, sur lequel est installé le récepteur d'image numérique, doit respecter les exigences de la Partie XII du Règlement sur les dispositifs émettant des radiations en vigueur. Les récepteurs d'image numériques doivent être installés sur les systèmes à rayons X qui ont une commande d'exposition automatique. Le système doit être calibré pour refléter la sensibilité du récepteur numérique.
2.5 Exigences détaillées concernant les appareils de mammographie
La mammographie doit être réalisée uniquement avec de l'équipement spécialement conçu pour la mammographie. L'équipement radiographique conçu pour l'usage général ou les procédures spéciales nonmammographiques sont interdits en mammographie. Cette interdiction vise les systèmes qui ont été modifiés ou qui sont équipés de pièces connexes pour la mammographie.
Les exigences d'information, d'étiquetage, de construction et de fonctionnement énoncés dans la présente section sont fondés sur le Règlement sur les dispositifs émettant des radiations, partie XII (Appareil de radiodiagnostic) et la norme de la Commission électrotechnique internationale (CEI), 60601-2-45 (IEC 2011). Ces exigences sont applicables aux fabricants de tous les appareils de mammographie et sont présentées ici dans le but de promouvoir la sensibilisation aux normes des équipements pour les personnes impliquées dans l'acquisition d'équipements de mammographie. Le Règlement sur les dispositifs émettant des radiations et les normes CEI applicables aux appareils de mammographie devrait servir de référence pour des informations complètes et détaillées sur les normes d'équipement.
2.5.1 Exigences relatives aux renseignements sur l'appareil
- L'appareil à mammographie doit être accompagné des renseignements suivants, fournis par le fabricant:
- les instructions de montage;
- l'adresse du fabricant;
- les instructions sur les mesures de sécurité radiologique et les précautions supplémentaires à prendre en raison de toute particularité de l'appareil;
- les instructions d'entretien de l'appareil afin d'assurer la conformité aux exigences de la Partie XII du Règlement sur les dispositifs émettant des radiations;
- les procédures de contrôle de la qualité à réaliser sur l'équipement, y compris:
- la fréquence des essais,
- les critères d'acceptation;
- le niveau minimal de rendement requis des autres équipements nécessaires pour présenter les images acquises par l'équipement à des fins diagnostiques;
- dans le cas de l'équipement avec récepteur d'images à rayons X intégré:
- l'identification de la version du traitement d'images appliqué aux données originales,
- une description du format de transfert de fichiers des images acquises par l'appareil et de toute donnée associée aux images;
- les instructions d'utilisation, y compris:
- la méthode d'inspection et l'utilisation sûre de toutes les plaques de compression qui sont fournies avec l'équipement mammographique,
- les méthodes visant à déterminer et à résoudre les problèmes avec des artefacts,
- dans le cas de l'équipement avec récepteur d'images intégré:
- la manutention et la maintenance propres au récepteur d'images à rayons X,
- la procédure de contrôle de la qualité du récepteur d'images à rayons X,
- les exigences relatives à la présentation de l'image,
- la méthode visant à déterminer et à identifier les éléments de détecteur défectueux présents dans le récepteur d'image à rayons X numérique,
- la méthode visant à déterminer le niveau de rendement du récepteur d'images suivant le remplacement des données provenant d'éléments défectueux du détecteur, et
- la méthode visant à déterminer si le niveau d'homogénéité de l'image est acceptable;
- la tension de secteur nominale, son courant de secteur maximal et la plage de régulation de sa tension de secteur permettant le fonctionnement au courant de secteur maximal;
- les paramètres de charge qui constituent la condition de courant de secteur maximal pour le générateur radiologique;
- pour chaque gaine équipée, les tailles nominales du foyer et la méthode qui a servi à les déterminer, les courbes de refroidissement de l'anode et de la gaine, les tables de capacité du tube radiogène, et la méthode permettant de déterminer la distance foyer-récepteur d'image;
- ses cycles de service, son type de redressement et la capacité du générateur;
- l'état de charge minimale nécessaire à la mise en fonctionnement s'il fonctionne à pile;
- la plage des valeurs de fonctionnement des tensions radiogènes et l'écart maximal que peut présenter la haute tension radiogène pour toute valeur sélectionnée dans cette plage;
- si l'appareil ne fonctionne pas exclusivement en mode automatique d'exposition, les limites de précision de la minuterie, les limites de précision du courant du tube radiogène, et les limites de précision du produit courant-temps;
- s'il fonctionne en mode automatique d'exposition, les limites de précision de la commande automatique d'exposition; et
- les conditions d'application des renseignements visés aux points n) à p).
2.5.2 Exigences relatives à l'étiquetage
- Tout appareil de mammographie doit porter, aux endroits indiqués ci-après et de manière lisible, permanente et visible, les renseignements suivants :
- sur la surface externe du poste de commande principal:
- un énoncé interdisant toute utilisation non autorisée de l'appareil et avertissant qu'il émet des rayons X dangereux lorsqu'il est en marche,
- le symbole de mise en garde contre les rayons X, qui doit être de deux couleurs contrastantes, être bien visible et reconnaissable à une distance de 1 m, n'avoir aucune dimension extérieure qui est inférieure à 2 cm, porter la mention «ATTENTION: RAYON X - CAUTION: X-RAYS», et être conforme à un des modèles suivants:

- une étiquette, accessible visuellement, qui indique, quant au générateur radiologique, le nom du fabricant, la désignation du modèle, le numéro de série, la date de fabrication, et le pays de fabrication;
- sur la surface externe de la gaine, une étiquette, accessible visuellement, qui indique, quant à la gaine équipée, le nom du fabricant, la désignation du modèle, le numéro de série, la date d'installation du tube radiogène dans la gaine, le pays du fabricant, et la filtration permanente minimale dans le faisceau de rayonnement X émis par la gaine équipée, exprimée en millimètres d'équivalent en aluminium à une haute tension radiogène donnée;
- sur la surface externe de la gaine, ou sur toute autre structure adéquate fixée en permanence à la gaine, un indicateur permettant d'évaluer à 2 % près la distance foyer-récepteur d'image et, si le tube radiogène et le générateur radiologique ne sont pas enveloppés dans une même gaine, des inscriptions indiquant clairement les bornes de l'anode et de la cathode sur la gaine et sur le générateur haute tension;
- sur la surface externe de tout dispositif de limitation du faisceau qui permet de filtrer davantage le faisceau de rayonnement X, une étiquette indiquant la filtration permanente totale que peut assurer ce dispositif, exprimée en millimètres d'équivalent en aluminium à une haute tension radiogène donnée.
- sur la surface externe du poste de commande principal:
- Les commandes, compteurs, avertisseurs lumineux et autres indicateurs exigés par la Partie XII du Règlement sur les dispositifs émettant des radiations doivent être clairement étiquetés quant à leur fonction.
2.5.3 Exigences relatives à la construction
- L'appareil de mammographie doit être fabriqué de façon à comprendre:
- un mécanisme permettant de compenser, selon le type de redressement applicable, les variations de tension du tube radiogène causées par les fluctuations de la tension de secteur;
- un indicateur visuel ou sonore qui se déclenche lorsque la variation de la tension de secteur dépasse la limite spécifiée ou un mécanisme qui, dans ces circonstances, empêche l'émission de rayons X;
- Sur le poste de commande:
- un avertisseur lumineux indiquant que l'appareil est prêt à être mis sous tension,
- un second avertisseur lumineux qui indique l'émission de rayons X,
- un indicateur visuel indiquant que le mode automatique d'exposition est sélectionné, et
- s'il le mode d'exposition automatique n'est pas sélectionné, des commandes et des indicateurs visuels qui permettent à l'opérateur de sélectionner, avant le déclenchement de l'irradiation, les paramètres de charge;
- un mécanisme qui déclenche et arrête l'irradiation;
- un signal sonore qui indique la fin de l'irradiation;
- un dispositif de limitation du faisceau;
- une commande automatique d'exposition; et
- dans le cas d'un appareil qui fonctionne dans la plage visée à la colonne 1 du tableau 3, des filtres de rayonnement qui permettent d'obtenir une couche de demi-transmission d'aluminium, measuré sans la plaque de compression en place, d'une valeur au moins égale à l'une des valeurs suivantes:
- pour toute valeur de tension radiogène figurant à la colonne 2, la valeur de la couche de demi-transmission prévue à la colonne 3,
- pour tout autre cas, la valeur de la couche de demi-transmission obtenue par la formule suivante:

Formule pour la calcule de la couche minimale de demi-transmission d'aluminium, mesuré sans la plaque de compression en place.
La couche minimale de demi-transmission d'aluminium, mesuré sans la plaque de compression en place, calculée au moyen de l'équation « CDT (mm d'aluminium) supérieure ou égale à la tension du tube à rayons X divisée par 100 ».
| Colonne 1 Plage de fonctionnement normal (kV) |
Colonne 2 Tension du tube radiogène (kV) |
Colonne 3 Couches de demi-transmission en aluminium (mm) |
|---|---|---|
| 50 ou moins | (a) 30 | 0,30 |
| (b) 40 | 0,40 | |
| (c) 50 | 0,50 |
2. Toute commande d'irradiation d'un appareil à mammographie ne doit pas permettre l'émission de rayons X que si l'opérateur y exerce une pression continue;
3. Toute minuterie d'un appareil à mammographie doit:
- arrêter automatiquement l'irradiation lorsqu'est atteinte l'une des valeurs sélectionnées du temps d'irradiation, du produit courant-temps, ou d'un nombre d'impulsions donné;
- permettre à l'opérateur d'arrêter l'irradiation à tout moment;
- revenir automatiquement à la position de réglage original ou à zéro à la fin de l'irradiation;
- empêcher le déclenchement d'une irradiation lorsqu'elle est réglée à zéro, à la position d'arrêt ou à une position non marquée.
4. Lorsqu'une table-support (total des couches, à l'exclusion de la grille) est placée entre le patient et le récepteur d'image radiologique, son équivalent en aluminium ne doit pas excéder 0,3 mm, d'après un faisceau de rayonnement X qui:
- est émis à une haute tension radiogène de 30 kV;
- a un taux d'oscillation maximal de la tension radiogène de 10%;
- a une couche de demi-transmission d'aluminium de 0,3 mm; et
Le capteur permettant le réglage automatique de l'exposition est considéré comme un élément du récepteur d'image radiologique.
5. L'appareil à mammographie doit être fabriqué de façon que:
- le tube radiogène soit fixé solidement à la gaine et bien aligné à l'intérieur de celle-ci;
- les filtres de rayonnement soient fixés solidement à l'orifice de sortie de la gaine et au dispositif de limitation du faisceau, ou à l'un des deux; et
- l'ensemble radiogène maintient la position ou le mouvement requis sans dévier ni vibrer pendant le fonctionnement.
6. L'appareil à mammographie muni d'une commande automatique d'exposition doit être fabriqué de façon à:
- comprendre un dispositif qui arrête automatiquement les irradiations lorsque le produit courant-temps dépasse 800 mAs par irradiation; et
- lorsqu'une exposition minutée automatiquement prend fin parce que les limites prévues à l'alinéa (a) ont été atteintes, être muni d'un indicateur visuel ou d'un signal sonore, qui signale la fin de l'exposition, et d'une commande de remise à zéro qui doit être réglée manuellement avant que toute autre exposition ne puisse être minutée automatiquement.
7. Tout appareil à mammographie doit comporter :
- un dispositif de support du récepteur d'image muni d'un blindage de protection radiologique qui limite le rayonnement résiduel, et conçu pour s'avancer jusqu'au thorax du patient, et dépasse le champ de rayon X d'au moins 1 % de la distance foyer-récepteur d'image sur les autres côtés;
- un support du patient conçu de manière que, en mode de non-agrandissement, la distance maximale entre le bord de la surface réceptrice de l'image devant jouxter la paroi thoracique du patient et le bord adjacent du support du patient, lorsqu'il est projeté sur le support du patient, doit être inférieure à 5 mm;
- un dispositif de limitation du faisceau qui restreint le faisceau de rayonnement X de façon que, en mode de non-agrandissement, le champ de rayon X:
- du côté situé près de la paroi thoracique du patient, ne s'étende pas au-delà de 2 mm du support du patient,
- pour l'équipement de mammographie sans balayage, doit s'étendre au-delà du bord de la surface de réception de l'image qui est conçue pour être adjacente à la paroi thoracique du patient, et
- pour l'équipement de mammographie sans balayage, ne s'étende pas de plus de 2 % de la distance focale directe au-delà de tous les autres côtés de la surface de réception de l'image efficace;
- une plaque de compression du sein qui:
- est actionnée par une commande au pied afin de commencer et contrôler la compression,
- comporte un mécanisme permettant d'ajuster avec précision le mouvement durant la compression,
- comporte un mécanisme de décompression rapide,
- peut être actionné par une commande mains libres (des pédales par exemple) qui sont accessibles de chaque coté du patient,
- comporte des plaques transparentes, de sorte que la peau du patient demeure visible lorsque elle est en contact avec elles, et
- permet d'amener la partie de la plaque de compression en contact avec le sein à 10 mm près de la surface de support;
- lors des situations de perte de courant, la compression est maintenue pendant la biopsie et autre opération de marquage avec cependant une décompression manuel qui doit être prévu dans la conception de l'équipement.
- un dispositif qui illumine le champ de rayon X dans des conditions ambiantes d'éclairage à l'intérieur d'une salle de radiographie; et
- un mécanisme indiquant à 2% près la distance foyer-récepteur d'image.
8. L'équipement mammographique à dispositif de limitation du faisceau amovible à ouverture fixe doit afficher sur la surface externe les dimensions de la surface réceptrice de l'image et la distance foyer-récepteur d'image à utiliser.
2.5.4 Exigences relatives au fonctionnement
1. Reproductibilité de la puissance du rayonnement
Pour toute combinaison de la tension radiogène, du courant dans le tube radiogène et de la durée d'irradiation, ou pour toute exposition sélectionnée au récepteur d'image radiologique, lorsque la tension de secteur pour chaque mesure correspond à la valeur moyenne de la tension de toutes les mesures à 1% près, et que toutes les commandes variables pour les paramètres de charge sont réglées à d'autres valeurs et ramenées à la valeur d'essai avant chaque mesure:
- le coefficient de variation de 10 mesures consécutives du kerma dans l'air ou de l'exposition, effectuées au même point sur l'axe du faisceau de rayons X, 4cm au-dessus du support du patient, pendant une heure, doit être d'au plus 0,05; et
- la valeur de chacune des 10 mesures effectuées conformément à l'alinéa (a) doit correspondre, à 15% près, à la valeur moyenne de ces mesures.
2. Précision des paramètres de charge
Le paramètre de charge indiqué à la colonne 1 du tableau 4 ne doit pas varier, par rapport à la valeur sélectionnée et pour toute combinaison de paramètres de charge, de plus de la valeur figurant à la colonne 2.
| Article | Colonne 1 | Colonne 2 |
|---|---|---|
| Facteur de charge | Variation maximale par rapport à la valeur sélectionnée | |
| 1. | Tension du tube radiogène | ±5% |
| 2. | Temps d'irradiation | ±(10% plus 1 ms) |
| 3. | Courant dans le tube radiogène | ±20% |
| 4. | Produit courant-temps | ±(10% plus 0,2 mAs) |
3. Minuterie et système automatique d'exposition
- Dans le cas des appareils de mammographie sans récepteur d'image intégré, si le système automatique d'exposition est choisi, la variation dans la densité optique établie à l'alinéa (b) doit être déterminée à l'aide d'objets qui sont constitués de matériaux équivalents à des tissus humains et qui ont une épaisseur qui est représentative de la plage réelle de l'épaisseur du corps des patients.
- Dans le cas d'un appareil de mammographie avec récepteur d'image film-écran, le dispositif de commande du système automatique d'exposition doit fonctionner de manière à ce que l'écart dans la densité optique sur les radiogrammes résultants ne dépasse pas ±0,15 de la densité optique moyenne lorsque l'épaisseur du matériau équivalent au tissu mammaire est variée de 2 à 7 cm et que la tension du tube et les combinaisons de filtre d'anode sont variées de manière appropriée pour tenir compte de l'épaisseur. Si cette exigence ne peut pas être atteinte, un tableau doit être élaboré pour indiquer quels facteurs de charge correspondent aux différentes épaisseurs de tissus mammaires et quelles combinaisons doivent être utilisées pour qu'une densité optique se situant à ±0,15 autour des conditions de commande automatique d'exposition soit produite.
- Dans le cas de l'équipement de mammographie muni d'un récepteur d'image numérique intégré, le rendement du système automatique d'exposition doit être évalué en tenant compte simultanément de la qualité de l'image, telle que mesurée par le rapport du contraste au bruit, dans des conditions spécifiques, et de la dose reçue par le patient, qui est caractérisée par la dose glandulaire moyenne; on doit ensuite comparer les résultats aux spécifications fournies par le fabricant.
- Pour ce qui est des appareils de mammographie à récepteur d'image intégré, la reproductibilité du système automatique d'exposition doit être évaluée en imageant à répétition un fantôme dans des conditions spécifiques, et en mesurant l'écart de l'une des quantités suivantes:
- chargement du tube (mAs),
- kerma dans l'air dans une position fixe entre la source de rayons X et le récepteur d'image, ou
- valeur moyenne des valeurs de pixel linéarisées dans une zone d'intérêt de l'image du fantôme.
La valeur mesurée de la quantité sélectionnée ne doit pas différer de plus de ±15% de la valeur moyenne des applications de charge d'essai ou, si approprié, de la spécification du fabricant.
4. Linéarité de la puissance du rayonnement
Formule pour la calcule de la linéarité
Linéarité, calculée au moyen de l'équation « valeur absolue, parenthèse ouverte, de X indice 1 moins X indice 2, parenthèse fermée, inférieure ou égale à 0,1 multiplié par, parenthèse ouverte, X indice 1 plus X indice 2, parenthèse fermée ».
- Pour toute valeur de tension radiogène sélectionnée dans la plage déterminée selon le l'alinéa (b), les quotients de la mesure moyenne du kerma dans l'air, ou de la mesure moyenne de l'exposition, par le produit courant-temps, qui sont obtenus aux deux positions de réglage applicables prévues à l'alinéa (c), ne peuvent présenter un écart supérieur à 0,10 fois leur somme, déterminée selon la formule suivante:
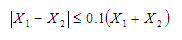
où X1 et X2 représentent les quotients de la mesure moyenne du kerma dans l'air ou de la mesure moyenne de l'exposition par le produit courant-temps;
- La tension du tube radiogène mentionnée à l'alinéa (a) doit être réglée à 30 kV ou à la tension de tube radiogène disponible la plus proche spécifiée par le fabricant pour l'appareil mammographique; et
- Les quotients mentionnés à l'alinéa (a) doivent être déterminés à deux positions de réglage quelconques du produit courant-temps dans le tube radiogène, sur la plage totale de la sélection des produits courants-temps disponible, de manière à ce que:
- La valeur la plus faible de la première paire, doit correspondre au réglage le plus faible disponible du produit courant-temps ,
- Le rapport des valeurs des réglages du produit courant-temps sélectionnés dans chaque paire doit être aussi proche que possible de 2, sans dépasser cette valeur,
- La valeur la plus élevée des réglages du produit courant-temps dans chaque paire à mesurer, doit être utilisée comme la valeur la plus faible de la paire suivante des réglages du produit courant-temps, et
- La valeur la plus élevée de la dernière paire doit correspondre au réglage du produit courant-temps le plus élevé disponible, et la valeur la plus faible doit s'élever à la moitié ou proche de la moitié de la valeur correspondant au réglage du produit courant-temps le plus élevé disponible.
5. Indications relatives à la dosimétrie
Dans le cas des appareils de mammographie munis d'un récepteur d'image numérique intégré, la dose glandulaire moyenne doit être indiquée pour chaque image acquise.
6. Rayonnement résiduel derrière le récepteur d'image
Aucun appareil à mammographie ne peut produire, derrière le dispositif de support du récepteur d'image, de rayonnement résiduel supérieur à un kerma dans l'air de 1,0 µGy ou à une exposition de 0,115 mR par irradiation lorsque l'appareil fonctionne:
- à son champ de rayon X maximal et à sa distance foyer-récepteur d'image minimale;
- à sa tension radiogène maximale et selon son produit courant-temps maximal.
La mesure du kerma dans l'air ou la mesure de l'exposition se détermine par le calcul de sa moyenne selon une surface de détection de 100 cm2 n'ayant aucune dimension linéaire supérieure à 20 cm et dont le centre est placé à 5 cm de toute surface accessible au dispositif de support du récepteur d'image.
7. Débit minimal de la puissance de rayonnement
Tout appareil à mammographie sans récepteur d'image radiolgoique intégré doit avoir un débit de rayonnement de sortie minimal de 7,0 mGy/s ou de 802 mR/s lorsque l'appareil fonctionne,
- avec une anode de molybdène et un filtre de molybdène;
- sans la plaque de compression du sein placée entre la source radiogène et le détecteur;
- à une tension radiogène de 28 kV en mode standard pour toute distance foyer-récepteur d'image; et
- le débit de rayonnement de sortie minimal doit être:
- mesuré à 4,0 cm au-dessus du support du patient et à 6,0 cm du côté de la paroi thoracique du patient sur la ligne centrale;
- réparti sur une période d'irradiation de 3,0 s.
8. Fuite de rayonnement lorsque l'appareil est en mode de chargement
Le rayonnement de fuite de l'ensemble radiogène de tout appareil de radiodiagnostic ne peut excéder un débit de kerma dans l'air de 1,0 mGy/h ou un débit d'exposition de 115 mR/h, lorsque l'appareil fonctionne à une tension nominale avec application d'une charge correspondant à l'apport énergétique maximal déterminé qui est produit pendant une heure.
La mesure du débit se détermine par le calcul de sa moyenne selon une surface de détection de 100 cm2 n'ayant aucune dimension linéaire supérieure à 20 cm et dont le centre est placé à une distance de 1 m du foyer.
9. Fuite de rayonnement lorsque l'appareil n'est pas en mode de chargement
L'ensemble radiogène de tout appareil de radiodiagnostic ne doit pas, si une haute tension peut apparaître dans le tube radiogène, émettre un rayonnement dont le débit de kerma dans l'air est supérieur à 20,0 µGy/h ou dont le débit d'exposition est supérieur à 2,3 mR/h lorsque le dispositif de limitation du faisceau est complètement ouvert, et la commande automatique d'exposition ou la commande d'irradiation n'a pas été actionnée.
La mesure du débit se détermine par le calcul de sa moyenne selon une surface de détection de 10 cm2 n'ayant aucune dimension linéaire supérieure à 5 cm et dont le centre est placé à 5 cm de toute surface accessible de l'élément.
10. Rayonnement provenance d'autres composants
Le débit de kerma dans l'air ou le débit d'exposition émis par tout élément d'un appareil de radiodiagnostic, autre que l'ensemble radiogène, ne peut, quelles que soient les conditions d'utilisation, dépasser 20,0 µGy/h et 2,3 mR/h respectivement.
La mesure du débit se détermine par le calcul de sa moyenne selon une surface de détection de 10 cm2 n'ayant aucune dimension linéaire supérieure à 5 cm et dont le centre est placé à 5 cm de toute surface accessible de l'élément.
3.0 Systèmes de traitement d'images
Le traitement d'images inclut à la fois le traitement des films et le traitement numérique des images radiologiques. Les systèmes de traitement des films ont été beaucoup utilisés par le passé. Récemment, avec les progrès de la technologie numérique, les systèmes de traitement numérique d'image ont été utilisés dans de nombreux établissements radiologiques. Quel que soit le type de système utilisé, l'optimisation de la qualité d'image à une dose acceptable pour le patient est une priorité pour les établissements radiologiques. Ceci est réalisé en s'assurant que le traitement d'image fait partie intégrante du programme d'assurance de la qualité de l'établissement.
3.1 Systèmes de mammographie film-écran
L'aptitude à produire un mammogramme de qualité diagnostique satisfaisante, à une dose acceptable au patient, dépend de la technique utilisée en effectuant l'examen, la sélection de paramètres de charge appropriée, l'écran-film employé, la manipulation et le traitement du film, et les conditions d'observation de l'image. Une bonne qualité d'image nécessite des techniques de chambre noire adaptées, la surveillance régulière du contrôle de la qualité du développeur, et le strict respect des instructions des fabricants du film et du système de développement.
3.1.1 Film radiographique
Les films radiographiques sont sensibles à la lumière, la chaleur, l'humidité, la pollution chimique, la contrainte mécanique et les rayons X. Un film non exposé doit être stocké de façon à être protégé des rayonnements parasites, des vapeurs chimiques et de la lumière. Le niveau de densité optique venant du matériau de base et du voile du film résultant de toutes les causes ne doit pas être supérieur à 0,30 DO. Généralement, les films radiographiques devraient être stockés verticalement, dans une zone à l'abri des vapeurs chimiques, à des températures dans la plage de 10°C à 21°C et une humidité entre 30% et 60%. Les instructions des fabricants de films doivent être observées. Il faut laisser les emballages de film hermétiques atteindre la température ambiante avant leur ouverture, pour éviter d'avoir de la condensation sur les films. Les cassettes chargées doivent être stockées dans une zone blindée contre l'exposition au rayonnement. Les expositions au rayonnement de films stockés doivent être limitées à 0,1 mGy et, pour les cassettes chargées, à 0,5 µGy. Cette zone est habituellement dans ou près d'une salle de radiographie. L'emplacement des cassettes chargées et non exposées doit être clairement marqué. La zone doit être assez grande pour recevoir le nombre de cassettes requis nécessaire pendant le fonctionnement de l'établissement.
3.1.2 Cassette et écran
Des cassettes ou écrans en mauvais état diminuent la qualité diagnostique de l'image. Les problèmes sont causés par les écrans sales ou détériorés, les cassettes faussées, la fatigue de matériau de compression en mousse ou de mécanisme de fermeture, les fuites de lumière, et un mauvais contact écran-film. L'usure et la propreté des cassettes doivent être vérifiées régulièrement et toute cassette détériorée doit être remplacée. Le nettoyant d'écran recommandé par les fabricants devrait être utilisé. Pour éviter les artefacts causés par la saleté et la poussière, les écrans renforçateurs et les cassettes doivent être nettoyés au moins une fois par mois. Les écrans renforçateurs doivent être inspectés avec une lampe à ultraviolet pour trouver les particules de poussière. Les outils de nettoyage comprennent un nettoyant d'écran avec une solution antistatique, des chiffons non pelucheux, de l'air comprimé et une brosse en poil de chameau. Les cassettes et les écrans doivent être numérotés pour identification et appariement, à la fois à l'intérieur de la cassette et sur l'extérieur de la cassette.
3.1.3 Chambre noire
À l'exception des appareils automatiques de développement d'image à la lumière du jour, qui ne nécessitent pas de chambre noire, les développeurs de films automatiques nécessitent des chambres noires bien conçues. Alors que des détails spécifiques peuvent varier d'une installation à l'autre, toutes les chambres noires doivent comprendre certaines caractéristiques de base, énumérées ci-dessous. Des renseignements détaillés sur la conception des chambres noires sont fournis par L'Agence internationale de l'énergie nucléaire (AIEA, 2009):
- La pièce doit être étanche à la lumière. Il faut faire particulièrement attention aux joints de la porte et au montage du développeur de films si l'introduction du film dans l'appareil s'effectue à travers un mur. La chambre noire doit comprendre une porte verrouillable ou des doubles portes pour assurer l'étanchéité à la lumière quand on manipule des films non développés. Une bande de film exposée à une densité optique de 1,2 unité ne doit pas présenter une augmentation de la densité optique supérieure à 0,05 unité en deux minutes d'exposition à l'environnement lumineux de la chambre noire;
- Si la chambre noire est contiguë à une salle de radiographie, le conteneur de stockage des films doit être blindé de façon adéquate pour s'assurer qu'il ne se produise pas d'exposition excessive du film aux rayons X. Un blindage suffisant doit être installé pour réduire le niveau de rayonnement sur le film à 0,1 mGy et sur les cassettes chargées à 0,5 μGy;
- Un voyant lumineux doit se trouver à l'extérieur de la chambre noire, à l'entrée, pour indiquer quand la pièce est en service. Ce voyant lumineux n'est pas nécessaire si la porte est verrouillée quand elle est fermée;
- Les lampes inactiniques, équipées d'ampoules d'intensité non supérieure à 15 watts, doivent être prévues au-dessus des plans de travail à l'intérieur de la chambre noire. La lampe inactinique doit avoir des filtres appropriés aux spécificités du film utilisé et doit être placée à des distances de plus de 1 mètre des zones de travail, afin de minimiser le voile des films;
- L'air vicié de l'appareil à développer devrait être évacué directement à l'extérieur de l'installation. Le ventilateur de l'appareil à développer devrait marcher sans arrêt, même lorsque l'appareil à développer n'est pas en fonction. Cela sert à empêcher des dommages à l'appareil à développer tel que l'accumulation des vapeurs des solutions de développement et la corrosion des parties; et
- La chambre noire doit être sous pression positive, afin que la poussière ne soit pas aspirée dans la pièce quand on ouvre la porte. Le nombre de renouvellements d'air doit être suffisamment élevé pour que l'appareil à développer fonctionne correctement et ne génère pas de situation dangereuse pour le personnel.
La propreté de la chambre noire et des écrans et cassettes est essentielle. Il est important de maintenir l'atmosphère la plus propre possible afin de minimiser tout artefact entraîné par la saleté, la poussière ou la manipulation incorrecte de film. Une lampe à ultraviolet devrait être utilisée pour trouver les zones poussiéreuses autour de la chambre noire. Personne ne doit manger ni boire dans la zone de la chambre noire. Toutes les surfaces de travail, dessus de comptoirs et le sol devraient être nettoyés régulièrement, au moins une fois par jour. Le dessus des armoires, les évents, les appareils d'éclairage et toute autre zone qui peut ramasser de la poussière devraient être nettoyés régulièrement. Le système de ventilation devrait être vérifié pour s'assurer qu'il n'introduit pas de poussière à l'intérieur de la chambre noire; tout filtre devrait être remplacé régulièrement. À l'exception des mélangeurs automatique, les produits chimiques ne devraient pas être mélangés à l'intérieur de la chambre noire, car cette opération peut entraîner des éclaboussures de produits sur les appareils ou les plans de travail. Le personnel devrait porter des effets de protection personnelle (gants, masques, etc.) en manipulant les produits chimiques.
Pour éviter de mettre des empreintes de doigt sur le film et de salir les écrans, il est important de se laver souvent les mains avec du savon qui ne laisse aucun résidu. Les lotions pour les mains et les crèmes peuvent aussi laisser des empreintes de doigt sur les films. Il faut supprimer le fouillis qui peut accumuler de la poussière. Les boîtes en carton ondulé qui contiennent les boîtiers de films, les produits chimiques et autres fournitures, ne devraient n'être ni stockés ni ouverts à l'intérieur de la chambre noire, car ils produiront beaucoup de poussière. Les boîtes devraient être ouvertes à l'extérieur de la chambre noire, et les films et fournitures transportés à l'intérieur. Dans la chambre noire, on ne devrait pas porter de vêtement fait de fibres lâches ou qui génèrent de l'électricité statique, comme la laine, la soie, certains cotons ou tissus de mélange de coton, ou alors il faut le recouvrir d'une blouse de laboratoire.
3.1.4 Développement des films
Un développement incorrect ou bâclé des films radiographiques exposés peut se traduire par des films de faible qualité d'image diagnostique et, par conséquent, augmenter la possibilité de mauvais diagnostic ou de reprise d'examens aux rayons X. Pour réaliser un développement complet, le film doit être traité dans du révélateur chimiquement frais, à la bonne température, et pendant un temps suffisant pour s'assurer que l'argent des cristaux d'halogénure d'argent de l'émulsion de film est complètement réduit. Si ce n'est pas le cas, le noircissement du film ne sera pas optimal et la tendance sera d'augmenter l'exposition au rayonnement pour obtenir une densité d'image correcte.
D'autres facteurs peuvent également affecter la qualité des films développés. Parmi ceux-ci, la propreté du système de développement, le temps d'immersion du film, et l'efficacité du rinçage. Pour assurer un développement correct des films, il faut suivre certaines procédures de base:
- La seule méthode acceptable pour contrôler le fonctionnement d'une développeuse de film automatisé est l'utilisation d'un sensitomètre pour produire une exposition à la lumière reproductible du film et l'utilisation d'un densitomètre pour vérifier la sensitométrie du film développé. Le contrôle de la développeuse de film doit être effectué chaque jour d'utilisation, avant de développer des radiographies de patient, une fois l'appareil mis en route et stabilisé, et à d'autres moments, après son nettoyage et après l'ajout de produits chimiques frais.
- Les instructions des fabricants concernant la concentration de la solution, la température et la durée doivent être suivies pour assurer un développement optimal.
- Les solutions de développement doivent être remplies lorsque c'est nécessaire, et doivent être changées ou recyclées régulièrement, selon les besoins. Ceci devrait être fait assez souvent pour éviter l'oxydation des solutions de développement. Même inutilisé, le révélateur se détériore avec le temps. Les produits chimiques de développement doivent être protégés du gel. Il faut suivre les instructions des fabricants en matière de stockage des produits chimiques, pour éviter l'oxydation. Les produits chimiques présentant des signes d'oxydation ou de sédimentation ne doivent pas être utilisés.
- Le fixateur doit être éliminé de façon adéquate des films développés. Il faut suivre les instructions des fabricants pour le lavage des films. Des essais de rétention de fixateur doivent être effectués régulièrement. Le fixateur est responsable de l'arrêt du processus de développement en éliminant les cristaux d'halogénure d'argent restant sur le film. Un lavage insuffisant des films pour éliminer le fixateur entraînera des taches sur les films et compromettra la durée de stockage des films.
- La propreté est extrêmement importante pour réduire les artefacts de film. Les mécanismes de transport des films des développeurs de films doivent être nettoyés fréquemment. Il ne faut jamais utiliser des chiffons ou des nettoyants abrasifs sur les appareils de développement.
- Les développeurs de film doivent être entretenus régulièrement, suivant les instructions des fabricants. La précision du thermomètre du développeur doit être vérifiée régulièrement par rapport à un thermomètre non mercure. Le thermomètre numérique du développeur doit être précis à 0,5°C près.
- Quand le volume de développement de film est inférieur à 50 films par jour, il peut ne pas être possible de contrôler de façon adéquate les concentrations et l'activité chimiques. Dans cette situation, il faut utiliser la régénération pour mieux contrôler les concentrations chimiques.
- Quand le volume de développement de film est d'au moins 50 films par jour, un système de remplissage du volume est généralement utilisé, qui remplit de solutions de traitement chaque fois qu'un film est introduit dans le développeur. Il est recommandé de suivre les spécifications des fabricants pour le remplissage de solutions de développement.
Le développement des films radiographiques génère des déchets contenant de l'argent. Les produits chimiques contenant de l'argent ne doivent pas être éliminés directement dans les égouts. Ces produits chimiques doivent être récupérés et remis à l'organisme approprié de gestion des déchets, pour élimination et/ou recyclage. La gestion des déchets contenant de l'argent doit être effectuée conformément aux exigences provinciales et municipales.
3.1.5 Négatoscope
Il faut vérifier régulièrement l'état des négatoscopes ainsi que les conditions dans lesquelles les radiologistes et les autres professionnels de la santé examinent les mammogrammes, car ceci peut influer sur la précision du diagnostic. Il faut corriger les problèmes d'éclairage causés par les différences de luminosité des tubes fluorescents ou à la dégradation et la décoloration de la surface de visionnement.
3.2 Systèmes de mammographie numérique
Un nombre croissant de centres de mammographie passe de la technologie film-écran à la technologie numérique. Différentes technologies de mammographie numérique sont disponibles utilisant différents types de détecteur pour produire une image numérique. En général nous caractérisons ses technologies en deux groupes distincts.
- Des systèmes CR comprennent une cassette, une plaque d'imagerie qui contient du phosphore photostimulable et un lecteur de plaque d'imagerie. La cassette de CR, chargée avec une plaque d'imagerie, est positionnée dans le système de mammographie, comme on le fait avec les cassettes de film. À l'exposition aux rayons X, la plaque d'imagerie stocke l'image latente. La plaque d'imagerie est ensuite lue et une image numérique est produite.
- Des systèmes DR sont construits avec un détecteur de rayon X numérique intégré. Trois principaux types de détecteur de rayon X numérique intégré sont présents dans les systèmes de mammographie. Ce sont les détecteurs à panneau plat au CsI sur une matrice de photodiodes, les détecteurs à panneau plats au sélénium amorphe sur une matrice d'électrodes et les détecteurs par balayage avec compteur de photons (AIEA, 2011). Dans les systèmes DR le signal électronique contenant l'information d'image est converti et l'image apparait presque instantanément.
Les essais de contrôle de la qualité des systèmes d'imagerie numériques sont essentiels. La vérification du bon fonctionnement des appareils d'imagerie radiographique ainsi que la sélection appropriée de la technique et des paramètres de charge restent essentiels pour obtenir une image satisfaisante à une dose minimale pour le patient. Pour les systèmes numériques, il faut également effectuer des essais de contrôle de la qualité spécifiques des systèmes d'acquisition, de stockage, de communication et d'affichage de l'image. À la section C du Code de sécurité, les essais généraux de contrôle de la qualité ont été inclus pour les systèmes d'imagerie numériques. Outre ces essais, il faut également effectuer tous les essais spécifiques aux appareils, spécifiés par les fabricants. Les centres sous juridiction provinciale ou territoriale sont sujets à d'autres exigences concernant les essais.
3.2.1 Plaques d'imagerie de CR
Les plaques d'imagerie de CR sont réutilisables et peuvent être exposées, lues et effacées à plusieurs reprises. Pour cette raison, il est nécessaire d'évaluer régulièrement l'état de ces plaques. En utilisation normale, l'accumulation de poussière, saletés, rayures et fissures peut réduire la qualité de l'image. L'exposition à des agents chimiques, tels que des nettoyants d'écran non approuvés, la manipulation avec les mains sales ou mouillées, ou le contact avec des lotions pour les mains sont toutes des causes possibles de détérioration des écrans. Il est recommandé de tenir à jour un registre traçant l'état physique des plaques d'imagerie et des ensembles de cassettes. La fréquence de nettoyage dépend du volume de patients, de la manipulation des plaques et de la fréquence à laquelle des artefacts sont perçus. Une inspection visuelle hebdomadaire de la propreté des systèmes d'imagerie doit être effectuée pour qu'il n'y ait ni poussière ni saleté. Les plaques d'imagerie doivent être nettoyées mensuellement suivant les procédures recommandées par les fabricants et en utilisant les nettoyants qu'ils préconisent. Le nettoyant ne doit pas être versé directement sur les plaques, car cela provoquerait des taches.
3.2.2 Cassette de CR
Dans les conditions normales d'utilisation, de la poussière et de la saleté peuvent s'accumuler sur les cassettes. Il est recommandé de tenir à jour un registre traçant l'état physique de toutes les cassettes et écrans. En général, un contrôle d'aspect hebdomadaire, pour vérifier l'absence de poussière et de saletés, est recommandé, ainsi qu'un nettoyage mensuel des cassettes et écrans CR suivant les procédures recommandées par les fabricants et en utilisant les nettoyants qu'ils recommandent. L'extérieur de la cassette peut se nettoyer facilement avec de l'eau et du savon ou un nettoyant non agressif. Mais il ne faut pas nettoyer l'intérieur à l'eau et au savon, car des résidus de savon peuvent rester sur le revêtement de protection après le nettoyage.
Les cassettes CR, chargées avec une plaque d'imagerie, qui ne sont pas en usage, doivent être emmagasinées dans un endroit dont les niveaux d'exposition aux rayonnements se limitent à 0,5 μGy.
3.2.3 Tomosynthèse du sein
La tomosynthèse numérique du sein (TNS) est une nouvelle technique d'imagerie du sein en 3D. Les équipements TNS acquièrent des images de projection du sein selon différents angles tomographiques. Le nombre d'images acquises et les angles diffèrent selon les fabricants des équipement TNS. Des algorithmes de reconstruction sont alors utilisés pour produire des images des planes tomographiques à travers le sein. Typiquement les épaisseurs des planes varient de 0,5mm à 3 mm. Les images peuvent être visualisées, sur un moniteur, une à une ou en séquence pour tout le sein.
La TNS a le potentiel d'améliorer la visualisation des tissus mammaires en surmontant les limites des techniques actuelles de mammographie résultant du chevauchement de tissu mammaire en une seule image bidimensionnelle. La dose administrée au patient pendant un balayage de tomosynthèse est comparable à un examen mammographique (Gennaro 2010). Les avantages potentiels de la TNS incluent l'amélioration de la sensibilité du dépistage, l'amélioration de la taille des lésions à la détection, l'amélioration de la caractérisation et la diminution des taux de rappel (Helvie 2010).
Pour les équipements TNS, les procédures d'assurance de la qualité et les procédures de contrôle de la qualité, ainsi que la dosimétrie doivent être effectuées conformément à la recommandation du fabricant.
3.2.4 Poste de travail mammographique/Dispositif de visualisation électronique
Afin de réaliser le potentiel réel de la mammographie numérique, il est important de s'assurer que les dispositifs de visualisation électroniques sur lesquels sont affichés les mammogrammes à interpréter permettent une visualisation optimale des tissus mammaires. Le poste de travail d'interprétation mammographique doit respecter les exigences suivantes, à tout le moins (AAPM 2005, CAR PAM 2011, Van Ongeval 2010):
- Il doit y avoir au moins deux moniteurs monochromes ayant une résolution spatiale de 5 mégapixels.
- Les dispositifs d'affichage doivent être conformes au profil d'image mammographique IHE.
- Une luminance minimum de 250 cd/m2 est requise. (Une luminance de 450 cd/m2 est recommandée.)
- Le rapport de la luminance maximale à la luminance minimale doit se situer entre 250 et 650 (incluant la lumière ambiante).
- Une résolution de luminance minimale de 8 bits est requise.
- Le bruit de l'affichage doit être aussi faible que possible (préférablement 2 à 2,5%).
- Les écrans doivent être étalonnés à l'aide de la fonction d'affichage de gris normalisée DICOM (NEMA 2011).
Un moniteur monochrome avec une résolution spatiale d'au moins 3 mégapixels devrait être utilisé pour les technologues et autres professionnels de la santé qui doivent consulter les mammographies.
Il est important de s'assurer que le dispositif d'affichage soit soumis à des essais d'acceptation et que son rendement soit vérifié lors d'essais de routine et de contrôles de la qualité chaque année. Des renseignements détaillés sur les essais d'acceptation et sur le contrôle de la qualité sont disponibles auprès de l'American Association of Physicists in Medicine (AAPM, 2005). La propreté de la surface de visualisation doit être bien entretenue. Il faut utiliser les nettoyants et procédures de nettoyage recommandés par les fabricants. La performance du poste de visualisation doit être vérifiée à l'aide des mires destinées à évaluer les différentes caractéristiques des éléments de visualisation. Une évaluation globale devrait être effectuée quotidiennement avant utilisation clinique. Une évaluation visuelle doit être effectuée hebdomadairement par un technologue et une évaluation détaillée doit être effectuée annuellement par un physicien médical. La Section C de ce document fournit une description des évaluations de contrôle de la qualité. Lors de la réalisation de ces évaluations de contrôle de la qualité, une attention particulière doit être accordée aux conditions de visualisation dans la salle ou les mammogrammes sont lus.
3.2.5 Système d'archivage et de transmission d'images (PACS)
En imagerie numérique, il faut qu'un système soit en place pour gérer les images de patient, pour permettre un stockage sûr et une récupération en temps utile des images. Le système d'archivage et de transmission d'images, PACS (de l'anglais Picture Archiving and Communications System) est un système qui est largement utilisé en radiologie. Un PACS d'établissement d'imagerie relie les dispositifs d'acquisition d'images numériques à un système qui peut stocker, retrouver et afficher des images numériques à l'intérieur et à l'extérieur de l'établissement. La transition au PACS nécessite une somme importante de planification, temps et ressources. Une fois établi, un PACS offre un certain nombre d'avantages, tels qu'une amélioration de la productivité, un accès étendu, simultané, aux images et à la manipulation d'images. Cependant, il faut s'assurer que les images de patient sont maintenu et que les renseignements sur les patients ne sont pas perdus ni modifiés involontairement. De telles situations peuvent mener à la reprise d'examens radiologiques et à des erreurs de diagnostic des patients.
3.2.6 Mise en œuvre du PACS
Lorsque l'on s'interroge sur l'éventualité de mettre en œuvre un PACS, il faut aborder un certain nombre de problèmes clés. Un PACS est un très gros investissement en capitaux. Il nécessite des ressources pour le matériel, le logiciel, et du personnel supplémentaire tel qu'un administrateur de PACS et tous les consultants qui peuvent être nécessaires. Dès le début des étapes de planification d'un PACS, il faudrait consulter les parties de tous les domaines qui seront affectés par les changements. Ceci doit inclure les administrateurs départementaux, les spécialistes de PACS, les physiciens médicaux, les radiologistes, les technologues, les médecins orienteurs, et tout personnel des technologies de l'information (TI) existant. Les renseignements obtenus pendant la consultation doivent servir à faire une analyse intensive des coûts/profits avant de prendre une décision. Le fait de consulter tôt toutes les parties concernées facilitera l'acceptation clinique du système. Quand on décide des spécifications d'un PACS, il faut tenir compte des composantes clés suivantes.
- Insister sur la conformité avec la norme DICOM (Digital Imaging and Communications in Medicine) et la conformation avec toutes les capacités nécessaires des équipements. Les capacités nécessaires dépendent des types d'équipements qui seront connectés avec le PACS. Ces capacités se traduiront éventuellement par une liste des classes de parité objet-service, SOP(de l'anglais Service-Object Pair) et des identificateurs uniques (UID) pouvant être pris en charge.
- S'assurer que tous les systèmes peuvent être intégrés, sur la base de la conception désirée de l'architecture du système. Ceci inclut les systèmes tels que le système d'information hospitalier (SIH), le système d'information radiologique (SIR), le PACS, les appareils d'acquisition d'image, les imprimantes, et tout système de compte rendu. Pour assurer la facilité d'intégration entre systèmes, les appareils devraient être adaptés au cadre technique de l'IHE (Integrating the Healthcare Enterprise). L'IHE est une initiative de promotion et de soutien de l'intégration des systèmes d'information dans l'entreprise de soins de santé, pour améliorer le déroulement du travail en facilitant la communication entre systèmes provenant de fournisseurs différents. Les données transmises d'un système à un autre supprimeront le besoin de les saisir à nouveau indépendamment dans chaque système et éviteront ainsi les incohérences, les redondances et l'indisponibilité des données (se reporter à la section 3.2.7). Lors de l'achat d'un équipement, il est important de spécifier la conformité désirée du IHE.
- La sécurité des renseignements de patient doit être une priorité. Seules les personnes autorisées doivent pouvoir accéder aux données et images des patients. Des mesures de sécurité doivent être établies pour contrôler l'accès aux renseignements sur les patients, ainsi que pour suivre toutes les activités qui sont exécutées sur ces données. Ceci inclut de surveiller qui accède aux renseignements, quand il y a accès à ces renseignements, et quelles modifications y sont apportées. Les utilisateurs autorisés du système doivent comprendre l'importance de garder les mots de passe du système confidentiels.
- Des caractéristiques automatisées devraient être incluses dans la conception du système pour faciliter un déroulement de travail plus rapide. Par exemple, une caractéristique devrait être disponible qui puisse faire une pré-recherche des études antérieures de l'individu pour permettre la comparaison avec l'étude en cours d'interprétation, lorsque les études applicables existent.
- Le système devrait s'adapter à l'utilisateur. Par exemple, l'interface graphique du logiciel client du PACS devrait s'adapter aux préférences de l'individu qui se connecte au système.
- Il faut avoir en place un système de détection et de correction rapide et efficace d`erreurs. Celui-ci devrait régulièrement, réconcilier la liste d`études prévues pour une période de travail aux études effectuées pour certaines modalités ainsi qu`aux études interprétées par les radiologistes. Cela permettra de détecter toute anomalie, réduire les cas perdus et s'assurer que les données mal classées sont rapidement identifiables.
- Tolérance des anomalies. En travaillant avec un système numérique d'imagerie et de compte rendu, il faut veiller à assurer la disponibilité du système. Les données critiques de patient doivent être disponibles chaque fois que nécessaire. Ceci est particulièrement important dans les salles d'opération et les services des urgences. Quand on achète du matériel pour des systèmes d'imagerie ou d'information, les fournisseurs doivent garantir le niveau de disponibilité de leur système. Les temps d'indisponibilité de système, pour mises à niveau et entretien, doivent être bien planifiés, de façon à ne pas interférer avec le déroulement du travail de l'établissement. En fonction du type d'établissement et de la charge de travail, les acheteurs peuvent exiger que le fournisseur garantisse une disponibilité jusqu'à 99,999%. Il faut déterminer s'il est acceptable que tout le système ou des parties du système soient indisponibles à un moment quelconque et pour combien de temps. Les pénalités et conditions pour non tenues des garanties de temps de disponibilité et d'indisponibilité doivent être clairement établis et approuvés avec le vendeur.
- Reprise après sinistre. L'établissement devrait établir un plan de reprise après sinistre en cas de panne de composant ou d'événements catastrophiques. Ce plan devrait comporter des directives et procédures documentées identifiant les responsabilités des personnes principales et les renforts et une description des actions nécessaires pour rétablir les fonctionnements. Une composante critique de reprise après sinistre est la sauvegarde et la tenue à jour des données à un emplacement hors site.
- Pour qu'un PACS fonctionne, il doit reposer sur, et être conçu pour refléter, un déroulement de travail éprouvé efficace. Les acheteurs devraient s'assurer que le fournisseur comprend le déroulement de travail de l'établissement, et fournit un système qui ne perturbe pas ce déroulement de travail. Les systèmes PACS qui reposent sur des déroulements de travail imparfaits répercuteront tous les problèmes existants.
- Quand on décide des exigences de réseau et de stockage d'un système d'imagerie ou d'information, il est important de ne pas limiter les systèmes aux seuls besoins actuels de l'établissement. Le système doit être évolutif pour permettre sa croissance future. La capacité du système devrait être basée sur les points suivants:
- les modalités actuelles à partir desquelles sont acquises les études;
- le nombre moyen d'images par étude par modalité;
- le nombre de films numérisés à être stockés;
- le nombre de pixels et la profondeur de bits de l'image;
- la croissance moyenne de procédures en fonction de taille des données;
- le nombre d'études effectuées chaque année;
- le volume de la croissance de procédures projetée;
- les modalités à ajouter à l'avenir;
- la possibilité de compression des données par modalité ou le PACS; et
- les autres sites/établissements à ajouter au système dans l'avenir.
3.2.7 Profils d'intégration de l'IHE (Integrating the Healthcare Enterprise)
L'IHE (Integrating the Healthcare Enterprise ou L'intégration de l'entreprise de soins de santé) est une initiative visant à rendre disponible aux prestataires toute l'information nécessaire à propos d'un patient afin de s'assurer que les meilleurs soins possibles lui sont donnés. L'IHE définit les profils d'intégration qui utilisent les normes des technologies de l'information (TI) établies pour intégrer les systèmes de différents vendeurs afin de favoriser une interopérabilité et une charge de travail efficaces dans les activités quotidiennes des professionnels de la santé (utilisateurs) en utilisant de l'information tirée des dossiers des patients. (IHE 2007)
Le profil d'intégration IHE décrit avec précision comment résoudre des problèmes cliniques réels grâce à des composants de système fonctionnels, appelés acteurs IHE. Ceux-ci sont basés sur des normes, comme la norme DICOM (Digital Imaging and Communication in Medicine) et la norme Health Level 7 (HL7). Les profils d'intégration ne créent pas de normes; ils définissent plutôt de manière précise comment les «acteurs», ou dispositifs, utilisent les normes établies de manière non ambiguë pour être interopérables et travailler ensemble (par ex. comment acquérir et afficher des images de mammographie numérique).
Profil d'image mammographique IHE (MAMMO)
Le Profil d'image mammographique IHE, lorsqu'il est appliqué à la modalité de mammographie numérique, permet de s'assurer que les images mammographiques numériques acquises contiennent toute l'information pertinente qui est nécessaire pour traiter les images, appliquer la détection assistée par ordinateur (DAO), stocker, examiner et imprimer les images. Ce profil est absolument nécessaire pour générer un contenu d'images mammographiques numériques correct afin d'assurer la présentation optimale des images dans un poste de travail d'interprétation en mammographie.
Les installations de mammographie devraient demander que le profil MAMMO soit supporté par la modalité de mammographie numérique. Cela donnera les avantages suivants a l'entreprise de soins de santé:
- Réduire les erreurs et amélioration de soins à donner au patient
- S'assurer que l'information sur le patient et l'information technique adéquate et cohérente est générée;
- Veiller à ce que les images acquises contiennent les données nécessaires pour identifier le patient et la technologie, et à ce que le traitement de l'image et l'examen soient corrects et significatifs, notamment en :
- Mettant l'image à l'échelle, de sorte que les images d'un même patient, acquises sur différents détecteurs peuvent être affichés dans les mêmes dimensions ou imprimés en taille réelle;
- Stocker l'information, à la modalité, sur le contraste de manière à ce que les ajustements du contraste ne diminuent pas la qualité des images affichées;
- Fournir une définition claire des tissus mammaires et de l'air ambiant, de manière à ce que la zone foncée soit maintenue s'il y a des ajustements du contraste effectués durant l'interprétation des images, afin d'obtenir une visualisation optimale de la structure du sein.
Le Profil d'image mammographique IHE, lorsqu'il est appliqué à un poste de travail pour examen de diagnostic en mammographie, permet de s'assurer que les images mammographiques numériques acquises sont affichées correctement, et qu'elles concordent avec les images ayant un profil semblable. Ce profil est absolument nécessaire pour permettre d'afficher les images de mammographie numérique de différents vendeurs de manière semblable à celles qui avaient été réalisées antérieurement en mammographie analogique.
Le Profil d'image mammographique IHE assure l'orientation adéquate de l'image, et fournit une justification, un affichage des contrastes et les dimensions de l'image. Ce profil dicte les dispositions menant à un affichage exact des marques DAO et à l'affichage de toute l'information technique pertinente et de l'information sur l'identification des éléments sur une base d'image. Le profil exige également que les écrans soient étalonnés correctement pour examiner l'image de manière optimale.
Le Profil d'image mammographique IHE a besoin d'un poste de travail pour diagnostic en mammographie qui permet d'afficher les images mammographiques de plusieurs manières normalisées: ajustée à la fenêtre d'observation, dimensions réelles, dimensions identiques et voir les pixels réels. L'affichage ajusté au hublot d'observation a pour but d'afficher simultanément jusqu'à huit images sur la paire d'affichages, principalement à des fins de comparaison temporelle. L'écran dimensions réelles est utile pour les biopsies percutanées, la planification des chirurgies ou la comparaison en taille avec des films antérieurs. L'écran dimensions identiques permet une comparaison facile des images de mammographie numérique acquises à différentes résolutions ou utilisant des tailles de détecteur différentes. L'écran voir les pixels réels permet d'afficher toutes les données saisies ou la «résolution complète», qui est particulièrement utiles au moment d'évaluer les microcalcifications ou les masses subtiles.
Cependant, le Profil d'image mammographique IHE ne définit pas les protocoles en suspens ou la manière dont les images des différentes matrices devrait être dimensionnées les unes par rapport aux autres.
Les installations de mammographie devraient demander que le profil MAMMO par le poste de travail en mammographie de diagnostic soit supporté. Voici les avantages qui en découlent:
- Réduction des erreurs et amélioration de soins à donner au patient:
- Veiller à ce que les images soient orientées et marquées correctement afin d'éviter les erreurs d'interprétation qui sont dues à une manipulation inadéquate de l'image.
- S'assurer que la ligne de peau est détectée, de manière à ce que l'on puisse maintenir la noirceur en cas d'ajustement du contraste pour l'interprétation du mammogramme au poste de travail; sans cela, le fond deviendra plus clair à mesure que la largeur et le niveau de la fenêtre seront augmentés, ce qui risque de rendre l'interprétation difficile.
- Permettre une mise à l'échelle de l'image sur le poste de travail, de manière à ce que les images d'un même patient, prises à différents moments sur des détecteurs différents puissent être affichées dans les mêmes dimensions ou même en dimensions réelles. Les densités peuvent ainsi être évaluées et cela permet au radiologiste d'évaluer les changements de dimensions dans des lésions connues lors des comparaisons temporelles avec des images numériques antérieures ou même avec des mammogrammes sur film.
- Permettre l'affichage de tous les renseignements cliniques et techniques pertinents. Par exemple, les dates d'acquisition peuvent être utilisées pour afficher en ordre les études actuelles ou antérieures ou des facteurs techniques peuvent être utilisés pour identifier des problèmes liés à la qualité de l'image.
- Veiller à ce que la présentation de l'image originale et à ce que les ajustements du contraste subséquents soient réalisés à le poste de travail suite à l'acquisition. Sans cela, la présentation de l'image et les ajustements du contraste effectués au poste de travail peuvent détériorer la qualité de l'image affichée.
- Accroissement du taux de traitement
- Réduire le temps requis pour lire les images. Les images seront affichées sur l'écran orienté de manière à ce que la zone importante soit automatiquement affichée dans l'orientation et les dimensions correctes.
- Amélioration de la qualité de l'image
- Améliorer la qualité et l'impression des images en incluant les données pertinentes dans les images.
- Veiller à ce que tous les paramètres d'acquisition techniques soient disponibles pour examen.
- S'assurer que les images peuvent être orientées, justifiées et dimensionnées correctement pour une interprétation adéquate et expéditive.
- S'assurer que les images de la présentation peuvent être utilisées de manière uniforme dans différentes poste de travail en mammographie.
- Utiliser automatiquement les réglages d'affichage du contraste créés à la modalité d'acquisition.
- Conserver la luminosité de l'arrière-plan tout en réglant le contraste des tissus du sein en maintenant la suppression du bruit de fond (air).
- Réduction des coûts opérationnels
- Éliminer considérablement les coûts d'impression, de gestion et de stockage des films, car le poste de travail affiche les images en permettant des examens des images d'écran, à la fois grâce au PACS de l'établissement ou en échangeant des CD d'imagerie avec les professionnels de la santé ou chirurgiens traitants.
Profil d'intégration Portable Data for Imaging (PDI) IHE (IHE 2009)
Le Profil Portable Data for Imaging (PDI) IHE permet de créer des CD d'images DICOM à la modalité. En demandant que le profil PDI soit supporté par la modalité de mammographie numérique, votre organisation bénéficiera des avantages suivants :
- Réduction des coûts opérationnels
- Diminue les films non nécessaires en chirurgie, pour l'orientation vers d'autres sites ou vers les médecins traitants--des images numériques sur CD, qui peuvent être transmises aux autres utilisateurs, sont créées par la modalité ou le poste de travail
- Accroissement du taux de traitement
- Épargne le temps de numérisation des films antérieurs provenant d'autres sites--le poste de travail prend en charge l'importation d'études sur CD. Le temps de lecture peut également être diminué de manière significative n'ayant pas à faire face à un environnement mixte où le film et les images numériques doivent être considérés ensemble.
- Améliore la disponibilité et l'accessibilité des données.
- Amélioration de l'accès à des données provenant d'autres sites--le poste de travail permet d'importer l'information des patients et de diagnostic d'un CD à partir d'autres institutions et cliniques.
- Améliore l'accès aux données par les autres fournisseurs de soins (par exemple, les médecins traitants, deuxième opinion radiologues) --le poste de travail exporte l'information du patient et du diagnostic sur CD.
3.2.8 Détection et diagnostic assistée par ordinateur (DAO)
La détection assistée par ordinateur (DAOe) et le diagnostic assisté par ordinateur (DAOx) sont des méthodes d'analyse d'images utilisées pour aider les radiologistes lors de l'interprétation des images mammographiques. Les techniques DAOe impliquent l'utilisation d'algorithmes informatiques pour localiser (en utilisant les signes distinctifs comme un triangle, un cercle, un carré ou autres) et identifier les régions suspectes d'une image avec l'objectif ultime d'améliorer la détection du cancer. Les techniques DAOx impliquent l'utilisation d'algorithmes informatiques pour indiquer la probabilité qu'une lésion connue soit maligne.
L'utilisation de la détection assistée et du diagnostic assisté par ordinateur doivent être gérés avec soin afin d'évaluer les effets sur la qualité globale du programme de mammographie. Différentes approches peuvent être utilisées pour évaluer les systèmes DAOe et DAOx (Bick 2010). Les déterminants de la qualité tels que la spécificité, la sensibilité, la valeur prédictive positive et la valeur globale devraient faire partie de l'évaluation des systèmes assistés pas ordinateur pour la détection et le diagnostic.
3.2.9 Télémammographie
La télémammographie est la transmission électronique d'images mammographique, d'un endroit à un autre, aux fins d'interprétation et/ou de consultation. Grâce à la télémammographie, on peut accéder aux images numériques et aux renseignements de patients depuis de multiples sites simultanément. Les avantages de la télémammographie comprennent une délivrance plus efficace des soins aux patients et la capacité à fournir des services radiologiques aux établissements de zones éloignées qui n'ont pas de radiologiste disponibles sur site. Comme la télémammographie implique l'acquisition et l'interprétation des images de patients à des sites différents, il est important que des directives et procédures soient en place à tous les endroits pour s'assurer que la qualité de l'image est optimisée et comparable entre tous les établissements ayant accès aux images de patients. Cela est particulièrement important quand des interprétations écrites authentifiées officielles sont effectuées au moyen de la télémammographie. Toutes les postes de travail utilisées pour l'interprétation des images de télémammographie doivent être incluses dans le programme d'assurance de la qualité de l'installation, afin de s'assurer que le rendement respecte les exigences minimales relatives aux postes de travail mammographique. Toutes les postes de travail télémammographique doivent avoir le même niveau de rendement et être soumises aux mêmes essais de contrôle de la qualité que ceux de l'installation où les images ont été acquises. Les essais de contrôle de la qualité pertinents de le poste de travail indiqués à la Section C doivent être réalisés aux fréquences requises. Les renseignements dans cette section sont basés sur les Normes de la CAR en matière de téléradiologie (CAR 2008).
1. Assurance de la qualité de la télémammographie - Quand il est utilisé pour fournir l'interprétation authentifiée officielle d'images, le lieu récepteur doit répondre aux exigences suivantes:
- L'ensemble de données d'image produit à l'aide de la technique de numérisation doit être transmis en totalité au système PAC/téléradiologie, ce qui signifie que la taille de la matrice et la profondeur de pixel doivent être conservées. La norme DICOM doit être respectée. Le logiciel de visualisation doit permettre à l'utilisateur d'effectuer un panoramique de toute l'image quand elle est affichée dans sa taille de matrice complète.
- Les images obtenues par post-traitement de l'image d'origine ne doivent pas être utilisées pour l'interprétation, à l'exclusion des images d'origine elles-mêmes. Elles ne doivent servir qu'à appuyer le processus d'interprétation.
- Seuls les mammogrammes obtenus au moyen d'appareils de mammographie numérique doivent être utilisés pour l'interprétation. Les mammogrammes obtenus par balayage d'images de film doivent être utilisés uniquement à des fins de comparaison. Il est recommandé que ces dispositifs aient une résolution spatiale minimale de 50 µm et une acquisition minimum de 12 bits échelle de gris.
2. Gestion de l'image - En télémammographie, il convient d'avoir recours à la gestion de l'image pour un rendement optimal. Tous les systèmes doivent inclure:
- Un mécanisme de vérification de l'intégrité, soit en logiciel soit à l'aide d'un procédé manuel, pour s'assurer que toutes les données transmises depuis le site d'origine sont reçues intactes par le site récepteur.
- Le stockage des images au site émetteur ou récepteur ainsi que la transmission doivent être arrangés de façon à ce que la confidentialité des patients soit maintenue et que le système soit sécurisé.
- Le prestataire doit s'assurer que la qualité de l'image est la même au site d'émission et au(x) site(s) de réception.
3. Transmission d'images et de données du patient - Les protocoles de communication et les formats de fichier doivent être conformes aux normes actuelles de réseau DICOM 3.0 et aux normes canadiennes IHE pour tous les nouveaux équipements d'acquisition. Le respect de ces normes doit être envisagée lorsque la mise à niveau des équipements existants. .
4. Capacités de visualisations - Les postes de visualisation utilisés pour la télémammographie doivent respecter les exigences de la section B3.2.4.
5. Base de données de patients - Pour les images transmises par télémammographie, il faut disposer d'une base de données, à la fois aux sites émetteur et récepteur, pour servir de base aux vérifications d'intégrité et aux audits à venir. Cette base de données doit comprendre:
- le nom du patient, le numéro d'identification et la date;
- le type d'examen;
- les types d'images;
- le nombre d'images;
- les sites d'acquisition et d'envoi des images (s'ils sont différents); et
- la date et l'heure de la transmission.
6. Sécurité - Les systèmes de télémammographie doivent fournir un réseau et/ou des protocoles logiciels protégeant la confidentialité de(s) enregistrement(s), image(s), interprétation(s) et autres donnés du patient, et assurer que le système est sécurisé et n'est utilisé que dans la mesure nécessaire par ceux autorisés par le patient, conformément à la législation provinciale ou territoriale et aux directives de l'Association médicale canadienne sur le caractère privé des renseignements.
7. Stockage des enregistrements - Les exigences légales de stockage et conservation des images et comptes rendus varieront de province à province et les prestataires du service de télérmammographie seront responsables du respect de ces exigences. Les images stockées à l'un ou l'autre site doivent répondre aux exigences juridictionnelles du site émetteur. Les images interprétées hors site ne nécessitent pas d'être stockées à l'établissement récepteur si elles sont stockées au site émetteur. Cependant, si les images sont conservées au site récepteur, il faut également respecter la période de conservation de cette juridiction. La politique de conservation des enregistrements doit être sous forme écrite.
8. Documentation - La communication est une composante essentielle de la télémammographie. Les médecins interprétant les examens de télémammographie doivent fournir des comptes rendus conformément aux Normes de communication de la CAR.
9. Contrôle de la qualité de la télémammographie - Les images au site récepteur doivent être aussi bonnes que les images au site d'envoi. Il est impératif qu'un radiologiste se trouve régulièrement au site émetteur pour s'assurer que l'équipement fonctionne correctement, et que les technologues sont supervisés et formés de façon adéquate. Les sites émetteurs et récepteurs doivent avoir des politiques et procédures documentées pour contrôler et évaluer la gestion efficace, la sécurité, le bon fonctionnement des équipements d'imagerie, de transmission, de réception et de visualisation.
10. Amélioration de la qualité - L'utilisation de la télémammographie ne réduit pas les responsabilités de gestion et de supervision de la médecine radiologique. Les procédures doivent être contrôlées et évaluées systématiquement dans le cadre du programme général d'amélioration de la qualité de l'établissement. Le contrôle doit comprendre l'évaluation de la précision des interprétations ainsi que de l'opportunité des examens. La fréquence des complications et des événements contraires doit être examinée pour identifier les occasions d'améliorer les soins aux patients.
L'utilisation de la télémammographie doit être documentée. Il faut effectuer des revues périodiques de la justesse, des problèmes et de la qualité des données transmises. Les données doivent être recueillies d'une manière conforme aux exigences statutaires et réglementaires.
3.2.10 Compression d'images numériques
Des quantités accrues de données sont générées par l'équipement de mammographie numérique. Les coûts relatifs au stockage et à la transmission de ces données ont soulevé beaucoup d'intérêt pour la compression des images numériques. Il existe deux types de compression: la compression réversible (ou «sans pertes») et la compression irréversible («avec pertes»). Les images de compression sans pertes peuvent être compressées et décompressées et il n'y a aucune modification des données d'image originales. L'utilisation de la compression de données avec pertes entraîne la modification des images décompressées et leur valeur en pixels peut être différente des valeurs originales.
En attendant que des études scientifiques fournissent des preuves à l'effet que la compression avec pertes ne compromette pas le diagnostic précis des patients, la compression avec pertes des images de mammographie numérique ne doit pas être utilisée.
4.0 Équipement d'essai
Il faut choisir avec soin l'équipement d'essai qui servira à assurer un bon rendement de l'équipement mammographique et des accessoires connexes et à assurer la radioprotection de l'installation.
- Le fonctionnement et le rendement de tout le matériel utilisé pour les essais d'acceptation et de contrôle de la qualité doivent être évalués régulièrement.
- Tout l'équipement sensitométrique et densitométrique, les dosimètres, les kVp-mètres et les photomètres doivent être étalonnés régulièrement, conformément aux recommandations des fabricants. Les instruments de mesure du kerma dans l'aire ou le débit de kerma dans l'aire doivent être étalonnés chaque deux ans et lorsqu'ils sont réparés. L'étalonnage de l'instrument doit être traçable à un étalon national et étalonné avec une précision de ± 6 pour cent (niveau de confiance 95 pour cent) dans la gamme d'énergie de mammographie. Pour les instruments qui ne sont pas certifiés par le fabricant pour les combinaisons cible-filtre W/Ag ou W/Rh, les corrections nécessaires doivent être obtenues lorsque les mesures des faisceaux de rayons x qui sont générés avec les combinaisons W/Ag ou W/Rh.
- Tous les fantômes et autres appareils utilisés pour l'évaluation de la qualité de l'image, de la dose et du rendement du système doivent être vérifiés contre tout dommage ou tout état qui pourrait affecter leur utilisation.
- Le matériel d'essai doit être stocké à l'abri de la chaleur, de la lumière directe du soleil et d'une humidité élevée, et doit être utilisé selon les recommandations des fabricants.
5.0 Enquêtes de radioprotection
Une enquête de radioprotection est une évaluation, conduite par l'autorité réglementaire, de la radioprotection d'un établissement de mammographie. L'enquête vise à s'assurer que l'installation de mammographie est conforme au présent code de sécurité, à démontrer que les appareils de mammographie et auxiliaires fonctionnent correctement et conformément aux normes applicables, et que ces appareils sont installés et utilisés d'une façon qui offre aux opérateurs, aux patients, et aux autres, la radioprotection maximale. Il convient de noter que les enquêtes décrit dans cette section servent essentiellement à l'instruction et à l'orientation des personnes employées dans les ministères et organismes fédéraux de la fonction publique, de même que celles qui sont régies par le Code canadien du travail. Les établissements de juridiction provinciale ou territoriale peuvent être soumis à des exigences spécifiques associées à leurs statuts. On doit communiquer avec les organismes énumérés à l'annexe I pour obtenir des détails sur les exigences des lois de chaque province et territoire.
Lors de l'enquête, l'autorité réglementaire peut demander les rapports de contrôle de la qualité réalisé par le physicien ou les technologues, les mesures de sécurité telles que le matériel de protection et le blindage sont aussi examinées pour s'assurer qu'ils sont présents et apportent la protection nécessaire. Il est donc important que les établissements radiologiques soient enquêtés à intervalles réguliers.
5.1 Procédures générales
Le fonctionnement habituel de toute nouvelle installation ou d'une installation qui a subi des modifications devrait être retardé jusqu'à ce qu'un expert ait fait une enquête complète. L'expert est une personne qui est qualifiée par éducation et expérience pour exécuter des procédures avancées ou complexes de radioprotection qui sont généralement au-delà des capacités de la plupart des personnes au sein de l'établissement. Ces procédures incluent l'évaluation de la conception de l'installation pour s'assurer qu'un blindage adéquat est en place, le contrôle et l'évaluation du rendement des appareils et accessoires radiographiques, et l'évaluation et la recommandation des programmes de radioprotection. Le propriétaire de l'établissement (ou un autre membre du personnel délégué, comme l'agent de radioprotection) doit contacter les autorités réglementaires appropriées pour vérifier les procédures d'essais relatives à l'inspection et à l'acceptation qui ont cours dans cette juridiction. Certaines juridictions peuvent exiger que l'établissement soit déclaré conforme à la réglementation gouvernementale applicable, avant les opérations.
Pour une nouvelle installation, il est particulièrement avantageux de faire des contrôles visuels pendant la construction, pour s'assurer de la conformité aux spécifications et déceler des possibles défectuosités des matériaux ou de la construction, car il est possible de remédier aux défauts de façon plus économiquement à ce stade que par la suite.
Pour les installations en service, il faut effectuer une enquête après tout changement apporté qui pourrait entraîner un danger de rayonnement. Ceci inclut l'altération des écrans de protection, la modification et le remplacement de l'appareil, les changements de modes opératoires, ou une augmentation de la charges de travail.
Finalement, les enquêtes de radioprotection doivent être effectuées à intervalles réguliers programmés pendant les opérations de routine pour détecter les problèmes dus à une panne de l'appareil ou toute tendance à long terme de diminution du niveau de sécurité de rayonnement. Les établissements doivent contacter l'autorité réglementaire compétente pour établir le calendrier des enquêtes.
Les résultats de ces enquêtes, y compris les conclusions tirées par l'autorité réglementaire, doivent être soumis au propriétaire ou à l'utilisateur responsable dans un rapport écrit. Le rapport devrait être déposé dans les 30 jours suivant l'enquête. Tous ces rapports doivent être conservés par le propriétaire ou l'utilisateur responsable. Dans le cas des établissements fédéraux, les rapports doivent être conservés pendant cinq (5) ans et des registres de dosimétrie pour le personnel doivent être conservés pendant toute la durée de vie de l'établissement.
5.2 Rapport d'enquête
Le rapport d'enquête doit présenter, de façon systématique claire, les détails et les résultats des mesures effectuées, ainsi que les conclusions tirées et les recommandations faites par l'enquêteur. Toutes constatations inhabituelles de l'appareil lui-même, l'établissement ou les modes opératoires, qui pourraient affecter la sécurité des opérateurs ou d'autres personnes à proximité de l'établissement radiologique, doivent être clairement identifiées.
Le rapport d'enquête doit comprendre ce qui suit:
- un croquis de l'établissement, montrant l'emplacement de l'appareil radiologique et du panneau de contrôle dans l'établissement, ainsi que la nature et l'occupation des zones contiguës à l'établissement;
- l'identification de l'appareil de mammographie (c.-à-d., le nom du fabricant, la désignation du modèle et le numéro de série de la génératrice, la commande, le tube radiogène équipé, etc., selon le cas) et la date, ou au moins la date approximative, de fabrication;
- l'observation des conditions opérationnelles (à la fois électriques et mécaniques) de l'appareil de mammographie au moment de l'enquête;
- la charge de travail totale, réelle ou estimée, de l'établissement, ainsi que la charge de travail répartie en différents sens du faisceau de rayons X et procédures utilisées, etc.;
- les résultats des mesures de rayonnement effectuées à l'intérieur et à l'extérieur de la zone contrôlée dans des conditions d'exploitation «typiques»;
- les endroits où les mesures ont été faites;
- une évaluation du rendement des appareils radiographiques et du rendement de l'imagerie et des diagnostics (ceci peut comprendre l'exécution des essais de contrôle de la qualité applicables, des sections C3.1 à C3.6);
- un résumé des paramètres de charge utilisés et une mesure de la filtration totale du faisceau de rayons X;
- une évaluation des techniques radiologiques du point de vue de la sécurité de rayonnement et une évaluation de la dose glandulaire moyenne. Il faut attirer l'attention sur toute pratique qui est ou pourrait être préjudiciable au patient ou au personnel travaillant dans l'établissement. Dans ces cas-là, il faut faire des recommandations de techniques améliorées ou plus sûres;
- un examen du programme d'assurance de la qualité de l'installation, pour s'assurer qu'il existe et est tenu à jour, y compris les enregistrements d'essais de contrôle de la qualité; et
- les recommandations concernant le besoin d'une enquête de suivi.
6.0 Élimination des appareils radiographiques
Quand un appareil radiographique est considéré pour le rebut, une évaluation devrait être effectuer pour savoir s'il peut être remis à neuf et/ou recyclé. Il est recommandé de communiquer avec le fabricant ou le fournisseur des appareils ou des composants des appareils pour voir si ceux-ci peuvent être recyclés ou leurs êtres retournés. Il faut aussi prendre en considération que, si l'appareil contient des données de patients, ces données doivent être éliminées adéquatement.Une fois prise la décision de mettre au rebut un appareil radiographique, il faut effectuer une évaluation pour déterminer si tout composant de l'appareil contient des matières dangereuses. Pour s'assurer que l'appareil n'est pas manœuvré dangereusement après élimination, il faudrait le rendre inexploitable avant de le rebuter. Il faudrait débrancher et enlever les câbles qui alimentent l'appareil et les autres raccordements électriques. Il est recommandé aux établissements radiologiques sous juridiction provinciale ou territoriale de contacter l'organisme responsable dans leur province ou territoire respectif pour de plus amples renseignements. Une liste de ces organismes responsables est fournie à l'annexe I.
Section C: Programme d'assurance de la qualité
1.0 Introduction
Tous les établissements de mammographie doivent développer et tenir un programme d'assurance de la qualité. L'assurance de la qualité en mammographie se définit comme les actions planifiées et organisées nécessaires destinées à donner la certitude suffisante que les appareils de mammographie et leurs composants associés produisent de façon fiable des mammogammes de qualité satisfaisante avec des doses minimales aux patients et aux membres du personnel. Un programme d'assurance de la qualité comprend des procédures de contrôle de la qualité pour la surveillance et l'évaluation des appareils de mammographie et leurs composants associés, ainsi que les méthodologies administratives permettant de s'assurer que les actions de surveillance, d'évaluation et de correction sont effectuées adéquatement. Le propriétaire d'un établissement radiologique est responsable de l'établissement d'un programme d'assurance de la qualité qui examine toutes les pratiques de l'établissement ayant des conséquences sur les aspects suivants:
- Qualité de l'information - s'assurer que toute l'information diagnostique produite permet une évaluation clinique précise;
- Efficacité clinique - s'assurer que toutes les étapes qui aboutissent à un diagnostic et une intervention précis sont effectuées et que l'information est disponible en temps opportun aux médecins du patient ou à ses principaux professionnels médicaux; et
- Dose - s'assurer que l'examen radiologique est effectué avec la dose de rayonnement la plus faible possible au patient, le personnel et les autres, en tenant compte des exigences d'imagerie clinique.
1.1 Buts du programme d'assurance de la qualité
Le but ultime d'un programme d'assurance de la qualité est de garantir un diagnostic et un traitement opportuns avec une dose minimale au patient et aux membres du personnel. Afin d'avoir un programme d'assurance de la qualité réussi, il est essentiel que les appareils soient en bon état de fonctionnement et que tous les membres du personnel comprennent les buts du programme et soient engagés à sa mise en oeuvre par leur entière participation.
L'information fournie par les appareils de mammographie doit être de la plus haute qualité afin de garantir un diagnostic et un traitement précis. S'il manque des éléments essentiels ou si des artefacts ont été ajoutés aux images, l'image est considérée comme étant de mauvaise qualité. L'information sur un mammogramme de mauvaise qualité a pour conséquence un diagnostic incorrect qui entraîne une répétition des procédures mammographique, des doses aux patients inutiles, un traitement en retard ou inadéquat et des coûts accrus.
1.2 Coûts et avantages d'un programme d'assurance de la qualité
La mise en application initiale et le fonctionnement général d'un programme d'assurance de la qualité comporteront des coûts à la fois en termes d'argent et de temps du personnel. Cependant, les économies réalisées du fait du fonctionnement du programme compenseront certains des coûts. On peut observer une réduction des coûts de fonctionnement globaux pour certaines installations.
1.2.1 Coûts d'un programme d'assurance de la qualité
Voici certains des coûts liés au programme d'assurance de la qualité:
- Personnel - Le personnel devra effectuer de nouvelles tâches, dont la production de test d'images pour les appareils de radiographie et l'archivage;
- Équipement d'essai - Il est nécessaire d'avoir l'équipement d'essai pour procéder à des essais de contrôle de la qualité, comme les fantômes. Le coût d'un tel équipement est toutefois peu élevé par rapport au coût de l'équipement d'imagerie et il peut être utilisé pour plusieurs systèmes de mammographie. Il ne serait pas nécessaire d'acquérir tous les équipements d'essai si l'installation décide de confier certains essais de contrôle de la qualité à une organisation indépendante ou à un particulier, qui serait alors responsable de fournir son propre équipement d'essai;
- Images d'essai - En ce qui a trait aux systèmes utilisant des films, et aux systèmes CR et de DR utilisant des imprimantes laser, une installation peut avoir à utiliser de deux à cinq pour cent des films pour effectuer une sensitométrie, une imagerie du fantôme, des essais de l'équipement et en imagerie; et
- Organisations indépendantes - Si les installations n'ont pas la capacité d'effectuer à l'interne tous les essais de contrôle de la qualité, elles peuvent choisir d'avoir recours aux services d'une organisation indépendante ou d'un particulier pour effectuer certains de ces essais et les évaluations de l'équipement. En outre, l'établissement peut retenir les services d'un physicien médical qui jouera le rôle de conseiller pendant la mise en oeuvre initiale du programme et de consultant pendant l'exécution de celui-ci.
1.2.2 Avantage d'un programme d'assurance de la qualité
Outre l'amélioration de la qualité diagnostique, voici certaines des économies liées au programme d'assurance de la qualité
- Films et solution de développement - En ce qui a trait aux systèmes utilisant des films et aux systèmes CR et DR utilisant des imprimantes laser, une diminution du nombre de reprises peut amener une réduction du nombre de films et de solution de développement utilisés.
- Appareil - La diminution du nombre de reprises réduira les charges de travail ce qui, en retour, exigera moins de l'appareil mammographique et des processeurs d'images. Les problèmes avec l'équipement peuvent être diagnostiqués plus tôt, avant même que des problèmes plus graves et plus coûteux se produisent, réduisant ainsi le temps d'arrêt et les coûts de service de l'équipement.
- Flux de patients - La réduction du nombre de reprises ainsi qu'une meilleure qualité de l'image permettront une utilisation efficace du temps pour les opérateurs de l'équipement radiologique. Ainsi l'horaire des rendez-vous sera mieux respecté et le débit de patient pourrait augmenter.
1.3 Mise en œuvre du programme d'assurance de la qualité
La mise en œuvre du programme d'assurance de la qualité n'a pas à être compliquée. Il suffit d'établir des procédures de contrôle de la qualité pour l'équipement ainsi qu'une méthodologie administrative pour en assurer la surveillance, l'évaluation et pour garantir que les mesures correctives soient prises adéquatement.
1.3.1 Élaboration de politiques et de directives
Une étape utile consiste à élaborer une série de politiques et de directives portant sur divers thèmes. La liste qui suit présente certaines de ces politiques et directives. Chaque installation peut avoir besoin de différents ensembles de politiques et de directives selon le genre de travail accompli et la structure organisationnelle de l'installation. Ces politiques devraient être établies par la direction avec la collaboration du personnel. Il est recommandé que toutes les politiques, procédures et que tous les processus soient révisés par un comité mixte sur la santé et la sécurité. Les politiques devraient être regroupées dans un manuel sur l'assurance de la qualité. Les renseignements qui suivent devraient être faciles à obtenir pour le personnel en radiologie :
- Personnel en radiologie
- Une liste des membres du personnel et un aperçu de leurs tâches, de leurs niveaux d'autorité et de leurs responsabilités.
- Politiques concernant la réduction de l'exposition du personnel et des opérateurs d'appareils radiographiques
- Politique concernant la réduction de l'exposition des travailleuses enceintes.
- Politique concernant l'immobilisation des patients.
- Politique concernant la présence de personnes dans les salles de radiologie au cours des procédures.
- Politique concernant le programme de formation ou d'orientation destiné aux opérateurs d'appareils radiographiques.
- Politique concernant la bonne utilisation de l'appareil de radiographie.
- Politique concernant la surveillance des doses de rayonnement pour le personnel.
- Politique concernant l'utilisation de dispositifs et d'équipement de protection contre les rayonnements.
- Politique concernant l'entretien et l'essai des dispositifs de protection contre les rayonnements.
- Politiques concernant la réduction de l'exposition des patients à la radioexposition
- Politique concernant la pratique d'examens radiologiques.
- Politique concernant l'examen radiologique de patientes enceintes.
- Politique concernant l'utilisation de dispositifs et d'équipement de protection contre les rayonnements.
- Politique concernant le positionnement du patient (manuel sur le positionnement).
- Politique concernant les paramètres de charge (fiches techniques).
- Politique concernant la qualité acceptable des images radiographiques.
- Politique concernant une analyse de rejet des images radiographiques.
- Directives pour les essais de contrôle de la qualité de l'équipement
- Lignes directrices énumérant les appareils radiographiques et les composantes du système qui doivent subir les essais.
- Lignes directrices pour tous les paramètres d'appareil à mesurer et la fréquence de suivi (calendrier) pour chaque système radiographique et composante de système.
- Lignes directrices pour les normes de performance de chaque équipement testé et limites de tolérance pour le rendement attendu de chaque essai de contrôle de la qualité.
- Lignes directrices pour la mesure de chaque paramètre et enregistrement des données.
- Lignes directrices pour l'évaluation des données de l'essai et pour prendre les mesures correctives pour maintenir le rendement optimal de l'appareil.
- Lignes directrices pour les mesures de dose du patient (dose glandulaire moyenne).
- Directives pour l'étalonnage et l'entretien des équipements de mesure de rayonnement et autres équipements d'essai.
- Politiques pour l'achat d'un nouvel équipement d'imagerie radiologique
- Politique pour une analyse des besoins.
- Politique pour la rédaction des spécifications de l'équipement.
- Politique pour l'essai d'acceptation de l'équipement.
- Politique pour l'évaluation et le remplacement de l'équipement.
- Politique concernant la conservation des registres
- Politique pour la révision du programme de l'assurance de la qualité.
- Politique pour la révision des procédures de contrôle de la qualité.
- Politique pour la conservation des documents (renseignements sur le patient, résultats de l'essai de contrôle de la qualité, rapports d'expertise, rapports de dosimétrie personnelle).
- Politique pour graver des images sur CD (IHE 2009 Radiology Technical Framework Supplement Extensions to the Portable Data for Imaging (PDI) Integration Profile
1.3.2 Mise en place de procédures de contrôle de la qualité
Les quatre étapes qui suivent doivent être comprises dans la mise en place de procédures de contrôle de la qualité
- Fonctionnement de l'équipement - Il est primordial que l'appareil mammographique, l'appareil de traitement des images et les dispositifs d'affichage électronique fonctionnent adéquatement avant de lancer un programme d'assurance de la qualité. Les fabricants et les fournisseurs devraient fournir les caractéristiques de fonctionnement de leur équipement. Les systèmes utilisant des films, les films et le traitement doivent répondre aux exigences du fabricant quant à la vitesse et au contraste. Pour les systèmes CR, le système d'imagerie doit être étalonné comme il se doit, selon les systèmes radiographiques. Ceci peut comprendre le remplacement, la réparation, la mise à niveau ou l'étalonnage de l'équipement.
- Rendement de référence - Les valeurs du rendement de références des appareils mammographique et du système de traitement d'images doivent être établies après s'être assuré que les appareils fonctionnent adéquatement. Les images utilisées pour déterminer les valeurs du rendement de références doivent être obtenues en utilisant une méthode de routine pour un tissu mammaire compressé (ex. pour une épaisseur de 4.2 cm à 4.5 cm). Le rendement de référence sera utilisé pour diagnostiquer tout changement dans le rendement des appareils. Il est important de conserver des registres des données de fonctionnement des appareils et des mesures du rendement de référence. Ces registres seront requis pour diagnostiquer tout changement dans la qualité de l'image. Les valeurs du rendement de références doivent être déterminées lorsque le nouvel équipement est installé, lorsqu'il y a des changements dans les composantes qui ont une incidence sur la qualité de l'image et sur la dose au patient et également lorsque l'équipement d'essai est changé.
- Image de référence - Pour évaluer la qualité de l'image, une image de référence est nécessaire. Cette image de référence est crée en utilisant l'appareil mammographique, le système de traitement de l'image, le fantôme de contrôle de la qualité et sera utilisée pour comparer les images d'essai de contrôle de la qualité.
- Évaluation des résultats et niveaux d'intervention - Un programme de vérification aux fins du contrôle de la qualité comprend non seulement un calendrier de vérification de la qualité régulier, l'enregistrement et l'archivage des données, mais également l'évaluation du résultat de la vérification, dont la détermination de limites acceptables et non acceptables du fonctionnement de l'équipement doublé d'une liste des mesures correctives qui pourraient être nécessaires. Un ensemble de limites devrait être établi indiquant le niveau de fonctionnement hors duquel le système ou la fonction devrait être étroitement surveillé, mais où aucune intervention immédiate n'est requise. Un autre ensemble de limites devrait également être établi lorsqu'une mesure corrective immédiate doit être prise.
1.3.3 Mise en place de procédures administratives
Les procédures administratives qui suivent doivent être comprises dans la mise en place d'un programme d'assurance de la qualité efficace.
- Responsabilité - Bien que le propriétaire de l'installation soit en dernier ressort responsable de la mise en place et du déroulement du programme d'assurance de la qualité, pour obtenir le niveau optimal de radioprotection et l'information sur la qualité du diagnostic, il est essentiel qu'une bonne collaboration existe entre toutes les parties concernées. Le personnel peut être affecté à des tâches relatives à la surveillance de l'appareil, à la tenue des registres et aux activités connexes au programme d'assurance de la qualité. Il est essentiel que le niveau de responsabilités et de participation du propriétaire et du personnel soit clairement déterminé, communiqué à tous et bien compris.
- Registres - Il est essentiel que les mesures et les renseignements recueillis dans le cadre du programme d'assurance de la qualité soient clairement documentés et que l'on puisse les consulter en vue d'une évaluation.
- Le rapport de physicien médical doit circuler parmi le personnel en mammographie et être conservé sur les lieux.
- Dans la mesure du possible, les données enregistrées doivent être indiquées comme des points de contrôle sur un graphique de contrôle chaque jour où la mesure est prise. Il est alors plus facile de tirer des conclusions. Un registre ou une autre méthode d'enregistrement facilement identifiable doit être utilisé et conservé pendant au moins 3 ans.
- Les graphiques de contrôle de qualité du développement doivent être conservés pendant un an.
- Les films de sensitométrie des derniers 30 jours de traitement des films mammographique doivent être conservés.
- Un échantillon mensuel d'image du fantôme de contrôle de qualité (sur film ou image numérique) doit être conservé pendant au moins 3 ans.
Certaines provinces ou certains territoires peuvent prescrire des exigences sur la conservation des registres différents. Une consultation avec l'organisme approprié de l'Annexe I peut être nécessaire pour déterminer les exigences en vigueur dans cette juridiction.
- Évaluation des données - Les données enregistrées doivent être évaluées immédiatement et les mesures nécessaires prises.
- Limites d'acceptabilité des données - Les limites supérieures et inférieures d'acceptabilité des données enregistrées doivent être déterminées et documentées. Lorsque ces limites sont atteintes, des mesures correctives doivent être prises. Par example, il peut s'agir de l'échelle des températures acceptables pour l'équipement à développer des films. Ces limites doivent être établies de façon à ce qu'elles soient dans l'échelle permise avant que des modifications importantes lors d'examens diagnostiques soient évidentes. Elles ne devraient pas être trop restrictives pour surpasser la capacité de l'appareil ou que des mesures correctives fréquentes soient prises sans qu'il y ait évidence de problèmes. Ces limites devraient être révisées à l'occasion, particulièrement lorsque des composantes majeures du système radiographique sont remplacées ou réparées.
- Fréquence des essais - La fréquence des essais doit faire en sorte qu'il y ait un équilibre entre le coût des essais, l'interruption du fonctionnement des installations et les activités d'entretien pour maintenir la qualité. La fréquence des essais devrait être accrue si l'équipement montre des changements importants entre les essais de contrôle de la qualité prévus, ou si l'équipement est utilisé pour un volume exceptionnellement élevé de procédures. Des essais additionnels devraient être effectués si les résultats se situent à l'extérieur des limites d'acceptabilité pour les essais, ou après l'application de toute mesure correctrice. L'équipement doit être remis à l'essai après l'entretien de toute pièce pouvant affecter la densité ou la qualité de l'image, ou le débit de rayonnement du tube radiogène. Le programme de contrôle de la qualité ne doit pas être interrompu si les résultats indiquent un rendement de l'équipement relativement stable. Le but du programme de contrôle de la qualité est de contrôler la qualité et il est essentiel d'effectuer des mesures périodiques du rendement de l'équipement. Quand il existe des différences entre les recommandations du fabricant concernant des essais et les exigences des lois et politiques applicables, il est recommandé de suivre les exigences les plus strictes pour assurer la conformité avec les lois/politiques et pour conserver certaines garanties du fabricant sur les équipements.
- Mesures correctives - Des procédures de réparation et d'étalonnage ayant pour but de traiter les problèmes importants doivent être établies. Un arbre de décision doit être élaboré pour donner une orientation lorsqu'il s'agit de traiter des événements comme les défaillances de l'équipement, et pour gérer les situations où le rendement de l'équipement s'écarte des limites établies. Une liste des individus ayant l'autorité d'arrêter le fonctionnement de l'équipement radiologique doit être établie. L'arbre de décision doit comprendre les étapes suivantes:
- Répéter l'essai pour confirmer ;
- ce qu'il faut faire si le nouvel essai confirme une défaillance du rendement;
- ce qu'il faut faire si l'essai connaît une légère défaillance;
- ce qu'il faut faire si l'essai indique un historique de défaillances; et
- ce qu'il faut faire si l'essai connaît une défaillance importante.
1.4 L'agrément en mammographie
L'agrément est un processus officiel selon lequel une installation de mammographie peut démontrer que la qualité de la mammographie réalisée dans l'établissement respecte les normes reconnues. L'agrément exige à la fois une auto-évaluation par l'installation de mammographie et une évaluation externe par un organisme de contrôle. Elle comprend une évaluation des compétences du personnel, des politiques et des procédures, de la conception et du rendement de l'équipement, et du programme d'assurance de la qualité incluant des essais de contrôle de la qualité. Grâce à l'agrément, l'installation de mammographie peut fournir une assurance additionnelle à l'effet qu'elle s'engage à offrir des soins de qualité.
Au Canada, des programmes d'agrément en mammographie ont été établis par les organismes provinciaux, et au niveau national, par l'Association canadienne des radiologistes. Il est fortement recommandé que toutes les installations de mammographie canadiennes soient accréditées par une norme reconnue, comme celle de l'Association canadienne des radiologistes ou l'équivalent.
2.0 Essais d'acceptation
L'essai d'acceptation est un processus de vérification de la conformité aux spécifications de performance des appareils radiographiques tel que stipulé dans le contrat d'achat; il permet de vérifier également si le rendement des appareils est conforme aux règlements fédéraux, provinciaux ou territoriaux. Les essais d'acceptation doivent être effectués avant toute utilisation clinique de l'appareil L'essai d'acceptation doit être effectué par, ou sous la supervision directe d'un physicien médical, ou par d'autres personnes qui ont une connaissance approfondie de cet équipement particulier de rayons X et des règlements pertinents avant l'utilisation clinique de l'équipement. Le propriétaire doit s'assurer que l'essai d'acceptation soit effectué par une personne ou une organisation qui n'a pas de lien avec le fabricant.
Les essais d'acceptation d'un système médical à rayons X comportent plusieurs étapes importantes. Ce sont:
- la vérification que les composants ou les systèmes livrés correspondent à ce qui a été commandé;
- la vérification de l'intégrité et de la stabilité de la mécanique du système, notamment des mécanismes de sécurité, la libération automatique du patient, les commandes motrices, le verrouillage des commandes;
- la vérification que les inspections appropriées des installations électriques ont été effectuées, y compris la sécurité de l'électricité et la fluctuation de la ligne de transport électrique;
- la vérification du rendement des rayons X; et
- la vérification des performances de l'imagerie ou du diagnostic.
Les résultats de l'essai d'acceptation devraient servir de valeurs de base et de limites d'acceptation pour le rendement opérationnel des appareils radiographiques. Ces valeurs et ces limites de référence sont essentielles au programme d'assurance de la qualité.
2.1 Évaluation de l'essai d'acceptation
Les essais d'acceptation pour les appareils de mammographie devraient permettre d'évaluer au moins les points figurant au tableau B2.1. Tout l'équipement ne sera pas nécessairement soumis à tous les essais. Le type d'équipement et sa configuration détermineront les séries d'essais à effectuer.
| Éléments sous évaluation pour l'essai d'acceptation | FS | CR | DR |
|---|---|---|---|
| 1.0. Identification | |||
| 1.1 Première inspection et inventaire | X | X | X |
| 1.2 Inspection de la documentation | X | X | X |
| 2.0. Essais visuels et fonctionnels | |||
| 2.1 Propriétés mécaniques | X | X | X |
| 2.2 Systèmes de sécurité | X | X | X |
| 3.0. Évaluation du rendement - Générateur de rayons X et contrôle | |||
| 3.1 Dimensions du foyer | X | X | X |
| 3.2 Distance source-image | X | X | X |
| 3.3 Tension du tube radiogène | X | X | X |
| 3.4 Produit courant-temps | X | X | X |
| 3.5 Temps de charge | X | X | X |
| 3.6 Limitation et indication du faisceau | X | X | X |
| 3.7 Filtration du faisceau radiogène | X | X | X |
| 3.8 Commande automatique d'exposition | X | X | X |
| 3.9 Puissance du rayonnement | X | X | X |
| 3.10 Fuite de rayonnement | X | X | X |
| 4.0. Évaluation du rendement - Compression | |||
| 4.1 Force de compression | X | X | X |
| 4.2 Précision de l'indicateur de la force de compression | X | X | X |
| 5.0. Évaluation du rendement - Acquisition d'images | |||
| 5.1 Fonction de réponse | X | X | |
| 5.2 Bruit | X | X | |
| 5.3 Tissu manquant du côté de la paroi thoracique | X | X | |
| 5.4 Uniformité | X | X | |
| 5.5 Élément du détecteur défectueux | X | ||
| 5.6 Éléments défectueux du détecteur non corrigés | X | ||
| 5.7 Sensibilité interplaques | X | ||
| 5.8 Influence d'autres sources de rayonnement | X | ||
| 5.9 Affaiblissement de l'image latente | X | ||
| 5.10 Écart intercassettes | X | ||
| 5.11 Contact film/ecran | X | ||
| 5.12 Conformité avec le profil d'image mammographique IHE (Acquisition Modality Actor) | X | X | |
| 6.0. Évaluation du rendement - Traitement du film | |||
| 6.1 Température du processeur | X | ||
| 6.2 Temps de traitement | X | ||
| 6.3 Sensitométrie | X | ||
| 6.4 Artefacts | X | ||
| 6.5 Lumière dans la chambre noire | X | ||
| 6.6 Lumière de sécurité | X | ||
| 7.0. Évaluation du rendement (Qualité d'images) | |||
| 7.1 Détectabilité du contraste | X | X | X |
| 7.2 Résolution spatiale (Fonction de transfert de modulation (FTM) et Spectre de puissance du bruit (SPB) dans le cas des systèmes numériques) | X | X | X |
| 7.3 Temps d'irradiation | X | X | X |
| 7.4 Distorsion géométrique et artefacts | X | X | X |
| 7.5 Image résiduelle dans le détecteur numérique (image fantôme) | X | X | |
| 8.0. Évaluation du rendement - Dosimétrie | |||
| 8.1 Dose glandulaire moyenne | X | X | X |
| 9.0 Présentation de l'image - Postes de travail d'interprétation | |||
| 9.1 Lumière ambiante | X | X | |
| 9.2 Visibilité de contraste | X | X | |
| 9.3 Résolution | X | X | |
| 9.4 Artefacts d'affichage | X | X | |
| 9.5 Gamme de luminance | X | X | |
| 9.6 Fonction d'affichage d'une échelle de gris standardisée | X | X | |
| 9.7 Uniformité de la luminance | X | X | |
| 9.8 Conformité avec le profil d'image mammographique IHE (Image Display Actor) | X | X | |
| 10.0. Présentation de l'image - Négatoscope | |||
| 10.1 Lumière ambiante | X | X | X |
| 10.2 Luminance | X | X | X |
| 10.3 Uniformité de la luminance | X | X | X |
| 10.4 Homogénéité de la luminance (entre les négatoscopes) | X | X | X |
| 11.0. Présentation de l'image - Imprimantes | |||
| 11.1 Distorsion géométrique | X | X | |
| 11.2 Visibilité de contraste | X | X | |
| 11.3 Résolution | X | X | |
| 11.4 Artefacts de l'imprimante | X | X | |
| 11.5 Fonction d'affichage d'une échelle de gris standardisée | X | X | |
| 11.6 Uniformité de la densité optique | X | X | |
| 12.0. Présentation de l'image - Numériseurs de films | |||
| 12.1 Qualité globale de l'image | X | ||
| 12.2 Bruit | X | ||
| 12.3 Résolution | X | ||
| 12.4 Densité optique | X | ||
3.0 Procédures et équipement d'essai de contrôle de la qualité
Les essais de contrôle de la qualité doivent être effectués au cours des activités courantes d'une installation mammographique. Cette section établit les essais de contrôle de la qualité nécessaires et recommandés, l'équipement d'essai associé et la fréquence des essais.
L'essai de contrôle de la qualité d'un système mammographique comprend de nombreuses étapes importantes, dont:
- la vérification de l'intégrité et de la stabilité de la mécanique du système, notamment des mécanismes de sécurité, la libération automatique du patient, les commandes motrices et le verrouillage des commandes;
- la vérification du rendement de l'équipement auxiliaire, notamment des appareils à développer les films et des dispositifs d'affichage électronique;
- la vérification du rendement des rayons X; et
- la vérification du rendement de l'imagerie ou de diagnostic, comprenant les évaluations des doses.
Des essais de contrôle de la qualité sont réalisés, soit quotidiennement ou jusqu'à une fois tous les six mois, par les technologues en radiologie. L'équipement requis pour ces essais doit être disponible sur place pour les personnes ayant la responsabilité de réaliser ces essais. Tout l'équipement d'essai doit être étalonné et vérifié pour fonctionner de manière précise. Les personnes qui effectuent les essais de contrôle de la qualité doivent recevoir la formation adéquate sur l'équipement d'essai et sur la façon d'effectuer les essais. Les essais annuels de contrôle de la qualité doivent être réalisés par un physicien médical qualifié.
Dans les sections qui suivent, les descriptions de chaque essai indiquent si l'exécution de l'essai est nécessaire ou recommandée. En outre, ce n'est pas tout l'équipement qui doit subir toute la gamme des essais décrits dans les sections qui suivent. La catégorie de système d'imagerie, que ce soit film/ecran (FE), CR, DR auquel le contrôle de la qualité s'applique est identifiée. Les essais de contrôle de qualité et les procédures d'essai prévues dans la présente section sont basées sur des normes des équipements mammographiques existante pour le film / écran et le numérique, établies par les organisations internationales, les organisations nationales et provinciales (AIEA 2009) (AIEA 2011) (CAR PAM 2011) (MSSS 2001) (MSSS 2006) (Mawdsley et. al 2011). D'autres essais peuvent être effectués en remplacement de ceux précisés si on peut prouver que l'essai peut vérifier le paramètre ou le rendement nécessaire. Pour certains essais, les procédures recommandées par les fabricants et les critères d'acceptation sont reconnus. Toutefois, les procédures d'essai équivalentes, telles que celles qui sont reconnues par un organisme responsable de la qualité en mammographie peuvent être acceptable. Les protocoles d'essai harmonisés sont souhaitables, car cela permet de comparer les performances et les essais normalisés.
3.1 Essais quotidiens de contrôle de la qualité
Les essais quotidiens de contrôle de la qualité doivent être effectués au début de chaque journée, avant de commencer les examens mammographiques des patients et le traitement des images.
| Procédure de contrôle de la qualité des appareils sous evaluation |
F/E | CR | DR |
|---|---|---|---|
| Essais quotidiens de contrôle de la qualité | |||
| Réchauffage de l'équipement | Q1 | Q1 | Q1 |
| Fonctionnement des compteurs | Q2 | Q2 | Q2 |
| État de l'équipement | Q3 | Q3 | Q3 |
| Propreté de la chambre noire | Q4 | ||
| Propreté des dispositifs d'affichage électronique et évaluation de conditions de visualisation | Q5 | Q5 | Q5 |
| Fonctionnement de l'appareil de traitement des films | Q6 | ||
| Évaluation de la qualité de l'image - Systèmes film-écran | Q7 | ||
| Évaluation de la qualité de l'image - Systèmes numérique | Q8 | Q8 | |
| Évaluation visuelle globale des dispositifs d'affichage électroniques | Q9 | Q9 | |
| Évaluation visuelle globale des imprimantes | Q10 | ||
Q1. Réchauffage de l'équipement - La procédure de réchauffage recommandée par le fabricant de l'équipement doit être respectée. La procédure de réchauffage doit être répétée si l'équipement ne fonctionne pas pendant une période de temps prolongée. Il faut noter que toutes les composantes du système d'imagerie qui sont utilisées de façon courante doivent être réchauffées, dont les écrans d'ordinateur et les imprimantes.
Q2. Fonctionnement des compteurs - Les compteurs ainsi que les indicateurs visuels et sonores devraient être vérifiés pour en assurer le bon fonctionnement.
Q3. État de l'équipement - L'état des appareils radiographiques devrait être inspecté visuellement pour s'assurer qu'il n'y a pas de composantes lâches ou brisées et pour en vérifier la propreté. L'ensemble radiogène doit être vérifié pour s'assurer qu'il ne bouge pas et ne vibre pas en cours d'utilisation. Il faut également procéder à une inspection visuelle de toutes les autres composantes des systèmes d'imagerie.
Q4. Propreté de la chambre noire - Pour que toutes les surfaces de travail de la chambre noire demeurent propres, les dessus des comptoirs et les planchers devraient être lavés tous les jours. La poussière et les saletés peuvent être plus facilement détectées au moyen d'une lampe UV-B.
Q5. Propreté des dispositifs d'affichage électronique et évaluation de conditions de visualisation - Une inspection visuelle de tous les dispositifs d'affichage électronique utilisés pour l'interprétation des images devrait être effectuée pour détecter la saleté et ceux ci doivent être nettoyés au besoin. On doit évaluer l'environnement dans lequel les mammogrammes sont lus. Le niveau de lumière ambiant doit être faible et constant, particulièrement lorsque les négatoscopes et les dispositifs d'affichage électronique sont présents dans la même salle. Une évaluation devrait également être faite pour déterminer la température, le bruit et les caractéristiques ergonomiques de la salle afin d'assurer des conditions de visualisation uniformes.
Q6. Fonctionnement de l'appareil de traitement des films - La fonctionnement de l'appareil de traitement des films doit être évaluée chaque matin avant les examens cliniques, après que l'appareil de traitement les films ait été mis en marche et qu'il ait atteint la température de traitement requise; et à d'autres moments, au besoin, comme après un changement du taux de réapprovisionnement.
- Les niveaux des solutions de traitement du film doivent être vérifiés pour s'assurer qu'ils sont conformes au niveau de fonctionnement de base recommandé par le fabricant pour un appareil de traitement des films particulier et pour un type de film particulier, pour un nombre donné de films traités quotidiennement.
- La température affichée de la solution de traitement de film doit être vérifiée pour s'assurer qu'elle respecte le niveau de la ligne de base recommandé par le fabricant pour l'appareil à traitement particulier et le film utilisé. Lors de la sensitométrie quotidienne si l'on constate des anomalies dans le processus de la vérification des conditions de développement, il peut être nécessaire de vérifier le pH des solutions de traitement, le temps de traitement, la densité relative des solutions et le taux de réapprovisionnement.
- Le traitement à bandes sensitométriques doit être effectué de manière à surveiller le rendement du système de traitement des films.
- L'indice de voile doit se situer à l'intérieur de +0,03 du niveau de fonctionnement établi.
- L'indice de vitesse doit se situer à l'intérieur de ±0,15 du niveau de fonctionnement établi.
- L'indice de contraste doit se situer à l'intérieur de ±0,15 du niveau de fonctionnement établi.
Q7. Évaluation de la qualité de l'image - Systèmes film-écran - Dans le cas des systèmes film-écran, une évaluation de la qualité de l'image doivent être effectués. Un fantôme uniforme représentant l'épaisseur d'un sein moyen devrait être utilisé de manière courante pour surveiller et maintenir la densité de l'image pour assurer une densité optique correcte, l'absence de phénomènes parasites excessifs, et un réglage uniforme du produit courant/temps. La densité optique du film au centre d'une image de fantôme doit être d'au moins 1,40 lorsque le film est exposé dans une condition clinique typique. Il est fortement recommandé que la densité optique soit supérieure à 1,60. La densité optique du film au centre de l'image de fantôme ne doit pas changer de plus de ±0,20 par rapport aux niveaux de fonctionnement établi.
Q8. Évaluation de la qualité de l'image - Systèmes numérique - Une évaluation de la qualité d'une image à champ plat doit être effectuée. Utiliser un fantôme uniforme représentant l'épaisseur moyenne du sein et évaluer sur l'image si des objets importants peuvent interférer avec les interprétations cliniques. Sur le poste d'acquisition afficher l'image « for presentation » en utilisant une largeur et un niveau de la fenêtre appropriés. Les images doivent avoir une apparence uniforme sans artefacts significatifs. Notez que pour les images CR, il est important de contrôler les images cliniques acquises tout au long de la journée pour assurer que des artefacts ne se développent pas en raison de l'accumulation de poussière.
Q9. Évaluation visuelle globale des dispositifs d'affichage électroniques - Le rendement des dispositifs d'affichage électronique utilisés pour interpréter les mammogrammes doit être évalué. Les essais quotidiens de contrôle de la qualité recommandés par l'American Association of Physicists in Medicine (AAPM, 2005), y compris les mires TG18, et les procédures d'essais et critères d'acceptation devraient être utilisés. Le système d'affichage doit être réchauffé avant les essais. Une attention particulière doit être accordée à la salle pour s'assurer que les niveaux de lumière ambiants sont appropriés (moins de 40 lux) et qu'ils sont représentatifs des conditions dans lesquelles les images cliniques sont visualisées. Une distance de 30 cm est recommandée pour regarder les images. En affichant l'image d'une mire, une évaluation de la qualité générale de l'image et de détection d'artefacts doit être effectuée. Les mires TG18-QC peuvent servir à cet essai et devraient être affichées au moyen du logiciel habituellement utilisé pour afficher les images cliniques.
- Distorsion géométrique -Les bords de la mire doivent être visibles, les lignes doivent être droites et la zone active d'affichage doit être centrée sur l'écran.
- Visibilité de contraste - Tous les coins doivent être visibles et les plages de luminance de 5% et 95% doivent être clairement visibles.
- Artefacts d'affichage - Il ne doit pas y avoir d'artefacts incommodants visibles.
Q10. Évaluation visuelle globale des imprimantes - Le rendement des imprimantes doit être vérifié, lorsqu'elles seront utilisées. À noter que toutes les imprimantes utilisées pour imprimer des images cliniques, que ce soit sur place ou dans une installation éloignée, doivent être soumises à ce test. Imprimer et inspecter la mire TG18 QC et inspecter dans le but de détecter:
- Distorsion géométrique - Les bords de la mire doivent être visibles et droits;
- Visibilité de contraste - Tous les coins doivent être visibles et les plages de luminance de 5% et 95 % doivent être clairement visibles; et
- Artefacts d'imprimante - Il ne doit pas y avoir d'artefacts incommodants visibles.
| Item | Équipment | Système | Référence |
|---|---|---|---|
| 1 | Fantôme uniforme représentant l'épaisseur d'un sein moyen (ex. PMMA 45 ± 0.5 mm) (si nécessaire en fonction des recommandations du fabricant pour le réchauffement de l'appareil |
FE, DR, CR | Q1, Q7, Q8 |
| 2 | Lampe ultraviolette | FE | Q4 |
| 3 | Sensitomètre (21 échelons avec des densités qui varient de 0.00 à 4.80 par échelon de de densité optique de 0.15) Précision: ± 0.02 log unité d'exposition Reproducibilité: ± 0.02 log unité d'exposition |
FE | Q6 |
| 4 | Thermomètre (sans-mercure) | FE | Q6 |
| 5 | Densitomètre Précision: ± 0.02 O.D. at 1.0 O.D. Reproductibilité: ± 0.01 O.D. at 1.0 O.D. |
FE | Q6, Q7 |
| 6 | Mire (TG18-QC) | DR, CR | Q9, Q10 |
| 7 | Négatoscope | FE | Q10 |
3.2 Essais hebdomadaires de contrôle de la qualité
| Procédure de Contrôle de la qualité des appareils sous évaluation |
FE | CR | DR |
|---|---|---|---|
| Essais hebdomadaires de contrôle de la qualité | |||
| Propreté et état de l'écran | H1 | ||
| Propreté et état des cassettes | H2 | H2 | |
| Inspection visuelle de la propreté des systèmes d'imagerie | H3 | H3 | H3 |
| Conditions de lumière dans la chambre noire | H4 | ||
| Température et humidité de la chambre noire | H5 | ||
| État des négatoscopes | H6 | H6 | H6 |
| Image fantôme pour les systèmes film-écran | H7 | ||
| Évaluation de la qualité de l'image numérique | H8 | H8 | |
| Performance du dispositif d'affichage électronique | H9 | H9 | H9 |
| Artefacts de l'imprimante laser | H10 | H10 | H10 |
H1. Propreté et état de l'écran - Il faut veiller à ce que les écrans soient exempts de taches et de dommages. Le nettoyant d'écran recommandé par le fabricant devrait être utilisé. Une inspection des particules de poussière devrait être faite avec de la lumière ultraviolette.
H2. Propreté et état des cassettes - Les aspects suivants des cassettes devraient être vérifiés: propreté, usure, gauchissement, perte d'élasticité de la mousse, fonctionnement du mécanisme de fermeture, pénétration de la lumière. Il est recommandé de s'assurer que le tunnel de support de la cassette soit exempt de poussière et de saleté. La fréquence et la méthode de nettoyage doivent être conformes aux recommandations du fabricant.
H3. Inspection visuelle de la propreté des systèmes d'imagerie - Les systèmes d'imagerie doivent être inspectés pour qu'il n'y ait ni poussière ni saleté sur ou près de la zone de réception de l'image où elles pourraient nuire à la qualité de l'image. Dans le cas des systèmes CR, les plaques doivent être inspectées. Le mécanisme de chargement et de déchargement de la plaque doit être nettoyé et lubrifié au besoin. Les récepteurs d'images des systèmes de mammographie numérique directe doivent être propres en tout temps, sans poussière, saleté ou tout autre élément qui pourrait les toucher. La propreté des numériseurs de balayage au laser doit également être vérifiée.
H4. Conditions de lumière dans la chambre noire - Un essai visuel doit être effectué dans la chambre noire pour s'assurer que la chambre est étanche à la lumière et que d'autres sources de lumière telles que les interrupteurs lumineux et alimentations informatiques ne causent pas de voile au film. Il faut faire particulièrement attention aux joints de la porte et au montage du développeur de films si l'introduction du film dans l'appareil s'effectue à travers un mur. L'évaluation des conditions de lumière dans la chambre noire doit être faite après une période de 10 à 15 minutes d'adaptation aux conditions de noirceur, l'éclairage inactinique étant fermé.
H5. Température et humidité de la chambre noire - Une vérification de la température et de l'humidité de la chambre noire devrait être effectuée. La température devrait se situer entre 15°C et 23°C et l'humidité entre 40 et 60%.
H6. État des négatoscopes - Les négatoscopes devraient être inspectés visuellement pour déterminer si elles sont propres, s'il y a décoloration de la zone et si l'éclairage est adéquat. À noter que ces essais sont également applicables aux situations où les images acquises au moyen de systèmes de mammographie numériques sont imprimées.
H7. Image fantôme pour les systèmes film-écran - Un fantôme, avec des objets d'évaluation de la qualité de l'image, doit être utilisé pour mettre à l'essai la performance de l'imagerie de l'appareil mammographique. Pour le fantôme en mammographie RMI-156, au moins quatre grandes fibres, les trois plus grands groupes de microcalcification et les trois plus grandes masses doivent être visibles.
- Le nombre d'objets d'essai de chaque type de groupe (fibres, microcalcification et masses) visible dans l'image fantôme ne doit pas diminuer de plus de la moitié.
- La densité optique de l'image fantôme en arrière-plan doit être entre 1,5 et 1.9 DO et ne doit pas varier de plus de ±0,20 par rapport au niveau de fonctionnement.
- La différence de densité due à un disque en acrylique de 4,0 mm doit être au moins de 0,40 et ne doit pas varier de plus de ±0,05 du niveau de fonctionnement établi.
H8. Évaluation de la qualité de l'image numérique - Dans le cas des systèmes CR et DR, une évaluation de la qualité de l'image numérique doit être faite. Acquérir une image d'un fantôme uniforme représentant l'épaisseur moyenne du sein et un objet de contraste (typiquement un disque acrylique de 2,5 cm de diamètre et 1 mm d'épaisseur). Les critères suivants doivent être atteints :
- Le produit courant temps. La valeur des mAs ne doit pas s'écarter de la valeur de base établie de plus de ± 10%
- Rapport de la différence du signal sur bruit (SDNR, de l'anglais Signal Difference to Noise Ratio). Le SDNR est une mesure de la différence entre un signal et le fond divisé par le bruit. La variation dans le SDNR doit être inférieure à ±10%;
- Pour les systèmes CR: Lorsque l'indice d'exposition est un S #, la valeur de l'indice S # doit correspondre à ± 10% de la valeur de base établie, lorsque l'indice d'exposition est un nombre SAL, SALlog ou PVIlog, la valeur doit être comprise dans ± 5%, ± 430, ± 580 de la valeur de base établie respectivement, lorsque l'indice d'exposition est une valeur EI, la valeur doit être dans les 40 unités de la valeur de référence établie;
- Les images doivent paraître uniformes;
- Aucun élément du détecteur défectueux; et
- Aucun artefact pouvant interférer avec l'interprétation clinique.
H9. Performance du dispositif d'affichage électronique - Le rendement de tous les dispositifs d'affichage électronique utilisés pour voir les images des systèmes numériques, de même que celles obtenues par le balayage des radiographies doivent être vérifiées. Cela comprend les dispositifs d'affichage utilisés pour l'acquisition et l'interprétation d'images. Pour ce test, il est recommandé d'utiliser une mire TG18-QC modifiée qui simule les images produites par chaque modèle de système de mammographie numérique dans l'établissement, ou qui pourrait être interprété à ce poste de travail (c.-à-d. a les mêmes dimensions x-y, nombre de bits, et un en-tête DICOM contenant des valeurs appropriées de tous les tags pertinents). Notez que lors de l'évaluation du poste de travail du radiologue, les conditions d'observation doivent être vérifiées. Les critères suivants doivent être remplis:
- La plage de luminance de 5% doit être visible dans la zone de luminance de 0%;
- La plage de luminance de 95% doit être visible dans la zone de luminance de 100%;
- Les 16 plages de luminance doivent être visible une par rapport à l'autre.
- Sur les moniteurs utilisés pour l'interprétation, les lettres "QUALITY CONT" doivent être visibles dans chacune des trois régions de l'image TG18-QC.
- Les images doivent apparaître de façon uniforme dans tous les postes de travail d'interprétation.
- Aucun artéfact à l'inspection visuelle.
H10. Artefacts de l'imprimante laser - Lorsqu'une imprimante est utilisée pour l'impression d'images numérique, la qualité des images obtenues doit être vérifiée pour voir s'il y a des artefacts. Assurez-vous que le négatoscope utilisé pour évaluer les films imprimés a suffisamment de luminance. Imprimez une image d'une mire uniforme (c.-à-d., mire TG18-UNL80). L'image imprimée de la mire doit répondre aux critères suivants:
- La densité optique doit être uniforme; et
- aucun artefact pouvant interférer avec l'interprétation clinique.
| Item | Equipment | Système | Référence |
|---|---|---|---|
| 1 | Lampe Ultraviolette | FE | H1 |
| 2 | Nettoyeur d'écran (selon les recommandations du fabricant) |
FE | H1 |
| 3 | Hygromètre Thermomètre (sans mercure) Précision: ± 0.3 °C Reproductibilité: ± 0.1 °C |
FE | H5 |
| 4 | Fantôme RMI 156 avec des objets pour évaluer la qualité | FE | H7 |
| 5 | Négatoscope | FE | H7 |
| 6 | Densitomètre Précision : ± 0.02 O.D. à 1.0 O.D. Reproductibilité: ± 0.01 O.D. à 1.0 O.D. |
FE, CR, DR | H7, H10 |
| 7 | Disque acrylique (4 mm d'épaisseur) | FE | H7 |
| 8 | Disque acrylique (ex. 2.5 cm de diamètre et 1mm d'épaisseur) | CR, DR | H8 |
| 9 | Fantôme uniforme représentant une épaisseur de sein moyen (ex. PMMA de 45 ± 0.5 mm) | CR, DR | H8 |
| 10 | Mire(s) pour l'évaluation des moniteurs de visualisation et pour l'imprimante laser (ex. TG18-QC, TG-18 UNL80) | CR, DR | H9, H10 |
| 11 | Loupe (4x à 5x grossissement) | CR, DR | H9, H10 |
3.3 Essais mensuels de contrôle de la qualité
| Procédure de contrôle de la qualité des appareils sous évaluation |
FE | CR | DR |
|---|---|---|---|
| Essais mensuels de contrôle de la qualité | |||
| Inspection mécanique, électrique et globale du système | M1 | M1 | M1 |
| Nettoyage de la cassette, de l'écran et de la plaque d'imagerie | M2 | M2 | |
| La précision du thermomètre du développeur | M3 | ||
| Taux de réapprovisionnement | M4 | ||
| Évaluation étendue de la présence d'artefacts | M5 | M5 | |
| Sensitométrie des imprimantes laser | M6 | M6 | |
M1. Inspection mécanique, électrique et globale du système - Une inspection de sécurité doit être effectuée les éléments suivants.
- La température ambiante dans la salle d'acquisition de mammographie;
- L'intégrité structurale des composants du système de mammographie y compris la protection de l'opérateur;
- Le mouvement du système;
- la performance des indicateurs, des commutateurs et des compteurs
- le bon fonctionnement des dispositifs de verrouillage;
- l'annotation correcte sur les images affichées et imprimées;
- autres éléments associés à la spécificité du système ou de l'établissement.
M2. Nettoyage de la cassette, de l'écran et de la plaque d'imagerie - Les cassettes, les écrans et les plaques d'imagerie doivent être vérifiés et inspectés pour s'assurer qu'ils ne sont pas endommagés. Les produits et procédures de nettoyage recommandés par le fabricant devraient être utilisés.
M3. Précision du thermomètre du développeur - La précision du thermomètre du développeur doit être vérifiée régulièrement par rapport à un thermomètre non mercure. La température du révélateur doit être exacte à 0,5°C près.
M4. Taux de réapprovisionnement - Le taux de réapprovisionnement doit être comparé au niveau de la ligne de base recommandée par le fabricant pour un appareil de traitement des films et un type de film particulier, pour le nombre donné de films traités quotidiennement et pour la méthode de traitement.
M5. Évaluation étendue de la présence d'artefacts - Une évaluation de qualité d'une image uniforme doit être effectuée en utilisant tous les foyers applicables, les filtres et les modes de grossissement. L'image d'un fantôme uniforme permet d'évaluer la présence d'artefacts importants qui pourraient interférer avec l'interprétation clinique. Visualisez l'image « for processing » en utilisant une largeur et un niveau de la fenêtre appropriés. Les images doivent avoir un aspect uniforme, sans artefacts significatifs.
M6. Sensitométrie des imprimantes laser - Une évaluation de la stabilité de la performance de l'imprimante laser doit être réalisée. L'impression d'une bande sensitométrique sur le film laser devrait permettre d'atteindre les critères suivants :
- Densité maximum Dmax - Dmax est la densité la plus foncée. Le Dmax doit être supérieure ou égal à la valeur de référence établie moins 0.15 DO ou 3.5DO, selon la moindre de ces valeurs.
- Différence de densité, DD - DD est l'échelon le plus proche de 2,20 DO moins l'échelon le plus proche, mais pas moins que 0.45 DO. La DD doit se situer à l'intérieur de ±0,15 du niveau de fonctionnement établi.
- Densité moyenne DM - DM est l'échelon le plus proche, mais pas au-dessus de 1.20 DO ou la densité optique de travail. La DM doit se situer à l'intérieur de ±0,15 du niveau de fonctionnement établi.
- Base plus le voile (B + V) - La B + V doit être plus petit ou égal à la valeur de référence plus 0.03 DO.
Notez que cet essai doit être effectué à chaque mois pour les imprimantes laser à développement sec. Cependant, pour les imprimantes laser à développement humide, cet essai doit être effectué chaque jour, avant de commencer le traitement des images cliniques.
| Item | Équipement | Système | Référence |
|---|---|---|---|
| 1 | Thermomètre (sans mercure) Précision: ± 0.3 °C Reproductibilité: ± 0.1 °C |
FE | M1, M3 |
| 2 | Nettoyeur d'écran (selon les recommandations du fabricant) |
FE | M2 |
| 3 | Lampe Ultraviolette | FE | M2 |
| 4 | Fantôme uniforme représentant une épaisseur de sein moyen (ex. PMMA de 45 ± 0.5 mm) | CR, DR | M5 |
| 5 | Densitomètre | CR, DR | M6 |
| 6 | Film avec barre de sensitométrie produite par l'imprimante laser ou par le poste d'acquisition. | CR, DR | M6 |
| 7 | Loupe (4x à 5x grossissement) | CR, DR | M6 |
3.4 Essais trimestriels de contrôle de la qualité
| Procédure de Contrôle de la qualité des appareils sous évaluation |
FE | CR | DR |
|---|---|---|---|
| Essais trimestriels de contrôle de la qualité | |||
| Analyse de la rétention du fixateur | T1 | ||
| Analyse des registres de reprise | T2 | T2 | T2 |
| Résolution spatiale/Évaluation de la Fonction de Transfer de Modulation de l'équipement CR | T3 | ||
| Qualité de l'imprimante laser | T4 | T4 | T4 |
| Rendement du numériseur de films | T5 | ||
T1. Analyse de la rétention du fixateur - Des essais de rétention du fixateur doivent être effectués pour s'assurer que le fixateur est retiré correctement des films traités conformément au niveau de de base établi. La quantité de fixateur résiduel ne doit pas dépasser 0,05 g/m2.
T2. Analyse des registres de reprise - Pour la mammographie film écran et la mammographie numérique, une analyse des registres de reprise doit être faite pour identifier et corriger toutes les tendances ou erreurs répétées. Les reprises sont définies comme des images reprises en raison de la qualité insuffisante. Ceci n'inclut pas les images prises à des fins de contrôle de la qualité, des images prises pour acquérir des vues supplémentaires, ou des images prises pour inclure les tissus qui n'ont pas pu être imagées en raison de la taille des seins. Le registre des reprises et leur analyse doivent être conservés séparément pour chaque système de mammographie. Les installations doivent tenir des registres de toutes les reprises, incluant la raison de la reprise et les mesures correctrices, immédiatement après que l'image de reprise ait été effectuée. Si les images contiennent des renseignements sur le diagnostic du patient, elles doivent être gardées dans le dossier du patient. Le taux de reprise doit être inférieur à 5 pour cent et de préférence moins de 2%.
mammographie CR doit être effectuée. La résolution spatiale peut être évaluée soit par l'imagerie d'un dispositif d'essai MTF et en utilisant un logiciel approprié, conformément aux procédures d'essai du fabricant ou utilisant une autre méthode jugée acceptable par un physicien qualifié. Le MTF doit se situer dans les spécifications du fabricant et à l'intérieur des niveaux de base établis. Cet essai peut aussi être réalisé sur les systèmes DR mais il est peu probable que la résolution des systèmes DR varie de façon significative sur une période trimestrielle.
T4. Qualité de l'imprimante laser - Une évaluation des images imprimées doit être réalisée. Pour cet essai, il est recommandé d'utiliser une mire TG18-QC modifiée qui simule les images produites par chaque modèle de système de mammographie numérique dans l'établissement, ou qui pourrait être interprétées à ce poste de travail (c.-à-d. a les mêmes dimensions x-y, nombre de bits, et un en-tête DICOM contenant des valeurs appropriées de tous les tags pertinents). Annoter la mire TG18-QC modifiée en ajoutant les règles de 5cm horizontale et verticale. Notez que les images doivent être imprimées à partir des postes d'acquisition et d'interprétation. Examiner les images sur un négatoscope. Les critères suivants doivent être remplis:
- La zone de 5% doit être visible dans la zone de 0%.
- La zone de 95% doit être visible dans la zone de 100%.
- Les fines lignes horizontales et verticales doivent être visibles dans les 4 coins.
- Les échelons de résolutions (noir sur blanc) doivent être distincts.
- Les lignes doivent être droites et égales.
- Aucun artéfact à l'inspection visuelle.
- Les lignes de 5 cm doivent mesurée 5 ± 0.3cm.
T5. Rendement du numériseur de films - Une évaluation du numériseur de films doit être réalisée pour s'assurer que la qualité des images numérisées est comparable à celle du film. La résolution des images numérisées doit correspondre à la résolution nominale du numériseur.
| Item | Equipement | Systèmes | Référence |
|---|---|---|---|
| 1 | Trousse de rétention du fixateur | FE | Q1 |
| 2 | Dispositf d'essai de MTF et logiciel approprié de calcul de MTF | CR | Q3 |
| 3 | Mire pour évaluer les moniteurs et les imprimantes lasers (ex. TG18 ) | CR, DR | Q4, Q5 |
| 4 | Loupe (4x à 5x grossissement) | FE,CR, DR | Q4 |
| 5 | Règle | FE,CR,CR | Q4 |
3.5 Essais semestriels de contrôle de la qualité
| Procédure de Contrôle de la qualité des appareils sous évaluation |
FE | CR | DR |
|---|---|---|---|
| Essais semestriel de contrôle de la qualité | |||
| Dispositif de compression du sein | SA1 | SA1 | SA1 |
| Vérification du voile en chambre noire | SA2 | ||
| Contact film/écran | SA3 | ||
| Variation de la sensibilité interplaques des plaques d'imagerie | SA4 | ||
SA1. Dispositif de compression du sein - Le dispositif de compression du sein doit être évalué afin de vérifier la force de compression, l'alignement des plaques de compression et la précision de l'indicateur de l'épaisseur du sein.
- La variation dans la force de compression mesurée et affichée doit se situer à l'intérieur ±2.0kg (±4.5lb, ±20 N).
- La force maximale de compression manuelle doit être inférieure à 300N. La force de compression maximale pour la compression motorisée initiale se situe entre 11,4 kg (25 lb, 111N) et 20,5 kg (45 lb, 200N). L'appareil à mammographie doit être capable de maintenir une force de compression jusqu'à ±1 kg (2.2 lb, 9.8N) pendant au moins 1 minute.
- La pelote de compression doit être plate et parallèle à a surface du récepteur d'image, et elle ne doit pas dévier de l'axe parallèle de plus de 1 cm en n'importe quel point de la surface de la pelote de compression lorsque la force est appliquée. Les appareils qui sont conçus de manière à ne pas être plats et parallèles doivent respecter les spécifications du fabricant et les exigences relatives aux spécifications du fabricant et à l'entretien.
- L'indicateur d'épaisseur du sein doit être précis à ±8 mm de l'épaisseur d'un bloc de PMMA et devrait être à ±5 mm de l'épaisseur d'un bloc de PMMA.
SA2. Vérification du voile en chambre noire - Une image d'un fantôme de mammographie exposée à une densité optique de 1,4 unité ne doit pas présenter une augmentation de la densité optique supérieure à 0,05 unité en deux minutes d'exposition à l'environnement lumineux de la chambre noire.
SA3. Contact écran/film - Toutes les cassettes utilisées en mammographie doivent être testées pour vérifier le contact écran/film à l'aide d'un écran de cuivre de 16 mailles/cm (40 mailles/po). Les cassettes présentant une surface de mauvais contact d'un diamètre supérieur à 5 mm et un faible contact qui n'est pas éliminé par le nettoyage de l'écran et qui demeure dans le même emplacement durant les essais subséquents doivent être remplacées. Les surfaces de mauvais contact d'un diamètre supérieur à 2 mm au bord de la paroi thoracique ne sont pas acceptables.
SA4. Variation de la sensibilité interplaques des plaques d'imagerie - Dans le cas de l'équipement CR, une évaluation de la sensibilité interplaques doit être réalisée.
- La variation du rapport signal-bruit dans une région de référence ne doit pas dépasser ±15% entre toutes les plaques d'imagerie utilisées cliniquement.
- La variation du kerma dans l'air de la surface d'entrée enregistré ou dans le chargement du tube (mAs) ne doit pas dépasser ±10%.
- Il ne doit pas y avoir d'inhomogénéité importante sur les images.
| Item | Équipement | Systèmes | Référence |
|---|---|---|---|
| 1 | Dispositif d'essai de la force de compression (Pèse-personne à cadran conventionnel) | FE, CR, DR | SA1 |
| 2 | Serviettes ou des coussins (masse spécifique environ:30 mg/cm3) | FE, CR, DR | SA1 |
| 3 | Ruban à mesurer | FE, CR, DR | SA1, SA3 |
| 4 | Feuille d'épaisseur uniforme de PMMA | FE, CR, DR | SA1, SA4 |
| 5 | Densitomètre Précision: ± 0.02 O.D. at 1.0 O.D. Reproductibilité: ± 0.01 O.D. at 1.0 O.D. |
FE | SA2 |
| 6 | Minuterie | FE | SA2 |
| 7 | Grille de contact film/écran pour la mammographie (40 mailles/po) | FE | SA3 |
3.6 Essais annuels de contrôle de la qualité
| Procédure de Contrôle de la qualité des appareils sous évaluation |
FE | CR | DR |
|---|---|---|---|
| Essais annuels de contrôle de la qualité | |||
| Précision de la haute tension radiogène | A1 | A1 | A1 |
| Reproductibilité de la haute tension radiogène | A2 | A2 | A2 |
| Puissance de rayonnement (kerma dans l'air) Reproductibilité et linéarité | A3 | A3 | A3 |
| Puissance de rayonnement normalisé | A4 | A4 | A4 |
| Filtration du faisceau de rayons X | A5 | A5 | A5 |
| Collimation | A6 | A6 | A6 |
| Alignement du champ lumineux et du champ de rayon X | A7 | A7 | A7 |
| Commande automatique d'exposition (AEC) | A8 | A8 | A8 |
| Rendement du récepteur d'image | A9 | A9 | A9 |
| Qualité de l'image | A10 | A10 | A10 |
| Dosimétrie | A11 | A11 | A11 |
| Négatoscope | A12 | A12 | A12 |
| Performance du dispositif d'affichage électronique | A13 | A13 | |
| Imprimantes | A14 | A14 | |
| Entretien général de prévention | A16 | A16 | A16 |
A1. Précision de la haute tension radiogène - Pour toute combinaison des paramètres de charge, la haute tension radiogène ne doit pas dévier de la valeur sélectionnée de plus de 5%.
A2. Reproductibilité de la haute tension radiogène - Aux tensions radiogènes couramment utilisées cliniquement, le coefficient de variation du kVp doit être égal ou inférieur à 0,02 sur la base de 5 mesures à chaque réglage de la haute tension radiogène. Noter cependant que, si la différence de pourcentage entre les deux premières mesures n'est pas supérieure à 5%, seulement deux mesures à chaque réglage de la tension du tube est acceptable.
A3. Reproductibilité et linéarité de la puissance de rayonnement (kerma dans l'air) - La reproductibilité et la linéarité du kerma dans l'air doit être évaluée en utilisant le mode manuel, une combinaison cible/filtre Mo/Mo et une haute tension radiogène de 28kVp. Sélectionner 3 valeurs des réglages du mAs qui sont typiquement utilisées. Le coefficient de variation de n'importe quelles cinq mesures consécutives du kerma dans l'air ne doit pas être supérieur à 0,05. Noter cependant que, si la différence de pourcentage entre les deux premières mesures n'est pas supérieure à 5%, seules deux mesures à chaque réglage du mAs sélectionné est acceptable. À chaque réglage du mAs, calculer la valeur moyenne de kerma dans l'air et la valeur de sortie de rayonnement (Y) en divisant chaque valeur moyenne de kerma dans l'air par la valeur correspondante de mAs. Pour des paires consécutives de réglage du mAs, calculer la linéarité comme L = 100 (Y1-Y2) / (Y1 + Y2). La linéarité doit être inférieure à ± 10%.
A4. Puissance de rayonnement normalisé- En utilisant les valeurs de sortie (Y) obtenues à l'essai A3, calculer la sortie normalisée en appliquant une correction de la loi du carré inverse pour obtenir la sortie à 1.0m. La sortie normalisée à 28kVp avec une combinaison Mo / Mo cible/filtre doit être supérieure à 30µGy/mAs.
A5. Filtration du faisceau de rayons X - La première couche de demi transmission (CDT) doit être déterminée pour toutes les réglages des tensions du tube radiogène utilisées cliniquement de manière courante et pour toutes les combinaisons cible/filtre. La première couche de demi transmission de l'aluminium, mesurée avec la plaque de compression en place, doit se situer dans la plage définie par.

Formule pour la détermination de la plage des valeurs de la première couche de demi-transmission d'aluminium, mesurée avec la plaque de compression en place.
La plage des valeurs de la première couche de demi-transmission de l'aluminium, mesurée avec la plaque de compression en place, déterminée par l'équation « Tension du Tube Radiogène (kV) divisé par 100, plus 0.03, égale ou inférieure à CDT (mm d'aluminium), égale ou inférieure à Tension du Tube Radiogène (kV) divise par 100, plus C ».
Où:
C =
0.12 pour Mo/Mo
0.19 pour Mo/Rh
0.22 pour Rh/Rh
0.30 pour W/Rh
0.32 pour W/Ag
0.25 pour W/Al
A6. Collimation - Une évaluation doit être faite de la collimation pour assurer une exposition complète du récepteur d'image et l'alignement de la palette de compression avec le bord de la paroi thoracique du récepteur d'image. Le champ de rayon X:
- doit recouvrir complètement la surface du récepteur d'image;
- doit s'étendre jusqu'au bord du support de patient qui est conçu pour être adjacente à la paroi thoracique du patient et ne pas s'étendre au-delà de ce bord de plus de 5 mm, et
- ne doit pas s'étendre de plus de 2 pour cent de la distance foyer-récepteur d'image de tous les autres bords du récepteur d'image.
L'alignement de la plaque de compression doit être telle que le bord de la plaque de compression:
- ne doit pas être visible dans l'image, et
- ne doit pas dépasser le bord de la paroi thoracique du récepteur d'image de plus de 5 mm.
A7. Alignement du champ lumineux et du champ de rayon X - Une évaluation devrait être faite de l'alignement du champ lumineux et du champ du rayon - x. La séparation entre le périmètre du champ illuminé et celui du champ de rayon X ne doit pas dépasser 2 pour cent de la distance foyer-récepteur d'image.
A8. Commande automatique d'exposition (AEC, de l'anglais Automatic Exposure Control) - Le rendement de l'AEC doit être évalué.
- AEC - Reproductibilité de l'AEC et Temps d'exposition maximal pour les systèmes film/écran - Pour les systèmes film/écran la reproductibilité de l'AEC doit être évaluée. En utilisant une épaisseur de 4,5 cm de PMMA et un espaceur approprié (8 mm d'épaisseur, radiotransparent-U polystyrène expansé) acquérir quatre expositions. Enregistrez les mAs pour chaque exposition. Le coefficient de variation des mAs ne doit pas dépasser 0,05. Le temps d'exposition maximal doit être inférieur ou égale à 2 secondes en mode contact et inférieur ou égal à 3 secondes dans le mode d'agrandissement.
- AEC - La constance de la densité optique pour les systèmes film/écran - La densité optique des films de mammographie doit être correctement mise en place et rester constante. La densité optique doit être d'au moins 1,4 DO et devrait être de l'ordre de 1,6 DO à 1,9 DO. En utilisant une épaisseur de 4,5 cm de PMMA et un espaceur approprié (8 mm d'épaisseur, radiotransparent en forme de U en polystyrène expansé), acquérir une image avec le contrôle de la densité à la position "0" ou "normal". Traiter le film et mesurer la densité optique à 40 mm à partir du bord de la paroi thoracique, centrée latéralement. Les valeurs mesurées de densité optique doivent être de ± 0,15 DO à partir de la valeur de base établie
- AEC - Compensation pour l'épaisseur pour les systèmes film/écran - Le dispositif d'AEC doit fonctionner, de telle sorte que la variation de densité optique dans les images produites ne dépasse pas ± 0,15 de la densité optique moyenne lorsque l'épaisseur uniforme d'un matériau équivalent de tissu mammaire varie dans une plage de 2 à 7 cm et la tension du tube radiogène, et les combinaisons de filtres anodes sont variées de manière appropriée dans la gamme utilisée en clinique dans l'établissement. Si cette exigence ne peut être satisfaite, un graphique technique doit être développée montrant les paramètres de charge appropriés pour l'épaisseur du sein et de compositions différentes qui doivent être utilisés de telle sorte que la densité optique demeure à l'intérieur de ± 0,15 de la moyenne lorsque le contrôle de l'exposition automatique est utilisé.
- AEC - Le réglage de contrôle de la densité optique pour les systèmes films/écran - La variation de la densité optique des images produites en faisant varier la commande de densité optique doit être de l'ordre de 0,05DO à 0,25 DO.
- AEC - Reproductibilité des systèmes numériques -Pour les systèmes CR et DR, les exigences énoncées dans le test A (8) (a) pour les systèmes film-écran s'appliquent également.
- AEC - Compensation d'épaisseur de l'objet et Temps d'exposition maximal pour les systèmes numériques - La performance de l'AEC doit être évalué conjointement avec la qualité d'image, tel que mesuré par la différence de rapport signal sur bruit dans des conditions spécifiées, et la dose au patient, telle qu'elle est caractérisée par la dose glandulaire moyenne, et en les comparant aux valeurs de base établies. La différence du rapport signal sur bruit est déterminée en utilisant des plaques d'épaisseur uniformes de PMMA sur une plage de 2 à 7 cm, les espaceurs appropriés et un objet de contraste (ex. disque de 1 mm d'épaisseur, 25 mm de diamètre de PMMA). Les valeurs recommandées pour les différences de rapport signal sur bruit pour évaluer l'AEC de systèmes de mammographie numériques sont disponibles auprès de l'AIEA (AIEA, 2011). Tout en assurant que les limites de la dose glandulaire moyenne sont respectées (voir A11), les valeurs de la différence de rapport signal sur bruit doivent répondre à des critères de référence établis et devraient respecter les valeurs recommandées par l'AIEA. En mode contact, le temps d'exposition doit être inférieur ou égal à 2 secondes pour 45 mm de PMMA et inférieur ou égal à 4 secondes pour 70 mm de PMMA.
- AEC - Étapes de contrôle d'exposition pour les systèmes numériques (si disponible) - La variation de la charge du tube radiogène (mAs) réalisée en variant les étapes du contrôle d'exposition devrait fournir environ de ± 10% à ± 25% de variation en mAs par étape.
- AEC - Correspondance entre les capteurs AEC - Dans le cas des systèmes mammographiques ayant des capteurs AEC indépendants multiples, le rendement de chaque capteur individuel devrait être évalué. La capacité à sélectionner correctement chaque capteur devrait être vérifiée. La variation dans la densité optique entre tous les capteurs AEC devrait se situer à l'intérieur de 0,20 DO. Pour les systèmes CR, la variation en mAs entre les capteurs AEC ne devrait pas dépasser ± 15%.
- AEC - Minuterie de secours - La performance de la minuterie de secours doit être vérifiée pour assurer une performance sécuritaire de l'équipement. Pour un appareil à mammographie, le produit courant-temps ne doit pas dépasser 800 mAs.
A9. Rendement du récepteur d'image - Le rendement du récepteur d'image doit être vérifié en consultant les spécifications du fabricant et les valeurs de référence établies.
- Uniformité de la vitesse écran/film - L'uniformité de la vitesse radiographique de tous les récepteurs d'image utilisés de manière courante doit être évaluée. La différence entre les densités optiques minimales et maximales des films ne doit pas dépasser 0,25 DO.
- Image fantôme pour un système film/écran - Un fantôme avec des objets, permettant l'évaluation de la qualité d'image, doit être utilisé pour voir la performance du système de mammographie. Pour le fantôme en mammographie RMI-156, au moins quatre plus grandes fibres, les trois plus grands groupes de micocalcification et les trois plus grandes masses doivent être visibles.
- Le nombre d'objets d'essau de chaque type de groupe (fibres, microcalcifications et masses) visible dans l'image fantôme ne doit pas diminuer de plus de la moitié.
- La densité optique de fond de l'image fantôme en arrière-plan devrait être entre 1,5DO et 1,9 DO et ne doit pas varier de plus de ± 0,20 par rapport au niveau de fonctionnement.
- La différence de densité due à un disque 4,0 mm en acrylique de 4,0 mm doit être d'au moins 0,40 et ne doit pas varier de plus de ± 0,05 du niveau de fonctionnement établi.
- Linéarité spatiale et Distorsion géométrique - Pour les systèmes comportant des pièces mobiles, telles que CR ou systèmes de balayage, une évaluation de la linéarité spatiale et la distorsion géométrique doivent être effectuées. Utiliser un dispositif d'essai de distorsion géométrique placé directement sur la surface du support du patient, acquérir une image en mode AEC. Placer le dispositif d'essai sur la table de grossissement et faire l'acquisition d'une seconde image. Évaluer les images pour s'assurer que:
- La taille de l'image ne doit pas dépasser 10% de la taille nominale mentionnée par le constructeur.
- Les dimensions, de l'élément détecteur efficace (del) de largeur et de longueur (x et y), ne doivent pas dépasser 5% un de l'autre.
- Les distances mesurées à l'aide de l'outil d'annotation sur le poste de travail doivent être inférieures à 5% de la taille réelle.
- Il devrait y avoir moins de 2% de déviation au centre du champ, sur une ligne droite d'une longueur de 100 mm.
- Fonction de réponse et Évaluation du bruit - La réponse du détecteur et les caractéristiques du bruit selon différents chargement du tube (produit courant-temps) doivent être évalués. En mode manuel, un bloc d'essai uniforme avec un objet de contraste doit être imagé avec au moins 4 différentes chargements du tube (mAs) couvrant la gamme habituellement utilisée en clinique. Enregistrez le chargement du tube. Pour les systèmes CR sans la capacité de faire l'analyse de la région d'intérêt (ROI), l'indice d'exposition doit aussi être enregistré.
À l'aide des images brutes, mesurer la valeur moyenne des valeurs de pixel dans une région d'intérêt (de l'ordre de 80 mm2) situé entièrement à l'intérieur de l'objet de contraste (A) et, dans une région d'intérêt de référence située juste à côté de l'objet de contraste, mesurer la valeur moyenne des valeurs de pixel (B) et l'écart type (C). Calculer le rapport de la différence du signal sur bruit comme SDNR = |A-B|/C. Pour les systèmes linéaires, tracer la valeur moyenne des valeurs de pixel de dans l'objet de contraste, la variance (i.e. l'écart type carré) et le SDNR en fonction du mAs. Pour les systèmes logarithmiques, il peut être nécessaire de tracer la valeur moyenne des valeurs de pixel et la variance en fonction 1/mAs. Notez que pour les systèmes où une valeur de pixel de décalage (Bo) a été appliquée, cette valeur doit être calculée et soustraite. Calculer la valeur de (B-Bo)/mAs pour toutes les valeurs de chargement du tube et de la valeur moyenne globale.
Pour les systèmes linéaires:- Utiliser les graphiques de la valeur moyenne des valeurs de pixel (B) et de la variance C2 pour calculer le carré du coefficient de corrélation (R2). R2 doit être supérieur ou égal à 0,95.
- Toutes les valeurs de (B-Bo)/mAs doivent se situer à l'intérieur de 10% de la valeur moyenne de ce rapport.
- La valeur de (B-Bo) doit se situer à l'intérieur de ± 10% de la valeur de base établie.
- La valeur de C doit se situer à l'intérieur de ± 5% de la valeur de base établie.
- La valeur du SDNR doit se situer à l'intérieur de ± 5% de la valeur de base établie.
- Tracer l'indice d'exposition en fonction des mAs et calculer R2. R2 doit être supérieur ou égal à 0,95.
- Lorsque l'indice d'exposition est un S #, la valeur de l'indice S # doit se situer à l'intérieur de ± 10% de la valeur de base établie.
- Lorsque l'indice d'exposition est un nombre SAL, SALlog ou PVIlog, la valeur doit se situer à l'intérieur de ± 5%, ± 430, ± 580 de la valeur de base établie respectivement.
- Lorsque l'indice d'exposition est une valeur EI, la valeur doit se situer à l'intérieur de 40 unités de la valeur de base établie.
- Homogénéité de l'image et Évaluation des artefacts - En utilisant un bloc d'essai uniforme de 45 mm d'épaisseur, couvrant l'ensemble du détecteur, obtenir des images avec des réglages cliniques pour un sein d'une épaisseur équivalente, pour toutes les combinaisons cibles filtre utilisé. Enregistrer les paramètres d'exposition. En utilisant le support d'agrandissement et un bloc d'essai uniforme de PMMA 25mm, acquérir des images avec des réglages cliniques pertinents et les combinaisons de filtre cibles. Enregistrer le réglage d'exposition. Examiner toutes les images non-traitées en utilisant les réglages de la largeur et le niveau de la fenêtre appropriés pour la visualisation d'artefacts. Enregistrez les réglages de la largeur et du niveau de la fenêtre.
- Il ne doit pas y avoir d'artefacts assez importants qui pourraient interférer avec l'interprétation des images (pixels morts visibles, lignes ou colonnes manquantes).
- Il ne doit pas y avoir de bruits structurés visuellement dérangeants.
- Il ne doit pas y avoir de régions de densité visiblement différente.
- Il ne doit pas y avoir de régions ayant une variation d'amplitude de bruit inattendue. Il est à noter qu'avec les systèmes CR la non-uniformité associée à l'effet talon ne peut être supprimée.
- Défaillance d'un élément de détection - Pour les systèmes de radiographie directe, une inspection doit être faite des éléments défectueux du détecteur (carte pixels morts) du récepteur de l'image numérique. Le nombre d'éléments défectueux du détecteur doit être conforme aux spécifications du fabricant.
- Image résiduel /fantôme du détecteur - Une évaluation doit être faite afin d'évaluer le niveau d'image résiduelle résultant d'une image précédente. Placer un bloc d'essai uniforme couvrant la moitié droite du récepteur d'image. En utilisant une technique manuelle, acquérir une image selon les techniques cliniques typiques pour un sein moyen. Repositionner le bloc d'essai, aligné et centré le long du côté de la paroi thoracique. Dès que le système est prêt, acquérir une autre image. Il est important que le temps écoulé entre les images soit court (1 minute ou moins) pour être compatible avec des temps entre les images typiques en usage clinique. Notez que pour les systèmes CR, ce sera le temps de traiter et de récupérer la même cassette (plaque d'imagerie). Utiliser l'outil d'analyse de région d'intérêt, et déterminer les valeurs moyennes des valeurs de pixels de A et B et l'écart-type, C, selon le schéma indiqué. Calculer le SDNR, comme SNDR =|(A-B)|/C. La valeur de SNDR doit être inférieure ou égale à 2,0. En variante, l'affichage de la seconde image (version "for processing") selon les réglages de la fenêtre utilisée cliniquement, l'image du bloc d'essai dans la première position ne doit pas être visible.

Plan schématique du récepteur d'image et le bloc d'essai pour l'évaluation de l'image résiduel/fantôme du détecteur numérique.
Un schéma d'un rectangle gris horizontal représentant le support du sein avec un deuxième plus petit rectangle horizontal blanc a l'intérieur aligné avec le bord inférieur du support du sein représentant un bloc d'essai, et un troisième rectangle vertical transparent représentant la première position du bloc d'essai qui couvre le coté droit du support du sein et qui divise en deux le deuxième rectangle le long d'un ligne D. Dans le deuxième rectangle, à gauche de la ligne D, il y un cercle avec la lettre A indiquée à l'intérieur. Dans le deuxième rectangle, à droite de la ligne D, il y a un cercle avec les lettres C et D indiquées à l'intérieur.
A10. Qualité de l'image - La qualité des images mammographiques doit être vérifiée par rapport aux spécifications du fabricant et les niveaux de base établis.
- Résolution spatiale des systèmes film/écran - Lors de l'utilisation d'une barre de mire de résolution, la résolution spatiale minimum mesurée du système de mammographie doit être:
- 11 paires de ligne/mm quand la mire de résolution à contraste élevée est orientée de telle manière que les barres sont perpendiculaires à l'axe anode-cathode, et
- 13 paires de ligne/mm lorsque les barres sont parallèles à l'axe.
- Fonction de transfert de modulation (FTM) - Systèmes numériques - Une évaluation quantitative doit être faite de la résolution spatiale des systèmes de mammographie numérique. La FTM est mesurée par un dispositif d'essai FTM, placé sur un bloc de PMMA de 45 mm, selon la technique utilisée cliniquement pour un sein moyen. Utiliser l'image « pour traitement » et le logiciel de FTM pour calculer la FTM.
- Les fréquences spatiales auxquelles la FTM tombe à 50% et 20% ne doivent pas être inférieures aux niveaux établis. Les valeurs de fréquences acceptables au cours de laquelle la FTM tombe à 50% et 20% sont disponibles à partir de l'AIEA (AIEA 2011).
- La FTM à 2,5, 5 et 7,5 cycles/mm ne doit pas changer plus de 10% par rapport aux valeurs de référence établies.
A11. Dosimétrie - Une évaluation du kerma dans l'air à la surface d'entrée et de la dose glandulaire moyenne doit être faite.
- Kerma dans l'air à la surface - Pour les appareils de mammographie film/écran, le kerma dans l'air à la surface d'entrée (sans rétrodiffusion) doit être mesuré en utilisant des facteurs d'expositions établies sélectionnées pour exposer un fantôme équivalent à un sein standard (45 ± 0,5 mm). Le kerma dans l'air à la surface d'entrée doit être associé à une densité optique de 1,6 à 2,0. Pour les appareils de mammographie numérique, le kerma dans l'air à la surface d'entrée (sans rétrodiffusion) doit être mesurée à l'aide des facteurs d'exposition établis qui produisent des niveaux de signal acceptables (SDNR) pour exposer le PMMA à des épaisseurs de 20 mm, 45 mm et 70 mm. Les valeurs mesurées du kerma dans l'air à la surface d'entrée doivent se situer dans les niveaux de référence établis.
- Calcul de la dose glandulaire moyenne (DGM) pour un sein standard - La DGM d'un sein standard ne doit pas dépasser 3.0 mGy et ne devrait pas exéder 2.5mGy. Pour les systèmes de mammographie numérique, la DGM doit être évalué pour les seins représentés par 20 mm, 45 mm et 70 mm de PMMA. Les lignes directrices sur l'estimation des DGM apparaissent à l'annexe III.
Alternativement, les fantômes du sein comme le RMI-156 ou le NA # 18-220 qui représentent un sein composé de 50 pourcent de gras et 50 pourcent tissu glandulaire et comprimé à 42 mm d'épaisseur, peut être utilisé pour déterminer la représentation de la dose glandulaire moyenne pour un sein de composition similaire. La dose glandulaire moyenne ne doit pas dépasser 2,5 mGy et doit être dans le niveau de référence établi.
A12. Négatoscope - Tous les négatoscopes utilisés pour l'interprétation des mammogrammes doivent être soumis à des essais visant à déterminer leur conformité avec les exigences suivantes. Soyez certain que tous les négatoscopes sont mis en marche au moins 30 minutes avant d'obtenir des mesures.
- Luminance - La luminance du négatoscope devrait être d'au moins 3 500 nits (cd/m2).
- Uniformité de la luminance - La luminance du négatoscope devrait offrir une uniformité ne devant pas varier de plus de 10 pour cent.
- Homogénéité de la luminance - L'homogénéité de la luminance enter toutes les négatoscopes utilisés pour les mammogrammes doit être uniforme jusqu'à 15 pour cent de la moyenne.
- Contrôle de la lumière ambiante - La lumière ambiante dans la salle de lecture doits être inférieure à 40 lux.
A13. Performance du dispositif d'affichage électronique - La performance du dispositif d'affichage électronique utilisé au poste de travail d'interprétation des images et au poste d'acquisition doit être vérifiée. Les essais annuels de contrôle de la qualité recommandés par l'American Association of Physicists in Medicine (AAPM 2005), comprenant l'utilisation des mires TG18, les procédures d'essai et les critères d'acceptation devraient être utilisés. Pour cet essai, il est recommandé d'utiliser les mire modifiées qui simulent les images produites par chaque modèle de système de mammographie numérique dans l'établissement, ou qui pourrait être interprétées à ce poste de travail (c.-à-d. ont les mêmes dimensions x-y, nombre de bits, et un en-tête DICOM contenant des valeurs appropriées de tous les tags pertinents). Le système d'affichage doit être mis en marche avant de procéder à l'essai et il faut porter attention à ce que le niveau de lumière ambiante soit approprié (20 - 40 lux) et corresponde aux conditions dans lesquelles les images cliniques sont lues. Une distance de 30 cm est recommandée pour regarder les images.
- La différence de la luminance maximale entre les moniteurs appartenant à un poste d'affichage ne doit pas dépasser 10%.
- Plage de luminance - Le rapport de la luminance maximale à la luminance minimale doit être supérieur ou égal à 250 pour ce qui est des dispositifs primaires, et supérieur ou égal à 100 pour ce qui est des dispositifs secondaires.
- Fonction d'affichage d'une échelle de gris standardisée (GSDF) - Les mammogrammes numériques doivent apparaître de manière uniforme sur les différents dispositifs d'affichage électronique. Afficher et mesurer la luminance au centre de l'écran des mires de luminance de la TG18 (TG18-LN12-01 à TG18-LN12-18). À noter que cet essai ne vise pas les dispositifs d'affichage des postes d'acquisition. Utiliser le logiciel pour déterminer la conformité à la fonction d'affichage de l'échelle de gris standardisé DICOM. La fonction de contraste calculée doit se situer à ±10% de la réponse de contraste GSDF pour ce qui est de l'affichage des classes primaires et à ±20% de la réponse de contraste GSDF pour ce qui est des affichages secondaires.
- Uniformité de la luminance - La visibilité du contraste doit être uniforme dans toutes les régions du dispositif d'affichage. Afficher les séquences d'essai TG18, TG18-UNL10 et TG18-UNL80, mesurer la luminance de l'écran à cinq emplacements pour chaque moniteur. L'écart maximal de luminance, calculé par (Lmax-Lmin)/Lcentre, doit être inférieur à 0,3 (30%).
A14. Imprimantes - Le rendement des imprimantes doit être évalué de manière à s'assurer que la qualité de l'impression est uniforme et que la qualité des images imprimées est comparable à celle des images figurant sur le moniteur. Pour cet essai, il est recommandé d'utiliser une mire TG18-QC modifiée qui simule les images produites par chaque modèle de système de mammographie numérique dans l'établissement, ou qui pourrait être interprétées à ce poste de travail (c.-à-d. a les mêmes dimensions x-y, nombre de bits, et un en-tête DICOM contenant des valeurs appropriées de tous les tags pertinents).
- Distorsion géométrique - Les lignes doivent paraître droites et la déviation spatiale maximale ne doit pas dépasser 2%.
- Fonction d'affichage d'une échelle de gris standardisée - Il faut déterminer si une imprimante est conforme à la norme DICOM GSDF. Imprimer la mire TG18-PQC, mesurer la densité optique des régions marquées des 18 barres. La GSDF est déterminée par la luminance correspondant à la densité optique. Utiliser le logiciel pour déterminer la conformité avec le DICOM GSDF. La fonction de contraste calculée doit se situer à l'intérieur de ±10% de la réponse de contraste de la GSDF.
- Uniformité de la densité - La densité optique doit être uniforme dans toutes les régions de l'image imprimée. L'écart maximal de densité optique, calculé par (Lmax-Lmin)/Lcentre, doit être inférieur à 0,1 (10%).
A15. Entretien général de prévention - L'entretien général de prévention pour l'équipement et les accessoires radiogènes est nécessaire pour prolonger la durée de l'équipement. Une inspection annuelle doit être effectuée pour assurer l'intégrité structurelle, la propreté, la liberté de mouvement de toutes composantes et toute autre procédure recommandée par les fabricants.
| Item | Equipment | Systèmes | Référence |
|---|---|---|---|
| 1 | Voltmètre radiogène non-invasif Précision : ± 1,5 kV Reproductibilité: ± 0,5 kV |
FE, CR, DR | A1, A2 |
| 2 | Dosimètre Précision: ± 5 % Reproductibilité: ± 1 % |
FE, CR, DR | A3, A4, A5, A11 |
| 3 | Filtre d'Aluminium (> 99.9 % pur) Précision: 1 % épaisseur |
FE, CR, DR | A5 |
| 4 | Plaque métallique pour protéger le détecteur de rayons X (ex. : 1 mm acier, 5 mm Aluminium, > 0.1 mm plomb) |
FE, CR, DR | A5 |
| 5 | Règle(s) ou ruban à mesurer | FS, CR, DR | A5, A6, A7 |
| 6 | Deux règles radiographiques | CR, DR | A6 |
| 7 | Marqueur écran phosphorescents (environ 20mm x50 mm) | CR, DR | A6 |
| 8 | Objets de Contraste (feuille ou marqueurs métalliques, pièces de monnaie, disque de PMMA) | FE, CR, DR | A6, A7, A8 |
| 9 | Plusieurs plaques d'atténuateur uniformes et équivalent au tissu (ex. un ensemble de plaque de PMMA d'épaisseurs de 10 mm couvrant complètement la surface du détecteur et capable de fournir des épaisseurs de 20, 45 et 70 mm) ou fantôme uniforme représentant l'épaisseur moyenne du sein (ex. PMMA d'épaisseur de 45 ± 0.5 mm) | FE, CR, DR | A6, A8, A9, A10, A11 |
| 10 | Espaceurs appropriés (ex. : forme en « U » de polystyrène expansé rigide radiotransparent) |
FE, CR, DR | A8, A11 |
| 11 | Minuterie | FE, CR, DR | A8, A10 |
| 12 | Densitomètre Précision: ± 0,02 O.D. à 1,0 O.D. Reproductibilité: ± 0,01 O.D. à 1,0 O.D. |
FE | A8, A9, A10, A14 |
| 13 | Fantôme, avec des objets d'évaluation de la qualité de l'image (ex. RMI-156) | FE | A9, A11 |
| 14 | Loupe (4x à 5x grossissement) | FE, CR, DR | A9, A10, A14 |
| 15 | Outil d'analyse des ROI ou logiciel de CQ pour l'analyse d'image | CR, DR | A9 |
| 16 | Dispositif d'essai de distorsion géométrique | CR | A9 |
| 17 | Dispositif d'essai de résolution spatiale (ex. mire de resolution jusqu'à 20 pl/mm) |
FE, CR, DR | A10 |
| 18 | Dispositif d'essai FTM et un logiciel pour calculer FTM | CR, DR | A10, |
| 19 | Photomètre (pour luminance et éclairement) Précision : ± 10 % Reproducibilité: ± 5 % |
FE, CR, DR | A12, A13 |
| 16 | Mire(s) pour évaluer les dispositif d'affichage électronique et les imprimantes lasers (ex. TG18 ou SMPTE) | CR, DR | A13, A14 |
| 21 | Règle transparent | CR, DR | A14 |
Annexe I: Organismes fédéraux/provinciaux/territoriaux de sécurité radiologique
Gouvernement fédéral
Bureau de la protection contre les rayonnements des produits cliniques et de consommation
Santé Canada
P.L. 6301A
775, chemin Brookfield
Ottawa (Ontario) K1A 1C1
Direction des produits thérapeutiques
Bureau des matériels médicaux
2934, chemin Baseline, Tower B
Indice de l'adresse : 3403A
Ottawa (Ontario) K1A 0K9
Colombie-Britannique
WorkSafe BC
6951 route Westminster
Richmond, Colombie-Britannique
V7C 1C6
Alberta
Workplace Policy and Standards Development Branch
Alberta Employment, Immigration and Industry
8th floor, 10808-99th Avenue
Edmonton (Alberta)
T5K 0G5
Saskatchewan
Radiation Safety
Ministry of Labour Relations and Workplace Safety
1870 Albert Street
Regina, Saskatchewan
S4P 4W1
Manitoba
Services de radioprotection
Department of Medical Physics
CancerCare Manitoba
675 McDermot Avenue
Winnipeg (Manitoba)
R3E 0V9
Ontario (pour les questions liées à la sécurité des patients et du public)
Ministère de la Santé et des soins de longue durée de l'Ontario
Service d'inspection des installations radiologiques
55 St Clair Avenue West, 8e étage
Toronto (Ontario)
M4V 2Y7
Ontario (pour les questions liées à la sécurité des employés)
Ministère du Travail
Service de radioprotection
81A, chemin Resources
Weston (Ontario)
M9P 3T1
Québec
Direction générale de la santé publique
Le Ministère de la Santé et des Services sociaux
1075, chemin Sainte-Foy, 11e étage
Ste-Foy (Québec)
G1S 2M1
Nouveau-Brunswick
Services de radioprotection
Ministère de la Santé et des Services communautaires
B.P. 5100, Carleton Place
3e étage
Fredericton (Nouveau-Brunswick)
E3B 5G8
Nouvelle-Écosse
Occupational Health and Safety Division
Nova Scotia Department of Environment and Labour
P. 697
Halifax (Nouvelle-Écosse)
B3J 2T8
Île-du-Prince-Édouard
Department of Health and Wellness
Gouvernement de l'Île-du-Prince-Édouard
P. 2000
Charlottetown (Île-du-Prince-Édouard)
C1A 7N8
Terre-Neuve et Labrador
Department of Labour
West Block, 4e étage, edifice Confederation
B.P. 8700
St. John (Terre-Neuve)
A1B 4J6
Territoires du Nord-Ouest
Occupational Health and Safety
Government of the Northwest Territories
B.P. 1320
Yellowknife (Territoires du Nord-Ouest)
X1A 2L9
Territoire du Yukon
Occupational Health and Safety
Yukon Workers' Compensation Health and Safety Board
401, rue Strickland
Whitehorse (Territoire du Yukon)
Y1A 5N8
Annexe II : Limites de dose de rayonnement ionisant dans un contexte professionnel
Pour les besoins de ce Code de sécurité, on peut classer les individus selon deux catégories : (1) les travailleurs sous rayonnements, c'est-à-dire les personnes qui sont, de par leur profession, exposées au rayonnement et (2) le public. Les doses limites sont indiquées pour toutes les deux catégories au tableau AII.1. Ces doses limites sont fondées sur les recommandations les plus récentes de la Commission Internationale de Protection Radiologique (CIPR) comme spécifié dans la publication 103 de la CIPR (2007).
Les doses limites pour les travailleurs exposés au rayonnement s'appliquent uniquement à l'irradiation résultant directement de leur profession et elles n'incluent pas l'exposition au rayonnement provenant d'autres sources, comme le diagnostic médical et le rayonnement de fond.
| Organe ou tissu touché | Travailleurs sous rayonnements | Membres du public |
|---|---|---|
| Tout le corps | Dose efficace moyenne de 20 mSv par année reçue sur une période de cinq ans et de 50 mSv sur une période d'un an. | Dose efficace de 1 mSv |
| Cristallin | Dose équivalente moyenne de 20 mSv par année reçue sur une période de cinq ans et de 50 mSv sur une période d'un an. |
Dose équivalente de 15 mSv |
| Peau | Dose équivalente de 500 mSv | Dose équivalente de 50 mSv |
| Mains et pieds | Dose équivalente de 500 mSv | - |
|
||
Formule pour la calcule de la dose efficace
La dose efficace égale à E plus 20 fois la somme de I divise par ALI, inferieure ou égale à 4mSv.
- Tout appareil de mammographie doit porter, aux endroits indiqués ci-après et de manière lisible, permanente et visible, les renseignements suivants :
- sur la surface externe du poste de commande principal:
- un énoncé interdisant toute utilisation non autorisée de l'appareil et avertissant qu'il émet des rayons X dangereux lorsqu'il est en marche,
- le symbole de mise en garde contre les rayons X, qui doit être de deux couleurs contrastantes, être bien visible et reconnaissable à une distance de 1 m, n'avoir aucune dimension extérieure qui est inférieure à 2 cm, porter la mention «ATTENTION: RAYON X - CAUTION: X-RAYS», et être conforme à un des modèles suivants:
- une étiquette, accessible visuellement, qui indique, quant au générateur radiologique, le nom du fabricant, la désignation du modèle, le numéro de série, la date de fabrication, et le pays de fabrication;
- sur la surface externe de la gaine, une étiquette, accessible visuellement, qui indique, quant à la gaine équipée, le nom du fabricant, la désignation du modèle, le numéro de série, la date d'installation du tube radiogène dans la gaine, le pays du fabricant, et la filtration permanente minimale dans le faisceau de rayonnement X émis par la gaine équipée, exprimée en millimètres d'équivalent en aluminium à une haute tension radiogène donnée;
- sur la surface externe de la gaine, ou sur toute autre structure adéquate fixée en permanence à la gaine, un indicateur permettant d'évaluer à 2 % près la distance foyer-récepteur d'image et, si le tube radiogène et le générateur radiologique ne sont pas enveloppés dans une même gaine, des inscriptions indiquant clairement les bornes de l'anode et de la cathode sur la gaine et sur le générateur haute tension;
- sur la surface externe de tout dispositif de limitation du faisceau qui permet de filtrer davantage le faisceau de rayonnement X, une étiquette indiquant la filtration permanente totale que peut assurer ce dispositif, exprimée en millimètres d'équivalent en aluminium à une haute tension radiogène donnée.
- sur la surface externe du poste de commande principal:
- Les commandes, compteurs, avertisseurs lumineux et autres indicateurs exigés par la Partie XII du Règlement sur les dispositifs émettant des radiations doivent être clairement étiquetés quant à leur fonction.
- Il faut souligner que toute irradiation comporte un certain degré de risque et que les doses recommandées dans la présente annexe sont les valeurs maximales recommandées. Ces doses doivent être les plus basses que l'on peut raisonnablement atteindre pour éviter toute irradiation inutile.
- La CIPR ne fait pas de différence entre les hommes et les femmes en âge de procréer si la dose est reçue à un débit régulier.
- Dans le cas des travailleuses exposées aux rayonnements, dès que la grossesse est déclarée, le foetus doit être protégé d'une exposition durant le reste de la grossesse. Pour les femmes exposées de par leur profession, une limite de dose efficace de 4 mSv doit être appliquée, pendant le reste de la période de grossesse, à partir de toutes les sources de rayonnement. Ceci est calculé de la manière suivante:
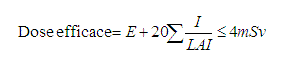
où
E est la partie de dose efficace, en millisievert- reçue par une personne de sources situées à l'extérieur du corps; et
- reçue par une personne, et engagée à son égard, de sources situées à l'intérieur du corps, mesurées directement ou dans des excréments,
I est l'activité, exprimée en becquerels, de tout radionucléide incorporé dans le corps, à l'exclusion de tout produit de filiation du radon et de tout autre radionucléide dont l'activité est prise en compte dans la détermination de E, et
LAI ou limite annuelle d'incorporation, est l'activité en becquerel, d'un radionucléide qui produira une dose effective de 20 mSv pendant 50 ans après l'absorption du radionucléide par une personne de 18 ans ou plus, ou pendant la période débutant avec l'absorption et se terminant à 70 ans si elle a été absorbée par une personne de moins de 18 ans.
Dans le champ d'application du présent document, les expositions professionnelles des employées en état de grossesse sont dues principalement aux rayonnements diffusés. Dans ce cas, la méthode la plus efficace pour surveiller les expositions du foetus est de mesurer la dose équivalente à la surface de l'abdomen à l'aide d'un dosimètre personnel de radiation.
- Pour les techniciens en stage de formation et les étudiants, les limites de dose recommandées devraient être les mêmes que pour le public en général.
- La CIPR ne recommande pas de limites spécifiques pour chacun des organes. La Commission estime que l'on peut prévenir les effets déterministes en imposant une limite annuelle de dose équivalente de 500 mSv pour tous les tissus à l'exception du cristallin de l'oeil, pour lequel elle recommande une limite de 20 mSv par an.
- La dose équivalente pour la peau est calculée en prenant la moyenne sur toute sa surface. Dans les situations où les effets déterministes sont probables, la limite de dose équivalente est 500 mSv et est calculée en prenant la moyenne sur des surfaces inférieures à 1 cm2. Cette limite s'applique à la peau du visage et des mains.
- Certaines provinces ou certains territoires peuvent prescrire pour certains travailleurs des doses limites équivalentes recommandées différentes de celles indiquées dans cette annexe. Une consultation avec l'organisme approprié peut être nécessaire pour déterminer les valeurs en vigueur dans cette province.
Annexe III : Calcul de la dose glandulaire moyenne
Méthode 1: Calcul de la dose glandulaire moyenne pour des seins simulés avec le polyméthacrylate de méthyle (PMMA) d'épaisseurs de 20, 45 et 70 mm.
La dose glandulaire moyenne, DGM, qui est la dose absorbée par les tissus glandulaires dans le sein peut être déterminée en utilisant la formule suivante :
DGM = Ki,t × gt × ct × s
Ki,t est le kerma dans l'air d'entrée (sans rétrodiffusion) à la surface supérieure d'une épaisseur de PMMA de t mm qui simule un sein standard
gt est le facteur de conversion du kerma dans l'air à DGM pour un sein composé de 50 % de tissue glandulaire, et de 50% de tissue adipeux à t mm d'épaisseur (présenté dans le tableau AIII.1)
ct est le facteur de conversion pour des différences dans la composition du sein a partir de 50% de tissue glandulaire d'un sein standard à t mm d'épaisseur (présenté dans le tableau A111.2)
s est le facteur de correction pour l'utilisation des combinaisons de la cible/filtres d'autres que Mo/Mo (présenté dans les tableaux AIII.3 et AIII.4)
Noter que les facteurs gt and ct varient en fonction de la valeur de CDT du faisceau radiogène mesuré avec la plaque de compression dans le faisceau. La valeur mesurée de CDT du système de mammographie doit être utilisée. Mesures typiques de CDT pour différentes tensions du tube radiogène et combinaisons cible / filtre sont présentes dans le tableau A111.5
| Épaisseur de PMMA (mm) | Épaisseur équivalente du sein (mm) | Facteurs-g (mGy/mGy) CDT (mm Al) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.25 | 0.30 | 0.35 | 0.40 | 0.45 | 0.50 | 0.55 | 0.60 | ||
| 20 | 21 | 0.329 | 0.378 | 0.421 | 0.460 | 0.496 | 0.529 | 0.559 | 0.585 |
| 30 | 32 | 0.222 | 0.261 | 0.294 | 0.326 | 0.357 | 0.388 | 0.419 | 0.448 |
| 40 | 45 | 0.155 | 0.183 | 0.208 | 0.232 | 0.258 | 0.285 | 0.311 | 0.339 |
| 45 | 53 | 0.130 | 0.155 | 0.177 | 0.198 | 0.220 | 0.245 | 0.272 | 0.295 |
| 50 | 60 | 0.112 | 0.135 | 0.154 | 0.172 | 0.192 | 0.214 | 0.236 | 0.261 |
| 60 | 75 | 0.088 | 0.106 | 0.121 | 0.136 | 0.152 | 0.166 | 0.189 | 0.210 |
| 70 | 90 | 0.086 | 0.098 | 0.111 | 0.123 | 0.136 | 0.154 | 0.172 | |
| 80 | 103 | 0.074 | 0.085 | 0.096 | 0.106 | 0.117 | 0.133 | 0.149 | |
| Épaisseur de PMMA (mm)) | Épaisseur équivalente du sein (mm) | Glandularité du sein équivalent |
Facteur-c CDT (mm Al) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.30 | 0.35 | 0.40 | 0.45 | 0.50 | 0.55 | 0.60 | |||
| 20 | 21 | 97 | 0.889 | 0.895 | 0.903 | 0.908 | 0.912 | 0.917 | 0.921 |
| 30 | 32 | 67 | 0.940 | 0.943 | 0.945 | 0.946 | 0.949 | 0.952 | 0.953 |
| 40 | 45 | 41 | 1.043 | 1.041 | 1.04 | 1.039 | 1.037 | 1.035 | 1.034 |
| 45 | 53 | 29 | 1.109 | 1.105 | 1.102 | 1.099 | 1.096 | 1.091 | 1.088 |
| 50 | 60 | 20 | 1.164 | 1.16 | 1.151 | 1.15 | 1.144 | 1.139 | 1.134 |
| 60 | 75 | 9 | 1.254 | 1.245 | 1.235 | 1.231 | 1.225 | 1.217 | 1.207 |
| 70 | 90 | 4 | 1.299 | 1.292 | 1.282 | 1.275 | 1.270 | 1.260 | 1.249 |
| 80 | 103 | 3 | 1.307 | 1.299 | 1.292 | 1.287 | 1.283 | 1.273 | 1.262 |
|
|||||||||
| Combinaisons cible / filtre | Épaisseur du filtre (µm) | Facteur-s |
|---|---|---|
| Mo/Mo | 30 | 1.000 |
| Mo/Rh | 25 | 1.017 |
| Rh/Rh | 25 | 1.061 |
| W/Rh | 50-60 | 1.042 |
| W/Ag | 50-75 | 1.042 |
| Épaisseur de PMMA (mm) | Épaisseur équivalente du sein (mm) | Facteur-s |
|---|---|---|
| 20 | 21 | 1.075 |
| 30 | 32 | 1.104 |
| 40 | 45 | 1.134 |
| 45 | 53 | 1.149 |
| 50 | 60 | 1.160 |
| 60 | 75 | 1.181 |
| 70 | 90 | 1.198 |
| 80 | 103 | 1.208 |
| CDT (mm Al) pour différentes combinaisons cible filtre | |||||
|---|---|---|---|---|---|
| kV | Mo + 30 μm Mo | Mo +25 μm Rh | Rh +25 μm Rh | W +50 μm Rh | W +0.45 μm Al |
| 25 | 0.33 ± .02 | 0.40 ± .02 | 0.38 ± .02 | 0.52 ± .03 | 0.31 ± .03 |
| 28 | 0.36 ± .02 | 0.42 ± .02 | 0.43 ± .02 | 0.54 ± .03 | 0.37 ± .03 |
| 31 | 0.39 ± .02 | 0.44 ± .02 | 0.48 ± .02 | 0.56 ± .03 | 0.42 ± .03 |
| 34 | 0.47 ± .02 | 0.59 ± .03 | 0.47 ± .03 | ||
| 37 | 0.50 ± .02 | 0.51 ± .03 | |||
|
|||||
Méthode 2 : Calcul de la dose glandulaire moyenne pour des seins composé de 50 % de tissue glandulaire et de 50% de tissue adipeux simulé par un fantôme d'épaisseur de 4.2cm.
La dose glandulaire moyenne (en millirad) est déterminée par le produit de l'exposition d'entrée dans l'aire (en Roentgen) et le facteur de conversion qui dépend sur la tension du tube radiogène (kVp) et la couche de demi-transmission pour une combinaison cible filtre donnée, selon la formule suivante:
DGM = K x FC
K est l'exposition dans l'aire à l'entrée en Roentgen
FC est le facteur de conversion en millirad par Roentgen (présenté dans les tableaux AIII.6, AIII.7 et AIII.8)
| Tension du tube radiogène (kVp) | W/Al Combinaison cible filtre |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CDT | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | |
| 0.23 | 116 | |||||||||||
| 0.24 | 121 | 124 | ||||||||||
| 0.25 | 126 | 129 | 131 | |||||||||
| 0.26 | 130 | 133 | 135 | 138 | ||||||||
| 0.27 | 135 | 138 | 140 | 142 | 143 | |||||||
| 0.28 | 140 | 142 | 144 | 146 | 147 | 149 | ||||||
| 0.29 | 144 | 146 | 148 | 150 | 151 | 153 | 154 | |||||
| 0.30 | 149 | 151 | 153 | 155 | 156 | 157 | 158 | 159 | 170 | |||
| 0.31 | 154 | 156 | 157 | 159 | 160 | 161 | 162 | 163 | 164 | 175 | ||
| 0.32 | 158 | 160 | 162 | 163 | 164 | 166 | 167 | 168 | 168 | 170 | 171 | 180 |
| 0.33 | 163 | 165 | 166 | 168 | 169 | 170 | 171 | 173 | 173 | 174 | 175 | 185 |
| 0.34 | 168 | 170 | 171 | 172 | 173 | 174 | 175 | 176 | 177 | 178 | 179 | 190 |
| 0.35 | 174 | 175 | 176 | 177 | 178 | 179 | 180 | 181 | 182 | 183 | 194 | |
| 0.36 | 179 | 181 | 182 | 183 | 184 | 185 | 185 | 186 | 187 | 199 | ||
| 0.37 | 185 | 186 | 187 | 188 | 189 | 190 | 191 | 191 | 204 | |||
| 0.38 | 190 | 191 | 192 | 193 | 194 | 195 | 195 | 208 | ||||
| 0.39 | 196 | 197 | 198 | 198 | 199 | 200 | 213 | |||||
| 0.40 | 201 | 202 | 203 | 201 | 204 | 217 | ||||||
| 0.41 | 206 | 207 | 208 | 208 | 221 | |||||||
| 0.42 | 211 | 212 | 212 | 225 | ||||||||
| 0.43 | 215 | 216 | 230 | |||||||||
| 0.44 | 220 | 234 | ||||||||||
| 0.45 | 238 | |||||||||||
|
||||||||||||
| Voltage du tube à rayons X (kVp) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CDT | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 |
| 0.28 | 149 | 151 | 154 | ||||||||
| 0.29 | 154 | 156 | 158 | 159 | |||||||
| 0.30 | 158 | 160 | 162 | 162 | 162 | 163 | |||||
| 0.31 | 163 | 164 | 166 | 166 | 166 | 167 | 167 | ||||
| 0.32 | 167 | 169 | 171 | 171 | 171 | 171 | 172 | 172 | |||
| 0.33 | 171 | 173 | 175 | 176 | 176 | 176 | 176 | 177 | |||
| 0.34 | 176 | 178 | 179 | 179 | 180 | 180 | 180 | 181 | 181 | ||
| 0.35 | 180 | 181 | 183 | 183 | 184 | 185 | 185 | 186 | 187 | ||
| 0.36 | 185 | 186 | 187 | 187 | 188 | 188 | 189 | 190 | 191 | 191 | |
| 0.37 | 189 | 190 | 191 | 191 | 192 | 193 | 193 | 194 | 195 | 195 | |
| 0.38 | 193 | 194 | 196 | 196 | 197 | 197 | 197 | 198 | 199 | 199 | 200 |
| 0.39 | 198 | 199 | 200 | 200 | 201 | 201 | 202 | 202 | 203 | 203 | 204 |
| 0.40 | 202 | 203 | 204 | 204 | 205 | 205 | 206 | 207 | 208 | 208 | 208 |
| 0.41 | 206 | 207 | 208 | 208 | 209 | 209 | 210 | 211 | 212 | 212 | 212 |
| 0.42 | 211 | 211 | 212 | 212 | 213 | 213 | 214 | 215 | 216 | 216 | 217 |
| 0.43 | 215 | 216 | 217 | 217 | 218 | 218 | 219 | 219 | 220 | 220 | 221 |
| 0.44 | 220 | 220 | 221 | 221 | 222 | 222 | 223 | 223 | 224 | 224 | 225 |
| 0.45 | 224 | 224 | 225 | 225 | 226 | 226 | 227 | 227 | 228 | 228 | 229 |
| 0.46 | 228 | 229 | 229 | 230 | 231 | 231 | 232 | 233 | 233 | 234 | |
| 0.47 | 233 | 233 | 234 | 235 | 235 | 236 | 237 | 237 | 238 | ||
| 0.48 | 238 | 238 | 239 | 240 | 240 | 241 | 241 | 242 | 242 | ||
| 0.49 | 242 | 243 | 243 | 244 | 244 | 245 | 245 | 246 | |||
| 0.50 | 247 | 247 | 248 | 248 | 249 | 250 | 251 | ||||
| 0.51 | 251 | 252 | 253 | 254 | 254 | 255 | |||||
| 0.52 | 257 | 257 | 258 | 258 | 259 | ||||||
| 0.53 | 261 | 261 | 262 | 263 | 264 | ||||||
| 0.54 | 265 | 266 | 267 | 268 | |||||||
| 0.55 | 269 | 270 | 271 | 272 | |||||||
| 0.56 | 275 | 276 | 276 | ||||||||
| 0.57 | 279 | 280 | 281 | ||||||||
| 0.58 | 284 | 285 | |||||||||
| 0.59 | 288 | 289 | |||||||||
| 0.60 | 293 | ||||||||||
| Voltage du tube à rayons X(kVp) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CDT | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 |
| 0.28 | 150 | 155 | 159 | ||||||||
| 0.29 | 155 | 160 | 164 | 168 | |||||||
| 0.30 | 160 | 164 | 168 | 172 | 176 | ||||||
| 0.31 | 168 | 168 | 172 | 174 | 180 | 182 | |||||
| 0.32 | 169 | 173 | 177 | 181 | 184 | 186 | 188 | ||||
| 0.33 | 174 | 178 | 181 | 185 | 188 | 190 | 192 | ||||
| 0.34 | 179 | 183 | 186 | 190 | 193 | 195 | 196 | 199 | |||
| 0.35 | 184 | 187 | 190 | 194 | 197 | 199 | 201 | 203 | |||
| 0.36 | 189 | 192 | 195 | 198 | 201 | 204 | 205 | 207 | 209 | ||
| 0.37 | 193 | 196 | 199 | 202 | 205 | 207 | 209 | 211 | 213 | ||
| 0.38 | 198 | 201 | 204 | 207 | 209 | 211 | 213 | 215 | 217 | 219 | 221 |
| 0.39 | 203 | 206 | 208 | 211 | 214 | 216 | 217 | 219 | 221 | 223 | 224 |
| 0.40 | 208 | 211 | 213 | 216 | 218 | 220 | 221 | 223 | 224 | 226 | 228 |
| 0.41 | 213 | 215 | 217 | 220 | 222 | 224 | 225 | 227 | 228 | 230 | 232 |
| 0.42 | 218 | 220 | 222 | 224 | 226 | 228 | 229 | 231 | 232 | 234 | 236 |
| 0.43 | 222 | 224 | 226 | 228 | 230 | 232 | 233 | 235 | 236 | 238 | 240 |
| 0.44 | 227 | 229 | 231 | 233 | 235 | 237 | 238 | 239 | 240 | 242 | 243 |
| 0.45 | 232 | 234 | 235 | 237 | 239 | 241 | 242 | 243 | 244 | 246 | 247 |
| 0.46 | 239 | 241 | 243 | 245 | 246 | 247 | 248 | 250 | 251 | ||
| 0.47 | 247 | 249 | 250 | 251 | 252 | 254 | 255 | ||||
| 0.48 | 251 | 253 | 254 | 255 | 256 | 258 | 259 | ||||
| 0.49 | 257 | 258 | 259 | 260 | 261 | 262 | |||||
| 0.50 | 261 | 262 | 263 | 264 | 265 | 266 | |||||
| 0.51 | 266 | 267 | 268 | 269 | 270 | ||||||
| 0.52 | 270 | 271 | 272 | 273 | 274 | ||||||
| 0.53 | 275 | 276 | 276 | 277 | 278 | ||||||
| 0.54 | 279 | 280 | 280 | 281 | |||||||
| 0.55 | 283 | 284 | 284 | 285 | |||||||
| 0.56 | 288 | 288 | 289 | ||||||||
| 0.57 | 292 | 293 | |||||||||
| 0.58 | 296 | 297 | |||||||||
| 0.59 | 300 | ||||||||||
| 0.60 | 304 | ||||||||||
Annexe IV : Guides de blindage pour le stockage des films radiographiques
Le tableau suivant indique l'épaisseur de plomb requise pour réduire le niveau de rayonnement (film et cassettes chargées) à 1,75 µGy (0,2 mR) pour une charge de travail hebdomadaire de 1 000 mA-min à 35 kilovolts (max).
| Distance entre le tube radiogène et le film stocké | |||||
|---|---|---|---|---|---|
| moins de 1 m | 0,2 m | plus de 3 m | 4 m | 0,5 m | |
| 1 jour | 0,4 mm | 0,3 mm | 0,2 mm | 0,2 mm | 0,1 mm |
| 1 semaine | 0,5 mm | 0,4 mm | 0,4 mm | 0,3 mm | 0,3 mm |
| 1 mois | 0,6 mm | 0,5 mm | 0,4 mm | 0,4 mm | 0,4 mm |
| 1 an | 0,7 mm | 0,6 mm | 0,6 mm | 0,6 mm | 0,5 mm |
Il est à noter que les cassettes de CR chargées avec une plaque d'imagerie ont un taux d'utilisation plus élevé que des cassettes chargées avec le film et ont donc un période de stockage plus courte (NCRP 147). Pour le stockage des cassettes CR chargées avec une plaque d'imagerie, les spécifications des fabricants concernant le niveau du blindage doivent être suivies.
Annexe V: Unités de mesure du rayonnement
Exposition
Suivant l'exemple de la Commission électrotechnique internationale, le kerma dans l'air (en Grays, Gy) remplace l'exposition (en Roentgens, R) comme mesure l'irradiation. La relation entre ces deux unités est la suivante :
- 1 Gy ~ 115 R
- 1 mGy ~ 115 mR
- 1 R ~ 8,73 mGy
- 1mR ~ 8,73 µGy
Dose absorbée
Le Gray (Gy) remplace le rad (rad) comme unité de dose absorbée. La relation entre ces deux unités est la suivante :
- 1 Gy ~ 100 rad
- 1mGy ~ 100 mrad
- 1 rad ~ 10 mGy
- 1 mrad ~ 10 µGy
Dose équivalente
Le Sievert (Sv) remplace le rem (rem) comme unité de dose équivalente. La relation entre ces deux unités est la suivante :
- 1 Sv ~ 100 rem
- 1 mSv ~ 100 mrem
- 1 rem ~ 10 mSv
- 1mrem ~ 10 µSv
Nota : m = milli = 10-3; µ = micro = 10-6
Annexe VI LexiqueFootnote 1
« artéfact »
toute structure ou caractéristique visible dans l'image qui ne fait pas partie de l'objet étudié
«atténuation»
réduction d'une grandeur liée au rayonnement lors du passage de ce rayonnement à travers la matière résultant de tous les types d'interaction avec cette matière
«cassette»
boîte étanche à la lumière, dans lequel l'écran renforçateur et le film ou une plaque CR sont placés.
«champ de rayon x»
dans l'intersection d'un faisceau de rayon x et d'une surface, lieu des points où l'intensité de rayonnement dépasse un niveau spécifique ou spécifié
«champ lumineux»
la zone lumineuse dans le plan du récepteur d'image qui simule le faisceau de rayonnement
«coefficient de variation»
le rapport entre l'écart type et la valeur moyenne d'une série de mesures, calculé selon l'équation suivante :

Équation pour le calcul du coefficient de variation
Le coefficient de variation, calculé au moyen de l'équation « C égal S divisé par X barre égal à, parenthèse carrée ouverte, la somme de i égal 1 à n, parenthèse ouverte, de X indice i moins X barre, parenthèse fermée, au carré, le tout divisé par n moins un, parenthèse carrée fermée, à la puissance d'un demi ».
où
Xi = la valeur de la ie mesure
X = la valeur moyenne des mesures
S = l'écart type estimé
n = le nombre de mesures
C = le coefficient de variation
«commande automatique d'exposition»
dans un appareil à rayonnement x, mode de fonctionnement dans lequel un ou plusieurs paramètres de change sont commandés de manière automatique pour obtenir, à un emplacement présélectionné, une quantité de rayonnement désirée
«couche de demi-transmission» ou «CDT»
épaisseur d'un matériau spécifié, qui transmet dans les conditions du faisceau étroit un rayonnement x avec un spectre particulier en réduisant le débit de kerma dans l'air, le débit d'exposition ou le débit de dose absorbée à la moitié de la valeur que l'on mesurerait en l'absence du matériau considéré.
«densitomètre»
instrument de mesure de la densité optique ou le niveau de noircissement du film
«dispositif de limitation du faisceau»
dispositif destiné à limiter le champ de rayonnement
«dose absorbée»
l'énergie moyenne déposée par un rayonnement ionisant à un volume de matière, divisée par la masse de ce volume. L'unité de mesure est le gray (Gy).
«dose efficace»
mesure de dose visant à rendre compte de l'ampleur du détriment radiologique. La dose efficace est obtenue par la somme des produits des doses équivalentes aux tissus ou aux organes, multipliées par leurs facteurs de pondération tissulaires respectifs. L'unité de mesure est le sievert (Sv).
«dose équivalente»
mesure de la dose au tissu ou à l'organe, visant à rendre compte de l'ampleur du dommage causé au tissu ou à l'organe. La dose équivalente est obtenue en multipliant la dose absorbée par un facteur de pondération radiologique pour tenir compte de l'efficacité biologique de différents rayonnements à causer des dommages au tissu ou à l'organe considéré. L'unité de mesure est le sievert (Sv). «dose glandulaire moyenne» dose moyenne absorbée par les tissus glandulaires (à l'exception de la peau) d'un sein uniformément comprimé de composition tissulaire connue, en utilisant une méthode de calcul spécifiée
«fantôme»
dispositif destiné à simuler des parties du patient à des fins d'essais
«filtre»
matériel ou dispositif qui modifie les caractéristiques du faisceau de rayonnement lorsque le faisceau le traverse
«gaine équipée»
ensemble d'une gaine avec un tube radiogène incorporé
«kerma dans l'aire»
l'énergie déposée par une unité de masse de l'aire. L'unité de mesure de kerma dans l'aire est le gray (Gy). Pour les rayons x dont l'énergie est moins que 300 kilo-électron volts (keV), l'ampleur de kerma dans l'aire est la dose absorbée sont équivalentes.
«paramètres de charge»
paramètre ayant une influence, de par sa valeur, sur la charge du tube radiogène, par exemple courant dans le tube radiogène, temps de charge, puissance anodique continue, haute tension radiogène et taux d'oscillation de celle-ci
«PMMA»
polyméthacrylate de méthyle, est également connu sous le nom générique acrylique et le nom de marque Plexiglas, Acrylate, Lucite, Perspex
«poste de commande»
partie d'un appareil ayant pour but de commander la totalité ou une partie des fonctions de l'appareil. Le poste de commande peut comporter des dispositifs indiquant ou visualisant des paramètres de fonctionnement.
«rayonnement de fuite»
rayonnement ionisant ayant traversé la barrière de protection radiologique de la source de rayonnement, ainsi que pour certains groupes radiogènes, le rayonnement ayant traversé la fenêtre avant et après l'application de la charge
«récepteur d'image»
dispositif destiné à transformer l'image radiologique potentielle en une autre forme, à partir
de laquelle une image visible est obtenue directement ou indirectement. L'image radiologique potentielle est l'information contenue dans un faisceau de rayonnement x dont la distribution de l'intensité a été modulée par l'objet traverse.
«sensitomètre»
appareil permettant d'exposer de manière reproductible un film à des niveaux différents de lumière
«voile»
le signal non désiré ajouté à l'image par l'exposition du récepteur d'image à la lumière, le rayonnement ou la chaleur entre les expositions des patients
Référence
AAPM (2005). American Association of Physicists in Medicine. Assessment of Display Performance for Medical Imaging Systems, AAPM On-Line Report No. 03 (American Association of Physicists in Medicine Task Group 18 Imaging Informatics Subcommittee).
ACR (1999). American College of Radiologists. Mammography Quality Control Manual 1999.
AIEA(2009) Agence international de l'énergie atomique. Human Health Series No.2. Quality Assurance Programme for Screen Film Mammography [en anglais seulement]
http://www-pub.iaea.org/MTCD/publications/PDF/Pub1381_web.pdf
AIEA (2011) Agence international de l'énergie atomique. Human Health Series No.17. Quality Assurance Programme for Digital Mammography
http://www-pub.iaea.org/books/iaeabooks/8560/Quality-Assurance-Programme-for-Digital-Mammography
Bick, U., Diekmann, F., Digital Mammography, Medical Radiology - Diagnostic Imaging and Radiation Oncology 2010, 88-102.
CAR (2008). L'Association canadienne des radiologistes, Normes de la CAR en matière de Téléradiologie. http://www.car.ca/uploads/standards%20guidelines/normes_teleradiologie_fr.pdf
CAR (2011). CAR Position Statement on the Use of Thyroid Shields. http://www.car.ca/uploads/about/201106_ps_car_thyroid_shield_fr.pdf
CAR (2012). L'Association canadienne des radiologistes, Lignes directrices relatives aux demandes d'examen en radiologie. http://www.car.ca/fr/standards-guidelines/guidelines.aspx
CAR PAM (2011). L'Association canadienne des radiologists Programme d'agrément en mammographie. http://www.car.ca/fr/accreditation/map.aspx
Coldman, A.J., Major, D., Doyle, G.P., et coll. Organized Breast Screening Programs in Canada: Effect of Radiologist Reading Volumes on Outcomes [en anglais seulement]. Radiology 2006;238(3): 809-815. http://radiology.rsnajnls.org/cgi/content/abstract/238/3/809)
CIPR (2007). Commission internationale de protection radiologique. Recommendation 2007 de la Commission internationale de protection radiologique. CIPR Publication 103, Annals de la CIPR 37 (2-4)1-332.
CIPR (2011). Commission internationale de protection radiologique. Statement on Tissue Reactions [en anglais seulement], 21 avril, 2011. http://www.icrp.org/docs/ICRP%20Statement%20on%20Tissue%20Reactions.pdf
CEI (2007). Commission électrotechnique internationale. Essais d'évaluation et de routine dans les services d'imagerie médicale - Partie 3-2:Essais d'acceptation - Performance d'imagerie des appareils de mammographie à rayonnement X, 2e éd., CEI 61223-3-2.
CEI (2008). Commission électrotechnique internationale. Appareils électromédicaux - Partie 1-3: Exigences générales pour la sécurité de base et les performances essentielles - Norme collatérale: Radioprotection dans les appareils à rayonnement X de diagnostic, 2e éd., CEI 60601-1-3.
CEI (2011). Commission électrotechnique internationale. Appareils électromédicaux - Partie 2-45: Exigences particulières pour la sécurité de base et les performances essentielles des appareils de mammographie à rayonnement X et des appareils mammographiques stéréotaxiques, 3e éd., CEI 60601-2-45.
D.R. DANCE et autres, Additional factors for the estimation of mean glandular breast dose using the UK mammography dosimetry protocols , Physics in medicine and biology, vol. 45, nº 11, novembre 2000, p. 3237.
Gennaro, G.et al. Digital breast tomosynthesis versus digital mammography: a clinical performance study Eur Radiol (2010) 20: 1545-1553 DOI 10.1007/s00330-009-1699-5
Helvie, M.A. "Digital Mammography Imaging: Breast Tomosynthesis and Advanced Applications", Radiologic Clinics of North America, Volume 48, Issue 5, September 2010, Pages 917-929, ISSN 0033-8389, 10.1016/j.rcl.2010.06.009.
IHE (2007). Integrating the Healthcare Enterprise Radiology: Mammography User's Handbook Revision 1.0. IHE, Chicago. http://www.ihe.net/Resources/handbook.cfm
IHE (2009). Integrating the Healthcare Enterprise Radiologie : Technical Framework Supplement
Extensions to the Portable Data for Imaging (PDI) Integration Profile Revision 1.0. IHE. http://www.ihe.net/technical_framework/index.cfm
MSSS (2001). Manuel de contrôle de la qualité en mammographie - Volume 1 - Technologue en radiologie http://msssa4.msss.gouv.qc.ca/fr/document/publication.nsf/
fb143c75e0c27b69852566aa0064b01c/fe422dad3353003985256ad300698cca?OpenDocument
MSSS (2006). Manuel de contrôle de la qualité pour la mammographie et la biopsie guidée par stéréotaxie (PQDCS) - Volume 2 http://msssa4.msss.gouv.qc.ca/fr/document/publication.nsf/
4b1768b3f849519c852568fd0061480d/1b95146d6a98fa1d8525719400538e57?OpenDocument
NCRP (2004). National Council on Radiation Protection and Measurement. Structural Shielding Design for Medical X-ray Imaging Facilities, NCRP Report No. 147 (National Council on Radiation Protection and Measurements, Bethesda, Maryland).
NEMA (2001). National Electrical Manufacturers Association. Digital Imaging and Communications in Medicine (DICOM) Part 14:Grayscale Standard Display Function, National Electrical Manufacturers Association, Roslyn, VA.
Loi sur les dispositifs émettant des radiations, R. S., C.34.
Règlement sur les dispositifs émettant des radiations, C.R.C., c. 1370, s. 3, Partie XII Appareils de radiodiagnostic.
SC (1995) Santé Canada. http://www.hc-sc.gc.ca/ewh-semt/pubs/radiation/Code-29/index-fra.php
Stanton, L., et autres. Dosage evaluation in mammography, Radiology, no 150, 1984, p. 577-584.
Van Engen, R., Van Wouldenberg, S., Bosmans, H., Young, K., Thijssen, M. European Protocol for the Quality Control of the Physical and Technical Aspects of Mammography Screening - Screen-Film Mammography. In: European Guidelines for Quality Assurance in Breast Cancer Screening and Diagnosis, 4th ed. European Commission, Luxembourg, (2006a).
Van Engen, R., Young, K., Bosmans, H., Thijssen, M. European Protocol for the Quality Control of the Physical and Technical Aspects of Mammography Screening - Digital Mammography. dans: European Guidelines for Quality Assurance in Breast Cancer Screening and Diagnosis, 4th ed. European Commission, Luxembourg, (2006b). Van Ongelval, C., Bellon, E., Deprez, T., Van Steel, A., Marchal, G., "Digital Workflow, PACS, and Telemammography", Digital Mammography, (BICK, U., DIEKMANN, F., Eds), Springer-Verlag, Berlin-Heidelberg-New York, (2010).
WU, X. Breast dosimetry in screen-film mammography, dans BARNES, G. T., et G.T. FREY, Screenfilm Mammography: Imaging Consideration and Medical Physics Responsibilities, Madison (Wisconsin), Medical Physics Publishing, (1991).
WU, X., et autres. Normalized average glandular dose in molybdenum target-rhodium filter and
rhodium target-rhodium filter mammography, Radiology, vol. 193, n° 1, octobre (1994), p. 83-89.