
The Georgia Basin-Puget Sound Airshed Characterization Report 2014: chapter 8
8. Particulate Matter
Roxanne Vingarzan (Environment Canada), Robert Kotchenruther (Environmental Protection Agency Region 10), Rebecca Saari, Sarah Hanna and Rita So (Environment Canada)
Particulate Matter (PM) consists of airborne particles in liquid or solid form which can have many different origins and diverse chemical properties. PM is usually characterized by its physical size or diameter, ranging from nanometers (nm) to several tens of micrometers (µm). Of particular interest to this report are particles less than 2.5 µm, which are commonly referred to as PM2.5 or fine particulate matter. These fine particles are of special concern for two reasons. First, these particles are able to penetrate deep into the human respiratory system and can be absorbed into the blood where they have been shown to cause biological effects. Second, these particles scatter light very efficiently and therefore play a major role in visibility impairment, a subject addressed in Chapter 9. Some forms of PM are emitted directly into the air (primary PM), including fugitive dust, sea spray, soot from wood smoke, forest fires and fossil fuel combustion. PM can also form in the atmosphere (secondary PM), as a result of reactions involving compounds such as nitrogen oxides (NOx), sulphur dioxide (SO2), volatile organic compounds (VOCs) and ammonia (NH3). This chapter reviews the various constituents that make up PM, the contribution of natural sources and background levels to ambient PM concentrations, the trends and variations in PM levels and identifies areas of concern in the airshed.
8.1 Classification of Ambient PM by Particle Size
As previously mentioned, particulate matter is usually characterized by its physical size and composition. Figure 8.1 illustrates a distribution of fine particulate matter by number count and mass. Particles with a diameter less than 10 μm are classified as PM10, which, for reference, is smaller than a human hair. Particles with diameters between PM10 and PM2.5 are defined as the coarse fraction. PM2.5 includes all particles with diameters less than 2.5 μm, also known as fine particles. The smallest particles, ultrafines, have diameters <0.1 um. Ultrafines, which are the most numerous but have the smallest mass and volume, are the subject of recent investigations into health effects.
The residence time of particulate matter in the atmosphere depends on its size. Coarse particles settle out very quickly by gravity. Smaller particles have atmospheric lifetimes on the order of a few days and are eventually removed from the atmosphere by wet deposition in rain, snow or fog, or by dry deposition through gravity or surface turbulence (see Chapter 12, “Atmospheric Deposition and Ecological Effects”). During their residency in the atmosphere, particles are transported away from their sources and undergo various physical and chemical transformation processes.
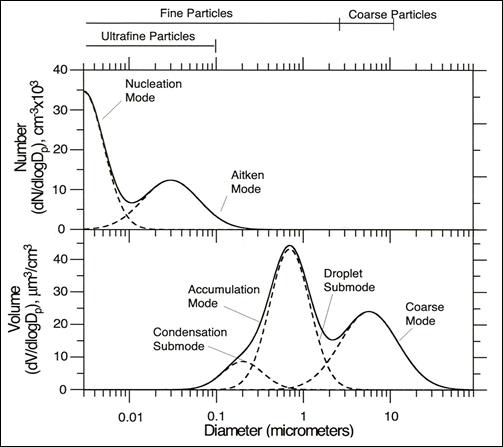
Figure 8.1. Distribution of fine particulate matter by number count and mass (from NARSTO, 2004)
Description of Figure 8.1
Figure 8.1 has two panels, the top showing fine particle number distribution and the bottom showing fine particle volume distribution. In both cases the x-axis is a log scale showing diameter from 0.003 to 90 microns. Bars along the top of the plots indicate that ultrafine particles are those less than 0.1 microns in diameter, fine particles are those less than 2.5 microns in diameter, and coarse particles are those from 2.5 to 10 microns in diameter.
In the top panel the y-axis is the normalized number concentration (dN/dlogDp) from 0 to 40 cm-3 x 103. Two log-normally distributed modes appear in the number concentration. One is the nucleation mode which peaks around 0.003 microns at 35 cm-3 x 103. The other is the Aitken mode which peaks between 0.02 and 0.03 microns at approximately 14 cm-3 x 103. These two modes overlap between 0.004 and 0.02 microns with the minimum between them occurring at 0.01 microns and approximately 6 cm-3 x 103.
In the bottom panel the y-axis is the normalized volume concentration (dV/dlogDp) from 0 to 50 µm/cm3. Two modes, the accumulation mode and the coarse mode appear in the volume concentration. The accumulation mode can be further decomposed into the condensation submode and the droplet submode. Both of these submodes and the coarse mode are log-normally distributed. The condensation submode peaks around 0.02 microns at approximately 10 µm/cm3. It extends from 0.005 to 1 micron. The droplet submode peaks at approximately 0.7 microns at 45 µm/cm3. It extends from 0.15 to 3 microns. The droplet submode makes up the bulk of the accumulation mode with the condensation submode appearing as a small shoulder. The coarse mode peaks around 6 microns at approximately 25 µm/cm3. It extends from 0.5 to 50 microns.
Nucleation takes place when it is more energetically feasible for a molecule to be in the liquid or solid phase and when there is an insufficient number of particles to scavenge the molecule of interest. This can occur when hot gases cool and condense, when reactions in the gas phase produce species of lower volatility or species are very hygroscopic. In each of these cases clusters of molecules lead to the production of a new particle, in a process known as gas-to-particle conversion. Most Aitken nuclei start as primary particles and grow by condensation or by coagulation. Coagulation occurs as a result of collisions between particles to form larger particles. Coagulation effectively increases the mass of particles while depleting smaller particles, resulting in a shift in the aerosol-size distribution toward larger particle sizes. An example of particles which form by gas-phase reactions is the reaction of H2SO4 with H2O to form hydrates of sulphuric acid (H2SO4·NH2O), thus transferring gaseous material to the particulate phase (Seinfeld and Pandis, 1998).
Particles may also grow through condensation by coming into contact with reactive vapours which condense onto the surface of the particle to form a larger secondary particle. In the presence of high relative humidity, water vapour will condense onto small particles to form cloud droplets. This aqueous phase enables chemical reactions such as the conversion of SO2 to SO4= to take place (NARSTO, 2004).
The volume or mass distribution in Figure 8.1 is dominated by two modes, the accumulation mode (from around 0.1 to around 2 μm) and the coarse mode (from around 2 to around 50 μm). The distribution shows a maximum around 0.6 to 0.8 μm in diameter, with a second peak near 6 μm. Accumulation-mode particles result from primary emissions, formation of secondary sulphate and nitrate aerosols, condensation of organics from the gas phase and coagulation of smaller particles. Particles in the coarse mode are usually produced by mechanical processes, such as wind or erosion (dust, sea salt, pollens, etc.) (NARSTO, 2004).
8.2 Sources and Formation of Secondary PM
The formation and composition of fine particulate matter is related to its emission sources. Primary particles are emitted directly into the atmosphere, whereas secondary particles are formed within the atmosphere.
Primary particulate matter is emitted by sea spray, blowing dust, tree pollens, forest fires, fossil fuel combustion and construction activities. These primary particles are found in both the fine (PM2.5) and the coarse fractions (PM2.5 to PM10). Primary particles of natural origin tend to be in the coarse fraction, while anthropogenic primary particles can be either fine or coarse.
Most secondary particles in the PM2.5 fraction are formed from gaseous precursors through physiochemical oxidation processes. The gases most commonly involved are SO2, NOx, NH3 and VOCs and the most common oxidants are OH, O3, and NO3. This section describes the key sources and formation mechanisms of particulates associated with SO2, NOx, NH3 and VOC precursors.
8.2.1 SO2
The oxidation of sulphur dioxide (SO2) to sulphate (SO4=) is an important chemical transformation in the formation of fine particulate matter (NARSTO, 2004). Natural sources of SO2 include volcanoes, marine bacteria and wetlands, although the dominant source is the combustion of fossil fuels (Makar, 2001). Sulphate is usually created through the chemical reaction of SO2 with the hydroxyl radical (OH) in air, aqueous oxidation reactions after the dissolution of SO2 in cloud, fog or rain water, and the oxidation of SO2 in reactions within the aerosol particles themselves. The first process dominates in the daytime but is negligible at night (NARSTO, 2004). The aqueous-phase oxidation that produces SO4= in cloud water can occur very quickly, often converting all of the available SO2. It has been reported that cloud and fog water can be responsible for producing as much as half of the SO4= (McHenry and Dennis, 1994; Langner and Rodhe, 1991), causing elevated concentrations of sulphate (Pandis et al., 1992). Particles are formed in this aqueous process after the cloud water has evaporated, leaving particulate sulphate in the form of sulphuric acid (H2SO4). Sulphuric acid is very acidic and is one of the main components of acid precipitation. Ammonia (NH3), the only basic gas phase species, will readily combine with H2SO4 to form ammonium sulphate ((NH4)2SO4). This is the fully neutralized form of the particle; however, if insufficient NH3 is available, ammonium bisulphate (NH4HSO4) is formed, resulting in a particle that is still acidic.
8.2.2 NOx
Oxides of nitrogen (NO, NO2) are formed biologically in soil, by lightning, and through reactions with oxygen in high-temperature combustion. The nitrate radical (NO3), ozone and the OH radical are involved in reactions during the daytime. Although NOx (NO+NO2) reacts with ozone to produce the NO3radical throughout the day, much of it is quickly destroyed via photolysis during the daytime (Makar, 2001). However the lack of photochemistry allows the buildup of NO3 at night. This reaction is important in the overnight reduction of ozone concentrations. These reactions alter the concentrations of the three species (NO3, O3, OH) that are the main oxidizing agents leading to the formation of aerosols (Makar, 2001). Another important chemical reaction involves the conversion of NOx (NO+NO2) to nitric acid (HNO3). This occurs in reactions between NO2 and OH during the day to form HNO3, and between NO2 and the NO3 radical to form dinitrogen pentoxide (N2O5) at night. N2O5 undergoes hydrolysis to form HNO3. HNO3 equilibrium in the atmosphere between the gaseous and liquid phase is mostly a function of ambient temperature, where warmer temperatures results in more off-gassing of aerosol HNO3. Nitric acid will react with NH3, sea salt and dust, although the production of NH4NO3 is the most likely outcome when sufficient NH3 is available (NARSTO, 2004). The latter is also enhanced by lower temperatures and high relative humidity. Particles resulting from the NH3 pathway are in the fine fraction, whereas particles formed with sea salt or dust can also be found in the coarse fraction. More detailed information on the chemical reactions forming particulate matter and aerosols is provided in Makar (2001).
8.2.3 NH3
Nitrogen in the form of ammonia (NH3) reacts with both HNO3 and H2SO4 to produce fine particulate matter. These reactions have been described previously. It is important to point out that the reaction between NH3 and HNO3 forming NH4NO3 depends on the relative concentrations of NH3 and SO4=. Generally the available ammonia preferentially reacts with SO4= first, since it is more acidic than HNO3 (although it does not always completely neutralize all available SO4=), before any excess becomes available to the NO3-neutralization reaction (Blanchard and Hldy, 2003). The nitric acid pathway prefers lower temperatures and higher relative humidities to enhance particle formation (NARSTO, 2004). Ammonia emissions play an important role in the temporal and spatial distribution of fine particulate matter (Barthelmie and Pryor, 1998).
8.2.4 Organics
The organic portion of fine particulate matter is a complex combination of hundreds of different organic compounds (NARSTO, 2004). Primary organic aerosols are emitted directly into the atmosphere from both natural and anthropogenic sources. Compounds that are important to the formation of organic aerosols are gaseous, semi-volatile and non-volatile organic compounds (Makar, 2001). The reactions and processes that lead to the formation of aerosols is complex. To move from the gaseous to the liquid phase, the organic compounds must be either semi- or non-volatile. Gaseous organics can produce products from oxidation reactions that are semi or non-volatile. Hence, many organic compounds may participate in the formation of particles. Particles that are formed in the atmosphere from organics are called secondary organic aerosols (SOAs). The ability of VOCs to form SOAs depends on many of the same conditions required to produce other secondary particles: temperature, relative humidity, concentration of individual VOCs, reactivity, availability of oxidants and surface area, and the volatility of products (NARSTO, 2004).
8.2.5 Natural Sources of PM
Windblown dust is the most common natural source of primary particles. However, sea spray, volcanoes, vegetative pollen and wildfires are also natural sources of primary particles. Organic compounds released from vegetation (biogenics) may be in the form of primary particles or they may react with other compounds to produce SOAs, as described earlier. This process, in which biogenic organic compounds produce fine particles, is very important in areas that are dominated by vegetation and can account for a large portion of the sub-micrometre particles.
Marine environments, volcanoes and wetlands are natural sources of sulphur and other chemical compounds that enter into reactions to form secondary particles. The saltwater areas within Georgia Basin/Puget Sound are examples of a natural source producing sulphur. A study by Norman et al. (2004) found that 30 per cent of the non-sea salt sulphate captured in precipitation over the southern Gulf Islands and up to 13 per cent of the aerosol sulphate in the Lower Fraser Valley (LFV) came from the oxidation of dimethyl sulphide from algal emissions. This estimate is in agreement with the biogenic contribution estimated for the Georgia Basin during peak phytoplankton bloom periods (Sharma et al., 2003). These natural sources provide a source of sulphate that reacts with other air pollutants, NH3 in particular, to form fine PM.
A portion of ambient PM10 and PM2.5 present in the Georgia Basin/Puget Sound airshed originates from long-range transport and is comprised of both soil dust and anthropogenic pollution. On average, this contribution is estimated to be in the order of 1.5 - 2 µg/m3 of the annual average PM2.5 concentrations (McKendry, 2006). Occasionally Asian dust events can cause regional PM episodes. In 1998, an Asian dust event produced PM10 concentrations in excess of 65 μg/m3 over the entire area from late April to early May, when normal levels would be 20 μg/m3(U.S. EPA, 2002). Chilliwack, in the eastern portion of the LFV, recorded PM10concentrations of 120 μg/m3 and PM2.5 concentrations of 44 μg/m3 (Suzuki and Taylor, 2003) during this period. Large scale long-range transport dust events have covered 15 to 30% of the area of the continental United States and have been shown to increase PM2.5levels by 8.7±2.3 μg/m3 on average during such events (U.S. EPA, 2002).
8.2.6 Background PM
Background levels of particulate matter describe the portion of the PM from sources that are not within the control of local jurisdictions. McKendry (2006) reviewed studies of background PM relevant to British Columbia and identified the following major candidate sources:
- Wind-blown local crustal material. The most well-known are the winter outflow events in the eastern Lower Fraser Valley when strong winds (8-10 m/s) mobilize riverbed materials (McKendry, 2000).
- Local forest fires (predominantly during summer) that affect mainly interior regions of B.C. but can have an impact on coastal air quality and neighbouring areas (including Washington State).
- Trans-Pacific transport of dust (McKendry et al. 2001) arising from severe spring dust storms in Asia. Due to the long distance from source, the mean particle diameter for this material is approximately 2.5 μm and can thus contribute significantly to PM2.5 (Husar et al. 2001).
The impact of episodic events of these sources on background PM levels are summarized in Table 8.1. The contribution of long-range transport will be explored further in Chapter 11,”Transboundary Transport”.
| Episodic sources | Maximum Ambient PM2.5 Concentration | Duration of Event | Episodic Contribution to Ambient Levels | Spatial Extent | Frequency |
|---|---|---|---|---|---|
| Kelowna August 2003 Forest Fires |
284 µg/m
3
|
7 days
|
100%
|
Regional
|
Inter-annual
|
| April 1998 Asian Dust Event1 |
44 µg/m
3
|
days
|
50%
2
|
Continental
|
Possibly decadal
|
| Chilliwack local dust event (7 Jan 2005) |
PM2.5 at 2-5 µg/m
3 (
PM
10 at 50 µg/m
3)
|
days
|
0-50%
|
Local
|
Annual
|
Notes:
1 McKendry et al, 2001; Husar et al. 2001
2 U.S. EPA (2002) suggest an increase of 8.7± 2.3 μg/m3 during dust events, with mean maximum dust contributions of 19.7± 8.4 μg/m3
Description of Table 8.1
Table 8.1 presents the impact of three episodic sources on PMbackground levels in British Columbia.
The first row of the table contains the headers “Episodic sources”, “Maximum Ambient PM2.5 Concentration”, “Duration of Event”, “Episodic Contribution to Ambient”, “Spatial Extent”, and “Frequency”. The first column shows the different episodic sources being considered. These are:
- Kelowna August 2003 Forest Fires
- April 1998 Asian Dust Event (references: McKendry et al, 2001; Husar et al. 2001)
- Chilliwack local dust event (7 Jan 2005)
The second through sixth columns give the details of how each episodic source impacts ozone in British Columbia.
For the Kelowna August 2003 forest fires maximum ambient PM2.5concentration is 284 µg/m3, duration of event is 7 days, episodic contribution to ambient is 100%, spatial extent is regional, and frequency is inter-annual.
For the April 1998 Asian Dust Event maximum ambient PM2.5 concentration is 44 µg/m3, duration of event is days, episodic contribution to ambient is 50% (U.S. EPA (2002) suggest an increase of 8.7± 2.3 μg/m3 during dust events, with mean maximum dust contributions of 19.7± 8.4 μg/m3), spatial extent is continental, and frequency is possibly decadal.
For the Chilliwack local dust event maximum ambient PM2.5 concentration is 2-5 µg/m3 (PM10 at 50 µg/m3), duration of event is days, episodic contribution to ambient is 0-50%, spatial extent is local, and frequency is annual.
Several studies have estimated PM background levels in regions within or near the Georgia Basin/Puget Sound airshed. Dann et al.(2011) reported mean and median values of 3.6 and 3.2 µg/m3, respectively, for the Saturna Island regional site in the Georgia Basin. This was based on an analysis of air parcel trajectory clusters associated with the lowest 6-hour average PM2.5concentrations at the 95th percentile between 2003 and 2006. McKendry (2006) estimated a mean background concentration in air masses arriving in British Columbia from north Pacific trajectories to be 1.5 - 2 µg/m3. Also on the Pacific west coast, Jaffe et al. (2005) estimated a combined marine/Asian PM2.5 background of 1.5 µg/m3 (median) and 2.1 µg/m3 (mean) at Crater Lake, Oregon. Vingarzan (2004) reported a western North American background range of 1-4 µg/m3. Long-term temporal trends in background PM are not available due to lack of data.
8.3 Annual, Seasonal and Diurnal Concentrations of Ambient PM2.5
8.3.1 PM2.5 Concentrations and Temporal Cycles
The annual average PM2.5 mass concentrations in rural portions of the Georgia Basin/Puget Sound airshed vary from 2 to 4 μg/m3. Near the major urban centres of Seattle, Victoria and Vancouver, PM2.5 annual averages are 7 to 8 μg/m3. Annual average PM2.5 TEOM-derived concentrations in the Lower Fraser Valley range between 5-6 μg/m3 (NAPS, 2006).
In the Lower Fraser Valley, summertime PM2.5 episodes sometimes occur during stagnant meteorological conditions conducive to secondary aerosol formation. In addition, transported smoke from forest fires in British Columbia and elsewhere can also contribute to locally observed PM2.5 episodes.
Cold season PM2.5 episodes occur in the fall or winter months when emissions from wood burning and space heating add to the existing PM load, coupled under stagnant meteorological conditions and lower mixing heights. Elevated PM2.5 levels at some locations in the LFV, including Vancouver Airport and Pitt Meadows can be caused by wood smoke in nearby residential areas (Larson et al., 2007). Similarly, peak average annual concentrations (98th percentile) in Seattle and Tacoma are usually observed in the fall and winter, reaching levels of 28-31 μg/m3 in Seattle and 43-46 μg/m3 in Tacoma.
During episodic conditions, 24 hour maxima typically exceed 15 μg/m3 and occasionally exceed 20 μg/m3 in the Lower Fraser Valley (Brook et al., 2011). Although rare, there have been occasional episodes where PM2.5 concentrations have reached extreme levels. The most recent such event occurred at the time of the Burns Bog fire in the summer of 2005. On September 19, a blanket of smoke covered the LFV, causing hourly PM2.5 concentrations to exceed 274 μg/m3 at Burnaby South and 24 hour average PM2.5concentrations to exceed 40 μg/m3 in some locations. As a result of forest fires, summertime smog, winter inversions and localized fireworks activity PM2.5 levels occasionally exceed Metro Vancouver’s 24 hour objective of 25 μg/m3 (Metro Vancouver, 2010).
Weekly variation in PM2.5 in the Georgia Basin is generally small (Suzuki and Taylor, 2003). The same is true of daily PM2.5, although several sites show evening peaks, and some show traffic-related peaks. Suzuki and Taylor (2003) found a diurnal pattern consisting of small peaks in the morning and evening, and mass concentrations varying only a few μg/m3 from the mean daily value. More recently, for the year 2006, Brook et al. (2011) reported a morning rush hour peak and a more pronounced evening peak at the Kitsilano site, which is located in a dense urban area near Vancouver’s downtown. The morning PM2.5 peak was not observed at the more rural sites of Hope and Langley. The shallow nocturnal boundary layer kept average hourly PM2.5 levels elevated overnight at all sites by concentrating local emissions (Brook et al., 2011).
8.4 Ambient PM2.5 Levels Compared to Standards and Objectives
Ambient standards for PM2.5 provide a regulatory baseline for assessing ambient concentrations. PM2.5 concentrations in the Georgia Basin/Puget Sound are usually below national objectives and standards in most regions, with a notable exception in the Puget Sound.
Figure 8.2 shows that Tacoma PM2.5 has been exceeding the 24-hour NAAQS of 35 μg/m3 for the entire period of record. For Marysville, PM2.5 exceeded the 24-hour NAAQS but has declined and reached borderline attainment since 2005. Regional average levels in Pierce County also exceed this standard; its highest 98th percentile 24-hour PM2.5 levels were over 41 μg/m3 between 2005 and 2008 (U.S. EPA, 2010). On December 14, 2009, EPA formally designated the Tacoma-Pierce County as a non-attainment area for the 24-hour PM2.5 NAAQS (WA DOE, 2010a).
Figure 8.2. US. EPA PM2.5 standard metric for Marysville, Seattle and Tacoma in the Puget Sound
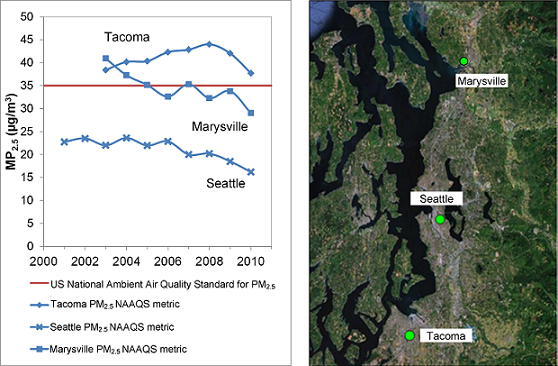
Notes: 3 year running means of highest 98th percentile from daily data (AIR Station 53-061-1007 for Marysville)
(AIRS Station 53-033-0080 for Seattle) (AIRS Station 53-053-0029 for Tacoma)
Description of Figure 8.2
Figure 8.2 has two parts. On the right is a satellite image showing the Puget Sound area with the locations of Marysville, Seattle, and Tacoma marked by circles. On the left is a plot with the years 2000 through 2011 on the x-axis and PM2.5concentration from 0 to 50 µg/m3 on the y-axis. A horizontal line at 35 ppb marks the current US National Ambient Air Quality Standard for PM2.5. The data shown are 3-year running means of highest 98th percentile from daily data (AIRS Station 53-061-1007 for Marysville, AIRS Station 53-033-0080 for Seattle, and AIRS Station 53-053-0029 for Tacoma).
For Marysville there is data for 2003 through 2010. The 2003 concentration was just above 40 µg/m3 , this fell steadily to approximately 32 µg/m3 in 2006 and remained between 30 and 35 µg/m3 through 2009. In 2010 the concentration was between 25 and 30 µg/m3.
For Seattle there is data for 2001 to 2010. From 2001 to 2006 the concentration remained between 20 and 25 µg/m3. After 2006 the concentration fell steadily to approximately 15 µg/m3 in 2010.
For Tacoma there is data for 2003 to 2010. From 2003 to 2008 the concentration roe steadily from approximately 37 µg/m3 to just below 45 µg/m3. After 2008 the concentration fell back to approximately 37 µg/m3 in 2010.
Other areas in the Georgia Basin/Puget Sound remain well below their respective national PM2.5 standards. Figure 8.2 shows Seattle to be in attainment of the U.S. National Ambient Air Quality Standard. Similarly, PM2.5 levels at four sites in the Lower Fraser Valley (Kitsilano, Pitt Meadows, Abbotsford, and Chilliwack) are well below the Canada-Wide Standard (Figure 8.3).
8.5 PM2.5 Trends
National-scale PM2.5 monitoring efforts are relatively recent in both countries, so the period of record is too short to develop long-term trends at many sites. Trend analysis is further complicated by changes to PM monitoring methods, namely the addition of dryers to TEOMs. Nonetheless, PM2.5 concentrations at most sites appear to be on a slow decline (Figure 8.2 and Figure 8.3), with the exception of Tacoma. At Mount Rainier in the Puget Sound (not shown), WA DOE (2002) found a statistically significant decreasing trend in PM2.5of 0.32 μg/m3 per year from 1988-1999. Similarly, 24-hour average PM2.5 data from Rocky Point in the Georgia Basin (not shown) indicate a statistically significant (p<0.05) decreasing trend of 0.42 μg/m3per year between 1993 and 2004. Recent analyses of particle backscatter (a surrogate for PM2.5 measurements) in the Lower Fraser Valley also indicate statistically significant declining trends since 2002 (So and Vingarzan, 2010).
Figure 8.3 Canada Wide Standard PM2.5 standard metric for four sites in the Lower Fraser Valley.
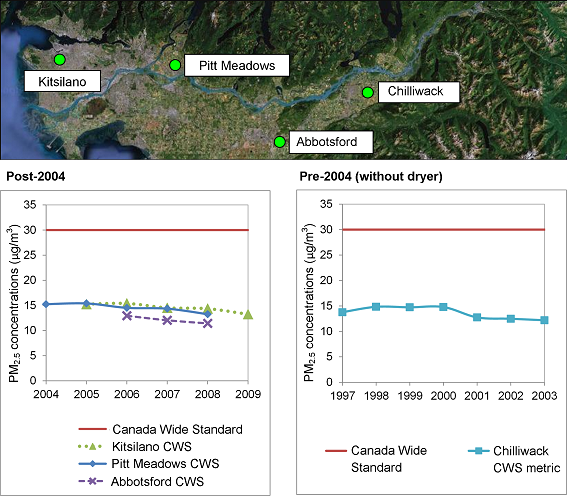
Notes: 3 year running means of highest 98th percentile based on 24-hour average data (NAPS Stations, west to east: 100118, 101202,101004, 101101)
Pre-2004 data was collected without a dryer and is thus shown separately.
Description of Figure 8.3
Figure 8.3 is composed of three parts. The upper panel is a satellite image of the Lower Fraser Valley with the locations of the Kitsilano, Pitt Meadows, Abbotsford, and Chilliwack air quality monitoring stations marked by circles. Two plots, one on the lower left and one the lower right, show PM2.5concentrations (in µg/m3) at these stations versus time. The lower left plot shows the years from 2004 through 2009. Pre-2004 data was collected without a dryer and is thus plotted separately in the lower right panel which shows the years 1997 to 2003. Both plots have a horizontal line at 30 µg/m3 marking the Canada Wide Standard. The data shown is 3-year running means of the highest 98th percentile based on 24-hour average data (NAPS Stations, west to east: 100118, 101202,101004, 101101)
For the Kitsilano station there is data for 2005 to 2009. In 2005 the PM2.5concentration was approximately 15 µg/m3. This declined slowly to between 10 and 15 µg/m3 in 2009.
For the Pitt Meadows station there is data for 2004 to 2008. In 2005 the PM2.5concentration was approximately 15 µg/m3. This declined slowly to between 10 and 15 µg/m3 in 2009.
For the Abbotsford station there is data for 2006 to 2008. In 2005 the PM2.5concentration was between 10 and 15 µg/m3. This declined slowly to approximately 10 µg/m3 in 2009.
The Chilliwack station is the only one for which there is pre-2004 data and in this case the record is from 1997 to 2003. In 1997 the PM2.5concentration was just below 15 µg/m3. It remained stable through 2000 before declining to approximately 12 µg/m3 in 2001 where it remained stable through 2003.
8.6 Compositional Analysis of fine PM
Understanding the composition of PM is the key to understanding its impacts on human health and the environment. Particulate matter can carry toxins and metals and variably affect the climate, ecology and visibility. Thus, particle composition can be as important as concentration when it comes to understanding and assessing ambient PM2.5impacts.
In Figure 8.4 , the relative concentrations of ammonium, sodium, sulphate and nitrate in PM2.5 are shown along with the measured particle size for two sites in the WISE (note that particle size decreases from left to right). Sodium nitrate dominates the coarse fraction (>2.5 um), whereas ammonium sulphate and, to a lesser degree, ammonium nitrate appear in the finer fraction <2.5 um). This is primarily due the formation of coarse mode sodium nitrate following the reaction of nitric acid and sea salt (NaCl).
Figure 8.4. Chemical composition and size fraction of PM2.5 at two locations in the Georgia Basin airshed (Pryor, 2003)
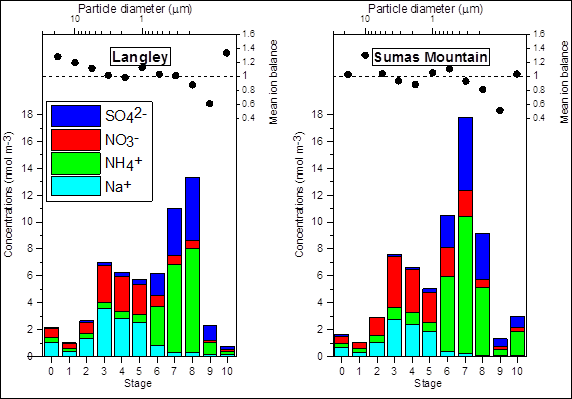
Notes: (1) Ten-stage micro-orifice uniform deposit impactors (MOUDI-110) were deployed at Langley and Sumas Mountain. Each stage has a successively smaller cut-point diameter to enable particle size classification. (2) Ion balance is a means of checking the accuracy of analytical results. Departures from a ratio of anions to cations of 1.0 is a measure of error in the analysis or a missing ion.
Description of Figure 8.4
Figure 8.4 has two panels, one for Langley and one for Sumas Mountain. In each panel there is a stacked bar chart of showing the concentration of ammonium, sodium, sulphate and nitrate (in mol/m3) for 11 different particle sizes. The particles were collected with ten-stage micro-orifice uniform deposit impactors (MOUDI-110). Each stage has a successively smaller cut-point diameter to enable particle size classification. The particle diameters for each of the stages 0 through 10 are plotted as 20, 10, 6, 3.5, 2, 1, 0.6, 0.35, 0.15, and 0.125 microns. For each size/stage the mean ion balance is also given. Ion balance is a means of checking the accuracy of analytical results. Departures from a ratio of anions to cations of 1.0 is a measure of error in the analysis or a missing ion.
The data for Langley is as follows:
- Stage 0 had approximately 1 mol/m3 sodium, 0.25 mol/m3 ammonium, 0.5 mol/m3 nitrate, and less than 0.1 mol/m3 sulphate. The mean ion balance was 1.4.
- Stage 1 had approximately 0.25 mol/m3 sodium, 0.1 mol/m3 ammonium, 0.25 mol/m3 nitrate, and less than 0.1 mol/m3 sulphate. The mean ion balance was 1.2.
- Stage 2 had approximately 1.1 mol/m3 sodium, 0.25 mol/m3 ammonium, 0. 5 mol/m3 nitrate, and less than 0.1 mol/m3 sulphate. The mean ion balance was 1.1.
- Stage 3 had approximately 3.5 mol/m3 sodium, 0.5 mol/m3 ammonium, 2.5 mol/m3 nitrate, and 0.25 mol/m3 sulphate. The mean ion balance was 1.0.
- Stage 4 had approximately 3 mol/m3 sodium, 0.5 mol/m3 ammonium, 3 mol/m3 nitrate, and 0.25 mol/m3 sulphate. The mean ion balance was 1.0.
- Stage 5 had approximately 3 mol/m3 sodium, 0.5 mol/m3 ammonium, 2 mol/m3 nitrate, and 0.25 mol/m3 sulphate. The mean ion balance was 1.1.
- Stage 6 had approximately 1 mol/m3 sodium, 2.5 mol/m3 ammonium, 1 mol/m3 nitrate, and 2 mol/m3 sulphate. The mean ion balance was 1.0.
- Stage 7 had approximately 0.25 mol/m3 sodium, 6.5 mol/m3 ammonium, 1 mol/m3 nitrate, and 4 mol/m3 sulphate. The mean ion balance was 1.0.
- Stage 8 had approximately 0.25 mol/m3 sodium, 8 mol/m3 ammonium, 0.75 mol/m3 nitrate, and 4 mol/m3 sulphate. The mean ion balance was 0.9.
- Stage 9 had approximately 0.1 mol/m3 sodium, 1 mol/m3 ammonium, 0.1 mol/m3 nitrate, and 1 mol/m3 sulphate. The mean ion balance was 0.6.
- Stage 10 had approximately 0.1 mol/m3 sodium, 0.1 mol/m3 ammonium, 0.1 mol/m3 nitrate, and 0.1 mol/m3 sulphate. The mean ion balance was 1.3.
The data for Sumas Mountain is as follows:
- Stage 0 had approximately 0.75 mol/m3 sodium, 0.25 mol/m3 ammonium, 0.5 mol/m3 nitrate, and 0.25 mol/m3 sulphate. The mean ion balance was 1.0.
- Stage 1 had approximately 0.25 mol/m3 sodium, 0.25 mol/m3 ammonium, 0.25 mol/m3 nitrate, and less than 0.1 mol/m3 sulphate. The mean ion balance was 1.3.
- Stage 2 had approximately 1.0 mol/m3 sodium, 0.5 mol/m3 ammonium, 1. 5 mol/m3 nitrate, and less than 0.1 mol/m3 sulphate. The mean ion balance was 1.0.
- Stage 3 had approximately 3 mol/m3 sodium, 1 mol/m3 ammonium, 4 mol/m3 nitrate, and less than 0.1 mol/m3 sulphate. The mean ion balance was 0.9.
- Stage 4 had approximately 2.5 mol/m3 sodium, 1 mol/m3 ammonium, 2.5 mol/m3 nitrate, and less than 0.1 mol/m3 sulphate. The mean ion balance was 0.8.
- Stage 5 had approximately 2 mol/m3 sodium, 1 mol/m3 ammonium, 2 mol/m3 nitrate, and 0.25 mol/m3 sulphate. The mean ion balance was 1.1.
- Stage 6 had approximately 0.25 mol/m3 sodium, 5.5 mol/m3 ammonium, 2 mol/m3 nitrate, and 2 mol/m3 sulphate. The mean ion balance was 1.1.
- Stage 7 had approximately 0.25 mol/m3 sodium, 10 mol/m3 ammonium, 2 mol/m3 nitrate, and 6 mol/m3 sulphate. The mean ion balance was 0.9.
- Stage 8 had less than 0.1 mol/m3 sodium, 5 mol/m3 ammonium, 0.5 mol/m3 nitrate, and 4 mol/m3 sulphate. The mean ion balance was 0.8.
- Stage 9 had less than 0.1 mol/m3 sodium, 0.5 mol/m3 ammonium, 0.25 mol/m3 nitrate, and 0.5 mol/m3 sulphate. The mean ion balance was 0.5.
- Stage 10 had less than 0.1 mol/m3 sodium, 2 mol/m3 ammonium, 0.25 mol/m3 nitrate, and 1 mol/m3 sulphate. The mean ion balance was 1.0.
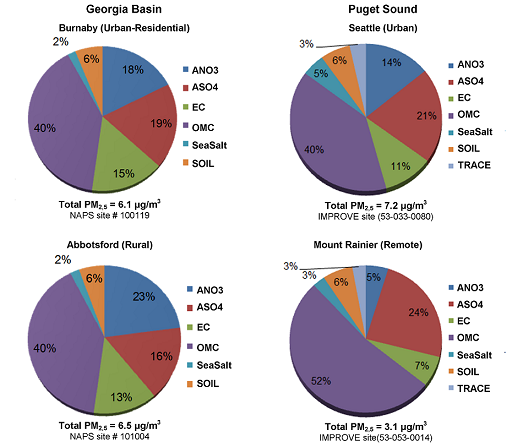
A compositional analysis of PM2.5 in the Georgia Basin/Puget Sound airshed is depicted in Figure 8.5. Organic carbon, ammonium nitrate and ammonium sulphate are the major components at all sites. To compare the four sites, which range from the remote Mount Rainier to urban Seattle, first consider the range in annual average PM2.5 concentrations. Seattle has the highest annual PM2.5 level (7.2 μg/m3), and Mount Rainier has half the level of any other site (3.1 μg/m3). Urban-residential Burnaby and rural Abbotsford have commensurate levels, which are similar to but less than those of Seattle.
There are some notable differences among the composition of PM2.5 at these sites. Ammonium sulphate is most important at the Seattle site, possibly due to higher local industrial and marine emissions. Mt. Rainier has a high sulphate component as well, which is likely attributable to transported sulphate and precursors; however, its total sulphate levels are much lower than Seattle’s, on a mass basis (0.8 μg/m3 compared to 1.6 μg/m3).
The total mass of sulphate at the Burnaby and Abbotsford sites are very similar however due to the presence of high concentrations of NH4, due to intense agricultural activity, the Abbotsford site has considerably higher nitrate present. High ammonia levels enhance ammonium nitrate formation, which in this airshed is limited by the availability of oxidized nitrogen species, not ammonia.
Figure 8.5. Percentage contribution to reconstructed total mass (RTM) for fine particulate matter from six PM2.5 constituents in Georgia Basin and Puget Sound.
Notes for Puget Sound RTM:
Average for all days from 2004-2008
ANO3 = ammonium nitrate = 1.29*NO3
ASO4 = ammonium sulphate = 1.375*SO4
EC = elemental carbon
OMC = organic matter = 1.8*OC (for rural locations) or 1.4*OC (for urban locations)
Sea Salt = sodium chloride = 1.8*(Chloride)
Soil = 2.2*Al + 2.49*Si + 1.63*Ca + 2.42*Fe + 1.94*Ti
Trace = sum of all other trace constituents
Description of Figure 8.5
Figure 8.5 shows data from two stations in the Georgia Basin (Burnaby which is an urban-residential site and Abbotsford which is a rural site) and data from two stations in the Puget Sound (Seattle which is an urban site and Mount Rainier which is a remote site). For each site there is a pie chart showing percentage contribution to reconstructed total mass (RTM) for fine particulate matter from six PM2.5 constituents. These constituents are ANO3 , ASO4, EC, OMC, Sea Salt, Trace and Soil.
There are the following notes for Georgia Basin RTM.
- Average for all days from 2003-2008
- ANO3 = ammonium nitrate = 1.29*NO3
- ASO4 = ammonium sulphate = 1.375*SO4
- EC = elemental carbon
- OMC = organic matter = 1.6*OC (as site is mixed urban/rural)
- Sea Salt = sodium chloride = 1.8*(Chloride)
- Soil = 3.48*Si+1.63*Ca+1.5*Fe+1.41*K+1.94*Ti
- Soil calculation does not include aluminum because aluminum data were not robust.
- The RTM was greater than the PM2.5 mass, indicating that one or more of the reconstructed constituent masses were overestimated. This also results in a negative value for Trace elements, which was therefore not included in the plots above.
There are the following notes for the Puget Sound RTM:
- Average for all days from 2004-2008
- ANO3 = ammonium nitrate = 1.29*NO3
- ASO4 = ammonium sulphate = 1.375*SO4
- EC = elemental carbon
- OMC = organic matter = 1.8*OC (for rural locations) or 1.4*OC (for urban locations)
- Sea Salt = sodium chloride = 1.8*(Chloride)
- Soil = 2.2*Al + 2.49*Si + 1.63*Ca + 2.42*Fe + 1.94*Ti
- Trace = sum of all other trace constituents
For Burnaby the percentages are ANO3 18% , ASO4 19%, EC 15%, OMC 40%, Sea Salt 2%, and Soil 6%. Total PM2.5 = 6.1 μg/m3.
For Abbotsford the percentages are ANO3 23% , ASO4 16%, EC 13%, OMC 40%, Sea Salt 2%, and Soil 6%. Total PM2.5 = 6.5 μg/m3.
For Seattle the percentages are ANO3 14% , ASO4 21%, EC 11%, OMC 40%, Sea Salt 5%, Soil 6%, and Trace 3%. Total PM2.5 = 7.2 μg/m3.
For Mount Rainier the percentages are ANO3 5% , ASO4 24%, EC 7%, OMC 52%, Sea Salt 3%, Soil 6%, and Trace 3%. Total PM2.5 = 3.1 μg/m3.
8.6.1 Seasonal Variations in PM2.5 Composition
The annual average compositions shown in Figure 8.5 also vary seasonally. These variations are shown in Figure 8.6 and Figure 8.7for the Puget Sound and the Georgia Basin, respectively. The seasonal patterns in the urban and rural sites in Figure 8.6 differ noticeably, both with regard to total mass and composition. Total mass peaks in the fall and winter for urban locations like Seattle, reflecting the increased frequency of reduced mixing depths and stagnant air episodes coupled with higher levels of direct PMemissions from wood smoke. Residential wood smoke is an important source of PM2.5in Seattle (responsible for 51% of King County’s emissions in 2005), and its emissions are known to peak during the colder months (WA DOE, 2007). Burnaby South, in the Georgia Basin, is an urban-residential site showing a similar but less pronounced pattern with PM2.5 concentrations peaking in autumn and winter (Figure 8.7). Conversely, rural Mt. Rainier has higher PM2.5 concentrations in summer, likely reflecting biomass burning (e.g. forest fire plumes) and SOA production from oxidation of biogenic VOCs.
Abbotsford, a site located in an agricultural area of the LFV, differs in its seasonal pattern from the urban and remote sites already mentioned. At Abbotsford, daily PM2.5 concentrations are lowest in the winter, are higher in spring and reach a maximum in the fall with a notable increase in the organic matter fraction (Figure 8.7). Note that this site experiences distinctly different wind patterns in the winter, when winds are mostly northeasterly and pass across an upwind residential area. In the summer, winds are southwesterly and blow from the direction of agricultural fields and Whatcom County in Washington State.
Among the four sites shown in Figure 8.6 and Figure 8.7, several seasonal patterns in PM composition are apparent. Sulphate concentrations peak in the summer due to higher rates of photochemical conversion of SO2 to sulphate, increased biogenic sulphur emissions and increased boating/cruise vessel activities. At the more urban sites of Burnaby and Seattle, maximum nitrate concentrations occur in winter because nitrate deposition to particles is favoured under colder temperatures and higher humidity. At Abbotsford, nitrate deposition to particles increases in the summer months, likely due to higher ammonia availability from large emissions during the summer from agriculture.
Figure 8.6. Urban (Seattle) versus rural (Mt. Rainier) annual and seasonal average reconstructed total mass (RTM).
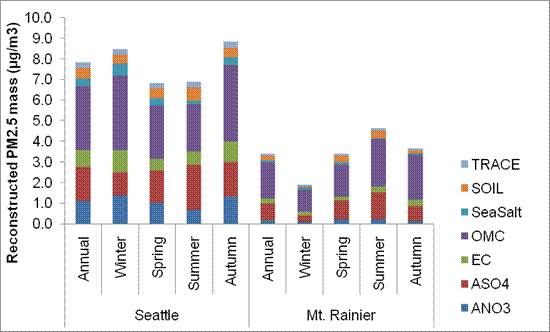
Notes: Based on 24-hour average concentrations from 1-in-3 day sampling for the years 2004-2008. IMPROVE station for Seattle (53-033-0080) and Mt. Rainier (53-053-0014). See notes for Figure 8.5 for reconstruction method and definition of constituents.
Description of Figure 8.6
Figure 8.6 is a stacked bar chart showing the reconstructed PM2.5 mass in µg/m3 for Seattle and Mt Rainier broken down by season. There is a note that the data is based on 24-hour average concentrations from 1-in-3 day sampling for the years 2004-2008. It is from the IMPROVE stations for Seattle (53-033-0080) and Mt. Rainier (53-053-0014). See notes for Figure 8.5 for reconstruction method and definition of constituents.
For Seattle the annual reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.0, ASO4 1.75, EC 0.75, OMC 3.0, Sea Salt 0.25, Soil 0.5, and Trace 0.25. The total reconstructed PM2.5 masses is approximately 8 µg/m3. The winter reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.25, ASO4 1.0, EC 1.0, OMC 4.0, Sea Salt 0.5, Soil 0.5, and Trace 0.25. The total reconstructed PM2.5 masses is approximately 8.5 µg/m3. The spring reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.0, ASO4 2.0, EC 0.5, OMC 2.5, Sea Salt 0.25, Soil 0.5, and Trace 0.25. The total reconstructed PM2.5 masses is approximately 7 µg/m3. The summer reconstructed PM2.5 masses in µg/m3 are approximately ANO3 0.75, ASO4 3.0, EC 0.5, OMC 2.5, Sea Salt 0.1, Soil 1.0, and Trace 0.25. The total reconstructed PM2.5 masses is approximately 7.5 µg/m3. The autumn reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.25, ASO4 2.0, EC 1.0, OMC 3.5, Sea Salt 0.25, Soil 0.5, and Trace 0.25. The total reconstructed PM2.5 masses is approximately 8.75 µg/m3.
For Mt Rainier the annual reconstructed PM2.5 masses in µg/m3 are approximately ANO3 0.1, ASO4 1.0, EC 0.25, OMC 2.0, Sea Salt 0.1, Soil 0.1, and Trace 0.1. The total reconstructed PM2.5 masses is approximately 3.25 µg/m3. The winter reconstructed PM2.5 masses in µg/m3 are approximately ANO3 0.1, ASO4 0.25, EC 0.1, OMC 1.0, Sea Salt 0.1, Soil 0.1, and Trace 0.1. The total reconstructed PM2.5 masses is approximately 2 µg/m3. The spring reconstructed PM2.5 masses in µg/m3 are approximately ANO3 0.25, ASO4 1.0, EC 0.25, OMC 1.5, Sea Salt 0.1, Soil 0.25, and Trace 0.1. The total reconstructed PM2.5 masses is approximately 3.5 µg/m3. The summer reconstructed PM2.5 masses in µg/m3 are approximately ANO3 0.25 , ASO4 1.5, EC 0.25, OMC 2.0, Sea Salt 0.1, Soil 0.25, and Trace 0.1 The total reconstructed PM2.5 masses is approximately 4.5 µg/m3. The autumn reconstructed PM2.5 masses in µg/m3 are approximately ANO3 0.1 , ASO4 1.0, EC 0.25, OMC 2.0, Sea Salt 0.1, Soil 0.1, and Trace 0.1 The total reconstructed PM2.5 masses is approximately 3.5 µg/m3.
Figure 8.7. Urban-residential (Burnaby South) versus rural-agricultural (Abbotsford) annual and seasonal average reconstructed total mass (RTM) for all days.
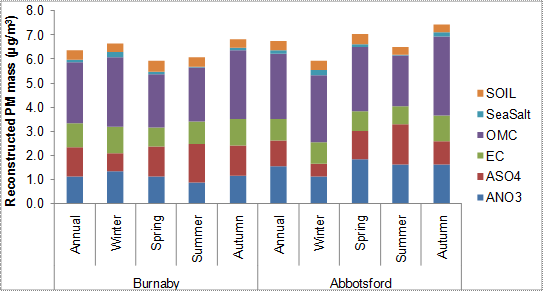
Notes: Based on 24-hour average concentrations from 1-in-3 day sampling for the years 2003-2008. NAPS station #100119 (Burnaby) and #101004 (Abbotsford). See notes for Figure 8.5 for reconstruction method and definition of constituents.
Description of Figure 8.7
Figure 8.7 is a stacked bar chart showing the reconstructed PM2.5 mass in µg/m3 for Burnaby South and Abbotsford broken down by season. There is a note that the data is based on 24-hour average concentrations from 1-in-3 day sampling for the years 2003-2008. It is from NAPS stations #100119 (Burnaby) and #101004 (Abbotsford). See notes for Figure 8.5 for reconstruction method and definition of constituents.
For Burnaby South the annual reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.0, ASO4 1.25, EC 1.0, OMC 2.25, Sea Salt 0.1, and Soil 0.25. The total reconstructed PM2.5 mass is approximately 6 µg/m3. The winter reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.25, ASO4 0.75, EC 1.0, OMC 2. 5, Sea Salt 0.25, and Soil 0.25. The total reconstructed PM2.5 mass is approximately 6.5µg/m3. The spring reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.0, ASO4 1.0, EC 0.75, OMC 2.0, Sea Salt 0.1, and Soil 0.5. The total reconstructed PM2.5 mass is approximately 6 µg/m3. The summer reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.0, ASO4 2.0, EC 1.0, OMC 2.0, Sea Salt 0.1, and Soil 0.5. The total reconstructed PM2.5 mass is approximately 6 µg/m3. The autumn reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.0, ASO4 1.0, EC 1.0, OMC 3.0, Sea Salt 0.1, and Soil 0.5. The total reconstructed PM2.5 mass is approximately 6.5µg/m3.
For Abbotsford the annual reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.5, ASO4 1.0, EC 1.0, OMC 2.5, Sea Salt 0.1, and Soil 0.5. The total reconstructed PM2.5 mass is approximately 6.5 µg/m3. The winter reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.0, ASO4 0.5, EC 1.0, OMC 2.5, Sea Salt 0.25, and Soil 0.25. The total reconstructed PM2.5 mass is approximately 6 µg/m3. The spring reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.5, ASO4 1.25, EC 1.0, OMC 2.5, Sea Salt 0.1, and Soil 0.5. The total reconstructed PM2.5 mass is approximately 6.5 µg/m3. The summer reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.5, ASO4 1.5, EC 1.0, OMC 2.0, Sea Salt 0.1, and Soil 0.5. The total reconstructed PM2.5 mass is approximately 6.5 µg/m3. The autumn reconstructed PM2.5 masses in µg/m3 are approximately ANO3 1.5, ASO4 1.0, EC 1.25, OMC 2.5, Sea Salt 0.25, and Soil 0.5. The total reconstructed PM2.5 mass is approximately 7.5µg/m3.
The seasonal variations in PM2.5 concentrations and composition shown in Figure 8.6 and Figure 8.7 are merely demonstrative and are not representative of the entire Georgia Basin/Puget Sound airshed. Seasonal patterns are likely to vary throughout the airshed depending on nearby sources and can be better understood for a given site through PMsource appointment.
8.7 PM Source Apportionment
Once the chemical composition of particulate matter has been analysed, the relative importance of the various sources involved in its formation can be determined through statistical methods such as Positive Matrix Factorization (PMF), Principal Component Analysis (PCA) and the Conditional Probability Function (CPF).
Several recent applications of PMF and CPF have been undertaken using speciated PM2.5 data collected by speciation samplers from NAPS and IMPROVE stations in the Georgia Basin/Puget Sound airshed. This includes two sites in the Georgia Basin, Burnaby South and Abbotsford (So et al., 2010) and two in the Puget Sound, Tacoma (WA DOE, 2010b), and Mount Rainier (Rose, 2006). Burnaby South, an urban-residential site in Metro Vancouver, is impacted by traffic, mixed industry and marine emissions. Abbotsford, a semi-rural site in central Lower Fraser Valley, is impacted by significant ammonia emissions, vehicle traffic and growing urbanization. Mount Rainier is a remote site in the Puget Sound, impacted by the transport of anthropogenic sulphur emissions, biogenic VOCs and biomass burning. Tacoma is an urban residential site neighbouring a major freeway and is influenced by downtown Tacoma, the Port of Tacoma, an Air Force base, an Army Base and several major industries (WA DOE, 2010b).
8.7.1 PM Sources in the Georgia Basin
Figure 8.8 and Figure 8.9 present PMF source factor contributions to PM2.5 mass for both summer and winter seasons for Burnaby South and Abbotsford, respectively (So et al., 2010). Dominant sources contributing to PM2.5 are motor vehicles, nitrate (linked to mixed combustion, but dominated here by transportation and agriculture) and sulphate (industrial, shipping and marine biogenic sources). In the Greater Vancouver area, SO2 emissions from marine traffic and oil refining contribute to particulate sulphate.
Source contributions vary by season at both sites. Wood burning, secondary nitrate, and vehicle emissions are more important in the winter, while fuel oil and marine biogenic influences are more important in the summer. Note that the marine oil combustion fraction (driven by the 3:1 Ni/V ratio tracer) is larger in the summer, during the cruise ship season. Secondary sulphate is higher during the summer due to a combination of increased SO2oxidation and biogenic emissions. Marine biogenic emissions in the form of dimethyl sulphide are also significant during the spring and summer, contributing as much as 26% of total sulphur emissions in the airshed during algal bloom periods (Sharma et al., 2003). Most of the nitrate in the urban core comes from oxidation of NOx from vehicle emissions, marine and industrial sources. Secondary nitrate contributions are lower in the summer due to preferential uptake on the particle of ammonium sulphate over ammonium nitrate, the uptake of nitric acid on coarse mode sea-salt particles and ammonium nitrate aerosol equilibrium being favoured under cold temperatures and high humidity.
Figure 8.8. Seasonal PMF factor contributions to PM2.5 mass at Burnaby.
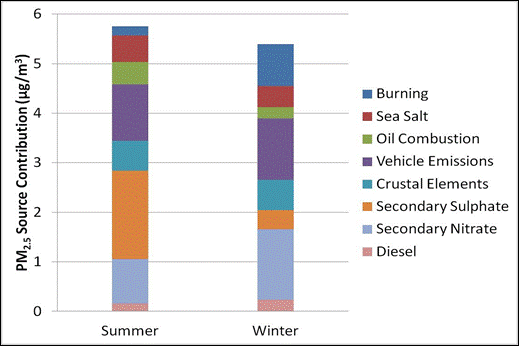
Notes: Based on PMF analysis with EPA PMF 3.0 of 24-hour average concentrations from 1-in-3 day sampling for the years 2003-2008. NAPS station #100119. (So et al., 2010)
Description of Figure 8.8
Figure 8.8 is a stacked bar chart showing PM2.5 source contributions in µg/m3 for summer and winter at Burnaby. The data are based on PMF analysis with EPA PMF 3.0 of 24-hour average concentrations from 1-in-3 day sampling for the years 2003-2008. NAPS station #100119. (So et al., 2010)
For summer the source contributions in µg/m3 are approximately as follows. Diesel 0.2, secondary nitrate 0.9, secondary sulphate 1.75, crustal elements 0.5, vehicle emissions 1.0, oil combustion 0.5, sea salt 0.6, and burning 0.2.
For winter the source contributions in µg/m3 are approximately as follows. Diesel 0.25, secondary nitrate 1.5, secondary sulphate 0.4, crustal elements 0.5, vehicle emissions 1.25, oil combustion 0.2, sea salt 0.3, and burning 0.75.
Figure 8.9. Seasonal PMF factor contributions to PM2.5 mass at Abbotsford.
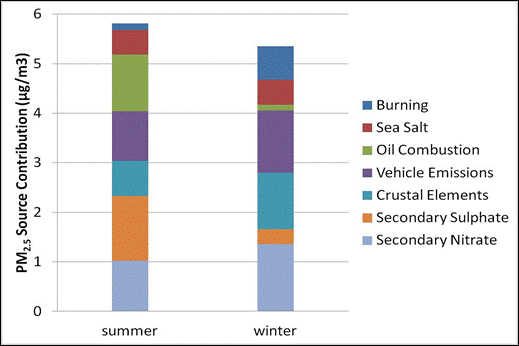
Notes: Based on PMF analysis with EPA PMF 3.0 of 24-hour average concentrations from 1-in-3 day sampling for the years 2003-2008. NAPS station #101004. (So et al., 2010).
Description of Figure 8.9
Figure 8.9 is a stacked bar chart showing PM2.5 source contributions in µg/m3 for summer and winter at Abbotsford. The data are based on PMF analysis with EPA PMF 3.0 of 24-hour average concentrations from 1-in-3 day sampling for the years 2003-2008. NAPS station #101004. (So et al., 2010).
For summer the source contributions in µg/m3 are approximately as follows. Secondary nitrate 1.0, secondary sulphate 1.25, crustal elements 0.75, vehicle emissions 1.0, oil combustion 1.2, sea salt 0.5, and burning 0.2.
For winter the source contributions in µg/m3 are approximately as follows. Secondary nitrate 1.3, secondary sulphate 0.25, crustal elements 1.1, vehicle emissions 1.25, oil combustion 0.1, sea salt 0.5, and burning 0.5.
At Abbotsford (Figure 8.9), secondary sulphate and oil combustion are dominant in the summer, and secondary nitrate and vehicle emissions dominate in the winter. Contributions from wood burning and crustal elements are also more important in the winter. Note that sea salt is overestimated at both sites due to factor separation uncertainties.
A source analysis of speciated data from Abbotsford for the period April 1994 to May 1995 estimated mobile sources at 39% (diesel 3%; gasoline 36%), secondary nitrate at 29% and secondary sulphate at 13% (Ostermann, 2002). For the more recent period of 2003-2008, So et al. (2010) estimated vehicle emissions at 21%, secondary nitrate at 24% and secondary sulphate at 12%. Methodological differences and uncertainties must be considered when comparing Ostermann’s results to So et al. (2010). Nonetheless, their results are similar, and the decrease in the relative contribution from vehicles in 2003-2008 (So et al., 2010) compared to 1994-1995 (Ostermann, 2002) is in line with the decrease in smog-forming emissions from vehicles as shown in Figure 5.9 (Chapter 5, “Emissions”).
8.7.2 PM Sources in Tacoma in the Puget Sound
Tacoma
WA DOE (2010b) performed a PMF study for Tacoma using similar methods to So et al. (2010). The data were collected from January 2006 to May 2009 using the Speciation Trends Network (STN) sampling protocols in South Tacoma, Washington. The source apportionment analysis was conducted using Positive Matrix Factorization (PMF) (Paatero and Tapper, 1994; Paatero, 1997) and the Conditional Probability Function (CPF) (Ashbaugh et al., 1985).
The sources contributing to PM2.5 at the Tacoma site, from most to least significant, were as follows: wood smoke (45%), secondary particles (25%), motor vehicles (13%), industrial emissions (6%), fugitive dust (4%), fresh and processed sea salt (4%), fireworks (2%), and oil combustion and ships (1%). Wood smoke was the single most important source of PM2.5 in Tacoma, contributing about half of the PM2.5 on most days in the winter, as well as on days when PM2.5 levels were the highest. Since outdoor burning is prohibited near the site, the smoke origin is likely from residential wood stoves.
Pronounced seasonal variations were observed for wood smoke, fireworks, secondary nitrate, motor vehicles, industrial emissions and fresh sea salt. Most of the fugitive dust and fireworks contributions occurred in the summer, while most of the wood smoke contributions occurred in the winter. Average contributions from motor vehicles and industry were highest in the winter probably due to the low atmospheric mixing heights. The increased number of cold-starts may also be a factor in the winter.
Although the annual average contributions from wood smoke and local industry appear to have declined each year since 2006, a similar decline was not observed with other sources or with the annual 98th percentile contributions from wood smoke.
Mount Rainier
At Mount Rainier, the dominant components of PM2.5 are organics and sulphate (Figure 8.5), which Rose (2006) attributed largely to biomass and secondary sulphate, respectively. Rose’s analysis compared the periods of 1991-1995 and 2000-2003 to explore multi-year trends and seasonal variations in PM sources at the remote site. The compositional analysis of the average and 90 percentile daily PM2.5 from Mount Rainier for the 1991-1995 and 2000-2003 time periods is presented in Figure 8.10. Biomass (organic carbon) contributed the largest amount of fine particulates for both time periods (35%-40% of the reconstructed total mass). This biomass contribution was likely composed of two main sources: (1) local or transported biomass smoke (from outdoor burning, wood-stoves, wild fires and other sources); and (2) secondary organic aerosols (SOAs) produced by oxidation of biogenic volatile organic compounds (VOCs) (Rose, 2006) (WA DOE, 2002). Between the two time periods, average biomass concentrations at Mt. Rainier decreased by 42% and the concentrations from secondary sulphate source, contributing the second highest amount of fine particulate (~30% of the reconstructed total mass), decreased by 37%. A possible reason could be that emission controls were added to a large power plant upwind from Mount Rainier National Park in 2001, which lead to a significant reduction in sulfur dioxide emissions, from 87,756 tons in 1999 to 3,355 tons by 2005 (Clow and Campbell, 2008).
Seasonal trends were identified in the secondary sulphate, secondary nitrate, soil and marine sources. The months of the year during which each source made its highest contribution to PM2.5 at Mount Rainier are shown in Table 8.2.
Figure 8.10. PMF factor contributions to PM2.5 mass at Mount Rainier
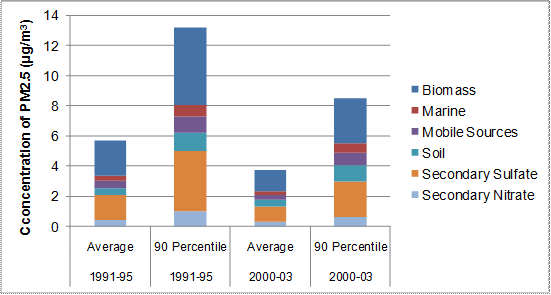
Notes: Based on PMF analysis with EPA PMF 3.0 of mean 24-hour average concentrations, and 90th percentile 24-hour concentrations, from 1-in-3 day sampling for the years 1991-1995 and 2000-2003. IMPROVE station (53-053-0014). (Rose, 2006).
Description of Figure 8-10
Figure 8.10 is a stacked bar chart showing PM2.5 source contributions in µg/m3 for 1991-95 and 2000-03 at Mt Rainier. The data for each time period are broken down into average and 90th percentile cases. They are based on PMF analysis with EPA PMF 3.0 of mean 24-hour average concentrations and 90thpercentile 24-hour concentrations from 1-in-3 day sampling IMPROVE station (53-053-0014). (Rose, 2006).
For 1991-95 the average source contributions in µg/m3 are approximately as follows. Secondary nitrate 0.5, secondary sulphate 1.5, soil 0.5, mobile sources 0.5, marine sources 0.25, and biomass 2.25. The 90th percentile source contributions in µg/m3 are approximately as follows. Secondary nitrate 1.0, secondary sulphate 4.0, soil 1.5, mobile sources 1.25, marine sources 0.5, and biomass 5.0.
For 2000-2003 the average source contributions in µg/m3 are approximately as follows. Secondary nitrate 0.5, secondary sulphate 1.0, soil 0. 5, mobile sources 0.25, marine sources 1.25, and biomass 2.25. The 90th percentile source contributions in µg/m3 are approximately as follows. Secondary nitrate 0.5, secondary sulphate 2.25, soil 1.0, mobile sources 0.75, marine sources 0.5, and biomass 3.25.
| PM2.5 Source |
Peak Contribution
|
|---|---|
| Biomass |
October-March
|
| Marine |
March-October
|
| Mobile Sources |
No pattern
|
| Soil |
March-October
|
| Secondary Sulfate |
April-October
|
| Secondary Nitrate |
April-October
|
Description of Table 8.2
Table 8.2 has two columns with the headers “PM2.5Source” and “Peak Contribution”. The sources and their peak contributions are as follows:
- Biomass: October-March
- Marine: March-October
- Mobile Sources: No pattern
- Soil - March: October
- Secondary Sulfate: April-October
- Secondary Nitrate: April-October
Seattle
A source apportionment study by Wu et al. (2007) using PM2.5 and VOC data from Beacon Hill in Seattle indicated that wood burning (24%-31%), secondary sulphate (20%-24%) and secondary nitrate (15%-20%) were the main contributors to PM2.5. Figure 8.11 shows the source contribution estimates from the Multi-linear Engine (ME-2) models with PM2.5 data only (left) and with both PM2.5and VOCs data (right) The study concluded that adding the VOC data to the speciated PM2.5 data in source apportionment modelling resulted in more accurate source contribution estimates for combustion related sources.
Figure 8.11. PMF factor contributions to PM2.5 mass at Beacon Hill (Seattle).
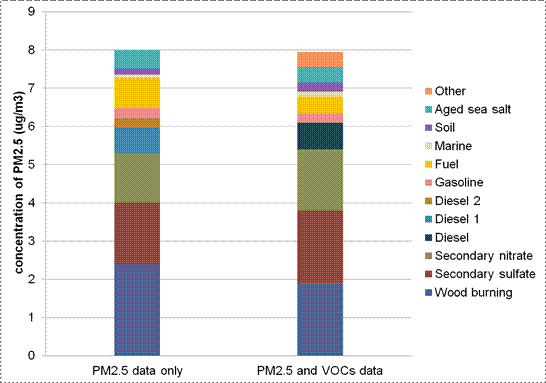
Notes: Based on PMF analysis with Multi-linear Engine (ME-2) of mean 24-hour average concentrations, from 1-in-3 day sampling for the years 2000-2004. The left stacked bar indicates results generated by the PM2.5 data only model with 100% constraint. The right stacked bar indicates results generated from the combined PM2.5 and VOCs data model with 100% constraint. IMPROVE station (53-033-0080). (Adapted from Wu et al., 2007).
Description of Figure 8.11
Figure 8.11 is a stacked bar chart showing PM2.5 source contributions in µg/m3 for Beacon Hill in Seattle. The data are broken down into PM2.5 only on the left and PM2.5 + VOCs on the right. There is a note that the data are based on PMF analysis with Multi-linear Engine (ME-2) of mean 24-hour average concentrations from 1-in-3 day sampling for the years 2000-2004. The left stacked bar indicates results generated by the PM2.5 data only model with 100% constraint. The right stacked bar indicates results generated from the combined PM2.5 and VOCs data model with 100% constraint. Beacon Hill (Seattle) is IMPROVE station (53-033-0080). (Adapted from Wu et al., 2007).
For the PM2.5only case the source contributions in µg/m3 are approximately as follows. Wood burning: 2.4, secondary sulphate: 1.6, secondary nitrate: 1.3, diesel 1: 0.7, diesel 2: 0.25, gasoline: 0.25, fuel: 0.75, marine: 0.1, soil: 0.15, and aged sea salt: 0.5.
For the PM2.5+ VOCs case the source contributions in µg/m3 are approximately as follows. Wood burning: 1.9, secondary sulphate: 2.0, secondary nitrate: 1.5, diesel: 0.8, gasoline: 0.25, fuel: 0.5, marine: 0.1, soil: 0.25, aged sea salt: 0.3, and other: 0.3.
8.7.3 PM Composition and Source Apportionment of the Lower Fraser Valley during the Pacific 2001 Air Quality Study
The Pacific 2001 Air Quality Study was undertaken to characterize the physical and chemical properties of particulate matter in the Canadian Lower Fraser Valley (LFV) and to provide detailed data for air quality model development (Vingarzan and Li, 2006). Five main ground sites were used during the August 13-31 study period, as illustrated in Figure 8.12. They included: Cassiar Tunnel in urban Vancouver, Slocan, an urban-suburban site in Vancouver; Langley, a rural site in the south-central LFV, Sumas, a semi-rural site in the eastern LFV, and Golden Ears, an elevated site on the north shore mountains. A large number of ground-level measurements were collected during the study, as well as LIDAR scans, vertical profiling, radiosonde and aircraft measurements.
Figure 8.12. WISE station locations during the Pacific 2001 Study (Vingarzan and Li, 2006)
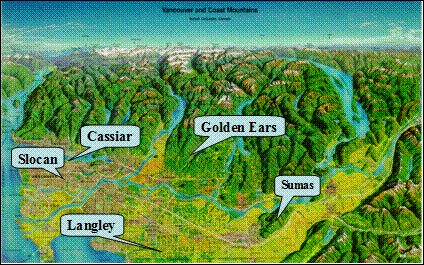
Description of Figure 8.12
Figure 8.12 is a relief map of the Lower Fraser Valley north of the 49th parallel and including the North Shore Mountains. The locations of the stations at Cassiar Tunnel, Slocan, Langley, Sumas, and Golden Ears are indicated by arrows. As described in the text, Cassiar Tunnel is in urban Vancouver, Slocan is an urban-suburban site in southeast Vancouver, Langley is a rural site in the south-central LFV, Sumas is a semi-rural site in the eastern LFV, and Golden Ears is an elevated site on the North Shore Mountains between Pitt and Stave Lakes.
Several chemical regimes were found to be influencing air quality in the WISE region. At Cassiar Tunnel, ambient aerosols were strongly dominated by gasoline and diesel emissions, the latter evidenced by elevated black carbon levels. At Slocan, the composition of ambient aerosols indicated an emission-controlled regime dominated by organics, of both anthropogenic and biogenic origin. Anthropogenic emissions were strongly influenced by vehicles and fugitive emissions of petroleum fuels. Plumes from ships operating around the area or at berth were observed to impact the city on a regular basis (Lu et al., 2006). At the rural site of Langley, the PM2.5 regime was controlled by secondary aerosol formation. Here the average particle composition was dominated by inorganics, with contributions from sea salt, sulphate, nitrogen species and aged organics. Elevated ammonia and biomass burning markers reflected the influence of agricultural sources. At the semi-rural site of Sumas, the particle mass composition indicated a mixed emission-formation regime dominated by urban, agricultural and biogenic sources. At the forested site of Golden Ears, the particle composition was dominated by biogenic organics. The aerosol chemical composition at this site was impacted by anthropogenic sources due to upslope flow, while biogenically-derived secondary organic aerosols (SOA) were transported into the WISE by downslope flow.
Particle composition studies at many of the sites indicated that organic carbon comprised approximately half of the PM1.0 mass, with the rest being comprised by inorganic species. Sulphate dominated the inorganic fraction of the fine particle mode at all sites. Sea salt chemistry was found to be important in the formation of coarse-mode sodium nitrate aerosols. The fine particle fraction was dominated by ammonium sulphate aerosols, with occasional peaks in ammonium nitrate. Overall, elemental carbon comprised a minor part of the fine particle fraction. Isoprene and monoterpenes were the dominant biogenic hydrocarbons detected, and their presence at all sites indicated the broad influence of biogenic organics throughout the LFV.
Source analysis using isotopic methods suggested that the WISE was influenced by a well dispersed source of sulphate, consistent with gasoline, diesel and gas combustion. Biogenic contributions were found to be significant, with approximately 30% of the sulphate in PM2.5 aerosols estimated to be derived from the oxidation of dimethyl sulphide (DMS). Organic aerosols were found to have significant contributions from terrestrial (plant material) sources during the summer months and from marine, biogenic and fossil fuel sources during the spring months. Biomass burning generally had a small, but measurable impact on the composition of ambient aerosols in the LFV. The importance of biogenics and fuel combustion (especially by cars) as determined by the Pacific 2001 Study in the WISE is similar to Wu’s (2007) findings for Seattle.
Particle formation and growth was observed to occur via several mechanisms. Nucleation was observed on cleaner days while, on more polluted days condensation and coagulation dominated particle growth. Ozone episodes were associated with an increase in both particle size and mass. Sulphate growth events were associated with photochemical production from advected SO2 from industrial sources in north-west Washington State. Within the City of Vancouver, sulphate peaks were observed to be associated with ship plume emissions. Fine particle nitrate events were associated with advection of HNO3 into the eastern WISE reacting with local sources of ammonia. Organic particle growth events were associated with stagnant conditions and advection of air over urban Vancouver. The extent of processing of air masses was found to increase in a west to east direction, although photochemically aged pollutants from the area where the Strait of Juan de Fuca converges with the Strait of Georgia could reverse this gradient. Both biogenic and anthropogenic species were observed to be chemically reactive and contribute to secondary aerosol formation.
8.8 Chapter Summary
Fine particulate matter (PM2.5) is a health concern because it is in a size range that is easily inhaled by the lungs. Elevated levels of PM2.5 are also responsible for visibility degradation. The principal contaminants responsible for the secondary formation of PM2.5 include NOx, SO2, NH3 and organic carbon. In the atmosphere, these contaminants are transformed by chemical and physical processes to ammonium nitrate, ammonium sulphate and organic compounds. Elemental carbon (from combustion of organic material), sea salt, and soil also contribute to fine particulate matter.
Concentrations of PM2.5 in the Georgia Basin/Puget Sound airshed can either peak in fall or winter (due to heating and wood combustion emissions and reduced dispersion under lower boundary layer heights) or in the late summer (due to enhanced photochemical conditions). Daily cycles include rush-hour peaks at urban sites and elevated levels in the evening and overnight, associated with low mixing heights. PM2.5 concentrations in the airshed are typically below national objectives and standards, with the notable exception of the Tacoma-Pierce County nonattainment area in Washington State. Outside the non-attainment area, PM2.5 shows evidence of a slight decline, although data records are too short to calculate robust trends.
Although PM2.5 composition varies by location, organic carbon was found to be the dominant component in the airshed followed by ammonium sulphate and ammonium nitrate. Contributions from sulphate peak in the summer due to increased biogenic sulphur emissions and higher rates of photochemical conversion of sulphur dioxide to sulphate. In contrast, maximum nitrate contributions occur at different times of the year in different locations due to local chemistry, including availability of ammonia. Where ammonia is abundant, ammonium nitrate production is favoured during the agriculturally active seasons.
Key sources contributing to ambient PM2.5 in the Canadian Lower Fraser Valley are motor vehicles, marine transport, agriculture and industry. Spatial differences include a stronger influence from marine transportation in the western part of the airshed. Seasonal influences include wood burning, secondary nitrate, and vehicle emissions which are more important in the winter, and increased cruise vessel activities and biogenic influences which are more important in the summer. In Seattle, wood burning, secondary sulphate and secondary nitrate are the three main contributors to PM2.5. At Tacoma, wood burning, secondary particles and motor vehicles dominate the PM2.5 composition. At Mount Rainier, biomass from natural and anthropogenic combustion sources contributes the largest amount to fine particulate composition. Between the early 1990s and the early 2000s, average biomass concentrations at Mt. Rainier decreased by 42% and the concentrations from secondary sulphate sources decreased by 37%, the latter reflecting implementation of industrial emission controls during this period.
In summary, PM2.5 levels in the Georgia Basin/Puget Sound airshed generally comply with national standards and regional objectives, but episodes of elevated particulate matter continue to affect certain areas. In most areas, ambient PM2.5concentrations have been declining slightly over the past decade. Future PM levels will depend on changes in background levels, climate and local and transported emissions.
8.9 References
Ashbaugh, L.L., Malm, W.C., and Sadeh, W.Z.,1985. A Residence Time Probability Analysis of Sulfur Concentrations at Grand Canyon National Park. Atmospheric Environment 19: 1263-1270.
Barthelmie, R.J. and Pryor, S.C., 1998. Implications of ammonia emissions for fine aerosol formation and visibility impairment - a case study from the Lower Fraser Valley, British Columbia. Atmospheric Environment 32 (3): 345-353.
Blanchard, C. and Hldy, G., 2003. Effects of changes in sulfate, ammonia, and nitric acid on particulate nitrate concentrations in the southeastern United States. Air & Waste Management Association 53: 283-290.
Brook, J.R., Farrell, C., Friesen, K., Langley, L., Kellerhals, M., Morneau, G., Reid, N., Vingarzan, R., Waugh, D., and Wiens, B., 2011. "Chapter 7. Air quality at the regional and local scale: The what, where, why and how of concentration variations". In:Canadian Smog Science Assessment. Volume 1. Atmospheric Science and Environmental Effects. Environment Canada and Health Canada.(Executive summary) (Full report available upon request) Environment Canada, Science and Technology Branch, 4905 Dufferin St, Downsview, Ontario, M3H 5T4
Clow, D.W., and Campbell, D.H., 2008. Atmospheric deposition and surface-water chemistry in Mount Rainier and North Cascades National Parks, U.S.A., water years 2000 and 2005-2006: U.S. Geological Survey Scientific Investigations Report 2008-5152, 37 p.
Dann, T., Vingarzan, R., Chan, E., Vet, B., Brook, J., Martinelango, K., Shaw, M., Dabek, E., Wang, D., Herod, D., Mignacca, D., Anlauf, K., Graham, M., O’Brien, J., 2011. “Chapter 3, Ambient Measurements and Observations”. In: Canadian Smog Science Assessment. Volume 1. Atmospheric Science and Environmental Effects. Environment Canada and Health Canada.(Executive summary) (Full report available upon request) Environment Canada, Science and Technology Branch, 4905 Dufferin St, Downsview, Ontario, M3H 5T4
Husar R., et al., (28 coauthors) 2001. The Asian Dust Events of April 1998. Journal of Geophysical Research106 (D16): 18,317-18,330, doi:10.1029/2000JD900788.
Langner, J., Rodhe, H., 1991. A global three dimensional model for the tropospheric sulfur cycle. Journal of Atmospheric Chemistry 13, 225-263.
Larson, T., Su, J., Baribeau, A.M., Buzzelli, M., Setton, E., and Michael Brauer, 2007. A Spatial Model of Urban Winter Woodsmoke Concentrations. Environmental Science & Technology 41 (7): 2429-2436.
Makar, P.A., 2001. Processes linking NOx, SO2, NH3, and VOCs to secondary particle formation, Chapter 2: Precursor Contributions to Ambient Fine Particulate Matter in Canada. Meteorological Service of Canada. ISBN: 0-662-30650-3. May 2001, 237 pp.
McHenry, J.N., Dennis, R.L., 1994. The relative importance of oxidation pathways and clouds to atmospheric ambient sulfate production as predicted by the Regional Acid Deposition Model. Journal of Applied Meteorology 33: 890-905.
McKendry, I.G., 2000. PM10 Levels in the Lower Fraser Valley, British Columbia, Canada: An Overview of Spatiotemporal Variations and Meteorological Controls. Journal of Air & Waste Management Association 50: 443-445.
McKendry, I.G., J.P. Hacker, R. Stull, S. Sakiyama, D. Mignacca and K. Reid, 2001. Long-range Transport of Asian Dust to the Lower Fraser Valley, British Columbia, Canada. Journal of Geophysical Research 106: 18 361-18 370.
McKendry, I. G., 2006. Background concentrations of PM2.5 and Ozone in British Columbia, Canada. University of British Columbia report prepared for the British Columbia Ministry of the Environment, Vancouver, B.C.
NARSTO, 2004. Particulate Matter Science for Policy Makers: A NARSTO Assessment. P. McMurry, M. Shepherd, and J. Vickery, eds. Cambridge University Press, Cambridge, England. ISBN 0 52 184287 5.
Norman, A., Belzer W., and Barrie, L., 2004. Insights into the biogenic contribution to total sulphate in aerosol and precipitation in the Fraser Valley afforded by isotopes of sulphur and oxygen. Journal of Geophysical Research D. 109, D05311, doi:10.1029/2002JD003072.
Ostermann, K., 2002. Source apportionment of particulate matter by Positive Matrix Factorisation in the Lower Fraser Valley of British Columbia. Prepared for the B.C. Ministry of Water, Lands and Air Protection, Nov. 2002, 89 pp.
Paatero, P. and Tapper, U., 1994. Positive Matrix Factorization: A Non-Negative Factor Model with Optimal Utilization of Error Estimates of Data Values. Environmetrics 5, 111-126.
Paatero, P., 1997. Least Squares Formulation of Robust Non-Negative Factor Analysis. Chemometrics and Intelligent Laboratory Systems 37, 23-35.
Pandis, S.N., Seinfeld, J.H., Pilinis, C., 1992. Heterogeneous Sulfate Production in an Urban Fog. Atmospheric Environment 26: 2509 - 2522.
Seinfeld J. H. and Pandis S. N., 1998. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 1st edition, J. Wiley, New York.
Sharma, S., Vingarzan, R., Barrie, L.A., Norman, A., Sirois, A., Henry, M., and di Cenzo, C., 2003. Concentrations of dimethyl sulphide in the Strait of Georgia and its impact on the atmospheric sulphur budget of the Canadian West Coast. Journal of Geophysical Research D. 108, D15, 4459.
So, R., Vingarzan, R. and Meyn, S. 2010. Source Apportionment of PM2.5 at Burnaby South and Abbotsford. Environment Canada. Environment Canada, Meteorological Services, #201-401 Burrard, Vancouver, BC, V6S 3S5.
U.S. EPA (Environmental Protection Agency), 2002. Air Quality Criteria for Particulate Matter. Volume 1. Third External Review (April 2002). National Center for Environmental Assessment - RTP Office, Office of Research and Development, U.S. Environmental Protection Agency, Research Triangle Park, N.C.EPA/600/P-99/002aC
Vingarzan, R., 2004. Ambient Particulate Matter Concentrations in Canada and Background Levels. Environment Canada, Environmental Conservation Branch, Aquatic and Atmospheric Sciences Division, #201-401 Burrard Street, Vancouver, British Columbia, V6C 3S5
Vingarzan R. and Li, S-M., 2006. The Pacific 2001 Air Quality Study - synthesis of findings and policy implications. Atmospheric Environment 40: 2637-2649.
WA DOE (Washington State Department of Ecology), 2007. Washington State Base Year 2005 County Inventories. WA DOE internal document; personal communication via Sally Otterson.
Wu, C., Larson, T., Wu, S, Williamson, J., Westberg, H., Liu, L-J., 2007. “Source apportionment of PM2.5 and selected hazardous air pollutants in Seattle”, Science of The Total Environment 386: 42-52.