
Clinical Trials Modernization: Consultation Paper
Table of Contents
- Part 1: Introduction
- Part 2: Proposals for Consideration
- Part 3: Anticipated Outcomes for Stakeholders
- Annex 1: Overview of the Existing Canadian Clinical Trial Regulatory Framework
- Annex 2: Specific Considerations of Clinical Trial Modernization by Product
- Annex 3: Consultation Questions
Part 1: Introduction
As one of the five key pillars of the Government of Canada's Regulatory Innovation Agenda for health products, the modernization of clinical trial regulations is an important goal for Health Canada, and Canadians more broadly. Health Canada, the federal regulator of the health products that Canadians rely upon in their daily lives, is responsible for the regulation of clinical trials involving a wide range of products, including drugs and biologics, natural health products (NHPs) and medical devices. Clinical trials testing products for the relief of aches and pains, all the way to those used in critical care interventions involving the insertion of a heart stent, are all required to safeguard participant safety and produce reliable information while upholding ethical principles for testing in humans.
With the continually accelerating advances in technology and the development of new types of promising health products, as well as the advent of new clinical trial types and designs, the need for a modernization of Canada's clinical trials regulatory framework has become clear. It is important that the regulatory framework for clinical trials in Canada supports the adoption of promising new therapies in the health care system and does not inadvertently stifle innovations that could help to improve the health of Canadians. Personalized health products, gene therapies and products intended for the treatment of rare diseases each pose their own unique requirements regarding the conduct of clinical trials. Meeting these requirements is critical in order to build a knowledge base that would support the introduction of novel safe and effective therapies to the Canadian market, improving the lives of Canadians while providing them with access to the information they need to make informed decisions about their health.
Additionally, the COVID-19 pandemic has further highlighted the need for change to the clinical trials regulatory framework in Canada. The Government of Canada was quick to respond to the pandemic by issuing the Interim Order respecting clinical trials for medical devices and drugs relating to COVID-19 (CT-IO) and the subsequent Interim Order No.2 respecting clinical trials for medical devices and drugs relating to COVID-19 (CT-IO 2),and their associated Guidance documents. These Interim Orders have and continue to provide greater flexibilities for the conduct of clinical trials in the context of the pandemic. However, these flexibilities only apply to drugs and devices intended for the diagnosis, treatment, mitigation or prevention of COVID-19. Although the measures under the CT-IOs are temporary, they have also provided an important proof of concept for some of the elements of clinical trial modernization that were already under consideration.
As committed to in Health Canada's Forward Regulatory Plan for 2021-2023, the department plans to modernize the clinical trials framework to introduce a coherent risk-based approach, afford greater flexibility in the safe development of innovative therapies, streamline processes toward greater efficiency and clarity, and align with international best practices regarding oversight and public access to information. With any changes proposed, Health Canada would continue to uphold the underlying core principles common to the regulation of all clinical trial types, including the protection of participant safety, support for innovation, proportional oversight, and international alignment.
To this end, Health Canada is pleased to introduce an ambitious vision for the modernization of Canada's clinical trials regulatory framework that will better serve the needs of Canadian stakeholders while continuing to protect patient safety in a modern healthcare ecosystem. A single clinical trials framework for all health products is being proposed as the foundation for the new regulatory regime that would provide proportional risk-based oversight, new regulatory agilities over the lifecycle of the trial, greater transparency through registration and public disclosure of results, and a modernized compliance and enforcement regime (see figure 1). A separate consultation on a proposed new regulatory framework to enable clinical trials involving foods for a special dietary purpose is currently underway, with a final deadline for submitting feedback of June 12, 2021. If you are interested in receiving the consultation document for this process, please send your request in an email to hc.bns-bsn.sc@canada.ca.
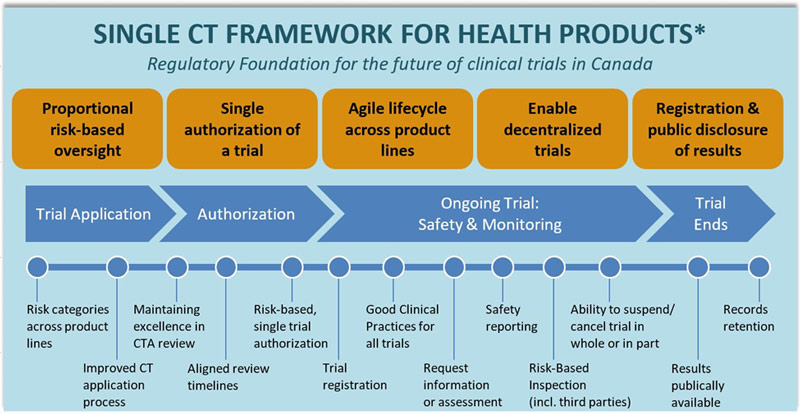
Figure 1 - Text Description
The regulatory foundation for the future of clinical trials in Canada is based on proposing a single framework for all health products. The new regulatory regime would enable proportional risk-based oversight, a single authorization of a trial, agile lifecycle approach across product lines, decentralized trials and registration and public disclosure results.
The clinical trial lifecycle is depicted in 4 stages, with the key new regulatory elements aligned to these stages:
- Trial Application
- Risk categories across product lines
- Improved CT application process
- Authorization
- Maintaining excellence in CTA review
- Aligned review timelines
- Risk-based, single trial authorization
- Ongoing Trial: Safety & Monitoring
- Trial registration
- Good Clinical Practices for all trials
- Request information or assessment
- Safety reporting
- Risk-based inspection (including third parties)
- Ability to suspend/cancel trial in whole or in part
- Trial Ends
- Results publically available
- Records retention
* There is currently no existing regulatory framework for clinical trials involving foods for a Special Dietary Purpose (FSDP). The new framework will enable FSDP trials, which would be aligned with health product clinical trials on key life cycle stages.
Within this new single clinical trials framework, there are several distinct key themes that will be further discussed in this consultation paper. These are:
- Agile Life Cycle
- Risk-based approach
- Transparency
- Modernization of Compliance and Enforcement
Proposal Feedback
The primary intent of this discussion paper is to provide Canadians with an opportunity to review policy proposals for the modernization of Canada's clinical trials regulatory framework and provide feedback prior to the department finalizing policy decisions and developing the regulations. Additional feedback from stakeholders will provide Health Canada with a better understanding of: how the clinical trials ecosystem will evolve, including emerging trends and innovative technologies that could impact the conduct of trials; and whether the proposals presented would help enable clinical trials that best serve the needs of Canadians, with the goal of allowing them access to innovating healthcare technologies while continuing to protect their health and safety. Written comments are welcomed until July 4, 2021 via responding to an online questionnaire, or by sending a written response by email to hc.policy.bureau.enquiries.sc@canada.ca.
Background
Canadian context
Canadian stakeholders have reported an increasing need to conduct clinical trials using novel designs to gain efficiency and have called for changes to increase regulatory coherence across the distinct drug, natural health product (NHP) and medical device clinical trial frameworks. While the existing approach to the regulation of health products for the purposes of a clinical trial has served Canadians well, it lacks sufficient agility to address the innovative types of clinical trials that are emerging. Furthermore, the regulatory frameworks for drug, NHP and medical device clinical trials have evolved separately over time, leading to significant differences between the regimes (refer to Annex 1: "Overview of the Canadian Clinical Trial Regulatory Framework" for more details). These differences have led to inefficiencies and unnecessary burden for both Health Canada and sponsors.
Health Canada recognizes the need for an integrated regulatory framework that would also provide greater transparency, flexibility and efficiency gains for stakeholders. Helping to enable more non-conventional trial designs would have the added benefit of ensuring that Canada remains an attractive place to conduct clinical trials while continuing to uphold high standards for the protection of the health and safety of participants.
Legislative changes to enable clinical trial modernization
As part of the Budget Implementation Act, 2019, necessary legislative provisions were introduced to the Food and Drugs Act to support the modernization of the clinical trial regulations. The changes were made in response to the findings of the Health and Bioscience Regulatory Review that "clinical trial regulations are limiting growth". Over the course of 2019 and 2020, Health Canada engaged with stakeholders on clinical trial modernization objectives. Along with the stakeholder feedback received in response to this consultation document, as well as from more targeted engagement sessions with key stakeholders, the public will also have the opportunity to comment on regulatory proposals following their pre-publication in Canada Gazette, Part I, which is targeted for 2022.
International context
Given the global nature of healthcare and technological innovation, Canada is not alone in recognizing a need for change. While there are differing international approaches to authorizing and regulating clinical trials, other international regulators are increasingly recognizing the need to adopt a more agile and risk-based approach that could better accommodate innovative trials and health products. For example, the European Medicines Agency (EMA) has published a forward-looking strategic document titled "EMA Regulatory Science to 2025: Strategic Reflection" which, among other topics, explores different ways to foster innovation in clinical trials and to develop a regulatory framework for emerging clinical data generation. Similarly, in March 2019, the US Food and Drug Administration (FDA) Commissioner released a statement on new strategies to modernize clinical trials. The statement confirms that the FDA "has worked closely with stakeholders, including the Clinical Trial Transformation Initiative, to identify innovative trial designs, evaluate the role of decentralized clinical trials and mobile technologies, and help validate novel endpoints that can enable trials to generate reliable evidence needed to assess product safety and efficacy more efficiently." The statement also points to several new guidance documents for industry that were released to help stakeholders achieve these goals while navigating the regulatory landscape.
Previous Consultations and What Was Heard
In July 2019, Health Canada released the discussion paper "Agile regulations for advanced therapeutic products and clinical trials", with a consultation period that closed in September 2019. In addition to this public consultation, the department has held a number of targeted meetings with a broad range of stakeholders.
Over the course of these consultations, stakeholders confirmed that having a flexible, enabling clinical trials framework would draw in key companies and investments, and support research and innovation in Canada. They also recommended that Health Canada should consider expanding in the following areas:
- Enabling innovative trials designed to simultaneously test a range of therapeutic interventions, companion diagnostics, and/or indications through greater streamlining of requirements across product lines;
- Making it easier to implement multi-site trials and to add sites to those already started;
- Supporting the use of real-world evidence and real-time collection and analysis of data to rapidly and continually assess and update benefit-harm-uncertainty profiles and ongoing patient care;
- Enabling flexibility for clinicians in special circumstances, including those conducting adaptive trials, so they are better able to conduct research while adapting treatments based on patient needs;
- Facilitating the connection and sharing of information so that researchers can build and support each other's work; and
- Supporting the inclusion of under-represented populations in studies and testing of population specific differences, where appropriate.
Part 2: Proposals for Consideration
The modernization of Canada's clinical trials framework is a large multifaceted endeavour that is expected to involve significant changes to different aspects of the existing regulations and policies. While the proposals introduced below share many of the same goals, they have been presented within several key themes to better structure the conversation and gather feedback.
Agile Life Cycle
The regulatory life cycle of a clinical trial involves the clinical trial application (CTA) process for a clinical trial authorization, including a pre-CTA meeting that may be held upon request, the oversight and reporting during the course of a trial, and post-trial requirements such as longer-term records retention. The establishment of a more agile life cycle approach to the regulation of clinical trials would provide Health Canada with the tools necessary to oversee the safe conduct of a trial in its entirety, while better enabling sponsors to conduct innovative types of trials. Health Canada would have the ability to authorize a trial rather than just the sale or importation of an investigational product, authorize a trial with multiple product types through a single authorization, and streamline the application requirements across all product lines for greater efficiency, while maintaining the protection of the health and safety of participants.
Proposed Approaches
Clinical Trial Authorization
Health Canada is proposing regulatory changes that would transition from an authorization of the sale or importation of the health product used in the clinical trial to an authorization of the clinical trial as a whole, including the sale or importation of the product(s).
This would provide Health Canada with the authority and oversight necessary to take action in a more nuanced and targeted way when a safety issue arises with an aspect of the clinical trial. It would also allow Health Canada to better regulate new types of innovative trial designs and future variations, such as:
- trials studying multiple therapies within a single clinical trial (i.e., master protocols); and
- novel adaptive trial designs which allow for planned changes to the study protocol to occur at pre-specified times during the life cycle of a trial (i.e. adaptive trials).
Master protocols are designed with multiple sub-studies and involve coordinated efforts to evaluate one or more investigational products in one or more indications within the overall trial structure. Types of master protocols include:
- Basket trials, which investigate the safety/efficacy/effect of an investigational product across a variety of indications
- Umbrella trials, which investigate the safety/efficacy/effects of several investigational products in a single indication
- Platform trials, which investigate several investigational products in one or multiple indications in a highly dynamic design
Additional regulatory changes that influence how Health Canada authorizes, amends, monitors, and suspends or cancels a trial would help to support this agile life cycle approach. Such regulatory changes would provide Health Canada oversight over the conduct of innovative trials through the entire trial process, with the goals of enabling innovation while protecting participant safety.
Single Authorization of a Trial with Multiple Product Types
In moving to a modernized clinical trials regulatory framework, Health Canada is proposing to allow for the single authorization of a clinical trial involving multiple health products from different categories, such as drugs, NHPs, and medical devices. This would significantly increase efficiencies for the application, amendment, and authorization processes for clinical trials involving multiple health products, further streamlining Health Canada's interactions with the sponsor throughout the trial.
Stakeholders have previously identified challenges with having to file separate drug and/or NHP CTAs and/or medical device Investigational Testing Applications (ITA) for a single trial. With broader authorities to regulate both clinical trials and the investigational products, maintaining the status quo of requiring separate product-based authorizations under different regulatory frameworks when more than one product is being used or tested in a single trial would be inefficient and cumbersome. The requirement for multiple authorizations with different review target times can lead to delays in opening the trial, reducing the timeliness of Canadians' access to potentially beneficial health products. A single authorization pathway would help to address these concerns, and would be accompanied by further aligning certain regulatory requirements across product lines, where appropriate. Specific details on the requirements of a single authorization pathway for trials involving multiple products would also be described in guidance.
Streamlining Requirements Across Product Types
Although product-specific requirements would continue to be necessary to contribute to the safety of the product within the trial, there are areas where Health Canada could align clinical trial regulatory requirements across the product lines of drugs, NHPs, and medical devices. Changes would be made to address any gaps and provide sponsors with a more streamlined and efficient experience when submitting an application or while conducting their trial.
Such changes would also enable greater agility in how Health Canada authorizes and oversees the safety of more innovative trials over their life cycle. In addition to the ability to authorize the trial and its product(s), Health Canada would expand its ability to monitor the safety of ongoing trials, and if warranted, to suspend or cancel these trials on a complete or partial basis.
Agile Oversight of the Life Cycle of a Clinical Trial

Figure 2 - Text Description
The lifecycle of a clinical trial includes the following elements: application, authorization/amendment, conduct of the clinical trial, safety monitoring and study end. The proposals for the agile oversight of the lifecycle of a clinical trial is mapped across the aforementioned elements.
In particular, Health Canada is proposing the following changes:
Clinical Trial Application
The application requirements for a clinical trial would be amended for greater agility to better regulate modern trial types and be further aligned across health product lines. For example, some provisions under the Interim Order for COVID-19 trials (such as the definition of a Research Ethics Board (REB) and the timing of submission for REB approval) would be reviewed, and may be incorporated when modernizing the regulations, as appropriate.
For medical devices, Health Canada is proposing to expand who can sponsor a clinical trial and file a CTA to include independent investigators (such as a researcher, clinician or health care facility) in addition to the manufacturers and importers of medical devices. This would bring Health Canada into greater alignment with the regulatory requirements of other international jurisdictions.
Review Timelines for Authorization/Amendments
Changes to the authorization scheme for the trial and products, as discussed above, may affect review timelines. The timelines would be defined by policy and aligned across product lines to provide greater coherence and predictability for sponsors.
Conduct of the Trial
Sponsors would be required to adhere to internationally recognized standards for Good Clinical Practices (GCP) throughout the conduct of a trial, regardless of product type, which would be a new regulatory requirement for medical device trials. This would better align the oversight models across health product lines, structuring the requirements for each product line to help them conform to international GCP standards, and serve as a basis for the future modernization of Health Canada's compliance and enforcement program for the clinical trials of health products.
Safety Monitoring
Adverse events and medical device incidents would continue to be reported on a per-product basis to ensure the appropriate safety of products being tested. Health Canada is proposing to clarify and broaden its existing authorities to request information, including the ability to request an analysis of safety data while the trial is ongoing. This could include a request for an assessment of safety signals to determine if the benefit/risk balance has changed during the conduct of the clinical trial.
Trial Suspension and Cancellation
Existing authorities related to Health Canada's ability to cancel or suspend a clinical trial authorization would be modified to allow for the suspension or cancellation of a trial in whole or in part (e.g., suspending only an arm of a trial, a site, study enrolment, or the use of a particular product). This would allow Health Canada to react more precisely if an aspect of the trial has demonstrated a lack of efficacy or poses a safety concern without compromising the trial as a whole, allowing the rest of the trial to remain open and continue to enrol new trial participants. Additionally, Health Canada is reviewing its existing requirements across the different health product regulations for reporting on the commencement and discontinuance of a study and what is required to resume a study.
Risk-based Approach
Health Canada is proposing to modernize the regulation of clinical trials through the establishment of a common risk-based approach for trials involving any type of health product to provide greater flexibility and consistency in the regulation of clinical trials. The introduction of proportional oversight based on the level of risk to study participants would help to reduce burden while enabling Health Canada to shift resources to the areas in need of greater oversight.
Proposed Approaches
Aligning the regulatory requirements for clinical trials regardless of product type would introduce:
- Categories of proportional oversight with stratified and tailored regulatory requirements for drugs and NHPs; and
- New flexible regulatory tools to manage risks over the life cycle of a trial.
Health Canada would enable a comprehensive risk-based approach for the regulation of clinical trials by moving from a one-size-fits-all oversight model for drugs and NHPs, while introducing new flexibilities and maintaining the pre-existing risk-based device classification scheme for medical devices. The proposed model would continue to support participant safety, while introducing greater efficiencies where appropriate. The model would also reflect international best practices and provide greater predictability for stakeholders. Any new requirements and obligations for proportional oversight would be based on the risk, uncertainty and known information about the product and its intended use within the clinical trial.
For drugs and NHPs, this approach would align with the Organization for Economic Co-operation and Development (OECD) recommendations where appropriate.Footnote 1 For medical devices, the current risk-based classification scheme would be maintained, given that the classification scheme already conforms to an internationally recognized system that has worked well for stakeholders.
The alignment between drugs, NHPs and medical devices in the new framework would help support moving towards a single authorization scheme for the conduct of the trial when multiple products of different types are used within a single trial.
Risk Categories and Requirements
Health Canada is proposing that clinical trials which fall under the scope of Food and Drugs Act and its regulations would be placed into one of the following three categories:
- Category A: Exemption from clinical trial authorization;
- Category B: Clinical trial authorization with tailored requirements; or
- Category C: Clinical trial authorization with full requirements.
These categories represent the level of risk, uncertainty and safety information available for each product used in the trial. A single trial involving multiple products would be classified according to the product with the highest level of risk. Through its review of the clinical trial application, Health Canada would continue to assess all other relevant risk factors, such as the use of non-traditional trial design, highly vulnerable study populations, or invasive procedures.
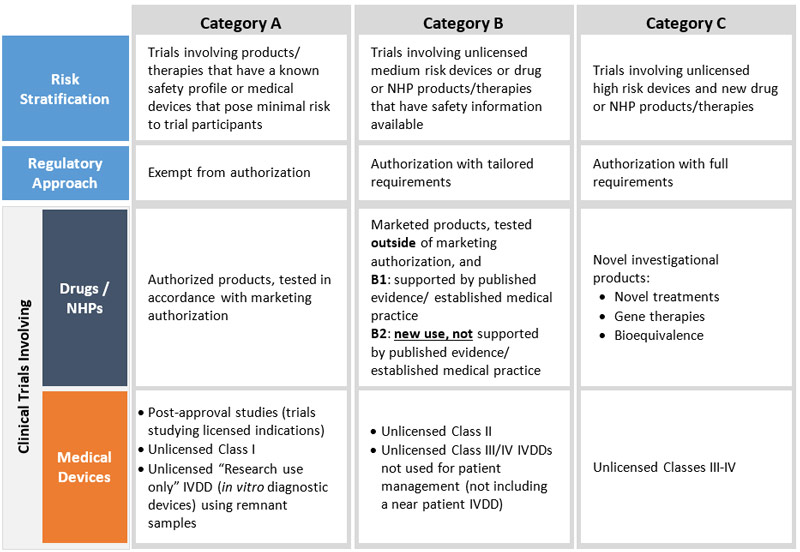
Figure 3 - Text Description
Health Canada is proposing a risk-based approach for drugs, medical devices and natural health products (NHP) which is intended to provide proportional oversight by placing the trials into one of three categories (A, B, or C) and two sub-categories for Category B (B1 and B2).
Category A will include trials involving products/therapies that have a known safety profile or medical devices that pose minimal risk to trial participants. These trials will be exempt from authorization. For drugs and NHPs, this category will include trials involving authorized products, tested in accordance with marketing authorization. For medical devices, this category will include trials involving: post-approval studies (trials studying licensed indications), unlicensed Class I devices, unlicensed "Research use only" IVDD (in vitro diagnostic devices using remnant samples).
Category B will involve unlicensed medium risk devices or drug or NHP products/therapies that have safety information available. These trials will require authorization, but with tailored requirements. For drugs and NHPs, this category will include trials involving marketed products, tested outside of a marketing authorization. Sub-category B1 applies to trials studying use of marketed drugs that is supported by published evidence/ established medical practice. Trials studying new use that is not supported by published evidence/established medical practice fall under Sub-category B2. For medical devices, this category will include trials involving: unlicensed Class II devices, unlicensed Class III/IV IVDDs not used for patient management (not including a near patient IVDD).
Category C will include trials involving unlicensed high risk devices and new drug or NHP products/therapies. These trials will require authorization with full requirements. For drugs and NHPs, this category will include trials involving novel investigational products (such as novel treatments, gene therapies, bioequivalence). For medical devices, this category will include trials involving unlicensed Classes III-IV.
Stratified Clinical Trial Requirements:
Category A:
This category would include clinical trials involving products or therapies that have a known safety profile and that pose minimal additional risk to trial participants compared to usual medical practice. These include marketed health products used as per their marketing authorization (all health product lines), clinical trials involving unlicensed Class I medical devices, and "research use only" IVDDs (in vitro diagnostic devices) using remnant samples that are not used for diagnosis or treatment of a patient.
Application and Authorization
For these trials, Health Canada would not require the submission of a CTA, which is consistent with what Health Canada has in place under the existing regulations.
Other requirements
For all product lines, many existing requirements would continue to apply. For trials involving drug and/or NHP products, this would include adherence to GCP, obtaining REB approval, and the reporting of adverse reactions (AR) by market authorization holders. For unlicensed Class I devices and "research use only" IVDDs using remnant samples that are not used for diagnosis or treatment of a patient, this would include labelling, advertising, distribution records, complaint handling and recalls, and mandatory problem reporting. In addition, Health Canada is proposing two new requirements for these medical devices: (1) for sponsors to adhere to GCP principles, aligned to ISO 14155 GCP, which is currently recommended in Health Canada's guidance to industry, and; (2) to explicitly require REB approval prior to trial commencement. For clinical trials studying marketed devices as per the approved indications, current licensing requirements would continue to apply in addition to GCP and REB approval. For all product types, sponsors would still be required to maintain certain information as part of the record-keeping requirements set out in regulation.
Category B:
This category would include clinical trials involving medium risk unlicensed medical devices or health products that have well-established safety information available, such as drug products marketed in Canada that are being tested for uses outside the authorized indication.
For drugs and NHPs, Health Canada is proposing for this category to be further stratified into sub-categories B1 and B2 depending on the level of evidence available to support testing outside the authorized indication. Category B1 would include trials studying unapproved indications, populations or dosage regimens that are supported by strong published evidence and/or established medical practice, whereas Category B2 would include trials studying anew use that is not supported by published evidence and/or established medical practice.
For medical devices, the following products would be incorporated in this category:
- unlicensed Class II devices, and
- unlicensed Class III or IV IVDDs that are not used for patient management, not including near-patient IVDDs.
Application and Authorization
For Category B trials, the submission of a CTA would be required, but with some tailored requirements based on the level of risk of the product or trial type.
For drugs and NHPs, one example of a tailored requirement in this Category would involve allowing sponsors to refer to an approved product monograph and/or provide information on how the proposed use of the product aligns with established medical practice. This information would be used to demonstrate specific aspects of what would typically be required as part of a full application, such as information that would normally be part of the Investigator's Brochure.
For medical devices, as with the requirements for Class I devices, the applicant would be required to possess records that contain all the information and documents that meet the regulatory requirements, consistent with current Health Canada requirements. Health Canada is also exploring reduced application requirements for medical devices marketed in Canada that are being investigated for use outside the approved indications. For example, Health Canada may not require the submission of a product's marketing history or the results of previous research conducted with a device.
Post-Authorization
For drugs and NHPs, trials studying indications that are supported by evidence (Category B1) would follow GCP requirements, while other requirements would be reduced, such as:
- labelling requirements;
- AR reporting. For example, when the safety profile of a drug is well-characterised, requirements could be reduced around the collection of non-serious adverse events or other safety data if outlined in the protocol. This would be aligned with the draft guidance from the International Council for Harmonization E19; and
- record keeping requirements, such as the temperature monitoring records for the health product.
Trials studying a new use that is not supported by published evidence and/or established medical practice (Category B2) would be required to follow GCP, would have reduced labelling requirements, and may have reduced AR reporting and reduced record keeping requirements as determined by Health Canada upon review. These approaches are significant as they reduce some burden on the sponsor without increasing risk to clinical trial participants.
For medical devices, the current requirements in place would continue to apply for unlicensed Class II devices or Class III or IV IVDDs that are not used for patient management, not including near-patient IVDDs, with the addition of adhering to GCP principles and explicitly requiring REB approval prior to beginning a trial. Health Canada is exploring reduced post-authorization requirements for medical devices marketed in Canada, but which are being used in the trial outside the approved indications. This may include potential labelling flexibility when specific information is provided to Health Canada for assessment (e.g., proposed off-label use of the device, rationale, and the risks, effectiveness or uncertainty the device and its off-label use).
Category C:
This category would include trials involving unlicensed high risk medical devices and/or new drugs, NHPs or therapies, as well as bioequivalence studies which typically involve testing on healthy participants.
Application and Authorization
Category C clinical trials would require the submission of a full CTA, which would remain consistent with what Health Canada currently requires for trials involving drugs and natural health products and unlicensed Class III and IV devices (excluding those mentioned in Category B).
Post-Authorization
The full set of post-authorization requirements, consistent with the current regulations, would apply for Category C clinical trials involving drugs, NHPs and/or unlicensed Class III and IV devices.
Use of New Authorities – Terms and Conditions
To better manage risk across the life cycle of a clinical trial, Health Canada would have the authority to impose terms and conditions (Ts & Cs) on a clinical trial authorization before or during the clinical trial in a predictable and agile manner that is consistent across product lines. Ts & Cs would be imposed on a case-by-case basis to address significant uncertainties or mitigate significant risks related to the product(s) being tested, or to the conduct of the trial.
This authority would enable the authorization of a broader range of trials where tailored measures are required to initiate or continue a trial.
Some examples of when the use of Ts & Cs would be appropriate could include:
- more frequent safety reporting (e.g. copies of Development Safety Update Report (DSUR), Data Safety Monitoring Board (DSMB) reports);
- Monitoring of specific populations because of potential increased risk (e.g. children, pregnancy); and
- Additional information to characterise and mitigate newly identified risks.
Company Z submits an application for a trial to test a new gene therapy for muscular dystrophy in pediatric populations. The gene therapy is intended to treat a rare disease in a vulnerable population. There are long-term consequences in the target population and the product's novel mechanism of action introduces many unknowns. To allow the trial to go forward, Health Canada could require the long-term safety monitoring and reporting beyond the period of study treatment through the application of a term or condition on the authorization.
Other clinical trial scenarios necessitating Ts & Cs may include: novel clinical trial designs, first-in-class products, or high risk study populations.
Decentralized Clinical Trials
With over 30% of the Canadian population residing outside of the large- to medium-sized urbanized areas, recruitment of participants from these more remote populations into clinical trials presents a logistical and financial challenge. While some types of therapies and medical interventions necessitate physical visits to hospitals or centers with high-tech equipment (such as MRI machines or linear accelerators) and specialized physicians, emerging technologies are beginning to enable the potential for patients residing in a remote rural location to be enrolled in a clinical trial that is being overseen centrally from a major urban center, allowing for a more patient-centered approach to trials.
Traditional clinical trials are conducted at designated trial sites where a Qualified Investigator and study staff perform the procedures related to the study. Decentralized clinical trials (DCTs) (sometimes called remote or virtual clinical trials) are clinical trials conducted with study participants located outside of clinical research centres for all of the required trial procedures. In a DCT, the study takes place remotely, without a physical visit to a trial site, after the necessary technology has been installed and explained to the patient. Study participants may use videoconferences with investigators, be visited at home by study personnel, have internet-based tools for data collection and reporting, and use mobile technology such as biosensor devices.
While the conduct of DCTs is not prohibited by the current regulations, Health Canada is proposing to help enable and encourage their use through several changes to the regulations that could make them more practical for sponsors. These regulatory proposals would include:
- Changing the wording of the requirements in the regulations from "written informed consent" to "documented informed consent". Further interpretation would be described in guidance, where "documented" in this case would be interpreted to mean an electronic signature, a video or audio recording of the consent process when a verbal confirmation is provided by the participant;
- Allowing for a witness to attest that informed consent was given if necessary under exceptional circumstances, similar to the current allowance under the CT-IOs;
- Defining "trial site" in regulation as "The location(s) where trial-related activities are actually conducted", where "trial related activities" could include recruitment, informed consent, monitoring and visits that are done in a virtual manner.
- Allowing greater flexibility for the types of health professionals that can be a Qualified Investigator. The type of qualification required for the Qualified Investigator would be determined on a case-by-case basis through the review of a trial protocol at the approval stage and/or by the REB prior to the start of the trial, or alternatively be defined in regulation.
Transparency
Currently, through its Clinical Trials Database, Health Canada provides the public with a listing of specific information relating to Phase I, II, and III clinical trials conducted in patients and involving human pharmaceutical and biologic drugs. Health Canada also encourages sponsors to register their trials in publicly accessible registries such as Clinical Trials.gov and ISRCTN, however, there remains a need to increase transparency for trials involving other interventions, such as medical devices and NHPs, and to provide greater access to all clinical trial information for Canadians.
The 2019 Budget Implementation Act provided authorities related to increasing the transparency of clinical trial information. In support of these authorities, as well as the Government of Canada's commitment to openness, Health Canada is examining options for how to best design and implement measures to promote registration of Canadian clinical trials in public registries as well as public disclosure of the results (results reporting). These measures may include the development of new policies and regulations, while leveraging information technology such as artificial intelligence (AI) to make clinical trial information more accessible to the public.
These transparency measures would help the public (e.g., patients and caregivers, researchers, decision-makers, health professionals, etc.) locate clinical trial information relevant to them and provide other wide-reaching benefits, such as:
- facilitating equity in patient access to clinical trials;
- improving public confidence in the safety of health products available in Canada;
- preventing unnecessary duplication of research, therefore lowering the risks for trial participants;
- facilitating the rapid transfer of knowledge from clinical trials into meaningful clinical practice guidelines resulting in improved health care for Canadians; and
- improving the quality of the available knowledge base for decision making by reducing publication bias.
Over the years, international efforts to improve clinical trial transparency have helped raise the bar with respect to the validity of the scientific evidence made available to the public. Health Canada has hosted a Clinical Trials Database for drugs since 2013 and encouraged the clinical trial registration in international registries for drugs and devices for a number of years. While compliance with this voluntary registration has been quite high, it may not provide a complete picture of all trials taking place, recruiting, completed, or discontinued.
One approach being considered would be to develop new policy that would guide sponsors in recommended steps and processes related to registration and results reporting. This would lead to future regulations for the same processes in a later phase of the clinical trials modernization project. Alternatively, the department could begin with new regulatory requirements for registration in the initial phase, with policy measures to address results reporting. Some of the elements of registration and results reporting under consideration are outlined below.
Registration
Health Canada is exploring policy and regulatory options for the registration of authorized Canadian clinical trials investigating drugs, medical devices and NHPsFootnote 2. All trials conducted in Canada and authorized by Health Canada (or with at least one Canadian site) could be in scope, with the exception of phase IV drug trials (unless required by the department with the placement of a term or condition on the market authorization), bioequivalence trials, trials for individual patients as well as Class I device, research use IVDD with remnant samples, and post-market medical device studies. Canadian trial sponsors may be asked to register their trials in an existing, international registry that has been deemed eligible by the department within a prescribed timeframe. The trial sponsor might also be asked to notify Health Canada with proof of their registration (e.g., registry link and trial identifying number), within a certain timeframe after completing registration.
Registry eligibility would be determined by using criteria that align with the International Committee of Medical Journal Editors (ICMJE) requirements and World Health Organization (WHO) criteria for a primary registerFootnote 3. More specifically, an eligible registry would be:
- publicly accessible without charge;
- open to all prospective registrants;
- managed by a not-for-profit organization;
- have a mechanism to ensure the validity of the registration data;
- electronically searchable; and
- include the minimum WHO 24-item trial registration dataset.
Dr. Jones is looking for a clinical trial for her patient. Currently, she relies on a variety of tools and has an easier time finding foreign trials than Canadian ones. Under regulatory modernization, Dr. Jones and her patients would be able to access the most up to date trial information on Canadian trials in the official language of their choice.
Eligible registries would also be required to accept interventional studies for pharmaceuticals, biologics, medical devices and natural health products (including dietary supplements), and to accept clinical trials conducted in Canada.
Public Disclosure of Results
Health Canada is also exploring policy and regulatory options for the requirement of public disclosure or reporting of trial results. This would mean that Canadian trial sponsors would be asked to publicly report trial results, and may be asked to notify Health Canada within prescribed timeframes once the results have been made public.
Health Canada website information
With enhanced transparency measures, more information about Canadian clinical trials would be available from international registries. However, Health Canada proposes to play a role in making this information more accessible. Improving access means making information easy to find, available in both official languages and using technology that would allow access on various platforms and mobile devices. The public should be able to locate this information and perform searches (using filters for various criteria) to find the information that is relevant to them in the official language of their choice.
Modernization of Compliance and Enforcement
As the nature of clinical trials continues to evolve, a modernized approach to compliance and enforcement (C&E) is necessary for Health Canada to maintain oversight over the conduct of clinical trials to protect the rights, safety and well-being of trial participants, to verify and validate the integrity of trial data, and to strengthen public confidence in trial outcomes. Factors such as the increased globalization of the healthcare industry, the growing complexity of trial designs and an expansion of the roles of Qualified Investigators (QIs), Contract Research Organizations (CROs) and Site Management Organizations (SMOs) in clinical trial management have further highlighted the need to modernize Health Canada's approach to C&E.
Health Canada's current approach to the C&E of clinical trials is focused on inspecting clinical trials at QI sites. In the future, Health Canada would focus more on sponsors of clinical trials by implementing a cyclical risk-based inspection approach of all clinical trial sponsors, CROs and SMOs in Canada. Health Canada would seek additional authority to compel third parties involved in the conduct of a clinical trial on behalf of a sponsor, to take action when an issue of non-compliance arises. Health Canada would also expand its C&E approach to include clinical trials using medical devices or NHPs, and has already initiated pilot inspections of trials involving medical devices as a first step to implement a full C&E approach for trials involving medical devices. Currently, there are no inspections of trials involving NHPs.
Modernizing Health Canada's C&E approach is expected to have several beneficial effects, such as allowing stakeholders to keep pace with best practices, maintaining the protection of trial participants, improving the overall compliance with the regulatory requirements and GCP, and further enabling Canada to remain competitive in a global clinical trials market sector.
Proposed Approaches
Health Canada is exploring several new approaches to the conduct of its C&E activities, which are described further below. However, a final C&E framework proposal will be advanced once the new framework for clinical trials has been determined.
Expanded Oversight Across Product Lines
While Health Canada currently has the authority to conduct inspections on clinical trial activities under Section 23 of the Food and Drugs Act, its activities have been limited to drugs.
Health Canada is proposing to support the modernization of clinical trials for medical devices and NHPs by extending C&E oversight to those product lines. Health Canada would inspect against the Act and its associated regulations, including international GCP standards, such as the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use's (ICH) ICH-E6(R2) and/or the International Standards Organization's (ISO) ISO 14155 standard. When identifying which trials to inspect, Health Canada would apply a risk-based and pro-active approach that considers the trial design and other factors, such as the medical condition under study, target population, or the risk classification of the investigational product (drug, NHP, or medical device). Similarly, when identifying which sponsors, CROs or SMOs to inspect, Heath Canada would implement a cyclical risk-based inspection approach that considers the compliance history and the volume of activities. Highly compliant entities would be inspected less frequently while less compliant ones would be inspected more frequently.
These new C&E activities being proposed would be consistent with other international jurisdictions such as the Food and Drug Administration of the US, the UK's Medicines and Healthcare Products Regulatory Agency (MHRA), and the EMA, which have risk-based inspection programs and standards built into their regulatory frameworks. It would also help to promote regulatory consistency between product lines and within multi-product trials, while working to enhance the safety of trial participants, public confidence in trial outcomes, and the integrity of clinical trial data and results.
Compliance and Enforcement of Third Parties
Under the present clinical trials regulatory regime, Health Canada has direct oversight authority over sponsors of clinical trials, but not over third parties that may be involved in the conduct of clinical trials, such as the QIs, CROs, or SMOs. Sponsors often rely on these third parties for the conduct of significant parts of a trial, and they are a large and growing part of the clinical trials sector.Footnote 4
Currently, Health Canada may not always be aware of third parties involved in a trial, given that there is no requirement for sponsors to provide information on the involvement of CROs and SMOs to Health Canada, although information on QIs is supplied as part of the clinical trials site information (CTSI) form or as part of the Investigational Testing Application for medical devices. This is an issue because these third parties may be responsible for non-compliance issues, which can affect the safety of participants during a clinical trial. While Health Canada can compel a trial sponsor to take corrective action and hold them accountable for the activities of these third parties, the department has no direct authority over the third parties.
Health Canada is proposing to amend the regulations so that any third party that conduct all or part of a clinical trial for a sponsor would be legally responsible for those activities it conducts on behalf of the sponsor, and therefore be subject to regulatory action in the event of non-compliance. This proposed amendment would result in additional regulatory burden for third parties involved in CTs, but would contribute to increased safety of clinical trial participants as Health Canada would be able to direct the appropriate parties to take actions to correct non-compliance in a timely manner. In addition, it would better align with the C&E approach in the United States, which allows sponsors the ability to transfer part of their legal responsibility to CROs.
Part 3: Anticipated Outcomes for Stakeholders
Health Canada's vision for the modernization of Canada's clinical trial regulatory framework presented above would help encourage clinical trials in Canada by creating an environment that supports the safe innovation of health products. Health Canada is only one of many stakeholders in Canada's clinical trial and health ecosystem, and there are many factors that influence and shape the future of clinical trials in Canada. A properly designed regulatory framework would provide a solid foundation for positive change. Informed by stakeholder feedback, it is anticipated that a modernized and properly designed framework would:
- Provide greater agility in oversight for the safe development of innovative therapies;
- Contribute to ensuring that administrative and regulatory requirements on sponsors are more proportional to risk;
- Offer Canadians access to safe clinical trials and trial information;
- Create a more streamlined approach that is better aligned internationally; and
- Leverage partnerships and provide leadership in Canada's health ecosystem to further facilitate clinical trials in Canada.
All stakeholders are encouraged to help shape the future of clinical trial regulation in Canada by submitting their responses to the online questionnaire or alternatively, by sending their written feedback by email to hc.policy.bureau.enquiries.sc@canada.ca using the questions provided in Annex 3 below as a guide. The responses received will be taken into consideration as Health Canada strives to build a more agile, risk-based and transparent regulatory regime, ultimately improving the lives of Canadians in the process.
Annex 1: Overview of the Existing Canadian Clinical Trial Regulatory Framework
The clinical trial regulations in Canada for the different product lines were developed at different points in time, leading to some significant differences in the respective frameworks. A short overview and description of the current state of each framework is provided below.
Drugs
Under the current regulatory framework for clinical trials involving the use of pharmaceutical and biologic drugs in humans, Health Canada authorizes the sale and importation of drugs in Canada that are proposed to be used in a trial. With the exception of Phase IV studies, sponsors of drug trials must submit a clinical trial application (CTA) to Health Canada prior to the initiation of a trial, and obtain approval from a Research Ethics Board (REB). As prescribed in regulation, there is a 30-day default review period for CTAs. Health Canada will issue a No Objection Letter (NOL) if the trial is authorized or a Non Satisfactory Notice (NSN) if the CTA is rejected within 30 days of receiving a complete CTA. If no response is sent within the 30 days, then the trial is authorized to commence by default.
This "default authorization" is necessary for both drugs that are not currently authorized in Canada, as well as market-authorized drugs that are being investigated for potential uses outside the parameters of their market authorization.
The conduct of clinical trials involving the administration of drugs are governed by The Food and Drugs Act and the Food and Drug Regulations (FDR),and the sponsors must follow Good Clinical Practices (GCP) as set out in section C.05.010 of the FDR.
Medical Devices
The Medical Devices Regulations (MDR) in Canada classify medical devices into four categories (I – IV), from lowest to highest risk. Health Canada authorizes the sale and importation of medical devices of Classes II - IV that are intended for investigational testing involving human participants. The conduct of these clinical trials are governed by The Food and Drugs Act and the MDR and the manufacturers and importers are encouraged to follow GCP as described by the standard ISO 14155 – Clinical investigation for medical devices for human subjects.
The manufacturer or importer of the medical device to be used in a trial must submit an Investigational Testing Authorization (ITA) application to Health Canada to obtain an authorization for the sale or importation of the device for investigational testing. Manufacturers undertaking the research must obtain approval from both Health Canada (with the exception of Class I devices) and a REB prior to the initiation of a clinical trial, following a risk-based approach. Class II medical devices are subject to reduced application requirements and Class III and IV medical devices must adhere to the full set of application requirements as described in Part 3 of the MDR. Currently, Health Canada applies a 30-day review period for medical device clinical trials from the point in time that a complete application is received. Health Canada may issue a "Letter of Authorization" for investigational testing of Class II, III, and IV medical devices if the submitted information is deemed to satisfy the requirements of the regulations. Alternatively, if information is missing, Health Canada may issue a request for additional information. There is no 30-day default review period for ITAs for medical devices.
Natural Health Products
Health Canada regulates the sale or importation of certain natural health products (NHPs) to be used for the purposes of clinical trials involving human participants. The conduct of clinical trials for NHPs are governed by The Food and Drugs Act and the Natural Health Products Regulations (NHPR) and must be conducted in accordance with GCP as set out in section 74 of the NHPR. In accordance with Part 4 of the NHPR, sponsors undertaking the research must obtain approval from both Health Canada (except for phase IV trials) and a REB prior to the initiation of a clinical trial. The requirements of a clinical trial with a NHP are very similar to the requirements for a clinical trial of a conventional drug. Currently, Health Canada follows a performance standard defined in policy as up to 90-days for NHP clinical trials review and approval. Health Canada will issue a Notice of Authorization if the sale or importation of an NHP for the purposes of a clinical trial is approved.
Annex 2: Specific Considerations of Clinical Trial Modernization by Product
Along with the broad changes outlined in this consultation document for clinical trial modernization, Health Canada recognizes that there are some elements of the proposal that represent unique changes for the different product lines. Some of these considerations and the anticipated impacts are described below to provide stakeholders with a comprehensive overview of what they could expect, and what the proposed changes would mean for them.
Drugs
Stakeholders have previously identified the lack of a risk-based approach to clinical trial regulation as an impediment to conducting research in Canada, and some have requested that Health Canada look to models of risk stratification used within other jurisdictions as a model for change to the regulation of drug clinical trials. In 2019, Health Canada published a Notice to Stakeholders: Statement on the Investigational Use of Marketed Drugs in Clinical Trials that, under certain conditions, could reduce some regulatory requirements for the off-label use of marketed drugs in a clinical trial when these drugs are not the subject of the investigation in the trial. The 2020 and 2021 CT-IOs incorporated this risk-based approach to reduce requirements for both investigational and non-investigational uses of already marketed drugs in a clinical trial.
Where the safety profile of a drug is well-characterised, as in the case of currently marketed drugs, Health Canada is proposing to reduce requirements for the collection of non-serious adverse events or other safety data under certain conditions. Decreasing unnecessary burden related to adverse event collection, if outlined in the protocol and approved by Health Canada, could result in more cost-effective clinical trials by lessening administrative burden.
Another major change for drug clinical trials would involve moving from a default "no objection" authorization of the sale or import of the drug product in the clinical trial, to a more comprehensive authorization of the trial as well as the products being tested. This proposed change, along with Health Canada's proposal to issue a single authorization for a trial involving multiple product lines, would empower Health Canada to better regulate the range of innovative trial types that are increasingly being used by sponsors, such as adaptive trials or master protocols. It would also help to facilitate and streamline the regulatory process for sponsors, which can be difficult to navigate for more complex trial types.
For drug trials, this proposed shift to trial authorization would also remove the current 30-day default "no objection" authorization from the regulations. This would mean that sponsors would have to receive an authorization from Health Canada before proceeding with a trial. The timelines for review of a CTA would be defined in policy and be aligned across product lines. For bioequivalence trials, Health Canada would remove the 7-day administrative review target, aligning this timeline with that of other reviews for a more equitable treatment among sponsors. Health Canada could revise the timelines for additional information requests to sponsors made during a CTA review, which are currently subject to a 2-day timeline by regulation.
Under the proposal, agile risk-based tools would be used to maintain participants' safety without necessarily affecting the progress of an entire clinical trial. Health Canada would have the authority to require a sponsor to perform an analysis of certain information and provide that analysis to Health Canada, which would also help ensure adequate monitoring of trials throughout the course of their life cycle. For drug trials, a new authority for Health Canada to add terms and conditions to a clinical trial approval could be used, for example, to require the establishment of a data monitoring committee (DMC), if not already part of an application, and request reports from this DMC at specific points during the study.
The ability to suspend one arm of a trial based on a safety signal without denying the participants in the unaffected arm(s) the access to potential therapies would also be a key agility for drug trials. Health Canada is also proposing that once a study has been terminated, the sponsor would have to request authorization to restart the study (at present, a study can be restarted with only a notification to Health Canada).
In addition to the changes noted above, Health Canada is considering referencing the ICH E6 guideline on GCP more clearly in the regulations. Health Canada is also assessing the ability to apply several concepts introduced through the CT-IOs more broadly, such as enabling alternate means of obtaining informed consent, and enabling a broader range of health care professionals who are permitted conduct a trial.
Medical Devices
Medical device investigational testing refers to the clinical study undertaken to assess the clinical performance, clinical safety and/or effectiveness of a medical device. Studies could also involve the evaluation of marketed medical devices for new intended uses, new populations, new materials or design changes.
With the 2018 Action Plan on Medical Devices (MDAP), Health Canada committed to reviewing longstanding stakeholder concerns with respect to how the current regulatory framework for medical devices might be unintentionally limiting investigational testing activity in Canada. This commitment focused on potential regulatory changes that could encourage more clinical research in Canada, while continuing to focus on the protection of patient safety. In 2019 and early 2020, the department held targeted stakeholder consultations to gather input on possible changes to the ITA framework. Stakeholders were identified from the following groups: investigators/researchers; industry; patient groups; physician groups; Research Ethics Board (REB) representatives; and granting agencies. The comments received during the MDAP consultations on the ITA framework have been used to inform policy development. These are further discussed below.
New flexibility for independent investigators
At present, manufacturers and importers must meet the regulatory requirements under Part 3 of the MDR to receive authorization from Health Canada to sell a Class II, III or IV device to a qualified investigator for the purpose of conducting investigational testing. Trials testing only Class I devices do not require authorization but they do need to meet all applicable regulatory requirements. Health Canada issues an authorization if it determines that the device can be safely used, that the testing is not contrary to the best interests of patients, and the objectives of the testing will be met.
Dr. Dylan is a cardiologist who would like to evaluate a new unlicensed stent as part of a clinical trial. Currently only manufacturers and importers of medical devices would be able to apply for authorization. Dr. Dylan would have to work with a manufacturer to have them submit an application for authorization on his behalf. Under regulatory modernization, Dr. Dylan could submit an application directly to Health Canada.
The current MDR permits only manufacturers and importers of medical devices to apply for an ITA. However, through policy described in guidanceFootnote 5, Health Canada provides a delegation mechanism through which an independent investigator (such as a researcher, clinician or health care facility) may be authorized by a device manufacturer to be a regulatory correspondent. Despite this, the authorized investigator cannot file the ITA application without express consent from the device manufacturer and has no associated legal responsibilities under the MDR. Amendments to the Regulations are being proposed to enable an investigator, independent of the device manufacturer, to apply for a Clinical Trial Authorization* in addition to a manufacturer and importer.
*Note: The use of "investigational testing" is inconsistent with the new enabling authorities in the Food & Drugs Act (F&DA), which refer specifically to "clinical trial." For this reason and for alignment with all product lines, investigational testing will be referred to as clinical trials in the revised regulations.
Modernized Regulatory Requirements
The current approach to classifying a medical device as one of four classes is risk-based, with tailored regulatory requirements that allow an acceptable level of uncertainty through which Health Canada can apply fewer requirements to lower risk products involved in clinical trials (refer to section 3.2 Risk-based approach for further details). Health Canada is considering modifying this regulatory approach in a few select areas.
Clinical trials of IVDDs using remnant samples when not used for patient management and for research use only purposes do not involve patients and therefore present no risk to patient safety. However, these devices may present some risks to the user with respect to the electrical safety of the instruments. Similarly, magnetic resonance imaging "work-in-progress" pulse sequence trials when certain conditions are met, pose little to no risk to patients and users. Health Canada is exploring exempting these low-risk products from requiring authorization prior to starting a trial.
With the new legislative authority under the Food and Drugs Act, all clinical trials will require authorization unless otherwise specified. Current Health Canada guidance directs manufacturers to submit an application to use a licensed device in a study intended to generate data to support a new indication (off-label use). However, off-label use of licensed medical devices used in clinical investigations and not sponsored by the manufacturer do not require an authorization. Going forward, Health Canada will be exploring whether there are opportunities for reduced requirements for clinical trials studying off-label use of medical devices licensed in Canada, while ensuring that patient safety remains a priority for both manufacturer and independent investigator-led trials.
Company Y has developed a test kit to detect Hepatitis B surface antigen. The company would like to perform a study to evaluate the performance of the test kit using remnant blood samples obtained from blood banks. The samples will be de-identified and the test results obtained will not be shared with the patients and will not be used for patient management. Currently, they would need to submit an application to obtain an investigational testing authorization to use the device in a trial. Under the regulatory modernization, they would be exempt from the requirements to obtain an authorization but other regulatory requirements would be maintained (e.g., incident reporting, complaint handling, recalls, REB approval, GCP).
The possibility of reduced regulatory requirements for the testing of a device within a single corporate entity (in-house trials) is being explored. Regulatory requirements for these in-house trials where no sale has occurred were previously not applicable, however, regulatory modernization would provide Health Canada with oversight of these trials.
Formalizing Current Medical Device Policy into Regulation
Health Canada provides Guidance documents to help interpret expectations for compliance with the regulations. Some topics currently in guidance have been identified as benefitting from formalization in regulation including Research Ethics Board (REB) approval, Good Clinical Practices (GCP), and revisions to a CTA.
REB Approval
The current regulations as written may be interpreted that Health Canada requires REB approval as part of the ITA application package for Class III and IV devices (REB is also required for Class I and II devices but these do not need to be submitted to Health Canada). However, Health Canada has provided direction within its guidance document to advise that REB approval may be provided after authorization but prior to the start of a trial. This different interpretation between the regulations and guidance has caused some confusion for stakeholders. Moving forward Health Canada proposes that this language be made clearer in regulation to explicitly require REB approval prior to the commencement of a trial. As is the current case, for clinical trials involving Class III and IV devices, the study cannot be initiated until REB approval is received and communicated to Health Canada. The proposed changes would also bring Canada in line with its international partners, such as the United States and the European Union.
Company X would like to conduct a clinical trial using a Class III medical device at a hospital in Ontario. They are aware that REB approval at the institution must be obtained as well as the authorization from Health Canada. The REB at the institution would not approve a clinical trial until they receive evidence of Health Canada authorization. At the same time, Health Canada requirements for REB approval are not clear for Company X as the regulations specify that written REB approval are to be provided prior to authorization whereas the guidance provides a different instruction. Company X is confused and does not know in what submission order they must proceed. To avoid this confusion, the modernized regulations would include clear language that permits REB approval of the clinical trial to occur after the issuance of the clinical trial authorization by Health Canada but before trial initiation.
GCPs
It is internationally recognized that research in humans should be conducted according to generally accepted principles of GCP, as stated in the ISO 14155 standard - Clinical investigation of medical devices for human subjects. These principles provide assurance that the data and reported results are credible and accurate, and that the rights, safety, and well-being of clinical trial participants are protected.
The MDR currently does not contain a requirement for GCP compliance, however, conformity to ISO 14155 is recommended in Guidance. The inclusion of a requirement in regulation for a trial sponsor to comply with GCP principles would provide greater assurance that patients remain protected during investigational studies. It would also better align the clinical trial requirements for medical devices and human drugs in Canada, and with our international partners (European Union, United Kingdom, United States, Australia).
Revisions to an Investigational Testing Application
Under the current regulatory framework, there are no provisions that allow Health Canada to authorize revisions to an ITA. As such, any changes made during a trial must be submitted as a new ITA application.
However, Health Canada allows manufacturers to submit some revisions to previously authorized ITAs in an abbreviated manner through a policy approach. Available Guidance also outlines an additional process by which more administrative modifications can be submitted to Health Canada through a notification. Health Canada is proposing to formalize this policy in regulation, allowing minor and significant changes to be made to an authorization without a trial sponsor having to submit a new application.
A device manufacturer based in Canada received authorization to conduct a clinical trial on a new type of ventilator (a Class III device) during the pandemic. The manufacturer decides that a software change is needed to add a new ventilation mode. Currently, there are no provisions in the regulations to authorize revisions to an authorization – a full new application would need to be submitted (however, guidance allows manufacturers to submit revisions in an abbreviated manner). Under a modernized clinical trial framework, regulatory flexibility would be in place for authorization holders to submit and obtain authorization for amendments to the clinical trial authorization if there are any significant changes to the information submitted in the original application.
Trial Commencement, Discontinuance and Resumption
There are no provisions in the MDR requiring a manufacturer to report to Health Canada when it commences, discontinues or resumes an investigational study, either in whole or in part. However, the Health Canada Guidance Document does address these scenarios. With the new enabling authorities to regulate the entire lifecycle of the trial, new requirements are being proposed for reporting the commencement of a study, or the discontinuance or resumption of a study, either trial-wide or site specific, to Health Canada.
Natural Health Products
Health Canada is proposing to modernize the regulations for the clinical trials of NHPs to be in line with the updated regulations for trials with other health products. The intention is for all proposals described in the Agile Life Cycle, Risk-Based Approach, and Transparency themes to be applied equally, while taking into account the unique needs of NHPs.
Changes would include updating the review timelines for a CTA of a NHP trial. The current timeline is a 90-day review, but this is likely to be reduced to be consistent with the requirements for other product lines, and as Health Canada considers the feedback received from this consultation process. Furthermore, in accordance with current NHP Regulations, Health Canada is proposing to maintain its current requirement that one member on a Research Ethics Board is knowledgeable in complementary or alternative care, ensuring expert knowledge relevant to NHPs during the ethics review process.
By introducing a risk-based approach based on the available information on the risk, benefit and uncertainty of a NHP, Health Canada can tailor requirements proportionately, rather than apply a one-size-fits-all approach that may not be appropriate for many NHPs. There would also be greater flexibility in regulations, allowing Health Canada to address different aspects of the trial in an appropriate manner over the course of the life cycle of the trial. Ensuring alignment with other health products, such as drugs, is critical as this process moves forward.
Canadian Company LifeChange plans to conduct a clinical trial that studies the efficacy of a combination therapy that includes Product X (a drug) and Product Y (a natural health product). Previously, LifeChange was required to submit two separate applications – one for the drug, and one for the natural health product – and wait for authorization under two approval streams with different review timelines. After CT modernization, LifeChange would be able to submit a single clinical trial application and receive a single approval for a clinical trial involving products X and Y. Internally, this CTA would be reviewed by both TPD and NNHPD, but the single window approach with the applicant would streamline the process and reduce their administrative burden.
Annex 3: Consultation Questions
To be more user-friendly, Health Canada has embedded the following questions within an online consultation. Health Canada encourages respondents to provide their feedback through this online format, which is a combination of multiple choice questions as well as text boxes in which you can provide your views or further details on multiple choice selections. If you choose to submit comments by email or through the traditional mail system, please feel free to do so. We would encourage you to use the following questions as a guide.
To safeguard privacy, you should ensure that any written comments you may provide are sufficiently general that you cannot be identified as the author and that individual identities are not disclosed.
Demographic questions:
- Please indicate which type of stakeholder you represent/identify with the most:
- Industry
- Academics / research institutes
- Health care practitioners
- Health care organizations not including those listed as researchers
- Patient advocacy
- Research ethics boards and the Canadian Association of Research Ethics Boards
- Provincial or territorial governments, please specify
- Health Canada
- Other federal government department, please specify
- What describes your clinical trial activity (select all that apply):
- Sponsor / Applicant Industry
- Sponsor / Applicant Academia
- Contract Research Organisation
- Site Management Organisation
- Qualified Investigator
- Study Coordinator / Study Team member
- Clinical Research Association
- Clinical Trial Participant
- Other, please specify
- What health product line(s) do you or your organization represent or conduct trials for or are interested in? (select all that apply)
- Drugs
- Medical devices
- Natural health products
- Do not conduct clinical trials
- Other, please specify
Questions for all respondents (please elaborate in your responses where appropriate):
- Do you anticipate any benefits or impacts of the following proposals for your organization?
- The agile life-cycle approach
- The single authorization for multiple products
- The risk-based approach
- The use of terms and conditions on a clinical trial authorization
- Decentralized trials
- Based on your experience and knowledge, would the proposals in the Consultation Paper meet Health Canada's goal of enabling innovative clinical trials in Canada?
- Are there innovations or other future considerations that Health Canada should account for when modernizing the clinical trials framework?
- Are there other factors Health Canada should consider when implementing this proposal to streamline regulatory requirements across product lines to better enable a single authorization?
- Are there factors Health Canada should take into consideration in the implementation of a risk-based approach?
- Are there other areas where burden can be reduced that will better enable your organization to conduct clinical trials without compromising patient safety?
- Are there areas where Health Canada could go further in modernizing the Canadian clinical trials regulatory framework beyond what is proposed in the consultation paper?
- Do you see value in implementing a new policy and/or regulation for registration and reporting of results for clinical trials conducted in Canada, investigating drugs, medical devices and natural health products?
- Do you typically register your trials with an international registry?
- Do you experience any challenges with keeping your registration up to date in the international registries, including the trial status?
- Do you currently outsource activities related to clinical trials or investigational tests to third parties (e.g., contract research organization or site management organization)? If so, what activities do you outsource? If you were no longer legally responsible for those activities you had outsourced, what impact would that have on your business?
- Are activities being outsourced to you for clinical trials involving drugs, NHPs or medical devices? If so, how many clinical trials/investigational tests are you involved in per year in Canada on average? If you were to be legally responsible for those activities that have been outsourced what impact would this have on your business?
- Are you currently or have you ever been involved in clinical trials/investigational testing in the United States?
- Based on how you conduct your activities, how would you define the end of a clinical trial?
- Are there any challenges to implementation associated with clarifying that retention of records should begin following completion or discontinuation of the clinical trial?
If you or your organization is involved in clinical trials with drugs:
- Given the increasing complexity of clinical trials and the need to establish a harmonized regulatory framework across product lines, would you support extending application review timelines? Please explain how many days you feel would be reasonable for the clinical trial application review time, and under which conditions.
- If other professionals are allowed to become investigators, should there still be a requirement that a designated physician be responsible for the overall medical care of participants?
If you or your organization is involved in clinical trials with natural health products:
- Based on your knowledge and experience, do you see a benefit in reduced clinical trial review timeline from 90 days on the conduct of your clinical trial?
- Should adverse NHP reaction reporting requirements be modified to align natural health products and drugs requirements?
- What challenges do you face in complying with the Good Clinical Practices provisions of the Natural Health Products Regulations/ ICH-E6(R2) Good Clinical Practice?
If you or your organization is involved in investigational testing with medical devices:
- Health Canada is maintaining the risk-based classification scheme (Class I-IV) for medical devices in Canada. However, the new framework proposes to further sub-divide medical device clinical trials into three (3) categories (A – No authorization; B – Tailored Requirements; C – Full Requirements) for increased alignment with drugs. Do you support this proposed framework? Please explain what advantages or disadvantages you can foresee with this new classification scheme.
- Should the following low-risk products be exempt from requiring authorization (other requirements would still need to be met e.g., incident reporting, Research Ethics Board approval):
- In Vitro diagnostic devices (IVDD) using remnant samples (when not used for patient management and for research use only purposes)?
- Magnetic resonance imaging "work-in-progress" pulse sequence trials (when certain conditions are met- please refer to Guidance document Section 2.3.2.3)?
- For manufacturer-initiated trials, should there be tailored regulatory requirements for in-house clinical trials, where there is no sale of a device?
- Should there be reduced regulatory requirements when a new use of a licensed medical device is being studied (off-label use)?
- While keeping patient safety in mind, should there be a difference in regulatory requirements depending on whether the off-label use of a medical device is the subject of the investigation, or whether it is being used as a comparator/for support (e.g., to deliver the investigational device).
- Do you foresee any risks related to the off-label use of a marketed medical device in a clinical trial?
- Please describe any concerns you may have with the Research Ethics Board approval of the trial occurring after issuance of the clinical trial authorization but before the study initiation.
- Would you have any concerns with Health Canada creating the following three pathways for clinical trial Authorization amendments?
- Significant changes (based on the Guidance for the Interpretation of Significant Change of a Medical Device), which would require clinical trial authorization revision and Health Canada approval.
- Notifiable changes, which would need to be filed with Health Canada but would not require approval.
- Non-significant or administrative changes, which would need to be documented only in an applicant's internal files.
- What types of amendments should fall under each category and why?
- Do you have any additional recommendations to increase efficiencies to the clinical trial authorization revision process without compromising on patient safety?
- What challenges would you anticipate if an investigator, independent of the manufacturer, wishes to apply for authorization to Health Canada to conduct a clinical trial of a medical device (either on or off-label use)? Do you have any recommendations that could help to overcome these challenges?
- How should challenges be overcome in situations where manufacturers are not willing to share medical device information with the investigator or with Health Canada to support an independent investigator's application?
- Are any investigator-led trials being held at your site? In general, what types of clinical trials are being conducted at your site? (Select all that apply):
- Medical device pre-market studies: First in Human Study
- Medical device pre-market studies: Feasibility / Pilot study
- Medical device pre-market studies: Pivotal study
- Medical device post-market studies: Marketed device as per approved indication of use
- Medical device post-market studies: Off-label use of marketed device
- Clinical trial using an unlicensed medical device not subject to investigations
- Other: Please specify
- In general, what is the purpose of the medical device clinical trials you are conducting in Canada? To generate data to support a future medical device licensing application for marketing, for research purposes only, or other?
- For clinical trials that you have conducted and that did not require an authorization, what types of safeguards were put in place to protect patient safety in addition to getting research ethics board approval?
- Do you foresee any challenges associated with complying with Good Clinical Practices as aligned to ISO 14155 requirements?