
Modernisation de la réglementation portant sur les essais cliniques: Document de consultation
Table des matières
- Partie 1 : Introduction
- Partie 2 : Propositions pour examen
- Partie 3 : Résultats attendus pour les parties prenantes
- Annexe 1 : Vue d'ensemble du cadre réglementaire canadien actuel sur les essais cliniques
- Annexe 2 : Considérations propres à la modernisation d'essais cliniques, par produit
- Annexe 3 : Questions de la consultation
Partie 1 : Introduction
En tant que l'un des cinq piliers clés du programme d'innovation de la réglementation du gouvernement du Canada pour les produits de santé, la modernisation de la réglementation portant sur les essais cliniques est un objectif important pour Santé Canada et, au sens plus large, pour les Canadiens. Santé Canada, l'organisme fédéral de réglementation des produits de santé sur lesquels les Canadiens comptent dans leur vie quotidienne, est responsable de la réglementation des essais cliniques portant sur un large éventail de produits, y compris les médicaments et les produits biologiques, les produits de santé naturels (PSN) et les instruments médicaux. Les essais cliniques portant sur des produits destinés à soulager les douleurs et les courbatures, jusqu'à ceux utilisés lors d'interventions de soins intensifs impliquant l'insertion d'une endoprothèse cardiaque, doivent tous garantir la sécurité des participants et produire des données fiables tout en respectant les principes éthiques des essais sur les humains.
Avec les progrès continuellement accélérés de la technologie et la création de nouveaux types de produits de santé prometteurs, ainsi que l'avènement de nouveaux types et conceptions d'essais cliniques, la nécessité de moderniser le cadre réglementaire des essais cliniques au Canada est devenue évidente. Il est important que le cadre réglementaire des essais cliniques au Canada favorise l'adoption de nouveaux traitements prometteurs dans le système de soins de santé et n'étouffe pas par inadvertance les innovations qui pourraient contribuer à améliorer la santé des Canadiens. Les produits de santé personnalisés, les thérapies géniques et les produits destinés au traitement de maladies rares posent chacun des exigences particulières en matière de conduite d'essais cliniques. Il est essentiel de satisfaire à ces exigences afin de constituer une base de connaissances qui soutiendrait l'introduction de nouveaux traitements sûrs et efficaces sur le marché canadien, améliorant ainsi la vie des Canadiens tout en leur donnant accès aux renseignements dont ils ont besoin pour prendre des décisions éclairées concernant leur santé.
En outre, la pandémie de COVID-19 a mis davantage en évidence la nécessité de modifier le cadre réglementaire des essais cliniques au Canada. Le gouvernement du Canada a réagi rapidement à la pandémie en publiant l'arrêté d'urgence sur les essais cliniques d'instruments médicaux et de drogues en lien avec la COVID-19 (AU-EC) et l'arrêté d'urgence no2 sur les essais cliniques d'instruments médicaux et de drogues en lien avec la COVID-19 (AU-EC 2), ainsi que leurs lignes directrices connexes. Ces arrêtés d'urgence ont fourni et continuent de fournir, une plus grande souplesse pour mener des essais cliniques dans le contexte de la pandémie. Toutefois, ces assouplissements ne s'appliquent qu'aux médicaments et aux instruments destinés au diagnostic, au traitement, à l'atténuation ou à la prévention de la COVID-19. Bien que les mesures prises dans le cadre des arrêtés d'urgence sur les essais cliniques (AU-EC) soient temporaires, elles ont également fourni une importante preuve de concept pour certains des éléments de la modernisation des essais cliniques qui étaient déjà à l'étude.
Comme il s'y est engagé dans le Plan prospectif de la réglementation 2021-2023 de Santé Canada, le Ministère prévoit moderniser le cadre des essais cliniques pour introduire une approche cohérente fondée sur le risque, offrir une plus grande souplesse dans le développement sécuritaire de traitements innovants, rationaliser les processus pour plus d'efficacité et de clarté, et s'aligner sur les pratiques exemplaires internationales en matière de surveillance et d'accès du public à l'information. Avec tous les changements proposés, Santé Canada continuerait à maintenir les principes fondamentaux sous-jacents communs à la réglementation de tous les types d'essais cliniques, notamment la protection de la sécurité des participants, le soutien à l'innovation, la surveillance proportionnelle et l'harmonisation à l'échelle internationale.
À cette fin, Santé Canada est heureux de présenter une vision ambitieuse de la modernisation du cadre réglementaire des essais cliniques au Canada, qui répondra mieux aux besoins des intervenants canadiens tout en continuant à protéger la sécurité des patients dans un écosystème de soins de santé moderne. Un cadre unique d'essais cliniques pour tous les produits de santé est proposé comme base du nouveau régime réglementaire qui fournirait une surveillance proportionnelle basée sur le risque, de nouvelles agilités réglementaires tout au long du cycle de vie de l'essai, une plus grande transparence par l'enregistrement et la divulgation publique des résultats, et un régime modernisé de conformité et d'application de la loi (voir figure 1). Une consultation distincte sur une proposition de nouveau cadre réglementaire visant à permettre la réalisation d'essais cliniques sur des aliments à des fins diététiques spéciales est actuellement en cours, avec une date limite de soumission des commentaires fixée au 12 juin 2021. Si vous souhaitez recevoir le document de consultation pour ce processus, veuillez envoyer votre demande par courrier électronique à hc.bns-bsn.sc@canada.ca.
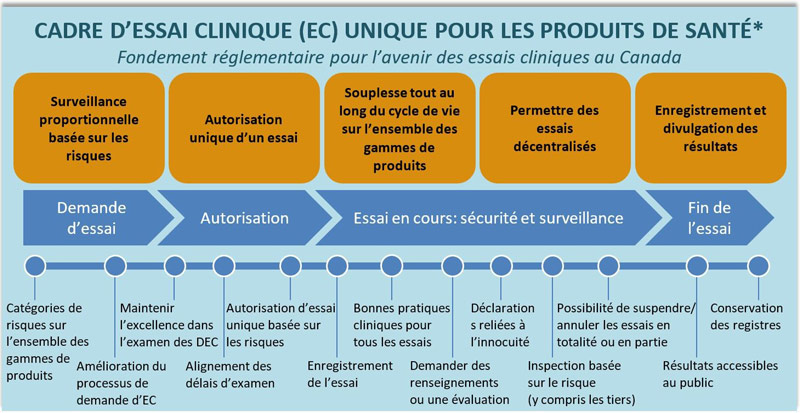
Figure 1 - Description textuelle
La fondation réglementaire de l'avenir des essais cliniques au Canada repose sur la proposition d'un cadre unique pour tous les produits de santé. Le nouveau régime réglementaire permettrait une surveillance proportionnelle basée sur les risques, une autorisation unique d'un essai, une approche agile du cycle de vie pour toutes les gammes de produits, des essais et des enregistrements décentralisés et des résultats divulgués au public.
Le cycle de vie des essais cliniques est décrit en 4 étapes, avec les nouveaux éléments réglementaires clés alignés sur ces étapes:
- Demande d'essai
- Catégories de risques sur l'ensemble des gammes de produits
- Amélioration du processus de demande d'EC
- Autorisation
- Maintenir l'excellence dans l'examen des DEC
- Alignement des délais d'examen
- Autorisation d'essai unique basée sur les risques
- Essai en cours: sécurité et surveillance
- Enregistrement de l'essai
- Bonnes pratiques cliniques pour tous les essais
- Demander des renseignements ou une évaluation
- Déclarations reliées à l'innocuité
- Inspection basée sur le risque (y compris les tiers)
- Possibilité de suspendre/ annuler les essais en totalité ou en partie
- Fin de l'essai
- Résultats accessibles au public
- Conservation des registres
* Il n'existe actuellement aucun cadre réglementaire pour les essais cliniques concernant les aliments à des fins diététiques spéciales (AFDS). Le nouveau cadre permettra des essais des AFDS, qui seront harmonisés avec les essais cliniques de produits de santé aux étapes clés du cycle de vie.
Dans ce nouveau cadre unique pour les essais cliniques, plusieurs thèmes clés distincts seront abordés plus en détail dans ce document de consultation. Voici ces thèmes :
- Souplesse tout au long du cycle de vie
- Approche basée sur le risque
- Transparence
- Modernisation de la surveillance de la conformité et de l'application de la loi
Commentaires sur la proposition
L'objectif principal de ce document de travail est de donner aux Canadiens l'occasion d'examiner les propositions de politiques pour la modernisation du cadre réglementaire des essais cliniques du Canada et de fournir des commentaires avant que le ministère finalise les décisions de politique et élabore la réglementation. Les commentaires supplémentaires des intervenants permettront à Santé Canada de mieux comprendre la manière dont l'écosystème des essais cliniques va évoluer, y compris les tendances émergentes et les technologies novatrices qui pourraient avoir une incidence sur le déroulement des essais, et de savoir si les propositions présentées permettraient des essais cliniques qui répondent le mieux aux besoins des Canadiens, dans le but de leur permettre d'accéder à des technologies de soins de santé novatrices tout en continuant à protéger leur santé et leur sécurité. Les commentaires sont acceptés jusqu'au 4 juillet 2021 en envoyant une réponse au questionnaire en ligne ou en envoyant une réponse par courriel à hc.policy.bureau.enquiries.sc@canada.ca.
Contexte
Contexte canadien
Les intervenants canadiens ont fait état d'un besoin croissant de mener des essais cliniques en utilisant de nouvelles conceptions pour gagner en efficacité et ont demandé des changements pour accroître la cohérence réglementaire entre les cadres d'essais cliniques distincts des médicaments, des produits de santé naturels (PSN) et des instruments médicaux. Bien que l'approche actuelle de la réglementation des produits de santé aux fins d'un essai clinique ait bien servi les Canadiens, elle n'est pas suffisamment souple pour répondre aux nouveaux types d'essais cliniques qui apparaissent. De plus, les cadres réglementaires des essais cliniques des médicaments, des PSN et des instruments médicaux ont évolué séparément au fil du temps, ce qui a entraîné des différences importantes entre les régimes (voir l'annexe 1 : « Vue d'ensemble du cadre réglementaire canadien des essais cliniques » pour plus de détails). Ces différences ont entraîné des inefficacités et un fardeau inutile tant pour Santé Canada que pour les promoteurs.
Santé Canada reconnaît la nécessité d'un cadre réglementaire intégré qui offrirait également une plus grande transparence, une plus grande souplesse et des gains d'efficacité aux intervenants. Le fait de permettre de mener un plus grand nombre d'essais non conventionnels aurait l'avantage supplémentaire de faire en sorte que le Canada demeure un endroit attrayant pour la réalisation d'essais cliniques, tout en continuant à respecter des normes élevées en matière de protection de la santé et de la sécurité des participants.
Des changements législatifs pour permettre la modernisation des essais cliniques
Dans le cadre de la Loi d'exécution du budget de 2019, des dispositions législatives nécessaires ont été introduites dans la Loi sur les aliments et les drogues en vue d'appuyer la modernisation de la réglementation des essais cliniques. Ces changements ont été apportés en réponse aux conclusions de l'examen de la réglementation du secteur des sciences de la santé et des sciences biologiques, qui indiquent que « la réglementation relative aux essais cliniques limite la croissance ». Au cours des années 2019 et 2020, Santé Canada s'est mobilisé auprès des intervenants sur les objectifs de modernisation des essais cliniques. En plus des commentaires reçus en réponse au présent document, ainsi que lors de séances de consultation plus ciblées avec les principaux intervenants, le public aura également l'occasion de commenter les propositions réglementaires après leur publication préalable dans la Partie I de la Gazette du Canada, qui est prévue en 2022.
Contexte international
Compte tenu de la nature mondiale de l'innovation technologique et en matière de soins de santé, le Canada n'est pas le seul à reconnaître la nécessité d'un changement. Bien qu'il existe des approches internationales différentes en matière d'autorisation et de réglementation des essais cliniques, d'autres organismes de réglementation internationaux reconnaissent de plus en plus la nécessité d'adopter une approche plus souple et fondée sur le risque qui pourrait mieux s'adapter aux essais et aux produits de santé innovants. Par exemple, l'Agence européenne des médicaments (AEM) a publié un document stratégique prospectif intitulé « EMA Regulatory Science to 2025 : Strategic Reflection » qui, parmi d'autres sujets, explore différentes manières de favoriser l'innovation dans les essais cliniques et d'encadrer l'émergence de nouvelles façons de générer des données cliniques. De même, en mars 2019, la commissaire de la Food and Drug Administration (FDA) américaine a publié une déclaration sur les nouvelles stratégies visant à moderniser les essais cliniques. La déclaration confirme que la FDA « a travaillé en étroite collaboration avec les intervenants, notamment la Clinical Trial Transformation Initiative, afin de définir des modèles d'essais innovants, d'évaluer le rôle des essais cliniques décentralisés et des technologies mobiles, et d'aider à valider de nouvelles variables d'évaluation qui peuvent permettre aux essais de générer des données probantes fiables nécessaires pour évaluer plus efficacement l'innocuité et l'efficacité des produits. » La déclaration fait également état de plusieurs nouveaux documents d'orientation pour l'industrie qui ont été publiés pour aider les intervenants à atteindre ces objectifs tout en naviguant dans le paysage réglementaire.
Consultations précédentes et ce qui a été entendu
En juillet 2019, Santé Canada a publié le document de réflexion intitulé « Réglementation souple concernant les produits thérapeutiques de pointe et les essais cliniques », avec une période de consultation qui s'est terminée en septembre 2019. En plus de cette consultation publique, le département a organisé un certain nombre de réunions ciblées avec un large éventail d'intervenants.
Au cours de ces consultations, les intervenants ont confirmé que le fait de permettre un cadre souple et propice aux essais cliniques allait attirer des entreprises et des investissements clés, et soutenir la recherche et l'innovation au Canada. Ils ont également recommandé que Santé Canada envisage une expansion dans les domaines suivants :
- Permettre des essais innovants conçus pour analyser simultanément une série d'interventions thérapeutiques, de tests diagnostiques complémentaires ou d'indications grâce à une plus grande rationalisation des exigences dans toutes les gammes de produits;
- Faciliter la mise en œuvre d'essais multisites et l'ajout de sites à ceux déjà lancés;
- Soutenir l'utilisation de données probantes du monde réel ainsi que la collecte et l'analyse de données en temps réel pour évaluer et mettre à jour rapidement et continuellement les profils avantages-méfaits-incertitudes et les soins continus aux patients;
- Permettre une certaine souplesse aux cliniciens dans des circonstances particulières, notamment ceux qui mènent des essais adaptatifs, afin qu'ils soient mieux à même de mener des recherches tout en adaptant les traitements en fonction des besoins des patients;
- Faciliter la mise en relation et l'échange d'information afin que les chercheurs puissent renforcer et soutenir leurs travaux mutuels;
- Soutenir l'inclusion des populations sous-représentées dans les études et analyser les différences propres aux populations, le cas échéant.
Partie 2 : Propositions pour examen
La modernisation du cadre des essais cliniques au Canada est une vaste entreprise à multiples facettes qui devrait entraîner des changements importants dans différents aspects des règlements et des politiques existants. Bien que les propositions présentées ci-dessous ont bon nombre des mêmes objectifs en commun, elles ont été présentées selon plusieurs thèmes clés afin de mieux structurer la conversation et de recueillir des commentaires.
Souplesse du cycle de vie
Le cycle de vie réglementaire d'un essai clinique comprend le processus de demande d'essai clinique (DEC) pour une autorisation d'essai clinique, y compris une réunion préalable à la DEC qui peut être organisée sur demande, la surveillance et l'établissement de rapports au cours d'un essai, et les exigences post-essai telles que la conservation des dossiers à plus long terme. La mise en place d'une approche plus souple tout au long du cycle de vie pour la réglementation des essais cliniques fournirait à Santé Canada les outils nécessaires pour superviser le déroulement sûr d'un essai dans son intégralité, tout en permettant aux promoteurs de mieux mener des types d'essais novateurs. Santé Canada aurait la possibilité d'autoriser un essai plutôt que de se contenter d'autoriser la vente ou de l'importation d'un produit faisant l'objet de la recherche, d'autoriser un essai portant sur plusieurs types de produits au moyen d'une seule autorisation, et de rationaliser les exigences relatives aux demandes pour toutes les gammes de produits afin de gagner en efficacité, tout en maintenant la protection de la santé et de la sécurité des participants.
Approches proposées
Autorisation d'essai clinique
Santé Canada propose des changements réglementaires qui feraient passer l'autorisation de la vente ou de l'importation du produit de santé utilisé dans l'essai clinique à une autorisation de l'essai clinique dans son ensemble, y compris la vente ou l'importation des produits.
Cela donnerait à Santé Canada l'autorité et la surveillance nécessaires pour prendre des mesures plus nuancées et plus ciblées lorsqu'un problème de sécurité se pose avec un aspect de l'essai clinique. Cela permettrait également à Santé Canada de mieux réglementer les nouveaux types de conceptions d'essais novateurs et leurs variations futures, par exemple :
- les essais étudiant plusieurs traitements au sein d'un seul essai clinique (c'est-à-dire les protocoles maîtres);
- les nouvelles conceptions d'essais adaptatifs qui permettent d'apporter des changements planifiés au protocole d'étude à des moments précisés au préalable pendant le cycle de vie d'un essai (c'est-à-dire les essais adaptatifs).
Les protocoles maîtres sont conçus avec de multiples sous-études et impliquent des efforts coordonnés pour évaluer un ou plusieurs produits expérimentaux dans une ou plusieurs indications au sein de la structure globale de l'essai. Voici quelques types de protocoles maîtres :
- les essais panier, qui étudient l'innocuité, l'efficacité et l'effet d'un produit expérimental dans diverses indications;
- les essais parapluies, qui étudient l'innocuité, l'efficacité et l'effet de plusieurs produits expérimentaux dans une seule indication;
- les essais plateforme, qui étudient plusieurs produits expérimentaux dans une ou plusieurs indications selon un schéma très dynamique;.
Des changements réglementaires supplémentaires qui influencent la façon dont Santé Canada autorise, modifie, surveille et suspend ou annule un essai contribueraient à soutenir cette approche souple tout au long du cycle de vie. Ces changements réglementaires permettraient à Santé Canada de superviser le déroulement d'essais novateurs tout au long du processus d'essai, dans le but de favoriser l'innovation tout en protégeant la sécurité des participants.
Autorisation unique d'un essai avec plusieurs types de produits
Dans le cadre de l'adoption d'un cadre réglementaire modernisé pour les essais cliniques, Santé Canada propose de permettre l'autorisation unique d'un essai clinique portant sur plusieurs produits de santé de différentes catégories, comme les médicaments, les PSN et les instruments médicaux. Cela permettrait d'accroître considérablement l'efficacité des processus de demande, de modification et d'autorisation des essais cliniques portant sur plusieurs produits de santé, ce qui simplifierait davantage les interactions de Santé Canada avec le promoteur tout au long de l'essai.
Les intervenants ont noté précédemment des difficultés liées à l'obligation de déposer des demandes d'essais cliniques (DEC) de médicaments ou de PSN et des demandes d'essai expérimental (DEE) d'instruments médicaux distinctes pour un seul essai. Avec des pouvoirs plus étendus pour réglementer à la fois les essais cliniques et les produits expérimentaux, le maintien du statu quo consistant à exiger des autorisations distinctes par produit dans des cadres réglementaires différents lorsque plus d'un produit est utilisé ou mis à l'essai dans un seul essai serait inefficace et laborieux. L'exigence d'autorisations multiples avec des délais d'examen différents peut entraîner des retards dans l'ouverture de l'essai, réduisant ainsi la rapidité de l'accès des Canadiens à des produits de santé potentiellement bénéfiques. Une voie d'autorisation unique permettrait de répondre à ces préoccupations et s'accompagnerait d'une harmonisation plus approfondie de certaines exigences réglementaires entre les gammes de produits, le cas échéant. Des détails précis sur les exigences d'une voie d'autorisation unique pour les essais impliquant plusieurs produits seraient également décrits dans les orientations.
Rationalisation des exigences entre les types de produits
Bien que des exigences propres à un produit continuent d'être nécessaires pour contribuer à l'innocuité du produit dans le cadre de l'essai, il y a des domaines où Santé Canada pourrait harmoniser les exigences réglementaires des essais cliniques entre les gammes de produits des médicaments, des PSN et des instruments médicaux. Des changements seront apportés pour combler les lacunes éventuelles et offrir aux promoteurs une expérience plus rationnelle et plus efficace lors de la soumission d'une demande ou pendant la réalisation de leur essai.
De tels changements permettraient également une plus grande souplesse dans la façon dont Santé Canada autorise et supervise la sécurité des essais plus novateurs au cours de leur cycle de vie. Outre la capacité d'autoriser l'essai et ses produits, Santé Canada élargirait sa capacité de surveiller la sécurité des essais en cours et, si cela est justifié, de suspendre ou d'annuler ces essais de façon complète ou partielle.
Supervision souple du cycle de vie d'un essai clinique

Figure 2 - Description textuelle
Le cycle de vie d'un essai clinique comprend les éléments suivants: demande, autorisation / modification, conduite de l'essai clinique, surveillance de la sécurité et fin de l'étude. Les propositions pour la surveillance agile du cycle de vie d'un essai clinique sont réparties à travers les éléments susmentionnés.
En particulier, Santé Canada propose les changements suivants :
Demande d'essai clinique
Les exigences relatives à la demande d'un essai clinique seraient modifiées pour plus de souplesse afin de mieux réglementer les types d'essais modernes et d'être davantage harmonisées par rapport aux gammes de produits de santé. Par exemple, certaines dispositions de l'arrêté d'urgence sur les essais liés à la COVID-19 (telles que la définition d'un comité d'éthique pour la recherche (CER) et le moment de la soumission pour l'approbation du CER) seraient examinées et pourraient être intégrées lors de la modernisation de la réglementation, le cas échéant.
Pour les instruments médicaux, Santé Canada propose d'élargir qui peut parrainer un essai clinique et déposer une DEC pour inclure les chercheurs indépendants (comme un chercheur, un clinicien ou un établissement de soins de santé) en plus des fabricants et des importateurs d'instruments médicaux. Cela permettrait de mieux adapter Santé Canada aux exigences réglementaires d'autres instances internationales.
Examen des délais pour les autorisations/modifications
Les modifications apportées au régime d'autorisation de l'essai et des produits, comme indiqué ci-dessus, peuvent influencer les délais d'examen. Les délais seraient définis par la politique et harmonisés par rapport aux gammes de produits afin d'offrir une plus grande cohérence et prévisibilité aux promoteurs.
Déroulement de l'essai
Les promoteurs seraient tenus d'adhérer aux normes internationalement reconnues en matière de bonnes pratiques cliniques (BPC) tout au long du déroulement d'un essai, quel que soit le type de produit, ce qui constituerait une nouvelle exigence réglementaire pour les essais d'instruments médicaux. Cette adhésion permettrait de mieux harmoniser les modèles de surveillance par rapport à l'ensemble des gammes de produits de santé, en structurant les exigences de chaque gamme de produits pour les aider à se conformer aux normes internationales liées aux BPC, et elle servirait de base à la modernisation future du programme de conformité et d'application de Santé Canada pour les essais cliniques des produits de santé.
Surveillance de l'innocuité
Les événements indésirables et les incidents liés aux instruments médicaux continueraient à être signalés par produit afin de garantir l'innocuité appropriée des produits mis à l'essai. Santé Canada propose d'éclaircir et d'élargir ses pouvoirs actuels en matière de demande d'information, y compris la possibilité de demander une analyse des données d'innocuité pendant que l'essai est en cours. Il peut s'agir d'une demande d'évaluation des signaux d'innocuité afin de déterminer si le rapport avantages-risques a changé au cours de l'essai clinique.
Suspension et annulation de l'essai
Les pouvoirs existants liés à la capacité de Santé Canada d'annuler ou de suspendre l'autorisation d'un essai clinique seraient modifiés pour permettre la suspension ou l'annulation d'un essai en tout ou en partie (p. ex., la suspension d'un seul volet d'un essai, d'un site, du recrutement ou de l'utilisation d'un produit particulier). Cela permettrait à Santé Canada de réagir plus précisément si un aspect de l'essai a démontré un manque d'efficacité ou pose un problème d'innocuité sans compromettre l'essai dans son ensemble, permettant ainsi au reste de l'essai de rester ouvert et de continuer à recruter de nouveaux participants. De plus, Santé Canada est en train de revoir les exigences existantes dans les différents règlements sur les produits de santé en ce qui concerne les notifications sur le commencement et l'arrêt d'une étude et les conditions requises pour reprendre une étude.
Approche basée sur le risque
Santé Canada propose de moderniser la réglementation des essais cliniques par l'établissement d'une approche commune basée sur le risque pour les essais portant sur tout type de produit de santé, afin d'offrir plus de souplesse et de cohérence dans la réglementation des essais cliniques. L'introduction d'une surveillance proportionnelle basée sur le niveau de risque pour les participants à l'étude contribuerait à réduire le fardeau tout en permettant à Santé Canada de transférer des ressources vers les domaines nécessitant une plus grande surveillance.
Approches proposées
L'harmonisation des exigences réglementaires pour les essais cliniques, quel que soit le type de produit, introduirait ce qui suit :
- Des catégories de surveillance proportionnelle avec des exigences réglementaires stratifiées et adaptées pour les médicaments et les PSN;
- De nouveaux outils réglementaires souples pour gérer les risques tout au long du cycle de vie d'un essai.
Santé Canada permettrait d'adopter une approche globale basée sur le risque pour la réglementation des essais cliniques en abandonnant un modèle de surveillance unique pour les médicaments et les PSN, tout en introduisant de nouveaux facteurs de souplesse et en maintenant le système préexistant de classification des instruments médicaux en fonction du risque. Le modèle proposé continuerait à soutenir la sécurité des participants, tout en introduisant des gains d'efficacité là où c'est approprié. Ce modèle refléterait également les pratiques exemplaires à l'échelle internationale et offrirait une plus grande prévisibilité aux intervenants. Toutes les nouvelles exigences et obligations en matière de surveillance proportionnelle seraient basées sur le risque, l'incertitude et les renseignements connus sur le produit et son utilisation prévue dans le cadre de l'essai clinique.
Pour les médicaments et les PSN, cette approche s'adapterait aux recommandations de l'Organisation de coopération et de développement économiques (OCDE), le cas échéant.Note de bas de page 1 Pour les instruments médicaux, le système actuel de classification basé sur le risque serait maintenu, étant donné que le système de classification est déjà conforme à un système internationalement reconnu qui a bien fonctionné pour les intervenants.
L'harmonisation entre les médicaments, les PSN et les instruments médicaux dans le nouveau cadre contribuerait à favoriser le passage à un régime d'autorisation unique pour la réalisation de l'essai lorsque plusieurs produits de types différents sont utilisés dans un seul essai.
Catégories de risques et exigences
Santé Canada propose que les essais cliniques qui relèvent du champ d'application de la Loi sur les aliments et drogues et de ses règlements soient placés dans l'une des trois catégories suivantes :
- Catégorie A : Exemption d'autorisation d'essai clinique;
- Catégorie B : Autorisation d'essai clinique avec des exigences adaptées;
- Catégorie C : Autorisation d'essai clinique avec toutes les exigences.
Ces catégories représentent le niveau de risque, d'incertitude et d'information de sécurité disponibles pour chaque produit utilisé dans l'essai. Un essai unique impliquant plusieurs produits serait classé en fonction du produit présentant le niveau de risque le plus élevé. Dans le cadre de son examen de la demande d'essai clinique, Santé Canada continuera d'évaluer tous les autres facteurs de risque pertinents, comme l'utilisation d'une conception d'essai non traditionnelle, de populations d'étude très vulnérables ou de procédures invasives.
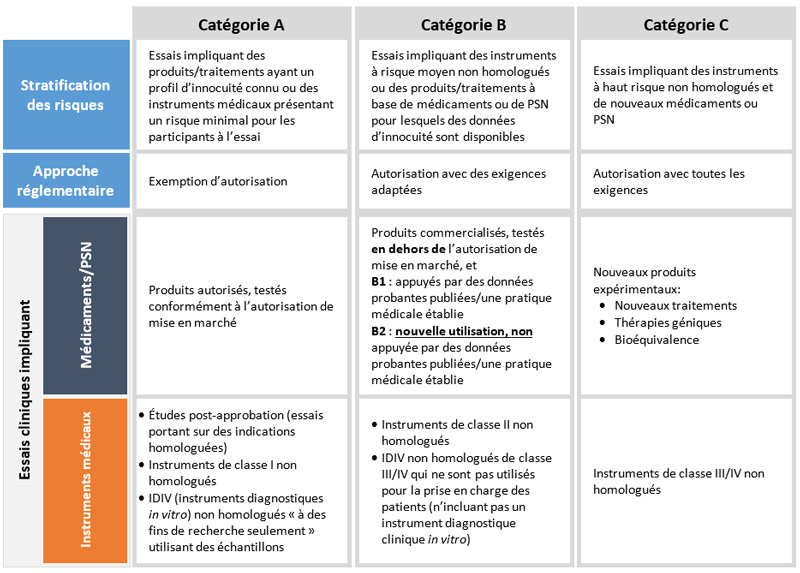
Figure 3 - Description textuelle
Santé Canada propose une approche fondée sur les risques pour les médicaments, les instruments médicaux et les produits de santé naturels (PSN) qui vise à fournir une surveillance proportionnelle en plaçant les essais dans l'une des trois catégories (A, B ou C) et deux sous-catégories pour la catégorie B (B1 et B2).
La catégorie A comprendra les essais impliquant des produits/thérapies ayant un profil d'innocuité connu ou des instruments médicaux présentant un risque minimal pour les participants aux essais. Ces essais seront dispensés d'autorisation. Pour les médicaments et les PSN, cette catégorie comprendra les essais impliquant des produits autorisés, testés conformément à l'autorisation de mise sur le marché. Pour les instruments médicaux, cette catégorie comprendra des essais impliquant: des études post-approbation (essais étudiant des indications homologuées), des instruments de classe I non homologués, des dispositifs de diagnostic in vitro utilisant des échantillons résiduels (IDIV) non homologués « à usage de recherche uniquement ».
La catégorie B comprendra des instruments à risque moyen non homologués ou des médicaments ou des PSN produits/thérapies pour lesquels des renseignements sur l'innocuité sont disponibles. Ces essais nécessiteront une autorisation, mais avec des exigences adaptées. Pour les médicaments et les PSN, cette catégorie comprendra les essais impliquant des produits commercialisés, testés en dehors d'une autorisation de mise sur le marché. La sous-catégorie B1 s'applique aux essais portant sur l'utilisation de médicaments commercialisés étayés par des preuves publiées/une pratique médicale établie. Les essais portant sur une nouvelle utilisation non étayée par des preuves publiées/une pratique médicale établie relèvent de la sous-catégorie B2. Pour les instruments médicaux, cette catégorie comprendra des essais portant sur: des dispositifs de classe II non homologués, des IDIV de classe III / IV non homologués et non utilisés pour la prise en charge des patients (sans inclure un IDIV proche du patient).
La catégorie C comprendra des essais portant sur des dispositifs à haut risque non homologués et de nouveaux médicaments ou PSN produits/thérapies. Ces essais nécessiteront une autorisation avec toutes les exigences. Pour les médicaments et les PSN, cette catégorie comprendra des essais portant sur de nouveaux produits de recherche (tels que des nouveaux traitements, des thérapies géniques, la bioéquivalence). Pour les instruments médicaux, cette catégorie comprendra les essais portant sur les classes III-IV sans licence.
Exigences stratifiées relatives aux essais cliniques :
Catégorie A :
Cette catégorie comprendrait les essais cliniques portant sur les produits ou traitements dont le profil d'innocuité est connu et qui présentent un risque supplémentaire minime pour les participants à l'essai par rapport à la pratique médicale habituelle. Il s'agit des produits de santé commercialisés utilisés conformément à leur autorisation de mise en marché (toutes les gammes de produits de santé), des essais cliniques impliquant des instruments médicaux de classe I non homologués et des IDIV (instruments diagnostiques in vitro) « à des fins de recherche seulement » utilisant des échantillons résiduels qui ne sont pas utilisés pour le diagnostic ou le traitement d'un patient.
Demande et autorisation
Pour ces essais, Santé Canada n'exigerait pas la soumission d'une DEC, ce qui est conforme à ce que le Ministère a mis en place en vertu des règlements existants.
Autres exigences
Pour toutes les gammes de produits, de nombreuses exigences existantes continueraient de s'appliquer. Pour les essais impliquant des médicaments ou des PSN, il s'agit notamment du respect des BPC, de l'obtention de l'approbation du CER et de la déclaration des effets indésirables (EI) par les titulaires d'une autorisation de mise en marché. Pour les instruments de classe I non homologués et les IDIV « à des fins de recherche seulement » utilisant des échantillons résiduels qui ne sont pas utilisés pour le diagnostic ou le traitement d'un patient, cela comprendrait l'étiquetage, la publicité, les registres de distribution, le traitement des plaintes et les rappels, ainsi que la notification obligatoire des problèmes. En outre, Santé Canada propose deux nouvelles exigences pour ces instruments médicaux : (1) que les promoteurs adhèrent aux principes des BPC, conformément aux BPC ISO 14155, ce qui est actuellement recommandé dans les directives de Santé Canada à l'industrie, et (2) qu'ils exigent explicitement l'approbation du CER avant le début de l'essai. Pour les essais cliniques portant sur des instruments commercialisés selon les indications approuvées, les exigences actuelles en matière d'homologation continueraient de s'appliquer en plus de l'approbation des BPC et du CER. Pour tous les types de produits, les promoteurs seraient toujours tenus de conserver certains renseignements dans le cadre des exigences de tenue de registres prévues par la réglementation.
Catégorie B :
Cette catégorie comprendrait les essais cliniques portant sur des instruments médicaux non homologués à risque moyen ou des produits de santé pour lesquels on dispose de données d'innocuité bien établies, tels que les produits pharmaceutiques commercialisés au Canada qui sont mis à l'essai pour des utilisations en dehors de l'indication autorisée.
Pour les médicaments et les PSN, Santé Canada propose que cette catégorie soit encore stratifiée en sous-catégories B1 et B2 selon le niveau de preuve disponible pour soutenir les essais en dehors de l'indication autorisée. La catégorie B1 comprendrait les essais étudiant des indications, des populations ou des posologies non approuvées qui sont étayées par des données probantes solides publiées ou la pratique médicale établie, tandis que la catégorie B2 comprendrait les essais étudiant une nouvelle utilisation qui n'est pas appuyée par des données probantes publiées ou la pratique médicale établie.
Pour les instruments médicaux, les produits suivants seraient incorporés dans cette catégorie :
- les instruments de classe II non homologués;
- les IDIV de classe III ou IV non homologués qui ne sont pas utilisés pour la prise en charge des patients, à l'exclusion des instruments diagnostiques cliniques in vitro.
Demande et autorisation
Pour les essais de catégorie B, la soumission d'une DEC serait requise, mais avec des exigences adaptées en fonction du niveau de risque du produit ou du type d'essai.
Pour les médicaments et les PSN, un exemple d'exigence adaptée dans cette catégorie consisterait à permettre aux promoteurs de se référer à une monographie de produit approuvée ou de fournir des renseignements sur la conformité de l'utilisation proposée du produit par rapport à la pratique médicale établie. On utiliserait ces renseignements pour démontrer des aspects précis de ce qui serait normalement requis dans le cadre d'une demande complète, comme les renseignements qui feraient normalement partie de la brochure du chercheur.
Pour les instruments médicaux, comme pour les exigences relatives aux instruments de classe I, le demandeur serait tenu de posséder des dossiers contenant tous les renseignements et documents répondant aux exigences réglementaires, conformément aux exigences actuelles de Santé Canada. Santé Canada étudie également la possibilité de réduire les exigences en matière de demande pour les instruments médicaux commercialisés au Canada et dont l'utilisation est étudiée en dehors des indications approuvées. Par exemple, Santé Canada peut ne pas exiger la présentation de l'historique de commercialisation d'un produit ou des résultats de recherches antérieures menées avec un instrument.
Après l'autorisation
En ce qui a trait aux médicaments et aux PSN, les essais portant sur des indications qui sont étayées par des données probantes (catégorie B1) suivraient les exigences des BPC, tandis que d'autres exigences seraient réduites, telles que :
- les exigences en matière d'étiquetage;
- les exigences de déclaration des effets indésirables. Par exemple, lorsque le profil d'innocuité d'un médicament est bien caractérisé, les exigences peuvent être réduites en ce qui concerne la collecte d'événements indésirables non graves ou d'autres données d'innocuité, si elles sont décrites dans le protocole. Cela serait conforme à l'ébauche de la directive de la Conférence internationale d'harmonisation E19;
- les exigences en matière de tenue de registres, comme les registres de contrôle de la température de certains produits de santé.
Les essais portant sur une nouvelle utilisation qui n'est pas soutenue par des données déjà publiées ou une pratique médicale établie (catégorie B2) seraient tenus de suivre les BPC, auraient des exigences réduites en matière d'étiquetage et pourraient être assujettis à des exigences réduites en matière de déclaration des effets indésirables et de tenue des dossiers, tel que déterminé par Santé Canada après examen. Ces approches sont importantes, car elles réduisent le fardeau du promoteur sans accroître le risque pour les participants aux essais cliniques.
En ce qui a trait aux instruments médicaux, les exigences actuelles continueraient de s'appliquer aux instruments de classe II non homologués ou aux IDIV de classe III ou IV qui ne sont pas utilisés pour la prise en charge des patients, à l'exclusion des IDIV in vitro, avec l'ajout de l'adhésion aux principes des BPC et la nécessité explicite d'obtenir l'approbation du CER avant de commencer un essai. Santé Canada étudie la possibilité de réduire les exigences post-autorisation pour les instruments médicaux commercialisés au Canada, mais dont l'usage envisagé dans l'essai n'est pas une indication approuvée. Cela peut inclure une flexibilité potentielle en matière d'étiquetage lorsque des renseignements particuliers sont fournis à Santé Canada aux fins d'évaluation (p. ex., l'utilisation proposée de l'instrument, la justification, ainsi que les risques, l'efficacité ou l'incertitude de l'instrument et de son utilisation non indiquée sur l'étiquette).
Catégorie C :
Cette catégorie comprendrait les essais impliquant des instruments médicaux à haut risque non homologués et/ou de nouveaux médicaments, PSN ou thérapies, ainsi que les études de bioéquivalence qui impliquent généralement des essais sur des participants en santé.
Demande et autorisation
Les essais cliniques de la catégorie C exigeraient la soumission d'une DEC complète, ce qui resterait conforme à ce que Santé Canada exige actuellement pour les essais impliquant des médicaments et des produits de santé naturels et des instruments non homologués de classe III et IV (à l'exception de ceux mentionnés dans la catégorie B).
Après l'autorisation
Toutes les exigences post-autorisation conformes à la réglementation actuelle s'appliqueraient aux essais cliniques de catégorie C impliquant des médicaments, des PSN et/ou des instruments non autorisés de classe III et IV.
Application des nouveaux pouvoirs – Conditions
Pour mieux gérer les risques tout au long du cycle de vie d'un essai clinique, Santé Canada aurait le pouvoir d'imposer des conditions à une autorisation d'essai clinique avant ou pendant l'essai clinique d'une manière prévisible et agile qui est cohérente entre les gammes de produits. Les conditions seraient imposées au cas par cas pour répondre à des incertitudes importantes ou atténuer des risques importants liés aux produits testés ou à la conduite de l'essai.
Ce pouvoir permettrait d'autoriser un plus large éventail d'essais lorsque des mesures adaptées sont nécessaires pour lancer ou poursuivre un essai.
Voici quelques exemples de situations dans lesquelles des conditions seraient appropriées :
- des rapports plus fréquents sur l'innocuité (par exemple, des copies du Rapport de mises à jour périodiques sur la pharmacovigilance relatif aux produits en développement [RPP-D], des rapports du comité de surveillance des données et de la sécurité [CSDS]);
- la surveillance de populations particulières en raison d'un risque potentiel accru (p. ex., enfants, grossesse); et
- des renseignements supplémentaires pour caractériser et atténuer les risques nouvellement signalés.
La société Z soumet une demande d'essai pour tester une nouvelle thérapie génique pour la dystrophie musculaire dans des populations pédiatriques. La thérapie génique est destinée à traiter une maladie rare dans une population vulnérable. Il y a des conséquences à long terme dans la population cible et le nouveau mécanisme d'action du produit introduit de nombreuses inconnues. Pour permettre à l'essai d'aller de l'avant, Santé Canada pourrait exiger la surveillance et la déclaration des effets à long terme au-delà de la période de traitement de l'étude par l'application d'une condition liée à l'autorisation.
D'autres scénarios d'essais cliniques nécessitant des conditions peuvent inclure : des conceptions d'essais cliniques nouvelles, premier produit dans une nouvelle classe thérapeutique, ou des populations d'étude à haut risque.
Essais cliniques décentralisés
Puisque plus de 30 % de la population canadienne réside à l'extérieur des zones urbaines de grande ou moyenne taille, le recrutement de participants de ces populations plus éloignées pour des essais cliniques représente un défi logistique et financier. Alors que certains types de thérapies et d'interventions médicales nécessitent des visites dans des hôpitaux ou des centres dotés d'équipement de haute technologie (tels que des appareils d'IRM ou des accélérateurs linéaires) et de médecins spécialisés, les technologies émergentes commencent à permettre aux patients résidant dans une zone rurale éloignée de participer à un essai clinique supervisé depuis un grand centre urbain, ce qui permet une approche des essais plus centrée sur le patient.
Les essais cliniques traditionnels sont menés à des sites d'essai désignés où un chercheur qualifié et le personnel de l'étude effectuent les procédures liées à l'étude. Les essais cliniques décentralisés (ECD), parfois appelés essais cliniques à distance ou virtuels, sont des essais cliniques menés avec des participants à l'étude situés en dehors des centres de recherche clinique pour toutes les procédures d'essai requises. Dans un ECD, l'étude se déroule à distance, sans visite physique à un site d'essai, après que la technologie nécessaire eut été installée et expliquée au patient. Les participants à l'étude peuvent communiquer par vidéoconférence avec les chercheurs, recevoir des visites à domicile du personnel de l'étude, disposer d'outils accessibles sur Internet pour la collecte et la transmission des données et utiliser des technologies mobiles telles que des biocapteurs.
Bien que la réalisation d'ECD ne soit pas interdite par le règlement actuel, Santé Canada propose de faciliter et d'encourager leur utilisation en apportant plusieurs modifications au règlement qui pourraient les rendre plus pratiques pour les promoteurs. Ces propositions réglementaires comprendraient :
- un changement de la formulation des exigences dans le règlement, de « consentement éclairé écrit » à « consentement éclairé documenté ». Une interprétation plus poussée serait décrite dans un document d'orientation, dans lequel « documenté » serait interprété comme signifiant une signature électronique, un enregistrement vidéo ou audio du processus de consentement lorsqu'une confirmation verbale est fournie par le participant;
- le fait de permettre à un témoin d'attester que le consentement éclairé a été donné si cela s'avère nécessaire dans des circonstances exceptionnelles, à l'instar de ce qui se fait actuellement dans le cadre des essais cliniques menés sous l'arrêté d'urgence;
- la définition du terme « site d'essai » dans la réglementation comme « les lieux où les activités liées à l'essai sont effectivement menées », où les « activités liées à l'essai » pourraient englober le recrutement, le consentement éclairé, le suivi et les visites qui sont effectuées de manière virtuelle;
- une plus grande flexibilité pour les types de professionnels de la santé pouvant agir à titre de chercheurs qualifiés. Le type de qualifications requises pour le chercheur qualifié serait déterminé au cas par cas lors de l'examen du protocole d'essai à l'étape de l'approbation et/ou par le CER avant le début de l'essai, ou encore défini dans la réglementation.
Transparence
Actuellement, grâce à sa base de données sur les essais cliniques, Santé Canada fournit au public une liste de renseignements déterminants relatifs aux essais cliniques de phase I, II et III menés sur des patients et portant sur des médicaments pharmaceutiques et biologiques à usage humain. Santé Canada encourage également les promoteurs à enregistrer leurs essais dans des registres accessibles au public, tels que Clinical Trials.gov et ISRCTN. Toutefois, il demeure nécessaire d'accroître la transparence des essais portant sur d'autres interventions, telles que les instruments médicaux et les PSN, et d'offrir aux Canadiens un meilleur accès à toute l'information relative aux essais cliniques.
La loi d'exécution du budget de 2019 a conféré des pouvoirs liés à une transparence accrue en matière d'information sur les essais cliniques. À l'appui de ces pouvoirs, ainsi que de l'engagement du gouvernement du Canada à l'égard de l'ouverture, Santé Canada examine les options quant à la meilleure façon de concevoir et de mettre en œuvre des mesures visant à promouvoir l'enregistrement des essais cliniques canadiens dans des registres publics ainsi que la divulgation publique des résultats (communication des résultats). Ces mesures peuvent englober l'élaboration de nouvelles politiques et règlements, tout en exploitant les technologies de l'information telles que l'intelligence artificielle (IA) pour rendre l'information sur les essais cliniques plus accessibles au public.
Ces mesures de transparence aideraient le public (par exemple, les patients et les soignants, les chercheurs, les décideurs, les professionnels de la santé, etc.) à trouver l'information sur les essais cliniques qui les concerne et apporteraient d'autres avantages de grande portée, par exemple :
- faciliter l'équité dans l'accès des patients aux essais cliniques;
- améliorer la confiance du public dans l'innocuité des produits de santé disponibles au Canada;
- éviter la duplication inutile des recherches, et ainsi réduire les risques pour les participants;
- faciliter le transfert rapide des connaissances issues des essais cliniques vers des guides de pratique clinique, et ainsi améliorer les soins de santé offerts aux Canadiens;
- améliorer la qualité de la base de connaissances disponible pour la prise de décision en réduisant les biais de publication.
Au fil des ans, les efforts internationaux visant à améliorer la transparence des essais cliniques ont contribué à relever le niveau de validité des données scientifiques mises à la disposition du public. Santé Canada héberge une base de données sur les essais cliniques pour les médicaments depuis 2013 et encourage depuis plusieurs années l'enregistrement des essais cliniques des médicaments et des instruments dans les registres internationaux. Bien que la conformité à cet enregistrement volontaire soit assez élevée, elle ne fournit pas nécessairement un portrait complet de tous les essais en cours, en cours de recrutement, terminés ou interrompus.
L'une des approches envisagées consisterait à élaborer une nouvelle politique qui guiderait les promoteurs dans les étapes et processus recommandés en matière d'enregistrement et de communication des résultats. Cela conduirait à de futurs règlements pour les mêmes processus dans une phase ultérieure du projet de modernisation des essais cliniques. Par ailleurs, le ministère pourrait commencer par élaborer de nouvelles exigences réglementaires pour l'enregistrement dans la phase initiale, et des mesures stratégiques pour aborder la communication des résultats. Certains des éléments de l'enregistrement et de la communication des résultats envisagés sont décrits ci-dessous.
Enregistrement
Santé Canada explore des options stratégiques et réglementaires pour l'enregistrement des essais cliniques canadiens autorisés portant sur des médicaments, des instruments médicaux et des PSNNote de bas de page 2. Tous les essais menés au Canada et autorisés par Santé Canada (ou avec au moins un site canadien) peuvent être inclus dans le champ d'application, à l'exception des essais de médicaments de phase IV (à moins que le ministère ne l'exige en posant une condition sur l'autorisation de mise en marché), des essais de bioéquivalence, des essais auprès de patients individuels ainsi que des instruments de classe I, des IDIV utilisés à des fins de recherche avec des échantillons résiduels et des études sur les instruments médicaux après leur mise en marché. Les promoteurs d'essais canadiens peuvent être invités à enregistrer leurs essais dans un registre international existant qui a été jugé admissible par le ministère dans un délai prescrit. On peut également demander au promoteur de l'essai de communiquer à Santé Canada la preuve de son enregistrement (par exemple, le lien vers le registre et le numéro d'identification de l'essai), dans un certain délai après avoir terminé l'enregistrement.
L'admissibilité du registre serait déterminée en utilisant des critères conformes aux exigences de l'International Committee of Medical Journal Editors (ICMJE) et aux critères de l'Organisation mondiale de la Santé (OMS) pour un registre primaireNote de bas de page 3. Plus précisément, un registre admissible :
- serait accessible au public sans frais;
- serait ouvert à tous les promoteurs désirant l'utiliser;
- serait géré par une organisation à but non lucratif;
- disposerait d'un mécanisme permettant de garantir la validité des données d'enregistrement;
- pourrait être consulté électroniquement;
- comprendrait l'ensemble minimal de données d'enregistrement des essais de l'OMS en 24 points.
Dr Jones cherche un essai clinique pour son patient. Actuellement, elle s'appuie sur une variété d'outils et a plus de facilité à trouver des essais étrangers que des essais canadiens. Grâce à la modernisation de la réglementation, Dr Jones et ses patients seraient en mesure d'accéder à l'information la plus récente sur les essais canadiens dans la langue officielle de leur choix.
Les registres admissibles seraient également tenus d'accepter les études d'intervention pour les produits pharmaceutiques, les produits biologiques, les instruments médicaux et les produits de santé naturels (y compris les compléments alimentaires), et d'accepter les essais cliniques réalisés au Canada.
Divulgation publique des résultats
Santé Canada étudie également les options stratégiques et réglementaires concernant l'obligation de divulguer ou de communiquer publiquement les résultats des essais. Cela signifierait que les promoteurs d'essais canadiens seraient invités à communiquer publiquement les résultats des essais, et qu'ils pourraient être invités à informer Santé Canada dans les délais prescrits une fois que les résultats ont été rendus publics.
Information sur le site Web de Santé Canada
Grâce à l'amélioration des mesures de transparence, les registres internationaux fourniraient davantage d'information sur les essais cliniques canadiens. Cependant, Santé Canada se propose de jouer un rôle pour rendre cette information plus accessible. Améliorer l'accès signifie rendre l'information facile à trouver, disponible dans les deux langues officielles et utiliser une technologie qui permettrait un accès sur diverses plateformes et appareils mobiles. Le public doit pouvoir trouver l'information et effectuer des recherches (en utilisant des filtres pour divers critères) afin de trouver l'information qui lui est utile dans la langue officielle de son choix.
Modernisation de la surveillance de la conformité et de l'application de la loi
Étant donné que la nature des essais cliniques continue d'évoluer, une approche modernisée de la surveillance de la conformité et de l'application de la loi est nécessaire pour que Santé Canada maintienne la surveillance de la conduite des essais cliniques afin de protéger les droits, la sécurité et le bien-être des participants, et afin de vérifier et de valider l'intégrité des données et de renforcer la confiance du public dans les résultats des essais. Des facteurs tels que la mondialisation accrue de l'industrie des soins de santé, la complexité croissante de la conception des essais et l'expansion des rôles des chercheurs qualifiés, des organismes de recherche sous contrat et des organismes de gestion des sites dans la gestion des essais cliniques ont mis en évidence la nécessité de moderniser l'approche de surveillance de la conformité et de l'application de la loi de Santé Canada.
L'approche actuelle de Santé Canada en matière de surveillance de la conformité des essais cliniques et d'application de la loi est axée sur l'inspection des sites d'essais cliniques. À l'avenir, Santé Canada se concentrera davantage sur les promoteurs d'essais cliniques en mettant en œuvre une approche cyclique d'inspection basée sur les risques de tous les promoteurs d'essais cliniques, des organismes de recherche sous contrat et des organismes de gestion des sites. Santé Canada solliciterait des pouvoirs supplémentaires pour obliger les tiers participant à la réalisation d'un essai clinique au nom d'un promoteur à prendre des mesures lorsqu'un problème de non-conformité se pose. Santé Canada élargirait également son approche d'examen et d'évaluation pour englober les essais cliniques utilisant des instruments médicaux ou des PSN, et a déjà lancé des inspections pilotes des essais impliquant des instruments médicaux comme première étape de la mise en œuvre d'une approche complète de la surveillance de la conformité et de l'application de la loi pour les essais impliquant des instruments médicaux. Actuellement, il n'existe aucune inspection des essais impliquant des PSN.
La modernisation de l'approche de Santé Canada en matière de surveillance de la conformité et d'application de la loi devrait avoir plusieurs effets bénéfiques, notamment en permettant aux parties prenantes de rester à jour relativement aux meilleures pratiques, en maintenant la protection des participants, en améliorant la conformité globale aux exigences réglementaires et aux BPC, et en permettant au Canada de rester concurrentiel sur le marché mondial des essais cliniques.
Approches proposées
Santé Canada explore plusieurs nouvelles approches pour la conduite de ses activités de surveillance de la conformité et d'application de la loi qui sont décrites plus loin. Cependant, une proposition définitive sera avancée une fois que le nouveau cadre pour les essais cliniques aura été déterminé.
Une surveillance élargie à toutes les gammes de produits
Bien que Santé Canada ait actuellement le pouvoir de mener des inspections des activités d'essais cliniques en vertu de l'article 23 de la Loi sur les aliments et drogues, ses activités ont été limitées aux médicaments.
Santé Canada propose de soutenir la modernisation des essais cliniques pour les instruments médicaux et les PSN en étendant la surveillance de la conformité et l'application de la loi à ces gammes de produits. Santé Canada procéderait à une inspection en fonction de la Loi et du règlement connexe, y compris les normes internationales de BPC, comme la norme ICH (Conférence internationale sur l'harmonisation des exigences techniques relatives à l'homologation des produits pharmaceutiques à usage humain) ICH-E6(R2) et/ou la norme ISO (Organisation internationale de normalisation) ISO 14155. Au moment de déterminer les essais à inspecter, Santé Canada appliquerait une approche proactive basée sur les risques qui tiendrait compte de la conception de l'essai et d'autres facteurs, comme la condition médicale à l'étude, la population cible ou la classification des risques du produit de recherche (médicament, PSN ou instrument médical). De même, lorsqu'il s'agira de déterminer quels promoteurs, organismes de recherche sous contrat ou organismes de gestion des sites à inspecter, Santé Canada mettra en œuvre une approche cyclique d'inspection basée sur le risque qui tiendra compte de l'historique de conformité et du volume d'activités. Les entités très conformes seraient inspectées moins fréquemment, tandis que les entités moins conformes seraient inspectées plus fréquemment.
Ces nouvelles activités de surveillance de la conformité et d'application de la loi proposées seraient cohérentes avec celles d'organismes d'autres juridictions telles que la Food and Drug Administration des États-Unis (FDA), la Medicines and Healthcare Products Regulatory Agency (MHRA) du Royaume-Uni et l'Agence européenne des médicaments (EMA), qui ont des programmes et des normes d'inspection basés sur les risques intégrés à leur cadre réglementaire. Cela contribuerait également à promouvoir la cohérence réglementaire entre les gammes de produits et au sein des essais multiproduits, tout en renforçant la sécurité des participants, la confiance du public dans les résultats et l'intégrité des données et des résultats des essais cliniques.
Conformité et application de la loi chez des tiers
Dans le cadre du régime actuel de réglementation des essais cliniques, Santé Canada a une autorité de surveillance directe sur les promoteurs d'essais cliniques, mais pas sur les tiers qui peuvent être impliqués dans la conduite des essais cliniques, comme les chercheurs qualifiés, les organismes de recherche sous contrat et les organismes de gestion des sites. Les promoteurs font souvent appel à ces tiers pour la réalisation de parties importantes d'un essai, et ils représentent une part importante et croissante du secteur des essais cliniques.Note de bas de page 4
Actuellement, Santé Canada n'est pas toujours au courant que des tiers sont impliqués dans un essai, étant donné que les promoteurs ne sont pas tenus de fournir à Santé Canada de l'information sur la participation des organismes de recherche sous contrat et des organismes de gestion des sites, bien que l'information sur les chercheurs qualifiés soit fournie dans le formulaire d'information sur le lieu de l'essai clinique (ILEC) ou dans le cadre de la demande d'essai expérimental pour les instruments médicaux. C'est problématique, car ces tiers peuvent être responsables de problèmes de non-conformité, ce qui peut mettre en péril la sécurité des participants pendant un essai clinique. Bien que Santé Canada puisse obliger un promoteur d'essai à prendre des mesures correctives et le tenir responsable des activités de ces tiers, le ministère n'a aucune autorité directe sur ces derniers.
Santé Canada propose de modifier le règlement de façon à ce que toute tierce partie qui mène l'ensemble ou une partie d'un essai clinique pour un promoteur soit légalement responsable des activités qu'elle mène au nom du promoteur, et soit donc soumise à des mesures réglementaires en cas de non-conformité. Cette modification proposée entraînerait un fardeau réglementaire supplémentaire pour les tiers participant aux essais cliniques, mais contribuerait à accroître la sécurité des participants, car Santé Canada serait en mesure de demander aux parties concernées de prendre des mesures pour corriger la non-conformité en temps opportun. En outre, elle s'alignerait mieux sur l'approche de surveillance de la conformité et d'application de la loi des États-Unis, qui permet aux promoteurs de transférer une partie de leur responsabilité juridique aux organismes de recherche sous contrat.
Partie 3 : Résultats attendus pour les parties prenantes
La vision de Santé Canada de la modernisation du cadre réglementaire des essais cliniques du Canada présentée ci-dessus contribuerait à encourager les essais cliniques au Canada en créant un environnement qui favorise l'innovation sécuritaire des produits de santé. Santé Canada n'est qu'un des nombreux intervenants dans l'écosystème des essais cliniques et de la santé au Canada, et de nombreux facteurs influencent et façonnent l'avenir des essais cliniques au Canada. Un cadre réglementaire bien conçu constituerait une base solide pour un changement positif. Éclairé des commentaires des parties prenantes, il est prévu qu'un cadre modernisé et correctement conçu permette :
- d'assurer une plus grande souplesse dans la surveillance du développement sécuritaire de thérapies innovantes;
- de contribuer à faire en sorte que les exigences administratives et réglementaires imposées aux promoteurs soient plus proportionnelles aux risques;
- d'offrir aux Canadiens un accès à des essais cliniques sécuritaires et à de l'information sur ces essais;
- de créer une approche plus rationnelle et mieux harmonisée au niveau international;
- de tirer parti des partenariats et d'assurer un leadership dans l'écosystème de la santé au Canada pour faciliter davantage les essais cliniques.
Tous les intervenants sont encouragés à contribuer à façonner l'avenir de la réglementation des essais cliniques au Canada en envoyant leurs réponses au questionnaire en ligne ou alternativement en envoyant leurs commentaires écrits par courriel à hc.policy.bureau.enquiries.sc@canada.ca en utilisant les questions fournies à l'Annexe 3 ci-dessous comme guide. Les réponses reçues seront prises en considération alors que Santé Canada s'efforce de mettre en place un régime de réglementation plus souple, basé sur le risque et transparent, pour en fin de compte améliorer la vie des Canadiens.
Annexe 1 : Vue d'ensemble du cadre réglementaire canadien actuel sur les essais cliniques
La réglementation des essais cliniques au Canada régissant les différentes gammes de produits a été élaborée à des moments différents, ce qui a entraîné des différences importantes dans les cadres respectifs. Un aperçu et une description de l'état actuel de chaque cadre sont présentés ci-dessous.
Médicaments
En vertu du cadre réglementaire actuel des essais cliniques impliquant l'utilisation de médicaments pharmaceutiques et biologiques pour usage chez l'humain, Santé Canada autorise la vente et l'importation au Canada des médicaments que l'on se propose d'utiliser dans un essai. À l'exception des études de phase IV, les promoteurs d'essais de médicaments doivent soumettre une demande d'essai clinique (DEC) à Santé Canada avant le début de l'essai et obtenir l'approbation d'un comité d'éthique de la recherche (CER). Comme le prévoit la réglementation, il existe une période d'examen par défaut de 30 jours des demandes d'essais cliniques. Santé Canada délivrera une lettre de non-objection (LNO) si l'essai est autorisé ou un avis de non-satisfaction (ANS) si la DEC est rejetée dans les 30 jours suivant la réception d'une demande complète. Si aucune réponse n'est envoyée dans les 30 jours, l'essai est autorisé par défaut.
Cette « autorisation par défaut » est nécessaire pour les médicaments qui ne sont pas actuellement autorisés au Canada, ainsi que pour les médicaments autorisés sur le marché qui font l'objet d'une étude pour des utilisations potentielles débordant les paramètres de leur autorisation de mise en marché.
La conduite d'essais cliniques impliquant l'administration de médicaments est régie par la Loi sur les aliments et drogues et le Règlement sur les aliments et drogues (RAD), et les promoteurs doivent suivre les bonnes pratiques cliniques (BPC) telles que définies à l'article C.05.010 du RAD.
Instruments médicaux
Le Règlement sur les instruments médicaux au Canada classe les instruments médicaux en quatre catégories (I – IV), du risque le plus faible au plus élevé. Santé Canada autorise la vente et l'importation des instruments médicaux des classes II à IV qui sont destinés à des essais expérimentaux sur des participants humains. La conduite de ces essais cliniques est régie par la Loi sur les aliments et drogues et le Règlement sur les instruments médicaux, et les fabricants et importateurs sont encouragés à suivre les BPC telles que décrites par la norme ISO 14155 – Investigation clinique des dispositifs médicaux pour sujets humains.
Le fabricant ou l'importateur de l'instrument médical à utiliser dans un essai doit présenter une demande d'autorisation d'essai expérimental (AEE) à Santé Canada afin d'obtenir une autorisation pour la vente ou l'importation de l'instrument à des fins d'essai expérimental. Les fabricants qui entreprennent la recherche doivent obtenir l'approbation de Santé Canada (à l'exception des instruments de classe I) et d'un CER avant le début d'un essai clinique, selon une approche basée sur le risque. Les instruments médicaux de classe II sont soumis à des exigences de demande réduites et les instruments médicaux de classe III et IV doivent respecter l'ensemble des exigences de demande décrites dans la partie 3 du Règlement sur les instruments médicaux. Actuellement, Santé Canada applique une période d'examen de 30 jours pour les essais cliniques d'instruments médicaux à partir du moment où une demande complète est reçue. Santé Canada peut délivrer une « lettre d'autorisation » pour les essais expérimentaux des instruments médicaux de classe II, III et IV si l'information soumise est jugée conforme aux exigences du règlement. Par ailleurs, s'il manque des renseignements, Santé Canada peut formuler une demande d'information supplémentaire. Il n'y a pas de période d'examen par défaut de 30 jours pour les demandes d'essai expérimental relatives aux instruments médicaux.
Produits de santé naturels
Santé Canada réglemente la vente ou l'importation de certains produits de santé naturels (PSN) destinés à être utilisés dans le cadre d'essais cliniques sur des participants humains. La conduite d'essais cliniques pour les PSN est régie par la Loi sur les aliments et drogues et le Règlement sur les produits de santé naturels(RPSN) et doit être menée conformément aux BPC telles que définies à l'article 74 du RPSN. Conformément à la partie 4 du RPSN, les promoteurs qui entreprennent la recherche doivent obtenir l'approbation de Santé Canada (sauf pour les essais de phase IV) et d'un CER avant le début d'un essai clinique. Les exigences d'un essai clinique avec un PSN sont très semblables à celles d'un essai clinique d'un médicament conventionnel. Actuellement, Santé Canada suit une norme de rendement définie dans la politique comme pouvant aller jusqu'à 90 jours pour l'examen et l'approbation des essais cliniques des PSN. Santé Canada délivrera un avis d'autorisation si la vente ou l'importation d'un PSN aux fins d'un essai clinique est approuvée.
Annexe 2 : Considérations propres à la modernisation d'essais cliniques, par produit
En plus des changements importants décrits dans ce document de consultation pour la modernisation des essais cliniques, Santé Canada reconnaît que certains éléments de la proposition représentent des changements uniques pour les différentes gammes de produits. Certaines de ces considérations et les impacts anticipés sont décrits ci-dessous afin de fournir aux parties prenantes un aperçu complet de ce à quoi elles peuvent s'attendre et de ce que les changements proposés signifieraient pour elles.
Médicaments
Les intervenants ont déjà indiqué que l'absence d'une approche basée sur le risque en matière de réglementation des essais cliniques constitue un obstacle à la réalisation de recherches au Canada, et certains ont demandé à Santé Canada de s'inspirer des modèles de stratification des risques utilisés dans d'autres pays pour modifier la réglementation des essais cliniques de médicaments. En 2019, Santé Canada a publié un Avis aux intervenants : Déclaration sur l'utilisation expérimentale de médicaments commercialisés lors d'essais cliniques qui, dans certaines conditions, pourrait réduire certaines exigences réglementaires relatives à l'utilisation non indiquée sur l'étiquette de médicaments commercialisés dans le cadre d'un essai clinique lorsque ces médicaments ne font pas l'objet de l'étude dans l'essai. Les arrêtés d'urgence de 2020 et 2021 ont intégré cette approche basée sur le risque pour réduire les exigences relatives aux utilisations expérimentales et non expérimentales de médicaments déjà commercialisés dans un essai clinique.
Lorsque le profil d'innocuité d'un médicament est bien caractérisé, comme c'est le cas pour les médicaments actuellement commercialisés, Santé Canada propose de réduire les exigences relatives à la collecte d'événements indésirables non graves ou d'autres données sur l'innocuité dans certaines conditions. La réduction du fardeau inutile lié à la collecte d'événements indésirables, si elle est prévue dans le protocole et approuvée par Santé Canada, pourrait se traduire par des essais cliniques plus rentables en réduisant le fardeau administratif.
Un autre changement important pour les essais cliniques de médicaments consisterait à passer d'une autorisation par défaut « sans objection » de la vente ou de l'importation du médicament faisant l'objet de l'essai clinique, à une autorisation plus complète de l'essai ainsi que des produits testés. Ce changement proposé, ainsi que la proposition de Santé Canada de délivrer une seule autorisation pour un essai portant sur plusieurs gammes de produits, permettrait à Santé Canada de mieux réglementer la gamme de types d'essais novateurs auxquels les promoteurs ont de plus en plus recours, comme les essais adaptatifs ou les protocoles maîtres. Il contribuerait également à faciliter et à simplifier le processus réglementaire pour les promoteurs, qui peut être difficile à suivre pour les types d'essais les plus complexes.
Pour les essais de médicaments, cette proposition de modification de l'autorisation de l'essai supprimerait également l'actuelle autorisation par défaut « sans objection » de 30 jours de la réglementation. Cela signifie que les promoteurs devraient recevoir une autorisation de Santé Canada avant de procéder à un essai. Les délais d'examen d'une demande d'essai clinique (DEC) seraient définis dans la politique et seraient alignés entre les gammes de produits. Pour les essais de bioéquivalence, Santé Canada supprimerait l'objectif d'examen administratif de sept jours, alignant ce délai sur celui des autres examens pour un traitement plus équitable entre les promoteurs. Santé Canada pourrait revoir les délais pour les demandes de renseignements supplémentaires aux promoteurs faites au cours d'un examen de DEC, qui sont actuellement soumises à un délai de deux jours par règlement.
En vertu de la proposition, des outils flexibles basés sur le risque seraient utilisés pour préserver la sécurité des participants sans nécessairement gêner le déroulement de l'entièreté d'un essai clinique. Santé Canada aurait le pouvoir d'exiger d'un promoteur qu'il effectue une analyse de certains renseignements et qu'il lui fournisse cette analyse, ce qui contribuerait également à assurer une surveillance adéquate des essais tout au long de leur cycle de vie. Pour les essais liés aux médicaments, la nouvelle capacité de Santé Canada d'ajouter des conditions à l'approbation d'un essai clinique pourrait être utilisée, par exemple, pour exiger la création d'un comité de surveillance des données (CSD), s'il ne fait pas déjà partie d'une demande, et demander des rapports de ce CSD à des moments précis de l'étude.
La possibilité de suspendre un volet d'un essai sur la base d'un signal d'innocuité sans priver les participants des autres volets non touchés de l'accès à des thérapies potentielles serait également un atout majeur pour les essais liés aux médicaments. Santé Canada propose également qu'une fois qu'une étude a été interrompue, le promoteur demande l'autorisation de la relancer (actuellement, une étude peut être relancée sur simple notification à Santé Canada).
En plus des changements mentionnés ci-dessus, Santé Canada considère faire plus clairement référence à la ligne directrice ICH E6 sur les BPC dans le règlement. Santé Canada évalue également la capacité d'appliquer plus largement plusieurs concepts introduits par l'intermédiaire des arrêtés d'urgence sur les essais cliniques (AU-EC), comme permettre d'autres moyens d'obtenir un consentement éclairé, et permettre à un plus grand nombre de professionnels de la santé d'être autorisés à mener un essai.
Instruments médicaux
L'essai expérimental lié à un instrument médical désigne l'étude clinique entreprise pour évaluer la performance clinique, l'innocuité clinique ou l'efficacité d'un instrument médical. Les études peuvent également comprendre l'évaluation d'instruments médicaux commercialisés pour de nouvelles utilisations prévues, de nouvelles populations, de nouveaux matériaux ou des changements de conception.
Avec le Plan d'action sur les instruments médicaux (PAIM) de 2018, Santé Canada s'est engagé à examiner les préoccupations de longue date des intervenants quant à la façon dont le cadre réglementaire actuel des instruments médicaux pourrait limiter involontairement les activités d'essais expérimentaux au Canada. Cet engagement s'est concentré sur les changements réglementaires potentiels qui pourraient encourager davantage de recherche clinique au Canada, tout en continuant à se concentrer sur la protection de la sécurité des patients. En 2019 et au début de 2020, le Ministère a organisé des consultations ciblées avec les intervenants afin de recueillir des commentaires sur les modifications possibles du cadre d'autorisation des demandes d'essais expérimentaux (DEE). Les intervenants ont été déterminés à partir des groupes suivants : chercheurs, industrie, groupes de patients, groupes de médecins, représentants des Comités d'éthique pour la recherche (CER) et organismes subventionnaires. Les commentaires reçus lors des consultations en vertu du PAIM sur le cadre des DEE ont été utilisés pour guider l'élaboration de la politique. Ces questions sont examinées plus en détail ci-dessous.
Nouvelle flexibilité pour les chercheurs indépendants
À l'heure actuelle, les fabricants et les importateurs doivent satisfaire aux exigences réglementaires de la partie 3 du Règlement sur les instruments médicaux pour recevoir l'autorisation de Santé Canada de vendre un instrument de classe II, III ou IV à un chercheur qualifié dans le but d'effectuer des essais expérimentaux. Les essais testant uniquement des instruments de classe I ne nécessitent pas d'autorisation, mais ils doivent répondre à toutes les exigences réglementaires applicables. Santé Canada délivre une autorisation s'il détermine que l'instrument peut être utilisé en toute sécurité, que les essais ne vont pas à l'encontre des intérêts fondamentaux des patients et que les objectifs des essais seront atteints.
Le Dr. Dylan est un cardiologue qui souhaite évaluer une nouvelle endoprothèse non homologuée dans le cadre d'un essai clinique. Pour le moment, seuls les fabricants et les importateurs d'instruments médicaux pourraient demander une autorisation. Dr. Dylan devrait travailler avec un fabricant pour qu'il soumette une demande d'autorisation en son nom. Dans le cadre de la modernisation de la réglementation, Dr. Dylan pourrait soumettre une demande directement à Santé Canada.
Le Règlement sur les instruments médicaux actuel permet uniquement aux fabricants et aux importateurs d'instruments médicaux de soumettre une DEE. Toutefois, grâce à la politique décrite dans le document d'orientationNote de bas de page 5, Santé Canada offre un mécanisme de délégation par lequel un chercheur indépendant (chercheur, clinicien ou établissement de soins de santé) peut être autorisé par un fabricant d'instruments à être un correspondant réglementaire. Malgré cela, le chercheur autorisé ne peut pas déposer la DEE sans le consentement exprès du fabricant d'instruments et n'a aucune responsabilité légale associée dans le cadre du Règlement sur les instruments médicaux. Des modifications au règlement sont proposées pour permettre à un chercheur, indépendant du fabricant d'instruments, de demander une autorisation d'essai clinique* en plus d'un fabricant et d'un importateur.
*Remarque : l'utilisation de l'expression « essai expérimental » est incompatible avec les nouveaux pouvoirs de la Loi sur les aliments et drogues (LAD), qui font précisément référence à « essai clinique ». Pour cette raison et par souci d'harmonisation avec toutes les gammes de produits, les essais expérimentaux seront appelés essais cliniques dans le règlement révisé.
Modernisation des exigences réglementaires
L'approche actuelle pour classer un instrument médical dans l'une des quatre classes est basée sur le risque, avec des exigences réglementaires adaptées qui permettent un niveau acceptable d'incertitude grâce auquel Santé Canada peut appliquer moins d'exigences aux produits à plus faible risque soumis à des essais cliniques (voir la section 3.2 Approche basée sur le risque pour plus de détails). Santé Canada envisage de modifier cette approche réglementaire dans quelques domaines choisis.
Les essais cliniques des IDIV utilisant des échantillons résiduels, lorsqu'ils ne sont pas utilisés pour la gestion des patients et aux fins de recherche uniquement, n'impliquent pas les patients et ne présentent donc aucun risque pour leur sécurité. Toutefois, ces instruments peuvent présenter certains risques pour l'utilisateur en ce qui concerne la sécurité électrique des instruments. De même, les essais de séquences d'impulsions en cours de production pour les appareils d'imagerie par résonance magnétique, lorsque certaines conditions sont remplies, ne présentent que peu ou pas de risques pour les patients et les utilisateurs. Santé Canada envisage d'exempter ces produits à faible risque de l'obligation d'obtenir une autorisation avant de commencer un essai.
Avec la nouvelle autorité législative conférée par la Loi sur les aliments et drogues, tous les essais cliniques devront être autorisés, sauf indication contraire. Selon les directives actuelles de Santé Canada, les fabricants doivent soumettre une demande d'utilisation d'un instrument homologué dans le cadre d'une étude visant à générer des données à l'appui d'une nouvelle indication (utilisation hors indication sur l'étiquette). Toutefois, l'utilisation hors indication sur l'étiquette d'instruments médicaux homologués utilisés dans le cadre d'études cliniques et non parrainés par le fabricant ne nécessite pas d'autorisation. À l'avenir, Santé Canada examinera s'il est possible de réduire les exigences relatives aux essais cliniques portant sur l'utilisation hors indication sur l'étiquette d'instruments médicaux homologués au Canada, tout en veillant à ce que la sécurité des patients demeure une priorité pour les essais menés par les fabricants et les chercheurs indépendants.
L'entreprise Y a élaboré une trousse d'essai pour détecter l'antigène de surface de l'hépatite. L'entreprise souhaite réaliser une étude pour évaluer le rendement de la trousse d'essai en utilisant des échantillons de sang résiduel obtenus auprès de banques de sang. Les échantillons seront dépersonnalisés et les résultats des tests obtenus ne seront pas communiqués aux patients et ne seront pas utilisés pour la gestion des patients. À l'heure actuelle, ils doivent soumettre une demande pour obtenir une autorisation d'essai expérimental afin d'utiliser l'instrument dans un essai. Dans le cadre de la modernisation de la réglementation, ils seraient exemptés de l'obligation d'obtenir une autorisation, mais d'autres exigences réglementaires seraient maintenues (par exemple, la déclaration des incidents, le traitement des plaintes, les rappels, l'approbation du Comité d'éthique de la recherche, les bonnes pratiques cliniques).
La possibilité de réduire les exigences réglementaires pour l'essai d'un instrument au sein d'une seule et même entreprise (essais internes) est actuellement étudiée. Les exigences réglementaires pour ces essais internes où aucune vente n'a eu lieu ne s'appliquaient pas auparavant; cependant, la modernisation de la réglementation permettrait à Santé Canada de superviser ces essais.
Formalisation de la politique actuelle en matière d'instruments médicaux dans la réglementation
Santé Canada fournit des documents d'orientation pour aider à interpréter les attentes en matière de conformité au règlement. Certains sujets faisant actuellement l'objet d'une politique bénéficieraient d'une formalisation dans le règlement, notamment l'approbation du Comité d'éthique de la recherche (CER), les bonnes pratiques cliniques (BPC) et les modifications d'essai clinique.
Approbation du CER
La formulation du règlement actuel laisse entendre que Santé Canada exige l'approbation du CER dans le cadre du dossier de DEE pour les instruments de classe III et IV (l'approbation du CER est également requise pour les instruments de classe I et II, mais n'a pas besoin d'être soumise à Santé Canada). Cependant, Santé Canada a fourni des directives dans son document d'orientation pour indiquer que l'approbation du CER peut être fournie après l'autorisation, mais avant le début d'un essai. Cette interprétation différente entre le règlement et le document d'orientation a provoqué une certaine confusion chez les parties prenantes. À l'avenir, Santé Canada propose de clarifier la formulation du règlement afin d'exiger explicitement l'approbation du CER avant le début d'un essai. Comme c'est le cas actuellement, pour les essais cliniques portant sur des instruments de classe III et IV, l'étude ne peut être initiée avant d'avoir reçu et communiqué l'approbation du CER à Santé Canada. Les changements proposés permettraient également d'aligner le Canada sur ses partenaires internationaux, tels que les États-Unis et l'Union européenne.
L'entreprise X souhaite réaliser un essai clinique utilisant un instrument médical de classe III dans un hôpital de l'Ontario. Ils sont conscients que l'approbation du CER de l'établissement doit être obtenue ainsi que l'autorisation de Santé Canada. Le CER de l'établissement n'approuvera pas un essai clinique tant qu'il n'aura pas reçu la preuve de l'autorisation de Santé Canada. De plus, les exigences de Santé Canada concernant l'approbation du CER ne sont pas claires pour la société X, car le règlement précise que l'approbation écrite du CER doit être fournie avant l'autorisation, alors que le document d'orientation donne une instruction différente. Ce n'est pas clair pour l'entreprise X, qui ne sait pas dans quel ordre de soumission elle doit procéder. Pour éviter ces incertitudes, la modernisation de la réglementation comprendrait un langage clair permettant l'approbation de l'essai clinique par le CER après la délivrance de l'autorisation de l'essai clinique par Santé Canada, mais avant le début de l'essai.
BPC
Il est reconnu à l'échelle internationale que la recherche sur les humains doit être menée conformément aux principes de BPC généralement acceptés, comme le stipule la norme ISO 14155 – Investigation clinique des dispositifs médicaux pour sujets humains. Ces principes donnent l'assurance que les données et les résultats rapportés sont crédibles et exacts, et que les droits, la sécurité et le bien-être des participants des essais cliniques sont protégés.
Le Règlement sur les instruments médicaux ne contient actuellement aucune exigence de conformité aux BPC, mais la conformité à la norme ISO 14155 est recommandée dans le document d'orientation. L'inclusion dans le règlement d'une obligation pour un promoteur d'essai de se conformer aux principes des BPC donnerait une plus grande assurance que les patients restent protégés pendant les études expérimentales. Elle permettrait également de mieux harmoniser les exigences en matière d'essais cliniques pour les instruments médicaux et les médicaments à usage humain au Canada et avec nos partenaires internationaux (Union européenne, Royaume-Uni, États-Unis, Australie).
Modification d'une demande d'essai expérimental
Dans le cadre réglementaire actuel, aucune disposition ne permet à Santé Canada d'autoriser la modification d'une demande d'essai expérimental (DEE). Ainsi, tout changement apporté au cours d'un essai doit être soumis comme une nouvelle DEE.
Cependant, Santé Canada permet aux fabricants de soumettre certaines modifications à des DEE déjà autorisées de manière abrégée par l'intermédiaire de lignes directrices. Le document d'orientation disponible décrit également un processus supplémentaire par lequel davantage de modifications administratives peuvent être soumises à Santé Canada par l'intermédiaire d'une notification. Santé Canada propose d'officialiser cette politique dans le règlement, permettant ainsi d'apporter des changements mineurs et importants à une autorisation sans qu'un promoteur d'essai n'ait à soumettre une nouvelle demande.
Un fabricant d'instruments médicaux établi au Canada a été autorisé à tenir un essai clinique sur un nouveau type de ventilateur (un instrument de classe III) durant la pandémie. Le fabricant décide qu'une modification au logiciel est nécessaire pour ajouter un nouveau mode de ventilation. À l'heure actuelle, le règlement ne prévoit aucune disposition autorisant la modification d'une autorisation; une nouvelle demande complète devrait être présentée (toutefois, une ligne directrice permet aux fabricants de présenter des modifications de manière abrégée). La flexibilité réglementaire conférée par un cadre d'essai clinique modernisé permettrait aux détenteurs d'autorisation de présenter et de faire autoriser des modifications à un essai clinique en cas de changements importants aux renseignements contenus dans la demande initiale.
Début, interruption et reprise de l'essai
Le Règlement sur les instruments médicaux ne comporte aucune disposition obligeant un fabricant à signaler à Santé Canada le début, l'interruption ou la reprise d'une étude expérimentale, en tout ou en partie. Cependant, le document d'orientation de Santé Canada aborde ces scénarios. Les nouvelles autorités étant habilitées à réglementer l'ensemble du cycle de vie de l'essai, de nouvelles exigences sont proposées pour signaler à Santé Canada le début, l'interruption ou la reprise d'une étude, que ce soit pour l'ensemble de l'essai ou pour un site particulier.
Produits de santé naturels
Santé Canada propose de moderniser le règlement relatif aux essais cliniques des produits de santé naturels (PSN) afin de les rendre conformes au règlement mis à jour pour les essais d'autres produits de santé. L'intention est que toutes les propositions décrites dans les thèmes de souplesse tout au long du cycle de vie flexible, d'approche basée sur le risque et de transparence soient appliquées de manière égale, tout en tenant compte des besoins uniques des PSN. Les changements comprendraient la mise à jour des délais d'examen d'une DEC concernant un essai relatif aux PSN. Le délai actuel est de 90 jours, mais il sera probablement réduit pour être conforme aux exigences relatives aux autres gammes de produits, et considérant les commentaires reçus dans le cadre de ce processus de consultation et examinés par Santé Canada. De plus, conformément au règlement actuel sur les PSN, Santé Canada propose de maintenir son exigence actuelle selon laquelle un membre d'un Comité d'éthique pour la recherche doit avoir des connaissances en matière de soins de santé complémentaires ou de médecine douce, assurant ainsi des connaissances spécialisées concernant les PSN pendant le processus d'évaluation éthique.
En introduisant une approche basée sur le risque, appuyée sur l'information disponible sur le risque, les avantages et l'incertitude liés à un PSN, Santé Canada peut adapter les exigences de façon proportionnelle, plutôt que d'appliquer une approche unique qui pourrait ne pas convenir à de nombreux PSN. Le règlement offrirait également une plus grande souplesse, ce qui permettrait à Santé Canada d'aborder différents aspects de l'essai de manière appropriée au cours du cycle de vie de l'essai. Il est essentiel de veiller à l'alignement sur d'autres produits de santé, tels que les médicaments, à mesure que ce processus avance.
L'entreprise canadienne LifeChange prévoit mener un essai clinique qui étudie l'efficacité d'une thérapie combinée comprenant le produit X (un médicament) et le produit Y (un produit de santé naturel). Auparavant, LifeChange devait soumettre deux demandes distinctes – l'une pour le médicament, l'autre pour le produit de santé naturel – et attendre l'autorisation dans le cadre de deux filières d'approbation avec des délais d'examen différents. Après la modernisation des essais cliniques, LifeChange serait en mesure de soumettre une seule demande d'essai clinique et de recevoir une seule approbation pour un essai clinique mené sur les produits X et Y. À l'interne, cette DEC serait examinée à la fois par la Direction des produits thérapeutiques (DPT) et la Direction des produits de santé naturels et sans ordonnance (DPSNSO), mais l'approche de guichet unique avec le demandeur simplifierait le processus et réduirait leur fardeau administratif.
Annexe 3 : Questions de la consultation
Pour une plus grande convivialité, Santé Canada a intégré les questions suivantes dans une consultation en ligne. Santé Canada encourage les participants à offrir leur rétroaction par ce format en ligne, qui allie des questions à choix multiples et des zones texte dans lesquelles vous pouvez donner votre point de vue ou bien des détails supplémentaires concernant la réponse choisie. Si vous préférez envoyer vos commentaires par courriel ou par courrier classique, n'hésitez pas à le faire. Dans ce cas, nous vous recommandons d'utiliser les questions suivantes comme guide.
Afin de protéger votre vie privée, veuillez vous assurer que tous les commentaires écrits que vous fournissez sont de nature suffisamment générale pour que l'on ne puisse pas vous identifier comme étant leur auteur et qu'aucun nom ne soit divulgué.
Questions d'ordre démographique :
- Veuillez indiquer le type d'intervenant que vous représentez ou auquel vous vous identifiez le plus :
- Industrie
- Milieu universitaire/instituts de recherche
- Praticiens de la santé
- Organismes de soins de santé, à l'exception de ceux figurant en tant que chercheurs
- Groupe de défense des intérêts des patients
- Comités d'éthique pour la recherche et Association canadienne des comités d'éthique de la recherche
- Gouvernements provinciaux ou territoriaux, veuillez préciser
- Santé Canada
- Autre ministère du gouvernement fédéral, veuillez préciser
- Qu'est-ce qui décrit votre activité en matière d'essais cliniques? (Veuillez sélectionner toutes les réponses qui s'appliquent) :
- Promoteur/demandeur – industrie
- Promoteur/demandeur – milieu universitaire
- Organisme de recherche sous contrat
- Organisme de gestion de site
- Chercheur qualifié
- Coordonnateur de l'étude/membre de l'équipe de l'étude
- Association de recherche clinique
- Participant aux essais cliniques
- Autre, veuillez préciser
- Quelles sont les gammes de produits de santé que vous ou votre organisation représentez, pour lesquelles vous menez des essais ou qui vous intéressent? (Veuillez sélectionner toutes les réponses qui s'appliquent)
- Médicaments
- Instruments médicaux
- Produits de santé naturels
- Ne mène aucun essai clinique
- Autre, veuillez préciser
Questions pour tous les répondants (veuillez détailler vos réponses, le cas échéant) :
- Croyez-vous que les propositions suivantes auront des avantages ou des répercussions pour votre organisation?
- Souplesse tout au long du cycle de vie
- Autorisation unique pour plusieurs produits
- Approche basée sur le risque
- Utilisation de conditions dans le cadre d'une autorisation d'essai clinique
- Essais décentralisés
- D'après votre expérience et vos connaissances, les propositions contenues dans le document de consultation permettraient-elles à Santé Canada d'atteindre son objectif de favoriser la tenue d'essais cliniques novateurs au Canada?
- Veuillez expliquer toute innovation ou autre considération future dont Santé Canada devrait tenir compte dans son projet de modernisation de la réglementation.
- Quels autres facteurs Santé Canada devrait-il prendre en compte lors de la mise en œuvre de cette proposition visant à rationaliser les exigences réglementaires pour l'ensemble des gammes de produits afin de mieux permettre une autorisation unique?
- Quels facteurs Santé Canada devrait-il prendre en considération dans la mise en œuvre d'une approche basée sur le risque?
- Existe-t-il d'autres domaines dans lesquels le fardeau peut être réduit et qui permettront à votre organisation de mieux mener les essais cliniques sans compromettre la sécurité des patients?
- Y a-t-il des domaines où Santé Canada pourrait aller plus loin dans la modernisation du cadre réglementaire canadien des essais cliniques au-delà de ce qui est proposé dans le document de consultation?
- Voyez-vous un intérêt à mettre en œuvre une nouvelle politique ou un nouveau règlement pour l'enregistrement et la communication des résultats des essais cliniques menés au Canada (essais qui portent sur des médicaments, des instruments médicaux et des produits de santé naturels)?
- Enregistrez-vous généralement vos essais auprès d'un registre international?
- Rencontrez-vous des difficultés pour tenir à jour votre enregistrement dans les registres internationaux, y compris le statut de l'essai?
- Sous-traitez-vous actuellement des activités liées aux essais cliniques ou aux tests expérimentaux à des tiers (par exemple, organisme de recherche sous contrat ou organisme de gestion de site)? Le cas échéant, quelles sont les activités que vous sous-traitez? Si vous n'étiez plus légalement responsable des activités que vous avez sous-traitées, quel impact cela aurait-il sur votre entreprise?
- Des activités vous sont-elles confiées en sous-traitance pour des essais cliniques portant sur des médicaments, des PSN ou des instruments médicaux? Le cas échéant, à combien d'essais cliniques/expérimentaux participez-vous chaque année en moyenne au Canada? Si vous deviez être légalement responsable des activités qui ont été sous-traitées, quel impact cela aurait-il sur votre entreprise?
- Participez-vous actuellement, ou avez-vous déjà participé, à des essais cliniques ou à des tests expérimentaux aux États-Unis?
- En fonction de la façon dont vous menez vos activités, comment définiriez-vous la fin d'un essai clinique?
- Y a-t-il des difficultés de mise en œuvre liées à la clarification du fait que la conservation des dossiers doit commencer après l'achèvement ou l'interruption de l'essai clinique?
Si vous ou votre organisation participez à des essais cliniques sur des médicaments :
- Compte tenu de la complexité croissante des essais cliniques et de la nécessité d'établir un cadre réglementaire harmonisé pour toutes les gammes de produits, seriez-vous favorable à l'allongement des délais d'examen des demandes? Veuillez indiquer combien de jours, selon vous, constitueraient un délai raisonnable pour l'examen de la demande d'essai clinique, et dans quelles conditions.
- Si d'autres professionnels sont autorisés à devenir chercheurs, faut-il encore exiger qu'un médecin désigné soit responsable de l'ensemble des soins médicaux des participants?
Si vous ou votre organisation participez à des essais cliniques sur des produits de santé naturels :
- Sur la base de vos connaissances et de votre expérience, voyez-vous un avantage à réduire le délai d'examen de l'essai clinique de 90 jours sur la conduite de votre essai clinique?
- Les exigences relatives à la déclaration des réactions indésirables aux PSN devraient-elles être modifiées afin d'harmoniser les exigences relatives aux produits de santé naturels et aux médicaments?
- Quels défis devez-vous relever pour respecter les dispositions relatives aux bonnes pratiques cliniques du Règlement sur les produits de santé naturels/ICH E6 (R2)?
Si vous ou votre organisation participez à des essais expérimentaux sur des instruments médicaux :
- Santé Canada maintient le système de classification basé sur le risque (classes I à IV) pour les instruments médicaux au Canada. Toutefois, le nouveau cadre propose de subdiviser davantage les essais cliniques des instruments médicaux en trois (3) catégories (A – Aucune autorisation; B – Exigences adaptées; C – Exigences complètes) pour une meilleure harmonisation avec les médicaments. Soutenez-vous ce cadre proposé? Veuillez expliquer quels avantages ou inconvénients vous pouvez prévoir avec ce nouveau système de classification.
- Les produits à faible risque suivants devraient-ils être exemptés de l'obligation d'obtenir une autorisation (d'autres exigences devraient tout de même être respectées, par exemple, le signalement des incidents, l'approbation du Comité d'éthique pour la recherche):
- Instruments diagnostiques in vitro (IDIV) utilisant des échantillons résiduels (lorsqu'ils ne sont pas utilisés pour la gestion des patients et aux fins de recherche uniquement)?
- Essais de séquences d'impulsions en cours de production pour les appareils d'imagerie par résonance magnétique (lorsque certaines conditions sont remplies – veuillez consulter la section 2.3.2.3 du document d'orientation)?
- Pour les essais mis en œuvre par le fabricant, serait-il possible d'avoir des exigences réglementaires adaptées pour les essais cliniques internes lorsqu'il n'y a pas de vente d'instrument?
- Les exigences réglementaires devraient-elles être réduites lorsqu'une nouvelle utilisation d'un instrument médical homologué est étudiée (utilisation non spécifiée sur l'étiquette)?
- Tout en gardant à l'esprit la sécurité du patient, veuillez expliquer s'il doit y avoir une différence dans les exigences réglementaires selon que le produit utilisé hors indication fait l'objet de l'enquête ou qu'il est utilisé à titre de comparateur ou aux fins de soutien (par exemple, pour livrer l'instrument expérimental)?
- Prévoyez-vous des risques liés à l'utilisation hors indication sur l'étiquette d'un instrument médical commercialisé dans un essai clinique?
- Veuillez expliquer toute préoccupation que vous pourriez avoir concernant l'approbation de l'essai par le Comité d'éthique pour la recherche après la délivrance de l'autorisation d'essai clinique, mais avant le début de l'étude.
- Veuillez expliquer toute préoccupation que vous pourriez avoir concernant la création par Santé Canada des trois voies suivantes pour les modifications des autorisations d'essais cliniques :
- Des modifications importantes (basées sur la Directive sur l'interprétation d'une modification importante d'un instrument médical), qui nécessiteraient une révision de l'autorisation d'essai clinique et l'approbation de Santé Canada.
- Des préavis de modification, qui devraient être déposés auprès de Santé Canada, mais ne nécessiteraient pas d'approbation.
- Des modifications non importantes ou administratives, qui devraient être documentées uniquement dans les dossiers internes du demandeur.
- Quels types de modifications devraient être classés dans chaque catégorie et pourquoi?
- Avez-vous d'autres recommandations pour accroître l'efficacité du processus de modification des autorisations d'essais cliniques sans compromettre la sécurité des patients?
- Quels défis prévoyez-vous si un chercheur, indépendant du fabricant, souhaite demander à Santé Canada l'autorisation d'effectuer l'essai clinique d'un instrument médical (pour une utilisation prévue ou hors indication)? Quelles sont, le cas échéant, vos recommandations pour surmonter ces difficultés?
- Dans les situations où les fabricants ne souhaitent pas communiquer les renseignements au sujet des instruments médicaux au chercheur ou à Santé Canada pour appuyer la demande d'un chercheur indépendant, comment cette difficulté peut-elle être surmontée?
- Y a-t-il des essais cliniques effectués à l'initiative d'un chercheur indépendant (clinicien ou universitaire) dans votre organisation? (Veuillez sélectionner toutes les réponses qui s'appliquent) :
- Études préalables à la mise en marché des instruments médicaux :
- Première étude menée chez l'humain
- Étude de faisabilité/pilote
- Étude pivot
- Études post-commercialisation des instruments médicaux :
- Instrument commercialisé selon l'indication d'utilisation approuvée
- Utilisation non spécifiée sur l'étiquette de l'instrument commercialisé
- Essai clinique utilisant un instrument médical non homologué et non soumis à des enquêtes
- Autre : veuillez préciser
- Études préalables à la mise en marché des instruments médicaux :
- En général, quel est le but des essais cliniques liés aux instruments médicaux que vous réalisez au Canada? Générer des données à l'appui d'une future demande d'homologation d'un instrument médical aux fins de commercialisation, de recherche uniquement, ou dans un autre but?
- Pour les essais cliniques que vous avez menés et qui ne nécessitaient pas d'autorisation, quels types de garanties ont été mis en place pour protéger la sécurité des patients en plus de l'obtention de l'approbation du Comité d'éthique pour la recherche?
- Pour les instruments médicaux, prévoyez-vous des difficultés liées à la conformité avec les bonnes pratiques cliniques alignées sur les exigences de la norme ISO 14155?