
Canada's Access to Medicines Regime: Application Process for Drugs for Export to Developing and Least Developed Countries
December 6, 2006
Notice
Our file number: 06-127652-79
Release of Canada's Access to Medicines Regime: Application Process for Drugs for Export to Developing and Least Developed Countries
This Guidance Document is intended to provide direction to manufacturers when submitting an application to the Therapeutic Products Directorate (TPD) for a patented drug for export to developing and least developed countries under Canada's Access to Medicines Regime (CAMR). It outlines how TPD manages the information and material submitted by sponsors in accordance with the Regulations Amending the Food and Drug Regulations (FDR) (1402 - Drugs for Developing Countries).
Comments on this Guidance Document are welcome in the context of the Government of Canada's Review of Canada's Access to Medicines Regime, announced on November 24, 2006. The Guidance Document will be amended where applicable as part of the statutory review currently underway. Further information on the review of CAMR can be found at Canada's Access to Medicines Regime
Your comments and questions should be directed to:
Bureau of Policy, Science and International Programs
Therapeutic Products Directorate
Health Canada
Telephone: (613) 946-9491
Fax: (613) 941-1812
E-mail: bpsip_info_bpspi@hc-sc.gc.ca
Guidance Document
Canada's Access to Medicines Regime: Application Process for Drugs for Export to Developing and Least Developed Countries
Published by authority of the
Minister of Health
Date Adopted: 2006/12/06
Effective Date: 2006/12/06
Health Products and Food Branch
Our mission is to help the people of Canada maintain and improve their health.
Health Canada
HPFB's Mandate is to take an integrated approach to managing the health-related risks and benefits of health products and food by:
- minimizing health risk factors to Canadians while maximizing the safety provided by the regulatory system for health products and food; and,
- promoting conditions that enable Canadians to make healthy choices and providing information so that they can make informed decisions about their health.
Health Products and Food Branch
© Minister of Public Works and Government Services Canada 2006
Également disponible en français sous le titre : Le Régime canadien d'accès aux médicaments : Processus de demande pour l'exportation de médicaments aux pays en développement et aux pays les moins avancés
Foreword
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable guidance documents.
Table of Contents
- 1. Introduction
- 2. Guidance for Implementation
- 2.1 Roles and Responsibilities
- 2.2 Definitions
- 2.3 Filing Procedures
- 2.4 Pre-Submission Information
- 2.5 Process for the Domestic Submission
- 2.6 Process for the Division 7 Application
- 2.7 Review Finalization
- 2.8 Fee Remission
- 2.9 Renewal of Authorization Under Section 21.04 of the Patent Act
- 2.10 Subsequent Applications for Additional Products, Countries and Quantities
- 2.11 Product Ceases to Meet the Requirements of the Food and Drugs Act and Regulations
- Appendix A: Division 7 Application Certification
- Appendix B: Process and Criteria for Assessing Significant Difference in Colour
- Appendix C: Diagram of CAMR Process with TPD
1. Introduction
1.1 Policy Objectives
This document provides direction to manufacturers for submitting an application to the Therapeutic Products Directorate (TPD) for a patented drug for export to developing and least developed countries under Canada's Access to Medicines Regime (CAMR). It outlines how TPD manages the information and material submitted by sponsors in accordance with the Regulations Amending the Food and Drug Regulations (FDR) (1402 - Drugs for Developing Countries).
1.2 Policy Statement
Drugs exported to least-developed and developing countries under Canada's Access to Medicines Regime will meet the same rigorous standards for safety, efficacy and quality as those available to Canadians.
1.3 Scope and Application
This guidance document is intended to cover the application process within TPD for drugs which are eligible under CAMR and which are listed on Schedule 1 to the Patent Act. Medical devices and biologic therapies are not encompassed by this document.
1.4 Background
An Act to amend the Patent Act and the Food and Drugs Act (The Jean Chrétien Pledge to Africa), formerly Bill C-9, and its accompanying regulations came into force on May 14, 2005. The Bill was introduced to facilitate timely access to less expensive versions of patented pharmaceutical products urgently needed by least-developed and developing countries to fight HIV/AIDS, malaria, tuberculosis and other epidemics.
This legislative framework amended both the Canadian Patent ActFootnote 1 and the Food and Drugs Act in order to implement an August 30, 2003 decision of the World Trade Organization (WTO) that waived certain obligations set out in the Agreement on the Trade-Related Aspects of Intellectual Property Rights (TRIPS).Footnote 2 The amendments to the Food and Drugs Act ensure that pharmaceutical products exported under CAMR meet the same rigorous standards for safety, efficacy and quality as those for products sold in Canada. Therefore, products exported under CAMR must meet the requirements of the Food and Drugs Act and its Regulations. This involves the review of submissions against Canadian standards for safety, efficacy and quality. An additional review is undertaken pursuant to the new Division 7 of the FDR to ensure that the product can be distinguished from the patented version on the Canadian market as required by the World Trade Organization's General Council Decision of August 30, 2003.
2. Guidance for Implementation
2.1 Roles and Responsibilities
Therapeutic Products Directorate (TPD):
TPD's roles and responsibilities for therapeutic product reviews under the Canada's Access to Medicines Regime are the same as its roles and responsibilities for equivalent therapeutic products destined for the Canadian market. Pharmaceutical products under this regime must continue to meet the Canadian standards and manufacturers must be in compliance with good manufacturing practices.
2.2 Definitions
Authorization under section 21.04 of the Patent Act: This is an authorization to use a patented invention which is granted by the Commissioner of Patents at the Canadian Intellectual Property Office (CIPO) under section 21.04 of the Patent Act. The authorization is often referred to as a "Compulsory License".
CIPO Hold: Manufacturers are required under section 21.04 of the Patent Act to file an application with the Commissioner of Patents in the Canadian Intellectual Property Office (CIPO) as part of the CAMR process. A copy of this application must be sent to TPD by the manufacturer. When the Division 7 Application (see definition below) has been reviewed by TPD and is considered acceptable in all other respects, it may be placed on an administrative hold termed "CIPO Hold" until a copy of the application to CIPO is received from the manufacturer.
Division 7 Application: The Division 7 Application is submitted by a manufacturer to the Therapeutic Products Directorate for authorization to sell a drug under Division 7 of the FDR (as outlined in section C.07.003). This represents one portion of the application process for patented drugs for export and will contain the documents and data to support differences in the export product marking and labelling to comply with section C.07.008 of the FDR. Separate Division 7 Applications are required for each country for export.
Division 7 Application Hold: The Division 7 Application makes reference to the Domestic Submission (defined below). Therefore, if the Domestic Submission has not been recommended for approval when the review of the information in the Division 7 Application is complete, the Division 7 Application will be placed on Division 7 Application Hold until the final recommendation for the Domestic Submission is completed.
Domestic Submission: The Submission / Application described in section C.07.003 (b) and (c) of the FDR which may be one of the following:
New Drug Submission (NDS),
Supplement to a New Drug Submission (SNDS),
Abbreviated New Drug Submission (ANDS),
Supplement to a Abbreviated New Drug Submission (SANDS) or
Drug Identification Number Application (DINA).
The Domestic Submission will follow the process and timelines outlined in the Management of Drug Submissions Guidance (MDSG). If the new drug submission is found to comply with the FDR, the product will be granted a Notice of Compliance (NOC) once the regulatory requirements of the Patented Medicines (Notice of Compliance) Regulations are met. In the interim, the submission will be placed on "patent hold". If the DIN application is found to comply with the FDR then a Drug Identification Number (DIN) will be issued.
XCL: The acronym "XCL" refers to "eXport under Compulsory License". This acronym is permanently marked on solid dosage form products and on the label of all products intended for export under CAMR.
Export Tracking Number (ETN): The ETN is an alpha-numeric code generated by TPD for each approved Division 7 Application product and printed on the label of the products for export. The code consists of "XCL", 7 digits and a 2 letter country code.
2.3 Filing Procedures
2.3.1 Filing Scenarios
There are two distinct components required to apply to sell and export a drug under Canada's Access to Medicines Regime:
- the Domestic Submission; and
- the Division 7 Application.
These will have separate supporting data packages, forms and timelines. The Domestic Submission must contain all of the information required to attain authorization to market the drug product in Canada and will follow the normal process and time lines applicable for that Submission/Application type (defined in the MDSG). The Division 7 Application will contain the documents and data to support differences in the export product marking and labelling in order to comply with section C.07.008 of the Food and Drug Regulations. Separate submission numbers will be assigned to the Domestic Submission and Division 7 Application.
A Division 7 Application may be filed at the same time as the Domestic Submission, during the review period of the Domestic Submission, or after the review of the Domestic Submission is complete.
Refer to Section 9 for information on filing requirements for renewals of Authorization under section 21.04 of the Patent Act and subsequent applications for additional products, quantities and countries.
2.3.2 Domestic Submission Package Requirements
The Domestic Submission for a new drug must comply with sections C.08.002, C.08.002.1 or C.08.003 as applicable, and C.08.005.1 of the FDR and should follow the guidance Preparation of New Drug Submissions in the CTD Format. The domestic submission for a drug that is not a new drug must comply with section C.01.014.1 and should follow the guidance Preparation of Drug Identification Number Submissions. There are no differences from the normal submission or application requirements. Since the establishment of Good Manufacturing Practices (GMP) status can be a prolonged process, sponsors need to ensure that GMP compliance has been established for drug establishment sites prior to submitting.
2.3.3 Division 7 Application Package Requirements
The following components should be provided in the Division 7 Application. Note that all components are to be provided in duplicate, with the exception of the data described in 6b where one copy is adequate:
- a Cover Letter including a statement that the manufacturer has or intends to file an application with the Commissioner of Patents under section 21.04 of the Patent Act;
- a Division 7 Application Certification Form (Appendix A);
- the Drug Submission Application Form (HC-SC3011);
- the Submission Number and date of filing of the domestic submission, if previously filed;
- the Drug Identification Number (DIN) of the domestic submission if one has been assigned;
- for a drug in a solid dosage form,
- a description of the manner in which the drug is permanently marked with "XCL" in accordance with section C.07.008(a) of the FDR and a description of the manner in which the drug is significantly different from the colour of the version of the drug sold in Canada in accordance with section C.07.008(b);
- data to support a change in product colour and product markings relative to the Domestic Submission product should be provided as outlined in the guidance Changes in Product Colour or Markings. The data should be provided in the format outlined in the guidance;
- evidence must be provided to demonstrate that the product intended for export is a significantly different colour from the version of the drug sold in Canada. For example, sponsors may submit samples of the two products or a colour photograph clearly showing the product intended for export next to the version of the drug sold in Canada (ie. in the same photograph) to allow for an assessment of whether or not they are significantly different in colour. The photograph should show the products as actual size or larger. Appendix B describes the criteria and process which will be used to assess significant difference in colour. Where an assessment cannot be made based on a photograph, samples of the products may be requested and should be submitted within seven calendar days of the request;
- a description of the manner in which the drug is permanently marked with "XCL" in accordance with section C.07.008(a) of the FDR and a description of the manner in which the drug is significantly different from the colour of the version of the drug sold in Canada in accordance with section C.07.008(b);
- for a drug in a dosage form which is not solid, a description of how the immediate container permanently bears the mark "XCL" in accordance with section C.07.008(a);
- a sample of the label for the drug which permanently bears the mark "XCL" in capital letters followed by a place holder for the Export Tracking Number and the words in capital letters "FOR EXPORT UNDER THE GENERAL COUNCIL DECISION. NOT FOR SALE IN CANADA." or the words in capital letters "POUR EXPORTATION AUX TERMES DE LA DÉCISION DU CONSEIL GÉNÉRAL. VENTE INTERDITE AU CANADA.". All of this information should be present on the main panel of the label. Since no Drug Identification Number (DIN) will be issued for products intended for export no DIN placeholder is required on the label.
An Application for authorization under section 21.04 of the Patent Act is filed by the manufacturer with the Commissioner of Patents. The manufacturer is required to provide a copy of that application to TPD. The copy may be sent after the Division 7 Application is filed, but must be received in order for TPD to notify the Commissioner of Patents, as set out in C.07.004 of the FDR, that the manufacturer's drug meets the requirements of the Food and Drugs Act and Regulations. For information on the process of applying to the Commissioner of Patents for authorization under section 21.04 of the Patent Act refer to the CIPO website.
2.4 Pre-Submission Information
2.4.1 Pre-Submission / Application Meeting
Sponsors are encouraged to hold a pre-submission meeting with TPD prior to filing a submission or application under CAMR. Both the Domestic Submission and Division 7 Application may be discussed. The MDSG outlines the process and requirements for pre-submission meetings.
2.5 Process for the Domestic Submission
2.5.1 Submission / Application Process and Timelines
The Domestic Submission will follow the process and timelines outlined in the MDSG. Appendix C contains a process diagram describing the CAMR process with TPD.
2.5.2 Processing
The Domestic Submission will be processed according to the normal procedure as outlined in the MDSG.
2.5.3 Screening
The Domestic Submission will be screened according to the normal process as outlined in the MDSG. If received together with a Division 7 Application, the Domestic Submission will be assigned the normal screening performance target (usually 45 days) but handled in a separate CAMR workload queue for screening which means that screening will begin without delay.
2.5.4 Review
The Domestic Submission will be reviewed according to the normal process as outlined in the MDSG. Once associated with a Division 7 Application, the Domestic Submission will retain the normal review performance target (usually 180 days for an ANDS and 300 days for a NDS) but be handled in a separate CAMR workload queue for review. Therefore, review will begin without delay. However, if the Division 7 Application is found to be Not Satisfactory following review or is cancelled by the manufacturer, the associated Domestic Submission will revert to its original position in the regular workload queue.
2.6 Process for the Division 7 Application
2.6.1 Application Process and Timelines
The assessment of the information contained in the Division 7 Application will be assigned a single performance standard of 60 calendar days which will incorporate both the screening and review functions. However, the final decision for the Division 7 Application is dependent on the Domestic Submission being considered acceptable and in compliance with the Food and Drugs Act and its Regulations. Therefore once the review of the Division 7 Application is complete and found to be acceptable, it may be placed on Division 7 Application Hold until a determination has been made that the Domestic Submission complies with the Food and Drugs Act and its Regulations.
If the associated Domestic Submission receives a Clarification Request (Clarifax) or non-final negative decision (Screening Deficiency Notice, Notice of Deficiency, Notice of Noncompliance), the Division 7 Application will continue to be reviewed without delay.
If the associated Domestic Submission is cancelled by the manufacturer or receives a final negative decision (Screening Rejection, Notice of Deficiency Withdrawal or Notice of Noncompliance Withdrawal in accordance with the MDSG), the Division 7 Application will be considered not satisfactory and a Not Satisfactory Notice will be issued to the manufacturer.
Appendix C contains a process diagram describing the CAMR process with TPD.
2.6.2 Processing
A separate submission number will be assigned to each Division 7 Application and an acknowledgement letter will be sent to the manufacturer.
2.6.3 Screening / Review
The screening and review functions will be combined for the Division 7 Application and handled in a separate CAMR workload queue. A Clarification Fax with a 15 calendar day response time may be issued to the manufacturer to request additional information during the review process.
2.7 Review Finalization
2.7.1 Domestic Submission Finalization
When the review has been completed for the Domestic Submission and a final recommendation has been made for approval, it will be placed on Patent Hold by the Office of Patented Medicines and Liason (OPML) as appropriate. A DIN will be generated for the Domestic Submission product however it will not be issued to the manufacturer until the Domestic Submission is taken off Patent Hold.
2.7.2 Division 7 Application Finalization
Following the final recommendation for approval for the Domestic Submission, the Division 7 Application Hold will be removed from the Division 7 Application if applicable.
If a copy of the Application for authorization under section 21.04 of the Patent Act has not been received from the manufacturer, the Division 7 Application will be placed on CIPO Hold until a copy of the Application for authorization under section 21.04 of the Patent Act is received.
When the Division 7 Application is recommended for approval, an Export Tracking Number (ETN) will be assigned for the Division 7 Application product. No separate DIN will be issued for the product for export. The Director General of TPD will issue a letter to the Commissioner of Patents (CIPO) and the manufacturer in the form of a "Notification to the Commissioner of Patents" for the purposes of paragraph 21.04(3)(b) of the Patent Act, indicating that the drug meets the requirements of the Food and Drugs Act and Regulations. A copy of the Export Tracking Number Form will also be included with the letter.
When the Division 7 Application is not recommended for approval due to deficiencies in the Division 7 Application or a final negative decision in the associated Domestic Submission (Screening Rejection, Notice of Deficiency Withdrawal or Notice of Noncompliance Withdrawal in accordance with the MDSG) the Director General of TPD will issue a letter to the Commissioner of Patents (CIPO) and the manufacturer in the form of a Not Satisfactory Notice indicating that the drug is not considered to meet the requirements of the Food and Drugs Act and Regulations.
2.7.3 Issuance of the Authorization under section 21.04 of the Patent Act by the Canadian Intellectual Property Office (CIPO)
In accordance with C.07.005 of the FDR, the manufacturer may only sell the drug after the following requirements have been met:
- TPD has notified the Commissioner of Patents that the drug meets the requirements of the Act and these Regulations, and
- the manufacturer has received authorization from the Commissioner of Patents under section 21.04 of the Patent Act.
2.8 Fee Remission
2.8.1 Remission of Fees
Fees normally associated with the review of a Domestic Submission will be remitted by TPD to the manufacturer upon Treasury Board approval. In order to be eligible for remission, the sponsor must provide a copy of the Authorization under section 21.04 of the Patent Act and file a claim for remission to TPD:
- within 45 days after issuance of the Authorization under section 21.04 of the Patent Act by CIPO; or,
- for a Supplement to a New Drug Submission or Supplement to a Abbreviated New Drug Submission, within 45 days after issuance of the notice stating that the examination of the supplement has been completed.
There are no fees associated with the assessment of a Division 7 Application by TPD.
2.9 Renewal of Authorization Under Section 21.04 of the Patent Act
2.9.1 Renewal of authorization under section 21.04 of the Patent Act
An Authorization under section 21.04 of the Patent Act is granted for a two year period for a specific quantity of drug to be exported to a specific country. Pursuant to section 21.12 of the Patent Act, the Commissioner of Patents shall renew the Authorization if the person certifies under oath in the renewal application that the quantities authorized for export were not exported before the Authorization ceases to be valid. The authorization may be renewed once for up to a further two years and the Application for renewal must be made within 30 days immediately before authorization ceases.
No application is required to TPD and a new Export Tracking Number is not issued. However TPD will monitor the renewal process to ensure that the Export Tracking Number Database information is correct with respect to the expiry date of the Authorization.
2.10 Subsequent Applications for Additional Products, Countries and Quantities
A separate Division 7 Application is required for each product (including different dosage forms and strengths), each destination country and a maximum specified quantity of each pharmaceutical product to be exported under the Authorization. The quantity and country are part of the Application for authorization under section 21.04 of the Patent Act which is submitted to CIPO.
Subsequent Division 7 Applications may be filed while previous Division 7 Applications are under review or after their review is complete. The Division 7 Application may be filed before or after an Application for authorization under section 21.04 of the Patent Act has been filed with the Commissioner of Patents.
2.10.1 Additional products
Where Authorization under section 21.04 of the Patent Act is being sought for an additional product, a new Domestic Submission and Division 7 Application must both be filed and reviewed to ensure the new product complies with the FDR. If the Domestic Submission for the new product has already been reviewed, the Division 7 Application may reference this Submission. A new ETN will be issued and the Director General of TPD will notify the Commissioner of Patents (CIPO) and the manufacturer that the product is in compliance with the FDR.
2.10.2 New contract for the same product and country (ie. a different quantity)
A new Division 7 Application is required. The cover letter for the Division 7 Application should identify the Domestic Submission being referenced (including the Submission Number), the Submission Number(s) of the previous Division 7 Application(s) and their status, and should indicate if the information in the subsequent Division 7 Application is the same as the previous Application(s). Any differences should be clearly identified and identical content may be cross-referenced to previously approved Applications.
2.10.3 New contract for a quantity of the same product for a different country
A new Division 7 Application is required. The cover letter for the Division 7 Application should identify the Domestic Submission being referenced (including the Submission Number), the Submission Numbers of the previous Division 7 Application(s) and their status, and should indicate if the information in the subsequent Division 7 Application is the same as the previous Application(s). Any differences should be clearly identified and identical content may be cross-referenced to previously approved Applications.
2.11 Product Ceases to Meet the Requirements of the Food and Drugs Act and Regulations
2.11.1 Notice to the Commissioner of Patents
Where a drug authorized to be sold under the CAMR process is determined to no longer meet the requirements of the Food and Drugs Act and Regulations the Director General will issue a letter to the Commissioner of Patents (CIPO) and the manufacturer in the form of a "Notice to the Commissioner of Patents" indicating that the drug has ceased to meet the requirements of the Food and Drugs Act and Regulations.
Appendix A: Division 7 Application Certification
Division 7 Application Certification Form
We certify that, to the best of our knowledge and belief, with reference to the application pertaining to (Name of product) submitted by (Name of Manufacturer)
- All the information and material included in the application and solicited information are accurate and complete, and that the summary documents correctly represent the information and material referred to in the application. No information is false or misleading, no omissions have been made that may affect its accuracy and completeness.
- All aspects of the product represented in this application and intended for export under Canada's Access to Medicines Regime are identical to that described in the Domestic Submission for (Name of product), (Control Number, if known), submitted to TPD on (date), including labelling, sites and methods of manufacture, packaging and testing except for the following:
- for a solid dosage form, changes required to mark and colour the product in accordance with section C.07.008 of the Food and Drug Regulations;
- for a dosage form that is not solid, changes to the immediate container label in accordance with section C.07.008;
- for all products, changes to the labelling in accordance with section C.07.008.
- Any changes to the product once an Authorization under section 21.04 of the Patent Act has been received will be conducted in accordance with the Health Canada policy "Changes to Marketed New Drug Products". This would include the filing of the appropriate application (e.g., Supplement to a New Drug Submission, Notifiable Change) if applicable, and maintaining the appropriate supporting documentation.
Senior Executive Officer in Canada:
Date:
Senior Medical / Scientific Officer:
Date
Appendix B: Process and Criteria for Assessing Significant Difference in Colour
Pure colours which are a distinctly different hue or shade (e.g. red, orange, yellow, green, blue, violet, white, black, grey, brown) will be considered significantly different. Red will be considered significantly different from yellow and yellow significantly different from white, for example.
Where pure hues are tinted with other colours they may be significantly different if none of the colours being combined are the same. For example, red orange would be considered different than blue green however red orange may or may not be considered significantly different from yellow orange.
Different lightness or paleness of the same hue (eg. light blue versus dark blue or red versus pink) may or may not be considered significantly different.
In cases where it is not clear if a proposed solid product colour is acceptable, a Colour Review Panel will be convened to make this assessment. The basis for this decision will be what an average person could be reasonably expected to consider a significantly different colour.
The assessment of whether a solid dosage form submitted with a Division 7 Application is a significantly different colour from the colour of the version of the drug sold in Canada will be made at the start of the screening/review period since failure to meet this requirement will result in a Not Satisfactory decision for the Division 7 Application.
Appendix C: Diagram of CAMR Process with TPD Domestic Submission review completed and on patent hold when Division 7 Application filed:
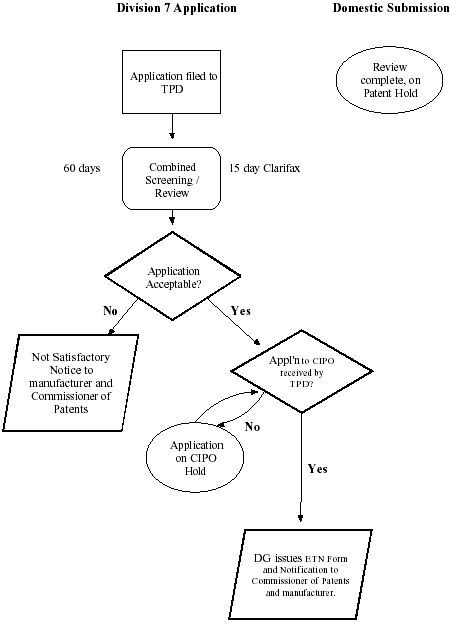
Domestic Submission review completed and on patent hold when Division 7 Application filed - Text Description
Division 7 Application:
Application Filed to Therapeutic Products Directorate (TPD). Combined Screening/review: 60 days, 15 day clarifax. Application acceptable? No, not satisfactory notice to manufacturer and Commissioner of Patents. Yes, Application to the Canadian Intellectual Property Office (CIPO) received by TPD? No, Application on CIPO hold. Yes, director general issues export tracking number (ETN) form and notification to Commissioner of Patents and manufacturer.
Domestic Submission:
review complete, on patent hold.
Domestic Submission filed at the same time as the Division 7 Application:
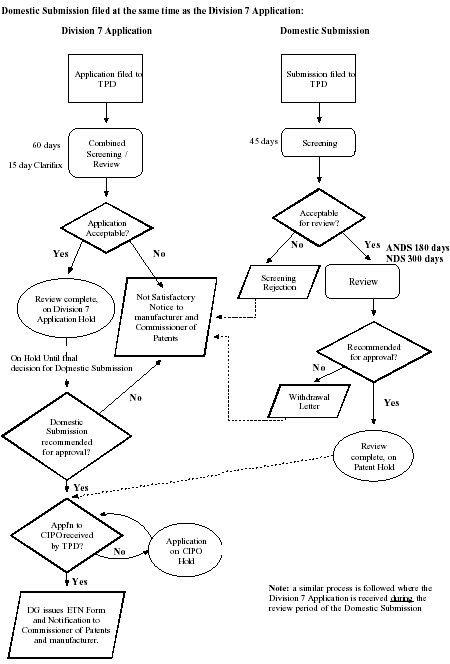
Footnotes
Domestic Submission filed at the same time as the Division 7 Application
Division 7 Application:
Application filed to Therapeutic Products Directorate (TPD). Combined screening/review: 60 days, 15 day clarifax. Application acceptable? No, not satisfactory notice to manufacturer and Commissioner of Patents. Application acceptable? No, not satisfactory notice to manufacturer and Commissioner of Patents. Yes, Review complete, on division application hold. On hold until final decision for domestic submission. Domestic submission recommended for approval? Yes, application to Canadian Intellectual Property Office (CIPO) received by TPD? Yes, director general issues export tracking number (ETN) form and notification to Commissioner of Patents and manufacturer. No, application on CIPO hold. Application to CIPO received by TPD.
Domestic Submission:
Submission filed to Therapeutic Products Directorate (TPD). Screening, 45 days. Acceptable for review? No, screening rejection, not satisfactory notice to manufacturer and Commissioner of Patents. Yes, review (abbreviated new drug submission 180 days, new drug submission 300 days). Recommended for approval? No, withdrawal letter. Yes, review complete, on patent hold. Yes, application to Canadian Intellectual Property Office (CIPO) received by TPD? Yes, director general issues export tracking number (ETN) form and notification to Commissioner of Patents and manufacturer. No, Application on CIPO hold. Application to CIPO received by TPD.
Note: a similar process is followed where the Division 7 application is received during the review period of the domestic submission.
- Footnote 1
-
Section 21 of the Patent Act
- Footnote 2
-
WTO members on 6 December 2005 approved changes to the intellectual property agreement making permanent a decision on patents and public health originally adopted in 2003. The decision directly transforms the 30 August 2003 "waiver" into a permanent amendment of the TRIPS Agreement.