
Guidance Document: Blood Regulations

Date Adopted: 2014-05-12
Effective Date: 2014-10-23
Modified Date: 2023-02-21
Foreword
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable guidance documents.
Table of Contents
- 1. Introduction
- 1.1 Purpose/Overview
- 1.2 Scope and Application
- 1.3 Policy objectives
- 1.4 Background
- 1.4.1 CSA Blood Standard
- 1.5 Definitions
- 2. Guidance for Implementation
- Section 1 Interpretation
- Sections 2-3 Application
- Sections 5-16 Authorizations
- Section 5 Authorization
- Section 6 Application for an authorization
- Section 7 Issuance
- Section 8 Refusal
- Section 9 Significant Changes
- Section 10 Emergency Changes
- Section 11 Administrative changes - notice
- Section 12 Other changes - annual report
- Section 13 New or amended terms and conditions
- Section 14 Suspension
- Section 15 Reinstatement
- Section 16 Cancellation
- Sections 17-29 Establishment Licences
- Section 17 Establishment licence required
- Section 18 Application for establishment licence
- Section 19 Inspection
- Section 20 Issuance
- Section 21 Refusal
- Section 22 Changes requiring application to amend licence
- Section 23 Administrative changes - notice
- Section 24 Changes requiring amendment of licence by Minister
- Section 25 New or amended terms and conditions
- Section 26 Additional information
- Section 27 Suspension
- Section 28 Reinstatement
- Section 29 Cancellation
- Sections 30-37 Registration
- Sections 38-58 Processing
- Sections 59-68 Labelling
- Section 59 Non-application - pre-assessed donors
- Section 60 Language requirement
- Section 61 General requirements
- Section 62 Circular of information
- Section 63 Donation code
- Section 64 Contents of label
- Section 65 Aliquots
- Section 66 Designated donations
- Section 67 Directed donations
- Section 68 Label verification
- Sections 69-72 Storage
- Sections 73-76 Distribution
- Sections 77-80 Transformation
- Sections 81-85 Exceptional Distribution
- Sections 86-91 Pre-Assessed Donor Programs
- Section 92 Importation in Urgent Circumstances
- Sections 93-123 Quality Management
- Sections 93-94 Quality Management System
- Sections 95-97 Operating Procedures
- Sections 98-102 Personnel, facilities, equipment and supplies
- Sections 103-108 Error and Accident Investigation and Reporting
- Sections 109-116 Adverse Reaction Investigation and Reporting
- Sections 117-123 Records
- Section 124 Powers of Inspectors
- Section 125 Consequential Amendment
- Sections 126-128 Transitional Provisions
- Section 129 Coming into force
- Appendix A: Glossary
- Appendix B: Summary Table of Annual Reporting Requirements for Blood Establishments
- Appendix C: Pre-Registration Self-Assessment Tool for Establishments applying for a Blood Establishment Registration
- Appendix D: Repealed Food and Drug Regulations C.04.400-C.04.423 Human Plasma Collected by Plasmapheresis
- Appendix E: Health Canada Guidance Documents and Directives superseded by the Guidance Document: Blood Regulations
Introduction
1.1 Purpose/Overview
The Blood Regulations are intended to promote the protection of the safety of Canadian blood donors and recipients in connection with the safety of blood for transfusion or for further manufacture into a drug for human use. See section 1, the Interpretation section of this guidance document, for the definition of safety.
The Blood Regulations contain requirements for human safety and the safety of blood with respect to the following activities related to human blood and blood components for transfusion: processing (donor suitability assessment, collection, testing, and blood component preparation); transforming (washing, pooling and irradiating); labelling; storing; record keeping; importing; distributing; and error, accident and adverse reaction investigation and reporting.
The Blood Regulations contain requirements for human safety and the safety of blood with respect to the following activities related to human blood and blood components for further manufacture: processing (donor suitability assessment, collection, testing, and blood component preparation); labelling; storing; record keeping; distributing; adverse donor reaction investigation and reporting; and error and accident investigation and reporting.
The Blood Regulations are administered by the Health Products and Food Branch, Health Canada. Any questions concerning the Blood Regulations or this guidance document can be sent to brddopic-bpcidmbr@hc-sc.gc.ca.
1.2 Scope and application
The Blood Regulations only apply to human blood that is collected for transfusion or for further manufacture into a drug for human use. Manufacturing of drug products using blood or blood components is outside the scope of the Blood Regulations and is regulated under the Food and Drug Regulations. Blood product fabricators are referred to in this guidance in respect of the chain of distribution and for blood safety communication purposes. See 1.5 Definitions, blood product fabricator.
The Blood Regulations fall under the authority of the Food and Drugs Act and apply to all persons or establishments that process, label, store, distribute or transform blood for transfusion or for further manufacture, including establishments that import blood for transfusion. The Food and Drugs Act and the current version of the National Standard of Canada, CAN/CSA Z902, Blood and blood components (CSA Blood Standard), published by the CSA Group, should be read in conjunction with the Blood Regulations.
It is the responsibility of the establishment to ensure that they follow the requirements of the most recent version of the Blood Regulations and the clauses of the CSA Blood Standard incorporated by reference into the Blood Regulations. The CSA Blood Standard clauses incorporated by reference into the Blood Regulations are regulatory requirements that must be met, while CSA Blood Standard clauses that are only referred to in this guidance document are recommended best practices. In the case of a discrepancy between the CSA Blood Standard that is not incorporated into the Blood Regulations and a requirement in the Blood Regulations, the regulatory requirements take precedence as they are the legislative rules enacted by the Governor in Council.
This guidance document replaces some of Health Canada's blood regulatory guidance documents (see Appendix E). This guidance document should be read in parallel with the Blood Regulations. In the event of any perceived inconsistency or conflict, the Blood Regulations take precedence over this guidance document.
In this guidance document, "must" is used to express a requirement, i.e. a provision of the Blood Regulations that the establishment is obliged to satisfy in order to comply with the regulatory requirements; "should" is used to express a recommendation or that which is advised but not required; and "may" is used to express an option or that which is permissible within the limits of the guidance document.
Where this guidance document indicates number of days for notification or further action required by an establishment or Health Canada, unless it is otherwise specified, the days are counted as calendar days.
1.3 Policy objectives
Under the Food and Drugs Act, the Blood Regulations introduce specific regulations for blood and its components intended for transfusion or for further manufacture into drugs for human use. This guidance document interprets the requirements of the Blood Regulations to provide necessary information for establishments that process, label, distribute, transform, or store blood and blood components for transfusion or for further manufacture, and establishments that import blood and/or blood components for transfusion, to comply with the requirements of the Blood Regulations.
1.4 Background
The Blood Regulations were developed to
- complete Health Canada's response to the Krever Commission recommendations;
- add specific safety requirements for whole blood and its components to the federal regulations;
- consolidate and clarify the existing regulations for blood safety that are contained in various divisions of the Food and Drug Regulations into standalone regulations specific to blood safety;
- address the specific needs of blood as a unique therapeutic product rather than applying general drug regulations to blood; and
- deal with fast changing technologies, emerging diseases, and blood shortages in urgent circumstances.
Establishments are regulated, under the Blood Regulations, based on the degree of risk that their activity poses to the safety of Canada's blood for transfusion or for further manufacture.
An establishment must apply to Health Canada for an Authorization and an Establishment Licence if it intends to conduct processing activities described under the Blood Regulations with respect to human allogeneic blood and blood components for transfusion, including plasma for further manufacture. The preparation of the circular of information of allogeneic blood or blood components for transfusion and the labelling of allogeneic units of blood prior to distribution must be conducted in accordance with an Authorization. Blood and/or blood components that are imported for transfusion must be associated with an Authorization and the importing establishment must have an Establishment Licence.
The requirements of the Food and Drug Regulations C.04.400-C.04.423 Human Plasma Collected by Plasmapheresis are provided in Appendix D for reference only. These requirements were the baseline of the authorized criteria for licensed establishments previously held to these requirements prior to the repeal of these sections of the Food and Drug Regulations.
An establishment must register with Health Canada if they collect autologous blood, have a Pre-Assessed Donor Program, or transform blood.
All establishments that store and transfuse blood and/or blood components need to meet specific requirements described in the Blood Regulations. Note: Labelling, after the blood and/or blood components are determined safe for distribution, is an activity that applies to establishments that transform or transfuse blood or blood components.
Some sections of the Blood Regulations reference specific clauses in the CSA Blood Standard that are within Health Canada's scope of authority. When a specific section, clause or table in the CSA Blood Standard is incorporated by reference into these regulations, it becomes a mandatory regulatory requirement. The CSA Blood Standard, as amended from time-to-time, is incorporated in this way. Clauses or tables in the CSA Blood Standard, not referenced in the Blood Regulations, remain voluntary.
1.4.1 CSA Blood Standard
The CSA Blood Standard covers the lifecycle of blood for transfusion and is widely regarded as industry best practices.
The CSA Blood Standard was developed and is amended through a consensus-driven process by a technical committee of experts in the field of blood safety, user groups, and federal and provincial and territorial governments. The CSA undertakes consultations on revisions to the CSA Blood Standard as part of their standard development process.
All establishments require access to the current version of the CSA Blood Standard, since some provisions of the Blood Regulations are standards-based. The CSA Blood Standard is available by ordering it through the Canadian Standards Association website (www.shopcsa.ca/onlinestore/welcome.asp) or by calling 1-800-463-6727. Information on how to receive updates or amendments to the standard is available on the "CSA Standards Update Service" page of the CSA Blood Standard.
The CSA Blood Standard is also available for view access by registering to the CSA's community website: https://community.csagroup.org/community/health-care-safety-and-accessibility/blood-and-transplants-standards-view-access.
All stakeholders play a key role in keeping the CSA Blood Standard up-to-date. The CSA Blood Standard contains a Proposal for Change Form that stakeholders may use to submit proposals for change directly to the CSA. The CSA recommends that stakeholders supply the following information, in addition to the appropriate contact information, to facilitate the evaluation of the proposed changes:
- standard/publication number;
- relevant Clause, Table, and /or Figure number(s);
- wording of the proposed change; and
- rationale for the change.
References to the CSA Blood Standard in the Blood Regulations are ambulatory, i.e. as amended from time-to-time. Health Canada will review any changes to clauses of the CSA Blood Standard, referenced in the Blood Regulations, with respect to risk and the potential impact on the safety of blood and its components.
1.5 Definitions
The additional definitions provided below are to assist in the interpretation of this guidance document.
"apheresis" means the process of withdrawing blood from a donor, separating specific components from the blood, and returning some or all of the remaining blood components to the donor.
"blood product fabricator" refers to the manufacturer of blood products from plasma for further manufacture. Innovation could bring about new blood products, so this term is not restricted to plasma within this guidance document.
"Clarifax" is a communication tool used to request information or to request clarification of information already filed.
"ISBT 128" is an international information standard for use in the labelling of blood for transfusion, blood components intended for use in the manufacture of a drug for human use, and products intended for transplantation that is managed and promoted by the International Council for Commonality in Blood Banking Automation (ICCBBA).
"lookback" is the process of identifying
- previous donations (and related blood components) from a donor who, on subsequent testing, is confirmed positive for a transfusion-transmissible infectious agent; and
- recipients who received blood components from a donor who is confirmed positive for a transfusion-transmissible infectious agent
The Medical Devices Directorate of the Health Products and Food Branch of Health Canada is the Canadian federal regulator responsible for licensing medical devices in accordance with the Food and Drugs Act and the Medical Devices Regulations. The Medical Devices Active Licence Listing (MDALL) is a database containing all licensed Class II, III and IV Medical Devices for sale in Canada. It can be found on the Health Canada website at https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices/licences/medical-devices-active-licence-listing.html.
"novel blood component" means a blood component that is not routinely processed or transfused in Canada. A novel blood component either provides a production benefit or is equivalent or superior to a reference product or fulfills an unmet clinical need.
"opportunity to be heard" means that an establishment can respond in writing to Health Canada in response to an action taken by Health Canada regarding the establishment's Authorization, Establishment Licence or Registration. In some cases, a face-to-face, virtual meeting or teleconference may occur.
"physician" means a person who is entitled to practice the profession of medicine under the laws of the province or territory in which the person provides medical service.
"physician substitute" means a person who
- acts under the general supervision and direction of a physician; and
- is authorized to provide the services that may be provided by a physician according to the applicable laws of the province or territory in which the person provides any of those services.
"pooling" includes mixing.
"quarantine" prevents suspected or confirmed non-conforming units of blood from being used for transfusion, further manufacture or distribution.
The term "senior executive officer" refers to an individual holding a position that has an assigned level of responsibility for activities the establishment conducts under the Blood Regulations. The term senior executive officer refers to a function within the establishment and is not necessarily a specific position title.
"traceback" is the process of investigating a report of a suspected transfusion-associated infection in order to identify a potential implicated donor. The purpose of the traceback investigation is to
- determine whether any donor who contributed to the transfusion is infected with, or positive for, serologic markers of the implicated infectious agent;
- trigger a recall of in date blood components contributed by that donor; or
- notify consignees and recipients of components collected from that donor.
2. Guidance for Implementation
Chart 1. The application of the Blood Regulations to different types of establishments
The purpose of this chart is to identify the sections of the regulations that apply to establishments who must hold an Authorization, Establishment Licence and/or a Registration because of the activities they conduct. See the Authorization (5-16), Establishment Licences (17-29) and Registration (30-37) sections of this guidance to learn more about the level of regulatory oversight required for the types of activities that your establishment conducts.
- A
- Establishment holding an Authorization
- EL
- Licensed Establishment
- R-Auto
- Registered Establishment that conducts autologous activities
- R-PADP
- Registered Establishment that has a Pre-Assessed Donor Program
- R-TWPI
- Registered Establishment that conducts transformation activities (washing, pooling, irradiating)
| Blood Regulations Section | Section Title | A/EL | R-Auto | R-PADP | R-TWPI |
|---|---|---|---|---|---|
| 1 | Interpretation | Yes | Yes | Yes | Yes |
| Application | |||||
| 2 | Scope of Regulations | Yes | Yes | Yes | Yes |
| 3 | Non-application | Yes | Yes | Yes | Yes |
| 4 | Prohibitions | Yes | Yes | Yes | Yes |
| Authorizations | |||||
| 5 | Authorization | Yes | n/a | n/a | n/a |
| 6 | Application for authorization | Yes | n/a | n/a | n/a |
| 7 | Issuance | Yes | n/a | n/a | n/a |
| 8 | Refusal | Yes | n/a | n/a | n/a |
| 9 | Significant changes | Yes | n/a | n/a | n/a |
| 10 | Emergency changes | Yes | n/a | n/a | n/a |
| 11 | Administrative changes - notice | Yes | n/a | n/a | n/a |
| 12 | Other changes - annual report | Yes | n/a | n/a | n/a |
| 13 | New or amended terms and conditions | Yes | n/a | n/a | n/a |
| 14 | Suspension | Yes | n/a | n/a | n/a |
| 15 | Reinstatement | Yes | n/a | n/a | n/a |
| 16 | Cancellation | Yes | n/a | n/a | n/a |
| Establishment Licences | |||||
| 17(1) | Establishment licence required | Yes | n/a | n/a | n/a |
| 17(2) | Test Labs | Yes (EL only for PADP) | n/a | n/a | n/a |
| 18 | Application for establishment licence | Yes | n/a | n/a | n/a |
| 19 | (1) Inspection |
Yes | n/a | n/a | n/a |
| 20 | Issuance | Yes | n/a | n/a | n/a |
| 21 | Refusal | Yes | n/a | n/a | n/a |
| 22 | Changes requiring application to amend licence | Yes | n/a | n/a | n/a |
| 23 | Administrative changes - notice | Yes | n/a | n/a | n/a |
| 24 | Changes requiring amendment of licence by Minister | Yes | n/a | n/a | n/a |
| 25 | New or amended terms and conditions | Yes | n/a | n/a | n/a |
| 26 | Additional information | Yes | n/a | n/a | n/a |
| 27 | Suspension | Yes | n/a | n/a | n/a |
| 28 | Reinstatement | Yes | n/a | n/a | n/a |
| 29 | Cancellation | Yes | n/a | n/a | n/a |
| Registration | |||||
| 30 | Requirement to register | n/a | Yes | Yes | Yes |
| 31 | Application for registration | n/a | Yes | Yes | Yes |
| 32 | Registration | n/a | Yes | Yes | Yes |
| 33 | Changes - notice | n/a | Yes | Yes | Yes |
| 34 | Amendment by Minister | n/a | Yes | Yes | Yes |
| 35 | Annual statement of compliance | n/a | Yes | Yes | Yes |
| 36 | Additional information | n/a | Yes | Yes | Yes |
| 37 | Cancellation | n/a | Yes | Yes | Yes |
| Processing - Donor Suitability Assessment | |||||
| 38 | Non-application - autologous donations | n/a | Yes | n/a | n/a |
| 39 | Licensed establishments | Yes | n/a | n/a | n/a |
| 40 | Past unsuitability | Yes | n/a | Yes | n/a |
| 41 | Donor screening | Yes | n/a | Yes | n/a |
| 42 | Exclusion criteria | Yes | n/a | Yes | n/a |
| 43 | When donor determined unsuitable | Yes | n/a | Yes | n/a |
| 44 | (1)When donor determined suitable |
Yes | n/a | Yes | n/a |
| Processing - Collection | |||||
| 45 | Licensed establishments | Yes | n/a | n/a | n/a |
| 46 | Donor identification code | n/a | Yes | n/a | n/a |
| 47 | Donation code | Yes | Yes | n/a | n/a |
| 48 | Labelling of containers | Yes | Yes | n/a | n/a |
| 49 | Collection procedures | Yes | Yes | Yes | n/a |
| 50 | Samples | Yes | Yes | Yes | n/a |
| 51 | Autologous donations | n/a | Yes | n/a | n/a |
| Processing - Testing | |||||
| 52 | Authorization | Yes | n/a | n/a | n/a |
| 53 | Autologous donations -transmissible disease testing | n/a | Yes | n/a | n/a |
| 54 | Autologous donations - ABO and Rh | n/a | Yes | n/a | n/a |
| 55 | Medical devices | n/a | Yes | See also S.17(2) | n/a |
| 56(1) | Test results - allogeneic blood | Yes | n/a | n/a | n/a |
| 56(2) | Test results - autologous blood | n/a | Yes | n/a | n/a |
| Blood Component Preparation | |||||
| 57 | Licensed establishments | Yes | n/a | n/a | n/a |
| 58 | Registered establishments | n/a | Yes | n/a | n/a |
| Labelling | |||||
| 59 | Non-application - pre-assessed donors | n/a | n/a | Yes | n/a |
| 60 | Language requirement | Yes | Yes | n/a | Yes |
| 61 | General requirements | Yes | Yes | n/a | Yes |
| 62 | Circular of information | Yes | n/a | n/a | n/a |
| 63 | Donation code | Yes | Yes | n/a | n/a |
| 64(1) | Contents of label - blood for transfusion | Yes | Yes | n/a | n/a |
| 64(2) | Contents of label - Autologous blood | n/a | Yes | n/a | n/a |
| 64(3) | Contents of label - blood for use in manufacture of drug for human use | Yes | n/a | n/a | n/a |
| 65 | Aliquots | Yes | Yes | Yes | Yes |
| 66(1) | Designated donations | Yes | n/a | n/a | n/a |
| 66(2) | Change of use | Yes | n/a | n/a | n/a |
| 67 | Directed donations | Yes | n/a | n/a | n/a |
| 68 | Label verification | Yes | Yes | n/a | Yes |
| Storage | |||||
| 69(1)(a) | Criteria - collecting establishment, licensed | Yes | n/a | n/a | n/a |
| 69(1)(b) | Criteria - collecting establishment, registered | n/a | Yes | Yes | n/a |
| 69(2) | Criteria - receiving establishment | Yes | Yes | n/a | Yes |
| 70 | Storage location | Yes | Yes | Yes | Yes |
| 71 | Segregation - autologous, designated and directed donations | Yes | Yes | n/a | Yes |
| 72 | Segregation - untested or positive or reactive test results | Yes | Yes | Yes | Yes |
| Distribution | |||||
| 73(1) | Determination of safety - allogeneic blood | Yes | n/a | Yes | n/a |
| 73(2) | Determination of safety - autologous blood | n/a | Yes | n/a | n/a |
| 74 | (1) Verification |
Yes | Yes | Yes | Yes |
| 75 | Shipping containers | Yes | Yes | Yes | Yes |
| 76 | Storage during transportation | Yes | Yes | Yes | Yes |
| Transformation | |||||
| 77 | Transformation methods | n/a | n/a | n/a | Yes |
| 78 | Washing | n/a | n/a | n/a | Yes |
| 79 | Pooling | n/a | n/a | n/a | Yes |
| 80 | Irradiation | n/a | n/a | n/a | Yes |
| Exceptional Distribution | |||||
| 81 | Conditions | Yes | Yes | Yes | Yes |
| 82 | (1) Notice of exceptional distribution |
Yes | n/a | n/a | n/a |
(3) Notice to be forwarded |
n/a | Yes | Yes | Yes | |
| 83 | Labelling | Yes | n/a | n/a | n/a |
| 84(1) | Follow-up | Yes | n/a | n/a | n/a |
| 84(2) | Results to be forwarded | n/a | Yes | Yes | Yes |
| 85 | When blood is not transfused | n/a | Yes | Yes | Yes |
| Pre-Assessed Donor Programs | |||||
| 86 | Program characteristics | n/a | n/a | Yes | n/a |
| 87 | Donor identification code | n/a | n/a | Yes | n/a |
| 88 | (1) Regular donor assessment and testing |
n/a | n/a | Yes | n/a |
| 89 | At each collection | n/a | n/a | Yes | n/a |
| 90 | Labelling | n/a | n/a | Yes | n/a |
| 91 | When blood not transfused | n/a | n/a | Yes | n/a |
| Importation in Urgent Circumstances | |||||
| 92 | Importation in urgent circumstances | Yes | n/a | n/a | n/a |
| Quality Management System | |||||
| 93 | (1) Organizational structure |
Yes | Yes | Yes | Yes |
| 94 | Requirements | Yes | Yes | Yes | Yes |
| Operating Procedures | |||||
| 95 | Operating procedures required | Yes | Yes | Yes | Yes |
| 96 | Requirements | Yes | Yes | Yes | Yes |
| 97 | Documented evidence | Yes | Yes | Yes | Yes |
| Personnel, Facilities, Equipment and Supplies | |||||
| 98 | (1) Personnel |
Yes | Yes | Yes | Yes |
| 99 | Facilities | Yes | Yes | Yes | Yes |
| 100 | Equipment | Yes | Yes | Yes | Yes |
| 101 | Storage equipment | Yes | Yes | Yes | Yes |
| 102 | Supplies | Yes | Yes | Yes | Yes |
| Error and Accident Investigation and Reporting | |||||
| 103 | Error or accident of another establishment | Yes | Yes | Yes | Yes |
| 104 | Establishment's own error or accident | Yes | Yes | Yes | Yes |
| 105 | Requirement to cooperate | Yes | Yes | Yes | Yes |
| 106 | Investigation results | Yes | Yes | Yes | Yes |
| 107 | Reports to Minister | Yes | Yes | Yes | Yes |
| 108 | Annual Report | Yes | Yes | Yes | Yes |
| Adverse Reaction Investigation and Reporting | |||||
| Adverse Donor Reactions | |||||
| 109 | Adverse Donor Reactions - Notice to Minister | Yes | Yes | Yes | n/a |
| Adverse Recipient Reactions | |||||
| 110 | Required Action | Yes | Yes | Yes | Yes |
| 111 | Autologous Donations | n/a | Yes | n/a | n/a |
| 112 | Requirement to cooperate | Yes | Yes | Yes | Yes |
| 113 | Notice to Minister | Yes | Yes | Yes | Yes |
| 114 | (1) Results of investigation |
Yes | Yes | Yes | Yes |
| 115 | Final report to Minister | Yes | Yes | Yes | Yes |
| 116 | Annual Report | Yes | Yes | Yes | Yes |
| Records | |||||
| 117 | Record quality | Yes | Yes | Yes | Yes |
| 118 | Donation code part of all records | Yes | Yes | Yes | Yes |
| 119 | (1) Retention periods - allogeneic blood |
Yes | n/a | Yes | n/a |
| 120 | (1) Retention periods - autologous blood |
n/a | Yes | n/a | n/a |
| 121 | (1) Retention periods - transformation |
n/a | n/a | n/a | Yes |
| 122 | (1) Retention periods — transfusion |
n/a | Yes | Yes | Yes |
| 123 | Storage of records | Yes | Yes | Yes | Yes |
| 124 | Powers of Inspectors | Yes | Yes | Yes | Yes |
| 125 | Consequential Amendment | Yes | n/a | n/a | n/a |
| Transitional Provisions | |||||
| 126 | Deemed authorization | Yes | n/a | n/a | n/a |
| 127 | Licence continued | Yes | n/a | n/a | n/a |
| 128 | (1) Delayed registration |
n/a | Yes | Yes | Yes |
| Coming Into Force | |||||
| 129(1) | One year after publication | Yes | Yes | Yes | Yes |
| 129(2) | Subsections 4(4) to (6) and paragraph 64(1)(b) | n/a | Yes | Yes | Yes |
| Note: “Blood” includes whole blood and blood components. | |||||
Chart 2. Application of the Blood Regulations to establishments that are not required to obtain an Authorization, an Establishment Licence or a Registration
Some establishments do not conduct activities for which an Authorization, an Establishment Licence or a Registration is required. However, these establishments must still meet the applicable sections of the Blood Regulations for the activities they conduct. These sections are identified in the chart below.
| Blood Regulations Section | Section Title |
|---|---|
| 1 | Interpretation |
| Application | |
| 2 | Scope of Regulations |
| 3 | Non-application |
| 4 | Prohibitions |
| Labelling | |
| 60 | Language requirement |
| 61 | General requirements |
| 65 | Aliquots |
| 68 | Label verification |
| Storage | |
| 69(2) | Criteria - receiving establishment |
| 70 | Storage location |
| 71 | Segregation - autologous, designated and directed donations |
| 72 | Segregation - untested or positive or reactive test results |
| Distribution | |
| 74 | (1) Verification |
| 75 | Shipping containers |
| 76 | Storage during transportation |
| Transformation | |
| 77 | Transformation methods *applies to establishments that only pool cryoprecipitate |
| 79 | Pooling *applies to establishments that only pool cryoprecipitate |
| Exceptional Distribution | |
| 81 | Conditions |
| 82 | (1) Notice to be forwarded |
| 84(2) | Results to be forwarded |
| 85 | When blood not transfused |
| Operating Procedures | |
| 95 | Operating procedures required |
| 96 | Requirements |
| 97 | Documented evidence *applies to establishments that only pool cryoprecipitate |
| Personnel, Facilities, Equipment and Supplies | |
| 98 |
|
| 101 | Storage equipment |
| Error and Accident Investigation and Reporting | |
| 103 | Error or accident of another establishment |
| 104 | Establishment's own error or accident |
| 105 | Requirement to cooperate |
| 106 | Investigation results |
| 107 | Reports to Minister |
| 108 | Annual Report |
| Adverse Reaction Investigation and Reporting | |
| Adverse Recipient Reactions | |
| 110 | Required Action |
| 112 | Requirement to cooperate |
| 113 | Notice to Minister |
| 114 | (1) Results of investigation |
| 115 | Final report to Minister |
| 116 | Annual Report |
| Records | |
| 117 | Record quality |
| 118 | Donation code part of all records |
| 121 | Retention Periods - transformation *applies to establishments that only pool cryoprecipitate |
| 122 | (1) Retention periods - transfusion |
| 123 | Storage of records |
| 124 | Powers of Inspectors |
| Coming Into Force | |
| 129(1) | One year after publication |
Section 1 Interpretation
The statements quoted below (enclosed in the boxes) are sections taken directly from the Blood Regulations.
Definitions
1. The following definitions apply in these Regulations.
"accident"
« accident »
"accident" means an unexpected event that is not attributable to a deviation from the operating procedures or applicable laws and that could compromise human safety or the safety of blood.
"Act"
« Loi »"Act" means the Food and Drugs Act.
"adverse reaction"
« effet indésirable »
"adverse reaction" means an undesirable response that is associated with
(a) in the case of a donor, the collection of blood; and
(b) in the case of a recipient, the safety of the transfused blood.
"allogeneic"
« allogénique »
"allogeneic", in respect of blood or a blood donation, means that the blood is collected from an individual either for transfusion into another individual or for use in the manufacture of a drug for human use.
The collection of allogeneic blood for distribution into the general blood supply either for transfusion or for use in the manufacture of a drug for human use requires an Authorization and an Establishment Licence.
The collection of allogeneic blood from a pre-assessed donor for an emergency transfusion to a specific patient requires a Registration, while the testing of allogeneic blood from a pre-assessed donor requires an Establishment Licence.
"authorization"
« homologation »
"authorization", in respect of any blood or process, means an authorization that is issued under section 7.
"autologous"
« autologue »
"autologous", in respect of blood or a blood donation, means that the blood is collected from an individual for transfusion into the same individual at a later time.
Autologous blood must only be used for transfusion to the same individual from whom it was collected.
Autologous blood that is collected for manufacture into a drug that is subject to the Food and Drug Regulations is not subject to the Blood Regulations. For example collection of the starting material for CAR-T cells and Expanded T cells are outside the scope of the Blood Regulations.
The scope of autologous blood collection, under the Blood Regulations, excludes the following because this type of blood is not considered to be collected for use at a later time:
- peri-operative blood that is collected and remains in the clinical patient care area, for example:
- collection just prior to surgery (e.g., acute normovolemic hemodilution);
- collection throughout surgery from the surgical site or an extracorporeal circuit (intraoperative); or
- collection following surgery or trauma from body cavities, joint spaces, and other closed surgical or trauma sites (post-operative).
- blood that is collected to be radio-labelled for diagnostic purposes.
See section 71 for storage segregation requirements.
"blood"
« sang »
"blood" means human blood that is collected either for transfusion or for use in the manufacture of a drug for human use, and for greater certainty, it includes whole blood and blood components.
The scope of blood, under the Blood Regulations, excludes blood products or blood derivatives.
Examples of blood components include red blood cells, plasma, platelets, and granulocytes. Blood components do not include products manufactured from plasma for further manufacture.
See section 2 of this guidance for the scope of application of the Blood Regulations.
"circular of information"
« document d'information »
"circular of information" means a document that describes all of the following in relation to blood:
(a) the composition and properties of the blood;
(b) directions for storage and for use; and
(c) indications for use, contraindications, warnings and a list of possible adverse reactions.
"critical"
« essentiel »
"critical", in respect of equipment, supplies and services, means that the equipment, supply or service could, if it does not meet its specifications, compromise human safety or the safety of blood.
The term critical applies to equipment, supplies and services used in any activities that are regulated under the Blood Regulations. Examples of critical equipment, supplies and services include, but are not limited to, those that are used in the collection of blood, the testing of blood, blood component preparation, storage, and transformation. The following examples are provided for your guidance and are not exhaustive.
Note: “Blood” includes whole blood and blood components.
Examples of critical equipment
- apheresis equipment;
- automated blood extractors/presses;
- automated blood processors;
- automated blood testing systems and/or transmissible disease test equipment;
- automated dockers/sealers;
- manual sealers;
- automated microbial detection systems;
- irradiator
- pathogen reduction device
- cell counters or hematology analyzers used in blood or blood component assessment;
- cell washers/deglycerolization;
- centrifuge used for the processing of blood component units or to prepare samples for testing;
- electrophoresis devices;
- fast freezers;
- freezers used to store blood (units or samples);
- nucleic acid testing (NAT) instruments, including extractors or pipettors;
- platelet agitators and incubators;
- refrigerators;
- thermometers and temperature probes (any type);
- balances to weigh blood;
- back-up generators;
- temperature-controlled vehicles to transport blood;
- blood thawing devices;
- containers used to store blood, including during transport; and
- Controlled temperature conditioning equipment (e.g. refrigerators/freezers/incubators used to store phase change material used during transportation of blood)
Critical equipment also includes critical software.
Examples of critical software
- software for transferring data between automated devices; and
- software that analyzes data regarding the suitability of blood for transfusion or for further manufacture.
Examples of critical supplies
- blood group or phenotype testing reagents;
- irradiation indicator labels;
- collection sets (bags and tubing);
- filters;
- labels;
- reagents for transmissible disease test kits; and
- transfer bags.
Examples of critical services
- calibration and maintenance of critical equipment;
- validation/qualification of critical equipment;
- laboratory testing;
- quality control;
- quality management;
- testing services; and
- training on critical equipment by vendor.
Examples of non-critical equipment or supplies
- scales to weigh donors;
- blood bag shaker;
- cell washers of the centrifuge type not used for deglycerolization;
- centrifuges not used for component separation or preparation of blood samples for testing;
- circulating bath;
- haemoglobinometers;
- incubators (except platelet agitator/incubator);
- manual extractors for blood component preparation;
- microhaematocrit centrifuges;
- pipettes (except nucleic acid testing pipettors);
- thermosealers for making blood tubing segments for blood sampling;
- timers (any type); and
- weights (any type).
"designated donation"
« don désigné »
“designated donation” means a blood donation that is made by a donor who is selected for medical reasons to make the donation for a specific recipient.
"directed donation"
« don dirigé »
“directed donation” means a blood donation that is made by a donor who is known by the recipient and selected for medical reasons by the recipient’s physician.
"distribute"
« distribution »
"distribute" does not include to transfuse.
Many requirements throughout the Blood Regulations are associated with the distribution of blood and blood components. This guidance document further explains the activity of distribution in various instances. Also refer to section 4, Prohibitions; section 73, Determination of safety; sections 81–85, Exceptional Distribution; section 92, Importation in Urgent Circumstances.
The following are some examples of distribution of blood and blood components under the Blood Regulations. In each of these instances, when blood and/or blood components are to be distributed to another establishment or to the operating theatre or the ward, the establishment must perform the additional verification steps required by section 74 of the Blood Regulations.
Example 1, Distribution of allogeneic blood for transfusion
Allogeneic blood is processed by a licensed establishment for transfusion. After determining the blood is safe for transfusion, it is placed into inventory. Distribution occurs when the establishment sends the blood or blood components to another establishment. Distribution also occurs when the transfusion medicine laboratory sends the blood or blood components to the operation theatre or the ward regardless if it is already matched to a patient or not.
Distribution can also occur if Hospital A receives a request for allogeneic units of blood or blood components for transfusion from Hospital B. Distribution occurs when Hospital A sends the blood or blood components to Hospital B.
Example 2, Distribution of autologous blood
Autologous blood is processed for transfusion by a registered establishment. Before the autologous units of blood can be distributed, they must be determined safe for autologous transfusion. Distribution occurs when the establishment sends the autologous unit(s) of blood to the hospital.
Distribution can also occur within the same establishment if a registered establishment processes autologous blood for transfusion at the same establishment where it will be transfused. Before the autologous units of blood can be distributed, they must be determined safe for autologous transfusion. Distribution occurs when the transfusion medicine laboratory sends the blood or blood components to the operation theatre or the ward.
If an establishment sends blood from a mobile clinic to the processing facility, distribution has not taken place because the blood has not yet been declared safe for distribution for transfusion or for further manufacture.
Example 3, Distribution of blood for further manufacture
Blood (plasma) is processed by a licensed establishment for the purpose of manufacturing into a drug for human use. After determining the blood is safe for distribution for further manufacture, it may be stored at the collection site. Distribution occurs when the establishment sends the blood and/or blood components to the blood product fabricator.
Example 4, Distribution of Red Blood Cells for immunization (iRBCs)
Blood is processed by a licensed establishment for the purpose of immunization. Before the iRBCs are placed into inventory they must be determined safe for distribution. Distribution occurs from the location where the inventory is stored to the location where the immunization of the donor occurs. This can be within the same establishment.
"donation code"
« code d'identification du don »m
“donation code” means the unique group of numbers, letters or symbols, or combination of any of them, that an establishment assigns to a unit of blood at the time of collection.
"donor identification code"
« code d'identification du donneur »
“donor identification code” means the unique group of numbers, letters or symbols, or combination of any of them, that an establishment assigns to a donor.
"donor suitability assessment"
« évaluation de l'admissibilité du donneur »
"donor suitability assessment" means an evaluation of a donor that is based on all of the following criteria:
(a) the donor's medical history;
(b) the results of any donor tests and physical examination; and
(c) the donor's social history, to the extent that it is relevant in determining the presence of risk factors for diseases transmissible by blood.
- A donor's medical history refers to:
- conditions that could pose a risk to the donor; and
- vaccinations, medications and transmissible diseases that could pose a risk to the recipient.
- A physical examination is one of the methods of qualifying a donor as acceptable for donating blood and is based on the establishment's authorized criteria; and
- A donor's social history refers to the prior activities of a donor that could put the donor and recipient(s) at risk for infection with transmissible disease(s).
"error"
« manquement »
“error” means a deviation from the operating procedures or applicable laws that could compromise human safety or the safety of blood.
"establishment"
« établissement »
"establishment" means a person that conducts any of the following activities in respect of blood:
(a) importation;
(b) processing;
(c) distribution;
(d) transformation; or
(e) transfusion.
(e) transfusion
Although blood transfusion itself is not regulated under the Blood Regulations, establishments that transfuse blood and blood components must meet the requirements of the Blood Regulations that apply to the activities that they conduct, such as storing blood and blood components.
"human safety"
« sécurité humaine »
"human safety" means the safety of donors and recipients of blood, in so far as it relates to the safety of the blood.
Whenever the Blood Regulations or this guidance document refer to human safety, this means the safety of blood donors, or the safety of recipients of blood as long as human safety is associated with the safety of the blood processed and distributed under these Blood Regulations.
"medical director"
« directeur médical »
“medical director”, in respect of an establishment, means a physician who is entitled under the laws of a province to practise the profession of medicine and who is responsible for all medical procedures carried out by the establishment and for the application of the operating procedures that relate to them.
"operating procedures"
« procedures opérationnelles »
“operating procedures”, in respect of an establishment, means the component of the establishment’s quality management system that is composed of instructions that set out the processes to follow in conducting its activities.
"pre-assessed donor"
« donneur pré-évalué »
“pre-assessed donor” means a donor who has been accepted into a pre-assessed donor program described in sections 86 to 91 from whom blood is taken in an emergency to be transfused before completion of the testing.
The term pre-assessed donor is used in the Blood Regulations to describe what was formerly referred to as a “walking donor.” See sections 86–91 of the Blood Regulations for requirements.
"processing"
« traitement »
"processing" means any of the following activities:
(a) donor suitability assessment;
(b) collection;
(c) testing; or
(d) blood component preparation.
An establishment processes blood and blood components for transfusion or for further manufacture if it carries out any of the following activities: donor suitability assessment, collection, testing, and blood component preparation. The scope of processing cannot extend beyond this interpretation.
Blood component preparation does not include transformation or dividing blood into aliquots.
"safety"
« sécurité »
“safety”, in respect of blood, means that the blood has been determined safe for distribution or for autologous transfusion, as the case may be, in accordance with section 73, and includes
(a) in the case of blood for transfusion, its quality and efficacy; and
(b) in the case of blood for use in the manufacture of a drug for human use, its quality.
Whenever the Blood Regulations or this guidance document refer to the safety of blood, this means (a) the safety, quality and efficacy of blood and blood components for transfusion; and (b) the safety and quality of blood and blood components for use in the manufacture of a drug for human use.
Blood safety and quality, in the case of blood and blood components for transfusion or for further manufacture, are determinants of whether the blood and/or blood components are safe for distribution. Blood safety is the degree to which the blood and blood components for transfusion or for use in the manufacture of a drug for human use is free of harmful substances or infectious agents. Blood quality is defined by quality assurance procedures and is determined by the specifications set for blood and blood components. Blood safety and quality includes policies for mandatory testing, donor selection, collection procedures, testing methods, donation handling, storage, transportation, and distribution.
Blood efficacy, in the case of blood and blood components for transfusion, is a determinant of whether the blood is safe for distribution. Blood efficacy is the capacity to produce a desired or intended result or effect in blood recipients.
"serious adverse reaction"
« effet indésirable grave »
"serious adverse reaction” means an adverse reaction that results in any of the following consequences for the donor or recipient:
(a) their in-patient hospitalization or its prolongation;
(b) persistent or significant disability or incapacity;
(c) medical or surgical intervention to preclude a persistent or significant disability or incapacity;
(d) a life-threatening condition; or
(e) death.
"standard"
« norme »
“standard” means National Standard of Canada CAN/CSA-Z902 published by the Canadian Standards Association and entitled Blood and blood components, as amended from time to time.
Throughout this guidance document, the standard is referred to as the CSA Blood Standard.
"transformation"
« transformation »
“transformation”, in respect of blood components, means washing, pooling and irradiation that are performed after blood has been determined safe for transfusion.
The definition of transformation states activities included within its scope: washing, pooling, and irradiation.
The scope cannot extend beyond this interpretation.
The definition of transformation in the Blood Regulations does not include pathogen reduction technologies.
Transformation does not include dividing blood into aliquots.
"unexpected adverse reaction"
« effet indésirable imprévu »
“unexpected adverse reaction” means an adverse reaction that is not identified among the possible adverse reactions either in the circular of information or in any other information provided to the recipient.
Unexpected adverse reaction means an adverse reaction whose nature, severity or outcome is not consistent with the circular of information or in any other information provided to the recipient.
Sections 2-3 Application
Section 2 Scope of the Regulations
Scope of Regulations
2. These Regulations apply to blood that is collected for transfusion or for use in the manufacture of a drug for human use.
The scope of the Blood Regulations applies to human blood that is collected from donors
- for the purpose of transfusion;
- as a raw material for further manufacture into blood products; and
- for the immunization of donors of plasma for further manufacture (e.g., red blood cells for immunization).
Included within this scope is
- the safety of blood donors, in so far as it relates to the safety of the blood;
- the safety of the blood collected and processed from these donors; and
- the safety of blood recipients.
Section 3 Non-application
Non-application - various therapeutic products
3. (1) These Regulations do not apply to any of the following therapeutic products:
(a) cord blood and peripheral blood that are for use in lymphohematopoietic cell transplantation and that are regulated under the Safety of Human Cells, Tissues and Organs for Transplantation Regulations;
(b)blood that is the subject of clinical trials under Division 5 of Part C of the Food and Drug Regulations; or
(c)blood that is imported for use in the manufacture of a drug for human use.Non-application - regulations
(2)Except for section A.01.045 of the Food and Drug Regulations, no other regulation made under the Act applies to blood that is the subject of these Regulations.Non-application - imported rare phenotypes
(3) Sections 4 to 124 do not apply to blood that is of a rare phenotype if it is imported pursuant to a prescription.
3(1) Table 1 describes blood that the Blood Regulations do not apply to.
| Blood or Blood Component | Applicable Regulation(s) |
|---|---|
| 1. Cord blood and peripheral blood for use in lymphohematopoietic cell transplantation | See the Safety of Human Cells, Tissues and Organs for Transplantation Regulations |
| 2. Blood that is the subject of a clinical trial | See sections C.04.230– C.04.241 and Division 5 of Part C of the Food and Drug Regulations |
| 3. Plasma for further manufacture after an establishment distributes the plasma to the blood product fabricator | See Part C, Divisions 1, 1A, 2, C.04.001. – C.04.020., C.04.230.–C.04.241. and 8 of the Food and Drug Regulations |
| 4. Autologous blood for further manufacture | See the Food and Drug Regulations |
| 5. Autologous Platelet Rich Plasma (PRP) | See Health Canada’s Information Update on PRP |
6. Blood products, such as plasma derivatives, and blood product manufacturing - examples of blood products include: coagulation factors, immune globulins, hyperimmune globulins, and albumin |
See the Food and Drug Regulations |
| 7. Blood for further manufacture collected outside Canada | Regulated by the foreign jurisdiction from which it is imported See the Food and Drug Regulations for the importation of blood for further manufacture |
| 8. Blood that is of a rare phenotype — not available in Canada — and that is imported in accordance with a prescription | Proof of prescription must be provided at port of entry |
Section 4 Prohibitions
Allogeneic blood
4. (1) Subject to subsections (2) and (3), an establishment must not import, distribute or transfuse allogeneic blood unless it is processed by an establishment in accordance with an authorization and determined safe for distribution under subsection 73(1).Exception - pre-assessed donor programs
(2) Subsection (1) does not apply if the processing is conducted as part of a pre-assessed donor program.
Exception - urgent circumstances
(3) An establishment may, in urgent circumstances,
(a) import, in accordance with section 92, allogeneic blood that has not been processed in accordance with an authorization; and
(b) distribute or transfuse such blood if the importer imported it in accordance with section 92.Pre-assessed donors
(4) An establishment must not transfuse allogeneic blood that is collected from a pre-assessed donor unless the establishment has complied with the requirements of sections 86 to 91.Transformations
(5) An establishment must not distribute or transfuse blood that has been transformed unless the transformation is conducted by a registered establishment.Autologous blood
(6) An establishment must not distribute or transfuse autologous blood unless it has been processed by a registered establishment and determined safe for autologous transfusion under subsection 73(2).Investigations
(7) An establishment must not distribute or transfuse blood in either of the following circumstances:
(a) while the blood is in quarantine; or
(b) when the results of an investigation into a suspected error or accident or an unexpected adverse reaction or serious adverse reaction are inconclusive or indicate that there has been a compromise to the safety of the blood.
4(1) Allogeneic blood must be processed in accordance with an Authorization, in order for an establishment to import, distribute or transfuse the blood and blood components.
When allogeneic blood and blood components are the subject of exceptional distribution, establishments must comply with the requirements in sections 81 through 85 of the Blood Regulations. Exceptional distribution is not the same as importation in urgent circumstances.
4(2) If an establishment conducts processing on allogeneic blood as part of a Pre-Assessed Donor Program, an Authorization is not required. See subsection 4(4) for the requirement to register.
4(3) In urgent circumstances, a licensed establishment may import blood and/or blood components that have not been processed in accordance with an Authorization. The establishment must have a licence to import blood and/or blood components in urgent circumstances as required in paragraph 18(1)(l) and subsection 92(1).
See section 92 regarding the information a licensed establishment must provide the Minister before Health Canada will allow the importation of blood in urgent circumstances.
4(4) In order to operate a Pre-Assessed Donor Program, an establishment must register under subsection 30(1) of the Blood Regulations and comply with specific requirements in sections 86 to 91.
An establishment that tests blood and/or blood components collected in a Pre-Assessed Donor Program must hold an Establishment Licence as required by subsection 17(2) of the Blood Regulations.
4(5) The Blood Regulations list specific transformation activities that may be conducted within the scope of a Registration. See section 1 (Interpretation, transformation), subsection 30(1) and sections 77 through 80 of the Blood Regulations.
4(6) An establishment must not process autologous blood unless it is registered under subsection 32(1) of the Blood Regulations.
4(7)(a) An establishment must quarantine blood and blood components as stated in the requirements in section 56, paragraphs 103(1)(b), 104(1)(b), 110(1)(b), subsection 110(3) and section 111. See sections 70 through 72 of this guidance document for guidance concerning the segregation of blood.
4(7)(b) An establishment must consider the prohibition in paragraph 4(7)(b) when notifying other establishments of the results of an investigation into a suspected error or accident or an unexpected adverse reaction or serious adverse reaction and any action required to be taken. See subsections 106(1) or 114(2).
Sections 5-16 Authorizations
Section 5 Authorization
Authorization - processing
5. (1) Except for an establishment that only tests blood, an establishment that processes allogeneic blood must have an authorization to do so.Exception - pre-assessed donor programs
(2) Subsection (1) does not apply if the processing is conducted as part of a pre-assessed donor program.Authorization - importation
(3) Subject to section 92, an establishment that imports blood must have an authorization to do so, unless the blood is already the subject of another establishment’s authorization.
What is an Authorization?
An Authorization is permission from Health Canada for an establishment to process allogeneic blood: i.e. conduct donor suitability assessments, collect blood from donors, test blood, and prepare blood components.
A new establishment obtains an Authorization by applying to the Biologic and Radiopharmaceutical Drugs Directorate (BRDD) for an Authorization and filing information for review. Licensed establishments apply for an amendment to their Authorization whenever they intend to make a significant change to their processing activities.
See section 1, the Interpretation section of this guidance document, for the definitions of “allogeneic” and “processing.”
What is the difference between an Authorization and an Establishment Licence?
Authorization
An Authorization gives an establishment the authority to process blood by describing
- the processes related to blood at the establishment; and
- the blood components prepared using these processes.
The BRDD reviews the evidence and the information provided by the establishment on the processes that the establishment intends to conduct. An on-site evaluation may complement the review process to confirm that the documents filed for review correspond with what the establishment proposes to implement. A Notice of Authorization or a Notice of Authorization Amendment with or without Terms and Conditions is issued if the Minister, as represented by Health Canada’s BRDD, is satisfied that the processes do not pose an unacceptable risk to human safety or the safety of the blood and blood components.
Establishment Licence
An Establishment Licence is a licence issued to an establishment in Canada allowing them to conduct activities requiring a licence in a building which has been assessed and is compliant with the Blood Regulations and the Authorization.
Most establishments requiring an Establishment Licence will also require an Authorization. An exception is an establishment that only tests allogeneic blood and/or blood components. See subsections 5(1) and 17(2) of the Blood Regulations.
The Minister, as represented by Health Canada’s Regulatory Operations and Enforcement Branch (ROEB), issues the Establishment Licence. See sections 17–29 for Establishment Licence requirements.
5(2) An Authorization is not required to process blood from pre-assessed donors. An establishment must have a Registration to process allogeneic blood in a Pre-Assessed Donor Program. See sections 30–37 for requirements specific to Registration. Chart 1 — The application of the Blood Regulations to different types of establishments — provides information about which sections of the Blood Regulations would apply to pre-assessed donor programs.
5(3) The term “import” in this and other sections applies only to the importation of blood and blood components for transfusion.
Blood and blood components imported into Canada for transfusion falls within the scope of an Authorization. The importer or the foreign establishment can apply for the Authorization. See paragraph 18(1)(k) for licensing requirements with respect to the importation of blood and/or blood components for transfusion.
Section 6 Application for an authorization
Application for authorization
6. (1) An establishment must file with the Minister an application for an authorization in the form established by the Minister. The application must be dated and signed by a senior executive officer and contain all of the following information:
(a)the applicant’s name and civic address, and its postal address if different, and the civic address of each building in which it proposes to conduct its activities;
(b)the name and telephone number, fax number, email address or other means of communication of a person to contact for further information concerning the application;
(c) the name and telephone number of a person to contact in an emergency, if different from the person mentioned in paragraph (b);
(d) a statement of whether the establishment proposes to import whole blood or blood components;
(e) a list of the whole blood and blood components that the establishment proposes to process or import;
(f) a list of the processing activities that are proposed to be conducted in each building;
(g) a description of the establishment’s facilities, including its buildings and all critical equipment, supplies and services that it proposes to use in the conduct of its activities;
(h) a description of the processes that the establishment proposes to use or to have used on its behalf in respect of blood and each blood component in the conduct of its activities;
(i) a draft of each proposed label and circular of information;
(j) evidence that any foreign establishment that it proposes to have conduct any of its processing activities is licensed in the foreign jurisdiction; and
(k) sufficient evidence to demonstrate that the proposed processes will not compromise human safety and will result in blood that can be determined safe for distribution.Site inspection
(2) During the review of an application, the Minister may inspect the establishment’s facilities to evaluate on site the information provided in the application.Information on request
(3) An establishment must provide the Minister, on written request, with any information that the Minister determines is necessary to complete the Minister’s review of the application, by the date specified in the request.
6(1) The application for an Authorization must contain sufficient information to enable the Minister to assess human safety and to demonstrate that the processes will result in blood and blood components that can be determined safe for distribution. An establishment must send their application for an Authorization and evidence requirements to the BRDD’s Office of Regulatory Affairs (ORA):
Office of Regulatory Affairs
Biologic and Radiopharmaceutical Drugs Directorate
Health Products and Food Branch, Health Canada
Address Locator #0601C
100 Eglantine Driveway
OTTAWA ON K1A 0K9
Tel (613) 957-1722
Fax (613) 946-9520
Email BRDD.ORA@hc-sc.gc.ca
Any questions concerning the Authorization should be communicated to the ORA, prior to the submission of the application, when possible.
The Application Process
Pre-Application Blood Establishment Meetings
Prior to applying for an Authorization, an establishment should consider a meeting or a teleconference to discuss application requirements with the BRDD. Meetings or teleconferences provide an opportunity to define the purpose of the application and to discuss various application requirements.
A written request for a pre-application meeting should be received by the BRDD no less than 3 months prior to the proposed meeting date and should include the following information:
- the purpose of the meeting;
- a brief description of the issues to be discussed at the meeting;
- At least three proposed dates and times for the meeting; and
- a note indicating if participation by other areas of Health Canada might facilitate the discussion (e.g., Medical Devices Directorate or ROEB).
In preparation for a pre-application meeting, the establishment should provide a meeting package 30 days prior to the date of the meeting containing the following:
- purpose of the meeting;
- agenda;
- list of establishment participants;
- background information;
- presentation to be made, if applicable; and
- a list of any questions/issues to be discussed with Health Canada.
Applicants will be requested to submit a Pre-Submission meeting package to the ORA for distribution within Health Canada.
Receipt of Application
The BRDD assigns a control number to each submission. A control number is a unique tracking number that is assigned by the BRDD to each submission at the time it is initially received. This number is referenced in all correspondence concerning the submission.
The BRDD assigns a document number to the initial application package and to each additional installment of information. Document numbers are also assigned to letters and other correspondence related to an application issued by the BRDD to the establishment.
A teleconference or meeting can be requested by either the BRDD or the establishment to discuss the application at any point during the application process.
Screening of Application
The BRDD screens all application-related materials for acceptability including, but not limited to, quality and completeness and to determine if the proposed processes or changes to processes are in compliance with the Blood Regulations. The screening process is separate from the review process and occurs prior to the review process. During the screening process, the BRDD also identifies the type of application:
Types of applications
- Regular - not a rolling or accelerated application
- Rolling — an application with multiple parts that the establishment files at different times; generally, a rolling application is required when the need for a pilot project and/or production trial is anticipated. All applications for the authorization of a new establishment are rolling applications. An establishment should have a discussion with Health Canada prior to filing the rolling submission.
- Accelerated — accelerated applications are those where the establishment and the BRDD agree that the application will be screened and reviewed in a shortened time frame due to safety or operational concerns. The establishment must communicate these concerns in writing, prior to filing to Health Canada, and share their critical timeline for implementation with BRDD. The establishment must provide adequate rationale for the accelerated status and the critical timeline.
In the case where Health Canada issues a directive requiring the rapid implementation of a change, the accelerated status is assumed and no written rationale is required.
The BRDD reviews accelerated applications ahead of others already filed or under review. Review times will depend upon the circumstances and the BRDD will communicate the review timeline to the establishment in a timely manner.
Solicited Information During Screening
The BRDD may request additional information or clarification from the establishment through various forms of communication, including information requests known as screening clarifaxes. The establishment must provide a rationale if they do not deem it necessary to provide a response. The response to the clarifax (solicited information) must be provided within 15 calendar days from the date of issuance of the screening clarifax or by the date indicated in the clarifax; however, the establishment may request an extension.
Screening Deficiencies
The BRDD may also issue a Screening Deficiency Notice if there are significant omissions or inadequacies that prevent a review from being performed. The Screening Deficiency Notice outlines the deficiencies and indicates the date after which the BRDD will not accept a response to the Screening Deficiency Notice. The establishment may
- withdraw the application; or
- provide a response to the Screening Deficiency Notice, containing the identified information, by the indicated response date:
- after receipt of the response to the Screening Deficiency Notice, a new screening period commences.
If the establishment fails to provide all requested information by the indicated response date, a Notice of Refusal to Issue an Authorization or a Notice of Refusal to Amend an Authorization may be issued.
Following a withdrawal or a Notice of Refusal, the establishment may re-file the entire application.
- This application will be processed as a new application and assigned a new control number;
- The re-filed application must cross-reference the original control number; and
- Relevant information and material filed in the original application, and that remains unchanged, must be re-submitted and should be clearly identified.
If the application is found to be acceptable for review, the BRDD issues an Acceptable Screening Letter to the establishment confirming the date the application becomes part of the review workload.
Review of Application
Once the BRDD accepts an application for review, the BRDD evaluates the information and supporting evidence to determine if there is an unacceptable risk to human safety or the safety of blood and blood components.
In cases where the BRDD receives extensive information (e.g. evidence of successful validation) from the establishment, either as an instalment in a multipart application or as solicited information, the review period begins on the day that the last of such information is screened and deemed acceptable for review by the BRDD.
Once all the necessary information and material has been received and reviewed, the BRDD’s decision to accept or reject the application is issued. See section 7 for guidance on Issuance of an Authorization.
Content - Application for an Authorization
An application for an Authorization must contain sufficient information and evidence to enable the Minister to assess the risk to human safety and to demonstrate that the proposed processes will result in blood and blood components that can be determined safe for distribution.
6(1)(a), (b) and (c) Administrative Information
The establishment must provide the administrative information listed in paragraphs 6(1)(a)–(c). The person named as a contact concerning the application in paragraph 6(1)(b) may be the same person to contact in an emergency in paragraph 6(1)(c).
6(1)(d) Importation
The establishment must state whether they are proposing to import whole blood or blood components, as applicable.
6(1)(e) List of blood components
The establishment must provide a list of the names of the allogeneic blood and blood components that they propose to process (including blood for use in the manufacture of a drug for human use) and a list of allogeneic blood and blood components for transfusion the establishment is proposing to import. The names must fully describe listed components, e.g., Platelets (leukoreduced)-apheresis-CMV negative-IgA deficient.
6(1)(f) List of processing activities
The establishment must provide a list of the processing activities with respect to allogeneic blood and blood components that it proposes to conduct in each building. These activities must correspond to the activities that are listed on the establishment’s licence.
Examples of processing activities include the following:
- donor suitability assessment of donors of blood for transfusion;
- apheresis collection of plasma for the manufacture of a drug for human use;
- nucleic acid testing of whole blood for viral transmissible disease agents; and
- blood component preparation by the Buffy Coat method.
6(1)(g) Facilities - Buildings and Critical Equipment, Supplies, and Services
The establishment must provide the following:
- for each building the establishment must provide:
- a building floor plan, including locations of built-in equipment such as walk-in freezers; and
- evidence that all facility systems (e.g. electrical, ventilation, water, security, temperature monitoring, etc.) are commissioned or validated successfully.
- a description of all critical equipment, supplies, and services, including the functions of each.
See section 1, the Interpretation section of this guidance, for the definition of critical and for examples of critical equipment, supplies, and services.
The application must contain adequate evidence of successful validation of the equipment performed at the establishment, including installation qualification, operational qualification and performance qualification, as applicable.
Equipment used in the processing of blood and blood components is often a medical device requiring a medical device licence issued by the Medical Devices Directorate of the Health Products and Food Branch at Health Canada. The applicant should contact the Medical Devices Directorate (meddevices-instrumentsmed@hc-sc.gc.ca) if they have any questions concerning the licensing of medical devices. The establishment should include in their application evidence of a current, valid medical device licence for all critical equipment and supplies that are also medical devices.
6(1)(h) Proposed processes
An establishment’s application for an Authorization must include a description of and all relevant evidence for the processes that the establishment proposes to implement in order to process allogeneic blood and/or blood components, from the time the establishment assesses the donor until the establishment determines that the unit of blood or blood components is safe for distribution. This includes the storage and labelling of allogeneic blood components during processing activities but prior to and at the point of determination that the blood and blood components are safe for distribution. See subsection 73(1) for the determination of safety of allogeneic blood for distribution.
Allogeneic blood and blood components processed in accordance with an Authorization must have the processing establishment’s licence number on its label (see paragraph 64(1)(b)). See section 1, the Interpretation section of this guidance document, for the definition of processing.
Note: An establishment must apply for a registration if they conduct autologous blood processing activities, transformation activities or have a Pre-Assessed Donor Program. See section 30.
Contract Establishments: An establishment that holds an Authorization may contract out processing activities to an establishment in Canada licensed under the Blood Regulations for those activities; for example, testing of blood samples for viral markers.
A contract establishment that only tests blood samples and does no other processing activities under the Blood Regulations does not have to apply for an Authorization if they are under contract to an establishment that holds an Authorization for testing. See subsection 5(1). The tests on blood samples performed by the contract establishment must accord with those specified in the Authorization. The establishment that holds the Authorization assumes the responsibility for testing and is required to apply to Health Canada for Authorization amendments for any significant changes to that process.
For contracts with foreign establishments, see paragraph 6(1)(j).
6(1)(i) Labels
For each allogeneic blood component that the establishment proposes to process, the establishment must provide Health Canada with the proposed final blood container label. In the case of allogeneic blood for transfusion, the establishment must provide the draft text of the circular of information. Labels for autologous blood, labels added to units of blood following transformation (transformation labels) and labels for blood collected in a Pre-Assessed Donor Program do not need to be provided to Health Canada for review.
6(1)(j) Foreign establishment: contracted processes
An establishment that proposes to contract out any of its activities to a foreign establishment must provide evidence to Health Canada that the foreign establishment has a current licence in the foreign jurisdiction. The establishment in Canada that holds the Authorization for the contracted processes assumes the responsibility for these processes and is required to apply to Health Canada for Authorization amendments for that activity.
Paragraph 6(1)(j) also applies if an establishment imports blood or blood components from a foreign establishment. If an establishment imports blood or blood components for transfusion — including the importation of red blood cells for the immunization of source plasma donors — the blood and blood components must meet the importation requirements described in the establishment’s Authorization. See also section 92, Importation in Urgent Circumstances.
6(1)(k) Sufficient evidence
An establishment must provide sufficient evidence to Health Canada when applying for an Authorization, including the following:
- all required data and information about the safety of each type of allogeneic blood component that it proposes to process;
- all required data and information to demonstrate that the processes it uses will result in allogeneic blood that is safe, including:
- donor suitability assessment;
- post-donation information;
- testing;
- blood component preparation;
- labelling prior to distribution;
- circular of information;
- storage prior to distribution; and
- exceptional distribution.
- evidence of successful validation of equipment including documented executed protocols; the validation of equipment should include the validation of any software associated with the equipment; and
- evidence that the components of its computer system are validated and that it has operating processes in place.
On-site evaluation
6(2) Health Canada may carry out an on-site evaluation (OSE) to complement the review of an application. The scheduling of the on-site evaluation will be done in consultation with the establishment and documented in writing. One or more staff members from Health Canada may participate in the OSE.
The issuance of an Authorization may be conditional on a successful on-site evaluation. An on-site evaluation may be used to
- review evidence, documentation or data;
- observe processes; and
- view a demonstration of new equipment.
The blood establishment should make every effort to make available all relevant documents and materials and ensure that subject specialists are available to answer questions.
Results of the on-site evaluation will be verbally summarized for the establishment prior to completing the visit. All actions to be taken by the establishment will be documented in writing at the close of the OSE, if possible, or within 15 calendar days subsequent to the OSE. Time frames for responses will be identified.
Solicited information during review
6(3) During the review of an application, the BRDD may request that the establishment provide additional material if the initial information submitted is found to be insufficient or unclear. This type of information is referred to as solicited information and is used to determine human safety or the safety of the blood and blood components.
The BRDD may request solicited information during a teleconference or meeting; through a clarifax; or in the form of a Notice of Deficiency. A clarifax is transmitted by an email in standard format. An establishment must provide the BRDD with the solicited information within 15 calendar days from the date of issuance of the clarifax or by the date specified in the request; however, the establishment may request an extension. A response is complete if:
- all clarifications or questions identified in the Clarification Request have been addressed; or
- the sponsor provided a sound scientific rationale as to why the requested information is not necessary.
Notice of Deficiency
If an application has deficiencies precluding further evaluation or if an establishment’s response to a review clarifax is inadequate, the BRDD issues a Notice of Deficiency (NOD). The NOD identifies the deficiencies and indicates the date after which the BRDD will not accept a response to the NOD. The establishment may provide a response to the NOD referencing the original application control number and containing the identified information. The establishment may also choose to file a letter to cancel the application; the BRDD will issue a Cancellation Acknowledgement Letter.
The establishment has 90 days to respond to the NOD. The review is inactive until the BRDD receives a response. Following receipt of the response to the NOD, a new screening period commences.
If the establishment does not provide a response to the NOD within the specified time frame and does not cancel their application, or if the application remains deficient, the BRDD will issue a NOD-Withdrawal Letter.
If the establishment is unable to meet the response date specified in the NOD, they should provide the BRDD with a written request and justification for an extension prior to the end of the 90 day period.
Following a cancellation of an application by the establishment or when a NOD-Withdrawal Letter is issued by the BRDD, the establishment may re-file the application (see section 8).
Section 7 Issuance
Issuance
7. On completion of the review of an application, the Minister must issue an authorization, with or without terms or conditions, if she or he determines that the establishment has provided sufficient evidence to demonstrate that issuance of the authorization will not compromise human safety or the safety of blood.
Notice of Authorization
When the BRDD completes the review of the information and evidence in the establishment’s application and finds it acceptable, the BRDD issues a Notice of Authorization that allows the establishment to proceed with implementation and summarizes the following information:
- a. name of the establishment;
- b. blood components that the establishment may process;
- c. processing activities that the establishment may perform;
- d. civic address of each building and the processing activities to be conducted at each location; and
- e. any terms and conditions on the Authorization.
The Notice of Authorization may also list the following:
- f. laboratories or other facilities with whom the establishment has contracted processing activities:
- domestic; or
- foreign;
- g. approved control documents;
- h. establishment computer systems and major software applications and their version numbers, such as electronic donor screening software applications, donor or blood management systems, or laboratory information systems;
- i. test kits used to qualify donors, e.g. transmissible disease, blood group or red blood cell antibodies (including the Medical Devices Bureau licence number and the date approved); and
- j. medical devices in use, e.g. major testing platforms (including the Medical Devices Directorate licence number and the date approved).
The BRDD issues a Notice of Authorization or a Notice of Authorization with Terms and Conditions following the review and approval of an establishment’s original application for an Authorization. All subsequent notices following review and approval of applications to amend an Authorization are Notices of Authorization Amendment (section 9) or Notices of Authorization Amendment with Terms and Conditions (section 13).
Section 8 Refusal
Refusal
8. The Minister may refuse to issue an authorization if she or he determines that the information provided by the establishment in its application is inaccurate or incomplete.
Notice of Insufficient Information
During the review, if the BRDD finds that the establishment’s information in the application is insufficient to demonstrate that issuance of the Authorization will not compromise human safety or the safety of blood and blood components, the BRDD may issue a Notice of Insufficient Information explaining why the information in the application is insufficient.
The establishment has 90 days to respond to the Notice of Insufficient Information. The review is inactive until the BRDD receives a response to the Notice of Insufficient Information. Following receipt of the response, a new screening period commences.
If the establishment fails to respond within 90 days, or the BRDD determines that the application remains non-compliant, the BRDD will issue a Notice of Insufficient Information Withdrawal Letter. The establishment may also choose to file a letter to cancel the application in which case the BRDD will issue a Cancellation Acknowledgement Letter.
If the establishment is unable to respond within the 90-day period, they should provide the BRDD with a written request and justification for an extension prior to the end of the 90-day period.
Following cancellation of an application by the establishment or when a Notice of Insufficient Information Withdrawal Letter is issued by the BRDD, the establishment may re-file the application.
Notice of Refusal
The BRDD may issue a Notice of Refusal to Issue an Authorization or a Notice of Refusal to Issue an Authorization Amendment in any of the following circumstances:
- if the establishment provided inadequate evidence to support the proposed processes;
- if there were inaccuracies in the application;
- if false or misleading statements were made; or
- if the processes reviewed were found to pose an unacceptable risk to human safety or to the safety of blood and blood components
The Notice of Refusal delineates the reasons for the refusal and provides the establishment with the opportunity to be heard in writing. The establishment may choose to re-file the application.
Re-filing
When an application is re-filed
- the re-filed application will be processed as a new application and assigned a new control number;
- the application should cross-reference the original control number; and
- the information and material filed in the original application, and that remains unchanged, should be clearly identified.
Section 9 Significant Changes
Significant changes
9. (1) Before making a significant change, an establishment must file with the Minister an application to amend its authorization and include with it all relevant information to enable the Minister to determine whether the change or the way in which it is implemented could compromise human safety or the safety of blood.Applications to amend
(2) Sections 6 to 8 apply to an application to amend an authorization, with any necessary modifications.Meaning of "significant change"
(3) In this section and sections 10 and 12, “significant change” means any of the following changes:
(a) the addition of blood or a blood component to the list required by paragraph 6(1)(e);
(b) the deletion of or a change to any authorized process;
(c) the addition of a process described in paragraph 6(1)(h); or
(d) a change to the description of the establishment's facilities referred to in paragraph 6(1)(g).
9(1) An establishment must file an application with the BRDD to amend their Authorization if they intend to make a significant change to their Authorization. The meaning of significant change is described in subsection 9(3) of the Blood Regulations.
Application to amend an Authorization - Minimum requirements
Applications for all significant changes have the same minimum information requirements:
- detailed description of the proposed changes;
- rationale for the proposed changes;
- risk assessment; and
- contact information, as required by paragraphs 6(1)(b)and 6(1)(c)of the Blood Regulations.
Additional requirements, where applicable
- Medical Device Directorate approval for medical devices;
- clinical data;
- scientific or technical data;
- a detailed description of new or modified critical equipment, supplies, or services if its operation or function differs from its authorized approval;
- impact on the blood management computer system;
- a description of changes to blood and/or blood components labels, including the Circular of Information (include new or revised labels);
- training plan;
- validation plan;
- validation results; and
- implementation schedule.
The application to amend the Authorization must be dated and signed by the senior executive officer stating that all of the information in the application is accurate and complete. See Subsection 6(1).
Questions regarding significant changes, new information that could trigger an amendment and the Authorization amendment process, should be directed to the ORA at the BRDD. See subsection 6(1) of this guidance for contact information.
9(2) When an establishment applies to amend an Authorization, sections 6 through 8 of the Blood Regulations apply. See sections 6 through 8 of this guidance.
The BRDD may require an on-site evaluation of the establishment following a review of the submitted documentation, depending on the nature of the change and the supporting evidence. See subsection 6(2).
The BRDD aims to screen an application to amend an Authorization within 15 calendar days and to review the application within 90 days following the date the application is accepted into review. An establishment is prohibited from implementing changes filed in an application to amend an Authorization prior to approval from Health Canada, even though the review may extend beyond 90 days.
Division of an application to amend an Authorization
Occasionally, an establishment files an application pertaining to two or more unrelated topics. The BRDD may divide the original application into two or more applications and assign a separate control number to each application. This may occur during the screening or review of the application. When the BRDD divides an application, the establishment will receive notice of the division in writing requesting the submission of the electronic data as separate files.
Notice of Authorization Amendment
When the BRDD completes the review of an application to amend an Authorization and finds it acceptable, the BRDD issues a Notice of Authorization Amendment that allows the establishment to proceed with implementation of the significant change.
The sum of the Notice of Authorization and all of the Notices of Authorization Amendment constitutes the establishment’s Authorization.
9(3)(a) Addition of blood or a blood component
A significant change includes the addition of blood or a blood component or a novel blood component to the list of blood or blood components that the establishment proposes to process or import. See 1.5 Definitions, novel blood component.
It is strongly recommended that an establishment consult with the BRDD prior to applying for an amendment to an Authorization when the establishment proposes to introduce
- a medical device that has the potential to produce an effect on a blood component; or
- a novel blood component developed in conjunction with a medical device manufacturer.
9(3)(b) Deletion or change to any authorized process
The meaning of significant change includes deleting or changing any authorized process.
9(3)(c) Addition of a process
An establishment must file an application with a detailed description of any new process that the establishment proposes to use in respect of each type of blood and blood component in the conduct of its activities.
9(3)(a), 9(3)(b) and 9(3)(c) Pilot studies and/or production trials
Pilot studies or production trials are sometimes required to provide the necessary evidence to demonstrate that changes to processes or blood components do not negatively impact human safety or the safety of blood and blood components. When an establishment wishes to implement a new or amended process or a new blood component, they should make their application to amend their Authorization for this change before they start the pilot study or production trial. Having prior input on the design of the study from the BRDD is strongly recommended to facilitate the application process.
The establishment is encouraged to contact the BRDD for any questions about pilot studies or production trials.
9(3)(d) Changes to facilities (buildings and critical equipment, supplies, and services)
New or renovated buildings
If an establishment proposes to add or renovate a building where processing (donor suitability assessment, collection, testing or blood component preparation) will take place, it must file an application to amend its Authorization.
It is not necessary for an establishment to include in their application information and evidence for processes and equipment that have already been validated by the establishment and authorized by the BRDD as long as a similar validation approach is being used.
If applicable, the application should include a statement indicating that there are no changes to the previously authorized processes and equipment, that these have been validated, and that a similar validation approach will be used for processes and equipment already authorized by the BRDD. This statement will confirm that the new facility will operate in compliance with the currently approved processes.
In addition to the minimum information requirements in subsection 9(1) above, the application for a change to the establishment’s facilities should include the following information:
- The civic address of the facility where the significant change is proposed to take place. [Paragraph 6(1)(a)]
- A list of the processing activities proposed to be conducted in each building where the significant change is proposed to take place.[Paragraph 6(1)(f)]
- A detailed description of changes to the facility, including:
- a building floor plan, including the locations of built-in equipment such as walk-in freezers; and
- a list of all critical equipment, supplies, and services that it proposes to use in the conduct of its activities.[Paragraph 6(1)(g)]
- A detailed description of any new or modified critical equipment, supplies, or services if its operation or function differs from its current Authorization. [Paragraph 6(1)(g)]
- Evidence that all new processes related to facility systems (e.g. electrical, ventilation, water, security, temperature monitoring, etc.) and equipment are commissioned/validated successfully.
As per section 22, an establishment must apply for an amendment to its Establishment Licence when proposing to add or make a change to a building in its list of facilities. Health Canada may inspect a new building after all changes have been made and prior to the start of operations (i.e. the establishment has not begun to accept donors for blood collection).
Critical equipment, supplies, and services
Significant changes to critical equipment are determined based on evaluating the impact to authorized processes and the subsequent impact on human safety or the safety of blood and blood components. Equipment includes software. See the definition of critical in section 1, the Interpretation section of this guidance, for examples of critical equipment, supplies, and services.
A new device or technology
An establishment must apply for an authorization amendment for significant changes to medical devices or for a new device or technology that has the potential to have an impact on human safety or the safety of blood and blood components.
In the following examples of changes, the establishment should consider these changes to be significant if they have the potential to have an impact on human safety or the safety of blood and blood components:
- Replacement of a device with another device that has the same indication or functionality but not the same make;
- Replacement of a device with another device that has a different indication or functionality;
- Replacement of a device with a new or different model from the same manufacturer; or
- Upgrade to a part of the device with new software or functionality.
Changes to a medical device licensed by Health Canada
Changes made to medical devices by the device manufacturer are deemed either significant or non-significant by Health Canada as per the Guidance for the Interpretation of Significant Change of a Medical Device. If an establishment intends to use a device that has had a change made to it, they should evaluate the impact of the change on human safety or the safety of blood and blood components and follow the reporting structure in the following 3 points:
- If Health Canada has determined the change to the device as “non-significant,” and there is no potential impact on human safety or the safety of blood and blood components, the establishment is not required to file this change either as an annual report or as an application to amend an Authorization;
- If the change is “non-significant” but there is a potential impact on safety, then the blood establishment should file this change in an annual report;
- If the change is “significant” and there is a potential impact on safety, the establishment should file the change as an application to amend their Authorization.
Information Technology
When an establishment that holds an Authorization plans to make changes to processes that have a significant information technology component, the establishment must file an application to amend their Authorization to the ORA at the BRDD.
Examples of significant changes to information technology include the following:
- adding a computerized system, such as:
- a new donor management system;
- a new electronic questionnaire platform;
- a new laboratory information system; or
- migrating to a newer version of the application software or data management system currently installed and validated;
- corresponding changes to operating processes when making significant changes to a computerized system;
- changes to the software configuration currently installed and validated; and
- additions to the functionality of the software application already installed and validated — including additions introduced in the course of system maintenance.
If an establishment is unsure whether a change to any validated components of a currently installed computerized system is considered significant, it is strongly recommended that the establishment consult with the BRDD for guidance prior to filing.
Guidance is essential when an establishment implements a new computerized system that performs one or more of the following functions:
- use of software for transferring data between automated devices where translation and/or reformatting is required;
- use of data to make decisions regarding the suitability of blood or blood components for transfusion or for further manufacture; or
- use of data to trace a unit of blood or a blood component from the source to its final disposition.
An establishment should direct questions about proposed changes to an existing computer system to the ORA at the BRDD. See subsection 6(1) of this guidance for contact information.
See section 12 of this guidance for changes to be filed in an annual report.
Section 10 Emergency Changes
Emergency changes
10. (1) In an emergency, if it becomes necessary for an establishment to implement a significant change before filing an application to amend its authorization, the establishment may do so if the change is necessary to prevent a compromise to human safety or the safety of blood.Notice and application
(2) The establishment must notify the Minister in writing of any significant change that it implements under subsection (1) no later than the day after implementing it and file an application to amend its authorization within 15 days after the day on which that notice is given.
10(1) Emergency Changes
Section 10 allows an establishment to make a significant change to an authorized process in an emergency situation. For example, an emergency situation may necessitate emergency changes to work around (i) an error in blood management software or (ii) a malfunction in equipment used in testing.
10(2) Notice and application
In an emergency, if it becomes necessary to implement a significant change to prevent a compromise to human safety or the safety of blood and blood components. The establishment should follow the two steps reporting process:
- The establishment must provide a written initial report to the BRDD within a day of implementing the emergency change. The initial report should contain the following information:
- definition of the issue and the emergency;
- decisions made;
- actions taken; and
- significant change made or that will be made by the establishment.
- Within 15 calendar days of notifying the BRDD, the establishment must apply to amend its Authorization with respect to these changes. The BRDD may apply additional terms and conditions to the Authorization as a result of emergency changes. (See section 13.)
Section 11 Administrative changes - notice
Administrative changes — notice
11. An establishment must notify the Minister in writing of any change to the information provided under paragraphs 6(1)(a) to (c) as soon as possible after the change is made, and the Minister must amend the authorization accordingly.
An establishment must notify the BRDD in writing of any change to information in the establishment’s application for an Authorization provided under paragraphs 6(1)(a) to (c). See subsection 6(1) of this guidance for contact information.
The BRDD will revise the establishment’s Authorization upon receipt of the notification and issue a Notice of Authorization Amendment.
Section 12 Other changes - annual report
Other changes - annual report
12. (1) An establishment must file with the Minister an annual report that describes any changes made in the year that are not described in section 9 or 11 and that could compromise human safety or the safety of blood.Amendment by Minister
(2) On receipt of the report, the Minister must amend the establishment’s authorization accordingly.When changes determined significant
(3) If the Minister determines that a change that was included in a report under subsection (1) is a significant change, the Minister must notify the establishment in writing to that effect and may require the establishment to cease or reverse the implementation of the change.Application to amend authorization
(4) On receipt of the notice, the establishment must file an application to amend its authorization.
12(1) When an establishment makes other changes to its processes that were not filed under section 9 (significant) or section 11 (administrative) — and these changes could compromise human safety or the safety of blood and blood components — the establishment must describe these changes in an annual report to Health Canada.
An establishment should contact the BRDD if there is any question about whether an intended change to an authorized process may be included in an annual report. Annual reports should be signed by a senior executive officer and sent to the ORA at the BRDD. See subsection 6(1) of this guidance for contact information.
The following are examples of the types of changes that an establishment may describe in an annual report:
- Changes to the List of Unacceptable Medications
Changes to the List of Unacceptable Medications used to assess donors of blood for transfusion.
- Maintenance changes for blood management information technology systems
Changes to blood management information technology systems due to maintenance activities to enhance functionality with no operational changes (e.g. enhancing memory) or to restore functionality (e.g. bug fixes).
When preparing an annual report, an establishment should ensure that all maintenance changes for the blood management information technology system have been described in the annual report, and that no new unauthorized processes have been inadvertently introduced.
Reporting requirements for annual reports
The establishment must provide the following information in an annual report:
- description of the change;
- rationale for the change;
- authorized processes involved, if applicable;
- information technology systems implicated in the change, if applicable;
- related errors or accidents, if applicable;
- licence number of a current and valid medical device licence, if applicable;
- implementation date; and
- other information necessary to describe the change or the impact of the change.
Before filing an annual report, the establishment should confirm that complete supporting information or data for the changes is available upon request.
Changes that do not require annual reporting or an application to amend an Authorization
The following types of changes are considered to have minimal potential to have an adverse effect on human safety or the safety of blood and blood components.
Donors
- Changes to donor consent procedures;
- Changes to the existing donor screening areas;
- Changes to the format, colour or layout of the establishment’s donor screening manual and questionnaire, and circular of information, where there is no change to authorized processes;
- Changes to the malaria endemic areas in the establishment’s manual for screening donors as long as they are the same as those areas identified by the Public Health Agency of Canada (PHAC) or the United States Centers for Disease Control and Prevention (CDC);
- Changes to infectious disease testing used solely for counselling purposes;
Equipment
- Changes to non-critical equipment or supplies. See section 1 of this guidance for the definition of critical and a list of non-critical equipment;
- Label changes that do not impact the content and that pertain to the orientation or placement of information;
- Provided that the establishment uses the same validation approach that was reviewed by the BRDD and used in the validation of the already approved device:
- replacement of a device with one of the same make and model;
- replacement of a part of such a device; or
- addition or removal of a unit of such a device.
- Changes to the general validation approach unrelated to validation protocols used in the validation of blood processing;
- Changes to versions of software in devices used for quality control testing provided there is no impact on blood component specifications or processes used in blood component preparation;
- Changes to existing computer operating systems;
- Changes to existing computer hardware;
- Changes required to update virus scanning software or to install daylight savings time operating system patches;
- Changes to “off-the-shelf” (non-configurable) software packages, e.g. Microsoft Office, provided there is no impact on authorized processes;
Buildings
- Changes to existing building or room ventilation or air conditioning systems as long as the environmental parameters remain within the authorized range and do not affect blood component specific requirements;
- Changes to the environmental data monitoring system. This is a system which accumulates environmental data and demonstrates that environmental parameters of areas housing critical processes are met; the system triggers an alarm when conditions are no longer within specifications;
- Changes to existing building security;
Quality management system
Blood inventory management
- Changes to blood inventory management;
Transportation, shipping, and shipping packaging
- Changes to transportation of blood or blood components;
- Changes to shipping and packaging for shipping of blood components;
Transformation activities
- Changes to transformation activities performed on blood components, i.e. washing, pooling, gamma irradiating, see sections 77–80 of this guidance;
Processes related to investigation and reporting management
- Changes to processes related to investigation and reporting management of the following, where there is no change to authorized processes:
- errors and accidents;
- adverse donor reactions;
- adverse transfusion reactions;
- lookback/traceback investigations; and
- post-donation information.
Section 13 New or amended terms and conditions
New or amended terms and conditions
13. (1) The Minister may add terms and conditions to an establishment’s authorization or amend its terms and conditions in either of the following circumstances:
(a) the Minister has reasonable grounds to believe that it is necessary to do so to prevent a compromise to human safety or the safety of blood; or
(b) the establishment fails to provide the Minister, on written request, with sufficient evidence to demonstrate that its processes will not compromise human safety and will result in blood that can be determined safe for distribution, by the date specified in the request.Notice
(2) Before adding terms or conditions to an authorization or amending its terms or conditions, the Minister must send the establishment a notice at least 15 days before the proposed terms and conditions are to take effect that sets out the Minister’s reasons and that gives the establishment a reasonable opportunity to be heard concerning them.Urgent circumstances
(3) Despite subsection (2), the Minister may immediately add terms and conditions to an authorization or amend its terms and conditions if she or he has reasonable grounds to believe that it is necessary to do so to prevent a compromise to human safety or the safety of blood.Urgent circumstances - notice
(4) When the Minister adds or amends terms or conditions under subsection (3), the Minister must send the establishment a notice that sets out the reasons for the new or amended terms and conditions and that gives the establishment a reasonable opportunity to be heard concerning them.Removal of terms and conditions
(5) The Minister may, by notice in writing, remove a term or condition from an authorization if she or he determines that the term or condition is no longer necessary to prevent a compromise to human safety or the safety of blood.
New or Amended Terms and Conditions placed on an Authorization
13(1) The BRDD may place or amend terms or conditions on an Authorization in order to address an identified safety risk, either within the context of an application for an Authorization or an Application to Amend an Authorization, or outside of the context of an application.
A Notice of Authorization (or Authorization Amendment) with Terms and Conditions permits an establishment to implement the processes proposed in its application but with certain restrictions. Terms and Conditions may be placed on a Notice of Authorization or Authorization Amendment in the following example situations:
- to allow a pilot project or production trial to proceed during which data is collected for later review by the BRDD in support of the proposed changes;
- to place conditions on an amendment, such as the requirement to provide post implementation data — see examples of terms and conditions below;
- to require the establishment to provide additional data for an implementation with multiple stages;
- to require subsequent studies to be performed;
- when actions are needed to address an emergent pathogen; and
- when a safety concern at the establishment has been identified.
Terms and Conditions may include any of the following:
- addition of a new donor screening test;
- addition of a new donor screening question;
- processing of a minimum number of donations as described in the application for an Authorization or the application for an Amendment to an Authorization;
- limitations on the type of donations collected;
- limitations on the type of activities that can be conducted;
- limitations on the distribution of all or specific blood components;
- provision of donor safety monitoring data;
- provision of quality control data on components processed;
- provision of additional stability studies;
- provision of additional information, as necessary;
- implementation of a corrective action due to an identified error or accident, following communications or interactions with Health Canada’s ROEB; or
- implementation of an improvement to component processing.
13(2) When Health Canada places terms or conditions on an Authorization, the Minister issues a Notice of Authorization Amendment with Terms and Conditions. The Minister must give the establishment 15 calendar days to respond to the notice. After 15 calendar days, the Terms and Conditions in the notice take effect if there is no response from the establishment or the response does not adequately address the risk. Discussion with the establishment can occur prior to the application of terms and conditions.
13(3) When urgent situations arise that put human safety or the safety of blood and blood components at risk, Health Canada may immediately add or amend terms and conditions to an Authorization. An example of an urgent situation is the emergence of a new pathogen that has the potential to be transmitted by blood and/or blood components.
Health Canada may also place urgent terms or conditions on an Authorization if an establishment has not taken corrective actions to address an issue that has the potential to impact the safety of the blood and blood components such as the potential to cause a serious adverse reaction.
13(5) When the establishment addresses the identified safety risk to Health Canada’s satisfaction, through the implementation of changes at the establishment or providing sufficient evidence to the Minister, Health Canada will remove the terms or conditions by communicating in writing. Where the Terms and Conditions were applied in the context of an application for Authorization or Authorization Amendment, an Application Closed Letter will be issued when the safety risk has been adequately addressed.
Section 14 Suspension
Suspension
14. (1) The Minister may suspend all or part of an authorization in either of the following circumstances:
(a) information provided by the establishment under section 6 or 9 proves to be inaccurate or incomplete; or
(b) the establishment fails to provide the Minister, on written request, with sufficient evidence to demonstrate that its processes will not compromise human safety and will result in blood that can be determined safe for distribution, by the date specified in the request.Notice
(2) Before suspending an authorization, the Minister must send the establishment a notice that
(a) sets out the reasons for the proposed suspension and the effective date;
(b) if applicable, indicates that the establishment must take corrective action and specifies the date by which it must be taken; and
(c) gives the establishment a reasonable opportunity to be heard concerning the suspension.Urgent circumstances
(3) Despite subsection (2), the Minister may immediately suspend all or part of an authorization if she or he has reasonable grounds to believe that it is necessary to do so to prevent a compromise to human safety or the safety of blood.Urgent circumstances - notice
(4) When the Minister suspends an authorization under subsection (3), the Minister must send the establishment a notice that
(a) sets out the reasons for the suspension; and
(b) gives the establishment a reasonable opportunity to be heard concerning the suspension.
Section 15 Reinstatement
Reinstatement
15. (1) Subject to subsection (2), the Minister must reinstate an authorization if the establishment provides the Minister with sufficient evidence to demonstrate that its processes will not compromise human safety and will result in blood that can be determined safe for distribution.Partial reinstatement
(2) If the Minister does not reinstate any part of an authorization that was suspended, the Minister must amend the authorization to remove that part.
Section 16 Cancellation
Cancellation
16. (1) The Minister must cancel an authorization in either of the following circumstances:
(a) the establishment fails to provide the Minister with the evidence described in paragraph 14(1)(b) within a reasonable period after the authorization was suspended; or
(b) the establishment's licence is cancelled under section 29.Notice
(2) When the Minister cancels an authorization, she or he must send the establishment a notice that sets out the reasons for the cancellation and the effective date.
Sections 17-29 Establishment Licences
Section 17 Establishment licence required
Establishment licence required
17. (1) An establishment that processes allogeneic blood — except, subject to subsection (2), blood from a pre-assessed donor — or that imports blood must have an establishment licence to do so.Test labs
(2) An establishment that tests blood from a pre-assessed donor for transmissible diseases or disease agents must have an establishment licence to do so.
Processing allogeneic blood and importation
An establishment must obtain an Establishment Licence if they process or import allogeneic blood and/or blood components. Processing activities include donor suitability assessment, collection, testing, and blood component preparation. An establishment must obtain an Establishment Licence if it intends to conduct any processing activity on behalf of another establishment.
Processing allogeneic blood by Pre-Assessed Donor Programs
Establishments that process allogeneic blood and/or blood components from a Pre-Assessed Donor Program do not require an Establishment Licence, but are required to be registered with Health Canada pursuant to section 30 of the Blood Regulations.
Testing
Every establishment in Canada that performs testing of allogeneic blood, including the blood and blood components collected by a Pre-Assessed Donor Program, requires an Establishment Licence.
Testing by foreign establishments
If testing is conducted by a foreign establishment on behalf of an establishment in Canada, the testing establishment must be listed on the Establishment Licence of the establishment in Canada. Guidance on section 18 of the Blood Regulations provides further details on the requirements for foreign establishments conducting activities on behalf of establishments in Canada.
Authorization before Establishment Licence
An Authorization issued by the BRDD must be granted prior to the issuance of an Establishment Licence, with the exception of testing conducted for a Pre-Assessed Donor Program. See sections 5–16 for further guidance regarding an Authorization. (See section 20, Issuance)
An establishment may apply for both an Authorization and an Establishment Licence simultaneously (i.e. an establishment does not have to wait to hold an Authorization before it applies for an Establishment Licence). However, in such a case, the Establishment Licence will not be issued until the Authorization is approved.
An Authorization is not required prior to obtaining a Registration.
Summary of Requirements (also available under Section 30)
| Establishment Type/Activities | Establishment Licence | Registration |
|---|---|---|
| Processing allogeneic blood | Required | Not Required |
| Importing Blood | Required | Not Required |
| Pre-Assessed Donor Program | Not Required | Required |
| Testing laboratory for Pre-Assessed Donor Program | Required | Not Required |
| Autologous Program | Not Required | Required |
| Transformation of Blood | Not Required | Required |
| Note: “Blood” includes whole blood and blood components. | ||
Section 18 Application for establishment licence
Application for establishment licence
18. (1) An establishment must file with the Minister an application for an establishment licence in the form established by the Minister. The application must be dated and signed by a senior executive officer and contain all of the following information:
- the applicant’s name and civic address, and its postal address if different;
- the civic address of each building in which records will be stored;
- in the case of an establishment that previously conducted its activities under another name, that other name;
- the name and telephone number, fax number, email address or other means of communication of a person to contact for further information concerning the application;
- the name and telephone number of a person to contact in an emergency, if different from the person mentioned in paragraph (d);
- a list of the establishment’s activities;
- a list of the whole blood and blood components in respect of which the activities are proposed to be conducted;
- the civic address of every building in which it proposes to conduct its activities and a list of the activities that are proposed to be conducted in each building;
- the name, civic address and licence number, if any, of any other establishment that it proposes to have conduct any of its activities;
- sufficient evidence to demonstrate that the establishment can conduct its activities in accordance with its quality management system and the requirements of these Regulations and that its activities will not compromise human safety or the safety of blood;
- in the case of an importer or an establishment that proposes to have any of its testing conducted by a foreign establishment, the information described in paragraphs (a) and (f) to (j) with respect to every foreign establishment that processes or distributes the blood that they propose to process or import; and
- in the case of an establishment that proposes to import blood in urgent circumstances, all of the information required by subsection 92(1).
Information on request
(2) An establishment must provide the Minister, on written request, with any information that the Minister determines is necessary to complete the Minister’s review of the application, by the date specified in the request.
Section 18 of the Blood Regulations specifies the information that must be provided in a Blood Establishment Licence Application Form. In the context of the Establishment Licences section of the Blood Regulations, the Minister’s Blood Establishment Licensing powers are exercised by the Biological Product Compliance Program (BPCP) of the ROEB.
Where to find the application form
The Blood Establishment Licence Application Form (FRM-0354), along with instructions, can be obtained by sending a request to: roeb.blood-sang.dgoral@hc-sc.gc.ca
It is the responsibility of the applicant to ensure that its Blood Establishment Licence Application is accurate and complete in accordance with the requirements of section 18 of the Blood Regulations before filing it with the BPCP of the ROEB. This will help prevent confusion, errors, and delays in processing.
Where to file the application form
By email: roeb.blood-sang.dgoral@hc-sc.gc.ca
Buildings in Canada
Paragraph 18(1)(h) requires the applicant to provide the civic address of each building in Canada in which the applicant proposes to conduct any activities and a list of those activities with respect to blood and blood components.
Establishments that solely store blood or blood components do not require an Establishment Licence. All establishments who store blood or blood components must do so in accordance with the Blood Regulations.
Establishments conducting activities on behalf of the applicant
Paragraph 18(1)(i) requires the applicant to provide the name, civic address and licence number (if applicable) of any other establishment, including foreign establishments*, that (will) conduct activities on their behalf.
*Note: Foreign establishments, conducting activities on behalf of an establishment in Canada, do not require an Establishment Licence under Canada’s Blood Regulations; however, the foreign establishment must be listed on the Establishment Licence of the establishment in Canada for whom they are performing activities.
It is the responsibility of the applicant to ensure that appropriate written agreements are in place for all activities contracted out to other establishments.
Evidence to demonstrate compliance with the Blood Regulations
Applicants are required to have sufficient evidence to demonstrate their compliance with the Blood Regulations. Health Canada may inspect the establishment during the review of an application. (See paragraph 18(1)(j) and section 19 of the Blood Regulations.)
Evidence required for foreign establishments
In accordance with section 18(1), the applicant must provide sufficient evidence to demonstrate that the foreign establishments meet the requirements of the Blood Regulations. If approved, these foreign establishment(s) will then be listed on the Establishment Licence of the establishment in Canada.
Sufficient evidence must be submitted with a completed Blood Establishment Licence Application (FRM-0354). Sufficient evidence includes, but is not limited to, the following:
- The most recent (within the last 4 years) inspection report issued by a regulatory authority which is a member of the Pharmaceutical Inspection Cooperation Scheme (PIC/S) (e.g. United States Food and Drug Administration Establishment Inspection Report); or
- In the absence of an inspection report, a copy of:
- The inspection observations (e.g. United States Food and Drug Administration Form 483); and
- The corrective actions taken as a result of the inspection, signed by a responsible official of the foreign establishment.
- In the absence of an inspection report, a copy of:
- A copy of the most recent, signed and dated quality agreement between the Canadian and foreign establishments. The agreement should clearly identify the parties involved and their responsibilities to ensure compliance with the Blood Regulations;
- A copy of the most recent:
- Signed and dated Site Master File; or
- Equivalent document.
If any of the above documents has not changed since the last submission, establishments may indicate this in the cover letter or email, in lieu of submitting a duplicate copy. Establishments are requested to clearly indicate the date which this information was previously submitted.
Further, if any of the required documents are not available, establishments are encouraged to discuss with Health Canada the reason why the required documents cannot be submitted and alternate evidence that may be submitted, prior to submitting an application.
New Evidence Required By (NERBY) Date
For foreign establishments assigned a C rating, a “New Evidence Required By” (NERBY) date will be assigned. The NERBY date is the date by which new evidence is required to be submitted to Health Canada as part of an application to renew a foreign establishment listed on an Establishment Licence.
The NERBY date will be determined using a risk‐based approach. The NERBY date will generally be 4 years, calculated from the start date of the foreign inspection. However, Health Canada may issue a shortened or extended NERBY date for a foreign establishment based on several factors including, but not limited to, the foreign establishment’s compliance history and the type of evidence submitted.
When renewing a NERBY date, as long as an application is submitted with complete and updated evidence by the establishment’s NERBY date, the foreign establishment will continue to be considered compliant and listed on the Establishment Licence.
In certain circumstances, where updated evidence is not available to renew a foreign establishment’s NERBY date (e.g. a recent inspection by a regulatory authority has not taken place or an inspection by a regulatory authority has taken place, but there is a delay in the issuance of the inspection report), establishments should contact the BPCP of the ROEB at roeb.blood-sang.dgoral@hc-sc.gc.ca before the NERBY date has passed to discuss information that may be submitted in these cases.
Foreign establishment evidence screening and assessment
Upon receipt of an Establishment Licence application with appropriate foreign establishment evidence, Health Canada will send a Screening Acceptance Notice to notify the Establishment Licence holder that the evidence has been screened and accepted for further assessment. The act of filing alone does not guarantee that the foreign establishment evidence will be considered acceptable and additional information may be requested during the application screening and/or assessment period (e.g. information on the scope and depth of an inspection conducted by a regulatory authority or regulatory authority responses to foreign establishment’s proposed corrective actions).
During the evidence assessment, establishments may continue to import from or have testing conducted by the foreign establishment, in accordance with the Establishment Licence and the Blood Regulations. If the evidence is deemed unacceptable or incomplete at any time in the assessment process, the foreign establishment may be removed from the Establishment Licence. Once a foreign establishment is removed from the Establishment Licence, the establishment is no longer authorized to import or to have any testing conducted by the foreign establishment and the Establishment Licence holder will be notified, as such. The Establishment Licence holder must submit a complete application to Health Canada to add the foreign establishment back onto their Establishment Licence.
Importation in urgent circumstances
An establishment in Canada that wishes to import blood and/or blood components in urgent circumstances must provide the following documents to Health Canada, in advance of the importation:
- A completed Blood Establishment Licence Application (FRM-0354);
- A valid copy of the foreign establishment’s licence or registration, issued by the regulatory authority in the foreign jurisdiction (e.g. United States Food and Drug Administration Blood Establishment Registration and Product Listing).
Upon approval, the foreign establishment will be listed in the Establishment Licence Other Establishment(s) Annex with Terms and Conditions for importation only in urgent circumstances, pursuant to section 92 of the Blood Regulations.
See guidance under section 92 for details on additional information required to be filed prior to the importation.
Information on request
As per subsection 18(2) Health Canada may request additional information, when necessary, to complete the review of an establishment’s application. The applicant must provide the additional information in writing by the date specified in the request.
Section 19 Inspection
Inspection
19. (1) During the review of an application for an establishment licence, the Minister may inspect the establishment’s facilities and equipment to assess whether the applicant’s activities are conducted in accordance with its proposed authorization and with these Regulations.Information on request
(2) An establishment must provide the Minister, on written request, with any information that she or he determines is necessary to complete the inspection, by the date specified in the request.
Establishments may be inspected prior to the issuance of the Establishment Licence and, therefore, must be prepared for the possibility of an inspection when filing the application for an Establishment Licence. Upon review of the completed application form, an inspection will be scheduled as required and the inspection results will be communicated to the establishment.
During an inspection, an establishment is inspected for their compliance with the requirements of the Blood Regulations and their Authorization. If a Compliant Rating is issued, the Establishment Licence will be issued to the establishment. An establishment shall not commence their activities until they have obtained an Establishment Licence to do so.
Section 20 Issuance
Issuance
20. On completion of the review of an application, the Minister must issue an establishment licence, with or without terms or conditions, if both of the following requirements are met:
- an authorization has been issued with respect to the blood — except blood from a pre-assessed donor — that is proposed to be processed or imported under the licence; and
- the Minister determines that the application provides sufficient evidence to demonstrate that issuance of the licence will not compromise human safety or the safety of blood.
Once the BPCP of the ROEB processes the establishment’s application, an Establishment Licence can be issued if the following requirements are met:
- an Authorization has already been issued in respect of the blood; and
- the Minister is satisfied that issuance of the Establishment Licence will not compromise human safety or the safety of blood and blood components.
Information related to licensed establishments
The information related to licensed establishments in Canada, including terms and conditions of buildings in Canada, is publicly available on the Drug and health product inspections database.
Establishment Licence expiry
An Establishment Licence issued under the Blood Regulations will not expire. However, licensed establishments will be subject to regular inspections to assess their continued compliance with the Blood Regulations at the frequency described in the Inspection Approach for Blood Establishments (POL-0039).
Section 21 Refusal
Refusal
21. The Minister may refuse to issue a licence if she or he determines that any of the information provided by the establishment in its application is inaccurate or incomplete.
It is the applicant’s responsibility to ensure that a complete and accurate application is filed with the Minister as represented by the BPCP of the ROEB of Health Canada. Failure to file a complete application may result in the refusal of the application. If the application is refused, the establishment will be informed in writing.
Section 22 Changes requiring application to amend licence
Changes requiring application to amend licence
22. (1) Before making any change that affects the information provided under any of paragraphs 18(1)(f) to (i), (k) and (l), the establishment must, subject to paragraph 23(b), file with the Minister an application to amend the licence.Applications
(2) Sections 18 to 21 apply to an application to amend a licence, with any necessary modifications.
Amendments (including additions, removals, modifications, and corrections) to information provided under any of paragraphs 18(1)(f) to (i), (k) and (l) must be filed with the BPCP of the ROEB using the Blood Establishment Licensing Application Form (FRM-0354). The email or letter accompanying the application should include the purpose of the application and any additional information to describe the change . Please see the form for further instructions. You can request a copy of the form by emailing roeb.blood-sang.dgoral@hc-sc.gc.ca.
If the application to amend the licence meets the requirements of the Blood Regulations, an updated Establishment Licence will be issued to reflect the amendment(s).
Note: Temporary cessation of activities does not require an amendment or notification.
Section 23 Administrative changes - notice
Administrative changes - notice
23. An establishment must notify the Minister in writing of the following changes:
(a) as soon as possible after any change is made to the information provided under any of paragraphs 18(1)(a) to (e); and
(b) within 30 days after the cessation of any licensed activity.
The Blood Establishment Licence Application Form (FRM-0354) is used when notifying the BPCP of the ROEB of administrative changes. See section 18 of this guidance for information on filing the application form.
Section 24 Changes requiring amendment of licence by Minister
Changes requiring amendment of licence by Minister
24. The Minister must amend an establishment licence in any of the following circumstances:
- an authorization is amended in a way that affects the information provided by the establishment under any of paragraphs 18(1)(f) to (k);
- the Minister receives a notice from the establishment under paragraph 23(a) concerning a change to the information provided under paragraph 18(1)(a);
- the Minister receives a notice from the establishment under paragraph 23(b) that it has ceased one or more but not all of its licensed activities; or
- an authorization is cancelled, and the cancellation affects the information provided by the establishment under any of paragraphs 18(1)(f) to (k).
Change to Authorization before Establishment Licence
If a licensed establishment intends to add an activity to their Establishment Licence, they must first add the processing or importation activities to their Authorization.
Section 25 New or amended terms and conditions
New or amended terms and conditions
25. (1) The Minister may add terms and conditions to an establishment licence or amend its terms and conditions in either of the following circumstances:
- the Minister has reasonable grounds to believe that it is necessary to do so to prevent a compromise to human safety or the safety of blood; or
- the establishment fails to provide the Minister, on written request, with sufficient evidence to demonstrate that the activities it conducts are in compliance with these Regulations, by the date specified in the request.
Notice
(2) Before adding terms or conditions to a licence or amending its terms or conditions, the Minister must send the establishment a notice at least 15 days before the day on which the proposed terms and conditions are to take effect that sets out the Minister’s reasons and that gives the establishment a reasonable opportunity to be heard concerning them.Urgent circumstances
(3) Despite subsection (2), the Minister may immediately add terms and conditions to a licence or amend its terms and conditions if she or he has reasonable grounds to believe that it is necessary to do so to prevent a compromise to human safety or the safety of blood.Urgent circumstances — notice
(4) When the Minister adds or amends terms or conditions under subsection (3), the Minister must send the establishment a notice that sets out the reasons for the new or amended terms and conditions and that gives the establishment a reasonable opportunity to be heard concerning them.Removal of terms and conditions
(5) The Minister may, by notice in writing, remove a term or condition from a licence if she or he determines that the term or condition is no longer necessary to prevent a compromise to human safety or the safety of blood.
Outlining terms and conditions
If assigned, an establishment must comply with all terms and conditions set out by the Minister. These terms and conditions are outlined in the terms and conditions annex to the Establishment Licence.
Removing terms and conditions
The Minister may remove a term or condition from the Establishment Licence if she or he determines that it is no longer necessary. In such a case, an amended Establishment Licence will subsequently be issued to the establishment.
Section 26 Additional information
Additional information
26. An establishment must provide the Minister, on written request, with any additional relevant information to demonstrate that the activities it conducts are in compliance with these Regulations, by the date specified in the request.
Section 27 Suspension
Suspension
27. (1) The Minister may suspend all or part of an establishment licence in any of the following circumstances:
- information provided by the establishment under section 18 or 22 proves to be inaccurate or incomplete;
- the establishment fails to provide the Minister, on written request, with sufficient evidence to demonstrate that the activities it conducts are in compliance with these Regulations, by the date specified in the request; or
- the establishment is not in compliance with these Regulations.
Notice
(2) Before suspending a licence, the Minister must send the establishment a notice that
- sets out the reasons for the proposed suspension and the effective date;
- if applicable, indicates that the establishment must take corrective action and specifies the date by which it must be taken; and
- gives the establishment a reasonable opportunity to be heard concerning the suspension.
Urgent circumstances
(3) Despite subsection (2), the Minister may immediately suspend all or part of a licence if she or
he has reasonable grounds to believe that it is necessary to do so to prevent a compromise to human safety or the safety of blood.Urgent circumstances — notice
- (4) When the Minister suspends a licence under subsection (3), the Minister must send the establishment a notice thatsets out the reasons for the suspension; and
- gives the establishment a reasonable opportunity to be heard concerning the suspension.
An establishment may not conduct any activities that the Minister has suspended on their Establishment Licence.
Section 28 Reinstatement
Reinstatement
28. (1) Subject to subsections (2) and (3), the Minister must reinstate an establishment licence if the establishment provides the Minister with sufficient evidence to demonstrate that it is in compliance with these Regulations.Exception — compliance history
(2) The Minister may refuse to reinstate an establishment’s licence if its compliance history demonstrates an inability to consistently conduct its activities in accordance with these Regulations.Partial reinstatement
(3) If the Minister does not reinstate any part of a licence that was suspended, the Minister must amend the licence to remove that part.
The Minister may refuse to reinstate all or part of an establishment’s licence if the establishment does not consistently comply with the Blood Regulations or the Food and Drugs Act. If an establishment does not demonstrate willingness or consistently refuses to implement corrective actions for non-compliance or the corrective actions fall short of rectifying the non-compliance, the Minister will not reinstate the establishment’s licence.
Section 29 Cancellation
Cancellation
29. (1) The Minister must cancel an establishment licence in any of the following circumstances:
- the establishment notifies the Minister under paragraph 23(b) that it has ceased all activities under the licence;
- the establishment fails to provide the Minister with the evidence described in paragraph 27(1)(b) within a reasonable period after the licence was suspended;
- the establishment’s compliance history demonstrates an inability to consistently conduct its activities in accordance with these Regulations; or
- no authorization under which the establishment processes blood remains in effect.
Notice
(2) On the cancellation of a licence, the Minister must send the establishment a notice that sets out the reasons for the cancellation and the effective date.
An establishment is not allowed to conduct any activities requiring a licence at a building for which it does not hold an Establishment Licence, including those where the licence has been cancelled. If an establishment intends to commence activities requiring a licence at a site for which its Establishment Licence was cancelled, it must file a new Blood Establishment Licence Application Form (FRM-0354). You can request a copy of the form by emailing roeb.blood-sang.dgoral@hc-sc.gc.ca.
Sections 30-37 Registration
Section 30 Requirement to register
Requirement to register
30. (1) An establishment that processes autologous blood, that transforms blood or that has a pre-assessed donor program must be registered under these Regulations to do so.Exceptions
(2) Subsection (1) does not apply to an establishment that only tests autologous blood or to an establishment whose only transformation activity is to pool cryoprecipitate.
Who is required to register?
The Blood Regulations require each of the following establishments to be registered:
A. Establishments that collect autologous blood, except for those that only test autologous blood samples
Establishments that conduct collection or component preparation of autologous blood are required to register with Health Canada. If another establishment conducts the testing on behalf of the establishment who collects autologous blood, the collecting applicant must list them on their application. The establishment who collects autologous blood is responsible for the testing activity whether it is conducted by them or by another establishment on their behalf.
Establishments that only test autologous blood samples on behalf of an establishment that holds a Registration to process autologous blood are not required to register.
Contract establishments are listed as Other Establishments on the Registration application and registration certificate of the registrant.
B. Establishments that have a Pre-Assessed Donor Program
C. Establishments that transform blood
Based on the definition of transformation, if an establishment pools, irradiates or washes whole blood or any components, then they are required to register with Health Canada for those activities. Pooling includes mixing two or more different components and therefore an establishment that mixes blood must register for that activity. For example, mixing red blood cells with plasma for the purpose of exchange transfusion is considered a transformation activity under the Blood Regulations.
Establishments whose only transformation activity is to pool cryoprecipitate are not required to register.
In addition to the washing of red blood cells to remove any trace of plasma proteins and anticoagulant, the washing of thawed red blood cells to remove the cryoprotectant (deglycerolization) is also included in transformation activities. Plasma reduction, saline replacement and volume reduced platelets are manipulation of the unit that are regulated but not currently considered transformation under the Blood Regulations and therefore do not require a registration. See chart 2 for information related to which sections of the Blood Regulations apply to non-registered establishments.
Although Establishments whose only transformation activity is to pool cryoprecipitate are not required to register, pooling of cryoprecipitate is still a transformation activity. Safe and effective methods must be used to ensure the safety of the blood and relevant sections of the Blood Regulations still apply. See Chart 2 at the beginning of this guidance document for information on which sections of the Blood Regulations apply to establishments that are not required to obtain a licence or registration.
See Appendix C for a Pre-Registration Self-Assessment Tool for Establishments applying for a Blood Establishment Registration.
Summary of Requirements (also available under Section 17)
| Establishment Type/Activities | Establishment Licence | Registration |
|---|---|---|
| Processing allogeneic blood | Required | Not Required |
| Importing Blood | Required | Not Required |
| Pre-Assessed Donor Program | Not Required | Required |
| Testing laboratory for Pre-Assessed Donor Program | Required | Not Required |
| Autologous Program | Not Required | Required |
| Transformation of Blood | Not Required | Required |
| Note: “Blood” includes whole blood and blood components. | ||
Section 31 Application for registration
Application for registration
31. (1) An establishment must file with the Minister an application for registration in the form established by the Minister that contains all of the following information:
- the applicant’s name and civic address, and its postal address if different;
- in the case of an establishment that previously conducted its activities under these Regulations under another name, that other name;
- the name and telephone number, fax number, email address or other means of communication of a person to contact for further information concerning the application;
- the name and telephone number of a person to contact in an emergency, if different from the person mentioned in paragraph (c);
- a list of the processing activities that the establishment proposes to conduct in respect of autologous blood and a list of the whole blood and blood components that it proposes to process;
- a list of the transformation activities that the establishment proposes to conduct and a list of all the whole blood and blood components that it proposes to transform;
- a statement of whether the establishment has a pre-assessed donor program;
- the civic address of every building in which it proposes to conduct its activities and a list of the activities that are proposed to be conducted in each building;
- the name and civic address of any other establishment that it proposes to have conduct any of its activities; and
- a statement, dated and signed by a senior executive officer, that certifies both of the following:
- that the establishment has sufficient evidence to demonstrate that it is in compliance with these Regulations, and
- that all of the information in the application is accurate and complete.
Information on request
(2) An establishment must provide the Minister, on written request, with any information that the Minister determines is necessary to complete the Minister’s review of the application, by the date specified in the request.
Section 31 of the Blood Regulations specifies the information that must be provided in the Blood Establishment Registration Application Form (FRM-0353).
Where to find the application form
The Blood Establishment Registration Application Form (FRM-0353), along with instructions, can be obtained by sending a request to roeb.blood-sang.dgoral@hc-sc.gc.ca.
It is the responsibility of the applicant to ensure that the Blood Establishment Registration Application Form is accurate and complete in accordance with the requirements of section 31 of the Blood Regulations before filing it with the BPCP of the ROEB. This will help prevent delays in processing.
Where to file the application form
By email: roeb.blood-sang.dgoral@hc-sc.gc.ca
Section 32 Registration
Registration
32. (1) On completion of the review of an application for registration, if the Minister determines that the information provided in the application is complete, the Minister must register the establishment and issue a registration number.Refusal
(2) The Minister may refuse to register an establishment if she or he determines that the information provided by the establishment in its application is incomplete or if she or he has reasonable grounds to believe that issuance of the registration could compromise human safety or the safety of blood.
Inspection
Health Canada may inspect establishments prior to and/or after the issuance of a registration number.
Information related to registered establishments
The information related to registered establishments in Canada is publicly available on the drug and health product inspections database.
Registration expiry
Subject to section 37, there is no expiry for a Registration; however, an annual statement of compliance is required as per section 35. Additionally, an updated registration certificate will be issued if any amendments are made to its corresponding Registration.
See section 35 for additional details on how to submit the Annual Statement of Compliance
Section 33 Changes - notice
Changes — notice
33. An establishment must notify the Minister in writing of any change to the information provided under section 31, within 30 days after the day on which the change is made, and in the case of a change to the information provided under any of paragraphs 31(1)(e) to (i), include in the notice another statement described in paragraph 31(1)(j).
Notifications and amendments must be sent to the BPCP of the ROEB. Establishment should use the Blood Establishment Registration Application Form (FRM-0353). The email or letter accompanying the form should include the purpose of the application and any additional information to describe the change. Please see the form for further instructions. FRM-0353 can be obtained by sending a request to roeb.blood-sang.dgoral@hc-sc.gc.ca.
It is recommended that establishments notify the BPCP of the ROEB of any changes as early as possible, to allow these changes to be processed and reflected on their registration certificates in a timely manner.Section 34 Amendment by Minister
Amendment by Minister
34. The Minister may amend an establishment’s registration to remove from it any activity or building if she or he has reasonable grounds to believe that it is necessary to do so to prevent a compromise to human safety or the safety of blood.
Removed activities
An establishment is not permitted to conduct any activities requiring a Registration that have been removed from or do not appear on their Registration. The establishment will be informed in writing upon removal of activities from its Registration, and will receive a revised registration certificate.
Removed buildings
An establishment is not permitted to conduct any activities requiring a Registration in a building that has been removed from or does not appear on their Registration.
Re-addition of removed activities/buildings
If a registered establishment would like to add removed activities/buildings to their Registration, it must file an application to Health Canada (as per section 31 of the Blood Regulations). The establishment may be subject to an inspection and/or the provision of supporting documents to verify the compliance of the requested activities/buildings.
Section 35 Annual statement of compliance
Annual statement of compliance
35. An establishment must, by April 1 of each year, provide the Minister with a statement dated and signed by a senior executive officer that certifies that the establishment has sufficient evidence to demonstrate that it is in compliance with these Regulations.
Section 35 only applies to establishments who are required to register. Although an establishment’s registration number does not expire, the establishment must renew their statement of compliance every year before April 1st in order for their Registration number to remain valid.
A registered establishment may renew their annual statement of compliance by using the Blood Establishment Registration Application Form (FRM-0353). Additional instructions are provided with the form. FRM-0353 can be obtained by sending a request to roeb.blood-sang.dgoral@hc-sc.gc.ca. Establishments will not receive a new registration certificate every year unless they also notify Health Canada of a change requiring modification to the information on the certificate.
If an establishment does not renew their annual statement of compliance, the Minister may have reason to believe the establishment is not in compliance with the Blood Regulations and may cancel the Registration as stated in section 37.
Section 36 Additional information
Additional information
36. An establishment must provide the Minister, on written request, with any additional relevant information to demonstrate that the activities it conducts are in compliance with these Regulations, by the date specified in the request.
Section 37 Cancellation
Cancellation
37. (1) The Minister may cancel a registration in any of the following circumstances:
- the Minister receives a notice under section 33 that the establishment has ceased all of its activities that are the subject of the registration;
- information provided by the establishment under section 31 proves to be false or misleading;
- the establishment has not complied with a request for additional information made under section 36;
- the establishment fails to take any corrective action within the required period; or
- the Minister has reasonable grounds to believe that the establishment is not in compliance with these Regulations or that human safety or the safety of blood could be compromised.
Notice
(2) Before cancelling a registration, the Minister must send the establishment a notice that
- sets out the reasons for the proposed cancellation and the effective date;
- if applicable, indicates that the establishment must take corrective action and specifies the date by which it must be taken; and
- gives the establishment a reasonable opportunity to be heard concerning the cancellation.
Urgent circumstances
(3) Despite subsection (2), the Minister may immediately cancel a registration if she or he has reasonable grounds to believe that it is necessary to do so to prevent a compromise to human safety or the safety of blood.Urgent circumstances — notice
(4) When the Minister cancels a registration under subsection (3), the Minister must send the establishment a notice that
- sets out the reasons for the cancellation;
- if applicable, indicates that the establishment must take corrective action and specifies the date by which it must be taken; and
- gives the establishment a reasonable opportunity to be heard concerning the cancellation.
Action by establishment on cancellation
(5) On the cancellation of its registration for any reason set out in paragraphs (1)(b) to (e), the establishment must immediately notify any establishment to which it distributed blood that it processed or transformed during the period set out in the notice that its registration has been cancelled and the effective date of the cancellation.
An establishment is not permitted to conduct any activities requiring a Registration at a building that is not registered or for which its Registration is cancelled. If an establishment intends to conduct activities requiring a Registration, it must file a new Blood Establishment Registration Application Form (FRM-0353). FRM-0353 can be obtained by sending a request to roeb.blood-sang.dgoral@hc-sc.gc.ca.
Sections 38-58 Processing
Sections 38-44 Donor Suitability Assessment
Section 38 Non-application - autologous donations
Non-application - autologous donations
38. Sections 39 to 44 do not apply to an autologous donation.
The donor suitability assessment of an autologous blood donor is not within the scope of the Blood Regulations.
Section 39 Licensed establishments
Licensed establishments
39. A licensed establishment that collects allogeneic blood must, before the collection, assess the donor's suitability to donate against the establishment's authorized criteria.
Authorized criteria for donor suitability assessment
Section 39 only applies to licensed establishments that collect allogeneic blood for transfusion or for further manufacture.
A registered establishment that collects blood from a pre-assessed donor is not required to have authorized criteria, therefore, section 39 does not apply. See subsection 5(2) for the Pre-Assessed Donor Program exception.
A physician or physician substitute should assess a plasmapheresis donor’s suitability to donate.
See 1.5 Definitions, physician and physician substitute.
Criteria included in a donor suitability assessment
A donor suitability assessment includes donor screening and donor deferral criteria. A deferral occurs when a donor is temporarily or indefinitely unsuitable to donate blood. See section 1, the Interpretation section, for guidance concerning the definition of donor suitability assessment.
Requirement for operating procedures
Donor suitability assessment criteria authorized by Health Canada protects human safety and the safety of blood and blood components. The establishment must develop and maintain operating procedures describing in detail the criteria and the methods for assessing donor suitability. The establishment’s operating procedures must also specify frequency of donation and donor deferral time frames. See section 95 for further guidance concerning the requirement for operating procedures.
Requirement to file significant changes to donor suitability assessment criteria
An establishment must apply to the BRDD to amend their Authorization prior to implementing significant changes to its donor suitability assessment criteria. See section 9 for guidance on significant changes to authorized processes.
Requirement to provide foreign establishment's donor suitability assessment criteria if importing blood
If a licensed establishment proposes to import blood or blood components, they must provide the foreign establishment’s donor suitability assessment criteria with their application for an Authorization or an amendment to their Authorization. This must occur prior to the establishment importing the foreign blood or blood components.
Section 40 Past unsuitability
Past unsuitability
40. In conducting a donor suitability assessment, an establishment must verify whether the donor has been previously determined unsuitable, and the reason why and the duration, if applicable.
This section applies to licensed establishments that collect allogeneic blood as well as to registered establishments that have a Pre-Assessed Donor Program.
Accessibility of donor deferral records
A licensed or registered establishment must have a system for retaining and accessing donor deferral records. An establishment should have a system to check the donor deferral records of other licensed establishments in Canada.
Considering the suitability of a donor by checking donor deferral records
When considering the suitability of a donor, a licensed or registered establishment must
- confirm the donor’s identity;
- use the donor’s identity to check its donor deferral system; and
- record the reason for the deferral and the duration, if applicable.
As stated in subsection 88(1), a regular donor suitability assessment, including past unsuitability, must occur every 3 months for pre-assessed donors.
Requirement to keep records of determinations of donor unsuitability
See items 5 and 6 in the Table to section 119, Records and retention periods, for the requirement to keep records of determinations of donor unsuitability.
Section 41 Donor screening
41. In conducting a donor suitability assessment, an establishment must take both of the following steps:
- obtain information from the donor by use of a questionnaire or other similar means about their identity and medical history, and their social history to the extent that it is relevant in determining the presence of risk factors for diseases transmissible by blood; and
- provide the donor with information about the risks associated with donating blood and the risks to the recipient of contracting a transmissible disease.
41(a) and (b) Subsections 41(a) and (b) apply to licensed establishments that collect allogeneic blood as well as to registered establishments that have a Pre-Assessed Donor Program.
General donor screening requirements for allogeneic blood donors
41(a) On the day of donation, a licensed or registered establishment must assess the donor in accordance with all of the requirements in section 41. The establishment must conduct donor screening in an area that provides privacy. The establishment must provide the donor with opportunities to ask questions and to exclude themselves from donating.
A licensed establishment must base donor suitability on the following Health Canada authorized criteria:
- frequency of donation;
- donor deferral criteria, see section 39;
- laboratory test results, see section 56;
- donor medical history and/or physical examination; and
- donor social history.
Screening a pre-assessed donor
Registered establishments that have a Pre-Assessed Donor Program should have a donor screening process that reflects the Health Canada authorized criteria listed on a licensed establishment’s donor screening questionnaire. A pre-assessed donor should meet the same donor suitability requirements as an allogeneic blood donor whose blood is destined for the general blood supply. See section 42 of this guidance document for details concerning exclusion criteria.
Donor medical history and social history
A donor’s medical history refers to (1) conditions that could pose a risk to the donor, and (2) vaccinations, medications and transmissible diseases that could pose a risk to the recipient.
A donor’s social history refers to the prior activities of a donor that could put the donor and recipient(s) at risk for infection with transmissible disease(s).
See section 1, the Interpretation section, for general guidance about medical history and social history in the context of a donor suitability assessment.
Requirement to provide information about risks
41(b) A licensed or registered establishment must inform the donor of the following:
- any potential risks to the donor’s health arising from donating blood;
- any potential risks to the recipient of contracting a transmissible disease; and
- any other information that is necessary for the donor to make an informed decision to donate blood.
Establishments must have documentation, including paper or electronic documents, that communicates all of the risks in plain language that a donor can easily understand. A donor must have the opportunity to change their decision to donate at any time.
Requirement to keep records of donor suitability assessment
See item 4 in the Table to section 119, Records and retention periods, for the requirement to keep records of donor suitability assessment.
Section 42 Exclusion criteria
Exclusion criteria
42. An establishment must determine that a donor is unsuitable to donate if any of the information obtained under sections 39 to 41 indicates that human safety or the safety of blood could be compromised.
Section 42 applies to licensed establishments that collect allogeneic blood as well as to registered establishments that have a Pre-Assessed Donor Program.
A licensed or registered establishment’s donor suitability assessment process must identify and manage conditions and factors that could affect human safety or the safety of blood and blood components.
Requirement to defer a donor - licensed establishment
A licensed establishment must defer a donor if the donor does not meet the establishment’s authorized donor suitability assessment criteria. A donor deferral must also occur for any other medical reason that could affect human safety or the safety of blood and blood components. The donor’s temporary or indefinite deferral depends on the criteria that they did not meet.
Requirement to defer a donor - registered establishment with a pre-assessed donor program
A registered establishment with a Pre-Assessed Donor Program must determine the donor's suitability to donate based on the most current criteria for allogeneic blood donors. It is recommended that the Pre-Assessed Donor Program contact the licensed establishment in their jurisdiction, to obtain the applicable criteria and the donor screening questionnaire. The donor’s temporary or indefinite deferral depends on the criteria that they did not meet, in accordance with the deferral periods of the licenced establishment in the jurisdiction of the Program.
Section 43 When donor determined unsuitable
When donor determined unsuitable
43. If a donor is determined unsuitable to donate, the establishment must not collect blood from that donor and must inform the donor of the reasons why they are not suitable to donate and indicate the date, if any, when the donor will again be suitable to donate.
Section 43 applies to licensed establishments that collect allogeneic blood as well as to registered establishments that have a Pre-Assessed Donor Program.
Requirement to inform the donor of their deferral information
The licensed or registered establishment must inform the donor of the reason(s) why they must not donate blood during the deferral period. When communicating deferral information to the donor, a licensed or registered establishment must make sure that the donor clearly understands the date, if any, when the donor is eligible to return and be assessed to donate blood. An establishment may inform the donor of the deferral either in person or in writing.
Requirement to defer an unsuitable donor in a pre-assessed donor program
As stated in section 88, a regular donor suitability assessment must occur every 3 months for pre-assessed donors. A registered establishment may determine a pre-assessed donor as unsuitable to donate either during the regular assessment or immediately prior to collection. A registered establishment must defer an unsuitable pre-assessed donor either indefinitely or temporarily. During the deferral period, the registered establishment must not collect blood from the donor. See section 42 for guidance concerning Requirement to defer a donor — registered establishment with a pre-assessed donor program.
Requirement to keep records of determinations of donor unsuitability
See items 5 and 6 in the Table to section 119, Records and retention periods, for the requirement to keep records of determinations of donor unsuitability, also known as donor deferral records.
Section 44 When donor determined suitable
When donor determined suitable
44. (1) If a donor is determined suitable to donate, the establishment must take both of the following steps:
- assign a donor identification code to the donor, if the donor does not already have one; and
- instruct the donor to inform the establishment in either of the following situations:
- the donor develops, within the periods set out in the establishment’s operating procedures, an illness or condition that may potentially compromise the safety of donated blood, or
- after the donation the donor has any reason to believe that their blood should not be used.
Reassessment
(2) On receipt of any post-donation information under paragraph (1)(b), the establishment must evaluate the information to reassess the safety of the current and any other donation made by that donor and the donor’s suitability for future donations.Notice
(3) If the reassessment shows that the safety of the blood may have been compromised and the establishment has already distributed the blood, it must notify every person to which it distributed the blood to that effect, and if the person is an establishment, specify in the notice that the blood must not be distributed or transfused.
44 Subsections 44(1) and (2) apply to licensed establishments that collect allogeneic blood as well as to registered establishments that have a Pre-Assessed Donor Program.
Requirement for an establishment to assign a donor identification code
44(1)(a) An establishment must assign a donor identification code to a donor, if the donor is determined suitable to donate and if the donor does not already have one. Registered establishments should refer to subsection 89(b) for guidance concerning donor identification codes for pre-assessed donors.
Post-donation information - licensed establishment
44(1)(b) A licensed establishment must inform the donor about when to provide the establishment with post-donation information. This includes any information provided by the donor that may affect the safety of the blood they donated, such as the following:
- the donor discovers or develops an illness, disease or condition;
- the donor recalls any information or history they believe was omitted during the screening process; or
- the donor has any other reason for why the establishment must not use their blood.
44(2) When a licensed establishment receives post-donation information, this information must be taken into account when determining the safety of a donor’s blood for transfusion or blood for further manufacture into a drug for human use. A licensed establishment must consider the safety of both current and previous donations when it receives a report of post-donation information.
Furthermore, upon receipt of post-donation information, a licensed establishment must reassess the donor’s future suitability to donate blood for transfusion or for further manufacture.
Post-donation information - registered establishment with a pre-assessed donor program
44(1)(b)(i) A registered establishment should follow clause 5.1.7 of the CSA Blood Standard when instructing a pre-assessed donor about the reporting of post-donation information related to the development of an illness or condition that could affect the safety of the blood they donated.
44(1)(b)(ii) Blood collected from a pre-assessed donor is used immediately in an emergency situation. However, a registered establishment must still instruct pre-assessed donors to inform the establishment if, after their blood has been collected, they have any reason to believe that their blood should not have been transfused.
44(2) With respect to post-donation information, a registered establishment with a Pre-Assessed Donor Program should follow clauses 19.1.2 through 19.1.6 of the CSA Blood Standard.
Lookback Procedure for a licensed or registered establishment
A licensed or registered establishment must perform a lookback procedure on previous donations from an allogeneic blood donor whose blood or blood components have evidence of confirmed infection for at least any of the following:
- HIV 1 and 2
- HCV
- HBV
- HTLV I/II
- WNV
Licensed establishments should refer to section 52 and subsection 56(1) of this guidance for clarification of testing requirements.
A report of post-donation information affects the suitability of the current donation and must also be considered for previous donations, depending on the type of information reported. Post-donation information triggers a lookback procedure when there is a nucleic acid positive test result or serology tests are reactive and confirmed positive for the transmissible diseases or disease agents listed above.
The medical director should be consulted, as needed, during a lookback procedure.
See 1.5 Definitions, lookback. See also subsection 56(1) for guidance on lookback procedures.
Requirement if a reassessment shows the safety of the blood may have been compromised
44(3) When a licensed establishment determines through its reassessment of the post-donation information that the safety of the blood and/or blood components might have been compromised, it must notify every establishment and person (e.g. blood product fabricator) to whom it distributed the blood or blood components.
When a licensed establishment notifies any establishment(s) to whom it distributed blood or blood components for transfusion the notice must say that the blood must not be further distributed or transfused.
Sections 45-51 Collection
Section 45 Licensed establishments
Licensed establishments
45. A licensed establishment that collects allogeneic blood must do so in accordance with its authorization.
Requirement to collect allogeneic blood in accordance with an Authorization
A licensed establishment must collect allogeneic blood in accordance with its Authorization.
Requirement to file significant changes to collection processes
An establishment must file for review and authorization by Health Canada any significant changes to its collection processes. See section 9 for guidance on significant changes to an authorized process.
Section 46 Donor identification code
Donor identification code
46. An establishment that collects autologous blood must assign a donor identification code to the donor.
Section 47 Donation code
Donation code
47. An establishment that collects blood must assign a donation code to every unit of blood that it collects and link the code in its records to the donor identification code.
Section 47 applies to licensed establishments that collect allogeneic blood and registered establishments that collect autologous blood.
Requirement to have a donation code assigned at the time of the blood donation
Each unit of blood must have a donation code assigned at the time of the blood donation. Record-keeping procedures must allow for a link between the donation code and the donor identification code. For traceability purposes, an establishment must be able to identify the donor of a specific donation and all other donations from the same donor. The donation code must link the donor, applicable samples collected, unit of blood, time or date of collection, and donor suitability assessment records. Donation code and donor identification code are defined in section 1, the Interpretation section, of the Blood Regulations.
Registered establishment - Pre-Assessed Donor Program
A registered establishment with a Pre-Assessed Donor Program should refer to subsection 89(b).
Section 48 Labelling of containers
Labelling of containers
48. Subject to section 59, an establishment that collects blood must ensure that every container is labelled in accordance with section 63 at the time of the collection.
Labelling of containers in accordance with section 63
Establishments that collect blood, with the exception of blood collected from a pre-assessed donor, must label every container at the time of collection in accordance with section 63. A registered establishment with a Pre-Assessed Donor Program should refer to section 90 for specific labelling requirements.
Section 49 Collection procedures
Collection procedures
49. (1) An establishment that collects blood must conduct the collection in the following way:
- use aseptic methods;
- use collection equipment that is licensed under the Medical Devices Regulations;
- use containers that are licensed under the Medical Devices Regulations and free from defects or damage; and
- record the container lot number in the records and link it to the donation code.
Reuse of containers prohibited
(2) An establishment must ensure that the containers that it uses are used only once.
Blood donation collection procedures
49 All of the requirements in section 49 must be met by licensed establishments that collect allogeneic blood and by registered establishments that collect autologous blood or blood from a pre-assessed donor. A registered establishment with a Pre-Assessed Donor Program must also meet the Pre-Assessed Donor Program collection requirements in section 89.
49(1)(b) A licensed or registered establishment must use collection equipment licensed by Health Canada under the Medical Devices Regulations.
Use of an automated apheresis device to collect autologous blood
In addition to the requirement for collection equipment to be licensed a registered establishment should meet the requirements below if it uses an automated apheresis device to collect autologous blood.
- In order to ensure human safety and the safety of the blood and blood components, a registered establishment should follow collection protocols and procedures specific to the apheresis device. The registered establishment’s operating procedures should specify, for each type of blood component or combination, all requirements and criteria to achieve these goals.
- The requirements and criteria to achieve the goals stated above should be based on clinical and scientific evidence and the most up-to-date scientific knowledge for the chosen criteria.
Container lot number
49(1)(d) After recording the container lot number, a licensed or registered establishment may over-label the lot number barcode with the blood component label. In cases where a licensed or registered establishment over labels the lot number bar code, the establishment should leave the lot number text as eye-readable on the container label. The establishment must have a system to trace the specific container lot number associated with each donation.
Reuse of containers prohibited
49(2) A licensed or registered establishment must only use a container once to collect blood. The sterility of the container must not be breached.
Section 50 Samples
Samples
50. An establishment that collects blood must obtain samples of blood for testing at the same time as the collection in a way that avoids contamination of the donated blood and the samples.
Blood sample collection requirement for establishments that collect blood
Section 50 applies to licensed establishments that collect allogeneic blood and registered establishments that collect autologous blood or blood from a pre-assessed donor.
Additional requirement for a registered establishment with a pre-assessed donor program
In addition to meeting the requirement in section 50, when collecting a sample of blood, a registered establishment must also comply with the requirement in subsection 89(c) after collecting a sample of blood from a pre-assessed donor.
Section 51 Autologous donations
Autologous donations
51. An establishment that collects autologous blood must
- comply with the criteria set out in section 12.2.1 of the standard; and
- when appropriate, adjust the volume of the blood collected and the volume of anticoagulant based on the donor’s weight.
Autologous donations - volume of blood and volume of anticoagulant
51(b) When considering the volume of blood to collect from an autologous blood donor and the volume of anticoagulant needed, a registered establishment should refer to clauses 6.2.4 and 12.1.4 of the CSA Blood Standard.
Sections 52-56 Testing
Section 52 Authorization
Authorization
52. A licensed establishment that tests allogeneic blood — except blood from a pre-assessed donor — must do so in accordance with an authorization.
Requirement to test allogeneic blood in accordance with an Authorization
The establishment that holds the Authorization assumes the responsibility for the testing activity and is required to apply to Health Canada for an Authorization or for an Authorization amendment for that activity.
Exception - Testing of allogeneic blood from a pre-assessed donor
The testing of blood and/or blood components from a pre-assessed donor must be conducted by a licensed establishment as stated in subsection 17(2). A licensed establishment that tests blood and/or blood components from a pre-assessed donor must conduct the testing in accordance with sections 55 b), 88 and 89.
Contracting testing activities to another establishment
An establishment that holds an Authorization may contract out processing activities to an establishment in Canada or a foreign establishment; for example, testing of blood samples for viral markers. Health Canada does not require the contract establishment that tests the blood samples to apply for an Authorization as long as they do no other allogeneic blood processing activities. Health Canada does require a contract establishment in Canada to file an application with Health Canada for an Establishment Licence (see section 17). Health Canada does not require the foreign testing establishment to hold an Establishment Licence in Canada. See sections 17 and 18 of this guidance for more information for testing contracted to foreign establishments.
If an establishment contracts the testing to another establishment, the testing must be conducted in accordance with the contracting establishment’s Authorization. See subsection 5(1), paragraphs 6(1)(h), 6(1)(j) and 6(1)(k).
Tests that Health Canada considers appropriate and effective for testing allogeneic blood
All allogeneic blood donors must be tested and found negative or non-reactive for transmissible diseases and disease agents using appropriate and effective tests performed on the sample obtained from each donation. See sections 88 and 89 for further details concerning testing blood samples from pre-assessed donors.
A test kit used by a laboratory in Canada is considered appropriate and effective if the following requirements are met:
- It is licensed for the detection of the transmissible disease agent or marker in accordance with the licensing requirements indicated under the Food and Drugs Act and the Medical Devices Regulations; and
- The establishment uses a test kit:
- in accordance with the test kit manufacturer’s instructions;
- in accordance with their Authorization for the detection of a transmissible disease agent or marker; and
- that is equivalent or exceeds the specificity and sensitivity that is required.
Testing of samples from each allogeneic blood donation for further manufacture
Health Canada requires that an establishment test samples from each allogeneic blood donation for further manufacture for transmissible disease agents or markers, including:
- antibodies to the human immunodeficiency virus, type 1 and type 2 (anti- HIV-1 and anti-HIV-2);
- hepatitis B surface antigen (HBsAg);
- antibodies to hepatitis C virus (anti-HCV); and
- nucleic acid testing (NAT) for HIV-1, HCV, and HBV.
Syphilis testing of samples from allogeneic blood donation or plasma donation for plasma fractionation is not required. However, if it is identified as part of the establishment’s Authorization, a licensed establishment must test a sample for syphilis using a non‐treponemal or treponemal‐specific assay as per the frequency specified in their Authorization.
When licensed in vitro diagnostic devices are unavailable
If no in vitro diagnostic device (which may include both the testing platform and the test kit) — licensed in Canada — is available to test for a particular disease agent or marker, a licensed establishment may
- use an in vitro diagnostic device that has received Special Access or Investigational Testing authorization by the Medical Devices Directorate, Health Products and Food Branch, Health Canada; or,
- apply for an amendment to their Authorization to use an in-house test kit.
For an in vitro diagnostic device that has received Special Access or Investigational Testing authorization by the Medical Devices Directorate, the licensed establishment must follow the in vitro diagnostic device manufacturer’s instructions, including the following:
- the collection, handling, and storage of blood specimens;
- the time frame within which samples must be tested, if applicable;
- the procedure for testing; and
- the interpretation of the test results, including the interpretation of repeat reactive or positive results.
The licensed establishment applying for an amendment to their Authorization to use the in-house diagnostic device must include the instructions required and itemized in the list a–d above, in addition to the full set of validation data supporting the use of the in-house diagnostic device in their processing activities.
When testing is performed by a laboratory outside of Canada
If testing is performed outside Canada, the following information must be provided to Health Canada as part of the Authorization:
- details of transmissible disease agents or markers and serology testing to be employed in blood screening testing;
- algorithms to be used for each marker, in case of initial reactive tests;
- a list of all test kits currently in use at the facility;
- certification that the kit is approved by the United States Food and Drug Administration, or Health Canada's Medical Devices Directorate, and confirmation that the tests are performed in accordance with the test kit manufacturer’s instructions; or alternatively, approval to use the kit must be obtained from Health Canada’s Biologics and Genetic Therapies Directorate;
- evidence of the laboratory accreditation of the testing facility; (i.e., valid accreditation certificates indicating the laboratory is accredited for the activities being performed)
Bacteriological testing of platelets
A licensed establishment that collects or prepares platelets must have a method, authorized by Health Canada under the Blood Regulations, to detect bacterial contamination of platelets. Completion of bacteriological testing is not necessary prior to release of blood components for transfusion. Alternatively, a pathogen reduction technology approved for use in Canada may be used when a licensed establishment has an authorization to do so.
A licensed establishment’s quality management system must have protocols in place for the management of platelet units and associated components for which there is a reactive result. See section 94 for quality management system requirements.
Quality control testing
See paragraph 94(1)(b) of this guidance for quality control testing.
Section 53 Autologous donations - transmissible disease testing
Autologous donations — transmissible disease testing
53. An establishment that collects autologous blood must test a sample of the blood using appropriate and effective tests for transmissible diseases and disease agents in accordance with section 12.3.1.2 of the standard.
Frequency of transmissible disease testing - autologous donations
When a registered establishment collects more than one donation from an autologous blood donor over a 42-day period, testing is only required on the first donation for transmissible disease agents listed in clause 12.3.1.2 of the CSA Blood Standard. Once a new 42-day period begins, the establishment must test the donor’s first autologous donation for that new period.
Appropriate and effective tests for transmissible diseases and disease agents
Health Canada considers tests for the following infectious disease markers to be appropriate and effective in order to comply with clause 12.3.1.2 of the Standard:
- antibodies to the human immunodeficiency virus, type 1 and type 2 (anti-HIV-1 and anti-HIV-2);
- hepatitis B surface antigen (HBsAg);
- antibodies to hepatitis C virus (anti-HCV); and
- antibodies to human T-lymphotropic virus type I and type II (anti-HTLV-I and anti-HTLV-II).
Nucleic acid testing and syphilis testing of autologous donors is not required.
Section 54 Autologous donations - ABO and Rh
Autologous donations — ABO and Rh
54. (1) An establishment that collects autologous blood must test a sample of the blood at the time of each donation to identify both of the following:
- the ABO group; and
- the Rh factor, including weak D testing when appropriate.
Comparison of results
(2) The establishment must compare the results of the tests conducted under paragraphs (1)(a) and (b) with the last available results, if any, for that donor.Discrepancies
(3) If the comparison indicates a discrepancy, the establishment must repeat the tests and must not transfuse the blood until the discrepancy is resolved.
Section 55 Medical devices
Medical devices
55. When testing autologous blood or blood that is collected from a pre-assessed donor, an establishment must use medical devices that are licensed under the Medical Devices Regulations for the following purposes:
- either for diagnosis or for screening donors, in the case of autologous blood; and
- for screening donors, in the case of blood that is collected from a pre-assessed donor.
Test kit requirements for testing autologous blood
55(a) Subsection 55(a) applies to registered establishments that test autologous blood.
Test kits licensed by Health Canada as diagnostic assays or as screening assays must be used when testing autologous donations. The use of unlicensed test kits — including in-house tests — is prohibited.
The registered establishment must follow the test kit manufacturer’s instructions including the following:
- the collection, handling, and storage of blood specimens;
- the time frame within which samples must be tested, if applicable;
- the procedure for testing; and
- the interpretation of the test results.
Registered establishments must have operating procedures for transmissible disease testing that conform with the manufacturer’s instructions. When a contract laboratory or another establishment tests the samples, the establishment must ensure that the operating procedures of the testing laboratory conform with the test kit manufacturer’s instructions. See section 95 for guidance concerning operating procedures.
Test kit requirements for testing allogeneic blood from a pre-assessed donor
55(b) A licensed establishment must test allogeneic blood and blood components from a pre-assessed donor for transmissible disease agents and markers using test kits licensed for donor screening by Health Canada. An establishment must not use test kits licensed for diagnostic use to test allogeneic blood or blood components for transmissible diseases or disease agents. Health Canada considers test kits licensed for donor screening as more appropriate for screening allogeneic blood donors.
Note: Donor screening test kits are licensed based on testing that has been conducted in a population with a low disease prevalence (e.g. healthy blood donors), with an emphasis on test sensitivity. In contrast, diagnostic test kits are licensed based on testing conducted in a symptomatic population, with an emphasis on test specificity.
Section 56 Test results
Test results — allogeneic blood
56. (1) An establishment that collects allogeneic blood must immediately take all of the following actions if a donor’s blood is positive or repeat reactive for a transmissible disease agent or marker listed in its authorization as a contraindication to use:
- quarantine any blood that was collected from that donor at that donation;
- identify and quarantine any other implicated blood from the same donor in the establishment’s possession; and
- notify every person to which it distributed any of the implicated blood from the same donor of the test results and, if the person is an establishment, specify in the notice that the blood must not be distributed or transfused.
Test results — autologous blood
(2) An establishment that collects autologous blood must inform the donor’s physician of any of the test results described in section 12.3.1.6 of the standard..
Test results that are a contraindication to use allogeneic blood
56(1) A licensed establishment must not distribute allogeneic blood or plasma for transfusion or for further manufacture if any test results for transmissible disease agents or markers, as required by their Authorization, are positive or repeat reactive. Any test results for transmissible disease agents or markers — that are a contraindication to use the blood and/or blood components — must be negative.
In the case of a repeat reactive or positive test for a transmissible disease agent or marker listed in the establishment’s Authorization as a contraindication to use, the establishment must notify as soon as possible every establishment and person (e.g., blood product fabricator) to which it distributed any implicated blood or blood components from the same donor.
Exception - Cytomegalovirus testing
A licensed establishment that collects allogeneic blood for transfusion may choose to test certain donors for cytomegalovirus (CMV). Health Canada recommends that a donor who previously tested negative for cytomegalovirus be retested at each donation, if the establishment intends to label and distribute the unit of blood and/or blood components as CMV negative. See clause 8.6.5.3 of the CSA Blood Standard. If a unit of blood and/or blood components is CMV positive, it does not require any special treatment or labelling. An establishment may distribute CMV positive blood and blood components.
Interpretation of Transmissible Disease Test Results - Allogeneic donation
- All test results must be negative in order for a donor to be suitable (exception CMV);
- If test results are initially reactive, the establishment must repeat the testing of the sample as per the package insert instructions;
- If the donor’s specimen is repeatedly reactive or positive for a transmissible disease agent or marker listed in the establishment’s Authorization, the establishment must not release the blood and blood components for transfusion or for further manufacture;
- If a donor’s specimen tests positive for a transmissible disease agent or marker the donor must be deferred in accordance with the criteria listed in the establishment’s Authorization. In the case of a positive donor specimen from a pre-assessed donor, see section 42 for guidance concerning Requirement to defer a donor — registered establishment with a pre-assessed donor program;
- When an allogeneic unit of blood or a blood component is repeat reactive or positive for a transmissible disease agent or marker, an establishment must inform other establishments to whom it distributed any blood or blood components from the same donor;
- A licensed establishment must include the interpretation of the transmissible disease test results, according to the test kit manufacturer’s instructions, when determining if blood and blood components are safe for distribution.
Donor Re-entry Criteria
A donor re-entry algorithm specifies the processes, including donor testing and waiting period, that a licensed establishment must follow in order for a previously deferred donor to be considered for re-entry as a suitable donor. Donor re-entry algorithms must be authorized by Health Canada as part of an establishment’s Authorization.
If a licensed establishment intends to use donor re-entry algorithms for the transmissible disease agents or markers listed in their Authorization, they must file an application for an Authorization amendment providing algorithms to be used for each marker, including confirmatory testing, with supporting scientific evidence and rationale.
Lookback Procedure
A licensed establishment must carry out the lookback procedure as required by their Authorization and may choose to conduct a lookback procedure for other disease agents that are not listed in their Authorization. A licensed establishment that collects blood must initiate a lookback procedure when it receives any of the following results from donor testing, as applicable:
- Positive Nucleic Acid Test result for HIV-1, HIV-2, HCV, HBV, or WNV;
- Confirmatory positive HIV-1, HIV-2 , HCV, HBsAg or HTLV test result following a repeat reactive serology test;
- Notification of confirmatory positive test results of a donor from any of the following:
- Physician;
- Establishment, such as a hospital, a licensed or a registered establishment;
- Public Health Authority;
- Information from a Traceback investigation; or
- Donor
- Lookback Investigation (Recipient tracing).
Note: The establishment conducting the lookback procedure should receive a report containing all of the test results if it receives information from an external source.
See 1.5 Definitions, lookback.
Test results - autologous blood
56(2) A registered establishment must inform the autologous blood donor’s physician of any abnormal test results for the diseases and disease agents specified in clause 12.3.1.2 of the CSA Blood Standard. See also section 53.
Sections 57-58 Blood Component Preparation
Section 57 Licensed establishments
Licensed establishments
57. A licensed establishment must prepare allogeneic blood components in accordance with its authorization.
Section 58 Registered establishments
Registered establishments
58. A registered establishment must prepare autologous blood components in accordance with sections 7.1.3, 7.2, 7.3.1, 7.3.2, 7.5.1.1 (without regard to the reference to Table 3), 7.5.1.2 and 7.5.1.5, paragraphs 7.5.2.1(a) to (c) and section 7.5.2.2 of the standard.
Sections 59-68 Labelling
Section 59 Non-application - pre-assessed donors
Non-application — pre-assessed donors
59. Sections 60 to 68 do not apply to the labelling of blood collected from a pre-assessed donor.
See section 90 for labelling requirements that apply to blood collected from a pre-assessed donor.
Section 60 Language requirement
Language requirement
60. All of the information that is required by these Regulations to appear on a label or circular of information must be in English or French.
Section 61 General requirements
General requirements
61. A label must meet all of the following requirements:
- all information on the label must be accurate and must be presented clearly and legibly;
- it must be made using only adhesives and inks that will not permeate the container;
- it must be permanently affixed to the container; and
- in the case of a tag, it must be firmly attached to the container.
61 The label on a unit of blood and blood components must provide accurate information about the contents of the container. See clause 8.6.3.3 of the CSA Blood Standard for instances when a label may be obscured, altered or removed.
61(d) When an establishment attaches a supplementary tag to a container, this is also considered a label. Likewise, a tag must have accurate, clear and legible text.
Section 62 Circular of information
Circular of information
62. (1) An establishment that collects allogeneic blood for transfusion must prepare a circular of information in accordance with the authorization and must ensure that it makes the circular available to every establishment to which the blood is distributed and to any other person who requests a copy of it.Exception
(2) Subsection (1) does not apply if the blood is transfused in the same establishment where it is collected.
62(1) See paragraphs 6(1)(h), 6(1)(i) and 6(1)(k) for Authorization requirements that pertain to labelling, including the circular of information.
A licensed establishment that collects allogeneic blood for transfusion must prepare a circular of information in accordance with the Authorization and must ensure that it makes the circular available to every establishment to which the blood and blood components are distributed and to anyone who requests a copy of it.
62(2) An example of when it is not necessary to prepare a circular of information is an establishment that collects iRBCs for the immunization of plasma donors at the same establishment.
A registered establishment that collects autologous blood is not required to prepare a circular of information. Circular of information is defined in section 1, the Interpretation section, of the Blood Regulations.
Section 63 Donation code
Donation code
63. An establishment that collects blood must ensure that every container into which blood is collected has a label on it on which the donation code is permanently marked at the time of the collection.
The container must have a label with the donation code at the time of collection. If the donation code is missing or illegible the establishment must not distribute the blood and/or blood components for transfusion or for further manufacture. See paragraph 74(2)(a).
Section 64 Contents of label
Contents of label — blood
64. (1) An establishment that collects blood for transfusion must ensure that all of the following information appears on the label of the blood:
- the establishment’s name and civic address;
- the establishment’s licence number, if it has one, or its registration number;
- the donation code;
- a statement of whether the donation is whole blood or a blood component, and if it is a component, its name;
- when appropriate, the ABO group and Rh factor of the blood;
- except in the case of apheresis, the approximate volume of the whole blood collected;
- the approximate volume of the contents of the container;
- the name of any anticoagulant or additive in the container;
- the recommended storage temperature;
- the expiry date and, if applicable, the time;
- in the case of blood for transfusion, a warning that the blood could transmit infectious agents; and
- in the case of allogeneic blood for transfusion, a direction to refer to any applicable circular of information for indications, contraindications, warnings and a list of possible adverse reactions
Autologous blood
(2) In addition to the information required by subsection (1), the establishment must ensure that all of the following information appears on the label of autologous blood:
- the statement “For Autologous Use Only”;
- if the test results indicate that the blood is positive for a transmissible disease or disease agent listed in section 12.3.1.2 of the standard, a symbol or words to indicate that the blood is a biohazard; and
- if the blood has not been tested for the transmissible diseases and disease agents listed in section 12.3.1.2 of the standard, an indication to that effect.
Contents of label — blood for use in manufacture of drug for human use
(3) An establishment must ensure that all of the following information appears on the label of blood that is for use in the manufacture of a drug for human use:
- the name, civic address and licence number of the establishment that collected the blood;
- the donation code; and
- the statement “Caution: For Manufacturing Use Only”.
64 When labelling allogeneic blood and blood components for transfusion, an establishment must meet the requirements in 64(1)(a)–(l). The establishment may also indicate on the label if the blood tested negative for cytomegalovirus.
A licensed establishment attaches the final label to the container at the end of processing and prior to transformation or distribution.
64(1)(a) The civic address on the label may be the address of the head office of an organization.
64(1)(b) Allogeneic blood and blood components for transfusion, processed in accordance with an Authorization, must have the processing establishment’s licence number on its label. In the case of an establishment with a number of collection or production sites, the Establishment Licence number can be a single number assigned by Health Canada to the establishment and its sites.
Some establishments may have an Establishment Licence number and a Registration number. These establishments have the option of using either their Establishment Licence number or Registration number on the label of autologous blood and blood components. If an establishment collects blood for transfusion and does not have an Establishment Licence number, they must ensure that their Registration number appears on the label of any autologous units of blood that they collect.
64(1)(d) The label must have the name of the blood or blood component in eye-readable text. The name of the component includes the blood component preparation method, when appropriate, (e.g. ACD Fresh Frozen Plasma Apheresis). The naming convention in the ISBT 128 Standard is recommended.
A registered establishment must also indicate on the label if a blood component has been transformed. Transformation refers to the washing, pooling (including the pooling of cryoprecipitate), and irradiation of blood components after they have been determined safe for transfusion. It does not include blood component preparation or pathogen reduction technologies that are considered part of blood component preparation. See subsections 78(2), 79(2) and section 80 for requirements specific to labelling transformed blood.
64(1)(g) Unless otherwise indicated on the label or in Circular supplements, the contents or volume are as described in the Circular of Information or blood and blood component information documents. Examples of blood component information are information bulletins or other forms of interim documentation.
Note: The circular of information (circular supplement) is an example of supplementary information bulletins or other interim documentation that can be attached to the blood bags.
Circular of Information (circular supplement) can be an extension of the blood component label, providing information regarding component composition, packaging, storage and handling, indications, warnings and precautions, adverse events, dose and administration, and other important information about the blood component. There are separate Circulars of Information for each of the blood component groupings i.e. Red Blood Cells, Platelets, and Plasma. They conform to the applicable regulations issued by the Health Canada.
64(1)(h) The label must include the name of any additive or anticoagulant in the container. This requirement includes any anticoagulant or other additive used in the preparation of the blood or blood components. The label must also include any sedimenting agent used during cytapheresis, if applicable.
64(1)(i) The label must include the recommended storage temperature. This requirement includes the temperature range for storing the blood or blood component.
64(1)(j) The label must include the expiry date. If an expiration time is not indicated, the unit of blood expires at 23:59 on the expiry date. Expiry labels for products with a shelf-life of 72 hours or less must include the time of expiry.
For most blood components, the licensed or registered establishment may choose to include the collection date on the label.
64(2) When labelling autologous blood or blood components for transfusion, an establishment must meet the requirements in 64(1)(a)–(k) in addition to those in 64(2). The autologous blood donor/patient name may also appear on the label of the autologous unit.
Additional machine readable code should be added, if possible and for the following:
- Collecting establishment’s name;
- Donation code;
- Whole blood or the name of the blood component;
- ABO and Rh group
For registered establishments that label autologous units of blood or blood components, table 3 summarizes the required label information for verification. The asterisks indicate when additional machine readable code should be added, if possible. The autologous blood donor/patient name may also appear on the label of the autologous unit.
| Item | Required Information | Machine Readable Code |
|---|---|---|
| 1. | Collecting establishment's name | * |
| 2. | Collecting establishment's civic address | n/a |
| 3. | Collecting establishment's Registration number or Establishment Licence number | n/a |
| 4. | Donation code | * |
| 5. | Whole blood or the name of the blood component | * |
| 6. | ABO and Rh group | * |
| 7. | Volume of the whole blood collection, except in the case of apheresis | n/a |
| 8. | Approximate volume of the container contents | n/a |
| 9. | Recommended storage temperature | n/a |
| 10. | Expiry date | n/a |
| 11. | "This product may transmit infectious agents." Optional: See circular of information for indications, contraindications, cautions and methods of infusion, if applicable. |
n/a |
| 12. | Biohazard text or label, if the donor tests positive for a transmissible disease agent for which testing is required | n/a |
| 13. | "For autologous use only" | n/a |
| 14. | If a subsequent unit of blood, within a 42-day period, the statement "Untested for HIV, HBV, HCV, HTLV I/II" as appropriate | n/a |
| 15. | Name of intended transfusing establishment, if known | n/a |
| Note: “Blood” includes whole blood and blood components. | ||
64(2)(b) If an autologous blood donor tests positive for a transmissible disease or disease agent, the label on the autologous unit of blood and blood components must have a biohazard symbol or words to indicate that the blood and its components are biohazardous.
64(3) Blood for use in the manufacture of a drug for human use should clearly state the name of the component on the label.
Section 65 Aliquots
Aliquots
65. Except for purposes of immunization, an establishment that divides blood into aliquots for transfusion must ensure that all of the following information appears on the label on each aliquot
container:
- the donation code;
- the name of the blood component;
- a code that identifies the aliquot;
- when appropriate, the ABO group and Rh factor of the blood; and
- the expiry date.
65 Aliquoting is a regulated activity but not a transformation activity. Therefore, establishments conducting this activity must have an operating procedure in place for performing it.
For transfusion purposes, the following containers are considered suitable for dividing blood into aliquots:
- a transfer pack or a series of transfer packs using open or closed system technology;
- sterile vials; or
- syringes.
65(e) The establishments must have an operating procedure in place (as required by section 95 and 96 of the Blood Regulations) that defines appropriate expiration dates for aliquots. If an establishment divides blood into aliquots using an open system, aseptic techniques must be used, which may include, but are not limited to, the following:
- Low traffic area with limited air currents away from any aerosols that could contaminate the blood (e.g. sinks, centrifuges, entries, etc.), if possible;
- Defined cleaning regimen, including disinfection (both before and after use);
- Use of sterile supplies including sterile gloves;
- Storage of supplies (eg. transfer bags) in a way that prevents their contamination.
In addition, the establishment needs to adjust the expiry date for both the aliquot and the parent bag. Section 7.2.1 of the CSA Blood Standards indicates that a reduced expiration date of 24 hours be assigned to blood that is prepared in an open system and stored at 1-6° C and 4 hours if stored at 20-24° C.
If an establishment divides blood into aliquots using a closed system, then the expiry date on the original label is maintained.
The expiry date is also dependent upon storage temperatures and the type of blood component. See Table 2 of the CSA Blood Standard for storage temperatures and expiration criteria. Additionally, it is important to note that when determining the expiry date, the establishment must also consider other transformation activities that may have been performed on the blood.
Section 66 Designated donations
Designated donations
66. (1) In addition to the information required by subsection 64(1), an establishment that collects blood for designated use must ensure that the identity of the intended recipient appears on the label.Change of use
(2) The establishment must remove from the label the mention of the identity of the intended recipient when the blood is no longer intended for designated use.
66(2) Designated donations may be moved into the general allogeneic blood inventory if the following requirements for general allogeneic blood and blood components are met:
- the donor meets all donor suitability criteria; and
- the label meets the labelling requirements.
Section 67 Directed donations
Directed donations
67. In addition to the information required by subsection 64(1), an establishment that collects blood for directed use must ensure that the expression “Directed Use Only” and the identity of the intended recipient appear on the label.
A directed donation may only be used for the intended recipient. Directed donations must never be relabelled for any other use.
Health Canada acknowledges ISBT 128 and the provision of international consistency that it supports for the labelling of blood and blood components for transfusion.
Section 68 Label verification
Label verification
68. An establishment that labels blood must verify that all of the information that it adds to the label is accurate and complete.
This requirement applies to any establishment that adds information to the label.
Labelling, after the blood and blood components are determined safe for distribution, is an activity that applies to establishments that transform and/or transfuse blood and blood components. In addition to meeting this requirement, see subsections 78(2), 79(2) and section 80 for requirements specific to labelling transformed blood. See table 4 for the breakdown of labelling requirements by activity for blood for transfusion.
S.64(1) Final label requirements |
S. 79 Pooling |
Pooling – using washed components |
S.78 Washing (original bag)- will add to label |
S.78 Washing (new bag) |
S.80 Irradiation - add to label |
S.80 Irradiation (new label) |
S.65 Aliquoting |
Aliquoted then irradiated |
Aliquoted then washed |
|
|---|---|---|---|---|---|---|---|---|---|---|
Establishment name and address |
X |
X |
X |
|||||||
Establishment's licence number, or registration number |
X |
X |
X |
|||||||
Donation code |
X |
X |
X |
X |
X |
X |
||||
Name of the blood component |
X |
X |
X |
X |
X |
X |
X |
X |
||
Abo/Rh (as applicable) |
X |
X1 |
X1 |
X |
X |
X |
X |
X |
||
Approx. volume of whole blood collected |
X |
X |
X |
|||||||
Approx. volume of contents |
X |
X2 |
X2 |
X2 |
X |
|
||||
Name of anticoagulant/additive |
X |
X3 |
X |
|||||||
Recommended storage temperature |
X |
X |
X |
|||||||
Expiry date of the component |
X |
X2 |
X2 |
X2 |
X2 |
X2 |
X2 |
X2 |
X2 |
X2 |
Warning that blood could transmit infectious agents |
X |
X |
X |
|||||||
Refer to circular of information (allogeneic) |
X |
X |
X |
|||||||
# units in pool |
X |
X |
||||||||
Name of the facility preparing |
|
X |
X |
4 |
4 |
X |
X |
X |
||
Unique identifier (pool, aliquot) |
X |
X |
X |
X |
X |
|||||
Mention of the washing |
X |
X |
X |
X |
||||||
Mention that product was irradiated |
X |
X |
X |
|||||||
1 Rh is not required for pooled cryoprecipitate or plasma components. 2 The volume of the final blood and its new expiry is required, not the volume or expiry of the original component 3 The name of anticoagulant/additive should be included in cases where the expiry time is extended beyond 24 hours 4 This is not a requirement in the Blood Regulations, but should be a recommendation to the establishments |
||||||||||
Sections 69-72 Storage
Section 69 Criteria
Criteria — collecting establishment
69. (1) An establishment that collects blood must store the blood in accordance with the following:
- in the case of a licensed establishment, its authorization; and
- in the case of a registered establishment, the storage and expiration criteria specified in Table 2 of the standard.
Criteria — receiving establishment
(2) An establishment that receives blood from another establishment must store it in accordance with the directions on its label and with any other directions that are specified in writing by the establishment that collected it.
69(1)(a) Criteria — collecting establishment — licensed
A licensed establishment that stores allogeneic blood and/or blood components must file an application with the BRDD to amend its Authorization if it intends to make a change to the storage and expiration criteria required by its Authorization for the allogeneic blood it has collected. Please refer to section 9 for guidance regarding an application to amend an Authorization.
69(1)(b) Criteria — collecting establishment — registered
A registered establishment that collects autologous blood or allogeneic blood from pre-assessed donors must follow the requirements for the storage temperatures and expiration criteria of that blood as specified in Table 2 of the CSA Blood Standard.
69(2) Criteria — receiving establishment
An example of other directions specified in writing is the circular of information.
Section 70 Storage location
Storage location
70. An establishment that stores blood must do so in a location that has appropriate environmental conditions that maintain the safety of the blood and that is secure against the entry of unauthorized persons.
Guidelines and procedures must be in place and implemented for storage conditions (such as temperature, humidity, ventilation, lighting controls, component rotation, sanitation, and any other precautions needed to maintain the safety of the blood and blood components).
To ensure the safety of the blood and blood components, all blood must be stored in a location that is secure against the entry of unauthorized persons, as defined in relevant operating procedures.
Environmental parameters for storage, such as temperature must be controlled and monitored. Temperature monitoring probes or devices should be located at points that represent extreme temperature areas, as determined by a temperature mapping study to assess temperature distribution. When conducting temperature mapping studies, consideration should be given to using empty and full loads, as applicable. Parameters such as lighting, humidity and ventilation should be appropriate and controlled to safely store blood and blood components.
Storage practices and loading configurations should not obstruct air circulation. An establishment that stores blood or blood components must keep documentation as evidence that units of blood and blood components were maintained under the appropriate environmental conditions. This documentation must be available upon request.
If the storage area has an alarm system with audible signals, alarm activation points should be set at temperatures that allow time for appropriate corrective actions before the units of blood or blood components reach unacceptable temperatures. The alarm warning should signal in a location that is continually monitored or staffed so that corrective action can be taken immediately.
An establishment that stores blood or blood components must have operating procedures describing the corrective actions to be taken in the event of a deviation from established storage criteria. Such an event must be appropriately investigated and documented.
Access to storage areas must be restricted to designated personnel (e.g., authorized card access, lock and key, etc.). Where physical quarantine areas are used, they must be marked appropriately with access restricted to designated personnel. Where electronic quarantine is used, electronic access must be restricted to designated personnel.
Blood and/or blood components that have been distributed from a controlled storage location must not be accepted back into inventory unless there is evidence that the blood and blood components continued to be stored under appropriate environmental conditions or that the blood and blood components, except for platelets, were not outside of a controlled environment for greater than 60 minutes. Procedures to assess returned blood before placing it back into inventory must be followed.
Note: Establishments must have a process in place for assessing platelet suitability to return to inventory that is based on an appropriate rationale.
Section 71 Segregation - autologous, designated and directed donations
Segregation — autologous, designated and directed donations
71. An establishment that stores blood must ensure that blood that is intended for autologous, designated or directed use is segregated from blood that is intended for other allogeneic use.
Autologous, designated and directed units of blood and blood components must be clearly labeled and segregated from blood and blood components that are intended for other allogeneic use either by physical segregation and/or by using a validated electronic segregation system.
Section 72 Segregation - untested or positive or reactive test results
Segregation — untested or positive or reactive test results
72. An establishment that stores blood must segregate all of the following blood from blood that has been determined safe for distribution or autologous transfusion under section 73:
- blood that is untested;
- blood for which the testing is incomplete or for which all of the test results are not yet available; and
- blood for which the test results on blood samples are positive or repeat reactive for transmissible disease agents or markers.
The units of blood and blood components should be clearly labelled and must be controlled by a system that ensures the segregation between the units that have been tested and determined safe for distribution or for autologous transfusion from the following:
- untested;
- testing is incomplete;
- the testing results are not yet available; or
- blood with test results that are positive or repeat reactive.
This can be achieved by either physical segregation and/or the use of a validated electronic segregation system.
Sections 73-76 Distribution
Section 73 Determination of safety
Determination of safety — allogeneic blood
73. (1) An establishment that collects allogeneic blood must, before distributing it for transfusion or for use in the manufacture of a drug for human use, determine that it is safe for distribution once the establishment is satisfied that the blood has been processed in accordance with these Regulations.Determination of safety — autologous blood
(2) An establishment that collects autologous blood must, before distributing it for transfusion, determine that it is safe for autologous transfusion once the establishment is satisfied that the blood has been processed in accordance with these Regulations.
See section 1, the Interpretation section, of this guidance document for the interpretation of distribute in the Blood Regulations.
73(1) Determination of safety — allogeneic blood
Allogeneic blood and blood components must meet the safety requirements of the Blood Regulations prior to distribution, including specific processing requirements within an establishment’s Authorization. A licensed establishment that collects allogeneic blood is responsible for determining that the blood and blood components are safe for distribution.
Section 74 Verification
Verification
74. (1) Before distributing blood for transfusion or for use in the manufacture of a drug for human use, an establishment must examine the container to verify all of the following:
- the information on the label is legible;
- the integrity of the container is intact;
- there are no signs of deterioration or contamination of the blood; and
- any frozen blood components show no signs of thawing.
Prohibition — distribution
(2) An establishment must not distribute blood for transfusion or for use in the manufacture of a drug for human use if the verification carried out under subsection (1) indicates any of the following:
- the donation code is missing or illegible;
- any information — other than the donation code — that is required by these Regulations to appear on the label of blood is missing or is illegible, unless the missing or illegible information can be retrieved from the establishment’s records;
- the container is defective or damaged to the extent that it does not protect the blood against external conditions; or
- there are signs of deterioration or contamination of the blood.
Each container must be visually examined for damage or evidence of contamination prior to release into available inventory; before the released blood or blood component is distributed; and prior to further distribution. When any defect, improper labelling or abnormal appearance is observed, the component must be immediately quarantined and properly dispositioned. The verification should be conducted under lighting conditions that allows a thorough visual examination of the blood. The employees conducting this verification must be adequately trained and aware of the required criteria. Establishments should verify that employees are capable of conducting the visual examination effectively.
An establishment must segregate returned units of blood and/or blood components until these units are deemed suitable for transfusion. Returned units of blood and blood components must not be redistributed unless they meet all of the requirements in sections 70 and 74 of the Blood Regulations.
74(1) An establishment must meet the requirements in paragraphs 74(1)(a) to (d) prior to distributing blood or blood components for transfusion or for further manufacture. Steps 74(1)(a) to (d) should also occur throughout the processing of blood and blood components.
74(1)(c) and 74(2)(d) Examples of deterioration and/or contamination of the blood or blood components may include hemolysis, clots, fibrin strands, cellular aggregates, particulate matter or discoloration.
74(2)(a) and (b) If the donation code is missing or becomes illegible on the label of the blood or blood components, the establishment must not distribute it and must discard the blood. For any other missing or illegible information, if the collecting establishment can retrieve the information from their records, they may distribute the blood and blood components. The establishment must have and follow a procedure for retrieving the missing information (e.g. reprinting the label or adding it to the label). See section 61 for general labelling requirements applicable to each unit of blood and blood components.
Section 75 Shipping containers
Shipping containers
75. An establishment that ships blood must
- examine the blood containers before shipping to verify the integrity of the container and the legibility of the labels; and
- use shipping containers that are capable of resisting damage and maintaining the safety of the blood.
75(b) Shipping blood to another establishment or between different sites of the same establishment
During shipping to another establishment or between different sites of an establishment, if the blood or blood components are transported by someone other than an employee of the establishment, the shipping container must maintain the safety of the blood and blood components to ensure no tampering occurred that could affect the safety of the blood and blood components. A tamper-evident seal is one way of maintaining and verifying the integrity of the container. If a tamper-evident seal is used, the receiving establishment must verify the integrity of the tamper-evident seal upon receipt of the container.
Section 76 Storage during transportation
76. An establishment that ships blood for transfusion must ensure that the blood is stored during transportation in accordance with the criteria specified in Table 2 of the standard.
Section 76 applies to all establishments that ship blood or blood components for transfusion. The shipping containers must be validated for their intended use, unless another method is used (e.g. data logger) for ensuring storage conditions have been met and appropriately documented.
Sections 77-80 Transformation
Section 77 Transformation methods
Transformation methods
77. An establishment that transforms blood must do so using safe and effective methods.
The guidance in this section is intended for registered establishments that transform blood components, and non-registered establishments that pool cryoprecipitate. Transformation activities under the Blood Regulations include washing, pooling and irradiating, after the blood has been determined safe for transfusion.
Note: Transformation activities are not included within the scope of blood component preparation. Pathogen inactivation technologies are not included within the scope of transformation.
Establishments that transform blood components must have validated operating procedures for washing, pooling or irradiating blood components as required in sections 95, 96 and 97 of the Blood Regulations. If a biological safety cabinet or laminar flow hood is used when pooling or washing blood, it must be used according to the manufacturer’s instructions. During transformation of blood using an open system, aseptic techniques must be used, which may include, but are not limited to, the following:
- Low traffic area with limited air currents away from any aerosols that could contaminate the blood (e.g. sinks, centrifuges, entries, etc.), if possible;
- Defined cleaning regimen, including disinfection (both before and after use);
- Use of sterile supplies including sterile gloves;
- Storage of supplies (e.g. transfer bags) in a way that prevents their contamination.
Records of washing, pooling and irradiating must be kept in accordance with sections 117, 118, and 121 of the Blood Regulations.
Prior to washing, pooling or irradiating, the components to be transformed must be inspected for evidence of leaking. Each component must be visually inspected to determine if the component is suitable for transfusion. If the component’s appearance is abnormal, the establishment must follow procedures as defined by their quality management system.
Washing
A registered establishment must meet the requirements in section 78, in addition to the following safe and effective methods.
Platelets - Washed
Registered establishments that wash platelets must develop and maintain operating procedures that describe the wash procedure. It is recommended that platelets be washed in sterile normal saline solution and used within 4 hours after washing.
Red Blood Cells, Washed
Red blood cells may be washed before transfusion and must be suspended in a Health Canada approved additive solution or saline. A registered establishment must validate and document the washing process.
See section 94(1)b for quality control requirements of washed red blood cells. Registered establishments that wash red blood cells should follow the quality control specifications for “Red blood cells – washed” in table 3 of the CSA Blood Standard.
Red Blood Cells, Thawed and Washed
Red blood cells that are frozen with a cryoprotectant agent must be washed before transfusion and suspended in a Health Canada approved additive solution. A registered establishment must validate and document the thawing and washing process. See section 94(1)b for quality control requirements of washed red blood cells. Registered establishments that wash red blood cells should follow the quality control specifications for “Red blood cells – frozen (deglycerolized)” in table 3 of the CSA Blood Standard.
Pooling
An establishment must meet the requirements in section 79, in addition to following safe and effective methods. An establishment that pools blood components must do so in an environment that is suitable for this purpose. Precautions must be taken by the establishment to prevent contamination of the ports of the blood unit being handled. See 1.5 Definitions pooling: pooling includes mixing.
Cryoprecipitate, pooled
Units of cryoprecipitate are prepared by licensed establishments that hold an Authorization. Pooling of cryoprecipitate is a transformation activity that does not require a Registration. Establishments that pool cryoprecipitate must use safe and effective methods, including using equipment validated for this purpose. Examples of equipment that may be used include, but are not limited to bio safety cabinets, thawing devices, sterile docking devices etc. All equipment must be used and maintained in accordance with the manufacturer’s instructions. When pooling in an open system, aseptic technique must be used as described above.
Irradiation
Irradiation requirements, within the scope of transformation activities, are specific to gamma irradiation. A registered establishment must meet the requirements in section 80, in addition to the following safe and effective methods. Health Canada recommends dedicated irradiation equipment be used when irradiating blood components. If a registered establishment intends to use radiotherapy machines to irradiate blood components, equivalent validated operating procedures are required for the use of this equipment for this purpose. The irradiation equipment must be maintained as required in section 100 of the Blood Regulations. Irradiation dosage measurements must be monitored and documented by the establishment.
Platelets, irradiated
A registered establishment may irradiate platelets at any time during their seven-day storage period. Once the platelets are irradiated, they may continue to be stored up to their standard expiry date.
Granulocytes, irradiated
When granulocytes are to be irradiated, a registered establishment should irradiate them as soon as possible following component preparation. Irradiated granulocytes should be transfused as soon as possible.
Section 78 Washing
Washing
78. (1) An establishment that washes blood must do so in accordance with sections 7.5.2.3 and 7.5.3 of the standard.Labels
(2) An establishment that washes blood must amend the label to add to it a mention of the washing and any new expiry date and time.
78(1) Red blood cells — washed in an open system — must be stored in accordance with clause 7.5.3.4 of the CSA Blood Standard.
If a closed system is used, the red blood cells must be stored in accordance with a defined validated period. A closed system has little or no interaction with external environmental conditions that could lead to the contamination of the blood component. An establishment may use sterile connecting devices to avoid contamination of the blood component during the washing process.
An establishment that washes red blood cells must meet the storage requirements stated in Table 2 of the CSA Blood Standard.
78(2) If the washed red blood cells are transferred into a new blood container, the new label must contain the information from the original label, including the donation code, in addition to the name of the washed red blood cell component and the new expiry date.
Section 79 Pooling
Pooling
79. (1) An establishment that pools blood components must do so in accordance with sections 7.11.1 and 7.11.3 of the standard.Labels
(2) An establishment that pools blood components must ensure that all of the information specified in sections 10.8.2 and 10.8.3 of the standard appears on the label of the pooled components.
Section 79 applies to all pooling activities. Although pooling of cryoprecipitate does not require a registration, establishments that pool cryoprecipitate must do so in accordance with this section.
Section 7.11.3 of the CSA Blood Standards describes the expiration times for pooled or mixed platelets or cryoprecipitate prepared in an open system. For other components, prepared in an open system, section 7.2 of the CSA Blood Standards describes the necessary expiration times.
Section 80 Irradiation
Irradiation
80. An establishment that irradiates blood must do so in accordance with sections 7.12.2 to 7.12.6 of the standard.
In accordance with clause 7.12.3 of the CSA Blood Standard, a registered establishment must have a validated method in place to ensure that the blood component has received the required dosage of irradiation. An establishment can monitor the irradiation of blood components by using a radiation sensitive label or device and documenting the blood component dosimetry results in its records. An establishment should be able to demonstrate compliance of its component labelling and release procedures with respect to irradiation. Expiration time of irradiated blood must follow the requirements of section 7.12.6 of the CSA Blood Standard.
Sections 81-85 Exceptional Distribution
Section 81 Conditions
Conditions
81. An establishment may distribute or transfuse allogeneic blood for transfusion for which the test results for ABO group, Rh factor and transmissible diseases or disease agents are not yet available if both of the following conditions are met:
- blood that has been determined safe for distribution is not immediately available; and
- the recipient’s physician requests the blood for use in the emergency treatment of their patient.
This section only permits blood or blood components otherwise processed in accordance with an Authorization to be exceptionally distributed for transfusion. The blood or blood components that are the subject of the exceptional distribution will therefore be allogeneic blood pursuant to an Authorization but that has not been fully tested in accordance with the Authorization.
Exceptional distribution occurs as an emergency treatment for a single patient on a case-by-case basis, and when the two conditions in 81(a) and (b) are met.
Allogeneic blood donors must meet the donor suitability requirements of the Blood Regulations. The exceptional distribution section of the Blood Regulations allows for the transfusion of allogeneic blood or blood components to a single patient when all test results are not yet available for the unit(s) of blood or blood components. An establishment may release blood components for transfusion prior to the completion of bacteriological testing. Please refer to section 52, bacteriological testing of platelets, of this guidance.
Section 82 Notice of exceptional distribution
Notice of exceptional distribution
82. (1) An establishment that distributes blood under section 81 must complete a notice of exceptional distribution that contains all of the following information:
- the name of the establishment and the signature of the medical director;
- the donation code;
- a statement of whether the blood was whole blood or a blood component, and if it was a component, its name;
- a list of the test results that were not available at the time of the distribution;
- the name and signature of the recipient’s physician;
- the justification for the distribution;
- the name of the establishment to which it distributed the blood; and
- the date and time of the distribution.
Notice in establishments’ records
(2) The establishment must keep the notice in its records and send a copy of it to the establishment to which it distributed the blood.
Notice to be forwarded(3) If the establishment to which the blood is distributed does not perform the transfusion, it must send a copy of the notice to the establishment where the transfusion is performed.
Notice in recipient’s file
(4) The establishment where the transfusion is performed must keep the notice in the recipient’s file.
82(1)(d) The notice must contain information about all test results that were not available at the time of exceptional distribution.
82(2) Notice of exceptional distribution in the establishment's records
An establishment that holds an Authorization and makes an exceptional distribution of blood or blood components must keep a copy of the notice in its records. The notice of exceptional distribution must be accessible. Similarly, follow-up assessment and donor testing results must be available in the records of the establishment that made the exceptional distribution.
82(3) Notice of exceptional distribution to be forwarded
If the intended recipient of the blood or blood component that is the subject of exceptional distribution is transferred to another establishment, the establishment must forward the notice of exceptional distribution to the establishment where the transfusion is performed.
82(4) Notice of exceptional distribution in the recipient's file
The establishment, where the transfusion was performed, must keep a copy of the notice in the recipient’s file. Similarly, follow-up assessment and testing results of the donor are to be added to the recipient’s file.
The notice of exceptional distribution must be accessible upon request.
Section 83 Labelling
Labelling
83. An establishment that distributes blood under section 81 must label it to indicate that the testing required by these Regulations is incomplete or that all of the test results are not yet available, as the case may be.
Section 84 Follow-up
Follow-up
84. (1) An establishment that distributes blood under section 81 either before the testing is complete or before the test results are all available must, after the distribution, conduct any remaining testing and provide the establishment to which it distributed the blood with all of the relevant test results as soon as they become available.Results to be forwarded
(2) If the establishment to which the blood was distributed did not perform the transfusion, it must send a copy of the test results to the establishment where the transfusion was performed.
An establishment that holds an Authorization and distributes blood and/or blood components under the conditions of exceptional distribution must complete all testing and conduct any other appropriate follow-up testing. The establishment that distributed the blood or blood components under section 81 must notify the establishment where the blood or blood components were distributed of the test results as soon as they are available.
Section 85 When blood not transfused
When blood not transfused
85. If blood that is the subject of an exceptional distribution is not transfused into the intended recipient in the emergency, the establishment that was to perform the transfusion must not store the blood or transfuse it into another recipient.
See item 7 of the Table to section 122, Records and retention periods, for record-keeping requirements regarding the disposition of unused allogeneic blood or blood components for transfusion.
Sections 86-91 Pre-Assessed Donor Programs
Section 86 Program characteristics
Program characteristics
86. An establishment that has a pre-assessed donor program must ensure that the program has
both of the following characteristics:
- it is carried out under the supervision of a medical director; and
- it is used only when
- no other alternative source of blood appropriate for the recipient is available, and
- the recipient’s physician requests the blood for use in the emergency treatment of their patient.
Section 87 Donor identification code
Donor identification code
87. An establishment that has a pre-assessed donor program must assign a donor identification code at the time of the donor’s acceptance into the program.
The donor identification code for a pre-assessed donor is specific to their participation in the Pre-Assessed Donor Program.
Section 88 Regular donor assessment and testing
Regular donor assessment and testing
88. (1) An establishment that has a pre-assessed donor program must take both of the following steps every three months:
- assess the suitability of every donor in the program in accordance with sections 40 to 44; and
- take blood samples from every donor and test them for all of the following:
- the transmissible diseases and disease agents listed in sections 8.4.1 and 8.4.2 of the standard;
- the ABO group;
- the Rh factor, including weak D testing when appropriate; and
- clinically significant antibodies.
Comparison of results
(2) The establishment must compare the results of the tests conducted under subparagraphs (1)(b)(ii) and (iii) with the last available results, if any, for that donor.Discrepancies
(3) If the comparison indicates a discrepancy, the establishment must repeat the tests and must not collect any blood from that donor until the discrepancy is resolved.
88(1) Regular donor assessment and testing
88(1)(a) See sections 40 to 44 for donor suitability assessment guidance.
88(1)(b) Blood samples taken from a donor every three months must be tested for the infectious disease agents listed in clauses 8.4.1 and 8.4.2 of the CSA Blood Standard. Health Canada considers tests for the following infectious disease markers to be appropriate and effective in order to comply with clauses 8.4.1 and 8.4.2 of the Standard:
- antibodies to the human immunodeficiency virus, type 1 and type 2 (anti-HIV-1 and anti-HIV-2);
- hepatitis B surface antigen (HBsAg);
- total antibody to hepatitis B core antigen (anti-HBc, IgG and IgM);
- antibodies to hepatitis C virus (anti-HCV);
- antibodies to human T-lymphotropic virus type I and type II (anti-HTLV-I and anti-HTLV-II);
- syphilis using a non-treponemal or treponemal specific assay;
- WNV NAT
- during times in the year when WNV is potentially transmissible to humans in Canada; and
- for donors who have travelled to WNV endemic areas in the preceding 56 days.
88(1)(b)(ii), (iii), (iv) Blood samples taken from a donor every three months must be tested to determine the donor’s blood type (i.e. ABO group and Rh type) and clinically significant red cell antibodies.
88(1)(b) Blood samples taken from a donor every three months may be tested to evaluate or provide information about the blood itself (e.g. red blood cell phenotyping) or to determine the human leukocyte antigen (HLA) type.
Section 89 At each collection
At each collection
89. An establishment that collects blood from a pre-assessed donor must take all of the following
steps at each collection:
- assess the suitability of the donor;
- assign a donation code to the blood collected and link the code in its records to the donor identification code; and
- take a sample of blood from the donor and test it within 72 hours for all of the following:
- the transmissible diseases and disease agents listed in sections 8.4.1 and 8.4.2 of the standard,
- the ABO group,
- the Rh factor, including weak D testing when appropriate, and
- clinically significant antibodies.
89(a) See sections 40 to 44 for donor suitability assessment guidance.
89(b) Each unit of blood and blood components must have a donation code assigned at the time of collection. Record-keeping procedures must allow for a link between the donation code and the donor identification code. For traceability purposes, the registered establishment must be able to quickly identify the donor of a specific donation and all other donations from the same donor. See section 1, the Interpretation section, for the definitions of donation code and donor identification code.
89(c) A registered establishment that collects blood from a pre-assessed donor must ensure that a blood sample is taken from the donor at the time of donation and is tested within 72 hours for the infectious disease markers specified under paragraph 88(1)(b).
If any test results for transmissible disease agents or markers are positive or repeat reactive from a pre-assessed donor, it is critical that the licensed establishment that tested the blood must immediately notify the registered establishment that assessed the implicated donor.
Section 90 Labelling
Labelling
90. An establishment that collects blood from a pre-assessed donor must ensure that at least the donation code and the ABO group and, when appropriate, the Rh factor appear on the label of the blood.
Section 91 When blood not transfused
When blood not transfused
91. If blood that is collected from a pre-assessed donor is not transfused into an intended recipient in the emergency, the establishment that was to perform the transfusion must comply with the requirements of section 16.2.5 of the standard.
Section 92 Importation in Urgent Circumstances
Information — before importation
92. (1) An establishment may, in urgent circumstances, import allogeneic blood that was not processed in accordance with an authorization if it provides the Minister with all of the following information before the importation:
- the information required by paragraphs 6(1)(a) and (j) with respect to each foreign establishment that processes blood that it proposes to import;
- a copy of the circular of information for the blood that is proposed to be imported, or an equivalent document;
- a copy of the donor screening questionnaire that is used by each foreign establishment that processes blood that it proposes to import, including a document that indicates how that questionnaire differs from the one referred to in section 41;
- a description of how post-donation information described in paragraph 44(1)(b) is evaluated in the foreign jurisdiction;
- a description of the conditions of storage and transportation of the blood that is proposed to be imported, both before and after its importation;
- a description of how the establishment proposes to identify the blood as having been imported in urgent circumstances; and
- a description of how errors, accidents and adverse reactions are investigated and reported in the foreign jurisdiction.
Information — at each importation
(2) At the time of each importation described in subsection (1), the establishment must provide the Minister with the following information:
- a written justification that demonstrates the existence of urgent circumstances; and
- a description of any further processing or labelling that may need to be done to the blood before its transfusion.
Meaning of “urgent circumstances”
(3) In this section, “urgent circumstances” means that there is an insufficiency of allogeneic blood in Canada that poses an immediate and substantial risk to public health.
92(1) Information - before importation
If an establishment intends to include the importation of blood or blood component in urgent circumstances as a part of its emergency contingency plan, the establishment must meet requirements specific to this type of importation prior to the occurrence of the urgent circumstance.
92(1)(a) Health Canada requires the importing establishment to provide information about the foreign establishment, as required by paragraphs 6(1)(a) and (j), to the ORA at the BRDD, Health Canada. See subsection 6(1) of this guidance for contact information.
92(1)(b) The importing establishment must provide Health Canada with the following:
- the foreign establishment’s circular of information; or
- equivalent information about the blood components it proposes to import in urgent circumstances.
92(1)(c) The importing establishment must provide Health Canada with the following:
- the donor screening questionnaire from each foreign establishment from whom it proposes to import blood and blood components in urgent circumstances; and
- a document that describes the differences between each foreign establishment’s donor screening questionnaire and the importing establishment’s authorized donor screening questionnaire.
92(1)(d) The importing establishment must provide Health Canada with a description of how the foreign establishment evaluates post-donation information in its jurisdiction. The requirement for the evaluation of post-donation information in Canada is described in subsection 44(2) of the Blood Regulations.
92(1)(e) When planning for the importation of blood or blood components in urgent circumstances, the importing establishment must provide Health Canada with a description of the conditions of storage and transportation of the blood and blood components both before its importation and after its importation.
The conditions of storage and transportation include temperature, expiration and segregation.
92(1)(f) The importing establishment must describe to Health Canada the means the establishment will use to distinguish blood and blood components that are imported in urgent circumstances from its regular inventory. This includes how the establishment will identify and trace the blood and blood components imported in urgent circumstances when it distributes it to transfusing establishments.
92(1)(g) The importing establishment must provide Health Canada with a description of how errors, accidents and adverse reactions are investigated and reported in the foreign jurisdiction. Any errors, accidents or adverse reactions that occur as a result of processing by the foreign establishment should be reported according to the requirements of the foreign jurisdiction. If the foreign establishment is conducting an investigation into a serious error or accident with respect to blood or blood components that were imported into Canada, the importer in Canada should report the investigation to Health Canada.
92(2) Information - at each importation
When an establishment imports blood or blood components in urgent circumstances, the following information must be provided to Health Canada for each importation:
- written documentation describing the urgent circumstances and why there is insufficient allogeneic blood or blood components in Canada; and
- a description of any further processing that the establishment may need to conduct before the blood or blood components may be transfused in Canada.
92(3) Meaning of urgent circumstances
Urgent circumstances make it impossible for a licensed establishment in Canada to rely on its own allogeneic blood supply or that of other establishments in Canada. The absence of a domestic solution to the situation brings about the justification to import blood or blood components in urgent circumstances. Note: urgent circumstances do not include blood for immunization of donors of plasma for further manufacture.
Sections 93-123 Quality Management
Sections 93-94 Quality Management System
Section 93 Organizational structure
Organizational structure
93. (1) A licensed or registered establishment must have an organizational structure that sets out the responsibility of management for all activities that the establishment conducts.Oversight
(2) The establishment must have an effective quality management system, and must name an individual who has responsibility for it.Periodic review
(3) The establishment must review its quality management system at regular intervals that are specified in the operating procedures, to ensure its continuing suitability and effectiveness.
93(1) Licensed and registered establishments must identify the hierarchical structure of the establishment with clear delineation of the areas of responsibility and lines of authority in an organizational chart that is kept up-to-date. These establishments must have an individual responsible for the quality management system. In addition, key personnel could include a Medical Director (as defined in section 1 of the Blood Regulations), and staff responsible for operations (e.g. processing, transformation, distribution, etc.), as applicable. The titles and areas of responsibility of all key personnel must be documented for all activities related to blood and blood components.
93(2) Licensed and registered establishments must ensure that their activities comply with the regulatory requirements. To ensure compliance, these establishments must design and implement a quality management system that includes all elements listed in subsection 94(1).
The quality management system is an integrated system of quality assurance and quality control that includes all matters that individually or collectively maximize the safety of blood and blood components. The quality management system must encompass the following:
- be defined, documented, implemented, periodically reviewed and kept up-to-date by the establishment;
- include elements that enable the detection, tracking, investigation, prevention and correction of deficiencies that may compromise the safety of the blood and blood components;
- include an organizational structure that defines and documents the personnel responsible for all activities under these Regulations; and
- ensure that current written policies, processes and procedures that cover the regulated activities are available and communicated to all relevant personnel.
The establishment must appoint an individual responsible for the quality management system, and this individual is responsible for ensuring that defined quality objectives are met. The attainment of the quality objectives requires the participation and commitment of personnel in many different departments and at all levels within the establishment.
The individual responsible for the quality management system may delegate duties and responsibilities to qualified personnel in accordance with subsection 98(1) of the Blood Regulations, but remains accountable for those delegated duties and responsibilities.
93(3) Licensed and registered establishments must review all elements of the quality management system listed in subsection 94(1) at specified intervals defined by the establishment to ensure its continued suitability and effectiveness. Any deficiencies or areas requiring improvement must be addressed and corrected, and a plan that includes goals, objectives and action plans should be developed and utilized.
Section 94 Requirements
Requirements
94. (1) The quality management system must include all of the following elements:
- a quality assurance unit;
- a quality control program;
- a change control system;
- a process control program, within the meaning of section 3.1 of the standard;
- a system for process improvement through complaint monitoring and the implementation of corrective and preventive actions;
- a system for the identification and investigation of post-donation information, errors, accidents and adverse reactions, including the implementation of corrective action and the conduct of recalls;
- a program for the training and competency evaluation of personnel;
- a proficiency testing program for the evaluation of the accuracy and reliability of test results;
- a document control and records management system;
- an internal audit system;
- emergency contingency plans;
- a system that uniquely identifies all critical equipment and supplies;
- written specifications for all critical equipment, supplies and services;
- a program for the preventive maintenance of critical equipment; and
- a program for process validation.
Separation of functions
(2) The establishment’s quality assurance unit must be a distinct organizational unit that functions and reports to management independently of any other functional unit.Exception
(3) Subsection (2) does not apply in the case of a licensed establishment that only tests allogeneic blood or a registered establishment if the establishment ensures that any individual who conducts an internal audit does not have direct responsibility for the activities being audited
94(1)(a) Quality Assurance Unit
Quality assurance includes planned and performed actions to provide confidence that all systems and elements that impact the safety of blood and blood components are working as expected, individually and collectively. A quality assurance unit consists of one or more individuals, with defined authority and responsibility, to ensure compliance to the Blood Regulations.
94(1)(b)Quality Control program
The quality control program is a component of the quality management system that includes the activities and controls used to determine the acceptance of the establishment’s products, supplies, and equipment, based on their specifications and requirements of the Blood Regulations. Quality control activities must be conducted as per operating procedures.
Section 100 and 102 of the Blood Regulations further specifies quality control requirements for critical equipment and critical supplies, respectively, applicable for both licensed and registered establishments.
Licensed establishments
A quality control program, that assesses the quality of blood and blood components, must be followed by every licensed establishment that collects allogeneic blood for transfusion. The following must be defined by each licensed establishment and authorized by Health Canada:
- the frequency of quality control testing, expressed as a percent of overall production;
- the minimum number of tests required specified over a period of time; and
- the acceptable criteria for quality control testing of each type of component.
The results of quality control testing must be analysed on an ongoing basis and appropriate corrective actions taken when test results deviate from defined acceptable limits.
Registered establishments
Registered establishments that wash red blood cells must conduct quality control testing of the blood. The following must be defined in operating procedures:
- the type of tests that must be completed;
- the acceptance criteria for each test; and
- the quantity of units to be tested.
Registered establishments should follow Table 3 of the CSA Blood Standard for this purpose.
94(1)(c) Change control system
A change control system must be established and maintained to identify, document, review, approve and control all processes. Any changes to the processes, supplies, equipment and facilities that may impact the safety of blood and/or blood components must be properly documented, thoroughly evaluated, properly implemented and approved by the appropriate senior personnel as determined by the establishment’s organizational structure based on the scope of the change. Any significant change may necessitate revalidation in accordance with the requirements of paragraphs 94(1)(d) and 94(1)(o).
These approvals are in addition to those required by Health Canada for licensed establishments as part of their Authorization.
94(1)(d) Process control program, within the meaning of clause 3.1 of the standard
Establishments must have a process control program that covers all stages of their regulated activities. Clause 3.1 of the CSA Blood Standard defines process control as “the management of processes and procedures that affect the quality of products and services, with the goal of ensuring that processes and procedures are performed consistently and as they were intended to be performed in order to produce predictable output.”
Policies and operating procedures must be in place to ensure all processes are conducted under controlled and defined conditions by qualified personnel. Process control can include process validation and monitoring, the establishment of acceptable results or parameters and personnel training.
94(1)(e) A system for process improvement through complaint monitoring and the implementation of corrective and preventive actions
The system for process improvement includes the following two components:
- responding to and tracking complaints;
- implementation of appropriate corrective and preventive actions, when required.
Complaints
The establishment must have policies, processes and operating procedures for the handling of complaints that could impact the safety of the blood and blood components. All complaints must be reviewed, assessed by the appropriate department, documented and investigated in accordance with the establishment’s operating procedures, including identifying and implementing corrective and preventive actions, as applicable. All decisions and follow-up actions, taken as a result of a complaint investigation, must be recorded.
Corrective and preventive actions
Corrective action focuses on eliminating causes of existing nonconformities or incidents in order to prevent recurrence whereas preventive action focuses on eliminating the causes of potential nonconformities in order to prevent occurrence.
The establishment must also have a system for implementing corrective and preventive actions, in a timely manner, whenever required. Some examples include but are not limited to the following:
- after the receipt of a complaint or post-donation information;
- during lookback or traceback investigations;
- to correct deficiencies identified after an internal audit;
- to correct deficiencies identified during an inspection by Health Canada;
- during the investigation of an error or accident or an adverse reaction; and
- quality control results not meeting pre-established criteria.
As part of the establishment’s system for process improvement, if corrective and/or preventive actions are required, they must be implemented and monitored to reduce the likelihood of a recurrence and to improve current processes. Once the corrective and/or preventive actions are implemented, the effectiveness of these actions must be evaluated.
94(1)(f) System for Identification and Investigation of Post-Donation Information, Errors/Accidents, Adverse Reactions and Conduct of Recalls
Establishments must have defined processes and operating procedures to identify, gather information, and address any post-donation information (section 44 of the Blood Regulations), errors and accidents (sections 103 to 108 of the Blood Regulations), and adverse reactions (sections 109 to116 of the Blood Regulations) that occur and for lookback/traceback programs. These processes and operating procedures must include the decision-making processes used in determining whether an investigation is warranted, steps to conduct a thorough investigation of the incidents that warrant an investigation and the implementation of any corrective and preventive actions, as appropriate. For more details on the steps to conduct an investigation, refer to section 104.
Investigations and Recalls
When an incident occurs that has the potential to affect the safety of the blood and/or blood components, the establishment where the incident took place must determine if an investigation is warranted.
Every incident will require an initial evaluation to determine if regulated activities have been implicated and whether the safety of the blood and/or blood components has been affected.
If the initial evaluation determines that the incident is not related to regulated activities performed by the establishment, this determination must be documented and no further actions are required by the establishment under the Blood Regulations.
When an investigation is warranted, it should consider the following:
- The type of incident (i.e. post-donation information, errors/accidents and/or an adverse reaction)
- relevant processes/procedures;
- regulated activities involved;
- what the impact to the safety of the blood was or could have been;
- root cause analysis; and
- corrective and preventive actions or other risk mitigation measure to prevent reoccurrence.
Establishments must have a system to effectively conduct prompt recalls of blood and blood components. With regards to the recall of blood distributed for further manufacturing into a drug for human use, this falls under the Food and Drug Regulations. Operating procedures must be in place to define steps for an effective removal of any implicated blood or blood components from distribution or use. Records must be kept to allow for the prompt identification and location of implicated blood. The procedures must identify the position(s) within the establishment responsible for (i) obtaining information on the implicated blood and blood components; (ii) initiating the recall; and (iii) reviewing distribution records necessary for recall coordination. The operating procedure should also outline the communication method to be used to notify all establishments to which the blood and/or blood components were distributed and, as such, are involved in the recall. All recalled blood and blood components must be identified and placed in quarantine until their disposition is determined.
Relevant procedures must also describe reporting requirements for errors and accidents and adverse reactions to Health Canada as required in the Blood Regulations under sections 107, 108, 109, 113, 115, and 116. Further guidance on investigations for these types of incident can be found in the respective sections mentioned above.
Establishments must document and retain records related to all investigations and recalls as per the requirements for record retention of investigations in sections 119 to 122 of the Blood Regulations.
94(1)(g) A program for the training and competency evaluation of personnel
Establishments must have a written training program for all relevant personnel, related to activities regulated under the Blood Regulations, as defined in operating procedures. The training program must include a formal competency evaluation for personnel. Personnel must receive initial and on-going training appropriate to their job responsibilities as defined in the training program. For further details on training and competency evaluation programs, refer to subsection 98(2).
94(1)(h) Proficiency testing program for the evaluation of accuracy and reliability of test results
Proficiency testing monitors a testing laboratory’s ability to manage and perform activities defined by its specific program within the predetermined acceptable limits of detection and accuracy through the analysis of unknown specimens.
The establishment must ensure that all personnel involved in testing required by the Blood Regulations participate in a proficiency testing program (e.g. on a rotational basis) using the establishment’s routine testing procedures.
The results of proficiency testing must be reviewed by management and examined to identify trends that signal a systemic issue. When required, an establishment must apply corrective or preventive actions in order to rectify identified issues.
Records related to proficiency testing must include test results, trend analyses and corrective or preventive actions taken. These records must be maintained as per the requirements for record retention of proficiency testing in sections 119 and 120 of the Blood Regulations.
94(1)(i) Document control and records management system
Document Control
Establishments must define, document and maintain operating procedures to control all quality documents and information relevant to the activities they conduct with respect to the Blood Regulations.
The distribution and maintenance of operating procedures and other quality documents, e.g. policies, forms, etc., must be controlled, so that only the current versions are available for use. Previous versions of quality documents must be removed, archived, and replaced with the current approved version. Obsolete quality documents must be removed and archived. A copy of every version of the operating procedures that was implemented must be retained in accordance with sections 119–122 of the Blood Regulations.
Records Management
Records must be accurate, complete and legible and consistently maintained in a manner to preserve their completeness and integrity over time. Section 117 provides additional details on records management and quality.
94(1)(j)Internal Audit System
Internal audits must be performed on all regulated activities, at intervals specified in the establishment’s operating procedures, to verify compliance with the requirements of the Blood Regulations. The audit program must include an assessment of whether the operating procedures are being followed and the activities conducted consistently lead to the expected results. The audit plan must include an assessment of all regulated activities. Typically, these audits are conducted every two years, at a minimum. These audits must be conducted in accordance with an established program and operating procedures.
Audits can be performed by trained personnel, or by an external auditor (qualified third party) who is performing the audit on behalf of the establishment and is knowledgeable in the subject matter being audited. Auditors must not have direct responsibility for the procedures or processes they are auditing. For example, a supervisor responsible for component preparation must not audit any component preparation activities conducted by their own department.
Any establishment that contracts another establishment to perform a regulated activity on its behalf (e.g. testing) is responsible for establishing processes to periodically verify that the performance of those activities comply with the Blood Regulations and applicable operating procedures. For example, the establishment can assess another establishment’s compliance by performing an audit on the other establishment or by reviewing audit reports that are provided by that other establishment. Establishments must also verify that services provided by a third party comply with the requirements of the Blood Regulations. For example, transportation and equipment maintenance services.
The findings from audits and follow-up actions required must be documented and subsequently reviewed by the individual responsible for the quality management system. Preventive and corrective actions must be implemented in a timely manner. Records of internal audits, including preventive and corrective actions, and audits of contracted establishments must be retained in accordance with sections 119–121 of the Blood Regulations.
94(1)(k) Emergency Contingency Plans
Licensed and registered establishments must have emergency contingency plans in the event that processes are interrupted, such as in the case of a power outage or natural disaster.
The emergency plans must include:
- manual contingency procedures to conduct relevant regulated activities and maintain related records during an emergency;
- manual contingency procedure to issue blood or blood components from released inventory to hospitals and to all applicable areas where blood or blood components are needed;
- details for maintaining the safety of blood and blood components in storage.
Traceability requirements must continue to be met during an emergency event (e.g. when a computerized inventory system and/or its back-up system have malfunctioned). The emergency contingency plans must be reviewed periodically for their effectiveness. If applicable, the emergency power supply must be maintained and tested periodically for its readiness.
94(1)(l) A system that uniquely identifies all critical equipment and supplies
See section 1, the Interpretation section of this guidance document, for the definition of critical and examples of critical equipment and supplies.
As part of the quality system, an establishment must have a system to identify, document and track all critical equipment and supplies. Within this system, each piece of equipment must have a unique identifier.
94(1)(m) Written specifications for all critical equipment, supplies and services
See section 1, the Interpretation section of this guidance document, for the definition of critical and examples of critical equipment, supplies and services. Written specifications must be available for all critical equipment, supplies and services. Establishments must have defined processes and ensure that in the event of any changes to regulatory requirements or technology, the specifications continue to meet the applicable requirements of the Blood Regulations.
In cases where the specifications are not met, an establishment must have a system in place to ensure prompt effective remedial action, which could include the timely reporting of complaints, deviations or product defects to their supplier or service provider. Sections 100 - 102 provide additional requirements for critical equipment and supplies.
94(1)(n) A program for preventive maintenance of critical equipment
See section 1, the Interpretation section of this guidance document, for the definition of critical and examples of critical equipment. Critical equipment must consistently meet its specifications in order to produce blood and blood components that are safe. Establishments must have a preventive maintenance program to keep the function of all critical equipment within required performance specifications.
The preventive maintenance program must have defined processes which include a predetermined schedule of technical services for regular maintenance and calibration to verify that specifications identified in the manufacturer’s manual and any additional specifications required by the establishment’s quality system are being met. The processes must include the method to be used, frequency of maintenance and calibration and actions to be taken when equipment performance deviates from defined limits. This requirement applies to all equipment, instruments and measuring devices critical to ensuring that blood and blood components conforms to the Blood Regulations. Preventive maintenance and calibration must be conducted by qualified personnel.
The preventive maintenance schedule must be followed and all records and reports of validation, qualification, calibration, maintenance and repair, including actual test results and test equipment used with evidence of appropriate calibration, must be retained as described in sections 119-122 of the Blood Regulations. Sections 100 and 101 of the Blood Regulations describe the requirements for cleaning, validation and calibration of critical equipment.
94(1)(o)A program for process validation
Establishments must have a program in place to demonstrate that all processes related to processing and transforming activities are capable of achieving planned results and meeting predetermined specifications with a high degree of assurance.
A written validation plan should include testing methods, equipment to be used, qualified personnel conducting the validation, validation procedures (i.e. the steps that need to be accomplished to meet the validated state), acceptance criteria and supporting documentation.
The need for revalidation must be assessed when changes are made to a validated process. Depending on the nature and extent of the changes, i.e. changes that could affect the original validation, process characteristics and/or safety of the blood and blood components, a revalidation may be necessary. Documentation requirements will be the same for the initial validation of the process. Section 97 provides further details on validation of processes.
94(2) Separation of functions
An individual may have more than one function, but the quality assurance unit or individual must function and report to management independently of the individual(s) responsible for operations.
94(3) Exception
Licensed establishments that only test allogeneic blood or blood components, or establishments that conduct activities requiring registration are not required to have a quality assurance unit that is a distinct organizational unit that functions and reports to management independently of any other unit, as long as the individual who conducts an internal audit does not have direct responsibility for the activities being audited. In addition, individuals with dual roles (conducting operational activities and quality assurance activities) should not be reviewing their own work.
Sections 95-97 Operating Procedures
Section 95 Operating procedures required
Operating procedures required
95. An establishment must have operating procedures for all of the activities that it conducts with respect to human safety and the safety of blood.
Operating procedures are composed of instructions that set out the steps for an establishment to follow in conducting its activities. Operating procedures provide personnel with instructions or directions, so that activities are performed and documented consistently and in compliance with regulatory requirements.
All establishments must develop and maintain written operating procedures describing the significant steps for each regulated activity that it conducts. For example, the establishment must have operating procedures in place to outline the process to manage critical equipment, supplies and/or services used in any activity regulated under the Blood Regulations. The procedures must also cover revalidation and recalibration after a repair, move or a change to critical equipment.
With regards to the non-critical equipment, supplies and/or services used in any regulated activity, the procedures should describe their management.
Section 96 Requirements
Requirements
96. The operating procedures must meet all of the following requirements:
- be in a standardized format;
- be approved by a senior executive officer;
- be readily accessible at all locations where the activities to which they relate are conducted; and
- be kept up to date.
Refer to section 1 for the definition of “senior executive officer”.
The format of each operating procedure should include:
- the title and purpose of the procedure;
- the unique number or code identifying the document and indicating the version;
- the date of implementation and the last revision date;
- the signature of the authorizing person and the date of authorization (either written or electronic);
- appropriate page numbers;
- clear instructions to be followed that correspond to the tasks required to perform the activity and may include the completion of worksheets, forms or electronic fields;
- the department responsible for performing the operating procedure;
- references to publications cited, regulations, guidance document and standards, if applicable; and,
- references to related operating procedures and forms.
Operating procedures must be kept up-to-date. The procedures should be reviewed and/or revised periodically at a minimum every two years. The operating procedures must be reviewed by a knowledgeable person(s) and changed, as applicable: (i) after any amendment to the Blood Regulations or the referenced CSA Blood Standard; (ii) in response to audit or inspection findings; or (iii) as a result of corrective or preventive actions identified following an error, accident or adverse reaction.
All personnel responsible for carrying out a procedure must be trained prior to performing any task associated with a new or revised operating procedure. Operating procedures must be accessible, electronically or in hard copy, at the location where individuals are conducting the activities.
Hard copies of procedures necessary to operate during an emergency (i.e. downtime procedures) (see section 94(1)(k) for information related to the Emergency Contingency Plan) must be kept up-to-date and available to applicable staff.
In an urgent situation, a deviation from a current operating procedure is allowed if approved by a senior executive officer or designate, and the deviation is documented, signed and dated. The reason for the deviation from the procedure must also be documented. For licensed and registered establishments, the deviation must be managed in accordance with the quality management system of the establishment. For deviations that result in permanent changes, procedures must be revised in a timely manner.
The distribution and maintenance of operating procedures must be controlled, so that only the current versions are available for use. Previous versions of procedures must be removed, archived and replaced with the current approved version. Obsolete procedures must be removed and archived. Every version of the operating procedures that were implemented must be retained in accordance with the requirements for record retention found in sections 119–122 of the Blood Regulations.
Section 97 Documented evidence
Documented evidence
97. An establishment must have documented evidence that demonstrates that the operating procedures that it uses in processing and transforming blood will consistently lead to the expected results.
An establishment’s activities, processes and technical operating procedures used in the processing and transformation of blood and blood components must meet the following:
- validated by the establishment; or, as appropriate:
- established in standards developed by recognized and relevant professional organizations, based on established practice; or
- supported by current and relevant information available in the scientific literature.
Sections 98-102 Personnel, facilities, equipment and supplies
Section 98 Personnel
Personnel
98. (1) An establishment must have sufficient personnel, who must be qualified by their education, training or experience to perform their respective tasks, to conduct the establishment’s activities.Competency
(2) An establishment must have a program for the orientation and training, both initial and ongoing, of personnel and for the evaluation of their competency.
An establishment must prepare and maintain a current organizational chart with clear delineation of the lines of responsibility. A sufficient number of qualified personnel must be available to perform the tasks required. Personnel qualifications, roles and responsibilities must be documented.
All personnel performing, or responsible for regulated activities, must be qualified in accordance with the establishment’s policies, and have the necessary combination of education and/or experience. They must also receive training appropriate to their duties.
Personnel must receive initial and ongoing training, including remedial and retraining as necessary and appropriate for their duties. Training provided must be given by qualified personnel who have knowledge related to the activities involved. Training must be given in accordance with a training program for all personnel involved in activities carried out with respect to blood or blood components. Training must be provided prior to the initiation of job duties or performing the tasks outlined in a new procedure or any revision of an existing procedure. Personnel involved in conducting regulated activities must perform their duties in compliance with the Blood Regulations. All personnel conducting regulated activities should be aware of the requirements in the Blood Regulations and this Guidance Document, as applicable to their job responsibilities.
An establishment must have and maintain a program for the evaluation of the competency of personnel. The elements of a competency program may include, but are not limited to
- direct observation of performance;
- monitoring of records;
- written tests;
- assessment of knowledge of operating procedures and theory; and
- for personnel who normally perform testing required by the Blood Regulations, an assessment of performance through proficiency tests.
Records of the qualifications, training and continuing competency of personnel must be maintained as described in sections 119-122 of the Blood Regulations. Training must be documented and include the following:
- the date(s) on which the training was conducted and/or completed;
- the mode of training; and
- identification of the trainer(s) and trainee(s).
Section 99 Facilities
Facilities
99. A licensed or registered establishment must have facilities that permit all of the following:
- the conduct of all of its activities;
- the performance by personnel of their respective tasks using proper hygiene;
- the cleaning of the facilities in a way that maintains sanitary conditions;
- environmental controls that are appropriate to all areas where its activities are conducted;
- controlled access to all areas where its activities are conducted; and
- donor screening to be conducted in privacy.
Premises must be designed, constructed and adapted to suit the activities to be conducted. Their design and furnishing must minimize the risk of errors and accidents. Buildings must be maintained in good order.
Facilities must have a donor screening area that allows for privacy when determining donor suitability. In addition, facilities must control access to all areas where its activities are conducted, as appropriate, and the entrance to the building should be monitored.
Premises should be designed to align with the process flow, so that operations can proceed in an orderly manner and include the following for control and security:
- a blood collection area, set up for safe blood withdrawal from donors;
- storage of critical supplies, including reagents and test kits, prior to and post quality assurance acceptance;
- an area for handling blood components and reagents not suitable for use, quarantined or that have been recalled;
- designated area for preparing blood components and laboratory testing;
- designated area for labelling and releasing of components into inventory;
- designated area for transforming and manipulating blood;
- storage areas as described in sections 69–72 of the Blood Regulations and this guidance document; and
- segregation control and appropriate disposition of biological waste.
Facilities must permit the conduct of activities using proper hygiene, with an emphasis on hand hygiene. Personnel hygiene practices must also include the use of appropriate protective clothing based on the activity being conducted.
The interior surfaces of the processing areas must be free of any cracks or holes and any porous surfaces must be sealed to allow efficient cleaning. The premises are to be maintained in a clean and sanitary condition. A written sanitation program should be available that addresses good housekeeping practices. An accidental spill clean-up procedure should be available and include instructions to dispose of blood and blood components spills as biohazardous material. The choice of cleaning supplies used in the processing or transformation areas must not have any negative effects on the safety of the blood and blood components. In addition, a pest control program must be in place.
Where necessary, the building must be equipped with an appropriate HVAC (heating, ventilation and air conditioning) system to maintain temperature and air flow control.
Environmental controls must be in place where transformation or manipulation of blood takes place to avoid contamination of the blood. If a biological safety cabinet or laminar flow hood is used, it must be used according to the manufacturer’s instructions. During transformation or manipulation of blood using an open system, aseptic technique must be used as described in section 65 e).
The following considerations must be given when blood collection is conducted in a mobile clinic:
- appropriate environmental conditions (e.g. temperature, humidity);
- general cleanliness;
- provision of a secure supply of water and electricity;
- adequate space to enable the collection of blood from donors;
- adequate control of access to blood, records and equipment; and
- an area for donor screening to be conducted in privacy.
Section 100 Equipment
Equipment
100. (1) A licensed or registered establishment must ensure that the critical equipment that it uses is cleaned and maintained and, as appropriate, validated for its intended purpose and calibrated.Repair or change
(2) The establishment must, whenever necessary after it repairs or makes any change to critical equipment, revalidate and recalibrate the equipment, as appropriate.
See section 1, the Interpretation section of this guidance document, for the definition of critical and for examples of critical equipment.
Equipment should be situated in a location that facilitates cleaning and maintenance. Cleaning must be performed according to established schedules to prevent contamination and maintain the safety of the blood and blood components. Cleaning procedures must ensure that cleaning product residues do not impact the safety of blood or blood components. The procedures must also include the cleaning and decontamination of any blood or blood components spills, as applicable.
To ensure the equipment consistently operates within established specifications, it must be validated and calibrated according to the manufacturer’s instructions and any additional specifications required by the establishment’s quality system. Schedules and procedures for the maintenance and calibration of equipment must be maintained and followed according to the specifications in the equipment manual. These procedures must include the frequency of calibration and include the actions to be taken when equipment performance deviates from defined limits. This requirement applies not only to the equipment, but also to all instruments and measuring devices used (e.g. scales to weigh blood or blood components, thermometers, temperature probes, etc.) that are critical to ensuring that the blood and blood components conforms to the Blood Regulations. Paragraph 94(1)(n) also describes the preventive maintenance of critical equipment.
If equipment has been repaired, moved, or modified, then re-calibration and/or revalidation must be conducted in accordance with the manufacturer’s manual and/or the establishment’s operating procedures before further use. In addition, where appropriate, measures should be taken to prevent unintended adjustments on the equipment or instrument that may impact its calibration settings.
All records and reports of validation, qualification, calibration, maintenance and repair must be retained as described in sections 119-122 of the Blood Regulations. This includes actual test results and test equipment used with evidence of appropriate calibration. If the preventive maintenance is conducted by another entity, the records of maintenance must be accessible, as required. However, the establishment must verify that the maintenance is conducted appropriately and within the required timeframe, as per the established schedule in the procedures. This verification should be documented.
If a licensed or registered establishment uses a computer system for regulated activities, it must be validated. All non-registered establishments conducting activities regulated under the Blood Regulations should also ensure that the computer system used for regulated activities is validated. Data integrity is an important consideration for all relevant computer systems. Data integrity is the maintenance and the assurance of accuracy and consistency of data over its entire life-cycle, and is a critical aspect to the design, implementation and usage of any system which stores, processes, or retrieves data. There should be processes and operating procedures to support the maintenance and security of computer systems and their data. Controls must be in place to limit access to the computer system data to ensure unauthorized changes or deletions are not made to software or data. Additionally, there must be a way to track changes made to the electronic data (i.e. audit trails must be enabled and reviewed).
For an establishment that holds an Authorization, any modifications to the computer system must be authorized and documented as per the requirements in sections 9, 10 and 12 of the Blood Regulations.
A registered and non-registered establishment’s program for the validation of computer systems should have a system acceptance test to address the following points:
- system functionality;
- system performance;
- critical parameters; and
- operating procedures.
The tests should ensure that the computer operates as indicated and meets the user requirements.
Data of a critical computer system must be backed up regularly and securely stored for data recovery. Computer validation records must be maintained and used as a reference for any system updates, changes and data recovery in case of system failures. Evidence must be presented that the equipment is performing to its specifications prior to the return to its regular use.
Any modifications, repairs and system updates to critical electronic equipment/system must be assessed for a re-validation.
Section 101 Storage equipment
Storage equipment
101. An establishment must use equipment to store blood that enables the establishment to meet the requirements of sections 69 to 72.
The storage equipment must be qualified and calibrated to demonstrate it can continuously maintain the required temperature and any other appropriate storage conditions (e.g. agitation). A predetermined schedule for equipment maintenance must be established and adhered to in order to safeguard the safety of the blood and blood components being stored.
Storage equipment used to store blood or blood components, such as refrigerators, freezers and incubators must have measures in place to ensure continuous monitoring. Monitoring devices must be qualified, calibrated and maintained appropriately. Documentation that the blood and blood components were maintained under the appropriate environmental conditions must be retained according to sections 117 to 122 of the Blood Regulations. The storage equipment must also be secure against the entry of unauthorized persons.
The storage equipment should have an automated alarm system with audible signals for monitoring the required environmental conditions. For temperature monitoring, alarm activation points should be set at temperatures that allow sufficient time for appropriate corrective actions before the blood or blood components reaches unacceptable temperatures. The alarm warning should signal in a location that is continually monitored or staffed so that corrective actions can be taken immediately. If a manual system is employed, adequate measures must be in place for monitoring temperature and agitation devices to ensure that the safety of the blood and blood components is maintained.
The establishment must have operating procedures in place to maintain the above-mentioned equipment and a continuous temperature monitoring program for the stored blood or blood components. The operating procedures must describe the actions to be taken in the event of deviations from established temperature ranges or failure of agitation. Such events must be appropriately documented and investigated.
The activity of storage under the Blood Regulations includes storage outside of the blood bank. All refrigerators or incubators/agitators used to store blood or blood components outside of the Transfusion Medicine Laboratory or blood bank must be continually monitored to ensure that the blood and blood components are stored safely. If the blood bank is responsible for monitoring these “satellite” blood storage equipment, then they must have the documentation to show that blood and blood components have been stored in accordance with the Blood Regulations. If blood or any component is stored temporarily in transport containers, the establishment must ensure storage conditions (time and temperature) have been met:
- in accordance with its validation; or
- by using another appropriate method for monitoring temperature (e.g. data logger).
The blood bank must follow procedures in place to assess returned blood or blood components from these equipment before placing it back into their inventory. For storage equipment that do not have proper monitoring documentation or are not monitored by the blood bank, the blood and blood components must be discarded if returned.
Section 102 Supplies
Supplies
102. A licensed or registered establishment must ensure that the critical supplies that it uses are validated or qualified, as applicable, for their intended use and must store them under appropriate environmental conditions.
See section 1, the Interpretation section of this guidance document, for the definition of critical and for examples of critical supplies.
The establishment must ensure that the critical supplies are validated or qualified, as applicable, for their intended use. Every time new shipments of supplies are received, the quality release of the supply is based on established specifications and may include visual examination, lot release testing, and review of certificates of analysis. If the manufacturer has a certificate of analysis (or similar document) for the critical supplies, then the establishment must verify these documents as part of the lot release of critical supplies. Only supplies that meet the established specifications must be released for use. The conditions of use and storage of each supply must meet the manufacturer’s specifications. The establishments must have an operating procedure to monitor and strictly follow expiry dates of critical supplies.
Sections 103–108 Error and Accident Investigation and Reporting
All establishments regulated under the Blood Regulations must comply with the regulatory requirements for errors and accidents (E/A), including investigation, reporting and record keeping. This not only applies to establishments that are licensed and/or registered with Health Canada, but also to any establishment, hospital, blood bank, laboratory or center that conducts activities that fall under the Blood Regulations. The requirements with respect to the handling of errors and accidents are set out in sections 103–108 of the Blood Regulations.
- Errors and accidents under the Blood Regulations are defined as:
- Error: means a deviation from the operating procedures or applicable laws that could compromise human safety or the safety of blood.
- Accident: means an unexpected event that is not attributable to a deviation from the operating procedures or applicable laws and that could compromise human safety or the safety of blood.
These definitions are also found in the Interpretation section of the Blood Regulations. The E/As referenced in the Blood Regulations and this guidance document are limited to those that occurred during a regulated activity and as stated in the definition of error and accident, those that may compromise the safety of the blood or blood components.
Establishments may suspect that an E/A occurred during an activity that they conducted, or during an activity conducted by another establishment. In all cases, the following actions must be taken as applicable:
- All involved establishments communicate to ensure all affected establishments are aware of the E/A and any results of an investigation;
- When notifying other establishments with respect to E/A, any verbal communications must be documented and written notices must be sent as soon as possible as per sections 103(4) and 104(6);
- When required to do so by the Blood Regulations, establishments must identify and quarantine any implicated blood or blood components, to prevent the transfusion or further distribution of the implicated blood and ensure it is segregated from all other blood and blood components;
- All establishments must maintain records of distribution, as per sections 119, 120 and 122;
- At a minimum, distribution records must contain the donation codes of the blood, to permit the rapid identification and location of all blood units, as per section 118.
Incidents Outside the Scope of the Blood Regulations
The Blood Regulations do not apply to incidents involving drugs manufactured from blood (e.g., blood products such as albumin, immunoglobulins or coagulation factors). Refer to the Food and Drug Regulations for blood product reporting requirements.
Furthermore, incidents that involve practice of transfusion medicine are not in scope of the Blood Regulations. Provincial and territorial reporting requirements may apply. The following are examples of incidents that are outside the scope of the Blood Regulations:
- mix-up during the collection of pre-transfusion specimens from patients;
- cross matching/pre-transfusion recipient testing (group and screen);
- ABO/Rh retesting errors conducted at hospitals;
- blood bank sending the wrong blood to the ward (blood type or component);
- collecting establishment sends the wrong blood to the hospital;
- blood sent to the ward for the wrong patient;
- ward ordered irradiated blood component(s) and blood bank sent non-irradiated blood component(s) (correctly labeled as non-irradiated) Note: if blood component(s) was not irradiated but was labeled as irradiated, it would be an error subject to these requirements;
- transfusing the unit of blood or blood component in the wrong patient;
- transfusing the blood or blood components too slowly or too fast;
- service delivery standards not met;
- nurse decides to put a unit of blood or blood component for a few hours in a fridge not monitored and not intended to store blood or blood component and then proceeds to transfusion (e.g. medication fridge);
- transfusing expired blood or blood components. Note: if the blood or blood components were already expired and unknowingly distributed by the blood bank, it would be an error subject to Blood Regulations.
Sections 103-104 Errors and Accidents
Section 103 Error or accident of another establishment
Error or accident of another establishment
103. (1) An establishment that has reasonable grounds to believe that the safety of blood may have been compromised by the occurrence of an error or accident during an activity conducted by another establishment must immediately take all of the following actions:
- determine the donation codes of the implicated blood;
- identify and quarantine any implicated blood in its possession; and
- notify all of the following establishments:
- the establishment that collected the implicated blood,
- the establishment from which it received the implicated blood, if different from the establishment mentioned in subparagraph (i), and
- any establishment to which it distributed implicated blood.
Contents of notice
(2) The notice must include all of the following information:
- the donation codes of the implicated blood;
- a statement of whether the implicated blood is whole blood or blood components, and the names of the implicated blood components; and
- the reason for the establishment’s belief that the safety of the blood may have been compromised.
Action on receipt of notice
(3) An establishment that is notified under subparagraph (1)(c)(iii) or under this subsection must
immediately notify to the same effect every establishment to which it distributed implicated blood and quarantine all implicated blood in its possession.Written notice
(4) If a notice under this section is given verbally, a confirmatory written notice must be sent as soon as possible afterwards.
An establishment that has reasonable grounds to believe, based on the information available, that the safety of blood or blood components may have been compromised because of an E/A that occurred during an activity conducted by another establishment (e.g. transport, storage, packaging, transformation, processing, etc.) must immediately follow the actions listed in subsection 103(1).
The establishment must
- identify and quarantine any blood or blood components implicated (known or suspected) in the E/A;
- notify all relevant establishments without delay.
In some cases, further investigation into an E/A may lead to the discovery of fewer or additional blood components that are impacted.
Under paragraph 103(2)b, the names of implicated blood components must be included in the notice. For example, red blood cells, apheresis plasma and pooled platelets. In all cases, if a notice was provided verbally, a written notice must follow as soon as possible.
Under subsection 103(3), all establishments that receive the notice, must
- identify and quarantine all of the implicated (known or suspected) blood and blood components in their possession; and
- forward the notice to any establishment to which they further distributed any implicated (known or suspected) blood or blood components.
Section 104 Establishment's own error or accident
Establishment’s own error or accident
104. (1) An establishment that receives a notice under subparagraph 103(1)(c)(i) or (ii) or suspects that an error or accident that occurred during an activity it conducted may have compromised the safety of blood must immediately take all of the following actions:
- determine the donation codes of the implicated blood;
- identify and quarantine any implicated blood in its possession; and
- determine whether there is sufficient evidence to warrant proceeding to an investigation into the suspected error or accident.
When no investigation — notice
(2) If the establishment determines that an investigation is not warranted, it must notify the establishment that sent it the notice under subparagraph 103(1)(c)(i) or (ii) that it will not be conducting an investigation and provide its reasons for that decision.Action on receipt of notice
(3) An establishment that is notified under subsection (2) or under this subsection must immediately notify to the same effect every establishment to which it distributed implicated blood.Notice of investigation
(4) If the establishment determines that an investigation is warranted, it must begin the investigation, notify every establishment and other person to which it distributed implicated blood, and include the following information in the notice:
- the donation codes of all implicated blood; and
- a description of the suspected error or accident and an explanation of how the safety of the implicated blood may have been compromised.
Action on receipt of notice
(5) An establishment that is notified under subsection (4) or under this subsection must immediately notify to the same effect every establishment to which it distributed implicated blood and quarantine all implicated blood in its possession.Written notice
(6) If a notice under this section is given verbally, a confirmatory written notice must be sent as soon as possible afterwards.
Section 104 addresses situations where establishments suspect that an error or accident (E/A) occurred during an activity that they conducted, or when an establishment receives a notice that an E/A could have occurred at their establishment.
Under subsection 104(1), upon receipt of the notice or when an establishment suspects that an E/A occurred during an activity it conducted, the establishment must immediately:
- determine the donation codes of the implicated blood or blood components and whether any other units of blood may be implicated in the same E/A (e.g. other components with the same donation code, any other units that were subject to the same suspected processing, transformation or storage conditions, etc.);
- identify and quarantine any implicated blood and blood components in its possession; and
- determine whether there is sufficient evidence that the E/A occurred at their establishment in order to initiate an investigation.
Every incident requires an assessment to determine whether an E/A actually occurred and whether it occurred during activities conducted at their establishment. If an assessment determines that an E/A is not related to regulated activities performed by the establishment, this determination must be documented.
Conducting an investigation
When an E/A affects the safety of the blood and/or blood components, the establishment must immediately:
- identify and quarantine any implicated blood and blood components in inventory;
- determine if any other blood and blood components are implicated and their status (distributed, quarantined or transfused); and
- determine if there are any other affected establishments to whom blood and/or blood components were further distributed.
When an investigation is initiated, the establishment must:
- notify all establishments or other persons (including blood product fabricators) to which it distributed known or suspected implicated blood or blood components that it is conducting an investigation.
In the notice, the establishment must:
- list the known or suspected donation codes of the potentially implicated blood and blood components;
- describe the suspected E/A; and
- include an explanation of how the safety of the implicated blood or blood components may have been compromised.
If a notice was provided verbally, a written notice must follow as soon as possible.
Known and suspected implicated blood and blood components must remain in quarantine until the investigation is completed and the final disposition for the blood and blood components have been determined (i.e. release back into inventory, recall, or discard).
In addition to the immediate quarantine or recall of the implicated blood and blood components, the E/A investigation must include:
- a root cause analysis;
- consideration of the potential impact of the E/A on the safety of the blood or blood components and the recipient if the blood or blood components had been transfused (if it was not); and
- development of appropriate and corrective actions.
Even if the E/A was caught by another establishment before transfusion, the initial establishment where the E/A occurred is required to investigate and take appropriate corrective and preventive actions.
Every investigation requires an initial assessment to determine if regulated activities were implicated and whether the safety of the blood or any component has been compromised. An investigation may be very simple or very complex depending on the nature and severity of the incident, the size and complexity of the establishment and/or its activities. This may require an examination of multiple aspects in order to identify the full extent of what happened:
- relevant processes/procedures;
- what regulated activities were involved; and
- what the impact to safety of the blood was or could have been.
Details of all actions taken during the investigation must be documented.
The investigation must determine what contributed to the occurrence of the incident to identify the root cause(s) so appropriate corrective and preventive actions can be implemented to effectively mitigate the risk and prevent reoccurrence of the incident. Corrective and preventive actions will be dependent on the nature and severity of the incident, personnel, processes, equipment, materials involved, and the identified causes that allowed the incident to occur. Corrective and preventive actions could include, but are not limited to the following:
- retraining staff;
- revisions to procedures;
- adding verification steps; and
- changes to one or more system(s).
Corrective and preventive actions must be implemented in a timely manner and must be evaluated for their effectiveness.
The type of incident must be identified (i.e. error/accident and/or adverse reaction) in order to determine the course of the investigation. Establishments should be aware that some E/A may cause adverse reactions and would require investigation for both the adverse reaction and the E/A. There should be a clear link between procedures for E/As and adverse reactions to ensure that if E/As lead to adverse reactions, then these cases are appropriately addressed.
When an investigation is not warranted
An establishment that receives a notice of a suspected error or accident:
There are several scenarios where an establishment may receive a notice of a suspected E/A and determine an investigation is not warranted. For example,
- establishment B suspects an E/A occurred at establishment A and notifies them;
- upon receipt of the notice, establishment A is able to confirm that the suspected E/A is not valid.
Another example could be:
- establishment Y is notified of a suspected E/A by establishment Z to whom they distributed blood;
- on examination of the facts, establishment Y does not believe it is due to an activity they conducted. In other words, the nature of the E/A suggests it occurred at the collecting establishment X, then an investigation may not be warranted by establishment Y or Z.
Under subsection 104(2), in cases where an establishment that collected or distributed implicated blood or blood components receives a notice, and does not have reasonable grounds to believe that the safety of the blood and any component has been compromised by the suspected E/A during an activity it conducted, it must notify the establishment from which it received the notice that it will not be conducting an investigation. The establishment must provide the reasons for not conducting an investigation and retain a documented, detailed rationale for that decision in its records. This is to ensure that all suspected E/As are acknowledged and assessed.
In the event that an establishment sent a notice to multiple establishments (under subparagraphs 103(1)(c)(i) and (ii)), the final decision on the disposition of the blood must be based on the assessments of all affected establishments.
Under subsection 104(3), any establishment that receives such a notice must forward this notice to every establishment to which they distributed any implicated blood or blood components. If a verbal notice is provided, a written notice must be sent as soon as possible afterwards.
An establishment that suspects that an error or accident occurred during a regulated activity it conducted:
If an establishment suspects an E/A occurred during an activity it conducted that could compromise the safety of the blood or blood components, but then determines that an investigation is not warranted, it must document the decision. The documented decision with a detailed rationale must be retained in its records as described in sections 119-122 of the Blood Regulations. For example, the operating room notifies the blood bank of a suspected E/A. The blood bank assesses the blood and blood components and is able to confirm that the suspected E/A is not valid.
Sections 105-108 Investigation and Reporting
Section 105 Requirement to cooperate
Requirement to cooperate
105. (1) An establishment must, on request, provide any establishment that is conducting an investigation with any relevant information in its possession in respect of blood that it distributed or transfused.Communication
(2) When more than one establishment is affected by an error or accident or the investigation of one, each establishment must ensure that every other establishment that is so affected is kept informed of all relevant information and of all developments and issues that arise during the investigation.
Establishments must cooperate with any establishment that is conducting an investigation and provide any relevant information, as requested. This information includes, but is not limited to, an inventory list of implicated blood and blood components with their disposition (e.g. distributed, transfused, quarantined) and the names of establishments to which the implicated blood or any component has been distributed.
In accordance with the Blood Regulations, it is critical that all involved establishments communicate to ensure that all affected establishments receive relevant information regarding the investigation. Therefore, it is expected that establishments notify the appropriate establishments, including any establishment to which they sent implicated blood or blood components, of any investigation of a suspected E/A and of any developments and issues that arise during the investigation.
The establishment conducting an investigation must have processes in place to communicate with all establishments that may have been impacted by the E/A in a timely and accurate manner. If an establishment sends a notice to multiple establishments (103(1)(c)(i) and (ii)), the final decision on the disposition of the blood must be based on the assessments of all affected establishments. If none of the notified establishments proceed to an investigation, all affected establishments must communicate to determine the disposition of the blood and blood components.
Section 106 Investigation results
Investigation results
106. (1) An establishment that is conducting an investigation must notify in writing every establishment and other person to which it distributed implicated blood of the results of the investigation and of any action that is required to be taken.Notice to be forwarded
(2) An establishment that is notified under subsection (1) or under this subsection must send a copy of the notice to every establishment to which it distributed implicated blood.
The establishment must notify in writing all relevant establishments or other persons (including blood product fabricators) of the results of the investigation. Where the results of the investigation indicate that the safety of the implicated blood or blood components has not been compromised, the investigating establishment may make recommendations with regards to the disposition of the blood and blood components.
As stated in paragraph 4(7)(b) of the Prohibition section of the Blood Regulations, where the results of the investigation show that the safety of the implicated blood or blood components has been compromised, or the results are inconclusive, the implicated blood and blood components must not be distributed or transfused.
Upon receipt of the notice under subsection 106(1) or subsection 106(2), the establishment must send a copy of the written notice to every establishment to which they further distributed the implicated blood or blood components.
Section 107 Reports to Minister
Reports to Minister
107. (1) An establishment that is conducting an investigation into a suspected error or accident that is thought to have occurred during an activity that it conducted and that is identified after the blood is distributed or transfused must file the reports described in subsection (2) with the Minister if there is a reasonable probability that the error or accident could lead to a serious adverse reaction.Contents and timing
(2) The reports must include the following information and be filed at the following times:
- a preliminary report that includes all relevant information that is available, within 24 hours after the start of the investigation; and
- a written update on any new information about the suspected error or accident, on the progress made in the investigation since the last report and on the steps taken to mitigate further risks:
- within 15 days after the start of the investigation; and
- on request of the Minister at any time after the preliminary report.
Written notice
(3) If the report under paragraph (2)(a) is given verbally, a written report must be filed as soon as
possible afterwards.Final report to Minister
(4) On completion of an investigation, the establishment must file a final report with the Minister
that contains all of the following information:
- the results of the investigation;
- the final disposition of the blood that was the subject of the investigation and the reasons for that disposition; and
- any corrective actions taken and any other changes that are recommended to be made to relevant processes.
Section 107 applies to all establishments that distribute blood or blood components and establishments that transfuse blood or blood components who are investigating suspected E/As for a regulated activity they conducted.
The establishment conducting an investigation must file a preliminary report with Health Canada within 24 hours after the start of the investigation, if all three of the criteria below are met.
- the E/A is thought to have occurred during a regulated activity they conducted;
- the E/A is identified after the blood or blood components are distributed or transfused; and
- there is a reasonable probability that the E/A could lead to a serious adverse reaction had the blood or blood components been transfused.
See further explanations below for each criteria:
- If the E/A occurred during a regulated activity conducted at a different establishment than the one who discovered it, it is the responsibility of the establishment where the E/A occurred to report to Health Canada. Incidents related to practice of transfusion medicine are not included in the scope of these Regulations. See examples of exclusions under Sections 103–108 Error and Accident Investigation and Reporting.
- The term distribution under the Blood Regulations, is explained in section 1 Interpretation in this guidance document. The second criteria is whether or not the blood was already distributed when the E/A was discovered. In general, blood is distributed prior to transfusion. Therefore, an E/A may still be reportable if the blood was distributed even if it was not yet transfused, as the second criteria has been met. However, as an exception, blood collected from a pre-assessed donor program is not distributed before transfusion. In this case, criterion 2 would only be met for an identified E/A if the blood was transfused.
- The third criteria is to assess the potential impact of the E/A. Even if the blood or blood components were not transfused, the establishment (in consultation with the medical director or specialist in the field) is required to assess the potential impact it could have had on the recipient if it had been transfused. If the blood or any components were transfused and there was no adverse reaction, the third criterion could still be met, if there is probability that a serious adverse reaction could have occurred. The medical director or specialist in the field should be consulted to assess if the third criterion is met.
If an E/A is discovered during the investigation of an adverse reaction, the E/A must also be reported (separately from the adverse reaction that is reported under section 113) to the contact information provided below.
The preliminary report must include any information regarding the suspected E/A available at that time. The information provided in the preliminary report could consist of, but is not limited to:
- a description of the E/A;
- risk assessments;
- number and types (i.e. RBCs, platelets, plasma, etc.) of implicated units of blood and/or blood components;
- corrections taken to date (including any recall or notifications sent to establishments that received the implicated blood and/or blood components); and,
- any anticipated corrective and preventive actions.
Note: Corrections are the immediate actions taken to eliminate a detected nonconformity. Corrective actions focus on eliminating causes of existing nonconformities or incidents in order to prevent reoccurrence, whereas preventive action focuses on eliminating the causes of potential nonconformities in order to prevent occurrence.
In the case of blood or blood components that have been imported into Canada, where the foreign establishment is conducting an E/A investigation with respect to blood or blood components that were distributed to Canada — and if there is a reasonable probability that the E/A could lead to a serious adverse reaction — the importer in Canada must report the investigation to Health Canada, as the foreign establishment is conducting the activities on its behalf. E/As discovered before the distribution or transfusion of the blood or blood components are not required to be reported, but still need to be investigated by the appropriate establishment(s) (i.e. foreign establishment and/or importer) and documented by the importer in an annual report. See section 108 Annual Report.
Reporting Errors and Accidents
All establishments are to provide E/A reports and any information required under section 107 of the Blood Regulations to Health Canada by:
Email (preferred method): bpcp-pcpb@hc-sc.gc.ca with the subject line “[Establishment Name] – Blood E/A Report”
or
Fax: 613-960-2156
An Error or Accident Investigation Preliminary Reporting Form (FRM-0337) is available on the Health Canada website.
Preliminary Report
It is acknowledged that all information may not be available at the time of initial reporting.
Written Updates
Following the preliminary report, the establishment is to provide a written update on any new information about the suspected E/A within 15 calendar days after the start of the investigation.
The update must include information regarding the:
- status of all implicated blood units;
- number of affected establishments contacted;
- progress made in the investigation since the last report;
- corrections, immediate corrective actions and steps taken to mitigate further risks, such as conducting a recall and changes made to relevant processes; and
- relevant dates of all actions taken.
Health Canada may also request an update at any time after the preliminary report and/or order a recall based on the information received.
Final Report
Upon completion of the investigation, the establishment is to notify and provide a final report to Health Canada.
The final report must include:
- the results of the investigation, including the root cause analysis;
- name of any infectious agent(s) involved, if applicable;
- results of any tests performed;
- follow-up and corrective actions taken (i.e. actions taken to prevent reoccurrence);
- details of the final disposition of the blood and blood components (e.g. number of units distributed, transfused, quarantined, returned to inventory and discarded), and the reasons for that disposition;
- any changes recommended to be made to relevant processes;
- relevant dates of all actions taken; and
- any additional information not previously shared with Health Canada.
Scenarios
The following scenarios are for illustrative purposes only and provide further guidance on the necessary steps to be followed by an establishment when a particular E/A is suspected.
The examples of scenarios given below must be reported to Health Canada within 24 hours of the start of the investigation, as all of the three criteria stated under section 107 are considered to be met.
Examples
- The floor ordered irradiated red blood cells (RBC). The Transfusion Medicine Laboratory or blood bank transformed the unit in the electronic system and re-labeled the unit as “Irradiated RBCs”, however the RBC unit was never actually irradiated. The blood was distributed to the floor for transfusion and the nurse noticed the Rad-Sure indicator was not of the appropriate color. She contacted the blood bank and returned the unit. It was confirmed that the unit was indeed not irradiated.
- A unit of RBC was left outside of its storage refrigerator for 120 minutes in the blood bank. The blood bank’s procedure states the maximum time allowed outside of a temperature controlled environment is 60 minutes, as indicated in the CSA blood standard. The unit was subsequently distributed without confirmation of the temperature of the blood.
- Two units of RBCs were issued to the operating room. At the end of each day, a blood bank employee retrieves the blood from the operating room fridge and brings it back to the blood bank. However, three days later, these two units were found in the operating room fridge with no records to demonstrate where they were stored in the last three days. The two RBC units were brought back to the blood bank, but not quarantined as required. Instead, they are put back into inventory and redistributed.
- For transporting RBCs, the ice packs were stored colder than the required specification. Blood arrived below acceptable temperature limits and was deemed not safe for transfusion. The receiving establishment must notify the establishment that sent them the blood. The establishment who shipped the blood is responsible to report this E/A to Health Canada as they are responsible for packaging the blood in a way to ensure that it remains within the required temperature range during transport.
- The collecting establishment sent a notice of component recall and withdrawal to a hospital requesting that 2 RBC units be quarantined because the corresponding units of platelets associated with the RBC units tested positive for bacterial growth using the BacT system. The hospital retrieved the 2 RBC units and placed them in the quarantine section of the fridge but misread one of the unit numbers and quarantined the incorrect RBC unit. The recalled unit was transfused after the recall notice was processed by the hospital and it was only after the units were returned to the collecting establishment that the error was recognized. The hospital is required to report this E/A to Health Canada.
- The collecting establishment conducted a lookback for a source plasma donor who tested positive for a transmissible disease. However, one implicated unit was not properly quarantined and was distributed to the blood product fabricator.
Note: At any time, if it is discovered that a previously unreported E/A should have been reported, then that E/A must immediately be reported. For instance, this could happen during the preparation of the Annual report (section 108), quality review, internal audit, regulatory inspection, etc.
Section 108 Annual Report
Annual report
108. (1) An establishment must prepare an annual report that summarizes all of the error and accident investigations that it conducted in the previous 12 months, including a concise critical analysis of those investigations, and must file it with the Minister on request.When to notify Minister
(2) If the analysis reveals a previously unidentified risk to the safety of blood, the establishment
must notify the Minister immediately.Additional reports
(3) An establishment must, on the Minister’s request, file additional reports described in subsection (1) in respect of the period specified in the request.
The requirements, in section 108, apply to all establishments that are regulated under the Blood Regulations including establishments that do not require an establishment licence or a registration.
All establishments must prepare an annual report that includes all the E/A investigations conducted by their establishment in the previous 12 months. This includes all E/As that fall under the scope of the Blood Regulations, identified before and after the distribution or transfusion of the blood and blood components.
The report could be separated into categories — e.g. types of E/As, types of blood components, sites where they occurred, activities, areas, risk, distributed vs not distributed, etc. The establishment must include an analysis of the investigations that clearly identifies any recurring issues and trends. Further corrective actions may be necessary if recurring issues and trends are identified. This process must be clearly outlined in the establishment’s procedure(s). These reports must be filed with Health Canada, at any time upon request. The annual reports may also be requested and reviewed prior to or during an inspection by Health Canada. In addition, Health Canada may request additional reports to be prepared and submitted by the establishment, for a specified time period.
If during the analysis of the investigations, any E/A that should have been reported to Health Canada is discovered during the preparation of the report, the establishment must notify Health Canada immediately. Written notifications should be sent to the following address:
Email (preferred method): bpcp-pcpb@hc-sc.gc.ca or Fax: 613-960-2156Sections 109-116 Adverse Reaction Investigation and Reporting
Section 109 Adverse Donor Reactions
Section 109 Notice to Minister
Notice to Minister
109. (1) An establishment that has reasonable grounds to believe that a donor has experienced a
serious adverse reaction during a donation or within 72 hours after a donation must notify the Minister of the adverse reaction within 24 hours after it learns of the death of the donor or within 15 days after it learns of the adverse reaction in any other case.Contents of notice
(2) The notice must contain all of the following information:
- a description of the adverse reaction;
- any actions that were taken to address it; and
- the outcome.
Written notice
(3) If a notice under this section is given verbally, a confirmatory written notice must be sent as soon as possible afterwards.
109(1) A serious donor adverse reaction may occur as a result of a whole blood or apheresis blood donation. When a serious donor adverse reaction occurs during a donation or within 72 hours after a donation and the reaction could impose a risk to the safety of the blood or blood components, the establishment that collected the blood must notify the Minister within 15 calendar days. If a donor dies during a blood donation or within 72 hours after the donation, the establishment that collected the blood must notify the Minister within 24 hours after it learns of the death of the donor. A licensed or registered establishment should refer to section 1, the Interpretation section, for the definitions of adverse reaction, and serious donor adverse reaction when determining what must be reported as an adverse donor reaction.
109(2) When a licensed or registered establishment reports a serious donor adverse reaction, the notice must contain, at a minimum, a description of the following:
- the adverse reaction;
- any actions that were taken to address it; and
- the outcome.
The licensed or registered establishment must describe any actions taken to address the serious donor adverse reaction, including treatment of the donor. The notice must include the outcome of the serious donor adverse reaction, i.e. whether the outcome is no deferral or temporary or indefinite deferral. The identity of the donor is not required in the notice.
The notice should also contain all of the following information:
- donor identification code;
- donation code;
- donor’s age and sex; and
- a description of the reaction, including:
- date, time, place;
- donor type (allogeneic vs. autologous);
- donation history (repeat vs. first time);
- donation type (whole blood, plasmapheresis, cytapheresis);
- clinical symptoms;
- assessment of the reaction and relationship to the donation; and
- sequelae.
In the event that all of the information above is not available at the time of reporting, a notice with all the information should be submitted to Health Canada as soon as possible after the initial report.
109(3) When a licensed or registered establishment provides verbal notice to Health Canada about a serious donor adverse reaction, the establishment must also provide written notice to Health Canada without delay.
An establishment may use any adverse reaction or event reporting form to provide written notice to Health Canada as long as all reporting requirements are met.
The completed form must be faxed or emailed to Health Canada’s Biologic and Radiopharmaceutical Drugs Directorate:
Office of Regulatory Affairs
Biologic and Radiopharmaceutical Drugs Directorate
Health Products and Food Branch, Health Canada
Tel (613) 957-1722
Fax (613) 946-9520
An establishment may email password protected scanned images of the form to BRDD.ORA@hc-sc.gc.ca
Sections 110-111 Adverse Recipient Reactions
Under the Blood Regulations, an unexpected or serious recipient adverse reaction must be reported to Health Canada if it is an undesirable response in the recipient to the transfused blood or blood components that indicates that the safety of the blood or blood components may be compromised and there is a risk to human safety. An establishment should refer to section 1, the Interpretation section, for the definitions of adverse reaction, serious adverse reaction, and unexpected adverse reaction when determining what must be reported to Health Canada. An adverse reaction caused by a blood labelling error that compromises the safety of the blood or blood components and leads to an adverse reaction in a recipient is an example of a reportable adverse reaction.
Reportable adverse recipient reactions only apply to human blood collected from donors for the purpose of transfusion or for the immunization of source plasma donors (e.g. red blood cells for immunization for production of anti-D RH factor). Adverse reaction reporting requirements under the Blood Regulations do not apply to blood products manufactured by a blood fabricator (often referred to as plasma protein products, plasma derivatives, fractionated plasma products, or fractionated blood products).
Section 110 Required action
Required action
110. (1) Subject to section 111, an establishment that has reasonable grounds to believe that a recipient has experienced an unexpected adverse reaction or a serious adverse reaction must immediately take all of the following actions:
- determine the donation codes of all implicated blood;
- identify and quarantine any implicated blood in its possession;
- if a preliminary inquiry indicates that the root cause of the adverse reaction is attributable to an activity that it carried out, conduct an investigation into the adverse reaction and notify any establishment to which it distributed implicated blood; and
- if a preliminary inquiry indicates that the root cause of the adverse reaction is attributable to an activity carried out by another establishment, notify all of the following establishments:
- the establishment that collected the implicated blood;
- the establishment from which it received the implicated blood, if different from the establishment mentioned in subparagraph (i); and
- any establishment to which it distributed implicated blood.
Contents of notice
(2) The notice required by paragraphs (1)(c) and (d) must contain all of the following information:
- a description of the adverse reaction;
- an explanation of how the safety of the implicated blood may have been compromised, if known;
- the donation codes of all implicated blood;
- a statement of whether the implicated blood is whole blood or blood components, and the names of the implicated blood components; and
- the name of any suspected transmissible disease or disease agent, if known.
Quarantine
(3) An establishment that is notified under subsection (1) or under this subsection must immediately notify to the same effect every establishment and other person to which it distributed implicated blood and quarantine any implicated blood in its possession.Investigation
(4) An establishment that is notified under subparagraph (1)(d)(i) or (ii) must, if a preliminary
inquiry indicates that the root cause of the adverse reaction is attributable to an activity that it carried out, conduct an investigation into the adverse reaction.Written notice
(5) If a notice under this section is given verbally, a confirmatory written notice must be sent as soon as possible afterwards.
110(1) The Investigation Requirements Flow Chart (Flow Chart A), found in subsection 110(4) of this guidance, describes the steps to be taken once a transfusion reaction has occurred and a preliminary inquiry is done to determine if an unexpected or a serious recipient adverse reaction has occurred.
Please note that section 110 is in reference to unexpected adverse reaction(s) and serious adverse reaction(s), even when referenced simply as adverse reaction(s). Action is required in all instances referenced above.
A transfusing establishment that has reasonable grounds to believe that a recipient has experienced an unexpected or a serious adverse reaction must immediately determine the donation codes, identify the implicated blood and blood components, and quarantine the implicated blood (Flow Chart A (1)–(2)). The transfusing blood establishment must then conduct a preliminary inquiry to determine if the root cause suggests the adverse reaction is attributable to an activity it carried out (Flow Chart A (3)). When an establishment has reasonable grounds to believe an unexpected or a serious adverse reaction has occurred due to an activity it carried out, the establishment must conduct an investigation and notify any establishment to which it distributed the implicated blood or blood components (Flow Chart A (12)–(13)). Preliminary reports and final reports of the adverse reaction investigation to Health Canada must then be sent within the time frames outlined under section 113.
If the transfusing blood establishment has other implicated blood or blood components in its possession /inventory, they must quarantine it (for example the hospital may have the companion of a double apheresis platelet or plasma in its inventory that it will need to quarantine). In most cases, the risk mitigation actions will be carried out by the blood establishment that provided the transfused component. Therefore, this establishment must be advised promptly of the occurrence of the adverse reaction. However, there may also be risk mitigation actions that will need to be carried out by the transfusing establishment.
When the unexpected or serious adverse reaction is due to an activity carried out by another establishment, the establishment must notify all of the establishments listed in 110(1)(d)(i)–(iii) (Flow Chart A (4)–(6)).
110(2) When notifying an establishment of an unexpected or a serious recipient adverse reaction, the notifying establishment must ensure that the notice to the establishments listed in paragraph 110(1)(c) or (d) contains all of the required information in subsection 110(2). The notice should also include all of the following information if known and applicable:
- recipient’s date of birth and sex;
- hospital identification;
- diagnosis, medical history;
- ABO and Rh blood group, antibody screen;
- date, time, and place and indication for transfusion;
- component transfused, donation code(s), ABO and Rh blood group, collection date/pooling date, date, time and location of any transformation (irradiation, washing etc.), infusion start/stop time, blood and blood component type and ISBT component code (www.isbtweb.org);
- description of adverse reaction, vital signs, treatment and course in hospital, investigation results including: culture of the recipient’s blood and of the component transfused; chest X-ray, direct antiglobulin test, complete blood count, echocardiogram, electrocardiogram, HLA typing and HLA/human neutrophil antigen antibody screen, and IgA levels as needed for the investigation of the adverse reaction;
- assessment and conclusion by transfusing establishment physician;
- hospital contact information, establishment physician;
- outcome; and
- any other relevant information.
An establishment may use any adverse reaction or event reporting form to provide written notice as long as all notification requirements in subsection 110(2) of the Blood Regulations are met.
110(3) An establishment that receives a notice under subsection 110 (1), about an unexpected or a serious adverse recipient reaction to transfused blood or blood component, must immediately notify every other establishment and other person (including blood product fabricators) to which it distributed implicated blood and blood components (Flow Chart A (7)). An establishment may meet this requirement by forwarding the notice from the establishment that collected the implicated blood to the establishment that now has the implicated blood or blood components. The establishment must quarantine any implicated blood components in its possession immediately. (Flow Chart A (8)).
Establishments must cooperate with one another and provide any relevant information to the investigating establishment. Cooperation between establishments (transfusing establishment where the AR occurred and blood establishment who supplied the blood components) enables the investigating establishment (usually the collecting establishment) to determine the cause of the unexpected or serious adverse reaction when it is not clear whether the blood or an activity the investigating establishment conducted on the blood or blood components led to the adverse reaction in the transfusion recipient (Flow Chart A (9)). Refer to section 110(2) and 112 of the Blood Regulations regarding the requirement to cooperate and share pertinent information in a timely manner.
110(4) An establishment that receives a notice, under 110(1)(d)(i) or (ii), about an unexpected or a serious adverse reaction to transfused blood or blood components must establish if the adverse reaction was attributable to an activity it conducted (Flow Chart A (10)). If so, it becomes the investigating establishment for the adverse reaction.
In order to carry out this preliminary inquiry into the root cause, all relevant information about the adverse reaction must be communicated to this establishment and/or efforts made to acquire it. This will enable a more timely determination that it is the investigating establishment (and expedite reporting of the adverse reaction to Health Canada). The investigating establishment must now report the adverse reaction to Health Canada and conduct a full investigation into the adverse reaction (Flow Chart A (12)–(13)). Preliminary reports and final reports of the adverse reaction investigation to Health Canada must then be sent within the periods outlined under section 113.
If it is unclear whether the root cause of the adverse reaction was an activity that the establishment carried out or an activity carried out by another establishment, the establishment should communicate with other establishments involved in the collection and distribution and transfusion of the implicated blood or blood components in order to share information and assist in this determination.
Flow Chart A: Investigation Requirements
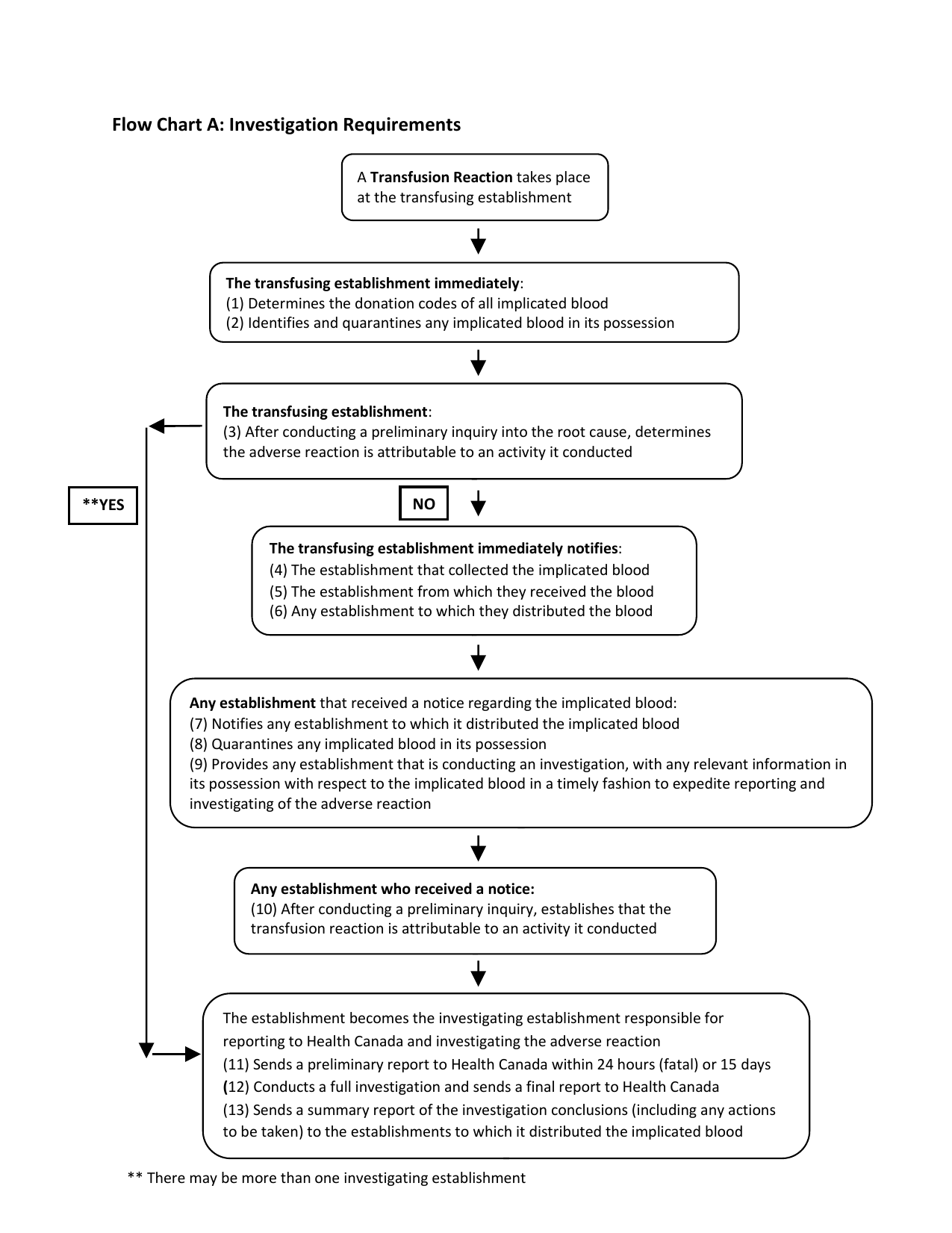
Text description
The Investigation Requirements Flow Chart (Flow Chart A), found in subsection 110(4) of this guidance, describes the steps to be taken once a transfusion reaction takes place at the transfusing establishment. The flow chart consists of a series of boxes that span the length of the page. Each box contains a description of one or more actions to take place where each action is listed as a step, 1 through 13. The boxes are connected by arrows that indicate the next set of steps that must be performed. At times more than one arrow will stem from a box. The choice of arrow to follow will be based on the outcome of the previous steps.
The first box in the flow chart reads - A transfusion reaction takes place at the transfusing establishment.
There is an arrow that leads from the first box pointing to the second box, found below the first box. The second box reads – The transfusing establishment immediately: (1) Determines the donation codes of all implicated blood and (2) Identifies and quarantines any implicated blood in its possession.
There is an arrow that leads from the second box pointing to the third box found below the second box. The third box reads - The transfusing establishment: (3) After conducting a preliminary inquiry into the root cause, determines the adverse reaction is attributable to an activity it conducted.
There are two arrows that stem from the third box. The first arrow, labelled "YES", leads from the bottom-left side of the third box to the seventh box.
The second arrow, labelled "NO", leads from the center of the third box to a fourth box which reads - The establishment notifies: (4) The establishment that collected the implicated blood, (5) The establishment from which they received the blood and (6) Any establishment to which they distributed the blood.
There is an arrow that leads from the fourth box pointing to the fifth box, found below the fourth box. The fifth box reads - Any establishment that received a notice regarding the implicated blood: (7) Notifies any establishment to which it distributed the implicated blood, (8) Quarantines any implicated blood in its possession, (9) Provides any establishment that is conducting an investigation, with any relevant information in its possession with respect to the implicated blood in a timely fashion to expedite reporting and investigating of the adverse reaction
There is an arrow that leads from the fifth box pointing to the sixth box, found below the fifth box. The sixth box reads - Any establishment who received a notice: (10) After conducting a preliminary inquiry, establishes that the transfusion reaction is attributable to an activity it conducted.
There is an arrow that leads from the sixth box pointing to the seventh box, found below the sixth box. The seventh box reads - The establishment becomes the investigating establishment responsible for reporting to Health Canada and investigating the adverse reaction: (11) Sends a preliminary report to Health Canada within 24 hours (fatal) or 15 days, (12) Conducts a full investigation and sends a final report to Health Canada, (13) Sends a summary report of the investigation conclusions (including any actions to be taken) to the establishments to which it distributed the implicated blood. The flow chart ends here.
At the end of the page, there is a note that indicates ** there may be more than one investigating establishment.
Traceback Investigation
The purpose of the traceback investigation is to identify the associated/implicated donor(s), retrieve available in-date components from those donors, and notify other consignees and recipients of those blood components. See 1.5 Definitions, traceback.
Establishments should conduct traceback investigation and reporting of unexpected or serious adverse reactions that are suspected to be transfusion-associated viral infections, such as HBV, HCV, HIV 1 and 2, HTLV I/II. Other transfusion-related infections may also trigger a traceback investigation and reporting. The reporting requirements are the same as other adverse reactions once an associated/implicated donor is identified and there is a reasonable probability that the transmission was due to the transfusion.
The establishment conducting an investigation into the unexpected or serious adverse reaction associated with the implicated blood or blood components initiates a traceback investigation when it identifies any of the following:
- infection via transfusion is consistent with the timing of the recipient's diagnosis;
- the recipient did not originate from a lookback procedure;
- in the case of HTLV I/II infection, the recipient received cellular components; or
- in the case of HCV infection, when the recipient is not a hemophiliac or thalassemic patient transfused prior to May 1992.
The establishment conducting the investigation into the unexpected or serious adverse reaction associated with the implicated blood or blood components should initiate a traceback investigation of a suspected transfusion-related infection when it receives a report of a positive recipient from any of the following:
- Physician;
- Establishment, such as a hospital, a licensed or a registered establishment;
- Public Health Authority;
- Information from a lookback procedure;
- Compensation programme;
- Transfusion recipient*.
* If a transfusion recipient reports a transfusion-related infection, the establishment should have a copy of the test results to proceed with the traceback investigation.
A traceback investigation includes procedures for the following:
- determine donor status;
- notify donor;
- monitor donor, tests, and traceback investigation; and
- close traceback investigation.
During a traceback investigation, the establishment assesses the available information to determine the likelihood of transmission by the transfused components of a transfusion-associated viral infection. The medical director or the senior executive officer should be consulted, as needed, during an investigation. The establishment must defer the donor, when the outcome of confirmatory testing yields indeterminate or positive results, and consult the medical director or the senior executive officer for further action.
The medical director or senior executive officer should also be consulted in the following situations:
- if a report is received from a source other than those listed above;
- if a transfusion-related infection is other than HBV, HCV, HIV, HTLV; or
- if there are other clear risks for infection.
Note: When a traceback investigation identifies a donor who is confirmed positive for a transfusion-transmissible infectious agent, a lookback procedure should be carried out. A copy of the test results should be included in the documentation for the lookback procedure. (See section 56 and paragraph 94(1)(h) of this guidance document.) The establishment must record an indefinite deferral code in the donor’s suitability assessment file. See subsections 44(2) and 56(1) for lookback procedure guidance. This should be included in the final adverse recipient reaction investigation report sent to Health Canada.
110(5) When an establishment provides verbal notice to another establishment about an unexpected or serious recipient adverse reaction, the establishment must also provide written notice immediately in accordance with information requirements in subsection 110(2).
Section 111 Autologous donations
Autologous donations
111. An establishment that both collects and transfuses the same autologous blood must, if it has reasonable grounds to believe that a recipient has experienced an unexpected adverse reaction or a serious adverse reaction, immediately quarantine any other blood from that donor in its possession and conduct an investigation into the adverse reaction and the implicated blood.
When the recipient of an autologous blood transfusion experiences an unexpected or a serious adverse reaction, the registered establishment that both collected and transfused the autologous blood must immediately take the following actions:
- quarantine any other blood or blood components from the affected autologous blood donor in its possession; and
- investigate the adverse reaction.
The investigation should determine the cause of the unexpected or serious adverse reaction, including the possibility of an error or accident. See sections 103–108 of this guidance for error and accident investigation and reporting requirements.
Sections 112-116 Investigation and Reporting of Adverse Recipient Reactions
Section 112 Requirement to cooperate
Requirement to cooperate
112. An establishment must, on request, provide every establishment that is conducting an investigation with any relevant information in its possession in respect of blood that it distributed or transfused.
The establishment conducting the investigation is the establishment that after a preliminary inquiry has determined that the root cause of the adverse recipient reaction was attributable to an activity that they carried out (Flow Chart A (3) and (12)).
The establishment conducting the investigation may request relevant information from other establishments that distributed or transfused the implicated blood or blood components. It is expected that this relevant information be provided in a timely manner as it impacts risk mitigation measures and reporting timelines to Health Canada.
On request, an establishment must provide the investigating establishment with any information relevant to the investigation if they transfused or distributed blood and/or blood components that were later implicated in an adverse recipient reaction’s investigation. This information includes, but is not limited to, an inventory list of the implicated blood and blood components and its disposition (e.g. distributed, transfused, quarantined). (Flow Chart A (9)).
Section 113 Notice to Minister
Notice to Minister
113. (1) An establishment that is conducting an investigation must notify the Minister of the adverse reaction within 24 hours after it learns of the death of a recipient or within 15 days after it learns of any other unexpected adverse reaction or serious adverse reaction.Written notice
(2) If a notice under this section is given verbally, a confirmatory written notice must be sent as soon as possible afterwards.
113(1) Once an establishment has conducted their preliminary inquiry and identifies itself as the investigating establishment (section 110(1) or 110 (4)) it must notify Health Canada of the adverse reaction. This preliminary report is expected to be sent to Health Canada within the next 24 hours after learning of the death, if the AR resulted in a death and within 15 calendar days if the adverse reaction is unexpected or serious. If the investigating establishment is unable to report to Health Canada within 24 hours after learning of the death because it took longer than 24 hours after learning of the death for the establishment to confirm it is an adverse reaction attributable to a regulated activity it conducted, the establishment must report to Health Canada without further delay and all relevant documents in their records must demonstrate that an ongoing preliminary inquiry into the root cause was occurring during this time.
Clinical judgement should be exercised by a qualified health care professional from the transfusing and investigating blood establishments to determine if the transfusion reaction is related to the safety of the transfused blood. In the case of death, the transfusion has to at least be suspected to have contributed to the factors leading to the death or be the direct cause of the death.
Examples of serious adverse recipient reactions include anaphylaxis, transfusion-related acute lung injury (TRALI), sepsis from transfusion transmitted infection (e.g. bacterial, viral, or parasitic), transfusion-associated graft versus host disease (e.g. ineffective irradiation and faulty labelling), and severe hemolytic reactions (e.g. incorrect typing or labelling), requiring intervention and/or admission to hospital.
A transfusion reaction that is unexpected or serious can happen during or after a transfusion of blood components and may be related to the safety of the blood component transfused i.e., an adverse reaction. The casual link between the transfusion and the adverse reaction is a matter of clinical judgement specific to the patient’s situation (underlying disease and concurrent medication/ treatment, etc.).
Errors or accidents could lead to an adverse recipient reaction. When an adverse recipient reaction may have been caused by an error or accident that occurred during a regulated activity, the establishment who carried out the regulated activity of the implicated blood or blood component must investigate and report any unexpected or serious adverse reaction to Canada Vigilance, as well as an error and accident investigation report to the ROEB of Health Canada. Refer to section 107 for more details on how to report errors and accidents to ROEB.
Two examples where the error or accident must be reported to ROEB and the adverse reaction must be reported to Canada Vigilance are:
- a transformation error or accident such as contamination of the blood during open washing procedure, which results in an unexpected and serious adverse reaction;
- a storage error is suspected to have resulted in a serious or unexpected adverse reaction, such as a severe hemolysis
The Reporting of an Adverse Reaction Flow Chart (Flow Chart B), found at the end of section 116, describes the steps to be undertaken once an establishment determines that they must conduct an investigation into an unexpected adverse reaction or a serious adverse reaction.
113(2) If an establishment provides verbal notice to Health Canada about an unexpected or a serious recipient adverse reaction, the establishment must also provide written notice to Health Canada without delay.
The written notice may include preliminary, follow-up and final reports. Preliminary and final investigation reports can be filed at the same time if the investigation is completed at the time of the preliminary report.
As long as all reporting requirements are met, an establishment may use any adverse reaction reporting form to provide mandatory written notice to Health Canada about an unexpected or a serious adverse reaction in a blood transfusion recipient.
Blood establishments must send adverse recipient reaction investigation reports to the Canada Vigilance Program of the HPSEB. The preferred method of securely submitting adverse reaction reports is by secure File Transfer Protocol (sFTP).
For enquiries on how to register for submission using sFTP, please contact tpmo-bgpc@hc-sc.gc.ca.
Fax is an alternate secure method to facilitate transmission of adverse reaction reports for establishments.
Canada Vigilance Program
Health Products Surveillance and Epidemiology Bureau (HPSEB)
Marketed Health Products Directorate
Tunney's Pasture
Address Locator: 1908C
Ottawa, Ontario
K1A 0K9
Fax: 613-957-0335
E-mail for enquiries: canada.vigilance.blood-sang@hc-sc.gc.ca
Section 114 Results of investigation
Results of investigation
114. (1) The establishment that is conducting an investigation must notify in writing every establishment and other person to which it distributed implicated blood of the results of the investigation and of any action that is required to be taken.Notice to be forwarded
(2) An establishment that is notified under subsection (1) or under this subsection must send a copy of the notice to every establishment to which it distributed implicated blood.
114(1) The blood establishment investigating the adverse recipient reaction must give written notification of the results of the investigation, as well as, any required actions to all the blood establishments to which it had distributed the implicated blood or blood components (including manufacturers of the blood products). This notice should reference the original notification that came from the transfusing establishment.
114(2) When an establishment receives the results of an investigation, they must forward the results to any other establishment to which they distributed implicated blood or blood components. It is not necessary for the establishment to add any additional information to the results of the investigation notification.
Section 115 Final report to Minister
Final report to Minister
115. On completion of the investigation, the establishment must file a final report with the Minister that contains all of the following information:
- the results of the investigation;
- the final disposition of the blood that was the subject of the investigation and the reasons for that disposition; and
- any corrective actions taken and any other changes that are recommended to be made to relevant processes.
The investigating establishment must file a final report to Health Canada concerning the recipient’s adverse reaction investigation containing all of the information required in section 115. When the information is inconclusive, the establishment may also provide comments based on their evaluation (Flow Chart B, step 3).
Section 116 Annual Report
Annual report
116. At the end of each year, an establishment must prepare an annual report that summarizes all of the final reports that it filed in the year, including a concise critical analysis of the investigations that were the subjects of those reports, and must file it with the Minister on request.
Establishments that conduct adverse reaction investigations must prepare an Annual Adverse Reaction Report (Flow Chart B, step 4) summarizing all of the final reports concerning unexpected or serious recipient adverse reactions that were filed during the year, including a concise critical analysis of the final adverse reaction investigation reports.
The annual report should include the following:
- an executive summary;
- the established degree of relationship of adverse reaction to the transfused blood;
- a detailed analysis and assessment of any new safety signals;
- an overall summary analysis of the adverse reactions reported in the period that considers blood or blood component use;
- a cumulative analysis of the adverse reactions reported that includes a trend analysis over time;
- traceback and lookback annual summary statistical reports; and
- overall conclusions and opportunities for improvement.
When requested by the Health Canada, an establishment may file an annual adverse reaction report that was prepared for other purposes, such as an annual hemovigilance report, as long as it includes the information required and described in section 116 above.
When requested by the Health Canada the annual report must be sent to the Canada Vigilance Program of the HPSEB.
Further information on how to provide the document to be provided at time of request.
E-mail for enquiries: canada.vigilance.blood-sang@hc-sc.gc.ca
Appendix B provides a summary of all annual reporting requirements for blood establishments.
Flow Chart B: Reporting an Adverse Recipient Reaction (ARR) to Health Canada

Text description
The Reporting of an Adverse Recipient Reaction to Health Canada Flow Chart (Flow Chart B), found at the end of section 116, describes the steps to be undertaken once an establishment determines that they must conduct an investigation into an adverse recipient reaction. The flow chart consists of a series of boxes that span the length of the page. Each box contains a description of one or more actions to take place where each action is listed as a step, 1 through 4. The boxes are connected by arrows that indicate the next set of steps that must be performed.
The first box in the flow chart reads - An establishment conducts an investigation of an Adverse Recipient Reaction (ARR), the word 'investigation' is bolded and capitalized.
There is an arrow that leads from the first box pointing to the second box found below the first box. The second box reads - Step 1 - The investigating establishment notifies the Minister:[bullet] Within 24hrs, if there is a death, and [bullet] Within 15 calendar days for any other unexpected or serious ARR.
There is an arrow that leads from the second box pointing to the third box found below the second box. The third box reads - Step 2 - The investigating establishment notifies every establishment or fabricator to which it distributed implicated blood:[bullet] The results of the investigation, and [bullet] Any action required to be taken.
There is an arrow that leads from the right hand side of the third box pointing to a box found to the right and below of the first three boxes, and to the right of the fourth and fifth boxes which reads - Every establishment that receives a notice of the results of an investigation sends a copy of the notice to every establishment to which it distributed implicated blood, where all of the words are italicized.
There is another arrow that leads from the third box pointing to the fourth box, found below the third box. The fourth box reads - Step 3 - The investigating establishment files a final report with the Minister on completion of the investigation that contains: [bullet] The results of the investigation, [bullet] The final disposition of the blood, and [bullet] any corrective actions taken.
There is an arrow that leads from the fourth box pointing to the fifth box, found below the fourth box. The fifth box reads - Step 4 - The investigating establishment prepares an annual report at the end of each year to file with the Minister upon request that: [bullet] Summarizes all final reports for the year, and [bullet] Includes a concise critical analysis.
Sections 117-123 Records
Establishments must retain records, including the ones produced prior to the coming into force of the Blood Regulations, in accordance with the regulatory requirements as set out in the Blood Regulations.
Section 117 Record quality
Record quality
117. Records kept by an establishment must be accurate, complete, legible, indelible and readily retrievable.
Records provide documented evidence of compliance. Records must be accurate, complete and legible. Records must be maintained concurrently with the performance of each significant step in the processing, importation, transformation, storage, distribution (including exceptional distribution), investigation of errors and accidents and adverse reactions of blood and/or blood components, so that all steps can be clearly associated with the person who conducted the step time/date and location (as applicable) of such activities. In addition, for records associated with the processing, transformation or any regulated activity, the lot number of critical supplies and the identity of the critical equipment associated with the activities must be part of the records.
All records must identify the person who conducted the activities and the dates of the various entries. Establishments must ensure that the records are accurate. For example, all manual transcriptions of test results must be independently verified in situations where the transcribed document is the permanent record.
Any handwritten entry of information must be made using indelible ink. Any correction, entry of information, or notation made after the original date of record completion must be clearly crossed out, initialled or signed and dated to indicate a change has been made to the original information.
All establishments must retain records in an easily understandable and retrievable format. Records must be accessible at all times. All establishments must be able to quickly and efficiently retrieve blood traceability information.
Records must consistently be maintained in a manner to preserve their completeness and integrity over time. The establishment must verify the accuracy and integrity of the information transferred to other media used to retain information. This verification should be conducted by someone other than the individual who transferred the information.
An establishment that keeps electronic records must have an electronic system validated for its intended use to ensure the maintenance of the data integrity of those records. All electronic records must be backed up regularly and securely stored for data recovery. Any changes to the electronic system must be evaluated, documented and approved prior to implementation to ensure the integrity of the data and that the records can be retrieved during the required retention period. A history of any changes to electronic records must be available in an audit trail.
An establishment must be able to retrieve and print a hard copy of information that is stored in an electronic record.
One standardized format for dates (e.g. YYYY-MM-DD or MM-DD-YYYY) should be used for all records. Where this is not possible, records should clearly indicate the date format if not readily apparent.
Section 118 Donation code part of all records
Donation code part of all records
118. An establishment must ensure that the donation code is a component of all of its records that relate to the processing, distribution, transformation and transfusion of blood.
Each unit of blood has a donation code that uniquely identifies it. The donation code enables the traceability of a given unit of blood or blood component and any associated information about that unit throughout any processing, transformation steps, chain of distribution and transfusion or final disposition. The donation code must be a part of all records related to the processing, distribution, transformation and transfusion of the unit of blood. A blood component is identified by the specific component product code (i.e. code for red blood cells, plasma, platelets) associated with the donation code. If an establishment is assigning a new code for a pooled unit, the donation code of all pooled components must be traceable in the establishment’s records.
Section 119 – 122 Retention Periods
An establishment that conducts more than one type of activity (e.g. transformation and transfusion) must comply with the retention periods in all applicable sections below.Section 119 Retention periods - allogeneic blood
Retention periods — allogeneic blood
119. (1) An establishment that collects allogeneic blood must keep the records set out in column 1 of the table to this section for the period set out in column 2.Calculation of record retention period
(2) The record retention period begins on the day on which the record is created, except for the personnel records set out in item 28 of the table, in which case the period begins on the last day on which the employee was last employed by the establishment.
All establishments that collect allogeneic blood must ensure that records are retained according to the Table to section 119, Records and Retention Periods. See clause 20.2.5 of the CSA Blood Standard for guidance concerning the reconciliation of donation codes (item 3 of the Table to section 119
For records of lookback investigations, if the lookback was triggered by an Adverse Reaction or Error/Accident investigation, the record must be kept for 10 years. However, in the situation whereby this investigation resulted in a “Determination of donor unsuitability” either “indefinite” or “temporary”, these records must be kept for 50 and 10 years respectively. Traceback investigation records are part of the adverse reaction investigation and therefore must be kept 10 years.
| Item | Column 1 Records |
Column 2 Retention period |
|---|---|---|
| 1. | Donor identification code | 50 years |
| 2. | Donation code | 50 years |
| 3. | Reconciliation of donation codes | 10 years |
| 4. | Donor suitability assessment | 5 years |
| 5. | Determinations of donor unsuitability - indefinite | 50 years |
| 6. | Determinations of donor unsuitability - temporary | 10 years |
| 7. | Collection - date of donation | 50 years |
| 8. | Collection - donor apheresis | 5 years |
| 9. | Collection - record of donation | 5 years |
| 10. | Lot number and name of manufacturer of container and other critical supplies for each donation | 1 year |
| 11. | Test results for transmissible disease testing, ABO group and Rh factor, and clinically significant antibody testing | 50 years |
| 12. | Blood component preparation | 10 years |
| 13. | Blood storage temperature monitoring | 5 years |
| 14. | Destruction or other disposition of blood | 50 years |
| 15. | Distribution | 50 years |
| 16. | Shipping documents | 1 year |
| 17. | Exceptional distribution | 50 years |
| 18. | Importation in urgent circumstances | 50 years |
| 19. | Post-donation information | 10 years |
| 20. | Complaints and their investigation | 5 years |
| 21. | Internal audit reports | 5 years |
| 22. | Quality control testing | 5 years |
| 23. | Maintenance, validation, qualification and calibration of critical equipment | 3 years |
| 24. | Critical supplies, including their qualification | 3 years |
| 25. | Proficiency testing | 5 years |
| 26. | Every version of the operating procedures that was implemented, other than those related to donor suitability assessments | 10 years |
| 27. | Every version of the operating procedures related to donor suitability assessments | 50 years |
| 28. | Personnel qualifications, training and competency evaluation | 10 years |
| 29. | Investigations and reports of errors and accidents | 10 years |
| 30. | Investigations and reports of adverse reactions | 10 years |
Section 120 Retention periods - autologous blood
Retention periods — autologous blood
120. (1) An establishment that collects autologous blood must keep the records set out in column 1 of the table to this section for the period set out in column 2.Calculation of record retention period
(2) The record retention period begins on the day on which the record is created, except for the personnel records set out in item 18 of the table, in which case the period begins on the last day on which the employee was employed by the establishment.
All establishments that collect autologous blood must ensure that records are retained according to the Table to section 120, Records and Retention Periods.
| Item | Column 1 Records |
Column 2 Retention period |
|---|---|---|
| 1. | Donor identification code | 10 years |
| 2. | Donation code | 10 years |
| 3. | Collection - donor record | 5 years |
| 4. | Lot number and name of manufacturer of container and other critical supplies for each donation | 1 year |
| 5. | Test results for transmissible disease testing, ABO group and Rh factor | 10 years |
| 6. | Blood component preparation | 10 years |
| 7. | Blood storage temperature monitoring | 5 years |
| 8. | Destruction or other disposition of blood | 10 years |
| 9. | Distribution | 10 years |
| 10. | Shipping documents | 1 year |
| 11. | Complaints and their investigation | 5 years |
| 12. | Internal audit reports | 5 years |
| 13. | Quality control testing | 5 years |
| 14. | Maintenance, validation, qualification and calibration of critical equipment | 3 years |
| 15. | Critical supplies, including their qualification | 3 years |
| 16. | Proficiency testing | 5 years |
| 17. | Every version of the operating procedures that was implemented | 10 years |
| 18. | Personnel qualifications, training and competency evaluation | 10 years |
| 19. | Investigations and reports of errors and accidents | 10 years |
| 20. | Investigations and reports of adverse reactions | 10 years |
Section 121 Retention periods - transformation
Retention periods — transformation
121. (1) An establishment that transforms blood must keep the records set out in column 1 of the
table to this section for the period set out in column 2.Calculation of record retention period
(2) The record retention period begins on the day on which the record is created, except for the personnel records set out in item 10 of the table, in which case the period begins on the last day on which the employee was employed by the establishment.
All establishments that transform blood or blood components must ensure that records are retained according to the Table to section 121, Records and Retention Periods - transformation. Section 121 also applies to establishments that only pool cryoprecipitate and do not require a registration.
The retention periods indicated in the Table to section 121 refer to the retention times of the records produced while performing transformation activities. For example, records that identify the transformed blood components by using the donation code must be retained for 10 years. In contrast, the table to section 119 refers to the retention times of records produced by an establishment that collects allogeneic blood. For example, records that identify the collected blood by using the donation code must be retained for 50 years.
| Item | Column 1 Records |
Column 2 Retention period |
|---|---|---|
| 1. | Donation code | 10 years |
| 2. | Records of washing, pooling and irradiation of blood | 10 years |
| 3. | Lot number and name of manufacturer of critical supplies for each transformation | 1 year |
| 4. | Complaints and their investigation | 5 years |
| 5. | Internal audit reports | 5 years |
| 6. | Quality control testing | 5 years |
| 7. | Maintenance, validation, qualification and calibration of critical equipment | 3 years |
| 8. | Critical supplies, including their qualification | 3 years |
| 9. | Every version of the operating procedures that was implemented | 10 years |
| 10. | Personnel qualifications, training and competency evaluation | 10 years |
| 11. | Investigations and reports of errors and accidents | 10 years |
| 12. | Investigations and reports of adverse reactions | 10 years |
Section 122 Retention periods - transfusion
Retention periods — transfusion
122. (1) An establishment that transfuses blood must keep the records set out in column 1 of the
table to this section for the period set out in column 2.Calculation of record retention period
(2) The record retention period begins on the day on which the record is created, except for the personnel records set out in item 11 of the table, in which case the period begins on the last day on which the employee was employed by the establishment.
All establishments that transfuse blood and/or blood components must ensure that records are retained according to the Table to section 122, Records and Retention Periods - transfusion.
For establishments who further distribute blood and blood components (e.g. Hospital A sends blood or blood components to Hospital B), distribution records provide information on the chain of distribution of the unit such that any unit of blood or blood components can be traced from its initial source to its final disposition. Shipping documents are records associated with the mode of transport used for distributing blood (e.g. waybills, courier receipts, etc.).
| Item | Column 1 Records |
Column 2 Records Retention period |
|---|---|---|
| 1. | Donation code - allogeneic blood | 50 years |
| 2. | Donation code - autologous blood | 10 years |
| 3. | Shipping documents | 1 year |
| 4. | Blood storage temperature monitoring | 5 years |
| 5. | Distribution | 50 years |
| 6. | Exceptional distribution | 50 years |
| 7. | Record of transfusion or disposition of allogeneic blood, including identification of recipient | 50 years |
| 8. | Record of transfusion or disposition of autologous blood | 10 years |
| 9. | Complaints and their investigation | 5 years |
| 10. | Every version of the operating procedures that was implemented | 10 years |
| 11. | Personnel qualifications, training and competency evaluation | 10 years |
| 12. | Investigations and reports of errors and accidents | 10 years |
| 13. | Investigations and reports of adverse reactions | 10 years |
Section 123 Storage of records
Storage of records
123. An establishment must store records in a location that has appropriate environmental conditions and that is secure against the entry of unauthorized persons.
Record storage areas must maintain the integrity of the records. Environmental parameters for storage, such as temperature, must be appropriate and controlled to the extent necessary in order to safeguard the integrity of the type of records being stored. The humidity should also be controlled as appropriate and as required. Access to the storage area must be restricted to authorized persons.
If records are copied or stored off-site, the establishment must have a signed contract with the service provider. The contract must include specific requirements, such as transport to the site, copy quality, retrieval information, and storage conditions. Where relevant, the contract must describe specific requirements for the destruction of the original document.
Section 124 Powers of Inspectors
Making visual recordings
124. An inspector may, in the administration of these Regulations, take photographs and make recordings of any of the following:
- any article that is referred to in subsection 23(2) of the Act;
- any place where the inspector believes on reasonable grounds any article referred to in paragraph (a)is processed, transformed or stored; and
- anything that the inspector believes on reasonable grounds is used or is capable of being used in the conduct of an establishment’s activities.
Section 125 Consequential Amendment
Provisions listed under this section are no longer applicable.
125. Section 18 of the Regulations Amending the Food and Drug Regulations (1475 — Good Manufacturing Practices)1 is replaced by the following:
18. The Food and Drug Regulations, as they read immediately before the coming into force of these Regulations, continue to apply in respect of whole blood and blood components until the day before the day on which subsection 3(2) of the Blood Regulations comes into force.
Sections 126-128 Transitional Provisions
Section 126 Deemed Authorization
Provisions listed under this section are no longer applicable.
Deemed authorization
126. The information that is required by section 6 to be included in an application for an authorization and that was filed with and accepted by the Minister under sections C.01A.005 to C.01A.007 and C.01A.014 of the Food and Drug Regulations before the day on which these Regulations come into force is deemed to be an authorization issued by the Minister under section 7 of these Regulations.
Section 127 Licence continued
Provisions listed under this section are no longer applicable.
Licence continued
127. If an establishment files an application for a licence under section 18 — without regard to paragraphs (1)(j) and (k) — within three months after the day on which these Regulations come into force, any licence that was issued to the establishment under section C.01A.008 of the Food and Drug Regulations before that day is continued until a licence is either issued under section 20 or refused under section 21 of these Regulations.
Section 128 Delayed registration
Provisions listed under this section are no longer applicable.
Delayed registration
128. (1) An establishment that, before the day on which these Regulations come into force, conducts any of the activities mentioned in section 30 may continue to do so without a registration if it files an application for registration under section 31 within three months after that day.Duration
(2) Subsection (1) applies until the determination of the application under section 32.
Section 129 Coming into force
Provisions listed under this section are no longer applicable.
One year after publication
129. (1) These Regulations — except subsections 4(4) to (6), paragraph 64(1)(b) as it applies to registration numbers, and section 125 — come into force one year after the day on which they are published in the Canada Gazette, Part II.Subsections 4(4) to (6) and paragraph 64(1)(b)
(2) Subsections 4(4) to (6) and paragraph 64(1)(b), as it applies to registration numbers, come into force six months after the day on which these Regulations come into force.Section 125
(3) Section 125 comes into force on the day on which these Regulations are registered.
Appendices
Appendix A: Glossary
- BPCP
- Biologic Product Compliance Program
- BRDD
- Biologic and Radiopharmaceutical Drugs Directorate (formerly the Biologics and Genetic Therapies Directorate)
- CMV
- Cytomegalovirus
- CSA
- CSA Group/ Canadian Standards Association
- E/A
- Error or Accident
- HBsAg
- Hepatitis B surface antigen
- HBV
- Hepatitis B virus
- HCV
- Hepatitis C virus
- HIV
- Human Immunodeficiency Virus
- HLA
- Human leukocyte antigen
- HTLV I/II
- Human T-lymphotropic virus-I/Human T-lymphotropic virus-II
- IgA
- Immunoglobulin A
- ISBT
- International Society of Blood Transfusion
- NERBY
- New Evidence Required By
- NOD
- Notice of Deficiency
- ORA
- Office of Regulatory Affairs
- OSE
- On-Site Evaluation
- PADP
- Pre-Assessed Donor Program
- RBC
- Red Blood Cells
- RH
- Rhesus factor
- ROEB
- Regulatory Operations and Enforcement Branch (formerly the Health Product and Food Branch Inspectorate)
- US-FDA
- United States Food and Drug Administration
- WNV
- West Nile virus
Appendix B: Summary Table of Annual Reporting Requirements for Blood Establishments
| Report Title | Blood Regulations section | Who prepares the report | When to file report | Where to file report |
|---|---|---|---|---|
| Authorization - Other Changes - Annual Report | 12 | Establishments who hold an Authorization | To be decided in consultation with Health Canada | Blood Establishment Regulation Unit, Office of Regulatory Affairs, Biologics and Genetic Therapies Directorate, Health Canada |
| Annual Report of Error and Accident Investigations | 108 | At the end of a 12 month period, establishments who have conducted an investigation into Errors and Accidents throughout the year | Upon request by the Minister | Indicated in request by Minister |
| Annual Report of Adverse Reactions in Recipients | 116 | At the end of each year, establishments who have conducted an investigation into a serious or unexpected adverse reaction in a blood transfusion recipient | Upon request by the Minister | Indicated in request by Minister |
Appendix C: Pre-Registration Self-Assessment Tool for Establishments applying for a Blood Establishment Registration
Establishments should refer to section 30 of the Blood Regulations to determine if they are conducting activities that require them to register with Health Canada.
Before applying for a Registration number, Health Canada strongly recommends that establishments complete this self-assessment tool to determine whether their practices meet the requirements of the Blood Regulations. This tool is designed to facilitate the identification of areas that the establishment may need to address in order to be compliant with the Blood Regulations. Establishments are not required to file this self-assessment with Health Canada nor will it be reviewed during an inspection. The completion of this form alone is not considered as a record of compliance with the internal audit requirements under paragraph 94(1)(j).
Please note that this tool does not supersede the requirements of the Blood Regulations. Each section of this document should be read in conjunction with the relevant sections of the Blood Regulations. For an interpretation of the sections listed below, please refer to the appropriate sections in this Guidance Document. Several terms used in this self-assessment tool are defined in the Blood Regulations. Please refer to the definitions in section 1, the Interpretation section, of the Blood Regulations.
| Blood Regulations Section | Requirements | Assessment |
|---|---|---|
| A. Processing Activities for Autologous Blood | ||
| Collection | ||
| 46 | Is there a donor identification code assigned to each donor? | Yes / No |
| 47 | Is there a donation code assigned to every unit of blood collected? | Yes / No |
| Is the donation code linked in the records to the donor identification code? | Yes / No | |
| 48 | Is it ensured that the blood containers are labelled as per section 63 of the Blood Regulations at the time of the collection? | Yes / No |
| 49(1) | Is blood being collected in the following way? | n/a |
| (a) use of aseptic methods | Yes / No | |
| (b) use of collection equipment that is licensed under the Medical Devices Regulations | Yes / No | |
| (c) use of containers that are licensed under the Medical Devices Regulations and free from defects or damage | Yes / No | |
| (d) lot number of the container is being documented and linked to the donation code | Yes / No | |
| 49(2) | Is each container used only once? | Yes / No |
| 50 | At the time of collection, are samples for testing obtained in a way that avoids contamination of the donated blood and the samples? | Yes / No |
| 51 | (a) Does the blood collection comply with the criteria set out in clause 12.2.1 of the CSA Blood Standard? | Yes / No |
| (b) Is the volume of the blood collected and volume of the anticoagulant adjusted based on the donor's weight, when appropriate? | Yes / No | |
| Testing | ||
| 53 | Is the blood tested using appropriate and effective tests for the transmissible diseases and disease agents specified in clause 12.3.1.2 of the CSA Blood Standard? | Yes / No |
| 54(1) | At the time of each donation, is the blood being tested to identify both of the following: | n/a |
| (a) the ABO group; and | Yes / No | |
| (b) the Rh factor, including weak D testing when appropriate? | Yes / No | |
| 54(2) | Are the results from paragraphs 54(1)(a) and (b) compared with the last available results for that donor? | Yes / No |
| 54(3) | If the comparison in subsection 54(2) indicates a discrepancy, are the tests described in 54(1) repeated and the blood is not transfused until the discrepancy is resolved? | Yes / No |
| 55(a) | Is the blood being tested with test kits that are licensed under the Medical Devices Regulations either for diagnosis or screening? | Yes / No |
| 56(2) | Is the donor's physician informed of the test results described in clause 12.3.1.6 of the CSA Blood Standard by the establishment that collects the blood? | Yes / No |
| 58 | Are the blood components prepared in accordance with clauses 7.1.3, 7.2, 7.3.1, 7.3.2, 7.5.1.1 (without regard to the reference Table 3 of the CSA Blood Standard), 7.5.1.2 and 7.5.1.5, clause 7.5.2.1 (a) to (c) and clause 7.5.2.2 of the CSA Blood Standard? | Yes / No |
| Labelling | ||
| 60 | Is the information appearing on the label of blood containers or the circular of information printed in either English or French? | Yes / No |
| 61 | Do the labels meet the following requirements? | n/a |
| (a) all information is accurate, and presented clearly and legibly | Yes / No | |
| (b) they are made using only adhesives and inks that are non-permeable to the blood container | Yes / No | |
| (c) they are permanently affixed to the blood container | Yes / No | |
| (d) tags are firmly attached to the blood container | Yes / No | |
| 63 | At the time of collection, is the donation code permanently marked on the label of every blood container? | Yes / No |
| 64(1) | Does the following information appear on the label of the blood? | n/a |
| (a) establishment's name and civic address |
Yes / No | |
| (b) the establishment's licence number, if it has one, or its registration number | Yes / No | |
| (c) donation code | Yes / No | |
| (d) a statement of whether the donation is whole blood or a blood component, and if it is a component, its name | Yes / No | |
| (e) ABO group and Rh factor, when appropriate | Yes / No | |
| (f) approximate volume of the whole blood collection | Yes / No | |
| (g) approximate volume of the contents of the container | Yes / No | |
| (h) name of the anticoagulant or additive in the container | Yes / No | |
| (i) recommended storage temperature | Yes / No | |
| (j) expiry date and, if applicable, the time | Yes / No | |
| (k) warning that the blood could transmit infectious agents, in the case of blood for transfusion | Yes / No | |
| 64(2) | Does the following additional information appear on the label of autologous blood? | n/a |
| (a) the statement "For Autologous Use Only" | Yes / No | |
| (b) a symbol or words to indicate the blood is a biohazard if the donor tested positive for a disease or disease agent listed in clause 12.3.1.2 of the CSA Blood Standard | Yes / No | |
| (c) if the unit of blood was not tested in accordance with clause 12.3.1.2 of the CSA Blood Standard, an indication to that effect | Yes / No | |
| 65 | Is the establishment that divides blood into aliquots for transfusion, ensuring that the following appears on the label of each aliquot container: | n/a |
| (a) donation code; | Yes / No | |
| (b) name of the blood component; | Yes / No | |
| (c) a code that identifies the aliquot; | Yes / No | |
| (d) when appropriate, the ABO group and Rh factor; and | Yes / No | |
| (e) expiry date? | Yes / No | |
| 68 | Is it verified that the information added to the label is accurate and complete? | Yes / No |
| Comments for Autologous Activities: | ||
| B. Storage | ||
| 69(1)(b) | Is the collected blood stored in accordance with the storage and expiration criteria specified in Table 2 of the CSA Blood Standard? | Yes / No |
| 69(2) | Is the blood received from another establishment stored in accordance with the directions on the label and with any directions that are specified in writing by the establishment that collected it? | Yes / No |
| 70 | Does the storage location have appropriate environmental conditions and is it secure against the entry of unauthorized persons? | Yes / No |
| 71 | Is blood intended for autologous, designated or directed use segregated from the blood intended for other allogeneic use? | Yes / No |
| 72 | Is blood that is untested, incompletely tested, tested positive or repeat reactive, segregated from blood that has been determined safe for distribution or autologous transfusion? | Yes / No |
| Comments for Storage: | ||
| C. Distribution | ||
| 73(2) | Before distributing blood for transfusion, does the establishment that collected autologous blood determine that it is safe for transfusion if it is satisfied that the blood has been processed in accordance with the Blood Regulations? | Yes / No |
| 74(1) | Prior to distributing blood for transfusion, is there an examination of the container to verify that | n/a |
| (a) the information on the label is legible; | Yes / No | |
| (b) the integrity of the blood container is intact; | Yes / No | |
| (c) there are no signs of deterioration or contamination of the blood; and | Yes / No | |
| (d) any frozen blood components show no signs of thawing? | Yes / No | |
| 74(2) | Is the blood prevented from distribution for transfusion if | n/a |
| (a) the donation code is missing or illegible; | Yes / No | |
| (b) any information - other than the donation code - that is required by the Blood Regulations to appear on the label of blood is missing or is illegible, unless the missing or illegible information can be retrieved from the establishment's records; | Yes / No | |
| (c) the container is defective or damaged to the extent that it does not protect the blood against external conditions; or | Yes / No | |
| (d) there are signs of deterioration or contamination of the blood? | Yes / No | |
| 75 | When the blood is shipped: | n/a |
| (a) Are the blood containers examined before shipping to verify the integrity of the container and the legibility of the labels? | Yes / No | |
| (b) Are the shipping containers used capable of resisting damage and maintaining the safety of the blood? | Yes / No | |
| 76 | Is it ensured that blood for transfusion being shipped is stored during transportation in accordance with the criteria specified in Table 2 of the CSA Blood Standard? | Yes / No |
| Comments for Distribution: | ||
| D. Transformation Activities of Allogeneic or Autologous Blood | ||
| 77 | Are the transformation methods used by the establishment safe and effective? | Yes / No |
| 78(1) | Is the washing of blood done in accordance with clauses 7.5.2.3 and 7.5.3 of the CSA Blood Standard? | Yes / No |
| 78(2) | For washed blood, is the label amended to add the mention of the washing and any new expiry date and time? | Yes / No |
| 79(1) | Is the pooling of blood components done in accordance with clauses 7.11.1 and 7.11.3 of the CSA Blood Standard? | Yes / No |
| 79(2) | Does the information specified in clauses 10.8.2 and 10.8.3 of the CSA Blood Standard appear on the label of the pooled blood components? | Yes / No |
| 80 | Is the irradiation of blood done in accordance with clauses 7.12.2 to 7.12.6 of the CSA Blood Standard? | Yes / No |
| Comments for Transformation Activities: | ||
| E. Pre-Assessed Donor Program (PADP) Processing Activities | ||
| 86 | (a) Is the PADP carried out under the supervision of a medical director? | Yes / No |
| (b) Is it only used when (i) no other alternative source of blood appropriate for the recipient is available, and (ii) the recipient's physician requests the blood for use in the emergency treatment of their patient? | Yes / No | |
| 87 | Is a donor identification code assigned to every donor at the time of acceptance into the PADP? | Yes / No |
| 88(1) | Are the following occurring every three months? | n/a |
| (a) donors are assessed as per sections 40 to 44 of the Blood Regulations | Yes / No | |
| (b) blood samples from every donor are tested for all of the following: | Yes / No | |
| (i) the transmissible diseases or disease agents listed in clauses 8.4.1 and 8.4.2 of the CSA Blood Standard | Yes / No | |
| (ii) ABO group | Yes / No | |
| (iii) the Rh factor including weak D testing when appropriate | Yes / No | |
| (iv) clinically significant antibodies | Yes / No | |
| 88(2) | Does the establishment ensure that the results of the tests conducted under subparagraphs 88(1)(b)(ii) and (iii) are compared with the last available results, if any, for that donor? | Yes / No |
| 88(3) | If the comparison in subsection 88(2) indicates a discrepancy, are these tests repeated and any blood not collected from that donor until the discrepancy can be resolved? | Yes / No |
| 89 | Are the following occurring at each collection? | n/a |
| (a) suitability of the donor is assessed | Yes / No | |
| (b) a donation code is assigned to the blood collected and linked to the donor's identification code in the records. | Yes / No | |
| (c) a blood sample is taken from the donor and tested within 72 hours for the following: | Yes / No | |
| (i) transmissible diseases or disease agents specified in clauses 8.4.1 and 8.4.2 of the CSA Blood Standard | Yes / No | |
| (ii) the ABO group | Yes / No | |
| (iii) the Rh factor, including weak D testing when appropriate | Yes / No | |
| (iv) clinically significant antibodies | Yes / No | |
| 90 | Does the donation code, ABO group and, when appropriate, Rh factor always appear on the label of the blood? | Yes / No |
| 91 | If the blood collected from a pre-assessed donor is not transfused into an intended recipient in the emergency, are the requirements in clause 16.2.5. of the CSA Blood Standard being followed? | Yes / No |
| Donor Suitability Assessment | ||
| 40 | In conducting the donor suitability assessment, does the establishment verify the following:
|
Yes / No |
| 41 | Are the following steps being performed during a donor suitability assessment? | n/a |
| (a) obtain information from the donor by use of a questionnaire or similar means about their identity, their medical history, and their social history to the extent that it is relevant in determining the presence of risk factors for diseases transmissible by blood | Yes / No | |
| (b) provide the donor with information about the risks associated with donating blood and the risks to the recipient of contracting a transmissible disease | Yes / No | |
| 42 | Is the donor deemed unsuitable to donate when any of the information obtained under sections 39 to 41 of the Blood Regulations indicates that human safety or the safety of blood could be compromised? | Yes / No |
| 43 | If a donor is determined unsuitable to donate: | n/a |
|
Yes / No | |
|
Yes / No | |
|
Yes / No | |
| 44(1) | Once the donor is determined suitable, does the establishment take the following steps: | n/a |
|
Yes / No | |
|
Yes / No | |
| 44(2) | On receipt of any post-donation information under paragraph (1)(b), is the information evaluated by the establishment to reassess the safety of the current and any other donation made by the donor and to the donor's suitability for future donations? | Yes / No |
| 44(3) | If the reassessment shows that the safety of the blood may have been compromised and the blood has already been distributed, does the establishment notify every person to which the blood was distributed to that effect, and if the person is an establishment, does the notice specify that the blood must not be distributed or transfused? | Yes / No |
| Collection | ||
| 47 | Is there a donation code assigned to every unit of blood collected? |
Yes / No |
| Is the donation code linked in the records to the donor identification code? | Yes / No | |
| 49(1) | Does the establishment ensure that the collection of blood is being conducted in the following way: | n/a |
| (a) use of aseptic methods; | Yes / No | |
| (b) use of collection equipment that is licensed under the Medical Devices Regulations; | Yes / No | |
| (c) use of containers that are licensed under the Medical Devices Regulations and free from defects or damage; and | Yes / No | |
| (d) lot number of the container is documented in the establishment's records and linked to the donation code? | Yes / No | |
| 49(2) | Is each blood container used only once? | Yes / No |
| 50 | At the time of collection, are samples for testing obtained in a way that avoids contamination of the donated blood and the samples? | Yes / No |
| Testing | ||
| 17(2) | Has the establishment ensured that the testing laboratory possesses an establishment licence from Health Canada to test blood for transmissible diseases or disease agents? | Yes / No |
| 55 b) | Is the blood being tested with test kits that are licensed under the Medical Devices Regulations for donor screening? | Yes / No |
| Comments for Pre-Assessed Donor Program Activities: | ||
| F. Quality Management System (applicable to all registered establishments) | ||
| 93(1) | Is there an organizational structure that sets out the responsibility of management for all activities that the establishment conducts? | Yes / No |
| 93(2) | Does the establishment have an effective quality management system and name an individual who has responsibility for it? | Yes / No |
| 93(3) | Does it review the quality management system at regular intervals specified in the operating procedures, to ensure its continuing suitability and effectiveness? | Yes / No |
| 94(1) | Is there a quality management system in place that includes the following elements: | n/a |
| (a) a quality assurance unit; | Yes / No | |
| (b) a quality control program; | Yes / No | |
| (c) a change control system; | Yes / No | |
| (d) a process control program, within the meaning of clause 3.1 of the CSA Blood Standard; | Yes / No | |
| (e) a system for process improvement through complaint monitoring and the implementation of corrective and preventive actions; | Yes / No | |
| (f) a system for the identification and investigation of post-donation information, errors and accidents and adverse reactions, including the implementation of corrective action and the conduct of recalls; | Yes / No | |
| (g) a personnel training and competency-evaluation program; | Yes / No | |
| (h) a proficiency testing program to evaluate the accuracy and reliability of test results; | Yes / No | |
| (i) a document control and records management system; | Yes / No | |
| (j) an internal audit system; | Yes / No | |
| (k) emergency contingency plans; | Yes / No | |
| (l) a system that uniquely identifies all critical equipment and supplies; | Yes / No | |
| (m) written specifications for all critical equipment, supplies and services; | Yes / No | |
| (n) a program for preventive maintenance of critical equipment; | Yes / No | |
| (o) a program for process validation? | Yes / No | |
| 94(3) | Unless any individual who conducts an internal audit does not have direct responsibility for the activities being audited, is the establishment's quality assurance unit a distinct organizational unit that functions and reports to management independently of any other functional unit? | Yes / No |
| 95 | Are there operating procedures for all of the activities the establishment conducts with respect to human safety and the safety of blood? | Yes / No |
| 96 | Do the operating procedures meet all of the following requirements? | n/a |
| (a) in a standardized format | Yes / No | |
| (b) approved by a senior executive officer | Yes / No | |
| (c) readily accessible at all locations where the relevant activities are conducted | Yes / No | |
| (d) kept up-to-date | Yes / No | |
| 97 | Is there documented evidence that demonstrates that the operating procedures used in the processing and transforming of blood will consistently lead to the expected results? | Yes / No |
| 98(1) | Are there sufficient personnel who are qualified by education, training or experience to perform their respective tasks to conduct the establishment's activities? | Yes / No |
| 98(2) | Is there a program for the orientation and training, both initial and ongoing, of personnel and for the evaluation of their competency? | Yes / No |
| 99 | Do the facilities permit all of the following: | n/a |
| (a) the conduct of all of the activities; | Yes / No | |
| (b) the performance by personnel of their respective tasks using proper hygiene; | Yes / No | |
| (c) the cleaning of the facilities in a way that maintains sanitary conditions; | Yes / No | |
| (d) environmental controls appropriate to all areas where its activities are conducted; | Yes / No | |
| (e) controlled access to all areas where activities are conducted; and | Yes / No | |
| (f) privacy for donor screening? | Yes / No | |
| 100(1) | Is the critical equipment cleaned and maintained and, as appropriate, validated for its intended purpose and calibrated? | Yes / No |
| 100(2) | After repairs or any changes are made to critical equipment, is the equipment revalidated and recalibrated, as appropriate? | Yes / No |
| 101 | Does the equipment used to store blood allow compliance with the storage requirements of sections 69 to 72 of the Blood Regulations? | Yes / No |
| 102 | Are the critical supplies validated or qualified, as applicable, for their intended use and stored under appropriate environmental conditions? | Yes / No |
| Comments for Quality Management System: | ||
| G. Error and Accident Investigation and Reporting (applicable to all establishments) | ||
| 103-108 | Has the establishment read and understood the requirements of error and accident investigation and reporting? | Yes / No |
| Comments for Error and Accident Investigation and Reporting: | ||
| H. Adverse Reaction Investigation and Reporting (applicable to all establishments) | ||
| 109-116 | Has the establishment read and understood the requirements of adverse reaction investigation and reporting? | Yes / No |
| Comments for Adverse Reaction Investigation and Reporting: | ||
| I. Records (applicable to all registered establishments) | ||
| 117 | Are the records accurate, complete, legible, indelible and readily retrievable? | Yes / No |
| 118 | Is the donation code a component of all the records related to the processing, distribution, transformation and transfusion of the blood? | Yes / No |
| Records for establishments that collect allogeneic blood | ||
| 119(1) | Are the following records relating to allogeneic blood, including PADP activities, retained for the amount of time specified from the date they were created? | n/a |
| 1. Donor identification code for 50 years | Yes / No | |
| 2. Donation code for 50 years | Yes / No | |
| 3. Reconciliation of donation codes for 10 years | Yes / No | |
| 4. Donor suitability assessment for 5 years | Yes / No | |
| 5. Determination of indefinite donor unsuitability for 50 years | Yes / No | |
| 6. Determination of temporary donor unsuitability for 10 years | Yes / No | |
| 7. Collection - date of donation for 50 years | Yes / No | |
| 8. Collection - donor apheresis for 5 years | Yes / No | |
| 9. Collection - record of donation for 5 years | Yes / No | |
| 10. Lot number and name of manufacturer of container and other critical supplies for each donation for 1 year | Yes / No | |
| 11. Test results for transmissible disease testing, ABO group and Rh factor, and clinically significant antibody testing for 50 years | Yes / No | |
| 12. Blood component preparation for 10 years | Yes / No | |
| 13. Blood storage temperature monitoring for 5 years | Yes / No | |
| 14. Destruction or other disposition of blood for 50 years | Yes / No | |
| 15. Distribution for 50 years | Yes / No | |
| 16. Shipping documents for 1 year | Yes / No | |
| 17. Post-donation information for 10 years | Yes / No | |
| 18. Complaints and their investigation for 5 years | Yes / No | |
| 19. Internal audit reports for 5 years | Yes / No | |
| 20. Quality control testing for 5 years | Yes / No | |
| 21. Maintenance, validation, qualification and calibration of critical equipment for 3 years | Yes / No | |
| 22. Records related to critical supplies, including their qualification for 3 years | Yes / No | |
| 23. Proficiency testing for 5 years | Yes / No | |
| 24. Every version of the operating procedures that was implemented, other than those related to donor suitability assessments, for 10 years | Yes / No | |
| 25. Every version of the operating procedures related to donor suitability assessment for 50 years | Yes / No | |
| 26. Personnel qualifications, training and competency evaluation for 10 years | Yes / No | |
| 27. Investigations and reports of errors and accidents for 10 years | Yes / No | |
| 28. Investigations and reports of adverse reactions for 10 years | Yes / No | |
| 119(2) | Are the records related to personnel qualifications, training and competency evaluation being stored for 10 years from the last date on which the employee was last employed? | Yes / No |
| Records for establishments that collect autologous blood | ||
| 120(1) | Are the following records relating to autologous donation retained for the amount of time specified from the date they were created? | n/a |
| 1. Donor identification code for 10 years | Yes / No | |
| 2. Donation code for 10 years | Yes / No | |
| 3. Collection - donor record for 5 years | Yes / No | |
| 4. Lot number and name of manufacturer of container and other critical supplies for each donation for 1 year | Yes / No | |
| 5. Test results for transmissible disease testing, ABO group and Rh factor for 10 years | Yes / No | |
| 6. Blood component preparation for 10 years | Yes / No | |
| 7. Blood storage temperature monitoring for 5 years | Yes / No | |
| 8. Destruction or other disposition of blood for 10 years | Yes / No | |
| 9. Distribution for 10 years | Yes / No | |
| 10. Shipping documents for 1 year | Yes / No | |
| 11. Complaints and their investigation for 5 years | Yes / No | |
| 12. Internal audit reports for 5 years | Yes / No | |
| 13. Quality control testing for 5 years | Yes / No | |
| 14. Maintenance, validation, qualification and calibration of critical equipment for 3 years | Yes / No | |
| 15. Critical supplies, including their qualification for 3 years | Yes / No | |
| 16. Proficiency testing for 5 years | Yes / No | |
| 17. Every version of the operating procedures that was implemented for 10 years | Yes / No | |
| 18. Personnel qualifications, training and competency evaluation for 10 years | Yes / No | |
| 19. Investigations and reports of errors and accidents for 10 years | Yes / No | |
| 20. Investigations and reports of adverse reactions for 10 years | Yes / No | |
| 120(2) | Are the records related to personnel qualifications, training and competency evaluation being stored for 10 years from the last date on which the employee was last employed | Yes / No |
| Records for establishments that transform blood | ||
| 121(1) | Are the following records relating to transformation retained for the amount of time specified from the date they were created? | n/a |
| 1. Donation code for 10 years | Yes / No | |
| 2. Records of washing, pooling and irradiation of blood for 10 years | Yes / No | |
| 3. Lot number and name of manufacturer of critical supplies for each transformation for 1 year | Yes / No | |
| 4. Complaints and their investigation for 5 years | Yes / No | |
| 5. Internal audit reports for 5 years | Yes / No | |
| 6. Quality control testing for 5 years | Yes / No | |
| 7. Maintenance, validation, qualification and calibration of critical equipment for 3 years | Yes / No | |
| 8. Critical supplies, including their qualification for 3 years | Yes / No | |
| 9. Every version of the operating procedures that was implemented for 10 years | Yes / No | |
| 10. Personnel qualifications, training and competency evaluation for 10 years | Yes / No | |
| 11. Investigations and reports of errors and accidents for 10 years | Yes / No | |
| 12. Investigations and reports of adverse reactions for 10 years | Yes / No | |
| 121(2) | Are the records related to personnel qualifications, training and competency evaluation being stored for 10 years from the last date on which the employee was last employed by the establishment? | Yes / No |
| 123 | Are the records stored in a location that has appropriate environmental conditions and that is secure against the entry of unauthorized persons? | Yes / No |
| Comments for Records: | ||
Appendix D: Repealed Food and Drug Regulations C.04.400-C.04.423 Human Plasma Collected by Plasmapheresis
This appendix provides the requirements of the Food and Drug Regulations C.04.400-C.04.423 Human Plasma Collected by Plasmapheresis. These requirements are the baseline of the authorized criteria for licensed establishments previously held to these requirements prior to the repeal of these sections of the Food and Drug Regulations. These baseline requirements will change once an application for an amendment to an Authorization is submitted by an establishment and approved by Health Canada.
Human Plasma Collected by Plasmapheresis
Interpretation
C.04.400. The following definitions apply in this section and in sections C.04.401 to C.04.423.
"accident" means an unexpected event that is not attributable to a deviation from a fabricator's procedures or applicable laws and that could adversely affect the safety of a donor or the safety, efficacy or quality of plasma. (accident)
"donor" means a person aged 17 years or older who has given their name to a fabricator for the purpose of participating in plasmapheresis with that fabricator. (donneur)
"error" means a deviation from a fabricator's procedures or applicable laws that could adversely affect the safety of a donor or the safety, efficacy or quality of plasma. (manquement)
"fabricator" means a person who is the holder of an establishment licence issued under these Regulations that authorizes the person to fabricate source plasma. (manufacturier)
"personal identifier" means a unique group of letters, numbers or symbols, or any combination of them, that is assigned to a donor by a fabricator. (identificateur personnel)
"physician" means a person who is entitled to practice the profession of medicine under the laws of the province in which the person provides medical service in connection with plasmapheresis or specific immunization. (médecin)
"physician substitute" means a person who
- (a) acts under the general supervision and direction of a physician; and
- (b) is authorized to provide the services that may be provided by a physician substitute under sections C.04.401 to C.04.423, according to the applicable laws of the province in which the person provides any of those services. (substitut)
"plasmapheresis" means a process during which:
- (a) blood is taken from a donor from which plasma is separated; and
- (b) red blood cells and formed elements from the blood are returned to the donor. (plasmaphérèse)
"plasmapheresis session" means a meeting between a fabricator and a donor held for the purpose of proceeding with plasmapheresis. (séance de plasmaphérèse)
"serious adverse reaction" means an unexpected and undesirable response in a donor, associated with plasmapheresis or specific immunization, that results in any of the following consequences for the donor:
- (a) hospitalization;
- (b) persistent or significant disability or incapacity;
- (c) a medical or surgical intervention to preclude a persistent or significant disability or incapacity;
- (d) a life-threatening condition; or
- (e) death. (effet indésirable grave)
"source plasma" means human plasma collected by plasmapheresis that is intended for use in producing a drug for human use. (plasma destiné au fractionnement)
"specific immunization" means the administration of an immunogen to a donor with the intention of eliciting an immune response in their blood for the purpose of plasmapheresis. (immunisation spécifique)
"unique identifier" means a unique group of letters, numbers or symbols, or any combination of them, that is assigned by a fabricator to source plasma or red blood cells to be used in specific immunization. (identificateur unique)
SOR/78-545, s. 1; SOR/85-1022, s. 1; SOR/2006-353, s. 1.
Prohibitions
C.04.401. No person shall
- (a) sell source plasma unless it has been fabricated, tested, packaged/labelled and stored in accordance with sections C.04.402 to C.04.423; or
- (b) fabricate source plasma from blood collected from a person who is not suitable to participate in plasmapheresis according to sections C.04.402 to C.04.423.
SOR/78-545, s. 1; SOR/85-1022, s. 2; SOR/2006-353, s. 1.
Fabricator's Responsibility
C.04.402. (1) A fabricator shall ensure that a person who provides services to them in connection with plasmapheresis or specific immunization is qualified by education and by training or experience to provide the services.
(2) The fabricator shall ensure that the premises used for donor screening, plasmapheresis or specific immunization are designed, constructed and maintained in a manner that permits medical information to be communicated in confidence.
SOR/78-545, s. 1; SOR/85-1022, s. 2; SOR/97-12, s. 47; SOR/2006-353, s. 1.
Consent and Preliminary Evaluation
C.04.403. (1) A fabricator shall not begin plasmapheresis with a donor unless
- (a) the fabricator has informed the donor of what is involved with plasmapheresis, including the risks to the donor's health associated with plasmapheresis and with participating in plasmapheresis more frequently than once every eight weeks; and
- (b) after paragraph (a) has been satisfied, the fabricator obtains from the donor
- (i) a written acknowledgement that the information specified in paragraph (a) has been provided to them, and
- (ii) in accordance with the applicable laws governing consent, written informed consent to participate in plasmapheresis.
(2) A fabricator shall not begin the specific immunization of a donor unless
- (a) a physician has selected the immunogen to be administered to the donor and informed the donor of
- (i) the name and nature of the selected immunogen,
- (ii) the proposed frequency and the maximum number of specific immunization injections the donor is expected to receive, and
- (iii) what is involved with specific immunization, including the risks to the donor's health associated with specific immunization and with receiving the selected immunogen; and
- (b) after paragraph (a) has been satisfied, the fabricator obtains from the donor
- (i) a written acknowledgement that the information specified in paragraph (a) has been provided to them, and
- (ii) in accordance with the applicable laws governing consent, written informed consent to receive the selected immunogen.
SOR/78-545, s. 1; SOR/2006-353, s. 1.
C.04.404. (1) A fabricator shall not proceed with plasmapheresis or specific immunization unless a physician or physician substitute has determined the donor's suitability to participate in plasmapheresis more frequently than once every eight weeks based on the donor's medical history and a medical examination of the donor.
(2) If the donor is determined to be suitable, the fabricator shall document the following information:
- (a) the fact that the donor is suitable to participate in plasmapheresis more frequently than once every eight weeks;
- (b) the donor's name and personal identifier;
- (c) the name and signature of the physician who makes the determination, or supervises the physician substitute making the determination; and
- (d) the date of the determination.
(3) The fabricator shall not proceed with plasmapheresis or specific immunization if the most recent determination under subsection (1) in respect of the donor was made more than
- (a) 30 days before the date set for the donor's first participation in plasmapheresis or specific immunization; or
- (b) one year before any other date set for the donor's participation in plasmapheresis or specific immunization.
SOR/78-545, s. 1; SOR/85-1022, s. 3; SOR/2006-353, s. 1.
Specific Immunization
C.04.405. (1) No one other than a physician or physician substitute shall administer an immunogen to a donor for the purpose of specific immunization.
(2) A physician shall monitor the donor's response to the immunogen to determine if the donor can continue to receive specific immunization.
(3) If the donor cannot continue to receive specific immunization, the fabricator shall cease to provide it to the donor until a physician determines that the donor can receive specific immunization using the same or another immunogen.
SOR/78-545, s. 1; SOR/85-1022, s. 3; SOR/2006-353, s. 1.
Evaluation Before Collection
C.04.406. (1) At the beginning of each plasmapheresis session, a physician or physician substitute shall determine if the donor is suitable to participate in plasmapheresis.
(2) If the donor is determined to be temporarily not suitable to participate in plasmapheresis based on the criteria set out in Table 1 or any other medical reason justifying a determination of temporary non-suitability, the fabricator shall cancel the session, inform the donor of the reason why they are temporarily not suitable and indicate the date when the donor may continue to participate in plasmapheresis.
(3) If the donor is determined to be not suitable to participate in plasmapheresis for an indefinite period based on the exclusion criteria set out in Table 2 or any other medical reason justifying a determination of indefinite non-suitability, the fabricator shall cancel the session and inform the donor of the reason why they are not suitable to participate in plasmapheresis for an indefinite period.
| Item | Criteria |
|---|---|
| 1. | Weight of less than 50 kg |
| 2. | Temperature outside of normal limits |
| 3. | Blood pressure above 100 mmHg diastolic or 180 mmHg systolic |
| 4. | Haemoglobin level of less than 125 g/L of blood or haematocrit value of less than 0.38 L/L of blood |
| 5. | Total protein level of less than 60 g/L of blood |
| 6. | Substantial blood loss |
| 7. | Prior donation of plasma or other blood components |
| 8. | Pregnancy |
| 9. | History of medical or surgical procedures |
| 10. | History of convulsions requiring medical treatment |
| 11. | Ability to answer questions compromised by alcohol or drug use |
| 12. | Prior transfusion of blood, blood components or a blood product, or prior transplantation of a cell, tissue or organ other than dura mater |
| 13. | Skin infection at the site of the phlebotomy |
| 14. | Sign or symptom of infection |
| 15. | Risk of infection with HIV, hepatitis B virus or hepatitis C virus based on, but not limited to, a history of acupuncture, skin piercing, tattooing, accidental needle-stick injury or occasional sexual relations with a person at risk of having any of those infections |
| 16. | Current or past use of medication that poses a risk to a recipient of a product manufactured from source plasma |
| 17. | Receipt of a live attenuated vaccine |
| 18. | Animal bite requiring prophylaxis for rabies or for which the need for post-exposure prophylaxis has not been assessed |
| Item | Exclusion Criteria |
|---|---|
| 1. | Abnormal cardiovascular function or serious or chronic cardiovascular disease |
| 2. | Abnormal respiratory function or serious or chronic respiratory disease |
| 3. | Bleeding disorder that poses a risk to the donor in relation to plasmapheresis |
| 4. | Serious disease or medical condition of the liver, kidneys, another organ, a system or blood |
| 5. | Persistent abnormal plasma proteins including monoclonal or polyclonal gammopathy |
| 6. | Current or past use of medication that poses an ongoing risk to a recipient of a product manufactured from source plasma |
| 7. | History of recurrent fainting associated with the donation of blood or plasma |
| 8. | History, signs or symptoms of injectable drug abuse such as skin punctures, scars or sharing needles to inject drugs |
| 9. | History, signs or symptoms of AIDS or HIV infection |
| 10. | Risk of HIV infection based on sexual practices |
| 11. | History, signs or symptoms of a chronic or persistent infection or parasitic disease transmissible by blood |
| 12. | History, signs or symptoms of hepatitis, other than hepatitis A |
| 13. | Cancer, other than non-melanoma skin cancer or insitu cervical cancer |
| 14. | Risk factor for Creutzfeldt-Jacob disease (CJD) or its variant (vCJD) based on, but not limited to, the receipt of dura mater transplant or a treatment using a human pituitary hormone |
| 15. | Positive test result for any transmissible disease agent |
SOR/78-545, s. 1; SOR/85-1022, s. 3; SOR/2006-353, s. 1.
Plasma Protein Composition
C.04.407. (1) Before beginning plasmapheresis with a donor, a fabricator shall take a blood sample from the donor to determine the plasma protein composition of the donor's blood by means of a serum protein electrophoresis test or an equivalent test.
(2) A blood sample shall be taken within seven days before the donor's first plasmapheresis session at which the fabricator proceeds with plasmapheresis.
(3) If 21 days have elapsed from the taking of the sample without a physician examining the test result, the fabricator may not proceed with plasmapheresis until a physician examines the test result.
(4) If a physician concludes that the plasma protein composition of the donor's blood is not within normal limits, the fabricator may not proceed with plasmapheresis until a physician determines that the plasma protein composition of the donor's blood is within normal limits.
(5) If the fabricator has not taken a blood sample from the donor as required under subsection (1) for more than four months, the fabricator may not proceed with plasmapheresis until the blood sample is taken from the donor.
SOR/78-545, s. 1; SOR/85-1022, s. 3; SOR/2006-353, s. 1.
Ongoing Review of Collection Records
C.04.408. (1) A physician shall determine if a donor is suitable to continue to participate in plasmapheresis more frequently than once every eight weeks, based on the test results and collection records for the donor that have been made or received by the fabricator within the preceding four months.
(2) The determination shall be made at least every four months after the date of the initial determination that the donor is suitable under section C.04.404.
(3) If the donor is determined to be temporarily not suitable to participate in plasmapheresis the fabricator shall inform the donor of the reason why they are temporarily not suitable and indicate the date when the donor may continue to participate in plasmapheresis.
(4) If the donor is determined to be not suitable for an indefinite period, the fabricator may not proceed with plasmapheresis and shall inform the donor of the reason why they are not suitable.
(5) If the requirement of subsection (2) is not met, the fabricator may not proceed with plasmapheresis until the determination is made.
SOR/78-545, s. 1; SOR/85-1022, s. 3; SOR/2006-353, s. 1.
Plasmapheresis Procedures
C.04.409. A fabricator who conducts a plasmapheresis session shall
- (a) use aseptic methods and a sterile collection system licensed under the Medical Devices Regulations;
- (b) ensure that all surfaces intended to come into contact with blood or plasma are pyrogen free;
- (c) ensure that the donor's skin where the phlebotomy is to be made is:
- (i) determined to be free from lesion, rash or other source of infection; and
- (ii) cleaned and disinfected; and
- (d) ensure that emergency medical personnel are capable of attending to the medical needs of the donor within 10 minutes after being contacted by the fabricator.
SOR/78-545, s. 1; SOR/85-1022, s. 4; SOR/2006-353, s. 1.
Maximum Volumes and Minimum Intervals
C.04.410. (1) A fabricator shall not collect plasma from a donor in a total amount, excluding anticoagulant solution, that exceeds
- (a) if the donor's weight is 50 kg or more but less than 68 kg:
- (i) 625 mL or 640 g in respect of a single plasmapheresis session; and
- (ii) 11.5 L in respect of all plasmapheresis sessions during the preceding six months;
- (b) if the donor's weight is 68 kg or more but less than 80 kg:
- (i) 750 mL or 770 g in respect of a single plasmapheresis session; and
- (ii) 15.5 L in respect of all plasmapheresis sessions during the preceding six months; and
- (c) if the donor's weight is 80 kg or more:
- (i) 800 Ml or 820 g in respect of a single plasmapheresis session; and
- (ii) 18.5 L in respect of all plasmapheresis sessions during the preceding six months.
(2) The fabricator shall have written procedures that describe
- (a) the minimum waiting period for a donor between donations of plasma and between a donation of plasma and a donation of blood or other blood components; and
- (b) the maximum number of plasma donations a donor may make in a given period.
SOR/78-545, s. 1; SOR/85-1022, s. 5; SOR/95-203, s. 1; SOR/2006-353, s. 1.
Anticoagulant Solution
C.04.411. (1) During plasmapheresis, the fabricator shall mix an anticoagulant solution with the blood collected from the donor.
(2) The anticoagulant solution shall have a valid drug identification number under these Regulations that indicates the solution is suitable for use in plasmapheresis.
SOR/78-545, s. 1; SOR/2006-353, s. 1.
Samples for Testing
C.04.412. (1) During a plasmapheresis session, the fabricator shall take a sample of blood or plasma in a manner that does not contaminate the sample or the source plasma.
(2) When the sample is taken, the fabricator shall clearly and permanently label the sample container with the unique identifier assigned to the source plasma.
(3) The fabricator shall ensure that the person who labels the sample container is the same person who labels the container holding the source plasma under subsection C.04.416(2).
SOR/78-545, s. 1; SOR/2006-353, s. 1.
C.04.413. (1) The fabricator shall test a sample taken under section C.04.412 to detect evidence of the following disease agents:
- (a) HIV types 1 and 2;
- (b) hepatitis B virus;
- (c) hepatitis C virus; and
- (d) syphilis.
(2) The fabricator shall retain the source plasma collected at the plasmapheresis session until all the test results are determined to be negative or non-reactive.
(3) In the case of a positive or reactive test result for any disease agent referred to in subsection (1), the fabricator shall
- (a) clearly and permanently label the container holding the source plasma collected at the session with:
- (i) the statement "Caution: Not for Manufacturing Use" or "Précaution : Non destiné à la fabrication"; and
- (ii) the hazard symbol for Biohazardous Infectious Material set out in Schedule II to the Controlled Products Regulations; and
- (b) segregate and dispose of the source plasma.
(4) In the case of a positive or reactive test result for syphilis, the fabricator may not proceed with plasmapheresis until a subsequent test shows that the donor is not infected with syphilis and a physician determines that the donor can continue to participate in plasmapheresis.
(5) In the case of a positive or reactive test result for a disease agent referred to in subsection (1), other than syphilis, the fabricator shall discontinue plasmapheresis and inform the donor of the reason why they are not suitable to participate in plasmapheresis for an indefinite period.
SOR/78-545, s. 1; SOR/97-12, s. 48; SOR/2006-353, s. 1.
Preservatives and Additives
C.04.414. No person shall add a preservative or additive to source plasma.
SOR/78-545, s. 1; SOR/85-1022, s. 6; SOR/2006-353, s. 1.
Containers
C.04.415. A fabricator shall place source plasma in a container
- (a) in respect of which a medical device licence has been issued under the Medical Devices Regulations for the purpose of collecting and storing plasma;
- (b) that permits visual, electronic or automated inspection of the plasma;
- (c) that has been visually inspected at the plasmapheresis session and found to be intact; and
- (d) that has not been previously used for any purpose, including holding source plasma from the same donor.
SOR/78-545, s. 1; SOR/85-1022, s. 6; SOR/2006-353, s. 1.
Labelling
C.04.416. (1) Sections C.01.004 and C.04.019 do not apply to source plasma.
(2) A fabricator shall clearly and permanently label the container used to hold source plasma with
- (a) the unique identifier assigned to the source plasma in the container;
- (b) the statement "Source Plasma" or "Plasma destine au fractionnement";
- (c) the statement "Caution: For Manufacturing Use Only" or "Précaution : À utiliser uniquement pour la fabrication";
- (d) the quantity of the source plasma;
- (e) the name and quantity of the anticoagulant solution used during the plasmapheresis;
- (f) the expiry date of the source plasma, expressed in an unambiguous format;
- (g) subject to subsection C.04.413(3), a statement indicating that the source plasma tests negative for the disease agents for HIV, hepatitis B and hepatitis C;
- (h) if the source plasma was collected from a donor who has received specific immunization, a statement indicating the immunogen that was used;
- (i) the name, address and establishment licence number of the fabricator; and
- (j) a statement indicating that the source plasma must be stored at a temperature of -20°C or colder.
(4) The unique identifier shall be placed on the container at the time of collection.
SOR/78-545, s. 1; SOR/85-1022, s. 7; SOR/2006-353, s. 1.
Storage
C.04.417. (1) In respect of the storage of source plasma, including storage during transportation, a fabricator shall ensure that the storage environment
- (a) is designed to maintain a temperature of -20°C or colder; and
- (b) remains consistently at a temperature of -20°C or colder.
(2) If the temperature of the environment rises above -20°C, the fabricator shall record the following information:
- (a) the reason for the elevated temperature;
- (b) the source plasma affected; and
- (c) the final disposition of the source plasma.
(3) If the temperature of the environment rises to between - 20°C and +10°C, the fabricator shall clearly and permanently label the container of the source plasma with the statement "Source Plasma - Salvaged" or "Plasma destiné au fractionnement - recyclé".
(4) Subsection (3) does not apply if the temperature of the environment rises to between -20°C and -5°C for a single period lasting less than 72 hours.
(5) If the temperature of the environment rises above +10°C, the fabricator shall dispose of the source plasma.
(6) Paragraph (1)(b) and subsections (2) to (5) do not apply in respect of the storage of source plasma during transportation, if the transportation is not conducted by the fabricator.
SOR/78-545, s. 1; SOR/85-1022, s. 8; SOR/2006-353, s. 1.
C.04.418. (1) A fabricator shall inspect each container of source plasma to determine if the container and its label are intact and if there are any indications that the source plasma has been subject to thawing.
(2) The fabricator shall dispose of the source plasma if the inspection shows that
- (a) the container is defective or damaged to the extent that it does not provide protection against external factors that could result in deterioration or contamination of the source plasma;
- (b) the unique identifier assigned to the source plasma is missing or illegible;
- (c) any information required under paragraphs C.04.416(2)(b) to (i) is missing or illegible, unless the missing or illegible information can be retrieved from the fabricator's records; or
- (d) the source plasma has been subject to thawing.
SOR/78-545, s. 1; SOR/2006-353, s. 1.
Records
C.04.419. (1) A fabricator shall use and maintain a recordkeeping system according to which the fabricator shall
- (a) assign a personal identifier to each donor;
- (b) keep on the donor's file a photograph of the donor or some other reliable means of identification; and
- (c) assign a unique identifier to the source plasma collected by the fabricator at each plasmapheresis session.
(2) The system shall be structured so that a fabricator may, based on a personal identifier or a unique identifier, identify the donor and retrieve sufficient records to permit the traceability and recall of source plasma.
(3) The fabricator shall keep the records referred to in subsection (2) indefinitely.
SOR/78-545, s. 1; SOR/85-1022, s. 9; SOR/2006-353, s. 1.
C.04.420. (1) For each donor, the fabricator shall keep
- (a) the original or a copy of the donor's acknowledgement and consent under paragraphs C.04.403(1)
- (b) and (2)(b), if any;
- (b) the original or a copy of any determinations, examinations, test results, reports and written notices made under sections C.04.401 to C.04.423;
- (c) for each specific immunization given by the fabricator to the donor, a record indicating
- (i) the date and location of the immunization;
- (ii) the physician or physician substitute who administered the immunogen; and
- (iii) for the immunogen injected, its name and manufacturer's name, the quantity and expiry date and either the immunogen's lot number and drug identification number or, if the immunogen is red blood cells, its unique identifier;
- (d) for each plasmapheresis session held by the fabricator for the donor, a record indicating
- (i) the date and location of the session;
- (ii) the volume of source plasma collected;
- (iii) the unique identifier assigned to the source plasma;
- (iv) the volume of red blood cells collected that was not returned to the donor, including the volume of red blood cells collected during sampling;
- (v) for the anticoagulant solution used, its name, its manufacturer's name and its lot number and drug identification number; and
- (vi) for the container used, the manufacturer's name and the container's lot number and expiry date.
(2) The fabricator shall maintain a summary of all accidents, errors, serious adverse reactions and recalls of source plasma involving the fabricator.
(3) The fabricator shall maintain temperature records made under subsection C.04.417(2).
SOR/78-545, s. 1; SOR/85-1022, s. 10; SOR/97-12, s. 61; SOR/2006-353, s. 1.
Information to the Minister
C.04.421. (1) A fabricator shall notify the Minister of any serious adverse reaction
- (a) within 24 hours after the fabricator becomes aware of the occurrence, in the case of a fatality; and
- (b) within 15 days after the fabricator becomes aware of the occurrence, in any other case.
(2) In the case of a verbal notice under subsection (1), the fabricator shall submit a written report of the serious adverse reaction to the Minister within 24 hours after submitting the notice.
(3) The notice, if in writing, or the written report shall include a description of the serious adverse reaction and any steps taken to address it.
SOR/78-545, s. 1; SOR/2006-353, s. 1.
C.04.422. If a fabricator recalls source plasma for a reason involving product safety, the fabricator shall provide the Minister with a written report stating the reason for the recall, the number of units involved and the location from which the units were recalled.
SOR/78-545, s. 1; SOR/2006-353, s. 1.
C.04.423. In order to prevent injury to the health and safety of donors and recipients of products manufactured from source plasma, a fabricator shall, on request, provide the Minister with a copy of any record pertaining to plasmapheresis, specific immunization or source plasma that is required by sections C.04.401 to C.04.422 to be kept by the fabricator.
SOR/78-545, s. 1; SOR/2006-353, s. 1.
Appendix E: Health Canada Guidance Documents and Directives superseded by the Guidance Document: Blood Regulations
Please note that other guidance documents and directives as well as any associated forms (not listed) that provide further interpretation of the Food and Drug Regulations, Part C, Division 1A, 2 and 4 no longer apply to blood that is the subject of the Blood Regulations.
- Guidance for Industry: Management of Blood Establishment Submissions
- Guidance Document: Human Plasma Collected by Plasmapheresis
- Annex 14 to the Current Edition of the GMP Guidelines - Schedule D Drugs, Human Blood and Blood Components (GUI-0032)
- Information Letter I.L. No. 816 November 1, 1995, 2. Health Canada Policy a) Donors considered to pose a risk of CJD b) Donor Deferral c) Withdrawal/quarantine of In-date Blood Products (November 1, 1995)
- D98-01: Implementation of Pre-storage Leukoreduction of Cellular Blood Components (November 2, 1998)
- D99-01: Donor Exclusion to Address Theoretical Risk of Transmission of variant CJD through the Blood Supply (August 17, 1999)
- D99-02: Donor Exclusion to Address Theoretical Risk of Transmission of variant CJD through the Use of Commercial Blood Products (August 17, 1999)
- D2000-01: Donor Exclusion to Address Theoretical Risk of Transmission of variant CJD through the Blood Supply (August 30, 2000)
- D2001-001: Donor Exclusion to Address Theoretical Risk of Transmission of variant Creutzfeldt-Jakob Disease (vCJD) through the Blood Supply: United Kingdom, France & Western Europe (August 30, 2001)
- Additional Donor Exclusion Measures to Address the Potential Risk of Transmission of variant Creutzfeldt-Jakob Disease (vCJD) through the Blood Supply (April 22, 2005)
- D2006-01: Implementation of blood donor screening measures to reduce the theoretical risk of transmission of simian foamy virus and possibly other yet unidentified simian viruses by transfusion (May 15, 2006)
- Information Letter Regarding Syphilis Testing and Deferral Requirements for History of Parasitic Disease for Donors of Source Plasma for Further Manufacturing (September 2, 2010)