
Ligne directrice : Règlement sur le sang

Date d'approbation : 2014-05-12
Date mis en vigueur : 2014-10-23
Date de modification : 2023-02-21
Avant-propos
Les documents d'orientation sont conçus pour apporter une aide à l'industrie et aux professionnels de la santé sur la façon de respecter la législation et la réglementation correspondantes. Ils fournissent par ailleurs aux membres du personnel de Santé Canada des renseignements sur la façon de mettre en œuvre le mandat et les objectifs du Ministère de manière équitable, uniforme et efficace.
Les lignes directrices sont des outils administratifs n'ayant pas force de loi, ce qui permet une certaine souplesse dans l'approche. Les principes et les pratiques énoncés dans le présent document pourraient être remplacés par d'autres approches, à condition que celles-ci s'appuient sur une justification scientifique adéquate. Il convient d'en discuter à l'avance avec le programme concerné pour s'assurer qu'elles respectent les exigences des lois et des règlements applicables.
Dans la foulée de ce qui précède, il importe également de mentionner que Santé Canada se réserve le droit de demander des renseignements ou du matériel supplémentaire, ou de définir des conditions dont il n'est pas explicitement question dans la présente ligne directrice afin que le Ministère puisse être en mesure d'évaluer adéquatement l'innocuité, l'efficacité ou la qualité d'un produit thérapeutique donné. Santé Canada s'engage à justifier de telles demandes et à documenter clairement ses décisions.
Ce document devrait être lu en parallèle avec l'avis qui l'accompagne et les sections pertinentes des autres lignes directrices applicables.
Table des matières
- 1. Introduction
- 1.1 Objectifs stratégiques
- 1.2 Portée et application
- 1.3 Contexte
- 1.4 Acronymes
- 1.5 Définitions
- 2. Ligne directrice de mise en œuvre
- Article 1 Définitions
- Articles 2-3 Champ d'application
- Article 4 Interdictions
- Articles 5-16 Homologation
- Article 5 Homologation
- Article 6 Demande d'homologation
- Article 7 Délivrance
- Article 8 Refus
- Article 9 Changement majeur
- Article 10 Changement en cas d'urgence
- Article 11 Changements administratifs - avis
- Article 12 Autres changements - rapport annuel
- Article 13 Ajout de conditions à l'homologation ou modification
- Article 14 Suspension
- Article 15 Rétablissement de l'homologation
- Article 16 Annulation de l'homologation
- Articles 17-29 Licence d'établissement
- Article 17 Obligation d'obtenir une licence
- Article 18 Demande de licence
- Article 19 Inspection
- Article 20 Délivrance
- Article 21 Refus
- Article 22 Changement des renseignements - demande de modification
- Article 23 Changement de nature administrative - avis
- Article 24 Modification de la licence par le ministre
- Article 25 Ajout de conditions ou modification
- Article 26 Renseignements et documents supplémentaires
- Article 27 Suspension
- Article 28 Rétablissement de la licence
- Article 29 Annulation
- Articles 30-37 Enregistrement
- Articles 38-58 Traitement
- Articles 59-68 Étiquetage
- Article 59 Non-application - donneurs pré-évalués
- Article 60 Exigences linguistiques
- Article 61 Exigences générales
- Article 62 Document d'information
- Article 63 Code d'identification du don
- Article 64 Texte de l'étiquette
- Article 65 Aliquotes
- Article 66 Don désigné
- Article 67 Don dirigé
- Article 68 Vérification des étiquettes
- Articles 69-72 Conservation
- Articles 73-76 Distribution
- Articles 77-80 Transformation
- Articles 81-85 Distribution Exceptionnelle
- Articles 86-91 Programme de donneurs pré-évalués
- Article 92 Importation dans des circonstances urgentes
- Articles 93-123 Gestion de la qualité
- Articles 93-94 Système de gestion de la qualité
- Articles 95-97 Procédures opérationnelles
- Articles 98-102 Personnel, installations, équipement, matériel et produits
- Articles 103-108 Enquêtes et rapports concernant les accidents et manquements
- Articles 109-116 Enquêtes et rapports concernant les effets indésirables
- Articles 117-123 Dossiers
- Article 117 Caractéristiques des documents
- Article 118 Codes d'identification du don
- Article 119 Période de rétention - don allogénique
- Article 120 Période de rétention - don autologue
- Article 121 Période de rétention - transformation
- Article 122 Période de rétention - transfusion
- Article 123 Entreposage des dossiers
- Article 124 Pouvoirs des inspecteurs
- Article 125 Modification corrélative
- Articles 126-128 Dispositions transitoires
- Article 129 Entrée en vigueur
- Annexe A: Glossaire
- Annexe B: Tableau récapitulatif des rapports annuels à fournir par les établissements de sang
- Annexe C : Outil d'autoévaluation pré-enregistrement pour les établissements de sang présentant une demande d'enregistrement
- Annexe D: Règlement sur les aliments et drogues C.04.400-C.04.423 Plasma humain prélevé par plasmaphérèse: Abrogés
- Annexe E: Les lignes directrices et directives de Santé Canada sont remplacées par la ligne directrice : Règlement sur le sang
1. Introduction
1.1 Aperçu et objectif
Le Règlement sur le sang a pour objet d’accroître la protection de la santé et de la sécurité des donneurs et des receveurs de sang au Canada par le maintien de la sécurité du sang destiné à la transfusion ou à une fabrication ultérieure. Veuillez vous référer à l’article 1, Définitions, de la présente ligne directrice de mise en œuvre pour la définition de « sécurité ».
Le Règlement sur le sang contient des dispositions sur la sécurité humaine et la sécurité du sang dans le cas des activités suivantes liées au sang et aux composants sanguins humains destinés à la transfusion : traitement (évaluation de l’admissibilité des donneurs, prélèvement, mise à l’essai et préparation de composants sanguins); transformation (lavage, mise en commun et irradiation du sang); étiquetage; conservation; gestion des dossiers; importation; distribution; enquêtes et rapports sur les manquements, les accidents et les effets indésirables.
Le Règlement sur le sang contient des dispositions sur la sécurité humaine et la sécurité du sang dans le cas des activités suivantes liées au sang et aux composants sanguins humains destinés à une fabrication ultérieure : traitement (évaluation de l’admissibilité des donneurs, prélèvement, mise à l’essai et préparation de composants sanguins); étiquetage; conservation; gestion des dossiers; distribution; enquêtes et rapports sur les manquements, les accidents et les effets indésirables.
Le Règlement sur le sang relève de la Direction générale des produits de santé et des aliments, Santé Canada. Toutes les questions concernant ce texte de loi ou la présente ligne directrice peuvent être envoyées à brddopic-bpcidmbr@hc-sc.gc.ca.
1.2 Portée et application
Le Règlement sur le sang ne s’applique qu’au sang humain destiné à la transfusion ou à la fabrication de drogues pour usage humain. La fabrication de médicaments utilisant le sang et les composants sanguins dépasse la portée du Règlement sur le sang et est régie par le Règlement sur les aliments et drogues. Les fabricants de produits sanguins sont mentionnés dans cette ligne directrice en ce qui a trait à la chaîne de distribution et aux fins des communications sur la sécurité du sang. Veuillez vous référer au paragraphe 1.5, Définitions, de la présente ligne directrice pour la définition de « fabricant de produits sanguins ».
Le Règlement sur le sang est établi en vertu de la Loi sur les aliments et drogues. Il s’applique à toute personne ou à tout établissement qui traite, étiquette, conserve, distribue ou transforme du sang destiné à la transfusion ou à une fabrication ultérieure, y compris à tout établissement qui importe du sang destiné à la transfusion. La Loi sur les aliments et drogues et la version en vigueur de la norme nationale canadienne CAN/CSA Z902, Sang et produits sanguins labiles (norme sur le sang de la CSA), publiée par le groupe CSA devraient être lues parallèlement à ce règlement.
Il incombe à l’établissement de s’assurer qu’il se conforme au Règlement sur le sang et aux articles de la norme sur le sang de la CSA qui y sont incorporés par renvoi, et ce, dans la version la plus récente de ces documents. Les articles de la norme sur le sang de la CSA incorporés par renvoi dans le Règlement sur le sang ont valeur d’exigences règlementaires et doivent être respectés. Les articles de la norme sur le sang de la CSA visés uniquement par cette ligne directrice ont valeur de pratiques exemplaires recommandées. En cas de divergence entre la norme sur le sang de la CSA qui n’est pas incorporée par renvoi dans le Règlement sur le sang et une exigence du Règlement sur le sang, les exigences règlementaires prévalent car elles constituent des règles législatives adoptées par le gouverneur en conseil.
La présente ligne directrice remplace certains documents d’orientation de Santé Canada sur le sang (veuillez vous référer à l’annexe E). Elle devrait être lue en parallèle avec le Règlement sur le sang. Dans l’éventualité de la perception d’une incohérence ou d’une contradiction, le Règlement a préséance sur la ligne directrice.
Le terme « doit » y indique une exigence, c.-à-d. une prescription du Règlement que l’établissement est légalement tenu de respecter afin de se conformer aux exigences réglementaires, le terme « devrait » indique une recommandation ou ce qu’il est conseillé, mais non obligatoire de faire, et le terme « peut » est utilisé pour exprimer une option ou ce qui est permis dans les limites de la ligne directrice.
Le nombre de jours du délai à respecter pour l’établissement ou Santé Canada relativement à un avis ou à une mesure est exprimé en jours civils, sauf indication contraire.
1.3 Énoncés de politique
Le Règlement sur le sang, en application de la Loi sur les aliments et drogues, prévoit des exigences précises réglementant le sang et les composants sanguins destinés à la transfusion ou à la transformation en drogues pour usage humain. La présente ligne directrice interprète ces exigences afin que les établissements qui traitent, étiquettent, distribuent, transforment ou conservent du sang et des composants sanguins à des fins de transfusion ou de fabrication ultérieure et les établissements qui importent du sang et/ou des composants sanguins destinés à la transfusion puissent disposer de renseignements qui les aident à se conformer au Règlement sur le sang.
1.4 Contexte
Le Règlement sur le sang a été élaboré dans le but de :
- finaliser la réponse de Santé Canada aux recommandations du rapport de la Commission Krever;
- ajouter à la réglementation fédérale des dispositions propres à la sécurité du sang et de ses composants;
- clarifier les dispositions relatives au sang des divers titres du Règlement sur les aliments et drogues et les regrouper dans un instrument séparé n’encadrant que la sécurité du sang;
- établir des exigences adaptées au sang en tant que produit thérapeutique particulier plutôt que d’assujettir le sang aux dispositions générales de la réglementation des drogues;
- disposer d’un instrument qui permettra de faire face à l’évolution rapide des technologies, aux maladies émergentes et aux pénuries de sang dans des circonstances urgentes.
Le Règlement sur le sang encadre les établissements selon le degré de risque que leurs activités présentent pour la sécurité du sang prélevé au Canada et destiné à la transfusion ou à une fabrication ultérieure.
L’établissement doit soumettre à Santé Canada une demande d’homologation et de licence d’établissement s’il entend exercer des activités de traitement décrites dans le Règlement sur le sang à l’égard de sang allogénique et de composants sanguins allogéniques destinés à la transfusion, y compris à l’égard de plasma destiné à une fabrication ultérieure. La rédaction du document d’information de sang allogénique ou de composants sanguins allogéniques destinés à la transfusion et l’étiquetage des unités de sang allogénique, avant la distribution, doivent se faire conformément à une homologation. Le sang et/ ou les composants sanguins importés à des fins de transfusion doivent être associés à une homologation, et l’établissement importateur doit détenir une licence d’établissement.
Les dispositions des articles C.04.400–C.04.423, à l’intertitre Plasma humain prélevé par plasmaphérèse du Règlement sur les aliments et drogues, sont présentées à l’annexe D à titre de référence seulement. C’est sur ces dispositions que sont fondés les critères approuvés des établissements titulaires d’une licence qui étaient assujettis à ces articles avant leur abrogation.
L’établissement doit s’inscrire auprès de Santé Canada s’il prélève du sang autologue, s’il a un programme de donneurs pré-évalués ou s’il transforme du sang.
Tout établissement qui conserve et transfuse du sang et/ou des composants sanguins doit satisfaire aux exigences applicables à ces activités qui sont décrites dans le Règlement sur le sang. Remarque : L’étiquetage, après que le sang et/ou les composants sanguins ont été jugés sécuritaires, est une activité qui concerne les établissements de transformation ou de transfusion.
Certains articles du Règlement sur le sang font référence à des dispositions précises de la norme sur le sang de la CSA qui relèvent de Santé Canada. Lorsqu’un article, une clause ou un tableau précis de la norme sur le sang de la CSA est incorporé par renvoi dans le présent règlement, il devient une exigence réglementaire obligatoire. La norme sur le sang de la CSA, telle que modifiée de temps à autre, est incorporée de cette façon. Les articles ou les tableaux de la norme sur le sang de la CSA, qui ne sont pas mentionnés dans le Règlement sur le sang, demeurent volontaires.
1.4.1 Norme sur le sang de la CSA
La norme sur le sang de la CSA couvre le cycle de vie du sang destiné à la transfusion et est largement considérée comme un recueil de pratiques exemplaires de l’industrie.
La norme sur le sang de la CSA a été élaborée et modifiée dans le cadre d’un processus consensuel par un comité technique formé d’experts dans le domaine de la sécurité du sang, de groupes d’utilisateurs et des gouvernements fédéral, provinciaux et territoriaux. La CSA entreprend des consultations sur les révisions de la norme sur le sang de la CSA dans le cadre de son processus d’élaboration de normes.
Tous les établissements doivent avoir accès à la version en vigueur de la norme sur le sang de la CSA, car certaines dispositions du Règlement sur le sang reposent sur cette norme. La norme peut être commandée par Internet, sur le site de l’Association canadienne de normalisation (www.shopcsa.ca/onlinestore/welcome.asp), ou par téléphone, au 1-800-463-6727. Des renseignements sur la façon d’obtenir les mises à jour ou les modifications sont publiées à la page « Service de mise à jour des normes CSA » sur le site de l’Association
La norme sur le sang de la CSA peut également être consultée en s’inscrivant sur le site Web communautaire de la CSA :
https://community.csagroup.org/community/health-care-safety-and-accessibility/blood-and-transplants-standards-view-access.
Tous les intervenants peuvent jouer un rôle important dans le maintien à jour de la norme sur le sang de la CSA. Celle-ci renferme d’ailleurs un formulaire de suggestions de changements, qui peut être soumis directement à la CSA et dans lequel les intervenants sont priés d’aider la CSA à évaluer les changements qu’ils proposent en fournissant, outre les coordonnées requises, tous les renseignements qui suivent.
- le numéro de la norme ou de la publication
- le numéro de l’article, du tableau ou de la figure visé
- la formulation proposée
- la raison de la modification
Les renvois à la norme sur le sang de la CSA dans le Règlement sur le sang ne sont pas statiques et peuvent donc être modifiés à l’occasion. Santé Canada examinera toute modification des articles de la norme cités par renvoi dans le Règlement pour vérifier les risques et les répercussions qu’elle pourrait avoir sur la sécurité du sang et des composants sanguins.
1.5 Définitions
Les définitions supplémentaires suivantes faciliteront la compréhension de la présente ligne directrice.
« aphérèse » Procédé de prélèvement sanguin consistant à séparer les composants du sang du donneur et à réinjecter, tous ou certains des composants qui restent, au donneur.
« Direction des instruments médicaux de la Direction générale des produits de santé et des aliments de Santé Canada » Chargé, à titre d’organisme fédéral de réglementation, de délivrer les homologations d’instruments médicaux conformément à la Loi sur les aliments et drogues et au Règlement sur les instruments médicaux. La Liste desinstruments médicaux homologués en vigueur (MDALL), base de données de tous les instruments homologués de classe, II, III ou IV en vente au Canada, est publiée sur le site Web de Santé Canada, à l’adresse :https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/instruments- medicaux/licences/liste-instruments-medicaux-homologues-vigueur.html
« clarifax » Outil de communication qui sert à demander des renseignements ou à demander des éclaircissements sur des renseignements déjà soumis.
« enquête sur les composants transfusés » Processus d’étude d’un rapport d’infection soupçonnée d’être associée à une transfusion qui permet d’identifier le donneur qui pourrait être en cause. Une telle enquête vise :
- à déterminer si le donneur qui a contribué à la transfusion est infecté par des marqueurs sérologiques de l’agent infectieux en cause ou s’il en est positif;
- à mener à un retrait/rappel des composants sanguins non périmés provenant de ce donneur; ou
- à aviser les destinataires et les receveurs des composants sanguins provenant de ce donneur.
« essais » est le terme utilisé dans le Règlement sur le sang et peut signifier « analyses » ou « tests ».
« étude des dons antérieurs » Processus dans le cadre duquel on retrouve :
- des dons antérieurs (et les produits sanguins connexes) d’un donneur qui s’est révélé, à la suite d’analyses ultérieures, infecté par un agent de maladie transmissible par transfusion; et
- les receveurs chez qui ont été transfusés des composants sanguins provenant d’un donneur qui, à la suite d’analyses ultérieures, a été confirmé positif à un agent infectieux transmissible par transfusion.
« fabricant de produits sanguins » Fabricant de produits sanguins à partir de plasma destiné à une fabrication ultérieure. Comme de nouveaux produits sanguins pourraient voir le jour à la suite d’innovations, l’utilisation de cette expression dans la présente ligne directrice n’est nullement restreinte au plasma.
On entend par « cadre supérieur » le titulaire d’un poste auquel a été conféré un niveau de responsabilité à l’égard d’activités menées par l’établissement dans le cadre du Règlement sur le sang. Veuillez noter que l’expression désigne avant tout une fonction au sein de l’établissement, et n’est pas nécessairement un titre de poste particulier.
« ISBT 128 » Norme internationale applicable à l’étiquetage de sang destiné à la transfusion, des produits destinés à la fabrication de drogues pour usage humain, et produits destinés à la transplantation et dont l’International Council for Commonality in Blood Banking Automation (ICCBBA) assure la gestion et la promotion.
« médecin » Personne autorisée à exercer la médecine en vertu des lois de la province ou le territoire où elle fournit des services médicaux.
« mise en commun » inclut le mélange.
« nouveau composant sanguin » Composant sanguin non systématiquement traité ou transfusé au Canada, et qui, soit présente un avantage sur le plan de la production, soit est équivalent ou supérieur à un produit de référence, soit répond à un besoin clinique non comblé.
« possibilité de se faire entendre » Possibilité pour un établissement de répondre par écrit à Santé Canada en réponse à une mesure ministérielle visant une homologation, une licence ou l’enregistrement de cet établissement. Dans certains cas, il peut y avoir une réunion en personne, une réunion virtuelle ou une téléconférence.
« quarantaine » Évite que les unités de sang dont on soupçonne ou confirme la non-conformité soient utilisées pour une transfusion, d’autres étapes de fabrication ou la distribution.
« substitut» Personne qui, à la fois :
- agit sous la supervision et la direction générales d’un médecin;
- est autorisée à fournir des services qui relèvent de la compétence du médecin selon les lois de la province ou le territoire où ces services sont fournis.
2. Directives sur la mise en œuvre
Diagramme 1. Application du Règlement sur le sang aux divers types d'établissements visés
Ce diagramme a pour objet d'indiquer les articles du règlement qui s'appliquent aux établissements tenus de posséder une homologation, une licence d'établissement et/ou un enregistrement en raison des activités qu'ils exercent. Veuillez vous référer aux articles 5-16 (homologation), 17-29 (licence d'établissement) et 30-37 (enregistrement) de la présente ligne directrice pour obtenir de plus amples renseignements sur le niveau de contrôle règlementaire que nécessitent les activités menées par votre établissement.
- H
- Établissement titulaire d'une homologation
- LE
- Établissement titulaire d'une licence
- E-Auto
- Établissement enregistré dont les activités concernent le sang autologue
- E-PDPE
- Établissement enregistré ayant un programme de donneurs pré-évalués
- E-TLMCI
- Établissement enregistré exerçant des activités de transformation (lavage, mise en commun, irradiation)
| Section du Règlement sur le sang | Titre de l'article | H/LE | E-Auto | E-PDPE | E-TLMCI |
|---|---|---|---|---|---|
| 1 | Définitions | Oui | Oui | Oui | Oui |
| Champ d'application | |||||
| 2 | Portée | Oui | Oui | Oui | Oui |
| 3 | Non-application | Oui | Oui | Oui | Oui |
| 4 | Interdictions | Oui | Oui | Oui | Oui |
| Homologation | |||||
| 5 | Homologation | Oui | s/o | s/o | s/o |
| 6 | Demande d'homologation | Oui | s/o | s/o | s/o |
| 7 | Délivrance | Oui | s/o | s/o | s/o |
| 8 | Refus | Oui | s/o | s/o | s/o |
| 9 | Changement majeur | Oui | s/o | s/o | s/o |
| 10 | Changement en cas d'urgence | Oui | s/o | s/o | s/o |
| 11 | Changements administratifs - avis | Oui | s/o | s/o | s/o |
| 12 | Autres changements - rapport annuel | Oui | s/o | s/o | s/o |
| 13 | Ajout de conditions à l'homologation ou modification | Oui | s/o | s/o | s/o |
| 14 | Suspension | Oui | s/o | s/o | s/o |
| 15 | Rétablissement | Oui | s/o | s/o | s/o |
| 16 | Annulation | Oui | s/o | s/o | s/o |
| Licence d'établissement | |||||
| 17(1) | Obligation d'obtenir une licence | Oui | s/o | s/o | s/o |
| 17(2) | Essais | Oui (LE seulement pour PDPE) | s/o | s/o | s/o |
| 18 | Demande de licence | Oui | s/o | s/o | s/o |
| 19 | (1) Inspection (2) Renseignements et documents complémentaires |
Oui | s/o | s/o | s/o |
| 20 | Délivrance | Oui | s/o | s/o | s/o |
| 21 | Refus | Oui | s/o | s/o | s/o |
| 22 | Changement des renseignements - demande de modification | Oui | s/o | s/o | s/o |
| 23 | Changement de nature administrative - avis | Oui | s/o | s/o | s/o |
| 24 | Modification de la licence par le ministre | Oui | s/o | s/o | s/o |
| 25 | Ajout de conditions ou modification | Oui | s/o | s/o | s/o |
| 26 | Renseignements et documents supplémentaires | Oui | s/o | s/o | s/o |
| 27 | Suspension | Oui | s/o | s/o | s/o |
| 28 | Rétablissement | Oui | s/o | s/o | s/o |
| 29 | Annulation | Oui | s/o | s/o | s/o |
| Enregistrement | |||||
| 30 | Obligation d'enregistrement | s/o | Oui | Oui | Oui |
| 31 | Demande d'enregistrement | s/o | Oui | Oui | Oui |
| 32 | Enregistrement | s/o | Oui | Oui | Oui |
| 33 | Avis de modification | s/o | Oui | Oui | Oui |
| 34 | Modification par le ministre | s/o | Oui | Oui | Oui |
| 35 | Attestation annuelle de conformité | s/o | Oui | Oui | Oui |
| 36 | Renseignements et documents supplémentaires | s/o | Oui | Oui | Oui |
| 37 | Annulation | s/o | Oui | Oui | Oui |
| Traitement - Évaluation de l'admissibilité des donneurs | |||||
| 38 | Non-application - dons autologues | s/o | Oui | s/o | s/o |
| 39 | Établissement titulaire d'une licence | Oui | s/o | s/o | s/o |
| 40 | Inadmissibilité antérieure | Oui | s/o | Oui | s/o |
| 41 | Évaluation préliminaire | Oui | s/o | Oui | s/o |
| 42 | Critères d'exclusion | Oui | s/o | Oui | s/o |
| 43 | Inadmissibilité | Oui | s/o | Oui | s/o |
| 44 | (1) Admissibilité (2) Nouvelle évaluation (3) Avis |
Oui | s/o | Oui | s/o |
| Traitement - Prélèvement | |||||
| 45 | Établissement titulaire d'une licence | Oui | s/o | s/o | s/o |
| 46 | Code d'identification du donneur | s/o | Oui | s/o | s/o |
| 47 | Code d'identification du don | Oui | Oui | s/o | s/o |
| 48 | Étiquetage des contenants | Oui | Oui | s/o | s/o |
| 49 | Procédures de prélèvement | Oui | Oui | Oui | s/o |
| 50 | Échantillons | Oui | Oui | Oui | s/o |
| 51 | Don autologue | s/o | Oui | s/o | s/o |
| Traitement - Essais | |||||
| 52 | Homologation | Oui | s/o | s/o | s/o |
| 53 | Don autologue - dépistage de maladies transmissibles | s/o | Oui | s/o | s/o |
| 54 | Don autologue - groupe ABO et facteur Rh | s/o | Oui | s/o | s/o |
| 55 | Instruments médicaux | s/o | Oui | (Oui) Voir également paragr.17(2) | s/o |
| 56(1) | Résultats des essais - don allogénique | Oui | s/o | s/o | s/o |
| 56(2) | Résultats des essais - don autologue | s/o | Oui | s/o | s/o |
| Préparation de composants sanguins | |||||
| 57 | Établissement titulaire d'une licence | Oui | s/o | s/o | s/o |
| 58 | Établissement enregistré | s/o | Oui | s/o | s/o |
| Étiquetage | |||||
| 59 | Non-application - donneurs pré-évalués | s/o | s/o | Oui | s/o |
| 60 | Exigences linguistiques | Oui | Oui | s/o | Oui |
| 61 | Exigences générales | Oui | Oui | s/o | Oui |
| 62 | Document d'information | Oui | s/o | s/o | s/o |
| 63 | Code d'identification du don | Oui | Oui | s/o | s/o |
| 64(1) | Texte de l'étiquette - sang destiné à la transfusion | Oui | Oui | s/o | s/o |
| 64(2) | Texte de l'étiquette - don autologue | s/o | Oui | s/o | s/o |
| 64(3) | Texte de l'étiquette - sang destiné à la fabrication de drogues pour usage humain | Oui | s/o | s/o | s/o |
| 65 | Aliquotes | Oui | Oui | Oui | Oui |
| 66 | (1) Don désigné (2) Changement de fin |
Oui | s/o | s/o | s/o |
| 67 | Don dirigé | Oui | s/o | s/o | s/o |
| 68 | Vérification des étiquettes | Oui | Oui | s/o | Oui |
| Conservation | |||||
| 69(1)a) | Exigences - établissement qui prélève, établissement titulaire d'une licence | Oui | s/o | s/o | s/o |
| 69(1)b) | Exigences - établissement qui prélève, établissement enregistré | s/o | Oui | Oui | s/o |
| 69(2) | Exigences - destinataire | Oui | Oui | s/o | Oui |
| 70 | Lieu de conservation | Oui | Oui | Oui | Oui |
| 71 | Mise à l'écart - fins différentes | Oui | Oui | s/o | Oui |
| 72 | Mise à l'écart - essais | Oui | Oui | Oui | Oui |
| Distribution | |||||
| 73(1) | Sécurité du sang - don allogénique | Oui | s/o | Oui | s/o |
| 73(2) | Sécurité du sang - don autologue | s/o | Oui | s/o | s/o |
| 74 | (1) Vérification (2) Interdiction de distribution |
Oui | Oui | Oui | Oui |
| 75 | Contenants d'expédition | Oui | Oui | Oui | Oui |
| 76 | Conservation pendant le transport | Oui | Oui | Oui | Oui |
| Transformation | |||||
| 77 | Transformation - méthodes | s/o | s/o | s/o | Oui |
| 78 | Lavage | s/o | s/o | s/o | Oui |
| 79 | Mise en commun | s/o | s/o | s/o | Oui |
| 80 | Irradiation | s/o | s/o | s/o | Oui |
| Distribution exceptionnelle | |||||
| 81 | Conditions | Oui | Oui | Oui | Oui |
| 82 | (1) Avis de distribution exceptionnelle (2) Dossier de l'établissement |
Oui | s/o | s/o | s/o |
(3) Mesures à prendre sur réception de l'avis |
s/o | Oui | Oui | Oui | |
| 83 | Étiquetage | Oui | s/o | s/o | s/o |
| 84(1) | Suivi | Oui | s/o | s/o | s/o |
| 84(2) | Mesures à prendre sur réception des résultats | s/o | Oui | Oui | Oui |
| 85 | Sang non‐transfusé | s/o | Oui | Oui | Oui |
| Programme de donneurs pré-évalués | |||||
| 86 | Exigences | s/o | s/o | Oui | s/o |
| 87 | Code d'identification du donneur | s/o | s/o | Oui | s/o |
| 88 | (1) Mesures à prendre régulièrement (2) Comparaison (3) Incohérence |
s/o | s/o | Oui | s/o |
| 89 | Mesures à prendre lors de chaque prélèvement | s/o | s/o | Oui | s/o |
| 90 | Étiquetage | s/o | s/o | Oui | s/o |
| 91 | Sang non-transfusé | s/o | s/o | Oui | s/o |
| Importation dans des circonstances urgentes | |||||
| 92 | Importation dans des circonstances urgentes | Oui | s/o | s/o | s/o |
| Système de gestion de la qualité | |||||
| 93 | (1) Structure organisationnelle (2) Supervision (3) Vérification périodique |
Oui | Oui | Oui | Oui |
| 94 | Exigences | Oui | Oui | Oui | Oui |
| Procédures opérationnelles | |||||
| 95 | Obligation - procédures opérationnelles | Oui | Oui | Oui | Oui |
| 96 | Exigences | Oui | Oui | Oui | Oui |
| 97 | Documentation | Oui | Oui | Oui | Oui |
| Personnel, installations, équipement, matériel et produits | |||||
| 98 | (1) Personnel (2) Compétence |
Oui | Oui | Oui | Oui |
| 99 | Installations | Oui | Oui | Oui | Oui |
| 100 | Équipement | Oui | Oui | Oui | Oui |
| 101 | Équipement de conservation | Oui | Oui | Oui | Oui |
| 102 | Matériel et produits | Oui | Oui | Oui | Oui |
| Enquêtes et rapports concernant les accidents et manquements | |||||
| 103 | Accident ou manquement - autre établissement | Oui | Oui | Oui | Oui |
| 104 | Accident ou manquement - établissement | Oui | Oui | Oui | Oui |
| 105 | Assistance à l'enquête | Oui | Oui | Oui | Oui |
| 106 | Résultats de l'enquête | Oui | Oui | Oui | Oui |
| 107 | Rapports au ministre | Oui | Oui | Oui | Oui |
| 108 | Rapport annuel | Oui | Oui | Oui | Oui |
| Enquêtes et rapports concernant les effets indésirables | |||||
| Effets indésirables chez le donneur | |||||
| 109 | Effets indésirables chez le donneur - Avis au ministre | Oui | Oui | Oui | s/o |
| Effets indésirables chez le receveur | |||||
| 110 | Mesures à prendre | Oui | Oui | Oui | Oui |
| 111 | Don autologue | s/o | Oui | s/o | s/o |
| 112 | Assistance à l'établissement qui enquête | Oui | Oui | Oui | Oui |
| 113 | Avis au ministre | Oui | Oui | Oui | Oui |
| 114 | (1) Résultats de l'enquête (2) Envoi d'une copie de l'avis |
Oui | Oui | Oui | Oui |
| 115 | Rapport d'enquête final au ministre | Oui | Oui | Oui | Oui |
| 116 | Rapport annuel | Oui | Oui | Oui | Oui |
| Dossiers | |||||
| 117 | Caractéristiques des documents | Oui | Oui | Oui | Oui |
| 118 | Codes d'identification du don | Oui | Oui | Oui | Oui |
| 119 | (1) Période de rétention - don allogénique (2) Calcul de la période de rétention |
Oui | s/o | Oui | s/o |
| 120 | (1) Période de rétention - don autologue (2) Calcul de la période de rétention |
s/o | Oui | s/o | s/o |
| 121 | (1) Période de rétention - transformation (2) Calcul de la période de rétention |
s/o | s/o | s/o | Oui |
| 122 | (1) Période de rétention — transfusion (2) Calcul de la période de rétention |
s/o | Oui | Oui | Oui |
| 123 | Entreposage des dossiers | Oui | Oui | Oui | Oui |
| 124 | Pouvoirs des inspecteurs | Oui | Oui | Oui | Oui |
| 125 | Modification corrélative | Oui | s/o | s/o | s/o |
| Dispositions transitoires | |||||
| 126 | Homologation | Oui | s/o | s/o | s/o |
| 127 | Licence | Oui | s/o | s/o | s/o |
| 128 | (1) Enregistrement (2) Durée |
s/o | Oui | Oui | Oui |
| Entrée en vigueur | |||||
| 129(1) | Un an après la publication | Oui | Oui | Oui | Oui |
| 129(2) | Paragraphes 4(4) à (6) et alinéa 64(1)b) | s/o | Oui | Oui | Oui |
Diagramme 2. Application du Règlement sur le sang aux établissements qui ne sont pas tenus d'obtenir une homologation, une licence d'établissement ou un enregistrement
Certains établissements n'exercent pas d'activités pour lesquelles une homologation, une licence d'établissement ou un enregistrement est requis. Cependant, ces établissements doivent quand même respecter les exigences des articles applicables du Règlement sur le sang pour les activités qu'ils exercent. Ces articles sont identifiés dans le diagramme ci-dessous.
| Section du Règlement sur le sang | Titre de l'article |
|---|---|
| 1 | Définitions |
| Champ d'application | |
| 2 | Portée |
| 3 | Non-application |
| 4 | Interdiction |
| Étiquetage | |
| 60 | Exigences linguistiques |
| 61 | Exigences générales |
| 65 | Aliquotes |
| 68 | Vérification des étiquettes |
| Conservation | |
| 69(2) | Destinataire |
| 70 | Lieu de conservation |
| 71 | Mise à l'écart - fins différentes |
| 72 | Mise à l'écart - essais |
| Distribution | |
| 74 | (1) Vérification (2) Interdiction de distribution |
| 75 | Contenants d'expédition |
| 76 | Conservation pendant le transport |
| Transformation | |
| 77 | Transformation - méthodes *s'applique aux établissements qui ne font que la mise en commun d'unités de cryoprécipité |
| 79 | Mise en commun *s'applique aux établissements qui ne font que la mise en commun d'unités de cryoprécipité |
| Distribution exceptionnelle | |
| 81 | Conditions |
| 82 | (1) Mesures à prendre sur réception de l'avis (2) Dossier du receveur |
| 84(2) | Mesures à prendre sur réception des résultats |
| 85 | Sang non-transfusé |
| Procédures opérationnelles | |
| 95 | Obligation - procédures opérationnelles |
| 96 | Exigences |
| 97 | Documentation *s'applique aux établissements qui ne font que la mise en commun d'unités de cryoprécipité |
| Personnel, installations, équipement, matériel et produits | |
| 98 | (1) Personnel (2) Compétence |
| 101 | Équipement de conservation |
| Enquêtes et rapports concernant les accidents et manquements | |
| 103 | Accident ou manquement - autre établissement |
| 104 | Accident ou manquement - établissement |
| 105 | Assistance à l'enquête |
| 106 | Résultats de l'enquête |
| 107 | Rapports au ministre |
| 108 | Rapport annuel |
| Enquêtes et rapports concernant les effets indésirables | |
| Effets indésirables chez le receveur | |
| 110 | Mesures à prendre |
| 112 | Assistance à l'établissement qui enquête |
| 113 | Avis au ministre |
| 114 | (1) Résultats de l'enquête (2) Envoi d'une copie de l'avis |
| 115 | Rapport d'enquête final au ministre |
| 116 | Rapport annuel |
| Dossiers | |
| 117 | Caractéristiques des documents |
| 118 | Codes d'identification du don |
| 121 | Période de rétention - transformation *s'applique aux établissements qui ne font que la mise en commun d'unités de cryoprécipité |
| 122 | (1) Période de rétention - transfusion (2) Calcul de la période de rétention |
| 123 | Entreposage des dossiers |
| 124 | Pouvoirs des inspecteurs |
| Entrée en vigueur | |
| 129(1) | Un an après la publication |
Article 1 Définitions
Les énoncés qui figurent dans les encadrés ont été extraits directement du Règlement sur le sang.
Définitions
1. Les définitions qui suivent s’appliquent au présent règlement.
« accident »
"accident"« accident » Événement imprévu qui n’est pas imputable à une inobservation des procédures opérationnelles ou des règles de droit applicables et qui risque de compromettre la sécurité humaine ou la sécurité du sang.
« allogénique »
"allogeneic"« allogénique » À l’égard d’un don de sang, se dit lorsque le sang prélevé d’un individu est destiné à la transfusion à un autre individu ou à la fabrication de drogues pour usage humain.
Le prélèvement de sang allogénique destiné à être distribué dans la réserve générale de sang à des fins de transfusion ou d’utilisation dans la fabrication d’une drogue pour usage humain exige une homologation et une licence d’établissement.
Le prélèvement de sang allogénique chez un donneur pré-évalué en vue de sa transfusion d’urgence à un patient spécifique exige un enregistrement, alors que la mise à l’essai du sang allogénique et des composants sanguins allogéniques provenant d’un donneur pré-évalué exige une licence d’établissement.
« autologue »
"autologous"« autologue » À l’égard d’un don de sang, se dit lorsque le sang prélevé d’un individu est destiné à la transfusion à ce même individu ultérieurement.
Le sang autologue ne peut être transfusé qu’à la personne chez qui il a été prélevé.
Le sang autologue qui est prélevé pour la fabrication d’une drogue en vertu du Règlementsur les aliments et droguesn’est pas assujetti au Règlement sur le sang. Par exemple, le prélèvement du matériel de départ pour les cellules CAR-T et les cellules T étendues sort du champ d’application du Règlementsur le sang.
La portée du prélèvement de sang autologue, en vertu du Règlement sur le sang, exclut ce qui suit parce que ce type de sang n'est pas considéré comme étant prélevé pour utilisation ultérieure :
- sang péri-opératoire qui est prélevé et qui demeure dans l'aire clinique de soins aux patients, par exemple :
- prélèvement immédiatement avant l’intervention chirurgicale (p. ex., hémodilution normovolémique aiguë) ;
- prélèvement tout au long de l’intervention chirurgicale (peropératoire), du site chirurgical ou d’un circuit extracorporel ; ou
- prélèvement après l’intervention chirurgicale (postopératoire) ou d’un traumatisme, à partir de cavités corporelles, d’espaces articulaires et d’autres sites de chirurgie ou de traumatisme fermés
- sang qui est prélevé pour radio-marquage à des fins diagnostiques
Veuillez vous référer à l’article 71 pour ce qui est des exigences relatives à la mise à l’écart du sang autologue pendant la conservation.
« code d'identification du don »
"donation code"« code d’identification du don » Code unique composé de chiffres, de lettres, de symboles ou de toute combinaison de ceux-ci et attribué par l’établissement à l’unité de sang lors de son prélèvement.
« code d'identification du donneur »
"donor identification code"« code d’identification du donneur » Code unique composé de chiffres, de lettres, de symboles ou de toute combinaison de ceux-ci et attribué par l’établissement au donneur.
« directeur médical »
"medical director"« directeur médical » Médecin d’un établissement qui est autorisé à exercer sa profession par les lois d’une province et qui est responsable des actes médicaux effectués par l’établissement ainsi que de l’application des procédures opérationnelles afférentes.
« distribution »
"distribute"« distribution » Ne vise pas la transfusion.
De nombreuses dispositions du Règlement sur le sang sont associées à la distribution du sang et des composants sanguins. La présente ligne directrice présente des renseignements supplémentaires sur l’activité de distribution dans divers contextes. Veuillez vous référer à l’article 4, Interdiction, à l’article 73, Sécurité du sang — don allogénique, aux articles 81–85, Distribution exceptionnelle, à l’article 92, Importation dans des circonstances urgentes.
Vous trouverez ci‑après un certain nombre d’exemples de distribution de sang et de composants sanguins conformes au Règlement sur le sang. Dans chacun des cas mentionnés, lorsque du sang et/ou des composants sanguins doivent être distribués à un autre établissement ou au bloc opératoire ou à l’étage, l’établissement doit procéder aux vérifications supplémentaires exigées à l’article 74 du Règlement sur le sang.
Exemple 1, distribution de sang allogénique à des fins de transfusion
Le sang allogénique est traité en vue de sa transfusion par un établissement titulaire d’une licence. Après avoir établi que le sang destiné à la transfusion est sécuritaire pour la transfusion, l’établissement le met en inventaire. Il y a distribution lorsque l’établissement envoie le sang ou les composants sanguins à un autre établissement. Il y a également distribution lorsque le laboratoire de médecine transfusionnelle envoie le sang ou les composants sanguins au bloc opératoire ou à l’étage qu’il ait été mis en compatibilité ou non avec un patient.
Il peut également y avoir distribution si l’hôpital A reçoit une demande de l’hôpital B pour des unités de sang allogénique ou de composants sanguins allogéniques destinées à la transfusion. La distribution a lieu lorsque l’hôpital A envoie le sang ou les composants sanguins à l’hôpital B.
Exemple 2, distribution de sang autologue
Le sang autologue est traité en vue de sa transfusion par un établissement enregistré. Avant que les unités de sang autologue puissent être distribuées, elles doivent être déterminées sécuritaires pour la transfusion autologue. Il y a distribution lorsque l’établissement envoie le sang autologue à l’hôpital.
Il peut également y avoir distribution à l’intérieur du même établissement si un établissement enregistré traite du sang autologue destiné à la transfusion au même établissement où il sera transfusé. Avant que les unités de sang autologue puissent être distribuées, elles doivent être déterminées sécuritaires pour la transfusion autologue. Il y a distribution lorsque le laboratoire de médecine transfusionnelle envoie le sang ou les composants sanguins au bloc opératoire ou à l’étage.
Si un établissement envoie du sang depuis une clinique mobile jusqu’à l’installation de traitement, il ne s’agit pas d’une distribution, puisque le sang n’a pas encore été jugé sécuritaire en vue de sa distribution à des fins de transfusion ou de fabrication ultérieure.
Exemple 3, distribution de sang à des fins de fabrication ultérieure
Le sang (plasma) est traité par un établissement titulaire d’une licence pour être transformé en une drogue pour usage humain. Après avoir déterminé que le sang destiné à la distribution à des fins de fabrication ultérieure est sécuritaire, l’établissement peut le conserver sur le lieu du prélèvement. Il y a distribution lorsque l’établissement envoie le sang et/ou les composants sanguins au fabricant de produits sanguins.
Exemple 4, distribution de globules rouges du sang à des fins d’immunisation
Le sang est traité aux fins d’immunisation par un établissement titulaire d’une licence. Avant d’être mis en inventaire, les globules rouges irradiés doivent être déterminés sécuritaires pour fin de distribution. Il y a distribution lorsque les globules rouges sont déplacés du lieu où l’inventaire est conservé au lieu où l’immunisation du donneur est pratiquée. Cela peut se produire à l’intérieur du même établissement.
« document d'information »
"circular of information"« document d'information » Document qui contient les renseignements suivants :
a) la composition et les propriétés du sang;
b) la manière de le conserver et de l'utiliser;
c) les indications, les contre-indications, les mises en garde et les réactions indésirables possibles.
« Document d'information » signifie aussi notice d'accompagnement.
« don désigné »
"designated donation"« don désigné » Don de sang qui est fait par un donneur choisi pour des raisons médicales et qui est destiné à un receveur particulier.
« don dirigé »
"directed donation"« don dirigé » Don de sang qui est fait par un donneur connu du receveur et choisi pour des raisons médicales par le médecin de celui-ci.
« donneur pré-évalué »
"pre-assessed donor"« donneur pré-évalué » Donneur qui a été accepté dans le programme de donneurs pré-évalués prévu aux articles 86 à 91 et dont le sang prélevé en raison d’une urgence n’a pas fait l’objet d’essais complets lors de sa transfusion.
Le terme « donneur pré-évalué » est utilisé dans le Règlement sur le sang en remplacement du terme « donneur ambulant ». Veuillez vous référer aux articles 86–91 du Règlement sur le sang pour les exigences.
« effet indésirable »
"adverse reaction"« effet indésirable » L'une des réactions suivantes :
a) dans le cas du donneur, réaction négative associée au prélèvement de sang;
b) dans le cas du receveur, réaction négative associée à l'aspect sécuritaire du sang transfusé.
« effet indésirable grave »
"serious adverse reaction"« effet indésirable grave » Effet indésirable qui entraîne l’une des conséquences suivantes pour le donneur ou le receveur :
a) son hospitalisation ou la prolongation de celle-ci;
b) une incapacité importante ou persistante;
c) la nécessité d'une intervention médicale ou chirurgicale visant à prévenir une telle incapacité;
d) une affection mettant sa vie en danger;
e) sa mort.
« effet indésirable imprévu »
"unexpected adverse reaction"« effet indésirable imprévu » Effet indésirable qui n’est pas mentionné parmi les effets indésirables possibles indiqués dans le document d’information ou communiqués par ailleurs au receveur.
Un « effet indésirable imprévu » est celui dont la nature, la gravité ou l’issue ne cadre pas avec l’information fournie dans le document d’information ou dans toute autre information fournie au receveur.
« essentiel »
"critical"« essentiel » Qualifie l’équipement, le matériel, les produits ou les services qui, s’ils ne sont pas conformes à leurs spécifications, risquent de compromettre la sécurité humaine ou la sécurité du sang.
Aux fins de cette ligne directrice, « essentiel » signifie critique.
Le terme « essentiel » s'applique à l'équipement, au matériel, aux produits et services qui sont utilisés dans les activités réglementées en vertu du Règlement sur le sang. Des exemples d'équipement, de matériel, produits et services essentiels comprennent, mais ne se limitent pas à ceux utilisés lors du prélèvement sanguin, d’essais sanguins, de la préparation des composants sanguins, de la conservation et de la transformation. Les listes non exhaustives qui suivent sont fournies à titre indicatif.
Remarque : Le « sang » comprend le sang total et les composants sanguins.
Exemples d’équipement essentiel
- équipement d’aphérèse;
- extracteurs/presses automatiques pour le sang;
- systèmes de traitement automatiques du sang;
- systèmes automatiques d’essais sanguins et/ou équipement servant à la réalisation des essais pour les maladies transmissibles;
- connecteurs automatiques/ scelleuses automatiques;
- scelleuses manuelles
- systèmes de détection microbienne automatisés;
- irradiateur
- appareil de réduction des pathogènes
- hématimètres (instruments de comptage cellulaire) ou hémato-analyseurs utilisés dans l’évaluation du sang ou des composants sanguins;
- appareils de lavage/déglycérolisation;
- centrifugeuse utilisée dans le traitement des unités de composants sanguins ou pour la préparation des échantillons de sang pour la mise à l’essai;
- instruments d’électrophorèse;
- congélateurs rapides;
- congélateurs servant à la conservation du sang (unités ou échantillons);
- instruments nécessaires à la réalisation des essais d’amplification des acides nucléiques (TAN), incluant les extracteurs ou les pipetteurs;
- agitateurs et incubateurs de plaquettes;
- réfrigérateurs;
- thermomètres et sondes de température (tout type);
- balances pour peser le sang;
- génératrices de secours;
- véhicules à température contrôlée pour le transport du sang; et
- appareils de décongélation du sang
- contenants utilisés pour conserver le sang, y compris pendant le transport
- Équipement de conditionnement à température contrôlée (p. ex. réfrigérateurs/congélateurs/incubateurs utilisés pour entreposer les plaques à changement de phase utilisées durant le transport du sang)
L’équipement essentiel comprend également les logiciels essentiels.
Exemples de logiciel essentiel
- logiciel de transfert des données entre équipements informatisés; et
- logiciel d’analyse des données servant à déterminer si le sang est propre à la transfusion ou à une fabrication ultérieure.
Exemples de matériel essentiel
- réactifs d’essai de groupe sanguin ou de phénotype;
- indicateurs d’irradiation;
- ensembles de prélèvement (poches et tubulures);
- filtres;
- étiquettes;
- réactifs pour des trousses de dépistage de maladies transmissibles; et
- sacs de transfert.
Exemples de service essentiel
- étalonnage et entretien de l’équipement essentiel;
- validation/qualification de l’ équipement essentiel;
- mise à l’essai de laboratoire;
- contrôle de la qualité;
- gestion de la qualité;
- services de mise à l’essai; et
- formation sur l’équipement essentiel donnée par le fournisseur.
Exemples d’équipement ou de matériel non essentiel
- balances pour peser les donneurs ;
- agitateur de poche de sang;
- laveurs de cellules de type centrifuge non utilisés pour la déglycérolisation;
- centrifugeuses non utilisées pour la séparation des composants ou la préparation des échantillons pour la mise à l’essai;
- bain à circulation;
- hémoglobinomètres;
- incubateurs (à l’exception des agitateurs/incubateurs de plaquettes);
- extracteurs manuels pour la préparation des composants sanguins;
- centrifugeuses à micro-hématocrite;
- pipettes ( à l’exception des pipetteurs pour les essais d’amplification des acides nucléiques);
- thermoscelleuses pour la segmentation de la tubulure aux fins d’échantillonnage du sang;
- chronomètres (tout type); et
- poids (tout type).
« établissement »
"establishment"« établissement » Personne qui exerce l'une des activités ci-après relativement au sang :
a) l'importation;
b) le traitement;
c) la distribution;
d) la transformation;
e) la transfusion.
e) transfusion
Même si la transfusion comme telle n’est pas visée par le Règlement sur le sang, les établissements de transfusion doivent satisfaire aux dispositions de ce règlement applicables à leurs activités, par exemple à la conservation du sang et des composants sanguins.
« évaluation de l'admissibilité du donneur »
"donor suitability assessment"« évaluation de l'admissibilité du donneur » Évaluation fondée sur les données ci-après relatives au donneur :
a) ses antécédents médicaux;
b) les résultats de tout essai effectué sur lui et de tout examen physique;
c) ses antécédents sociaux, dans la mesure où ils peuvent être utiles pour déterminer la présence d’un facteur de risque de maladies transmissibles par le sang.
- Les antécédents médicaux du donneur font référence aux :
- conditions pouvant présenter un risque pour celui-ci;
- vaccins, médicaments et maladies transmissibles pouvant présenter un risque pour le receveur.
- L’examen physique est l’une des méthodes permettant de juger de l’admissibilité d’un donneur au don de sang. Cet examen doit s’appuyer sur les critères homologués de l’établissement.
- Les antécédents sociaux du donneur concernent les activités antérieures de cette personne qui pourraient l’exposer, elle ou les receveurs, au risque de maladie infectieuse transmissible.
« homologation »
"authorization"« homologation » Homologation délivrée conformément à l'article 7 à l'égard du sang et des processus.
« Loi »
"Act"« Loi » La Loi sur les aliments et drogues.
« manquement »
"error"« manquement » Inobservation des procédures opérationnelles ou des règles de droit applicables qui risque de compromettre la sécurité humaine ou la sécurité du sang.
« norme »
"standard"« norme »La norme nationale du Canada CAN/CSA-Z902 de l’Association canadienne de normalisation, intitulée Sang et produits sanguins labiles, avec ses modifications successives.
Dans la présente ligne directrice, la « norme » est appelée norme sur le sang de la CSA.
« procedures opérationnelles »
"operating procedures"« procédures opérationnelles » Composante du système de gestion de la qualité constituée d’une série d’instructions énonçant les processus applicables aux activités de l’établissement.
« sang »
"blood"« sang »Sang humain prélevé à des fins de transfusion ou de fabrication de drogues pour usage humain, étant entendu que le sang total et les composants sanguins sont notamment visés.
Le sang, au sens du Règlement sur le sang, n’inclut pas les produits sanguins et les dérivés du sang.
Exemples de composants sanguins : globules rouges, plasma, plaquettes et granulocytes. Les produits fabriqués à partir de plasma en vue de leur fabrication ultérieure ne font pas partie des composants sanguins.
Veuillez vous référer à l’article 2 de la présente ligne directrice qui définit la portée du Règlement sur le sang.
« sécurité »
"safety"« sécurité » À l’égard du sang, se dit lorsque le sang a été, conformément l’article 73, jugé sécuritaire à des fins de distribution ou, dans le cas de sang provenant d’un don autologue, à des fins de transfusion et vise notamment les caractéristiques suivantes :
a) dans le cas de sang destiné à la transfusion, sa qualité et son efficacité;
b) dans le cas de sang destiné à la fabrication de drogues pour usage humain, sa qualité.
Lorsque le Règlement sur le sang ou la présente ligne directrice fait mention de la sécurité du sang, cela signifie (a) la sécurité, la qualité et l’efficacité du sang et des composants sanguins destinés à la transfusion et (b) la sécurité et la qualité du sang et des composants sanguins destinés à la fabrication d’une drogue pour usage humain.
La sécurité et la qualité du sang, dans le cas du sang et des composants sanguins destinés à la transfusion ou à la fabrication ultérieure, sont des facteurs déterminants qui permettent de juger de la sécurité du sang et/ou des composants sanguins à des fins de distribution. La sécurité du sang est la mesure dans laquelle le sang et les composants sanguins destinés à la transfusion ou à la fabrication d’une drogue pour usage humain sont exempts de substances nocives ou d’agents infectieux. La qualité du sang est définie au moyen de procédures d’assurance de la qualité et déterminée par les spécifications établies à l’égard du sang et des composants sanguins.
La sécurité et la qualité du sang comprennent des procédures sur les essais obligatoires, la sélection des donneurs, le prélèvement, les méthodes d’essais, la manipulation des dons, la conservation, le transport et la distribution des dons.
L’efficacité du sang, dans le cas du sang et des composants sanguins destinés à la transfusion, est un facteur déterminant qui permet de juger de la sécurité du sang à des fins de distribution. L’efficacité du sang s’entend de sa capacité de produire un résultat ou un effet escompté chez le receveur de sang.
« sécurité humaine »
"human safety"« sécurité humaine » Sécurité des donneurs et des receveurs dans la mesure où elle est associée à l’aspect sécuritaire du sang.
Lorsque le Règlement sur le sang ou la présente ligne directrice fait mention de la sécurité humaine, celle-ci désigne la sécurité des donneurs de sang ou la sécurité des receveurs de sang dans la mesure où la sécurité humaine soit associée à la sécurité du sang traité et distribué en vertu du Règlement sur le sang.
« traitement »
"processing"« traitement » L'une des activités suivantes :
a) l'évaluation de l'admissibilité du donneur;
b) le prélèvement;
c) la mise à l'essai;
d) la préparation de composants sanguins.
Un établissement traite du sang et de composants sanguins pour la transfusion ou la fabrication ultérieure s’il exerce l’une des activités suivantes : évaluation de l’admissibilité du donneur, prélèvement, mise à l’essai, et préparation de composants sanguins. Le champ d’application de la définition de traitement ne peut s’étendre au- delà des limites de la définition.
La transformation du sang ou sa division en aliquotes ne font pas partie de la préparation de composants sanguins.
« transformation »
"transformation"« transformation » Lavage, mise en commun ou irradiation de composants sanguins qui sont effectués après que le sang a été jugé sécuritaire à des fins de transfusion.
La définition de transformation précise les activités comprises dans son champ d’application : lavage, mise en commun et irradiation. Le champ d'application ne peut s'étendre au-delà de cette interprétation.
La « transformation », au sens du Règlement sur le sang, n’inclut pas l’utilisation d’une technologie de réduction des agents pathogènes.
La division du sang en aliquotes ne fait pas partie de la transformation.
Articles 2-3 Champ d'application
Article 2 Portée
Portée
2.Le présent règlement s’applique au sang prélevé qui est destiné à la transfusion ou à la fabrication de drogues pour usage humain.
Le Règlement sur le sang s’applique au sang humain prélevé chez un donneur :
- à des fins de transfusion;
- comme matière première pour la fabrication de produits sanguins;
- en vue de l’immunisation de donneurs de plasma destiné à une fabrication ultérieure (p. ex. globules rouges destinés à l’immunisation).
Cette portée s’étend :
- à la sécurité des donneurs dans la mesure où elle est associée à la sécurité du sang;
- à la sécurité du sang prélevé et traité chez ces donneurs;
- à la sécurité des receveurs.
Article 3 Non-application
Non-application à certains produits thérapeutiques
3. (1) Le présent règlement ne s’applique pas aux produits thérapeutiques suivants :
- le sang périphérique et le sang du cordon ombilical destinés à la transplantation de cellules lymphohématopoïétiques et visés par le Règlement sur la sécurité des cellules, tissus et organes humains destinés à la transplantation;
- le sang qui fait l’objet d’essais cliniques conformément au titre 5 de la partie C du Règlement sur les aliments et drogues;
- le sang importé qui est destiné à la fabrication de drogues pour usage humain.
Non-application — règlements
(2) Sous réserve de l’article A.01.045 du Règlement sur les aliments et drogues, les autres règlements pris en vertu de la Loi ne s’appliquent pas au sang visé par le présent règlement.
Non-application — sang de phénotype rare importé
(3) Les articles 4 à 124 ne s’appliquent pas au sang de phénotype rare qui est importé au titre d’une ordonnance.
3(1) Le tableau 1 décrit le sang auquel le Règlement sur le sang ne s’applique pas.
| Sang ou composant sanguin | Réglementation applicable |
|---|---|
1. Sang de cordon ombilical et sang périphérique destiné à la transplantation de cellules lymphohématopoïétiques |
Voir le Règlement sur la sécurité des cellules, tissus et organes humains destinés à la transplantation |
| 2. Sang faisant l’objet d’un essai clinique | Voir les articles C.04.230 à C.04.241 et le titre 5 de la partie C du Règlement sur les aliments et drogues |
| 3. Plasma destiné à une fabrication ultérieure après que l’établissement a distribué le plasma à un fabricant de produits sanguins | Voir les articles C.04.001. – C.04.020, C.04.230–C.04.241, titres 1, 1A et 2 de la partie C et le titre 8 du Règlement sur les aliments et drogues |
| 4. Sang autologue destiné à la fabrication de drogues | Voir le Règlement sur les aliments et drogues |
| 5. Plasma riche en plaquettes autologue | Voir la mise à jour de Santé Canada sur le plasma riche en plaquettes |
6. Produits sanguins, comme les dérivés plasmatiques et la fabrication de produits sanguins
|
Voir le Règlement sur les aliments et drogues |
| 7. Sang destiné à une fabrication ultérieure prélevé à l’étranger | Réglementé par l’autorité du pays exportateur Voir le Règlement sur les aliments et drogues pour ce qui est de l’importation de sang destiné à une fabrication ultérieure |
| 8. Sang de phénotype rare qui n’est pas disponible au Canada et qui est importé conformément à une ordonnance médicale | Une preuve d’ordonnance doit être fournie au point d’entrée |
Article 4 Interdictions
Don allogénique
4. (1) Sous réserve des paragraphes (2) et (3), l’établissement ne peut importer, distribuer ou transfuser du sang provenant d’un don allogénique que si le sang a été traité conformément à une homologation et jugé sécuritaire à des fins de distribution conformément au paragraphe 73(1).Exception — programme de donneurs pré-évalués
(2) Le paragraphe (1) ne s’applique pas aux activités de traitement effectuées dans le cadre d’un programme de donneurs pré-évalués.Exception — circonstances urgentes
(3) L’établissement peut, dans des circonstances urgentes, effectuer les activités suivantes :
- importer du sang provenant d’un don allogénique qui n’a pas été traité conformément à une homologation, s’il se conforme aux exigences prévues à l’article 92;
- distribuer ou transfuser ce sang, si l’importateur l’a importé conformément à l’article 92.
Don fait par un donneur pré-évalué
(4) L’établissement ne peut transfuser du sang prélevé d’un donneur pré-évalué faisant un don allogénique que s’il satisfait aux exigences prévues aux articles 86 à 91.Transformation
(5) L’établissement ne peut distribuer ou transfuser du sang ayant fait l’objet d’une transformation effectuée par un établissement non enregistré.
Don autologue(6) L’établissement ne peut distribuer ou transfuser du sang provenant d’un don autologue que si le sang a été traité par un établissement enregistré et a été jugé sécuritaire à des fins de transfusion à ce donneur conformément au paragraphe 73(2).
Enquête(7) L’établissement ne peut distribuer ou transfuser de sang dans les cas suivants :
- le sang fait l’objet d’une mise en quarantaine;
- les résultats d’une enquête concernant soit un accident ou manquement soupçonné, soit un effet indésirable grave ou effet indésirable imprévu démontrent que la sécurité du sang en cause a été compromise ou ne permettent pas de conclure qu’elle ne l’est pas.
4(1) Le sang allogénique doit être traité conformément à l’homologation pour qu’un établissement puisse l’importer, le distribuer ou le transfuser.
Quand le sang allogénique et les composants sanguins allogéniques font l’objet d’une distribution exceptionnelle, les établissements doivent respecter les dispositions des articles 81 à 85 du Règlement sur le sang. La distribution exceptionnelle n’équivaut pas à l’importation de sang dans des circonstances urgentes.
4(2) Une homologation n’est pas requise dans le cas d’un établissement qui traite du sang allogénique prélevé dans le cadre d’un programme de donneurs pré-évalués. Veuillez vous référer au paragraphe 4(4) en ce qui concerne l’enregistrement obligatoire.
4(3) L’établissement titulaire d’une licence peut, dans des circonstances urgentes, importer du sang et des composants sanguins non traités conformément à une homologation. L’alinéa 18(1)l) et le paragraphe 92(1) renferment les dispositions relatives à l’obligation de détenir une licence à laquelle est tenu l’établissement qui veut importer du sang et/ou des composants sanguins dans des circonstances urgentes.
Veuillez vous référer à l’article 92 pour ce qui est des renseignements que l’établissement titulaire d’une licence doit fournir au ministre avant que Santé Canada n’autorise l’importation de sang dans des circonstances urgentes.
4(4) Pour avoir un programme de donneurs pré-évalués, l’établissement doit s’enregistrer conformément au paragraphe 30(1) du Règlement sur le sang et satisfaire aux exigences prévues aux articles 86 à 91.
Un établissement qui effectue des essais sur du sang et/ou des composants prélevé dans le cadre d’un programme de donneurs pré-évalués doit détenir une licence d’établissement conformément au paragraphe 17(2) du Règlement sur le sang.
4(5) Le Règlement sur le sang cite les activités de transformation autorisées pour l’établissement enregistré. Veuillez vous référer à l’article 1 (Définition de « transformation »), le paragraphe 30(1) et les articles 77 à 80 du Règlement sur le sang.
4(6) Pour traiter du sang autologue, l’établissement doit être enregistré, conformément au paragraphe 32(1) du Règlement sur le sang.
4(7)a) Un établissement doit mettre du sang et des composants en quarantaine conformément à l'article 56, aux alinéas 103(1)b), 104(1)b), 110(1)b), au paragraphe 110(3) et à l'article 111. Veuillez vous référer aux articles 70 à 72 de la présente ligne directrice pour obtenir des directives relatives à la ségrégation du sang.
4(7)b) Un établissement doit prendre en considération l'interdiction à l’alinéa 4(7)b) lorsqu'il avise d'autres établissements des résultats d'une enquête sur un accident ou manquement soupçonné ou sur un effet indésirable imprévu ou un effet indésirable grave, ainsi que des mesures à prendre. Veuillez vous référer aux paragraphes 106(1) ou 114(2).
Articles 5-16 Homologation
Article 5 Homologation
Homologation — traitement
5. (1) L’établissement, à l’exception de celui qui n’effectue que des essais sanguins, obtient une homologation pour traiter du sang provenant d’un don allogénique.Exception — programme de donneurs pré-évalués
(2) Le paragraphe (1) ne s’applique pas aux activités de traitement effectuées dans le cadre d’un programme de donneurs pré-évalués.Homologation — importation
(3) Sous réserve de l’article 92, l’établissement obtient une homologation pour importer du sang à moins que celui-ci ne fasse déjà l’objet d’une homologation détenue par un autre établissement.
Qu'est-ce qu'une homologation?
Une homologation est l’autorisation donnée par Santé Canada à l’établissement pour traiter du sang allogénique, c’est-à-dire évaluer l’admissibilité des donneurs, prélever le sang de ces derniers, analyser le sang et préparer les composants sanguins.
Un nouvel établissement obtient une homologation en présentant une demande à la Direction des médicaments biologiques et radiopharmaceutiques (DMBR) et en fournissant des renseignements qui feront l'objet d'un examen. Les établissements titulaires d’une homologation doivent présenter une demande de modification chaque fois qu’ils souhaitent apporter des changements majeurs à leurs activités de traitement.
Consultez l’article 1 de la section Interprétation pour voir les définitions des termes « allogénique » et « traitement ».
Quelle est la différence entre une homologation et une licence d’établissement?
Homologation
L’homologation habilite l’établissement à traiter le sang selon la description qui y est donnée :
- des processus envisagés dans cet établissement; et
- des composants sanguins produits selon ces processus.
La DMBR examine les preuves et les renseignements sur les processus prévus que l’établissement lui a fournis. Une évaluation sur place peut compléter cet examen pour confirmer que les documents remplis aux fins d’examen correspondent bien à ce que l’établissement souhaite mettre en œuvre. Un avis d’homologation ou un avis de modification de l’homologation avec ou sans condition est délivré si le ministre, représenté par la DMBR de Santé Canada, conclut que les processus ne présentent pas de risques inacceptables pour la sécurité humaine ou pour la sécurité du sang et des composants sanguins.
Licence d’établissement
Une licence d'établissement est une licence accordée à un établissement au Canada lui permettant d'exercer des activités nécessitant une licence dans un bâtiment qui a été évalué et jugé conforme au Règlement sur le sang et à l’homologation.
La plupart des établissements tenus d’avoir une licence devront aussi obtenir une homologation. Parmi les exceptions figurent les établissements dont les activités se limitent aux essais relatifs au sang allogénique et/ou aux composants sanguins allogéniques. Voir à cet égard les paragraphes 5(1) et 17(2) du Règlement sur le sang.
Le ministre, représenté par Direction générale des opérations réglementaires et de l’application de la loi (DGORAL) de Santé Canada, délivre la licence d’établissement. Veuillez vous référer aux articles 17 à 29 pour ce qui est des exigences relatives aux licences d’établissement.
5(2) Il n’est pas nécessaire de détenir une homologation pour traiter du sang provenant de donneurs pré-évalués. Un établissement doit être enregistré pour traiter du sang allogénique dans le cadre d’un programme de donneurs pré-évalués. Veuillez vous référer aux articles 30 à 37 pour ce qui est des exigences applicables à l’enregistrement. Le diagramme 1 — Application du Règlement sur le sang aux divers types d’établissements visés — fournit des renseignements sur les articles de ce règlement qui s’appliquent aux programmes de donneurs pré-évalués.
5(3) Le terme « importation » dans cet article et dans d’autres ne s’applique qu’à l’importation de sang et de composants sanguins destiné à la transfusion. Le sang importé au Canada à des fins de transfusion s’inscrit dans le cadre d’une homologation. L’importateur ou l’établissement de l’étranger peut faire une demande d’homologation. Veuillez vous référer à l’alinéa 18(1)k) pour ce qui est des exigences relatives à l’importation de sang et/ou de composants sanguins destiné à la transfusion.
Article 6 Demande d'homologation
Demande d’homologation
6. (1) L’établissement présente au ministre, en la forme établie par celui-ci, une demande d’homologation, datée et signée par un cadre supérieur, qui contient les renseignements et documents suivants :
- ses nom et adresse municipale, son adresse postale si elle est différente, ainsi que l’adresse municipale de tout autre bâtiment où il envisage d’exercer ses activités;
- le nom de la personne à contacter pour toute question concernant la demande, ses numéros de téléphone et de télécopieur, son adresse de courriel ou toutes autres coordonnées;
- les nom et numéro de téléphone de la personne à contacter en cas d’urgence, si elle est différente de la personne visée à l’alinéa b);
- une mention indiquant qu’il envisage d’importer du sang total ou des composants sanguins;
- une liste indiquant ce qu’il envisage de traiter ou d’importer, à savoir sang total ou tel ou tel composant sanguin;
- la liste des activités de traitement qu’il envisage d’exercer dans chaque bâtiment;
- la description de ses installations, y compris ses bâtiments ainsi que l’équipement, le matériel et les produits essentiels qu’il envisage d’utiliser lors de l’exercice de ses activités ainsi que celle des services essentiels auxquels il envisage de recourir;
- la description des processus applicables au sang total et à chaque composant sanguin qu’il envisage d’utiliser lors de l’exercice des activités effectuées par lui ou en son nom;
- l’ébauche des étiquettes et des documents d’information proposés;
- toute preuve démontrant que l’établissement étranger auquel il envisage de recourir pour effectuer l’une de ses activités de traitement est autorisé à le faire par les lois du lieu où est situé cet établissement;
- toute preuve suffisante démontrant que les processus proposés ne compromettront pas la sécurité humaine et permettront de conclure à la sécurité du sang à des fins de distribution.
Inspection
(2) Le ministre peut, lors de l’examen de la demande, effectuer l’inspection des installations de l’établissement pour évaluer sur place l’exactitude des renseignements fournis dans la demande.Renseignements et documents complémentaires
(3) L’établissement fournit au ministre, au plus tard à la date précisée dans sa demande écrite à cet effet, tout renseignement ou document qu’il juge nécessaires pour compléter l’examen de la demande d’homologation.
6(1) La demande d’homologation doit contenir suffisamment de renseignements pour permettre au ministre d’évaluer la sécurité humaine et pour démontrer que les processus produiront du sang et des composants sanguins jugé sécuritaire à des fins de distribution. L’établissement doit envoyer sa demande d’homologation et les preuves à fournir au Bureau des affaires réglementaires (BAR) de la DMBR :
Bureau des affaires réglementaires
Direction des médicaments biologiques et radiopharmaceutiques
Direction générale des produits de santé et des aliments, Santé Canada
Indice de l’adresse 0601C
100, promenade Églantine
Ottawa (Ontario) K1A 0K9
Tél. : (613) 957-1722
Téléc. : (613) 946-9520
Courriel : BRDD.ORA@hc-sc.gc.ca
Toute question concernant l’homologation doit être communiquée au BAR avant la présentation de la demande, si possible.
Processus de demande
Réunions préalables à la demande avec les établissements de sang
Avant de présenter une demande d’homologation, un établissement devrait envisager la tenue d’une réunion ou téléconférence avec la DMBR pour discuter des exigences relatives aux demandes. Les réunions ou les téléconférences permettent de préciser l’objectif de la demande et d’aborder les différentes exigences qui y sont liées.
Une demande écrite pour une telle réunion devrait être reçue à la DMBR au moins trois mois avant la date proposée de la réunion. Elle devrait comprendre les renseignements qui suivent :
- l’objectif de la réunion;
- une courte description des sujets à aborder;
- au moins trois dates et heures proposées pour la réunion;
- une mention indiquant si des participants d’autres bureaux à Santé Canada, pourraient contribuer à la discussion (p. ex. La Direction des instruments médicaux ou DGORAL).
Pour se préparer à une réunion préalable à la demande, l’établissement devrait fournir trente jours avant la réunion une trousse contenant les renseignements suivants :
- objectif de la réunion;
- ordre du jour;
- liste des participants de l’établissement;
- contexte;
- exposé qui sera présenté, le cas échéant;
- liste de questions ou d’enjeux à aborder avec Santé Canada.
On demandera aux promoteurs de présenter une trousse de réunion avant la présentation au BAR aux fins de distribution à Santé Canada.
Réception de la demande
Au moment de la réception de la soumission, la DMBR attribue à cette dernière un numéro de contrôle (numéro de suivi unique attribué à chaque soumission). Ce numéro est indiqué dans toutes les correspondances portant sur la soumission.
La DMBR attribue un numéro de document à la trousse de demande initiale et à chaque nouvel envoi de renseignements. Des numéros de document sont attribués également aux lettres et autre correspondance émises par la DMBR liées à la demande.
À n’importe quel moment pendant le processus de demande, la DMBR ou l’établissement peut demander la tenue d’une téléconférence ou d’une réunion pour discuter de la demande.
Examen préliminaire des demandes
La DMBR évalue tous les documents relatifs à la demande pour en déterminer l’acceptabilité et, notamment, la qualité et l’exhaustivité. Cette étape sert aussi à déterminer si les processus ou les changements proposés à ces derniers se conforment au Règlement sur le sang. Le processus d’examen préliminaire diffère du processus d’examen et a lieu avant ce dernier. Pendant l’examen préliminaire, la DMBR détermine également le type de demande.
Types de demandes
Régulière — Pas une demande progressive ou accélérée.
Progressive — Demande comprenant diverses parties que l’établissement présente à différents moments. Généralement, une demande progressive est présentée lorsqu’un projet pilote ou un essai de production est prévu. Toutes les demandes d’homologation d’un nouvel établissement sont des demandes progressives. Tout établissement devrait avoir une discussion avec Santé Canada avant de déposer une demande progressive.
Accélérée — Les demandes accélérées sont celles dont l’examen préliminaire et l’examen sont accélérés pour des raisons de sécurité ou opérationnelles, conformément à ce qui a été convenu par l’établissement et la DMBR. Avant de soumettre sa demande à Santé Canada, l’établissement doit indiquer par écrit à la DMBR ses préoccupations et l’échéancier critique de mise en œuvre. Il doit clairement justifier pourquoi sa demande doit être traitée en accéléré selon un échéancier précis.
Si Santé Canada émet une directive exigeant la mise en œuvre rapide d’un changement, le traitement en accéléré est sous-entendu et il n’est pas nécessaire de fournir une justification.
La DMBR examine des demandes accélérées avant les autres demandes déjà déposées ou en cours d’examen. Les délais d’examen varieront selon les circonstances et la DMBR communiquera ce délai le plus rapidement possible à l’établissement.
Renseignements sollicités en examen préliminaire
La DMBR peut demander à l’établissement de fournir des renseignements supplémentaires ou des clarifications par l’entremise de divers types de communication, y compris des demandes d’information connues sous le nom de demande de clarification (clarifax) – examen préliminaire. L’établissement doit fournir une justification s’il juge qu’il n’est pas nécessaire d’envoyer une réponse. La réponse (renseignements sollicités) doit être fournie dans un délai de 15 jours civils après la date d’envoi du clarifax, ou avant la date indiquée sur le clarifax. Toutefois, l’établissement peut demander une prolongation de ce délai.
Avis d’insuffisance en examen préliminaire
La DMBR peut également envoyer un Avis d’insuffisance en examen préliminaire si des lacunes empêchent la réalisation de l’examen. L’Avis d’insuffisance en examen préliminaire décrit les lacunes et indique la date après laquelle la DMBR n’acceptera plus de réponse à l’avis. L’établissement peut faire ce qui suit :
- retirer la demande; ou
- fournir une réponse à l’Avis d’insuffisance en examen préliminaire, y inclus les renseignements identifiés avant la date indiquée
- une nouvelle période d’examen préliminaire commence après la réception de la réponse;
Si l’établissement ne parvient pas à fournir tous les renseignements demandés avant la date indiquée, un Avis de refus de délivrer une homologation ou un Avis de refus de modifier une homologation peut être émis:
Suite au retrait de la demande ou à un Avis de refus, l’établissement peut présenter de nouveau la demande entière.
- Cette demande sera traitée comme une nouvelle demande, et un nouveau numéro de contrôle sera attribué.
- Cette demande doit comprendre un renvoi au numéro de contrôle original.
- Les renseignements pertinents et les documents provenant de la demande originale, qui n’ont subi aucun changement, doivent être resoumis et devraient être clairement indiqués.
Si la demande est jugée acceptable aux fins d’examen, la DMBR envoie une Lettre d’admissibilité à l’évaluation à l’établissement pour confirmer la date à laquelle la demande fera partie du processus d’examen.
Examen des demandes
Lorsqu’elle accepte d’examiner une demande, la DMBR évalue les renseignements et les preuves connexes pour déterminer s’il y a un risque inacceptable pour la sécurité humaine ou pour la sécurité du sang et des composants sanguins.
Si la DMBR reçoit des renseignements détaillés (p. ex. preuve de validation réussie) de l’établissement, que ce soit un volet d’une demande en plusieurs parties ou des renseignements sollicités, la période d’examen commence à la date où ces renseignements ont été vérifiés et jugés acceptables aux fins d’examen par la DMBR.
Une fois que tous les renseignements et documents nécessaires ont été reçus et examinés, la DMBR indique si elle accepte ou refuse la demande. Veuillez vous référer à l’article 7 pour en savoir davantage sur la délivrance d’une homologation.
Contenu — Demande d’homologation
Une demande d’homologation doit contenir suffisamment de renseignements et de données probantes pour permettre au ministre d’évaluer le risque pour la sécurité humaine et pour montrer que les processus proposés produiront du sang et des composants sanguins jugés sécuritaire aux fins de distribution.
6(1)a), b) et c) Renseignements administratifs
L’établissement doit fournir les renseignements administratifs indiqués aux alinéas 6(1)a) à c). La personne à contacter pour toute question concernant la demande indiquée à l’alinéa 6(1)b) peut être la même que celle à contacter en cas d’urgence indiquée à l’alinéa 6(1)c).
6(1)d) Importation
L’établissement doit indiquer s’il a l’intention d’importer du sang total ou des composants sanguins, le cas échéant.
6(1)e) Liste des composants sanguins
L’établissement doit fournir une liste des types de sang et de composants sanguins allogéniques qu’il souhaite traiter (y compris le sang destiné à la fabrication de drogues pour usage humain) ainsi qu’une liste des types de sang allogénique et de composants sanguins allogéniques à des fins de transfusion qu’il souhaite importer. Les types doivent décrire en détail les composants figurant sur la liste, par exemple les plaquettes (partiellement déleucocytées) obtenues par aphérèse de donneurs CMV négatifs et déficients en IgA.
6(1)f) Liste des activités de traitement
L’établissement doit fournir une liste des activités de traitement relatives ausang allogénique et auxcomposants sanguins allogéniques qu’il prévoit exercer dans chaque bâtiment. Ces activités doivent correspondre à celles énumérées dans la licence d’établissement.
Voici des exemples d’activités de traitement :
- évaluation de l’admissibilité des donneurs de sang destiné à la transfusion;
- prélèvement de plasma par aphérèse destiné à la fabrication de drogues pour usage humain;
- essai d’amplification des acides nucléiques sur le sang total pour détecter la présence d’agents de maladies virales transmissibles;
- préparation de composants sanguins en utilisant la méthode de la couche leuco‑plaquettaire.
6(1)g) Installations — Bâtiments et équipement, produits et services essentiels
L’établissement doit fournir ce qui suit :
- pour chaque bâtiment, l’établissement doit fournir;
- un plan d’étage du bâtiment indiquant l’emplacement de l’équipement intégré, comme les congélateurs-chambres,
- une preuve d’approbation ou de validation réussie de tous les systèmes de l’installation (p. ex. électriques, de ventilation, d’eau, de sécurité, de contrôle de la température, etc.);
- une description de tout l’équipement et de tous les produits et services essentiels, y compris la fonction de chacun d’entre eux.
Consultez l’article 1 de la section Interprétation pour voir la définition du terme « essentiel » et des exemples d’équipement, de produits et de services essentiels.
La demande doit démontrer que la validation de l’équipement réalisée au sein de l’établissement, comprenant la qualification de l’installation, la qualification opérationnelle et la qualification de performance, est adéquate, le cas échéant.
L’équipement utilisé pour traiter le sang et les composants sanguins est souvent un instrument médical exigeant une homologation délivrée par la Direction des instruments médicaux de la Direction générale des produits de santé et des aliments de Santé Canada. Le demandeur devrait communiquer avec La Direction des instruments médicaux (meddevices-instrumentsmed@hc-sc.gc.ca) s’il a des questions au sujet de l’homologation des instruments médicaux. L’établissement devrait fournir des preuves montrant qu’une homologation d’un instrument médical valide et active existe pour tout l’équipement et tous les produits essentiels qui sont aussi des instruments médicaux.
6(1)h) Processus proposés
Dans la demande d’homologation, l’établissement doit fournir une description des processus et toutes les preuves pertinentes en ce qui concerne tous les processus qu’il souhaite mettre en œuvre pour traiter du sang allogénique et/ou des composants sanguins allogéniques, à partir du moment où il évalue le donneur jusqu’à ce qu’il juge le sang ou les composants sanguins sécuritaire à des fins de distribution. Ces processus comprennent l’entreposage et l’étiquetage des composants sanguins allogéniques réalisés pendant des activités de traitement mais avant et au moment de déterminer si le sang et les composants sanguins sont jugé sécuritaire à des fins de distribution. Consultez également le paragraphe 73(1) pour voir le critère de sécurité du sang allogénique à des fins de distribution.
Le numéro de la licence d’établissement doit figurer sur l’étiquette du sang allogénique et des composants sanguins allogéniques traité conformément à une homologation [voir l’alinéa 64(1)b)]. Consultez l’article 1 de la présente ligne directrice pour voir la définition du terme « traitement ».
Remarque : L’établissement doit être enregistré pour exercer des activités de traitement de sang autologue et de transformation ou avoir un programme de donneurs pré-évalués. Veuillez vous référer à l’article 30.
Établissement sous-traitant: L’établissement qui détient une homologation peut confier en sous‑traitance, à un établissement au Canada titulaire d’une licence en vertu du Règlement sur le sang, certaines activités de traitement, comme des tests de dépistage des marqueurs viraux.
S’il effectue seulement les essais des échantillons de sang et n’exerce pas d’autres activités de traitement assujetties au Règlement sur le sang, l’établissement sous-traitant n’a pas à soumettre une demande d’homologation si l’établissement contractant détient une homologation visant les essais [consultez le paragraphe 5(1)]. Les essais sur les échantillons de sang réalisés par l’établissement sous-traitant doivent être faits conformément à ceux visés dans l’homologation. L’établissement qui détient une homologation assume la responsabilité des essais et doit présenter une demande de modification d’homologation à Santé Canada pour tous les changements importants qu’il compte apporter à ce processus.
Pour obtenir d’autres renseignements sur la sous-traitance des établissements étrangers, consultez l’alinéa 6(1)j).
6(1)i) Étiquettes
Pour chaque composant sanguin allogénique qu’il prévoit traiter, l’établissement doit fournir à Santé Canada l’étiquette finale du contenant de sang proposée. Pour ce qui est du sang allogénique destiné à la transfusion, l’établissement doit fournir l’ébauche du document d’information. Il n’est pas nécessaire de fournir à Santé Canada, aux fins d’examen, les étiquettes pour le sang autologue, les étiquettes ajoutées aux unités après la transformation (étiquettes relatives à la transformation) et les étiquettes pour le sang prélevé dans le cadre d’un programme de donneurs pré-évalués.
6(1)j) Établissements étrangers : processus convenus dans les contrats
Un établissement qui prévoit confier une de ses activités en sous‑traitance à un établissement étranger doit fournir à Santé Canada des preuves indiquant que l’établissement étranger détient une licence active dans la juridiction étrangère. L’établissement au Canada qui est titulaire d’une homologation pour les processus convenus dans les contrats en assume la responsabilité et doit présenter à Santé Canada des demandes de modification de l’homologation pour cette activité.
L’alinéa 6(1)j) s’applique également si un établissement importe du sang ou des composants sanguins d’un établissement étranger. Si un établissement importe du sang ou des composants sanguins aux fins de transfusion - y compris l’importation de globules rouges pour l’immunisation de donneurs de plasma — le sang et des composants sanguins doivent respecter les exigences en matière d’importation figurant dans l’homologation de l’établissement. Veuillez vous référer également à l’article 92 « Importation dans des circonstances urgentes ».
6(1)k) Preuves suffisantes
Dans sa demande d’homologation, l’établissement doit présenter à Santé Canada des preuves suffisantes, notamment :
- toutes les données et tous les renseignements nécessaires au sujet de la sécurité de chaque type de composant sanguin allogénique qu’il souhaite traiter;
- toutes les données et tous les renseignements nécessaires pour démontrer que les processus qu’il utilise permettront de conclure à la sécurité du sang allogénique, y compris :
- l’évaluation de l’admissibilité du donneur;
- les renseignements obtenus après le don;
- les essais;
- la préparation de composants sanguins;
- l’étiquetage avant la distribution;
- le document d’information;
- l’entreposage avant la distribution;
- la distribution exceptionnelle;
- preuve de validation réussie de l’équipement, y compris des protocoles exécutés documentés; la validation de l’équipement devrait comprendre la validation de tout logiciel lié à celui-ci;
- preuve que les composantes du système informatique sont validées et que des procédures opérationnelles sont en place.
Évaluation sur place
6(2) Santé Canada peut effectuer une évaluation sur place (ESP) pour compléter l’examen d’une demande. La date de l’évaluation sur place sera fixée en consultation avec l’établissement et notée par écrit. Un ou plusieurs membres du personnel de Santé Canada peuvent participer à l’évaluation sur place.
La délivrance d’une homologation peut être conditionnelle à une évaluation sur place réussie. Une évaluation sur place peut être réalisée pour les raisons suivantes :
- examiner les preuves, la documentation et les données;
- observer les processus;
- observer une démonstration d’un nouvel équipement.
L’établissement devrait faire tous les efforts possibles pour que tous les documents et tout le matériel pertinents soient accessibles et s’assurer que des spécialistes en la matière puissent répondre aux questions.
Les résultats de l’évaluation sur place seront résumés de vive voix à l’établissement avant la fin de la visite. Toutes les mesures à prendre par l’établissement seront consignées par écrit à la fin de l’ESP, si possible, ou dans les 15 jours civils qui suivent l’ESP. Les délais de réponse seront indiqués.
Renseignements sollicités en cours d'examen
6(3) Pendant l’examen d’une demande, la DMBR peut demander à l’établissement de fournir du matériel supplémentaire si l’information qui a été soumise initialement est insuffisante ou n’est pas claire. Ce type d’information, appelé renseignements sollicités, est utilisé pour déterminer la sécurité humaine ou la sécurité du sang et des composants sanguins.
La DMBR peut demander des renseignements sollicités pendant une téléconférence ou une réunion; en envoyant un clarifax; ou sous la forme d’un avis d’insuffisance. Le clarifax peut être transmis par télécopieur ou par courriel dans un format standard. L’établissement doit fournir à la DMBR les renseignements sollicités dans les 15 jours suivant la date d’émission du clarifax ou à la date indiquée dans la demande. Toutefois, l’établissement peut demander une prolongation de ce délai. Une réponse est complète si :
- toutes les clarifications ou questions indiquées dans la demande d’éclaircissement ont été traitées, ou
- le promoteur a fourni une justification scientifique solide expliquant pourquoi les renseignements demandés ne sont pas nécessaires.
Avis d’insuffisance
Si la demande contient des lacunes empêchant une évaluation plus approfondie ou si la réponse de l’établissement à un clarifax d’examen n’est pas adéquate, la DMBR émet un avis d’insuffisance (AI). Cet avis décrit les lacunes et indique la date après laquelle la DMBR n’acceptera plus de réponse à l’AI. L’établissement peut fournir une réponse à l’AI en y incluant les renseignements identifiés sous le numéro de contrôle de la demande originale. L’établissement peut également décider d’annuler la demande; la DMBR émettra un accusé de réception de l’annulation.
L’établissement a 90 jours pour répondre à l’AI. L’examen est suspendu jusqu’à ce que la DMBR reçoive une réponse. Après avoir reçu la réponse, une nouvelle période d’examen préliminaire commence.
Si l’établissement ne répond pas à l’AI dans les délais prescrits et n’annule pas sa demande, ou si la demande demeure insuffisante, la DMBR émettra une lettre de retrait - AI.
Si l’établissement n’est pas en mesure de respecter le délai indiqué dans l’AI, il devrait fournir à la DMBR une demande et une justification écrites pour une prolongation avant la fin des 90 jours.
Suite à l’annulation de la demande par l’établissement ou lorsqu'une lettre de retrait - AI est émise par la DMBR, l’établissement peut présenter de nouveau la demande (consultez l’article 8).
Article 7 Délivrance
Délivrance
7.Le ministre, au terme de l’examen de la demande d’homologation, délivre l’homologation, avec ou sans conditions, s’il juge que la preuve fournie est suffisante pour démontrer que la délivrance de l’homologation ne compromettra ni la sécurité humaine ni la sécurité du sang.
Avis d'homologation
Lorsque la DMBR termine l’examen des renseignements et des preuves présentés dans la demande de l’établissement et que la DMBR les considère acceptables, elle émet un avis d’homologation qui permet à l’établissement de mettre en œuvre ses processus et qui résume les renseignements suivants :
- a. nom de l’établissement;
- b. composants sanguins que l’établissement peut traiter;
- c. activités de traitement que l’établissement peut exercer;
- d. adresse civique de chaque bâtiment et activités de traitement qui seront réalisées à chaque emplacement;
- e. toutes les conditions de l’homologation.
L’avis d’homologation peut également comprendre ce qui suit :
- f. laboratoires ou autres installations à qui l’établissement a confié des activités de traitement;
- i. domestiques ou
- ii. étrangers;
- g. documents de contrôle approuvés;
- h. systèmes informatiques et principaux logiciels de l’établissement ainsi que les numéros de version, comme les logiciels pour l’évaluation de l’admissibilité des donneurs, les systèmes de gestion du sang et des donneurs ou les systèmes informatiques des laboratoires;
- i. trousses d’essai utilisées pour qualifier les donneurs, par exemple maladies transmissibles, groupe sanguin ou anticorps associés aux globules rouges (y compris le numéro de licence attribué par le Bureau des matériels médicaux et la date d’approbation); et
- j. matériels médicaux utilisés, par exemple principales plateformes d’essai (y compris le numéro de licence attribué par la Direction des instruments et la date d’approbation).
La DMBR émet un avis d’homologation ou un avis d’homologation avec conditions après l’examen et l’approbation de la demande d’homologation originale d’un établissement. Tous les avis ultérieurs suivant l’examen et l’approbation des demandes de modification de l’homologation sont des avis de modification de l’homologation (article 9) ou des avis de modification de l’homologation avec conditions (article 13).
Article 8 Refus
Refus
8.Le ministre peut, s’il juge que des renseignements fournis par l’établissement dans sa demande d’homologation sont inexacts ou incomplets, refuser de délivrer l’homologation.
Avis de renseignement insuffisant
Pendant l’examen, si la DMBR considère que les renseignements dans l’application de l’établissement sont insuffisants pour démontrer que la délivrance de l’homologation ne compromettra pas la sécurité humaine ou la sécurité du sang et des composants sanguins, la DMBR peut délivrer un avis de renseignement insuffisant. Cet avis fournira les raisons pour lesquelles les renseignements fournis dans l’application ont été jugés insuffisants.
L’établissement a 90 jours pour répondre à l’avis de renseignement insuffisant. L’examen est suspendu jusqu’à ce que la DMBR reçoive une réponse à cet avis suivant laquelle une nouvelle période d’examen préliminaire sera établie.
Si l’établissement ne répond pas dans les 90 jours, ou la DMBR juge que la demande demeure non-conforme, la DMBR émettra une lettre de retrait-avis de renseignement insuffisant. L’établissement peut également choisir de fournir une lettre d’annulation de la demande; la DMBR émettra alors un accusé de réception de l’annulation.
Si l’établissement n’est pas en mesure de respecter le délai de 90 jours, il devrait fournir à la DMBR une demande et une justification écrites pour une prolongation avant la fin des 90 jours.
Suite à l’annulation de la demande par l’établissement ou lorsqu'une lettre de retrait-avis de renseignement insuffisant est émise par la DMBR, l’établissement peut présenter de nouveau la demande.
Avis de refus
La DMBR peut émettre un avis de refus de délivrer une homologation ou un avis de refus de délivrer une modification d’une homologation dans l’une des circonstances suivantes :
- si l’établissement fournit des preuves insuffisantes pour appuyer les processus proposés;
- s’il y a des inexactitudes dans la demande;
- si des énoncés faux ou trompeurs sont présentés; ou
- si le processus examiné présente un risque inacceptable pour la sécurité humaine ou la sécurité du sang et des composants sanguins.
L’avis de refus présente les raisons du refus et offre à l’établissement l’occasion d’exposer son point de vue par écrit. L’établissement peut choisir de présenter de nouveau la demande.
Présenter de nouveau
Lorsqu’une demande est présentée de nouveau, cette demande :
- sera traitée comme une nouvelle demande et un nouveau numéro de contrôle sera attribué;
- devrait comporter des renvois au numéro de contrôle original; et
- devrait clairement indiquer quels renseignements et documents de la demande originale demeurent inchangés.
Article 9 Changement majeur
Changement majeur
9. (1) L’établissement, avant d’effectuer un changement majeur, présente au ministre une demande de modification de son homologation accompagnée des renseignements et documents utiles pour permettre au ministre de juger si le changement ou sa mise en œuvre risque de compromettre la sécurité humaine ou la sécurité du sang.Demande de modification
(2) Les articles 6 à 8 s’appliquent à la demande de modification de l’homologation, avec les adaptations nécessaires.
Définition de « changement majeur »
(3) Pour l’application du présent article ainsi que des articles 10 et 12, « changement majeur » s’entend de l’un des changements suivants :
- l’ajout d’un élément à la liste fournie en application de l’alinéa 6(1)e);
- la modification ou le retrait d’un processus homologué;
- l’ajout d’un processus visé à l’alinéa 6(1)h);
- toute modification à la description des installations décrites en application de l’alinéa 6(1)g).
9(1) Un établissement doit présenter une demande de modification de l’homologation à la DMBR s’il a l’intention d’apporter des changements majeurs. L’expression « changement majeur » est définie au paragraphe 9(3) du Règlement sur le sang.
Demande de modification de l’homologation — Exigences minimales générales
Les demandes pour n’importe quel changement majeur ont les mêmes exigences minimales :
- description détaillée des changements proposés;
- justification des changements proposés;
- évaluation des risques; et
- les coordonnées de la personne-ressource, telles qu’elles sont demandées aux alinéas 6(1)b) et 6(1)c) du Règlement sur le sang.
Exigences supplémentaires, le cas échéant
- approbation des matériels médicaux du Bureau des matériels médicaux;
- données cliniques;
- données scientifiques ou techniques;
- une description détaillée d’équipement (nouveau ou modifié) et de tout produit ou service (nouveau ou modifié) si son fonctionnement diffère des homologations accordées
- incidences sur le système informatique de gestion du sang et/ou des composants sanguins;
- changements apportés aux étiquettes de sang, y compris le document d’information (inclure les nouvelles étiquettes ou étiquettes révisées)
- plan de formation;
- plan de validation;
- résultats de la validation; et
- calendrier de mise en œuvre.
La demande de modification de l’homologation doit être signée et datée par le cadre supérieur indiquant que tous les renseignements présentés dans la demande sont exacts et complets [consultez le paragraphe 6(1)].
Toutes questions concernant les changements majeurs, les nouveaux renseignements qui pourraient déclencher une modification et le processus de modification d’une homologation devraient être envoyées au BAR de la DMBR. Veuillez vous référer au paragraphe 6(1) de la présente ligne directrice pour obtenir les coordonnées.
9(2) Lorsqu’un établissement présente une demande de modification de l’homologation, les articles 6 à 8 du Règlement sur le sang s’appliquent. Veuillez vous référer aux articles 6 à 8 de la présente ligne directrice.
La DMBR peut exiger une évaluation sur place de l’établissement à la suite d’un examen de la documentation présentée, selon la nature du changement et des données à l’appui. Veuillez vous référer au paragraphe 6(2).
La DMBR vise à compléter l’examen préliminaire d’une demande de modification de l’homologation dans les 15 jours civils de la réception et l’examen de celle-ci dans les 90 jours suivant la date d’acceptation de la demande pour fins d’examen. Il est interdit à un établissement de mettre en œuvre les changements présentés dans la demande de modification de l’homologation avant d’avoir reçu l’autorisation de Santé Canada, même si le délai d’examen est prolongé au-delà des 90 jours.
Division d’une demande de modification de l’homologation
À l’occasion, il se peut qu’un établissement présente une demande portant sur deux sujets ou plus qui ne sont pas liés. Lors du processus d’examen préliminaire ou d’examen, la DMBR peut diviser la demande originale en deux demandes ou plus et attribuer un numéro de contrôle à chacune. Lorsque la DMBR divise une demande, l'établissement reçoit un avis écrit de la division demandant la soumission des données électroniques sous forme de fichiers distincts.
Avis de modification de l’homologation
Après que la DMBR a terminé d’examiner la demande de modification et qu’elle a déterminé qu’elle était acceptable, elle envoie un avis de modification de l’homologation permettant à l’établissement de mettre en œuvre les changements majeurs.
Ensemble, l’avis d’homologation et tous les avis de modification de l’homologation constituent l’homologation de l’établissement.
9(3)a) Ajout de sang ou de composants sanguins
Un changement majeur comprend l’ajout de sang ou de composants sanguins, ou encore d’un « nouveau composant sanguin » à la liste de sang ou de composants sanguins, que l’établissement souhaite traiter ou importer. Veuillez vous référer à l’article 1.5 « Définitions » de la présente ligne directrice pour la définition du terme « nouveau composant sanguin ».
Il est fortement recommandé que l’établissement consulte la DMBR avant de présenter une demande de modification de l’homologation lorsqu’il propose les ajouts suivants :
- matériel médical ayant le potentiel d’avoir une incidence sur un composant sanguin;
- nouveau composant sanguin développé en collaboration avec un fabricant de matériel médical.
9(3)b) Retrait ou modification des processus homologués
La signification de « changement majeur » comprend le retrait ou la modification de tout processus homologué.
9(3)c) Ajout d’un processus
L’établissement doit présenter une demande comprenant une description détaillée de tout nouveau processus qu’il propose d’utiliser pour chaque type de sang et de composant sanguin dans le cadre de ses activités.
9(3)a), 9(3)b) et 9(3)c) Études pilotes ou essais de production
Des études pilotes ou des essais de production peuvent être requis pour élaborer les preuves nécessaires pour démontrer que tous les changements aux processus ou aux composants sanguins n’ont aucune incidence indésirable sur la sécurité humaine ou la sécurité du sang et des composants sanguins. Lorsqu’un établissement souhaite mettre en œuvre un nouveau processus ou un processus modifié ou un nouveau composant sanguin, il devrait présenter une demande de modification de son homologation pour ce changement avant de commencer l’étude pilote ou l’essai de production. La contribution préalable de la DMBR sur la conception est fortement recommandée afin de faciliter le processus de la demande.
L’établissement est encouragé à communiquer avec la DMBR pour toute question sur des études pilotes ou des essais de production.
9(3)d) Modification des installations (bâtiments et équipement, produits et services essentiels)
Nouveaux bâtiments ou bâtiments rénovés
L’établissement doit présenter une demande de modification de l’homologation s’il propose d’ajouter ou de rénover un bâtiment où des activités de traitement (évaluation de l’admissibilité des donneurs, prélèvement, analyse ou préparation de composants sanguins) auront lieu.
Un établissement n’est pas tenu de fournir dans sa demande, des renseignements et des données à l’appui des processus et d’équipement qu’il a déjà validé et qui ont été homologués par la DMBR, en autant qu’une méthode de validation semblable soit utilisée.
Le cas échéant, la demande doit comprendre un énoncé qui indique qu’il n’y a aucun changement apporté aux processus et à l’équipement déjà homologués, qu’ils ont été validés et qu’une méthode de validation semblable sera utilisée pour les processus et l’équipement déjà homologués par la DMBR. Cette déclaration doit confirmer que le nouvel établissement respectera les processus déjà approuvés.
Outre les exigences d’informations minimales (consultez le paragraphe 9(1)), la demande de modification des installations de l’établissement devrait inclure les mentions suivantes :
- l’adresse municipale de l’installation où le changement majeur serait effectué [alinéa 6(1)a)];
- une liste des activités de traitement qui seraient effectuées dans chaque bâtiment où le changement majeur serait apporté [alinéa 6(1)f)];
- une description détaillée des changements apportés à l’installation, y compris :
- un plan d’étage du bâtiment indiquant l’emplacement de l’équipement intégré, comme des congélateurs-chambres, et
- une liste de l’équipement, des produits et des services essentiels qui seraient utilisés pour mener les activités [alinéa 6(1)g)];
- une description détaillée de tout équipement (nouveau ou modifié) et de tout produit ou service (nouveau ou modifié) si son fonctionnement diffère des homologations actuelles [alinéa 6(1)g)];
- Preuve que tous les nouveaux processus liés aux systèmes des installations (électricité, ventilation, eau, sécurité, surveillance de la température, etc.) et tout l’équipement sont mis en service et sont validés.
Conformément à l’article 22, l’établissement doit présenter une demande de modification de la licence d’établissement lorsqu’il souhaite ajouter un bâtiment à sa liste d’installations ou qu’il souhaite y apporter une modification. Santé Canada peut inspecter le nouveau bâtiment après que tous les changements ont été apportés, et avant le début des activités (c’est-à-dire que l’établissement n’a pas commencé à accepter les donneurs).
Équipement, produits et services essentiels
Les changements majeurs apportés à l’équipement essentiel sont définis en fonction de leur incidence sur les processus homologués et de leurs répercussions sur la sécurité humaine et la sécurité du sang et des composants sanguins. Le terme équipement comprend aussi tous les logiciels. Veuillez vous référer à la définition du terme « essentiel » à l’article 1 de la présente ligne directrice pour obtenir des exemples d’équipement, de produits et de services essentiels.
Un nouveau matériel/une nouvelle technologie
L’établissement doit présenter une demande de modification d’homologation lorsqu’il souhaite apporter des changements majeurs à du matériel médical ou utiliser un nouvel appareil ou une nouvelle technologie susceptible d’avoir une incidence sur la sécurité humaine ou la sécurité du sang et des composants sanguins.
Dans les exemples de changements suivants, déterminer si des changements importants peuvent ou non compromettre la sécurité des personnes ou la sécurité du sang et des composants sanguins:
- remplacement d’un appareil par un autre appareil qui possède la même indication ou fonctionnalité, mais dont la marque diffère;
- remplacement d’un appareil par un autre dont l’indication ou la fonctionnalité diffère;
- remplacement d’un appareil par un nouveau modèle ou un modèle différent provenant du même fabricant;
- mise à niveau d’une partie de l’appareil avec un nouveau logiciel ou une nouvelle fonctionnalité
Les modifications apportées à un matériel médical homologué par Santé Canada
Les modifications apportées aux matériels médicaux par le fabricant sont jugées significatives ou non par Santé Canada selon la Ligne directrice - Directive sur l'interprétation d'une modification importante d'un instrument médical. Si l’établissement a l’intention d’employer du matériel médical modifié, il devra évaluer l’incidence de ces modifications sur la sécurité humaine ou la sécurité du sang et des composants sanguins et présenter ces changements pour examen par la DMBR selon les exigences énumérées aux trois points suivants :
- Si la modification est jugée « non-significative » par Santé Canada, et qu’il n’y a pas d’incidence potentielle sur la sécurité humaine ou la sécurité du sang et des composants sanguins, l’établissement n’est pas tenu de présenter le(s) changement(s) dans un rapport annuel ou une demande de modification d’homologation.
- Dans le cas où la modification est « non-significative », mais qu’il y a une incidence potentielle sur la sécurité, l’établissement devra présenter les changements dans un rapport annuel.
- Dans le cas où la modification est « significative », et qu’il y a une incidence potentielle sur la sécurité, l’établissement devra présenter les changements par le biais d’une demande de modification d’homologation.
Technologie de l’information
Lorsque l’établissement qui détient une homologation prévoit apporter des changements à des processus qui ont une composante informatique importante, il doit déposer une demande de modification de son homologation auprès du BAR de la DMBR.
Voici des exemples de changements majeurs apportés aux technologies de l’information :
- l’ajout d’un système informatique, comme :
- un nouveau système de gestion des donneurs,
- une nouvelle plateforme de questionnaires électroniques,
- un nouveau système d’information de laboratoire; ou
- transition vers une nouvelle version du logiciel d’application ou du système de gestion des données déjà en place et validé;
- changements correspondants aux procédures opérationnelles lorsque des changements majeurs sont apportés à un système informatique;
- modifications apportées à la configuration du logiciel déjà en place et validé;
- ajouts à la fonctionnalité du logiciel d’application déjà en place et validé (y compris les ajouts effectués lors de l’entretien du système).
Si un établissement n’est pas sûr qu’un changement apporté à l’un des composants validés d’un système informatisé actuellement installé soit considéré comme important, il est fortement recommandé que l’établissement consulte la DMBR pour obtenir des conseils avant de déposer sa demande.
Des directives sont essentielles lorsqu’un établissement installe un nouveau système informatique qui remplit une ou plusieurs des fonctions suivantes :
- utilisation d’un logiciel pour le transfert de données entre des équipements automatisés lorsque de la traduction ou du reformatage est nécessaire;
- utilisation de données pour la prise de décisions sur la qualité du sang ou des composants sanguins destinés à la transfusion ou à une fabrication ultérieure; ou
- utilisation de données pour faire le suivi d’une unité de sang ou d’un composant sanguin, de la source à sa destination finale.
Toutes questions au sujet des changements à apporter à un système informatique existant devraient être envoyées au BAR de la DMBR. Veuillez vous référer au paragraphe 6(1) de la présente ligne directrice pour les coordonnées de la personne‑ressource.
Voir l’article 12 de la présente ligne directrice pour d’autres changements devant figurer dans le rapport annuel.
Article 10 Changement en cas d'urgence
Changement en cas d’urgence
10. (1) L’établissement peut, avant de présenter une demande de modification de son homologation, effectuer tout changement majeur qui s’impose, en cas d’urgence, pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises.Avis et demande
(2) Le cas échéant, il en avise le ministre par écrit au plus tard le jour suivant sa mise en œuvre et présente dans les quinze jours suivant la date de cet avis une demande de modification de l’homologation.
10(1) Changement en cas d’urgence
En vertu de l’article 10, l’établissement peut effectuer, en cas d’urgence, tout changement majeur qui s’impose à un processus autorisé. Par exemple, une situation d’urgence peut nécessiter la mise en œuvre de changements pour i) corriger une erreur dans le système informatique de la gestion du sang ou ii) remédier à un problème associé à de l’équipement défectueux utilisé dans le cadre d’essais.
10(2) Avis et demande
En cas d’urgence, s’il s’avère nécessaire de mettre en œuvre un changement important pour éviter de compromettre la sécurité humaine ou la sécurité du sang et des composants sanguins. L’établissement doit suivre le processus de déclaration en deux étapes :
- L’établissement doit fournir à la DMBR un rapport initial écrit dans la journée suivant la mise en œuvre du changement en cas d’urgence. Le rapport initial devrait contenir les renseignements suivants :
- définition du problème et de l’urgence;
- décisions prises;
- mesures prises; et
- changement majeur apporté ou qui sera apporté par l’établissement.
- Dans les 15 jours civils suivant la déclaration à la DMBR, l’établissement doit présenter une demande de modification de l’homologation à l’égard de ces changements. La DMBR peut fixer des conditions supplémentaires à l’homologation suite aux changements apporté en cas d’urgence (consultez l’article 13).
Article 11 Changements administratifs - avis
Changements administratifs -avis
11.L’établissement qui modifie tout renseignement fourni en application des alinéas 6(1)a) à c) en avise dès que possible par écrit le ministre qui modifie l’homologation en conséquence.
L’établissement doit aviser par écrit la DMBR s’il prévoit modifier les renseignements qu’il a présentés dans la demande d’homologation conformément aux alinéas 6(1)a) à c). Veuillez vous référer au paragraphe 6(1) de la présente ligne directrice pour obtenir les coordonnées.
La DMBR révisera l’homologation de l’établissement après avoir reçu l’avis et délivrera un avis de modification de l’homologation.
Article 12 Autres changements - rapport annuel
Autres changements — rapport annuel
12. (1) L’établissement présente au ministre un rapport annuel détaillé des changements apportés au courant de l’année qui ne sont pas visés aux articles 9 ou 11 et qui pourraient compromettre la sécurité humaine ou la sécurité du sang.Modification de l’homologation
(2) Le ministre modifie, sur réception du rapport, l’homologation de l’établissement en conséquence.Changement jugé majeur
(3) Le ministre, s’il juge qu’un changement indiqué dans le rapport est un changement majeur, en avise l’établissement par écrit et peut l’enjoindre d’interrompre la mise en œuvre du changement ou de rétablir l’état antérieur des choses.Demande de modification de l’homologation
(4) L’établissement qui reçoit l’avis présente au ministre une demande de modification de son homologation.
12(1) Lorsque l’établissement apporte d’autres changements à ses processus qui n'ont pas été présentés en vertu de l'article 9 (changement majeur) ou de l'article 11 (administratif) — et que ces changements pourraient compromettre la sécurité humaine ou la sécurité du sang et des composants sanguins — l'établissement doit décrire ces changements dans un rapport annuel à Santé Canada.
L’établissement devrait communiquer avec la DMBR pour savoir si un changement devant être apporté à un processus autorisé peut être inclus dans un rapport annuel. Les rapports annuels devraient être signés par un cadre supérieur et envoyés au BAR de la DMBR. Veuillez vous référer au paragraphe 6(1) de la présente ligne directrice pour obtenir les coordonnées.
Exemples des types de changements qu’un établissement peut inclure dans un rapport annuel :
- Changements apportés à la liste des médicaments inacceptables
Changements apportés à la liste des médicaments inacceptables utilisée lors de l’évaluation des donneurs de sang pour transfusion.
Changements relatifs à l’entretien des systèmes de technologies informatiques de gestion du sang
Changements apportés au système de technologie informatique de gestion du sang en raison d’activités d’entretien pour améliorer la fonctionnalité, sans changement opérationnel (p. ex. augmentation de la mémoire), ou restaurer la fonctionnalité (p. ex. correction de bogues).
Lorsqu’il rédige un rapport annuel, l’établissement devrait s’assurer que tous les changements relatifs à l’entretien du système informatique de gestion du sang ont été décrits dans le rapport annuel et qu’aucun nouveau processus non autorisé n’a été intégré par inadvertance.
Exigences en matière de présentation de rapports annuels
L’établissement doit fournir les renseignements ci-dessous dans son rapport annuel :
- description du changement;
- justification du changement;
- processus homologués touchés, le cas échéant;
- systèmes de technologies informatiques touchés par le changement, le cas échéant;
- accidents ou manquement connexes, le cas échéant;
- numéro de licence d’une homologation d’un instrument médical actuelle et valide, le cas échéant;
- date de mise en œuvre; et
- autres renseignements nécessaires pour décrire le changement ou les répercussions du changement.
Avant de présenter un rapport annuel, l’établissement devrait confirmer que des renseignements complets ou des données sur les changements sont disponibles sur demande.
Changements qui n’exigent pas la déclaration annuelle ni la présentation d’une demande de modification de l’homologation
Il a été déterminé que ces types de changements sont peu susceptibles d’entraîner des effets indésirables sur la sécurité humaine ou la sécurité du sang et des composants sanguins.
Donneurs
- Changements apportés aux procédures de consentement du donneur;
- Changements apportés aux aires de sélection des donneurs existantes;
- Changements apportés au format, à la couleur et à la mise en page de n’importe quel document, du manuel de sélection des donneurs, du questionnaire de sélection des donneurs et du document d’information de l’établissement, lorsqu’aucun changement n’est apporté aux processus homologués;
- Changements des régions impaludées dans le manuel de sélection des donneurs de l’établissement à condition qu’elles soient identiques à celles identifiées par l’Agence de la santé publique du Canada (ASPC) ou les Centers for Disease Control and Prevention (CDC) des États-Unis;
- Changements apportés aux essais pour les maladies infectieuses servant uniquement aux fins de conseils.
Équipement
- Changements apportés à de l’équipement ou à des produits non essentiels. Veuillez vous référer à l’article 1 de la présente ligne directrice pour la définition du terme « essentiel » et une liste de l’équipement non essentiel;
- Changements apportés aux étiquettes qui n’ont pas d’incidence sur le contenu et qui sont liés à l’orientation ou au placement de l’information;
- Sous réserve que l'établissement utilise les mêmes approches de validation qui ont été examinés par la DMBR et utilisés lors de la validation d’équipement déjà approuvé :
- remplacement d’équipement par un autre dont la marque et le modèle sont semblables;
- remplacement d’une partie d’un tel équipement; ou
- ajout ou retrait d’autres unités d’un tel équipement.
- Les changements à la méthode de validation générale n’ont aucun lien avec les protocoles de validation utilisés pour le traitement du sang;
- Changements dans les versions des logiciels des équipements utilisés pour les analyses de contrôle de la qualité, pour autant qu’ils n’engendrent aucun effet sur les spécifications des composants sanguins ou les processus employés pour la préparation des composants;
- Changements dans les systèmes d'exploitation informatiques existants;
- Changements apportés aux matériels informatiques;
- Changements nécessaires pour mettre à niveau le logiciel antivirus ou installer des rustines de changement d’heure au système d'exploitation informatique;
- Changements apportés à des programmes logiciels types disponibles dans le commerce (non configurables), par exemple Microsoft Office, pour autant qu’ils n’engendrent aucun effet sur les processus homologués.
Bâtiments
- Changements apportés aux systèmes de ventilation ou de climatisation centraux ou individuels déjà en place, pour autant que les paramètres environnementaux demeurent à l’intérieur des limites autorisées et n’aient aucun effet sur les exigences propres aux composants sanguins;
- Changements apportés au système de surveillance des données environnementales. Il s’agit d’un système qui accumule des données environnementales et démontre que les paramètres environnementaux des aires où des processus critiques sont rencontrés; le système déclenche une alarme lorsque les conditions ne rencontrent plus les spécifications;
- Changements apportés à la sécurité existante des bâtiments.
Système de gestion de la qualité
Gestion des stocks de sang
- Changements apportés au processus de gestion des stocks de sang.
Transport, envoi et expédition/emballage
- Changements apportés aux processus de transport du sang et des composants sanguins;
- Changements apportés au processus d’envoi et d’emballage des composants sanguins.
Activités de transformation
- Changements apportés aux activités de transformation réalisées sur les composants sanguins, c’est-à-dire lavage, mise-en-commun, irradiation gamma. Veuillez vous référer aux articles 77 à 80 de la présente ligne directrice.
Processus liés aux enquêtes et à la gestion des rapports
- Changements apportés aux processus liés aux enquêtes et à la gestion des rapports suivants où il n'y a pas de changement des processus homologués:
- accidents et manquement;
- effets indésirables chez les donneurs;
- effets indésirables;
- études des dons antérieurs et enquêtes sur les produits transfusés; et
- renseignements obtenus après le don.
Article 13 Ajout de conditions à l'homologation ou modification
Ajout de conditions à l’homologation ou modification
13. (1) Le ministre peut modifier les conditions de l’homologation d’un établissement ou en ajouter de nouvelles dans les cas suivants :
- il a des motifs raisonnables de le croire nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises;
- l’établissement ne lui fournit pas, au plus tard à la date précisée dans sa demande écrite à cet effet, de preuve suffisante démontrant que ses processus ne compromettront pas la sécurité humaine et permettront de conclure à la sécurité du sang à des fins de distribution.
Préavis
(2) Le ministre, avant de modifier les conditions de l’homologation ou d’en ajouter de nouvelles, envoie à l’établissement, au moins quinze jours avant la date de prise d’effet prévue des conditions, un préavis motivé l’informant qu’il peut présenter ses observations à cet égard.Cas d’urgence
(3) Malgré le paragraphe (2), le ministre peut, s’il a des motifs raisonnables de le croire nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises, modifier sans préavis les conditions de l’homologation ou en ajouter de nouvelles.Cas d’urgence — avis
(4) Le ministre, s’il modifie les conditions de l’homologation ou en ajoute de nouvelles conformément au paragraphe (3), envoie à l’établissement un avis motivé l’informant qu’il peut présenter ses observations à cet égard.Suppression d’une condition
(5) Le ministre peut, par avis écrit, supprimer toute condition de l’homologation qu’il ne juge plus nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises.
Modification des conditions de l'homologation ou imposition de nouvelles conditions
13(1) La DMBR peut imposer des conditions à l’homologation ou modifier les conditions d’une homologation afin de gérer un risque identifié pour la sécurité, soit dans le cadre d’une demande d’homologation ou d’une demande de modification de l’homologation ou en dehors du contexte d’une demande.
Un avis d’homologation (ou un avis de modification de l’homologation) avec conditions permet à l’établissement de mettre en œuvre les processus proposés dans sa demande mais avec certaines restrictions. Des conditions peuvent être imposées à l’homologation ou à la modification de l’homologation dans les exemples de situations suivants :
- permettre l’exécution d’un projet pilote ou d’un essai de production pendant lequel des données seront recueillies et serviront à l'évaluation (ou l’examen) par la DMBR pour appuyer les changements proposés;
- imposer des conditions à une modification, comme l’exigence de fournir des données après la mise en œuvre – voir des exemples de conditions plus bas;
- obliger l’établissement à fournir des données supplémentaires en vue d’une mise en œuvre en plusieurs étapes;
- demander la réalisation d’études ultérieures;
- lorsque des interventions sont nécessaires pour gérer un nouveau pathogène;
- lorsque des préoccupations entourant la sécurité ont été relevées à l’établissement.
Les conditions peuvent comprendre :
- l’ajout d’un nouvel essai pour les donneurs;
- l’ajout d’une nouvelle question d’admissibilité des donneurs;
- le traitement d’un nombre minimal de dons, tel qu’indiqué dans la demande d’homologation ou dans la demande de modification de l’homologation;
- les limites quant aux types de dons recueillis;
- les limites quant aux types d’activités qui peuvent être effectuées;
- les limites quant à la distribution de tous les composants sanguins ou de certains d’entre eux;
- la présentation de données sur la surveillance de la sécurité du donneur;
- la présentation de données sur le contrôle de la qualité des composants traités;
- la présentation d’études de stabilité supplémentaires;
- la présentation de renseignements supplémentaires, au besoin;
- la mise en œuvre d’une mesure corrective après l’observation d’un accident ou d’un manquement, après avoir communiqué avec la DGORAL de Santé Canada;
- la mise en œuvre d’une amélioration du processus relatif au traitement des composants sanguins.
13(2) Lorsque Santé Canada impose des conditions à une homologation, le ministre émet à l’établissement un Avis de modification d’une homologation avec conditions. Le ministre doit accorder à l’établissement un délai de 15 jours civils pour répondre à l’avis. Les conditions dans l’avis prennent effet après le délai de 15 jours civils s’il n’y a pas de réponse de l’établissement ou si la réponse n’adresse pas suffisamment le risque. Des discussions peuvent être engagées avec l’établissement avant que les conditions ne soient imposées.
13(3) En cas d’urgence menaçant la sécurité humaine ou la sécurité du sang et des composants sanguins, Santé Canada peut immédiatement ajouter des conditions ou modifier les conditions d’une homologation. Par exemple, l’émergence d’un nouveau pathogène susceptible d’être transmis par le sang et/ou les composants sanguins est considérée comme une situation d’urgence.
Santé Canada peut également imposer des conditions urgentes à une homologation si un établissement n’a pas pris de mesures correctives pour régler un problème susceptible d’affecter la sécurité du sang et des composants sanguins, comme la possibilité de causer un effet indésirable grave.
13(5) Lorsque l’établissement adresse le risque à la sécurité identifié à un niveau que Santé Canada juge satisfaisant grâce à la mise en œuvre de changements dans ses locaux ou après avoir soumis des preuves convaincantes au ministre, Santé Canada supprime les conditions par communication écrite. Dans le cas où les conditions ont été imposées dans le cadre d’une demande d’homologation ou de modification de l’homologation, une lettre de fermeture de la demande sera émise lorsque le risque à la sécurité aura été adressé adéquatement.
Article 14 Suspension
Suspension
14. (1) Le ministre peut suspendre complètement ou partiellement l’homologation d’un établissement dans les cas suivants :
- des renseignements fournis en application des articles 6 ou 9 se révèlent inexacts ou incomplets;
- l’établissement ne lui fournit pas, au plus tard à la date précisée dans sa demande écrite à cet effet, de preuve suffisante démontrant que ses processus ne compromettront pas la sécurité humaine et permettront de conclure à la sécurité du sang à des fins de distribution.
Préavis
(2) Le ministre, avant de suspendre l’homologation, envoie à l’établissement un préavis qui contient les précisions suivantes :
- les motifs de la suspension envisagée et sa date de prise d’effet;
- l’obligation qu’a l’établissement de prendre des mesures correctives, s’il y a lieu, au plus tard à la date précisée;
- le fait qu’il lui donne la possibilité de présenter ses observations à cet égard.
Cas d’urgence
(3) Malgré le paragraphe (2), le ministre peut, s’il a des motifs raisonnables de le croire nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises, suspendre sans préavis l’homologation complètement ou partiellement.Cas d’urgence — avis
(4) Le cas échéant, le ministre envoie à l’établissement un avis qui contient les précisions suivantes :
- les motifs de la suspension;
- le fait qu’il lui donne la possibilité de présenter ses observations à cet égard.
Article 15 Rétablissement de l'homologation
Rétablissement de l'homologation
15. (1) Sous réserve du paragraphe (2), le ministre rétablit l’homologation de l’établissement qui fournit une preuve suffisante démontrant que ses processus ne compromettront pas la sécurité humaine et permettront de conclure à la sécurité du sang à des fins de distribution.Rétablissement partiel
(2) Le ministre supprime de l’homologation toute partie suspendue qu’il ne rétablit pas.
Article 16 Annulation de l'homologation
Annulation de l’homologation
16. (1) Le ministre annule l’homologation de l’établissement dans les cas suivants :
- l’établissement ne lui fournit pas la preuve visée à l’alinéa 14(1)b) dans un délai raisonnable suivant la suspension;
- la licence de l’établissement a été annulée au titre de l’article 29.
Avis
(2) Le cas échéant, il envoie à l’établissement un avis motivé à cet effet indiquant la date de prise d’effet de l’annulation.
Articles 17-29 Licence d'établissement
Article 17 Obligation d'obtenir une licence
Obligation d’obtenir une licence
17. (1) L’établissement obtient une licence d’établissement pour traiter du sang provenant d’un don allogénique exception faite, sous réserve du paragraphe (2), du sang prélevé d’un donneur pré-évalué, ou pour l’importer.Essais
(2) L’établissement qui met à l’essai du sang prélevé d’un donneur pré-évalué à des fins de dépistage de maladies transmissibles ou de leurs agents obtient une licence à cette fin.
Traitement du sang allogénique et importation
Un établissement doit obtenir une licence d’établissement s’il traite ou importe du sang allogénique et/ou des composants sanguins allogéniques. Les activités de traitement regroupent l’évaluation de l’admissibilité des donneurs, le prélèvement du sang, les essais sur le sang et la préparation des composants du sang. Un établissement doit également obtenir une licence d’établissement s’il souhaite mener des activités de traitement pour un autre établissement.
Traitement du sang allogénique par des programmes de donneurs pré‑évalués
Les établissements qui traitent du sang allogénique et/ou des composants sanguins allogéniques provenant d’un programme de donneurs pré-évalués n’ont pas besoin d’obtenir une licence d’établissement, mais ils doivent être enregistrés auprès de Santé Canada conformément à l’article 30 du Règlement sur le sang.
Essais
Tout établissement au Canada qui effectue des essais sur du sang et des composants sanguins prélevé à des fins allogéniques, y compris le sang prélevé dans le cadre d’un programme de donneurs pré-évalués, doit détenir une licence d’établissement.
Essais réalisés par des établissements étrangers
Si les essais sont effectués par un établissement étranger pour le compte de l’établissement au Canada, l’établissement étranger doit être inscrit sur la licence de l’établissement au Canada. La directive sur l’article 18 du Règlement sur le sang explique plus en détails les exigences applicables aux établissements étrangers qui exercent des activités pour le compte d’établissements au Canada.
Délivrance d’une homologation avant la licence d’établissement
Une homologation doit être délivrée par la DMBR avant qu’une licence d’établissement ne soit accordée, sauf pour les essais effectués dans le cadre des programmes de donneurs pré-évalués. Voir les articles 5 à 16 pour d’autres renseignements sur les homologations (consultez l’article 20, Délivrance).
Un établissement peut présenter en même temps une demande d’homologation et une demande de licence d’établissement (c.-à-d. que l’établissement n’a pas à attendre d’obtenir une homologation pour présenter une demande de licence d’établissement). Toutefois, le cas échéant, la licence d’établissement ne sera accordée qu’après l’approbation de l’homologation.
Il n’est pas nécessaire de détenir une homologation avant la délivrance d’un enregistrement.
Résumé des exigences (également disponible à l’article 30)
| Type d'établissement / activités | Licence d'établissement | Enregistrement |
|---|---|---|
| Traitement de sang allogénique | Requise | Non requis |
| Importation de sang | Requise | Non requis |
| Programme de donneurs pré-évalués | Non requise | Requis |
| Laboratoire d'essai pour programme de donneurs pré-évalués | Requise | Non requis |
| Programme de dons autologues | Non requise | Requis |
| Transformation du sang | Non requise | Requis |
Article 18 Demande de licence
Demande de licence
18. (1) L’établissement présente au ministre, en la forme établie par celui-ci, une demande de licence d’établissement, datée et signée par un cadre supérieur, qui contient les renseignements et documents suivants :
- ses nom et adresse municipale, ainsi que, si elle est différente, son adresse postale;
- l’adresse municipale de chaque bâtiment où seront conservés ses dossiers;
- tout autre nom sous lequel l’établissement a exercé ses activités antérieurement à la demande;
- le nom de la personne à contacter pour toute question concernant la demande, ses numéros de téléphone et de télécopieur, son adresse de courriel ou toutes autres coordonnées;
- les nom et numéro de téléphone de la personne à contacter en cas d’urgence, si elle est différente de la personne visée à l’alinéa d);
- la liste de ses activités;
- une liste indiquant ce qu’il envisage de manipuler lors de l’exercice de ses activités, à savoir sang total ou tel ou tel composant sanguin;
- l’adresse municipale des bâtiments où il envisage d’exercer ses activités et, pour chacun, la liste des activités visées;
- les nom et adresse municipale ainsi que, s’il y a lieu, le numéro de licence de tout autre établissement auquel il envisage de recourir pour faire effectuer l’une de ses activités;
- toute preuve suffisante démontrant qu’il peut exercer ses activités conformément à son système de gestion de la qualité ainsi qu’aux exigences du présent règlement et que ses activités ne compromettront ni la sécurité humaine ni la sécurité du sang;
- dans le cas d’un importateur ou d’un établissement qui envisage de recourir à un établissement étranger pour faire effectuer des essais, les renseignements visés aux alinéas a) et f) à j) concernant tout établissement étranger qui traite ou distribue le sang qu’il envisage d’importer ou de traiter;
- dans le cas où il envisage d’importer du sang dans des circonstances urgentes, les renseignements prévus au paragraphe 92(1).
Renseignements et documents complémentaires
(2) L’établissement fournit au ministre, au plus tard à la date précisée dans sa demande écrite à cet effet, tout renseignement ou document qu’il juge nécessaire pour compléter l’examen de la demande de licence.
L’article 18 du Règlement sur le sang précise les renseignements qui doivent être présentés dans un formulaire de demande de licence d’établissement. Dans le cadre des dispositions sur les licences d’établissement du Règlement sur le sang, les pouvoirs des licences d’établissement du ministre sont exercés par le Programme de la conformité des produits biologiques (PCPB) de la DGORAL.
Où trouver le formulaire de demande?
Pour obtenir une copie du formulaire de demande de licence d'établissement pour les établissements de sang (FRM-0354), ainsi que les instructions, envoyez une requête à l'adresse suivante: roeb.blood-sang.dgoral@hc-sc.gc.ca
Pour éviter toute confusion, toute erreur ou tout retard dans le traitement, il incombe au demandeur de vérifier que sa demande de licence d’établissement est exacte et qu’elle a été dûment remplie conformément aux exigences de l’article 18 du Règlement sur le sang avant de la soumettre au PCPB de la DGORAL.
Comment envoyer le formulaire de demande?
Par courriel: roeb.blood-sang.dgoral@hc-sc.gc.ca
Bâtiments au Canada
En vertu de l’alinéa 18(1)h), le demandeur doit indiquer l’adresse municipale de chaque bâtiment au Canada dans lequel il se propose exercer des activités ainsi qu’une liste de ces activités concernant le sang et les composants sanguins.
Les établissements qui se limitent à conserver du sang ou des composants sanguins n’ont pas besoin d’obtenir une licence d’établissement. Tous les établissements qui conservent du sang ou des composants sanguins doivent le faire conformément au Règlement sur le sang.
Établissements menant des activités pour le compte du demandeur
Conformément à l’alinéa 18(1)i), le demandeur doit fournir le nom, l’adresse municipale et le numéro de licence (le cas échéant) de tout autre établissement, y compris les établissements étrangers*, qui mènera des activités en son nom.
*Remarque : En vertu du Règlement sur le sang, les établissements étrangers qui mènent des activités pour le compte d’un établissement au Canada n’ont pas besoin de détenir une licence d’établissement. Par contre, l’établissement étranger doit figurer sur la licence d’établissement de l’établissement au Canada pour le compte duquel il mène des activités.
Il incombe au demandeur de vérifier que toutes les ententes écrites pertinentes ont été conclues pour les activités confiées à d’autres établissements.
Preuves démontrant la conformité au Règlement sur le sang
Les demandeurs doivent avoir des preuves suffisantes pour démontrer qu’ils se conforment au Règlement sur le sang. Santé Canada peut mener des inspections lors de l’examen de la demande (consultez l’alinéa 18(1)j) et l’article 19 du Règlement sur le sang).
Preuves requises pour les établissements étrangers
Conformément à l’article 18(1), le demandeur doit fournir des preuves suffisantes pour démontrer que les établissements étrangers répondent aux exigences du Règlement sur le sang. S’ils sont approuvés, ces établissements étrangers seront alors inscrits sur la licence d’établissement de l’établissement au Canada.
Des preuves suffisantes doivent être présentées avec une demande de licence d’établissement de sang (FRM-0354) dûment remplie. Les éléments de preuve suffisants comprennent, sans s’y limiter, les éléments suivants :
- Le rapport d’inspection le plus récent (au cours des 4 dernières années) émis par une autorité réglementaire membre du Pharmaceutical Inspection Cooperation Scheme (PIC/S) (par exemple, le rapport d’inspection d’un établissement de la Food and Drug Administration des États-Unis), ou
- En l’absence d’un rapport d’inspection, une copie des documents suivants :
- Les observations consignées lors de l’inspection (p. ex. le formulaire 483 de la Food and Drug Administration des États-Unis), et
- Les mesures correctives prises à la suite de l’inspection, signées par un responsable de l’établissement étranger.
- En l’absence d’un rapport d’inspection, une copie des documents suivants :
- Une copie de l’accord de qualité le plus récent, signé et daté, établi entre l’établissement canadien et les établissements étrangers. Cet accord doit clairement identifier les parties concernées et leurs responsabilités afin d’assurer la conformité au Règlement sur le sang.
- Une copie la plus récente de l’un des documents suivants :
- Dossier d’établissement signé et daté, ou
- Document équivalent.
Si l’un des documents ci-dessus n’a pas changé depuis la dernière soumission, les établissements peuvent l’indiquer dans la lettre d’accompagnement ou dans le courriel, au lieu de soumettre une autre copie. Les établissements sont priés d’indiquer clairement la date à laquelle ces renseignements ont été soumis précédemment.
De plus, si l’un des documents requis n’est pas disponible, on encourage les établissements à discuter avec Santé Canada de la raison pour laquelle les documents requis ne peuvent être soumis et des autres éléments de preuve qui pourraient être soumis, avant de présenter une demande.
Veuillez noter qu’il incombe au titulaire de la licence d’établissement d’aviser Santé Canada de tout événement qui fait en sorte que l’établissement étranger contrevient à l’une des exigences applicables du Règlement sur le sang, lorsque cela peut avoir une incidence sur la sécurité humaine ou la sécurité du sang et des composants sanguins.
Date limite pour la présentation de nouvelles preuves (Date NERBY)
Pour les établissements étrangers qui se voient attribuer la cote C, une date limite pour la présentation de nouvelles preuves (date NERBY) sera attribuée. La date NERBY est la date avant laquelle de nouvelles preuves doivent être soumises à Santé Canada dans le cadre d’une demande de renouvellement d’un établissement étranger figurant sur une licence d’établissement.
La date NERBY sera déterminée au moyen d’une approche fondée sur le risque. La date NERBY sera généralement de 4 ans, calculée à partir de la date de début de l’inspection à l’étranger. Cependant, Santé Canada peut émettre une date NERBY raccourcie ou prolongée pour un établissement étranger en fonction de plusieurs facteurs, y compris, mais sans s’y limiter, l’historique de conformité de l’établissement étranger et le type d’éléments de preuve présentés.
Lors du renouvellement d’une date NERBY, tant qu’une demande est soumise avec des éléments de preuve complets et mis à jour avant la date NERBY de l’établissement, l’établissement étranger continuera à être considéré comme conforme et à figurer sur la licence d’établissement.
Dans certaines circonstances, quand des preuves mises à jour ne sont pas disponibles pour renouveler la date NERBY d’un établissement étranger (p. ex. il n’y a pas eu d’inspection récente par une autorité réglementaire, ou une inspection par une autorité réglementaire a eu lieu mais il y a un délai dans la délivrance du rapport d’inspection), les établissements devraient contacter le PCPB de la DGORAL au roeb.blood-sang.dgoral@hc-sc.gc.ca avant que la date NERBY soit dépassée afin de discuter l’information qui peut être soumise dans de tels cas.
Évaluation préliminaire et évaluation des preuves d’établissements étrangers
À la réception d’une demande de licence d’établissement accompagnée des éléments de preuve appropriés relatifs à l’établissement étranger, Santé Canada enverra un avis d’acceptation pour informer le titulaire de la licence d’établissement que les éléments de preuve ont fait l’objet d’une évaluation préliminaire et sont acceptés pour une évaluation plus approfondie. Le seul fait de déposer une demande ne garantit pas que la preuve de l’établissement à l’étranger sera considérée comme acceptable et des renseignements supplémentaires peuvent être demandés lors de l’évaluation préliminaire de la demande et/ou la période d’évaluation (p. ex. information sur la portée et l’étendue d’une inspection effectuée par une autorité réglementaire ou les réponses de l’autorité réglementaire aux actions correctives proposées par l’établissement étranger).
Pendant l’évaluation des éléments de preuve, les établissements peuvent continuer à importer de l’établissement étranger ou à faire effectuer des analyses par celui-ci, conformément à la licence d’établissement et au Règlement sur le sang. Si les éléments de preuve sont jugés inacceptables ou incomplets à tout moment du processus d’évaluation, l’établissement étranger peut se voir retirer de la licence d’établissement. Lorsqu’un établissement étranger est retiré de la licence d’établissement, il n’est plus autorisé à importer ou à faire effectuer des essais par l’établissement étranger et le titulaire de la licence d’établissement en est informé. Le titulaire de la licence d’établissement doit soumettre une demande complète à Santé Canada pour ajouter à nouveau l’établissement étranger sur sa licence d’établissement.
Importation dans des circonstances urgentes
Un établissement au Canada qui souhaite importer du sang et/ou des composants sanguins dans des circonstances urgentes doit fournir les documents suivants à Santé Canada, préalablement à l’importation :
- Une demande de licence d’établissement de sang remplie (FRM-0354).
- Une copie valide de la licence ou de l’enregistrement de l’établissement étranger, délivré par l’autorité de réglementation dans le pays étranger (p. ex. le document intitulé « Blood Establishment Registration and Product Listing » de la Food and Drug Administration des États-Unis).
Une fois approuvé, l’établissement étranger sera inscrit à l’annexe de la licence d’établissement (Autre(s) établissements), avec la condition que l’importation est seulement permise, dans des circonstances urgentes, en vertu de l’article 92 du Règlement sur le sang.
Veuillez vous référer aux directives à l’article 92 pour plus de détails sur les renseignements supplémentaires à fournir avant l’importation.
Informations sur demande
En vertu du paragraphe 18(2), Santé Canada peut demander des renseignements supplémentaires, lorsque nécessaire, pour compléter l’examen de la demande d’un établissement. Le demandeur doit fournir les renseignements supplémentaires par écrit avant la date indiquée dans la demande.
Article 19 Inspection
Inspection
19. (1) Le ministre peut, lors de l’examen de la demande de licence d’établissement, effectuer l’inspection des installations et de tout équipement de l’établissement pour déterminer la conformité de ses activités à l’homologation et au présent règlement.Renseignements et documents complémentaires
(2) L’établissement fournit au ministre, au plus tard à la date précisée dans sa demande écrite à cet effet, tout renseignement ou document qu’il juge nécessaires pour compléter son inspection.
L’établissement peut être inspecté avant la délivrance de sa licence et doit donc se préparer à une telle possibilité lorsqu’il présente sa demande. Après examen du formulaire dûment rempli, une inspection sera planifiée au besoin et les résultats de l’inspection seront communiqués à l’établissement.
L’inspection d’un établissement vise à s’assurer que ce dernier se conforme aux exigences du Règlement sur le sang et à vérifier son homologation. Si la cote Conforme est attribuée, une licence d’établissement sera délivrée à l’établissement. Un établissement ne doit pas commencer ses activités avant d’avoir obtenu une licence d’établissement pour le faire.
Article 20 Délivrance
Délivrance
20. Le ministre, au terme de l’examen de la demande de licence, délivre la licence d’établissement, avec ou sans conditions, si les conditions ci-après sont respectées :
- un établissement a déjà obtenu une homologation pour le sang, exception faite de celui prélevé d’un donneur pré-évalué, qu’il envisage de traiter ou d’importer au titre de la licence;
- il juge que la preuve fournie est suffisante pour démontrer que la délivrance de la licence ne compromettra ni la sécurité humaine ni la sécurité du sang.
Après que le PCPB de la DGORAL a traité la demande de l’établissement, une licence d’établissement peut être délivrée si les exigences suivantes sont respectées :
- une homologation a déjà été délivrée à l'égard du sang;
- le ministre juge que la délivrance d’une licence d’établissement ne compromettra pas la sécurité humaine ou la sécurité du sang et des composants sanguins.
Renseignements concernant les établissements titulaires d’une licence
Les informations portant sur les établissements titulaires d’une licence au Canada, incluant les conditions des bâtiments au Canada, sont disponibles publiquement dans la base de données Inspections des médicaments et des produits de santé.
Date d’expiration de la licence d’établissement
Une licence d’établissement délivrée en vertu du Règlement sur le sang n’expirera pas. Toutefois, les établissements titulaires d’une licence feront l’objet d’inspections régulières pour évaluer leur conformité continue au Règlement sur le sang, à la fréquence décrite dans l’Approche d’inspection des établissements de sang (POL-0039).
Article 21 Refus
Refus
21. Le ministre peut, s’il juge que des renseignements fournis par l’établissement dans sa demande de licence sont inexacts ou incomplets, refuser de délivrer la licence.
Il incombe au demandeur de veiller à ce que les renseignements contenus dans la demande qu’il présente au PCPB de la DGORAL de Santé Canada, agissant au nom du ministre, soient complets et exacts. La demande peut être refusée si les renseignements qu’elle contient ne sont pas complets. Le cas échéant, l’établissement en sera avisé par écrit.
Article 22 Changement des renseignements - demande de modification
Changement des renseignements — demande de modification
22. (1) L’établissement présente au ministre, sous réserve de l’alinéa 23b), une demande de modification de la licence avant d’apporter un changement ayant une incidence sur tout renseignement fourni en application des alinéas 18(1)f) à i), k) et l).Demande
(2) Les articles 18 à 21 s’appliquent à la demande de modification de la licence, avec les adaptations nécessaires.
Les changements (y compris les ajouts, les suppressions, les modifications et les corrections) apportés aux renseignements fournis en vertu des alinéas 18(1)f) à i), k) et l) doivent être soumis à du PCPB de la DGORAL au moyen du formulaire de demande de licence d’établissement de sang (FRM-0354) .Le courriel ou la lettre accompagnant la demande devrait inclure l’objet de la demande et toute information supplémentaire pour décrire le changement. Veuillez consulter le formulaire pour d’autres directives. Vous pouvez demander une copie du formulaire en envoyant un courriel à hc.roeb.blood-sang.dgoral.sc@canada.ca.
Si la demande de modification de la licence respecte les exigences du Règlement sur le sang, la licence d’établissement mise à jour sera délivrée pour refléter les modifications.
Remarque : La cessation temporaire des activités n’exige pas de modification ni d’avis.
Article 23 Changement de nature administrative - avis
Changement de nature administrative — avis
23. L’établissement avise par écrit le ministre des changements ci-après dans les délais suivants :
- tout changement aux renseignements fournis en application des alinéas 18(1)a) à e), dès que possible;
- la cessation d’une activité prévue dans sa licence, dans les trente jours qui suivent.
Le formulaire de demande de licence d’établissement de sang (FRM-0354) est utilisé pour aviser le PCPB de la DGORAL des changements administratifs. Veuillez consulter la section 18 de la présente ligne directrice pour obtenir des renseignements sur le dépôt du formulaire de demande.
Article 24 Modification de la licence par le ministre
Modification de la licence par le ministre
24. Le ministre modifie la licence d’établissement dans les cas suivants :
- la modification d’une homologation a une incidence sur tout renseignement fourni par l’établissement en application des alinéas 18(1)f) à k);
- il reçoit de l’établissement l’avis prévu à l’alinéa 23a) selon lequel un renseignement fourni en application de l’alinéa 18(1)a) a été changé;
- il reçoit de l’établissement l’avis prévu à l’alinéa 23b) selon lequel l’établissement a cessé d’exercer certaines des activités faisant l’objet de sa licence;
- l’annulation d’une homologation a une incidence sur tout renseignement fourni par l’établissement en application des alinéas 18(1)f) à k).
Changement apporté à l'homologation avant la licence d'établissement
Si un établissement titulaire d’une licence prévoit ajouter une activité à sa licence d’établissement, il doit d’abord ajouter les activités de traitement et d’importation à son homologation.
Article 25 Ajout de conditions ou modification
Ajout de conditions ou modification
25. (1) Le ministre peut modifier les conditions de la licence d’établissement ou en ajouter de nouvelles dans les cas suivants :
- il a des motifs raisonnables de le croire nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises;
- l’établissement ne lui fournit pas, au plus tard à la date précisée dans sa demande écrite à cet effet, de preuve suffisante démontrant la conformité de ses activités au présent règlement.
Préavis
(2) Le ministre, avant de modifier les conditions de la licence ou d’en ajouter de nouvelles, envoie à l’établissement, au moins quinze jours avant la date de prise d’effet prévue des conditions, un préavis motivé l’informant qu’il peut présenter ses observations à cet égard.Cas d’urgence
(3) Malgré le paragraphe (2), le ministre peut, s’il a des motifs raisonnables de le croire nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises, modifier sans préavis les conditions de la licence ou en ajouter de nouvelles.Cas d’urgence — avis
(4) Le ministre, s’il modifie les conditions de la licence ou en ajoute de nouvelles conformément au paragraphe (3), envoie à l’établissement un avis motivé l’informant qu’il peut présenter ses observations à cet égard.Suppression d’une condition
(5) Le ministre peut, par avis écrit, supprimer toute condition de la licence qu’il ne juge plus nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises.
Description des conditions
L’établissement doit se conformer à toutes les conditions établies par le ministre, le cas échéant. Ces conditions sont décrites dans une des annexes de la licence d’établissement.
Retrait des conditions
Le ministre peut abroger une condition d’une licence d’établissement s’il estime qu’elle n’est plus nécessaire. Le cas échéant, une licence d’établissement modifiée sera délivrée à l’établissement.
Article 26 Renseignements et documents supplémentaires
Renseignements et documents supplémentaires
26. L’établissement fournit au ministre, au plus tard à la date précisée dans sa demande écrite à cet effet, tout renseignement ou document supplémentaire utiles pour démontrer la conformité de ses activités au présent règlement.
Article 27 Suspension
Suspension
27. (1) Le ministre peut suspendre complètement ou partiellement la licence d’établissement dans les cas suivants :
- des renseignements fournis en application des articles 18 ou 22 se révèlent inexacts ou incomplets;
- l’établissement ne lui fournit pas, au plus tard à la date précisée dans sa demande écrite à cet effet, de preuve suffisante démontrant la conformité de ses activités au présent règlement;
- l’établissement ne se conforme pas au présent règlement.
Préavis
(2) Le ministre, avant de suspendre la licence, envoie à l’établissement un préavis qui contient les précisions suivantes :
- les motifs de la suspension envisagée et sa date de prise d’effet;
- l’obligation qu’a l’établissement de prendre des mesures correctives, s’il y a lieu, au plus tard à la date précisée;
- le fait qu’il lui donne la possibilité de présenter ses observations à cet égard.
Cas d’urgence
(3) Malgré le paragraphe (2), le ministre peut, s’il a des motifs raisonnables de le croire nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises, suspendre sans préavis la licence complètement ou partiellement.Cas d’urgence — avis
(4) Le cas échéant, le ministre envoie à l’établissement un avis qui contient les précisions suivantes :
- les motifs de la suspension;
- le fait qu’il lui donne la possibilité de présenter ses observations à cet égard.
L’établissement ne peut pas mener les activités indiquées sur la licence d’établissement que le ministre a suspendues.
Article 28 Rétablissement de la licence
Rétablissement de la licence
28. (1) Sous réserve des paragraphes (2) et (3), le ministre rétablit la licence d’établissement si l’établissement lui fournit une preuve suffisante démontrant sa conformité au présent règlement.Exception — contraventions précédentes
(2) Le ministre peut refuser de rétablir la licence si l’établissement a des antécédents de contravention qui démontrent son incapacité d’exercer ses activités conformément au présent règlement de façon constante.Rétablissement partiel
(3) Le ministre supprime de la licence toute partie suspendue qu’il ne rétablit pas.
Le ministre peut refuser de rétablir en tout ou en partie une licence si l’établissement ne se conforme pas de manière constante au Règlement sur le sang ou à la Loi sur les aliments et drogues. Le ministre ne rétablira pas la licence des établissements qui font preuve de mauvaise foi ou refusent systématiquement d’appliquer des correctifs pour éliminer une infraction ou appliquent des correctifs insuffisants.
Article 29 Annulation
Annulation
29. (1) Le ministre annule la licence d’établissement dans les cas suivants :
- il reçoit l’avis prévu à l’alinéa 23b) selon lequel l’établissement a cessé d’exercer toutes les activités faisant l’objet de sa licence;
- l’établissement ne lui fournit pas la preuve visée à l’alinéa 27(1)b) dans un délai raisonnable suivant la suspension;
- l’établissement a des antécédents de contravention qui démontrent son incapacité d’exercer ses activités conformément au présent règlement de façon constante;
- toutes les homologations en vertu desquelles l’établissement traite le sang sont annulées.
Avis
(2) Le cas échéant, il envoie à l’établissement un avis motivé à cet effet indiquant la date de prise d’effet de l’annulation.
L’établissement n'est pas autorisé à mener des activités assujetties à une licence dans un bâtiment pour lequel il ne détient pas de licence d’établissement, y compris dans ceux dont la licence a été annulée. Si un établissement prévoit mener des activités assujetties à une licence dans un site dont la licence d’établissement a été annulée, il doit remplir une nouvelle demande de licence d’établissement (FRM-0354). Vous pouvez demander une copie du formulaire en envoyant un courriel à roeb.blood-sang.dgoral@hc-sc.gc.ca.
Articles 30-37 Enregistrement
Article 30 Obligation d'enregistrement
Obligation d’enregistrement
30. (1) Tout établissement s’enregistre conformément au présent règlement pour traiter du sang provenant d’un don autologue, procéder à la transformation de sang ou se doter d’un programme de donneurs pré-évalués.
Exceptions
(2) Le paragraphe (1) ne s’applique pas à l’établissement qui ne met à l’essai que du sang provenant d’un don autologue ni à celui dont la seule activité de transformation qu’il exerce est la mise en commun d’unités de cryoprécipité.
Qui doit s'enregistrer?
Le Règlement sur le sang exige que chacun des établissements qui suivent soit enregistré.
A. Établissements qui traitent du sang autologue, sauf ceux qui se limitent à effectuer des essais sur des échantillons de sang autologue
Les établissements qui prélèvent du sang autologue ou préparent des composants de sang autologue sont tenus de s’enregistrer auprès de Santé Canada. Si un autre établissement effectue les essais pour le compte de l’établissement qui prélève du sang autologue, le demandeur qui prélève doit mentionner le nom de cet établissement dans sa demande d’enregistrement. L’établissement qui prélève du sang autologue est responsable des essais, que ceux-ci soient réalisés par lui ou par un autre établissement, pour son compte.
Les établissements qui se limitent à effectuer des essais sur des échantillons de sang prélevés à des fins autologues pour le compte d’un autre établissement qui détient un enregistrement pour le traitement de ce type de sang ne sont pas tenus de s’enregistrer.
Les établissements contractuels sont inscrits en tant qu’autres établissements sur la demande d’enregistrement et le certificat d’enregistrement du titulaire d’enregistrement.
B. Établissements qui sont dotés d’un programme de donneurs pré-évalués
C. Établissements qui transforment du sang
Selon la définition de transformation, l’établissement qui met en commun, irradie ou lave du sang est tenu de s’enregistrer auprès de Santé Canada pour ces activités. La mise en commun comprend le mélange de deux ou plusieurs composants différents et, par conséquent, un établissement qui mélange du sang doit s’enregistrer pour cette activité. Par exemple, le mélange de globules rouges avec du plasma en vue d’une transfusion d’échange est considéré comme une activité de transformation en vertu du Règlement sur le sang.
Les établissements dont la seule activité de transformation consiste à mettre en commun des cryoprécipités ne sont pas tenus de s’enregistrer.
Outre le lavage des globules rouges pour éliminer toute trace de protéines plasmatiques et d’anticoagulant, le lavage des globules rouges décongelées pour éliminer l’agent cryoprotecteur (déglycérolisation) est également inclus dans les activités de transformation. La réduction du plasma, le remplacement par une solution saline et la réduction du volume des plaquettes constituent des manipulations de l’unité qui sont réglementées, mais qui ne sont pas actuellement considérées comme des activités de transformation en vertu du Règlement sur le sang et qui ne nécessitent donc pas d’enregistrement. Veuillez vous référer au tableau 2 pour connaître les articles du Règlement sur le sang qui s’appliquent aux établissements qui ne sont pas tenus d’obtenir une licence ou de s’enregistrer.
Bien que les établissements dont la seule activité de transformation consiste à mettre en commun les cryoprécipités ne soient pas tenus de s’enregistrer, la mise en commun des cryoprécipités demeure une activité de transformation. Des méthodes sécuritaires et efficaces doivent être utilisées pour assurer la sécurité du sang, et les articles pertinents du Règlement sur le sang s’appliquent toujours. Veuillez vous référer au tableau 2 au début de la présente ligne directrice pour savoir quels articles du Règlement sur le sang s’appliquent aux établissements qui ne sont pas tenus d’obtenir une licence ou un enregistrement.
Veuillez vous référer à l’annexe C pour un outil d’autoévaluation préalable à l’enregistrement pour les établissements qui demande un enregistrement.
Résumé des exigences (également disponible à l’article 17)
| Type d'établissement / activités | Licence d'établissement | Enregistrement |
|---|---|---|
| Traitement de sang allogénique | Requise | Non requis |
| Importation de sang | Requise | Non requis |
| Programme de donneurs pré-évalués | Non requise | Requis |
| Laboratoire d'essai pour programme de donneurs pré-évalués | Requise | Non requis |
| Programme de dons autologues | Non requise | Requis |
| Transformation du sang | Non requise | Requis |
Remarque : Le « sang » comprend le sang total et les composants sanguins.
Article 31 Demande d'enregistrement
Demande d’enregistrement
31. (1) L’établissement présente au ministre, en la forme établie par celui-ci, une demande d’enregistrement qui contient les renseignements et documents suivants :
- ses nom et adresse municipale ainsi que, si elle est différente, son adresse postale;
- tout autre nom sous lequel l’établissement a, en vertu du présent règlement, exercé ses activités antérieurement à la demande;
- le nom de la personne à contacter pour toute question concernant la demande, ses numéros de téléphone et de télécopieur, son adresse de courriel ou toutes autres coordonnées;
- les nom et numéro de téléphone de la personne à contacter en cas d’urgence si elle est différente de la personne visée à l’alinéa c);
- une liste indiquant ce qu’il envisage de traiter, à savoir sang total ou tel ou tel composant sanguin, ainsi que la liste des activités de traitement de sang provenant d’un don autologue qu’il envisage d’exercer;
- une liste indiquant ce qu’il envisage de transformer, à savoir sang total ou tel ou tel composant sanguin, ainsi que la liste des activités de transformation qu’il envisage d’exercer;
- la mention selon laquelle il est doté d’un programme de donneurs pré-évalués;
- l’adresse municipale des bâtiments où il envisage d’exercer ses activités et, pour chacun, la liste des activités;
- les nom et adresse municipale de tout autre établissement auquel il envisage de recourir pour faire effectuer l’une de ses activités;
- une attestation, datée et signée par un cadre supérieur, qui certifie les faits suivants :
- l’établissement possède des preuves suffisantes démontrant sa conformité au présent règlement,
- les renseignements et documents fournis dans la demande sont exacts et complets.
Renseignements et documents complémentaires
(2) L’établissement fournit au ministre, au plus tard à la date précisée dans sa demande écrite à cet effet, tout renseignement ou document qu’il juge nécessaires pour compléter l’examen de la demande d’enregistrement.
L’article 31 du Règlement sur le sang précise les renseignements que doivent fournir les établissements de sang qui présentent une demande d’enregistrement (FRM-0353).
Où trouver le formulaire?
Le formulaire de demande d'enregistrement pour les établissements de sang (FRM-0353), incluant des instructions, peut être obtenu en envoyant une demande à roeb.blood-sang.dgoral@hc-sc.gc.ca.
Il incombe au demandeur de vérifier que le formulaire de demande d’enregistrement est exact et qu’il a été dûment rempli conformément aux exigences de l’article 31 du Règlement sur le sang avant de le soumettre au PCPB de la DGORAL. Cela permettra d’éviter les retards dans le traitement.
Comment envoyer le formulaire de demande?
Par courriel : roeb.blood-sang.dgoral@hc-sc.gc.ca
Article 32 Enregistrement
Enregistrement
32. (1) Le ministre, au terme de l’examen de la demande d’enregistrement, enregistre l’établissement et lui attribue un numéro d’enregistrement s’il juge que les renseignements sont complets.Refus
(2) Le ministre peut, s’il juge que des renseignements fournis par l’établissement dans sa demande sont incomplets ou s’il a des motifs raisonnables de croire que l’enregistrement pourrait compromettre la sécurité humaine ou la sécurité du sang, refuser d’enregistrer l’établissement.
Inspection
Santé Canada peut inspecter un établissement avant et/ou après l’attribution d’un numéro d’enregistrement.
Renseignements concernant les établissements enregistrés
Les informations portant sur les établissements enregistrés au Canada, sont disponibles publiquement dans la base de données Inspections des médicaments et des produits de santé.
Expiration de l’enregistrement
Sous réserve de l’article 37 il n’y a pas d’expiration pour un enregistrement; toutefois, une attestation annuelle de conformité est requise conformément à l’article 35. De plus, un certificat d’enregistrement mis à jour sera délivré si des modifications sont apportées à l’enregistrement correspondant.
Veuillez vous référer à l’article 35 pour plus de renseignements sur la façon de soumettre l’attestation annuelle de conformité.
Article 33 Avis de modification
Avis de modification
33. L’établissement avise le ministre par écrit de tout changement aux renseignements fournis en application de l’article 31 dans les trente jours suivant le changement et fournit à nouveau, si des renseignements visés aux alinéas 31(1)e) à i) sont changés, l’attestation visée à l’alinéa 31(1)j).
Les avis et les modifications doivent être envoyés au PCPB de la DGORAL. L’établissement devrait utiliser le formulaire de demande d’enregistrement d’un établissement de sang (FRM-0353).
Le courriel ou la lettre accompagnant le formulaire devrait inclure l’objet de la demande et toute information supplémentaire pour décrire le changement. Veuillez consulter le formulaire pour de plus amples instructions. Pour obtenir le formulaire FRM-0353, veuillez envoyer une demande à roeb.blood-sang.dgoral@hc-sc.gc.ca.
Il est recommandé que l’établissement présente dès que possible au PCPB de la DGORAL tous les changements qu’il souhaite apporter afin que ceux-ci soient traités et figurent rapidement sur les certificats d’enregistrement.
Article 34 Modification par le ministre
Modification par le ministre
34. Le ministre peut, s’il a des motifs raisonnables de le croire nécessaire pour éviter que la sécurité humaine ou la sécurité du sang ne soient compromises, modifier l’enregistrement de l’établissement pour y supprimer la mention d’une activité ou d’un bâtiment.
Activités supprimées
L’établissement n'est pas autorisé à mener des activités nécessitant un enregistrement qui ont été supprimées de l’enregistrement ou qui n’y figurent pas. L’établissement sera avisé par écrit du retrait de l’enregistrement de certaines activités et recevra un certificat d’enregistrement mis à jour.
Bâtiments supprimés
L’établissement ne peut pas mener des activités nécessitant un enregistrement dans un bâtiment qui a été retiré de l’enregistrement ou qui n’y figure pas.
Ajout d’activités ou de bâtiments précédemment retirés
Si un établissement enregistré souhaite ajouter des activités ou des bâtiments précédemment retirés de son enregistrement, il doit envoyer une demande à Santé Canada (conformément à l’article 31 du Règlement sur le sang). L’établissement peut être inspecté ou devoir fournir des documents aux fins de vérification de la conformité des activités et des bâtiments visés par la demande.
Article 35 Attestation annuelle de conformité
L'article 35 s'applique uniquement aux établissements qui sont tenus de s'inscrire.
Attestation annuelle de conformité
35. L’établissement fournit au ministre, au plus tard le 1er avril de chaque année, une attestation, datée et signée par un cadre supérieur, qui certifie que l’établissement possède une preuve suffisante démontrant sa conformité au présent règlement.
L’article 35 s’applique uniquement aux établissements qui sont tenus de s’enregistrer. Bien que le numéro d’enregistrement ne comporte pas de date d’échéance, l’établissement doit renouveler son attestation annuelle de conformité chaque année avant le 1er avril s’il souhaite que son numéro d’enregistrement demeure valide.
Un établissement enregistré peut renouveler son attestation annuelle de conformité en utilisant la demande d’enregistrement d’un établissement de sang (FRM-0353). Des instructions supplémentaires sont fournies avec le formulaire. Pour obtenir le formulaire FRM-0353, veuillez envoyer une demande à roeb.blood-sang.dgoral@hc-sc.gc.ca. Les établissements ne recevront pas de nouveau certificat d’enregistrement chaque année à moins qu’ils n’avisent également Santé Canada d’un changement nécessitant une modification des renseignements figurant sur le certificat.
Si l’établissement ne renouvelle pas son attestation annuelle de conformité, le ministre peut avoir des raisons de croire, que l’établissement n’est pas conforme au Règlement sur le sang et décider d’annuler l’enregistrement, comme il est indiqué à l’article 37.
Article 36 Renseignements et documents supplémentaires
Renseignements et documents supplémentaires
36. L’établissement fournit au ministre, au plus tard à la date précisée dans sa demande écrite à cet effet, tout renseignement ou document supplémentaire utiles pour démontrer la conformité de ses activités au présent règlement.
Article 37 Annulation
Annulation
37. (1) Le ministre peut annuler l’enregistrement de l’établissement dans les cas suivants :
- il reçoit l’avis prévu à l’article 33 indiquant que l’établissement a cessé d’exercer toutes les activités faisant l’objet de son enregistrement;
- des renseignements fournis en application de l’article 31 se sont révélés faux ou trompeurs;
- l’établissement n’a pas fourni les renseignements ou documents demandés au titre de l’article 36;
- l’établissement n’a pas pris les mesures correctives dans le délai fixé;
- le ministre a des motifs raisonnables de croire que l’établissement ne se conforme pas au présent règlement ou que la sécurité humaine ou la sécurité du sang pourraient être compromises.
Préavis
(2) Le ministre, avant d’annuler l’enregistrement, envoie à l’établissement un préavis qui contient les précisions suivantes :
- les motifs de la suspension envisagée et sa date de prise d’effet;
- l’obligation qu’a l’établissement de prendre des mesures correctives, s’il y a lieu, au plus tard à la date précisée;
- le fait qu’il lui donne la possibilité de présenter ses observations à cet égard.
Cas d’urgence
(3) Malgré le paragraphe (2), le ministre peut, s’il a des motifs raisonnables de croire que la sécurité humaine ou la sécurité du sang risquent d’être compromises, annuler l’enregistrement sans préavis.Cas d’urgence — avis
(4) Le cas échéant, le ministre envoie à l’établissement un avis qui contient les précisions suivantes :
- les motifs de la suspension envisagée et sa date de prise d’effet;
- l’obligation qu’a l’établissement de prendre des mesures correctives, s’il y a lieu, au plus tard à la date précisée;
- le fait qu’il lui donne la possibilité de présenter ses observations à cet égard.
Mesures consécutives à l’annulation
(5) L’établissement dont l’enregistrement est annulé pour l’une des raisons précisées aux alinéas (1)b) à e) avise aussitôt tout établissement auquel il a distribué du sang qu’il a traité ou transformé pendant la période précisée dans son avis, de l’annulation et de sa date de prise d’effet.
L’établissement n'est pas autorisé à mener des activités nécessitant un enregistrement dans un bâtiment qui n’est pas enregistré ou dont l’enregistrement a été annulé. Si l’établissement souhaite mener des activités nécessitant un enregistrement, il doit remplir une nouvelle demande d’enregistrement d’un établissement de sang (FRM-0353). FRM-0353 peut être obtenu en envoyant une demande à roeb.blood-sang.dgoral@hc-sc.gc.ca.
Articles 38-58 Traitement
Articles 38-44 Évaluation de l'admissibilité des donneurs
Article 38 Non-application - dons autologues
Non-application - dons autologues
38. Les articles 39 à 44 ne s'appliquent pas aux dons autologues.
L’évaluation de l’admissibilité d’un donneur faisant un don autologue n’est pas, à ce titre, assujettie au Règlement sur le sang.
Article 39 Établissement titulaire d'une licence
Établissement titulaire d’une licence
39. L’établissement titulaire d’une licence évalue, préalablement au prélèvement du sang d’un donneur faisant un don allogénique, l’admissibilité du donneur au regard des critères approuvés dans son homologation.
Critères approuvés pour l’admissibilité du donneur
L’article 39 ne s’applique qu’aux établissements titulaires d’une licence qui prélèvent du sang allogénique destiné à la transfusion ou à une fabrication ultérieure.
L’établissement enregistré qui prélève du sang d’un donneur pré-évalué n’est pas tenu d’avoir des critères approuvés, de sorte que l’article 39 ne s’applique pas. Veuillez vous référer au paragraphe 5(2) au sujet de l’exception prévue pour les programmes de donneurs pré-évalués.
Un médecin ou un substitut devrait évaluer l’admissibilité des donneurs à effectuer des dons de plasma par aphérèse. Veuillez vous référer au paragraphe 1.5 « Définitions » du présent document pour obtenir la définition de « médecin » et de « substitut » afin d’interpréter la présente directive.
Critères applicables à l’évaluation de l’admissibilité du donneur
Une évaluation de l’admissibilité du donneur comprend une évaluation préliminaire du donneur et des critères d’exclusion du donneur. Une interdiction temporaire ou permanente au don est appliquée à tout donneur jugé inadmissible à donner du sang. Veuillez vous référer à l’article 1 « Définitions » pour obtenir des directives sur la définition de « évaluation de l’admissibilité du donneur ».
Obligation de se doter de procédures opérationnelles
Les critères d’évaluation de l’admissibilité du donneur homologués par Santé Canada protègent la sécurité humaine et la sécurité du sang et des composants sanguins. L’établissement doit établir et tenir à jour des procédures opérationnelles décrivant en détail les critères et les méthodes d’évaluation de l’admissibilité du donneur. Les procédures opérationnelle de l’établissement doivent également préciser la fréquence des dons et les délais d’exclusion du donneur. Veuillez vous référer à l’article 95 qui porte sur les procédures opérationnelles.
Obligation de soumettre les changements majeurs touchant les critères d’évaluation de l’admissibilité du donneur
Un établissement doit déposer une demande auprès de la DMBR afin de modifier son homologation avant d’apporter des changements importants à ses critères d’évaluation préliminaire des donneurs. Veuillez vous référer à l’article 9 pour plus de renseignements sur les changements majeurs aux processus homologués.
Obligation de présenter les critères d’évaluation d’admissibilité de l’établissement étranger d’où proviendra le sang importé
L’établissement titulaire d’une licence qui prévoit importer du sang ou des composants sanguins doit présenter dans sa demande d’homologation, ou de modification de son homologation, les critères d’évaluation de l’admissibilité des donneurs en vigueur dans l’établissement étranger. Cela doit être fait avant l'importation par l'établissement du sang ou des composants sanguins étranger.
Article 40 Inadmissibilité antérieure
Inadmissibilité antérieure
40. L’établissement vérifie lors de l’évaluation de l’admissibilité du donneur si ce dernier a déjà été déclaré inadmissible et, le cas échéant, les raisons ainsi que la durée de l’inadmissibilité.
L’article 40 s’applique aux établissements titulaires d’une licence qui prélèvent du sang allogénique ainsi qu’aux établissements enregistrés qui ont un programme de donneurs pré-évalués.
Accessibilité aux dossiers d’exclusion de donneurs
Un établissement titulaire d’une licence ou un établissement enregistré doit avoir un système pour conserver et consulter les dossiers d’exclusion de donneurs. L’établissement devrait avoir un système permettant de vérifier les dossiers d’exclusion de donneurs d’autres établissements titulaires d’une licence au Canada.
Examen de l’admissibilité du donneur par la consultation des dossiers d’exclusion de donneurs
Lorsqu’il évalue l’admissibilité d’un donneur, un établissement titulaire d’une licence ou un établissement enregistré doit :
- confirmer l’identité du donneur;
- utiliser l’identité du donneur pour effectuer une vérification dans son système d’exclusion de donneurs; et
- consigner la raison et la durée de l’exclusion, s’il y a lieu.
Comme il est indiqué à l’article 88(1), une évaluation régulière de l’admissibilité du donneur, y compris l'inadmissibilité antérieure, doit être effectuée tous les 3 mois pour les donneurs pré-évalués.
Obligation de conserver les dossiers d’inadmissibilité de donneurs
Veuillez vous référer aux points 5 et 6 du « Tableau de l’article 119, Renseignements et documents – période de rétention » pour obtenir les exigences relatives à la conservation de dossiers d’inadmissibilité du donneur.
Article 41 Évaluation préliminaire
Évaluation préliminaire
41. L’établissement qui évalue de façon préliminaire l’admissibilité du donneur prend les mesures suivantes :
- il obtient du donneur, à l’aide d’un questionnaire ou tout autre moyen similaire, des renseignements sur son identité, ses antécédents médicaux ainsi que ses antécédents sociaux dans la mesure où ces derniers peuvent être utiles pour déterminer la présence d’un risque de maladies transmissibles par le sang;
- il fournit au donneur des renseignements sur les risques associés au don de sang et sur le risque que le receveur contracte une maladie transmissible.
41 a) et b) Les alinéas 41 a) et b) s’appliquent aux établissements titulaires d’une licence qui prélèvent du sang allogénique ainsi qu’aux établissements enregistrés qui ont un programme de donneurs pré-évalués.
Dispositions générales relatives à l’évaluation préliminaire du donneur faisant un don allogénique
41 a) Le jour du don, un établissement titulaire d’une licence ou un établissement enregistré doit évaluer l’admissibilité du donneur conformément à l’ensemble des exigences de l’article 41. L’établissement doit effectuer une évaluation préliminaire du donneur dans un endroit qui permet de protéger la vie privée. L’établissement doit permettre au donneur de poser des questions et de s’auto-exclure.
Un établissement titulaire d’une licence doit déterminer l’admissibilité d’un donneur en s’appuyant sur les critères de Santé Canada suivants :
- fréquence des dons;
- critères d’exclusion des donneurs (consultez l’article 39);
- résultats d’analyses de laboratoire (consultez l’article 56);
- antécédents médicaux et/ou examen physique du donneur; et
- antécédents sociaux du donneur.
Évaluation préliminaire d’un donneur pré-évalué
Les établissements enregistrés qui ont un programme de donneurs pré-évalués devraient avoir un processus d’évaluation des donneurs qui tient compte des critères homologués par Santé Canada et qui sont indiqués sur le questionnaire d’évaluation préliminaire du donneur d’un établissement titulaire d’une licence. Un donneur pré- évalué devrait satisfaire aux mêmes exigences d’admissibilité qu’un donneur dont le sang allogénique est destiné à l’approvisionnement en sang général. Veuillez vous référer à l’article 42 de la présente ligne directrice au sujet des critères d’exclusion.
Antécédents médicaux et sociaux
Les antécédents médicaux du donneur se rapportent 1) aux conditions pouvant présenter un risque pour celui-ci et 2) aux vaccins, aux médicaments et aux maladies transmissibles pouvant présenter un risque pour le receveur.
Les antécédents sociaux du donneur se rapportent aux activités antérieures du donneur qui pourraient l’exposer ou exposer le receveur à un risque de maladie infectieuse transmissible.
Veuillez vous référer à l’article 1 « Définitions » pour obtenir des directives générales sur les antécédents médicaux et les antécédents sociaux dans le contexte d’une « évaluation de l’admissibilité du donneur ».
Obligation de fournir des renseignements sur les risques
41 b) Un établissement enregistré ou titulaire d’une licence doit aviser le donneur de ce qui suit :
- tout risque possible pour la santé du donneur découlant du don de sang;
- tout risque possible pour le receveur de contracter une maladie transmissible; et
- tout autre renseignement requis pour que le donneur prenne une décision éclairée quant au don de sang.
Les établissements doivent avoir la documentation, papier ou en version électronique, qui fait part de tous les risques en langage clair et qu’un donneur peut facilement comprendre. Un donneur doit avoir l’occasion de changer d’idée à tout moment.
Obligation de tenir des dossiers d’évaluation de l’admissibilité du donneur
Veuillez vous référer au point 4 du « Tableau de l’article 119, Renseignements et documents – période de rétention » pour obtenir les exigences relatives à la conservation de dossiers d’évaluation de l’admissibilité du donneur.
Article 42 Critères d'exclusion
Critères d’exclusion
42. L’établissement conclut que le donneur est inadmissible si tout renseignement obtenu au titre des articles 39 à 41 révèle que la sécurité humaine ou la sécurité du sang pourraient être compromises
L’article 42 s’applique aux établissements titulaires d’une licence qui prélèvent du sang allogénique ainsi qu’aux établissements enregistrés qui ont un programme de donneurs pré-évalués.
Le processus d’évaluation de l’admissibilité du donneur d’un établissement titulaire d’une licence ou d’un établissement enregistré doit permettre de déterminer et de gérer les conditions et les facteurs qui pourraient compromettre la sécurité humaine ou la sécurité du sang et des composants sanguins.
Obligation d’exclure un donneur — établissement titulaire d’une licence
Un établissement titulaire d’une licence doit exclure un donneur s’il ne satisfait pas aux critères d’évaluation de l’admissibilité du donneur autorisés. Par ailleurs, il doit y avoir exclusion du donneur en cas de raisons médicales pouvant compromettre la sécurité humaine ou la sécurité du sang et des composants sanguins. L’exclusion temporaire ou permanente du donneur dépend des critères auxquels il ne satisfait pas.
Obligation d’exclure un donneur — établissement enregistré ayant un programme de donneurs pré-évalués
Un établissement enregistré ayant un programme de donneurs pré-évalués doit déterminer l’admissibilité du donneur à faire un don en se fondant sur les critères les plus récents applicables aux donneurs de sang allogénique. Il est recommandé aux responsables du programme de donneurs pré-évalués de communiquer avec l'établissement titulaire d'une licence de leur juridiction pour obtenir les critères applicables et le questionnaire d'évaluation préliminaire du donneur. La période d’exclusion temporaire ou indéfinie du donneur dépend des critères auxquels il n’a pas satisfait,conformément aux périodes d’exclusion de l’établissement titulaire d’une licence dans la juridiction du programme.
Article 43 Inadmissibilité
Inadmissibilité
43. L’établissement qui conclut que le donneur est inadmissible ne prélève pas de son sang et l’informe des raisons de son inadmissibilité ainsi que, le cas échéant, de la date prévue à laquelle il sera considéré de nouveau admissible.
L’article 43 s’applique aux établissements titulaires d’une licence qui prélèvent du sang allogénique ainsi qu’aux établissements enregistrés qui ont un programme de donneurs pré-évalués.
Obligation d’informer le donneur des raisons de son exclusion
L’établissement titulaire d’une licence ou l’établissement enregistré doit informer le donneur des raisons pour lesquelles il ne doit pas donner de sang pendant la période d’exclusion. Lorsqu’il informe un donneur de l’exclusion, l’établissement titulaire d’une licence ou l’établissement enregistré doit s’assurer que le donneur comprend bien la date, le cas échéant, à laquelle il pourra être admissible de nouveau au don de sang.
Obligation d’exclure un donneur inadmissible dans le cadre d’un programme de donneurs pré-évalués
Comme il est indiqué à l’article 88, les donneurs pré-évalués doivent se soumettre à une évaluation régulière de l’admissibilité des donneurs tous les trois (3) mois. Un établissement enregistré peut déterminer si un donneur pré-évalué est inadmissible à donner du sang soit pendant l'évaluation régulière ou immédiatement avant le prélèvement. Un établissement enregistré doit exclure, de façon permanente ou temporaire, tout donneur pré- évalué devenu inadmissible. Pendant la période d’exclusion, l’établissement enregistré ne doit pas prélever de sang du donneur concerné. Veuillez vous référer aux exigences relatives à l’Obligation d’exclure un donneur — établissement enregistré ayant un programme de donneurs pré-évalués, à l’article 42.
Obligation de conserver des dossiers relativement à l’inadmissibilité d’un donneur
Veuillez vous référer aux points 5 et 6 du « Tableau de l’article 119, Renseignements et documents – période de rétention » pour ce qui est des exigences relatives à la période de rétention de dossiers relatifs à l’inadmissibilité du donneur, également appelés dossiers d’exclusion de donneurs.
Article 44 Admissibilité
Admissibilité
44. (1) L’établissement qui conclut que le donneur est admissible prend les mesures suivantes :
- il lui attribue un code d’identification du donneur, si ce n’est déjà fait;
- il lui mentionne qu’il a l’obligation de l’informer s’il se trouve dans l’une des situations suivantes :
- il contracte au cours de la période prévue dans les procédures opérationnelles de l’établissement une maladie ou une affection susceptible de compromettre la sécurité de son don de sang,
- il a, ultérieurement à son don, des raisons de croire que le sang provenant de ce dernier ne devrait plus être utilisé.
Nouvelle évaluation
(2) L’établissement, dès qu’il reçoit des renseignements après le don au titre de l’alinéa (1)b), les prend en considération afin d’évaluer de nouveau la sécurité de ce don et de tout autre don provenant de ce donneur ainsi que l’admissibilité de ce dernier à d’autres dons.Avis
(3) L’établissement, si la nouvelle évaluation démontre que la sécurité du sang a pu être compromise, en avise toute personne à laquelle il a déjà distribué ce sang et, s’agissant d’un établissement, il mentionne également dans l’avis qu’il ne doit pas le distribuer ou le transfuser.
44 Les paragraphes 44(1) et (2) s’appliquent aux établissements titulaires d’une licence qui prélèvent du sang allogénique ainsi qu’aux établissements enregistrés qui ont un programme de donneurs pré-évalués.
Obligation pour l’établissement d’attribuer un code d’identification du donneur
44(1)a) L’établissement doit attribuer un code d’identification au donneur si celui-ci est admissible et qu’il n’en possède pas déjà un. Les établissements enregistrés devraient se reporter à l’alinéa 89b) pour connaître les directives sur le code d’identification du donneur pour les donneurs pré-évalués.
Renseignements après le don — établissement titulaire d’une licence
44(1)b) Un établissement titulaire d’une licence doit mentionner aux donneurs dans quels cas ils doivent lui fournir certains renseignements après le don. Il peut s’agir de renseignements permettant de croire que la sécurité du sang provenant des dons est compromise, notamment dans les cas suivants :
- le donneur contracte une maladie ou une affection ou obtient un diagnostic en ce sens;
- le donneur se souvient de renseignements ou d’antécédents qu’il pourrait avoir omis de divulguer pendant le processus d’évaluation;
- le donneur estime, pour quelque raison que ce soit, que l'établissement ne devrait pas utiliser son sang.
44(2) Si l’établissement titulaire d’une licence reçoit des renseignements après le don, il doit en tenir compte pour déterminer si le sang est sécuritaire à être transfusé ou utilisé dans la fabrication d’un médicament à usage humain. Ces renseignements doivent servir à établir la sécurité du don actuel ainsi que celle des dons antérieurs.
En outre, dès réception des renseignements après le don de sang, un établissement titulaire d’une licence doit réévaluer l’aptitude future du donneur à donner du sang pour une transfusion ou pour une transformation ultérieure.
Renseignements après le don — établissement enregistré ayant un programme de donneurs pré-évalués
44(1)b)i) Tout établissement enregistré devrait se conformer à l’article 5.1.7 de la norme sur le sang de la CSA lorsqu’il informe un donneur pré-évalué sur la déclaration de renseignements après le don relativement à une maladie ou à une affection qui pourrait affecter la sécurité du sang provenant du don.
44(1)b)ii) Le sang prélevé d’un donneur pré-évalué est utilisé immédiatement pendant une situation d’urgence. Toutefois, un établissement enregistré doit tout de même demander au donneur pré-évalué d’informer l’établissement, après le prélèvement du don, s’il a des raisons de croire que son sang ne devrait pas avoir servi à des transfusions.
44(2) En ce qui concerne les renseignements après le don, un établissement enregistré ayant un programme de donneurs pré-évalués devrait se conformer aux articles 19.1.2 à 19.1.6 de la norme sur le sang de la CSA.
Étude des dons antérieurs pour l’établissement titulaire d’une licence ou enregistré
Un établissement enregistré ou titulaire d’une licence doit étudier les dons antérieurs d’un donneur allogénique dont le sang ou les composants du sang ont été confirmés infectés par tout agent infectieux parmi les suivants :
- VIH 1 et 2
- VHC
- VHB
- HTLV I/II
- VNO
Les établissements titulaires d’une licence devraient se référer à l’article 52 et au paragraphe 56(1) de cette ligne directrice pour des précisions sur les exigences des essais.
Un rapport de renseignements après le don a des répercussions sur l’admissibilité du don actuel, et doit également être tenu en compte pour les dons antérieurs, selon le type de renseignements rapportés. Les renseignements obtenus après le don déclenchent une étude des dons antérieurs lorsque les résultats à l’essai d’amplification des acides nucléiques sont positifs ou que les tests sérologiques sont réactifs et confirmés positifs pour la présence d’une maladie transmissible ou d’un agent indiqué ci-dessus.
Le directeur médical devrait être consulté au besoin lors de l’étude des dons antérieurs.
Veuillez vous référer à l’article 1.5 « Définitions » « étude des dons antérieurs » Veuillez vous référer également au paragraphe 56(1) pour en savoir davantage sur les procédures d’étude des dons antérieurs.
Obligation en cas de résultats d’une nouvelle évaluation indiquant que la sécurité du sang a pu être compromise
44(3) Lorsqu’un établissement titulaire d’une licence détermine, à la suite d’une nouvelle évaluation en fonction des renseignements reçus après le don, que la sécurité du sang et/ou des composants sanguins a pu être compromise, il doit en aviser tout établissement et toute personne (y compris les fabricants de produits sanguins) à qui il a distribué le sang ou des composants sanguins.
Lorsqu’un établissement titulaire d’une licence envoie un avis à un établissement à qui il a distribué du sang ou des composants sanguins aux fins de transfusion, il doit indiquer dans l’avis que le sang ne doit plus être distribué ni transfusé.
Articles 45-51 Prélèvement
Article 45 Établissement titulaire d'une licence
Établissement titulaire d’une licence
45. L’établissement titulaire d’une licence qui prélève du sang d’un donneur faisant un don allogénique le fait conformément à son homologation.
Obligation d’effectuer le prélèvement du sang allogénique conformément à l’homologation
Un établissement titulaire d’une licence qui prélève du sang allogénique doit le faire conformément à son homologation.
Obligation de soumettre les changements importants apportés aux processus de prélèvement
L’établissement doit soumettre à l’homologation de Santé Canada les changements majeurs qu’il prévoit apporter à ses processus de prélèvement. Veuillez vous référer à l’article 9 qui concerne les changements importants apportés à un processus homologué.
Article 46 Code d'identification du donneur
Code d’identification du donneur
46. L’établissement qui prélève du sang d’un donneur faisant un don autologue lui attribue un code d’identification du donneur.
Article 47 Code d'identification du don
Code d’identification du don
47. L’établissement qui prélève du sang attribue à chaque unité de sang prélevé un code d’identification du don qu’il associe, dans ses dossiers, au code d’identification du donneur.
L’article 47 s’applique aux établissements titulaires d’une licence qui prélèvent du sang allogénique et aux établissements enregistrés qui prélèvent du sang autologue.
Obligation d’attribuer un code au don au moment où il est effectué
Au moment du prélèvement, il faut apposer un code d’identification du don sur chaque unité. Les procédures de tenue de dossiers doivent permettre d’établir un lien entre le code d’identification du don et le code d’identification du donneur. À des fins de traçabilité, un établissement doit être en mesure d’identifier le donneur qui a fait un don en particulier et tous les autres dons de ce donneur. Le code d’identification du don doit permettre d’établir un lien entre le donneur, les échantillons de sang applicables, l’unité de sang, l’heure ou la date du prélèvement et le dossier d’admissibilité du donneur. Les termes « code d’identification du don » et « code d’identification du donneur » sont définis dans l’article 1 « Définition » du Règlement sur le sang.
Établissement enregistré — Programme de donneurs pré-évalués
Un établissement enregistré ayant un programme de donneurs pré-évalués devrait se reporter à l’alinéa 89b).
Article 48 Étiquetage des contenants
Étiquetage des contenants
48. Sous réserve de l’article 59, l’établissement qui prélève du sang veille, lors du prélèvement, à apposer les étiquettes sur chaque contenant conformément à l’article 63.
Étiquetage des contenants conforme à l’article 63
L’établissement qui prélève du sang, sauf s’il s’agit du sang d’un donneur pré-évalué, doit étiqueter chaque contenant conformément à l’article 63. L’établissement enregistré ayant un programme de donneurs pré-évalués devrait consulter l’article 90 qui définit les exigences particulières en matière d’étiquetage.
Article 49 Procédures de prélèvement
Procédures de prélèvement
49. (1) L’établissement qui prélève du sang le fait conformément aux exigences suivantes :
- il recourt à des méthodes aseptiques;
- l’équipement qu’il utilise pour le prélèvement est homologué conformément au Règlement sur les instruments médicaux;
- les contenants qu’il utilise pour le prélèvement sont homologués conformément au Règlement sur les instruments médicaux et sont exempts de défaut ou de détérioration;
- il consigne dans ses dossiers le numéro du lot du contenant et l’associe au numéro d’identification du don.
Interdiction de réutiliser un contenant
(2) L’établissement s’assure que les contenants qu’il utilise n’ont jamais été utilisés auparavant.
Procédures de prélèvement du sang
49 Les établissements titulaires d’une licence qui prélèvent du sang allogénique et les établissements enregistrés qui prélèvent du sang autologue ou du sang de donneurs pré-évalués doivent se conformer à toutes les exigences énoncées à l’article 49. Un établissement enregistré ayant un programme de donneurs pré-évalués doit également se conformer aux exigences énoncées à l’article 89 concernant les prélèvements de sang de donneurs pré-évalués.
49(1)b) Un établissement titulaire d’une licence ou d’un enregistrement doit effectuer les prélèvements au moyen d’équipement homologué par Santé Canada conformément au Règlement sur les instruments médicaux.
Utilisation d’un appareil d’aphérèse automatisé pour le prélèvement du sang – don autologue
En plus des exigences relatives à l’utilisation d’équipement de prélèvement homologué, un établissement enregistré devrait se plier aux exigences ci-dessous s’il utilise un appareil d’aphérèse automatisé pour prélever du sang autologue.
- Afin d’assurer la sécurité humaine et la sécurité du sang et des composants sanguins, un établissement enregistré devrait respecter les protocoles et les procédures de prélèvement spécifiques à l’appareil d’aphérèse. Pour chaque type de composant sanguin ou de combinaison, un établissement enregistré devrait préciser dans ses procédures opérationnelles toutes les exigences et tous les critères permettant d’atteindre les buts susmentionnés.
- L’établissement devrait s’appuyer sur des données cliniques et scientifiques et les connaissances scientifiques les plus récentes pour établir les exigences et les critères en vue de réaliser les buts susmentionnés.
Numéro de lot du contenant
49(1)d) Après avoir inscrit le numéro de lot du contenant, un établissement titulaire d’une licence ou un établissement enregistré peut apposer l’étiquette de composant sanguin par-dessus le code à barres du numéro de lot mais ce, sans obstruer le numéro de lot, lequel devrait rester visible sur l’étiquette de base. L’établissement doit avoir un système en place pour retracer chaque don à l’aide du numéro de lot associé au contenant.
Interdiction de réutiliser un contenant
49(2) Un établissement titulaire d’une licence ou un établissement enregistré n’utilise un contenant qu’une seule fois pour prélever du sang. Le contenant doit être stérile et ne présenter aucun signe remettant en question sa stérilité.
Article 50 Échantillons
Échantillons
50. L’établissement qui prélève du sang prélève en même temps des échantillons de sang pour des essais et ce, de manière à éviter toute contamination croisée.
Obligation pour l’établissement qui prélève du sang de prélever également des échantillons
L’article 50 s’applique aux établissements titulaires d’une licence qui prélèvent du sang allogénique et aux établissements enregistrés qui prélèvent du sang autologue ou du sang de donneurs pré-évalués.
Autre obligation que doit respecter l’établissement ayant un programme de donneurs pré-évalués
L’établissement enregistré qui prélève du sang doit respecter l’article 50, ainsi que le paragraphe 89c) après avoir prélevé un échantillon de sang d’un donneur pré-évalué.
Article 51 Don autologue
Don autologue
51. L’établissement qui prélève du sang d’un donneur faisant un don autologue satisfait aux exigences suivantes :
- il respecte les exigences prévues à l’article 12.2.1 de la norme;
- il détermine le volume de sang à prélever et celui de l’anticoagulant à y mélanger en fonction du poids du donneur, s’il y lieu.
Don autologue — volume de sang et volume d’anticoagulant
51 b) Lorsqu’il évalue le volume de sang autologue à prélever et le volume d’anticoagulant qui doit y être mélangé, un établissement enregistré devrait se reporter aux articles 6.2.4 et 12.1.4 de la norme sur le sang de la CSA.
Articles 52-56 Essais
Article 52 Homologation
Homologation
52. L’établissement titulaire d’une licence qui met à l’essai du sang provenant d’un don allogénique, exception faite de celui provenant d’un donneur pré-évalué, le fait conformément à une homologation.
Obligation de mettre à l’essai le sang provenant d’un don allogénique, conformément à une homologation
L’établissement qui détient une homologation assume la responsabilité des essais et doit présenter une demande d’homologation ou de modification d’homologation à Santé Canada pour cette activité.
Exception — Mise à l’essai du sang provenant d’un donneur pré-évalué faisant un don allogénique
Le paragraphe 17(2) prévoit que la mise à l’essai du sang et/ou des composants sanguins d’un donneur pré-évalué faisant un don allogénique doit être effectuée par un établissement titulaire d’une licence. L’établissement titulaire d’une licence qui met à l’essai du sang et/ou des composants sanguins d’un donneur pré-évalué doit le faire conformément aux articles 55 b), 88 et 89.
Services d’essais confiés en sous-traitance à un autre établissement
L’établissement qui détient une homologation peut confier en sous-traitance, à un établissement au Canada ou étranger titulaire d’une licence, certaines activités, comme des tests essais pour les marqueurs viraux. Santé Canada n’exige pas que l’établissement sous-traitant présente une demande d’homologation, tant et aussi longtemps qu’il ne mène pas d’autres activités de traitement de sang allogénique. Santé Canada exige toutefois que l’établissement au Canada sous-traitant lui présente une demande de licence d’établissement (consultez l’article 17). Santé Canada n’exige pas que l'établissement étranger qui effectue des essais détienne une licence d’établissement au Canada. Veuillez vous référer aux articles 17 et 18 de la présente ligne directrice pour plus d'information concernant les essais délégués à des établissements étrangers.
Si un établissement confie les mises à l’essai en sous-traitance, ces mises à l’essai doivent être réalisées conformément à l’homologation de l’établissement qui les confie en sous-traitance. Veuillez vous référer au paragraphe 5(1), aux alinéas 6(1)h), 6(1)j) et 6(1)k).
Essais que Santé Canada juge appropriés et efficaces – sang provenant d’un don allogénique
Pour tous les donneurs de sang allogénique, des essais appropriés et efficaces pour les maladies transmissibles ou de leurs agents doivent être réalisés sur l’échantillon prélevé à chaque don, essais qui doivent obtenir un résultat négatif ou non réactif. Veuillez vous référer aux articles 88 et 89 pour obtenir d’autres renseignements sur la mise à l’essai des échantillons de sang prélevés chez des donneurs pré-évalués.
- elle est homologuée pour la détection d’agents ou de marqueurs de maladies transmissibles, conformément aux exigences en matière d’homologation énoncées par la Loi sur les aliments et drogues et le Règlement sur les instruments médicaux;
- l’établissement utilise une trousse d’essai :
- conformément aux instructions du fabricant de la trousse;
- conformément à son homologation pour la détection d’un agent ou d’un marqueur de maladie transmissible;
- qui est équivalente ou supérieure quant à la spécificité et à la sensibilité requises.
L’homologation de l’établissement mentionne les agents ou les marqueurs de maladie devant faire l’objet d’un essai pour les maladies transmissibles effectué sur du sang et des composants sanguins provenant d’un don allogénique destiné à la transfusion ou à une fabrication ultérieure.
Mise à l’essai d’échantillons pour chaque don allogénique de sang destiné à une fabrication ultérieure
Pour chaque don allogénique de sang destiné à une fabrication ultérieure, Santé Canada exige que l’établissement soumette des échantillons aux essais pour les agents ou marqueurs de maladies transmissibles, notamment :
- anticorps contre le virus d’immunodéficience humaine, type 1 et type 2 (anti-VIH 1 et anti-VIH 2);
- antigène de surface de l’hépatite B (AgHBs);
- anticorps contre le virus de l’hépatite C (anti-VHC); et
- essai d’amplification des acides nucléiques (TAN) pour le VIH 1, le VHC et le VHB.
Il n’est pas requis d’effectuer un essai pour la syphilis sur des échantillons provenant d’un don de sang allogénique ou de plasma destiné au fractionnement du plasma. Cependant, s’il est identifié dans l’homologation de l’établissement, l’établissement titulaire d’une licence doit effectuer un essai pour la syphilis sur un échantillon au moyen d’un essai non tréponémique ou tréponémique selon la fréquence précisée dans son homologation.
Instrument de diagnostic in vitro non disponible
Si aucun instrument de diagnostic in vitro (pouvant comprendre la plateforme d’essai et la trousse d’essai) homologué au Canada n’est disponible pour l’essai pour un agent infectieux ou marqueur d’une maladie donnée, l’établissement titulaire d’une licence peut :
- utiliser un instrument de diagnostic in vitro qui a reçu une autorisation d’accès spécial ou d’essai expérimental par la Direction des instruments médicaux de la Direction générale des produits de santé et des aliments de Santé Canada, ou
- demander une modification à son homologation pour l’utilisation d’une trousse d’essai interne.
Pour un instrument de diagnostic in vitro ayant reçu une autorisation d’accès spécial ou d'essai expérimental par la Direction des instruments médicaux, l’établissement titulaire d’une licence doit suivre les instructions du fabricant de l’instrument de diagnostic in vitro, notamment en ce qui a trait :
- au prélèvement, à la manipulation et à l’entreposage des échantillons de sang;
- au délai à l’intérieur duquel les échantillons doivent être analysés, s’il y a lieu;
- à la procédure d’analyse;
- à l’interprétation des résultats, y compris l’interprétation de résultats réactifs de façon répétée ou positifs.
L’établissement titulaire d’une licence qui demande une modification à son homologation afin d’utiliser l’instrument de diagnostic interne doit inclure les instructions requises et détaillées dans la liste ci-dessus, en plus de l’ensemble complet des données de validation appuyant l’utilisation de l’instrument de diagnostic dans leurs activités de traitement.
Essais effectués par un laboratoire à l’extérieur du Canada
Si les essais sont effectués à l’extérieur du Canada, les renseignements suivants doivent être fournis à Santé Canada conformément à l’homologation :
- détails sur les essais pour les agents ou marqueurs de maladies transmissibles et les essais sérologiques qui serviront à analyser le sang;
- algorithmes à utiliser pour chaque marqueur, en cas de résultat réactif au test initial;
- liste de toutes les trousses d’essais actuellement utilisées à l’installation ;
- certification que la trousse est approuvée par la Food and Drug Administration des États-Unis, ou la Direction des instruments médicaux de Santé Canada, et la confirmation que les essais sont effectués conformément aux instructions du fabricant de la trousse d’essai ou encore, l’approbation d’utiliser la trousse doit être obtenue auprès de la Direction des médicaments biologiques et radiopharmaceutiques (DMBR) de Santé Canada;
- preuve de l'accréditation du laboratoire de l’établissement effectuant l’essai (c.-à-d. certificats d'accréditation valides indiquant que le laboratoire est accrédité pour les activités exécutées).
Analyse bactériologique des plaquettes
Un établissement titulaire d’une licence qui prélève ou prépare des plaquettes doit avoir une méthode de détection des bactéries dans les plaquettes, méthode qui doit être homologuée par Santé Canada en vertu du Règlement sur le sang. Il n’est pas nécessaire que les analyses bactériologiques soient terminées avant la distribution des composants sanguins aux fins de transfusion. Par ailleurs, une technologie de réduction des agents pathogènes dont l’utilisation est approuvée au Canada peut être utilisée lorsqu’un établissement titulaire d’une licence dispose d’une homologation à cet égard.
Le système de gestion de la qualité d’un établissement titulaire d’une licence doit disposer de protocoles pour la gestion des unités de plaquettes et des composants connexes pour lesquels il y a un résultat positif. Veuillez vous référer à l’article 94 pour ce qui est des exigences relatives à un système de gestion de la qualité.
Essais de contrôle de la qualité
Veuillez vous référer à l’alinéa 94(1)b) de la présente ligne directrice au sujet des essais de contrôle de la qualité.
Article 53 Don autologue - dépistage de maladies transmissibles
Don autologue — dépistage de maladies transmissibles
53. L’établissement qui prélève du sang d’un donneur faisant un don autologue effectue sur un échantillon sanguin des essais appropriés et efficaces à des fins de dépistage de maladies transmissibles ou de leurs agents mentionnés à l’article 12.3.1.2 de la norme.
Fréquence des essais pour les maladies transmissibles — dons autologues
Si l’établissement enregistré prélève chez un donneur plus d’un don de sang autologue sur une période de 42 jours, seul le premier prélèvement doit être soumis à un essai pour les agents de maladies transmissibles énumérés à l’article 12.3.1.2 de la norme sur le sang de la CSA. Lorsqu’une nouvelle période de 42 jours débute, l’établissement doit effectuer des essais sur le premier don de sang autologue de cette nouvelle période.
Essais appropriés et efficaces pour les maladies et agents infectieux
Santé Canada considère que les essais pour les marqueurs de maladies infectieuses suivants sont appropriés et efficaces et respectent la clause 12.3.1.2 de la Norme :
- anticorps contre le virus d’immunodéficience humaine, type 1 et type 2 (anti-VIH 1 et anti-VIH 2);
- antigène de surface de l’hépatite B (AgHBs);
- anticorps contre le virus de l’hépatite C (anti-VHC);
- anticorps contre le virus T-lymphotrope humain type 1 et type 2 (anti-HTLV-I et anti-HTLV-II).
Il n’est pas nécessaire d’effectuer des essais d’amplification des acides nucléiques ou pour la syphilis sur les prélèvements autologues.
Article 54 Don autologue - groupe ABO et facteur Rh
Don autologue — groupe ABO et facteur Rh
54. (1) L’établissement effectue, lors de chaque prélèvement de sang provenant d’un don autologue, des essais aux fins ci-après sur un échantillon sanguin :
- détermination du groupe sanguin ABO;
- détermination du facteur Rh ainsi que, s’il y a lieu, dépistage de l’antigène D faible.
Comparaison
(2) L’établissement compare les résultats des essais prévus aux alinéas (1)a) et b) aux derniers résultats consignés au dossier du donneur, le cas échéant.Incohérence
(3) L’établissement, dans le cas où la comparaison révèle une incohérence, recommence les essais et ne peut transfuser le sang tant que l’incohérence n’est pas résolue.
Article 55 Instruments médicaux
Instruments médicaux
55. L’établissement qui met à l’essai du sang provenant d’un don autologue ou d’un don fait par un donneur pré-évalué utilise des instruments médicaux homologués conformément au Règlement sur les instruments médicaux aux fins suivantes :
- dans le cas du sang provenant d’un don autologue, pour l’évaluation préliminaire de donneurs ou à des fins diagnostiques;
- dans le cas du sang provenant d’un don fait par un donneur pré-évalué, pour l’évaluation préliminaire de donneurs.
Dispositions relatives aux trousses d’essais – sang provenant d’un don autologue
55 a) L’alinéa 55 a) s’applique aux établissements enregistrés qui effectuent les essais du sang autologue.
Les dons de sang autologues doivent être mis à l’essai au moyen de trousses d’essais homologuées de Santé Canada pour les essais de diagnostic ou les essais de dépistage. L’utilisation de trousses d’essais non homologuées — y compris les trousses internes — est interdite.
L’établissement enregistré doit suivre les instructions du fabricant de la trousse d’essai, notamment en ce qui a trait :
- au prélèvement, à la manipulation et à l’entreposage des échantillons de sang;
- au délai à l’intérieur duquel les échantillons doivent être mis à l’essai, s’il y a lieu;
- à la procédure de mise à l’essai;
- à l’interprétation des résultats des essais.
Les procédures opérationnelles des établissements enregistrés en matière d’essais pour les maladies transmissibles doivent être conformes aux instructions des fabricants. Si les essais sont effectués par un laboratoire sous-traitant ou un autre établissement, l’établissement enregistré doit veiller à ce que les procédures opérationnelles du laboratoire qui effectue les essais soient conformes aux instructions du fabricant de la trousse d’essai. Veuillez vous référer à l’article 95 de la présente ligne directrice qui fournit des directives au sujet des procédures opérationnelles.
Dispositions relatives aux trousses d’essais – sang provenant d’un don allogénique d’un donneur pré-évalué
55 b) L’établissement titulaire d’une licence doit mettre à l’essai le sang et les composants sanguins provenant d’un don allogénique d’un donneur pré-évalué afin de détecter des agents ou des marqueurs de maladies transmissibles au moyen de trousses d’essais homologuées par Santé Canada pour le dépistage. Un établissement ne doit pas utiliser de trousses d’essai homologuées à des fins de diagnostic pour détecter des maladies transmissibles ou leurs agents dans du sang allogénique ou des composants sanguins allogéniques. Santé Canada considère que les trousses d’essais homologuées pour le dépistage sont plus appropriées pour le dépistage des maladies chez les donneurs de sang allogénique.
Remarque : Les trousses pour les essais de dépistage sont homologuées en fonction d’essais menés dans une population où la prévalence de maladies est faible (p. ex. donneurs de sang en santé), l’accent étant mis sur la sensibilité de l’essai. À l’opposé, les trousses de diagnostic sont homologuées en fonction d’essais menés dans une population symptomatique, l’accent étant mis sur la spécificité de l’essai.
Article 56 Résultats des essais
Résultats des essais — don allogénique
56. (1) L’établissement qui prélève du sang d’un donneur faisant un don allogénique prend sans délai les mesures ci-après, dans le cas où l’échantillon sanguin de tout don qu’il met à l’essai se révèle réactif de façon répétée ou positif pour tout agent de maladies transmissibles ou marqueur virologique à l’égard duquel son homologation mentionne qu’il est contre-indiqué de l’utiliser :
- il met en quarantaine toute unité de sang prélevée du donneur concerné lors de ce don;
- il identifie et met en quarantaine toute autre unité de sang en cause qu’il a en sa possession et qui provient du donneur concerné;
- il avise des résultats des essais toute personne à laquelle il a distribué du sang en cause et, s’agissant d’un établissement, il mentionne également dans l’avis qu’il ne doit pas le distribuer ou le transfuser.
Résultats des essais — don autologue
(2) L’établissement qui prélève du sang d’un donneur faisant un don autologue avise le médecin du donneur concerné de tout résultat des essais visé à l’article 12.3.1.6 de la norme.
Résultats de tests indiquant qu’il est contre-indiqué d’utiliser le sang provenant d’un don allogénique
56(1) L’établissement titulaire d’une licence ne doit pas distribuer de sang ou de plasma allogénique destiné à la transfusion ou à une fabrication ultérieure si ce sang ou ce plasma se révèle positif ou réactif de façon répétée aux essais pour les agents ou marqueurs de maladies transmissibles requis par l’homologation de cet établissement. Les résultats aux essais pour les agents ou marqueurs de maladies transmissibles doivent toujours être négatifs, car la présence d’agents ou de marqueurs constitue une contre-indication à l’utilisation du sang et/ou des composants sanguins.
Dans le cas d’un résultat positif ou réactif de façon répétée à un essai pour un agent ou marqueur infectieux qui, dans l’homologation de l’établissement, figure parmi les contre-indications d’utilisation, l’établissement doit aviser dès que possible tous les établissements et personnes (p. ex. le fabricant de produits sanguins) auxquels il a distribué tout sang ou des composants sanguins concernés venant du même donneur.
Exception — Mise à l’essai pour le cytomégalovirus
Un établissement titulaire d’une licence qui prélève du sang allogénique aux fins de transfusion peut décider d’effectuer des essais pour le cytomégalovirus (CMV) pour certains donneurs. Dans le cas d’un donneur qui a déjà obtenu un résultat négatif à un essai pour le cytomégalovirus, si l’établissement prévoit étiqueter et distribuer le sang et/ou les composants sanguins comme produit CMV négatif, Santé Canada recommande que l’établissement effectue un essai pour le cytomégalovirus à chaque don. Veuillez vous référer à l’article 8.6.5.3 de la norme sur le sang de la CSA. Si une unité de sang et/ou de composants sanguins est positif pour le CMV, aucun traitement ni aucun étiquetage particulier n’est requis. Un établissement peut distribuer du sang et des composants sanguins CMV positif.
Interprétation de résultats d’essais pour les maladies transmissibles — dons à des fins allogéniques
- Pour qu’un donneur soit admissible, tous les résultats des essais doivent être négatifs (à l’exception du CMV).
- Si l’échantillon se révèle initialement réactif, l’établissement doit répéter l’essai conformément aux instructions de l’encart de la trousse d’essai.
- Si les résultats ultérieurs des essais pour les agents de maladies transmissibles ou marqueurs virologiques énumérés dans l’homologation de l’établissement indiquent que l’échantillon se révèle réactif de façon répétée ou positif, l’établissement ne doit pas mettre en circulation le sang et les composants sanguins destinés à la transfusion ou à une fabrication ultérieure.
- Si l’échantillon du sang prélevé chez le donneur se révèle positif ou réactif de façon répétée d’un essai pour un agent de maladies transmissibles ou marqueur virologique, le donneur doit être exclu, conformément aux critères énumérés dans l’homologation de l’établissement. S’il s’agit du sang d’un donneur pré-évalué, veuillez vous référer à l’article 42 Obligation d’exclure un donneur — établissement enregistré ayant un programme de donneurs pré-évalués.
- Si un échantillon d’une unité de sang allogénique ou d’un composant sanguin allogénique se révèle réactif de façon répétée ou positif à un essai pour un agent de maladies transmissibles ou marqueur virologique, l’établissement doit informer les autres établissements auxquels il a distribué le sang ou des composants sanguins du donneur en cause.
- L’établissement titulaire d’une licence doit inclure l’interprétation des résultats des essais pour les maladies transmissibles, conformément aux instructions du fabricant de la trousse d’essai, lorsqu’il détermine si le sang et les composants sanguins destinés à la distribution sont sécuritaires.
Critères de réintégration de donneurs
Un algorithme de réintégration de donneurs précise les processus, y compris les essais et la période d’attente, qu’un établissement titulaire d’une licence doit respecter pour qu’un donneur qui a déjà été exclu puisse être pris en compte pour réintégrer la liste des donneurs admissibles. Les algorithmes de réintégration des donneurs doivent être homologués par Santé Canada dans le cadre du processus d’homologation de l’établissement.
L’établissement titulaire d’une licence qui prévoit utiliser des algorithmes de réintégration des donneurs à la suite d’essais pour les agents de maladies transmissibles ou marqueurs virologiques énumérés dans son homologation doit présenter une demande de modification d’homologation fournissant, avec preuves scientifiques et justification à l’appui, les algorithmes qui seront utilisés pour chaque marqueur, y compris les essais de confirmation.
Étude des dons antérieurs
Un établissement titulaire d’une licence doit effectuer l’étude des dons antérieurs tel qu’exigé par son homologation et peut choisir de mener une étude des dons antérieurs pour d’autres agents infectieux non mentionnés dans l’homologation. Un établissement titulaire d’une licence qui prélève du sang doit entamer une étude des dons antérieurs lorsqu’il reçoit l’un des résultats suivants après des mises à l’essai de sang de donneurs, le cas échéant :
- résultat positif de l’essai d’amplification des acides nucléiques (VIH-1, VIH-2, VHC, VHB, VNO);
- confirmation des résultats d’essais positifs de VIH-1, du VIH-2, du VHC, de l’AgHB ou du HTLV après des essais sérologiques réactifs répétés;
- s’il reçoit un avis d’une des sources ci-dessous indiquant que les résultats d’essais du donneur sont positifs :
- médecin
- établissement, comme un hôpital, un établissement titulaire d’une licence ou d'un enregistrement
- enquête sur les composants transfusés, ou donneur
- si une demande d’étude des dons antérieurs (retraçage des receveurs) est présentée.
Remarque : L’établissement qui mène l’étude des dons antérieurs devrait obtenir un rapport contenant tous les résultats d’essais s’il reçoit l’information d’une source externe.
Veuillez vous référer à la section 1.5 « Définitions » « étude des dons antérieurs ».
Résultats des tests – sang de don autologue
56(2) Un établissement enregistré doit informer le médecin d’un donneur de sang autologue de tout résultat anormal à un essai pour les maladies et agents de maladie indiqués à l’article 12.3.1.2 de la norme sur le sang de la CSA. Veuillez également vous référer à l’article 53.
Articles 57-58 Préparation de composants sanguins
Article 57 Établissement titulaire d'une licence
Établissement titulaire d’une licence
57. L’établissement titulaire d’une licence qui prépare des composants sanguins provenant d’un don allogénique le fait conformément à son homologation.
Article 58 Établissement enregistré
Établissement enregistré
58. L’établissement enregistré qui prépare des composants sanguins provenant d’un don autologue le fait selon les articles 7.1.3, 7.2, 7.3.1, 7.3.2, 7.5.1.1, abstraction faite du renvoi au tableau 3, ainsi que 7.5.1.2, 7.5.1.5, les alinéas 7.5.2.1a) à c) et l’article 7.5.2.2 de la norme.
Articles 59-68 Étiquetage
Article 59 Non-application - donneurs pré-évalués
Non-application — donneurs pré-évalués
59. Les articles 60 à 68 ne s’appliquent pas à l’étiquetage de contenants de sang prélevé d’un donneur pré-évalué.
Veuillez vous référer à l’article 90 pour ce qui est des exigences relatives au sang prélevé d’un donneur pré-évalué.
Article 60 Exigences linguistiques
Exigences linguistiques
60. Les renseignements qui doivent, en application du présent règlement, figurer sur les étiquettes ou les documents d’information doivent être en français ou en anglais.
Article 61 Exigences générales
Exigences générales
61. Les étiquettes satisfont aux exigences suivantes :
- les renseignements qui y figurent sont exacts et sont présentés de façon claire et lisible;
- elles sont faites de manière à ce qu’aucun adhésif ni aucune encre ne puisse traverser le contenant de sang;
- elles sont apposées sur le contenant de façon permanente;
- s’agissant d’étiquettes volantes, elles sont solidement fixées au contenant.
61 L’étiquette d’une unité de sang et de composants sanguins doit fournir des renseignements précis sur le contenu du contenant. Consultez la disposition 8.6.3.3 de la norme sur le sang de la CSA, dans les cas où une étiquette est masquée, altérée ou retirée.
61d) Une étiquette volante supplémentaire fixée à un contenant par un établissement est aussi considérée comme une étiquette. Dans la même veine, les renseignements figurant sur l’étiquette doivent être précis, clairs et lisibles.
Article 62 Document d'information
Document d’information
62. (1) L’établissement qui prélève, d’un donneur faisant un don allogénique, du sang destiné à la transfusion élabore, conformément à son homologation, un document d’information et veille à le rendre accessible à tout établissement auquel il distribue ce sang et à toute autre personne qui en fait la demande.Exception
(2) Le paragraphe (1) ne s’applique pas à l’établissement qui non seulement prélève du sang mais le transfuse aussi.
62(1) Consultez les alinéas 6(1)h), 6(1)i) et 6(1)k) pour obtenir les exigences en matière d’homologation qui ont trait à l’étiquetage, y compris le document d’information.
Un établissement titulaire d’une licence qui prélève du sang allogénique destiné à la transfusion doit préparer un document d’information, en conformité avec l’homologation, et doit veiller à ce que le document soit accessible à tous les établissements à qui le sang et les composants sanguins sont remis et à quiconque en demande une copie.
62(2) Par exemple, dans le cas d’un établissement qui prélève des globules rouges irradiés destinés à l’immunisation de donneurs de plasma dans le même établissement, il n’est pas nécessaire de préparer un document d’information.
Un établissement enregistré prélevant du sang autologue n’est pas tenu de préparer un document d’information. L’expression « Document d’information » est définie à l’article 1, soit l’article des définitions, du Règlement sur le sang.
Article 63 Code d'identification du don
Code d’identification du don
63. L’établissement qui prélève du sang appose, lors du prélèvement, une étiquette sur chaque contenant de sang où figure de façon indélébile le code d’identification du don.
Le contenant doit posséder une étiquette accompagnée du code d’identification du don au moment du prélèvement. Si le code d’identification du don manque ou est illisible, l’établissement ne doit pas distribuer le sang et/ou les composants sanguins à des fins de transfusion ou à des fins de fabrication ultérieure. Consultez l’alinéa 74(2)a).
Article 64 Texte de l'étiquette
Texte de l’étiquette — sang destiné à la transfusion
64. (1) L’établissement qui prélève du sang destiné à la transfusion indique sur l’étiquette du contenant de sang les renseignements suivants :
- ses nom et adresse municipale;
- le numéro de sa licence ou, à défaut, celui de son enregistrement;
- le code d’identification du don;
- une mention indiquant que le contenu est du sang total ou un composant sanguin et le cas échéant, le nom du composant;
- le groupe sanguin ABO et le facteur Rh, s’il y a lieu;
- le volume approximatif de sang prélevé, sauf dans les cas d’aphérèse;
- le volume approximatif du contenu du contenant;
- le nom de l’anticoagulant ou de l’additif présent dans le contenant;
- la température de conservation recommandée;
- la date et, s’il y a lieu, l’heure de péremption;
- dans le cas de sang destiné à la transfusion, la mise en garde selon laquelle ce sang peut transmettre des agents infectieux;
- dans le cas de sang qui provient d’un don allogénique et qui est destiné à la transfusion, la mention de consulter les documents d’information pour les indications, les contre-indications, les mises en garde et les réactions indésirables possibles, s’il y a lieu.
Don autologue
(2) L’établissement, dans le cas de sang provenant d’un don autologue, ajoute aux renseignements figurant sur l’étiquette prévue au paragraphe (1) les renseignements suivants :
- la mention « Pour utilisation à des fins autologues seulement »;
- dans le cas où les résultats des essais démontrent que les échantillons sanguins mis à l’essai à des fins de dépistage des maladies transmissibles et agents mentionnés à l’article 12.3.1.2 de la norme sont positifs, une mention selon laquelle le sang présente un risque biologique ou un symbole à cet effet;
- dans le cas où l’établissement n’a pas effectué les essais pour les maladies et agents mentionnés à l’article 12.3.1.2 de la norme, une mention à cet effet.
Texte de l’étiquette — sang destiné à la fabrication de drogues pour usage humain
(3) L’établissement indique sur l’étiquette du contenant de sang destiné à la fabrication de drogues pour usage humain les renseignements suivants :
- les nom, adresse municipale et numéro de licence de l’établissement qui a prélevé le sang;
- le code d’identification du don;
- la mention « Précaution : À utiliser uniquement pour la fabrication ».
Lorsque l’établissement étiquette du sang allogénique et des composants sanguins allogéniques destinés à la transfusion, il doit respecter les exigences des alinéas 64(1)a) à l). L’établissement peut aussi indiquer sur l’étiquette si le sang s’est révélé négatif pour le cytomégalovirus.
Un établissement titulaire d’une licence attache l’étiquette finale au contenant à la fin du processus et avant la transformation ou la distribution.
64(1)a) L’adresse municipale indiquée sur l’étiquette peut être celle du siège social d’une organisation.
64(1)b) Le sang et les composants sanguins allogéniques destinés à la transfusion, traités conformément à une homologation, doivent avoir le numéro de licence de l’établissement traitant sur son étiquette. Dans le cas d’un établissement possédant plusieurs sites de prélèvement ou de production, le numéro de la licence d’établissement peut consister en un seul numéro attribué par Santé Canada à l’établissement et à ses sites.
Certains établissements peuvent avoir un numéro de licence d’établissement et un numéro d’enregistrement. Ces établissements peuvent apposer soit leur numéro de licence d’établissement soit leur numéro d’enregistrement sur l’étiquette du sang autologue et des composants sanguins autologues. Un établissement qui prélève du sang destiné à la transfusion et qui n’a pas de numéro de licence d’établissement doit s’assurer que son numéro d’enregistrement figure sur l’étiquette du sang autologue qu’il prélève.
64(1)d) L’étiquette doit avoir le nom du sang ou du composant sanguin dans un texte lisible à l’œil nu. Le nom du composant sanguin comprend, le cas échéant, la méthode de préparation du composant par exemple, (p. ex. Plasma frais congelé ACD prélevé par aphérèse). La règle d’affectation des noms dans la norme ISBT 128 est recommandée.
Un établissement enregistré doit aussi indiquer sur l’étiquette si un composant sanguin a été transformé. La transformation désigne le lavage, la mise en commun (y compris la mise en commun de cryoprécipité) et l’irradiation de composants sanguins après qu’ils ont été jugés sécuritaires à des fins de transfusion. Elle ne comprend pas la préparation de composants sanguins ou les techniques de réduction des agents pathogènes qui eux relèvent de la préparation de composants sanguins. Consultez les paragraphes 78(2) et 79(2) et l’article 80 pour obtenir les exigences précises concernant l’étiquetage du sang transformé.
64(1)g) À moins d’avis contraire sur l’étiquette ou dans les suppléments au document d’information (ou bulletins d’information), le contenu ou le volume respecte la description faite dans le document d’information ou dans tout autre document accompagnant le sang ou les composants sanguins. Une notice d’accompagnement est un exemple d’information accompagnant le sang ou les composants sanguins.
Remarque : Le supplément au document d’information ou à la notice d’accompagnement est un exemple de document intermédiaire qui peut être joint aux poches de sang.
Le document d’information peut être une extension de l’étiquette du composant sanguin, fournissant des renseignements concernant la composition du composant, l’emballage, la conservation et la manipulation, les indications, les avertissements et les précautions, les effets indésirables, la dose et l’administration, ainsi que d’autres renseignements importants sur le composant sanguin. Il existe des documents d’information distincts pour chacun des groupes de composants sanguins, à savoir les globules rouges, les plaquettes, et le plasma. Ils sont conformes aux règlements applicables de Santé Canada.
64(1)h) L’étiquette doit indiquer le nom de l’additif ou de l’anticoagulant utilisé. Cette exigence comprend tout anticoagulant ou autre additif utilisé dans la préparation du sang ou des composants sanguins. L’étiquette doit aussi comprendre tout agent de sédimentation utilisé durant la cytaphérèse, s’il y a lieu.
64(1)i) L’étiquette doit indiquer la température d’entreposage recommandée. Cette exigence comprend la plage de température permettant l’entreposage du sang et des composants sanguins.
64(1)j) L’étiquette doit comprendre la date de péremption. Si l’heure de péremption n’est pas indiquée, l’unité de sang sera périmée à 23 h 59 à la date de péremption. Les étiquettes des produits dont la durée de conservation est de 72 heures ou moins doivent indiquer l'heure de péremption.
Pour la plupart des composants sanguins, l’établissement titulaire d’une licence ou d’un enregistrement peut décider d’ajouter la date de prélèvement sur l’étiquette.
64(2) Pour étiqueter du sang autologue ou des composants sanguins autologues pour une transfusion, l’établissement doit satisfaire aux exigences des alinéas 64(1)a)–k) en plus de ceux de 64(2). Le donneur de sang autologue/nom du patient peut également apparaître sur l’étiquette de l’unité autologue.
Un code supplémentaire lisible par un lecteur d’étiquettes code à barres doit être ajouté, si possible et pour ce qui suit:
- nom d’établissement qui administre la collection du sang
- code de don
- sang entier ou le nom du composant sanguin
- groupe sanguin ABO et le facteur Rh
Le tableau 3 résume l’information requise concernant l’étiquetage que doivent vérifier les établissements enregistrés qui étiquettent les unités de sang ou de composants sanguins autologues. Les astérisques indiquent quand un code supplémentaire lisible par un lecteur d’étiquettes code à barres doit être ajouté, si possible.
Le nom du donneur de sang autologue ou du patient peut aussi être indiqué sur l’étiquette de l’unité de sang autologue.
| Point | Information nécessaire | Code à barres |
|---|---|---|
| 1. | Nom de l'établissement qui prélève du sang | * |
| 2. | Adresse municipale de l'établissement qui prélève du sang | s/o |
| 3. | Numéro d'enregistrement ou de licence de l'établissement de l'établissement qui prélève du sang | s/o |
| 4. | Code d'identification du don | * |
| 5. | Sang total ou nom du composant sanguin | * |
| 6. | Groupe sanguin ABO et facteur Rh | * |
| 7. | Volume de sang total prélevé, sauf dans les cas d'aphérèse | s/o |
| 8. | Volume approximatif du contenu du contenant | s/o |
| 9. | Température de conservation recommandée | s/o |
| 10. | Date de péremption | s/o |
| 11. | « Ce produit peut transmettre des agents infectieux. » Optionnel : Voir le document d'information pour obtenir les indications, les contre-indications, les précautions et les méthodes de perfusion. |
s/o |
| 12. | Texte ou étiquette indiquant l'existence d'un biorisque, si le donneur obtient un test indiquant qu'il a été infecté par un agent de maladie transmissible pour lequel un test de dépistage est requis. | s/o |
| 13. | « Pour utilisation à des fins autologues seulement » | s/o |
| 14. | S'il s'agit d'une unité de sang subséquente dans un délai de 42 jours, l'énoncé selon lequel aucun essai n'a été effectué concernant le VIH, le VHB, le VHC, le HTLV-1 et le HTLV-2 doit être présent, le cas échéant. | s/o |
| 15. | Nom de l'établissement envisagé pour la transfusion, s'il est connu. | s/o |
| Remarque : Le « sang » comprend le sang total et les composants sanguins. | ||
64(2)b) Si un donneur de sang autologue obtient un test indiquant qu’il est atteint d’une maladie transmissible ou qu’il est porteur d’un agent pathogène, l’étiquette de l’unité de sang autologue et de l’unité de composant sanguin autologue doit être accompagnée d’un symbole de risque biologique ou de mots indiquant que le sang et des composants sanguins sont un biorisque.
64(3) L’étiquette de sang destiné à la fabrication d’un médicament à usage humain devrait clairement indiquer le nom du composant.
Article 65 Aliquotes
Aliquotes
65. L’établissement qui prépare des aliquotes de sang à des fins de transfusion mais non d’immunisation indique sur l’étiquette des contenants des aliquotes les renseignements suivants :
- le code d’identification du don;
- le nom du composant sanguin;
- le code qui permet d’identifier l’aliquote;
- le groupe sanguin ABO et le facteur Rh, s’il y a lieu;
- la date de péremption.
65 L’aliquotage est une activité réglementée, mais pas une activité de transformation. Par conséquent, les établissements qui exercent cette activité doivent disposer d’une procédure opérationnelle pour la réaliser.
Aux fins de transfusion, les contenants suivants sont jugés acceptables pour la division du sang en aliquotes :
- poche de transfert ou une série de poches de transfert utilisant la technologie en système ouvert ou fermé;
- flacons stériles; ou
- seringues.
65 e) Les établissements doivent avoir une procédure opérationnelle en place (comme l’exigent les articles 95 et 96 du Règlement sur le sang) qui définit les dates de péremption appropriées pour les aliquotes. Si un établissement divise le sang en aliquotes en système ouvert, des techniques aseptiques doivent être utilisées ; elles peuvent inclure, sans s’y limiter, les éléments suivants :
- Zone de faible circulation avec des courants d’air limités, loin de tout aérosol susceptible de contaminer le sang (p. ex. éviers, centrifugeuses, entrées, etc.), si possible
- Régime de nettoyage défini, y compris la désinfection (avant et après utilisation)
- Utilisation de fournitures stériles, notamment de gants stériles
- Entreposage des fournitures (p. ex. les poches de transfert) de manière à éviter leur contamination
De plus, l’établissement doit ajuster la date de péremption de l’aliquote et de la poche parent. L’article 7.2.1 de la norme sur le sang de la CSA indique qu’une date de péremption réduite de 24 heures doit être attribuée au sang préparé dans un système ouvert et entreposé à une température de 1 à 6 °C, et de 4 heures s’il est entreposé à 20-24 °C.
Si un établissement divise le sang en aliquotes en système fermé, la date de péremption figurant sur l’étiquette originale est maintenue.
La date de péremption dépend également des températures de conservation et du type de composant sanguin. Consultez le tableau 2 de la norme sur le sang de la CSA pour les températures de conservation et les critères de péremption. De plus, il est important de noter que pour déterminer la date de péremption, l’établissement doit également tenir compte des autres activités de transformation qui peuvent avoir été effectuées sur le sang.
Article 66 Don désigné
Don désigné
66. (1) L’établissement qui prélève du sang d’un donneur faisant un don désigné ajoute aux renseignements figurant sur l’étiquette prévue au paragraphe 64(1) les renseignements permettant d’identifier le receveur à qui le sang est destiné.Changement de fin
(2) L’établissement, si le sang n’est plus destiné à ce receveur, supprime ces renseignements de l’étiquette.
66(2) Les dons de sang désignés peuvent être transférés dans l’inventaire général des dons faits à des fins allogéniques si les exigences générales suivantes concernant le sang et les composants sanguins provenant de dons faits à des fins allogéniques sont respectées :
- le donneur respecte tous les critères d’admissibilité; et
- l’étiquette respecte les exigences en matière d’étiquetage.
Article 67 Don dirigé
Don dirigé
67. L’établissement qui prélève du sang d’un donneur faisant un don dirigé ajoute aux renseignements figurant sur l’étiquette prévue au paragraphe 64(1) les renseignements permettant d’identifier le receveur à qui le sang est destiné et la mention « Pour utilisation dirigée seulement ».
Un don dirigé ne peut être utilisé que pour le receveur visé. Les dons dirigés ne doivent jamais être ré-étiquetés à d’autres fins.
Santé Canada reconnaît ISBT 128 et la disposition de l’uniformité internationale qu’elle supporte pour l’étiquetage du sang et des composants sanguins destinés à la transfusion.
Article 68 Vérification des étiquettes
Vérification des étiquettes
68. L’établissement s’assure que les renseignements qu’il indique sur l’étiquette du contenant de sang sont exacts et complets.
Cette exigence s’applique à tout établissement ajoutant de l’information sur l’étiquette.
L’étiquetage, après que le sang et les composants sanguins ont été jugés sécuritaire pour distribution, est une activité qui s’applique aux établissements qui transforment et/ou transfusent du sang et des composants sanguins. En plus de respecter cette exigence, voir les paragraphes 78(2) et 79(2) ainsi que l’article 80 pour connaître les exigences propres à l’étiquetage du sang transformé. Consultez le tableau 4 pour connaître la répartition des exigences d’étiquetage par activité pour le sang destiné à la transfusion.
A.64(1) Exigences de l'étiquette finale |
A. 79 Mise en commun |
Mise en commun – avec composants lavés |
A.78 Lavage (poche d'origine)- ajouter les renseignements à l'étiquette. |
A.78 Lavage (nouvelle poche) |
A.80 Irradiation - ajouter les renseignements à l'étiquette. |
A.80 Irradiation (nouvelle poche) |
A.65 Aliquotes |
Aliquoté et irradié |
Aliquoté et lavé |
|
|---|---|---|---|---|---|---|---|---|---|---|
Ses nom et adresse municipale |
X |
X |
X |
|||||||
le numéro de sa licence ou de son enregistrement |
X |
X |
X |
|||||||
le code d'identification du don |
X |
X |
X |
X |
X |
X |
||||
Le nom du composant du sang |
X |
X |
X |
X |
X |
X |
X |
X |
||
Abo/ facteur Rh (le cas échéant) |
X |
X1 |
X1 |
X |
X |
X |
X |
X |
||
le volume approximatif de sang prélevé |
X |
X |
X |
|||||||
le volume approximatif du contenu |
X |
X2 |
X2 |
X2 |
X |
|
||||
le nom de l'anticoagulant ou de l'additif |
X |
X3 |
X |
|||||||
la température de conservation recommandée |
X |
X |
X |
|||||||
la date de péremption |
X |
X2 |
X2 |
X2 |
X2 |
X2 |
X2 |
X2 |
X2 |
X2 |
la mise en garde selon laquelle ce sang pourrait transmettre des agents infectieux |
X |
X |
X |
|||||||
consulter les documents d'information (allogénique) |
X |
X |
X |
|||||||
le nombre de poches utilisées pour le pool |
X |
X |
||||||||
le nom de l'établissement qui a préparé |
X |
X |
4 |
4 |
X |
X |
X |
|||
le code unique du produit sanguin labile (pool, aliquote) |
X |
X |
X |
X |
X |
|||||
une mention au lavage |
X |
X |
X |
X |
||||||
Une mention que le produit a été irradié |
X |
X |
X |
|||||||
Remarques : 1 Le Rh n’est pas nécessaire pour les cryoprécipités ou les composants plasmatiques mis en commun. 2 Le volume du sang final et sa nouvelle date de péremption sont requis, et non le volume ou la date de péremption du composant original. 3 Le nom de l’anticoagulant ou de l’additif devrait être inclus dans les cas où le délai de péremption est prolongé au-delà de 24 heures. 4 Il ne s’agit pas d’une exigence du Règlement sur le sang, mais plutôt d’une recommandation aux établissements. |
||||||||||
Articles 69-72 Conservation
Article 69 Exigences
Exigences — établissement qui prélève
69. (1) L’établissement qui prélève du sang le conserve conformément aux exigences suivantes :
- s’il est titulaire d’une licence, selon son homologation;
- s’il est enregistré, selon les exigences concernant la conservation et la date de péremption prévues au tableau 2 de la norme.
Destinataire
(2) L’établissement qui reçoit un contenant de sang de tout autre établissement le conserve conformément aux instructions indiquées sur le contenant et, s’il y a lieu, à toute autre instruction précisée par écrit par l’établissement qui a prélevé le sang.
69(1)a) Exigences — Établissement qui prélève — Établissement titulaire d’une licence
Un établissement titulaire d’une licence qui conserve du sang allogénique et/ou des composants sanguins allogéniques doit présenter à la DMBR une demande de modification de l’homologation s’il souhaite modifier les températures de conservation et les critères de péremption du sang allogénique prélevé requis par son homologation. Consultez l’article 9 de la présente ligne directrice pour de plus amples renseignements sur les demandes de modification de l’homologation.
69(1)b) Critères — Établissement qui prélève — Établissement enregistré
Un établissement enregistré prélevant du sang provenant d’un don fait à des fins autologues ou du sang provenant d’un don fait à des fins allogéniques par des donneurs pré‑évalués doit suivre les exigences pour les températures de conservation et les dates de péremption de ce sang, conformément à ce qui est précisé au tableau 2 de la norme sur le sang de la CSA.
69(2) Exigences — destinataire
Le document d’information représente un exemple de d’autres instructions précisées par écrit.
Article 70 Lieu de conservation
Lieu de conservation
70. L’établissement qui conserve du sang le fait dans un lieu où les conditions ambiantes permettent d’assurer la sécurité du sang et dont l’accès est restreint aux seules personnes autorisées.
Des directives et des procédures doivent être mises en place et appliquées pour les conditions de conservation (telles que la température, l’humidité, la ventilation, les contrôles d’éclairage, la rotation des composants, le nettoyage et toute autre précaution nécessaire pour maintenir la sécurité du sang et des composants sanguins).
Pour assurer la sécurité du sang et des composants sanguins, tout le sang doit être conservé dans un endroit dont l’accès est restreint aux seules personnes autorisées, tel que défini dans les procédures opérationnelles pertinentes.
Les paramètres environnementaux de la conservation comme la température doivent être contrôlés et surveillés. Des sondes ou des dispositifs de surveillance de la température devraient être situés à des points représentant les zones de température extrême, conformément à ce qui a été déterminées par une étude de cartographie thermique afin d’évaluer la distribution de la température. Lors de la réalisation d’études de cartographie thermique, l’utilisation de charges vides et de charges pleines devrait être considérée, selon le cas. Les paramètres tels que l’éclairage, l’humidité et la ventilation devraient être appropriés et contrôlés pour conserver le sang et les composants sanguins en toute sécurité.
Les pratiques de conservation et les configurations de chargement ne doivent pas entraver la circulation de l’air. Un établissement qui conserve du sang ou des composants sanguins doit conserver une documentation prouvant que les unités de sang ou de composants sanguins ont été conservées dans les conditions environnementales appropriées. Cette documentation doit être fournie sur demande.
Si la zone de conservation dispose d’un système d’alarme comportant des signaux audibles, des points d’activation de l’alarme devraient être établis à des températures donnant du temps pour apporter les mesures correctives adéquates avant que les unités de sang n’atteignent des températures inacceptables. Le signal d’alarme devrait se trouver dans un lieu où la surveillance est constante afin que des mesures correctives puissent être apportées dans l’immédiat.
Un établissement qui conserve du sang ou des composants sanguins doit disposer d’une procédure opérationnelle où sont décrites les mesures correctives à prendre en cas d’écart par rapport aux critères de conservation établis. Un tel événement doit faire l’objet d’une enquête appropriée et être documenté adéquatement.
L’accès aux lieux de conservation doit être limité au personnel désigné (p. ex. accès autorisé par carte, serrure et clé, etc.). Quand les lieux de mise en quarantaine sont utilisés, ils doivent être indiqués adéquatement et leur accès doit être limité au personnel désigné. Quand la mise en quarantaine est effectuée à l’aide d’un système électronique, l’accès électronique doit être limité au personnel désigné.
Le sang et/ou les composants sanguins qui ont été distribués à partir d’un lieu de conservation contrôlé ne doivent pas être acceptés de nouveau dans l’inventaire, à moins qu’il n’existe une preuve que le sang et les composants sanguins ont continué à être conservés dans des conditions environnementales appropriées ou que le sang et les composants sanguins, à l’exception des plaquettes, n’ont pas été hors d’un environnement contrôlé pendant plus de 60 minutes. Les procédures pour évaluer le sang retourné avant son retour dans l’inventaire doivent être suivies.
Remarque : Les établissements doivent avoir un processus en place, basé sur un raisonnement approprié, pour déterminer si l’état des plaquettes est adéquat pour permettre leur retour à l’inventaire.
Article 71 Mise à l'écart - fins différentes
Mise à l’écart — fins différentes
71. L’établissement qui conserve du sang ségrègue les unités de sang provenant de dons autologues, désignés ou dirigés des autres unités provenant de dons allogéniques.
Des unités de sang et de sanguins composants provenant de dons autologues, désignés ou dirigés doivent être clairement étiquetées et ségréguées des autres unités du sang et des composants sanguins provenant de dons allogéniques, soit par séparation physique, soit au moyen d’un système de séparation électronique validé, ou les deux.
Article 72 Mise à l'écart - essais
Mise à l’écart — essais
72. L’établissement qui conserve du sang ségrègue les unités de sang ci-après des autres unités de sang jugées sécuritaires, conformément à l’article 73, à des fins de distribution ou, dans le cas de sang provenant d’un don autologue, à des fins de transfusion :
- celles à l’égard desquelles les essais n’ont pas encore été effectués;
- celles à l’égard desquelles les essais n’ont pas tous été effectués ou à l’égard desquelles les résultats des essais ne sont pas encore tous connus;
- celles à l’égard desquelles les échantillons sanguins se sont révélés réactifs de façon répétée ou positifs pour des maladies transmissibles ou pour leurs marqueurs virologiques.
Les unités de sang et de composants sanguins devraient être clairement étiquetées et doivent être contrôlées par un système qui veille à la séparation entre les unités qui ont été mises à l’essai et déterminées sécuritaires pour la distribution ou la transfusion autologue des éléments suivants :
- essais non effectués ;
- essais incomplets ;
- résultats des essais non encore connus ; ou
- résultats réactifs de façon répétée ou positifs.
Pour ce faire, une méthode de séparation physique et/ou un système de séparation électronique validé peuvent être utilisés.
Articles 73-76 Distribution
Article 73 Sécurité du sang
Sécurité du sang — don allogénique
73. (1) L’établissement qui prélève du sang d’un donneur faisant un don allogénique doit, avant de le distribuer à des fins de transfusion ou de fabrication de drogues pour usage humain, conclure à sa sécurité à des fins de distribution s’il est d’avis que le sang a été traité conformément au présent règlement.Sécurité du sang — don autologue
(2) L’établissement qui prélève du sang d’un donneur faisant un don autologue doit, avant de le distribuer à des fins de transfusion, conclure à sa sécurité à des fins de transfusion s’il est d’avis que le sang a été traité conformément au présent règlement.
Consultez l’article 1, soit la partie des définitions, de la présente ligne directrice pour obtenir la définition de « distribution » du Règlement sur le sang.
73(1) Sécurité du sang — don allogénique
Le sang allogénique et les composants sanguins allogéniques doivent respecter les exigences en matière de sécurité du Règlement sur le sang avant sa distribution, y compris les exigences particulières prévues par l’homologation de l’établissement. L’établissement titulaire d’une licence qui prélève du sang d’un donneur faisant un don à des fins allogéniques doit conclure à la sécurité du sang et des composants sanguins à des fins de distribution.
Article 74 Vérification
Vérification
74. (1) L’établissement, avant de distribuer du sang à des fins de transfusion ou de fabrication de drogues pour usage humain, examine le contenant de sang pour vérifier les éléments suivants :
- la lisibilité des renseignements figurant sur les étiquettes du contenant;
- l’intégrité du contenant;
- l’absence de tout signe de détérioration ou de contamination du sang;
- dans le cas de composants sanguins congelés, l’absence de tout signe de dégel.
Interdiction de distribution
(2) L’établissement ne peut distribuer à des fins de transfusion ou de fabrication de drogues pour usage humain les contenants de sang si l’examen effectué conformément au paragraphe (1) révèle l’un des faits suivants :
- le code d’identification du don manque ou est illisible;
- les renseignements, autres que le code d’identification du don, qui doivent figurer en application du présent règlement sur les étiquettes du contenant sont absents ou illisibles et ne peuvent être obtenus des dossiers;
- le contenant est défectueux ou endommagé et n’assure plus la protection du sang contre les conditions extérieures;
- des signes de détérioration ou de contamination du sang sont présents.
Chaque contenant doit faire l’objet d’un examen visuel afin de détecter tout dommage ou signe de contamination avant sa mise à l’inventaire, avant qu’il ne soit distribué et avant toute autre distribution. Lorsqu’un défaut, un étiquetage incorrect ou une apparence anormale est observé, le composant doit être immédiatement mis en quarantaine et éliminé de manière appropriée. La vérification devrait être effectuée dans des conditions d’éclairage permettant un examen visuel approfondi du sang. Les employés qui effectuent cette vérification doivent être formés de manière adéquate et connaître les critères requis. Les établissements devraient vérifier que les employés sont capables de réaliser efficacement l’examen visuel.
Les établissements devraient vérifier que les employés sont capables de réaliser efficacement l’examen visuel.
Un établissement doit séparer les unités de sang et/ou de composants sanguins retournées jusqu’à ce que ces unités soient jugées adéquates pour la transfusion. Les unités de sang et de composants sanguins retournées ne doivent pas être redistribuées à moins qu’elles ne répondent à toutes les exigences des articles 70 et 74 du Règlement sur le sang.
74(1) Avant de distribuer du sang ou des composants sanguins à des fins de transfusion ou de la fabrication ultérieure, un établissement doit satisfaire aux exigences dans les alinéas 74(1)a) à d). Les étapes 74(1)a) à d) devraient également être suivies tout au long du traitement du sang et des composants sanguins.
74(1)c) et 74(2)d) Les cas de détérioration ou de contamination du sang ou des composants sanguins peuvent comprendre : l’hémolyse, les caillots, les amas de fibrine, les agrégats cellulaires, les particules et l’altération de la couleur.
74(2)(a) et (b) Si le code de don est absent ou devient illisible sur l’étiquette du sang ou des composants sanguins, l’établissement ne doit pas le distribuer et doit jeter le sang. Si toute autre information est manquante ou illisible, mais que l’établissement de prélèvement peut retrouver l’information dans ses dossiers, il peut distribuer le sang et les composants sanguins. L’établissement doit avoir et suivre une procédure pour récupérer les renseignements manquants (p. ex. réimprimer l’étiquette ou l’ajouter à l’étiquette). Consultez l’article 61 pour les exigences générales d’étiquetage applicables à chaque unité de sang et de composants sanguins.
Article 75 Contenants d'expédition
Contenants d’expédition
75. L’établissement qui expédie des contenants de sang satisfait aux exigences suivantes :
- il vérifie, avant l’expédition, leur intégrité et la lisibilité de leurs étiquettes;
- il utilise des contenants d’expédition qui résistent aux dommages et permettent d’assurer la sécurité du sang.
75b) Expédition de sang vers un autre établissement ou entre des sites d’un même établissement
Si, pendant l’expédition vers un autre établissement ou entre deux sites différents de l’établissement, le sang et les composants sanguins sont transportés par une personne autre qu’un employé de l’établissement, le contenant d’expédition doit garantir la sécurité du sang et des composants sanguins et empêcher toute altération qui pourrait compromettre la sécurité du sang et des composants sanguins. Le recours à un sceau d’inviolabilité constitue un moyen de maintenir et de vérifier l’intégrité du contenant. Si un sceau inviolabilité est utilisé, l’établissement qui reçoit le sang doit vérifier l’intégrité du sceau d’inviolabilité à la réception du contenant.
Article 76 Conservation pendant le transport
Conservation pendant le transport
76. L’établissement qui expédie des contenants de sang destiné à la transfusion veille à ce que le sang soit conservé, pendant son transport, conformément aux exigences prévues au tableau 2 de la norme.
L’article 76 s’applique à tous les établissements qui expédient du sang ou des composants sanguins destiné à la transfusion. Les contenants d’expédition doivent être validés pour leur utilisation prévue, à moins qu’une autre méthode ne soit utilisée (p. ex. enregistreur de données) pour s’assurer que les conditions de conservation ont été respectées et dûment documentées.
Articles 77-80 Transformation
Article 77 Transformation - méthodes
Transformation — méthodes
77. L’établissement qui procède à la transformation de sang le fait selon des méthodes sécuritaires et efficaces.
Les directives de cet article sont destinées aux établissements enregistrés qui transforment des composants sanguins et aux établissements non enregistrés qui mettent en commun des cryoprécipités. Les activités de transformation visées par le Règlement sur le sang comprennent le lavage, la mise en commun et l’irradiation, après que le sang a été jugé sécuritaire pour la transfusion.
Remarque : Les activités de transformation ne sont pas comprises dans la portée de la préparation des composants sanguins. Les techniques d’inactivation des agents pathogènes ne sont pas incluses dans la portée des exigences des activités de transformation.
Les établissements qui transforment les composants sanguins doivent disposer de procédures opérationnelles validées pour le lavage, la mise en commun ou l’irradiation des composants sanguins, conformément à ce qui est exigé aux articles 95, 96 et 97 du Règlement sur le sang. Si, au moment de la mise en commun ou du lavage, une enceinte de sécurité biologique ou une hotte à flux laminaire est utilisée, elle doit être utilisée en fonction des instructions du fabricant. Pendant la transformation du sang en système ouvert, des techniques aseptiques doivent être utilisées ; elles peuvent inclure, sans s’y limiter, les éléments suivants :
- Zone de faible circulation avec des courants d’air limités, loin de tout aérosol susceptible de contaminer le sang (p. ex. éviers, centrifugeuses, entrées, etc.), si possible
- Régime de nettoyage défini, y compris la désinfection (avant et après utilisation)
- Utilisation de fournitures stériles, notamment de gants stériles
- Entreposage des fournitures (p. ex. les poches de transfert) de manière à éviter leur contamination
Les dossiers relatifs au lavage, à la mise en commun et à l’irradiation doivent être conservés, conformément aux articles 117, 118 et 121 du Règlement sur le sang.
Avant le lavage, la mise en commun ou l’irradiation, il faut vérifier les composants devant être transformés pour s’assurer de l’absence de fuite. Chaque composant doit être visuellement inspecté pour savoir s’il peut être transfusé. Si l’aspect du composant est anormal, l’établissement doit suivre ses procédures, conformément à son système de gestion de la qualité.
Lavage
Un établissement enregistré doit respecter les exigences de l’article 78, en plus des méthodes sécuritaires et efficaces qui suivent.
Plaquettes – Lavées
Les établissements enregistrés où s’effectue le lavage de plaquettes doivent établir et administrer des procédures opérationnelles décrivant la marche à suivre pour cette activité. Il est recommandé de laver les plaquettes avec une solution saline stérile et de les utiliser dans un délai de quatre heures après le lavage.
Globules rouges, lavés
Les globules rouges peuvent être lavés avant la transfusion et doivent être suspendus dans une solution additive ou une solution saline approuvée par Santé Canada. Un établissement enregistré doit valider et documenter le processus de lavage.
Consultez l’alinéa 94(1)b) pour les exigences de contrôle de qualité des globules rouges lavés. Les établissements enregistrés qui lavent des globules rouges devraient suivre les spécifications de contrôle de la qualité pour les « globules rouges — lavés » du tableau 3 de la norme sur le sang de la CSA.
Globules rouges, décongelés et lavés
Les globules rouges qui sont congelés avec un agent cryoprotecteur doivent être lavés avant la transfusion et mis en suspension dans une solution additive approuvée par Santé Canada. Un établissement enregistré doit valider et documenter le processus de décongélation et de lavage. Consultez l’alinéa 94(1)b) pour les exigences de contrôle de qualité des globules rouges lavés. Les établissements enregistrés qui lavent des globules rouges devraient respecter les spécifications de contrôle de la qualité pour les « globules rouges — congelés (déglycérolés) » du tableau 3, de la norme sur le sang de la CSA.
Mise en commun
Un établissement doit respecter les exigences de l’article 79, en plus de respecter les méthodes sécuritaires et efficaces. Un établissement qui met en commun des composants sanguins doit le faire dans un milieu indiqué à cet effet. L’établissement doit prendre des précautions pour éviter la contamination des ports de l’unité de sang manipulée. Consultez la section 1.5 « Définitions » «mise en commun»: mise en commun inclut le mélange.
Cryoprécipité, mis en commun
Les unités de cryoprécipité sont préparées par des établissements titulaires d’une licence et d’une homologation. La mise en commun de cryoprécipités est une activité de transformation qui ne requiert pas d’enregistrement. Les établissements qui font la mise en commun de cryoprécipités doivent le faire selon des méthodes sécuritaires et efficaces, notamment en utilisant des équipements validés à cette fin. Les exemples d’équipements qui peuvent être utilisés comprennent, sans toutefois s’y limiter, les enceintes de sécurité biologique, les dispositifs de décongélation, les connecteurs stériles etc. Tous les équipements doivent être utilisés et entretenus conformément aux instructions du fabricant. Lors de la mise en commun en système ouvert, une technique aseptique doit être utilisée comme décrite ci-dessus.
Irradiation
Les exigences relatives à l’irradiation, dans le cadre des activités de transformation, se rapportent spécifiquement à l’irradiation gamma. Un établissement enregistré doit répondre aux exigences de l’article 80, en plus de respecter les méthodes sécuritaires et efficaces qui suivent. Santé Canada recommande d’irradier les composants sanguins avec de l’équipement conçu spécifiquement à cette fin. Si un établissement enregistré compte utiliser des machines de radiothérapie pour irradier des composants sanguins, l’établissement doit avoir recours à des procédures opérationnelles validées et équivalentes pour utiliser l’équipement à cet effet. L’équipement conçu pour l’irradiation doit être entretenu en conformité avec l’article 100 du Règlement sur le sang. Les mesures relatives au dosage de l’irradiation doivent être surveillées et documentées par l’établissement.
Plaquettes, irradiées
Un établissement enregistré peut irradier des plaquettes en tout temps au cours des sept jours que dure leur conservation. Une fois les plaquettes irradiées, elles peuvent continuer d’être conservées jusqu’à leur date de péremption.
Granulocytes, irradiés
Quand les granulocytes doivent être irradiés, un établissement enregistré devrait les irradier le plus rapidement possible après la préparation du composant. Les granulocytes irradiés devraient être transfusés le plus rapidement possible.
Article 78 Lavage
Lavage
78. (1) L’établissement qui procède au lavage de sang le fait conformément aux articles 7.5.2.3 et 7.5.3 de la norme.Étiquetage
(2) L’établissement qui procède au lavage de sang veille à ce qu’une mention à cet effet figure sur l’étiquette du contenant de sang ainsi que, s’il y a lieu, toute nouvelle date ou heure de péremption.
78(1) Les globules rouges, lavés dans un circuit ouvert, doivent être entreposés en conformité avec la disposition 7.5.3.4 de la norme sur le sang de la CSA.
Si un circuit fermé est utilisé, les globules rouges doivent être entreposés pour une période validée et définie. Un circuit fermé n’a aucune interaction avec les conditions environnementales extérieures qui pourraient mener à une contamination du composant sanguin. Un établissement peut utiliser des connecteurs stériles pour éviter la contamination du composant sanguin pendant le processus de lavage.
Un établissement qui lave des globules rouges doit respecter les exigences en matière de conservation énoncées dans le tableau 2 de la norme sur le sang de la CSA.
78(2) Si les globules rouges lavés sont transférés dans un nouveau contenant de sang, la nouvelle étiquette doit contenir l’information de l’étiquette originale, notamment le code d’identification du don, en plus du nom du composant de globules rouges lavés et la nouvelle date de péremption.
Article 79 Mise en commun
Mise en commun
79. (1) L’établissement qui procède à la mise en commun de composants sanguins le fait conformément aux articles 7.11.1 et 7.11.3 de la norme.Étiquetage
(2) L’établissement qui procède à la mise en commun de composants sanguins veille à ce que les renseignements prévus aux articles 10.8.2 et 10.8.3 de la norme figurent sur l’étiquette du contenant des composants.
L’article 79 s’applique à toutes les activités de mise en commun. Bien que la mise en commun de cryoprécipités ne nécessite pas d’enregistrement, les établissements qui mettent en commun des cryoprécipités doivent le faire conformément au présent article.
L’article 7.11.3 de la norme sur le sang de la CSA décrit les délais de péremption des plaquettes ou des cryoprécipités mis en commun ou mélangés et préparés dans un système ouvert. Pour les autres composants, préparés dans un système ouvert, l’article 7.2 des normes sur le sang de la CSA décrit les délais de péremption nécessaires.
Article 80 Irradiation
Irradiation
80. L’établissement qui procède à l’irradiation de sang le fait conformément aux articles 7.12.2 à 7.12.6 de la norme.
En vertu de la disposition 7.12.3 de la norme sur le sang de la CSA, un établissement enregistré doit avoir une méthode validée pour veiller à ce que le composant sanguin reçoive la dose d’irradiation requise. Un établissement peut surveiller l’irradiation des composants sanguins au moyen d’une étiquette ou d’un dispositif sensible à la radiation et inscrire les résultats de dosimétrie du composant sanguin dans ses dossiers. Un établissement devrait être en mesure de démontrer sa conformité à l’étiquetage des composants et aux procédures de libération quant à l’irradiation. La date de péremption du sang irradié doit être conforme aux exigences relatives à la section 7.12.6 de la norme sur le sang de la CSA.
Articles 81-85 Distribution Exceptionnelle
Article 81 Conditions
Conditions
81. L’établissement peut distribuer ou transfuser du sang qui provient d’un don allogénique et qui est destiné à la transfusion même si les résultats des essais effectués pour en déterminer le groupe sanguin ABO et le facteur Rh ainsi que pour dépister des maladies transmissibles ou leurs agents ne sont pas encore connus, si les conditions ci-après sont réunies :
- il ne dispose pas, à ce moment-là, de sang jugé sécuritaire à des fins de distribution;
- un médecin requiert ce sang pour traiter d’urgence son patient.
Cet article permet la distribution exceptionnelle de sang ou de composants sanguins qui n’ont pas encore été entièrement mis à l’essai mais qui autrement ont été traités en conformité avec l’homologation d’un établissement. Le sang ou les composants sanguins faisant l’objet d’une distribution exceptionnelle est du sang allogénique traité conformément à l’homologation de l’établissement mais dont la mise à l’essai est incomplète en vertu de cette même homologation.
La distribution exceptionnelle a lieu quand un traitement urgent doit être donné à un patient au cas par cas et quand les deux conditions des paragraphes 81a) et b) sont respectées.
Les donneurs de sang allogénique doivent satisfaire aux exigences d'admissibilité énoncées dans le Règlement sur le sang. La partie sur la distribution exceptionnelle du Règlement autorise la transfusion du sang allogénique ou des composants sanguins allogéniques à un seul patient même si les résultats des essais de l’unité ou des unités ne sont pas tous encore connus. L’établissement peut distribuer des composants sanguins aux fins de transfusion avant la fin des essais bactériologiques. Veuillez consulter la section « Analyse bactériologique des plaquettes » de l’article 52 de la présente ligne directrice.
Article 82 Avis de distribution exceptionnelle
Avis de distribution exceptionnelle
82. (1) L’établissement qui distribue du sang au titre de l’article 81 prépare un avis de distribution exceptionnelle qui contient les renseignements suivants :
- son nom et la signature de son directeur médical;
- le code d’identification du don;
- une mention indiquant que le sang distribué est du sang total ou un composant sanguin et, le cas échéant, le nom du composant;
- la liste des essais effectués dont les résultats ne sont pas encore connus lors de la distribution;
- le nom du médecin du receveur et sa signature;
- les raisons de la distribution;
- le nom de l’établissement auquel il distribue ce sang;
- la date et l’heure de la distribution.
Dossier de l’établissement
(2) L’établissement verse l’avis à ses dossiers et en envoie copie à l’établissement auquel il a distribué le sang.Mesures à prendre sur réception de l’avis
(3) L’établissement qui reçoit l’avis mais qui n’effectue pas la transfusion en envoie copie à celui qui effectue la transfusion.Dossier du receveur
(4) L’établissement qui effectue la transfusion verse la copie de l’avis au dossier du receveur.
82(1)d) L’avis doit contenir de l’information sur tous les résultats des essais qui n’étaient pas disponibles au moment de la distribution exceptionnelle.
82(2) Avis de distribution exceptionnelle dans les dossiers de l’établissement
Un établissement titulaire d’une homologation qui procède à une distribution exceptionnelle de sang ou de composants sanguins doit conserver une copie de l’avis dans ses dossiers. L’avis de distribution exceptionnelle doit être accessible. Dans un même ordre d’idées, l’évaluation de suivi et les résultats des essais effectués auprès des donneurs doivent être accessibles dans les dossiers de l’établissement qui a fait la distribution exceptionnelle.
82(3) Avis de distribution exceptionnelle à faire suivre
Si le receveur du sang ou des composants sanguins faisant l’objet d’une distribution exceptionnelle est transféré dans un autre établissement, l’établissement doit faire suivre l’avis de distribution exceptionnelle à l’établissement où la transfusion aura lieu.
82(4) Avis de distribution exceptionnelle au dossier du receveur
L'établissement qui effectue la transfusion doit conserver une copie de l'avis au dossier du receveur, de même, le suivi et les résultats des essais effectués auprès du donneur doivent être ajoutés au dossier du receveur.
L’avis de distribution exceptionnelle doit être fourni sur demande.
Article 83 Étiquetage
Étiquetage
83. L’établissement qui distribue du sang au titre de l’article 81 indique sur l’étiquette du contenant de sang que les essais exigés par le présent règlement n’ont pas tous été effectués ou que les résultats des essais ne sont pas encore tous connus, selon le cas.
Article 84 Suivi
Suivi
84. (1) L’établissement qui distribue, au titre de l’article 81, du sang à l’égard duquel les essais n’ont pas tous été effectués ou à l’égard duquel les résultats des essais ne sont pas encore tous connus, effectue après la distribution tout essai non effectué, s’il y a lieu, et fournit tous les résultats des essais, dès qu’ils sont connus, à l’établissement à qui il a distribué ce sang.Mesures à prendre sur réception des résultats
(2) L’établissement qui reçoit les résultats des essais mais qui n’a pas effectué la transfusion en envoie copie à celui qui a effectué la transfusion.
Un établissement titulaire d’une homologation qui distribue du sang et/ou des composants sanguins dans les conditions de la distribution exceptionnelle doit terminer tous les essais et effectuer tout autre essai de suivi adéquat. L’établissement qui a distribué le sang ou les composants sanguins en vertu de l’article 81 doit aviser l’établissement où le sang ou les composants sanguins ont été distribués des résultats des essais dès qu’ils sont connus.
Article 85 Sang non-transfusé
Sang non-transfusé
85. L’établissement qui, lors de l’urgence, n’a pas transfusé le sang faisant l’objet de la distribution exceptionnelle au receveur à qui il était destiné ne peut le transfuser à un autre receveur ni le conserver.
Consultez le point 7 du « Tableau de l’article 122 : Renseignements et documents – période de rétention » en ce qui concerne les exigences en matière de tenue de dossiers relatives au sort du sang allogénique ou des composants sanguins allogéniques non utilisés destinés à la transfusion.
Articles 86-91 Programme de donneurs pré-évalués
Article 86 Exigences
Exigences
86. L’établissement qui a un programme de donneurs pré-évalués satisfait aux exigences suivantes :
- il effectue les activités reliées au programme sous la supervision d’un directeur médical;
- il ne donne accès à son programme que si les conditions ci-après sont respectées :
- du sang adéquat pour le receveur n’est pas, par ailleurs, disponible,
- un médecin requiert ce sang pour traiter d’urgence son patient.
Article 87 Code d'identification du donneur
Code d’identification du donneur
87. L’établissement qui a un programme de donneurs pré-évalués attribue au donneur un code d’identification du donneur lors de son acceptation dans le programme.
Le code d’identification est attribué à un donneur pré-évalué qui a pris part au programme de donneurs pré-évalués.
Article 88 Mesures à prendre régulièrement
Mesures à prendre régulièrement
88. (1) L’établissement qui a un programme de donneurs pré-évalués prend les mesures ci-après tous les trois mois :
- il effectue une évaluation de l’admissibilité au programme de chaque donneur conformément aux articles 40 à 44;
- il prélève des échantillons sanguins du donneur et les met à l’essai aux fins suivantes :
- dépistage de maladies transmissibles et agents mentionnés aux articles 8.4.1 et 8.4.2 de la norme,
- détermination du groupe sanguin ABO,
- détermination du facteur Rh ainsi que, s’il y a lieu, dépistage de l’antigène D faible,
- dépistage de la présence d’anticorps cliniquement significatifs.
Comparaison
(2) L’établissement compare les résultats des essais prévus aux sous-alinéas (1)b)(ii) et (iii) aux derniers résultats consignés au dossier du donneur, le cas échéant.Incohérence
(3) L’établissement, dans le cas où la comparaison requise révèle une incohérence, recommence les essais et ne peut prélever de sang de ce donneur tant que l’incohérence n’est pas résolue.
88 (1) Mesures à prendre régulièrement
88(1)a) Consultez les articles 40 à 44 pour obtenir les renseignements quant à l’évaluation de l’admissibilité des donneurs.
88(1)b) Des échantillons de sang prélevés sur des donneurs tous les trois mois doivent être mis à l’essai pour les agents infectieux énumérés dans les clauses 8.4.1 et 8.4.2 de la norme sur le sang de la CSA. Santé Canada considère que les essais pour les marqueurs de maladies infectieuses suivants sont appropriés et efficaces afin de se conformer aux clauses 8.4.1 et 8.4.2 de la norme :
- anticorps contre le virus d’immunodéficience humaine, type 1 et type 2 (anti-VIH 1 et anti-VIH 2);
- antigène de surface de l’hépatite B (AgHBs);
- tous les anticorps contre l’antigène capsidique de l’hépatite B (anti-HBc, IgG et IgM);
- anticorps contre le virus de l’hépatite C (anti-VHC);
- anticorps contre le virus T-lymphotrope humain type 1 et type 2 (anti-HTLV-I et anti-HTLV-II);
- syphilis, au moyen d’un essai non tréponémique ou d’un essai tréponémique; et
- essai d’amplification des acides nucléiques (TAN)
- pendant les périodes de l'année où le VNO est potentiellement transmissible à l'homme au Canada; et
- pour les donneurs qui ont voyagé dans des régions endémiques VNO dans les 56 jours précédents.
88(1)b)(ii), (iii), (iv) Des échantillons de sang prélevés tous les trois mois, d’un même donneur, doivent être mis à l’essai pour déterminer le type du sang du donneur (c.-à-d. groupe ABO et facteur Rh) et détecter les anticorps érythrocytaires cliniquement significatifs;
88(1)b) Des échantillons de sang prélevés tous les trois mois, d’un même donneur, peuvent être mis à l’essai pour évaluer ou fournir des renseignements sur le sang lui-même (p. ex. phénotypage des globules rouges) ou pour déterminer le type d’antigènes du système HLA (human leukocyte antigen).
Article 89 Mesures à prendre lors de chaque prélèvement
Mesures à prendre lors de chaque prélèvement
89. L’établissement qui prélève du sang d’un donneur pré-évalué prend, lors de chaque prélèvement, les mesures suivantes :
- il effectue une évaluation de l’admissibilité du donneur;
- il attribue à chaque unité de sang prélevé un code d’identification du don qu’il associe, dans ses dossiers, au code d’identification du donneur;
- il prélève un échantillon sanguin qu’il met à l’essai, dans les soixante-douze heures suivant le prélèvement, aux fins suivantes :
- dépistage des maladies transmissibles et agents mentionnés aux articles 8.4.1 et 8.4.2 de la norme,
- détermination du groupe sanguin ABO,
- détermination du facteur Rh ainsi que, s’il y a lieu, dépistage de l’antigène D faible,
- dépistage de la présence d’anticorps cliniquement significatifs.
89a) Consultez les articles 40 à 44 pour obtenir les renseignements quant à l’évaluation de l’admissibilité des donneurs.
89b) Chaque unité du sang et des composants sanguins doit avoir un code d’identification du don qui lui est attribué au moment du prélèvement. Les mécanismes de gestion de dossiers doivent permettre l’établissement d’un lien entre le code d’identification du don et le code d’identification du donneur. Aux fins de traçabilité, l’établissement enregistré doit pouvoir identifier rapidement le donneur d’un don précis et tous les autres dons provenant du même donneur. Consultez l’article 1, soit la partie des définitions, pour obtenir les définitions de « code d’identification du don » et « code d’identification du donneur ».
89c) Un établissement enregistré qui prélève du sang d’un donneur déjà évalué doit veiller à ce qu’un échantillon de sang soit recueilli du donneur au moment du don et qu’il soit mis à l’essai dans les 72 heures pour détecter les marqueurs de maladie infectieuse indiqués à l’alinéa 88(1)b).
Si le sang d’un donneur pré-évalué se révèle positif ou réactif de façon répétée à un essai pour les agents ou marqueurs de maladies transmissibles, il est essentiel que l’établissement qui a effectué l’essai doit immédiatement en aviser l’établissement enregistré qui a évalué le donneur en cause.
Article 90 Étiquetage
Étiquetage
90. L’établissement qui prélève du sang d’un donneur pré-évalué veille à ce qu’au moins le code d’identification du don, le groupe sanguin ABO et, s’il y a lieu, le facteur Rh figurent sur l’étiquette du contenant de sang.
Article 91 Sang non-transfusé
Sang non-transfusé
91. L’établissement qui, lors d’une urgence, n’a pas transfusé aux receveurs concernés le sang prélevé d’un donneur pré-évalué se conforme aux exigences prévues à l’article 16.2.5 de la norme.
Article 92 Importation dans des circonstances urgentes
Renseignements préalables à l’importation
92. (1) L’établissement peut, dans des circonstances urgentes, importer du sang qui provient d’un don allogénique mais qui a été traité autrement que conformément à une homologation, s’il fournit au ministre, préalablement à l’importation, les renseignements et documents suivants :
- les renseignements visés aux alinéas 6(1)a) et j) à l’égard de chaque établissement étranger qui traite le sang visé par l’importation;
- une copie du document d’information visant ce sang, ou son équivalent;
- une copie du questionnaire d’évaluation préliminaire des donneurs utilisé par chaque établissement étranger qui traite le sang visé ainsi qu’un document indiquant les différences entre ce questionnaire et celui visé à l’article 41;
- une description de la manière utilisée par chaque établissement étranger pour évaluer les renseignements obtenus après le don au titre de l’alinéa 44(1)b);
- une description des conditions de conservation et de transport de ce sang préalablement à son importation ainsi que celles envisagées par la suite;
- une description de la façon dont il entend indiquer que le sang visé a été importé dans des circonstances urgentes;
- une description de la manière utilisée par chaque établissement étranger pour effectuer les enquêtes et les rapports concernant les accidents, les manquements et les effets indésirables.
Renseignements — lors de chaque importation
(2) L’établissement, lors de l’importation visée au paragraphe (1), fournit au ministre les renseignements et documents suivants :
- une justification écrite démontrant l’existence de circonstances urgentes;
- les détails concernant tout traitement ou tout étiquetage subséquents qui pourraient être effectués avant la transfusion du sang visé.
Définition de « circonstances urgentes »
(3) Pour l’application du présent article, « circonstances urgentes » s’entend de l’insuffisance, au Canada, de sang provenant de dons allogéniques, insuffisance qui constitue un risque immédiat et important pour la santé publique.
92(1) Renseignements — avant l’importation
Si un établissement compte inclure dans son plan d’intervention d’urgence l’importation de sang ou des composants sanguins dans des circonstances urgentes, cet établissement doit respecter les exigences propres à ce type d’importation avant qu’il y ait urgence.
92(1)a) Santé Canada exige de l’établissement qu’il fournisse de l’information sur l’établissement étranger, conformément aux alinéas 6(1)a) et j), au BAR de la DMBR, Santé Canada. Consultez le paragraphe 6(1) de la présente ligne directrice pour obtenir les coordonnées.
92(1)b) L’établissement importateur doit fournir à Santé Canada ce qui suit :
- le document d’information de l’établissement étranger; ou
- l’information équivalente sur les composants sanguins proposés pour importation dans des circonstances urgentes.
92(1)c) L’établissement importateur doit fournir à Santé Canada les documents suivants :
- le questionnaire d’évaluation préliminaire des donneurs de chaque établissement étranger d’où il entend importer le sang et les composants sanguins dans des circonstances urgentes;
- un document qui décrit les différences entre le questionnaire d’évaluation préliminaire des donneurs de l’établissement étranger et le questionnaire d’évaluation préliminaire des donneurs autorisés de l’établissement importateur.
92(1)d) L’établissement importateur doit fournir à Santé Canada une description de la façon dont l’établissement étranger évalue les renseignements après le don. L’exigence relative à l’évaluation des renseignements après le don au Canada est décrite au paragraphe 44(2) du Règlement sur le sang.
92(1)e) Lorsqu’il planifie l’importation de sang ou de composants sanguins dans des circonstances urgentes, l’établissement importateur doit fournir à Santé Canada une description des conditions de conservation et de transport du sang et des composants sanguins tant avant son importation qu’après son importation.
Les conditions de conservation et de transport sont, entre autres, la température, la date de péremption et la mise à l’écart.
92(1)f) L’établissement importateur doit décrire à Santé Canada les moyens qu’il utilisera pour différencier le sang et les composants sanguins importés dans des circonstances urgentes à partir de son inventaire régulier. Il s’agit notamment de la manière dont l’établissement identifiera et retracera le sang et les composants sanguins importés dans des circonstances urgentes lorsqu’il les distribuera aux établissements de transfusion.
92(1)g) L’établissement importateur doit soumettre à Santé Canada une description de la manière dont l’établissement étranger effectuera les enquêtes sur les manquements, les accidents et les effets indésirables et en rendra compte. Tout manquement, accident ou effet indésirable qui découle du traitement par l’établissement étranger devrait être déclaré conformément aux exigences du pays étranger. Si l’établissement étranger fait enquête sur un manquement ou un accident grave par rapport à du sang ou des composants sanguins qui ont été importés au Canada, l’importateur au Canada devrait en aviser Santé Canada.
92(2) Renseignements — lors de chaque importation
Lorsqu’un établissement importe du sang ou des composants sanguins dans des circonstances urgentes, les renseignements suivants doivent être fournis à Santé Canada pour chaque importation :
- de la documentation écrite décrivant les circonstances urgentes et la raison pour laquelle il manque du sang ou de composants sanguins allogéniques au Canada;
- une description de tout traitement subséquent que l’établissement pourrait devoir effectuer avant que le sang ou les composants sanguins puissent être transfusés au Canada.
92(3) Signification de circonstances urgentes
Les circonstances urgentes réfèrent à une situation qui empêche un établissement titulaire d’une licence au Canada de compter sur sa propre réserve de sang allogénique ou sur celle provenant d’autres établissements au Canada. L’absence de solution au pays à cette situation vient justifier l’importation de sang ou de composants sanguins dans des circonstances urgentes. Il est à noter que le terme circonstances urgentes ne s’applique pas au sang servant à l’immunisation de donneurs de plasma destiné à une fabrication ultérieure.
Articles 93-123 Gestion de la qualité
Articles 93-94 Système de gestion de la qualité
Article 93 Structure organisationnelle
Structure organisationnelle
93. (1) L’établissement titulaire d’une licence ou enregistré se dote d’une structure organisationnelle qui reflète les responsabilités de la direction à l’égard de chaque activité exercée par l’établissement.Supervision
(2) L’établissement se dote également d’un système de gestion de la qualité efficace et nomme un individu qui en assume la responsabilité.Vérification périodique
(3) L’établissement revoit son système de gestion de la qualité, à intervalles réguliers prévus dans ses procédures opérationnelles, pour s’assurer qu’il demeure pertinent et efficace.
93(1) Les établissements titulaires d’une licence et les établissements enregistrés doivent désigner une structure organisationnelle dans un organigramme tenu à jour qui délimite clairement les domaines de responsabilité et les voies hiérarchiques. Dans ces établissements, il doit y avoir une personne responsable du système de gestion de la qualité. De plus, le personnel clé pourrait inclure un directeur médical (tel que défini dans l’article 1 du Règlement) ainsi que le personnel responsable des activités (p.ex. traitement, transformation, distribution etc.), le cas échéant. Les titres et les domaines de responsabilités du personnel clé doivent y être inscrits pour toutes les activités liées au sang et aux composants sanguins.
93(2) Les établissements titulaires d’une licence et les établissements enregistrés doivent s’assurer que leurs activités respectent les exigences réglementaires. Afin d’assurer la conformité, il est nécessaire que ces établissements conçoivent et mettent en œuvre un système de gestion de la qualité complet intégrant tous les éléments énumérés au paragraphe 94(1).
Le système de gestion de la qualité est un système intégré d’assurance de la qualité et du contrôle de la qualité qui comprend tous les aspects maximisant, individuellement ou collectivement, la sécurité du sang et des composants sanguins. Le système de gestion de la qualité doit :
- être défini, documenté, mis en œuvre, examiné périodiquement et tenu à jour par l’établissement;
- comprendre des éléments qui permettent la détection, le suivi, l’enquête, la prévention et la correction de toute lacune qui pourrait compromettre la sécurité du sang et des composants sanguins;
- inclure une structure organisationnelle dans laquelle le personnel responsable de toutes les activités visées par le Règlement est défini et consigné; et
- faire en sorte que les politiques, processus et procédures écrits en vigueur portant sur les activités réglementées sont disponibles et communiqués à tout le personnel concerné.
L’établissement doit désigner une personne responsable du système de gestion de la qualité. Cette personne doit s’assurer que les objectifs définis de qualité sont atteints. L’atteinte de ces objectifs de qualité exige la participation et l’engagement du personnel de différents départements à tous les niveaux de l’établissement.
La personne responsable du système de gestion de la qualité peut déléguer ses tâches et ses responsabilités à un membre du personnel qualifié en conformité avec le paragraphe 98(1) du Règlement, mais elle demeure responsable des tâches et des responsabilités ainsi déléguées.
93(3) Les établissements titulaires d’une licence et les établissements enregistrés doivent effectuer, aux intervalles précisés et définis par l’établissement, une vérification de tous les éléments de leur système de gestion de la qualité énumérés au paragraphe 94(1) pour en assurer la pertinence et l’efficacité continues. Toute lacune ou tout point à améliorer doit être traité et corrigé et un plan comprenant des buts, des objectifs et des plans d’action devrait être développé et utilisé.
Article 94 Exigences
Exigences
94. (1) Le système de gestion de la qualité comprend les éléments suivants :
- une division responsable de l’assurance de la qualité;
- un programme de contrôle de la qualité;
- un système de contrôle des changements;
- un programme de contrôle des processus au sens de l’article 3.1 de la norme;
- un système qui permet l’amélioration des processus par le suivi des plaintes et la prise de mesures correctives ou préventives;
- un système qui permet de recenser les renseignements obtenus après le don ainsi que ceux concernant les manquements, les accidents et les effets indésirables, de faire enquête à leur égard, de procéder à des retraits et de prendre des mesures correctives;
- un programme de formation et d’évaluation des compétences du personnel;
- un programme d’évaluation de l’exactitude et la fiabilité des résultats d’essais;
- un système de contrôle des documents et de gestion de dossiers;
- un système de vérification interne;
- des plans d’intervention d’urgence;
- un système qui permet d’identifier spécifiquement l’équipement, le matériel et les produits essentiels;
- des spécifications écrites concernant l’équipement, le matériel, les produits et les services essentiels;
- un programme d’entretien préventif de l’équipement essentiel;
- un programme de validation des processus.
Fonctions séparées
(2) La division responsable de l’assurance de la qualité relève de la direction et est indépendante des autres fonctions.Exception
(3) Le paragraphe (2) ne s’applique pas à l’établissement titulaire d’une licence qui ne fait que de la mise à l’essai de sang provenant d’un don allogénique ni à celui enregistré s’il veille à ce que l’individu qui effectue la vérification interne d’une activité ne soit pas directement responsable de l’activité visée.
94(1)a) Division responsable de l’assurance de la qualité
L’assurance de la qualité comprend les activités prévues et réalisées pour garantir que tous les systèmes et éléments qui ont une incidence sur la sécurité du sang et des composants sanguins fonctionnent comme prévu, individuellement et collectivement. Une division responsable de l’assurance de la qualité est constituée d’une ou de plusieurs personnes, disposant de pouvoirs et de responsabilités clairement définis, afin d’assurer la conformité au Règlement sur le sang.
94(1)b) Programme de contrôle de la qualité
Le programme de contrôle de la qualité est un élément du système de gestion de la qualité qui comprend les mesures et les contrôles utilisés pour établir l’acceptation des produits, du matériel et de l’équipement de l’établissement, selon leurs spécifications et les exigences du Règlement sur le sang. Les activités de contrôle de la qualité doivent être effectuées selon les procédures opérationnelles.
Les articles 100 et 102 du Règlement sur le sang précisent les exigences de contrôle de la qualité relatives à l’équipement essentiel, et le matériel et produits essentiels respectivement, applicables aux établissements titulaires d’une licence ou d’un enregistrement.
Établissements titulaires d’une licence
Un programme de contrôle de la qualité, servant à évaluer la qualité du sang et des composants sanguins, doit être appliqué par tout établissement titulaire d’une licence qui prélève du sang allogénique à des fins de transfusion. Les éléments suivants doivent être définis par chaque établissement titulaire d’une licence et autorisés par Santé Canada :
- la fréquence des essais de contrôle de la qualité, exprimée en pourcentage de la production globale;
- le nombre minimal d’essais requis, précisé au cours d’une période donnée;
- les critères acceptables quant aux essais de contrôle de la qualité de chaque type de composants.
Les résultats des essais de contrôle de la qualité doivent être analysés en continu, et des mesures correctives appropriées doivent être prises lorsque les résultats des essais s’écartent des limites acceptables définies.
Établissements enregistrés
Les établissements enregistrés qui procèdent au lavage de sang doivent réaliser des essais de contrôle de la qualité du sang. Les éléments suivants doivent être définis dans les procédures opérationnelles :
- le type des essais à effectuer
- les critères d’acceptation pour chaque essai
- nombre d’unités à mettre à l’essai
À cet effet, les établissements enregistrés devraient observer le tableau 3 de la norme sur le sang de la CSA.
94(1)c) Système de contrôle des changements
Un système de contrôle des changements doit être établi et tenu à jour pour permettre de déterminer, de consigner, d’examiner, d’approuver et de contrôler tous les processus. Tout changement aux processus, matériel, équipement et installations qui peuvent avoir une incidence sur la sécurité du sang et des composants sanguins doit être adéquatement consigné et évalué en profondeur, correctement mis en œuvre et approuvé par les cadres supérieurs appropriés, ainsi qu’il est déterminé par la structure organisationnelle de l’établissement, en fonction de la portée du changement. Tout changement majeur peut nécessiter une nouvelle validation conformément aux exigences des alinéas 94(1)d) et 94(1)o).
Ces approbations viennent s’ajouter à celles exigées par Santé Canada pour les établissements titulaires d’une licence dans le cadre de leur homologation.
94(1)d) Programme de contrôle des processus, au sens de l’article 3.1 de la norme
Les établissements doivent se doter d’un programme de contrôle des processus qui couvre tous les aspects de leurs activités réglementées. La section 3.1 de la norme sur le sang de la CSA définit comme suit le contrôle des procédés (ou processus) : « gestion des procédés et procédures touchant la qualité des produits et services. On désire ainsi veiller à ce que ces procédés et procédures soient appliqués uniformément et de la façon prévue pour produire des résultats prévisibles ».
Des politiques et procédures doivent être en place pour veiller à ce que tous les processus soient menés dans des conditions contrôlées et définies par du personnel qualifié. Le contrôle des processus peut comprendre la validation et la surveillance des processus, l’établissement de résultats ou de paramètres acceptables et la formation du personnel.
94(1)e) Système qui permet l’amélioration des processus par le suivi des plaintes et la prise de mesures correctives ou préventives
Le système d’amélioration des processus comprend les deux composantes suivantes :
- répondre aux plaintes et en assurer le suivi;
- mettre en œuvre des mesures correctives et préventives appropriées, le cas échéant.
Plaintes
L’établissement doit avoir des politiques, processus et procédures opérationnelles pour le traitement des plaintes qui risquent de compromettre la sécurité du sang et des composants sanguins. Toutes les plaintes doivent être examinées, évaluées par le département approprié, consignées, et faire l’objet d’une enquête selon les procédures opérationnelles de l’établissement, incluant l’identification et la mise en œuvre de mesures correctives et préventives, selon le cas. Toute décision et mesure de suivi prise à la suite d’une enquête sur la plainte doit être consignée.
Mesures correctives et préventives
Les mesures correctives sont axées sur l’élimination des causes de non-conformité existantes ou d’incidents afin d’empêcher la récurrence, tandis que les mesures préventives mettent l’accent sur l’élimination des causes de non-conformité potentielles pour éviter l’occurrence de celles-ci.
L’établissement doit également disposer d’un système permettant de mettre en œuvre des mesures correctives et préventives, en temps opportun, chaque fois que cela est nécessaire. En voici quelques exemples :
- après la réception d’une plainte ou de renseignements obtenus après le don;
- pendant les études de dons antérieurs ou d’enquêtes sur les composants transfusés;
- pour corriger les lacunes relevées à la suite d’une vérification interne;
- pour corriger les lacunes relevées lors d’une inspection de Santé Canada;
- lors de l’enquête sur un accident, un manquement ou un effet indésirable; et
- lorsque les résultats du contrôle de la qualité ne répondent pas aux critères préétablis.
Dans le cadre du système d’amélioration des processus de l’établissement, si des mesures correctives et/ou préventives s’avèrent nécessaires, elles doivent être mises en œuvre et faire l’objet d’un suivi dans le but de réduire le risque de récurrence et d’améliorer le processus actuel. Une fois les mesures correctives et/ou préventives mises en œuvre, l’efficacité de ces mesures doit être évaluée.
94(1)f) Système permettant l’identification et l’enquête des renseignements obtenus après le don, des manquements et accidents, des effets indésirables et la conduite des retraits.
Les établissements doivent avoir des processus et des procédures opérationnelles définis pour identifier, recueillir et traiter tout renseignement obtenu après le don (article 44 du Règlement sur le sang), tout manquement et accident (articles 103 à 108 du Règlement sur le sang) et tout effet indésirable (articles 109 à 116 du Règlement sur le sang) qui surviennent ainsi que pour les programmes d’étude des dons antérieurs et d’enquête sur les composants transfusés. Ces processus et ces procédures opérationnelles doivent inclure les processus décisionnels employés pour déterminer si une enquête est justifiée, les étapes pour mener une enquête approfondie sur les incidents qui l’exigent et la mise en œuvre de toute mesure corrective et préventive, le cas échéant. Pour plus de détails sur les étapes à suivre pour mener une enquête, consultez l’article 104.
Enquêtes et retraits
Lorsqu’un incident se produit et qu’il est susceptible de compromettre la sécurité du sang et/ ou des composants sanguins, l’établissement où il a eu lieu doit déterminer si une enquête est justifiée.
Pour chaque incident, il est nécessaire de faire une évaluation initiale afin de déterminer si des activités réglementées ont été mises en cause et si la sécurité du sang et/ou des composants sanguins a été touchée.
Si l’évaluation initiale permet de déterminer que l’incident n’est pas lié à des activités réglementées effectuées par l’établissement, cette conclusion doit être documentée et aucune autre mesure n’est requise par l’établissement en vertu du Règlement sur le sang.
Lorsqu’une enquête est justifiée, elle devrait tenir compte des éléments suivants :
- le type d’incident (c.-à-d. renseignements obtenus après le don, accident/manquement et/ou effet indésirable);
- les processus/procédures pertinents;
- les activités réglementées concernées;
- l’impact sur la sécurité du sang ou ce qu’ il aurait pu être;
- l’analyse des causes fondamentales;
- les mesures correctives et préventives ou d’autres mesures d’atténuation des risques pour éviter la récurrence.
Les établissements doivent disposer d’un système pour effectuer efficacement des retraits rapides du sang et des composants sanguins. En ce qui concerne le retrait de sang distribué à des fins de fabrication ultérieure en drogue pour usage humain, cela relève du Règlement sur les aliments et drogues. Des procédures opérationnelles doivent être en place afin de définir les étapes assurant le retrait efficace du sang ou des composants sanguins impliqués afin d’en éviter la distribution ou l’utilisation. Des dossiers doivent être conservés pour permettre d’identifier et de repérer rapidement le sang en cause. Les procédures doivent préciser le ou les poste(s) au sein de l’établissement responsable(s) i) d’obtenir de l’information sur le sang et les composants sanguins en cause, ii) d’effectuer le retrait et iii) d’examiner les dossiers de distribution nécessaires pour la coordination du retrait. La procédure opérationnelle devrait également décrire le mode de communication qui sera employé pour aviser tous les établissements auxquels le sang et/ou les composants sanguins ont été distribués et qui sont, par conséquent, visés par le retrait. Tout sang et composant sanguin ayant fait l’objet d’un retrait doit être identifié et mis en quarantaine jusqu’à ce que leur sort soit déterminé.
Les procédures applicables doivent également décrire les exigences liées à la déclaration à Santé Canada des manquements, des accidents et des effets indésirables conformément aux articles 107, 108, 109, 113, 115 et 116 du Règlement sur le sang. Des indications supplémentaires sur les enquêtes relatives à ces types d’incidents se trouvent dans les articles respectifs mentionnés ci-dessus.
Les établissements sont tenus de consigner et de conserver les dossiers concernant toutes les enquêtes et tous les retraits conformément aux exigences énoncées dans les articles 119 à 122 du Règlement sur le sang.
94(1)g) Un programme de formation et d’évaluation des compétences du personnel
Les établissements doivent avoir un programme de formation écrit pour tout le personnel concerné, pour les activités réglementées par le Règlement sur le sang, tel que défini dans les procédures opérationnelles. Le programme de formation doit comprendre une évaluation formelle des compétences du personnel. Le personnel doit recevoir une formation initiale et continue adaptée à ses responsabilités professionnelles tel que défini dans le programme de formation. Pour plus de détails sur les programmes de formation et d’évaluation des compétences, veuillez vous référer au paragraphe 98(2).
94(1)h) Programme d’évaluation de l’exactitude et de la fiabilité des résultats d’essais
Ce programme d’évaluation permet de surveiller la capacité d’un laboratoire d’essai de gérer et d’exécuter des activités définies par son programme spécifique dans les limites acceptables de détection et de précision prédéterminées par la mise à l’essai d’échantillons inconnus.
L’établissement doit veiller à ce que tout le personnel qui exécute les essais requis en vertu du Règlement sur le sang participe à un programme d’évaluation de l’exactitude et la fiabilité des résultats d’essais (p. ex., en rotation) au moyen des procédures de mise à l’essai de routine de l’établissement.
Les résultats de ce programme d’évaluation doivent être revus par la direction et examinés afin de cerner les tendances signalant un problème systémique. S’il y a lieu, l’établissement doit prendre des mesures correctives ou préventives afin de rectifier les problèmes détectés.
Les dossiers liés à ce programme d’évaluation doivent inclure les résultats d’essais, les analyses des tendances et les mesures correctives ou préventives qui ont été prises. Ces dossiers doivent être tenus conformément aux exigences relatives à la conservation des dossiers des programmes d’évaluation prévues aux articles 119 et 120 du Règlement sur le sang.
94(1)i) Système de contrôle des documents et de gestion de dossiers
Contrôle des documents
Les établissements doivent définir, consigner et tenir à jour des procédures opérationnelles visant à contrôler tous les documents de qualité et les renseignements applicables aux activités qu’ils mènent et relatives au Règlement sur le sang.
La distribution et la tenue à jour des procédures opérationnelles et d’autres documents de qualité, par exemple les politiques et les formulaires, doivent faire l’objet d’un contrôle afin que seules les versions à jour puissent être utilisées. Les versions précédentes des documents de qualité doivent être retirées, archivées et remplacées par la version approuvée actuelle. Les versions obsolètes des documents de qualité doivent être retirées et archivées. Cependant, conformément aux articles 119 à 122 du Règlement sur le sang, il faut conserver une copie de chaque version des procédures opérationnelles qui ont été mises en œuvre.
Gestion des dossiers
Les dossiers doivent être exacts, complets et lisibles et être tenus à jour de façon constante afin de préserver leur complétude et leur intégrité au fil du temps. L’article 117 fournit des détails supplémentaires sur la gestion et la qualité des documents.
94(1)j) Système de vérification interne
À la fréquence précisée dans les procédures opérationnelles de l’établissement, une vérification interne doit être effectuée pour toutes les activités réglementées afin de vérifier la conformité aux exigences du Règlement sur le sang. La programme de vérification doit comprendre une évaluation dans le but de déterminer si les procédures opérationnelles sont suivies et si les activités menées permettent d’atteindre de façon constante les résultats attendus. Le plan de vérification doit porter sur l’évaluation de toutes les activités réglementées. Généralement, une telle vérification a lieu au moins tous les deux ans. Les vérifications doivent être faites selon un programme établi et des procédures opérationnelles.
Les vérifications peuvent être réalisées par du personnel qualifié ou par un vérificateur externe (tierce partie qualifiée) qui réalise la vérification au nom de l'établissement et possède une connaissance de la matière vérifiée. Les vérificateurs ne doivent pas être directement responsables des procédures ou processus qu’ils vérifient. Par exemple, un superviseur responsable de la préparation des composants ne doit faire la vérification d’aucune activité liée à la préparation des composants réalisée par son propre département.
Tout établissement qui confie en sous-traitance à un autre établissement la réalisation en son nom d’une activité réglementée (p. ex. l’essai) est tenu d’établir un processus qui lui permet de vérifier périodiquement que celle-ci est exécutée en conformité avec le Règlement sur le sang et les procédures opérationnelles applicables. Par exemple, l’établissement peut évaluer la conformité d’un autre établissement en procédant à une vérification de celui-ci ou en examinant les rapports de vérification que cet autre établissement lui a fournis. Les établissements doivent également vérifier que les services fournis par un tiers sont conformes aux exigences du Règlement sur le sang (par exemple, les services de transport et d’entretien de l’équipement).
Les résultats des vérifications et les mesures de suivi requises doivent être consignés, puis examinés par la personne responsable du système de gestion de la qualité. Les mesures correctives et préventives doivent être appliquées en temps opportun. Les dossiers sur les vérifications internes, dont les mesures correctives et préventives, et les vérifications des établissements contractuels doivent être conservés conformément aux articles 119 à 121 du Règlement sur le sang.
94(1)k) Plans d’intervention d’urgence
Les établissements titulaires d’une licence ou d’un enregistrement doivent se doter des plans d’intervention d’urgence au cas où les processus seraient interrompus, comme dans le cas d’une panne de courant ou d’une catastrophe naturelle.
Les plans d’intervention d’urgence doivent comprendre :
- des procédures d’intervention d’urgence manuelles permettant de mener les activités réglementées pertinentes et de conserver les dossiers connexes dans une situation d’urgence;
- une procédure d’intervention d’urgence manuelle pour la distribution du sang ou des composants sanguins de l’inventaire aux hôpitaux et à tous les secteurs applicables où du sang ou des composants sanguins sont nécessaires;
- les renseignements permettant de maintenir la sécurité du sang et des composants sanguins conservés.
Les exigences en matière de traçabilité doivent continuer d’être respectées pendant une situation d’urgence (p. ex. lorsqu’un système d’inventaire informatisé ou son système de sauvegarde ont mal fonctionné). Il faut examiner périodiquement les plans d’intervention d’urgence pour leur assurer l’efficacité. S’il y a lieu, le circuit d’alimentation d’urgence doit être entretenu et mis à l’essai régulièrement pour que l’on puisse s’assurer de sa disponibilité.
94(1)l) Système permettant d’identifier spécifiquement l’équipement, le matériel et les produits essentiels
Le terme « essentiel » est défini à l’article 1 de la présente ligne directrice et y sont aussi inclus des exemples d’équipement de services et de matériels essentiels. Dans le cadre de son système qualité, un établissement doit se doter d’un mécanisme pour identifier, consigner et surveiller tout l’équipement, le matériel et les produits essentiels.
Dans le cadre de son système qualité, un établissement doit se doter d’un système pour identifier, consigner et retracer tout l’équipement, le matériel et les produits essentiels. Dans ce système, chaque équipement doit porter un code d’identification unique.
94(1)m) Spécifications écrites concernant l’équipement, le matériel, les produits et les services essentiels
Le terme « essentiel » est défini à l’article 1 de la présente ligne directrice et y sont aussi inclus des exemples d’équipement et de matériel, produits et services essentiels. Des spécifications écrites concernant l’équipement, le matériel, les produits et les services essentiels doivent être disponibles. Les établissements doivent se doter de processus définis et veiller à ce qu’en cas de tout changement aux exigences réglementaires ou à la technologie, les spécifications continuent de respecter les exigences applicables du Règlement sur le sang.
Si les spécifications ne sont pas respectées, un établissement doit avoir un système en place pour assurer la prise de mesures correctives rapides et efficaces, qui pourrait comprendre le signalement en temps opportun des plaintes, des déviations ou des défauts de produit à leur fournisseur ou au fournisseur de services. Les articles 100 à 102 prévoient des exigences supplémentaires en ce qui concerne l’équipement, le matériel et les produits essentiels.
94(1)n) Programme d’entretien préventif de l’équipement essentiel
Le terme « essentiel » est défini à l’article 1 de la présente ligne directrice et y sont aussi inclus des exemples d’équipement essentiel. L’équipement essentiel doit toujours rencontrer ses spécifications dans le but de produire du sang et des composants sanguins qui sont sécuritaires. Les établissements doivent se doter d’un programme d’entretien préventif pour s’assurer que tout l’équipement essentiel fonctionne selon les spécifications de rendement requises.
Le programme d’entretien préventif doit avoir des processus définis, dont des calendriers prédéterminés de service technique pour l’entretien régulier et l’étalonnage permettant de vérifier que les spécifications énoncées dans le manuel du fabricant et les spécifications supplémentaires exigées par le système qualité de l’établissement sont respectées. Les processus doivent préciser la méthode à utiliser, la fréquence d’entretien et d’étalonnage et les mesures à prendre lorsque le rendement de l’équipement s’écarte des limites définies. Cette exigence s’applique à tout équipement, instrument ou appareil de mesure essentiel pour garantir que le sang et les composants sanguins sont conformes au Règlement sur le sang. L’entretien préventif et l’étalonnage doivent être effectués par du personnel compétent.
Le calendrier d’entretien préventif doit être respecté, et tous les dossiers et rapports de validation, de qualification, d’étalonnage, d’entretien et de réparation, incluant les résultats réels des essais et l’équipement d’essai utilisé avec la preuve d’un étalonnage approprié, doivent être conservés tel que décrit aux articles 119-122 du Règlement sur le sang. Les articles 100 et 101 du Règlement sur le sang décrivent les exigences à respecter quant au nettoyage, à la validation et à l’étalonnage de l’équipement essentiel.
94(1)o) Programme de validation des processus
Les établissements doivent mettre en place un programme afin de démontrer que tous les processus liés aux activités de traitement et de transformation permettent de donner, avec un degré élevé de certitude, les résultats attendus et de rencontrer les spécifications prédéfinies.
Un plan de validation écrit devrait comprendre les méthodes d’essai, l’équipement à utiliser, le personnel qualifié qui effectue la validation, les procédures de validation (c.-à-d. les étapes qui doivent être accomplies pour respecter l’état validé), les critères d’acceptation et les documents à l’appui.
Quand des changements sont apportés à un processus validé, la nécessité de procéder à une revalidation doit être évaluée. La revalidation peut être nécessaire selon la nature et l’étendue des changements, i.e. des changements qui sont susceptibles d’influer la validation originale, les caractéristiques du processus et/ou la sécurité du sang et des composants sanguins. Les exigences en matière de documentation sont les mêmes que lors de la validation initiale du processus. L’article 97 fournit plus de détails sur la validation des processus.
94(2) Fonctions séparées
Une personne peut avoir plus d’une fonction, mais la division ou la personne responsable de l’assurance de la qualité doit fonctionner et relever de la direction et être indépendante des personnes responsables des opérations.
94(3) Exception
Les établissements titulaires d’une licence qui ne font que de la mise à l’essai de sang ou de composants sanguins allogéniques ou les établissements qui mènent des activités nécessitant un enregistrement ne sont pas tenus d’avoir une division responsable de l’assurance de la qualité qui agit comme division organisationnelle distincte et relevant de la direction de façon indépendante des autres divisions en autant que la personne qui effectue une vérification interne n’est pas directement responsable des activités vérifiées. De plus, les personnes ayant un double rôle (effectuant des activités opérationnelles et des activités d’assurance de la qualité) ne devraient pas examiner leur propre travail.
Articles 95-97 Procédures opérationnelles
Article 95 Obligation - procédures opérationnelles
Obligation — procédures opérationnelles
95. L’établissement se dote de procédures opérationnelles pour celles de ses activités comportant des aspects liés à la sécurité humaine et la sécurité du sang.
Les procédures opérationnelles sont composées d’instructions qui décrivent les étapes à suivre par l’établissement dans la conduite de ses activités. Elles contiennent des instructions ou directives à l’intention du personnel afin que les activités soient réalisées et consignées de façon constante et conformément aux exigences réglementaires.
Tous les établissements doivent élaborer et tenir à jour des procédures opérationnelles écrites décrivant les étapes importantes de chaque activité réglementée qu’ils mènent. Par exemple, l'établissement doit avoir des procédures opérationnelles en place qui décrivent le processus de la gestion de l’équipement, du matériel, des produits et des services essentiels utilisés dans toute activité réglementée en vertu du Règlement sur le sang. Les procédures doivent également couvrir la revalidation et le réétalonnage suite à une réparation, un déplacement ou un changement d’équipement essentiel.
Concernant tout équipement, matériel, produit et/ou service non essentiels utilisés dans les activités réglementées, les procédures devraient décrire le processus de la gestion de ceux-ci.
Article 96 Exigences
Exigences
96. Les procédures opérationnelles satisfont aux exigences suivantes :
- elles sont présentées selon un modèle normalisé;
- elles sont approuvées par un cadre supérieur;
- elles sont facilement accessibles à chaque endroit où l’établissement exerce les activités visées par les procédures;
- elles sont tenues à jour.
Consultez l'article 1 pour la définition de « cadre supérieur »
Le format de chaque procédure devrait contenir les renseignements suivants :
- le titre et le but de la procédure;
- le numéro ou le code unique identifiant le document et indiquant la version;
- la date de mise en œuvre et la date de la dernière révision;
- la signature de la personne autorisée et la date de l’autorisation (écrite ou électronique);
- la pagination;
- des directives claires à suivre qui correspondent aux tâches requises pour effectuer l’activité et qui peuvent comprendre la documentation des feuilles de travail, de formulaires ou de champs électroniques;
- les départements responsables de l’exécution des procédures opérationnelles;
- les références des publications citées, les règlements, les lignes directrices et les normes, le cas échéant; et
- les références aux procédures opérationnelles et aux formulaires connexes.
Les procédures opérationnelles doivent être tenues à jour. Elles devraient être examinées et/ou révisées périodiquement au moins tous les deux ans. Les procédures opérationnelles doivent être examinées par une ou plusieurs personnes compétentes et modifiées, le cas échéant, (i) après toute modification du Règlement sur le sang ou de la norme sur le sang de la CSA à laquelle il fait référence, (ii) pour donner suite aux résultats de vérifications ou d’inspections ou (iii) à la suite de mesures correctives ou préventives proposées à la suite d’un manquement, d’un accident ou d’un effet indésirable.
Tout le personnel responsable de mener une procédure doit être formé avant de faire toute tâche associée à une procédure opérationnelle nouvelle ou révisée. Les procédures opérationnelles doivent être accessibles, électroniquement ou en version papier, à l’endroit où des personnes mènent les activités.
Les copies papier des procédures nécessaires au fonctionnement en cas d’urgence (c.-à-d. les procédures utilisées durant la période de non-disponibilité) ( consultez l’alinéa 94(1)k) pour les renseignements relatifs au plan d’intervention d’urgence) doivent être tenues à jour et mises à la disposition du personnel concerné.
Lors d’une situation urgente, une déviation d’une procédure opérationnelle en vigueur est permise si celle-ci est approuvée par un cadre supérieur ou de son désigné et que la déviation est consignée, signée et datée. La raison de la déviation à la procédure doit aussi être consignée. Pour les établissements titulaires d’une licence ou d’un enregistrement, la déviation doit être gérée en conformité avec le système de gestion de la qualité de l’établissement. Pour les déviations entraînant des changements permanents, les procédures doivent être révisées en temps opportun.
La distribution et la tenue à jour des procédures opérationnelles doivent être contrôlées, afin que seules les versions à jour puissent être utilisées. Les versions précédentes des procédures doivent être retirées, archivées et remplacées par la version approuvée appropriée. Les procédures désuètes doivent être retirées et archivées. Chaque version des procédures opérationnelles qui ont été mises en œuvre doit être conservée conformément aux exigences de la période de rétention des dossiers et renseignements décrites aux articles 119 à 122 du Règlement sur le sang.
Article 97 Documentation
Documentation
97. L’établissement qui traite ou transforme du sang veille à ce que ses procédures opérationnelles visant ces activités permettent d’atteindre de façon constante les résultats attendus et conserve les documents justificatifs à cet effet.
Les activités, les processus et les procédures opérationnelles techniques de l’établissement visant le traitement et la transformation du sang et des composants sanguins doivent respecter les critères suivants :
- être validés par l’établissement; ou s’il y a lieu;
- être basés sur des normes fondées sur des pratiques établies et élaborées par des organisations professionnelles pertinentes et reconnues; ou
- appuyés par des renseignements à jour et pertinents tirés de publications scientifiques.
Articles 98-102 Personnel, installations, équipement, matériel et produits
Article 98 Personnel
Personnel
98. (1) L’établissement doit, pour exercer ses activités, avoir en nombre suffisant du personnel dont les membres sont qualifiés de par leurs études, leur formation ou leur expérience pour accomplir les tâches qui leur sont confiées.Compétence
(2) L’établissement offre à son personnel un programme d’orientation et de formation de base et continue et est doté d’un mécanisme d’évaluation des compétences.
L’établissement doit préparer et tenir à jour un organigramme présentant clairement les lignes de responsabilité. Il doit disposer d’un nombre suffisant d’employés qualifiés pour effectuer les tâches requises. Les qualifications, les rôles et les responsabilités du personnel doivent être consignés.
Tous les membres du personnel qui exécutent ou qui sont responsables des activités réglementées doivent détenir les qualifications conformes aux politiques de l’établissement et posséder la combinaison nécessaire de formation et/ou d’expérience. Ils doivent aussi recevoir une formation appropriée en regard de leurs fonctions.
Le personnel doit recevoir une formation initiale et continue, y compris une formation de rattrapage ou un renouvellement de formation, au besoin et selon leurs fonctions. La formation doit être donnée par du personnel qualifié qui a des connaissances liées aux activités concernées. La formation doit être donnée conformément à un programme pour tous les employés qui participent à des activités liées au sang ou aux composants sanguins. La formation doit être donnée avant le début des fonctions du poste ou l’exécution de tâches décrites dans une nouvelle procédure ou toute autre procédure existante révisée. Le personnel qui participe à la conduite d’activités réglementées doit exercer ses fonctions en conformité avec le Règlement sur le sang. Tous les membres du personnel exerçant des activités réglementées devraient connaître les exigences du Règlement sur le sang et de la présente ligne directrice, applicables à leurs responsabilités professionnelles.
L’établissement doit avoir et maintenir un programme d’évaluation des compétences du personnel. Les éléments d’un programme d’évaluation de compétence peuvent comprendre ce qui suit, sans s’y limiter :
- observation directe du rendement;
- surveillance des dossiers;
- examens écrits;
- évaluation de la connaissance des procédures opérationnelles et de la théorie;
- pour le personnel qui effectue normalement les essais requis par le Règlement sur le sang, une évaluation du rendement au moyen d’un programme d’évaluation de l’exactitude et la fiabilité des résultats d’essais tel que décrit à l’alinéa 94(1)h).
Il est nécessaire de tenir un dossier des qualifications, de la formation et de l’évaluation de compétence du personnel tel que décrit aux articles 119-122 du Règlement sur le sang. Les formations doivent être consignées et inclure ce qui suit :
- la ou les dates auxquelles la formation a été donnée et/ou complétée
- le mode de formation; et
- l'identification des formateurs et des personnes formées.
Article 99 Installations
Installations
99. L’établissement titulaire d’une licence ou enregistré se dote d’installations qui permettent ce qui suit :
- l’exercice des activités de l’établissement;
- l’observation de bonnes pratiques d’hygiène par le personnel dans l’exercice de ses fonctions;
- le nettoyage de façon à assurer la salubrité;
- le contrôle approprié des conditions ambiantes dans toutes les zones d’activités;
- le contrôle de l’accès aux zones d’activités;
- l’évaluation préliminaire de donneurs dans des conditions où la confidentialité est assurée.
Les installations doivent être conçues, construites et adaptées de façon à permettre la réalisation des activités. La conception et l’aménagement doivent minimiser le risque d’accidents et de manquements.
Les installations doivent avoir une aire d’évaluation préliminaire des donneurs qui assure leur intimité au moment de déterminer leur admissibilité.
De plus, les installations doivent contrôler l’accès à toutes les aires où leurs activités sont menées, le cas échéant, et l’entrée du bâtiment devrait être surveillée.
Les locaux devraient être conçus en fonction du déroulement du processus de manière à ce que les activités soient réalisées de façon ordonnée et comprendre ce qui suit à des fins de contrôle et de sécurité :
- une aire pour les prélèvements sanguins permettant d’effectuer le prélèvement des donneurs en toute sécurité;
- une aire pour la conservation du matériel et des produits essentiels, y compris les réactifs et les trousses d’essai, avant et après le contrôle de l’assurance de la qualité;
- une aire pour la manipulation des composants sanguins et des réactifs qui sont non conformes pour utilisation, mis en quarantaine, ou visés par un retrait;
- une aire désignée pour la préparation des composants sanguins et les essais de laboratoire;
- une aire désignée pour l’étiquetage et la mise en circulation des composants en inventaire;
- une aire désignée pour la transformation et la manipulation du sang;
- des aires de conservation décrites aux articles 69 à 72 du Règlement sur le sang et dans cette ligne directrice; et
- un contrôle sur la ségrégation et la disposition appropriée des déchets biologiques.
Les installations doivent permettre l’exécution des activités d’une façon propre et hygiénique en accordant une importance particulière à l’hygiène des mains. L’utilisation de vêtements de protection appropriés, selon l’activité menée, doit aussi faire partie des pratiques d’hygiène du personnel.
Les surfaces dans les aires de traitement ne doivent pas être fissurées ni perforées, et les surfaces poreuses doivent être étanchéifiées afin d’en faciliter le nettoyage. Les lieux doivent demeurer propres et hygiéniques. L’établissement devrait disposer d’un programme sanitaire écrit pour assurer le bon entretien des locaux ainsi que d’une procédure en cas de déversement accidentel. Cette dernière devrait comprendre des instructions afin d’éliminer les déversements de sang et de composants sanguins en tant que matière présentant un danger biologique. Le choix des produits de nettoyage utilisés dans les aires de traitement ou de transformation ne doit pas avoir d’effets néfastes sur la sécurité du sang et des composants sanguins. De plus, un programme de lutte antiparasitaire doit être en place.
Au besoin, le bâtiment doit être doté d’un système de chauffage, de ventilation et d’air climatisé (CVAC) approprié afin d’assurer le contrôle de la température et du débit d’air.
Des contrôles environnementaux doivent être mis en place dans les lieux de transformation ou de manipulation du sang pour éviter la contamination du sang. Si une enceinte de sécurité biologique ou une hotte à flux laminaire est utilisée, elle doit être utilisée en fonction des instructions du fabricant. Lors de la transformation ou de la manipulation du sang en système ouvert, une technique aseptique doit être utilisée comme il est décrit à l’alinéa 65e).
Les éléments suivants doivent être pris en considération quand le sang est prélevé dans une collecte mobile :
- conditions ambiantes appropriées (p. ex. température, humidité);
- propreté générale;
- approvisionnement sécuritaire en eau et en électricité;
- espace suffisant pour effectuer les prélèvements sanguins;
- contrôle adéquat de l’accès au sang, aux dossiers et à l’équipement;
- une aire pour l’évaluation préliminaire des donneurs qui assure leur intimité.
Article 100 Équipement
Équipement
100. (1) L’établissement titulaire d’une licence ou enregistré qui utilise de l’équipement essentiel le nettoie, l’entretient et, au besoin, le valide en fonction de son utilisation prévue et l’étalonne.Réparation ou modification
(2) L’établissement, si une réparation ou modification à l’égard de son équipement essentiel le rend nécessaire, doit valider et étalonner de nouveau celui-ci, s’il y a lieu.
Se référer à l’article 1 « Définitions » de la présente ligne directrice pour obtenir la définition du terme « essentiel » et des exemples d’équipement essentiel.
L’équipement devrait être situé dans un endroit qui facilite le nettoyage et l’entretien. Le calendrier établi pour le nettoyage doit être respecté afin de prévenir la contamination et d’assurer la sécurité du sang et des composants sanguins. Les procédures de nettoyage doivent permettre de s’assurer que des résidus de produit de nettoyage ne compromettent pas la sécurité du sang ou des composants sanguins. Les procédures doivent aussi inclure le nettoyage et la désinfection des déversements de sang, le cas échéant.
Pour que l’équipement fonctionne constamment selon les spécifications établies, celui-ci doit être validé et étalonné conformément aux instructions du fabricant et à toute autre spécification supplémentaire requise par le système qualité de l’établissement. Un calendrier et des procédures pour l’entretien et l’étalonnage de l’équipement doivent être tenus à jour et respectés, selon les spécifications précisées dans le manuel d’utilisation de l’équipement. Ces procédures doivent traiter de la fréquence d’étalonnage et inclure les mesures à prendre lorsque la performance de l’équipement s’écarte des limites définies. Cette exigence s’applique non seulement à l’équipement, mais aussi à tous les instruments et appareils de mesure utilisés (p. ex. balances pour peser le sang ou les composants sanguins, thermomètres, sondes de température, etc.) qui sont essentiels pour assurer la conformité du sang et des composants sanguins avec le Règlement sur le sang. L’alinéa 94(1)n) décrit également le programme d’entretien préventif de l’équipement essentiel.
Avant de pouvoir être utilisé, tout équipement qui a été réparé, déplacé ou modifié doit être validé et/ou étalonné de nouveau, conformément au manuel du fabricant et/ou aux procédures opérationnelles de l’établissement. De plus, s’il y a lieu, des mesures devraient être prises pour prévenir tout réglage non intentionnel de l’équipement ou de l’instrument qui pourrait avoir une incidence sur les paramètres d’étalonnage.
Tous les dossiers et rapports de validation, de qualification, d’étalonnage, d’entretien et de réparation doivent être conservés tel que décrit aux sections 119-122 du Règlement sur le sang. Cela inclut les résultats réels des essais effectués sur l’équipement et les appareils de mesure utilisés avec la preuve d’un étalonnage approprié. Si l’entretien préventif est effectué par une tierce partie, les dossiers d’entretien doivent être accessibles, au besoin. Toutefois, l’établissement doit vérifier que l’entretien est effectué correctement et dans les délais définis, selon le calendrier établi dans les procédures. Cette vérification devrait être documentée.
Si un établissement titulaire d’une licence ou d’un enregistrement utilise un système informatique pour des activités réglementées, ce système doit être validé. Tous les établissements non-enregistrés qui exercent des activités réglementées en vertu du Règlement sur le sang devraient également s’assurer que le système informatique utilisé pour les activités réglementées est validé. L’intégrité des données est une considération importante pour tous les systèmes informatiques concernés.
L’intégrité des données se traduit par le maintien et l’assurance de l’exactitude et de la cohérence des données tout au long de leur cycle de vie, et constitue un aspect essentiel de la conception, de la mise en œuvre et de l’utilisation de tout système qui emmagasine, traite ou extrait des données. Des processus et des procédures opérationnelles devraient être établis pour assurer l’entretien et la sécurité des systèmes informatiques et des données qu’ils contiennent. Des mesures de contrôle doivent être en place pour limiter l’accès aux données informatiques afin d’éviter que des changements ou suppressions non autorisés soient apportés aux logiciels ou aux données. De plus, il doit y avoir un moyen de suivre les changements apportés aux données électroniques (c.- à-d. que les pistes de vérification rétrospective doivent être activées et examinées).
Dans le cas d’un établissement qui détient une homologation, toute modification au système informatique doit être homologuée et documentée conformément aux exigences énoncées aux articles 9, 10 et 12 du Règlement sur le sang.
Le programme de validation des systèmes informatiques d’un établissement enregistré et non-enregistré devrait comprendre un essai d’acceptation du système qui porte sur les points suivants :
- fonctionnalité du système;
- rendement du système;
- paramètres essentiels;
- procédures opérationnelles.
Les essais devraient permettre de s’assurer que l’ordinateur fonctionne comme indiqué et qu’il satisfait aux exigences de l’utilisateur.
Une copie de sauvegarde des données des systèmes informatiques essentiels doit être effectuée régulièrement et conservée en lieu sûr pour la récupération des données. Les dossiers de validation des systèmes informatiques doivent être tenus à jour et être utilisés à titre de référence pour la mise à jour ou la modification des systèmes et la récupération des données en cas de défaillance de système. Avant le retour à son utilisation habituelle, il faut démontrer que l’équipement fonctionne conformément aux spécifications établies.
Toute modification, réparation ou mise à jour sur un système électronique essentiel doit être évaluée pour savoir si ce système doit être validé de nouveau.
Article 101 Équipement de conservation
Équipement de conservation
101. L’établissement utilise, pour conserver du sang, de l’équipement qui lui permet de se conformer aux articles 69 à 72.
L’équipement de conservation doit être qualifié et étalonné de manière à démontrer que la température requise peut être maintenue en permanence et que les autres conditions requises peuvent être satisfaites (p. ex. agitation). Un calendrier prédéterminé pour l’entretien de l’équipement doit être établi et respecté afin de garantir la sécurité du sang et des composants sanguins conservés.
Des mesures doivent être prises pour assurer la surveillance constante de l’équipement utilisé pour la conservation du sang ou des composants sanguins, tel que les réfrigérateurs, les congélateurs et les incubateurs. Les dispositifs de surveillance doivent être qualifiés, étalonnés et entretenus correctement. Il est nécessaire de tenir des documents attestant que le sang et les composants sanguins ont été conservés dans des conditions ambiantes appropriées conformément aux articles 117 à 122 du Règlement sur le sang. L’accès à l’équipement de conservation doit être restreint aux personnes autorisées.
L’équipement de conservation devrait être muni d’un système d’alarme automatisé avec signaux sonores pour surveiller les conditions ambiantes requises. Pour la surveillance de la température, les commandes de déclenchement de l’alarme devraient être réglées à des températures qui offrent un délai suffisant pour la prise de mesures correctives avant que le sang ou les composants sanguins n’atteigne des températures inacceptables. Les signaux d’alarme devraient se déclencher dans un endroit faisant l’objet d’une surveillance constante ou occupé par un employé afin que des mesures correctives puissent être prises immédiatement. Si le système utilisé est manuel, des mesures adéquates doivent être en place pour la surveillance de la température et des appareils d’agitation afin de garantir la sécurité du sang et des composants sanguins.
L’établissement doit mettre en place des procédures opérationnelles pour l’entretien de l’équipement susmentionné et un programme de surveillance continue de la température du sang ou des composants sanguins conservé. Les procédures opérationnelles doivent décrire les mesures à prendre en cas d’écart aux plages de température établies ou lors de défaillance de l’agitation. De telles situations doivent être consignées de façon appropriée et faire l’objet d’une enquête.
L’activité de conservation en vertu du Règlement sur le sang comprend la conservation à l’extérieur de la banque de sang. Tous les réfrigérateurs ou incubateurs/agitateurs utilisés pour conserver le sang ou les composants sanguins à l’extérieur de la banque de sang doivent faire l’objet d’une surveillance continuelle pour veiller à ce que le sang et les composants sanguins soient conservés de façon sécuritaire. Si la banque de sang est responsable de la surveillance de ces équipements « satellites » pour la conservation du sang, elle doit disposer des documents prouvant que le sang et les composants sanguins ont été conservés conformément au Règlement sur le sang. Si le sang ou tout composant est conservé temporairement dans des contenants de transport, l’établissement doit s’assurer que les conditions de conservation (durée et température) ont été respectées :
- conformément à sa validation, ou
- en utilisant une autre méthode appropriée pour surveiller la température (p. ex. un enregistreur de données).
La banque de sang doit suivre les procédures en place pour évaluer le sang ou les composants sanguins retournés qui proviennent de ces équipements avant de les remettre dans son inventaire. Pour les équipements de conservation qui ne disposent pas d’une documentation de surveillance appropriée ou qui ne sont pas surveillés par la banque de sang, le sang et les composants sanguins doivent être jetés s’ils sont retournés.
Article 102 Matériel et produits
Matériel et produits
102. L’établissement titulaire d’une licence ou enregistré qui utilise du matériel et des produits essentiels les qualifie ou les valide, selon le cas, en fonction de leur utilisation prévue et les conserve dans des conditions ambiantes adéquates.
Se référer à l’article 1 « Définitions » de la présente ligne directrice pour obtenir la définition du terme « essentiel » et des exemples de matériel et produits essentiels.
L’établissement doit s’assurer que le matériel et les produits essentiels sont validés ou qualifiés, selon le cas, en fonction de leur utilisation prévue. Chaque fois, qu’une nouvelle livraison de matériel ou produits est reçue, la vérification de la qualité du matériel et des produits avant leur mise en circulation est basée sur des spécifications établies et peut inclure un examen visuel, une analyse de mise en circulation des lots et un examen des certificats d’analyse. Si le fabricant possède un certificat d’analyse (ou un document similaire) pour le matériel et les produits essentiels, l’établissement doit alors vérifier ces documents dans le cadre de la mise en circulation des lots de matériel et produits essentiels. Seuls le matériel et les produits qui répondent aux spécifications établies doivent être mis en circulation aux fins d’utilisation.
Les conditions d’utilisation et d’entreposage de chaque matériel et produits doivent répondre aux spécifications du fabricant. Les établissements doivent disposer d’une procédure opérationnelle pour surveiller et respecter strictement les dates de péremption du matériel et des produits essentiels.
Articles 103-108 Enquêtes et rapports concernant les accidents et manquements
Tous les établissements régis par le Règlement sur le sang doivent se conformer aux exigences réglementaires en matière d’accidents et de manquements (A/M), y compris les enquêtes, les rapports et leur déclaration ainsi que la tenue des dossiers. Cette disposition vise non seulement les établissements enregistrés ou titulaires d’une licence auprès de Santé Canada, mais aussi tout établissement, hôpital, banque de sang, laboratoire ou centre qui réalise des activités visées par le Règlement sur le sang. Les exigences relatives au traitement des accidents et des manquements sont énoncées dans les articles 103 à 108 du Règlement sur le sang.
Les manquements et les accidents en vertu du Règlement sur le sang sont définis comme suit :
Manquement : s’entend de l’inobservation des procédures opérationnelles ou des règles de droit applicables qui risque de compromettre la sécurité humaine, ou la sécurité du sang.
Accident : s’entend d’un événement imprévu qui n’est pas imputable à une inobservation des procédures opérationnelles ou des règles de droit applicables et qui risque de compromettre la sécurité humaine, ou la sécurité du sang.
Ces définitions se trouvent aussi dans la section « Définitions » du Règlement sur le sang. Les A/M mentionnés dans le Règlement sur le sang et dans la présente ligne directrice sont limités à ceux qui se sont produits au cours d’une activité réglementée et, comme il est indiqué dans la définition des manquements et des accidents, à ceux qui peuvent compromettre la sécurité du sang ou des composants sanguins.
Les établissements peuvent soupçonner qu’un A/M s’est produit pendant une de ses activités ou pendant une activité réalisée par un autre établissement. Dans tous les cas, les mesures suivantes doivent être prises, le cas échéant :
- Tous les établissements concernés communiquent entre eux afin que tous soient au courant de l’A/M et des résultats d’une enquête.
- Lorsqu’un établissement avise les autres établissements concernés d’un A/M, toute communication de vive voix doit être consignée par écrit et des avis écrits doivent être envoyés le plus rapidement possible, conformément aux paragraphes 103(4) et 104(6).
- En vertu du Règlement sur le sang, l’établissement doit identifier et mettre en quarantaine le sang ou les composants sanguins en cause afin qu’il ne soit ni utilisé pour la transfusion ni distribué et s’assurer qu’il soit mis à l’écart des autres unités de sang et de composants sanguins.
- Comme il est indiqué aux articles 119, 120 et 122 du Règlement sur le sang, tous les établissements doivent tenir des documents relatifs à la distribution.
- Au minimum, les documents relatifs à la distribution doivent contenir les codes d’identification de don du sang, afin de permettre l’identification et la localisation rapides de toutes les unités de sang, conformément à l’article 118.
Incidents qui sont hors de la portée du Règlement sur le sang
Le Règlement sur le sang ne s’applique pas aux incidents mettant en cause des médicaments fabriqués à partir du sang (p. ex. des produits sanguins tels que l’albumine, les immunoglobulines ou les facteurs de coagulation). Pour connaître les exigences en matière de déclaration des produits sanguins, consultez le Règlement sur les aliments et drogues.
De plus, les incidents qui impliquent la pratique de la médecine transfusionnelle ne sont pas couverts dans la portée du Règlement sur le sang. Des exigences provinciales et territoriales en matière de déclaration peuvent s’appliquer. Voici des exemples d’incidents qui sont hors de la portée du Règlement sur le sang :
- mélange lors du prélèvement d’échantillons pré-transfusionnels auprès des patients;
- essai pré-transfusionnel/compatibilité croisée du receveur (groupe et dépistage);
- manquements dans la reprise des essais ABO/Rh dans les hôpitaux;
- la banque de sang envoie le mauvais sang au département (groupe sanguin ou composant);
- l’établissement de prélèvement envoie le mauvais sang à l’hôpital;
- le sang est envoyé au département pour le mauvais patient;
- le département a commandé des composants sanguins irradiés et la banque de sang a envoyé un composant sanguin non irradié (correctement étiqueté comme étant non irradié). Remarque : si le composant sanguin n’était pas irradié, mais était étiqueté comme étant irradié, il s’agirait d’un manquement assujetti à ces exigences;
- transfusion de l’unité de sang ou de composant sanguin au mauvais patient;
- transfusion du sang ou des composants sanguins trop lentement ou trop rapidement;
- les normes de prestation de services ne sont pas respectées;
- l’infirmière décide de placer une unité de sang ou de composant sanguin pendant quelques heures dans un réfrigérateur non surveillé et non destiné à conserver le sang ou le composant sanguin, puis procède à la transfusion (p. ex. réfrigérateur à médicaments);
La transfusion de sang ou de composants sanguins périmés. Remarque : si le sang ou les composants sanguins étaient déjà périmés et distribués sans le savoir par la banque de sang, il s’agirait d’ un manquement en vertu du Règlement sur le sang.
Articles 103-104 Accidents et manquements
Article 103 Accident ou manquement - autre établissement
Accident ou manquement — autre établissement
103. (1) L’établissement qui a des motifs raisonnables de croire que la sécurité du sang a pu être
compromise par un accident ou un manquement lors d’une activité exercée par un autre établissement prend aussitôt les mesures suivantes :
- il relève les codes d’identification du don associés au sang en cause;
- il repère et met en quarantaine le sang en cause qu’il a en sa possession;
- il avise les établissements suivants :
- celui qui a prélevé le sang en cause,
- celui duquel il a reçu le sang en cause, s’il diffère de celui visé au sous-alinéa (i),
- ceux auxquels il a distribué du sang en cause.
Contenu de l’avis
(2) L’avis contient les renseignements suivants :
- les codes d’identification du don associés au sang en cause;
- une mention indiquant que le sang en cause est du sang total ou tel ou tel composant sanguin;
- la raison pour laquelle l’établissement croit que la sécurité du sang a pu être compromise.
Mesure à prendre sur réception de l’avis
(3) L’établissement qui est avisé en application du sous-alinéa (1)c)(iii) ou par application du présent paragraphe en avise aussitôt tout établissement auquel il a distribué du sang en cause et met en quarantaine le sang en cause qu’il a en sa possession.Avis écrit
(4) L’établissement confirme par écrit dans les meilleurs délais tout avis fourni verbalement en application du présent article.
Un établissement qui a des motifs raisonnables de croire, selon les renseignements disponibles, que la sécurité du sang ou des composants sanguins peut avoir été compromise en raison d’un A/M qui s’est produit au cours d’une activité menée par un autre établissement (p. ex. transport, conservation, emballage, transformation, traitement etc.) doit immédiatement prendre sans tarder les mesures indiquées au paragraphe 103(1).
L’établissement doit :
- repérer et mettre en quarantaine tout sang ou composant sanguin en cause (connu ou soupçonné) dans l'A/M
- aviser sans délai tous les établissements concernés.
Dans certains cas, le déroulement de l’enquête d’un A/M peut conduire à la découverte de composants sanguins supplémentaires qui sont touchés.
En vertu du paragraphe 103(2)b), les noms des composants sanguins en cause doivent être inclus dans l’avis (par exemple, les globules rouges, le plasma d’aphérèse et les plaquettes mises en commun). Dans tous les cas, si un avis a été donné verbalement, un avis écrit doit suivre dès que possible.
En vertu du paragraphe 103(3), tous les établissements qui reçoivent l’avis, doivent
- identifier et mettre en quarantaine tout le sang et les composants sanguins en cause (connus ou soupçonnés) qu’ils ont en leur possession; et
- transmettre l’avis à tout établissement auquel ils ont distribué du sang ou des composants sanguins en cause (connus ou soupçonnés).
Article 104 Accident ou manquement - établissement
Accident ou manquement — établissement
104. (1) L’établissement qui reçoit un avis en application des sous-alinéas 103(1)c)(i) ou (ii) ou qui soupçonne que la sécurité du sang a pu être compromise par un accident ou un manquement lors d’une activité qu’il a exercée prend sans délai les mesures suivantes :
- il relève les codes d’identification du don associés au sang en cause;
- il repère et met en quarantaine le sang en cause qu’il a en sa possession;
- il décide s’il y a suffisamment de preuves pour justifier une enquête sur l’accident ou le manquement soupçonné.
Enquête non justifiée — avis
(2) L’établissement qui conclut que les preuves ne justifient pas une enquête en avise l’établissement qui l’a avisé en application des sous-alinéas 103(1)c)(i) ou (ii) et lui fournit les motifs de sa conclusion.Mesures à prendre sur réception de l’avis
(3) L’établissement qui est avisé en application du paragraphe (2) ou par application du présent paragraphe en avise aussitôt tout établissement auquel il a distribué du sang en cause.Enquête — avis
(4) L’établissement qui conclut qu’il y a suffisamment de preuves pour justifier une enquête y procède et en avise tout établissement ou toute autre personne auxquels il a distribué du sang en cause en précisant les renseignements suivants :
- les codes d’identification du don associés au sang en cause;
- des explications sur la nature de l’accident ou du manquement soupçonné et la manière dont la sécurité du sang en cause a pu être compromise.
Mesures à prendre sur réception de l’avis
(5) L’établissement qui est avisé en application du paragraphe (4) ou par application du présent paragraphe en avise aussitôt tout établissement auquel il a distribué du sang en cause et met en quarantaine le sang en cause qu’il a en sa possession.Avis écrit
(6) L’établissement confirme par écrit dans les meilleurs délais tout avis fourni verbalement en application du présent article.
L’article 104 traite des cas où un établissement soupçonne qu’un accident ou un manquement (A/M) s’est produit lors de l’une de ses activités ou lorsqu’il est avisé qu’un accident ou un manquement aurait pu se produire à son établissement.
En vertu du paragraphe 104(1), à la réception de l’avis, ou lorsqu’un établissement soupçonne qu’un A/M s’est produit au cours d’une activités qu’il a menée, l’établissement doit immédiatement :
- déterminer les codes d’identification du don associés au sang ou aux composants sanguins en cause et déterminer si d’autres unités de sang ont pu être affectées par cet A/M (p. ex. autres composants ayant le même code d’identification du don, autres unités soumises aux mêmes conditions de traitement, de transformation ou de conservation etc.);
- repérer et mettre en quarantaine le sang et les composants sanguins en cause qu’il a en sa possession; et
- déterminer s’il y a suffisamment de preuves que l’A/M s’est produit à son établissement afin de lancer une enquête.
Chaque incident nécessite une évaluation afin de déterminer si un A/M a réellement eu lieu et s’il s’est produit au cours des activités menées dans son établissement. Si une évaluation détermine qu’un A/M n’est pas lié aux activités réglementées réalisées par l’établissement, cette conclusion doit être consignée.
Mener une enquête
Lorsqu’un A/M affecte la sécurité du sang et/ou des composants sanguins, l'établissement doit immédiatement :
- identifier et mettre en quarantaine tout sang et composant sanguin en cause en inventaire;
- déterminer si d’autre sang et d’autres composants sanguins sont en cause et établir leur statut (distribués, en quarantaine ou transfusés); et
- déterminer s’il existe d’autres établissements concernés auxquels du sang et/ou des composants sanguins ont été distribués.
Lorsqu’une enquête est initiée, l’établissement doit :
- aviser tous les établissements ou autres personnes (y compris les fabricants de produits sanguins) auxquels il a distribué du sang ou des composants sanguins dont l’implication est connue ou soupçonnée qu’une enquête est en cours.
Dans l’avis, l’établissement doit :
- énumérer les codes de don connus ou soupçonnés du sang et des composants sanguins potentiellement en cause;
- décrire l’A/M soupçonné;
- inclure une explication de la manière dont la sécurité du sang ou des composants sanguins en cause a pu être compromise.
Si l’information a été communiquée de vive voix, un avis écrit doit suivre sans tarder.
Le sang et les composants sanguins connus et soupçonnés d’être en cause doivent rester en quarantaine jusqu’à ce que l’enquête soit terminée et que le sort final du sang et des composants sanguins ait été déterminé (c’est-à-dire remise en inventaire, retrait ou élimination).
En plus de la mise en quarantaine ou du retrait immédiat du sang et des composants sanguins concernés, l’enquête sur l’A/M doit comprendre :
- une analyse des causes fondamentales
- une considération de l’impact potentiel de l’A/M sur la sécurité du sang ou des composants sanguins et du receveur, si le sang ou les composants sanguins avaient été transfusés (même s’ils n’avaient pas été transfusés);
- une élaboration de mesures correctives appropriées.
Même si l’A/M a été détecté par un autre établissement avant la transfusion, l’établissement initial où l’A/M s’est produit est tenu d’enquêter et de prendre les mesures correctives et préventives appropriées.
Chaque enquête requiert une évaluation initiale pour déterminer si des activités réglementées sont en cause et si la sécurité du sang ou de tout composant sanguin a été compromise. Une enquête peut être très simple ou très complexe selon la nature et la gravité de l’incident, la taille et la complexité de l’établissement et/ou de ses activités. Il peut être nécessaire de procéder à l’examen de multiples aspects afin de déterminer l’étendue complète de ce qui s’est passé :les processus/procédures pertinents :
- les processus/procédures pertinents;
- les activités réglementées en cause;
- ce qu’a été l’impact sur la sécurité du sang ou ce qu’elle aurait pu être.
Les détails de toutes les mesures prises pendant l’enquête doivent être documentés.
L’enquête doit déterminer ce qui a contribué à la survenue de l’incident afin d’en déterminer les causes fondamentales, de sorte que des mesures correctives et préventives puissent être mises en œuvre pour atténuer efficacement le risque et d’empêcher que l’incident ne se reproduise. Les mesures correctives et préventives dépendront de la nature et de la gravité de l’incident, du personnel, des processus, de l’équipement, du matériel en cause et des causes identifiées ayant contribué à la survenue de l’incident. Les mesures correctives et préventives peuvent inclure, sans s’y limiter, ce qui suit :
- le renouvellement de la formation du personnel;
- les révisions des procédures;
- l’ajout d’étapes de vérification;
- les modifications apportées à un ou plusieurs systèmes.
Les mesures correctives et préventives doivent être mises en œuvre en temps opportun et leur efficacité doit être évaluée.
Le type d’incident doit être défini (c.-à-d. accident/manquement et/ou effet indésirable) afin de déterminer le déroulement de l’enquête. Les établissements devraient savoir que certains A/M peuvent provoquer des effets indésirables et qu’une enquête devrait être menée à la fois pour l’effet indésirable et l’A/M en question. Il devrait y avoir un lien clair entre les procédures relatives aux A/M et aux effets indésirables afin de garantir que, si les A/M entraînent des effets indésirables, ces cas sont traités de manière appropriée.
Lorsqu’une enquête n’est pas nécessaire
Lorsqu’un établissement reçoit un avis d’accident ou de manquement soupçonné :
Il existe plusieurs scénarios dans lesquels un établissement peut recevoir un avis d’un A/M soupçonné et déterminer qu’une enquête n’est pas justifiée. Par exemple :
- l’établissement B soupçonne qu’un A/M s’est produit dans l’établissement A et l’en avise
- à la réception de l’avis, l’établissement A est en mesure de confirmer que l’A/M soupçonné n’est pas valable.
Voici un autre exemple :
- l’établissement Y est avisé d’un A/M soupçonné par l’établissement Z auquel il a distribué du sang;
- après examen des faits, l’établissement Y ne croit pas que cela soit dû à une activité qu’il a menée. En d’autres termes, si la nature de l’A/M suggère qu’il s’est produit dans l’établissement de prélèvement X, alors une enquête peut ne pas être justifiée par l’établissement Y ou Z.
En vertu du paragraphe 104(2), dans les cas où un établissement qui a prélevé ou distribué du sang ou des composants sanguins en cause reçoit un avis et qu’il n’a aucun motif raisonnable de croire que la sécurité du sang et de tout composant a été compromise par l’A/M soupçonné survenu lors de l’une de ses activités, il doit aviser l’établissement qui lui a transmis l’avis qu’il ne procédera pas à une enquête. L’établissement doit fournir les motifs de sa décision de ne pas procéder à une enquête et conserver dans ses dossiers la justification détaillée. Cette mesure vise à garantir que tous les A/M soupçonnés ont été pris en considération et ont été évalués.
Dans le cas où un établissement a envoyé un avis à plusieurs établissements [en vertu des sous-alinéas 103(1)c)(i) et (ii)], la décision finale sur le sort du sang doit être fondée sur les évaluations de tous les établissements concernés.
En vertu du paragraphe 104(3), tout établissement qui reçoit un tel avis doit transmettre cet avis à tout établissement auquel il a distribué du sang ou des composants sanguins en cause. Si un avis verbal est donné, un avis écrit doit être envoyé dès que possible par la suite.
Lorsqu’un établissement soupçonne qu’un accident ou un manquement est survenu lors de l’une de ses activités :
Si un établissement soupçonne qu’un A/M s’est produit au cours d’une activité qu’il a menée et qui pourrait compromettre la sécurité du sang ou des composants sanguins, et détermine par la suite qu’une enquête n’est pas justifiée, il doit documenter cette décision avec une justification détaillée. La décision documentée doit être conservée, conformément aux articles 119-122 du Règlement sur le sang. Par exemple, le bloc opératoire avise la banque de sang d’un cas soupçonné d’A/M. La banque de sang évalue le sang et les composants sanguins et est en mesure de confirmer que le A/M soupçonné n’est pas valide.
Articles 105-108 Enquête et rapports
Article 105 Assistance à l'enquête
Assistance à l’enquête
105. (1) L’établissement fournit, sur demande de l’établissement qui effectue une enquête, les renseignements ou documents utiles qu’il a en sa possession concernant le sang qu’il a distribué ou transfusé.Communication des renseignements
(2) L’établissement concerné par un accident, un manquement ou une enquête à leur égard communique les renseignements ou documents utiles qu’il a en sa possession à tout autre établissement pareillement concerné et le tient au courant des progrès de l’enquête.
Les établissements doivent coopérer avec tout établissement qui effectue une enquête et fournir, sur demande, les renseignements pertinents. Cette information comprend, sans toutefois s’y limiter, une liste d’inventaire du sang et des composants sanguins en cause avec leur disposition (p. ex. distribué, transfusé, mis en quarantaine) ainsi que du nom des établissements auxquels le sang ou tout composant en cause a été distribué.
Conformément au Règlement sur le sang, il est essentiel que tous les établissements concernés communiquent entre eux afin que tous les établissements reçoivent l’information pertinente relative à l’enquête. Par conséquent, les établissements doivent aviser les établissements concernés, y compris ceux auxquels ils ont expédié du sang ou des composants sanguins en cause, de la tenue d’une enquête sur un accident ou un manquement soupçonné ainsi que de tout élément nouveau ou problème qui survient pendant l’enquête.
L’établissement qui mène une enquête doit avoir des processus en place pour communiquer avec tous les établissements qui peuvent avoir été touchés par l’A/M de manière opportune et précise.
Si un établissement envoie un avis à plusieurs établissements (103(1)c)(i) et (ii)), la décision finale sur le sort du sang doit être fondée sur les évaluations de tous les établissements concernés. Si aucun des établissements avisés ne procède à une enquête, tous les établissements concernés doivent communiquer pour déterminer du sort du sang et des composants sanguins.
Article 106 Résultats de l'enquête
Résultats de l’enquête
106. (1) L’établissement qui effectue une enquête avise par écrit tout établissement ou toute autre personne auxquels il a distribué du sang en cause des résultats de l’enquête et des mesures à prendre, le cas échéant.Envoi d’une copie de l’avis
(2) L’établissement qui reçoit l’avis en application du paragraphe (1) ou qui en reçoit copie par application du présent paragraphe en envoie copie à tout établissement auquel il a distribué du sang en cause.
L’établissement doit aviser par écrit tout établissement concerné ou toute autre personne (y compris les fabricants de produits sanguins) des résultats de l’enquête. Lorsque les résultats de l’enquête indiquent que la sécurité du sang ou des composants sanguins en cause n’a pas été compromise, l’établissement qui effectue l’enquête peut formuler des recommandations quant au sort du sang et des composants sanguins.
Comme il est indiqué à l’alinéa 4(7)b) de la section « Interdictions » du Règlement sur le sang, lorsque les résultats de l’enquête révèlent que la sécurité du sang ou des composants sanguins en cause a été compromise ou lorsque les résultats ne sont pas concluants, le sang et les composants sanguins en cause ne doivent pas être distribués ni transfusés.
À la réception de l’avis envoyé en vertu des paragraphes 106(1) ou 106(2), l’établissement doit envoyer une copie de l’avis écrit à chaque établissement auquel il a distribué le sang ou les composants sanguins en cause.
Article 107 Rapports au ministre
Rapports au ministre
107. (1) L’établissement qui effectue, dans le cas de sang déjà distribué ou transfusé, une enquête sur un accident ou un manquement soupçonné de s’être produit lors d’une activité qu’il a exercée et de pouvoir vraisemblablement entraîner un effet indésirable grave présente au ministre les rapports prévus au paragraphe (2).Contenu et délais
(2) Les rapports ci-après sont présentés dans les délais suivants :
- un rapport préliminaire contenant tout renseignement utile connu, dans les vingt-quatre heures suivant le début de l’enquête;
- un rapport écrit faisant état de tout nouveau renseignement concernant l’accident ou le manquement soupçonné, du progrès de l’enquête depuis le dernier rapport et des mesures prises pour limiter les risques pour l’avenir dans les délais suivants :
- dans les quinze jours suivant le début de l’enquête,
- à tout moment après le rapport préliminaire, sur demande du ministre.
Rapport écrit
(3) L’établissement qui présente verbalement le rapport prévu à l’alinéa (2)a) le présente ensuite par écrit dans les meilleurs délais.
Rapport d’enquête final au ministre
(4) L’établissement, au terme de son enquête, présente au ministre un rapport d’enquête final qui contient les renseignements et documents suivants :
- les conclusions de l’enquête;
- le sort réservé au sang en cause et les raisons l’ayant justifié;
- toute mesure corrective prise et toute modification recommandée aux processus visés.
L’article 107 s’applique à tous les établissements qui distribuent et aux établissements qui transfusent du sang ou des composants sanguins et qui font enquête sur des cas soupçonnés d’A/Ms survenus lors d’une activité réglementée qu’ils ont menée.
L’établissement qui mène une enquête doit déposer un rapport préliminaire auprès de Santé Canada dans les 24 heures suivant le début de l’enquête, si les trois critères ci-dessous sont remplis.
- L’A/M se serait produit au cours d’une activité réglementée qu’il a menée;
- L’A/M a été identifié après la distribution ou la transfusion du sang ou des composants sanguins
- Il existe une probabilité raisonnable que l’A/M aurait pu entraîner un effet indésirable grave si le sang ou les composants sanguins avaient été transfusés.
Consultez les explications additionnelles ci-dessous pour chaque critère :
- Si l’A/M s’est produit au cours d’une activité réglementée menée dans un établissement différent de celui qui l’a découvert, il incombe à l’établissement où l’A/M s’est produit de le déclarer à Santé Canada. Les incidents liés à la pratique de la médecine transfusionnelle ne sont pas inclus dans la portée du présent règlement. Consultez les exemples d’exclusions sous les articles 103 à 108 « Enquêtes et rapports concernant les accidents et les manquements ».
- Le terme « distribution » dans le Règlement sur le sang est expliqué, à l’article 1« Interprétation, de la présente ligne directrice ». Le deuxième critère consiste à savoir si le sang était déjà distribué ou non lorsque l’A/M a été découvert. En général, le sang est distribué avant la transfusion. Par conséquent, un A/M peut toujours être à déclarer si le sang a été distribué, même s’il n’a pas encore été transfusé, puisque le deuxième critère a été rempli. Toutefois, à titre exceptionnel, le sang prélevé dans le cadre d’un programme de donneurs pré-évalués n’est pas distribué avant la transfusion. Dans ce cas, le critère 2 ne serait rempli pour un A/M détecté que si le sang était transfusé.
- Le troisième critère consiste à évaluer l’impact potentiel de l’A/M. Même si le sang ou les composants sanguins n’ont pas été transfusés, l’établissement (en consultation avec le directeur médical ou un spécialiste dans le domaine) doit évaluer l’impact potentiel qu’ils auraient pu avoir sur le receveur s’ils avaient été transfusés. Si le sang ou un composant a été transfusé et qu’il n’y a pas eu d’effet indésirable, le troisième critère pourrait tout de même être rencontré, s’il existe une probabilité qu’un effet indésirable grave ait pu se produire. Le directeur médical ou un spécialiste dans le domaine devrait être consulté pour évaluer si le troisième critère est rempli.
Si un A/M est découvert au cours d’une enquête sur un effet indésirable, cet A/M doit également être déclaré (séparément de l’effet indésirable déclaré en vertu de l’article 113) aux coordonnées fournies ci-dessous.
Le rapport préliminaire doit inclure toute information disponible à ce moment-là concernant l’A/M soupçonné. Les renseignements fournis dans le rapport préliminaire peuvent inclure, sans toutefois s’y limiter :
- une description de l’ A/M,
- des évaluations du risque,
- le nombre et les types (c.-à-d. les globules rouges, les plaquettes, le plasma, etc.) des unités de sang et/ ou de composants sanguins en cause,
- les corrections prises à ce jour (y compris tout retrait ou avis envoyé aux établissements ayant reçu le sang et/ou les composants sanguins en cause) et
- toute mesure corrective et préventive anticipée.
Remarque : Les corrections correspondent aux mesures immédiates prises pour éliminer une non-conformité détectée. Les mesures correctives sont axées sur l’élimination des causes de non-conformité existantes ou d’incidents afin d’empêcher la récurrence, tandis que les mesures préventives mettent l’accent sur l’élimination des causes de non-conformité potentielles pour éviter l’occurrence.
Dans le cas de sang ou de composants sanguins importés au Canada, si l’établissement étranger effectue une enquête sur un manquement ou un accident concernant le sang ou les composants sanguins distribués au Canada, et s’il y a une probabilité raisonnable que l’A/M puisse entraîner un effet indésirable grave, l’importateur canadien doit déclarer à Santé Canada qu’une enquête est en cours, puisque l’établissement étranger mène les activités en son nom.
Les A/Ms découverts avant la distribution ou la transfusion de sang ou des composants sanguins n’ont pas à être déclarés, mais doivent tout de même faire l’objet d’une enquête par le ou les établissements appropriés (c.-à-d. l’établissement étranger et/ou l’importateur) et être consignés par l’importateur dans un rapport annuel. Consultez l’article 108 – Rapport annuel.
Déclaration des accidents et manquements
Tous les établissements doivent fournir à Santé Canada des rapports sur les A/M et toute information requise en vertu de l’article 107 du Règlement sur le sang par :
Courriel (méthode privilégiée) : bpcp-pcpb@hc-sc.gc.ca En objet « [Nom de l’établissement] – Rapport sur un A/M affectant le sang ».
ou
Télécopieur : 613-960-2156
Un formulaire de rapport d’enquête concernant un accident ou un manquement (FRM-0337) est disponible sur le site Web de Santé Canada.
Rapport préliminaire
Il est reconnu que tous les renseignements ne sont pas nécessairement disponibles au moment de la déclaration initiale.
Mises à jour écrites
À la suite du rapport préliminaire, l’établissement doit fournir, une mise à jour écrite de toute nouvelle information concernant l’A/M soupçonné dans les quinze jours civils suivant le début de l’enquête.
La mise à jour doit inclure des renseignements concernant :
- le statut de toutes les unités de sang en cause
- le nombre d’établissements concernés qui ont été avisés.
- les progrès réalisés dans l’enquête depuis le dernier rapport
- les corrections, les mesures correctives immédiates et les mesures prises pour atténuer d’autres risques, comme la conduite d’un retrait et les modifications apportées aux processus pertinents.
- les dates pertinentes de toutes les mesures qui ont été prises
Santé Canada peut également demander une mise à jour à tout moment après le rapport préliminaire et/ou ordonner un retrait sur la base des renseignements reçus.
Rapport final
À la fin de l’enquête, l’établissement doit aviser le Santé Canada et lui fournir un rapport final.
Le rapport final doit inclure :
- les résultats de l’enquête, y compris l’analyse des causes fondamentales le nom;
- le nom du ou des agents infectieux en cause, le cas échéant,;
- les résultats de tous les essais effectués;
- le suivi et les mesures correctives prises (c.-à-d. les mesures prises pour prévenir la récurrence );
- les détails du sort final du sang et des composants sanguins, (p.ex. le nombre d’unités distribuées, transfusées, mises en quarantaine, remises en inventaire et jetées), et les raisons de cette disposition;
- tout changement recommandé aux processus pertinents;
- les dates pertinentes de toutes les mesures prises;
- toute information supplémentaire non communiquée précédemment à Santé Canada.
Scénarios
Les exemples qui suivent sont fournis à titre indicatif seulement et fournissent de l’aide supplémentaire sur les étapes nécessaires à suivre par un établissement lorsqu’un A/M particulier est soupçonné.
Les types de situations données en exemple ci-dessous doivent être déclarés à Santé Canada dans les 24 heures suivant le début de l’enquête, car on considère que les trois critères, énoncés à l’article 107, sont rencontrés.
Exemples
- Le personnel de l’étage a commandé des globules rouges irradiés. Le laboratoire des services de médecine transfusionnelle ou la banque de sang a transformé l’unité dans le système électronique et l’a renommée « globules rouges irradiés » Cependant, l’unité n’a jamais été réellement irradiée. Le sang a été distribué à l’étage pour la transfusion et l’infirmière a remarqué que l’indicateur Rad-Sure n’était pas de la bonne couleur. Elle a communiqué avec la banque du sang et lui a retourné l’unité. Il a été confirmé que l’unité n’était effectivement pas irradiée.
- Une unité de globules rouges a été laissée à l’extérieur de son réfrigérateur de conservation pendant 120 minutes dans la banque de sang. La procédure de la banque de sang stipule que la durée maximale durant laquelle il est permis de laisser une unité à l’extérieur d’un environnement à température contrôlée est de 60 minutes, comme l’indique la norme CSA sur le sang. L’unité a ensuite été distribuée sans que la température du sang n’ait été confirmée.
- Deux unités de globules rouges ont été distribuées à la salle d’opération. À la fin de chaque journée, un employé de la banque de sang récupère le sang dans le réfrigérateur de la salle d’opération et le ramène à la banque de sang. Cependant, trois jours plus tard, ces deux unités ont été trouvées dans le réfrigérateur de la salle d’opération sans qu’aucun dossier ne démontre où elles avaient été entreposées au cours des trois derniers jours. Les deux unités de globules rouges ont été ramenées à la banque de sang, mais n’ont pas été mises en quarantaine comme il se doit. Elles ont plutôt été remises en inventaire et redistribuées.
- Pour le transport des globules rouges, les blocs réfrigérants étaient entreposés à une température inférieure à la spécification requise. Le sang est arrivé sous les limites de température acceptables et a été jugé non sécuritaire pour la transfusion. L’établissement qui a reçu le sang doit en informer l’établissement qui lui a envoyé le sang. L’établissement qui a expédié le sang est tenu de déclarer cet A/M à Santé Canada, car il est responsable de l’emballage du sang de manière à ce qu’il demeure dans les limites de température requise pendant le transport.
- L’établissement de prélèvement a envoyé un avis de retrait de composants à un hôpital demandant que deux unités de globules rouges soient mises en quarantaine parce que les unités de plaquettes correspondantes associées aux unités de globules rouges ont donné un résultat positif à l’essai de croissance bactérienne effectué à l’aide du système de la mise à l’essai bactériologique BacT. Le personnel de l’hôpital a récupéré les deux unités de globules rouges et les a placées dans la section de mise en quarantaine du réfrigérateur, mais a mal lu un des numéros de l’unité et a mis en quarantaine la mauvaise unité de globules rouges. L’unité faisant l’objet du retrait a été transfusée après que l’hôpital a traité l’avis de retrait et ce n’est qu’après que les unités ont été retournées à l’établissement de prélèvement que le manquement a été constaté. L’hôpital est tenu de déclarer cet A/M à Santé Canada.
- L’établissement de prélèvement a effectué une étude de dons antérieurs pour un donneur de plasma qui a été déclaré positif pour une maladie transmissible. Cependant, une unité en cause n’a pas été correctement mise en quarantaine et a été distribuée au fabricant de produits sanguins.
Remarque : En tout temps, si l’on découvre qu’un A/M qui n’a pas été déclaré aurait dû l’être, alors celui-ci doit immédiatement faire l’objet d’une déclaration. Par exemple, la constatation pourrait avoir lieu pendant la préparation du rapport annuel (article 108), l’examen de la qualité, la vérification interne, l’inspection réglementaire, etc.
Article 108 Rapport annuel
Rapport annuel
108. (1) L’établissement établit un rapport annuel qui, non seulement résume les enquêtes sur les accidents et manquements qu’il a effectuées au cours des douze derniers mois, mais encore en fait une analyse critique et concise, et le présente sur demande au ministre.Avis — ministre
(2) L’établissement, si l’analyse fait état de risques pour la sécurité du sang qui n’avaient pas été envisagés, en avise aussitôt le ministre.
Rapports additionnels
(3) L’établissement présente au ministre un rapport additionnel conforme au paragraphe (1) pour toute période qu’il précise dans sa demande.
Les exigences précisées à l’article 108 s’appliquent à tous les établissements qui sont régis en vertu du Règlement sur le sang y compris les établissements dont les activités ne requièrent pas l’obtention d’une licence ou d’un enregistrement.
Tous les établissements doivent préparer un rapport annuel qui comprend toutes les enquêtes sur les A/M menées par leur établissement au cours des 12 derniers mois. Cela comprend tous les A/M qui relèvent de la portée du Règlement sur le sang, identifiés avant et après la distribution ou la transfusion du sang et des composants sanguins.
Le rapport peut être divisé en catégories (p.ex., types d’A/M, types de composants sanguins, sites où ils se sont produits, activités, secteurs, risques, distribués ou non distribués etc.). L’établissement doit y inclure une analyse des enquêtes qui identifie clairement tout problème récurrent ou toute tendance. D’autres mesures correctives peuvent être nécessaires si des problèmes récurrents et des tendances sont observés. Ce processus doit être clairement décrit dans les procédures de l’établissement. Ces rapports doivent être soumis à Santé Canada, à tout moment sur demande. Les rapports annuels peuvent également être demandés et examinés avant ou pendant une inspection de Santé Canada. De plus, Santé Canada peut exiger que l’établissement prépare et soumet des rapports supplémentaires pour une période de temps précise. Si au cours de l’analyse des enquêtes, on découvre un A/M qui aurait dû être déclaré à Santé Canada, l’établissement doit en informer immédiatement Santé Canada. Les avis écrits devraient être envoyés à l’adresse suivante :
Courriel (méthode privilégiée) : bpcp-pcpb@hc-sc.gc.ca
Ou
Télécopieur : 613-960-2156
Articles 109-116 Enquêtes et rapports concernant les effets indésirables
Article 109 Effets indésirables chez le donneur
Article 109 Avis au ministre
Avis au ministre
109. (1) L’établissement qui a des motifs raisonnables de croire qu’un effet indésirable grave s’est produit chez le donneur lors du prélèvement de son sang ou dans les soixante-douze heures qui suivent, en avise le ministre au plus tard vingt-quatre heures après avoir pris connaissance de la mort du donneur ou au plus tard quinze jours après avoir pris connaissance de tout autre effet indésirable.Contenu de l’avis
(2) L’avis contient les renseignements suivants :
- la description de l’effet indésirable;
- toute mesure prise à son égard;
- l’issue.
Avis écrit
(3) L’établissement confirme par écrit dans les meilleurs délais tout avis fourni verbalement en application du présent article.
109(1) Un effet indésirable grave chez un donneur peut survenir à la suite d’un don de sang total ou par aphérèse. Lorsqu’un effet indésirable grave survient chez un donneur pendant un don ou dans les 72 heures après un don, et si l’effet peut poser un risque pour la sécurité du sang et des composants sanguins, l’établissement qui a prélevé le sang doit en aviser le ministre dans les 15 jours civils. Si le donneur décède pendant un don de sang ou dans les 72 heures après le don, l’établissement qui a prélevé le sang doit aviser le ministre dans les 24 heures après avoir eu connaissance du décès. Un établissement titulaire d’une licence ou un établissement enregistré devrait consulter l’article 1, Définitions, pour obtenir les définitions d’« effet indésirable » et d’ « effet indésirable grave » afin de déterminer ce qui doit être déclaré comme effet indésirable chez un donneur.
109(2) Lorsqu’un établissement titulaire d’une licence ou un établissement enregistré déclare un effet indésirable grave chez un donneur, l’avis doit contenir au moins une description des éléments suivants :
- l’effet indésirable;
- les mesures prises; et
- les conséquences
L’établissement titulaire d’une licence ou l’établissement enregistré doit décrire les mesures prises vis-à-vis de l’effet indésirable grave, y compris le traitement du donneur. L’avis doit comprendre les conséquences de l’effet indésirable grave du donneur, c.-à-d. si le résultat n’occasionne aucune exclusion du donneur ou occasionne une exclusion temporaire ou indéfinie de celui-ci. L’identité du donneur ne doit pas être inscrite dans l’avis.
L’avis devrait contenir tous les renseignements suivants :
- le code d’identification du donneur;
- le code du don;
- l’âge et le sexe du donneur;
- une description de l’effet, y compris :
- la date, l’heure et le lieu;
- le type de donneur (don fait à des fins allogéniques ou autologues);
- les antécédents du donneur (dons répétés ou premier don);
- le type de don (sang total, plasmaphérèse, cytaphérèse);
- les symptômes cliniques;
- l’évaluation de l’effet et son lien avec le don;
- les séquelles.
Dans l’éventualité où l’information ci-dessus ne soit pas disponible au moment de présenter un rapport, un avis contenant toute l’information devrait être soumis à Santé Canada le plus rapidement possible après la transmission du rapport initial.
109(3) Lorsque l’établissement titulaire d’une licence ou l’établissement enregistré déclare verbalement à Santé Canada un effet indésirable grave chez le donneur, il doit, dans les plus brefs délais, fournir un avis écrit.
Pour autant que toutes les exigences en matière de rapport soient respectées, un établissement peut utiliser n’importe quel formulaire de déclaration des effets ou des événements indésirables pour aviser Santé Canada par écrit.
Le formulaire rempli doit être envoyé par télécopieur ou par courriel à la Direction des médicaments biologiques et radiopharmaceutiques de Santé Canada :
Bureau des affaires réglementaires
Direction des médicaments biologiques et radiopharmaceutiques
Direction générale des produits de santé et des aliments, Santé Canada
Tél. : 613-957-1722
Téléc. : 613-946-9520
L’établissement peut envoyer par courriel une image numérisée protégée par mot de passe du formulaire, à BRDD.ORA@hc-sc.gc.ca.
Articles 110-111 Effets indésirables chez le receveur
En vertu du Règlement sur le sang, un effet indésirable imprévu ou grave chez le receveur doit être déclaré à Santé Canada s’il est lié à un effet indésirable chez le receveur au sang ou aux composants sanguins transfusés, qui indique que la sécurité du sang pourrait être compromise et qu’il y a un risque pour la sécurité humaine. Un établissement devrait consulter l’article 1, Interprétation, pour obtenir les définitions d’« effet indésirable », d’« effet indésirable grave » et d’« effet indésirable imprévu » pour déterminer ce qui doit être déclaré à Santé Canada. Un effet indésirable causé par un manquement d’étiquetage du sang qui compromet la sécurité du sang et des composants sanguins et mène à un effet indésirable chez un receveur est un exemple d’un effet indésirable devant être déclaré.
Les effets indésirables à déclaration obligatoire chez un receveur ne s’appliquent qu’au sang humain prélevé auprès de donneurs aux fins de transfusion ou d’immunisation de donneurs de plasma destiné au fractionnement (p. ex. globules rouges pour l’immunisation et production de facteur Rh ou anti-D). Les exigences relatives à la déclaration des effets indésirables en vertu du Règlement sur le sang ne s’appliquent pas aux produits sanguins fabriqués par un fabricant de produits sanguins (souvent appelés produits de protéines plasmatiques, dérivés plasmatiques, produits plasmatiques fractionnés ou produits sanguins fractionnés).
Article 110 Mesures à prendre
Mesures à prendre
110. (1) Sous réserve de l’article 111, l’établissement qui a des motifs raisonnables de croire qu’un effet indésirable grave ou un effet indésirable imprévu s’est produit chez le receveur prend sans délai les mesures suivantes :
- il relève les codes d’identification du don associés au sang en cause;
- il repère et met en quarantaine le sang en cause qu’il a en sa possession;
- si la cause fondamentale de l’effet indésirable est, selon un examen préliminaire, attribuable à une activité exercée par lui, il procède à une enquête et en avise tout établissement auquel il a distribué du sang en cause;
- si la cause fondamentale de l’effet indésirable est, selon un examen préliminaire, attribuable à une activité exercée par un autre établissement, il en avise les établissements suivants :
- celui qui a prélevé le sang en cause,
- celui duquel il a reçu le sang en cause, s’il diffère de celui visé au sous-alinéa (i),
- ceux auxquels il a distribué du sang en cause.
Contenu de l’avis
(2) L’avis exigé aux alinéas (1)c) et d) contient les renseignements suivants :
- la description de l’effet indésirable;
- l’explication de la manière dont la sécurité du sang en cause a pu être compromise, si elle est connue;
- les codes d’identification du don associés au sang en cause;
- une mention précisant que le sang en cause est du sang total ou tel ou tel composant sanguin;
- le nom de toute maladie transmissible soupçonnée ou de son agent, s’ils sont connus.
Mise en quarantaine
(3) L’établissement qui est avisé en application du paragraphe (1) ou par application du présent paragraphe en avise aussitôt tout établissement ou toute autre personne auxquels il a distribué du sang en cause et met en quarantaine le sang en cause qu’il a en sa possession.Enquête
(4) L’établissement qui est avisé en application du sous-alinéa (1)d) (i) ou (ii) doit, si la cause fondamentale de l’effet indésirable est, selon un examen préliminaire, attribuable à une activité exercée par lui, procéder à l’enquête.Avis écrit
(5) L’établissement confirme par écrit dans les meilleurs délais tout avis fourni verbalement en application du présent article.
110(1) L’organigramme sur les exigences en matière d’enquête (organigramme A), qui se trouve au paragraphe 110(4) de la présente ligne directrice, décrit les étapes à suivre lorsqu’ une réaction transfusionnelle s’est produite et qu’une enquête est effectuée pour déterminer si un effet indésirable imprévu ou grave s’est produit chez le receveur.
Veuillez noter que l'article 110 fait référence aux effets indésirables imprévus et effets indésirables graves, même lorsqu'ils sont simplement décrits comme effets indésirables. Une mesure est requise dans tous les cas mentionnés ci-dessus.
Un établissement de transfusion qui a des motifs raisonnables de croire qu’un effet indésirable imprévu ou grave s’est produit chez un receveur doit immédiatement déterminer les codes d’identification du don, identifier le sang et les composants sanguins en cause et mettre en quarantaine ce sang (organigramme A (1)–(2)). L’établissement qui effectue la transfusion doit effectuer une enquête préliminaire pour déterminer si la cause fondamentale suggère que l’effet indésirable est attribuable à une activité qu’il a réalisée (organigramme A (3)). Lorsqu’un établissement a des motifs raisonnables de croire qu’un effet indésirable imprévu ou grave s’est produit en raison d’une activité qu’il a réalisée, il doit entreprendre une enquête et aviser tout établissement auquel il a distribué le sang ou les composants sanguins en cause (organigramme A (12)–(13)). Les rapports préliminaires et les rapports finaux de l’enquête sur les effets indésirables doivent ensuite être envoyés à Santé Canada dans les délais prévus à l’article 113.
Si l’établissement de transfusion a en sa possession ou dans son inventaire d’autres composants sanguins en cause, il doit mettre en quarantaine (p. ex. l’hôpital peut avoir dans son inventaire l’autre unité de la double aphérèse de plaquettes ou de plasma qu’il devra mettre en quarantaine). Dans la plupart des cas, les mesures d’atténuation des risques seront réalisées par l’établissement de sang qui a fourni le composant transfusé. Par conséquent, cet établissement doit être informé rapidement de la manifestation de l’effet indésirable. Cependant, l’établissement de transfusion peut aussi devoir mener des actions d’atténuation des risques.
Lorsque l’effet indésirable imprévu ou grave est attribuable à une activité réalisée par un autre établissement, l’établissement doit aviser tous les établissements figurant aux sous-alinéas 110(1)d)(i)–(iii) (organigramme A (4)–(6)).
110(2) Lorsqu’un établissement avise un autre établissement d’un effet indésirable imprévu ou grave chez un receveur, l’établissement à l’origine de l’avis doit veiller à ce que l’avis aux établissements mentionnés à l’alinéa 110(1)c) ou d) contienne tous les renseignements requis au paragraphe 110(2). L’avis devrait également comprendre tous les renseignements suivants, s’ils sont connus et pertinents :
- la date de naissance et le sexe du receveur;
- l’identification de l’hôpital;
- le diagnostic et les antécédents médicaux;
- le groupe sanguin ABO et Rh, et les résultats du dépistage d’anticorps;
- e. la date, l’heure, le lieu et l’indication de la transfusion;
- le composant transfusé, le ou les codes du don, le groupe sanguin ABO et facteur Rh sanguin, la date du prélèvement ou de la mise en commun, l’heure et le lieu de toute transformation (irradiation, lavage, etc.), l’heure du début/et de la fin de l’infusion, le nom du type de sang et du composant sanguin et le code de composant ISBT (disponible en anglais seulement) (www.isbtweb.org);
- la description de l’effet indésirable, des signes vitaux, du traitement et le parcours à l’hôpital, les résultats de l’enquête, y compris : la culture du sang du receveur et du composant transfusé, radiographie pulmonaire, essai d’antiglobuline direct, hémogramme, échocardiogramme, électrocardiogramme, groupage HLA et dépistage des anticorps HLA/antigène neutrophile humain, et les taux d’IgA, au besoin, pour l’enquête sur l’effet indésirable;
- l’évaluation et les conclusions du médecin de l’établissement ayant effectué la transfusion;
- les coordonnées de l’hôpital, le nom du médecin de l’établissement;
- les conséquences; et
- tout autre renseignement pertinent.
Un établissement peut utiliser tout formulaire de déclaration d’un effet ou d’un événement indésirable pour fournir ses avis écrits, à condition que toutes les exigences entourant les avis cites au paragraphe 110(2) du Règlement sur le sang soient respectées.
110(3) Un établissement qui reçoit un avis en vertu du paragraphe 110(1) au sujet d’un effet indésirable imprévu ou grave suite à une transfusion chez un receveur doit immédiatement en aviser tous les autres établissements et les autres personnes (y compris les fabricants de produits sanguins) auxquels il a distribué le sang et les composants sanguins en cause (organigramme A (7)). Un établissement peut respecter cette exigence en transmettant l’avis de l’établissement qui a prélevé le sang en cause à l’établissement qui possède maintenant le sang ou les composants sanguins en cause. L’établissement doit immédiatement mettre en quarantaine tous les composants sanguins en cause qu’il a en sa possession (organigramme A (8)).
Les établissements doivent collaborer les uns avec les autres et fournir toute information pertinente à l’établissement qui effectue l’enquête. La coopération entre les établissements (l’établissement de transfusion où l’effet indésirable est survenu et l’établissement de sang qui a fourni les composants sanguins) permet à l’établissement qui effectue l’enquête (généralement l’établissement de prélèvement) de déterminer la cause de l’effet indésirable imprévu ou grave lorsqu’il n’est pas clair si le sang prélevé ou une activité réalisée avec le sang ou les composants sanguins par l’établissement effectuant l’enquête a donné lieu à un effet indésirable chez le receveur de la transfusion (organigramme A (9)). Se référer à l’article 110(2) et 112 du Règlement sur le sang concernant l’obligation de coopérer et de partager l’information pertinente en temps opportun.
110(4) Lorsqu’un établissement reçoit un avis, aux termes du sous-alinéa 110(1)d)(i) ou (ii), au sujet d’un effet indésirable imprévu ou grave associé au sang ou aux composants sanguins transfusés, il doit déterminer si la cause fondamentale de l’effet indésirable est attribuable à une activité qu’il a réalisée (organigramme A (10)). Si c’est le cas, il devient l’établissement qui enquête sur l’effet indésirable.
Afin de mener à bien cette enquête préliminaire sur la cause fondamentale, toutes les données pertinentes sur l’effet indésirable doivent être communiquées à cet établissement, et/ou des efforts doivent être faits pour les obtenir. Cela permettra de déterminer plus rapidement s’il s’agit de l’établissement chargé de l’enquête (et aussi d’accélérer la déclaration de l’effet indésirable à Santé Canada). L’établissement chargé de l’enquête doit maintenant déclarer l’effet indésirable à Santé Canada et mener une enquête complète sur l’effet indésirable (organigramme A [12]-[13]). Les rapports préliminaires et les rapports finaux de l’enquête sur les effets indésirables doivent ensuite être envoyés à Santé Canada dans les délais prévus à l’article 113.
Lorsqu’il n’est pas clair si la cause fondamentale de l’effet indésirable est une activité réalisée par l’établissement ou par un autre établissement, l’établissement devrait communiquer avec les autres établissements qui ont participé au prélèvement et à la distribution et transfusion du sang et des composants sanguins impliqués, afin de partager l’information et d’aider à déterminer la cause de l’effet indésirable.
Organigramme A: Exigences d’enquête
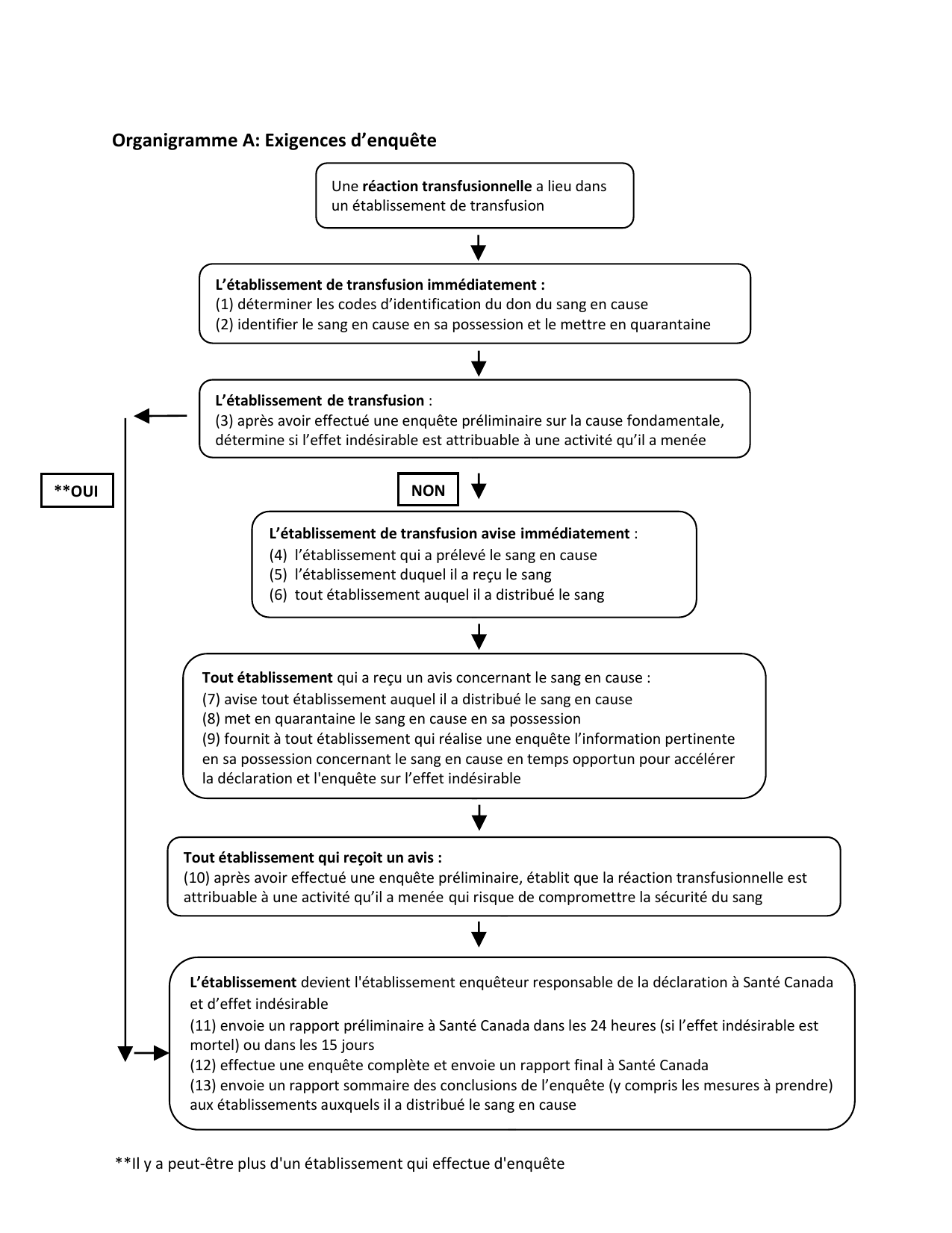
Texte descriptive pour Organigramme A
L'organigramme sur les Exigences d'enquête (organigramme A), qui se trouve sous l'article 110(4) de cette ligne directrice, décrit les étapes à prendre lorsqu'une réaction transfusionnelle a lieu dans un établissement de transfusion. L'organigramme est composé d'une série de boîtes qui couvrent la longueur de la page. Chaque boîte contient une description d'une ou plusieurs mesures à prendre où chaque mesure est énumérée comme une étape, de 1 à 13. Les boîtes sont reliées par des flèches indiquant les prochaines étapes qui doivent être exécutées. Parfois plus qu'une flèche proviendra d'une boîte. Le choix de flèche à suivre sera basé sur le résultat de l'étape précédente.
La première boîte dans l'organigramme indique - Une réaction transfusionnelle a lieu dans un établissement de transfusion.
Il y a une flèche qui provient de la première boîte et qui se dirige à la deuxième boîte, situé dessous la première boîte. La seconde boîte indique - L'établissement de transfusion immédiatement : (1) déterminer les codes d'identification du don de sang en cause et (2) identifier le sang en cause en sa possession et le mettre en quarantaine.
Il y a une flèche qui provient de la deuxième boîte et qui se dirige à la troisième boîte, situé dessous la deuxième boîte. La troisième boîte indique - L'établissement : (3) après avoir effectué une enquête préliminaire sur la cause fondamentale, détermine si l’effet indésirable est attribuable à une activité qu'il a menée.
Il y a deux flèches qui proviennent de la troisième boîte. La première flèche, étiqueté « OUI », mène à partir du côté gauche en bas de la troisième boîte jusqu'à la septième boîte.
La deuxième flèche, étiqueté « NON », provient du centre de la troisième boîte et se dirige à la quatrième boîte qui indique - L'établissement avise: (4) l'établissement qui a prélevé le sang en cause, (5) L'établissement duquel il a reçu le sang et (6) tout établissement auquel il a distribué le sang.
Il y a une flèche qui provient de la quatrième boîte et qui se dirige à la cinquième boîte, situé dessous la quatrième boîte. La cinquième boîte indique - Tout établissement qui a reçu un avis concernant le sang en cause : (7) avise tout établissement auquel il a distribué le sang en cause, (8) met en quarantaine le sang en cause en sa possession, (9) fournit à tout établissement qui réalise une enquête l’information pertinente en sa possession concernant le sang en cause en temps opportun pour accélérer la déclaration et l'enquête sur l’ effet indésirable.
Il y a une flèche qui provient de la cinquième boîte et qui se dirige à la sixième boîte, situé dessous la cinquième boîte. La sixième boîte indique - Tout établissement qui reçoit un avis : (10) après avoir effectué une enquête préliminaire, établit que la réaction transfusionnelle est attribuable à une activité qu’il a menée qui risque de compromettre la sécurité du sang.
Il y a une flèche qui provient de la sixième boîte et qui se dirige à la septième boîte, situé dessous la sixième boîte. La septième boîte indique - L’établissement devient l'établissement enquêteur responsable de la déclaration à Santé Canada et d’effet indésirable (11) envoie un rapport préliminaire à Santé Canada dans les 24 heures (si l’effet indésirable est mortel) ou dans les 15 jours, (12) effectue une enquête complète et envoie un rapport final à Santé Canada, (13) envoie un rapport sommaire des conclusions de l’enquête (y compris les mesures à prendre) aux établissements auxquels il a distribué le sang en cause. L'organigramme se termine ici.
A la fin de la page, il se trouve une phrase qui indique : **Il y a peut-être plus d'un établissement qui effectue d'enquête.
Enquêtes sur les composants transfusés
Le but de l’enquête est de trouver le ou les donneurs en cause, de récupérer les composants non périmés disponibles provenant de ces donneurs et d’aviser les autres destinataires et receveurs de ces composants sanguins. Consultez la section 1.5 « Définitions » « enquête sur les composants transfusés ».
Les établissements devraient effectuer une enquête sur les composants transfusés et préparer un rapport lors de la survenue d’effets indésirables imprévus ou graves soupçonnés d’être des infections virales associées à la transfusion tel que le VHB, le VHC, le VIH 1 et 2, le HTLVI/II. D’autres infections associées à la transfusion peuvent également déclencher une enquête sur les composants transfusés et leur déclaration. Les exigences en matière de déclaration sont les mêmes que pour les autres effets indésirables une fois qu’un donneur associé/impliqué est identifié et qu’il existe un doute raisonnable que la transmission était due à la transfusion.
L’établissement qui effectue l’enquête sur un effet indésirable imprévu ou grave associé au sang ou aux composants sanguins en cause doit effectuer une enquête sur les composants transfusés lorsqu’il établit l’un des éléments suivants :
- si l’infection par transfusion concorde avec le moment où le diagnostic du receveur a été posé;
- si le receveur n’a pas participé à une étude des dons antérieurs;
- dans les cas d’infection à HTLV I/II infection, le receveur a reçu des composants cellulaires;
- dans les cas d’infection au VHC, le receveur n’est pas un hémophile ou un thalassémique transfusé avant mai 1992.
L’établissement réalisant l’enquête concernant un effet indésirable imprévu ou grave associé au sang ou aux composants sanguins en cause devrait également lancer une enquête sur les composants transfusés soupçonnés d’être la cause d’une infection par transfusion lorsqu’il reçoit un rapport d’un résultat d’essai positif pour un receveur provenant de l’une des sources suivantes :
- médecin
- établissement, comme un hôpital, un établissement titulaire d’une licence ou enregistré
- autorité de santé publique
- information provenant d’une étude sur les dons antérieurs
- programme d’indemnisation
- receveur de la transfusion*
* Si le receveur de la transfusion déclare une infection par transfusion, l’établissement devrait obtenir une copie des résultats des essais avant d’effectuer une enquête sur les composants transfusés.
Une enquête sur les composants transfusés comprend les étapes suivantes :
- déterminer le statut du donneur ;
- aviser le donneur ;
- faire le suivi du donneur, des essais et de l’enquête sur les composants transfusés ;
- conclure l’enquête.
Pendant une enquête sur les composants transfusés, l’établissement analyse les renseignements dont il dispose pour déterminer la probabilité de transmission par les composants transfusés associés à une infection virale. Le directeur médical ou le cadre supérieur devrait être consulté, au besoin, pendant une enquête. L’établissement doit exclure le donneur, lorsque les tests de confirmation donnent des résultats indéterminés ou négatifs, et consulter le directeur médical ou le cadre supérieur pour obtenir d’autres directives.
L’établissement devrait également consulter le directeur médical ou cadre supérieur :
- si le rapport reçu provient d’une autre source que celles susmentionnées ;
- si les résultats des essais ont révélé la présence d’une maladie ou d’un agent autre que le VHB, le VHC, le VIH et le HTLV ;
- s’il y a d’autres risques évidents d’infection.
Remarque : Lorsqu’une enquête sur les composants transfusés identifie un donneur ayant reçu un résultat positif à un essai pour un agent infectieux transmissible par transfusion, une étude des dons antérieurs devrait être effectuée. Une copie des résultats des essais devrait figurer aux documents pour l’étude des dons antérieurs (consultez l’article 56 et l’alinéa 94(1) h) de la présente ligne directrice.) L’établissement doit attribuer au donneur un code d’exclusion indéfinie et l’inscrire au dossier d’évaluation de l’admissibilité du donneur de celui-ci. Consultez les paragraphes 44(2) et 56(1) pour de plus amples renseignements sur les études des dons antérieurs. Cela devrait être inclus dans le rapport final d’enquête sur les effets indésirables du receveur envoyé à Santé Canada.
110(5) Lorsqu’un établissement déclare verbalement à un autre établissement un effet indésirable imprévu ou grave chez un receveur, l’établissement doit également fournir immédiatement un avis écrit conformément aux exigences en matière de renseignements prévues au paragraphe 110(2).
Article 111 Don autologue
Don autologue
111. L’établissement qui prélève du sang d’un donneur faisant un don autologue et le transfuse procède sans tarder, s’il a des motifs raisonnables de croire qu’un effet indésirable grave ou un effet indésirable imprévu s’est produit chez le receveur, à la mise en quarantaine de toute autre unité de sang qu’il a en sa possession provenant de ce donneur ainsi qu’à l’enquête sur cet effet indésirable et sur le sang en cause.
Lorsque le receveur de sang autologue déclare un effet indésirable imprévu ou grave, l’établissement enregistré qui a recueilli et transfusé le sang doit immédiatement prendre les mesures suivantes :
- mettre en quarantaine toutes les autres unités de sang ou de composants sanguins provenant du donneur de sang autologue qu’il a en sa possession;
- effectuer une enquête sur l’effet indésirable.
L’enquête devrait permettre de déterminer la cause de l’effet indésirable imprévu ou grave y compris la possibilité d’un accident ou d’un manquement. Se référer aux articles 103 à 108 de la présente ligne directrice pour les exigences relatives à l’enquête sur les accidents ou manquements.
Articles 112-116 Enquêtes et rapports sur les effets indésirables chez le receveur
Article 112 Assistance à l'établissement qui enquête
Assistance à l’établissement qui enquête
112. L’établissement fournit, sur demande de l’établissement qui effectue une enquête, les renseignements ou documents utiles qu’il a en sa possession concernant le sang qu’il a distribué ou transfusé.
L’établissement qui effectue l’enquête est l’établissement qui, après une enquête préliminaire, a déterminé que la cause fondamentale de l’effet indésirable chez le receveur est attribuable à une activité qu’il a réalisée (organigramme A (3) et (12)).
L’établissement qui effectue l’enquête peut demander de l’information pertinente auprès d’autres établissements qui ont distribué ou transfusé le sang ou les composants sanguins en cause. Il est attendu que cette information pertinente soit fournie en temps opportun, car elle a une incidence sur les mesures d’atténuation des risques et les délais de déclaration à Santé Canada.
Un établissement doit, sur demande, fournir à l’établissement qui effectue l’enquête tous les renseignements pertinents à l’enquête s’il a transfusé ou distribué du sang et/ou des composants sanguins qui ont par la suite fait l’objet d’une enquête sur un effet indésirable chez le receveur. Cette information comprend, sans s’y limiter, une liste du sang et des composants sanguins en cause en inventaire et leur disposition (p. ex. distribués, transfusés, mis en quarantaine) (organigramme A (9)).
Article 113 Avis au ministre
Avis au ministre
113. (1) L’établissement qui effectue une enquête avise le ministre au plus tard vingt-quatre heures après avoir pris connaissance de la mort d’un receveur ou au plus tard quinze jours après avoir pris connaissance de tout autre effet indésirable grave ou effet indésirable imprévu.Avis écrit
(2) L’établissement confirme par écrit dans les meilleurs délais tout avis fourni verbalement en application du présent article.
113(1) Une fois qu’un établissement a effectué son enquête préliminaire et qu’il s’est défini comme étant l’établissement menant l’enquête (paragr. 110 [1] ou 110 [4]), il doit déclarer l’effet indésirable à Santé Canada. Ce rapport préliminaire doit être envoyé à Santé Canada dans les 24 heures suivant la prise de connaissance du décès si l’effet indésirable a entraîné un décès et dans les 15 jours civils si l’effet indésirable est imprévu ou grave. Si l’établissement enquêteur n’est pas en mesure de déclarer à Santé Canada dans les 24 heures suivant la prise de connaissance de la mort parce qu’il a fallu plus de 24 heures après avoir appris de la mort pour que l’établissement confirme qu’il s’agit d’un effet indésirable attribuable à une activité règlementée qu’il a réalisée, il doit faire rapport à Santé Canada sans plus tarder, et tous les documents pertinents dans leurs dossiers doivent démontrer qu’une enquête préliminaire sur la cause fondamentale était en cours pendant ce temps.
Le jugement clinique devrait être exercé par un professionnel de la santé qualifié de l’établissement de transfusion et l’établissement menant l’enquête afin de déterminer si la réaction transfusionnelle est liée à la sécurité du sang transfusé. En cas de décès, la transfusion doit au moins être soupçonnée d'avoir contribué aux facteurs ayant conduit au décès ou d'être la cause directe du décès.
Parmi les exemples d’effets indésirables graves chez le receveur, mentionnons l’anaphylaxie, le syndrome respiratoire aigu post-transfusionnel (TRALI), la septicémie causée par une infection transmise par transfusion (p. ex. bactérienne, virale ou parasitaire), la maladie du greffon contre l’hôte associée à la transfusion (p. ex. irradiation inefficace et étiquetage défectueux),et les réactions hémolytiques graves (p. ex. typage ou étiquetage incorrect) nécessitant une intervention et une admission à l’hôpital.
Une réaction transfusionnelle imprévue ou grave chez le receveur peut se produire pendant ou après une transfusion de composants sanguins et peut être liée à la sécurité du composant sanguin transfusé, c.-à-d. un effet indésirable. Le lien causal entre la transfusion et l’effet indésirable est une question de jugement clinique propre à la situation du patient (maladie sous-jacente et médication/traitement concomitant, etc.).
Des accidents ou manquements pourraient entraîner un effet indésirable chez un receveur. Lorsqu’un effet indésirable chez un receveur peut avoir été causé par un accident ou manquement survenu au cours d’une activité réglementée, l’établissement qui a mené l’activité réglementée impliquant le sang en cause doit effectuer une enquête et déclarer tout effet indésirable imprévu et grave au Programme Canada Vigilance, et soumettre un rapport d’enquête sur l’accident ou le manquement à la Direction générale des opérations réglementaires et de l’application de la loi (DGORAL) de Santé Canada. Consultez l’article 107 pour obtenir plus de renseignements sur la manière de déclarer les manquements et les accidents à la DGORAL.
Voici deux exemples où l'accident ou le manquement doit être déclaré à la DGORAL et l'effet indésirable doit être déclaré au Programme Canada Vigilance:
- un accident ou manquement lors de la transformation, comme la contamination du sang pendant une procédure de lavage en système ouvert, qui entraîne un effet indésirable grave et imprévu
- un manquement lors de la conservation est soupçonné d'avoir entraîné un effet indésirable grave ou imprévu, comme une hémolyse grave
L’organigramme sur la déclaration d’un effet indésirable (organigramme B), se trouvant à la fin de l’article 116, contient une description des étapes à suivre lorsqu’un établissement détermine qu’il doit effectuer une enquête sur un effet indésirable imprévu ou un effet indésirable grave.
113(2) Lorsque l’établissement déclare verbalement à Santé Canada un effet indésirable imprévu ou grave chez le receveur, il doit, dans les plus brefs délais, fournir un avis écrit.
L’avis écrit peut comprendre des rapports préliminaires, de suivi et finaux. Les rapports d’enquête préliminaires et finaux peuvent être soumis en même temps si l’enquête est terminée au moment du rapport préliminaire.
À condition que toutes les exigences de rapport soient respectées, un établissement peut utiliser tout formulaire de déclaration d’effet indésirable pour fournir un avis écrit obligatoire à Santé Canada au sujet d’un effet indésirable imprévu ou grave chez un receveur de transfusion sanguine.
Les établissements de sang doivent envoyer les rapports d’enquête sur les effets indésirables des receveurs au programme Canada Vigilance du BSPSE. La méthode privilégiée pour soumettre de manière sécurisée les rapports sur les effets indésirables est le protocole de transfert de fichiers sécurisé (sFTP).
Pour toute question sur la façon de s’inscrire pour la soumission à l’aide de sFTP, veuillez envoyer un courriel à tpmo-bgpc@hc-sc.gc.ca.
Le télécopieur est une autre méthode sécurisée pour faciliter la transmission des rapports d’effets indésirables aux établissements.
Programme Canada vigilance
Bureau de la surveillance des produits de santé et de l’épidémiologie
Direction des produits de santé commercialisés
Pré Tunney
Indice de l’adresse: 1908C
Ottawa, Ontario
K1A 0K9
Téléc. : 613-957-0335
Courriel pour les demandes de renseignements : canada.vigilance.blood-sang@hc-sc.gc.ca
Article 114 Résultats de l'enquête
Résultats de l’enquête
114. (1) L’établissement qui effectue une enquête avise par écrit tout établissement ou toute autre personne auxquels il a distribué du sang en cause des résultats de l’enquête et des mesures à prendre, le cas échéant.Envoi d’une copie de l’avis
(2) L’établissement qui reçoit l’avis en application du paragraphe (1) ou qui en reçoit copie par application du présent paragraphe en envoie copie à tout établissement auquel il a distribué du sang en cause.
L’établissement de sang qui enquête sur l’effet indésirable chez le receveur doit communiquer par écrit les résultats de l’enquête ainsi que toute mesure requise à tous les établissements de sang auxquels il a distribué le sang ou les composants sanguins en cause (y compris les fabricants de produits sanguins). Cet avis devrait faire référence à l’avis original qui a été envoyé par l’établissement de transfusion.
114(2) Lorsqu’un établissement reçoit les résultats d’une enquête, il doit les envoyer à tous les autres établissements auxquels il a distribué le sang en cause. Il n’est pas nécessaire d’ajouter d’autres renseignements aux résultats de l’enquête.
Article 115 Rapport d'enquête final au ministre
Rapport d’enquête final au ministre
115. L’établissement, au terme de son enquête, présente au ministre un rapport d’enquête final qui contient les renseignements et documents suivants :
- les conclusions de l’enquête;
- le sort réservé au sang en cause et les raisons l’ayant justifié
- toute mesure corrective prise et toute modification recommandée aux processus visés.
L’établissement qui effectue l’enquête doit fournir à Santé Canada un rapport final concernant l’enquête de l’effet indésirable chez le receveur qui contient tous les renseignements demandés à l’article 115. Lorsque l’information n’est pas concluante, l’établissement peut également fournir des commentaires fondés sur son évaluation (organigramme B, étape 3).
Article 116 Rapport annuel
Rapport annuel
116. L’établissement établit à la fin de chaque année un rapport qui, non seulement résume les rapports finals présentés au cours de l’année, mais encore fait une analyse critique et concise des enquêtes visées par ces rapports, et le présente sur demande au ministre.
L’établissement qui effectue les enquêtes sur les effets indésirables doit préparer un rapport annuel sur les effets indésirables (organigramme B, étape 4) dans lequel sont résumés tous les rapports finaux rédigés pendant l’année concernant les effets indésirables imprévus ou graves chez des receveurs, dont une analyse critique et concise des rapports finaux d’enquêtes sur les effets indésirables.
Le rapport annuel devrait inclure ce qui suit :
- un résumé;
- l’étendue du lien entre l’effet indésirable et le sang transfusé;
- une analyse détaillée et une évaluation portant sur tout nouveau signal concernant la sécurité;
- une analyse globale des effets indésirables déclarés au cours de la période qui tient compte de l’utilisation de sang ou de composants sanguins ;
- une analyse cumulative des effets indésirables déclarés, y compris une analyse des tendances au fil du temps;
- les rapports sommaires annuels des statistiques sur les études des dons antérieurs et les enquêtes des produits transfusés;
- des conclusions générales et des possibilités d’amélioration.
À la demande de Santé Canada, l’établissement peut soumettre un rapport annuel sur les effets indésirables qui a été préparé à d’autres fins, tel qu’un rapport annuel d’hémovigilance, en autant que les critères requis et décrits dans cette section soient rencontrés.
À la demande de Santé Canada, le rapport annuel doit être transmis au programme Canada Vigilance du BSEPS.
De plus amples renseignements sur la manière de soumettre le document seront fournis au moment de la demande.
Courriel pour les demandes de renseignements : canada.vigilance.blood-sang@hc-sc.gc.ca
L'annexe B contient la liste des rapports annuels à fournir par les établissements de sang.
Organigramme B : Déclaration sur les effets indésirables chez un receveur (EIR) a Santé Canada
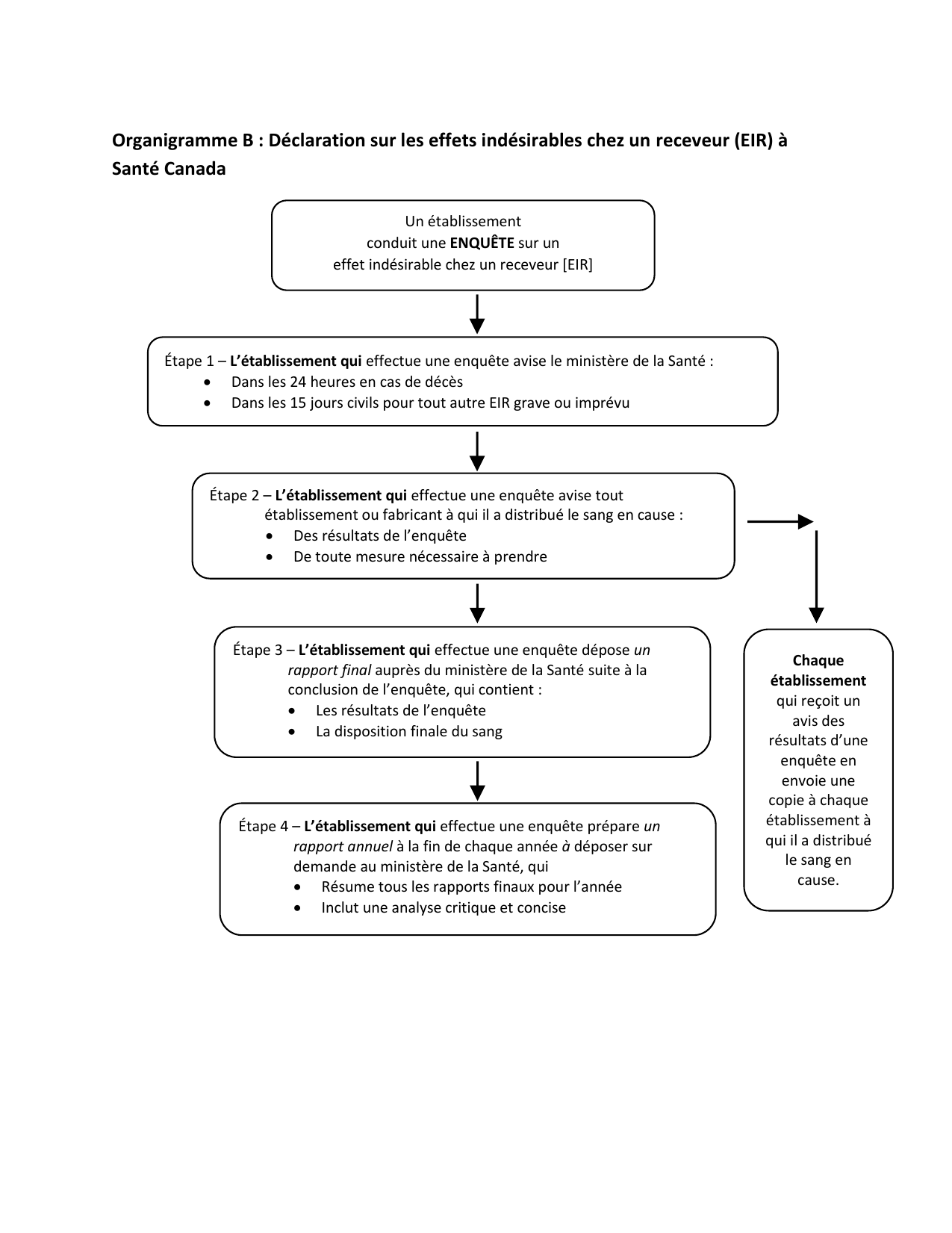
Texte descriptif
L'organigramme sur la Déclaration sur les effets indésirables chez un receveur a Santé Canada (organigramme B), qui se trouve à la fin de l'article 116, décrit les étapes à prendre lorsqu'un établissement détermine qu'il doit conduire une enquête sur un effet indésirablechez un receveur. L'organigramme est composé d'une série de boîtes qui couvrent la longueur de la page. Chaque boîte contient une description d'une ou plusieurs mesures à prendre où chaque mesure est énumérée comme une étape, de 1 à 4. Les boîtes sont reliées par des flèches indiquant les prochaines étapes qui doivent être exécutées.
La première boîte dans l'organigramme indique - Un établissement conduit une enquête sur un effet indésirable chez un receveur (EIR), où le mot "enquête" est en gras et en majuscule.
Il y a une flèche qui provient de la première boîte et qui se dirige à la deuxième boîte, situé dessous la première boîte. La seconde boîte indique - Étape 1 - L'établissement qui effectue une enquête avise le ministère de la Santé : [point] Dans les 24 heures en cas de décès et [point] Dans les 15 jours civils pour tout autre EIR grave ou imprévu.
Il y a une flèche qui provient de la deuxième boîte et qui se dirige à la troisième boîte, situé dessous la deuxième boîte. La troisième boîte indique - Étape 2 - L'établissement qui effectue une enquête avise tout établissement ou fabriquant à qui il a distribué le sang en cause : [point] Des résultats de l'enquête et [point] De toute mesure nécessaire à prendre.
Il y a une flèche qui provient du côté droit de la troisième boîte et qui se dirige vers une boîte qui se trouve à la droite et sous les premières trois boîtes, et à la droite des quatrièmes et cinquièmes boîtes qui indique - Chaque établissement qui reçoit un avis des résultats d'une enquête en envoie une copie à chaque établissement à qui il a distribué le sang en cause, où les tous mots sont en italique.
Il y a une autre flèche qui provient de la troisième boîte et qui se dirige à la quatrième boîte, situé dessous la troisième boîte. La quatrième boîte indique - Étape 3 - L'établissement qui effectue une enquête dépose un rapport final auprès du ministère de la Santé suite à la conclusion de l'enquête, qui contient : [point] Les résultat de l'enquête, [point] La disposition finale du sang, et [point] Toutes actions correctives entreprise.
Il y a une flèche qui provient de la quatrième boîte et qui se dirige à la cinquième boîte, situé dessous la quatrième boîte. La cinquième boîte indique - Étape 4 - L'établissement qui effectue une enquête prépare un rapport annuel à la fin de chaque année à déposer sur demande au ministère de la Santé, qui : [point] Résumé tous les rapports finaux pour l'année et [point] Inclut une analyse critique et concise.
Articles 117-123 Dossiers
Les établissements doivent conserver des documents et des renseignements aux dossiers, y compris ceux produits avant l’entrée en vigueur du Règlement sur le sang, conformément aux exigences précisées dans le Règlement sur le sang.
Article 117 Caractéristiques des documents
Caractéristiques des documents
117. Le contenu des dossiers tenus par l’établissement doit être complet, exact, lisible, indélébile et facilement accessible.
Les dossiers fournissent des preuves documentées de la conformité. Le contenu des dossiers doit être exact, complet et lisible. Les dossiers doivent être conservés simultanément à la réalisation de chaque étape importante du traitement, de l’importation, de la transformation, de la conservation, de la distribution (y compris la distribution exceptionnelle) et des enquêtes sur les accidents, les manquements, et les effets indésirables du sang et/ ou les composants sanguins, de sorte que toutes les étapes soient clairement associées à la personne qui a exécuté l’étape, l’heure/la date et le lieu (le cas échéant) où l’activité a été effectuée. De plus, en ce qui concerne les dossiers associés au traitement, à la transformation ou à toute autre activité réglementée, le numéro de lot du matériel et des produits essentiels et les données permettant d’identifier l’équipement essentiel associés aux activités doivent faire partie des dossiers.
Tous les dossiers doivent indiquer la personne qui a effectué les activités ainsi que les dates des différentes entrées. Les établissements veillent à l’exactitude des dossiers. Par exemple, toutes les transcriptions manuelles des résultats des essais doivent être vérifiées de façon indépendante, si le document transcrit constitue la documentation permanente.
Toute entrée manuelle de renseignements doit être faite à l’encre indélébile. Toute correction, entrée de renseignements ou note faite à la suite de la date originale de l’achèvement du dossier doit être raturée, parafée ou signée, et datée pour indiquer qu’un changement a été apporté aux renseignements originaux.
Tous les établissements doivent conserver les dossiers dans un format facilement compréhensible et récupérable. Les dossiers doivent être accessibles en tout temps. Tous les établissements doivent pouvoir récupérer rapidement et efficacement les renseignements sur la traçabilité du sang.
Les dossiers doivent être tenus de manière à préserver leur intégrité et leur exactitude au fil du temps. L’établissement doit vérifier l’exactitude et l’intégrité des renseignements transférés vers un autre support utilisé pour conserver l’information. Cette vérification devrait être réalisée par une personne autre que celle qui a transféré l’information.
Un établissement qui conserve des dossiers électroniques doit posséder un système électronique validé pour la fin à laquelle il est destiné, afin d’assurer l’intégrité des données de ces documents. Tous les dossiers électroniques doivent être sauvegardés régulièrement et entreposés en toute sécurité pour la récupération des données. Tout changement au système électronique doit être évalué, documenté et approuvé avant sa mise en œuvre, afin d’assurer l’intégrité de données et la récupération des dossiers pendant la période de rétention prescrite. Un historique des changements aux dossiers électroniques doit être accessible par traces de vérification.
Un établissement doit pouvoir récupérer et imprimer une copie papier des renseignements entreposés dans un dossier électronique.
Un format normalisé pour les dates (p. ex. AAAA-MM-JJ ou MM-JJ-AAAA) devrait être utilisé pour tous les dossiers. Quand ce n’est pas possible, les dossiers devraient indiquer clairement le format de la date s’il n’est pas facilement identifiable.
Article 118 Codes d'identification du don
Codes d’identification du don
118. L’établissement veille à ce que le code d’identification du don figure à chacun des dossiers relatifs au traitement, à la distribution, à la transformation et à la transfusion de sang.
Chaque unité de sang possède un code d’identification du don unique. Le code d’identification du don permet de retracer une unité de sang ou de composants sanguins donnée et tout renseignement associé à cette unité tout au long des étapes du traitement, de la transformation, de la chaîne de distribution et de la transfusion ou de la disposition finale du sang. Le code d’identification du don doit faire partie de tous les documents relatifs au traitement, à la distribution, à la transformation et à la transfusion de l’unité de sang. Un composant sanguin est identifié par le code de produit spécifique au composant (c.-à-d. le code pour les globules rouges, le plasma, les plaquettes) et associé au code d’identification du don. Si un établissement attribue un nouveau code pour une unité mise en commun, le code de don de tous les composants mis en commun doit être retraçable dans les dossiers de l’établissement.
Article 119 – 122 Période de rétention
Un établissement qui exerce plus d’un type d’activité (p. ex. transformation et transfusion) doit respecter les périodes de rétention indiquées dans toutes les sections applicables ci-dessous.
Article 119 Période de rétention - don allogénique
Période de rétention — don allogénique
119. (1) L’établissement qui prélève du sang d’un donneur faisant un don allogénique conserve dans ses dossiers les documents et renseignements figurant à la colonne 1 du tableau du présent article pendant la période prévue à la colonne 2.Calcul de la période de rétention
(2) La période de rétention débute à la date de la création du document au dossier, à l’exception des documents concernant tout employé de l’établissement qui figurent à l’article 28 du tableau pour lesquels la période débute à la date où l’employé a cessé de travailler pour l’établissement.
Tous les établissements qui prélèvent du sang allogénique veillent à ce que tous les dossiers soient conservés conformément au « Tableau de l’article 119 : Documents et renseignements– période de rétention ». Consultez la clause 20.2.5 de la Norme sur le sang de la CSA pour plus d’information sur le rapprochement des codes d’identification du don (point 3 du tableau de l’article 119).
Pour les dossiers des études des dons antérieurs, si l’étude a lieu à la suite d’une enquête sur un effet indésirable ou un accident/manquement, le dossier doit être conservé pendant 10 ans. Toutefois, dans le cas où cette enquête a mené à une « détermination de l’inadmissibilité d’un donneur », soit « permanente » ou « temporaire », ces dossiers doivent être conservés pendant 50 et 10 ans respectivement. Les dossiers des études des dons antérieurs font partie de l’enquête sur les effets indésirables et doivent donc être conservés pendant 10 ans.
| Article | Colonne 1 Documents et renseignements |
Colonne 2 Période de rétention |
|---|---|---|
| 1. | Code d'identification du donneur | 50 ans |
| 2. | Code d'identification du don | 50 ans |
| 3. | Rapprochement des codes d'identification du don | 10 ans |
| 4. | Évaluation de l'admissibilité du donneur | 5 ans |
| 5. | Inadmissibilité permanente du donneur | 50 ans |
| 6. | Inadmissibilité temporaire du donneur | 10 ans |
| 7. | Prélèvement - date du don | 50 ans |
| 8. | Prélèvement - données relatives à l'aphérèse | 5 ans |
| 9. | Prélèvement - renseignements au dossier du don | 5 ans |
| 10. | Nom du fabriquant et numéro de lot des contenants de sang ainsi que du matériel et des produits essentiels associés à chaque don | 1 an |
| 11. | Résultats des essais effectués à des fins de dépistage de maladies transmissibles ainsi qu'à des fins de détermination du groupe sanguin ABO, du facteur Rh et de la présence d'anticorps cliniquement significatifs | 50 ans |
| 12. | Préparation des composants sanguins | 10 ans |
| 13. | Contrôle de la température de conservation du sang | 5 ans |
| 14. | Sort des unités de sang, notamment leur destruction | 50 ans |
| 15. | Distribution | 50 ans |
| 16. | Documents d'expédition | 1 an |
| 17. | Distribution exceptionnelle | 50 ans |
| 18. | Importation de sang dans des circonstances urgentes | 50 ans |
| 19. | Renseignements obtenus après le don | 10 ans |
| 20. | Plaintes et enquêtes connexes | 5 ans |
| 21. | Rapports de vérification interne | 5 ans |
| 22. | Essais de contrôle de la qualité | 5 ans |
| 23. | Entretien, qualification, validation et étalonnage de l'équipement essentiel | 3 ans |
| 24. | Matériel et produits essentiels, y compris leur qualification | 3 ans |
| 25. | Évaluation de l'exactitude et la fiabilité des résultats d'essais | 5 ans |
| 26. | Chaque version des procédures opérationnelles mise en application, autre que celles concernant l'évaluation de l'admissibilité des donneurs | 10 ans |
| 27. | Chaque version des procédures opérationnelles concernant l'évaluation de l'admissibilité des donneurs | 50 ans |
| 28. | Qualifications, formation et évaluation des compétences des employés | 10 ans |
| 29. | Enquêtes sur les accidents ou manquements et rapports connexes | 10 ans |
| 30. | Enquêtes sur les effets indésirables et rapports connexes | 10 ans |
Article 120 Période de rétention - don autologue
Période de rétention — don autologue
120. (1) L’établissement qui prélève du sang d’un donneur faisant un don autologue conserve dans ses dossiers les documents et renseignements figurant à la colonne 1 du tableau du présent article pendant la période prévue à la colonne 2.Calcul de la période de rétention
(2) La période de rétention débute à la date de la création du document au dossier, à l’exception des documents concernant tout employé de l’établissement qui figurent à l’article 18 du tableau pour lesquels la période débute à la date où l’employé a cessé de travailler pour l’établissement.
Tous les établissements qui prélèvent du sang d’un donneur faisant un don autologue veillent à ce que tous les dossiers soient conservés conformément au « Tableau de l’article 120 : Documents et renseignements – période de rétention ».
| Article | Colonne 1 Documents et renseignements |
Colonne 2 Période de rétention |
|---|---|---|
| 1. | Code d'identification du donneur | 10 ans |
| 2. | Code d'identification du don | 10 ans |
| 3. | Prélèvement - dossier du don | 5 ans |
| 4. | Nom du fabriquant et numéro de lot des contenants de sang ainsi que du matériel et des produits essentiels associés à chaque don | 1 an |
| 5. | Résultats des essais effectués à des fins de dépistage de maladies transmissibles ainsi qu'à des fins de détermination du groupe sanguin ABO et du facteur Rh | 10 ans |
| 6. | Préparation des composants sanguins | 10 ans |
| 7. | Contrôle de la température de conservation du sang | 5 ans |
| 8. | Sort des unités de sang, notamment leur destruction | 10 ans |
| 9. | Distribution | 10 ans |
| 10. | Documents d'expédition | 1 an |
| 11. | Plaintes et enquêtes connexes | 5 ans |
| 12. | Rapports de vérification interne | 5 ans |
| 13. | Essais de contrôle de la qualité | 5 ans |
| 14. | Entretien, qualification, validation et étalonnage de l'équipement essentiel | 3 ans |
| 15. | Matériel et produits essentiels, y compris leur qualification | 3 ans |
| 16. | Évaluation de l'exactitude et la fiabilité des résultats d'essais | 5 ans |
| 17. | Chaque version des procédures opérationnelles mise en application | 10 ans |
| 18. | Qualifications, formation et évaluation des compétences des employés | 10 ans |
| 19. | Enquêtes sur les accidents ou manquements et rapports connexes | 10 ans |
| 20. | Enquêtes sur les effets indésirables et rapports connexes | 10 ans |
Article 121 Période de rétention - transformation
Période de rétention — transformation
121. (1) L’établissement qui fait de la transformation conserve dans ses dossiers les documents et renseignements figurant à la colonne 1 du tableau du présent article pendant la période prévue à la colonne 2.Calcul de la période de rétention
(2) La période de rétention débute à la date de la création du document au dossier, à l’exception des documents concernant tout employé de l’établissement qui figurent à l’article 10 du tableau pour lesquels la période débute à la date où l’employé a cessé de travailler pour l’établissement.
Tous les établissements qui transforment du sang ou des composants sanguins doivent s’assurer que les dossiers sont conservés conformément au « Tableau de l’article 121 : Documents et renseignements– période de rétention - transformation ». L’article 121 s’applique également aux établissements qui ne mettent que des cryoprécipités en commun et qui n’ont pas besoin d’un enregistrement.
Les périodes de rétention indiquées dans le Tableau de l’article 121 réfèrent au temps de rétention des documents et renseignements produits lors de la réalisation des activités de transformation. Par exemple, les dossiers qui identifient les composants sanguins transformés à l’aide du code d’identification du don doivent être conservés pour une période de 10 ans. Par contre, le Tableau de l’article 119 fait référence aux périodes de rétention des documents et renseignements produits par un établissement qui prélève du sang allogénique. Par exemple, les documents et renseignements qui identifient le sang prélevé à l’aide du code d’identification du don doivent être conservés pour une période de 50 ans.
| Article | Colonne 1 Documents et renseignements |
Colonne 2 Période de rétention |
|---|---|---|
| 1. | Code d'identification du don | 10 ans |
| 2. | Renseignements concernant le lavage, la mise en commun et l'irradiation | 10 ans |
| 3. | Nom du fabriquant et numéro de lot du matériel et des produits essentiels associés à chaque transformation | 1 an |
| 4. | Plaintes et enquêtes connexes | 5 ans |
| 5. | Rapports de vérification interne | 5 ans |
| 6. | Essais de contrôle de la qualité | 5 ans |
| 7. | Entretien, qualification, validation et étalonnage de l'équipement essentiel | 3 ans |
| 8. | Matériel et produits essentiels, notamment leur qualification | 3 ans |
| 9. | Chaque version des procédures opérationnelles mise en application | 10 ans |
| 10. | Qualifications, formation et évaluation des compétences des employés | 10 ans |
| 11. | Enquêtes sur les accidents ou manquements et rapports connexes | 10 ans |
| 12. | Enquêtes sur les effets indésirables et rapports connexes | 10 ans |
Article 122 Période de rétention - transfusion
Période de rétention — transfusion
122. (1) L’établissement qui fait de la transfusion conserve dans ses dossiers les documents et renseignements figurant à la colonne 1 du tableau du présent article pendant la période prévue à la colonne 2.Calcul de la période de rétention
(2) La période de rétention débute à la date de la création du document au dossier, à l’exception des documents concernant tout employé de l’établissement qui figurent à l’article 11 du tableau pour lesquels la période débute à la date où l’employé a cessé de travailler pour l’établissement.
Tous les établissements qui font de la transfusion de sang et/ou de composants sanguin veillent à ce que tous les dossiers soient conservés conformément au « Tableau de l’article 122 : Documents et renseignements – période de rétention ».
Pour les établissements qui distribuent du sang et des composants sanguins ultérieurement (p. ex. l’hôpital A envoie du sang ou des composants sanguins à l’hôpital B), les dossiers de distribution fournissent des renseignements sur la chaîne de distribution de l’unité de sorte que toute unité de sang ou de composants sanguins peut être retracée de sa source initiale à sa disposition finale. Les documents d’expédition sont des dossiers associés au mode de transport utilisé pour la distribution du sang (p. ex. bordereau d’expédition, reçus de messagerie, etc.).
| Article | Colonne 1 Documents et renseignements |
Colonne 2 Période de rétention |
|---|---|---|
| 1. | Code d'identification du don allogénique | 50 ans |
| 2. | Code d'identification du don autologue | 10 ans |
| 3. | Documents d'expédition | 1 an |
| 4. | Contrôle de la température de conservation du sang | 5 ans |
| 5. | Distribution | 50 ans |
| 6. | Distribution exceptionnelle | 50 ans |
| 7. | Renseignements concernant la transfusion ou le sort des unités de sang provenant de dons allogéniques, notamment ceux permettant d'identifier le receveur | 50 ans |
| 8. | Renseignements concernant la transfusion ou le sort des unités de sang provenant de dons autologues | 10 ans |
| 9. | Plaintes et enquêtes connexes | 5 ans |
| 10. | Chaque version des procédures opérationnelles mise en application | 10 ans |
| 11. | Qualifications, formation et évaluation des compétences des employés | 10 ans |
| 12. | Enquêtes sur les accidents ou manquements et rapports connexes | 10 ans |
| 13. | Enquêtes sur les effets indésirables et rapports connexes | 10 ans |
Article 123 Entreposage des dossiers
Entreposage des dossiers
123. L’établissement entrepose ses dossiers dans un lieu où les conditions ambiantes sont adéquates et dont l’accès est restreint aux seules personnes autorisées.
Les zones d’entreposage des dossiers doivent permettre de maintenir l’intégrité des documents. Les paramètres de l’environnement d’entreposage comme la température doivent être adéquats et contrôlés dans la mesure nécessaire afin de sauvegarder l’intégrité des types de dossiers entreposés. L’humidité devrait aussi être contrôlée au besoin et si nécessaire. L’accès aux zones d’entreposage doit être restreint aux personnes autorisées.
Si les dossiers sont copiés ou entreposés dans un endroit à l’extérieur du site, l’établissement doit avoir signé un contrat avec le fournisseur de services. Le contrat doit comprendre des exigences précises, comme le transport vers le site, la qualité des copies, les renseignements sur la récupération des documents et les conditions d’entreposage. Lorsque c’est pertinent, le contrat doit décrire les exigences précises quant à la destruction des documents originaux.
Article 124 Pouvoirs des inspecteurs
Enregistrements visuels
124. L’inspecteur peut, pour l’application du présent règlement, prendre des photographies ou effectuer des enregistrements de ce qui suit :
- tout article visé au paragraphe 23(2) de la Loi;
- tout lieu dont il a des motifs raisonnables de croire qu’un article visé à l’alinéa a) y est traité, transformé ou conservé;
- toute chose dont il a des motifs raisonnables de croire qu’elle sert ou peut servir lors de l’exercice d’une activité par l’établissement.
Article 125 Modification corrélative
125. L’article 18 du Règlement modifiant le Règlement sur les aliments et drogues (1475 — bonnes pratiques de fabrication)1 est remplacé par ce qui suit :
18. Le Règlement sur les aliments et drogues, dans sa version antérieure à l’entrée en vigueur du présent règlement, continue de s’appliquer à l’égard du sang entier et de ses composants jusqu’à l’entrée en vigueur du paragraphe 3(2) du Règlement sur le sang.
Articles 126-128 Dispositions transitoires
Les dispositions énumérées dans cet article ne sont plus applicables
Article 126 Homologation
Homologation
126. Les renseignements et documents exigés lors d’une demande d’homologation au titre de l’article 6 qui ont déjà été fournis au ministre en application des articles C.01A.005 à C.01A.007 et C.01A.014 du Règlement sur les aliments et drogues et qui ont été acceptés par lui avant l’entrée en vigueur du présent règlement sont réputés constituer l’homologation délivrée par le ministre en vertu de l’article 7 du présent règlement.
Article 127 Licence
Les dispositions énumérées dans cet article ne sont plus applicables
Licence
127. La licence que l’établissement a obtenue avant la date d’entrée en vigueur du présent règlement, en vertu de l’article C.01A.008 du Règlement sur les aliments et drogues, est maintenue jusqu’à la délivrance ou jusqu’au refus de délivrance d’une licence aux termes des articles 20 ou 21, selon le cas, du présent règlement s’il présente sa demande de licence conformément à l’article 18, exception faite des alinéas (1)j) et k), dans les trois mois suivant la date d’entrée en vigueur du présent règlement.
Article 128 Enregistrement
Les dispositions énumérées dans cet article ne sont plus applicables
Enregistrement
128. (1) L’établissement qui, avant la date d’entrée en vigueur du présent règlement, exerce des activités devant faire l’objet d’un enregistrement conformément à l’article 30 peut néanmoins continuer d’exercer ses activités s’il présente sa demande d’enregistrement conformément à l’article 31 dans les trois mois suivant cette date.Durée
(2) Le paragraphe (1) s’applique jusqu’à ce qu’il soit statué sur la demande conformément à l’article 32.
Article 129 Entrée en vigueur
Les dispositions énumérées dans cet article ne sont plus applicables
Un an après la publication
129. (1) Le présent règlement, sauf les paragraphes 4(4) à (6), l’alinéa 64(1)b) mais seulement dans le cas où le numéro d’enregistrement est visé, ainsi que l’article 125, entre en vigueur un an après la date de sa publication dans la Partie de la Gazette du Canada.Paragraphes 4(4) à (6) et alinéa 64(1)b)
(2) Les paragraphes 4(4) à (6) et, dans le cas du numéro d’enregistrement seulement, l’alinéa 64(1)b), entrent en vigueur six mois après la date d’entrée en vigueur du présent règlement.Article 125
(3) L’article 125 entre en vigueur à la date d’enregistrement du présent règlement.
Annexes
Annexe A : Glossaire
- AgHBs
- Antigène de surface de l’hépatite B
- AI
- Avis d’insuffisance
- A/M
- Accident ou manquement
- BAR
- Bureau des affaires réglementaires
- CSA
- Groupe CSA/Association canadienne de normalisation
- CMV
- Cytomégalovirus
- Date Limite
- Date limite pour la présentation de nouvelles preuves
- DGORAL
- Direction générale des opérations réglementaires et de l’application de la loi (anciennement l’inspectorat de la Direction générale des produits de santé et des aliments
- DMBR
- Direction des produits biologiques et des thérapies génétiques ESP Évaluation sur place
- ESP
- Évaluation sur place
- HLA
- Antigène d’histocompatibilité humain
- HTLV I/II
- Virus T-lymphotropique humain-I/ Virus T-lymphotropique humain-II
- IgA
- Immunoglobuline A
- ISBT
- Société international de transfusion sanguine
- PCPB
- Programme de conformité des produits biologiques
- PDPE
- Programme de donneurs pré-évalués
- Rh
- Facteur rhésus
- RBC
- Globules rouges
- US-FDA
- Food and Drug Administration des États-Unis
- VHB
- Virus de l’hépatite B
- VHC
- Virus de l’hépatite C
- VIH
- Virus de l’immunodéficience humaine
- VNO
- Virus du Nil occidental
Annexe B: Tableau récapitulatif des rapports annuels à fournir par les établissements de sang
| Titre du rapport | Article du Règlement sur le sang | Qui prépare le rapport | Quand déposer le rapport | Où soumettre le rapport |
|---|---|---|---|---|
| Homologation: - Autres modifications - Rapport annuel | 12 | Établissements qui détiennent une homologation | En consultation avec Santé Canada | Unité de la réglementation des établissements de sang, Bureau des affaires réglementaires, Direction des médicaments biologiques et radiopharmaceutiques, Santé Canada |
| Rapport annuel des enquêtes sur les manquements et les accidents | 108 | À la fin d'une période de 12 mois, les établissements qui ont mené une enquête sur les manquements et les accidents survenus pendant l'année. | À la demande du ministre | Tel qu'il est indiqué dans la demande du ministre |
| Rapport annuel sur les effets indésirables chez les receveurs | 116 | À la fin de chaque année, les établissements qui ont mené une enquête sur un effet indésirable grave ou inattendu chez un receveur de sang. | À la demande du ministre | Tel qu'il est indiqué dans la demande du ministre |
Annexe C : Outil d'autoévaluation pré-enregistrement pour les établissements de sang présentant une demande d'enregistrement
Les établissements devraient consulter l'article 30 du Règlement sur le sang afin de vérifier s'ils effectuent des activités qui nécessitent leur enregistrement auprès de Santé Canada.
Avant de demander un numéro d'enregistrement, Santé Canada recommande fortement que les établissements utilisent l'outil d'autoévaluation afin de déterminer si leurs activités sont conformes aux exigences du Règlement sur le sang. Cet outil est conçu afin d'identifier les éléments que les établissements pourraient devoir corriger afin de se conformer au Règlement sur le sang. Les établissements ne sont pas tenus de présenter cette auto-évaluation à Santé Canada, et elle ne sera pas examinée pendant une inspection. Le simple remplissage de ce formulaire n'est pas considéré comme un document démontrant la conformité aux exigences de la vérification interne de l'alinéa 94(1)j).
Veuillez noter que cet outil ne remplace pas les exigences du Règlement sur le sang. Chaque section du présent document devrait être lue en parallèle avec les articles pertinents du Règlement sur le sang. Pour des explications sur les articles ci-dessous, veuillez consulter les sections appropriées de la ligne directrice. Plusieurs termes utilisés dans l'outil d'autoévaluation sont des termes définis dans le Règlement sur le sang. Consultez l'article 1 « Définitions » du Règlement sur le sang.
| Section du Règlement sur le sang | Exigences | Evaluation |
|---|---|---|
| A. Activités de traitement reliées au sang d'un donneur faisant un don autologue | ||
| Prélèvement | ||
| 46 | Un code d'identification du donneur a-t-il été attribué à chaque donneur? | Oui / Non |
| 47 | Un code d'identification du don a-t-il été attribué à chaque unité de sang prélevé? | Oui / Non |
| Le code d'identification du don a-t-il été associé, dans les dossiers, au code d'identification du donneur? | Oui / Non | |
| 48 | Des étiquettes sont-elles apposées sur les contenants de sang au moment du prélèvement, conformément à l'article 63 du Règlement sur le sang? | Oui / Non |
| 49(1) | Le sang est-il prélevé de la manière suivante? | s/o |
| a) il recourt à des méthodes aseptiques | Oui / Non | |
| b) l'équipement qu'il utilise pour le prélèvement est homologué conformément au Règlement sur les instruments médicaux | Oui / Non | |
| c) les contenants qu'il utilise pour le prélèvement sont homologués conformément au Règlement sur les instruments médicaux et sont exempts de défaut ou de détérioration | Oui / Non | |
| d) il consigne dans ses dossiers le numéro du lot du contenant et l'associe au numéro d'identification du don | Oui / Non | |
| 49(2) | Les contenants n'ont jamais été utilisés auparavant? | Oui / Non |
| 50 | Lors du prélèvement, le sang prélevé et les échantillons de sang pour des essais sont prélevés de manière à éviter toute contamination croisée? | Oui / Non |
| 51 | a) Le prélèvement sanguin respecte-t-il les exigences prévues à l'article 12.2.1 de la norme sur le sang de la CSA? | Oui / Non |
| b) Le volume du sang prélevé et celui de l'anticoagulant à y mélanger sont-ils déterminés en fonction du poids du donneur s'il y a lieu? | Oui / Non | |
| Essais | ||
| 53 | Le sang fait-il l'objet d'essais appropriées et efficaces à des fins de dépistage des maladies transmissibles et de leurs agents mentionnés à l'article 12.3.1.2 de la norme sur le sang de la CSA? | Oui / Non |
| 54(1) | Lors de chaque prélèvement, les essais suivants sont effectués sur un échantillon sanguin afin de déterminer : | s/o |
| a) le groupe sanguin ABO; | Oui / Non | |
| b) le facteur Rh, y compris, s'il y a lieu, le dépistage de l'antigène D faible? | Oui / Non | |
| 54(2) | Est-ce que les résultats prévus aux alinéas 54(1)a) et b) sont comparés avec les plus récents résultats consignés au dossier du donneur? | Oui / Non |
| 54(3) | Dans le cas où la comparaison requise au paragraphe 54(2) révèle une incohérence, les essais prévus au paragraphe 54(1) sont-ils recommencés et le sang n'est pas transfusé tant que l'incohérence n'est pas résolue? | Oui / Non |
| 55 a) | L'essai du sang est-il effectué au moyen d'instruments médicaux homologués conformément au Règlement sur les instruments médicaux aux fins de l'évaluation préliminaire de donneurs ou à des fins diagnostiques? | Oui / Non |
| 56(2) | Le médecin du donneur est-il informé des résultats des essais décrits à l'article 12.3.1.6 de la norme sur le sang de la CSA par l'établissement ayant effectué le prélèvement du sang? | Oui / Non |
| 58 | Les composants sanguins sont-ils préparés conformément aux articles 7.1.3, 7.2, 7.3.1, 7.3.2, 7.5.1.1 (abstraction faite du renvoi au tableau 3 de la norme sur le sang de la CSA), ainsi que 7.5.1.2 et 7.5.1.5, les alinéas 7.5.2.1 a) à c) et à l'article 7.5.2.2 de la norme sur le sang de la CSA? | Oui / Non |
| Étiquetage | ||
| 60 | Les renseignements figurant sur l'étiquette des contenants de sang et les documents d'information sont-ils en français ou en anglais? | Oui / Non |
| 61 | Les étiquettes satisfont-elles aux exigences suivantes? | s/o |
| a) tous les renseignements sont exacts et présentés de façon claire et lisible | Oui / Non | |
| b) les étiquettes sont fabriquées de manière à ce qu'aucun adhésif ou encre ne puisse traverser le contenant de sang | Oui / Non | |
| c) les étiquettes sont apposées sur le contenant de façon permanente | Oui / Non | |
| d) les étiquettes volantes sont fermement fixées au contenant | Oui / Non | |
| 63 | Lors du prélèvement, une étiquette où figure de façon indélébile le code d'identification du don a-t-elle été apposée sur chaque contenant de sang? | Oui / Non |
| 64(1) | Les renseignements suivants se trouvent-ils sur l'étiquette du contenant de sang? | s/o |
| a) nom et adresse municipale de l'établissement | Oui / Non | |
| b) le numéro de licence ou, à défaut, celui de son d'enregistrement | Oui / Non | |
| c) code d'identification du don | Oui / Non | |
| d) mention indiquant s'il s'agit de sang total ou d'un composant sanguin, et le cas échéant, le nom du composant | Oui / Non | |
| e) le groupe sanguin ABO et le facteur Rh, s'il y a lieu | Oui / Non | |
| f) volume approximatif de sang prélevé, sauf dans les cas d'aphérèse | Oui / Non | |
| g) volume approximatif du contenu du contenant | Oui / Non | |
| h) nom de l'anticoagulant ou de l'additif présent dans le contenant | Oui / Non | |
| i) température de conservation recommandée | Oui / Non | |
| j) date et, s'il y a lieu, heure de péremption | Oui / Non | |
| k) mise en garde indiquant que le sang peut transmettre des agents infectieux dans le cas de sang destiné à la transfusion | Oui / Non | |
| 64(2) | Les renseignements supplémentaires suivants figurent-ils sur l'étiquette de sang de don autologue? | s/o |
| a) mention « Pour utilisation à des fins autologues seulement » | Oui / Non | |
| b) symbole indiquant que le sang présente un risque biologique ou un symbole à cet effet, si les résultats des essais démontrent que le donneur est porteur d'une maladie ou d'un agent pathogène mentionnés à l'article 12.3.1.2 de la norme sur le sang de la CSA | Oui / Non | |
| c) dans le cas où l'établissement n'effectue pas les essais pour les maladies et agents mentionnés à l'article 12.3.1.2 de la norme sur le sang de la CSA, mention à cet effet | Oui / Non | |
| 65 | L'établissement qui prépare des aliquotes de sang à des fins de transfusion s'assure-t-il que les renseignements suivants figurent sur l'étiquette de chaque contenant d'aliquote : | s/o |
| a) code d'identification du don; | Oui / Non | |
| b) nom du composant sanguin; | Oui / Non | |
| c) code qui permet d'identifier l'aliquote; | Oui / Non | |
| d) groupe sanguin ABO et facteur Rh, s'il y a lieu | Oui / Non | |
| e) date de péremption? | Oui / Non | |
| 68 | L'établissement a-t-il été vérifié que les renseignements indiqués sur l'étiquette sont exacts et complets? | Oui / Non |
| Commentaires au sujet des dons autologues : | ||
| B. Conservation | ||
| 69(1)b) | Le sang prélevé est-il conservé conformément aux exigences concernant la conservation et la date de péremption prévues dans le tableau 2 de la norme sur le sang de la CSA? | Oui / Non |
| 69(2) | Le sang reçu d'un autre établissement est-il conservé conformément aux instructions sur l'étiquette du contenant et à toute autre instruction précisée par écrit par l'établissement qui a prélevé le sang? | Oui / Non |
| 70 | Le sang est-il conservé dans un lieu dont les conditions ambiantes sont appropriées et dont l'accès est restreint aux personnes autorisées? | Oui / Non |
| 71 | Les dons autologues, désignés ou dirigés sont-ils ségrégués des autres unités provenant des dons allogéniques? | Oui / Non |
| 72 | Les unités de sang dont les essais n'ont pas été effectués, dont les essais n'ont pas tous été effectués ou dont les résultats ne sont pas encore tous connus, ou se sont révélés réactifs de façon répétées ou positifs, ont-elles été ségréguées des autres unités de sang jugées sécuritaires aux fins de distribution ou de transfusion autologue? | Oui / Non |
| Commentaires au sujet de la conservation : | ||
| C. Distribution | ||
| 73(2) | Avant de distribuer le sang à des fins de transfusion, l'établissement ayant prélevé le sang d'un don autologue conclut-il à sa sécurité pour des fins de transfusion s'il est d'avis que le sang a été traité conformément au Règlement sur le sang? | Oui / Non |
| 74(1) | Avant de distribuer le sang destiné à des fins de transfusion, l'établissement a-t-il examiné le contenant pour vérifier : | s/o |
| a) la lisibilité des renseignements figurant sur l'étiquette du contenant de sang; | Oui / Non | |
| b) l'intégrité du contenant; | Oui / Non | |
| c) l'absence de tout signe de détérioration ou de contamination du sang; | Oui / Non | |
| d) dans le cas de composants sanguins congelés, l'absence de tout signe de dégel? | Oui / Non | |
| 74(2) | L'établissement empêche-t-il la distribution du sang aux fins de transfusion s'il relève l'un des faits suivants : | s/o |
| a) le code d'identification du don manque ou est illisible; | Oui / Non | |
| b) les renseignements, autres que le code d'identification du don, qui doivent figurer en application du Règlement sur le sang sur les étiquettes du contenant sont absents ou illisibles et ne peuvent être obtenus des dossiers; | Oui / Non | |
| c) le contenant est défectueux ou endommagé et n'assure plus la protection du sang contre les conditions extérieures; | Oui / Non | |
| d) des signes de détérioration ou de contamination sont présents? | Oui / Non | |
| 75 | Lorsque le sang est expédié : | s/o |
| a) l'intégrité des contenants de sang et la lisibilité de leurs étiquettes sont-elles vérifiées avant leur expédition? | Oui / Non | |
| b) Les contenants d'expédition utilisés sont-ils résistants aux dommages et permettent-ils d'assurer la sécurité du sang? | Oui / Non | |
| 76 | S'assure-t-on que le sang expédié à des fins de transfusion est adéquatement conservé pendant le transport conformément aux exigences prévues au tableau 2 de la norme sur le sang de la CSA? | Oui / Non |
| Commentaires au sujet de la distribution : | ||
| D. Activités de transformation du sang provenant d'un don allogénique ou autologue | ||
| 77 | Les méthodes de transformation du sang utilisées sont-elles sécuritaires et efficaces? | Oui / Non |
| 78(1) | Le lavage de sang est-il fait conformément aux articles 7.5.2.3 et 7.5.3 de la norme sur le sang de la CSA? | Oui / Non |
| 78(2) | L'établissement qui procède au lavage de sang veille-t-il à ce qu'une mention à cet effet figure sur l'étiquette du contenant de sang ainsi que, s'il y a lieu, toute nouvelle date et heure de péremption? | Oui / Non |
| 79(1) | L'établissement qui procède à la mise en commun de composants sanguins le fait-il conformément aux articles 7.11.1, et 7.11.3 de la norme sur le sang de la CSA? | Oui / Non |
| 79(2) | Les renseignements prévus aux articles 10.8.2 et 10.8.3 norme sur le sang de la CSA figurent-ils sur l'étiquette du contenant des composants sanguins mis en commun? | Oui / Non |
| 80 | L'irradiation de sang est-il fait conformément aux articles 7.12.2 à 7.12.6 de la norme sur le sang de la CSA? | Oui / Non |
| Commentaires au sujet des activités de transformation : | ||
| E. Activités de traitement du Programme de donneurs pré-évalués (PDPE) | ||
| 86 | a) Le PDPE est-il sous la supervision d'un directeur médical? | Oui / Non |
| b) A-t-on recours au PDPE seulement (i) lorsque du sang adéquat pour le receveur n'est pas, par ailleurs, disponible et (ii) que le médecin du receveur requiert ce sang pour le traitement urgent de son patient? | Oui / Non | |
| 87 | Un code d'identification du donneur est-il attribué au donneur lors de son acceptation dans le PDPE? | Oui / Non |
| 88(1) | Les mesures suivantes sont-elles prises tous les trois mois? | s/o |
| a) une évaluation de l'admissibilité au programme de chaque donneur conformément aux articles 40 à 44 du Règlement sur le sang | Oui / Non | |
| b) il prélève des échantillons sanguins du donneur et les met à l'essai aux fins suivantes : | Oui / Non | |
| (i) dépistage de maladies transmissibles et agents mentionnés aux articles 8.4.1 et 8.4.2 de la norme sur le sang de la CSA | Oui / Non | |
| (ii) détermination du groupe sanguin ABO | Oui / Non | |
| (iii) détermination du facteur Rh ainsi que, s'il y a lieu, dépistage de l'antigène D faible | Oui / Non | |
| (iv) dépistage de la présence d'anticorps cliniquement significatifs | Oui / Non | |
| 88(2) | L'établissement s'assure-t-il que les résultats des essais prévus aux sous-alinéas 88(1)b)(ii) et (iii) sont comparés avec les derniers résultats consignés au dossier du donneur, le cas échéant? | Oui / Non |
| 88(3) | Si la comparaison requise en vertu du paragraphe 88(2) révèle une incohérence, les essais sont-ils recommencés et l'établissement ne prélève-t-il pas de ce donneur tant que l'incohérence n'est pas résolue? | Oui / Non |
| 89 | Les mesures suivantes sont-elles prises à chaque prélèvement? | s/o |
| a) L'évaluation de l'admissibilité du donneur est effectuée | Oui / Non | |
| b) Un code d'identification du don est attribué à chaque unité de sang prélevé qu'il associe dans les dossiers au code d'identification du donneur | Oui / Non | |
| c) Un échantillon sanguin est prélevé puis mis à l'essai dans les 72 heures suivant le prélèvement aux fins suivantes: | s/o | |
| (i) dépistage des maladies transmissibles et agents mentionnés aux articles 8.4.1 et 8.4.2 de la norme sur le sang de la CSA | Oui / Non | |
| (ii) détermination du groupe sanguin ABO | Oui / Non | |
| (iii) détermination du facteur Rh ainsi que, s'il y a lieu, dépistage de l'antigène D faible | Oui / Non | |
| (iv) dépistage de la présence d'anticorps cliniquement significatifs | Oui / Non | |
| 90 | Le code d'identification du don, le groupe sanguin ABO et, s'il y a lieu, le facteur Rh figurent-ils toujours sur l'étiquette du contenant de sang? | Oui / Non |
| 91 | Si, lors d'une urgence, le sang prélevé d'un donneur pré-évalué n'est pas transfusé aux receveurs concernés, les exigences prévues à l'article 16.2.5 de la norme sur le sang de la CSA sont-elles respectées? | Oui / Non |
| Évaluation de l'admissibilité des donneurs | ||
| 40 | Lorsqu'il vérifie l'évaluation de l'admissibilité du donneur, l'établissement vérifie-t-il ce qui suit :
|
Oui / Non |
| 41 | L'établissement qui évalue de façon préliminaire l'admissibilité du donneur prend-il les mesures suivantes? | s/o |
| a) il obtient du donneur, à l'aide d'un questionnaire ou tout autre moyen similaire, des renseignements sur son identité, ses antécédents médicaux ainsi que ses antécédents sociaux, dans la mesure où ces derniers peuvent être utiles pour déterminer la présence d'un risque de maladies transmissibles par le sang | Oui / Non | |
| b) il fournit au donneur des renseignements sur les risques associés au don de sang et le risque que le receveur contracte une maladie transmissible | Oui / Non | |
| 42 | Le donneur est-il déclaré inadmissible si tout renseignement obtenu au titre des articles 39 à 41 du Règlement sur le sang révèle que la sécurité humaine ou la sécurité du sang pourraient être compromises? | Oui / Non |
| 43 | Si l'établissement conclut que le donneur est inadmissible, prend-il les mesures suivantes : | s/o |
|
Oui / Non | |
|
Oui / Non | |
|
Oui / Non | |
| 44(1) | Une fois que le donneur est déclaré admissible, l'établissement prend-il les mesures suivantes : | s/o |
| a) Il lui attribue un code d'identification du donneur, si ce n'est déjà fait; | Oui / Non | |
| b) Il lui mentionne qu'il a l'obligation d'informer l'établissement (i) s'il contracte au cours de la période prévue dans les procédures opérationnelles de l'établissement une maladie ou une affection susceptible de compromettre la sécurité de son don de sang, ou (ii) s'il a, ultérieurement à son don, des raisons de croire que le sang provenant de ce dernier ne devrait plus être utilisé? | Oui / Non | |
| 44(2) | L'établissement, dès qu'il reçoit des renseignements après le don au titre de l'alinéa (1)b), prend-il en considération les renseignements afin d'évaluer de nouveau la sécurité de ce don et de tout autre don provenant de ce donneur ainsi que l'admissibilité de ce dernier à d'autres dons? | Oui / Non |
| 44(3) | Si la nouvelle évaluation démontre que la sécurité du sang a pu être compromise et que le sang a déjà été distribué, l'établissement en avise-t-il toute personne à laquelle le sang a été distribué et, s'agissant d'un établissement, il mentionne également dans l'avis qu'il ne doit pas le distribuer ou le transfuser? | Oui / Non |
| Prélèvement | ||
| 47 | Un code d'identification du don a-t-il été attribué à chaque unité de sang prélevé? | Oui / Non |
| Le code d'identification du don a-t-il été associé, dans ses dossiers, au code d'identification du donneur? | Oui / Non | |
| 49(1) | L'établissement qui prélève du sang s'assure-t-il de respecter les exigences suivantes : | s/o |
| a) il recourt à des méthodes aseptiques; | Oui / Non | |
| b) l'équipement qu'il utilise pour le prélèvement est homologué conformément au Règlement sur les instruments médicaux; | Oui / Non | |
| c) les contenants qu'il utilise pour le prélèvement sont homologués conformément au Règlement sur les instruments médicaux et sont exempts de défaut ou de détérioration; | Oui / Non | |
| d) il consigne dans ses dossiers le numéro du lot du contenant et l'associe au numéro d'identification du don? | Oui / Non | |
| 49(2) | Les contenants sont-ils utilisés qu'une seule fois? | Oui / Non |
| 50 | Lors du prélèvement, le sang prélevé et les échantillons de sang pour des essais sont prélevés de manière à éviter toute contamination croisée? | Oui / Non |
| Essais | ||
| 17(2) | L'établissement vérifie-t-il que l'établissement qui met à l'essai du sang prélevé d'un donneur pré-évalué à des fins de dépistage de maladies transmissibles ou de leurs agents obtient une licence à cette fin? | Oui / Non |
| 55 b) | Le sang est-il mis à l'essai au moyen d'instruments médicaux homologués conformément au Règlement sur les instruments médicaux pour l'évaluation préliminaire de donneurs? | Oui / Non |
| Commentaires au sujet des activités du programme de donneurs pré-évalués : | ||
| F. Système de gestion de la qualité (s'applique à tous les établissements) | ||
| 93(1) | Y a-t-il une structure organisationnelle qui reflète les responsabilités de la direction à l'égard de chaque activité exercée par l'établissement? | Oui / Non |
| 93(2) | L'établissement dispose-t-il d'un système de gestion de la qualité efficace et a-t-il nommé un individu qui en assume la responsabilité? | Oui / Non |
| 93(3) | L'établissement revoit-il son système de gestion de la qualité, à intervalles réguliers qui sont déterminés dans ses procédures opérationnelles, pour s'assurer de sa pertinence et efficacité? | Oui / Non |
| 94(1) | Le système de gestion de la qualité comprend-il les éléments suivants : | s/o |
| a) une division responsable de l'assurance de la qualité; | Oui / Non | |
| b) un programme de contrôle de la qualité; | Oui / Non | |
| c) un système de contrôle des changements; | Oui / Non | |
| d) un programme de contrôle des processus au sens de l'article 3.1 de la norme sur le sang de la CSA; | Oui / Non | |
| e) un système qui permet l'amélioration des processus par le suivi des plaintes et la prise de mesures correctives ou préventives; | Oui / Non | |
| f) un système qui permet de recenser les renseignements obtenus après le don ainsi que ceux concernant les manquements, les accidents et les effets indésirables, de faire enquête à leur égard, de procéder à des retraits et de prendre des mesures correctives; | Oui / Non | |
| g) un programme de formation et d'évaluation des compétences du personnel; | Oui / Non | |
| h) un programme d'évaluation de l'exactitude et la fiabilité des résultats d'essais; | Oui / Non | |
| i) un système de contrôle des documents et de gestion de dossiers; | Oui / Non | |
| j) un système de vérification interne; | Oui / Non | |
| k) des plans d'intervention d'urgence; | Oui / Non | |
| l) un système qui permet d'identifier spécifiquement l'équipement, le matériel et les produits essentiels; | Oui / Non | |
| m) des spécifications écrites concernant l'équipement, le matériel, les produits et les services essentiels; | Oui / Non | |
| n) un programme d'entretien préventif de l'équipement essentiel; | Oui / Non | |
| o) un programme de validation des processus? | Oui / Non | |
| 94(3) | À moins que l'individu qui effectue la vérification interne d'une activité ne soit pas directement responsable de l'activité visée, la division responsable de l'assurance de la qualité relève-t-elle de la direction et est-elle indépendante des autres fonctions? | Oui / Non |
| 95 | L'établissement s'est-il doté de procédures opérationnelles pour toutes les activités menées par l'établissement et comportant des aspects liés à la sécurité humaine et la sécurité du sang? | Oui / Non |
| 96 | Les procédures opérationnelles répondent-elles aux exigences suivantes? | s/o |
| a) Elles sont présentées selon un modèle normalisé. | Oui / Non | |
| b) Elles sont approuvées par un cadre supérieur. | Oui / Non | |
| c) Elles sont facilement accessibles à chaque endroit où l'établissement exerce les activités visées par les procédures. | Oui / Non | |
| d) Elles sont tenues à jour. | Oui / Non | |
| 97 | L'établissement veille-t-il à ce que ses procédures opérationnelles visant les activités de traitement et de transformation permettent d'atteindre de façon constante les résultats attendus et conserve les documents justificatifs à cet effet ? | Oui / Non |
| 98(1) | L'établissement dispose-t-il de suffisamment de personnel qualifié par leurs études, leur formation ou leur expérience pour accomplir les tâches qui leur sont confiées? | Oui / Non |
| 98(2) | L'établissement offre-t-il un programme d'orientation et de formation de base et continue à son personnel et est-il doté d'un mécanisme d'évaluation des compétences? | Oui / Non |
| 99 | Les installations permettent-elles : | s/o |
| a) l'exercice des activités de l'établissement; | Oui / Non | |
| b) l'observation de bonnes pratiques d'hygiène par le personnel dans l'exercice de ses fonctions; | Oui / Non | |
| c) le nettoyage de façon à assurer la salubrité; | Oui / Non | |
| d) le contrôle approprié des conditions ambiantes dans toutes les zones d'activités; | Oui / Non | |
| e) le contrôle de l'accès aux zones d'activités; | Oui / Non | |
| f) l'évaluation préliminaire de donneurs dans des conditions où la confidentialité est assurée? | Oui / Non | |
| 100(1) | L'équipement essentiel est-il nettoyé, entretenu et, au besoin, validé en fonction de son utilisation prévue, et étalonné? | Oui / Non |
| 100(2) | Après une réparation ou une modification, l'équipement essentiel est-il, validé et étalonné de nouveau, s'il y a lieu? | Oui / Non |
| 101 | L'équipement utilisé pour conserver du sang permet-il à l'établissement de se conformer aux articles 69 à 72 du Règlement sur le sang? | Oui / Non |
| 102 | Le matériel et les produits essentiels sont-ils qualifiés ou validés, selon le cas, en fonction de leur utilisation prévue et sont-ils conservés dans des conditions ambiantes adéquates? | Oui / Non |
| Commentaires au sujet du système de gestion de la qualité : | ||
| G. Enquêtes et rapports concernant les accidents et manquements (s'applique à tous les établissements) | ||
| 103-108 | L'établissement a-t-il lu et compris les exigences en matière d'enquêtes et de rapports concernant les accidents et manquements? | Oui / Non |
| Commentaires au sujet des enquêtes et des rapports concernant les accidents et les manquements : | ||
| H. Enquêtes et rapports concernant les effets indésirables (s'applique à tous les établissements) | ||
| 109-116 | L'établissement a-t-il lu et compris les exigences en matière d'enquêtes et de rapports concernant les effets indésirables? | Oui / Non |
| Commentaires au sujet des enquêtes et des rapports concernant les effets indésirables : | ||
| I. Dossiers (s'applique à tous les établissements) | ||
| 117 | Le contenu des dossiers est-il complet, exact, lisible, indélébile et facilement accessible? | Oui / Non |
| 118 | Le code d'identification du don figure-t-il à chacun des dossiers relatifs au traitement, à la distribution, à la transformation et à la transfusion de sang? | Oui / Non |
| Documents aux dossiers des établissements qui prélèvent du sang allogénique | ||
| 119(1) | Les documents et renseignements énumérés ci-dessous qui concernent un don allogénique, y compris les activités du PDPE, sont-ils conservés dans ses dossiers durant la période de rétention prévue débutant à la date de la création du document au dossier? | s/o |
| 1. Code d'identification du donneur (50 ans) | Oui / Non | |
| 2. Code d'identification du don (50 ans) | Oui / Non | |
| 3. Rapprochement des codes d'identification du don (10 ans) | Oui / Non | |
| 4. Évaluation de l'admissibilité du donneur (5 ans) | Oui / Non | |
| 5. Inadmissibilité permanente du donneur (50 ans) | Oui / Non | |
| 6. Inadmissibilité temporaire du donneur (10 ans) | Oui / Non | |
| 7. Prélèvement - date du don (50 ans) | Oui / Non | |
| 8. Prélèvement - données relatives à l'aphérèse (5 ans) | Oui / Non | |
| 9. Prélèvement - renseignements au dossier du don (5 ans) | Oui / Non | |
| 10. Nom du fabricant et numéro de lot des contenants de sang ainsi que du matériel et des produits essentiels associés à chaque don (1 an) | Oui / Non | |
| 11. Résultats des essais effectués à des fins de dépistage de maladies transmissibles ainsi qu'à des fins de détermination du groupe sanguin ABO, du facteur Rh et de la présence d'anticorps cliniquement significatifs (50 ans) | Oui / Non | |
| 12. Préparation des composants sanguins (10 ans) | Oui / Non | |
| 13. Contrôle de la température de conservation du sang (5 ans) | Oui / Non | |
| 14. Sort des unités de sang, notamment leur destruction (50 ans) | Oui / Non | |
| 15. Distribution (50 ans) | Oui / Non | |
| 16. Documents d'expédition (1 an) | Oui / Non | |
| 17. Renseignements obtenus après le don (10 ans) | Oui / Non | |
| 18. Plaintes et enquêtes connexes (5 ans) | Oui / Non | |
| 19. Rapports de vérification internes (5 ans) | Oui / Non | |
| 20. Essais de contrôle de la qualité (5 ans) | Oui / Non | |
| 21. Entretien, qualification, validation et étalonnage de l'équipement essentiel (3 ans) | Oui / Non | |
| 22. Matériel et produits essentiels, y compris leur qualification (3 ans) | Oui / Non | |
| 23. Évaluation de l'exactitude et la fiabilité des résultats d'essais (5 ans) | Oui / Non | |
| 24. Chaque version des procédures opérationnelles mise en application, autre que celles concernant l'évaluation de l'admissibilité des donneurs (10 ans) | Oui / Non | |
| 25. Chaque version des procédures opérationnelles concernant l'évaluation de l'admissibilité des donneurs (50 ans) | Oui / Non | |
| 26. Qualifications, formation et évaluation des compétences des employés (10 ans) | Oui / Non | |
| 27. Enquêtes sur les accidents ou manquements et rapports connexes (10 ans) | Oui / Non | |
| 28. Enquêtes sur les effets indésirables et rapports connexes (10 ans) | Oui / Non | |
| 119(2) | Les documents concernant les qualifications, la formation et l'évaluation des compétences des employés sont-ils conservés durant 10 ans, à partir de la date à laquelle l'employé a cessé de travailler pour l'établissement? | Oui / Non |
| Documents aux dossiers des établissements qui prélèvent du sang autologue | ||
| 120(1) | Les documents et renseignements énumérés ci-dessous qui concernent un don autologue sont-ils conservés dans ses dossiers durant la période de rétention prévue débutant à la date de la création du document au dossier? | s/o |
| 1. Code d'identification du donneur (10 ans) | Oui / Non | |
| 2. Code d'identification du don (10 ans) | Oui / Non | |
| 3. Prélèvement - dossier du don (5 ans) | Oui / Non | |
| 4. Nom du fabricant et numéro de lot des contenants de sang ainsi que du matériel et des produits essentiels associés à chaque don (1 an) | Oui / Non | |
| 5. Résultats des essais effectués à des fins de dépistage de maladies transmissibles ainsi qu'à des fins de détermination du groupe sanguin ABO et du facteur Rh (10 ans) | Oui / Non | |
| 6. Préparation des composants sanguins (10 ans) | Oui / Non | |
| 7. Contrôle de la température de conservation du sang (5 ans) | Oui / Non | |
| 8. Sort des unités de sang, notamment leur destruction (10 ans) | Oui / Non | |
| 9. Distribution (10 ans) | Oui / Non | |
| 10. Documents d'expédition (1 an) | Oui / Non | |
| 11. Plaintes et enquêtes connexes (5 ans) | Oui / Non | |
| 12. Rapports de vérification internes (5 ans) | Oui / Non | |
| 13. Essais de contrôle de la qualité (5 ans) | Oui / Non | |
| 14. Entretien, qualification, validation et étalonnage de l'équipement essentiel (3 ans) | Oui / Non | |
| 15. Matériel et produits essentiels, y compris leur qualification (3 ans) | Oui / Non | |
| 16. Évaluation de l'exactitude et la fiabilité des résultats d'essais (5 ans) | Oui / Non | |
| 17. Chaque version des procédures opérationnelles mise en application (10 ans) | Oui / Non | |
| 18. Qualifications, formation et évaluation des compétences des employés (10 ans) | Oui / Non | |
| 19. Enquêtes sur les accidents ou manquements et rapports connexes (10 ans) | Oui / Non | |
| 20. Enquêtes sur les effets indésirables et rapports connexes (10 ans) | Oui / Non | |
| 120(2) | Les documents aux dossiers concernant les qualifications, la formation et l'évaluation des compétences des employés sont-ils conservés dans ses dossiers durant 10 ans, à partir de la date où l'employé a cessé de travailler pour l'établissement? | Oui / Non |
| Documents aux dossiers des établissements qui font la transformation du sang | ||
| 121(1) | Les documents et renseignements énumérés ci-dessous qui concernent la transformation du sang sont-ils conservés durant la période de rétention prévue débutant à la date de la création du document au dossier? | s/o |
| 1. Code d'identification du don (10 ans) | Oui / Non | |
| 2. Renseignements concernant le lavage, la mise en commun et l'irradiation (10 ans) | Oui / Non | |
| 3. Nom du fabricant et numéro de lot du matériel et des produits essentiels associés à chaque transformation (1 an) | Oui / Non | |
| 4. Plaintes et enquêtes connexes (5 ans) | Oui / Non | |
| 5. Rapports de vérification interne (5 ans) | Oui / Non | |
| 6. Essais de contrôle de la qualité (5 ans) | Oui / Non | |
| 7. Entretien, qualification, validation et étalonnage de l'équipement essentiel (3 ans) | Oui / Non | |
| 8. Matériel et produits essentiels, notamment leur qualification (3 ans) | Oui / Non | |
| 9. Chaque version des procédures opérationnelles mise en application (10 ans) | Oui / Non | |
| 10. Qualifications, formation et évaluation des compétences des employés (10 ans) | Oui / Non | |
| 11. Enquêtes sur les accidents ou manquements et rapports connexes (10 ans) | Oui / Non | |
| 12. Enquêtes sur les effets indésirables et rapports connexes (10 ans) | Oui / Non | |
| 121(2) | Les documents concernant les qualifications, la formation et l'évaluation des compétences des employés sont-ils conservés durant 10 ans, à partir de la date où l'employé a cessé de travailler pour l'établissement? | Oui / Non |
| 123 | L'établissement entrepose-t-il ses dossiers dans un lieu où les conditions ambiantes sont adéquates et dont l'accès est restreint aux personnes autorisées? | Oui / Non |
| Commentaires au sujet des dossiers : | ||
Annexe D: Règlement sur les aliments et drogues C.04.400-C.04.423 Plasma humain prélevé par plasmaphérèse: Abrogés
Cette annexe fournit les exigences du Règlement sur les aliments et drogues C.04.400-C.04.423 Plasma humaine prélevé par plasmaphérèse. C'est sur ces dispositions que sont fondés les critères approuvés des établissements titulaires d'une licence qui étaient assujettis à ces articles avant leur abrogation. Ces exigences de base seront modifiées une fois qu'une demande de modification d'une homologation sera déposée par un établissement approuvé par Santé Canada
Plasma humain prélevé par plasmaphérèse
Interprétation
C.04.400. Les définitions qui suivent s'appliquent au présent article et aux articles C.04.401 à C.04.423.
« accident » Événement imprévu qui n'est pas imputable à une inobservation des procédures du manufacturier ou des règles de droit applicables et qui peut compromettre la sécurité du donneur ou l'innocuité, l'efficacité ou la qualité du plasma. (accident)
« donneur » Personne âgée d'au moins 17 ans qui a fourni son nom à un manufacturier en vue de participer à la plasmaphérèse avec celui-ci. (donor)
« effet indésirable grave » Réaction imprévue et indésirable du donneur relative à la plasmaphérèse ou à l'immunisation spécifique et qui entraîne chez lui l'une ou l'autre des conséquences suivantes :
- a) son hospitalisation;
- b) une incapacité persistante ou importante;
- c) une intervention médicale ou chirurgicale visant à prévenir une incapacité persistante ou importante;
- d) la mise en danger de sa vie;
- e) sa mort. (serious adverse reaction)
« identificateur personnel » Série unique de lettres, de chiffres ou de symboles, ou toute combinaison de ceux-ci, attribuée à un donneur par le manufacturier. (personal identifier)
« identificateur unique » Série unique de lettres, de chiffres ou de symboles, ou toute combinaison de ceux-ci, attribuée par le manufacturier au plasma destiné au fractionnement ou aux globules rouges qui seront utilisés pour l'immunisation spécifique. (unique identifier)
« immunisation spécifique » L'administration d'un immunogène à un donneur visant à induire la production d'anticorps précis dans son sang en vue de la plasmaphérèse. (specific immunization)
« manquement » Inobservation des procédures du manufacturier ou des règles de droit applicables pouvant compromettre la sécurité du donneur ou l'innocuité, l'efficacité ou la qualité du plasma. (error)
« manufacturier » Titulaire d'une licence d'établissement délivrée aux termes du présent règlement et l'autorisant à manufacturer du plasma destiné au fractionnement. (fabricator)
« médecin » Personne habilitée à exercer la médecine en vertu des lois de la province où cette personne fournit les soins médicaux relatifs à la plasmaphérèse ou à l'immunisation spécifique. (physician)
« plasma destiné au fractionnement » Plasma humain prélevé par plasmaphérèse et destiné à la fabrication de drogues pour usage humain. (source plasma)
« plasmaphérèse » Procédé comportant les étapes suivantes :
- a) est prélevé, du donneur, du sang duquel est séparé le plasma;
- b) les globules rouges et autres éléments figurés du sang sont retournés au donneur. (plasmapheresis)
« séance de plasmaphérèse » Rencontre entre le manufacturier et le donneur tenue dans le but de procéder à la plasmaphérèse chez celui-ci. (plasmapheresis session)
« substitut » Personne qui, à la fois :
- a) agit sous la surveillance et la direction générales d'un médecin;
- b) est autorisée à fournir les services pouvant être effectués par le substitut aux termes des articles C.04.401 à C.04.423 en vertu des lois applicables de la province où elle fournit ces services. (physician substitute)
DORS/78-545, art. 1; DORS/85-1022, art. 1; DORS/2006-353, art. 1.
Interdictions
C.04.401. Il est interdit à toute personne :
- a) de vendre du plasma destiné au fractionnement à moins que celui-ci n'ait été manufacturé, analysé, emballé-étiqueté et entreposé conformément aux articles C.04.402 à C.04.423;
- b) de fabriquer du plasma destiné au fractionnement à partir de sang prélevé d'une personne jugée inadmissible à participer à la plasmaphérèse au titre des articles C.04.402 à C.04.423.
DORS/78-545, art. 1; DORS/85-1022, art. 2; DORS/2006-353, art. 1.
Responsabilité du manufacturier
C.04.402. (1) Le manufacturier veille à ce que toute personne qui lui fournit des services relatifs à la plasmaphérèse ou à l'immunisation spécifique soit qualifiée de par son éducation ainsi que de par sa formation ou son expérience à fournir ces services.
(2) Le manufacturier veille à ce que les locaux utilisés pour la sélection des donneurs, la plasmaphérèse ou l'immunisation spécifique soient conçus, construits et entretenus de manière à permettre la communication de renseignements médicaux en toute confidentialité.
DORS/78-545, art. 1; DORS/85-1022, art. 2; DORS/97-12, art. 47; DORS/2006-353, art. 1.
Consentement et évaluation préliminaire
C.04.403. (1) Le manufacturier ne peut procéder à la plasmaphérèse chez un donneur que si les conditions suivantes sont réunies :
- a) le manufacturier informe le donneur de ce en quoi consiste la plasmaphérèse, notamment des risques que celle-ci présente pour la santé du donneur, dont ceux associés au fait d'y participer plus d'une fois toutes les huit semaines;
- b) après avoir respecté l'alinéa a), il obtient de lui par écrit :
- (i) la confirmation que les renseignements mentionnés à l'alinéa a) lui ont été communiqués,
- (ii) un consentement éclairé à participer à la plasmaphérèse, donné conformément aux règles de droit régissant les consentements.
(2) Le manufacturier ne peut procéder à l'immunisation spécifique d'un donneur que si les conditions suivantes sont réunies :
- a) un médecin choisit l'immunogène à être administré et informe le donneur :
- (i) du nom et de la nature de l'immunogène choisi,
- (ii) de la fréquence envisagée des injections et du nombre maximal d'injections que celui-ci pourrait recevoir,
- (iii) de ce en quoi consiste l'immunisation spécifique, notamment des risques que celle-ci présente pour sa santé, dont ceux associés au fait de recevoir l'immunogène choisi;
- b) après avoir respecté l'alinéa a), il obtient de lui par écrit :
- (i) la confirmation que les renseignements mentionnés à l'alinéa a) lui ont été communiqués,
- (ii) un consentement éclairé à recevoir l'immunogène choisi, donné conformément aux règles de droit régissant les consentements.
DORS/78-545, art. 1; DORS/2006-353, art. 1.
C.04.404. (1) Le manufacturier ne peut procéder à la plasmaphérèse ou à l'immunisation spécifique que si un médecin ou son substitut décide si le donneur est admissible à participer à la plasmaphérèse plus d'une fois toutes les huit semaines d'après ses antécédents médicaux et un examen médical.
(2) Si le donneur est jugé admissible, le manufacturier consigne les renseignements suivants :
- a) le fait que le donneur est admissible à participer à la plasmaphérèse plus d'une fois toutes les huit semaines;
- b) les nom et identificateur personnel du donneur;
- c) les nom et signature du médecin ayant pris la décision ou chargé de la surveillance du substitut qui l'a prise;
- d) la date de la décision.
(3) Le manufacturier ne peut procéder à la plasmaphérèse ou à l'immunisation spécifique si la dernière décision visée à l'alinéa (1) à l'égard du donneur a été prise au-delà des délais suivants :
- a) trente jours avant la date prévue pour sa première participation à la plasmaphérèse ou à l'immunisation spécifique;
- b) un an avant toute autre date prévue pour sa participation à la plasmaphérèse ou à l'immunisation spécifique.
DORS/78-545, art. 1; DORS/85-1022, art. 3; DORS/2006-353, art. 1.
Immunisation spécifique
C.04.405. (1) Seul le médecin ou son substitut peut administrer un immunogène à un donneur en vue de l'immunisation spécifique.
(2) Le médecin surveille la réponse du donneur à l'immunogène pour décider si celui-ci peut continuer de recevoir l'immunisation spécifique.
(3) Le manufacturier s'abstient de procéder à l'immunisation spécifique du donneur qui ne peut continuer de la recevoir jusqu'à ce qu'un médecin décide que le donneur peut la recevoir de nouveau, soit avec le même immunogène, soit avec un autre.
DORS/78-545, art. 1; DORS/85-1022, art. 3; DORS/2006-353, art. 1.
Évaluation avant le prélèvement
C.04.406. (1) Au début de chaque séance de plasmaphérèse, le médecin ou son substitut décide si le donneur est admissible à participer à la plasmaphérèse.
(2) Si le donneur est jugé temporairement inadmissible eu égard aux critères énumérés au tableau 1 ou pour toute autre raison médicale justifiant une telle décision, le manufacturier annule la séance et informe le donneur des motifs pour lesquels il est jugé temporairement inadmissible et de la date à laquelle il pourra continuer à participer à la plasmaphérèse.
(3) Si le donneur est jugé inadmissible pour une durée indéterminée eu égard aux critères d'exclusion énumérés au tableau 2 ou pour toute autre raison médicale justifiant une telle décision, le manufacturier annule la séance de plasmaphérèse et informe le donneur des motifs pour lesquels il est jugé inadmissible à participer à la plasmaphérèse pour une durée indéterminée.
| Article | Critères |
|---|---|
| 1. | Poids inférieur à 50 kg |
| 2. | Température anormale |
| 3. | Pression artérielle diastolique supérieure à 100 mmHg ou pression artérielle systolique supérieure à 180 mmHg |
| 4. | Niveau d'hémoglobine inférieur à 125 g par litre de sang ou valeur hématocrite inférieure à 0,38 L par litre de sang |
| 5. | Niveau total de protéines inférieur à 60 g par litre de sang |
| 6. | Perte importante de sang |
| 7. | Don antérieur de plasma ou d'autres composants sanguins |
| 8. | Grossesse |
| 9. | Antécédents d'interventions médicales ou chirurgicales |
| 10. | Antécédents de convulsions nécessitant un traitement médical |
| 11. | Faculté à répondre aux questions compromise par la consommation d'alcool ou de drogue |
| 12. | Transfusion antérieure de sang, d'un composant sanguin ou d'un dérivé sanguin ou transplantation antérieure de cellules, de tissus ou d'organes autres que la dure-mère |
| 13. | Infection de la peau à l'endroit où doit être pratiquée la phlébotomie |
| 14. | Signe ou symptôme d'infection |
| 15. | Risque d'une infection par le VIH, par le virus de l'hépatite B ou le virus de l'hépatite C compte tenu, entre autres, d'antécédents d'acupuncture, de perçage corporel, de tatouage, de piqûre accidentelle avec des aiguilles ou de relations sexuelles occasionnelles avec une personne présentant un risque d'être atteinte de l'une ou l'autre de ces infections |
| 16. | Prise de médicaments, présente ou passée, présentant un risque pour les receveurs de produits fabriqués à partir de plasma destiné au fractionnement |
| 17. | Réception d'un vaccin à virus vivant atténué |
| 18. | Morsure d'animal nécessitant une prophylaxie contre la rage ou pour laquelle la nécessité d'une prophylaxie post-exposition n'a pas été évaluée |
| Article | Critères d'exclusion |
|---|---|
| 1. | Fonction cardiovasculaire anormale ou cardiopathie grave ou chronique |
| 2. | Fonction respiratoire anormale ou maladie respiratoire grave ou chronique |
| 3. | Trouble de saignement présentant, pour le donneur, un risque associé à la plasmaphérèse |
| 4. | Maladie grave ou état pathologique du foie, des reins, d'un autre organe, d'un système ou du sang |
| 5. | Protéines plasmatiques anormales persistantes, y compris des gammopathies monoclonales ou polyclonales |
| 6. | Prise de médicaments, présente ou passée, présentant un risque continu pour les receveurs de produits fabriqués à partir de plasma destiné au fractionnement |
| 7. | Antécédents d'évanouissements récurrents associés au don de sang ou de plasma |
| 8. | Antécédents, signes ou symptômes d'abus de drogues injectables tels que traces de piqûres, cicatrices ou partage d'aiguilles pour l'injection de drogues |
| 9. | Antécédents, signes ou symptômes du SIDA ou d'une infection par le VIH |
| 10. | Risque d'une infection par le VIH compte tenu des pratiques sexuelles |
| 11. | Antécédents, signes ou symptômes d'une infection chronique ou persistante ou d'une maladie parasitaire transmissible par le sang |
| 12 | Antécédents, signes ou symptômes d'une hépatite autre que l'hépatite A |
| 13 | Cancer, autre qu'un cancer de la peau nonmélanocytique ou qu'un cancer cervical in situ |
| 14 | Facteur de risque pour la maladie de Creutzfeldt-Jakob ou sa variante parce que le donneur a, entre autres, reçu une transplantation de duremère ou subi un traitement avec une hormone pituitaire humaine |
| 15 | Résultat positif à un essai visant à dépister tout agent de maladie transmissible |
DORS/78-545, art. 1; DORS/85-1022, art. 3; DORS/2006-353, art. 1.
Composition en protéines plasmatiques
C.04.407. (1) Avant de procéder à la plasmaphérèse, le manufacturier prélève un échantillon du sang du donneur afin d'en déterminer la composition en protéines plasmatiques au moyen d'une électrophorèse des protéines sériques ou d'un essai équivalent.
(2) L'échantillon est prélevé dans les sept jours précédant la première séance de plasmaphérèse du donneur au cours de laquelle le manufacturier procède à la plasmaphérèse.
(3) S'il s'écoule vingt et un jours depuis le prélèvement de l'échantillon sans qu'un médecin n'examine les résultats de l'essai, le manufacturier ne peut procéder à la plasmaphérèse, et ce, jusqu'à ce qu'un médecin examine les résultats de l'essai.
(4) Si le médecin constate que la composition en protéines plasmatiques du sang du donneur ne se situe pas dans les limites normales, le manufacturier ne peut procéder à la plasmaphérèse, et ce, jusqu'à ce qu'un médecin constate le retour à la normale.
(5) Si aucun échantillon n'a été prélevé sur le donneur, comme l'exige le paragraphe (1), depuis plus de quatre mois, le manufacturier ne peut procéder à la plasmaphérèse, et ce, jusqu'à ce qu'un nouvel échantillon soit prélevé.
DORS/78-545, art. 1; DORS/85-1022, art. 3; DORS/2006-353, art. 1.
Examen permanent des dossiers
C.04.408. (1) Le médecin décide si le donneur est admissible à continuer à participer à la plasmaphérèse plus d'une fois toutes les huit semaines d'après les résultats des essais et les dossiers de prélèvement du donneur recueillis par le manufacturier dans les quatre mois précédents
(2) La décision est prise au moins tous les quatre mois suivant la date à laquelle le donneur a été jugé admissible pour la première fois au titre de l'article C.04.404.
(3) Si le donneur est jugé inadmissible à participer à la plasmaphérèse pour une durée temporaire, le manufacturier informe le donneur des motifs de son inadmissibilité temporaire et de la date à laquelle il pourra continuer à participer à la plasmaphérèse.
(4) Le manufacturier ne peut plus procéder à la plasmaphérèse si le donneur est jugé inadmissible à y participer pour une durée indéterminée; le cas échéant, il en informe le donneur, motifs à l'appui.
(5) En cas de manquement au paragraphe (2), le manufacturier ne peut procéder à la plasmaphérèse tant que la décision n'a pas été prise.
DORS/78-545, art. 1; DORS/85-1022, art. 3; DORS/2006-353, art. 1.
Procédures de plasmaphérèse
C.04.409. Le manufacturier prend les mesures ci-après lors de la séance de plasmaphérèse :
- a) il utilise des méthodes aseptiques et un système de prélèvement stérile homologué en vertu du Règlement sur les instruments médicaux;
- b) il veille à ce que toute surface destinée à entrer en contact avec le sang ou le plasma soit apyrogène;
- c) il veille à ce que la peau du donneur, à l'endroit où doit être pratiquée la phlébotomie, soit, à la fois :
- (i) exempte de toute lésion, éruption cutanée ou autre source d'infection,
- (ii) nettoyée et désinfectée;
- d) il veille à ce que du personnel médical d'urgence puisse procurer des soins médicaux au donneur en moins de dix minutes après avoir été avisé par le manufacturier.
DORS/78-545, art. 1; DORS/85-1022, art. 4; DORS/2006-353, art. 1.
Quantité maximale et intervalle minimal
C.04.410. (1) Le manufacturier ne peut prélever du donneur une quantité de plasma qui, sans comprendre la solution anticoagulante, excède les quantités totales suivantes :
- a) dans les cas où le poids du donneur est égal ou supérieur à 50 kg, mais inférieur à 68 kg :
- (i) 625 mL ou 640 g par séance de plasmaphérèse,
- (ii) 11,5 L pour l'ensemble des séances de plasmaphérèse au cours de la période précédente de six mois;
- b) dans les cas où le poids du donneur est égal ou supérieur à 68 kg, mais inférieur à 80 kg :
- (i) 750 mL ou 770 g par séance de plasmaphérèse,
- (ii) 15,5 L pour l'ensemble des séances de plasmaphérèse au cours de la période précédente de six mois;
- c) dans les cas où le poids du donneur est égal ou supérieur à 80 kg :
- (i) 800 mL ou 820 g par séance de plasmaphérèse,
- (ii) 18,5 L pour l'ensemble des séances de plasmaphérèse au cours de la période précédente de six mois.
(2) Le manufacturier se dote de procédures écrites exposant :
- a) la période minimale d'attente qu'un donneur doit respecter entre chaque don de plasma et entre un don de plasma et un don de sang ou d'autres composants sanguins;
- b) le nombre maximal de dons de plasma qu'un donneur peut faire au cours d'une période donnée.
DORS/78-545, art. 1; DORS/85-1022, art. 5; DORS/95-203, art. 1; DORS/2006-353, art. 1.
Solution anticoagulante
C.04.411. (1) Au cours de la plasmaphérèse, le manufacturier mélange une solution anticoagulante avec le sang prélevé du donneur.
(2) La solution anticoagulante doit porter une identification numérique de drogue valide attribuée aux termes du présent règlement qui indique que la solution convient à la plasmaphérèse.
DORS/78-545, art. 1; DORS/2006-353, art. 1.
Échantillons pour essais
C.04.412. (1) Au cours d'une séance de la plasmaphérèse, le manufacturier prélève un échantillon de sang ou de plasma de manière à ne pas contaminer l'échantillon ou le plasma destiné au fractionnement.
(2) Au moment de l'échantillonnage, le manufacturier étiquette clairement et de façon permanente le récipient contenant l'échantillon de sang ou de plasma avec l'identificateur unique assigné au plasma destiné au fractionnement.
(3) Le manufacturier veille à ce que la personne qui étiquette le récipient contenant l'échantillon soit la même qui étiquette le récipient contenant le plasma destiné au fractionnement au titre du paragraphe C.04.416(2).
DORS/78-545, art. 1; DORS/2006-353, art. 1.
C.04.413. (1) Le manufacturier soumet l'échantillon prélevé au titre de l'article C.04.412 à des essais visant à dépister les agents de maladie suivants :
- a) le VIH, types 1 et 2;
- b) le virus de l'hépatite B;
- c) le virus de l'hépatite C;
- d) la syphilis.
(2) Le manufacturier conserve tout plasma destiné au fractionnement prélevé lors de la séance de plasmaphérèse jusqu'à ce que tous les résultats des essais soient jugés négatifs ou non réactifs.
(3) Si le résultat d'un essai visant à dépister un agent de maladie énuméré au paragraphe (1) est positif ou réactif, le manufacturier prend les mesures suivantes :
- a) il étiquette clairement et de façon permanente le récipient contenant le plasma destiné au fractionnement prélevé lors de la séance de plasmaphérèse avec les renseignements suivants :
- (i) la mention « Précaution : Non destiné à la fabrication » ou « Caution : Not for Manufacturing Use »,
- (ii) le signal de danger pour les matières infectieuses prévu à l'annexe II du Règlement sur les produits contrôlés;
- b) il isole et élimine le plasma destiné au fractionnement.
(4) Si le résultat d'un essai visant à dépister l'agent de maladie pour la syphilis est positif ou réactif, le manufacturier ne peut procéder à la plasmaphérèse, et ce, jusqu'à ce qu'un essai subséquent démontre que le donneur n'est plus infecté par la syphilis et qu'un médecin décide que le donneur peut continuer à participer à la plasmaphérèse.
(5) Si le résultat d'un essai visant à dépister un agent de maladie énuméré au paragraphe (1), autre que la syphilis, est positif ou réactif, le manufacturier met fin à la plasmaphérèse du donneur et l'informe des motifs pour lesquels il est jugé inadmissible à participer à la plasmaphérèse pour une durée indéterminée.
DORS/78-545, art. 1; DORS/97-12, art. 48; DORS/2006-353, art. 1.
Agent de conservation et additive
C.04.414. Il est interdit d'ajouter un agent de conservation ou un additif au plasma destiné au fractionnement.
DORS/78-545, art. 1; DORS/85-1022, art. 6; DORS/2006-353, art. 1.
Récipients
C.04.415. Le manufacturier place le plasma destiné au fractionnement dans un récipient qui respecte les exigences suivantes :
- a) il est homologué en vertu du Règlement sur les instruments médicaux et destiné au prélèvement et à l'entreposage de plasma;
- b) il permet l'inspection visuelle, électronique ou automatisée du plasma;
- c) il a été inspecté visuellement à la séance de plasmaphérèse et a été jugé intact;
- d) il n'a pas servi à d'autres fins, dont celle d'avoir contenu du plasma destiné au fractionnement du même donneur.
DORS/78-545, art. 1; DORS/85-1022, art. 6; DORS/2006-353, art. 1.
Étiquetage
C.04.416. (1) Les articles C.01.004 et C.04.019 ne s'appliquent pas au plasma destiné au fractionnement.
(2) Le manufacturier étiquette clairement et de façon permanente le récipient utilisé pour contenir le plasma destiné au fractionnement avec les renseignements suivants :
- a) l'identificateur unique attribué au plasma destiné au fractionnement contenu dans le récipient;
- b) la mention « Plasma destiné au fractionnement » ou « Source Plasma »;
- c) la mention « Précaution : À utiliser uniquement pour la fabrication » ou « Caution : For Manufacturing Use Only »;
- d) la quantité de plasma destiné au fractionnement;
- e) le nom et la quantité totale de la solution anticoagulante utilisée au cours de la plasmaphérèse;
- f) la date limite d'utilisation du plasma destiné au fractionnement exprimée de façon claire et sans équivoque;
- g) sous réserve du paragraphe C.04.413(3), une mention indiquant que le plasma destiné au fractionnement ne réagit pas aux agents de maladie pour le VIH, le virus de l'hépatite B et le virus de l'hépatite C;
- h) si le plasma destiné au fractionnement est prélevé d'un donneur ayant reçu l'immunisation spécifique, une mention indiquant l'immunogène utilisé;
- i) les nom, adresse et numéro de licence d'établissement du manufacturier;
- j) une mention indiquant que le plasma destiné au fractionnement doit être conservé à une température de -20 °C ou moins.
(3) L'identificateur unique est ajouté sur l'étiquette du récipient au moment du prélèvement.
DORS/78-545, art. 1; DORS/85-1022, art. 7; DORS/2006-353, art. 1.
Entreposage
C.04.417. (1) S'agissant de l'entreposage du plasma destiné au fractionnement, y compris dans le cadre de son transport, le manufacturier est tenu aux obligations suivantes :
- a) s'assurer que l'environnement soit conçu pour maintenir la température à -20 °C ou moins;
- b) veiller à ce que la température ambiante soit constamment à -20 °C ou moins.
(2) Dans le cas où la température ambiante s'élève à plus de -20 °C, le manufacturier note les renseignements suivants :
- a) une explication de la température élevée;
- b) le plasma destiné au fractionnement touché;
- c) la disposition finale du plasma destiné au fractionnement.
(3) Si la température ambiante s'élève entre -20 °C et +10 °C, le manufacturier étiquette clairement et de façon permanente le récipient contenant le plasma destiné au fractionnement avec la mention « Plasma destiné au fractionnement - recyclé » ou « Source Plasma - Salvaged ».
(4) Le paragraphe (3) ne s'applique pas dans les cas où la température ambiante s'élève entre -20 °C et -5 °C pour une seule période de moins de soixante-douze heures.
(5) Si la température ambiante s'élève à plus de +10 °C, le manufacturier élimine le plasma destiné au fractionnement.
(6) L'alinéa (1)b) et les paragraphes (2) à (5) ne s'appliquent pas à l'entreposage du plasma destiné au fractionnement dans le cadre de son transport, si celui-ci n'est pas effectué par le manufacturier.
DORS/78-545, art. 1; DORS/85-1022, art. 8; DORS/2006-353, art. 1.
C.04.418. (1) Le manufacturier inspecte chaque récipient contenant du plasma destiné au fractionnement pour vérifier son intégrité, l'intégrité de l'étiquette et la présence de signes de dégel du plasma destiné au fractionnement.
(2) Le manufacturier élimine le plasma destiné au fractionnement dans l'un ou l'autre des cas suivants :
- a) le récipient est défectueux ou endommagé au point de ne plus fournir de protection contre les facteurs externes pouvant entraîner la détérioration ou la contamination du plasma destiné au fractionnement;
- b) l'identificateur unique du plasma destiné au fractionnement manque ou est illisible;
- c) un des renseignements visés aux alinéas C.04.416(2)b) à i) manque ou est illisible, à moins qu'il ne puisse être obtenu et extrait des dossiers du manufacturier;
- d) le plasma destiné au fractionnement présente des signes de dégel.
DORS/78-545, art. 1; DORS/2006-353, art. 1.
Documents
C.04.419. (1) Le manufacturier utilise et maintient un système de gestion de documents en vertu duquel :
- a) il attribue un identificateur personnel à chaque donneur;
- b) il place, au dossier du donneur, une photo servant à confirmer son identité ou y prévoit un autre moyen fiable d'identification;
- c) il attribue un identificateur unique au plasma destiné au fractionnement à chaque séance de plasmaphérèse.
(2) Le système est organisé de manière à ce que le manufacturier puisse, à partir de l'identificateur personnel ou de l'identificateur unique, identifier le donneur et récupérer suffisamment de documents pour permettre la traçabilité et le retrait du plasma destiné au fractionnement.
(3) Le manufacturier conserve pour une période indéterminée les documents visés au paragraphe (2).
DORS/78-545, art. 1; DORS/85-1022, art. 9; DORS/2006-353, art. 1.
C.04.420. (1) Pour chaque donneur, le manufacturier conserve les documents suivants :
- a) l'original ou une copie des confirmation et consentement visés aux alinéas C.04.403(1)b) et (2)b), le cas échéant;
- b) l'original ou une copie des décisions, examens médicaux, résultats des essais, rapports et avis écrits visés aux articles C.04.401 à C.04.423;
- c) pour chaque immunisation spécifique pratiquée chez le donneur par le manufacturier, un document indiquant :
- (i) les date et lieu de l'immunisation,
- (ii) le nom du médecin ou du substitut ayant administré l'immunogène,
- (iii) le nom de l'immunogène administré, le nom de son fabricant, la quantité utilisée, sa date limite d'utilisation et, soit ses numéro de lot et identification numérique de drogue, soit son identificateur unique s'il s'agit de globules rouges;
- d) pour chaque séance de plasmaphérèse avec le donneur, un document indiquant :
- (i) les date et lieu de la séance,
- (ii) le volume du plasma destiné au fractionnement prélevé,
- (iii) l'identificateur unique du plasma destiné au fractionnement,
- (iv) le volume de globules rouges prélevés qui ne lui a pas été retourné, notamment au cours de l'échantillonnage,
- (v) le nom de la solution anticoagulante utilisée, le nom de son fabricant et ses numéro de lot et identification numérique de drogue,
- (vi) le numéro de lot de tout récipient utilisé, le nom de son fabricant et sa date limite d'utilisation.
(2) Le manufacturier conserve un résumé de tout accident, manquement, effet indésirable grave et retrait de plasma destiné au fractionnement touchant le manufacturier.
(3) Le manufacturier conserve les données de température notées conformément au paragraphe C.04.417(2).
DORS/78-545, art. 1; DORS/85-1022, art. 10; DORS/97-12, art. 61; DORS/2006-353, art. 1.
Renseignements pour le ministre
C.04.421. (1) Le manufacturier avise le ministre de tout effet indésirable grave dans les délais suivants :
- a) dans les vingt-quatre heures du moment où il en prend connaissance dans le cas d'un décès;
- b) dans les quinze jours du moment où il en prend connaissance dans les autres cas.
(2) Le manufacturier fournit au ministre un rapport écrit concernant l'effet indésirable grave dans les vingt-quatre heures de l'avis donné conformément au paragraphe (1) si celui-ci a été donné verbalement.
(3) L'avis, s'il a été donné par écrit, ou le rapport écrit comprend la description de l'effet indésirable grave et de toute mesure corrective prise à la suite de sa survenance.
DORS/78-545, art. 1; DORS/2006-353, art. 1.
C.04.422. S'il retire du plasma destiné au fractionnement pour une raison qui a trait à la sécurité d'un produit, le manufacturier en avise le ministre par écrit en indiquant les motifs de retrait, le nombre d'unités touchées et le lieu d'où ces unités ont été retirées.
DORS/78-545, art. 1; DORS/2006-353, art. 1.
C.04.423. Afin de prévenir les risques pour la santé ou la sécurité des donneurs ou des receveurs de produits fabriqués à partir du plasma destiné au fractionnement, le manufacturier fournit au ministre, à la demande de celui-ci, une copie de tout document concernant la plasmaphérèse, l'immunisation spécifique ou le plasma destiné au fractionnement qu'il doit tenir en vertu des articles C.04.401 à C.04.422.
DORS/78-545, art. 1; DORS/2006-353, art. 1.
Annexe E: Les lignes directrices et directives de Santé Canada sont remplacées par la ligne directrice : Règlement sur le sang
À noter que toute autre ligne directrice et directive, ainsi que tout formulaire connexe (non mentionné) qui présente une autre interprétation du Règlement sur les aliments et drogues, Partie C, Division 1A, 2 et 4 ne s'applique plus au sang qui est assujetti au Règlement sur le sang.
- Ligne directrice pour la gestion des présentations des établissements de sang
- Ligne directrice : Plasma humain prélevé par plasmaphérèse
- Annexe 14 à l'édition actuelle des Lignes directrices sur les Bonnes pratiques de fabrication - Drogues visées à l'annexe D, sang et composants du sang humain (GUI-0032)
- Lettre de renseignements L.R. No 816 le 1er novembre 1995, 2. Politique de Santé Canada a) Donneurs jugés à risque pour ce qui est de la transmission de la MCJ b) Exclusion de donneurs c) Retrait/mise en quarantaine de produits sanguins non périmés (le 1er novembre 1995)
- D98-01: Directive: mise en oeuvre de la déleucocytation avant entreposage des éléments figurés (le 2 novembre 1998)
- D99-01: Directive en vue de l'exclusion des donneurs en raison du risque théorique de transmission de la variante de la vMCJ par l'approvisionnement en sang (17 août 1999)
- D99-02: Directive en vue de l'exclusion des donneurs en raison du risque théorique de transmission de la variante de la vMCJ par l'approvisionnement en sang par l'utilisation de produits sanguins commerciaux
- D2000-01: Directive d'exclusion des donneurs visant à réduire le risque théorique de transmission de la variante de la maladie de Creutzfeldt-Jocob (vMCJ) par le sang (30 août 2000)
- D2001-001:Directive d'exclusion des donneurs visant à réduire le risque théorique de transmission de la variante de la maladie de Creutzfeldt-Jacob (vMCJ) par le sang pour le Royaume-Uni, la France et l'Europe de l'Ouest (30 août 2001)
- Mesures supplémentaires d'exclusion des donneurs visant à réduire le risque théorique de transmission de la variante de la maladie de Creutzfeldt-Jacob (vMCJ) par le sang (le 22 avril 2005)
- D2006-01: Application de mesures de sélection des donneurs de sang pour réduire le risque théorique de transmission du virus spumeux simien ou d'éventuels virus simiens encore non identifiés par les transfusions (15 mai 2006)
- Lettre de renseignements sur le dépistage de la syphilis et les exigences en matière d'exclusion des donneurs de plasma destiné au fractionnement qui ont des antécédents de maladies parasitaires (le 2 septembre 2010)