
FDA guidance: Molecular diagnostic template for commercial manufacturers
Notice to Reader:
This is an archived FDA guidance. Please refer to the current version (Word document) of this guide.
Please note a French version has not been made available by the US FDA. Health Canada is committed to providing service in both official languages. Applicants who require guidance in the language of their choice are encouraged to contact us at devicelicensing-homologationinstruments@hc-sc.gc.ca.
On this page
- Before using the FDA guidance
- Template
- Purpose for submission
- Measurand
- Applicant
- Proprietary and established names
- Regulatory information
- Proposed intended use
- Device description and test principle
- Interpretation of results
- Product manufacturing
- Performance evaluation
- Unmet need addressed by the product
- Approved/cleared alternative products
- Benefits and risks
- Fact sheet for health care providers and patients
- Instructions for use, proposed labelling, package insert
- Record keeping and reporting information to the FDA
- Appendix A: Flex study design details, as applicable for the device
Before using the FDA guidance
Manufacturers following the FDA guidance for molecular and antigen tests for non-laboratory use should note that Health Canada expects them to follow the guidance for non-prescription testing. This is because the distinction made by the FDA between prescription and non-prescription testing does not exist in Canada.
The FDA guidances are in a template format and outline requirements that these products must meet.
The following FDA guidance document has been modified for accessibility, but the content is identical to the original document. Version July 28, 2020.
Template
This template (the “template”) provides FDA’s current recommendations concerning what data and information should be submitted to FDA in support of a pre-EUA/EUA submission for a molecular diagnostic for SARS-CoV-2.
As outlined in Section V.A. of the FDA guidance document Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised), FDA recommends that the following validation studies be conducted for a SARS-CoV-2 molecular diagnostic assay:
- limit of detection
- clinical evaluation
- inclusivity
- cross-reactivity
This template is intended to help manufacturers provide these validation data and other information to FDA, but alternative approaches can be used. It reflects FDA’s current thinking on the topic, and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word “should” means that something is suggested or recommended, but not required.
For more information about EUAs in general, please see the FDA guidance document Emergency Use Authorization of Medical Products and Related Authorities.
A. Purpose for submission
Emergency Use Authorization (EUA) request for distribution and/or use of the [test name] to [indicate labs, if applicable] for the in vitro qualitative detection of RNA from the SARS-CoV-2 in [add all claimed specimen types, such as nasopharyngeal/ oropharyngeal swabs, sputa, BAL] [select appropriate testing population, for example, from patients suspected of COVID-19 by a health care provider or for screening individuals without symptoms or other reasons to suspect COVID-19]. Additional testing and confirmation procedures should be performed in consultation with public health and/or other authorities to whom reporting is required. Test results should be reported in accordance with local, state and federal regulations.
If you plan to include a sample pooling protocol in your instructions for use, please include a brief description of the pooling strategy in your EUA request.
If you plan to request authorization to test specimens collected with a home specimen collection kit, please refer to the Home Specimen Collection Molecular Diagnostic Template and include any relevant information in this request.
B. Measurand
- Specific nucleic acid sequences from the genome of the SARS-CoV-2 (please specify the targeted gene(s) of the pathogen).
C. Applicant
Official name, address and contact information of applicant
D. Propriety and established names
Proprietary Name - [test name]
Established Name - [test name]
E. Regulatory information
Approval/clearance status:
The [test name] test is not cleared, CLIA waived, approved or subject to an approved investigational device exemption.
Product code:
- QJR
F. Proposed intended use
1) Intended use
The proposed intended use will be finalized based on the performance data and recommendations from public health authorities at the time of authorization. Example text is provided below for a qualitative molecular test that detects organism RNA but may be adapted according to the specific emergency situation addressed by the device.
[Test name] is a [specify test technology, such as real-time RT-PCR test] intended for the [presumptive] qualitative detection of RNA from the SARS-CoV-2 in [describe all the specimen types, such as nasopharyngeal, nasal and oropharyngeal swab specimens and lower respiratory tract, BAL or sputum]. [If your test is intended for testing multiple respiratory pathogens, please list the specific analytes detected by your test] [describe intended use population, for example, from individuals suspected of COVID-19 by their health care provider or for screening individuals without symptoms or other reasons to suspect COVID19 infection]. Testing is limited to [laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, to perform high complexity tests, or by similarly qualified non-U.S. laboratories]. [Describe the sample pooling approach and maximum number of specimens which can be pooled, as applicable.]
Results are for the identification of SARS-CoV-2 RNA. The SARS-CoV-2 RNA is generally detectable in [name specimen type, for example, upper respiratory] during the acute phase of infection. Positive results are indicative of the presence of SARS-CoV-2 RNA. Clinical correlation with patient history and other diagnostic information is necessary to determine patient infection status. Positive results do not rule out bacterial infection or co-infection with other viruses. The agent detected may not be the definite cause of disease. Laboratories within the United States and its territories are required to report all test results to the appropriate public health authorities.
The [test name] is intended for use by [include intended user, for example, qualified and trained clinical laboratory personnel specifically instructed and trained in the techniques of real-time PCR and in vitro diagnostic procedures]. The [test name] is only for use under the Food and Drug Administration’s Emergency Use Authorization.
[Depending on the performance data submitted and patient population included in the clinical evaluation, additional limitations may be recommended and/or your intended use may be modified to include the following, as applicable:
- Negative results do not preclude SARS-CoV-2 infection and should not be used as the sole basis for patient management decisions. Negative results must be combined with clinical observations, patient history and epidemiological information.
- Negative results from pooled samples should be treated as presumptive. If inconsistent with clinical signs and symptoms or necessary for patient management, pooled samples should be tested individually. Negative results do not preclude SARS-CoV-2 infection and must not be used as the sole basis for patient management decisions. Negative results must be considered in the context of a patient’s recent exposures, history, presence of clinical signs and symptoms consistent with COVID-19.
- Use of the [test name] in a general, asymptomatic screening population is intended to be used as part of an infection control plan, that may include additional preventative measures, such as a predefined serial testing plan or directed testing of high-risk individuals. Negative results should be considered presumptive and do not preclude current or future infection obtained through community transmission or other exposures. Negative results must be considered in the context of an individual’s recent exposures, history, presence of clinical signs and symptoms consistent with COVID-19.
If your test is intended for use at point of care settings, the following statement should be included:
- Testing is limited to laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, that meet the requirements to perform high, moderate or waived complexity tests. The [test name] is authorized for use at the Point of Care (POC), that is, in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance or Certificate of Accreditation.
2) Special conditions for use statements
- For Emergency Use Authorization (EUA) only
- For prescription use only
- For in vitro diagnostic use only
3) Special instrument requirements
The [test name] test is to be used with the [list all RT-PCR instruments, software requirements, automated extraction instruments].
G. Device description and test principle
Example text has been added under each of the sub-heading below for a fluorescence-based rRT-PCR test for detection of organism RNA. If a different test principle is used by the test for the detection of a specific analyte, please modify the description accordingly to capture the salient points in each of the sub-headings below. Please note that for new investigative technologies, FDA may request additional detailed information so we can adequately assess the risks and benefits associated with the device.
1) Product overview/test principle
Describe the technology of the test and how this technology works to identify the measurand, the instruments employed/required to perform the test from sample collection to result (include all claimed extraction and PCR detection instruments) and the specimen types for which you claim to have specific performance characteristics as described below. If applicable, list all primer and probe sets and briefly describe what they detect. Please include the nucleic acid sequences for all primers and probes used in the test. Please indicate if the test uses biotin-Streptavidin/avidin chemistry in any of the steps for coupling reagents. Please note that an alignment with available reference genomes for different strains of the target pathogen is requested as part of the inclusivity evaluation (Section J).
The [test name] is a real-time reverse transcription polymerase chain reaction (rRT -PCR) test. The SARS-CoV-2 primer and probe set(s) is designed to detect RNA from the SARS-CoV-2 in [list all the specimens] from patients suspected of COVID-19 by their health care provider.
2) Description of test steps
List and describe in detail all the steps of the test sequentially from specimen collection to detection.
Nucleic acids are isolated and purified from [specimens] using [please describe the method(s) of extraction (please specify the specimen input volume for extraction and/or test, the nucleic acid elution volume and whether isolation/purification is manual and/or automated)]. The purified nucleic acid is reverse transcribed using [enzyme mix/kits – please specify the input volume of purified nucleic acid added to the rRT-PCR reaction mix]into cDNA, which is then subsequently amplified in [please describe the instrument(s) and enzyme mix] . In the process, the probe anneals to a specific target sequence located between the forward and reverse primers. During the extension phase of the PCR cycle, the 5’ nuclease activity of Taq polymerase degrades the probe, causing the reporter dye to separate from the quencher dye, generating a fluorescent signal. With each cycle, additional reporter dye molecules are cleaved from their respective probes, increasing the fluorescence intensity. Fluorescence intensity is monitored at each PCR cycle by [please describe the detection instrument(s)].
3) Control material(s) to be used
List all control materials (provided with the test kit and/or required but not provided with the test kit and describe what they are, how they are expected to work, where in the testing process they are used and the frequency of use. If a control is commercially available, provide supplier’s name and catalog number or other identifier. If your device relies on external controls that are manufactured by a third party, please note that these controls should also be validated within your analytical and clinical studies described below in Section J.
Controls that will be provided with the test kit include:
- A “no template” (negative) control is needed to [describe need] and is used [describe use – please also specify frequency of use]
- A positive template control is needed to [describe need] and is used [describe use – please specify the concentration of the positive control relative to the LoD of your test (note that ideally the positive control concentration should be such that it is close to the LoD of your test) and also specify frequency of use]
- An extraction control [describe control] is needed to [describe need] and is used [describe use – please also specify frequency of use]. Please note that if the no template control and positive control are taken through the entire sample processing procedure, including the extraction, then a separate extraction control is not required.
- An internal control [describe control] is needed to [describe need] and is used [describe use].
Controls that are required but not provided with the test kit include [describe control; provide recommended sources of the control materials, either a separate control kit for purchase that you, the applicant, develops or a control material that can be purchased from a third party]. This/these control(s) is/are needed to [describe need] and is used [describe use – please also specify frequency of use].
Please note that any control recommended to be used with your device (provided with the kit or not) should be validated in the context of your analytical and clinical study (that is, you will need to run these controls as part of your studies). In instances where control material is not readily available through third-party vendors (which is often the case at the beginning of an outbreak), FDA may request that you include suitable control material with your device. Please note that external control materials are considered particularly important when GMP requirements are waived and reagent stability studies are limited.
H. Interpretation of results
All test controls should be examined prior to interpretation of patient results. If the controls are not valid, the patient results cannot be interpreted. Please describe if a Ct cutoff is used as part of your testing algorithm and/or if the end user is required to review fluorescent curves for weakly positive samples before final interpretation. Although not typical for molecular-based tests, if the test result involves the use of an algorithm/calculation (for example, a ratio value) when determining the final patient test result, please include a detailed description and any additional calibration materials that may be required.
1) [Test name] controls: positive, negative and internal
Describe in detail the expected results generated, including acceptance criteria, for all the controls described in detail in Section G above. Describe the measured values (if applicable) for valid and invalid controls and outline the recommended actions the laboratory should take in the event of an invalid control result.
2) Examination and interpretation of patient specimen results
Describe when clinical specimen test results should be assessed and outline the criteria for test validity.
Example text:
- Assessment of clinical specimen test results should be performed after the positive and negative controls have been examined and determined to be valid and acceptable. If the controls are not valid, the patient results cannot be interpreted.
Clearly indicate how to interpret numeric test values (if applicable) as positive or negative for presence of the SARS-CoV-2. Indicate if the end user is required to review fluorescent curves for weakly positive samples before final interpretation and how to identify indeterminate/inconclusive results (if they exist) results and how the user should resolve them(for example, if repeat testing may be required).
When applicable, provide a table clearly describing the possible combinations of test result values for each primer/probe set and how they should be combined into a final interpretation of the result for your test. If the test produces result that will be used as part of a CDC recommended testing algorithm, please indicate what follow-up testing/process should be conducted, if applicable.
I. Product manufacturing
1) Overview of manufacturing and distribution
The product will be manufactured at [manufacturer’s name and FDA registration number (if applicable)] by [manufacturer name] personnel consistent with practices for the production of [types of devices] based on [type of quality system*]. Material manufactured by [manufacturer’s name] may be bottled and kitted by [packager name] manufacturing facility.
The current manufacturing capabilities include the ability to manufacture approximately [please insert the approximate number of units/products that can currently be manufactured per week at the manufacturing facility] products per week. However, in the event of a surge in demand, this could be increased to [please insert the approximate maximum number of units/products that could potentially be manufactured per week at the manufacturing facility if there was a surge in demand] product per week within a [please specify in weeks/months the expected timeframe required to increase product production if required] timeframe.
The product will be distributed by [please describe the distribution plan for the product and list all current distributors].
*Under the Emergency Use Authorization (EUA), any of the 21 CFR Part 820 Quality System Regulation (QSR) requirements can be waived for the duration of the EUA but FDA recommends that developers follow comparable practices as much as possible if such requirements are waived. Among other things, FDA may consider previous compliance history when determining whether or not to waive certain QSR requirements for a specific product. Please note adverse events, as per 21 CFR Part 803, have to be reported for authorized devices (see Section P).
2) Components included with the test
Components manufactured by [manufacturer’s name and FDA registration number (if applicable)] and supplied with the test include:
- List all components and reagents provided for your test, including a description of the primers and probes, volumes, concentrations, quantities, buffer components and so on.
3) Components required but not included with the test
Components required but not included with the test:
- List all components and reagents not included with the test that must be supplied by the user to perform the test, with specific supplier names and catalog numbers or other identifiers for obtaining these components and reagents. Please include here all specific consumables that were validated for use with your device, that are not interchangeable with other products and that are needed to guarantee device performance as established in the EUA validation studies listed in Section J below.
4) Software validation
If you are introducing a system onto the market which has not been previously reviewed by FDA, we recommend providing evidence that the software has been validated to ensure that:
- the inputs and outputs of the software are appropriate to fulfill the system and assay requirements
- all expected inputs produce the expected outputs for all functions critical for system operation
- the system will be provided to the customer free of defects or defects will be known and mitigated
If this evidence is not available prior to authorization, they may be incorporated into the conditions of authorization. If changes which impact assay performance or safety and effectiveness of the system are needed to address validation failures post-authorization, then these may be required to be submitted as an EUA amendment in a condition of authorization. If no changes are needed or changes which do not impact assay performance or safety and effectiveness of the system are implemented, then the condition of authorization may require that validation data be kept on file.
We recommend you:
- perform electromagnetic compatibility (EMC) testing to International Electrotechnical Commission (IEC) 60601-1-2 Edition 4.0:2014
- evaluate cybersecurity of your system to ensure user and patient safety in the intended use environment
- complete validation of all systems and software to ensure that all functions of the system perform as labelled
For more information on system validation, please see the following FDA guidance documents and resources:
- Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices
- General Principles of Software Validation; Final Guidance for Industry and FDA Staff
- Off-The-Shelf Software Use in Medical Devices
- 21 CFR 820.30 Subpart C – Design Controls of the Quality System Regulation
5) Testing capabilities
Briefly describe current sample throughput capacity, total time required to perform the test (from clinical specimen collection, specimen transport, to result) and number of tests that can be performed per instrument run and per day.
6) Reagent stability
Briefly describe the stability test plan for reagents and include accelerated stability information, if available. Please note that reagent stability studies do not need to be completed at the time of EUA issuance. However, the study design should be agreed upon during interactive review and the stability studies started immediately following authorization, if not before.
You should consider the following recommendations when designing your stability study:
- For EUAs, you may follow the current FDA-recognized CLSI Standard EP25 – Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline when evaluating the suitability of stability study designs. If you are planning to pursue a De Novo/510(k) for your device, we recommend discussing in more detail your stability design to facilitate potential use of the EUA data in your regular premarket submission.
- We recommend testing a known positive diluted patient sample at 3x to 5x LoD rather than positive control material to establish reagent stability.
- If you are claiming multiple clinical specimen types in which similar LoDs are determined, you should use the most challenging clinical matrix for this study.
- We typically recommend your stability study design includes the evaluation of at least 5 replicates. You should also evaluate, if available, 3 different lots of reagents.
- You should design your study to provide data for a timeframe that is about 10% longer than the one to be claimed (for example, a claim of 18 months should be supported by stability data out to 20 months and a claim of 7 days should include stability data out to 8 days).
- FDA considers 15°C to 30°C to represent room temperature conditions. Ideally, you should evaluate stability at both 15°C and 30°C. However, for the purposes of the EUA evaluation, at 30°C is acceptable as the worse-case scenario.
- Shelf-life stability - unopened kit:
- You should evaluate real-time kit stability studies with unopened kits stored at the claimed storage temperature for your test.
- Accelerated stability evaluations for unopened kits is acceptable for EUA submissions while the real-time studies are ongoing. However, please note real-time stability data is required to support regular pre-market submissions and for the final claim of an EUA.
- Shipping stability - unopened kit:
- Study should evaluate the anticipated handling and shipping times and temperatures expected for unopened kits.
- In-use/opened kit stability:
- Depending on your device, your stability study design should also support in-use stability of the kit reagents once the kit has been opened (for example, storage at 2°C to 8°C for 7 days). This includes on-board stability once reagents have been placed on the instrument (if applicable).
- Inverted stability (if applicable):
- Study should support inverted stability for of kits.
- Freeze-thaw stability:
- If you recommend aliquoting the reagents to meet the end-users’ needs following the initial thaw, this recommendation should be supported by a freeze-thaw stability study, including the specific number of allowed freeze-thaw cycles.
- FDA analysis recommendations for real time stability studies are as follows:
- Baseline of the study (t=0 of stability study) should not exceed a month from bottling
- Clear baselines should be described (for example, a month from bottling) for each stability claim under each study
- Claims should be determined based on regression analysis. Any %change (%shift) from time zero (baseline) should be calculated between the target claim and the zero-time as (Ttest-Tbaseline)/ Tbaseline*100 with 95%CI using the regression equation obtained from plotting the mean values. When formulating your acceptance criteria for evaluating the shift from baseline, you should consider the reproducibility of your device. However, generally, that the shift at the target claim due to storage cannot exceed 10% to 15%. The target stability is the next to last tested point that was within +/- 10% of time zero.
- Acceptance criterion may be different, depending on the test samples analyte concentration distribution in the intended use population and the risk (in other words, the impact of false results to public health).
7) Sample stability
Please provide sample stability information, including the study design and results if the sample is shipped to a testing site from a location other than health care settings (for example, samples collected at home).
J. Performance evaluation
The following validation studies should be performed during your assay development:
1) Limit of detection (LoD) (analytical sensitivity)
You should determine the LoD of the test utilizing all components of the test system from sample preparation to detection. Testing quantified inactivated virus (for example, heat treated or irradiated virus) spiked into real clinical matrix (for example, BAL fluid, sputum, nasopharyngeal swab) for LoD determination is recommended since the inactivated virus most closely reflects live virus in a clinical sample. If you are unable to acquire inactivated virus, FDA believes that viral genomic RNA is the next best material to use to generated contrived samples for testing. As positive natural clinical specimens are increasingly becoming available, a quantified known positive clinical specimen as determined by an EUA-authorized test can also be used to create dilutions in clinical matrix for LoD determination. Respiratory swab matrix should derive from swab specimens collected from SARS-CoV-2 negative individuals.
FDA recommends that preliminary LoD be determined by testing a 2- to 3-fold dilution series of 3 replicates per concentration. The lowest concentration that gives positive results 100% of the time is defined as the preliminary LoD. The final LoD concentration should be confirmed by testing 20 individual extraction replicates at the preliminary LoD. FDA defines LoD as the lowest concentration at which 19/20 replicates are positive.
If multiple clinical matrices are intended for clinical testing, you should submit to FDA the results from 1 representative matrix of each claimed clinical matrix type. For example:
- If testing common upper respiratory tract specimens (for example, nasopharyngeal (NP) swabs, oropharyngeal (OP), swabs, nasal swabs, anterior nasal swabs, mid-turbinate nasal swabs, nasal aspirates and nasal washes), please submit results from the most challenging upper respiratory matrix. FDA considers nasopharyngeal (NP) swabs to be the most challenging upper respiratory matrix.
- If claiming common lower respiratory tract specimens (for example, tracheal aspirates, sputum), please submit results from the most challenging lower respiratory matrix. FDA considers sputum to be the most challenging lower respiratory matrix.
- If claiming both upper and lower respiratory matrixes, submitting results from sputum samples may suffice to support both upper and lower respiratory matrices.
- If claiming alternative respiratory specimens, such as saliva, oral fluid or buccal swab, please submit results from testing each of the claimed uncommon respiratory specimen type.
- If needed, FDA recommends that you follow the most current version of the CLSI standard, Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures (CLSI EP17).
Please describe your LoD study, the specific material used (for example, live or in-activated viral stocks, viral RNA), the specific clinical matrix used and the LoD (with appropriate units) for your assay.
2) Inclusivity (analytical sensitivity)
Laboratories should document the results of an inclusivity study that demonstrates the strains of SAR-CoV-2 that can be detected by the proposed molecular assay. It is acceptable to conduct an in silico analysis of published SARS-CoV-2 sequences using the assay’s primers and probes. FDA anticipates that 100% of published SAR-CoV-2 sequences will be detectable with the selected primers and probes.
Please describe your inclusivity study and confirm that there was 100% detection of all SARS-CoV-2 strains. If sequences with less than 100% homology with any of the primers and probes in your test are identified, please provide a risk assessment on how such mismatches may impact the performance of your test.
3) Cross-reactivity (analytical specificity)
Cross-reactivity studies are performed to demonstrate that the test does not react with related pathogens, high prevalence disease agents and normal or pathogenic flora that are reasonably likely to be encountered in a clinical specimen. For respiratory specimen claims excluding saliva and oral fluid, the recommended list of organisms to be analyzed in silico and by wet testing is provided in the table below. For non-respiratory matrices or saliva and oral fluid, an appropriate list of organisms should be tested (please see previous FDA decision summaries for recommended organisms or contact FDA for recommended organisms). For wet testing, concentrations of 106 CFU/ml or higher for bacteria and 105 pfu/ml or higher for viruses is recommended. In silico analyses alone may be acceptable for organisms that are difficult to obtain. FDA defines in silico cross-reactivity as greater than 80% homology between one of the primers/probes and any sequence present in the targeted microorganism.
| Other high priority pathogens from the same genetic family | High priority organisms likely present in a respiratory specimen |
|---|---|
| Human coronavirus 229E | Adenovirus (for example, C1 Ad. 71) |
| Human coronavirus OC43 | Human Metapneumovirus (hMPV) |
| Human coronavirus HKU1 | Parainfluenza virus 1-4 |
| Human coronavirus NL63 | Influenza A & B |
| SARS-coronavirus | Enterovirus (e.g. EV68) |
| MERS-coronavirus | Respiratory syncytial virus |
| Rhinovirus | |
| Chlamydia pneumoniae | |
| Haemophilus influenzae | |
| Legionella pneumophila | |
| Mycobacterium tuberculosis | |
| Streptococcus pneumoniae | |
| Streptococcus pyogenes | |
| Bordetella pertussis | |
| Mycoplasma pneumoniae | |
| Pneumocystis jirovecii (PJP) | |
| Pooled human nasal wash, to represent diverse microbial flora in the human respiratory tract | |
| Candida albicans | |
| Pseudomonas aeruginosa | |
| Staphylococcus epidermis | |
| Streptococcus salivarius |
Microbial interference studies
If in silico analysis reveals ≥80% homology between the cross-reactivity microorganisms and your test primers/ probe(s), we recommend that you either perform:
- a microbial interference study with SARS-CoV-2 and the microorganisms that your test primers/probe(s) have homology to or
- as an alternative to the microbial interference study, you may provide justification as to why (for example, amount of primer(s)/probe(s) included in your master mix) the performance of your test would not be impacted by the presence of a causative agent of a clinically significant co-infection or
- explain why the in silico results are clinically irrelevant (for example, low prevalence of MERS-CoV)
Competitive microbial interference testing should be conducted for multiplex panels. The study should assess the effects of clinically relevant co-infections by testing selected microorganisms commonly found in the claimed specimen matrix in the presence of SARS-CoV-2 at low concentration. The interference should be evaluated by testing with a minimum of 3 sample replicates spiked at a low (≤3x LoD) SARS-CoV-2 concentration and a high interferent level (preferably microorganisms), to represent the worst-case scenario. The interferent microorganisms can be tested individually or as a pool (of 4 or 5) in the presence of low concentration of SARS-CoV-2. Each microorganism of a pool should be tested individually, if that pool shows interference. If you plan to claim both upper and lower respiratory clinical specimens, the study should be performed in the most challenging specimen matrix (that is, sputum). If interference is observed at the level tested, an additional titration study should be performed to determine the highest microorganism interferent level your test can achieve the stated performance.
Endogenous interference substances studies
The extent of testing for endogenous interference substances depends on the matrix that is claimed for the device as well as on the technology of the device (for example, if a nucleic acid extraction procedure is performed prior to testing or not). If your test uses extraction methods not previous reviewed by FDA as part of premarket submission or the test does not use an extraction procedure, we recommend testing of potential interferents.
Please contact FDA to discuss the appropriate study designs.
4) Clinical evaluation
a) Claims for testing respiratory specimens from patients suspected of COVID-19 by their health care provider
FDA recommends using natural clinical specimens in the clinical evaluation. Please refer to the following table for additional information regarding clinical study design.
Note: Clinical study recommendations listed in the table below do not apply to claims for screening individuals without symptoms or other reasons to suspect COVID-19 and to saliva or other alternative respiratory specimen-type claims.
| Minimum number of positive specimens | A minimum of 30 individual natural (prospective or retrospective or leftover samples) positive clinical specimens should be collected from patients suspected of SARS-CoV-2 infection by a health care provider in COVID-19 disease endemic region(s). Samples can be a mixture of specimen types if you are seeking an upper respiratory claim (for example, nasopharyngeal (NP) swab, oropharyngeal (OP) swab, nasal swab (NS)). If you are seeking a sputum claim and any other respiratory specimen claim except alternative respiratory specimen types (for example, saliva), we recommend a combination of 15 NP and 15 sputum samples. Specimens collected from different anatomical sites from the same patient may be used to support claims for multiple specimen types. The use of frozen samples is acceptable. Specimens representing a wide range of viral load including low positive samples should be tested. The use of samples previously tested positive by another EUA RT-PCR assay may be acceptable without additional comparator testing. You should indicate the source of the samples, provide results for each tested sample, indicate specimen type, and initial test date. |
| Minimum number of negative specimens | A minimum of 30 individual negative samples acquired from the following sources are acceptable: (1) prospective samples from the individuals suspected of COVID-19 by their health care provider (2) archived/retrospective respiratory samples collected from patients with signs and symptoms of respiratory infection and (3) other subjects that are expected to be negative for SARS-CoV-2, such as specimens collected prior to COVID-19 pandemic in the U.S. |
| Recommended comparator method for percent agreement performance calculations | Positive percent agreement should be calculated in comparison to an EUA RT-PCR test. We recommend using only a high-sensitivity EUA RT-PCR assay, which uses a chemical lysis step followed by solid phase extraction of nucleic acid (for example, silica bead extraction). If available, FDA recommends selecting a comparator assay that has established high sensitivity with an internationally recognized standard or FDA SARS-CoV-2 Reference Panel. Please contact CDRH-EUA-Templates@fda.hhs.gov to discuss options to establish the sensitivity of your comparator method. Please see the web page for the most recent list of FDA-authorized 2019-nCoV tests. Negative result agreement may be calculated in comparison to an EUA RT-PCR test (prospectively collected samples) or as agreement with expected results if samples were collected from individuals known to be negative for SARS-CoV2 (for example, collected before December 2019). The comparator assay may have the same, or different, targets as your assay. False results can be investigated using an additional EUA RT-PCR assay and/or Sanger sequencing. The results of the discordant analysis can be footnoted in your final performance table but cannot be used to change the final performance calculations. |
| Acceptance criteria | FDA believes a minimum of 95% positive and negative agreement is acceptable clinical performance. |
| Natural clinical specimens IRB/informed consent note | Prospective collection of clinical specimens to support the EUA request should be done in accordance with regulations for human subject protection, including IRB approval and informed consent. Use of leftover de-identified samples may follow the policy outlined in the FDA Guidance on Informed Consent for In Vitro Diagnostic Device Studies Using Leftover Human Specimens that are not individually identifiable). |
| Testing approach note 1 | All clinical specimens tested in your study should be evaluated in accordance with your proposed diagnostic algorithm (that is, tested using the procedure in the instructions for use), including retesting when appropriate. The limited volume of natural specimens may preclude retesting. In instances where retesting is indicated but not performed, for the purposes of performance evaluation, initial results will be analyzed for performance, and equivocal/indeterminate/inconclusive results should count against your final performance. |
| Testing approach Note 2 | Specimens should be tested in a blinded fashion (for example, positive and negative samples should be presented to the end user in a blinded fashion). The end user should also be blinded to the results of any comparator method testing. |
b) Testing alternative specimens (that is, other than respiratory specimens) from patients suspected of COVID-19 by their health care provider
If you seek a claim for alternative specimens, such as saliva, oral fluid or buccal swabs, you should test 2 paired specimens from at least 30 positive and 30 negative patients. Consecutively collected specimens are preferred. Specimens representing a wide range of viral load including low positive samples should be tested. One specimen from each patient should be collected by a health care worker using a nasopharyngeal (NP) swab and tested with an assay authorized for use with NP specimens. FDA recommends selecting a comparator assay that has established high sensitivity with an internationally recognized standard or FDA SARS-CoV-2 Reference Panel. Please contact the FDA to discuss options to establish the sensitivity of your comparator method. The other specimen from each patient should be the alternative specimen and should be tested with your candidate EUA assay, provided it is authorized for testing of NP specimens, or using a previously authorized test with an NP swab claim. To minimize the occurrence of discordant results due to biological variability, both samples should be collected within a short time period. FDA believes ≥95% positive percent agreement with similar Ct values for the paired specimen types is acceptable performance.
Please provide detailed information regarding the type of collection device and transport media you propose to validate for use with your assay. Please note that some transport media may not be compatible with assays that do not use a nucleic acid extraction step. In addition, some transport medium may not be acceptable for use for at-home collection due to the presence of hazardous chemicals.
For additional information that may be needed to support at-home sample collection and transport, please review the Home Specimen Collection Molecular Diagnostic Template or contact the FDA.
c) Screening individuals without symptoms or other reasons to suspect COVID-19 with a previously unauthorized test
The recommendations below reflect FDA’s current thinking. The study design and recommendations may change as additional information becomes available regarding asymptomatic infections, including but not limited to viral titer dynamics and transmission rates in this population.
If you seek claims for screening individuals without symptoms or other reasons to suspect COVID-19, FDA recommends that you conduct a clinical study in the intended population. In the clinical study, you should compare results from for your assay and a comparator assay for each patient enrolled.
Please consider the following when designing your clinical validation study:
- The number of enrolled patients should be sufficient to ensure at least 20 positive samples are prospectively collected in the intended use population and be sufficient to demonstrate the following minimum performance:
- PPA ≥95% (lower bound of the 2-sided 95% confidence interval >76%)
- NPA ≥98% (lower bound of the 2-sided 95% confidence interval >95%)
- The total number of samples needed will depend on the prevalence of SARS-CoV-2 in the intended use population.
- Samples for the candidate test should be collected according to the instruction for use.
- Samples for comparator method testing should be health care provider-collected NP swabs. If an NP swab cannot be collected, a nasal swab may be used. However, both anterior nares should be sampled with the same swab. Sampling for the candidate test and comparator method should occur within a short timeframe to avoid biological variability in viral load.
- If available, FDA recommends selecting a comparator assay that has established high sensitivity with an internationally recognized standard or the FDA SARS-CoV-2 Reference Panel. Please contact the FDA to discuss options to establish the sensitivity of your test.
- In general, we recommend that you collect samples at a minimum of 3 geographically diverse sites, especially if you are planning to use the same data to support a subsequent De novo/510k submission. If this is not possible, FDA will consider samples collected at 1 or 2 sites in the context of an EUA.
- It may be possible to use archived samples that were collected from asymptomatic patients. We recommend you contact FDA to discuss such an approach prior to initiating your study.
d) Adding population screening of individuals without symptoms or other reasons to suspect COVID-19 to an authorized test
Alternative approaches may be acceptable for tests that have been previously authorized with clinical data for symptomatic patients.
For example:
- If your assay is highly sensitive as determined by testing with the FDA SARS-CoV-2 Reference Panel or a recognized international standard, a post-authorization study may be appropriate. We recommend testing a minimum of 20 consecutively collected asymptomatic positive specimens and at least 100 consecutively collected negative specimens based on the results of the candidate test. All specimens should then be tested with another EUA-authorized molecular assay. Using estimates of the predictive values and the percentage of positive results, this study can be used to establish the sensitivity (PPA) and specificity (NPA) of your test in a general, asymptomatic population, as this is an important performance metric for tests intended for screening of large populations without symptoms or other reasons to suspect COVID-19. The FDA expectation is that PPA should be >95% (lower bound of the 2-sided 95% confidence interval >76%) and NPA should be ≥98% (with a lower bound of the 2-sided 95% confidence interval >95%). If you do not have access to either the FDA SARS-CoV-2 Reference Panel or a recognized international standard, then please contact the FDA to discuss options.
- If you can demonstrate that performance of your assay in both populations is likely similar (that is, the percent of positive individuals with Ct values representing low viral loads are similar in individuals suspected and not suspected of COVID-19 by their health care provider), you may include both populations in your evaluation. We encourage the use of historic data (that is, existing or published data) for this evaluation.
FDA is open to considering additional alternative study designs to demonstrate that the performance of your assay is appropriate for screening individuals without symptoms or other reasons to suspect COVID-19. We recommend contacting FDA to discuss alternative study designs prior to beginning such a study.
e) Specimen pooling
The recommendations below reflect FDA’s current thinking. The study design and other recommendations may change as additional information becomes available. At this time, the need for testing remains greater than available resources. Combining multiple patient samples to create one pooled sample for testing could enable broader access to testing.
To establish performance of your test with pooling, FDA recommends conducting a clinical validation study in the intended use population that includes testing each sample individually and using your proposed pooling strategy.
Currently FDA recommends 2 approaches to patient specimen pooling:
- pooling aliquots of transport media, which each contain a single patient sample (sample/media pooling) or
- adding swabs from multiple patients into a single volume of transport media (swab pooling)
As more data become available and new approaches are identified, our recommendations may evolve.
Monitoring
Commercial test kit manufacturers should provide instructions for laboratories to incorporate ongoing monitoring of the pooling strategy by addressing the following in their instructions for use:
- Before implementation of pooling, evaluate existing test data in the testing population from the previous 7 to 10 days to estimate the initial positivity rate.
- When implementing a pooling strategy, continue to test a random sampling of patient samples without pooling to:
- evaluate the positivity rate and percent of weak positive samples in the testing population
- identify differences in positivity rate between those tested individually and those tested through pooling
- Calculate the percent of positive results after implementation of pooling using a moving average (such as a rolling average updated daily using data from the previous 7 to 10 days) to determine whether there is a change in the positivity rates between individual testing and pooled testing. Re-evaluate testing strategy if the moving average of the positivity rate for pooled samples starts trending in a positive or negative direction.
Finally, when resource availability is sufficient to meet testing demand, FDA recommends considering whether the risks of reduced test sensitivity with pooling continue to outweigh the benefits of resource conservation.
e.1) Sample/media pooling
A simple, or Dorfman, approach involves testing an “n-sample pool,” where n is the number of transport media samples included in the pool. A negative result implies that all samples in the pool are negative. A positive result indicates that at least one sample in the pool is positive. When an n-sample pool is positive, each sample within the pool must be individually tested to determine which is/are positive. When used effectively, n-sample pooling can generally enable testing of more individuals despite limited testing resources.
- When pooling transport media, rather than swabs, one individual sample is defined as a single specimen swab collected from a subject and placed in a specific volume of transport media. In this type of pooling, an aliquot of each individual sample is combined into non-overlapping pools of n samples and each n-sample pool is tested. Therefore, the volume of samples initially collected from an individual must be sufficient for both the pooled testing and individual follow-up testing, if needed.
- N-sample pooling should be considered in the context of the positivity rate of a test in the test population, analytical sensitivity of the test, and the percent of weak positive subjects in the tested population. Pooling of n samples reduces the analytical sensitivity of the test (increase in the LoD) because samples are diluted. The impact of decreased analytical sensitivity depends on the percent of subject specimens with viral genetic material concentrations close to the LoD (weak positives) in the tested population. Therefore, analytical sensitivity of the test with n-sample pools should be evaluated.
- FDA believes an n=5 is a reasonable starting point for validation of pooling for a high-sensitivity test in populations with a positivity rate of approximately 5% to 6%. In populations with lower prevalence, larger sample pools may be feasible. In populations with higher prevalence, smaller sample pools may be needed. FDA recommends that developers begin by validating their tests for pooling using an n=5. Tests validated and authorized for n=5 can then be used with any n≤5 depending on testing needs and taking into consideration local prevalence. In cases where a developer wants to validate an n>5 or is considering alternate pooling schemes, FDA recommends that developers reach out to the FDA or submit a pre-EUA to discuss their approach and validation plan.
- The table below presents calculated n-sample pool sizes with the maximal efficiency (a maximum increase in the number of tested patients because of n-sample pooling strategy) for different positivity rates P. This n with maximal efficiency (nmaxefficiency) should be a starting pool size for validation of pooling with positivity rate P. If the accuracy of the test with regard to missed positive patients because of nmaxefficiency samples pooling is not acceptable, n < nmaxefficiency should be considered and accuracy of pooling with this n should be evaluated.
| P, percent of positive subjects in the tested population | nmaxefficiency (n corresponding to the maximal efficiency) |
Efficiency of n-sample pooling (a maximum increase in the number of tested patients when Dorfman n-pooling strategy used) |
|---|---|---|
| 1% | 11 | 5.11 |
| 2% | 8 | 3.65 |
| 3% | 6 | 3.00 |
| 4% | 6 | 2.60 |
| 5% | 5 | 2.35 |
| 6% | 5 | 2.15 |
| 7% | 4 | 1.99 |
| 8% | 4 | 1.87 |
| 9% | 4 | 1.77 |
| 10% | 4 | 1.68 |
| 11% | 4 | 1.61 |
| 12% | 4 | 1.54 |
| 13% | 3 | 1.48 |
| 14% | 3 | 1.43 |
| 15% | 3 | 1.39 |
| 16% | 3 | 1.35 |
| 17% | 3 | 1.31 |
| 18% | 3 | 1.28 |
| 19% | 3 | 1.25 |
| 20% | 3 | 1.22 |
| 21% | 3 | 1.19 |
| 22% | 3 | 1.16 |
| 23% | 3 | 1.14 |
| 24% | 3 | 1.12 |
| 25% | 3 | 1.10 |
Because a single positive sample in a pool requires individual retesting of each sample in the pool, the efficiency of any pooling strategy depends on the positivity rate. The efficiency (F) of n-sample pooling for positivity rate (P) can be calculated with the following formula F=1/(1+1/n-(1-P)n). The efficiency (F) indicates how many more patients can be tested with n-sample pools compared to individual testing. For example, a 3-sample pooling strategy increases the number of tested patients by 1.48 times for positivity rate P of 13% (F=1.48) and by 1.22 times for positivity rate P of 20% (F=1.22). At F=1.48, 1,000 tests can cover testing of 1,480 patients. Likewise, at F=1.22, 1,000 tests can cover testing of 1,220 patients.
- A test validated for a specific n-sample pooling strategy is also considered to be validated for any number of pooled samples below n. For example, a test validated for a 5-sample pooling strategy can be performed for any n≤5.
- Different specimen types should not be pooled together.
- FDA recommends that your instructions for use specify a sample volume great enough to allow for individual and pooled testing so that, during clinical use, any samples in a positive pool can be re-tested without the need for a second sample collection.
- Due to the reduction in analytical sensitivity, a pooling strategy should include risk mitigations such as additional language in the report noting that pooling was used during testing.
Validation
Test developers should characterize the reduction in assay analytical sensitivity (that is, shift in Ct value for RT-PCR assays) with respect to the number (n) of samples to be pooled to ensure the selected n-sample pooling strategy will maintain appropriate sensitivity. This maximum number of samples acceptable to pool should be determined and validated using the recommendations below for each specimen type you intend to pool.
We strongly recommend that you develop and validate a system for deconvoluting pooled test data which is intended to accurately identify individual patient samples composing each pooled sample. If you plan to use a software solution intended to deconvolute pooled SARS-CoV-2 diagnostic test data, then we recommend providing validation data establishing that the software can achieve its intended use. For example, we recommend providing evidence that the software has been validated to ensure that:
- the inputs and outputs of the software are appropriate for the intended use of the assay
- all expected inputs produce the expected outputs for all functions critical for system operation
- the system will be provided to the customer free of defects or defects will be known and mitigated
A) Sample pooling: adding a pooling strategy to a previously authorized (EUA) test
When requesting to add an n-sample pooling strategy to the authorized uses and the authorized instructions for use for your own previously authorized assay, you should submit an EUA amendment request with the appropriate validation data as described below. To add a pooling strategy to a previously authorized test, you generally do not need to establish performance with a separate comparator assay.
You should conduct a clinical study with at least 20 individual positive samples, comparing the performance of the EUA-authorized assay when testing single specimens according to the authorized instructions for use to the performance of the assay when testing n-sample pools.
We strongly encourage you to work with your customers to gather existing data (for example, 100 Ct values from individually tested positive patient samples) and evaluate the percentage of samples with Ct values close to your assay LoD (that is, weak positives). A theoretical Ct shift of Log2(n) can be estimated for most RT-PCR tests (for example, for n=5, a Ct shift of 2.3 would be expected). Therefore, if a large percentage of positive patient samples are close to your assay LoD, you may want to consider a smaller n, which will reduce the observed Ct shift and maintain higher sensitivity.
Please consider the following when designing your clinical validation study with 20 individually tested positive samples:
- If archived individual samples are available and have enough volume for testing with n-sample pools, we recommend that you use at least 20 archived positive samples. If these samples are not available with sufficient volume, we recommend that you enroll enough patients to collect at least 20 positive samples and an appropriate number of individual negative samples from the intended use population. For example, for a 5-sample pooling strategy, a total of 80 unique comparator method negative samples are recommended in order to make up 20 5-sample pools with the 20 positive samples (20 positives + 4x20 negatives). Additionally, 100 comparator method negative samples are recommended to make up 20 5-sample negative pools (5x20 negatives) as described below. If there is sufficient volume, the same negative patient samples can be used to create positive and negative pooled samples.
- We recommend that at least 25% of the validation samples be within 2 Ct to 3 Ct of the cut off, and no more than within 2 Ct to 4 Ct.
- Samples should be collected according to the instructions for use, keeping in mind that additional sample volume will be needed to test using an n-sample pooling strategy (n-sample pooling will need 1+1/n times the volume needed for individual testing).
- All samples should be individually tested by your assay, either previously for archived specimens or prospectively, and have recorded Ct values if using an RT-PCR test.
- To characterize the performance of your assay when testing pooled samples, those samples with positive results when tested individually should each be pooled with n-1 (for example, where n=5, n-1=4) randomly selected negative samples. The resulting 20 pools, each consisting of 1 positive sample and n-1 negative samples, should be tested by your assay.
- To confirm that negative samples remain negative in n-sample pools, we recommend testing 20 pools each consisting of n (for example, n=5) negative samples. If there is sufficient volume, the same negative patient samples can be used to create positive and negative pooled samples.
Analysis of data
- You should report estimates of positive and negative percent agreement comparing the performance of your test for pooled samples to the expected result. With regard to positive percent agreement (PPA), using a study design with 20 positives, you should calculate the percent of pools (1 positive and n-1 negative) with positive results. It is anticipated that all samples that were identified individually as positive by your test should still be positive when tested in pools with n-1 negative samples (PPA=100%). Lower levels of PPA in the range of 85% to 90% may be acceptable depending on pooling efficiency and other factors. The n that allows a test to meet 85% or higher PPA should be validated for each test.
- Additionally, for RT-PCR tests, you should provide an analysis of Ct values for each target detected by your test. We recommend presenting the Ct values for the n-sample pools on the Y-axis and Ct values for the individually tested samples on the X-axis. The clinical validation study should demonstrate that individual positive samples with viral loads close to the assay’s LoD (that is, weak positives) are accurately detected by your test in a pool with (n-1) negative samples.
- We recommend that you provide an appropriate type of regression analysis with slope and intercept, along with 95% confidence interval. Using regression analysis, we recommend that you evaluate the shift in Ct values for the positive patient samples diluted with negative patient samples.
B) Sample pooling: new test (not previously authorized)
When requesting to include an n-sample pooling strategy for a new test, you should submit an EUA request with the appropriate validation data for individual testing in your proposed intended use population and for pooled testing, as described below. This should involve using a high-sensitivity comparator assay to characterize performance of your candidate test.
You should conduct a clinical study with at least 30 individual positive samples, as identified by the comparator assay, comparing the performance of the candidate assay both when testing single specimens and when testing n-sample pools to the performance of the comparator assay.
Please consider the following when designing your clinical validation study:
- The number of enrolled patient specimens should be sufficient to ensure at least 30 comparator method positive samples and an appropriate number of comparator method negative samples are collected from the intended use population. The number of comparator method negative samples depends on the pooling strategy. For instance, for a 5-sample pooling strategy, a total of 120 unique comparator method negative samples are recommended in order to make up 30 5-sample pools with the 30 positive samples (30 positives + 4x30 negatives). Additionally, 150 comparator method negative samples should make up 30 5-sample negative pools (5x30 negatives) as described below. If there is sufficient volume, the same negative patient samples can be used to create positive and negative pooled samples.
- Samples for comparator method testing should be health care provider-collected NP swabs. If an NP swab cannot be collected, a nasal swab can be used. However, both anterior nares should be sampled with the same swab. Sampling for the candidate test and comparator method should occur within a short timeframe, such as during the same visit, to avoid biological variability in viral load.
- If available, FDA recommends selecting a comparator assay that has established high sensitivity with an internationally recognized standard or the FDA SARS-CoV-2 Reference Panel. Please contact the FDA to discuss options to establish sensitivity.
- Samples for the candidate test should be collected according to the instructions for use. Depending on the sample volume required for your test, a single specimen collected from each study participant may be sufficient for individual and pooled sample testing.
- In general, we recommend that you collect samples at a minimum of 3 geographically diverse sites, especially if you are planning to use the same data to support a subsequent De novo/510k submission. If this is not possible, FDA recommends samples collected at 1 or 2 sites in the context of an EUA.
- It may be possible to use archived positive samples that were collected from the intended use population. We recommend you contact FDA to discuss such an approach prior to initiating your study. If archived samples are available, we recommend that at least 25% of the validation samples should be within 2 Ct to 3 Ct of the cutoff and no more than within 2 Ct to 4 Ct.
- All samples should be individually tested by the comparator assay and individually tested by the candidate assay to characterize the performance of your assay when testing individual samples.
- To characterize the performance of your assay when testing n-sample pools, those samples with positive results by the comparator method should each be pooled with n-1 (for example, where n=5, n-1=4) randomly selected comparator method negative samples. The resulting 30 pools, each consisting of 1 comparator method positive sample and n-1 comparator method negative samples, should be tested by your candidate assay.
- To confirm that samples with comparator method negative results remain negative in n-sample pools, we recommend testing 30 pools each consisting of n (for example, n=5) comparator method negative samples.
Analysis of data
- You should report estimates of positive and negative percent agreement comparing individual results from your test and the comparator test, as well as performance of pooled samples to the expected results (for instance, a pool which includes a comparator method positive sample is expected to remain positive when pooled). With regard to positive percent agreement (PPA), using a study design with 30 positives, you should calculate the percent of pools (1 positive and n-1 negative) with positive results. It is anticipated that all samples that were identified individually as positive should still be positive when tested in pools with n-1 negative samples (PPA=100%); lower levels of PPA in the range of 85% to 90% may be acceptable depending on pooling efficiency and other factors. The n that allows a test to meet 85% or higher PPA should be validated for each test.
- Additionally, for RT-PCR tests, you should provide an analysis of Ct values of each target detected by your test. We recommend presenting the Ct values for the n-sample pools on the Y-axis and Ct values for the individually tested samples on the X-axis. The clinical validation study should demonstrate that individual positive samples with viral load close to the assay’s LoD (that is, weak positives) are accurately detected by your test in a pool with (n-1) negative samples.
- We recommend that you provide an appropriate type of regression analysis with slope and intercept along with 95% confidence interval. Using regression analysis, we recommend that you evaluate the shift in Ct values for the positive patient samples diluted with negative patient samples.
C) Example of validation and data presentation
The information below is included as an example of how data can be presented to FDA in a pre-EUA or EUA request. It is for illustrative purposes only and is not reflective of data from any specific test nor the only way to present such information. This example is based on a 5-sample pooling strategy using an extraction method requiring a 500 uL sample.
- Used the candidate assay to individually test 500 uL aliquots of 30 comparator positive samples and 150 comparator negative samples.
Example of table for presenting calculation of PPA and NPA of the candidate test results for samples tested individually vs the comparator test results:
Samples tested individually Comparator method result Candidate test result Positive Negative Positive Insert content here Insert content here Negative Insert content here Insert content here - Created expected positive 5-sample pools by combining 100 uL of ) individual positive patient sample with 100 uL aliquots from each of 4\) unique comparator method negative patient samples. This was done for all positive patient samples thereby creating 30 5-sample pools (that is, a total of 30 positives combined with a total of 120 negatives).
- Created expected negative 5-sample pools by combining 100 uL of ) individual negative patient samples using a total of 150 unique negative samples. When there was sufficient volume, the same negative patient samples were used to create positive and negative pooled samples.
- Tested all 5-sample pools by following the instructions for use of the candidate test. All previous results were unknown to the user (that is, an individual other than the user performing the testing prepared the samples such that testing was performed “blinded”).
- Calculated the percent agreement of the pooled samples with respect to the expected results (for instance, if a positive patient sample was included in the 5-sample pools, the expected result was positive).
Example of table for presenting calculation of PPA and NPA of the candidate test results for samples tested in 5-sample pools vs expected results (where expected results are based on the individual testing):
Samples tested in 5-sample pool Expected result Pooled test result Positive Negative Positive Insert content here Insert content here Negative Insert content here Insert content here - If the candidate assay is an RT-PCR test and cycle threshold values (Ct value) are available, we recommend that you provide a data plot (example below) of the positive sample Ct values of an individual tested positive (that is, the Ct value of the individual positive sample used to create the positive pooled sample) and the positive pooled sample. We recommend that you include a diagonal line with a slope of 1 and a y-intercept of 0. We recommend that you provide an appropriate type of regression analysis with slope and intercept along with 95% confidence interval. Using the regression analysis, we recommend that you evaluate the shift in Ct values for the positive patient samples diluted with negative patient samples.
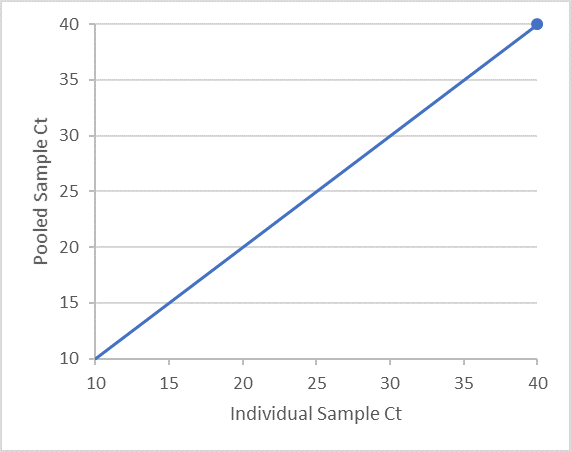
- Agreement should also be presented in a stratified manner so that performance over the range of Ct values can be evaluated. For example, if the cut-off for the candidate test is Ct = 40, then the following table should be provided:
Samples tested in a 5-sample pool Expected result
Individual samples with 37 <Ct ≤40Pooled test result Positive Negative Positive Insert content here Insert content here Negative Insert content here Insert content here N/A Expected result
Individual samples with 34 <Ct ≤37Positive Insert content here Insert content here Negative Insert content here Insert content here N/A Expected result
Individual samples with Ct ≤34Positive Insert content here Insert content here Negative Insert content here Insert content here N/A Expected result
All individual samplesPositive Insert content here Insert content here Negative Insert content here Insert content here
e.2) Swab pooling
Swab pooling is an approach which conserves transport media and has the potential to maintain sensitivity of the test. However, deconvolving which swab was positive cannot be done without collecting another specimen. This approach also results in a high concentration of swab specimen in transport media, therefore inhibition may be observed. The effects of inhibition due to high concentrations of swab specimens (for example, mucin) and high concentrations of virus when there are multiple positive swabs in the swab pool should be investigated.
We recommend performing swab pooling validation using the 2 studies described below using the highest number of swabs that is both desired and deemed feasible. If the data does not meet the acceptance criteria noted below we recommend evaluating a lower number of swabs until the recommended acceptance criteria are met. Laboratories can proceed testing with any number of pooled swabs up to the highest number of pooled swabs that was successfully validated.
In your instructions for use, you should provide a detailed procedure describing a method to combine swabs into a single volume of transport media. The procedure should:
- include recommendations to maximize the amount of specimen resuspended into the transport media from the swab and
- help ensure that the user performs sample and swab handling in a manner consistent with current infection control procedures, which should also reduce the chance of carryover between sample pools
The maximum number of swabs that can be pooled for maximum efficiency can be calculated the same way as the maximum number of samples as discussed above for Dorfman pooling.
To establish performance of your test with swab pooling, FDA recommends conducting a clinical validation study in the intended use population that includes testing each sample individually and using your proposed pooling strategy. Examples of clinical validation studies for adding pooling to a previously authorized (EUA) test or to include pooling in an EUA request for a new test are included in the sample/media pooling section above. These studies can be adjusted to validate a swab pooling strategy.
For n-swab pooling strategies, the 2 studies below should also be conducted:
- We recommend establishing performance related to test interference from multiple swab specimens in a single volume of transport media. N-swab samples containing the maximum number of swabs you intend to validate in the minimum volume of transport media you intend to validate should be tested with an analyte concentration of 2x to 3x LoD. The swabs should contain clinical matrix negative for SARS-CoV-2. The acceptable range of transport media volume should be noted in your instructions for use and interference performance should be validated by testing in the minimum recommended volume. We recommend testing replicates of 3 n-swab pooling samples at the same analyte concentration both with and without clinical matrix. Each n-swab pooling sample should contain maximum number of swabs you recommend pooling in your instructions for use.
For example, if you recommend pooling 3 swabs (n=3) then we recommend acquiring a total of 9 confirmed negative swabs from individual subjects and adding 3 unique swabs to 3 unique tubes of transport media, thereby making 3 n-swab pooling samples. Each n-swab pooling sample should be spiked with either positive patient sample (in transport media), live virus or inactivated virus at a concentration of 2x to 3x the LoD of your assay. We recommend testing a total of at least 20 replicates, which can be composed of equal numbers of aliquots taken from each n-swab pooling sample (that is, 7 replicates from each sample in this example). Ideally, negative n- swab sample matrix should be tested prior to spiking to ensure that the matrix is negative. Acceptance criteria should be at least 95% agreement with the expected results and an invalid rate of <5%. We recommend providing the Ct value line data (if applicable) for analysis.
- We recommend evaluating the effect of high viral concentrations on assay performance. It appears that patients with SARS-CoV-2 infection can exhibit unusually high viral loads. This, combined with the possibility of pooling multiple positive swabs into a single volume of transport media, could result in unexpectedly high viral titer in the pooled sample. We recommend evaluating existing data on viral loads in infected subjects and, in combination with your existing LoD data, propose a maximum expected viral titer per swab. Using this number, estimate the expected viral titer in transport media with at least 3 positive swabs. For instance, if you expect a maximum of 100,000x LoD per swab, we recommend spiking a single negative n-swab sample with 300,000x LoD target analyte and testing with 10 replicates. It is anticipated that all replicates are either positive or have an invalid rate of ≤5%.
Studies to support point-of-care indication
If your device is intended for near patient or point-of-care (POC) testing, please provide data to demonstrate that non-laboratory personnel can perform the test accurately in the intended use environment (for instance, a non-laboratorian health care provider accuracy study). Please also provide data to demonstrate the robustness of your device for near patient testing (for example, as applicable, studies to demonstrate the impact of adding different volumes of sample, different volumes of reagents, incorrect order of sample or reagent application). For assays intended for use with a test system that was previously CLIA-waived by the FDA, testing is generally only needed to establish the performance of the SARS-CoV-2 assay chemistry.
In general, additional test data is not needed to demonstrate that the system is simple enough for use at the point-of-care, unless there is a feature of the SARS-CoV-2 assay that would make performing the test more complicated than assays previously cleared for use on the test system.
1. Clinical evaluation
The clinical study design should mimic how the test will be used in clinical practice. It is expected that a test with “POC” designation will be widely used in CLIA-waived medical facilities (for example, physician office, outpatient clinic, ER), but also in less traditional settings, such as tents and schools, with health care worker oversight of testing. This clinical study design does not apply to testing sites where NO HCWs are present.
- Sites and test users (operators): You should select 1 or 2 non-laboratory sites in the United States to assure that the operators are representative of operators in the U.S. (for example, doctor’s office, ER, outpatient clinic, drive-through testing facility or another area in a medical facility outside the central laboratory where samples are collected and tested in real time). This would allow evaluation of the sample collection and handling, including addition into the sample port/well of the test, both of which may be significant sources of error. Four to 6 operators, representing health care professionals, but who are not laboratory-trained (such as nurses, nursing assistants and doctors) should participate in the study. Testing should be performed using only Quick Reference Instructions (QRI). Supplemental materials, such as a video or an app that can be easily accessed by the user, are encouraged but should not be used during the study (mimicking worst-case scenario).
- Comparator method: All patients tested during the clinical study with the POC device should also be tested by an FDA-authorized SARS-CoV-2 molecular assay. The comparator method selected should be one of the more sensitive EUAs on the FDA website (supported by peer-reviewed literature, comparative studies testing the FDA reference material and so on) that uses a chemical lysis step followed by solid phase extraction of nucleic acid. Ideally, the same comparator would be used for all samples. The comparator should be authorized for use with the specimen type and transport medium (if applicable) that is being tested. Typically, the standard of care specimen is collected first, so as not to compromise the medical care of the patient. After the standard of care specimen has been collected, swabs taken from the same area for the comparator and subject device (for example, nasal swabs, OP swabs) should be randomized to ensure that bias is not introduced due to an unequal distribution of viral materials. Randomizing collection when 2 distinct anatomical sites are being assessed may not be needed (for example, saliva compared to NP swabs).
- Clinical samples: A total of 30 prospectively collected positive (confirmed by an authorized test) and 30 negative natural clinical specimens should be tested (mock clinical samples are not acceptable). Testing should be conducted for at least 2 weeks. If an insufficient number of positive results is observed after such time (<30), you may collect samples at another site to ship to the testing site or use banked specimens to supplement your positive specimens. Banked specimens should not be pre-selected based on Ct value and should be presented blinded (mixed with negatives) to the testing site. Ideally, the same comparator method should be used for banked and prospectively collected specimens.
A molecular POC test should demonstrate positive and negative agreement of ≥95%. However, positive agreement of ≥80% may be considered with appropriate limitations added to the intended use that would mitigate the risk of false negative results. For example, negative results may be considered presumptive negative if the demonstrated PPA is lower than 95%.
- Notification of public health authorities: When setting up the study, you should have a clear strategy for reporting test results to the CDC and/or local public health authorities both during the study and after authorization.
2. Post-market clinical study
If prospective clinical study results are not available at the time of the test authorization, manufacturers of new POC tests should conduct a post-market prospective clinical study to collect at least 30 positive and 30 negative natural clinical specimens to demonstrate the assay performance relative to an FDA-authorized comparator method. You should propose a post-market study design in your EUA submission for FDA review and feedback. Generally, a final study design will be agreed upon before authorization and will be a condition of authorization.
3. Performance around LoD
You should conduct testing with samples prepared in clinical matrix with SARS-CoV-2 viral load near the LoD of your assay. The testing should be performed by inexperienced users at the clinical sites. The test samples should consist of 10 low positives (<2x LoD) and 10 negatives (matrix) per site. The blinded and randomized samples should be distributed among the operators. We recommend that each operator tests at least 3 low positive and 3 negative samples integrated into the site’s workflow with the clinical samples above.
4. Flex studies
Flex studies assess the robustness of an assay performed with the device in its final design/format and should be performed in-house by staff who have been trained in the use of the test. The flex studies should evaluate the most common or likely sources of error based on the use locations and test procedure. Flex studies should be conducted by testing negative sample and a low positive (at 1.5x to 2x LoD) samples, under each condition being evaluated. In general, the flex studies should be conducted to the point of failure to determine the maximum deviation that will still generate accurate results.
We recommend testing 3 replicates per condition per sample concentration. Line data for each condition evaluated should be provided. If erroneous results are observed during studies evaluating the robustness of the device, adequate mitigation(s) should be provided.
Each study should be performed using a pre-defined study protocol that includes the following:
- the objective of the study
- detailed test procedure
- materials used
Examples of conditions that may be evaluated as potential user errors and anticipated environmental stresses (temperature and humidity extremes) are shown below:
- 40°C and 95% RH (mimicking hot and humid climates) applicable to small portable devices that could be used outdoors (such as tents and mobile vans)
- delay in sample testing
- delay in operational steps
- delay in reading results
- sample volume variability
- buffer volume variability
- environmental stability of electronics (temperature and humidity, in combination)
- vibrations
- disturbance during analysis
- placement on non-level surface
- If hand-held, positioning at 90° angle
- sensitivity to power failures (for example, surge protection, battery power failure)
- error reporting and device failure handling instructions
- electrical interference testing (for example, validation of system functions in the presence of potential EM interference sources, including cell phones, Bluetooth, Wi-Fi radios and medical equipment expected in the intended environment)
Please see Appendix A for more in-depth flex study designs. Alternative sources of information on flex studies that may be applicable to your device can be found on the FDA CDRH web page containing CLIA waiver by application decision summaries.
Multi-analyte respiratory panels under EUA
An emergency declaration by the HHS Secretary allowing for the issuance of EUAs is typically specific for a pathogen/disease (that is, there is a publicly declared health emergency involving a particular etiologic agent). Therefore, for tests, the EUA pathway is generally only an option for testing patients for that single agent in a given emergency.
Given the overlap in signs and symptoms between SARS-CoV-2 and other respiratory viral infections, including influenza, FDA has authorized multi-analyte respiratory panels for the qualitative detection and differentiation of nucleic acid from multiple pathogens, including the SARS-CoV-2 virus. These panels are useful to efficiently detect and differentiate between multiple pathogens that are relevant to the event/disease outbreak that is the subject of the specific emergency declaration. They may also be useful in preserving critical testing resources during the public health emergency by reducing the number of tests, and therefore supplies, needed per patient.
When determining whether to issue an EUA for a multi-analyte respiratory panel, FDA takes into consideration:
- the use of the test (multi-analyte pathogen detection as an aid in differential diagnosis)
- clearance/approval status of IVDs for the other panel members
- whether the proposed intended use fits within the HHS emergency declaration and
- how the panel test would fit into current public health authority patient testing algorithm recommendations
If you are requesting an EUA for a multi-analyte respiratory panel, analytical and clinical evaluations for each target analyte should be provided. We recommend you contact the FDA at for specific feedback on this type of EUA request.
1) Addition of SARS-CoV-2 to previously FDA-cleared multi-analyte respiratory Panels
To add the SARS-CoV-2 target to respiratory panels previously cleared by the FDA where the SARS-CoV2 reagents are run in a separate well (or tube) and no modifications are required to the cleared portion of the assay, only studies for validation of the SARS-CoV-2 reagents described in this template are recommended.
To add the SARS-CoV-2 target to respiratory panels previously cleared by the FDA where the SARS-CoV-2 reagents are combined in the same well as the reagents for previously cleared analytes (in a multiplex reaction), the following studies should be conducted to validate the SARS-CoV-2 reagents and the modifications made to the cleared respiratory panel:
- studies described in this template to validate the SARS-CoV-2 reagents
- LoD confirmation of the previously cleared analytes by conducting side-by-side testing of 3 to 5 replicates of serially diluted viruses with modified and original versions of the test to show that the LoD is unchanged due to modifications
- testing 10 retrospective positive samples for each previously cleared analyte
- competitive inhibition study with clinically relevant titers of each analyte in the panel (viruses 105 PFU/mL, bacteria 106 CFU/mL)
2) Multi-analyte panels not previously cleared by the FDA
To support an EUA for a multi-analyte respiratory panel that was not previously cleared by FDA, analytical and clinical evaluations for each target analyte should be provided. The following analytical studies should be conducted and data provided to the FDA for review:
- limit of detection (analytical sensitivity)
- cross-reactivity/microbial interference
- inclusivity/analytical reactivity
- collection media equivalency (each claimed additional sample collection media not used in your clinical study should be validated, if appropriate for study designs)
- co-infection (competitive interference)
- interfering substances study (endogenous and exogenous)
- clinical specimen stability
- reagent stability testing protocol
- carry-over/cross-contamination, if a new instrument previously not reviewed by the FDA is used
- reproducibility and repeatability, if a new instrument previously not reviewed by the FDA is used
- fresh versus frozen (if you intend submit data testing archived frozen specimens in support of your EUA, please conduct an analytical study to demonstrate that preservation of samples, such as by freezing at ≤-70°C, does not affect the accuracy of test results compared to freshly collected samples)
Clinical performance
To evaluate the clinical performance of your multi-analyte test, a prospective clinical study should be conducted. Considering the public health needs in the current emergency, a clinical performance study in support of the EUA application may be conducted at 1 site testing archived positive and negative clinical samples with known specimen types. The pre-selection of archived positive samples should represent a range of viral load or Ct values, including low positive samples near the assay cut-off.
Since your device has not been FDA-cleared for the respiratory pathogens included in your test, and it is likely that your test would be used in patients with respiratory symptoms in lieu of an FDA-cleared respiratory panel, FDA generally intends to include a condition of authorization that you conduct a post-EUA prospective clinical study. The prospective clinical study should include a minimum of 3 sample collection sites and 3 testing sites, prospectively enrolling patients with general respiratory symptoms. You may consider conducting a prospective clinical study in Southern Hemisphere countries during their typical influenza/respiratory season to increase the likelihood of obtaining a sufficient number of positive samples (for example, for influenza at least 50 positive Flu A and 30 positive Flu B samples) in a timely fashion.
The FDA performance expectation for SARS-CoV-2 is that PPA and NPA should be ≥95% (with a lower bound of the 2-sided 95% confidence interval >85%). For Flu A/B and other respiratory viruses, PPAshould be ≥90% (with a lower bound of the 2-sided 95% confidence interval ≥80%) and the NPA should be ≥95% (with a lower bound of the 2-sided 95% CI ≥90%) in comparison to an EUA RT-PCR test. We recommend using only a high sensitivity EUA RT-PCR assay, which uses a chemical lysis step followed by solid phase extraction of nucleic acid (for example, silica bead extraction).
We recommend that you submit a pre-EUA with an outline of the studies that you plan to conduct to support the FDA-authorization or contact the FDA at for specific feedback.
Claiming multiple instruments and/or extraction methods
FDA recommends the following analytical and clinical validation for use of multiple instruments and/or extraction methods where the elution volumes from the extraction methods and PCR volumes on the different RT-PCR instruments are identical:
- Limit of detection (LoD): These studies should be repeated for each clinical matrix claimed in the intended use. Pick 1 RT-PCR instrument and determine the tentative LoD (using 5 replicates in 10-fold dilution) followed by the confirmatory LoD (20 replicates spiked at tentative LoD) for each extraction method on the chosen instrument. Note: If you detect 20/20 replicates in your confirmatory LOD study, you should test the next lower concentration, using a 3-fold dilution, until you achieve a hit rate of <20/20.
- If the different extraction methods yield the same LoD (≤3xLOD) on the RT-PCR instrument chosen for initial testing, pick 1 extraction method for further LoD determination on the remaining RT-PCR instruments and follow the recommendations below.
- If the extraction methods do not yield the same LoD on the chosen RT-PCR instrument, please choose the extraction method with the worst LoD for further comparison of the LoD on all RT-PCR instruments.
For all other RT-PCR instruments, you should use the following adaptive LoD study design:
- Please perform a refined tentative LoD study with 5 replicates at 0.5x, 1x and 1.5 to 2x LoD. If you detect 4/5 replicates as positive at all the tested levels, you need to include the next higher concentration (for instance, 3x LoD). If you obtain 5/5 replicates at 0.5x LoD, you need to test the next lower concentration (for instance, 0.25x LoD). You will test in this manner until you find the lowest concentration that gives you 5/5 positive results for the tested RT-PCR instrument. This concentration should be used for a confirmatory LoD study for the given RT-PCR instrument using 20 replicates.
Final reported LoD: Please list all RT-PCR instruments with their respective LoDs if different LoDs are obtained. LoDs are considered comparable if they are between 1x to 3x LoD. These studies should be repeated for each clinical matrix claimed in the intended use.
- Interference substances studies, if applicable: FDA recommends evaluating interfering substances with the extraction method and RT-PCR instrument combination that has the worst overall LoD.
- Inclusivity testing: FDA recommends evaluating inclusivity with the extraction method and RT-PCR instrument combination that has the worst overall LoD.
- Exclusivity testing: FDA recommends evaluating exclusivity with any extraction/instrument combination.
- Clinical study: If an LoD study confirms equivalency for all RT-PCR instruments (between 2x to 3x LoD), then the clinical study may be conducted with any RT-PCR instrument. If one or more RT-PCR instruments have different LoDs, we recommend conducting the clinical study with the extraction method/RT-PCR instrument combination with the worst LoD.
Note: If there are differences in the extraction input volume, extraction elution volume and PCR input volume (extracted nucleic acid), then the LoD should be confirmed for each.
K. Unmet need addressed by the product
This section will be completed by FDA.
L. Approved/cleared alternative products
Currently, no methods for the detection of the SARS-CoV-2 have been approved and/or cleared by FDA.
M. Benefits and risks
This section will be completed by FDA.
N. Fact sheet for health care providers and patients
Include proposed fact sheets for patients and health care providers (see examples for authorized EUA tests on our website). Templates will be made available.
O. Instructions for use, proposed labelling, package insert
Include instructions for use, box labels, vial labels and any other proposed labelling.
P. Record keeping and reporting information to FDA
[Manufacturer name] will track adverse events and report to FDA under 21 CFR Part 803. A website is available to report on adverse events, and this website is referenced in the Fact Sheet for Health Care Providers as well as through the [Manufacturer name] Product Support website: [include link to website]. Each report of an adverse event will be processed according to [Manufacturer name]’s rnon-conformance reporting requirements, and medical device eports will be filed with the FDA as required. Through a process of inventory control, [Manufacturer name] will also maintain records of device usage/purchase. [Manufacturer name] will collect information on the performance of the test, and report to FDA any suspected occurrence of false positive or false negative results of which [Manufacturer name] becomes aware. [Manufacturer name] will maintain records associated with this EUA and ensure these records are maintained until notified by FDA. Such records will be made available to FDA for inspection upon request.
Appendix A: Flex study design details, as applicable for the device
If incorrect results are observed under the test conditions, the sponsor should implement adequate mitigations to prevent reporting of erroneous results.
Reading time
Recommend evaluating test results at reading times 4 times below and 3 times above the recommended reading time. For example, for a test where the recommended read time is 20 minutes, reading time times would be performed to evaluate at least read times of 5, 10, 15, 20, 30 and 60 minutes.
Specimen volume
Recommend evaluating test results at specimen volumes 2 times below and 2 times above the recommended specimen volume, and the maximum possible added. For example, for a test where the recommended specimen volume is 10 µL, specimen volume testing should be performed to evaluate at least specimen volumes of 5, 10, 20 µL and maximum volume. If incorrect results are observed at either 5 or 20 µL, additional testing at 7.5 and/or 15 uL may be needed. The diluent/buffer amount added should be that specified in the instructions for use.
Sample diluent volume
Recommend evaluating test results at diluent/buffer volumes at 2 times below and 2 times above the recommended diluent/buffer volume and the maximum volume. For example, for a test where the recommended buffer/diluent volume is 2 drops, sample diluent volume testing would be performed to evaluate at least sample diluent volumes of 1, 2, 3, 4 drops and whole bottle. The sample volume added should be that specified in the instructions for use.
Sample elution
Recommend evaluating how mixing the swab in elution buffer (or other reagent) affects results. You should evaluate all extremes from not mixing to vigorous shaking, generating bubbles as well as intermediate mixing (swirling 1 or 2 times, instead of the prescribed number from the instructions).
Temperature and humidity
Recommend evaluating test results at temperature and humidity extremes that are likely to occur in the United States. For example, 40°C and 95% RH, mimicking hot and humid climates, and 5°C and 5% RH, mimicking cold and dry climates.
Light
Recommend evaluating of test results in different lighting conditions that would be expected during use of the device, for visually read devices. For example, fluorescent, incandescent and natural lighting mimicking the outside environment.
Disturbance during analysis
You should evaluate the effect on expected test results of moving the device while the test is running. This could include:
- dropping the test while it is being run
- moving the test to another surface
- unplugging the test
- receiving a phone call while the mobile app is running
Device orientation
Recommend evaluating unique device characteristics, as determined by a robust risk analysis. For example, if the device is intended to be run upright, evaluating test results if the device is used horizontally, or vice versa.