
Guidance Document: Use of a Foreign-sourced Reference Product as a Canadian Reference Product
Download the alternative format
(PDF format, 264 KB, 14 pages)
Foreword
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable guidance documents.
Table of Contents
1. Introduction
1.1 Policy Objectives
To provide sponsors of an Abbreviated New Drug Submission or an Abbreviated Extraordinary Use New Drug Submission who are seeking to demonstrate pharmaceutical equivalence and bioequivalence with a reference drug under paragraph (c) of the definition of Canadian reference product (CRP) in C.08.001.1 of the Food and Drug Regulations with guidance on how they may demonstrate that this reference drug is acceptable to the Minister as a CRP.
1.2 Policy Statements
The purpose of demonstrating pharmaceutical equivalence and bioequivalence against the CRP is to provide evidence that the safety and efficacy profiles of the subsequent-entry (generic) product will be equivalent to that of the innovative product which is marketed in Canada and for which safety and efficacy has been demonstrated clinically. In order to determine the acceptability of this evidence, the Minister must be satisfied that a reference product proposed under paragraph (c) of the definition of CRP appears to be the same as the innovative product marketed in Canada and that any potential undocumented differences between these two products would not be therapeutically significant.
1.3 Scope and Application
This guidance is intended to be applied to all Abbreviated New Drug Submissions and Abbreviated Extraordinary Use New Drug Submissions where the submission sponsor is seeking approval based upon pharmaceutical equivalence and bioequivalence to a reference drug under paragraph (c) of the definition CRP in C.08.001.1 of the Food and Drug Regulations. In particular this guidance applies to those submissions which involve demonstration of bioequivalence based upon bioavailability studies, pharmacodynamic studies, clinical studies, or a combination of thereof.
This document provides guidance only for the dosage forms covered in Section 2. Application of the principles of this guidance to other immediate-release dosage forms or immediate-release combination products are not addressed in this guidance and will be considered on a case by case basis. In such cases, it is recommended that the sponsor consult Health Canada prior to filing their submission and, if possible, prior to conducting any bioequivalence studies.
1.4 Background
In order to file an Abbreviated New Drug Submission or an Abbreviated Extraordinary Use New Drug Submission, the manufacturer must meet specified requirements, including pharmaceutical equivalence and bioequivalence, in comparison to the CRP. Section C.08.001.1 of the Food and Drug Regulations states the following:
"Canadian reference product" means:
- (a) a drug in respect of which a notice of compliance is issued under section C.08.004 or C.08.004.01 and which is marketed in Canada by the innovator of the drug,
- (b) a drug, acceptable to the Minister, that can be used for the purpose of demonstrating bioequivalence on the basis of pharmaceutical and, where applicable, bioavailability characteristics, where a drug in respect of which a notice of compliance has been issued under section C.08.004 or C.08.004.01 cannot be used for that purpose because it is no longer marketed in Canada, or
- (c) a drug, acceptable to the Minister, that can be used for the purpose of demonstrating bioequivalence on the basis of pharmaceutical and, where applicable, bioavailability characteristics, in comparison to a drug referred to in paragraph (a)".
This guidance document establishes criteria for the use of a drug marketed in a country other than Canada as a CRP under paragraph (c) of this regulation.
The purpose of demonstrating pharmaceutical equivalence and bioequivalence against the Canadian innovator's product is to provide evidence that the safety and efficacy profiles of the subsequent-entry (generic) product will be equivalent to that of the innovative product which is marketed in Canada and for which safety and efficacy has been demonstrated clinically.
With the increasing globalization of the pharmaceutical industry, many reference products are manufactured for other countries with the same formulations and under the same conditions as for the Canadian market. On occasion, these products are manufactured in only one location for worldwide distribution. In this situation, the acceptance by the Minister of a reference drug marketed in another country as a CRP, can avoid repetition of comparative studies in human subjects and thereby reduce the risks for study participants and the cost to manufacturers. This is most relevant for drug submissions which involve demonstration of bioequivalence based upon bioavailability studies, pharmacodynamics studies, clinical studies, or a combination thereof.
However, detailed information regarding the formulation and manufacture of products marketed outside of Canada is not generally available to Health Canada. This guidance therefore outlines criteria for acceptance by the Minister, of a drug as a CRP under C.08.001.1, including data requirements to demonstrate that the foreign-sourced reference product is the same as the corresponding Canadian innovative product and conditions under which any remaining undocumented differences are not expected to be therapeutically significant.
2. Guidance for Implementation
A drug product purchased in another country which complies with the following criteria may be considered acceptable to the Minister for use as a Canadian Reference Product (CRP):
- In order to ensure that the foreign-sourced drug product is of equivalent quality to the Canadian innovator drug product, it should be documented that the drug product is authorised for marketing by a regulatory authority of a country or region with drug assessment criteria comparable to those in Canada as required by Part C, Division 8 of the Food and Drug Regulations and interpreted in Health Canada guidance documents and policies (for example (e.g.), United States, Europe (that is (i.e.), a marketing application that has been authorised through the Centralised Procedure or the De-Centralised Procedure), Japan, Switzerland, Australia).
- In support of the assertion that the foreign-sourced reference product and the Canadian innovator product are the same, evidence should be provided in the drug submission confirming that the foreign-sourced reference product is marketed in the country or region of origin by the same innovator company or corporate entity which currently markets the identical amount(s) of the identical medicinal ingredient(s) in the identical dosage form in Canada.
- The following information should be provided on both the batch(es) of the innovator (reference) product marketed in Canada and on the batch(es) of the reference product purchased in another country that was(were) used in the comparativein vivo studies:
- Product labelling;
- Certificates of Analysis (analysed using the specifications proposed in the drug submission for the subsequent-entry product);
- Proof of purchase (batch number, date and place of purchase);
- Samples in their original container closure systems should be available upon request.
- For all comparative in-vitro testing, batches of the foreign-sourced reference product used in the comparative in vivo studies and the innovator product marketed in Canada should be analysed and the results provided. The testing should be conducted using suitably validated analytical procedures. Copies of the validation reports and, if used, House analytical procedures should be included in the drug submission.
- Since undocumented differences between the foreign-sourced reference product and the Canadian innovator product may result in unacceptable differences in the safety and efficacy profiles of the two drugs when they contain higher risk medicinal ingredients, the medicinal ingredient:
- should not be a "Critical Dose Drug" as defined in the Health Canada Guidance Document: Comparative Bioavailability Standards: Formulations Used for Systemic Effects;
- should not require patient monitoring in order to avoid the consequences of under- or over-treatment.
- The foreign-sourced reference product should satisfy the following criteria:
- For solid oral, immediate-release dosage forms (including orally disintegrating dosage forms):
- the medicinal ingredient(s) is considered to have "high solubility" as defined in Appendix 1;
- the foreign-sourced reference product is the same as the innovator product marketed in Canada with respect to colour, shape, size, weight, type of coating (e.g., uncoated, film-coated), and scoring configuration;
- the non-medicinal ingredients in the formulation of the innovator product marketed in Canada, when compared to the foreign-sourced reference product, should be qualitatively the same. If quantitative formulation information is available for these two products it should also show them to be the same.
- results from multi-point, comparative dissolution profiles should be provided, on the two reference products in at least three (3) media covering the physiological pH range (pH 1.2 - 6.8). Adequate sampling should be performed to provide an assessment of the dissolution phase of the profiles (e.g., at 5, 10, 15, 30, 45, 60, and 120 minutes) and should be continued until either 90% of drug substance from the drug product is dissolved or an asymptote is reached. All profiles should be conducted on at least 12 individual dosage units. Dissolution profiles should be considered similar using the equation that defines a similarity factor (f2) as described in Appendix 2. An f2 value between 50 and 100 suggests the two dissolution profiles are similar.
- For immediate-release oral suspensions:
- the medicinal ingredient(s) is considered "highly soluble" as defined in Appendix 1;
- the foreign-sourced reference product is the same as the innovator product marketed in Canada with respect to formulation. The non-medicinal ingredients in the formulation of the innovator product marketed in Canada, when compared to the foreign-sourced reference product, should be qualitatively and quantitatively the same. If the quantitative information on the reference products is not available, the sponsor for the subsequent-entry product should provide results from comparative analytical studies of the formulations of the innovator product marketed in Canada compared to the foreign-sourced reference product.
For the purposes of this document, quantitatively the same would be interpreted as the amount (or concentration) of each excipient in the innovator product marketed in Canada to be within ±5% of the amount (or concentration) of each excipient in the foreign-sourced reference product. Differences beyond this criterion should be scientifically justified. A side-by-side comparison of the qualitative and quantitative formulations for the two products should be provided;
- results from multi-point, comparative dissolution profiles should be provided on the two reference products in at least three (3) media covering the physiological pH range (pH 1.2 - 6.8). Adequate sampling should be performed to provide an assessment of the dissolution phase of the profiles (e.g., at 5, 10, 15, 30, 45, 60, and 120 minutes) and should be continued until either 90% of drug substance from the drug product is dissolved or an asymptote is reached. All profiles should be conducted on at least 12 individual dosage units. Dissolution profiles should be considered similar using the equation that defines a similarity factor (f2) as described in Appendix 2. An f2 value between 50 and 100 suggests the two dissolution profiles are similar.
- For immediate-release orally inhaled suspensions, immediate-release nasal suspensions and immediate-release orally inhaled solutions that require an in vivo demonstration of bioequivalence:
- The foreign-sourced reference product is the same as the innovator product marketed in Canada as determined by a comparison of the relevant pharmaceutical characteristics for: (i) formulation, (ii) physicochemical properties, and (iii) device attributes.
- The comparative studies for the relevant pharmaceutical characteristics should be conducted as described below:
(i) formulation - the non-medicinal ingredients in the formulation of the innovator product marketed in Canada, when compared to the foreign-sourced reference product, should be qualitatively and quantitatively the same. For the purposes of this document, quantitatively the same would be interpreted as the amount (or concentration) of each excipient in the innovator product marketed in Canada to be within ±5% of the amount (or concentration) of each excipient in the foreign-sourced reference product. Differences beyond this criterion should be scientifically justified. A side-by-side comparison of the qualitative and quantitative formulations for the two products should be provided.
(ii) physicochemical properties - when comparing the physicochemical properties, the results for the two products should be essentially the same. For the purposes of this document, essentially the same would be interpreted as the results of the two reference products are within ±10%. Differences beyond this criterion should be scientifically justified. Below is a summary of examples of the testing parameters that should be considered for the comparative physicochemical property study for solutions and suspensions:
- pH;
- Buffering capacity (for products containing a buffering agent);
- Viscosity;
- Specific gravity or density;
- Surface tension;
- Osmolality (mol/kg) / osmolarity (mol/L) (if tonicity is declared on the product labelling);
- Delivered dose uniformity (if packaged with a device for delivery);
- Droplet size or volume (if administered as drops);
- Droplet size distribution (if administered as a spray);
- Spray pattern and plume geometry (if administered as a spray).
In addition to the above recommended comparative tests, the following specific dosage forms should include additional comparative testing as indicated:
- Suspensions: drug substance properties (e.g., particle size, crystal structure), particle size distribution, dissolution profiles (where applicable).
- Suspensions for Nebulisation: drug substance properties (e.g., particle size, crystal structure), particle size distribution, nebulisation time, drug delivery rate and total drug delivered.
- Metered dose inhalers: vapour pressure, freezing point, refractive index, individual stage particle size distribution, fine particle mass.
(iii) device attributes - results of a qualitative and quantitative analysis of the physical and operating characteristics of the devices for the two products (e.g., dimensions, materials used) should be provided. Any differences beyond what may be considered as "normal manufacturing tolerances" would be considered significant.
- For immediate-release orally inhaled dry powders (Inhalation powders):
- The foreign-sourced reference product is the same as the innovator product marketed in Canada as determined by a comparison of the relevant pharmaceutical characteristics for: (i) formulation, (ii) physicochemical properties, and (iii) device attributes.
- The comparative studies for the relevant pharmaceutical characteristics should be conducted as described below:
(i) formulation - the non-medicinal ingredients in the formulation of the innovator product marketed in Canada, when compared to the foreign-sourced reference product, should be qualitatively and quantitatively the same. For the purposes of this document, quantitatively the same would be interpreted as the amount (or concentration) of each excipient in the innovator product marketed in Canada to be within ±5% of the amount (or concentration) of each excipient in the foreign-sourced reference product. Differences beyond this criterion should be scientifically justified. A side-by-side comparison of the qualitative and quantitative formulations for the two products should be provided.
(ii) physicochemical properties and in-vitro performance - when comparing the physicochemical properties and in-vitro performance, the results for the two products should be essentially the same. For the purposes of this document, essentially the same would be interpreted as the results of the two reference products are within ±10%. Differences beyond this criterion should be scientifically justified. Below is a summary of examples of the testing parameters that should be considered for the comparative physicochemical property study for inhalation powders:
- Drug substance properties (e.g., particle size, crystal structure);
- Drug product properties (e.g. particle size distribution of the carrier, if present), bulk and tapped density, particle morphology (shape, texture and surface properties), melting point, electrostatic charge, porosity, specific surface area, hygroscopicity and moisture content;
- Particle size distribution (individual stage and Fine Particle Mass; over the range of flow rates achievable by the intended patient population (through the delivery device), at constant volume);
- Delivered dose uniformity.
(iii) device attributes - results of a qualitative and quantitative analysis of the physical and operating characteristics of the devices for the two products (e.g., dimensions, materials used) should be provided. Any differences beyond what may be considered as "normal manufacturing tolerances" would be considered significant.
- For solid oral, immediate-release dosage forms (including orally disintegrating dosage forms):
If any of the above conditions are not met, the manufacturer should demonstrate the equivalence of the subsequent-entry product to the innovator's product marketed in Canada by the appropriate comparative in vivo study or studies.
3. Appendices
Appendix 1 - Determining Solubility
Test Conditions
The stability of the drug substances at the relevant pH range of 1.2 to 6.8 should be demonstrated. A profile of the equilibrium solubility of the drug substance should be developed for the physiologically relevant pH range of 1.2 - 6.8 employing the following conditions:
Conditions
- Solvent:
- At a minimum, aqueous buffer solutions of pH 1.2, 4.5, 6.8
- Temperature:
- 37 plus or minus (±) 1°C
- Replicates:
- Not less than three (3) at each pH tested
- Methodology:
- Shake-flask technique or similar method with justification
Additional information
The pH for each test solution should be confirmed before and after the addition of the drug substance in order to ensure pH stability of the buffered medium. The pH should be adjusted if necessary.
Classification
- High solubility:
- A drug substance is classified as highly soluble if the highest single therapeutic dose of the drug substance is completely soluble in 250 mL or less of aqueous media over the pH range of 1.2-6.8 at 37 ± 1°C, that is (i.e.), dose solubility volume (DSV) less than or equal to (≤) 250 mL over the pH range.
Appendix 2 - Calculation of Similarity Factor (f2)
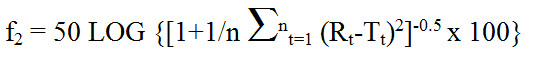
Text Equivalent
The similarity factor f subscript 2 is equal to 50 times the log of open bracket, open bracket, 1 plus 1 over n times the summation from t equals 1 to n of open bracket R subscript t minus T subscript t, close bracket, raised to the power of 2, close bracket, raised to the power of negative 0.5, times 100, close bracket; where R subscript t and T subscript t are the percent dissolved at each time point from 1 to n. An f subscript 2 value between 50 and 100 suggests the two dissolution profiles are similar.
where Rt (innovator's product marketed in Canada) and Tt (foreign-sourced reference product) are the percent dissolved at each time point. An f2 value between 50 and 100 suggests the two dissolution profiles are similar.
For a dissolution profile comparison:
- At least 12 units should be used for each profile determination. Mean dissolution values can be used to estimate the similarity factor, f2. To use mean data, the % coefficient of variation at the first time point should not be more than 20% and at other time points should not be more than 10%.
- The dissolution measurements of the two products should be made under the same test conditions. The dissolution time points for both profiles should be the same (e.g., 5, 10, 15, 30, 45 and 60 minutes).
- It is recommended that at least 3 points (other than zero), be utilised with only one point past the plateau of the profiles be used.
- If both products show more than 85% dissolution within 15 minutes, calculation of f2 values are not considered necessary.