
Ligne directrice : Utilisation d'un produit de référence étranger comme produit de référence canadien
Télécharger le rapport complet (Format PDF, 270 ko, 14 pages)
Avant-propos
Les lignes directrices sont des documents destinés à guider l'industrie et les professionnels de la santé sur la façon de se conformer aux politiques et aux lois et règlements qui régissent leurs activités. Elles servent également de guide au personnel lors de l'évaluation et de la vérification de la conformité et permettent ainsi d'appliquer les mandats d'une façon équitable, uniforme et efficace.
Les lignes directrices sont des outils administratifs n'ayant pas force de loi, ce qui permet une certaine souplesse d'approche. Les principes et les pratiques énoncés dans le présent document pourraient être remplacés par d'autres approches, à condition que celles-ci s'appuient sur une justification. Ces autres approches devraient être examinées préalablement en consultation avec le programme concerné pour s'assurer qu'elles respectent les exigences des lois et des règlements applicables.
Corollairement à ce qui précède, il importe également de mentionner que Santé Canada se réserve le droit de demander des renseignements ou du matériel supplémentaire, ou de définir des conditions dont il n'est pas explicitement question dans la ligne directrice, et ce, afin que le ministère puisse être en mesure d'évaluer adéquatement l'innocuité, l'efficacité ou la qualité d'un produit thérapeutique donné. Santé Canada s'engage à justifier de telles demandes et à documenter clairement ses décisions.
Ce document devrait être lu en parallèle avec l'avis d'accompagnement et les sections pertinentes des autres lignes directrices qui s'appliquent.
Ce document devrait être lu en parallèle avec l'avis d'accompagnement et les sections pertinentes des autres lignes directrices qui s'appliquent.
Table des matières
1 Introduction
1.1 Objectifs de la politique
Fournir aux promoteurs d'une présentation abrégée de drogue nouvelle (PADN) ou d'une présentation abrégée de drogue nouvelle pour usage exceptionnel (PADNUE) qui cherchent à démontrer l'équivalence pharmaceutique et la bioéquivalence de la drogue proposée par rapport à une drogue de référence visée par l'alinéa c) de la définition d'un produit de référence canadien (PRC) se trouvant à l'article C.08.001.1 du Règlement sur les aliments et drogues, des directives quant à la marche à suivre pour démontrer que cette drogue est un PRC jugé acceptable par le ministre.
1.2 Énoncé de politique
En démontrant l'équivalence pharmaceutique et la bioéquivalence par rapport à un PRC, on veut prouver que les profils d'innocuité et d'efficacité du produit de mise en marché subséquent (générique) seront comparables à ceux du produit innovateur qui est commercialisé au Canada, dont l'innocuité et l'efficacité ont été cliniquement démontrées. Pour déterminer l'admissibilité de cette preuve, le ministre doit être convaincu qu'un produit de référence proposé en vertu de l'alinéa c) de la définition d'un PRC semble être identique au produit innovateur commercialisé au Canada et que tout écart non appuyé par des données entre ces deux produits n'aura pas de conséquence thérapeutique.
1.3 Portée et application
La présente ligne directrice s'applique à toutes les présentations abrégées de drogues nouvelles et à toutes les présentations abrégées de drogues nouvelles pour usage exceptionnel dans le cadre desquelles un promoteur cherche à faire approuver une drogue en s'appuyant sur son équivalence pharmaceutique et sur sa bioéquivalence avec une drogue de référence visée par l'alinéa c) de la définition d'un PRC se trouvant à l'article C.08.001.1 du Règlement sur les aliments et drogues. Cette ligne directrice vise plus particulièrement les présentations où la bioéquivalence sera démontrée par le biais d'études de biodisponibilité, d'études pharmacodynamiques, d'études cliniques ou d'une combinaison de ces méthodes.
Le présent document vise uniquement les formes posologiques dont il est question à la section 2. L’application des principes énoncés à d’autres formes posologiques à libération immédiate ou à des produits mixtes à libération immédiate n’y est pas abordée et sera examinée au cas par cas. Dans de tels cas, il est recommandé que le promoteur consulte avec Santé Canada avant de déposer leur présentation et, dans la mesure du possible, avant de procéder aux études de bioéquivalence.
1.4 Contexte
Pour déposer une présentation abrégée de drogue nouvelle ou une présentation abrégée de drogue nouvelle pour usage exceptionnel, le fabricant doit satisfaire aux exigences précisées, en outre, démontrer l'équivalence pharmaceutique et la bioéquivalence de la drogue proposée par rapport au produit de référence canadien. Selon l'article C.08.001.1 du Règlement sur les aliments et drogues :
Un « produit de référence canadien » signifie :
- (a) une drogue pour laquelle un avis de conformité a été délivré aux termes de l'article C.08.004 (ou C.08.004.01) et qui est commercialisée au Canada par son innovateur;
- (b) une drogue jugée acceptable par le ministre qui peut être utilisée pour la détermination de la bioéquivalence d'après les caractéristiques pharmaceutiques et, le cas échéant, les caractéristiques en matière de biodisponibilité, lorsqu'une drogue pour laquelle un avis de conformité a été délivré aux termes de l'article C.08.004 (ou C.08.004.01) ne peut être utilisée à cette fin parce qu'elle n'est plus commercialisée au Canada;
- (c) une drogue jugée acceptable par le ministre qui peut être utilisée pour la détermination de la bioéquivalence d'après les caractéristiques pharmaceutiques et, le cas échéant, les caractéristiques en matière de biodisponibilité, par rapport à une drogue visée par l'alinéa a).
La présente ligne directrice énonce les critères d'utilisation d'une drogue commercialisée comme PRC dans un pays autre que le Canada en vertu de l'alinéa c) de ce règlement.
En démontrant l'équivalence pharmaceutique et la bioéquivalence d'une drogue par rapport au produit d'un innovateur canadien, on veut prouver que les profils d'innocuité et d'efficacité du produit de mise en marché subséquent (générique) seront comparables à ceux du produit innovateur qui est commercialisé au Canada, dont l'innocuité et l'efficacité ont été cliniquement démontrées.
En raison de la mondialisation de l'industrie pharmaceutique, on fabrique plusieurs produits de référence destinés à d'autres pays selon la même composition et dans les mêmes conditions que ceux destinés au marché canadien. Il arrive que des produits soient fabriqués en un seul endroit, mais distribués dans le monde entier. Le cas échéant, l'acceptation par le ministre d'une drogue de référence commercialisée dans un autre pays en tant que PRC permet d'éviter que les études comparatives sur des sujets humains ne doivent être répétées, ce qui réduit les risques encourus par les participants aux études et les coûts des fabricants. Cet argument est particulièrement vrai dans le cas des présentations de drogue où la bioéquivalence sera démontrée par le biais d'études de biodisponibilité, d'études pharmacodynamiques, d'études cliniques ou d'une combinaison de ces méthodes.
Toutefois, les renseignements détaillés sur la formulation et la fabrication des produits commercialisés à l'extérieur du Canada ne sont généralement pas répartis avec Santé Canada. La présente ligne directrice énonce donc les critères auxquels une drogue doit satisfaire pour que le ministre accepte qu'elle soit utilisée comme PRC en vertu de l'article C.08.001.1, y compris les données qui servent à démontrer que le produit de référence étranger est identique au produit innovateur canadien correspondant et les conditions dans lesquelles tout écart non appuyé par des données entre ces deux produits ne devrait pas avoir de conséquence thérapeutique.
2 Conseils pour la mise en oeuvre
Le ministre peut accepter qu'un produit pharmaceutique acheté dans un autre pays soit utilisé comme produit de référence canadien (PRC) s'il satisfait aux critères suivants :
- Pour s'assurer que le produit étranger est de qualité équivalente au produit innovateur canadien, il faut fournir des données attestant que la commercialisation du produit est autorisée par les autorités réglementaires d'un pays ou d'une région qui applique des critères d'évaluation des médicaments comparables à ceux du Canada, conformément au titre 8 de la partie C du Règlement sur les aliments et drogues et selon l'interprétation donnée dans les lignes directrices et les politiques de Santé Canada (par exemple, les États-Unis, l'Europe (c'est-à-dire une demande de commercialisation autorisée par le biais de la procédure centralisée ou de la procédure décentralisée), le Japon, la Suisse, l'Australie).
- Pour appuyer l'affirmation selon laquelle le produit de référence étranger et le produit innovateur canadien sont équivalents, il faut inclure, dans la présentation de drogue, des données confirmant que le produit de référence étranger est commercialisé dans le pays ou la région d'origine par la même entreprise innovatrice ou la société qui commercialise actuellement la même quantité de cet ingrédient médicinal sous la même forme posologique au Canada.
- On doit fournir les renseignements suivants pour tous les lots du produit innovateur (de référence) commercialisé au Canada et les lots du produit de référence acheté dans un autre pays qui ont servi à mener les études comparatives in vivo :
- l'étiquette du produit;
- les certificats d'analyse (analysés selon les caractéristiques proposées dans la présentation du produit de mise en marché subséquent);
- la preuve d'achat (numéro de lot, date et lieu d'achat);
- des échantillons dans leurs systèmes récipient-fermeture originaux devraient être disponibles sur demande.
- Dans le cadre de toutes les études comparatives in vitro, il faut analyser les lots du produit de référence étranger utilisés lors des études comparatives in vivo et les lots du produit innovateur commercialisé au Canada, et diffuser les résultats obtenus. L'étude doit reposer sur des méthodes d'analyse convenablement validées. La présentation de drogue doit comprendre des copies des rapports de validation et, le cas échéant, une description des méthodes d'analyse maison.
- Comme des écarts non appuyés par des données entre le produit de référence étranger et le produit innovateur canadien pourraient se traduire par des différences inacceptables dans les profils d'innocuité et d'efficacité des deux médicaments s'ils contiennent des ingrédients médicinaux à risque élevé, l'ingrédient médicinal :
- ne doit pas correspondre à un « médicament à dose critique » tel que défini dans la ligne directrice de Santé Canada Normes en matière d'études de biodisponibilité comparatives : Formes pharmaceutiques de médicaments à effets systémiques;
- ne doit pas engendrer une nécessité de surveiller le patient pour éviter les effets d'un sous-traitement ou d'un surtraitement.
- Le produit de référence étranger doit satisfaire aux critères suivants :
- Pour les formes pharmaceutiques orales solides à libération immédiate (y compris les formes orales qui se désintègrent) :
- les ingrédients médicinaux sont considérés comme « hautement solubles » aux termes de l'annexe 1;
- le produit de référence étranger a la même couleur, la même forme, la même grosseur, le même poids et le même type d'enrobage (par exemple, non enrobé, enrobé d'une pellicule) que le produit innovateur commercialisé au Canada et la même configuration des rainures;
- les ingrédients non médicinaux contenus dans la formulation du produit innovateur commercialisé au Canada, comparé au produit de référence étranger, doivent être qualitativement les mêmes. Les données quantitatives sur la formulation de ces deux produits, lorsqu'elles sont disponibles, doivent montrer qu'ils sont identiques;
- les valeurs des profils comparatifs de dissolution à de multiples points des deux produits doivent être déterminées dans au moins trois (3) milieux qui se situent à l'intérieur de la plage physiologique de pH (pH de 1,2 à 6,8). Un échantillonnage adéquat doit être effectué pour évaluer la phase de dissolution des profils (par exemple à 5, 10, 15, 30, 45, 60 et 120 minutes) jusqu'à ce que l'ingrédient actif du produit médicamenteux soit dissous à 90 % ou qu'une asymptote soit obtenue. Au moins 12 unités posologiques doivent être utilisées pour la détermination des profils. La similarité des profils de dissolution doit être établie au moyen de l'équation qui définit un facteur de similarité (f2), fournie à l'annexe 2. Si la valeur de f2 se situe entre 50 et 100, on peut considérer que les deux profils de dissolution sont similaires.
- Pour les suspensions orales à libération immédiate :
- les ingrédients médicinaux sont considérés comme « hautement solubles » aux termes de l'annexe 1;
- la formulation du produit de référence étranger est la même que celle du produit innovateur commercialisé au Canada. Les ingrédients non médicinaux contenus dans la formulation du produit innovateur commercialisé au Canada doivent être quantitativement et qualitativement les mêmes que ceux du produit de référence étranger. En l'absence de données quantitatives sur le produit de référence, le promoteur du produit de mise en marché subséquent doit fournir les résultats de l'analyse comparative des formulations du produit innovateur commercialisé au Canada et du produit de référence étranger.
Aux fins du présent document, quantitativement les mêmes signifie que la quantité (ou concentration) de chaque excipient du produit innovateur commercialisé au Canada équivaut à ±5% de la quantité (ou concentration) de chaque excipient du produit de référence étranger. Toute différence au-delà de ce critère doit être justifiée scientifiquement. Une comparaison côte à côte des formulations qualitative et quantitative des deux produits doit être présentée.
- les valeurs des profils comparatifs de dissolution à de multiples points des deux produits doivent être déterminées dans au moins trois (3) milieux qui se situent à l'intérieur de la plage physiologique de pH (pH de 1,2 à 6,8). Un échantillonnage adéquat doit être effectué pour évaluer la phase de dissolution des profils (par exemple, à 5, 10, 15, 30, 45, 60 et 120 minutes) jusqu'à ce que l'ingrédient actif du produit médicamenteux soit dissous à 90 % ou qu'une asymptote soit obtenue. Au moins 12 unités posologiques doivent être utilisées pour la détermination des profils. La similarité des profils de dissolution doit être établie au moyen de l'équation qui définit un facteur de similarité (f2), fournie à l'annexe 2. Si la valeur de f2 se situe entre 50 et 100, on peut considérer que les deux profils de dissolution sont similaires.
- Pour les suspensions orales administrées par inhalation à libération immédiate, les suspensions administrées par voie nasale à libération immédiate et les solutions administrées par inhalation à libération immédiate dont la bioéquivalence doit être démontrée par le biais d'un essai in vivo :
- Le produit de référence étranger est identique au produit innovateur commercialisé au Canada comme en atteste la comparaison des caractéristiques pharmaceutiques pertinentes des deux produits, c'est-à-dire (i) la formulation, (ii) les propriétés physico-chimiques et (iii) les attributs du dispositif.
- Des études comparatives portant sur caractéristiques pharmaceutiques doivent être effectuées conformément aux indications ci-dessous :
(i) la formulation - les ingrédients non médicinaux contenus dans la formulation du produit innovateur commercialisé au Canada, comparé au produit de référence étranger, doivent être qualitativement et quantitativement les mêmes. Aux fins du présent document, quantitativement les mêmes signifie que la quantité (ou concentration) de chaque excipient du produit innovateur commercialisé au Canada équivaut à ±5% de la quantité (ou concentration) de chaque excipient du produit de référence étranger. Toute différence au-delà de ce critère doit être justifiée scientifiquement. Une comparaison côte à côte des formulations qualitative et quantitative des deux produits doit être présentée.
(ii) les propriétés physico-chimiques - lors de la comparaison des propriétés physico-chimiques, les résultats des deux produits doivent être essentiellement les mêmes. Aux fins du présent document, essentiellement les mêmes signifie que les résultats des deux produits de référence se situent dans une limite de ±10%. Toute différence au-delà de ce critère doit être justifiée scientifiquement. Voici des exemples de paramètres d'essai qui doivent être considérés lors de l'étude des propriétés physico-chimiques des solutions et des suspensions :
- le pH;
- le pouvoir tampon (si le produit contient un tampon);
- la viscosité;
- la gravité spécifique ou densité;
- la tension superficielle;
- l'osmolalité (mol/kg) / osmolarité (mol/L) (si la tonicité est déclarée sur l'étiquette);
- l'uniformité de la dose administrée (si accompagné d'un dispositif pour l'administration);
- le volume ou la dimension des gouttes (si administré sous forme de gouttes);
- la distribution de la dimension des gouttes (si administré en tant qu'aérosol);
- la forme de dispersion du jet et la géométrie des panaches (si administré en tant qu'aérosol).
Outre les études comparatives recommandées ci-dessus, les formes posologiques déterminées suivantes doivent faire l'objet des études comparatives additionnelles mentionnées ci-dessous :
- Suspensions : propriétés des substances pharmaceutiques (par exemple, taille des particules, structure cristalline), distribution de la taille des particules, profils de dissolution (le cas échéant).
- Suspensions pour la nébulisation : propriétés des substances pharmaceutiques (par exemple, taille des particules, structure cristalline), distribution de la taille des particules, temps de nébulisation, vitesse de libération du médicament et quantité totale de médicament administrée.
- Aérosols-doseurs : pression de vapeur, point de congélation, indice de réfraction, distribution de la taille des particules, masse des particules fines.
(iii) les attributs du dispositif - les résultats de l'analyse qualitative et quantitative des caractéristiques physiques et des caractéristiques de fonctionnement des dispositifs des deux produits (par exemple, dimensions, matériaux utilisés) doivent être fournis. Toute différence au-delà de ce qui pourrait constituer des « tolérances normales de fabrication » sera considérée comme importante.
- Pour les poudres sèches administrées par inhalation à libération immédiate (poudres à inhaler) :
- Le produit de référence étranger est identique au produit innovateur commercialisé au Canada comme en atteste la comparaison des caractéristiques pharmaceutiques pertinentes des deux produits, c'est-à-dire (i) la formulation, (ii) les propriétés physico-chimiques et (iii) les attributs du dispositif.
- Des études comparatives portant sur caractéristiques pharmaceutiques doivent être effectuées conformément aux indications ci-dessous :
(i) la formulation - les ingrédients non médicinaux contenus dans la formulation du produit innovateur commercialisé au Canada, comparé au produit de référence étranger, doivent être qualitativement et quantitativement les mêmes. Aux fins du présent document, quantitativement les mêmes signifie que la quantité (ou concentration) de chaque excipient du produit innovateur commercialisé au Canada équivaut à ±5% de la quantité (ou concentration) de chaque excipient du produit de référence étranger. Toute différence au-delà de ce critère doit être justifiée scientifiquement. Une comparaison côte à côte des formulations qualitative et quantitative des deux produits doit être présentée.
(ii) les propriétés physico-chimiques et le rendement in vitro - lors de la comparaison des propriétés physico-chimiques et du rendement in vitro, les résultats des deux produits doivent être essentiellement les mêmes. Aux fins du présent document, essentiellement les mêmes signifie que les résultats des deux produits de référence se situent dans une limite de ±10%. Toute différence au-delà de ce critère doit être justifiée scientifiquement. Voici des exemples de paramètres d'essai qui doivent être considérés lors de l'étude des propriétés physico-chimiques des poudres à inhaler :
- les propriétés des substances pharmaceutiques (par exemple, taille des particules, structure cristalline);
- les propriétés du produit pharmaceutique (par exemple, distribution de la taille des particules du vecteur (le cas échéant), masse volumétrique et masse spécifique, morphologie des particules (forme, texture et propriétés superficielles), point de fusion, charge électrostatique, porosité, surface active spécifique, hygroscopicité et teneur en humidité;
- la distribution de la taille des particules (chaque degré et masse des particules fines, dans la plage des débits atteignable par la population de patients visée (au moyen du mécanisme de libération), à volume constant);
- l'uniformité de la dose administrée.
(iii) attributs du dispositif - les résultats de l'analyse qualitative et quantitative des caractéristiques physiques et des caractéristiques de fonctionnement des dispositifs des deux produits (par exemple, dimensions, matériaux utilisés) doivent être fournis. Toute différence au-delà de ce qui pourrait constituer des « tolérances normales de fabrication » sera considérée comme importante.
- Pour les formes pharmaceutiques orales solides à libération immédiate (y compris les formes orales qui se désintègrent) :
Si l'une des conditions susmentionnées n'est pas remplie, le fabricant doit démontrer que le produit de mise en marché subséquent est équivalent au produit innovateur commercialisé au Canada au moyen des études in vivo comparatives appropriées.
3 Annexes
Annexe 1 - Détermination de la solubilité
Conditions d'essai
La stabilité de la substance pharmaceutique dans la phase de pH adéquate de 1,2 à 6,8 doit être démontrée. Un profil de solubilité à l'équilibre de la substance pharmaceutique doit être établi pour la plage physiologique de pH adéquate (1,2 à 6,8) dans les conditions suivantes :
Conditions
- Solvant :
- au minimum, des solutions tampons aqueuses de pH 1,2, 4,5 et 6,8
- Température :
- 37 plus ou moins (±) 1°C
- Répétitions :
- au moins trois (3) pour chaque pH analysé
- Méthodologie :
- méthode par agitation en flacon ou méthode similaire avec justification
Renseignements supplémentaires
Le pH de chacune des solutions à l'essai doit être vérifié avant et après l'ajout de la substance pharmaceutique pour confirmer la stabilité du pH du milieu tamponné. Le pH doit être ajusté au besoin.
Classification
- Solubilité élevée :
- Une substance pharmaceutique est considérée comme hautement soluble si la dose thérapeutique maximale se dissout entièrement dans 250 mL ou moins de solution aqueuse à tous les pH de la plage physiologique (1,2 à 6,8, à 37 ± 1 °C, c'est-à-dire que la dose/volume de solubilité (DVS) est inférieur ou égal à (≤) 250 mL pour toute la plage de pH.
Annexe 2 - Calcul du facteur de similarité (f2)
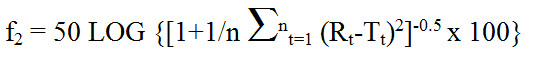
Équivalent du texte
Le facteur de similarité f indice 2 est égal à 50 fois le log de, ouverture de la parenthèse, ouverture de la parenthèse, 1 plus 1 sur n multiplié par la sommation lorsque T égal 1 à n de, ouverture de la parenthèse, R indice t moins T indice t, fermeture de la parenthèse, au carré, fermeture de la parenthèse, le tout exposant négatif 0.5, multiplié par 100, fermeture de la parenthèse, où R indice t et T indice t sont les pourcentages dissous à chaque point dans le temps de 1 à n. Un f indice 2 se situant entre 50 et 100 indique que les deux profils de dissolution sont similaires.
où Rt (produit innovateur commercialisé au Canada) et Tt (produit de référence étranger) sont les pourcentages dissous à chaque point dans le temps. Un f2 se situant entre 50 et 100 indique que les deux profils de dissolution sont similaires.
Pour comparer les profils de dissolution :
- Au moins 12 unités doivent être utilisées pour la détermination du profil de dissolution. On peut estimer le facteur de similarité f2 à l'aide des valeurs moyennes de dissolution. Si l'on utilise les données moyennes, le coefficient de variation (%) au premier temps d'échantillonnage ne doit pas dépasser 20 %, ni plus de 10 % aux autres temps d'échantillonnage.
- Les mesures de dissolution des deux produits devraient être effectuées dans les mêmes conditions d'essai. Les points dans le temps pour les deux profils de dissolution devraient être les mêmes (par exemple, 5, 10, 15, 30, 45 et 60 minutes).
- Il est recommandé d'utiliser au moins trois points (mis à part zéro), dont un seul se situerait au‑delà du plateau des profils.
- Si les deux produits sont dissous à plus de 85 % en 15 minutes, il n'est alors pas nécessaire de calculer les valeurs de f2.