
Draft Guidance Document - Pre-market Requirements for Medical Device Cybersecurity

This guidance document is being distributed for comment purposes only.
Draft date: 2018/12/07
Foreword
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
Table of Contents
- 1. Introduction
- 2. Guidance for Implementation
- 3. References
- Appendix A
- Manufacturers’ Cybersecurity Risk Management Framework
1. Introduction
Medical devices have evolved from largely analogue, non-networked and isolated hardware to networked devices incorporating remote access, wireless technology and complex software. Increasing levels of interconnectedness and data exchange between medical devices can have significant benefits to both patients and the healthcare system but can also leave devices vulnerable to unauthorized access. These vulnerabilities can negatively impact safety by causing diagnostic or therapeutic errors, or by affecting clinical operations.
The Food and Drugs Act sets out the legislative framework under which medical devices are regulated in Canada. Health Canada as the federal regulator of medical device safety and effectiveness, considers cybersecurity vulnerabilities in medical devices as a potential risk to patients that must be mitigated or eliminated by manufacturers of medical devices.
1.1 Policy Objectives
Health Canada considers the inclusion of cybersecurity measures an important consideration in issuing medical device licenses. Therefore, this guidance document provides medical device manufacturers advice on the practices, responses and mitigation measures which can improve the cybersecurity of their device. This guidance also outlines the information to be submitted as part of a medical device licence or licence amendment application to demonstrate that their medical device, consisting of or containing software, is sufficiently secure from intentional or unintentional unauthorized access.
1.2 Policy Statements
Health Canada considers cybersecurity a component of the medical device’s design and life-cycle that can impact safety and effectiveness. Manufacturers should consider cybersecurity when designing their medical device.
Risk management is required for all medical devices throughout their life-cycle. Manufacturers should incorporate cybersecurity into the risk management process for every device which consists of or contains software. Manufacturers are also encouraged to develop and maintain a framework for managing cybersecurity risks throughout their organizations.
All cybersecurity risk control measures should be successfully verified and validated against the device’s design requirements and/or design specifications. Manufacturers should be able to trace all verification and validation activities back to the device’s design requirements and/or design specifications.
1.3 Scope and Application
This guidance document applies to products that consist of or contain software and are regulated as medical devices (Class I to Class IV) under the Medical Devices Regulations.
This guidance document should be read in conjunction with the guidance documents on supporting evidence to be provided for medical device licence applications and licence amendment applications for Class III and Class IV medical devices. The content described in this guidance document is to be submitted for review in addition to the general data elements listed in Sections 32(3) and (4) of the Medical Devices Regulations.
The document provides guidance to manufacturers regarding the evidence to support Class III and Class IV medical device licence and licence amendments applications. Considerations related to the design, risk management, verification and validation testing and planning for future events are included in this guidance document. However, not all considerations will be applicable to every device type.
Although this document recommends that manufacturers demonstrate in their pre-market licence or licence amendment application that adequate provisions are in place to monitor, prevent, and respond to post-market cybersecurity events, this document does not provide guidance on post-market activities to be performed by the manufacturer.
1.4. Abbreviations and Definitions
1.4.1 Abbreviations
- AAMI
- Association for the Advancement of Medical Instrumentation
- ANSI
- American National Standards Institute
- BOM
- Bill of Materials
- IEC
- International Electrotechnical Commission
- IMDRF
- International Medical Device Regulators Forum
- ISO
- International Standards Organization
- MDB
- Medical Devices Bureau
- NIST
- National Institute of Standards and Technology
- TIR
- Technical Information Report
- TPD
- Therapeutic Products Directorate
- UL
- UL LLC
1.4.2 Definitions
authentication means verifying the identity of a user, process or device often as a prerequisite to allowing access to resources in an information system. [AAMI TIR57: 2016]
cybersecurity means the body of technologies, processes, practices, responses and mitigation measures designed to protect the medical device against unauthorized access, modification, misuse, or denial-of-use, and against the unauthorized use of information associated with a medical device.
device means an instrument, apparatus, contrivance or other similar article, or an in vitro reagent, including a component, part or accessory of any of them that is manufactured, sold or represented for use in diagnosis, treating, mitigating or preventing a disease, disorder or abnormal physical state, or any of their symptoms in human beings or animals. (instrument) [Food and Drugs Act]
malware means software designed with malicious intent to disrupt normal function, gather sensitive information, and/or access other connected systems.
risk means a combination of the probability of occurrence of harm and the severity of that harm. [ISO 13485: 2016]
system means a medical device comprising a number of components or parts intended to be used together to fulfill some or all of the device’s intended functions, and that is sold under a single name. (système)[Medical Devices Regulations]
software means a software system that has been developed for the purpose of being incorporated into the medical device being developed or that is intended for use as a medical device in its own right. [IEC 62304:2006]
validation means confirmation by examination and the provision of objective evidence that the requirements for a specific intended use have been fulfilled, as set out in the definition of validation in section 2.18 of International Organization for Standardization standard ISO 8402:1994, Quality management and quality assurance - Vocabulary, as amended from time to time. (validation) [Medical Devices Regulations]
threat means any circumstance or event with the potential to adversely impact health and safety via unauthorized access, destruction, disclosure, modification of information, and/or denial of service. [modified from AAMI TIR57:2016]
verification means confirmation through provision of objective evidence that specified requirements have been fulfilled. [IEC 62304:2006]
vulnerability means a weakness in an information system, system security procedures, internal controls, or implementation that could be exploited or triggered by a threat source. [AAMI TIR57:2016]
2. Guidance for Implementation
Medical device cybersecurity is a shared responsibility between the manufacturer, regulator, user and network administrator. Manufacturers are responsible for continuously monitoring, assessing, and mitigating potential cybersecurity risks throughout the lifecycle of their product.
Health Canada recommends manufacturers consider a methodology that addresses cybersecurity risk throughout their organization. The NIST document “Framework for Improving Critical Infrastructure Cybersecurity” (Version 1.0) is an established framework which may be utilized in whole or in part by the manufacturer. More information on how the framework may apply to medical devices is provided in Appendix A.
Additionally, a manufacturer must have a strategy to address the cybersecurity risk of a medical device (Class I to Class IV) that runs software code. This strategy should include the following elements.
- Secure design
- Risk management
- Verification and validation testing
- Planning for continued monitoring of and response to emerging risks and threats
During the evaluation of Class III and Class IV medical device licence and licence amendment applications, Health Canada will consider these elements in the assessment of the safety and effectiveness of the device. The elements listed above, and Health Canada’s expectations with respect to each element, are outlined in the subsequent sections of this guidance document.
2.1 Medical Device Cybersecurity Strategy
2.1.1 Secure Design
Manufacturers should consider cybersecurity early in the product life-cycle when design requirements are being developed. This includes:
- cybersecurity risks and controls when making design choices, and
- design choices that maximize device cybersecurity while not unduly affecting other safety-related aspects of the medical device (e.g., usability)
Design inputs captured in a requirement specification should include those related to cybersecurity. Where applicable, these cybersecurity requirements should be cross-referenced to specific device cybersecurity hazards if the requirements are mitigations to identified hazards. The manufacturer should also consider some design controls that allow the device to detect, resist, respond and recover from cybersecurity attacks. Some design control considerations are outlined in Table 1.
| Design Principle | Description |
|---|---|
Secure Communications |
The manufacturer should consider how the device will interface with other devices or networks. Interfaces may include hardwired connections and/or wireless communications. For each type of interface the manufacturer should determine the method the device will use to communicate with users (e.g., patients or healthcare professionals), other medical devices/sensors or healthcare systems. Examples of interface methods include Wi-Fi, Ethernet, Bluetooth and USB. |
| The manufacturer should consider how data transfer to and from the device will be secured to prevent unauthorized access. | |
Data Security |
The manufacturer should consider if data that is stored on or transferred to the device requires some level of encryption. |
| The manufacturer should consider design controls that take into account a device that communicates with a system and/or device that is less secure (e.g., a device connects to a home network or a legacy device with no device security controls). | |
| User Access | The manufacturer should consider user access controls that validate who can use the device. There may also be a requirement for authentication that grants privileges to different classes of users. Examples of authentication or access authorization include passwords, hardware keys or biometrics. |
Software Maintenance |
The manufacturer should consider how the software will be updated to secure the device against newly discovered cybersecurity threats. Consideration should be given to whether updates will require user intervention or be initiated by the device. |
| The manufacturer should determine what connections will be required to conduct updates. | |
| The manufacturer should consider how often a device will need to be updated. | |
| The manufacturer should consider how operating system software, third-party software (e.g., libraries) or open source software will be updated or controlled. | |
| Hardware or Physical Design | The manufacturer should consider controls to prevent an unauthorized person from making physical and software changes to the device in order to bypass security controls (e.g., disable a USB port that is not being used on device to prevent unauthorized access via USB key). |
| Reliability and Availability | The manufacturer should consider design controls that will allow the device to detect, resist, respond and recover from cybersecurity attacks. |
2.1.2 Device Specific Risk Management
Risk management is required for a medical device throughout its life-cycle. Manufacturers should incorporate medical device cybersecurity into each device’s risk management process, and should develop and maintain an organizational framework for managing cybersecurity risks.
Sound risk management principles, as described in ISO 14971-07:2007 Medical devices - Application of risk management (ISO 14971), should be incorporated throughout the life-cycle of a medical device. Health Canada recommends manufacturers extend these risk management principles to cybersecurity with additional considerations.
Generally, a manufacturer should:
- identify any cybersecurity hazard
- estimate and evaluate the associated risks
- control those risks to an acceptable level, and
- monitor the effectiveness of the risk controls
However, as shown in Figure 1, there are cybersecurity risks that may have an impact on the safety or effectiveness of the medical device.
A cybersecurity risk that reduces effectiveness, negatively affects clinical operations, or results in diagnostic or therapeutic errors should also be considered in the medical device’s risk management process. This consideration is reflected AAMI TIR57:2016 Principles for medical device security - Risk management which suggests that the risks associated with the cybersecurity of a device include harms to patient safety (as described in ISO 14971), and can be associated with indirect patients harm via cybersecurity security risks.
The Venn diagram below illustrates this concept of cybersecurity risk.
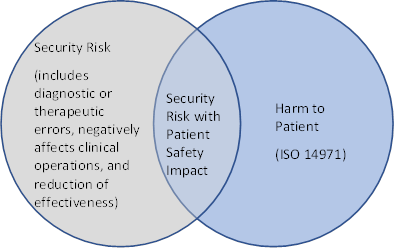
Long description
A Venn diagram illustrating the relationship between cybersecurity risk and safety risks as defined by ISO 14971 (AAMI TIR57).
Health Canada recommends that device-specific cybersecurity risk management processes be conducted in parallel to the safety risk management process described in ISO 14971. This parallel process is outlined in Figure 2 and is necessary because some cybersecurity risks may not have a safety impact.
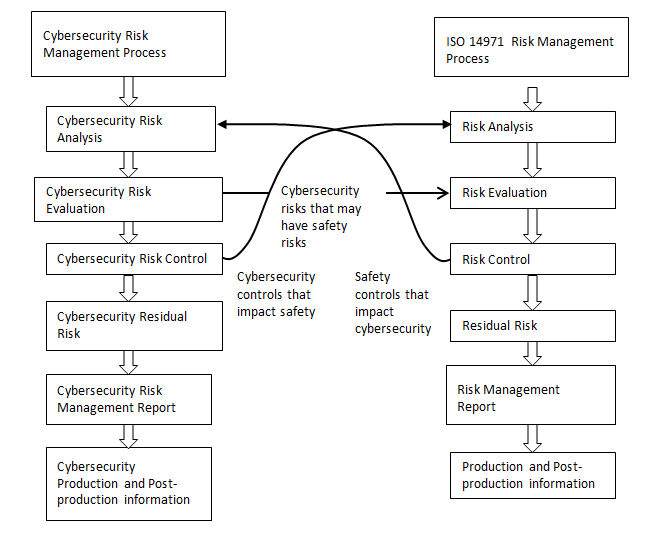
Long description
Illustrating the relationship between cybersecurity risk management process and safety risk management process as defined in ISO 14971 (AAMI TIR57:2016). TIR57 lists six steps involved in the cybersecurity risk management process: Cybersecurity risk analysis; Cybersecurity risk evaluation; Cybersecurity risk control; Evaluation of overall residual cybersecurity risk acceptability; Cybersecurity risk management report; Production and postproduction information.
The following table outlines four examples showing this relationship between cybersecurity risk management and patient safety management.
| Risk Relationship | Device | Security Harm | Safety Harm |
|---|---|---|---|
| Security Control | Safety Control | ||
| Security risk only. | A smart infusion pump and its remote control. | An unauthorized listening on wireless communications between the smart infusion pump and its remote control. | None. |
| Not Applicable. | Not Applicable. | ||
| Security risk with a safety impact. | A smart infusion pump with its remote control. | An unauthorized user gains access to wireless communications and issues a command to infuse insulin. | The smart infusion pump infuses more insulin than what was prescribed by an authorized user. |
| Not Applicable. | Not Applicable. | ||
| Security risk control with a safety impact. | An x-ray machine. | Not Applicable. | The device not readily accessible during an emergency because of the password requirement. |
| Requires password for access control to device. | Design an emergency mode to mitigate the safety risk. | ||
| Safety risk control with a security impact. | A smart infusion pump with a drug library. | An unauthorized user gains access to the drug library and makes changes to the limits. | The drug library requires an update to its limits to meet clinical needs. |
| Access to the drug library requires authentication of updates. | The smart infusion pump has hardwired or a wireless connection for library updates. |
Health Canada recommends the following standards to assist manufacturers conduct their cybersecurity risk management processes in parallel, and potentially iteratively, with their current established risk management process:
- AAMI TIR57:2016 - Principles for medical device security - Risk management
- ANSI/CAN/UL 2900-1:2017 - Standard for Software Security Network-Connectable Products, Part 1: General Requirements
- ANSI/CAN/UL 2900-2-1:2018 - Software Cybersecurity for Network Connectable Products
- IEC 80001-1: 2010 - Application of risk management for IT-networks incorporating medical devices
- NIST 800-30 Revision 1 Guide for Conducting Risk Assessments, September 2012
2.1.3 Verification and Validation Testing
All cybersecurity risk control measures should be successfully verified and validated against design specifications and/or design requirements. Manufacturers should be able to trace all verification and validation activities back to design specifications and/or design requirements.
Testing should include verification and validation of the functions, features and design elements that have been implemented to mitigate identified cybersecurity hazards. Health Canada recommends the UL 2900-1:2017 and UL 2900-2-1:2018 standards for guidance on cybersecurity testing.
The following table outlines the types of testing manufacturers may consider during the software verification and validation process.
| Test Category | Test Description |
|---|---|
| Vulnerabilities and Exploits Testing | Known Vulnerability Testing: Software code is tested against a database of known vulnerabilities such as the National Vulnerability Database. |
| Malware Testing: Malware tools are used to scan the code to determine if any known malware exists. | |
| Malformed Input Testing: The device is subjected to massive amounts of malformed (invalid or unexpected inputs) to observe if the device will behave in an unorthodox manner or if it will “crash”. | |
| Structured Penetration Testing: This type of testing requires a cybersecurity expert who is familiar with hacking techniques (i.e., white hat hacker). The cybersecurity expert attempts to circumvent the layers of defense that were designed into the device. | |
| Software Weakness Testing | Static Source Code Analysis: Utilization of a software tool to examine (i.e., debug) the source code without executing the software code. |
| Static Binary and Bytecode Analysis: Utilization of tools that will examine compiled code created from source code. |
2.1.4 Monitoring and Response to Emerging Risks
It is essential that manufacturers proactively monitor, identify and address vulnerabilities and exploits as part of their post-market management because cybersecurity risks to medical devices are continuously evolving. Manufacturers should demonstrate, in their pre-market licence application, that consideration has been given to address ongoing monitoring of and response to emerging cybersecurity threats to their device throughout its expected service life for both Class III and Class IV medical devices.
2.2 Medical Device Licence Applications: Cybersecurity Requirements
Medical device licence and licence amendment applications should include sufficient information for Health Canada to assess the following elements with respect to cybersecurity.
- Secure design
- Risk control activities
- Verification and validation testing
- The plan for on-going monitoring for and action against emerging threats
Details on the general data elements requirements for medical device licence and licence amendment applications are in Guidance Document: Guidance on supporting evidence to be provided for new and amended licence applications for Class III and Class IV medical devices, not including In Vitro Diagnostic Devices (IVDDs). The following data elements are relevant to cybersecurity:
- Device Labels, Package Label and Documentation
- Marketing History
- Risk Assessment
- Device Specific Quality Plan
- Safety and Effectiveness
2.2.1 Device Label, Package Label and Documentation
Manufacturers must provide a copy of all labelling, package inserts, product brochures and file cards to be used in connection of the device.
This includes the following information with respect to cybersecurity.
- The software BOM which lists all third-party or open source software components that were included in building the medical device software. The version and build of the components should be included in the software BOM. The manufacturer should also include a description of the tools used to create the software in the labelling.
- Any instructions:
- To the user and/or patient related to operation of the device that are part of the mitigation controls to reduce a cybersecurity risk(s).
- To the user on how to respond to and recover from a cybersecurity incident.
- Related to how the device will update its software as part of the mitigation of cybersecurity risks.
- To network system personnel on the proper IT environment to mitigate cybersecurity risks.
2.2.2 Marketing History
This section should include a summary of reported problems and details of any recalls associated with cybersecurity incidents (e.g., recall to address vulnerability discovered in a device).
2.2.3 Risk Assessment
A risk management report should include a risk analysis and evaluation of the risks inherent in the use of the device. The report should also include the risk reduction measures adopted to satisfy safety and effectiveness requirements as described in section 2.1.2 of this guidance document.
2.2.4 Device-Specific Quality Plan
Manufacturers are required to submit a quality plan for a Class IV licence application. The quality plan should demonstrate that a cybersecurity framework is part of the quality standards for the medical device being manufactured.
2.2.5 Safety and Effectiveness
Details of any cybersecurity studies that the manufacturer relied on to ensure that the device meets the safety and effectiveness requirements should be included in the Safety and Effectiveness section of the submission.
2.2.5.1 Standards
A list of all standards applied, in whole or in part, with respect to cybersecurity in the design and manufacture of the device should be included. For standards recognized by Health Canada, a Declaration of Conformity to cybersecurity related standards should still be accompanied by evidence that the proposed device is safe and effective from all identified cybersecurity risks.
2.2.5.2 Cybersecurity Testing
Cybersecurity testing evidence should be provided and include:
- For Class III and Class IV devices a detailed summary of testing that was conducted to verify and validate the security of the device.
- For Class IV devices, reports of cybersecurity testing.
2.2.5.3 Traceability Matrix
A traceability matrix should be included that maps all identified cybersecurity risks to:
- Requirement specification(s) (i.e., design inputs)
- Design specification(s) (i.e., design outputs), and
- Design verification and validation test(s)
2.2.5.4 Maintenance plan
A summary of the device’s maintenance plan should be included. The summary should describe:
- how software will be updated to maintain the safety and effectiveness of the device, and
- the post-market process(es) by which the manufacturer intends to ensure the continued safety and effectiveness of the device throughout its life-cycle
3. References
AAMI TIR57: 2016 Principles for medical device security - Risk management
ANSI/CAN/UK 2900-1: 2017 Software Cybersecurity for Network-Connectable Products, Part1: General Requirements
ANSI/CAN/UL 2900-2-1: 2018 Software Cybersecurity for Network-Connectable Products, Part 2-1: Particular Requirements for Network Connectable Components of Healthcare and Wellness Systems
IEC 62304 Medical Device Software - Software life cycle processes
IEC 62304 Amendment 1 Medical Device Software - Software life cycle processes
IMDRF Software as a Medical Device (SaMD): Key Definitions
ISO 14971 Medical devices - Application of risk management to medical devices
NIST: Framework for Improving Critical Infrastructure Cybersecurity, Version 1.1
NIST: Guide for Conducting Risk Assessments, September 2012
Appendix A
Manufacturers’ Cybersecurity Risk Management Framework
Manufacturers should consider the Framework for Improving Critical Infrastructure Cybersecurity (NIST, Version 1.1, April 2018) as a blueprint of best practices to guide their cybersecurity activities, including those related to risk management. Although this document is intended to improve cybersecurity risk management activities for critical infrastructure, Health Canada supports the framework as a way to improve and maintain the cybersecurity of medical devices. The framework can be applied to both the business and compliance processes (i.e., design controls) of a medical device company.
Heath Canada has focused on how the five core functions of the framework relate specifically to medical device design controls.
- Identify: The manufacturer should perform a risk analysis to identify cybersecurity risks in their product(s).
- Protect: Design controls should be implemented to limit the risk associated with the identified cybersecurity risks.
- Detect: Processes or measures should be in place to identify when the device has been compromised due to a cybersecurity event.
- Respond: A defined process or plan on should be developed on how the device, manufacturer or user will respond to a cybersecurity event.
- Recover: A plan describing the activities the device, manufacturer or user must undertake to restore the device to normal operating capacity following a cybersecurity event. The outcome of any investigations into previous recoveries may be used as feedback into the risk management process.
The framework is intended to complement the ISO 14971 risk management processes. A medical device manufacturer with a mature cybersecurity risk management process is encouraged to utilize the concepts of the framework to identify areas in its cybersecurity risk management processes that can be improved. A manufacturer that does not have an established cybersecurity risk management process may consider using the framework as a guide to establish organizational best practices in the cybersecurity of the devices that they manufacture.