
Ébauche de la ligne directrice - Exigences relatives à la cybersécurité des instruments médicaux avant leur mise en marché

La présente ligne directrice est publiée dans le seul but de recueillir des commentaires.
Date de l’ébauche : 2018/12/07
Avant-propos
Les lignes directrices sont des documents destinés à guider l’industrie et les professionnels de la santé sur la façon de se conformer aux lois et aux règlements en vigueur. Elles fournissent également aux membres du personnel des renseignements concernant la façon de mettre en œuvre le mandat et les objectifs de Santé Canada de manière juste, uniforme et efficace.
Les lignes directrices sont des outils administratifs n’ayant pas force de loi, ce qui permet une certaine souplesse d’approche. Les principes et les pratiques énoncés dans le présent document peuvent être remplacés par d’autres approches, à condition que celles-ci s’appuient sur une justification adéquate. Il faut tout d’abord discuter d’autres approches avec le programme concerné pour s’assurer qu’elles respectent les exigences des lois et des règlements applicables.
Dans la foulée de ce qui précède, il importe également de mentionner que Santé Canada se réserve le droit de demander des renseignements ou du matériel supplémentaire, ou de définir des conditions dont il n’est pas explicitement question dans le présent document, afin que le Ministère puisse être en mesure d’évaluer adéquatement l’innocuité, l’efficacité ou la qualité d’un produit thérapeutique donné. Santé Canada s’engage à justifier de telles demandes et à documenter clairement ses décisions.
Table des matières
- 1. Introduction
- 2. Ligne directrice concernant la mise en œuvre
- 2.1 Stratégie relative à la cybersécurité des instruments médicaux
- 2.2 Demandes d’homologation d’instruments médicaux : exigences relatives à la cybersécurité
- 3. Références
- Annexe A
- Cadre de gestion des risques pour la cybersécurité du fabricant
1. Introduction
Les instruments médicaux ont évolué d’appareils principalement analogiques, non reliés à un réseau et isolés vers des instruments en réseau qui incorporent des accès à distance, une technologie sans fil et un logiciel complexe. L’accroissement de l’interconnexion et de l’échange de données entre les instruments médicaux offrent de grands avantages aux patients et au système de soins de santé, mais peuvent laisser les instruments exposés aux accès non autorisés. Ces vulnérabilités peuvent avoir des répercussions négatives sur la sécurité en entraînant des erreurs de diagnostics ou thérapeutiques, ou en ayant des incidences sur les activités cliniques.
La Loi sur les aliments et drogues (LAD) précise le cadre législatif en vertu duquel les instruments médicaux sont réglementés au Canada. Santé Canada, en tant qu'organisme fédéral de réglementation de la sécurité et de l'efficacité des instruments médicaux, considère les vulnérabilités en matière de cybersécurité des instruments médicaux comme un risque potentiel pour les patients que les fabricants doivent atténuer ou éliminer.
1.1 Objectifs stratégiques
Santé Canada considère que l’inclusion de mesures de cybersécurité est un facteur important dans la délivrance des homologations d’instruments médicaux. Par conséquent, la présente ligne directrice fournit aux fabricants des instruments médicaux des conseils sur les pratiques, les interventions et les mesures d’atténuation qui sont susceptibles d’améliorer la cybersécurité de leur instrument. La présente ligne directrice décrit également l’information à présenter dans le cadre d’une demande d’homologation d’instrument médical ou de modification d’homologation pour démontrer que l’instrument médical, qui est ou qui contient un logiciel, est suffisamment protégé contre l’accès non autorisé intentionnel ou non intentionnel.
1.2 Énoncés de politique
Santé Canada considère la cybersécurité comme étant un élément de la conception et du cycle de vie de l’instrument médical pouvant avoir une incidence sur la sécurité et l’efficacité. Les fabricants doivent tenir compte de la cybersécurité lorsqu’ils conçoivent leur instrument médical.
La gestion des risques est requise pour tous les instruments médicaux durant tout leur cycle de vie. Les fabricants doivent intégrer la cybersécurité dans le processus de gestion des risques pour chaque instrument qui est ou qui contient un logiciel. On encourage également les fabricants à élaborer et à maintenir un cadre de gestion des risques pour la cybersécurité dans toutes leurs organisations.
Toutes les mesures de contrôle des risques pour la cybersécurité doivent être vérifiées et validées avec succès par rapport aux exigences de conception ou aux spécifications de conception de l’instrument. Les fabricants doivent être en mesure de retracer toutes les activités de vérification et de validation jusqu’aux exigences de conception ou aux spécifications de conception de l’instrument.
1.3 Portée et application
La présente ligne directrice s’applique aux produits qui sont ou qui contiennent un logiciel et qui sont réglementés comme des instruments médicaux (classes I à IV) en vertu du Règlement sur les instruments médicaux.
Le présent document devrait être lu en parallèle avec les lignes directrices sur les données à fournir pour étayer les demandes d’homologation des instruments médicaux de classe III et de classe IV et les demandes de modification. Le contenu décrit dans la présente ligne directrice doit être soumis pour examen, de même que les données générales énumérées aux paragraphes 32(3) et (4) du Règlement sur les instruments médicaux.
Le document fournit une orientation aux fabricants relativement aux données à fournir à l’appui des demandes d’homologation des instruments médicaux de classe III et de classe IV et des demandes de modification d’homologation. Les facteurs à considérer relativement à la conception, à la gestion des risques, aux tests de vérification et de validation et à la planification des futurs événements sont inclus dans le présent document. Cependant, les facteurs ne s’appliqueront pas tous à chaque type d’instrument.
Bien que le présent document recommande que les fabricants démontrent dans leur demande d’homologation ou de modification d’homologation avant la mise en marché que des dispositions adéquates sont en place pour surveiller ou prévenir les événements de cybersécurité après la mise en marché et intervenir au besoin, le présent document ne fournit pas de ligne directrice sur les activités suivant la mise en marché devant être effectuées par le fabricant.
1.4 Abréviations et définitions
1.4.1 Abréviations
- AAMI
- Association for the Advancement of Medical Instrumentation
- ANSI
- American National Standards Institute
- NMP
- Nomenclature des matériaux et produits
- CEI
- Commission électrotechnique internationale
- IMDRF
- International Medical Device Regulators Forum
- ISO
- Organisation internationale de normalisation
- BMM
- Bureau des matériels médicaux
- NIST
- National Institute of Standards and Technology
- RIT
- Rapport d’information technique
- DPT
- Direction des produits thérapeutiques
- UL
- UL LLC
1.4.2 Définitions
authentification : vérifier l’identité d’un utilisateur, d’un processus ou d’un instrument, souvent comme conditions préalables à l’autorisation de l’accès aux ressources dans un système d’information. [AAMI TIR57: 2016]
cybersécurité : l'ensemble des technologies, processus, pratiques, mesures d'intervention et d'atténuation dont la raison d'être est de protéger l’instrument médical contre l’accès non autorisé, la modification, le mésusage ou le refus d’utilisation, et contre l’utilisation non autorisée d’information associée à un instrument médical.
instrument : tout instrument, appareil, dispositif ou article semblable ou tout réactif in vitro, y compris tout composant, partie ou accessoire de l’un ou l’autre de ceux-ci, fabriqué ou vendu pour servir à l’une ou l’autre des fins ci-après ou présenté comme pouvant y servir : le diagnostic, le traitement, l’atténuation ou la prévention d’une maladie, d’un désordre ou d’un état physique anormal ou de leurs symptômes, chez l’être humain ou les animaux. (device) [Loi sur les aliments et drogues]
maliciel : logiciel conçu avec l'intention malveillante de perturber le fonctionnement normal, de rassembler des informations sensibles et / ou d'accéder à d'autres systèmes connectés.
risque : combinaison de la probabilité d'un dommage et de sa gravité. [ISO 13485: 2016]
système : instrument médical qui est formé de composants ou de parties destinés à être utilisés ensemble pour remplir certaines ou la totalité des fonctions auxquelles il est destiné et qui est vendu sous un seul nom. (system) [Règlement sur les instruments médicaux]
logiciel : système logiciel mis au point dans le but d’être intégré dans l’instrument médical en train d’être mis au point ou destiné à être utilisé comme un instrument médical de plein droit. [CEI 62304:2006]
validation : confirmation par examen et apport de preuves tangibles que les exigences particulières pour une utilisation donnée sont respectées, selon la définition figurant à l’article 2.18 de la norme ISO 8402:1994 de l’Organisation internationale de normalisation, intitulée Management de la qualité et assurance de la qualité - Vocabulaire, avec ses modifications successives. (validation) [Règlement sur les instruments médicaux]
menace : tout événement ou circonstance ayant le potentiel de porter atteinte à la santé et la sécurité par un accès non autorisé, une destruction, une divulgation, une modification de l’information et/ou un refus de service. [Définition modifiée de l’AAMI TIR57:2016]
vérification : confirmation par apport de preuves tangibles que des exigences particulières sont respectées. [CEI 62304:2006]
vulnérabilité : faiblesse dans un système d’information, des procédures de sécurité de système, des contrôles internes ou une mise en œuvre qui pourrait être exploitée ou déclenchée par une source de menace. [AAMI TIR57:2016]
2. Ligne directrice concernant la mise en œuvre
La cybersécurité des instruments médicaux est une responsabilité partagée entre le fabricant, l'organisme de réglementation, l'utilisateur et l'administrateur du réseau. Les fabricants ont la responsabilité de continuellement surveiller, évaluer et atténuer les risques potentiels liés à la cybersécurité tout au long du cycle de vie de leur produit.
Santé Canada recommande aux fabricants d’envisager une méthodologie qui aborde les risques pour la cybersécurité dans toute leur organisation. Le document du NIST intitulé « Framework for Improving Critical Infrastructure Cybersecurity » (Cadre d'amélioration de la cybersécurité des infrastructures essentielles) (Version 1.0), est un cadre établi qui peut être utilisé en tout ou en partie par le fabricant. De plus amples renseignements sur la façon dont le cadre pourrait s’appliquer aux instruments médicaux sont fournis dans l’annexe A.
De plus, un fabricant doit se doter d’une stratégie pour traiter les risques pour la cybersécurité d’un instrument médical (classes I à IV) qui exécute un code de logiciel. Cette stratégie doit inclure les éléments suivants :
- Conception sécuritaire
- Gestion des risques
- Tests de vérification et de validation
- Planification de la surveillance continue des nouveaux risques et menaces et de la réponse à ceux-ci
Lors de l’évaluation des demandes d’homologation d’instruments médicaux de classe III et de classe IV et des demandes de modification d’homologation, Santé Canada tiendra compte de ces éléments dans l’évaluation de la sécurité et de l’efficacité de l’instrument. Les éléments énumérés ci-dessus, et les attentes de Santé Canada à l’égard de chaque élément, sont brièvement décrits dans des sections ultérieures de la présente ligne directrice.
2.1 Stratégie relative à la cybersécurité des instruments médicaux
2.1.1 Conception sécuritaire
Les fabricants doivent tenir compte de la cybersécurité au début du cycle de vie du produit lors de l’élaboration des exigences de conception. Cela comprend :
- les risques et les contrôles associés à la cybersécurité lors des choix en matière de conception
- des choix en matière de conception qui maximisent la cybersécurité de l’instrument médical tout en ne nuisant pas indûment aux autres aspects liés à sa sécurité (p. ex. l’utilisabilité)
Les intrants de conception saisis dans une spécification des besoins doivent comprendre ceux liés à la cybersécurité. Le cas échéant, ces exigences en matière de cybersécurité doivent être recoupées avec les dangers pour la cybersécurité de l’instrument particulier si les besoins consistent à atténuer les dangers identifiés. De plus, le fabricant doit envisager certains contrôles de conception qui permettent à l’instrument de détecter des attaques à la cybersécurité, d’y résister, d’y répondre et de permettre la reprise après incident. Certains facteurs à considérer dans les contrôles de conception sont indiqués dans le tableau 1.
| Principe de conception | Description |
|---|---|
| Communications sécurisées | Le fabricant doit considérer la façon dont l’instrument interfacera avec d’autres instruments ou réseaux. Les interfaces peuvent comprendre des raccordements fixes et/ou des communications sans fil. Pour chaque type d’interface, le fabricant doit déterminer la méthode que l’instrument utilisera pour communiquer avec les utilisateurs (p. ex. les patients ou les professionnels de la santé), d’autres instruments/capteurs médicaux ou systèmes de soins de santé. À titre d’exemples de méthodes d’interface, citons Wi-Fi, Ethernet, Bluetooth et USB. |
| Le fabricant doit considérer la façon dont le transfert de données à destination et en provenance de l’instrument sera sécurisé pour empêcher l’accès non autorisé. | |
Sécurité des données |
Le fabricant doit vérifier si les données qui sont stockées dans l’instrument ou transférées vers celui-ci nécessitent un certain niveau de chiffrement. |
| Le fabricant doit envisager des contrôles de conception qui tiennent compte d’un instrument qui communique avec un système et/ou un dispositif qui est moins sécuritaire (p. ex. un dispositif qui se branche sur un réseau domestique ou un dispositif patrimonial sans contrôles de sécurité du dispositif). | |
| Accès utilisateur | Le fabricant doit envisager des restrictions d’accès qui valident qui peut utiliser l’instrument. Il pourrait aussi y avoir une exigence d’authentification qui accorde des privilèges à différentes classes d’utilisateurs. À titre d’exemples d’authentification ou d’autorisation de l’accès, citons les mots de passe, les clés matérielles ou la biométrie. |
Maintenance du logiciel |
Le fabricant doit considérer la façon dont le logiciel sera mis à jour pour protéger l’appareil contre les menaces à la cybersécurité nouvellement découvertes. Il doit déterminer si les mises à jour nécessiteront l’intervention de l’utilisateur ou si elles seront déclenchées par l’instrument. |
| Le fabricant doit déterminer quelles connexions seront requises pour effectuer les mises à jour. | |
| Le fabricant doit déterminer la fréquence à laquelle un instrument devra être mis à jour | |
| Le fabricant doit considérer la façon dont le logiciel d’exploitation, le logiciel tiers (p. ex. les bibliothèques) ou le logiciel ouvert seront mis à jour ou contrôlés. | |
| Conception matérielle ou physique | Le fabricant doit envisager des contrôles pour empêcher une personne non autorisée d’apporter des modifications physiques ou logicielles à l’instrument afin de passer outre aux contrôles de sécurité (p. ex. désactiver un port USB qui n’est pas utilisé sur l’instrument afin de prévenir son utilisation non-autorisée). |
| Fiabilité et disponibilité | Le fabricant doit envisager des contrôles de conception qui permettront à l’instrument de détecter les attaques à la cybersécurité, d’y résister, d’y répondre et de permettre la reprise après incident. |
2.1.2 Gestion des risques propres à l’instrument
La gestion des risques est requise pour un instrument médical pendant tout son cycle de vie. Les fabricants doivent intégrer la cybersécurité des instruments médicaux dans le processus de gestion des risques de chaque instrument et ils doivent élaborer et maintenir un cadre organisationnel de gestion des risques pour la cybersécurité.
De solides principes de gestion des risques, comme décrits dans la norme ISO 14971-07:2007 Dispositifs médicaux - Application de la gestion des risques (ISO 14971), doivent être intégrés pendant tout le cycle de vie d’un instrument médical. Santé Canada recommande que les fabricants étendent ces principes de gestion des risques à la cybersécurité avec des facteurs à considérer supplémentaires.
Généralement un fabricant doit :
- identifier tout danger pour la cybersécurité
- estimer et évaluer les risques connexes
- contrôler ces risques jusqu’à un niveau acceptable
- surveiller l’efficacité des contrôles des risques
Cependant, comme l’illustre la figure 1, il existe des risques pour la cybersécurité qui pourraient avoir une incidence sur la sécurité ou l’efficacité de l’instrument médical.
Un risque pour la cybersécurité qui réduit l’efficacité, a des effets négatifs sur les opérations cliniques ou entraîne des erreurs de diagnostic ou thérapeutiques doit également être pris en considération dans le processus de gestion des risques de l’instrument médical. Ce facteur est pris en considération dans la norme AAMI TIR57:2016 Principles for medical device security - Risk management (Principes pour la sécurité des instruments médicaux - Gestion des risques) qui laissent entendre que les risques associés à la cybersécurité d’un instrument comprennent l’atteinte à la sécurité des patients (comme décrit dans la norme ISO 14971), et peuvent être associés à des patients indirects par le biais des risques pour la sécurité et la cybersécurité.
Le diagramme de Venn ci-dessous illustre ce concept du risque pour la cybersécurité.
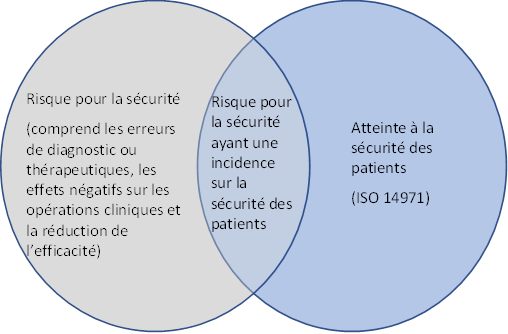
Équivalent textuel
Long description : Diagramme de Venn illustrant la relation entre un risque pour la cybersécurité et les risques pour la sécurité au sens de la norme ISO 14971 (AAMI TIR57).
Santé Canada recommande que des processus de gestion des risques pour la cybersécurité propres à l’instrument soient effectués en parallèle avec le processus de gestion des risques pour la sécurité décrits dans la norme ISO 14971. Ce processus parallèle est exposé à grands traits à la figure 2 et est nécessaire parce que certains risques pour la cybersécurité pourraient ne pas avoir une incidence sur la sécurité.
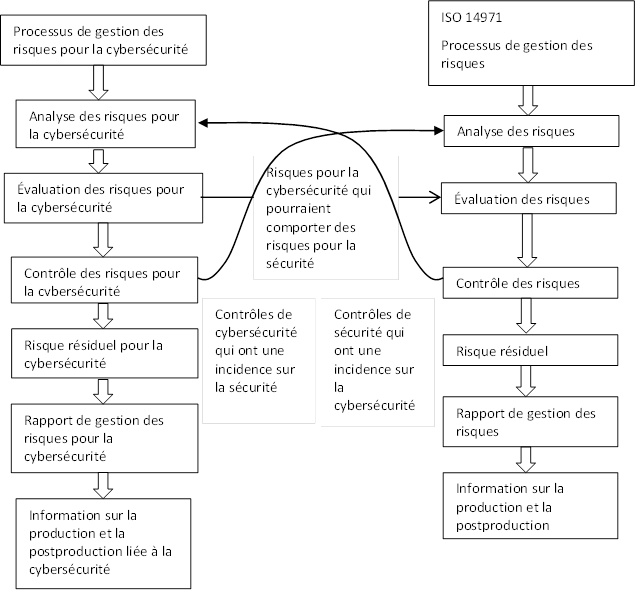
Équivalent textuel
Long description : Illustrer la relation entre le processus de gestion des risques pour la cybersécurité et le processus de gestion des risques pour la sécurité aux termes de la norme ISO 14971 (AAMI TIR57:2016). TIR57 indique les six étapes comprises dans le processus de gestion des risques pour la cybersécurité : analyse des risques pour la cybersécurité; évaluation des risques pour la cybersécurité; contrôle des risques pour la cybersécurité; évaluation de l’acceptabilité des risques résiduels globaux pour la cybersécurité; rapport sur la gestion des risques pour la cybersécurité; renseignements sur la production et la postproduction.
Le tableau suivant décrit brièvement quatre exemples montrant cette relation entre la gestion des risques pour la cybersécurité et la gestion de la sécurité des patients.
| Relation entre les risques | Instrument | Atteinte à la sécurité | Atteinte à la sécurité des patients |
|---|---|---|---|
| Contrôle de sécurité | Contrôle de sécurité des patients | ||
| Risques pour la sécurité seulement. | Une pompe à perfusion intelligente et sa télécommande. | Une écoute non autorisée sur des communications sans fil entre la pompe à perfusion intelligente et sa télécommande. | Aucun. |
| Sans objet. | Sans objet. | ||
| Risque pour la sécurité ayant une incidence sur la sécurité des patients. | Une pompe à perfusion intelligente et sa télécommande. | Un utilisateur non autorisé obtient l’accès aux communications sans fil et émet une commande de perfuser de l’insuline. | La pompe à perfusion intelligente perfuse plus d’insuline que la dose prescrite par un utilisateur autorisé. |
| Sans objet. | Sans objet. | ||
| Contrôle des risques pour la sécurité ayant une incidence sur la sécurité des patients. | Appareil de radiographie. | Sans objet. | L’instrument n’est pas facilement accessible en cas d’urgence à cause du mot de passe requis. |
| Mot de passe requis pour le contrôle d’accès à l’instrument. | Concevoir un arrêt en cas d’urgence pour atténuer le risque pour la sécurité. | ||
| Contrôle des risques pour la sécurité ayant une incidence sur la sécurité. | Une pompe à perfusion intelligente dotée d’une pharmacothèque. | Un utilisateur non autorisé obtient l’accès à la pharmacothèque et apporte des modifications aux limites. | La pharmacothèque requiert une mise à jour pour répondre aux besoins cliniques. |
| L’accès à la pharmacothèque requiert l’authentification des mises à jour. | La pompe à perfursion intelligente est dotée d’une connexion câblée ou sans fil pour les mises à jour de la pharmacothèque. |
Santé Canada recommande de consulter les normes suivantes pour aider les fabricants à mener leurs processus de gestion des risques pour la cybersécurité en parallèle, et possiblement de manière itérative, avec leur processus de gestion des risques actuel :
- AAMI TIR57:2016 - Principles for medical device security - Risk management
- ANSI/CAN/UL 2900-1:2017 - Standard for Software Security Network-Connectable Products, Part 1: General Requirements
- ANSI/CAN/UL 2900-2-1:2018 - Software Cybersecurity for Network Connectable Products
- IEC 80001-1: 2010 - Application of risk management for IT-networks incorporating medical devices
- NIST 800-30 Revision 1 Guide for Conducting Risk Assessments, septembre 2012
2.1.3 Tests de vérification et de validation
Toutes les mesures de contrôle des risques pour la cybersécurité doivent être vérifiées et validées avec succès par rapport aux spécifications de conception et/ou aux exigences de conception. Les fabricants doivent être en mesure de retracer toutes les activités de vérification et de validation jusqu’aux exigences de conception ou aux spécifications de conception de l’instrument.
Les tests doivent comprendre la vérification et la validation des fonctions, des caractéristiques et des éléments de conception qui ont été mis en œuvre pour atténuer les dangers identifiés pour la cybersécurité. Santé Canada recommande de consulter les normes UL 2900-1:2017 et UL 2900-2-1:2018 pour les tests concernant la cybersécurité.
Le tableau suivant décrit brièvement les types de tests que pourraient envisager les fabricants durant le processus de vérification et de validation de logiciel.
| Catégorie de test | Description du test |
|---|---|
| Test des vulnérabilités et des exploits | Test des vulnérabilités connues : Le code de logiciel est testé par rapport à une base de données des vulnérabilités connues, telle que la base de données nationales sur les vulnérabilités. |
| Test de maliciel : Des outils de maliciel sont utilisés pour scanner le code afin de déterminer si tout maliciel connu existe. | |
| Test d’entrée malformée : L’instrument est assujetti à des quantités massives d’entrées malformées (invalides ou inattendues) afin d’observer s’il se comportera d’une manière peu orthodoxe ou s’il « se plantera ». | |
| Test de pénétration structuré : Ce type de test requiert un expert en cybersécurité qui connaît bien les techniques de piratage (c.-à-d. chapeau blanc). L’expert en cybersécurité tente de contourner les couches de défense qui ont été conçues dans l’instrument. | |
| Test des faiblesses de logiciel | Analyse statique du code source : Utilisation d’un outil logiciel pour examiner (c.-à-d. déboguer) le code source sans exécuter le code de logiciel. |
| Analyse binaire et de code à octet statique : Utilisation d’outils qui examineront le code compilé créé à partir du code source. |
2.1.4 Surveillance des nouveaux risques et réponse à ceux-ci
Il est essentiel que les fabricants surveillent, identifient et abordent de manière proactive les vulnérabilités et les exploits dans le cadre de leur gestion suivant la mise en marché parce que les risques pour la cybersécurité des instruments médicaux évoluent sans cesse. Les fabricants doivent démontrer dans leur demande d’homologation avant la mise en marché qu’il a été envisagé d’aborder la surveillance continue des nouvelles menaces à la cybersécurité de leur instrument et la réponse à celles-ci pendant toute la durée de vie prévue de l’instrument; cela s’applique aux instruments médicaux de classe III et de classe IV.
2.2 Demandes d’homologation d’instruments médicaux : exigences relatives à la cybersécurité
Les demandes d’homologation d’instruments médicaux et les demandes de modification de l’homologation doivent comprendre suffisamment de renseignements pour que Santé Canada puisse évaluer les éléments suivants relativement à la cybersécurité.
- Conception sécuritaire
- Activités de contrôle des risques
- Tests de vérification et de validation
- Le plan de surveillance continue des nouvelles menaces et des mesures pour les contrer
Les détails sur les éléments de donnée généraux requis pour les demandes d’homologation d’instruments médicaux et les demandes de modification d’homologation se trouvent dans la Ligne directrice sur les données à fournir pour étayer les demandes d’homologation des instruments médicaux de classe III et de classe IV et les demandes de modification, à l’exception des instruments de diagnostic in vitro (IDIV). Les éléments de donnée suivants sont pertinents pour la cybersécurité :
- Étiquettes de l’instrument, étiquette de l’emballage et documentation
- Historique de marketing
- Évaluation des risques
- Plan de qualité propre à l’instrument
- Sécurité et efficacité
2.2.1 Étiquette de l’instrument, étiquette de l’emballage et documentation
Les fabricants doivent fournir un exemplaire des étiquettes, des notices, des brochures et des fiches à utiliser relativement à l’instrument.
Cela comprend les renseignements suivants en ce qui a trait à la cybersécurité.
- La NMP du logiciel qui énumère tous les composants de logiciel ouvert ou tiers qui ont été inclus dans le développement du logiciel de l’instrument médical. La version des composants doit être incluse dans la NMP du logiciel. Le fabricant doit également inclure dans l’étiquetage une description des outils utilisés pour créer le logiciel.
- Toute instruction :
- à l’utilisateur et/ou au patient se rapportant au fonctionnement de l’instrument et qui fait partie intégrante des mesures d’atténuation visant à réduire un ou plusieurs risques pour la cybersécurité
- à l’utilisateur sur la manière de répondre à un incident de cybersécurité et de reprendre les opérations
- ayant trait à la façon dont l’instrument mettra à jour son logiciel dans le cadre de l’atténuation des risques pour la cybersécurité
- au personnel du système réseau sur l’environnement de TI approprié pour atténuer les risques pour la cybersécurité
2.2.2 Historique de marketing
Cette section doit comprendre un résumé des problèmes signalés et les détails de tout rappel associé à des incidents de cybersécurité (p. ex. rappel pour traiter la vulnérabilité découverte dans un instrument).
2.2.3 Évaluation des risques
Un rapport de gestion des risques doit comprendre une analyse des risques et une évaluation des risques intrinsèques à l’utilisation de l’instrument. De plus, le rapport doit inclure les mesures de réduction des risques adoptées pour satisfaire aux exigences en matière de sécurité et d’efficacité telles qu’elles sont décrites à la section 2.1.2 de la présente ligne directrice.
2.2.4 Plan de qualité propre à l’instrument
Les fabricants sont tenus de présenter un plan de qualité pour une demande d’homologation de classe IV. Le plan de qualité doit démontrer qu’un cadre de cybersécurité fait partie intégrante des normes de qualité de l’instrument médical en train d’être fabriqué.
2.2.5 Sécurité et efficacité
Les détails de toute étude sur la cybersécurité sur laquelle a compté le fabricant pour s’assurer que l’instrument satisfait aux exigences de sécurité et d’efficacité doivent être inclus dans la section de la présentation portant sur la sécurité et l’efficacité.
2.2.5.1 Normes
Une liste de toutes les normes appliquées, en tout ou en partie, relativement à la cybersécurité dans la conception et la fabrication de l’instrument devrait être incluse dans la demande. Dans le cas des normes reconnues par Santé Canada, une Déclaration de conformité aux normes se rapportant à la cybersécurité doit encore être accompagnée de preuves indiquant que l’instrument proposé est sécuritaire et efficace contre tous les risques pour la cybersécurité identifiés.
2.2.5.2 Test de cybersécurité
Les preuves du test de cybersécurité devraient être incluses dans la demande et comprendre ce qui suit :
- Dans le cas des instruments de classe III et de classe IV, un résumé détaillé du test qui a été effectué pour vérifier et valider la sécurité de l’instrument.
- Dans le cas des instruments de classe IV, les rapports du test de cybersécurité.
2.2.5.3 Matrice de traçabilité
Une matrice de traçabilité devrait être inclused dans la demande. Cette matrice doit cartographier tous les risques pour la cybersécurité relatifs :
- aux spécifications des besoins (c.-à-d. les intrants de conception)
- aux spécifications de conception (c.-à-d. les extrants de conception)
- au(x) test(s) de vérification et de validation de la conception
2.2.5.4 Plan d’entretien
Un résumé du plan d’entretien de l’instrument doit être inclus. Le résumé doit décrire ce qui suit :
- la façon dont le logiciel sera mis à jour pour maintenir la sécurité et l’efficacité de l’instrument
- les processus après la mise en marché selon lesquels le fabricant compte assurer la sécurité et l’efficacité continues de l’instrument pendant tout son cycle de vie
3. Références
- AAMI TIR57: 2016 Principles for medical device security - Risk management (en anglais seulement)
- ANSI/CAN/UK 2900-1:2017 Software Cybersecurity for Network-Connectable Products, Part1: General Requirements (en anglais seulement)
- ANSI/CAN/UL 2900-2-1:2018 Software Cybersecurity for Network-Connectable Products, Part 2-1: Particular Requirements for Network Connectable Components of Healthcare and Wellness Systems (en anglais seulement)
- CEI 62304 Logiciels de dispositifs médicaux - Processus du cycle de vie du logiciel
- CEI 62304 Modification 1 de Logiciels de dispositifs médicaux - Processus du cycle de vie du logiciel
- Logiciels à titre d’instruments médicaux (LIM) : définitions clés de l’IMDRF
- ISO 14971 Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux
- NIST : Framework for Improving Critical Infrastructure Cybersecurity, Version 1.1 (en anglais seulement)
- NIST : Guide for Conducting Risk Assessments, septembre 2012 (en anglais seulement)
Annexe A
Cadre de gestion des risques pour la cybersécurité du fabricant
Les fabricants devraient considérer le Framework for Improving Critical Infrastructure Cybersecurity (NIST, version 1.1, avril 2018) comme un plan directeur des pratiques exemplaires pour guider leurs activités de cybersécurité, y compris celles liées à la gestion des risques. Bien que ce document vise à améliorer les activités de gestion des risques pour la cybersécurité pour les infrastructures essentielles, Santé Canada soutient le cadre comme moyen d’améliorer et de maintenir la cybersécurité des instruments médicaux. Le cadre peut être appliqué tant aux processus opérationnels qu’aux processus de conformité (c.-à-d. contrôles de conception) d’une entreprise d’instruments médicaux.
Santé Canada s’est concentré sur la façon dont les cinq fonctions de base du cadre se rapportent plus particulièrement aux contrôles de conception des instruments médicaux.
- Identifier : Le fabricant doit effectuer une analyse des risques pour identifier les risques pour la cybersécurité dans ses produits.
- Protéger : Des contrôles de conception doivent être mis en œuvre pour limiter le risque associé aux risques pour la cybersécurité identifiés.
- Détecter : Des processus ou mesures doivent être en place pour déterminer quand l’instrument a été compromis par suite d’un événement de cybersécurité.
- Répondre : Un processus ou plan défini doit être élaboré sur la façon dont l’instrument, le fabricant ou l’utilisateur répondra à un événement de cybersécurité.
- Reprendre : Un plan décrivant les activités que l’instrument, le fabricant ou l’utilisateur doit entreprendre pour remettre l’instrument en état normal de fonctionnement par suite d’un événement de cybersécurité. Le résultat de toute enquête sur des reprises antérieures pourrait être utilisé comme rétroaction dans le processus de gestion des risques.
Le cadre est conçu pour compléter les processus de gestion des risques de la norme ISO 14971. On encourage le fabricant d’instruments médicaux qui a un processus standard de gestion des risques pour la cybersécurité à utiliser les concepts du cadre pour repérer les points à améliorer dans ses processus de gestion des risques pour la cybersécurité. Le fabricant qui n’a pas un processus établi de gestion des risques pour la cybersécurité pourrait envisager d’utiliser le cadre comme un guide pour établir des pratiques exemplaires organisationnelles dans la cybersécurité des instruments qu’il fabrique.