
Submission of targeted risk management plans and follow-up commitments for prescription opioid-containing products - Guidance for industry
Effective date: May 2, 2018
Forward
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada's mandates and objectives should be implemented in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant programme area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable guidance documents.
This document should be read in parallel with Part C, Division 1 of the Food and Drug Regulations. In the event of any inconsistency or conflict, the regulations take precedence over this guidance documentFootnote 1.
Table of contents
- 1 Introduction
- 2 Terminology and acronyms
- 3 Guidance for implementation
- 3.1 General considerations
- 3.2 Mandatory Canadian Specific Opioid targeted Risk Management Plan sections
- 3.3 Canadian Specific Opioid targeted Risk Management Plan submission process and timeline
- 3.4 Implementation process and enforcement
- 3.5 Consultation period and status requests
- Appendix 1 – Contact information
- Reference
1 Introduction
Opioids are regulated under the Food and Drugs Act and the Controlled Drugs and Substances Act. A majority of the marketed prescription opioid products are used to treat pain, but some are also used to control moderate to severe cough, to control diarrhea, as anesthetics, and to treat opioid use disorder.
Although opioids offer effective pain relief for many patients, they pose potential harms, including their potential for causing opioid use disorder. To increase patient safety, Health Canada has identified a need for increased on-market regulatory oversight of prescription opioids to help mitigate their risks, address their uncertainties, and better inform patients of their potential harms.
In response to the growing public health crisis due to opioids, the Minister of Health announced the Federal Action on Opioids on June 17, 2016. One of the commitments made under the Federal Action on Opioids was mandatory Risk Management Plans (RMP) for prescription opioid-containing products. In November 2016, a Scientific Advisory Panel on Opioids (SAP-Opioids)Footnote 2 was convened by Health Canada to provide recommendations on the monitoring and managing of the risks related to opioids. Recommendations from this panel were taken into consideration for the development of this guidance document.
1.1 Policy objective
Under subsection C.01.014.21(1) of the Food and Drug Regulations, the Minister of Health has the authority to add or amend terms and conditions to the drug identification number (DIN) authorization of a prescription drug in Part B of the List of OpioidsFootnote 3. Health Canada intends to use the Minister's authority to impose or amend terms and conditions on a drug identification number (DIN) of a prescription drug in Part B of the List of (The List) OpioidsFootnote 3 to require Market Authorization Holders (MAHs) to submit a Canadian Specific Opioid targeted Risk Management Plan (CSO-tRMP) and to undertake the activities set out within it.
The issuance of a market authorization indicates that the manufacturer has provided sufficient data to demonstrate that the drug's benefits outweigh the potential harms at the time of market authorization. The application of terms and conditions and the implementation of CSO-tRMP provide greater on-market monitoring of the opioid drug and may be used to address outstanding data gaps or to further characterize uncertainties to ensure that the benefits of the product continue to outweigh its harms when used in the exposed population as a whole. In particular, the policy objectives are to:
- Standardize and strengthen the rigor of the post-market surveillance of prescription opioids, allowing better quantification and characterization of the risks associated with opioid-related harms in Canadian patients.
- Put in place targeted risk minimization activities to prevent or decrease prescription opioid-related harms in Canada.
1.2 Policy statements
All other Health Canada guidelines and policies designed to assist the preparation and filing of drug submissions as well as those pertaining to Risk Management Plans, remain applicable.
This guidance document should be used in conjunction with the following Health Canada guidance documents:
- Guidance Document – Submission of Risk Management Plans and Follow-up CommitmentsFootnote 4
- Clinical Assessment of Abuse Liability for Drugs with Central Nervous System ActivityFootnote 5
- The Distinction between Advertising and Other ActivitiesFootnote 6
Unlike RMPs currently in place for some products, CSO-tRMPs submitted under this policy are tailored to prescription opioid products.
A CSO-tRMP is not expected to cover all risks related to a prescription opioid; it focuses on opioid-related harms in Canadian patients. Other risks associated with a prescription opioid are expected to be included in a broad RMP that will be requested for some products as per Health Canada's Guidance Document – Submission of Risk Management Plans and Follow-up CommitmentsFootnote 4, which is outside the scope of this CSO-tRMP Guidance. The MAH has a choice to consolidate the contents of the two RMPs within the same document or to provide two separate RMPs.
The acceptability of the content of an individual CSO-tRMP will be considered on a case by case basis, and is based on the details and circumstances of each submission.
Any material regarding opioid use provided directly by the MAH to Health Care Professionals is expected to be submitted for review to a preclearance agency recognized by Health Canada, and found to be compliant with legislative and regulatory advertising provisions prior to dissemination (pre-cleared). This material should be clearly referenced and appended in the CSO-tRMP (Refer to section 3.2.3 of this Guidance for more details).
Health Canada, in collaboration with manufacturers, will periodically monitor the criteria set out in this guidance document for continued effectiveness of the CSO-tRMP in preventing/decreasing prescription opioid-related harms in Canada while maintaining patient access to treatment and minimizing potential burden on the health care system.
1.3 Scope and application
This document provides the market authorization holder with guidance on:
- how to comply with terms and conditions
- the recommended content of CSO-tRMPs
- the process and timelines by which terms and conditions will be imposed on a prescription opioid with DIN authorization.
The requirements to submit CSO-tRMPs are limited to prescription opioids included on Part B of the List, which is incorporated by reference into the RegulationsFootnote 3. The List names products by active ingredient so that authorization holders, health care practitioners, and the public will know with certainty to which products these provisions apply. The List is administered by Health Canada and published on the Government of Canada websiteFootnote 3. Stakeholders will be notified of any subsequent changes to the List and provided with an opportunity to comment, as per the Health Canada's policy on modifications to documents incorporated by reference.
Products currently not on the List – such as non-prescription opioids, opioid products used only in hospitals and new active pharmaceutical ingredients with a chemical structure or pharmacological action similar to opioids – may be reviewed for inclusion on Part B of the ListFootnote 3 as needed. Stakeholders will be notified of any subsequent changes to the List and provided with an opportunity to comment, as per Health Canada's policy on modification to documents incorporated by reference.
CSO-tRMPs are not required for opioid products (DINs) that are to be administered only under the supervision of a health care professional. Settings in which administration under the supervision of a health care professional would usually occur include: hospital wards for admitted patients, nursing homes, outpatient clinics, emergency departments and outpatient surgery settings.
2 Terminology and acronyms
- Opioid-related harms:
- For the purpose of this guidance, "opioid-related harms" is defined as any adverse event related to opioid use disorder resulting from therapeutic or non-therapeutic use of a drug product.
- Active surveillance:
- An active surveillance system, as defined by the World Health Organization, is the collection of case safety information as a continuous pre-organized process. Active surveillance can be: (1) drug based: identifying adverse events in patients taking certain products; (2) setting based: identifying adverse events in certain health care settings where they are likely to present for treatment; or (3) event based: identifying adverse events that are likely to be associated with medical productsFootnote 7, Footnote 8. An example of active surveillance is the follow-up of patients treated with a particular medicinal product through a risk management system. Patients who fill a prescription for this product may be asked to complete a brief survey and give permission to be contacted at a later stageFootnote 8.
- Case-control study:
- An epidemiological study design whereby cases of disease (or events) are identified and patients from the source population that gave rise to the cases but who do not have the disease or event of interest at the time of selection are then selected as controls. The odds of exposure are then compared between the two groupsFootnote 8.
- Cohort study:
- An epidemiological study design whereby a population-at-risk for an event of interest is followed over time for the occurrence of that event. Information on exposure status is known throughout the follow-up period for each study participant. Since the population exposure during follow-up is known, incidence rates can be calculatedFootnote 8.
- Misuse:
- For the purpose of this guidance, the term "misuse" refers to the intentional or unintentional use of a drug product in an inappropriate way (i.e. outside of the prescribed indication or dosing) for a therapeutic purposeFootnote 9.
- Non-interventional study:
- A study fulfilling cumulatively the following requirements: 1) the medicinal product is prescribed according to clinical practice; 2) the assignment of the patient to a particular therapeutic strategy is not decided in advance by a trial protocol but falls within current practice and the prescription of the medicine is clearly separated from the decision to include the patient in the study; 3) and no additional diagnostic or monitoring procedures are applied to the patients and epidemiological methods are used for the analysis of collected dataFootnote 10.
- Post-Authorization Safety Study (PASS):
- a study relating to an authorized medicinal product conducted with the aim of identifying, characterizing or quantifying a safety hazard, confirming the safety profile of the medicinal product, or of measuring the effectiveness of risk management measures. A PASS may be interventional or non-interventionalFootnote 8.
- Prescription monitoring program [PMP]:
- a proactive and comprehensive approach to collect and analyze information about prescription and dispensing of certain monitored drugsFootnote 11.
- Passive surveillance:
- A surveillance method that relies on health care providers and consumers to take the initiative in communicating suspicions of adverse drug reactions that may have occurred in individual patients to a spontaneous reporting systemFootnote 8.
- Terms and conditions:
- Terms and conditions on a DIN authorization are legally enforceable requirements that the MAH must comply with under s. 21.7 of the Food and Drugs Act. The Minister of Health has authority under section C.01.014.21of the Food and Drug Regulations to impose and amend terms and conditions for a Class B opioid.
- Psychological dependence:
- Refers to the experience of impaired control over drinking or drug useFootnote 12.
- Physical dependence:
- Refers to tolerance and withdrawal symptomsFootnote 11.
- Withdrawal syndrome:
- A group of symptoms of variable clustering and degree of severity which occur on cessation or reduction of use of a psychoactive substance that has been taken repeatedly, usually for a prolonged period and/ or in high doses. The syndrome may be accompanied by signs of physiological disturbance. A withdrawal syndrome is one of the indicators of a dependence syndrome. It is also the defining characteristic of the narrower psycho-pharmacological meaning of dependence. Opioid withdrawal is accompanied by rhinorrhoea (running nose), lacrimation (excessive tear formation), aching muscles, chills, gooseflesh, and, after 24-48 hours, muscle and abdominal cramps. Drug-seeking behaviour is prominent and continues after the physical symptoms have abatedFootnote 13.
- High-risk population:
- for the purpose of this guidance, high-risk population refers to patients who exhibit risk factors associated with the risk of opioid overdose or addiction (e.g., long-term opioid use (> 3 months) and substance-use disorder)Footnote 14.
3 Guidance for implementation
3.1 General considerations
- The format should follow the guidelines set out in the Health Canada Guidance Document - Submission of Risk Management Plans and Follow-up CommitmentsFootnote 4. However, it should be clear that in the case of CSO-tRMP, the submission of a Canadian addendum to an already prepared European Union RMP is not acceptable. The manufacturers should submit Canadian specific safety data, pharmacovigilance and risk minimization activities within a comprehensive CSO-tRMP.
- The scope of a single CSO-tRMP could include a single product or multiple products with the same active opioid ingredient.
- MAHs are encouraged to work together, where possible, to develop a common approach for pharmacovigilance studies and risk minimization measures for similar products (i.e., those sharing the same active opioid ingredient). This would support consistency among the various pharmacovigilance activities and minimize duplication.
- Delegated or shared initiatives (e.g., post-marketing studies) with other MAHs should be detailed in the CSO-tRMP (if applicable). An up-to-date list of collaborators and the responsibilities of each delegate should also be included (if applicable).
- Health Canada recognizes that alternative activities and/or data sources not outlined in this guidance document may need to be considered. While submission of CSO-tRMPs will be required through the terms and conditions, a careful consideration will be given to the complexity and feasibility of pharmacovigilance and risk minimization activities prior to amending the terms and conditions to require those studies.
- In situations where specific pharmacovigilance or risk minimizations measures are not applicable or not feasible for a specific product, a rationale should be provided to Health Canada for discussion during the consultation period prior to submission of the CSO-tRMP.
3.2 Mandatory Canadian Specific Opioid targeted Risk Management Plan sections
The Minister of Health, under the authority of section C.01.014.21 of the Food and Drug Regulations, imposes the terms and conditions on all prescription opioid products. The following are the terms and conditions relevant to the content of the CSO-tRMP:
- The CSO-tRMP must contain the following information:
- a detailed Safety Specification section that:
- describes, quantitatively and qualitatively, the occurrence of opioid-related harms associated with the use of DRUGNAME in Canada; and
- provides detailed information on the evidence gaps and uncertainties related to opioid-related harms that are associated with the use of DRUGNAME in Canada;
- a detailed Pharmacovigilance Plan section that:
- describes routine (passive surveillance) and additional (active surveillance) activities in place or that are to be put in place to monitor and characterize opioid-related harms and address uncertainties associated with the use of DRUGNAME in Canada; and
- provides the timelines for the conducting of those activities;
- a detailed Risk Minimization Plan section that:
- describes risk minimization activities (beyond the approved product label) that are designed to minimize or prevent the occurrence of opioid–related harms in Canadians using DRUGNAME;
- provides the timelines for the conducting of those activities;
- provides all materials that are - or will be - communicated and/or disseminated by the MAH to healthcare professionals with respect to DRUGNAME;
- demonstrates that the materials referred to in (iii) have been submitted to an independent advertising preclearance agency (APA) recognized by Health Canada for a determination as to whether the materials are promotional, and for those that are promotional, a determination as to whether the materials comply with the terms of the marketing authorization. The plan must include:
- the date the materials were submitted to the APA;
- the name of the APA;
- the date of the review; and
- the outcome of the APA's determinations referred to in (iii), above.
- a detailed Evaluation of the Risk Minimization section that:
- describes the activities that the MAH will carry out to assess the effectiveness of the risk minimization activities on health outcomes in Canadians who are using DRUGNAME; and
- provides the timelines for the conducting of those activities.
- a detailed Safety Specification section that:
- You must revise and re-submit to Health Canada the CSO-tRMP annually to:
- update the Safety Specification section by adding new evidence (quantitative or qualitative) related to opioid-related harms or uncertainties that is generated through the risk-monitoring/characterising activities conducted in Canada or internationally (e.g. pharmacoepidemiological studies or clinical trials undertaken to investigate DRUGNAME); and
- update the Pharmacovigilance and Risk Minimization sections to provide the status of the ongoing Pharmacovigilance and Risk Minimization activities and describe any changes to these sections.
The following sections provide Health Canada's recommendation on how to comply with the above noted terms and conditions.
3.2.1 Safety specification section
As per the terms and conditions imposed on the DIN for an opioid product, the MAH will be expected to provide a detailed Safety Specification section that:
- describes, quantitatively and qualitatively the occurrence of opioid-related harms associated with the use of DRUGNAME in Canada; and
- provides detailed information on the evidence gaps and uncertainties related to opioid-related harms that are associated with the use of DRUGNAME in Canada;
In order to improve comparability between the risks associated with different products and to inform decision making, the recommended elements of the Safety Specification section include, but are not limited to, the following:
- Analysis of opioid related harms based on both crude numbers and reporting/incidence rates of events in the context of worldwide and Canadian post-market exposure (if applicable).
- The Canadian trends of opioid-related harms relevant to the product should be provided (over time, age/sex, region, indication, concomitant use of other controlled substances, etc.). When more than one formulation is covered in the same CSO-tRMP (e.g., ER/IR), a break down according to the formulation should be provided whenever possible regarding opioid-related harms collected from active surveillance (e.g., completed post-authorisation safety study (PASS).
3.2.2 Pharmacovigilance plan section
As per the terms and conditions imposed on the DIN for an opioid product, the MAH will be expected to provide a detailed Pharmacovigilance Plan section that:
- describes routine (passive surveillance) and additional (active surveillance) activities put in place or that are to be put in place to monitor and characterize opioid-related harms and uncertainties associated with the use of DRUGNAME in Canada, and
- provides the timelines for the conducting of those activities;
For each type of surveillance, a search strategy (including the data lock point) or study protocol should be clearly documented. The most recent Adverse Reaction Terms (MedDRA Preferred Terms) should be used for spontaneous data extraction.
Passive adverse event surveillance alone is considered insufficient to fulfil the above requirements, because of well recognised problems related to underreporting and poor quality of the reports. Therefore one or more of the following pharmacovigilance active surveillance activities may be needed to fill the current knowledge gaps:
- A well-designed prospective non-interventional post-authorization safety study (PASS) estimating the incidence of opioid–related harms in the indicated patient population;
- A retrospective non-interventional PASS in high-risk populations; and/or
- A surveillance system capable of monitoring (longitudinal or cross-sectional), opioid-related harms discussed associated outcomes, among diverse populations in Canada.
Milestones:
- Final Protocol Submissions are expected to be included in the initial CSO-tRMP.
- Progress reports submitted in updated CSO-tRMPs which according to the terms and conditions must be provided to Health Canada on an annual basis.
Additional considerations
Study protocols should be prepared prior to initiating studies, with input from qualified methodologists and statisticians. The choice of risk factors should be made a priori, and with the anticipated direction of effect to reduce the risk of spurious findings. Further, factors that have already been established by the literature should be included. Further, significant associations for risk factors should be presented, when possible, as both relative and absolute effects.
The manufacturer's role should be restricted to sponsoring, reviewing of the protocol, and provision of non-binding feedback. Study design, implementation, or interpretation should not be unduly influenced by parties that stand to benefit financially from the results. Post-marketing studies should ideally be conducted by not-for-profit organizations.
When developing a CSO-tRMP, MAHs should seek early dialogue with Health Canada's Marketed Health Products Directorate regarding the applicability, feasibility, and design of the proposed studies. Feasibility of particular measures will be discussed and considered during the consultation periods.
3.2.2.1 Prospective Post-Authorization Safety Study
For a prospective PASS study, the recommended elements to be provided in the CSO-tRMP include, but are not limited to, the following:
- Design: Cohort study in the indicated patient population
- Primary objective: To estimate the incidence of opioid-related harms in Canada. The adverse event data should be stratified by intent (e.g., unintentional vs. intentional) wherever possible.
- Secondary objectives: To characterize and quantify the prevalence of risk factors (listed below) in the patient population and to evaluate their effects on the risk of opioid-related harms. Confounders should be identified.
- Patient demographics (e.g., age, sex, ethnicity);
- Active ingredient/formulation/route of administration;
- Dosage;
- Duration of opioid use;
- Prescriber specialty;
- Indication;
- Concomitant psychotropic medications;
- Personal or family history of substance use disorder;
- Personal or family history of psychiatric illness;
- Other relevant factors (e.g., medical history, geographical region, socio- economic factors).
- Data sources: The study (studies) should produce estimates of opioid-related harms that are nationally representative, or are based on data from multiple geographic regions that can reasonably be generalized to the national level. In the absence of national data, smaller or regional studies may be informative, but must be accompanied by a clear explanation of their representativeness and generalizability for appropriate interpretation.
- The study or studies must assess overall active ingredient and formulation-specific levels of opioid-related harms.
- Exposure to opioids should be ascertained using validated tools (e.g. drug screening tests) where possible and should not solely rely on patients' self-reporting.
- Power considerations: The projected study size, the precision sought for study estimates and any calculations of the sample size that can minimally detect a risk with a specified statistical precision should be pre-specified in the CSO-tRMP.
- The necessary duration of the study may depend on a variety of factors, including drug utilization and market share and changes in the market.
- Milestones:
- Final Protocol Submission: To be included in the CSO-tRMP.
- Interim Report Submission: At midpoint of the study (e.g., once 50% of the target exposures have been achieved).
- Final Report Submission: Within 48 months after terms and conditions are imposed on DINs.
3.2.2.2 Retrospective Post-Authorization Safety Study
For a Retrospective PASS study, the recommended elements to be provided in the CSO-tRMP include, but are not limited to, the following:
- Design: Observational study (e.g., cohort, case-control, etc.)
- Primary objective: To estimate the risk of opioid-related harms associated with the use of prescription opioids using Canadian databases. Data should be stratified by intent whenever possible.
- Secondary objectives: To quantify the prevalence of important variables/known risk factors in the patient population and to evaluate their effects on the risk of opioid-related harms. Confounders should be identified. Potential variables/risk factors include:
- patient demographics (e.g., age, sex, ethnicity);
- active ingredient/formulation/route of administration;
- dosage;
- duration of opioid use;
- prescriber specialty;
- indication;
- concomitant psychotropic medications;
- personal or family history of substance use disorder;
- personal or family history of psychiatric illness;
- other relevant factors (e.g., medical history, geographical region, socio-economic factors).
- Data sources: Canadian health information databases covering relevant patient populations.
- Because of the retrospective nature of the study, data sources may not always be able to distinguish between brand-name and generic products or among multiple generic products. Therefore, when both brand-name and generic versions of a product are marketed, all should be included in the study.
- Exposure and outcome measures that include self-reported opioid-related harms should be validated prior to the initiation of the study.
- Milestones:
- Final Protocol Submission: included in the CSO-tRMP.
- Study Completion: 24 months after terms and conditions are imposed on DINs.
- Final Report Submission: within 30 months after terms and conditions are imposed on DINs.
3.2.2.3 Longitudinal or cross-sectional multi-surveillance system
The aim of this surveillance system is to monitor diverse intended and/or high risk populations and capture numerator data on different facets of opioid-related harms in Canada.
For a longitudinal or cross-sectional multi-surveillance system, the recommended elements to be provided in the CSO-tRMP include, but are not limited to, the following:
An ongoing collection of data in a systematic fashion from sources such as:
- MAH-owned adverse events databases
- prescription Monitoring Programs
- hospital admissions
- pharmacies
- poison centers
- opioid treatment programs in Canada
- internet monitoring of drug use disorder and substance use disorder
- patient and/or general population surveys
Milestones:
- Final Protocol Submission: should be included in the CSO-tRMP.
- Annual Report Submission: starting 12 months after terms and conditions are imposed on DINs.
3.2.3 Risk minimization plan section
As per the terms and conditions imposed on the DIN for an opioid product, the MAH will be expected to provide a detailed Risk Minimization Plan section that:
- describes risk minimization activities (beyond the approved product label) that are designed to minimize or prevent the occurrence of opioid–related harms in Canadians using DRUGNAME;
- provides the timelines for the conducting of those activities;
- provides all materials that are - or will be - communicated and/or disseminated by the MAH to healthcare professionals with respect to DRUGNAME;
- demonstrates that the materials referred to in (iii) have been submitted to an independent advertising preclearance agency (APA) recognized by Health Canada for a determination as to whether the materials are promotional, and for those that are promotional, a determination as to whether the materials comply with the terms of the marketing authorization. The plan must include:
- the date the materials were submitted to the APA;
- the name of the APA;
- the date of the review; and
- the outcome of the APA's determinations referred to in (iii), above.
For routine risk minimization measures, refer to the Guidance Document – Submission of Risk Management Plans and Follow-up CommitmentsFootnote 4 for further guidance.
For additional risk minimization measures, the following discussion focuses on educational tools for healthcare professionals and patients as acceptable standard for all opioids that are subject to market authorisation terms and conditions. However, it is recognized that the risk minimization approach may evolve throughout the therapeutic product life cycle and a variety of tools could be implemented in the future. Moreover, this approach may be required for some products depending on their unique benefit/risk profile. Such additional measures could include for example implementation of a restricted distribution and prescribing, or establishment of performance-linked access programs/registries as defined by CIOMS IXFootnote 15.
When making decisions about the need for additional risk minimization measures, Health Canada will consider both the added positive effects of the product's benefit-risk balance as well as unintended consequences on the existing workflow and standard of care in various health care settings.
3.2.3.1 Educational program
Health care professional education is considered an important element to reduce or prevent opioid-related harms while preserving the highest standard of care for indicated patient populations. Therefore, the MAH must promote the use of educational programs developed by accredited Continuous Education (CE) providers.
A preferable educational curriculum incorporates interactive teaching methods along with experiential and didactic components and provides information on the following key components of opioid use:
- Evaluation of patients prior to deciding to initiate opioid therapy including:
- Comprehensive assessment, including pain assessment, medical history, psychiatric status, substance use history, and concomitant medications
- Addiction risk screening
- Evaluation of efficacy for the indication
- Evaluation of drug-seeking behaviour
- Initiation of therapy, conducting an opioid trial with the patient, modifying dosing, and discontinuing use of opioids
- The importance of including indication in each opioid prescription
- Appropriate selection of opioid and initial dosing
- Dose titration approach
- Early monitoring for effectiveness and risk of misuse, reassessment
- Rotating and tapering opioids
- Discontinuing use of opioids
- Explanation of potential benefits and risks of opioid therapy and obtain informed consent from the patient
- Monitoring long-term opioid therapy
- Treating specific populations (i.e., elderly, adolescents, pregnant women, co-morbid psychiatric illness)
- Recognising and managing opioid use disorder
- Product specific safety information, aligned with the current Canadian Product Monograph
- Promoting the use of Patient-Prescriber Agreement Form to facilitate conversation between the patient and prescriber and raise patient awareness about opioid-related harms.
The MAH should clearly identify providers of Continuous Education (CE) to health care professionals and targeted audience (e.g., nurses, physicians).
3.2.3.2 Review and Preclearance of Materials
Any material regarding opioid use provided directly by the MAH to Health Care Professionals is expected to be submitted for review to an advertising preclearance agency (APA) recognized by Health Canada. The APA will first determine whether the materials are promotional or not (i.e., the APA will provide an advisory opinion). For materials deemed promotional, the APA will determine whether the materials comply with legislative and regulatory advertising provisions prior to dissemination (pre-cleared). This review will ensure that materials to be disseminated are evidence-based, balanced and/or compliant with Health Canada's advertising regulatory framework. This material should be clearly referenced and appended in the CSO-tRMP. Health Canada is the national regulatory authority for health product advertising. In that capacity, Health Canada provides guidance to APAs and will review and render its decisions on any case where industry would disagree with the APAs advice. Both the APA and industry can refer or appeal to Health Canada.
3.2.3.3 Warning Sticker and Patient Information Handout
Additionally, Health Canada has developed a warning sticker and patient information handout with standardized, easy-to-understand information on safe use and associated risks of opioids. The sticker and handout were developed with input from an expert advisory panel, Health Canada experts, and patient user-testing, among other sources of input. The application of a warning sticker on the container of prescription opioids and the distribution of the handout will happen when the drug is dispensed to the patient, most commonly at a pharmacy. The MAH's role in this part of the educational program will be limited to the assessment of the effectiveness as described in the following section.
3.2.4 Evaluation of the effectiveness of the risk minimization activities
As per the terms and conditions imposed on the DIN for an opioid product, the MAH will be expected to provide a detailed section on Evaluation of the Effectiveness of Risk Minimization activities that:
- describes the activities that the manufacturer will carry out to assess the effectiveness of the risk minimization activities on health outcomes in Canadians who are using DRUGNAME, and
- provides the timelines for the conducting of those activities.
Overall, the strategies to assess the effectiveness of the risk minimization aim to determine:
- Whether the risk minimization measure achieve the desired level of risk management. Any objective (quantifiable) measures that aim to determine the success of the risk management are considered as outcome indicators.
- Whether the risk minimization has been successfully implemented. Any objective (quantifiable) measures that aim to evaluate the success of the implementation of the risk minimization measure are considered as process indicators.
Since the ultimate goal of a risk minimization activity is to improve the safety (i.e., to minimize or prevent harms), for the purpose of fulfilling the terms and conditions, the MAH must provide outcome indicators.
Acceptable examples of outcome indicators include:
- Frequencies or rates of opioid-related harms (pre- and post-implementation of the risk minimization);
- Any metrics related to changes in severity of the health outcomes (i.e., hospitalizations, or deaths) related to opioid-related harms (pre- and post-implementation of the risk minimization);
The following process indicators metrics may be used to assess performance of the educational programs:
- Number and proportion of health care professionals that have completed the education module;
- Physician and/or patient risk awareness metrics pre- and post‑implementation of the educational programs);
- Any metrics related to change in prescribing patterns such as the number of patients who have been prescribed opioids according to the recommended dosing/frequency schedule (pre- and post‑implementation of the educational programs);
- Measures of the distribution and/or receipt of patient educational material
The evaluation of the effectiveness of risk minimization is a rapidly evolving area with no universally accepted standards. Generally, the following tools are used:
- Surveillance studies of the key safety outcomes
- Drug Utilisation Studies
- Prescriber/patient surveys
Possible challenges that the MAH should discuss within CSO-tRMP when interpreting the data from evaluation of the effectiveness of risk minimization measures:
- As more than one risk minimization measure may be implemented at the same time as a result of numerous federal, provincial, local or institutional efforts, it may be difficult to isolate the impact of one particular risk minimization measure on health outcomes at the population level.
- Sufficient time may need to be determined to observe the impact of risk minimization on overall health outcomes. For instance, reducing the number of new prescriptions may not have an immediate impact on current opioid-related harms.
- How the desirable changes in process indicators may impact (positively or negatively) interpretation of outcome indicators. For instance, following the implementation of educational programs, a higher proportion of patients may be exposed to safer opioid dosing schedule (a process indicator). This may decrease the rate of hospitalization or poison center calls (outcome indicators). On the contrary, the implementation of educational programs could also lead to earlier recognition of problematic behavior (a process indicator) leading to augmentation of the hospitalization and poison center call rates.
3.3 Canadian Specific Opioid targeted Risk Management Plan submission process and timeline
The following diagram outlines the process and timeline for submission of the CSO-tRMP.
Process for CSO-tRMP
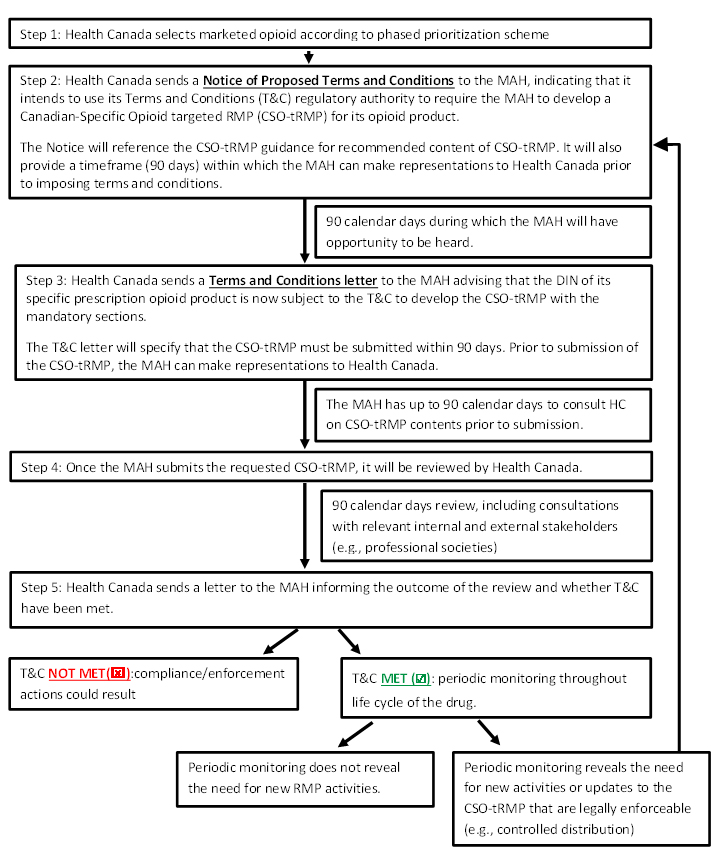
Process for CSO-tRMP - Text equivalent
Step 1: Health Canada selects marketed opioid according to phased prioritization scheme
Step 2: Health Canada sends a Notice of Proposed Terms and Conditions to the MAH, indicating that it intends to use its Terms and Conditions (T&C) regulatory authority to require the MAH to develop a Canadian-Specific Opioid targeted RMP (CSO-tRMP) for its opioid product.
The Notice will reference the CSO-tRMP guidance for recommended content of CSO-tRMP. It will also provide a timeframe (90 days) within which the MAH can make representations to Health Canada prior to imposing terms and conditions.
- 90 calendar days during which the MAH will have opportunity to be heard.
Step 3: Health Canada sends a Terms and Conditions letter to the MAH advising that the DIN of its specific prescription opioid product is now subject to the T&C to develop the CSO-tRMP with the mandatory sections.
The T&C letter will specify that the CSO-tRMP must be submitted within 90 days. Prior to submission of the CSO-tRMP, the MAH can make representations to HC.
- The MAH has up to 90 calendar days to consult HC on CSO-tRMP contents prior to submission.
Step 4: Once the MAH submits the requested CSO-tRMP, it will be reviewed by Health Canada.
- 90 calendar days review, including consultations with relevant internal and external stakeholders (e.g., professional societies)
Step 5: Health Canada sends a letter to the MAH informing the outcome of the review and whether T&C have been met.
- T&C NOT MET: compliance/enforcement actions could result, end this process; or
- T&C MET: periodic monitoring throughout life cycle of the drug.
- Periodic monitoring does not reveal the need for new RMP activities, Process ends; or
- Periodic monitoring reveals the need for new activities or updates to the CSO-tRMP that are legally enforceable (e.g., controlled distribution), Return to step 2
Health Canada will inform the MAH in writing of its intention to impose terms and conditions on the DIN of an opioid by way of a Notice of Proposed Terms and Conditions (Notice). The Notice will refer to this guidance document and provide submission timelines. Upon issuance of the Notice, a 90-calendar day consultation period will begin (first consultation period), during which time the MAH will have the opportunity to be heard with respect to the activities that would be required to be included as part of the CSO-tRMP.
At the end of the first consultation period, Health Canada will issue a Terms and Conditions Letter (TCL) to the MAH advising them that the product's DIN authorization is now subject to specific Terms and Conditions (i.e., to develop a CSO-tRMP) as per the regulatory requirements set out in C.01.014.21(2). Upon issuance of the TCL, another consultation period of up to 90 calendar days will begin (second consultation period), during which time the MAH will have another opportunity to be heard prior to submission of the CSO-tRMP at the end of the second consultation period.
As directed by the TCL, the MAH must prepare a CSO-tRMP and submit it to the Marketed Health Products Directorate (MHPD) of Health Canada within 90 calendar days of the date of the TCL. Upon receipt of the CSO-tRMP, the MHPD will start the review process. Once the review is completed, a CSO-tRMP Review Letter will be sent to the MAH communicating its findings to the MAH. This would typically occur within 90 calendar days of accepting the CSO-tRMP for review.
As per the TCL, the MAH is required to provide an updated CSO-tRMP to Health Canada on an annual basis. Health Canada will coordinate the review of the submitted CSO-tRMP with the review of the annual reports required under section C.01.018 of the Food and Drug Regulations.
Health Canada will monitor the MAH's compliance with the terms and conditions via a periodic assessment of submitted materials (e.g., updates to CSO-tRMPs and study reports). An amendment to the CSO-tRMP will be required by Health Canada whenever it is considered that the pharmacovigilance or risk management activities should be modified as a result of new information that may lead to a significant change to the benefits, harms or uncertainties of a drug in the Canadian context or when study results impact the overall safety profile of the drug (e.g., further characterization of a safety risk that will require modifications to various parts of the CSO-tRMP).
Health Canada would issue a new Notice leading to a TCL any time the department determines that a CSO-tRMP amendment or new activity needs to be imposed as terms and conditions on the DIN authorization of the prescription opioid.
In cases where a MAH wants to make a change to an approved CSO-tRMP, the MAH should consult with Health Canada for guidance on the type of change (e.g., amendment or notification) and supporting data requirements.
Note to sponsors who are planning to submit a New Drug Submission for an opioid product:
CSO-tRMPs:
The terms and conditions requiring a submission of a CSO-tRMP apply to products that are assigned a DIN. Therefore, no CSO-tRMP is required at the time of New Drug Submission in Canada.
However, as a pro-active measure, sponsors seeking drug approval of their opioid-containing product are encouraged to bring any potential questions related to the CSO-tRMP content to pre-submission meetings with Health Canada. Representatives from the MHPD who are present at the pre-submission meetings will provide appropriate guidance outlining Health Canada's expectations with respect to the appropriate content and format of the CSO-tRMP.
RMPs that cover non-opioid-related harms:
For an RMP that covers risks beyond the scope of the CSO-tRMP, refer to Health Canada's Guidance Document – Submission of Risk Management Plans and Follow-up CommitmentsFootnote 4 for the relevant process and timelines.
3.4 Implementation process and enforcement
The implementation of this procedure is primarily the responsibility of the review bureaus within the Marketed Health Products Directorate.
Implementation will follow a phased approach beginning with the highest priority opioids as determined by the Department, informed by feedback from the Scientific Advisory Panel on Opioids. The panel agreed that all opioids carry the risk of harm, but suggested that the Department prioritize those opioids that are currently implicated in the majority of opioid-related harms in Canada. These opioids are designated "high priority" opioids and are defined as any pharmaceuticals that contain as active ingredient a full µ-opioid receptor agonist with a morphine milligram equivalent (MME) conversion factor equal to or greater than 1 by at least one route of administration (e.g., fentanyl, hydrocodone, hydromorphone, methadone, morphine, normethadone, opium, oxycodone, and oxymorphone)Footnote 2.
Opioids that do not fulfil this criterion (e.g., tramadol, codeine) still present the risk of opioid-related harms. Therefore, CSO-tRMPs will be requested for the remaining prescription opioids at a later stage of the implementation process.
All terms and conditions will be enforceable under section 21.7 of the Food and Drugs Act. This means that if an authorization holder were not to respond to requests from the Department to comply with the terms and conditions imposed (i.e., failure to submit a CSO-tRMP within the specified time outlined in the TCL, or failure to include the mandatory sections) the Department could consider pursuing compliance and enforcement measures, as per its Compliance and Enforcement PolicyFootnote 16.
Other existing compliance and enforcement related regulatory provisions, such as stop-sale, label change, or cancellation of authorization, will continue to apply to opioids just as they apply to all marketed drugs.
3.5 Consultation period and status requests
The total duration of consultation period is up to 180 calendar days, including 90 days prior to and up to 90 days post TCL issuance.
During this consultation period the manufacturer may file representations to seek further clarifications or to justify the reasons why they believe certain elements proposed in this Guidance may not be feasible or are not warranted. The manufacturer's justifications should be accompanied by well-grounded evidence. Health Canada will give advice based on the best current scientific knowledge and expertise in that area and based on the documentation provided by the manufacturer. Health Canada may seek advice from experts in the relevant field (in cases where that expertise is not present in-house) to determine the most appropriate course of action. For example scientific expert advice may be sought to review the issues raised regarding the proposed study designs.
The program area will conduct a review to decide next steps based on the merit of the MAH's representations and information received from experts.
Manufacturers with questions related to their CSO-tRMPs should communicate with the regulatory project manager from MHPD; refer to Appendix 1 for specific contact information.
Appendix 1 – Contact information
MAHs are requested to submit CSO-tRMPs and follow-up commitments to:
Marketed Pharmaceuticals and Medical Devices Bureau
Regulatory Project Management Section
Health Canada
Address Locator 1912A
200 Eglantine Driveway
Ottawa, Ontario
K1A 0K9
E-mail: hc.mpmdb.rpm-bppmmc.gpr.sc@hc-sc.gc.ca
Facsimile: (613) 960-9754
Reference
- Footnote 1
Health Canada. Regulations Amending the Food and Drug Regulations (Opioids).
- Footnote 2
Health Canada. Scientific Advisory Panel on Opioids (SAP-Opioids).
- Footnote 3
Health Canada. Opioids that are subject to market authorization terms and conditions.
- Footnote 4
Health Canada. Submitting risk management plans guidance document.
- Footnote 5
Health Canada. Clinical Assessment of Abuse Liability for Drugs with Central Nervous System Activity.
- Footnote 6
Health Canada. The Distinction between Advertising and Other Activities.
- Footnote 7
CIOMS. Practical Aspects of Signal Detection in Pharmacovigilance: Report of CIOMS Working Group VIII.
- Footnote 8
European Medicines Agency. Guideline on good pharmacovigilance practices (GVP) Module VIII - Post-authorisation safety studies (Rev 3).
- Footnote 9
Smith SM, Dart RC, Katz NP et al. Classification and definition of misuse, abuse, and related events in clinical trials: ACTTION systematic review and recommendations. Pain 2013;154(11):2287-2296.
- Footnote 10
European Medicines Agency. Guideline on good pharmacovigilance practices (GVP): Annex I - Definitions (Rev 3).
- Footnote 11
Furlan AD, MacDougall P, Pellerin D et al. Overview of four prescription monitoring/review programs in Canada. Pain Res Manag 2014;19(2):102-106.
- Footnote 12
World Health Organization. Dependence syndrome, date cited, April 19, 2018.
- Footnote 13
World Health Organization. Withdrawal state, date cited, April 19,2018.
- Footnote 14
Volkow ND, McLellan AT. Opioid Abuse in Chronic Pain GÇö Misconceptions and Mitigation Strategies. N Engl J Med 2016;374(13):1253-1263.
- Footnote 15
CIOMS. Practical Approaches to Risk Minimisation for Medicinal Products: Report of CIOMS Working Group IX.
- Footnote 16