
Page 4: Guidance on Chloral Hydrate in Drinking Water
Chloral hydrate (2,2,2-trichloro-1,1-ethanediol) has a relative molecular mass of 165.4, a crystalline appearance, an aromatic and slightly acrid odour, and a slightly bitter taste (Reynolds and Prasad, 1982; Budavari, 1996). It is synthesized by the chlorination of ethanol (Reynolds and Prasad, 1982; Budavari, 1996; Verschueren, 2001).
Chloral hydrate has a melting point of 57°C, a boiling point of 96°C (Hansch et al., 1995), and a density/specific gravity of 1.91 g/cm³ at 20°C. It has an octanol/water partition coefficient (log Kow) of 0.99; therefore, bioconcentration is not an important factor. At 25°C, chloral hydrate has a vapour pressure of 2 kPa (Reynolds and Prasad, 1982; Hansch et al., 1995) and a water solubility of 9.3 × 106 mg/L (McEvoy, 1999). It slowly volatilizes when exposed to ambient air, and it decomposes when exposed to light (McEvoy, 1999).
Chloral hydrate is used as a sedative and hypnotic in human and veterinary medicine. It is also used in the manufacture of DDT (Budavari, 1996) and dichloroacetic acid (DCA) (Kirk-Othmer, 1991). In addition, chloral hydrate is used as an intermediate in the production of the insecticides methoxychlor, naled, trichlorfon, and dichlorvos, the herbicide trichloroacetic acid (TCA), and the hypnotic drugs chloral betaine, chloralose, and trichlorfos sodium (IARC, 1995). Chloral hydrate can be formed as a by-product of the chlorination of water containing organic precursor molecules, such as fulvic and humic acids. Chloral hydrate can also be released to the environment from wastewater treatment facilities, from the manufacture of pharmaceutical-grade chloral hydrate, and from the waste stream during the manufacture of insecticides and herbicides that use chloral hydrate as an intermediate (U.S. EPA, 2000).
Chloral hydrate can be transformed into trichloroethanol (TCOH) and TCA by the methanotrophic bacterium Methylosinus trichosporium. The transformation of chloral hydrate into chloroform occurs under abiotic conditions (pH 9.0 and 60°C) after 2.3 minutes. Formic acid is another decomposition product of chloral hydrate (Newman and Wackett, 1991).
No data are available on human exposure to chloral hydrate in air. The high water solubility and low volatility of chloral hydrate preclude significant exposure by inhalation from a water solution (U.S. EPA, 2000).
According to surveys conducted in Canada in 1995 and 1997, the mean level of chloral hydrate in drinking water ranged from 1.2 to 3.8 µg/L in winter and from 3.6 to 8.4 µg/L in summer, with a maximum level of 22.5 µg/L observed in winter from a sampling of 53 sites (Health Canada, 1995; Edsall and Charlton, 1997; Williams et al., 1997). Although slightly higher levels of chloral hydrate may be found in smaller treatment systems with limited ability to remove organic matter before adding the chlorine disinfectant, levels are still below any level of concern.
In the United States, median chloral hydrate concentrations in finished water have been reported to range from 1.7 to 2.5 µg/L, whereas maximum concentrations ranged from 22 to 46 µg/L (Krasner et al., 1989; U.S. EPA, 1992). The chloral hydrate concentration was higher in distribution systems of surface water plants (median 4.0 µg/L) than in groundwater (median 0.5 µg/L) and was generally higher than the concentration in the finished water (median 2.4 µg/L), suggesting that chloral hydrate concentrations increase across the distribution system.
No data are available on human exposure to chloral hydrate in food (IARC, 1995).
For adults, the usual hypnotic dose of chloral hydrate is 0.5-1 g, whereas the usual sedative dosage is 250 mg 3 times daily. When chloral hydrate is administered in the management of alcohol withdrawal symptoms, the usual dosage is 0.5-1 g repeated at 6-hour intervals if needed. Generally, single doses or daily dosages for adults should not exceed 2 g. For children, the hypnotic dose of chloral hydrate is 50 mg/kg bw or 1.5 g/m³, with a maximum dose of 1 g. The sedative dosage for children is 8 mg/kg bw or 250 mg/m³ 3 times daily, with a maximum dosage of 500 mg 3 times a day. As a premedication before electroencephalogram evaluation, children have been given chloral hydrate at a dose of 2-25 mg/kg bw (McEvoy, 1999).
Chloral hydrate is highly water soluble, has a log Kow of less than 10, and is known to occur in drinking water supplies as a DBP. There are no data on the levels of chloral hydrate in air, soil and food, and there is no indication that it would be present at significant levels in these environmental media. These characteristics suggest that drinking water would be the primary source of exposure to chloral hydrate for the general population; therefore, an allocation factor of 80% is used in the risk assessment. Occupational exposure during manufacturing (IARC, 1995) and exposure from pharmaceutical use of chloral hydrate may also occur.
Chloral hydrate was introduced into therapeutics more than 100 years ago and has been used as a sedative/hypnotic agent in children, adults, and animals since its introduction (Henderson et al., 1997). Insufficient data are available to determine a no-observed-adverse-effect level (NOAEL) in humans. The lowest-observed-adverse-effect level (LOAEL) is 10.7 mg/kg bw per day (assuming a body weight of 70 kg), based on the recommended dose as a sedative for an adult of 250 mg 3 times a day.
Oral administration of chloral hydrate at high doses causes gastric irritation, with nausea, vomiting, and diarrhoea as the most frequent adverse effects. Other adverse effects of chloral hydrate may include leukopenia, eosinophilia, and, rarely, ketonuria (McEvoy, 1999).
The toxic blood level and the lethal blood level for chloral hydrate were estimated to be 10 mg/100 mL and 25 mg/100 mL, respectively (Ellenhorn et al., 1997).
While a lethal oral dose of 10 g has been reported for adults, death has occurred after ingestion of 4 g, and some patients have survived ingestion of as much as 30 g (McEvoy, 1999). Ingestion of 20 g by a patient, who later became comatose, resulted in gastric perforation that was detected 4 days post-ingestion. Gastrointestinal haemorrhage followed by the development of oesophageal strictures has been observed with a dose of 18 g. Hepatic (jaundice, aminotransferase elevation) and renal (albuminuria) dysfunction may occur several days after ingestion, but is rarely serious or prolonged (Abbas et al., 1996).
A variety of adverse effects were reported in 1618 patients who had received chloral hydrate at various doses, although it was not clear if the patients had the identified clinical effects prior to being exposed to chloral hydrate or if they developed the clinical effects after being exposed to chloral hydrate. The results indicated that cirrhosis of the liver was the most common diagnosis (15%), whereas chronic obstructive respiratory tract disease (7%), carcinoma of the breast (7%), and congestive cardiac failure (7%) were also reported, although a causal association could not be determined. Other adverse reactions, including gastrointestinal symptoms (10 patients), depression of the central nervous system (20 patients), skin rash (5 patients), prolonged prothrombin time (1 patient), worsened hepatic encephalopathy (1 patient), and bradycardia (1 patient), disappeared soon after the end of chloral hydrate administration (Shapiro et al., 1969). Another review of medical records has shown central nervous system depression to be the preponderant effect following exposure to chloral hydrate in 5435 patients (Greenberg et al., 1991).
No long-term studies of chloral hydrate exposure in humans were available in the published literature.
The LD50 for chloral hydrate in mice was determined to be 1265 mg/kg bw for females and 1442 mg/kg bw for males. Rats were found to be more sensitive to chloral hydrate, with LD50s of 285 and 479 mg/kg bw for newborn pups and adults, respectively (Sanders et al., 1982).
Mice exposed to chloral hydrate at 603 mg/m³ for 6 hours by inhalation exhibited several changes in the lung, including vacuolization of Clara cells, alveolar necrosis, desquamation of the epithelium, and alveolar oedema (Odum et al., 1992).
The liver is the primary target organ of short-term exposure to chloral hydrate. In a 7-day study with 28 male Sprague-Dawley rats administered chloral hydrate in drinking water at dose levels of 5, 43, or 375 mg/kg bw per day, no NOAEL could be determined, as no histopathological changes were observed in the liver. However, other changes in the liver (e.g., increase in hepatic peroxisomal enzyme palmitoyl coenzyme A [CoA] oxidase, suppression of hepatic aldehyde dehydrogenase [ALDH] activity, decreases in liver cholesterol and liver triglyceride levels) suggested that the liver is the target organ of chloral hydrate exposure (Poon et al., 2000). In a 13-week study by the same investigators in which Sprague-Dawley rats (10 per sex per dose) were administered chloral hydrate in drinking water at 0, 0.2, 2, 20, or 200 mg/L (corresponding to dose levels of 0, 0.02, 0.19, 1.9, and 19.8 mg/kg bw per day for males and 0, 0.03, 0.24, 2.6, and 23.6 mg/kg bw per day for females), the no-observed-effect level (NOEL) was identified as 1.9 mg/kg bw per day in males and 2.6 mg/kg bw per day in females based on the decrease in ALDH activity in both sexes at the highest dose, the increase in aniline hydroxylase activity in both sexes at the highest dose, and the minimal vacuolation of the myelin sheath in males at the highest dose. The LOAEL for males in this study was 19.8 mg/kg bw per day, based on the mild vacuolation of the myelin sheath (Poon et al., 2002). (The authors stated that nervous tissue is particularly susceptible to inadequate fixation, with vacuolation being one of the most common histological artefacts.)
In a study in which CD-1 mice (48 per sex for control; 32 per sex for treatment groups) were administered chloral hydrate in drinking water for 90 days at concentrations of 70 or 700 mg/L (corresponding to dose levels of 16 and 160 mg/kg bw per day for males and 18 and 173 mg/kg bw per day for females), a LOAEL of 160 mg/kg bw per day and a NOAEL of 16 mg/kg bw per day were identified, based on changes observed in the liver of males, including increased liver weight, hepatomegaly, and microsome proliferation (Sanders et al., 1982). In a similar study, the authors determined a NOAEL of 16 mg/kg bw per day for decreased humoral immunity (assessed by verifying the number of splenic antibody-forming cells produced in response to sheep red blood cells and haemagglutination titres) and a LOAEL of 160 mg/kg bw per day (Kauffmann et al., 1982).
The liver was confirmed as the target organ in a study in which Sprague-Dawley rats (10 per sex per dose) were exposed to chloral hydrate for 90 days in drinking water at concentrations of 300, 600, 1200, or 2400 mg/L (corresponding to dose levels of 24, 48, 96, and 168 mg/kg bw per day for males and 33, 72, 132, and 288 mg/kg bw per day for females). Based on hepatotoxic effects (focal hepatocellular necrosis in males) and serum enzyme changes (observed in both sexes, but not dose related or toxicologically significant), the study identified a LOAEL of 96 mg/kg bw per day and a NOAEL of 48 mg/kg bw per day (Daniel et al., 1992b).
The liver has been confirmed as the primary target organ of chloral hydrate toxicity in several long-term and carcinogenicity studies.
In a drinking-water study in mice (Rijhsinghani et al., 1986), chloral hydrate at 5 or 10 mg/kg bw was given to 15-day-old male C57BL × C3HF1 mice as a single dose in distilled water. The control group was given distilled water only. To study long-term effects, animals were sacrificed when found moribund or at intervals up to 92 weeks. An increase in tumours was statistically significant (P < 0.05) only in the 10 mg/kg bw group. There was an increase in the relative weight of the liver in mice given chloral hydrate at 10 mg/kg bw, but not in mice given 5 mg/kg bw. Compared with the control group, there was also a significant increase in the incidence of hepatic nodules in mice given chloral hydrate at a dose of 10 mg/kg bw only. The hepatic nodular lesions ranged from hyperplastic foci of clear or acidophilic cells to hepato-cellular adenomas and trabecular carcinomas containing eosinophilic hepatocytes (Rijhsinghani et al., 1986). It is important to note that this study is more than 20 years old and that the protocol used was not based on Organisation for Economic Co-operation and Development (OECD) guidelines regarding the use of two sexes, a required number of animals, and a sufficient number of doses for a carcinogenicity study. The increased incidence of hepatic tumours in this study is believed to have been due to normal variation in mice and not a result of chloral hydrate treatment (NTP, 2002b).
In a key study, male B6C3F1 mice (72 per dose) were exposed to chloral hydrate in drinking water at concentrations of 0, 120, 580, or 1280 mg/L (corresponding to dose levels of 0, 13.5, 65, and 146.6 mg/kg bw per day) for 104 weeks (George et al., 2000). The prevalence of hepatocellular carcinomas was increased in the high-dose group (84.4%) compared with 54.8%, 54.3%, and 59.0% in the control, low-, and mid-dose groups. The prevalence of hepatoadenomas was significantly increased in all dose groups: 43.5%, 51.3%, and 50.0% for the low-, mid-, and high-dose chloral hydrate groups, respectively, compared with 21.4% in the control group. Serum lactate dehydrogenase (LDH), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and sorbitol dehydrogenase (SDH) activities and total antioxidant levels reflected the minimal degree of hepatocellular damage observed microscopically. None of these parameters in the chloral hydrate-treated groups was altered compared with the control values after 52 and 78 weeks of exposure. Palmitoyl CoA oxidase activities in the homogenates of livers were not significantly increased above the control value, indicating that chloral hydrate did not induce peroxisome proliferation. Enhanced liver neoplasia occurred at the lowest dose, 13.5 mg/kg bw per day; therefore, a NOAEL could not be determined. However, a LOAEL can be set at 13.5 mg/kg bw per day. Results for combined neoplasms were significantly increased in the mid- and high-dose groups for prevalence and in all dose groups for multiplicity. This study indicated that the incidence of hepatocellular adenomas was increased at all dose levels, but the incidence of hepatocellular carcinomas was increased at the high dose only (George et al., 2000). IPCS (2000) evaluated this study and set a NOAEL of 146.6 mg/kg bw per day for non-cancer effects in mice, justifying a NOAEL for non-cancer end-points because of the prevalence of proliferative lesions in the controls. It was also noted that there was no increase in the prevalence of neoplasia at sites other than the liver. The male mice showed an increase of proliferative lesions in the liver at all exposure levels (hyperplasia, adenoma, carcinoma, and combined adenoma and carcinoma).
In another component of the study (George et al., 2000), male F344 rats (78 per dose) were administered chloral hydrate in drinking water at concentrations of 0, 120, 580, or 2510 mg/L (corresponding to dose levels of 0, 7.4, 37.4, and 162.6 mg/kg bw per day) for 104 weeks. No measured parameters in any of the chloral hydrate-treated groups were altered compared with the control values after 52 and 78 weeks of exposure, and the NOAEL for this study was set at the highest dose (George et al., 2000). However, the U.S. EPA (2000) concluded that this study did not achieve the maximum tolerated dose, and we concur with this conclusion.
Two National Toxicology Program (NTP) studies (NTP, 2002a, 2002b) provide weak evidence of the carcinogenicity of chloral hydrate in male and female B6C3F1 mice. In the first study (NTP, 2002a), five groups of B6C3F1 mice received chloral hydrate in distilled water by gavage for 105 weeks in a complicated dosing regimen. Chloral hydrate doses ranging up to 100 mg/kg bw per day were administered 5 days per week (71.4 mg/kg bw per day adjusted for 7 days per week dosing). In the first group, the incidence of pituitary pars distalis adenomas occurred with a positive dose-related trend, and the incidence at the highest dose was significantly greater than that in controls; there was also a significant positive time-related trend in the incidence of adenomas and a significant increase in the severity of pars distalis hyperplasia in female mice administered 71.4 mg/kg bw per day for up to 24 months. The authors concluded that there was equivocal evidence of carcinogenic activity of chloral hydrate in female mice treated continuously for 2 years, based on increased incidences of pituitary gland pars distalis adenomas. No increased incidences of neoplasms were seen in female B6C3F1 mice that received a single dose of chloral hydrate at 15 or 28 days of age or in male B6C3F1 mice that received a single dose of chloral hydrate at 15 days of age. No hepatic carcinogenicity was seen under all dosing conditions. The NOAEL for non-neoplastic effects was determined to be 71.4 mg/kg bw per day (NTP, 2002a).
Haseman et al. (1998) reported significant discrepancies between the experimental and historical control in the NTP (2002a) study, as well as dose group incidences of pituitary pars distalis adenomas and hepatocellular neoplasms and adenomas/carcinomas. For example, the incidence of adenomas (12% at 71.4 mg/kg bw per day) exceeded the incidence in historical controls (historical incidence for control groups in National Center for Toxicological Research [NCTR] studies: 4.9%, range 0-6%). The incidence of pituitary pars distalis adenomas/ carcinomas reached 14.8% in the historical control (Haseman et al., 1998), which is higher than the incidence observed at the highest dose (12%) in the first two test groups. This type of adenoma was not observed in any other studies. In both cases (pituitary gland adenoma and hyperplasia), no dose-related effects can be inferred. The incidence of malignant lymphomas found at the low and high doses (33% at 10 mg/kg bw) (in the third group) is higher than the incidence found in both control groups for the NCTR (24.6%) and Haseman et al. (1998) (20.9%) studies. The incidences of malignant lymphomas were not affected by the duration of the treatment and were within the range of incidences in the historical controls, inferring that the incidences observed in the first and third test groups reflect the normal variation found in female B6C3F1 mice. The incidence of alveolar/bronchiolar adenomas was significantly higher (18%) in the second group after 12 months. However, no significant difference was observed after 2 years, suggesting that the incidence may not have been due to chloral hydrate. The incidence found in this study is within the historical range (5.9%, range 0-24%) for control female B6C3F1 mice fed NIH-07 (an open formula cereal-based diet, the NTP standard for chemical toxicity and carcinogenicity studies) (Haseman et al., 1998), but it is higher than that observed in the other NCTR study in female B6C3F1 mice (3.8%, range 2-6%). No other studies have reported this type of lesion with chloral hydrate. In the NTP (2002a) study, the incidence of hepatocellular neoplasms in females in the third and fourth test groups was quite low, ranging from 1% to 13% at up to 50 mg/kg bw; in males (in the fifth group), the incidence of hepatocellular neoplasms was quite high, even in the control group (50%). There were no neoplasms in mice that could be attributed to chloral hydrate treatment.
In the second NTP study (NTP, 2002b), a group of 120 male B6C3F1 mice was fed 0, 25, 50, or 100 mg chloral hydrate/kg bw per day, 5 days per week (0, 17.9, 35.7, or 71.4 mg/kg bw per day, adjusted for 7 days per week dosing), for 2 years. The male mice were divided into two groups of 60: one group received feed ad libitum, while the other received feed in a measured daily amount (gavage). Evaluation of 12 mice per dose group and diet was performed after 15 months, and the other 48 mice per dose group and diet were evaluated after 2 years. Histopathological changes were observed only in the liver. The incidence of hepatocellular adenomas or carcinomas was significantly greater in the 17.9 mg/kg bw per day group only in the ad libitum group. In the dietary controlled study, the incidence of hepatocellular carcinomas was significantly different at 71.4 mg/kg bw per day only. The NOAEL for non-neoplastic effects is 71.4 mg/kg bw per day (NTP, 2002b). The incidences of hepatocellular adenomas/ carcinomas for the ad libitum group in male B6C3F1 mice were 33%, 52%, 48%, and 46% at dose levels of 0, 17.9, 35.7, and 71.4 mg/kg bw per day, respectively, compared with 42% in the historical controls. In the controlled diet, the incidences of combined adenomas and carcinomas were lower than the historical control of 42.2% with the same strain of mice, at 23%, 23%, 29%, and 38% for dose levels of 0, 17.9, 35.7, and 71.4 mg/kg bw per day, respectively. No female mice were treated in this study.
In a study by Daniel et al. (1992a), 40 male B6C3F1 mice received 1 g chloral hydrate/L (166 mg/kg bw per day) in drinking water for 104 weeks. Only mild histopathological changes were observed in the liver, and no changes were noted in other organs. At the 60-week sacrifice, hepatocellular carcinomas were found in two chloral hydrate-treated mice, compared with zero in the controls. No lesions were found in the control group at this time. At the end of the study, 11 out of 24 surviving mice exposed to chloral hydrate had hepatocellular carcinomas, and 7 had hepatocellular adenomas. Hepatocellular lesions in controls included 2 animals with carcinomas and 1 with adenomas out of 20 survivors examined. The incidence of both adenomas and carcinomas was significantly greater than in the control group. The increase in the combined incidence of the two lesions, adenomas and carcinomas, was highly significant (Daniel et al., 1992a). In this study, a low incidence of hepatocellular tumours was observed in the control group (15%), compared with 42.2% for the historical control (Haseman et al., 1998). However, the study indicated that the liver is the target organ following exposure to chloral hydrate. Hepatocellular necrosis as well as tumours (carcinoma, adenoma) were observed in this study. However, this study would be considered inadequate based on OECD guideline standard protocols, because only one dose was used. The difference in the incidences of hepatocellular adenomas and carcinomas in the NTP (2002a) study and that by Daniel et al. (1992a) might be due to the higher dose used in the latter study.
A chronic bioassay was conducted with Sprague-Dawley rats (50 per sex per group) administered chloral hydrate at doses of 0 (untreated drinking water), 15, 45, or 135 mg/kg bw per day in drinking water for 124 weeks for males and 128 weeks for females. There was no evidence of an increased incidence of tumours in any organs. An increase in the incidence of hepatocellular hypertrophy was observed at the highest dose (28%) compared with the control (11%). The finding was characterized by diffuse liver cell enlargement with slightly eosinophilic cytoplasm. The increase in hepatocellular hypertrophy was seen only in the male rats and was graded as minimal to slight in severity. The type, incidence, and organ distribution of the neo-plastic lesions in the chloral hydrate-treated rats did not differ from those in the control rats, and the lesions were therefore regarded as random events. No change was observed in body weight or organ weight. A NOAEL of 45 mg/kg bw per day and a LOAEL of 135 mg/kg bw per day were set based on the increased incidence of hepatocellular hypertrophy (Leuschner and Beuschner, 1998). The U.S. EPA (2000) concluded that there are indications that the study did not achieve the maximum tolerated dose, and we concur with that conclusion.
There is equivocal evidence of the genotoxicity of chloral hydrate. Moore and Harrington-Brock (2000) found chloral hydrate and its metabolites to show evidence of some genotoxic activity, albeit at very high doses, indicating that chloral hydrate is a weakly genotoxic chemical.
In in vitro tests, positive results were reported for Salmonella typhimurium strains TA198 and TA100 in point mutation assays (Waskell, 1978; Bruce and Heddle, 1979; Bignami et al., 1980; NTP, 2002a), for sister chromatid exchanges (Bruce and Heddle, 1979; NTP, 2002a), for DNA strand breaks in human lymphocytes (Gu et al., 1981), for chromosomal aberrations in Chinese hamster ovary cells with or without S9 (Bruce and Heddle, 1979; NTP, 2002a), for aneuploidy and clastogenicity in several test systems using mammalian cells (Natarajan et al., 1993; Harrington-Brock et al., 1998), and for aneuploidy in various fungi in the absence of metabolic activation (U.S. EPA, 2000). Chloral hydrate induced aneuploidy in Chinese hamster embryonic fibroblasts in vitro without an exogenous metabolic system at 250 µg/mL (Harrington-Brock et al., 1998), in two Chinese hamster pulmonary lines at 250 µg/mL and 50 µg/mL (Warr et al., 1993), and in human peripheral blood lymphocytes at 50 µg/mL without an exogenous metabolic system (Sbrana et al., 1993).
Increases in micronuclei in mouse spermatids were observed when spermatogonia stem cells were exposed to chloral hydrate at 41, 83, or 165 mg/kg bw (Allen et al., 1994). In Drosophila melanogaster, chloral hydrate induced a small increase in the frequency of sex-linked recessive lethal mutations in germ cells of male flies fed 5500 mg/kg bw (Yoon et al., 1985).
Negative results were reported for S. typhimurium strain TA1535 in point mutation assays, for mitotic crossing-over in Aspergillus nidulans in the absence of metabolic activation, for reverse mutation in Saccharomyces cerevisiae, for DNA-protein crosslinks in rat liver nuclei, for DNA single strand breaks in rodent hepatocytes in primary culture and in CCRFCEM cells, a human lymphoblastic leukaemia cell line (Chang et al., 1992), for DNA repair in Escherichia coli (U.S. EPA, 2000), and for micronuclei induction and aneuploidy in mouse lymphoma cells (Natarajan et al., 1993; Harrington-Brock et al., 1998). Chloral hydrate also gave negative test results in studies with ICR mouse metaphase II oocytes (Mailhes et al., 1993).
In vivo, chloral hydrate was positive in the mouse bone marrow micronucleus test (U.S. EPA, 2000; NTP, 2002b). Chloral hydrate increased the frequency of chromosomal aberrations in mouse bone marrow, spermatogonia, and primary and secondary spermatocytes, but not in oocytes, after in vivo treatment (U.S. EPA, 2000).
The reproductive, embryo-fetotoxic, and teratogenic effects of chloral hydrate have been studied in several species. Effects have included neurodevelopmental toxicity, reduction in sperm motility, and an increased incidence of heart malformations.
Male and female CD-1 mice (four per cage, total number not specified) were exposed to chloral hydrate in drinking water at concentrations of 60 or 600 mg/L (equivalent to 21.3 or 204.8 mg/kg bw per day). All animals were exposed for 3 weeks prior to breeding. Females were also exposed during gestation and until pups were weaned at 21 days of age. At 204.8 mg/kg bw per day, at 23 days of age, pups showed impaired retention of passive avoidance learning in both 1-hour and 24-hour retention tests. This study identified a NOAEL for neurodevelopmental toxicity of 21.3 mg/kg bw per day. A reproductive and developmental effects NOAEL was also identified at 204.8 mg/kg bw per day (Kallman et al., 1984).
Male F344 rats (two per cage, total number not specified) were administered chloral hydrate in drinking water at concentrations of 0, 780, or 2700 mg/L (corresponding to dose levels of 0, 55, and 188 mg/kg bw per day) for 52 weeks to evaluate the effects of chloral hydrate on sperm morphology and motility. A reduction in sperm motility was observed in rats exposed to 188 mg/kg bw per day (58%) compared with controls (68%). A shift in the frequency distribution of the average straight-line velocities of sperm also occurred at this dose compared with the controls. A NOAEL for effects on sperm motility was set at 55 mg/kg bw per day, and a LOAEL of 188 mg/kg bw per day was identified (Klinefelter et al., 1995).
An in vitro embryotoxicity study was performed with embryos from Sprague-Dawley rats exposed to chloral hydrate at 0.5-2.5 mmol/L (83-414 mg/L) on gestational day 10 for 46 hours. All embryos died at the highest dose, but no deaths were observed at lower doses. Chloral hydrate caused concentration-dependent decreases in growth and differentiation and increases in the incidence of morphologically abnormal embryos. At 1.0 mmol/L, decreases in crown-rump length, somite (embryonic segments) numbers, and the protein or DNA content of embryos were observed. At 1.0, 1.5, and 2.0 mmol/L, 18%, 68%, and 100% of embryos, respectively, had malformations, malformations of the brain, eyes, and ears being among the most frequent developmental effects encountered. At 2.0 mmol/L, abnormalities were also observed in the trunk and in the optic and otic systems. At the higher concentrations, embryos exhibited severe alterations of the craniofacial region. Hypoplasia of the prosencephalon was also observed. Chloral hydrate caused pericardial dilation (45% of the embryos at 2 mmol/L). Chloral hydrate produced a step increase in embryolethality as concentrations increased. Based on this in vitro study, chloral hydrate was found to be more potent than TCA and DCA. A NOAEL of 0.5 mmol/L (83 mg/L) was identified for embryotoxicity (Saillenfait et al., 1995). Since this study is an in vitro study, it is difficult to compare the results with those of an in vivo study, nor is it possible to extrapolate the risk to human health. This study is unusual, because reproductive studies tend to involve exposure over the period of organogenesis (days 5-12) or over the entire gestation period.
Pregnant female Sprague-Dawley rats were exposed to chloral hydrate in drinking water from gestational day 1 to day 22 at 1.232 mg/mL (corresponding to an average exposure of 151 mg/kg bw per day). There was no evidence of maternal toxicity, no change in the number of live or dead fetuses, no change in placental or fetal weight, no change in crown-rump length, and no increase in the incidence of morphological changes. Heart malformations (not significant), such as atrial septal defects (2), mitral valve defects (2), ventricular septal defects (3), and pulmonary valve defects (1), were observed, compared with the control group, which had a total of 15 types of heart malformations. The chloral hydrate-exposed group had a 3.23% incidence of heart abnormalities compared with 2.15% for the control group (significantly different). A LOAEL of 151 mg/kg bw per day for chloral hydrate was identified in this study based on developmental toxicity, and no NOAEL was identified (Johnson et al., 1998).
In the Poon et al. (2002) subchronic toxicity study described above, it was postulated that the biological effects observed were attributable to TCA, a known peroxisomal proliferator. Triglyceride depression may also be a TCA effect. The presence of TCA in the serum and increased liver catalase lend support to a peroxisomal proliferation effect of chloral hydrate. However, this is of limited relevance in humans, since humans and other primates are less responsive than rats and mice in terms of peroxisomal proliferation. In contrast, hepatic hypotriglyceridaemia is of relevance to humans, because the hypolipidaemic effect of peroxisome proliferators is common to both experimental animals and humans.
In an in vitro study using male B6C3F1 mouse liver microsomes, it was found that chloral hydrate generated free radical intermediates that resulted in endogenous lipid peroxidation, thus forming malondialdehyde, formaldehyde, acetaldehyde, acetone, and propionaldehyde, substances that are known to be tumorigenic. Both TCA and TCOH also induced lipid peroxidation, but TCA had a stronger effect than TCOH, suggesting that TCA formation is the predominant pathway leading to lipid peroxidation. Cytochrome P-450 (possibly the isoenzyme CYP2E1) was the enzyme responsible for the metabolic activation of chloral hydrate and its metabolites to TCA and TCOH, leading to tumorigenic lipid peroxidation (Ni et al., 1996).
In humans, chloral hydrate is rapidly absorbed and then either oxidized to TCA (8%) or reduced to TCOH (92%), mainly by the liver, but also by the kidney. TCOH may be conjugated with glucuronic acid to form trichloroethanol glucuronide (TCOG), an inactive metabolite (Ogino et al., 1990; McEvoy, 1999). Additional TCA is formed during enterohepatic circulation of TCOH, such that 35% of the initial dose of chloral hydrate is converted to TCA (Allen and Fisher, 1993). The erythrocytes also metabolize chloral hydrate to TCOH, mainly via alcohol dehydrogenase.
Healthy male volunteers (n = 18) were administered a single dose of 250 mg of chloral hydrate in drinking water. Chloral hydrate, TCOH, and TCA were measured in the plasma. Chloral hydrate could be detected in only some of the plasma samples, 8-60 minutes after dosing. No concentration was reported, but the limit of detection was given as 0.1 mg/L. The maximum plasma concentrations of TCOH and TCA, 3 mg/L and 8 mg/L, respectively, were achieved 0.67 hour and 32 hours after dosing, respectively. The terminal half-lives were 9.3-10.2 hours for TCOH and 89-94 hours for TCA (Zimmermann et al., 1998).
The plasma half-lives in humans for therapeutic doses of chloral hydrate, TCOH, and TCA are about 4-5 minutes, 8-12 hours, and 67 hours, respectively (Ellenhorn and Barceloux, 1988).
In infants and children given chloral hydrate as a sedative, TCA, DCA, and TCOH were detected in plasma, and TCA's plasma half-life was shown to be very long (Henderson et al., 1997). There is evidence that the TCA may be converted to DCA in samples of blood taken for analysis unless appropriate steps are taken, raising concerns about whether the reported levels of DCA in humans are too high (Ketcha et al., 1996).
Chloral hydrate and TCOH do not accumulate in the human body (Gilman et al., 1985). As infants have an immature hepatic metabolism, particularly the glucuronidation pathway, with decreased glomerular filtration, they have a longer TCOH half-life compared with adults. In contrast, toddlers have a TCOH half-life similar to that of adults, indicating maturation of liver metabolism in toddlers (IPCS, 2000).
Chloral hydrate is rapidly metabolized by rats and mice, producing both TCOH and TCA as the major metabolites, with a higher concentration of TCA in mice than in rats. The metabolism of chloral hydrate, TCA, and TCOH was shown in vitro to give rise to free radical intermediates that caused lipid peroxidation and the formation of malondialdehyde (Beland, 1999). As with humans, chloral hydrate disappears rapidly from the blood of mice, and TCOH, TCOG, TCA, and DCA are identified as metabolites (Abbas et al., 1996). The half-lives of TCOH and TCOG appear to be significantly greater in rats than in mice (Beland et al., 1998). Lipscomb et al. (1996) found TCOH to be the first major metabolite of chloral hydrate in vivo in the blood and liver of Fischer 344 rats, B6C3F1 mice, and humans.
Chloral hydrate is an important metabolite of trichloroethylene (TCE) and an intermediate in the formation of TCA. Based on the results from a number of studies, a physiologically based pharmacokinetic (PBPK) model for TCE was developed. This model includes enterohepatic recirculation of its metabolites. The model quantitatively predicts quite well the uptake, distribution, and elimination of TCE, TCOH, TCOG, and TCA. The PBPK model clearly shows that the formation of TCA is delayed following the enterohepatic recirculation, accounting for the longer half-life of TCA observed in animal studies (Stenner et al., 1998). This is supported by the findings of Merdink et al. (1999), who found that some TCOH is converted back to chloral hydrate and oxidized to TCA.
Most chloral hydrate is excreted via the urine as TCOG, with small amounts excreted as free TCOH. The remainder is excreted as TCA (Butler, 1948; Marshall and Owens, 1954; Allen and Fisher, 1993). Chloral hydrate is not excreted unchanged (McEvoy, 1999).
No epidemiological or carcinogenic studies were found in humans that associated exposure to chloral hydrate with cancer, despite the fact that chloral hydrate has been used for many decades (and still is used) as a sedative and hypnotic drug in adults and children (specifically for dental procedures). The U.S. EPA (2000) derived an acute oral reference dose of 0.1 mg/kg bw per day based on the pharmacologically active dose (250 mg, equivalent to 10.7 mg/kg bw per day) in humans. This dose is said to be protective for any non-cancer health effects from chronic exposure. However, chloral hydrate has shown some evidence of carcinogenicity in two long-term drinking water bioassays in male mice and in a lifetime study following a single oral dose in male mice. In addition, chloral hydrate was found to be a weak mutagen and clastogen, suggesting that genotoxicity may play a role in the toxicity of chloral hydrate, but at concentrations higher than those expected to be found in the environment. The pharmacological dose of 10.7 mg/kg bw per day is not considered appropriate for the derivation of a health-based value for chloral hydrate in drinking water.
The International Agency for Research on Cancer classified chloral hydrate as Group 3, "not classifiable as to its carcinogenicity to humans," in 1995, based on inadequate evidence in humans and limited evidence in experimental animals (IARC, 1995). The U.S. EPA (2000) classified chloral hydrate as a possible human carcinogen, concluding that the most likely mode of action for the formation of tumours in mice involves interaction with cellular enzymes and proteins, in contrast to direct interaction with DNA. Health Canada has classified chloral hydrate in Group III.B -- possibly carcinogenic to humans (inadequate data in humans, limited data in animals), as defined in Health Canada (1994). There is equivocal evidence of genotoxicity for chloral hydrate.
For compounds that are "possibly carcinogenic to humans," a health-based value is based on a tolerable daily intake (TDI) derived by the division of the lowest NOAEL or LOAEL by appropriate uncertainty factors.
Two studies from the NTP (2002a, 2002b) provide weak evidence of carcinogenicity in B6C3F1 mice (both sexes). However, significant discrepancies exist between the experimental and historical control (Haseman et al., 1998) and dose group incidences of pituitary pars distalis adenomas and hepatocellular neoplasms and adenomas/carcinomas, making the studies unsuitable for derivation of a guideline. However, in these two studies, hepatocellular neoplasms developed at concentrations similar to those observed in the study chosen for the risk assessment (George et al., 2000), supporting the evidence of proliferative lesions at these concentrations.
The non-cancer end-point of histopathology in the liver as derived in George et al. (2000) was chosen for the risk assessment. Male B6C3F1 mice were treated in a lifetime study with chloral hydrate at concentrations of 0, 120, 580, or 1280 mg/L (corresponding to dose levels of 0, 13.5, 65, and 146.6 mg/kg bw per day). The prevalence of hepatocellular carcinomas was increased in the high-dose group (84.4%) compared with 54.8%, 54.3%, and 59.0% in the control, low-, and mid-dose groups. The prevalence of hepatoadenomas was significantly increased in all dose groups: 43.5%, 51.3%, and 50.0% for the low-, mid-, and high-dose chloral hydrate groups, respectively, compared with 21.4% in the control group. In this study, drinking water was used as a vehicle rather than gavage dosing 5 days per week as in the NTP (2002a, 2002b) studies, supporting the use of the George et al. (2000) study for this evaluation.
Although IPCS (2000) set a NOAEL of 1280 mg/L (146.6 mg/kg bw per day) for non-cancer end-points (based on the lack of evidence of hepatocellular necrosis at any exposure and only minimal changes in the levels of serum enzymes), the George et al. (2000) study showed that chloral hydrate induced an increase in the incidence of proliferative lesions (hyperplasia, adenoma, carcinoma, and combined adenoma and carcinoma) at all exposures, except for carcinoma at the two lower exposures. In the control groups, lesions (hyperplasia, adenoma, carcinoma, and combined adenoma and carcinoma) were also observed, but at lower percentages for the hyperplasia and hepatocellular adenoma. At 120 mg/L (13.5 mg/kg bw per day) and above, significant increases in the incidence of proliferative lesions were observed. This increase in proliferative lesions is an important end-point. Since these lesions were observed at all dose levels, no NOAEL could be derived; therefore, a LOAEL of 120 mg/L (13.5 mg/kg bw per day) was set to derive a TDI. An additional uncertainty factor of 3 was added to account for the limitations of the database in regards to evidence of carcinogenicity in animals.
The TDI is derived as follows:
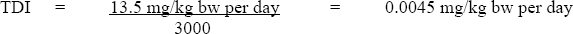
Figure 1 - Text Description
where
- 13.5 mg/kg bw per day is the LOAEL for B6C3F1 mice based on increased incidence of liver histopathology,
- 3000 is the uncertainty factor (×10 for interspecies variability; ×10 for intraspecies variability; ×10 to account for the use of a LOAEL instead of a NOAEL, as no NOAEL was observed in the relevant study; and ×3 to account for limitations of the database in regards to evidence of carcinogenicity).
Based on this TDI, a health-based value for chloral hydrate in drinking water can be derived as follows:
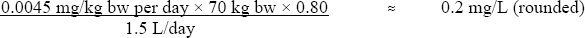
Figure 2 - Text Description
where:
- 0.0045 mg/kg bw per day is the TDI, as derived above,
- 70 kg is the average body weight of an adult,
- 0.80 is the proportion of total daily intake allocated to drinking water, since the exposure to chloral hydrate for the general population comes mostly from chlorinated drinking water,
- 1.5 L/day is the average daily consumption of drinking water for an adult.
The U.S. Environmental Protection Agency (EPA) recognizes and approves EPA Method 551.1 for the determination of chloral hydrate in drinking water. This method uses a solvent extraction procedure for the analysis of chloral hydrate, with methyl tert-butyl ether (MTBE) as the solvent (U.S. EPA, 1995). Chloral hydrate is analysed using gas chromatography/electron capture detection, and the method detection limit is 0.005 µg/L. The sampling protocol requires field pH adjustment (pH 4.8) with a phosphate buffer and use of sodium sulphite to quench the residual chlorine.
Standard Method 5710 D of the 21st edition of the Standard Methods for the Examination of Water and Wastewater is also used to analyse chloral hydrate (APHA et al., 2005). This method stipulates that chloral hydrate may be analysed with THMs using a sodium sulphite solution to quench the reaction. Chloral hydrate is then analysed using liquid-liquid extraction capillary column gas chromatography with electron capture detection.
Although chloral hydrate formation in water is largely a function of the amount of organic compounds in water and their contact time with chlorine, it is important to recognize that the use of chlorination and other disinfection processes has virtually eliminated waterborne microbial diseases. As with THMs and other chlorinated DBPs, it is important to characterize the source water to ensure that the treatment process is optimized for precursor removal in order to reduce chloral hydrate levels in the finished water.
The U.S. EPA has suggested that enhanced coagulation and softening will control chloral hydrate levels in drinking water by removing the DBP precursors (total organic carbon). Moving the point of disinfection to reduce the reaction between chlorine and DBP precursors and using chloramines instead of chlorine for residual disinfection have also been suggested as ways to reduce chloral hydrate production (U.S. EPA, 1998a). Controlling levels of total THMs and HAAs and using enhanced coagulation/softening for DBP precursor removal will control for chloral hydrate as well as other chlorination by-products (U.S. EPA, 1998b).
There are three approaches to limiting the concentrations of chloral hydrate in municipally treated drinking water:
- treatment of water to remove chloral hydrate precursors prior to disinfection;
- the use of alternative disinfectants and disinfection strategies; and
- treatment of water to remove chloral hydrate after its formation.
The majority of changes occurring in the water industry today focus on strategies to remove DBP precursors prior to disinfection and the use of alternative disinfectants and alternative disinfection strategies.
The removal of organic precursors is the most effective way to reduce the concentrations of all DBPs, including chloral hydrate, in finished water (U.S. EPA, 1999b; Reid Crowther & Partners Ltd., 2000). These precursors include synthetic organic compounds and natural organic matter, which can react with disinfectants to form chloral hydrate. Conventional municipal-scale water treatment techniques can reduce the amount of precursors, but are ineffective in removing chloral hydrate once it is formed. Granular activated carbon, membranes, and ozone biofiltration systems can also remove organic matter from water. The U.S. EPA has identified precursor removal technologies such as granular activated carbon (GAC) and membrane filtration as Best Available Technologies (BAT) for controlling disinfection by-products formation (U.S. EPA, 2005). However, membrane processes generate concentrated residuals, and their disposal can be expensive (Xie, 2004). Combinations of disinfectants, when optimized, can help control chloral hydrate formation.
Potassium permanganate can be used to oxidize organic precursors at the head of the treatment plant, thus minimizing the formation of DBPs at the disinfection stage (U.S. EPA, 1999a). The use of ozone for oxidation of precursors is currently being studied. Early work has shown that the effects of ozonation, prior to chlorination, depend on treatment design and raw water quality and thus are unpredictable. The key variables that seem to determine the effect of ozone are dose, pH, alkalinity, and the nature of the organic material in the water. Ozone has been shown to be effective at reducing precursors at low pH. Above pH 7.5, however, ozone may actually increase the production of chlorinated DBP precursors (U.S. EPA, 1999a).
The use of alternative disinfectants, such as chloramines (secondary disinfection only), ozone (primary disinfection only), and chlorine dioxide (primary disinfection only), is increasing. However, each of these alternatives has also been shown to form its own set of DBPs. Preozonation is feasible for water sources that have turbidity levels below 10 nephelometric turbidity units and bromide concentrations below 0.01 mg/L, to minimize the formation of bromate (Reid Crowther & Partners Ltd., 2000). Ultraviolet (UV) disinfection is also being used as an alternative disinfectant. Since UV disinfection is dependent on light transmission to the microbes, water quality characteristics affecting UV transmittance must be considered in the design of the system. Neither ozone nor UV disinfection leaves a residual disinfectant, and both must therefore be used in combination with a secondary disinfectant to maintain a residual in the distribution system.
It is recommended that any change made to the treatment process, particularly when changing the disinfectant, be accompanied by close monitoring of lead levels in the distributed water. A change of disinfectant has been found to affect the levels of lead at the tap, for example in Washington, DC, where a change from chlorine to chloramines resulted in significantly increased levels of lead in the distributed drinking water. When chlorine, a powerful oxidant, is used as the disinfectant, lead dioxide scales formed in distribution system pipes have reached a dynamic equilibrium in the distribution system. In Washington, DC, switching from chlorine to chloramines decreased the oxidation-reduction potential of the distributed water and destabilized the lead dioxide scales, which resulted in increased lead leaching (Schock and Giani, 2004). Subsequent laboratory experiments by Edwards and Dudi (2004) and Lytle and Schock (2005) confirmed that lead dioxide deposits could be readily formed and subsequently destabilized in weeks to months under realistic conditions of distribution system pH, oxidation-reduction potential and alkalinity.
Municipal treatment of drinking water is designed to reduce contaminants to levels at or below their guideline values. As a result, the use of residential-scale treatment devices on municipally treated water is generally not necessary but primarily based on individual choice. In cases where municipal treatment has produced low concentrations of chloral hydrate in drinking water, some residential-scale point-of-entry or point-of-use treatment devices may remove chloral hydrate from the water. Treatment device technologies that may remove chloral hydrate include reverse osmosis and adsorption media, such as activated carbon, although none is currently certified specifically for this use.
NSF International (NSF) has developed several standards for residential water treatment devices designed to reduce the concentrations of various types of contaminants in drinking water, but chloral hydrate is not currently included in any NSF standard. Research is ongoing in the private and public sectors to test and adopt efficient methods for the reduction of chloral hydrate levels in drinking water.
Devices can lose removal capacity through usage and time and need to be maintained and/or replaced. Consumers should verify the expected longevity of the adsorption media in their treatment devices as per the manufacturers' recommendations and service the media when required.
Health Canada has conducted a study on the effectiveness of a number of point-of-use drinking water treatment devices for the removal of chloral hydrate. Boiling water for 2-5 minutes in a pot or kettle was effective in removing chloral hydrate (~98% decrease). The efficiency of filters (pressure filters and gravity filters using granular activated carbon) for the removal of chloral hydrate largely depended on the brand and age of the filters (new filters, 28 to >99% decrease). Aging of the filter, even in the short term, significantly reduced its capacity to remove chloral hydrate (Benoit et al., 2000; LeBel et al., 2002).
Health Canada does not recommend specific brands of drinking water treatment devices, but it strongly recommends that consumers look for a mark or label indicating that the device has been certified by an accredited certification body to the appropriate NSF/American National Standards Institute (ANSI) drinking water materials standard. These standards have been designed to safeguard drinking water by helping to ensure the material safety and performance of products that come into contact with drinking water. Certification organizations provide assurance that a product conforms to applicable standards and must be accredited by the Standards Council of Canada (SCC). In Canada, the following organizations have been accredited by the SCC to certify drinking water devices and materials as meeting NSF/ANSI standards:
- Canadian Standards Association International;
- NSF International;
- Water Quality Association;
- Underwriters Laboratories;
- Quality Auditing Institute; and
- International Association of Plumbing and Mechanical Officials.
An up-to-date list of accredited certification organizations can be obtained from the SCC.
- Abbas, R.R., Seckel, C.S., Kidney, J.K. and Fisher, J.W. (1996) Pharmacokinetic analysis of chloral hydrate and its metabolism in B6C3F1 mice. Drug Metab. Dispos., 24(12): 1340-1346.
- Allen, B.C. and Fisher, J.W. (1993) Pharmacokinetic modeling of trichloroethylene and trichloroacetic acid in humans. Risk Anal., 13: 71-86.
- Allen, J.W., Collins, B.W. and Evansky, P.A. (1994) Spermatid micronucleus analyses of trichloroethylene and chloral hydrate effects in mice. Mutat. Res., 323(1-2): 81-88.
- APHA (American Public Health Association), AWWA (American Water Works Association) and WEF (Water Environment Federation) (2005) Standard methods for the examination of water and wastewater. 21st edition. Washington, DC. p. 5.70.
- Beland, F.A. (1999) NTP technical report on the toxicity and metabolism studies of chloral hydrate (CAS No. 302-17-0) administered by gavage to F344/N rats and B6C3F1 mice. National Toxicology Program, National Institutes of Health, Research Triangle Park, NC. pp. 1-136 (NTP Toxicity Report Series No. 59).
- Beland, F.A., Schmitt, T.C., Fullerton, N.F. and Young, J.F. (1998) Metabolism of chloral hydrate in mice and rats after single and multiple doses. J. Toxicol. Environ. Health, 54: 209-226.
- Benoit, F.M., LeBel, G.L. and Jay, B. (2000) Evaluation of remedial actions to reduce the levels of chlorinated disinfection by-products in drinking water at the point of use. Proceedings of the 9th National Conference on Drinking Water, Regina, Saskatchewan, May 16-18, 2000.
- Bignami, M., Conti, G., Conti, L., Crebelli, R., Misuraca, F., Puglia, A.M., Randazzo, R., Sciandrello, G. and Carere, A. (1980) Mutagenicity of halogenated aliphatic hydrocarbons in Salmonella typhimurium, Streptomyces coelicolor and Aspergillus nidulans. Chem. Biol. Interact., 30: 9-23.
- Bruce, W.R. and Heddle, J.A. (1979) The mutagenic activity of 61 agents as determined by the micronucleus, Salmonella, and sperm abnormality assays. Can. J. Gen. Cytol., 21: 319-334.
- Budavari, S. (1996) The Merck index. An encyclopedia of chemicals, drugs, and biologicals. 12th edition. Merck and Co. Inc., Whitehouse Station, NJ.
- Butler, T.C. (1948) The metabolic fate of chloral hydrate. J. Pharmacol. Exp. Ther., 92: 49-58.
- Chang, L.W., Daniel, F.B. and DeAngelo, A.B. (1992) Analysis of DNA strand breaks induced in rodent liver in vivo, hepatocytes in primary culture, and a human cell line by chlorinated acetic acid and chlorinated acetaldehydes. Environ. Mol. Mutagen., 20(4): 277-288.
- Daniel, F.B., DeAngelo, A.B., Stober, J.A., Olson, G.R. and Page, N.P. (1992a) Hepatocarcinogenicity of chloral hydrate, 2-chloroacetaldehyde, and dichloroacetic acid in the male B6C3F1 mouse. Fundam. Appl. Toxicol., 19(2): 159-168.
- Daniel, F.B., Robinson, M., Stober, J.A., Page, N.P. and Olson, G.R. (1992b) Ninety-day toxicity study of chloral hydrate in the Sprague-Dawley rat. Drug Chem. Toxicol., 15: 217-232.
- Edsall, T.A. and Charlton, M.N. (1997) Nearshore waters of the Great Lakes. State of the Lakes Ecosystem Conference 1996. U.S. Environmental Protection Agency and Environment Canada (EPA 905-R-97-015A; Catalogue No. En40-11/35-1-1-1997E).
- Ellenhorn, M.J. and Barceloux, D.G. (1988) Medical toxicology. Diagnosis and treatment of human poisoning. Elsevier Science Publishing Co., Inc., New York, NY.
- Ellenhorn, M.J., Schonwald, S., Ordog, G. and Wasserberger, J. (1997) Ellenhorn's medical toxicology: Diagnosis and treatment of human poisoning. 2nd edition. Williams and Wilkins, Baltimore, MD.
- George, M., Moore, T., Kilburn, S., Olson, G.R. and DeAngelo, A.B. (2000) Carcinogenicity of chloral hydrate administered in drinking water to the male F344/N rat and male B6C3F1 mouse. Toxicol. Pathol., 28: 610-618.
- Gilman, A.G., Goodman, L.S. and Gilman, A. (1985) Chloral hydrate. In: Goodman and Gilman's the pharmacological basis of therapeutics. Macmillan, New York, NY. pp. 352, 370.
- Greenberg, S.B., Faerber, E.N. and Aspinall, C.L. (1991) High-dose chloral hydrate sedation for children undergoing CT. J. Comput. Assist. Tomogr., 15: 467-469.
- Gu, Z.W., Sele, B., Jalbert, P., Vincent, M., Vincent, F., Marka, C., Chmara, D. and Faure, J. (1981) [Induction of sister chromatid exchange by trichloroethylene and its metabolites.] Toxicol. Eur. Res., 3(2): 63-67 (in French).
- Hansch, C., Leo, A. and Hoekman, D. (1995) Exploring QSAR -- Hydrophobic, electronic, and steric constants. American Chemical Society, Washington, DC.
- Harrington-Brock, K., Doerr, C.L. and Moore, M.M. (1998) Mutagenicity of three disinfection by-products: di- and trichloroacetic acid and chloral hydrate in L5178Y/TK +/!(!)3.7.2C mouse lymphoma cells. Mutat. Res., 413(3): 265-276.
- Haseman, J.K., Hailey, J.R. and Morris, R.W. (1998) Spontaneous neoplasm incidences in Fischer 344 rats and B6C3F1 mice in two-year carcinogenicity studies: A National Toxicology Program update. Toxicol. Pathol., 26(3): 428-441.
- Health Canada (1994) Canadian Environmental Protection Act. Human health risk assessment for priority substances. Minister of Supply and Services Canada, Ottawa, Ontario (Catalogue No. En40-215/41E).
- Health Canada (1995) A national survey of chlorinated disinfection by-products in Canadian water. Environmental Health Directorate, Health Canada, Ottawa, Ontario (95-EHD-197).
- Henderson, G.N., Yan, Z., James, M.O., Davydova, N. and Stacpoole, P.W. (1997) Kinetics and metabolism of chloral hydrate in children: identification of dichloroacetate as a metabolite. Biochem. Biophys. Res. Commun., 235: 695-698.
- IARC (International Agency for Research on Cancer) (1995) Chloral and chloral hydrate. IARC Monogr. Eval. Carcinogen. Risks Hum., 63: 245-269.
- IPCS (International Programme on Chemical Safety) (2000) Chloral hydrate. World Health Organization, Geneva (Concise International Chemical Assessment Document No. 25).
- Johnson, P.D., Dawson, B.V. and Goldberg, S.J. (1998) Cardiac teratogenicity of trichloroethylene metabolites. J. Am. Coll. Cardiol., 32: 540-545.
- Kallman, M.J., Kaempf, G.L. and Balster, R.L. (1984) Behavioral toxicity of chloral in mice: an approach to evaluation. Neurobehav. Toxicol. Teratol., 6: 137-146.
- Kauffmann, B.M., White, K.L., Sanders, V.M., Douglas, K.A., Sain, L.E., Borzelleca, J.F. and Munson, A.E. (1982) Humoral and cell-mediated status in mice exposed to chloral hydrate. Environ. Health Perspect., 44: 147-151.
- Ketcha, M.M., Stevens, D.K., Warren, D.A., Bishop, C.T. and Brashear, W.T. (1996) Conversion of trichloroacetic acid to dichloroacetic acid in biological samples. J. Anal. Toxicol., 20: 236-241.
- Kirk-Othmer (1991) Kirk-Othmer encyclopedia of chemical technology. 4th edition. Wiley Interscience, New York, NY.
- Klinefelter, G.R., Suarez, J.D., Roberts, N.L. and DeAngelo, A.B. (1995) Preliminary screening for the potential of drinking water disinfection by-products to alter male reproduction. Reprod. Toxicol., 9: 571-578.
- Krasner, S.W., McGuire, M.J., Jacangelo, J.G., Patania, N.L., Reagan, K.M. and Aieta, E.M. (1989) The occurrence of disinfection by-products in US drinking water. J. Am. Water Works Assoc., 81(8): 41-52.
- LeBel, G.L., Jay, B. and Benoit, F.M. (2002) Evaluation of filtration devices to reduce levels of chlorinated disinfection by-products (CDBPs) in drinking water at the point of use. Proceedings of the 10th National Conference on Drinking Water, Halifax, Nova Scotia, April 27-30, 2002.
- Leuschner, J. and Beuscher, N. (1998) Studies on the mutagenic and carcinogenic potential of chloral hydrate. Arzneimittel-Forschung, 48: 961-968.
- Lipscomb, J.C., Mahle, D.A., Brashear, W.T. and Garrett, C.M. (1996) A species comparison of chloral hydrate metabolism in blood and liver. Biochem. Biophys. Res. Commun., 227: 340-350.
- Mailhes, J.B., Aardena, M.J. and Marchetti, F. (1993) Investigation of aneuploidy induction in mouse oocytes following exposure to vintablastine-sulfate, pyrimethamine, diethylstibestrol diphosphate, or chloral hydrate. Environ. Mol. Mutagen., 22: 107-114.
- Marshall, E.K. and Owens, A.H. (1954) Absorption, excretion and metabolic fate of chloral hydrate and trichloroethanol. Bull. Johns Hopkins Hosp., 95: 1-18.
- McEvoy, G.K. (1999) American Hospital Formulary Service Drug Information 99. American Society of Health-System Pharmacists, Inc., Bethesda, MD.
- Merdink, J.L., Stenner, R.D., Stevens, D.K., Parker, J.C. and Bull, R.J. (1999) Effect of enterohepatic circulation on the pharmacokinetics of chloral hydrate and its metabolites in F344 rats. J. Toxicol. Environ. Health, 57: 357-368.
- Moore, M.M. and Harrington-Brock, K. (2000) Mutagenicity of trichloroethylene and its metabolites: implications for the risk assessment of trichloroethylene. Environ. Health Perspect., 108: 215-223.
- Natarajan, A.T., Duivenvooden, W.C., Meijers, M. and Zwanenburg, T.S. (1993) Induction of mitotic aneuploidy using Chinese hamster primary embryonic cells: Test results of 10 chemicals. Mutat. Res., 287(1): 47-56.
- Newman, L.M. and Wackett, L.P. (1991) Fate of 2,2,2-trichloroacetaldehyde (chloral hydrate) produced during trichloroethylene oxidation by methanotrophs. Appl. Environ. Microbiol., 57: 2399-2402.
- Ni, Y.C., Wong, T.Y., Lloyd, R.V., Heinze, T.M., Shelton, S., Casciano, D., Kadlubar, F.F. and Fu, P.P. (1996) Mouse liver microsomal metabolism of chloral hydrate, trichloroacetic acid, and trichloroethanol leading to induction of lipid peroxidation via a free radical mechanism. Drug Metab. Dispos., 24(1): 81-90.
- NTP (National Toxicology Program) (2002a) Toxicology and carcinogenesis studies of chloral hydrate in B6C3F1 mice (gavage studies). National Institutes of Health, Research Triangle Park, NC. 197 pp. (NTP Technical Report Series 502).
- NTP (National Toxicology Program) (2002b) Toxicology and carcinogenesis studies of chloral hydrate (ad libitum and dietary controlled) in male B6C3F1 mice (gavage study). National Institutes of Health, Research Triangle Park, NC. 218 pp. (NTP Technical Report Series 503).
- Odum, J., Foster, J.-R. and Green, T. (1992) A mechanism for the development of Clara cell lesions in the mouse lung after exposure to trichloroethylene. Chem.-Biol. Interact., 83(2): 135-153.
- Ogino, K., Hobara, T., Kobayashi, H. and Iwamoto, S. (1990) Gastric mucosal injury by chloral hydrate. Toxicol. Lett., 52(2): 129-133.
- Poon, R., Nadeau, B. and Chu, I. (2000) Biochemical effects of chloral hydrate on male rats following 7-day drinking water exposure. J. Appl. Toxicol., 20: 455-461.
- Poon, R., Nakai, J., Yagminas, A., Benoit, F., Moir, D., Chu, I. and Valli, V.E. (2002) Subchronic toxicity of chloral hydrate on rats -- a drinking water study. J. Appl. Toxicol., 22: 227-236.
- Reid Crowther & Partners Ltd. (2000) Canadian water treatment study: water treatment and disinfection byproducts. Edmonton, Alberta. September.
- Reynolds, J.E.F. and Prasad, A.B. (1982) Martindale -- The extra pharmacopia. 28th edition. The Pharmaceutical Press, London.
- Rijhsinghani, K.S., Abrahams, C., Swerdlow, M.A., Rao, K.V.N. and Ghose, T. (1986) Induction of neoplastic lesions in the livers of C57BL × C3HF1 mice by chloral hydrate. Cancer Detect. Prev., 9(3-4): 279-288.
- Saillenfait, A.M., Langonne, I. and Sabate, J.P. (1995) Developmental toxicity of trichloroethylene, tetrachloroethylene and four of their metabolites in rat whole embryo cultures. Arch. Toxicol., 70: 71-78.
- Sanders, V.M., Kauffman, B.M., White, K.L., Douglas, K.A., Barnes, D.W., Sain, L.E., Bradshaw, T.J., Borzelleca, J.F. and Munson, A.E. (1982) Toxicology of chloral hydrate in the mouse. Environ. Health Perspect., 44: 137-146.
- Sbrana, I., Di Sibio, A., Lomi, A. and Scarcelli, V. (1993) C-mitosis and numerical chromosome aberration analyses in human lymphocytes: 10 known or suspected spindle poisons. Mutat. Res., 287(1): 57-70.
- Shapiro, S., Stone, D., Lewis, G.P. and Jick, H. (1969) Clinical effects of hypnotics. II. An epidemiological study. J. Am. Med. Assoc., 209: 2016-2020.
- Stenner, R.D., Merdink, J.L., Fisher, J.W. and Bull, R.J. (1998) Physiologically-based pharmacokinetic model for trichloroethylene considering enterohepatic recirculation of major metabolites. Risk Anal., 18(3): 261-269.
- U.S. EPA (United States Environmental Protection Agency) (1992) Technical Support Division Disinfection By-Products Field Study Database. Technical Support Division, Office of Ground Water and Drinking Water, U.S. EPA, Cincinnati, OH.
- U.S. EPA (United States Environmental Protection Agency) (1995) Methods for the determination of organic compounds in drinking water -- Supplement III. U.S. EPA, Washington, DC (EPA-600/R-95/131).
- U.S. EPA (United States Environmental Protection Agency) (1998a) National Primary Drinking Water Regulations; disinfectants and disinfection byproducts: Notice of data availability. 40 CFR Parts 141 and 142. Fed. Regist., 63(145): 15674-15692.
- U.S. EPA (United States Environmental Protection Agency) (1998b) National Primary Drinking Water Regulations: disinfectants and disinfection byproducts. 40 CFR Parts 9, 141, and 142. Fed. Regist., 63: 6904.
- U.S. EPA (United States Environmental Protection Agency) (1999) EPA guidance manual -- Alternative disinfectants and oxidants. U.S. EPA, Washington, DC, April (EPA-815-R-99-014).
- U.S. EPA (United States Environmental Protection Agency) (2000) Toxicological review of chloral hydrate. In support of summary information on the Integrated Risk Information System (IRIS). U.S. EPA, Washington, DC (EPA/635/R-00/006).
- Verschueren, K. (2001) Handbook of environmental data on organic chemicals. 3rd edition. Van Nostrand Reinhold Co., New York, NY.
- Warr, T.J., Parry, E.M. and Parry, J.M. (1993) A comparison of two in vivo mammalian cell cytogenetic assays for the detection of mitotic aneuploidy using 10 known or suspect aneugens. Mutat. Res., 287(1): 29-46.
- Waskell, L. (1978) A study of the mutagenicity of anesthetics and their metabolites. Mutat. Res., 57(2): 141-153.
- Williams, D.T., LeBel, G.L. and Benoit, F.M. (1997) Disinfection by-products in Canadian drinking water. Chemo-sphere, 34: 299-316.
- Xie, Y.F. (2004) Disinfection byproducts in drinking water: Formation, analysis, and control. Lewis Publishers, CRC Press, Boca Raton, FL.
- Yoon, J.S., Mason, J.M., Valencia, R., Woodruff, R.C. and Zimmering, S. (1985) Chemical mutagenesis testing in Drosophila. IV. Results of 45 coded compounds tested for the National Toxicology Program. Environ. Mutagen., 7: 349-367.
- Zimmermann, T., Wehling, M. and Schulz, H.U. (1998) [The relative bioavailability and pharmacokinetics of chloral hydrate and its metabolites.] Arzneimittelforschung, 48: 5-12 (in German).
- ALDH
- aldehyde dehydrogenase
- ALT
- alanine aminotransferase
- ANSI
- American National Standards Institute
- AST
- aspartate aminotransferase
- bw
- body weight
- CoA
- coenzyme A
- DBP
- disinfection by-product
- DCA
- dichloroacetic acid
- DDT
- dichlorodiphenyltrichloroethane
- DNA
- deoxyribonucleic acid
- EPA
- Environmental Protection Agency (United States)
- HAA
- haloacetic acid
- K ow
- octanol/water partition coefficient
- LD 50
- median lethal dose
- LDH
- lactate dehydrogenase
- LOAEL
- lowest-observed-adverse-effect level
- MTBE
- methyl tert-butyl ether
- NCTR
- National Center for Toxicological Research (United States)
- NOAEL
- no-observed-adverse-effect level
- NOEL
- no-observed-effect level
- NSF
- NSF International
- NTP
- National Toxicology Program (United States)
- OECD
- Organisation for Economic Co-operation and Development
- PBPK
- physiologically based pharmacokinetic
- S9
- metabolic activation (9000 × g supernatant)
- SCC
- Standards Council of Canada
- SDH
- sorbitol dehydrogenase
- TCA
- trichloroacetic acid
- TCE
- trichloroethylene
- TCOG
- trichloroethanol glucuronide
- TCOH
- trichloroethanol
- TDI
- tolerable daily intake
- THM
- trihalomethane
- UV
- ultraviolet
- WHO
- World Health Organization