
Page 4: Guidance on Controlling Corrosion in Drinking Water Distribution Systems
Part B. Supporting information
Exposure to contaminants resulting from the internal corrosion of drinking water systems can be the result of corrosion in either the distribution system or the plumbing system, or both. The degree to which corrosion is controlled for a contaminant in a system can be assessed adequately by measuring the contaminant at the tap over time and correlating its concentrations with corrosion control activities.
Corrosion is defined as "the deterioration of a material, usually a metal, that results from a reaction with its environment" (NACE International, 2000). In drinking water distribution systems, the material may be, for example, a metal pipe or fitting, the cement in a pipe lining or a PVC pipe.
This document focuses primarily on the corrosion and leaching of lead-, copper- and iron-based materials. It also briefly addresses the leaching from PVC and cement pipes, but does not include microbiologically influenced corrosion.
The corrosion of metallic materials is electrochemical in nature and is defined as the "destruction of a metal by electron transfer reactions" (Snoeyink and Wagner, 1996). For this type of corrosion to occur, all four components of an electrochemical cell must be present: (1) an anode, (2) a cathode, (3) a connection between the anode and the cathode for electron transport and (4) an electrolyte solution that will conduct ions between the anode and the cathode. In the internal corrosion of drinking water distribution systems, the anode and the cathode are sites of different electrochemical potential on the metal surface, the electrical connection is the metal and the electrolyte is the water.
The key reaction in corrosion is the oxidation or anodic dissolution of the metal to produce metal ions and electrons:
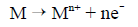
Figure 4 - Guidance on Controlling Corrosion in Drinking Water Distribution Systems - Text description
where:
- M is the metal
- e- is an electron
- n is the valence and the corresponding number of electrons.
In order for this anodic reaction to proceed, a second reaction must take place that uses the electrons produced. The most common electron acceptors in drinking water are dissolved oxygen and aqueous chlorine species.
The ions formed in the reaction above may be released into drinking water as corrosion products or may react with components present in the drinking water to form a scale on the surface of the pipe. The scale that forms on the surface of the metal may range from highly soluble and friable to adherent and protective. Protective scales are usually created when the metal cation combines with a hydroxide, oxide, carbonate, phosphate or silicate to form a precipitate.
The concentration of a specific metal in drinking water is determined by the corrosion rate and by the dissolution and precipitation properties of the scale formed. Initially, with bare metal, the corrosion rate far exceeds the dissolution rate, so a corrosion product layer builds over the metal's surface. As this layer tends to stifle corrosion, the corrosion rate drops towards the dissolution rate (Snoeyink and Wagner, 1996).
The materials present in the distribution system determine which contaminants are most likely to be found at the tap. The principal contaminants of concern that can leach from materials in drinking water distribution systems are aluminum, antimony, arsenic, bismuth, cadmium, copper, iron, lead, nickel, organolead, organotin, selenium, tin, vinyl chloride and zinc. It is important to assess whether these contaminants will be present at concentrations that exceed those considered safe for human consumption.
In Canada, copper plumbing with lead-tin solders (widely used until 1989) and brass faucets and fittings are predominant in domestic plumbing systems (Churchill et al., 2000).
Cast iron and ductile iron pipes account for more than two-thirds of the existing water mains in use across Canada (InfraGuide, 2001). In new installations, PVC pipes often replace copper tubing, lead service lines and distribution pipes. Cement-based materials are also commonly used to convey water in large-diameter pipes.
Lead may leach into potable water from lead pipes in old water mains, lead service lines, lead in pipe jointing compounds and soldered joints, lead in brass and bronze plumbing fittings, and lead in goosenecks, valve parts or gaskets used in water treatment plants or distribution mains. Lead was a common component of distribution systems for many years. All provinces and territories use the National Plumbing Code of Canada (NPC) as the basis for their plumbing regulations. The NPC allowed lead as an acceptable material for pipes (service lines) until 1975. Under the NPC, all fittings must comply to the American Society of Mechanical Engineers (ASME)/Canadian Standards Association (CSA) standard ASME 112.18.1/CSA B125.1 (formerly CSA B125.1) for plumbing supply fittings. This standard limited the lead content of solder to 0.2% in 1986. The NPC officially prohibited lead solders from being used in new plumbing or in repairs to plumbing for drinking water supplies in the 1990 version (NRCC, 2005). The most common replacements for lead solders are tin-antimony, tin-copper and tin-silver solders.
A new generation of reasonably priced brass alloys is now available for plumbing fittings and in-line devices. These "very low lead" brasses contain < 0.25% lead as an impurity, and bismuth or a combination of bismuth and selenium replaces the lead in the alloy (AwwaRF, 2007).
Copper is used in pipes and copper alloys found in domestic plumbing. Copper alloys used in potable water systems are brasses (in domestic fittings) and bronzes (in domestic plumbing valves). Brasses are basically alloys of copper and zinc, with other minor constituents, such as lead. Brass fittings are also often coated with a chromium-nickel compound. Bronzes (also referred to as red brass) are alloys of copper, tin and zinc, with or without lead. Most brasses contain between 2% and 8% lead.
In addition to lead that can be found in brass and bronze fittings such as faucets and valves, fixtures such as refrigerated water coolers and bubblers commonly used in schools and other non-residential buildings may contain lead. Selected components of water coolers such as soldered joints within the fixtures or the lining in the tank may contain alloys with lead (U.S. EPA, 2006b).
The following iron-based materials are the principal sources of iron in drinking water distribution systems: cast iron, ductile iron, galvanized iron and steel. The specific components that are likely to come in contact with drinking water in its transit from the treatment plant to the consumer include walls or working parts of well casings, pumps, mixing equipments, meters, pipes, valves and fittings. Iron may be released directly from iron-based materials or indirectly through the iron corrosion by-products, or tubercles, formed during the corrosion process.
Galvanized pipes will release zinc, since they are manufactured by dipping steel pipes in a bath of molten zinc. Galvanized pipes can also be sources of cadmium and lead, since these materials are present as impurities (Leroy et al., 1996). The NPC permitted the use of galvanized steel as an acceptable material for pipes for plumbing systems until 1980 (NRCC, 2005).
Cement-based materials used to convey drinking water include reinforced concrete pipes, cement mortar linings and asbestos-cement pipes. In addition to the aggregates (sand, gravel or asbestos), which constitute the basic structure of the cement, the binder, which is responsible for the cohesion and mechanical properties of the material, consists mostly of calcium silicates and calcium aluminates in varying proportions (Leroy et al., 1996). Degradation of cement-based materials can be a source of calcium hydroxide (lime) in the distributed water, which may result in an increase in pH and alkalinity. The degradation of cement-based materials can also be a source of aluminum and asbestos in drinking water.
According to the literature, cement-based materials rarely cause serious water quality problems. However, newly installed in situ mortar linings have been reported to cause water quality problems in dead ends or low-flow water conditions when water alkalinity is low (Douglas and Merrill, 1991).
PVC, polyethylene and chlorinated PVC pipes used in the distribution system have the potential to release organic chemicals into the distributed water. PVC mains manufactured prior to 1977 contain elevated levels of vinyl chloride, which they are prone to leaching (Flournoy et al., 1999). Stabilizers are used to protect PVC from decomposition when exposed to extreme heat during production. In Canada, organotin compounds are the most common stabilizers used in the production of PVC pipes for drinking water and have been found in drinking water distributed by PVC pipes. Chlorinated PVC pipes are made using stabilizers containing lead, which can then leach into the distributed water. It must be noted that fittings intended for PVC pipes can be made of brass, which contains lead and can be a potential source of lead where PVC pipes are used. Under the NPC, all plastic pipes must comply to the CSA B137 series of standards for plastic pipe, which require that pipes and the associated fittings comply with the NSF International (NSF)/American National Standards Institute (ANSI) Standard 61 requirements for leaching of contaminants.
There is no single, reliable index or method to measure water corrosivity and reflect population exposure to contaminants that are leached by the distribution system. Given that a major source of metals in drinking water is related to corrosion in distribution and plumbing systems, measuring the contaminant at the tap is the best tool to assess corrosion and reflect population exposure.
The literature indicates that lead, copper and iron are the contaminants whose levels are most likely to exceed guideline values owing to the corrosion of materials in drinking water distribution systems. The MAC for lead is based on health considerations for the most sensitive population (i.e., children). Guidelines for copper and iron are based on aesthetic considerations, such as colour and taste. An aesthetic objective of ≤ 1.0 mg/L has been established for copper in drinking water; copper is an essential element in humans and is generally considered to be nontoxic except at high doses, in excess of 15 mg/day. An aesthetic objective of ≤ 0.3 mg/L has been established for iron in drinking water; iron is also an essential element in humans. Based on these considerations, lead concentrations at the tap are used as the basis for initiating corrosion control programs (Health Canada, 1978, 1992).
A recent review of the literature (Schock, 2005) indicates that a number of contaminants can be accumulated in and released from the distribution system. Scales formed in distribution system pipes that have reached a dynamic equilibrium can subsequently release contaminants such as aluminum, arsenic, other trace metals and radionuclides. Changes made to the treatment process, particularly those that affect water quality parameters such as pH, alkalinity and oxidation-reduction potential (ORP), should be accompanied by close monitoring in the distributed water.
A national survey was conducted in 1981 to ascertain the levels of cadmium, calcium, chromium, cobalt, copper, lead, magnesium, nickel and zinc in Canadian distributed drinking water (Méranger et al., 1981). Based on the representative samples collected at the tap after 5 min of flushing at maximum flow rate, the survey concluded that only copper levels increased to a significant degree in the drinking water at the tap when compared with raw and treated water.
Concurrently, several studies showed that concentrations of trace elements from household tap water sampled after a period of stagnation can exceed guideline values (Wong and Berrang, 1976; Lyon and Lenihan, 1977; Nielsen, 1983; Samuels and Méranger, 1984; Birden et al., 1985; Neff et al., 1987; Schock and Neff, 1988; Gardels and Sorg, 1989; Schock, 1990a; Singh and Mavinic, 1991; Lytle et al., 1993; Viraraghavan et al., 1996).
A study on the leaching of copper, iron, lead and zinc from copper plumbing systems with lead-based solders in high-rise apartment buildings and single-family homes was conducted by Singh and Mavinic (1991). The study showed that for the generally corrosive water (pH 5.5-6.3; alkalinity 0.6-3.7 mg/L as calcium carbonate) of the Greater Vancouver Regional District, the 1st litre of tap water taken after an 8-h period of stagnation exceeded the Canadian drinking water guidelines for lead and copper in 43% (lead) and 62% (copper) of the samples from high-rise buildings and in 47% (lead) and 73% (copper) of the samples from single-family homes. Even after prolonged flushing of the tap water in the high-rise buildings, the guidelines were still exceeded in 6% of the cases for lead and in 9% of the cases for copper. In all cases in the single-family homes, flushing the cold water for 5 min successfully reduced levels of lead and copper below the guideline levels.
Subramanian et al. (1991) examined the leaching of antimony, cadmium, copper, lead, silver, tin and zinc from new copper piping with non-lead-based soldered joints exposed to tap water. The levels of antimony, cadmium, lead, silver, tin and zinc were below the detection limits even in samples that were held in pipes for 90 days. However, copper levels were found to be above 1 mg/L in some cases. The authors concluded that tin-antimony, tin-silver and tin-copper-silver solders used in copper pipes do not leach antimony, cadmium, lead, silver, tin or zinc into drinking water.
Samuels and Méranger (1984) conducted a study on the leaching of trace metals from kitchen faucets in contact with the City of Ottawa's water. Water was collected after a 24-h period of stagnation in new faucets not washed prior to testing. Cadmium, chromium, copper, lead and zinc were leached from the kitchen faucets in varying amounts depending on the type of faucet and the solutions used. In general, the concentrations of cadmium, chromium, copper and zinc in the leachates did not exceed the Canadian drinking water guideline values applicable at that time. However, levels well above the guideline value for lead were leached from the faucets containing lead-soldered copper joints.
Similar work by Schock and Neff (1988) revealed that new chrome-plated brass faucets can be a significant source of copper, lead and zinc contamination of drinking water, particularly upon stagnation of the water. The authors also concluded that faucets, as well as other brass fittings in household systems, provide a continuous source of lead, even when lead-free solders and fluxes are used in copper plumbing systems.
Studies have also examined lead concentrations in drinking water in non-residential buildings such as workplaces and schools. Maas et al. (1994) conducted a statistical analysis of water samples collected after an overnight stagnation period from over 12 000 water fountains, bubblers, chillers, faucets and ice makers. The analysis indicated that over 17% of the samples had lead concentrations above 15 µg/L. Further analysis indicated that the drinking water collected from bubblers, chillers and faucets had lead concentrations above 15 µg/L in over 25% of the samples. Other studies found that between 5% and 21% of drinking water fountains or faucets had lead concentrations above 20 µg/L following a period of stagnation greater than 8 h (Gnaedinger, 1993; Bryant, 2004; Sathyanarayana et al., 2006; Boyd et al., 2008a).
Studies conducted in Copenhagen, Denmark, found that nickel was leaching from chromium-nickel-plated brass after periods of water stagnation (Anderson, 1983). Nickel concentrations measured in the first 250 mL ranged from 8 to 115 µg/L. These concentrations dropped to 9-19 µg/L after 5 min of flushing. Similarly, large concentrations of nickel (up to 8700 µg/L in one case) were released from newly installed chromium-nickel-plated brass, nickel-plated parts and nickel-containing gunmetal following 12-h periods of water stagnation (Nielsen and Andersen, 2001). Experience with the U.S. EPA's Lead and Copper Rule also revealed that brass was a potential source of nickel at the tap (Kimbrough, 2001). Nickel was found in the 1st litre after a period of water stagnation (mean concentrations in the range of 4.5-9.2 µg/L, and maximum concentrations in the range of 48-102 µg/L). The results also indicated that almost all of the nickel was contained in the first 100 mL.
Since cast iron and ductile iron make up more than two-thirds of Canadian drinking water distribution systems, it is not surprising that red water is the most common corrosion problem reported by consumers. When the iron concentration exceeds the aesthetic objective of ≤ 0.3 mg/L established in the Guidelines for Canadian Drinking Water Quality, the iron can stain laundry and plumbing fixtures, produce undesirable taste in beverages and impart a yellow to red-brownish colour to the water.
In addition to aesthetic problems, iron tubercles may contain several types of microorganisms. Tuovinen et al. (1980) isolated sulphate reducers, nitrate reducers, nitrate oxidizers, ammonia oxidizers, sulphur oxidizers and unidentified heterotrophic microorganisms from iron tubercles. Similarly, Emde et al. (1992) isolated coliform species, including Escherichia coli, Enterobacter aerogenes and Klebsiella spp., from iron tubercles in Yellowknife's distribution system. High concentrations of coliforms (> 160 bacteria per gram of tubercles) were also detected in iron tubercles at a New Jersey utility that experienced long-term bacteriological problems in its distribution system, even though no coliforms were detected in the treatment plant effluents. The coliform bacteria identified were E. coli, Citrobacter freundii and Enterobacter agglomerans (Le Chevallier et al., 1988). Although most pipe surfaces in distribution systems are colonized with microorganisms, iron tubercles can especially favour microorganism growth. The nodular areas of the scale can physically protect bacteria from disinfection by providing rough-surfaced crevices in which the bacteria can hide (Le Chevallier et al., 1987).
Iron hydroxides may also adsorb and concentrate chemicals. The installation of chlorination at a Midwestern water system in the United States caused exceptionally high arsenic concentrations at the tap. Chlorination of the groundwater (whose arsenic concentrations never exceeded 10 µg/L) induced the formation of ferric hydroxide solids, which readily sorbed and concentrated arsenic present in the groundwater. The addition of chlorine also affected the scale formed on copper plumbing, resulting in the release of copper oxides, which in turn sorbed and concentrated arsenic. Arsenic concentrations as high as 5 mg/L were found in water samples collected (Reiber and Dostal, 2000). Furthermore, the scale may adsorb chemicals, such as arsenic, which can be later released if the quality of the water distributed is modified (Reiber and Dostal, 2000; Lytle et al., 2004). After finding arsenic concentrations in the range of 10-13 650 µg/L in iron pipe scales of 15 drinking water utilities, Lytle et al. (2004) concluded that distribution systems transporting water containing arsenic at concentrations below 10 µg/L could still produce dangerous levels of arsenic at the consumer's tap. Arsenic that accumulates in corrosion by-products found in the distribution system over time could be released back into the water, especially during changes in hydraulic regime and/or water quality.
High concentrations of aluminum were found in the drinking water of Willemstad, Curaçao, Netherlands Antilles, following the installation of 2.2 km of new factory-lined cement mortar pipes (Berend and Trouwborst, 1999). Aluminum concentrations in the distributed water increased from 5 to 690 µg/L within 2 months of the installation. More than 2 years later, aluminum continued to leach from the lining at concentrations above 100 µg/L. These atypical elevated aluminum concentrations were attributed to the high aluminum content of the cement mortar lining (18.7% as aluminum oxide), as well as to the low hardness (15-20 mg/L as calcium carbonate), low alkalinity (18-32 mg/L as calcium carbonate), high pH (8.5-9.5), long contact time (2.3 days) of the distributed water and use of polyphosphate as a corrosion inhibitor.
Aluminum was also found to leach from in situ portland cement-lined pipes in a series of field trials carried out throughout the United Kingdom in areas with different water qualities (Conroy, 1991). Aluminum concentrations above the European Community (EC) Directive of 0.2 mg/L were found for the first 2 months following installation in very low alkalinity water (around 10 mg/L as calcium carbonate) with elevated pH (> 9.5) and contact times of 6 h. Aluminum concentrations dropped below the EC Directive level after 2 months of pipe service. Furthermore, in water with slightly higher alkalinity (around 50 mg/L as calcium carbonate), aluminum was not found to exceed the EC Directive at any time. The Canadian guideline for aluminum in drinking water is an operational guidance value, which applies to treatment plants using aluminum-based coagulants in their treatment process. Because of the lack of "consistent, convincing evidence that aluminum in drinking water causes adverse health effects in humans," a health-based guideline has not been established for aluminum in drinking water (Health Canada, 1998).
Asbestos fibres have been found to leach from asbestos-cement pipes (Leroy et al., 1996). Although a Guideline Technical Document is available for asbestos in drinking water, it states that "there is no consistent, convincing evidence that ingested asbestos is hazardous. There is, therefore, no need to establish a maximum acceptable concentration for asbestos in drinking water." (Health Canada, 1989).
A study of organotin concentrations in Canadian drinking water distributed through newly installed PVC pipes was conducted in the winter and spring (28 sites) and autumn (21 sites) of 1996 (Sadiki and Williams, 1999). Approximately 29% and 40% of the samples of distributed water supplied through PVC pipes contained organotin compounds in the winter/spring and autumn surveys, respectively. The most commonly detected organotin compounds were monomethyltin and dimethyltin, at concentrations ranging from 0.5 to 257 ng tin/L. An additional study in the summer of 1996 of locations where the highest organotin levels were detected in the winter/spring survey indicated that organotin levels had decreased in 89% of the distributed water samples (tin concentrations ranging from 0.5 to 21.5 ng/L). There is no Canadian drinking water guideline for organotins.
Many factors contribute to the corrosion and leaching of contaminants from drinking water distribution systems. However, the principal factors are the type of materials used, the age of the plumbing system, the stagnation time of the water and the quality of the water in the system. The concentrations of all corrosive or dissolvable materials present in the distribution system will be influenced by some or all of these factors. However, the manner in which these factors will impact each contaminant will vary from one contaminant to another.
Factors influencing the corrosion and leaching of lead, copper, iron and cement are discussed here, since these materials are most likely to produce contaminants that exceed the Canadian drinking water guidelines, pose health risks to the public or be a source of consumer complaints. A list of the specific key factors and their main effects is provided in Section C.3.
Lead concentrations at the tap originating from lead solders and brass fittings decline with age (Sharrett et al., 1982; Birden et al., 1985; Boffardi, 1988, 1990; Schock and Neff, 1988; Neuman, 1995). Researchers have concluded that the highest lead concentrations appear in the 1st year following installation and level off after a number of years of service (Sharrett et al., 1982; Boffardi, 1988). However, unlike lead-soldered joints and brass fittings, lead piping can continue to provide a consistently strong source of lead after many years of service (Britton and Richards, 1981; Schock et al., 1996). In a field study in which lead was sampled in tap water, Maas et al. (1991) showed that homes of all ages were at a substantial risk of lead contamination.
Copper release into the drinking water largely depends on the type of scale formed within the plumbing system. It can be assumed that at a given age, a corrosion by-product governs the release of copper into the drinking water. A decrease in solubility in the following order is observed when the following scales predominate: cuprous hydroxide [Cu(OH)2] > bronchantite [Cu4(SO4)(OH)6] >> cupric phosphate [Cu3(PO4)2] > tenorite [CuO] and malachite [Cu2(OH)2CO3] (Schock et al., 1995). Copper concentrations continue to decrease with the increasing age of plumbing materials, even after 10 or 20 years of service, when tenorite or malachite scales tend to predominate (Sharrett et al., 1982; Neuman, 1995; Edwards and McNeill, 2002). In certain cases, sulphate and phosphate can at first decrease copper concentrations by forming bronchantite and cupric phosphate, but in the long run they may prevent the formation of the more stable tenorite and malachite scales (Edwards et al., 2002).
The age of an iron pipe affects its corrosion. In general, both iron concentration and the rate of corrosion increase with time when a pipe is first exposed to water, but both are then gradually reduced as the scale builds up (McNeill and Edwards, 2001). However, most red water problems today are caused by heavily tuberculated old unlined cast iron pipes that are subject to stagnant water conditions prevalent in dead ends. Sarin et al. (2003) removed unlined cast iron pipes that were 90-100 years old from distribution systems. The internal surface of these pipes was so heavily corroded that up to 76% of the cross-section of the pipes was blocked by scales. Such pipes are easily subject to scouring and provide the high surface areas that favour the release of iron.
A newly installed cement-based material will typically leach lime, which, in turn, will increase water pH, alkalinity and concentrations of calcium (Holtschulte and Schock, 1985; Douglas and Merrill, 1991; Conroy et al., 1994; Douglas et al., 1996; Leroy et al., 1996). Experiments by Douglas and Merrill (1991) showed that after 1, 6 and 12 years in low-flow, lowalkalinity water, lime continued to leach from cement mortar linings upon prolonged exposure. The rate of lime leaching, however, significantly decreased from the 6- and 12-year-old pipes when compared with the 1-year-old pipe. These observations were explained by the fact that the lime leaching rate naturally slows down as surface calcium becomes depleted. As well, the deposits formed after extensive exposure may serve to protect the mortar against further leaching.
Concentrations of lead and copper in drinking water from various sources of leaded material including lead service lines, leaded solder and brass fittings that contain lead, can increase significantly following a period of water stagnation of a few hours in the distribution system. Many factors, such as the water quality and the age, composition, diameter and length of the lead pipe, impact the shape of stagnation curves and the time to reach an equilibrium state (Lytle and Schock, 2000).
In reviewing lead stagnation curves drawn by several authors, Schock et al. (1996) concluded that lead levels increase exponentially upon stagnation, but ultimately approach a fairly constant equilibrium value after overnight stagnation. Lytle and Schock (2000) showed that lead levels increased rapidly with the stagnation time of the water, with the most critical period being during the first 20-24 h for both lead pipe and brass fittings. Lead levels increased most rapidly over the first 10 h, reaching approximately 50-70% of the maximum observed value. In their experiment, lead levels continued to increase slightly even up to 90 h of stagnation.
Kuch and Wagner (1983) plotted lead concentrations versus stagnation time for two different water qualities and lead pipe diameters. The lead concentrations in 1/2-inch (1.3-cm ) pipe where the pH of the water was 6.8 and the alkalinity was 10 mg/L as calcium carbonate (CaCO3) were significantly higher than lead concentrations stagnating in 3/8-inch (0.95-cm) pipe where the pH of the water was 7.2 and the alkalinity was 213 mg/L CaCO3. Additional data from Kuch and Wagner (1983) indicate that lead levels approach maximum or equilibrium concentrations at greater than 300 minutes (5 hrs) for 1/2-inch pipe (1.3-cm lead service line) and at greater than 400 min (6.7 h) for 3/8-inch (1.0-cm) pipe. The diameter of pipes or lead service lines in Canada ranges from 1/2-inch (1.3-cm) to 3/4-inch (1.9-cm) but is typically 5/8-inch (1.6-cm) to 3/4- inch (1.9-cm). In addition, lead concentrations have been demonstrated to be highly sensitive to stagnation time in the first 3 h of standing time for 1/2-inch (1.3-cm ) to 3/4-inch (1.9-cm) pipe. Depending on the water quality characteristics and pipe diameters, differences between 10% and 30% could be observed with differences in standing time as little as 30- 60 min (Kuch and Wagner, 1983; Schock, 1990a). Long lead or copper pipe of small diameter produces the greatest concentrations of lead or copper, respectively, upon stagnation (Kuch and Wagner, 1983; Ferguson et al., 1996).
Lead is also leached during no-flow periods from soldered joints and brass fittings (Birden et al., 1985; Neff et al., 1987; Schock and Neff, 1988). Wong and Berrang (1976) concluded that lead concentrations in water sampled in a 1-year-old household plumbing system made of copper with tin-lead solders could exceed 0.05 mg/L after 4-20 h of stagnation and that lead concentrations in water in contact with lead water pipes could exceed this value in 10-100 min. In a study examining the impact of stagnation time on lead release from brass coupons, Schock et al. (1995) observed that for brass containing 6% lead, lead concentrations increased slowly for the 1st hour but ultimately reached a maximum concentration of 0.08 mg/L following 15 h of stagnation. Following a 6-h stagnation period, the lead concentration was greater than 0.04 mg/L. The amount of lead released from brass fittings was found to vary with both alloy composition and stagnation time.
Copper behaviour is more complex than lead behaviour when it comes to the stagnation of the water. Copper levels will initially increase upon stagnation of the water, but can then decrease or continue to increase, depending on the oxidant levels. Lytle and Schock (2000) showed that copper levels increased rapidly with the stagnation time of the water, but only until dissolved oxygen fell below 1 mg/L, after which they dropped significantly. Sorg et al. (1999) also observed that in softened water, copper concentrations increased to maximum levels of 4.4 and 6.8 mg/L after about 20-25 h of standing time, then dropped to 0.5 mg/L after 72-92 h. Peak concentrations corresponded to the time when the dissolved oxygen was reduced to 1 mg/L or less. In non-softened water, the maximum was reached in less than 8 h, because the dissolved oxygen decreased more rapidly in the pipe loop exposed to non-softened water.
Cyclic periods of flow and stagnation were reported as the primary cause of red water problems resulting from iron corrosion of distribution systems (Benjamin et al., 1996). Iron concentration was also shown to increase with longer water stagnation time prevalent in dead ends (Beckett et al., 1998; Sarin et al., 2000).
Long contact time between distributed water and cement materials has been correlated with increased water quality deterioration (Holtschulte and Schock, 1985; Conroy, 1991; Douglas and Merrill, 1991; Conroy et al., 1994; Douglas et al., 1996; Berend and Trouwborst, 1999). In a survey of 33 U.S. utilities with newly installed in situ lined cement mortar pipes carrying low-alkalinity water, Douglas and Merrill (1991) concluded that degraded water quality was most noticeable in dead ends or where the flow was low or intermittent. Similar conclusions were reached by the Water Research Centre in the United Kingdom, where the longer the supply water was in contact with the mortar lining, the greater was the buildup of leached hydroxides, and hence the higher was the pH (Conroy, 1991; Conroy et al., 1994). Long residence times in new cement mortar pipes installed in Curaçao were also linked with elevated concentrations of aluminum in drinking water (Berend and Trouwborst, 1999), but these were due to the high aluminum content of the mortar (18.7% as aluminum oxide).
The effect of pH on the solubility of the corrosion by-products formed during the corrosion process is often the key to understanding the concentration of metals at the tap. An important characteristic of distributed water with higher pH is that the solubility of the corrosion by-products formed in the distribution system typically decreases.
The solubility of the main lead corrosion by-products (divalent lead solids: cerussite [PbCO3], hydrocerussite [Pb3(CO3)2(OH)2] and lead hydroxide [Pb(OH)2]) largely determines the lead levels at the tap (Schock, 1980, 1990b; Sheiham and Jackson, 1981; De Mora and Harrison, 1984; Boffardi, 1988, 1990; U.S. EPA, 1992; Leroy, 1993; Peters et al., 1999). From thermodynamic considerations, lead solubility of corrosion by-products in distribution systems decreases with increasing pH (Britton and Richards, 1981; Schock and Gardels, 1983; De Mora and Harrison, 1984; Boffardi, 1988; Schock, 1989; U.S. EPA, 1992; Singley, 1994; Schock et al., 1996). Solubility models show that the lowest lead levels occur when pH is around 9.8 (Schock and Gardels, 1983; Schock, 1989; U.S. EPA, 1992; Schock et al., 1996). However, these pH relationships may not be valid for insoluble tetravalent lead dioxide (PbO2) solids, which have been discovered in lead pipe deposits from several different water systems (Schock et al., 1996, 2001). Based on tabulated thermodynamic data, the pH relationship of lead dioxide may be opposite to that of divalent lead solids (e.g., cerussite, hydrocerrussite) (Schock et al., 2001; Schock and Giani, 2004). Lytle and Schock (2005) demonstrated that lead dioxide easily formed at pH 6-6.5 in water with persistent free chlorine residuals in weeks to months.
Unlike contamination from lead pipes and leaded copper alloys, which is mainly controlled by the solubility of the corrosion products, contamination from leaded solders is largely controlled by galvanic corrosion (Oliphant, 1983b; Schock, 1990b; Reiber, 1991; Singley, 1994). An increase in pH is associated with a decrease in galvanic corrosion of leaded solders (Oliphant, 1983b; Gregory, 1990; Reiber, 1991; Singley, 1994).
Utility experience has also shown that the lowest levels of lead at the tap are associated with pH levels above 8 (Karalekas et al., 1983; Lee et al., 1989; Dodrill and Edwards, 1995; Douglas et al., 2004). From 1999 to 2003, the City of Ottawa evaluated a number of chemical alternatives to control corrosion in their distribution system (Douglas et al., 2004). Based on bench- and pilot-scale experimental results and analysis of the impacts on a number of criteria, a corrosion control strategy was established whereby a pH of 9.2 and a minimum alkalinity target of 35 mg/L as calcium carbonate would be achieved through the use of sodium hydroxide and carbon dioxide. During the initial implementation phase, the switch to sodium hydroxide occurred while maintaining the pH at 8.5. However, subsequent to a request for lead testing by a client, the investigators found an area of the city with high levels of lead at the tap (10-15 µg/L for flowing samples). The problem was attributed to nitrification within the distribution system, which caused a reduction in the pH from 8.5 to a range of 7.8-8.2 and resulted in lead leaching from lead service lines. The pH was increased from 8.5 to 9.2 to address the nitrification issue and reduce the dissolution of lead. This increase in the pH almost immediately reduced lead concentrations at the tap in the problem area to a range of 6-8 µg/L for flowing samples. Ongoing monitoring has demonstrated that lead levels at the tap consistently ranged from 1.3 to 6.8 µg/L following the increase in pH, well below the regulated level (Ontario Drinking Water Standard) of 10 µg/L (Douglas et al., 2007).
Examination of utility data provided by 365 utilities under the U.S. EPA Lead and Copper Rule revealed that the average 90th-percentile lead levels at the tap were dependent on both pH and alkalinity (Dodrill and Edwards, 1995). In the lowest pH category (pH < 7.4) and lowest alkalinity category (alkalinity < 30 mg/L as calcium carbonate), utilities had an 80% likelihood of exceeding the U.S. EPA Lead and Copper Rule Action level for lead of 0.015 mg/L. In this low-alkalinity category, only a pH greater than 8.4 seemed to reduce lead levels at the tap. However, when an alkalinity greater than 30 mg/L as calcium carbonate was combined with a pH greater than 7.4, the water produced could, in certain cases, meet the U.S. EPA Lead and Copper Rule Action level for lead.
A survey of 94 water utilities conducted in 1988 to determine lead levels at the consumer's tap and to evaluate the factors that influence them showed similar results (Lee et al., 1989). In total, 1484 sites, including both non-lead and lead service lines, were sampled after an overnight stagnation of at least 6 h. The results of the study clearly demonstrated that maintaining a pH of at least 8.0 effectively controlled lead levels (< 10 µg/L) in the 1st litre collected at the tap. The Boston, Massachusetts, metropolitan area conducted a 5-year study to reduce lead concentrations in its drinking water distribution system (Karalekas et al., 1983). Fourteen households were examined for lead concentrations at the tap, in their lead service lines and in their adjoining distribution systems from 1976 to 1981. Average concentrations were reported for combined samples taken (1) after overnight stagnation at the tap, (2) after the water turned cold and (3) after the system was flushed for an additional 3 min. Even if alkalinity remained very low (on average 12 mg/L as calcium carbonate), raising the pH from 6.7 to 8.5 reduced average lead concentrations from 0.128 to 0.035 mg/L.
Although the hydrogen ion does not play a direct reduction role on copper surfaces, pH can influence copper corrosion by altering the equilibrium potential of the oxygen reduction half-reaction and by changing the speciation of copper in solution (Reiber, 1989). Copper corrosion increases rapidly as the pH drops below 6; in addition, uniform corrosion rates can be high at low pH values (below about pH 7), causing metal thinning. At higher pH values (above about pH 8), copper corrosion problems are almost always associated with non-uniform or pitting corrosion processes (Edwards et al., 1994a; Ferguson et al., 1996). Edwards et al. (1994b) found that for new copper surfaces exposed to simple solutions that contained bicarbonate, chloride, nitrate, perchlorate or sulphate, increasing the pH from 5.5 to 7.0 roughly halved corrosion rates, but further increases in pH yielded only subtle changes.
The prediction of copper levels in drinking water relies on the solubility and physical properties of the cupric oxide, hydroxide and basic carbonate solids that comprise most scales in copper water systems (Schock et al., 1995). In the cupric hydroxide model of Schock et al. (1995), a decrease in copper solubility with higher pH is evident. Above a pH of approximately 9.5, an upturn in solubility is predicted, caused by carbonate and hydroxide complexes increasing the solubility of cupric hydroxide. Examination of experience from 361 utilities reporting copper levels under the U.S. EPA Lead and Copper Rule revealed that the average 90th-percentile copper levels were highest in waters with pH below 7.4 and that no utilities with pH above 7.8 exceeded the U.S. EPA's action level for copper of 1.3 mg/L (Dodrill and Edwards, 1995). However, problems associated with copper solubility were also found to persist up to about pH 7.9 in cold, high-alkalinity and high-sulphate groundwater (Edwards et al., 1994a).
In the pH range of 7-9, both the corrosion rate and the degree of tuberculation of iron distribution systems generally increase with increasing pH (Larson and Skold, 1958; Stumm, 1960; Hatch, 1969; Pisigan and Singley, 1987). Iron levels, however, were usually reported to decrease with increasing pH (Karalekas et al., 1983; Kashinkunti et al., 1999; Broo et al., 2001; Sarin et al., 2003). In a pipe loop system constructed from 90- to 100-year-old unlined cast iron pipes taken from a Boston distribution system, iron concentrations were found to steadily decrease when the pH was raised from 7.6 to 9.5 (Sarin et al., 2003). Similarly, when iron was measured in the distribution system following a pH increase from 6.7 to 8.5, a consistent downward trend in iron concentrations was found over 2 years (Karalekas et al., 1983). These observations are consistent with the fact that the solubility of iron-based corrosion by-products decreases with increasing pH.
Water with low pH, low alkalinity and low calcium is particularly aggressive towards cement materials. The water quality problems that may occur are linked to the chemistry of the cement. Lime from the cement releases calcium ions and hydroxyl ions into the drinking water. This, in turn, may result in a substantial pH increase, depending on the buffering capacity of the water (Leroy et al., 1996). Pilot-scale tests were conducted to simulate low-flow conditions of newly lined cement mortar pipes carrying low-alkalinity water (Douglas et al., 1996). In the water with an initial pH of 7.2, alkalinity of 14 mg/L as calcium carbonate and calcium at 13 mg/L as calcium carbonate, measures of pH as high as 12.5 were found. Similarly, in the water with an initial pH of 7.8, alkalinity of 71 mg/L as calcium carbonate and calcium at 39 mg/L as calcium carbonate, measures of pH as high as 12 were found. The most significant pH increases were found during the 1st week of the experiment, and pH decreased slowly with aging of the lining. In a series of field and test rig trials to determine the impact of in situ cement mortar lining on water quality, Conroy et al. (1994) observed that in low-flow and low-alkalinity water (around 10 mg/L as calcium carbonate), pH increases exceeding 9.5 could occur for over 2 years following the lining.
A series of field trials carried out throughout the United Kingdom in areas with different water qualities found that high pH in cement pipes can render lead soluble. Lead levels increased significantly with increasing pH when pH was above 10.5. The concentration of lead ranged from just less than 100 µg/L at pH 11 to greater than 1000 µg/L above pH 12 (Conroy, 1991). This brings into question the accuracy of the solubility models for high pH ranges and the point at which pH adjustment may become detrimental.
Elevated pH levels resulting from cement leaching may also contribute to aluminum leaching from cement materials, since high pH may increase aluminum solubility (Berend and Trouwborst, 1999).
Alkalinity serves to control the buffer intensity of most water systems; therefore, a minimum amount of alkalinity is necessary to provide a stable pH throughout the distribution system for corrosion control of lead, copper and iron and for the stability of cement-based linings and pipes.
Alkalinity of the finished water is affected by the use of reverse osmosis and nanofiltration processes. These processes remove sodium, sulphate, chloride, calcium and bicarbonate ions and result in a corrosive finished water (Taylor and Wiesner, 1999). This underlines the importance of process adjustments such as addition of base and aeration of the permeate stream to recover alkalinity prior to distribution.
According to thermodynamic models, the minimum lead solubility occurs at relatively high pH (9.8) and low alkalinity (30-50 mg/L as calcium carbonate) (Schock, 1980, 1989; Schock and Gardels, 1983; U.S. EPA, 1992; Leroy, 1993; Schock et al., 1996). These models show that the degree to which alkalinity affects lead solubility depends on the form of lead carbonate present on the pipe surface. When cerussite is stable, increasing alkalinity reduces lead solubility; when hydrocerussite is stable, increasing alkalinity increases lead solubility (Sheiham and Jackson, 1981; Boffardi, 1988, 1990). Cerussite is less stable at pH values where hydrocerussite is stable and may form. Eventually, hydrocerussite will be converted to cerussite, which is found in many lead pipe deposits. Higher lead release was observed in pipes where cerussite was expected to be stable given the pH/alkalinity conditions. However, when these conditions are adjusted so that hydrocerussite is thermodynamically stable, lead release will be lower than in any place where cerussite is stable (Schock, 1990a).
Laboratory experiments also revealed that, at pH 7-9.5, optimal alkalinity for lead control is between 30 and 45 mg/L as calcium carbonate and that adjustments to increase alkalinity beyond this range yield little additional benefit (Schock, 1980; Sheiham and Jackson, 1981; Schock and Gardels, 1983; Edwards and McNeill, 2002) and can be detrimental in some cases (Sheiham and Jackson, 1981).
Schock et al. (1996) reported the existence of significant amounts of insoluble tetravalent lead dioxide in lead pipe deposits from several different water systems. However, the alkalinity relationship for lead dioxide solubility is not known, as no complexes or carbonate solids have been reported. The existence of significant amounts of insoluble lead dioxide in lead pipe deposits may explain the erratic lead release from lead service lines and poor relationship between total lead and alkalinity (Lytle and Schock, 2005).
Alkalinity is not expected to influence the release of lead from leaded solders, since this release is mostly dependent on the galvanic corrosion of the leaded solders as opposed to the solubility of the corrosion by-products formed (Oliphant, 1983a). However, Dudi and Edwards (2004) predicted that alkalinity could play a role in the leaching of lead from galvanic connections between lead- and copper-bearing plumbing. A clear relationship between alkalinity and lead solubility based on utility experience remains to be established. Trends in field data of 47 U.S. municipalities indicated that the most promising water chemistry targets for lead control were a pH level of 8-10 with an alkalinity of 30-150 mg/L as calcium carbonate (Schock et al., 1996). A subsequent survey of 94 U.S. water companies and districts revealed no relationship between lead solubility and alkalinity (Lee et al., 1989). In a survey of 365 utilities under the U.S. EPA Lead and Copper Rule, lead release was significantly lower when alkalinity was between 30 and 74 mg/L as calcium carbonate than when alkalinity was < 30 mg/L as calcium carbonate. Lower lead levels were also observed in utilities with alkalinities between 74 and 174 mg/L and greater than 174 mg/L when the pH was 8.4 or lower (Dodrill and Edwards, 1995).
Laboratory and utility experience demonstrated that copper corrosion releases are worse at higher alkalinity (Edwards et al., 1994b, 1996; Schock et al., 1995; Ferguson et al., 1996; Broo et al., 1998) and are likely due to the formation of soluble cupric bicarbonate and carbonate complexes (Schock et al., 1995; Edwards et al., 1996).
Examination of utility data for copper levels, obtained from 361 utilities under the U.S. EPA Lead and Copper Rule, also revealed the adverse effects of alkalinity and estimated that they were approximately linear and more significant at lower pH: a combination of low pH (< 7.8) and high alkalinity (> 74 mg/L as calcium carbonate) produced the worst-case 90th-percentile copper levels (Edwards et al., 1999).
However, low alkalinity (< 25 mg/L as calcium carbonate) also proved to be problematic under utility experience (Schock et al., 1995). For high-alkalinity waters, the only practical solutions to reduced cuprosolvency are lime softening, removal of bicarbonate or addition of rather large amounts of orthophosphate (U.S. EPA, 2003).
Lower copper concentrations can be associated with higher alkalinity when the formation of the less soluble malachite and tenorite has been favoured (Schock et al., 1995). A laboratory experiment conducted by Edwards et al. (2002) revealed the possible dual effect of high alkalinity. For relatively new pipes, at pH 7.2, the maximum concentration of copper released was nearly a linear function of alkalinity. However, as the pipes aged, lower releases of copper were measured at an alkalinity of 300 mg/L as calcium carbonate, at which malachite had formed, than at alkalinities of 15 and 45 mg/L as calcium carbonate, at which the relatively soluble cupric hydroxide prevailed.
Lower iron corrosion rates (Stumm, 1960; Pisigan and Singley, 1987; Hedberg and Johansson, 1987; Kashinkunti et al., 1999) and iron concentrations (Horsley et al., 1998; Sarin et al., 2003) in distribution systems have been associated with higher alkalinities.
Experiments using a pipe loop system built from 90- to 100-year-old unlined cast iron pipes taken from a Boston distribution system showed that decreases in alkalinity from 30-35 mg/L to 10-15 mg/L as calcium carbonate at a constant pH resulted in an immediate increase of 50-250% in iron release. Changes in alkalinity from 30-35 mg/L to 58-60 mg/L as calcium carbonate and then back to 30-35 mg/L also showed that higher alkalinity resulted in lower iron release, but the change in iron release was not as dramatic as the changes in the lower alkalinity range (Sarin et al., 2003). An analysis of treated water quality parameters (pH, alkalinity, hardness, temperature, chloride and sulphate) and red water consumer complaints was conducted in the City of Topeka, Kansas (Horsley et al., 1998). Data from the period 1989-1998 were used for the analysis. The majority of red water problems were found in unlined cast iron pipes that were 50-70 years old. From 1989 to 1998, the annual average pH of the distributed water ranged from 9.1 to 9.7, its alkalinity ranged from 47 to 76 mg/L as calcium carbonate and its total hardness ranged from 118 to 158 mg/L as calcium carbonate. The authors concluded that the strongest and most useful relationship was between alkalinity and red water complaints and that maintaining finished water with an alkalinity greater than 60 mg/L as calcium carbonate substantially reduced the number of consumer complaints.
Alkalinity is a key parameter in the deterioration of water quality by cement materials. When poorly buffered water comes into contact with cement materials, the soluble alkaline components of the cement pass rapidly into the drinking water. Conroy et al. (1994) observed that alkalinity played a major role in the deterioration of the quality of the water from in situ mortar lining in dead-end mains with low-flow conditions. When the alkalinity was around 10 mg/L as calcium carbonate, pH levels remained above 9.5 for up to 2 years, and aluminum concentrations were above 0.2 mg/L for 1-2 months following the lining process. However, when alkalinity was around 35 mg/L as calcium carbonate, the water quality problem was restricted to an increase in pH level above 9.5 for 1-2 months following the lining process. When the alkalinity was greater than 55 mg/L as calcium carbonate, no water quality problems were observed.
No simple relationship exists between temperature and corrosion processes, because temperature influences several water quality parameters, such as dissolved oxygen solubility, solution viscosity, diffusion rates, activity coefficients, enthalpies of reactions, compound solubility, oxidation rates and biological activities (McNeill and Edwards, 2002).
These parameters, in turn, influence the corrosion rate, the properties of the scales formed and the leaching of materials into the distribution system. The corrosion reaction rate of lead, copper and iron is expected to increase with temperature. However, the solubility of several corrosion by-products decreases with increasing temperature (Schock, 1990a; Edwards et al., 1996; McNeill and Edwards, 2001, 2002).
Seasonal variations in temperature between the summer and winter months were correlated with lead concentrations, with the warmer temperatures of the summer months increasing lead concentrations (Britton and Richards, 1981; Karalekas et al., 1983; Colling et al., 1987, 1992; Douglas et al., 2004). From 1999 to 2003, the City of Ottawa investigated a number of corrosion control options for their distribution system (Douglas et al., 2004). The investigators reported a strong seasonal variation in lead concentration, with the highest lead levels seen during the months of May to November.
Similarly, in a survey of the release of copper corrosion by-products into the drinking water of high-rise buildings and single-family homes in the Greater Vancouver Regional District, Singh and Mavinic (1991) noted that copper concentrations in water run through cold water taps were typically one-third of copper concentrations in water run through hot water taps. A laboratory experiment that compared copper release at 4, 20, 24 and 60°C in a soft, lowalkalinity water showed higher copper release at 60°C, but little difference in copper release between 4°C and 24°C (Boulay and Edwards, 2001). However, copper hydroxide solubility was shown to decrease with increasing temperature (Edwards et al., 1996; Hidmi and Edwards, 1999).
In a survey of 365 utilities under the U.S. EPA Lead and Copper Rule, no significant trend between temperature and lead or copper levels was found (Dodrill and Edwards, 1995). Red water complaints as a function of temperature were analysed by Horsley et al. (1998). Although no direct correlation was found between temperature and red water complaints, more red water complaints were reported during the warmer summer months. Corrosion rates, measured in annular reactors made of new cast iron pipes, were also strongly correlated with seasonal variations (Volk et al., 2000). The corrosion rates at the beginning of the study (March) were approximately 2.5 milli-inch per year [mpy] (0.064 mm per year ) at a temperature below 13°C. The corrosion rates started to increase in May and were highest during the months of July to September (5-7 mpy[0.13-0.18 mm per year] and > 20°C).
No information was found in the reviewed literature on the relationship between temperature and cement pipe degradation.
Traditionally, it was thought that calcium stifled corrosion of metals by forming a film of calcium carbonate on the surface of the metal (also called passivation). However, many authors have refuted this idea (Stumm, 1960; Nielsen, 1983; Lee et al., 1989; Schock, 1989, 1990b; Leroy, 1993; Dodrill and Edwards, 1995; Lyons et al., 1995; Neuman, 1995; Reda and Alhajji, 1996; Rezania and Anderl, 1997; Sorg et al., 1999). No published study has demonstrated, through compound-specific analytical techniques, the formation of a protective calcium carbonate film on lead, copper or iron pipes (Schock, 1989). Leroy (1993) even showed that in certain cases, calcium can slightly increase lead solubility. Furthermore, surveys of U.S. water companies and districts revealed no relationship between lead or copper levels and calcium levels (Lee et al., 1989; Dodrill and Edwards, 1995).
For iron, many authors have reported the importance of calcium in various roles, including calcium carbonate scales, mixed iron/calcium carbonate solids and the formation of a passivating film at cathodic sites (Larson and Skold, 1958; Stumm, 1960; Merill and Sanks, 1978; Benjamin et al., 1996; Schock and Fox, 2001). However, calcium carbonate by itself does not form protective scales on iron materials (Benjamin et al., 1996).
Calcium is the main component of cement materials. Calcium oxide makes up 38-65% of the composition of primary types of cement used for distributing drinking water (Leroy et al., 1996). Until an equilibrium state is reached between the calcium in the cement and the calcium of the conveyed water, it is presumed that calcium from the cement will be either leached out of or precipitated into the cement pores, depending on the calcium carbonate precipitation potential of the water.
Hypochlorous acid is a strong oxidizing agent used for the disinfection of drinking water and is the predominant form of free chlorine below pH 7.5. Free chlorine species (i.e., hypochlorous acid and hypochlorite ion) can also act as primary oxidants towards lead and thus increase lead corrosion (Boffardi, 1988, 1990; Schock et al., 1996; Lin et al., 1997). However, a pipe loop study on the effect of chlorine on corrosion demonstrated that a free chlorine residual (0.2 mg/L) did not increase lead concentrations (Cantor et al., 2003). A survey of 94 U.S. water companies and districts also revealed no relationship between lead levels and free chlorine residual concentrations (in the range of 0-0.5 mg/L) (Lee et al., 1989).
Significant lead dioxide deposits in scales were first reported by Schock et al. (1996) in pipes from several different water systems. Suggestions were made as to the chemical conditions that would favour these tetravalent lead deposits and the changes in treatment conditions (particularly disinfection changes) that could make the tetravalent lead scales vulnerable to destabilization. Schock et al. (2001) found deposits in lead pipes of the Cincinnati, Ohio, distribution system that contained lead dioxide as the primary protective solid phase. Subsequent to these findings, different attributes of the theoretical solubility chemistry of lead dioxide were expanded upon, particularly the association with high free chlorine residuals and low oxidant demand.
Following the discovery of elevated lead concentrations after sections of Washington, DC, converted to chloramination, Renner (2004) described the link of the disinfectant change to the previous U.S. EPA research on tetravalent lead scale formation (Schock et al., 2001). Schock and Giani (2004) reported the results of tap monitoring history and scale analysis from the Water and Sewer Authority system in Washington, DC, confirming lead dioxide as the primary starting material; this validated the hypothesis that the lowering of ORP by changing from high dosages of free chlorine to chloramination caused high rates of lead dissolution. The laboratory experiments of Edwards and Dudi (2004) and Lytle and Schock (2005) confirmed that lead dioxide deposits could be readily formed and subsequently destabilized in weeks to months under realistic conditions of distribution system pH, ORP and alkalinity. A more recent laboratory study by Switzer et al. (2006) demonstrated that water with free chlorine oxidized lead to insoluble lead dioxide deposits and that lead was almost completely dissolved in a chloramine solution. These study findings further support the hypothesis that a change from free chlorine to chloramine can cause lead dissolution.
When hypochlorous acid is added to a water supply, it becomes a dominant oxidant on the copper surface (Atlas et al., 1982; Reiber, 1987, 1989; Hong and Macauley, 1998). Free chlorine residual was shown to increase the copper corrosion rate at lower pH (Atlas et al., 1982; Reiber, 1989). Conversely, free chlorine residual was shown to decrease the copper corrosion rate at pH 9.3 (Edwards and Ferguson, 1993; Edwards et al., 1999). However, Schock et al. (1995) concluded that free chlorine species would affect the equilibrium solubility of copper by stabilizing copper(II) solid phases, which results in a substantially higher level of copper release. The authors did not observe any direct effects of free chlorine on copper(II) solubility other than the change in valence state and, hence, the indirect change in potential of cuprosolvency.
Several authors reported an increase in the iron corrosion rate with the presence of free chlorine (Pisigan and Singley, 1987; Cantor et al., 2003). However, a more serious health concern is the fact that iron corrosion by-products readily consume free chlorine residuals (Frateur et al., 1999). Furthermore, when iron corrosion is microbiologically influenced, a higher level of free chlorine residual may actually decrease corrosion problems (LeChevallier et al., 1993). No information was found in the literature correlating iron levels with free chlorine residuals.
No information was found in the literature correlating free chlorine residual with cement pipe degradation.
Chloramines have been reported to influence lead in drinking water distribution systems. As noted previously, in 2000, the Water and Sewer Authority in Washington, DC, modified its disinfection treatment to comply with the U.S. EPA's Disinfection Byproducts Rule. The utility started using chloramines instead of chlorine for the purpose of secondary disinfection. Following this change, more than 1000 homes in Washington, DC, exceeded the U.S. EPA's action level for lead of 0.015 mg/L, and more than 157 homes were found to have lead concentrations at the tap greater than 300 µg/L (Renner, 2004; U.S. EPA, 2007). Since chlorine is a powerful oxidant, the lead oxide scale formed over the years had reached a dynamic equilibrium in the distribution system. Switching from chlorine to chloramines reduced the oxidizing potential of the distributed water and destabilized the lead oxide scale, which resulted in increased lead leaching (Schock and Giani, 2004; Lytle and Schock, 2005). The work of Edwards and Dudi (2004) also showed that chloramines do not form a low-solubility solid on lead surfaces, resulting in a greater probability of lead leaching into drinking water. The ORP brought about by chloramination favours divalent lead solids. Generally, lead solubility and lead release are dependent on pH, alkalinity and corrosion inhibitor (orthophosphate) concentration (Schock et al., 2005b). Thermodynamic models suggest that lead dioxide is relatively insensitive to orthophosphate and alkalinity. For chloramines to have an impact on lead release through the mechanism of ORP, lead dioxide formation and stability must occur. A study by Treweek et al. (1985) also indicated that under some conditions, chloraminated water is more solubilizing than water with free chlorine, although the apparent lead corrosion rate is slower.
Little information has been reported in the literature about the effect of chloramines on copper or iron. Some authors reported that chloramines were less corrosive than free chlorine towards iron (Treweek et al., 1985; Cantor et al., 2003). Hoyt et al. (1979) also reported an increase in red water complaints following the use of chlorine residual instead of chloramines.
No information was found in the reviewed literature linking chloramines and cement pipe degradation.
Studies have shown the effect of chloride on lead corrosion in drinking waters to be negligible (Schock, 1990b). In addition, chloride is not expected to have a significant impact on lead solubility (Schock et al., 1996). However, Oliphant (1993) found that chloride increases the galvanic corrosion of lead-based soldered joints in copper plumbing systems.
Chloride has traditionally been reported to be aggressive towards copper (Edwards et al., 1994b). However, high concentrations of chloride (71 mg/L) were shown to reduce the rate of copper corrosion at pH 7-8 (Edwards et al., 1994a,b, 1996; Broo et al., 1997, 1999). Edwards and McNeill (2002) suggested that this dichotomy might be reconciled when long-term effects are considered instead of short-term effects: chloride would increase copper corrosion rates over the short term; however, with aging, the copper surface would become well protected by the corrosion by-products formed.
Studies have shown the effect of sulphate on lead corrosion in drinking water to be generally negligible (Boffardi, 1988; Schock, 1990b; Schock et al., 1996). Sulphate was found to stifle galvanic corrosion of lead-based solder joints (Oliphant, 1993). Its effect was to change the physical form of the normal corrosion product to crystalline plates, which were more protective.
Sulphate is a strong corrosion catalyst implicated in the pitting corrosion of copper (Schock, 1990b; Edwards et al., 1994b; Ferguson et al., 1996; Berghult et al., 1999). Sulphate was shown to decrease concentrations of copper in new copper materials; however, upon aging of the copper material, high sulphate concentrations resulted in higher copper levels in the experimental water (Edwards et al., 2002). The authors concluded that this was due to the ability of sulphate to prevent the formation of the more stable and less soluble malachite and tenorite scales. However, Schock et al. (1995) reported that aqueous sulphate complexes are not likely to significantly influence cuprosolvency in potable water.
A review of lead levels reported by 365 water utilities, following the implementation of the U.S. EPA Lead and Copper Rule, revealed that higher chloride to sulphate mass ratios were associated with higher 90th-percentile lead levels at the consumer's tap. The study showed that 100% of the utilities that delivered drinking water with a chloride to sulphate mass ratio below 0.58 met the U.S. EPA's action level for lead of 0.015 mg/L. However, only 36% of the utilities that delivered drinking water with a chloride to sulphate mass ratio higher than 0.58 met the U.S. EPA's action level for lead of 0.015 mg/L (Edwards et al., 1999). Dudi and Edwards (2004) also conclusively demonstrated that higher chloride to sulphate mass ratios increased lead leaching from brass due to galvanic connections. High levels of lead in the drinking water of Durham, North Carolina, were found to be the primary cause of elevated blood lead concentrations in a child. This was initially believed to be linked with a change in the secondary disinfectant from chlorine to chloramine. However, upon further investigation, it was determined that a concurrent change in coagulant from alum to ferric chloride increased the chloride to sulphate mass ratio, resulting in lead leaching from the plumbing system (Renner, 2006; Edwards and Triantafyllidou, 2007).
No clear relationship between chloride or sulphate and iron corrosion can be established from a review of the literature. The studies of Larson and Skold (1958) found that the ratio of the sum of chloride and sulphate to bicarbonate (later named the Larson Index) was important (a higher ratio indicating a more corrosive water). Other authors reported that chloride (Hedberg and Johansson, 1987; Velveva, 1998) and sulphate (Velveva, 1998) increased iron corrosion. When sections of 90-year-old cast iron pipes were conditioned in the laboratory with chloride at 100 mg/L, an immediate increase in iron concentrations (from 1.8 to 2.5 mg/L) was observed. Conversely, sulphate was found to inhibit the dissolution of iron oxides and thus yield lower iron concentrations (Bondietti et al., 1993). The presence of sulphate or chloride was also found to lead to more protective scales (Feigenbaum et al., 1978; Lytle et al., 2003). However, neither sulphate nor chloride was found to have an effect on iron corrosion (Van Der Merwe, 1988).
Rapid degradation of cement-based material can be caused in certain cases by elevated concentrations of sulphate. Sulphate may react with the calcium aluminates present in the hydrated cement, giving highly hydrated calcium sulpho-aluminates. These compounds have a significantly larger volume than the initial aluminates, which may cause cracks to appear and reduce the material's mechanical strength. The effect of sulphate may be reduced if chloride is also present in high concentrations (Leroy et al., 1996).
Natural organic matter (NOM) has apparently been implicated in increasing lead solubility, and some complexation of dissolved lead by organic ligands has also been demonstrated. Some organic materials, however, have been found to coat pipes, thus reducing corrosion; therefore, a reasonable prediction cannot be made about the effect of various NOM on plumbosolvency (Schock, 1990b).
Research in copper plumbing pitting has indicated that some NOM may alleviate the propensity of a water to cause pitting attacks (Campbell, 1954a,b, 1971; Campbell and Turner, 1983; Edwards et al., 1994a; Korshin et al., 1996; Edwards and Sprague, 2001). However, NOM contains strong complexing groups and has been shown to increase the solubility of copper corrosion products (Korshin et al., 1996; Rehring and Edwards, 1996; Broo et al., 1998, 1999; Berghult et al., 1999, 2001; Edwards et al., 1999; Boulay and Edwards, 2001; Edwards and Sprague, 2001). Nevertheless, the significance of NOM to cuprosolvency relative to competing ligands has not been conclusively determined (Schock et al., 1995; Ferguson et al., 1996).
Several authors have shown that NOM decreases iron corrosion rate (Larson, 1966; Sontheimer et al., 1981; Broo et al., 1999). However, experiments conducted by Broo et al. (2001) revealed that NOM increased the corrosion rate at low pH values, but decreased it at high pH values. The authors concluded that this opposite effect was due to different surface complexes forming under different pH conditions. NOM was also found to encourage the formation of more protective scales (Campbell and Turner, 1983). However, NOM can complex metal ions (Benjamin et al., 1996), which may lead to increased iron concentrations.
Little information was found in the reviewed literature on the relationship between NOM and cement pipe degradation.
As noted above, there is no direct and simple method to measure internal corrosion of drinking water distribution systems. Over the years, a number of methods have been put forward to indirectly assess internal corrosion of drinking water distribution systems. The Langelier Index has been used in the past to determine the aggressivity of the distributed water towards metals. Coupon and pipe rig systems were developed to compare different corrosion control measures. As the health effects of corrosion (i.e., leaching of metals in the distribution system) became a concern, measuring the metal levels at the tap became the most appropriate method to both assess population exposure to metals and monitor corrosion control results.
Corrosion indices should not be used to assess the effectiveness of corrosion control programs, as they provide only an indication of the tendency of calcium carbonate to dissolve or precipitate. They were traditionally used to assess whether the distributed water was aggressive towards metals and to control for corrosion. These corrosion indices were based on the premise that a thin layer of calcium carbonate on the surface of a metallic pipe controlled corrosion. Accordingly, a number of semi-empirical and empirical relationships, such as the Langelier Index, the Ryzner Index, the Aggressiveness Index, the Momentary Excess and the Calcium Carbonate Precipitation Potential, were developed to assess the calcium carbonate-bicarbonate equilibrium. However, a deposit of calcium carbonate does not form an adherent protective film on the metal surface. The work of Edwards et al. (1996) has even shown that under certain conditions, the use of corrosion indices results in actions that may increase the release of corrosion by-products. In light of significant empirical evidence contradicting the presumed connection between corrosion and the most common of the corrosion indices, the Langelier Index, the American Water Works Association Research Foundation recommended that the use of corrosion indices for corrosion control practices be abandoned (Benjamin et al., 1996).
Coupons and pipe rig systems are good tools to compare different corrosion control techniques prior to initiating system-wide corrosion control programs. They provide a viable means to simulate distribution systems without affecting the integrity of the full-scale system. However, even with a prolonged conditioning period for the materials in the water of interest, coupons used in the field or laboratory and pipe rig systems cannot give an exact assessment of the corrosion of larger distribution systems. Such tests cannot reliably reflect population exposure to distribution system contaminants, since too many factors influence contaminant concentration at the consumer's tap.
The selection of the most appropriate materials for the conditions under study is critical to achieve the most reasonable approximation. The use of new plumbing material in simulators (e.g., pipe rigs) must be deemed to be appropriate for the corrosion of concern. For instance, new copper is appropriate when a water system uses copper in new construction. Leaded brass faucets are appropriate when permitted under existing regulatory regimes and available to consumers. Conversely, new lead pipe is not appropriate when looking at a system that has old lead service lines or goosenecks/pig-tails with well-developed scales of lead and non-lead deposits. In fact, predicting the behaviour of these materials in response to different treatments or water quality changes may be erroneous if appropriate materials are not selected for the simulator. Although no standards exist for designing simulators, there are publications that can help guide researchers on complementary design and operation factors to be considered when these studies are undertaken (AwwaRF, 1990, 1994).
Coupons inserted in the distribution system are typically used to determine the corrosion rate associated with a specific metal; they provide a good estimate of the corrosion rate and allow for visual evidence of the scale morphology. There is currently no single standard regarding coupon geometry, materials or exposure protocols in drinking water systems (Reiber et al., 1996). The coupon metal used must be representative of the piping material under investigation. The coupons are typically inserted in the distribution system for a fixed period of time, and the corrosion rate is determined by measuring the mass loss rate per unit of surface area. The duration of the test must allow for the development of corrosion scales, which may vary from 3 to 24 months, depending on the type of metal examined (Reiber et al., 1996).
The major drawback of coupons is their poor reproducibility performance (high degree of variation between individual coupon measurements). This lack of precision is due both to the complex sequence of handling, preparation and surface restoration procedures, which provides opportunity for analysis-induced errors, and to the high degree of variability that exists in metallurgical properties or chemical conditions on the coupon surface during exposure (Reiber et al., 1996).
Pipe rig systems are more complex than coupons and can be designed to capture several water quality conditions. Laboratory experiments with pipe rig systems can also be used to assess the corrosion of metals. In addition to measuring mass loss rate per unit of surface area, electrochemical techniques can be used to determine the corrosion rate. Furthermore, pipe rig systems can simulate a distribution system and/or plumbing system and allow for the measurement of contaminant leaching, depending on which corrosion control strategy is used.
These systems, which can be made from new materials or sections of existing pipes, are conditioned to allow for the development of corrosion scales or passivating films that influence both the corrosion rate of the underlying metal and the metal release. The conditioning period must allow for the development of corrosion scales, which may vary from 3 to 24 months, depending on the type of metal examined. Owing to this variability, 6 months is recommended as the minimum study duration (Eisnor and Gagnon, 2003).
As with coupon testing, there is currently no single standard for the use of pipe rig systems in the evaluation of corrosion of drinking water distribution systems. Eisnor and Gagnon (2003) published a framework for the implementation and design of pilot-scale distribution systems to try to compensate for this lack of standards. This framework identified eight important factors to take into consideration when designing pipe rig systems: (1) test section style (permanent or inserts), (2) test section materials, (3) test section diameter, (4) test section length, (5) flow configuration, (6) retention time, (7) velocity and (8) stagnation time.
Population exposure to contaminants resulting from the internal corrosion of drinking water systems arises from the corrosion of both the distribution system and the plumbing system. Measuring the contaminant at the tap remains the best means to determine population exposure. The degree to which a system has minimized corrosivity for the contaminant can also be assessed adequately through measuring the contaminant at the tap over time and correlating it with corrosion control activities.
The U.S. EPA recognizes and approves the following four analytical methods for the determination of lead in drinking water: (1) EPA Method 200.8 (U.S. EPA, 1994b), (2) EPA Method 200.9 (U.S. EPA, 1994b), (3) Standard Method 3113B (APHA et al., 2005) and (4) American Society for Testing and Materials (ASTM) Method 3559-96D (ASTM, 1996). Triantafyllidou et al. (2007) recently showed that particulate lead may not be sufficiently dissolved for analytical purposes if it is not acidified for a long enough period of time. Holding times for a minimum of 16 h after acidification are necessary for lead analysis, and heat digestion may be required when particulate lead is present.
Atomic absorption is the most common method for the determination of lead in water, with detection limits ranging from about 0.0006 to 0.001 mg/L (0.6-1 µg/L); the practical quantitation limit (PQL) for these methods is stated as 0.005 mg/L (U.S. EPA, 2000, 2006a).
A proprietary differential pulse anodic stripping voltammetry method, Method 1001 (no detection limit stated), by Palintest Inc. of Kentucky (U.S. EPA, 2006a), is also approved for analysis of lead in drinking water.
This document defines the levels of lead at the tap as the only measure used to initiate or optimize a corrosion control program. Nevertheless, control measures for copper and iron are also described here, since both the corrosion and concentrations of these metals will be largely influenced by the corrosion control method chosen.
Corrosion of drinking water systems and the release of contaminants into the conveyed water depend on both the material that is subject to corrosion and the water that comes in contact with the material. The contact time of the water with the material greatly influences the level of metals present in the drinking water. Therefore, a first mechanism of defence to reduce exposure to contaminants from drinking water is to flush the plumbing materials prior to human consumption of the water.
Drinking water can also be made less corrosive by adjusting its pH or alkalinity or by introducing corrosion inhibitors. Corrosion inhibitors and pH or alkalinity adjustments to control lead, copper or iron levels in drinking water should be employed with caution. Pilot studies should be conducted to determine the effectiveness of the corrosion control method chosen for the particular conditions prevailing in the distribution system. Furthermore, even though a particular method is effective in reducing lead, copper or iron levels in pilot tests, it might not be effective in practice when it is exposed to the particular conditions of the distribution system. Thus, rigorous full-scale monitoring should also be conducted before, during and following the initiation or optimization of a system's corrosion control program.
Reducing exposure to heavy metals can also be achieved, as an interim measure, by the use of certified drinking water treatment devices.
The judicious selection of materials (i.e., materials that contain little lead, such as leadfree solders, low-lead fittings or in-line devices) is one of the possible means to reduce population exposure to the contaminants of concern. For example, the use of lead-free solders ensures that less lead is found in the drinking water as a result of solder corrosion. Since 1990, the NPC has prohibited lead solders from being used in new plumbing or in repairs to plumbing for drinking water supplies. Lead up to a maximum of 0.2% is still allowed in lead-free solders under the NPC (NRCC, 2005).
Full replacement of the lead service line can significantly reduce lead concentrations at consumers' taps. Partial lead service line replacements (e.g., replacing only the utility or consumer's portion) can also reduce lead concentrations. However, lead levels typically do not decrease as significantly as with full service line replacement (U.S. EPA, 2004a). Both full and partial lead service line replacement can, however, disturb or dislodge existing lead scales and result in a significant temporary increase in lead levels at the tap (U.S. EPA, 2004a; Renner, 2007). This increase can occur for 3 or more months after replacing the lead service line. Partial replacement may also induce galvanic corrosion at the site where new copper piping is attached to the remaining lead pipe. Generally, utilities should make every effort to encourage consumers to replace their portion of the lead service line. Corrosion control measures that include partial or full replacement of the lead service line should ensure that appropriate flushing is conducted after the replacement and that debris is subsequently cleaned from the screens or aerators of outlets. Good record keeping of partial replacements is also strongly recommended for future reference (U.S. EPA, 2004a). The water quality at the consumer's tap should be monitored closely following both full and partial replacement, especially for the 1st month after replacement. Appropriate flushing should be conducted after the replacement of the service line and when elevated levels of lead are observed. Reducing exposure to lead can also be achieved, as an interim measure, by the use of drinking water treatment devices. It must be noted that in situations where high levels of lead are possible after replacement, drinking water treatment devices may have reduced capacity and require more frequent replacement.
Health Canada recommends that, where possible, water utilities and consumers use drinking water materials that have been certified as conforming to the applicable NSF/ANSI health-based performance standard (NSF/ANSI Standard 61 applies to drinking water system components) (NSF International, 2007). These standards have been designed to safeguard drinking water by helping to ensure material safety and performance of products that come into contact with drinking water.
A recent study by Dudi et al. (2004) used the NSF/ANSI Standard 61 Section 8 testing protocol to assess the lead leaching of in-line devices such as meters and shut-off valves. The study showed that in-line devices exposed to non-aggressive tap water leached less lead than did in-line devices exposed to the protocol's pH 5 water. In-line devices leached at least 4 times less lead than did brass hose bibs and 3.5 times less lead than did pure lead pipes using the pH 5 water. This was attributed to the fact that the protocol's pH 5 water contains 20-100 times more phosphate (a corrosion inhibitor) than is usually added to water by utilities to control lead leaching from drinking water. The authors concluded that there is no guarantee that the current protocol used by NSF/ANSI Standard 61 to measure lead leaching from in-line devices has anything to do with real water exposures, especially over long-term exposures.
In response to the questions raised by Dudi et al. (2004), statements of clarification are being added to the NSF/ANSI Standard 61 protocol to indicate that it is intended to assess the leaching potential for a group of metals, including lead. The standard's testing protocol is conducted at pH 5 and pH 10 to account for a variety of metals; metal analysis for both conditions must be met in order for a device or component to be certified under this standard. In the case of lead, a pH of 10 is considered to be aggressive.
The adjustment of pH at the water treatment plant is the most common method for reducing corrosion in drinking water distribution systems and leaching of contaminants in the distributed water. Raising the pH remains one of the most effective methods for reducing lead and copper corrosion and minimizing lead, copper and iron levels in drinking water. Experience has shown that the optimal pH for lead and copper control falls between 7.5 and 9.5. The higher spectrum of this pH range would also be beneficial in reducing iron levels, but may favour iron corrosion and tuberculation. Although increasing alkalinity has traditionally been recommended for corrosion control, it is not clear if it is the best means to reduce levels of lead and copper in drinking water. The literature appears to indicate that the optimal alkalinity for lead and copper control falls between 30 and 75 mg/L as calcium carbonate. Higher alkalinity (> 60 mg/L as calcium carbonate) is also preferable for the control of iron corrosion, iron level and red water occurrences. Moreover, alkalinity serves to control the buffer intensity of most water systems; therefore, sufficient alkalinity is necessary to provide a stable pH throughout the distribution system for corrosion control of lead, copper and iron and for the stability of cement-based linings and pipes.
Two predominant types of corrosion inhibitors are available for potable water treatment: phosphate- and silicate-based compounds. The most commonly used inhibitors include orthophosphate, polyphosphate (typically, blended polyphosphates) and sodium silicate, each with or without zinc.
The successful use of corrosion inhibitors is very much based on trial and error and depends on both the water quality and the conditions prevailing in the distribution system. The effectiveness of corrosion inhibitors is largely dependent on maintaining a residual of inhibitors throughout the distribution system and on the pH and alkalinity of the water.
Measuring the concentration of inhibitors within the distribution system is part of any good corrosion control practice. Generally, direct correlations between the residual concentration of inhibitors in the distribution system and the levels of lead, copper or iron at the tap are not possible.
Health Canada recommends that, where possible, water utilities and consumers choose drinking water additives, such as corrosion inhibitors, that have been certified as conforming to the applicable NSF/ANSI health-based performance standard or equivalent. Phosphate- and silicate-based corrosion inhibitors are included in NSF/ANSI Standard 60, Drinking Water Treatment Chemicals-Health Effects (NSF International, 2005). These standards have been designed to safeguard drinking water by ensuring that additives meet minimum health effects requirements and thus are safe for use in drinking water.
Recently, the use of tin chloride as a corrosion inhibitor for drinking water distribution systems has been added to NSF/ANSI Standard 60. However, very few experimental data on this inhibitor exist. Under certain conditions, this inhibitor reacts with the metal present at the surface of the pipe or the corrosion by-products already in place to form a more insoluble deposit on the inside walls of the pipe. Since the deposits are less soluble, levels of metals at the tap are reduced.
Orthophosphate and zinc orthophosphate are the inhibitors most often reported in the literature as being successful in reducing lead and copper levels in drinking water (Bancroft, 1988; Reiber, 1989; Boffardi, 1993; Johnson et al., 1993; Dodrill and Edwards, 1995; Rezania and Anderl, 1995, 1997; Schock et al., 1995; Boireau et al., 1997; MacQuarrie et al., 1997; Churchill et al., 2000; Schock and Fox, 2001; Becker, 2002; Dudi and Edwards, 2004; Kirmeyer et al., 2004). Some authors reported that the use of orthophosphate may reduce copper levels in the short term, but that in the long term the formation of more stable scales such as malachite and tenorite may be prevented (Schock and Clement, 1998; Edwards et al., 2001; Cantor et al., 2003). There is evidence that ineffective treatment for lead and copper with phosphate was successful when higher dosages were applied or when pH and orthophosphate dosages were optimized (Schock et al., 1996; Schock and Fox, 2001). Schock and Fox (2001) demonstrated successful copper control in high-alkalinity water with orthophosphate when pH and alkalinity adjustments were not successful. Typical orthophosphate residuals are between 0.5 and 3.0 mg/L (as phosphoric acid) (Vik et al., 1996).
Solubility models for lead and copper indicate that the optimal pH for orthophosphate film formation is between 6.5 and 7.5 on copper surfaces (Schock et al., 1995) and between 7 and 8 on lead surfaces (Schock, 1989). A survey of 365 water utilities under the U.S. EPA Lead and Copper Rule also revealed that utilities using orthophosphate had significantly lower copper levels only when pH was below 7.8 and lower lead levels only when pH was below 7.4 and alkalinity was below 74 mg/L as calcium carbonate (Dodrill and Edwards, 1995).
Several authors reported that orthophosphate reduced iron levels (Benjamin et al., 1996; Lytle and Snoeyink, 2002; Sarin et al., 2003), iron corrosion rates (Benjamin et al., 1996; Cordonnier, 1997) and red water occurrences (Shull, 1980; Cordonnier, 1997). Phosphate-based inhibitors, especially orthophosphate, were also shown to reduce heterotrophic plate counts and coliform bacteria in cast iron distribution systems by controlling corrosion. It was observed in an 18-month survey of 31 water systems in North America that distribution systems using phosphate-based inhibitors had fewer coliform bacteria compared with systems that did not have corrosion control (LeChevallier et al., 1996). Similarly, orthophosphate treatment at the rate of 1 mg/L applied to a highly corroded reactor made of cast iron immediately reduced iron oxide release and bacterial count in the reactor's water (Appenzeller et al., 2001).
The chloride, sulphate and orthophosphate salts of zinc have been found to provide substantial protection of asbestos-cement pipe when proper concentrations and pH ranges are maintained throughout the distribution system (Leroy et al., 1996). Zinc coats the pipe and protects it against fibre release and water attack. It is postulated that the zinc initially reacts with the water to form a zinc-hydroxycarbonate precipitate such as hydrozincite [Zn5(CO3)2(OH)6]. The zinc solid may then react with the pipe surface. A study of lead and asbestos-cement pipes in a recirculation system demonstrated that orthophosphate salt containing zinc provided corrosion inhibition for both types of pipe materials at pH 8.2 (Leroy et al., 1996).
Several authors reported that the use of polyphosphate could prevent iron corrosion and control iron concentrations (McCauley, 1960; Williams, 1990; Facey and Smith, 1995; Cordonnier, 1997; Maddison and Gagnon, 1999). However, polyphosphate does not act towards iron as a corrosion inhibitor but as a sequestrant, causing a decrease in the visual observation of red water (Lytle and Snoeyink, 2002). According to McNeill and Edwards (2001), this led many researchers to conclude that iron by-products had decreased, when in fact the iron concentrations or the iron corrosion rates may have increased.
The use of polyphospha te was reported as being successful at reducing lead levels in some studies (Boffardi, 1988, 1990, 1993; Lee et al., 1989; Hulsmann, 1990; Boffardi and Sherbondy, 1991). However, it was also reported as being ineffective at reducing lead concentrations and even detrimental towards lead in some circumstances (Holm et al., 1989; Schock, 1989; Holm and Schock, 1991; Maas et al., 1991; Boireau et al., 1997; Cantor et al., 2000; Edwards and McNeill, 2002). McNeill and Edwards (2002) showed that polyphosphate significantly increased lead in 3-year-old pipes for both 8-h and 72-h stagnation times. Increases in lead concentrations by as much as 591% were found when compared with the same conditions without inhibitors. The authors recommended not using polyphosphate to control for lead. Only limited data are available on the impact of polyphosphate on copper solubility. In a case study of three water utilities, Cantor et al. (2000) reported that the use of polyphosphate increased copper levels at the tap. In a copper pipe rig study, Edwards et al. (2002) reported that although polyphosphate generally reduced soluble copper concentrations, copper concentrations significantly increased at pH 7.2 and alkalinity of 300 mg/L as calcium carbonate, since polyphosphates hinder the formation of the more stable malachite scales.
Only limited data are available on the impact of sodium silicate on lead and copper solubility. As sodium silicate is a basic compound, it is always associated with an increase in pH, making it difficult to attribute reductions in lead or copper concentrations to sodium silicate alone when an increase in pH may also result in a decrease in lead and copper concentrations.
A study conducted by Schock et al. (2005a) in a medium-sized utility was able to solve problems from iron in source water as well as lead and copper leaching in the plumbing system. The problems were solved simultaneously through the addition of sodium silicate with chlorination. Sodium silicate was added to the three wells that contained elevated levels of iron and manganese and that serviced homes containing lead service lines. A fourth well required only chlorination and pH adjustment with sodium hydroxide. At the three wells, an initial silicate dose of 25-30 mg/L increased the pH from 6.3 to 7.5 and immediately resulted in 55% and 87% reductions in lead and copper levels, respectively. An increase in the silicate dose to 45-55 mg/L increased the pH to 7.5 and resulted in an even greater reduction in the lead and copper levels (0.002 mg/L and 0.27 mg/L, respectively). It is also interesting to note that the quality of the water after treatment, as it relates to colour and iron levels, was equal or superior to that prior to treatment. However, the use of sodium silicate alone was not shown conclusively in the literature to reduce lead or copper concentrations.
Between 1920 and 1960, several authors reported reductions in red water occurrences when using sodium silicate (Tresh, 1922; Texter, 1923; Stericker, 1938, 1945; Loschiavo, 1948; Lehrman and Shuldener, 1951; Shuldener and Sussman, 1960). However, a field study conducted in the distribution network of the City of Laval, Quebec, in the summer of 1997 revealed no beneficial effects of using low levels of sodium silicate (4-8 mg/L; pH range of 7.5-8.8) to control iron concentrations in old cast iron and ductile iron pipes. A camera inserted inside a cast iron pipe (1) prior to the injection of sodium silicate, (2) prior to the injection of sodium silicate and immediately following the mechanical removal of the tubercles and (3) after 5 months of sodium silicate use revealed that no reductions in the degree of tuberculation or the prevention of the formation of tubercles were found using sodium silicates at these low concentrations (Benard, 1998). Although very few studies have proven the efficiency of sodium silicates as corrosion inhibitors or their true mechanism of action, manufacturers recommend that a large dose of sodium silicate be initially injected to form a passivating film on the surface of the pipe. Manufacturers recommend concentrations ranging from 20 to 30 mg/L; once the film is formed, concentrations from 4 to 10 mg/L are recommended to maintain this film on the surface of the pipes (Katsanis et al., 1986).
Experiments that studied effects of high levels of silica at different pH found that at pH 8, silica may play a role in the stabilization of cement pipe matrix by interfering with the formation of protective ferric iron films that slow calcium leaching (Holtschulte and Schock, 1985).
Since the level of trace metals increases upon stagnation of the water, flushing the water present in the plumbing system can significantly reduce the levels of lead and copper. In that respect, flushing can be seen as an exposure control measure. A study by Gardels and Sorg (1989) showed that 60-75% of the lead leached from common kitchen faucets appears in the 1st 125 mL of water collected from the faucet. They further concluded that after 200-250 mL, 95% or more of the lead has normally been flushed from faucets (assuming no lead contribution from other sources upstream of the faucet). In a study on contamination of tap water by lead solders, Wong and Berrang (1976) concluded that the first 2 L of water from cold water taps should not be used for human consumption if the water has been stagnant for a day. In Canadian studies, in which the cold water tap of homes was flushed for 5 min, no concentrations of trace metals exceeded their respective Canadian drinking water guidelines at that time (Méranger et al., 1981; Singh and Mavinic, 1991). However, flushing the cold water tap in buildings may not be sufficient to reduce the levels of lead and copper below the guidelines (Singh and Mavinic, 1991; Murphy, 1993). Murphy (1993) demonstrated that the median lead concentration in samples collected from drinking fountains and faucets in schools had increased significantly by lunchtime after a 10-min flush in the morning. The authors concluded that periodic flushing throughout the day would be necessary to adequately reduce lead concentrations.
When lead service lines are the source of lead, flushing the system until the water turns cold is not an appropriate measure, since it is generally the point at which the water from the service line reaches the consumer. Collection of lead profiles at representative sites by sampling several litres sequentially can provide significant insights into lead leaching. It can also determine if flushing alone will be successful in reducing lead concentrations and the length of time required for flushing.
As noted previously, in 2000, Washington, DC utilities switched from chlorine to chloramines as the residual disinfectant in the distribution system. This caused very high levels of lead to leach, primarily from the service lines. Data collected during this corrosion crisis revealed that lead levels were not at the highest level in the first-draw samples at some homes, but were sometimes highest after 1 min of flushing (Edwards and Dudi, 2004). Samples collected after flushing were found to contain lead concentrations as high as 48 mg/L. In some cases, the concentration of lead in samples did not return to safe levels even after 10 min of flushing. In the end, Washington, DC, utilities advised their consumers to flush their water for 10 min prior to consumption and provided them with filters to remove lead (Edwards and Dudi, 2004). Flushing strategies included showering, laundry, toilet flushing or dishwashing prior to consuming the water first thing in the morning. It should be noted that in some cases flushing may not be sufficient to reduce lead concentrations at the tap. Therefore, utilities should conduct the appropriate monitoring to ensure that flushing is an appropriate measure before recommending flushing to consumers.
Good practice also calls for the flushing of larger distribution systems on a regular basis, especially in dead ends, to get rid of loose corrosion by-products and any attached microorganisms.
Maintenance activities, such as the routine cleaning of debris from aerators or screens on faucets, may also be important for reducing lead levels at the tap. Debris on aerators or screens can include particulate lead, which can be abraded and pass through the screen during periods of water use. This can result in a significant increase of particulate lead in the water from the tap, which can be variable and sporadic. Depending on the type of particulate lead present and the preparation method that is used for sample analysis, elevated lead concentrations can also be difficult to accurately measure (Triantafyllidou et al., 2007). For these reasons, it is also important to ensure that sampling is done with the aerator or screen in place so that potential particulate lead contributions may be detected.
Drinking water treatment devices can be installed at the point of entry or point of use in both residential and non-residential settings to further reduce contaminant concentrations. Since the concentrations of lead, copper and iron may increase in plumbing systems, and because exposure to these contaminants from drinking water is only a concern if the contaminants are ingested (i.e., inhalation and dermal absorption are not significant routes of exposure), point-of use treatment devices installed at drinking water taps are considered to be the best approach to reduce concentrations to safe or aesthetic levels immediately before consumption.
Health Canada does not recommend specific brands of drinking water treatment devices, but it strongly recommends that consumers look for a mark or label indicating that the device or component has been certified by an accredited certification body as meeting the appropriate NSF/ANSI drinking water material standards. These standards have been designed to safeguard drinking water by helping to ensure the safety and performance of materials that come into contact with drinking water. Certification organizations provide assurance that a product conforms to applicable standards and must be accredited by the Standards Council of Canada (SCC). Certification bodies can certify treatment devices for reduction of lead, copper and iron to the relevant NSF/ANSI standards. These standards list lead and copper as health-based contaminants, whereas iron is listed as an aesthetic-based contaminant. In Canada, the following organizations have been accredited by the SCC to certify drinking water devices and materials as meeting NSF/ANSI standards:
- Canadian Standards Association International (www.csa-international.org);
- NSF International (www.nsf.org);
- Water Quality Association (www.wqa.org);
- Underwriters Laboratories Inc. (www.ul.com);
- Quality Auditing Institute (www.qai.org);
- International Association of Plumbing & Mechanical Officials (www.iapmo.org).
An up-to-date list of accredited certification organizations can be obtained from the SCC (www.scc.ca).
Table 2 illustrates the water treatment technologies used in treatment devices that are capable of reducing lead, copper and iron concentrations in drinking water.
| Contaminant | Treatment technology | NSF/ANSI Standard |
Reduction claim
|
|
|---|---|---|---|---|
| Influent (mg/L) |
Effluent (mg/L) |
|||
| Lead | Adsorption (i.e., carbon/charcoal) | 53 | 0.15 | 0.010 |
| Reverse osmosis | 58 | 0.15 | 0.010 | |
| Distillation | 62 | 0.15 | 0.010 | |
| Copper | Adsorption (i.e., carbon/charcoal) | 53 | 3 | 1.3 |
| Reverse osmosis | 58 | 3 | 1.3 | |
| Distillation | 62 | 4 | 1.3 | |
| Iron | Filtration | 42 (aesthetic) |
3-5 | 0.3 |
The sampling protocols and action levels for the monitoring protocols presented below are based on an understanding of the variations in lead concentrations observed at the tap, which depend on the period of stagnation, the age and source of lead, and other factors, such as analytical limitations.
Previous residential monitoring programs conducted in the United States and Europe have demonstrated that lead levels at the tap vary significantly both across a system and within one site (Karalekas et al., 1978; Bailey and Russell, 1981; AwwaRF, 1990; Schock, 1990a,b; U.S. EPA, 1991a). As discussed in Section B.2.2, the concentration of lead at the tap depends on a variety of chemical and physical factors, including water quality (pH, alkalinity, temperature, chlorine residual, etc.), stagnation time, as well as the age, type, size and extent of the lead-based materials. Water use and the volume of water collected have also been identified as important factors affecting the concentration of lead at the tap. Statistically, the greater the variability, the larger the sample population size must be to obtain results that are representative of a system. In addition, when monitoring is conducted to assess the effectiveness of changes in a treatment approach to corrosion control, it is important to reduce the variability in the lead levels at the tap (AwwaRF, 1990). Monitoring programs must, therefore, include controls for the causes of variability in order to obtain results that are representative and reproducible (Schock, 1990a; AwwaRF, 2004; European Commission, 1999).
For residential monitoring programs, sampling considerations should include ensuring that sampling is done at the kitchen tap, with the aerator or screen on and at flow rates typically used (approximately 4-5 L/min) by consumers (van den Hoven and Slaats, 2006). These steps help to ensure that the sample collected is representative of the typical lead concentrations from the tap. In addition, it is recommended that the 4-L samples be taken in 1-L aliquots, since this lead profile will help provide the best information on the source of lead.
B.5.2 Determination of sampling protocols and action level for residential monitoring program: option 1 (two-tier protocol)
The objectives of the residential monitoring program: option 1 (two-tier protocol) are to identify and diagnose systems in which corrosion of lead from a variety of materials is an issue; to assess the potential for consumers to be exposed to elevated concentrations of lead; and to assess the quality and effectiveness of corrosion control programs. Consideration of the sampling protocols used in various studies of lead levels at the tap as well as studies on the factors that affect the variability of lead concentrations was given in the selection of the residential monitoring program: option 1. A two-tier approach was determined to be an effective method for assessing system-wide corrosion and identifying the highest potential levels of lead. It is also effective in providing the appropriate information for selecting the best corrective measures and evaluating the effectiveness of corrosion control for residential systems in Canada.
In some cases, the responsible authority may wish to collect samples for both tiers during the same site visit. This step eliminates the need to return to the residence if the action level for Tier 1 is not met. The analyses for the second tier are then done only on the appropriate samples, based on the results of the Tier 1 samples.
The first-tier sampling protocol determines the contribution of lead at the consumer's tap from the internal plumbing following a period of stagnation and from the transitory contact with the lead service line. A first-draw 1-L sample is taken at the consumer's cold drinking water tap (without removing the aerator or screen) after the water has been stagnant for a minimum of 6 h. When more than 10% of the sites (defined as the 90th percentile) have a lead concentration greater than 0.015 mg/L (lead action level), it is recommended that utilities take corrective measures, including conducting additional sampling following the Tier 2 sampling protocol.
The Tier 1 sampling protocol has been widely used for assessing system-wide lead levels and has been demonstrated to be an effective method for identifying systems both with and without lead service lines that would benefit from implementing corrosion control (AwwaRF, 1990; U.S. EPA, 1990, 1991a). The U.S. EPA conducted an analysis of extensive system-wide data and determined that if sufficient sites are sampled, then a Tier 1 sampling protocol can accurately represent lead concentrations across a system, as well as trigger corrective measures when needed (U.S. EPA, 1991a).
Each component of a sampling protocol, such as the stagnation time, the volume of water collected and the action level, has important implications to the overall assessment of corrosion in a system. Selection of a 6-h stagnation period is based on reducing the variability in lead levels at the tap as well as ensuring that high lead levels are detected if they occur in a system. The accurate detection of the high lead levels in a system is important so that the appropriate corrective measures can be implemented and it can be demonstrated that corrosion control has been optimized.
A recent review of variability in lead levels at the tap identified stagnation time as the most important physical factor in the consideration of a monitoring program (AwwaRF, 2004). The standard deviation of lead concentrations at the tap typically ranges between 50% and 75% (Schock et al., 1990a). Sampling following periods of stagnation that are closest to equilibrium has been demonstrated to reduce the variability in lead levels at the tap (Bailey et al., 1986; U.S. EPA, 1991a). Similarly, U.S. EPA experiments found that the lowest standard deviations of lead levels over a variety of stagnation times in lead pipe occurred in samples with a standing time greater than 6 h (AwwaRF, 1990).
Reducing the variability in lead concentrations at the tap is particularly important in systems where lead levels are close to the action level (0.015 mg/L) and in systems that need to demonstrate optimization. Selection of a stagnation time that is most likely to allow the highest lead concentrations to be measured is important to be able to demonstrate that a reduction in lead concentrations is due to treatment changes (or other corrective measures) rather than other factors that affect the variability of lead concentrations. Demonstrating that a significant reduction in elevated lead concentrations is due to corrosion control is subsequently important in evaluating whether a system has been optimized. In particular, as lead levels decline in a system, the variability also needs to be reduced so that there is an adequate degree of confidence that the decreases in lead levels are due to treatment changes (AwwaRF, 2004).
Lead has also been shown to leach, during no-flow periods, from soldered joints and brass fittings (Neff et al., 1987; Schock and Neff, 1988). Studies examining sources of lead at the tap have found leaded solder and brass fittings to be significant sources of elevated lead concentrations following a period of stagnation (Lee et al., 1989; Singh and Mavinic, 1991; AwwaRF, 2004; U.S. EPA, 2007). Depending on the age and type of the material, the concentrations of lead from brass fittings have been shown to increase significantly following stagnation periods between 4 and 20 h (Lytle and Shock, 2000). To increase the likelihood that high lead levels resulting from the corrosion of lead-based materials (leaded solder, brass fittings and in-line devices) are detected in a system, it is important to sample following a period of stagnation that approaches an equilibrium value (i.e., after more than 6 h).
The overnight stagnation period can vary considerably between sites; however, the mean overnight stagnation period has been reported to be 7.3 h (Bailey and Russell, 1981). The most conservative standing time prior to sampling is between 8 and 18 h, since it reflects peak concentrations of lead. However, this standing time might be difficult to achieve from an operational standpoint. It was shown that only negligible differences in lead concentrations exist between standing times of 8 and 6 h (Lytle and Schock, 2000). Six hours is therefore selected as the minimum standing time prior to sampling lead at the tap for residential monitoring program: option 1. It should be noted that it is not a requirement for the 6-h stagnation time to occur overnight; in order to facilitate the collection of Tier 1 samples from multiple residences in one day, utilities may consider collecting samples from residences that are vacant for 6 h during other periods of the day.
Sample volume is also considered to be an important factor in determining lead concentrations at consumers' taps (AwwaRF, 1990, 2004). Elevated lead concentrations have been observed in samples collected from the tap ranging from the 1st litre to the 12th litre, depending on the source and location of the lead-containing material in the distribution system. Recent studies (Campbell and Douglas, 2007; Huggins, 2007; Kwan, 2007; Sandvig, 2007; U.S. EPA, 2007; Craik et al., 2008) have indicated that the highest concentration of lead at the tap generally occurs in samples that are representative of water that has been stagnant in a lead service line. However, collection of multiple litres of water from each site is generally considered onerous; therefore, a smaller volume of water is often collected for conducting system-wide corrosion assessments. Several studies have indicated that collection of a 1-L sample following a 6-h stagnation period is effective at assessing system-wide corrosion levels (Frey, 1989; Lee et al., 1989; U.S. EPA, 1991a). Collection of 1 L determines the lead concentration resulting from contact with plumbing material that may contain lead, such as brass fittings and lead solder, following a period of stagnation, as well as the lead concentration in water that has been in transitory contact with a lead service line, if present. Data collected from several utilities in the United States indicate that lead concentrations in the 1st litre of water can be significantly elevated as a result of contributions from both the internal plumbing as well as the lead service line. The data indicated that the concentration of lead in the 1st litre of water was significantly higher in residences with lead service lines than in residences without lead service lines and was dependent on the water quality (Karalekas et al., 1978; U.S. EPA, 1990, 1991a). A recent study conducted by a Canadian utility similarly demonstrated that lead concentrations in the 1st litre are significantly higher in residences with lead service lines than in residences with non-lead service lines (Craik et al., 2008). Therefore, a 1-L sample volume is selected for the Tier 1 sampling protocol, since it provides informative data for assessing corrosion and is also practical for sample collection and analysis.
The action level for the Tier 1 sampling protocol is based on the concentration of lead in the site representing the 90th-percentile value, as measured during a monitoring event. Additional information on how to determine the 90th percentile is provided in Section C.2. A 90th-percentile value of 0.015 mg/L is selected on the basis of an analysis of lead concentrations across many systems conducted by the U.S. EPA (1990, 1991a). In the promulgation of the final Lead and Copper Rule, the U.S. EPA analysed data on lead levels at the tap collected from 39 medium-sized systems (over 100 000 sampling sites) representing a variety of water qualities. The data indicated that over 80% of the systems without lead service lines were able to achieve 90th-percentile lead concentrations of 0.015 mg/L with pH adjustment or the addition of corrosion inhibitors. Systems with lead service lines had substantially higher lead levels and only 25% of the systems were able to meet an action level of 0.015 mg/L; however, in these cases, additional corrective measures, such as lead service line replacement, were considered appropriate.
Selection of a 90th-percentile value of 0.015 mg/L for the Tier 1 action level is also based on analytical limitations. A 90th-percentile value of 0.015 mg/L is chosen because it does not require assumptions concerning values less than the PQL of the analytical methods of 0.005 mg/L. As data on lead concentrations in drinking water are generally a lognormal distribution, a large number of lead values from a system would be expected to be below the PQL for lead. If an average value were to be used, the analytical uncertainties and assumptions associated with values below the PQL could influence the results. The 90th-percentile value of 0.015 mg/L is also chosen as the most conservative value, since it corresponds approximately to an average lead concentration of 0.005 mg/L (U.S. EPA, 1991a). Concentrations of lead of 0.005 mg/L are considered to be maximum background levels found in water leaving the drinking water treatment plant. Lead concentrations greater than 0.005 mg/L indicate that lead is being contributed from brass fittings or meters, lead solder in plumbing or lead service lines present in the distribution or internal plumbing system.
The Tier 1 action level is intended to trigger corrective measures, including conducting additional sampling. If less than 10% of sites (defined as the 90th percentile) have lead concentrations above 0.015 mg/L (lead action level), utilities should provide customers in residences with lead concentrations above 0.010 mg/L with information on methods to reduce their exposure to lead. These measures can include flushing the appropriate volume of water prior to consumption following a period of stagnation, checking screens/aerators for debris that may contain lead, such as lead solder, and replacing their portion of the lead service line. It is also recommended that utilities conduct follow-up sampling for these sites to assess the effectiveness of the corrective measures undertaken by the consumer.
This sampling protocol will provide utilities with the water quality information needed to protect the most sensitive populations from unsafe concentrations of lead, by determining whether consumers need to be educated to flush their drinking water systems after periods of stagnation. The samples collected are also used from an operational standpoint to determine whether or not the water distributed has a tendency to be corrosive towards lead and, if so, to help determine the next steps that should be taken in implementing a corrosion control program. The Tier 1 sampling technique is considered to be the most informative when compared with other routine sampling techniques and should be used to increase the likelihood that system-wide problems with lead will be correctly identified, including the occurrence of elevated concentrations of lead resulting from an overnight stagnation period in contact with a variety of leaded materials.
Tier 2 sampling is required only when the first-tier sampling identified more than 10% of sites (defined as the 90th percentile) with lead concentrations above 0.015 mg/L (lead action level). Sampling is conducted at 10% of the sites sampled in Tier 1, specifically the sites at which the highest lead concentrations were measured. For smaller systems (i.e., serving 500 or fewer people), a minimum of two sites should be sampled to provide sufficient lead profile data for the system.
Four consecutive 1-L samples are taken at the consumer's cold drinking water tap (without removing the aerator or screen) after the water has been stagnant for a minimum of 6 h. Each 1-L sample is analysed individually to obtain a profile of lead contributions from the faucet, plumbing (lead in solder, brass and bronze fittings, brass water meters, etc.) and a portion or all of the lead service line. Alternatively, utilities that choose to collect four 1-L samples during the site visits for Tier 1 sampling can proceed with analysis of the remaining three 1-L samples once the analysis of the samples identifies the appropriate residences.
The objectives of Tier 2 sampling are to provide information on the source and potentially highest levels of lead, which will help utilities select the best corrective measures. It will also provide the best information for assessing the effectiveness and optimization of the corrosion control program.
In order to obtain information on the potentially highest levels of lead, sampling after a period of stagnation is important. In particular, the Tier 2 protocol is intended to capture water that has been stagnant not only in the premise plumbing but also in a portion or all of the lead service line (if present). Similarly to other leaded materials (i.e., leaded solder and brass fittings), lead concentrations in water that has been stagnant in lead pipe also increase significantly with time up to 8 h. Several factors affect the slope of the stagnation curves for lead pipe in drinking water. Generally, the concentration of lead increases rapidly in the first 300 min. The typical stagnation curve for lead pipe is very steep for stagnation times shorter than 6 h; therefore, small differences in the amount of time that water is left to stagnate may cause considerable variability in the lead concentration (Kuch and Wagner, 1983; AwwaRF, 1990, 2004; Schock, 1990a).
Another important factor that contributes to lead levels at the tap is the volume of water that has been in contact with the lead service line following a period of stagnation. Lead profiling studies conducted in Canada and the United States have indicated that the highest concentration of lead at the tap in residences with lead service lines occurs in samples that are representative of the water that has stagnated in the lead service line (Campbell and Douglas, 2007; Huggins, 2007; Kwan, 2007; U.S. EPA, 2007; Craik et al., 2008). Data from these studies indicate that when water is stagnant in the lead service line for 6 h, the maximum concentration of lead can be found between the 4th and 12th litres of sample volume. Generally, substantially elevated lead concentrations were observed in the 4th, 5th or 6th litres of sample volume in a number of studies (Campbell and Douglas, 2007; Douglas et al., 2007; Sandvig, 2007; Craik et al., 2008). Extensive profiling of lead levels in homes with lead service lines in Washington, DC, following a switch to chloramination demonstrated that the average mass of lead release (concentration adjusted for actual volume) attributed to the lead service line was 470 µg (73 µg/L) compared with 26 µg (26 µg/L) in the 1st-litre sample and 72 µg (31 µg/L) in samples from the remaining home piping and components prior to the lead service line (U.S. EPA, 2007).
Determining the potential for elevated concentrations of lead from water that has been stagnant in lead service lines is, therefore, an important component of a sampling protocol for assessing corrosion in residential distribution systems and subsequent corrosion control optimization. Comparing samples with the highest lead concentrations before and after corrosion control implementation will provide utilities with essential data in evaluating whether treatment has been optimized. This will ultimately help demonstrate that the highest lead levels have been reduced to the greatest extent possible. It is estimated that, in Canada, collection of a minimum of four 1-L samples following a period of stagnation of 6 h will increase the likelihood that the highest concentrations of lead will be detected. Since the volume of sample needed to obtain water that has been stagnant in the lead service line will depend on the plumbing configuration at each site, utilities should conduct a broad characterization of the types of high-risk sites to estimate if collection of four 1-L samples will be sufficient.
Collection of four 1-L samples to be analysed individually is selected, since this will provide a profile of the lead contributions from the faucet, the interior plumbing of the home and, in many cases, all or a portion of the lead service line. Previous studies have indicated that 95% of the lead contributed from faucets is flushed in the 1st 200-250 mL. In addition, the contribution from leaded solder can generally be found in the 1st 2 litres of water flushed from the plumbing system. Collection of four 1-L samples to be analysed individually will, therefore, provide the water supplier with information on both the highest potential lead levels at the tap as well as the source of the lead contamination. This information can then be used to determine the best corrective measures for the system and to provide data to help assess whether corrosion control has been optimized.
B.5.3 Determination of sampling protocols and action levels for residential monitoring program: option 2 (lead service line residences)
This protocol is intended to provide an alternative tool for jurisdictions in which sampling after a 6-h stagnation time is not practical or regulatory obligations restrict the use of the two-tier approach outlined above. Option 2 for residential monitoring programs measures the concentration of lead in water that has been in contact with the lead service line as well as with the interior plumbing (e.g., lead solder, leaded brass fittings) for a transitory and short period of time (30 min). Four consecutive 1-L samples are taken at the consumer's cold drinking water tap (without removing the aerator or screen) after the water has been fully flushed for 5 min and the water has then been left to stagnate for 30 min. Each 1-L sample is analysed individually to obtain a profile of lead contributions from the faucet, plumbing (lead in solder, brass and bronze fittings, brass water meters, etc.) and a portion or all of the lead service line. If the average lead concentration from the four samples taken at each site is greater than 0.010 mg/L at more than 10% of the sites (90th percentile) during one monitoring event, it is recommended that utilities take corrective measures.
Selection of this protocol as an alternative method for residential monitoring is based on adaptation of a sampling protocol used in a variety of European studies that were intended to estimate the average weekly exposure of consumers to lead at the tap (Baron, 1997, 2001; European Commission, 1999). Although the protocol was used in these studies for estimating the average weekly exposure, it may also be useful for obtaining information on the corrosivity of water towards lead pipe. It is, therefore, presented as a tool that can be used to identify residential sites with lead service lines that may have elevated lead concentrations. As discussed in detail below, the protocol has been adapted so that it can be used as a tool for assessing corrosion; however, it its intended to apply to residences with lead service lines only, owing to the limitations inherent to this protocol.
The European studies evaluated different sampling protocols to identify methods that could be used to estimate the average weekly concentration of lead at a consumer's tap (Baron, 1997, 2001; European Commission, 1999). In the European Commission (1999) study, common sampling methodologies (random daytime, fully flushed and fixed stagnation time) were compared with the composite proportional methodology to determine which one was the most representative of a weekly average amount of lead ingested by consumers. These studies identified the average lead concentration from two 1-L samples collected after the water had been fully flushed for 5 min and then left to stagnate for 30 min as an effective method for estimating the average lead concentration at a consumer's tap. The use of a 30-min stagnation time is also considered representative of the average inter-use stagnation time of water in a residential setting (European Commission, 1999; Baron, 2001; van den Hoven and Slaats, 2006).
The European studies from which this protocol was adapted also analysed the data to determine if a 30-min stagnation protocol could be used for identifying areas in a system that may have elevated lead concentrations. The authors concluded that if a sufficient number of samples were taken to provide statistically valid data, then collection of two consecutive 1-L samples of water following a 30-min stagnation period could potentially be used for conducting assessments of systems or areas in a system in which corrosion control should be implemented (European Commission, 1999; van den Hoven and Slaats, 2006).
An important aspect of these studies is that a large percentage of the sites that were monitored had internal lead plumbing or a lead service line, and the authors concluded that 80% of the properties that were not accurately detected as problem properties had lead service pipes and more than 5 m of non-lead internal plumbing (copper, galvanized steel or plastic). The studies indicated that collection of two consecutive 1-L samples of water would underestimate the average weekly exposure of consumers from these types of sites and that the collection of a larger volume of water would be needed to detect problem properties, but a volume was not specified. In addition, since 60% of the sites sampled in the European study had lead service lines or internal lead piping, sampling water that had been stagnant for 30 min in lead pipes was identified as a critical aspect of accurately identifying properties in which elevated lead concentrations may occur using this protocol.
Results of lead profiling from two Canadian utilities indicate that the concentrations of lead in the 4th, 5th or 6th litre of sample volume (lead service line) following a period of stagnation of 30 min are higher than the concentrations in the 1st and 2nd litres (Campbell and Douglas, 2007; Douglas et al., 2007; Craik et al., 2008). Increasing the volume of water collected that was used in the European studies to four consecutive 1-L samples should enable utilities to obtain a better understanding of systems in which corrosion of lead service lines may be an issue.
Data on lead levels at the tap in two Canadian cities, determined using a variety of sampling protocols, support the use of a protocol in which four consecutive 1-L samples are collected following a 30-min stagnation period as a tool for identifying residences in which elevated lead concentrations may occur as a result of corrosion from a lead service line (Douglas et al., 2007; Craik et al., 2008). These studies found that, in general, contributions from the lead service line can be detected following a 30-min stagnation time in the samples ranging from the 3rd to 6th litre. Therefore, this protocol is presented as an option for utilities that wish to conduct a residential monitoring program for sites with lead service lines. This method has not, however, been verified as being effective for assessing corrosion of distribution system materials other than lead service lines or internal lead plumbing. There are no data to indicate that it would be effective in assessing corrosion problems where only lead solder, brass fittings containing lead, piping or other leaded materials are present in the plumbing system.
The action level for residential monitoring program: option 2 is based on the guideline value for lead, the MAC of 0.010 mg/L. If the average concentration of lead in the four samples from the same site is greater than 0.010 mg/L at more than 10% of the sites (90th percentile), then responsible authorities need to implement corrective actions. The average concentration of lead is used for comparison with 0.010 mg/L, since it is believed that this will more accurately identify systems that will benefit from the implementation of corrosion control. Analysis of limited data from a Canadian utility (Campbell and Douglas, 2007) that has implemented corrosion control suggests that comparison of each individual sample with 0.010 mg/L may result in inappropriate conclusions regarding the need for treatment. An average value is, therefore, believed to be a more appropriate value for comparison with the action level. However, analysis of each individual litre of water collected is recommended so that a better understanding of the source of lead and the potentially higher lead concentrations can be obtained and used to select the appropriate corrective measures for the system.
If less than 10% of sites have average lead concentrations above 0.010 mg/L, utilities should provide consumers in residences with individual sample lead concentrations above 0.010 mg/L with information on methods to reduce their exposure to lead. It is also recommended that utilities conduct follow-up sampling for these sites to assess the effectiveness of the corrective measures undertaken by the consumer.
Since this protocol is being used for assessing corrosion after a short period of stagnation, contact with the lead service line is critical for increasing the likelihood that elevated lead concentrations will be detected if they occur. In cases where a utility is aware that the lead service line may be captured only with a greater sample volume, collection of four 1-L samples should be considered a minimum. Responsible authorities should be aware of the limitations of this protocol and should incorporate the practice of assessing potentially higher lead levels through sampling after a 6-h stagnation. A reduced subset of 6-h stagnation samples should be taken before and after corrosion control measures are implemented to ensure an accurate assessment of corrosion in the system and optimization of corrosion control.
In general, the objectives of a residential monitoring program are to identify and diagnose systems in which corrosion of lead from a variety of materials is an issue; to assess the potential for consumers to be exposed to elevated concentrations of lead; and to assess the quality and effectiveness of corrosion control programs. The residential monitoring program: option 2 for residences with lead service lines has not been assessed for these purposes; rather, it is intended as a tool for identifying elevated lead concentrations at residences with lead service lines. It is important to note that this sampling protocol has not been evaluated to determine its effectiveness for detecting corrosion of other plumbing materials, nor does it measure the potentially higher levels of lead that may be present in water stagnating for longer periods in the household plumbing and lead service lines.
A study by Kuch and Wagner (1983) indicates that concentrations of lead approach an equilibrium value after 5-7 h of stagnation, depending on the diameter of the pipes (correlating to 1/2-inch and 3/8-inch [1.3-cm and 1.0-cm]). In addition, the concentration of lead increases exponentially in the first 300 min of stagnation in lead pipe. Lead contributions from other materials, such as leaded brass fittings and lead solder, have also been found to increase significantly following 4-20 h of stagnation. There are limited field data comparing lead levels at the tap following different periods of stagnation; therefore, it is difficult to evaluate if a 30-min stagnation period is accurate for assessing corrosion. Limited studies suggest that lead concentrations following a period of stagnation of 30 min are substantially lower in the equivalent sample volume than those measured at the same tap following 6 h of stagnation (AwwaRF, 1990; Douglas et al., 2007; Craik et al., 2008). Therefore, the possibility of underestimating the highest concentration of lead at consumers' taps may be significant when using a stagnation time of 30 min.
In 1990, the U.S. EPA analysed extensive system-wide studies of lead levels at the tap determined by collection of a 1-L sample following 6 h of stagnation (first-draw) (U.S. EPA, 1990, 1991a). The analysis indicated that 1-L first-draw sampling was effective at identifying elevated lead concentrations from materials other than lead service lines in utilities that maintained a pH lower than 8 in the distribution system or that were not adding corrosion inhibitors. In analysing the data in the U.S. EPA document (U.S. EPA, 1991a), it is apparent that a 6-h stagnation period is an essential component of the sampling protocol. A large number of systems had lead concentrations between 15 and 30 µg/L; at these lower concentrations, a shorter stagnation period may not have detected the problems in these systems. A similar observation can be made from 1-L samples collected after a minimum of 6 h stagnation from lead service lines in 11 different systems. The U.S. EPA analysis of these data determined that 48% of these systems had 90th-percentile lead concentrations below 30 µg/L, and 25% of the systems had concentrations between 10 µg/L and 30 µg/L (U.S. EPA, 1991a). These data suggest that a longer stagnation time is important for accurately identifying systems that would benefit from corrosion control in the lower lead level range.
Several recent studies indicate the important contribution that leaded materials other than lead service lines can make to lead levels at the tap (Kimbrough, 2001; U.S. EPA, 2004a). Lead profiling studies conducted in Washington, DC, following a change from chlorination to chloramination indicated that elevated concentrations of lead from plumbing components such as brass fittings and tin-lead solder can occur when changes are made to the water quality. The average first-draw concentrations of lead from these profiles were 26 µg/L for the 1st litre and 31 µg/L for the premise plumbing (U.S. EPA, 2007). If sampling had occurred after a shorter stagnation period, it is possible that corrosion would not have been accurately assessed under these conditions.
One of the important aspects of a corrosion control program is to assess the quality and effectiveness of corrosion control treatment when it is implemented. Many studies have been conducted that have evaluated the effectiveness of corrosion control treatments by measuring the concentrations of first-draw lead before and after treatment is initiated. By correlating first-draw lead levels and the values of different water quality parameters, such as pH, alkalinity and inhibitor concentration, the effectiveness of the treatment can be evaluated (Karalekas et al., 1983; U.S. EPA, 1991a). Comparison of lead concentrations that are closest to maximum values is considered to be essential in order to determine if treatment has been optimized. In particular, systems in which the lead concentrations are only slightly above the action level (0.010 mg/L) may have difficulty demonstrating optimization unless the variability in lead levels in the system is small. Collection of samples after a longer stagnation period when the lead concentrations are likely to be higher may provide more statistically valid data for demonstrating optimization.
The European study from which the 30-min stagnation period protocol was adapted recommends various sampling protocols for evaluating the effectiveness of corrosion control treatment compared with the effectiveness of measures taken at individual properties such as lead pipe replacement. A 30-min stagnation period sampling protocol was identified as a suitable protocol for measuring the effectiveness of lead pipe replacement; however, it was not identified as a suitable protocol for assessing corrosion control treatment effectiveness (European Commission, 1999).
The suggested number of monitoring sites for different system sizes presented in Table 2 is adapted from the U.S. EPA Lead and Copper Rule (U.S. EPA, 1991a). In the development of the Lead and Copper Rule, the U.S. EPA estimated the minimum number of monitoring sites that would be required to adequately characterize the distribution of lead levels at the tap across a system. As part of this analysis, the U.S. EPA considered the need to ensure that the number of monitoring sites was reasonable for utilities as well as representative of system-wide lead levels at the tap. Data from studies of lead levels at the tap conducted by several utilities in the United States were used as the basis for a statistical analysis of system-wide lead levels. The data used in the analysis were based on first-draw 1-L samples following a minimum stagnation period of 6 h (AwwaRF, 1990).
The system-wide studies indicated that lead levels at the tap have a lognormal distribution, and between 25% and 40% of the locations sampled had lead concentrations greater than 0.015 mg/L. An analysis of the data was conducted using statistical sampling methods such as those used in quality control applications. The results of the analysis determined that the number of monitoring sites provided in Table 2 will correctly identify, with approximately 90% confidence, systems serving more than 100 people in which the 90th-percentile lead concentration is greater than 0.015 mg/L (U.S. EPA, 1991b). It should be noted that the U.S. EPA evaluation was conducted using data collected using a 6-h stagnation sampling protocol and an action level of 0.015 mg/L. Data using a 30-min stagnation period and an action level of 0.010 mg/L were not evaluated in this context. Therefore, the level of confidence that systems with lead concentrations above 0.010 mg/L will be accurately detected using this protocol is not known and cannot be easily estimated without doing a similar analysis with a much larger data set.
Given the variability of lead levels at the tap, additional measures are considered appropriate to increase the level of confidence that the suggested number of monitoring sites will detect elevated lead levels at the tap. Targeting high-risk locations such as those with lead service lines within the suggested number of monitoring sites as well as conducting sampling during periods when water temperatures are higher are expected to increase the level of confidence that the entire system has been adequately represented. The U.S. EPA indicated that monitoring two times per year and targeting high-risk residences are necessary to increase this level of confidence. This was noted as being particularly important for systems serving 100 or fewer people.
In the statistical analysis conducted by the U.S. EPA, it was noted that a 90% confidence level is not achieved with the suggested number of monitoring sites for systems serving fewer than 100 people; however, collecting a larger number of samples was not considered practical for these types of systems. The U.S. EPA determined that sampling the suggested number of sites at high-risk locations would, however, be reasonable for representing lead levels in the system (U.S. EPA, 1991b).
The objectives of the sampling protocols and action levels for non-residential sites, such as child care centres, schools and office buildings, are to locate specific lead problems within the buildings and identify where and how to proceed with remedial actions. The intention is to minimize lead concentrations at the cold drinking water outlets (i.e., fittings/fixtures such as faucets and fountains) used for drinking and cooking and therefore protect occupants from exposure to lead. The sampling protocols and action levels are based on an understanding of the variations in lead concentrations observed at outlets in a non-residential building resulting from sources of lead within the plumbing and water use patterns.
In some cases, responsible authorities may want to collect Tier 1 and Tier 2 samples at the same time to eliminate the need to return to the site. In this case, authorities should be aware that the confidence in some sample results will decrease, since flushing water through one outlet may compromise the flushed samples taken from other outlets that are located in close proximity.
A first-draw 250-mL sample is taken at the locations identified in the sampling plan after the water has been stagnant for a minimum of 8 h, but generally not more than 24 h. To ensure that representative samples are collected, the aerator or screen on the outlet should not be removed prior to sampling. If the lead concentration exceeds 0.020 mg/L (lead action level) at any of the monitoring locations, corrective measures should be taken.
The Tier 1 sampling protocol has been used in non-residential settings for locating specific lead issues, determining how to proceed with remedial measures and demonstrating that remediation has been effective. Numerous studies have been published on extensive sampling programs for measuring lead concentrations at the tap, conducted in schools and other nonresidential buildings. These studies demonstrated that collection of 250-mL samples following a period of stagnation of a minimum of 8 h, but generally not more than 24 h, is effective at identifying outlets with elevated lead concentrations (Gnaedinger, 1993; Murphy, 1993; Maas et al., 1994; Bryant, 2004; Boyd et al., 2008a,b). Using this sampling method, several studies were able to determine the source of lead within schools and develop a remediation plan (Boyd et al., 2008a,b; U.S. EPA, 2008).
As with residential monitoring programs, each component of a sampling protocol in nonresidential settings, such as the stagnation time, the volume of water collected and the action level, has important implications as to the usefulness of the data collected. Since the objectives of conducting sampling in non-residential buildings are different from those in residential settings, the volume of water collected and the lead action levels are also different.
The Tier 1 and Tier 2 sampling protocols for non-residential sites are based on collection of a 250-mL sample volume. Studies have demonstrated that to evaluate the amount of lead leaching from outlets such as kitchen faucets, more than 95% of the lead can be found in the 1st 200-250 mL of water from the faucet (Gardels and Sorg, 1989). Lead levels in non-residential buildings have generally been found to decrease significantly following flushing of the outlet for 30 s. This suggests that the fountain or faucet and the connecting plumbing components can be major contributors to elevated lead concentrations at outlets in non-residential buildings (Bryant, 2004; Boyd et al., 2008a,b). Collection of a larger volume of water, such as 1 L, would include a longer line of plumbing prior to the outlet. This plumbing may contain valves, tees and soldered joints that could contribute to the lead concentration in the 1-L sample; however, it would not be possible to identify which material was releasing the lead. In addition, it is suggested that collecting such a large volume from a drinking water fountain might dilute the initial high concentrations observed in the outlet. This is not desirable, since water collected from sections of plumbing farther from the outlet typically have lower lead concentrations (U.S. EPA, 2004b). Therefore, collection of a sample volume that is smaller (250 mL) than those typically used to assess corrosion (1 L and greater) in residential systems is considered important for sampling in non-residential buildings. A 250-mL sample volume is selected for sampling in non-residential buildings, as it represents water from the fitting (fountain or faucet) and a smaller section of plumbing and is therefore more effective at identifying the source of lead at an outlet (U.S. EPA, 1994a, 2006b).
As discussed in Section B.5.2.1, studies examining sources of lead at the tap have found leaded solder and brass fittings to be significant sources of elevated lead concentrations following a period of stagnation (Lee et al., 1989; Singh and Mavinic, 1991; AwwaRF, 2004; U.S. EPA, 2007). Depending on the age and type of the material, the concentrations of lead from brass fittings have been shown to increase significantly following stagnation periods between 4 and 20 h (Lytle and Schock, 2000). As a result, the water use pattern in a building is an important factor in determining lead concentrations at the tap. Since water use patterns are often intermittent in non-residential buildings, such as day care centres, schools and office buildings, it is important to sample following a period of stagnation. The most conservative standing time prior to sampling is between 8 and 18 h, since it is most likely to result in the measurement of peak concentrations of lead. Therefore, first-flush samples should be collected following a minimum period of stagnation of 8 h, but not greater than 24 h, so that they are representative of the longer periods in which outlets are not used for drinking during most days of the week in a non-residential building.
An action level of 0.020 mg/L is based on the premise that a first-draw sample is being collected that has a smaller volume (250 mL) than those typically collected to assess corrosion at residential sites, which are generally 1 L or greater. A direct comparison of the action levels for the residential protocols with the action level for the non-residential protocols is not possible. However, if additional volumes of water were collected following the initial 250-mL sample (i.e., 250-1000 mL), the result from this larger volume may correspond to a lower concentration when calculated as a 1-L sample. This is due to the fact that the subsequent volumes would most likely contain lower concentrations of lead than that seen in the initial 250-mL sample and result in a dilution effect (U.S. EPA, 2004b).
Tier 1 sampling is used to identify which outlets in a building may be contributing to elevated lead concentrations. When the action level of 0.020 mg/L is exceeded, interim corrective measures should be taken to protect the health of sensitive populations in situations with exposure patterns such as those found in non-residential buildings. Occupants of the building and other interested parties such as parents should be informed of the results of any sampling conducted in the building.
In order to help identify the source of lead at outlets that exceed the Tier 1 action level, follow-up samples are taken of the water that has been stagnant in the upstream plumbing but not in the outlet itself. The results can then be compared to assess the sources of elevated lead and to determine the appropriate corrective measures. In order to be able to compare the results, a second 250-mL sample is collected following the same period of stagnation. To obtain water that has been stagnant in the plumbing prior to the outlet, a 250-mL sample is taken after a period of stagnation of a minimum of 8 h, but generally not more than 24 h, followed by a 30-s flush. Water fountains and cold water outlets exceeding the Tier 1 action level are resampled in the same year and in the same season. The Tier 2 action level is also established at 0.020 mg/L, since the sample volume is 250 mL. Thirty-second flushing was selected, since it should normally eliminate the water present in the outlet.
If the lead concentration in the second 250-mL sample decreases below 0.020 mg/L (lead action level), then it can be concluded that the water fountain, the cold drinking water outlet or the plumbing in the immediate vicinity is the source of the lead. If concentrations of lead above 0.020 mg/L (lead action level) are found in the Tier 2 samples, then the lead sources may include the plumbing materials that are behind the wall, a combination of both the outlet and the interior plumbing or contributions of lead from the service connection. When lead concentrations exceed the Tier 2 action level, immediate corrective measures should be taken, the lead sources should be determined and remediation measures should be implemented.
The results of Tier 1 and Tier 2 sampling should be interpreted in the context of the plumbing profile so that an assessment of the lead contributions can be made and the appropriate interim and long-term corrective measures can be taken. Competent authorities can develop the plumbing profile using the questions provided in Section C.6. Information on other sampling that can be conducted to help determine the source of lead if it has not been identified as well as detailed information on the interpretation of Tier 1 and Tier 2 sampling results can be obtained from other reference material (U.S. EPA, 2006b).
In general, the level of lead in drinking water entering non-residential buildings from a distribution system is low. It is recommended that at each monitoring event, samples be taken from an outlet close to the point where the water enters the non-residential building. This will determine the concentration of lead contributed by either the service line or the main water distribution system (water main). Ideally, samples should be collected after an appropriate period of flushing so that they are representative of water from the service line and from the water main. The volume of water to flush will depend on the characteristics of the building plumbing system (i.e., the distance between the service line and the water main). In some situations (e.g., where there is a lead service line to the building), it may be difficult to obtain a sample that is representative of water from the water main as a result of contributions of lead from the service line. In this case, an alternative sampling location may need to be selected.
The occurrence of elevated lead concentrations within buildings such as schools is typically the result of leaching from plumbing materials and fittings and water use patterns (U.S. EPA, 2006b; Boyd et al., 2007; Pinney et al., 2007). Studies evaluating lead levels at drinking water fountains and taps in schools in Canada and the United States have demonstrated that levels can vary significantly within buildings and can be randomly distributed (Boyd et al., 2007; Pinney et al., 2007). An evaluation of lead levels in schools in Seattle, Washington, found that 19% of drinking fountains had concentrations of lead above 0.020 mg/L (lead action level) in the first-draw 250-mL samples (Boyd et al., 2008a). The lead was attributed to galvanized steel pipe, 50:50 lead-tin solder, brass components such as bubbler heads, valves, ferrules and flexible connectors. As a result, it is important to measure lead levels at fountains and outlets used for consumption in non-residential buildings to determine if elevated lead levels may be present and identify where corrective measures are required to protect the health of the occupants.
Although limited information is available on the variability of lead levels at individual fountains and outlets within non-residential buildings, studies have shown that it is not possible to predict elevated levels. The number of monitoring sites that should be sampled in a nonresidential building should be based on the development of a sampling plan. A plumbing profile of the building should be completed to assess the potential for lead contamination at each drinking water fountain or cold drinking water or cooking outlet. Competent authorities can develop the plumbing profile using the questions provided in Section C.6. Information in the plumbing profile can then be used to develop a sampling plan that is appropriate for the type of building that is being sampled (e.g., child care centres, schools, office buildings).
Authorities that are responsible for maintaining the water quality within non-residential buildings will need to do more extensive sampling at individual outlets based on the sampling plan developed for the building. The sampling plan should prioritize drinking water fountains and cold water outlets used for drinking or cooking based on information obtained in the plumbing profile, including, but not limited to, areas containing lead pipe, solder or brass fittings and fixtures, areas of stagnation and areas that provide water to consumers, including infants, children and pregnant women.
When sampling at kitchen taps in non-residential buildings, the aerators and screens should be left in place, and typical flow rates should be used (approximately 4-5 L/min). However, for other types of outlets, such as water fountains, lower flow rates are typical and should be used when sampling. These steps help to ensure that the sample collected is representative of the average water quality consumed from the type of outlet being sampled. It is also important to note that opening and closing shut-off valves to fittings and fixtures (i.e., faucets and fountains) prior to sampling have been shown to significantly increase lead concentrations (Seattle Public Schools, 2005). After opening a shut-off valve, outlets should be completely flushed and then allowed to stagnate for the appropriate period of time.