
Rapport final - Audit du recouvrement des coûts des produits de santé - juin 2015
Table des matières
- Sommaire
- A - Introduction
- B - Constatations, recommandations et réponses de la direction
- C - Conclusion
- Annexe A - Champs d'enquête et critères
- Annexe B - Grille d'évaluation
- Annexe C - Frais d'utilisation et activités connexes
- Annexe D - Normes de service, frais et résultats de rendement relativement à l'évaluation de médicaments
- Annexe E - Normes de service, frais et résultats de rendement concernant les évaluations d'instruments médicaux, les licences d'exploitation et le droit de vente
- Annexe F - Organigramme des activités de recouvrement des coûts
- Annexe G - Structure de gouvernance
- Annexe H - Répartition des revenus tirés des frais d'utilisation pour 2013-2014, par direction générale et catégorie de frais (en milliers de dollars)
Version traduite. En cas de divergence entre le présent texte et le texte anglais, la version anglaise a préséance.
Sommaire
Santé Canada (le Ministère) est le régulateur fédéral responsable d'évaluer la sécurité, la qualité et l'efficacité des médicaments, des instruments médicaux et des autres produits thérapeutiques, conformément au mandat qui lui est donné en vertu de la Loi sur le ministère de la Santé et de la Loi sur les aliments et drogues et le Règlement sur les aliments et drogues. Le financement des activités réglementées touchant les médicaments pour usage humain et les instruments médicaux dont les coûts sont recouvrables provient d'une combinaison de fonds publics (crédits) et de revenus obtenus auprès du secteur privé par le paiement de frais d'utilisation des services. La mise en place du cadre réglementaire pour les produits de santé relève de l'activité du Programme sur les produits de santé de Santé Canada et la Direction générale des produits de santé et des aliments est responsable de ce dossier.
L'objectif de cet audit était d'évaluer l'efficacité du cadre de contrôle de gestion en place, pour appuyer les activités de recouvrement des coûts relatives aux produits de santé et assurer la conformité avec les lois et les règlements applicables. La portée de l'audit comprend les activités de recouvrement des coûts liées au Règlement sur les prix à payer à l'égard des drogues et instruments médicaux. L'audit a été mené selon les Normes relatives à la vérification interne au sein du gouvernement du Canada et les Normes internationales pour la pratique professionnelle de l'audit interne.
L'audit a permis de conclure que le cadre de contrôle de gestion en place pour appuyer les activités de recouvrement des coûts pour les produits de santé et assurer la conformité avec les lois et règlements nécessite quelques améliorations.
La direction générale dispose d'une structure de gouvernance qui lui permet de veiller à la surveillance adéquate du recouvrement des coûts liés aux produits de santé à usage humain. Les rôles et les responsabilités sont définis et communiqués, et la haute direction reçoit des renseignements complets et opportuns pour l'appuyer dans la prise de décisions quotidienne. Cependant, il serait bénéfique de fournir au Ministère, sur une base annuelle, de l'information détaillée sur les coûts; la direction pourrait ainsi déterminer le moment du renouvellement des frais d'utilisation. En mars 2015, la direction générale a mis en place une nouvelle structure de gouvernance dans le but de simplifier et d'améliorer le processus de prise de décisions par le Comité exécutif de la direction générale (CEDG).
En ce qui concerne la gestion des risques, les risques opérationnels et financiers quotidiens liés aux activités de recouvrement des coûts sont communiqués au moyen de tableaux de bord et de rapports sur les écarts financiers. La gestion des questions opérationnelles, réglementaires et politiques qui nécessitent des solutions à plus long terme pourrait être renforcée par l'examen et la mise à jour périodiques d'un registre permanent des risques.
Dans l'ensemble, les systèmes de suivi et de production de rapports fournissent des renseignements complets, exacts et opportuns sur les revenus générés grâce aux frais d'utilisation et les activités d'imposition de frais d'utilisation. Les mesures de contrôle internes pourraient être rehaussées davantage par la mise en place d'un processus d'admissibilité à une remise des frais; par la modification de la date de renouvellement de la licence d'exploitation (LE) dans la réglementation, afin qu'elle soit harmonisée avec le cycle opérationnel; et par l'examen de la structure des frais d'utilisation exigés pour l'obtention d'une licence d'exploitation pour les produits pharmaceutiques (LEPP), afin qu'ils correspondent de plus près au modèle de coût connexe, de manière à ce que les frais d'utilisation liés exigés pour la LEPP puissent être clairement identifiés et expliqués dans les prochaines initiatives de renouvellement des frais d'utilisation.
Dans l'ensemble, l'information sur le rendement fait l'objet d'une surveillance et de rapports en regard des normes relatives aux frais d'utilisation des services. Les responsabilités concernant le tri et le premier examen des demandes d'homologation d'instruments médicaux de classes III et IV devraient être déléguées à des tiers indépendants. En ce qui concerne la gestion des ressources financières, les revenus perçus ont été répartis aux sous-programmes des médicaments et instruments médicaux en fonction des activités correspondantes relatives aux frais d'utilisation.
La direction est d'accord avec les six recommandations et a fourni un plan d'action qui permettra de consolider le cadre de contrôle de gestion à l'appui des activités de recouvrement des coûts pour les produits de santé.
A - Introduction
1. Contexte
Santé Canada (le Ministère) est le régulateur fédéral responsable d'évaluer la sécurité, la qualité et l'efficacité des médicaments, des instruments médicaux et des autres produits thérapeutiques, conformément au mandat qui lui est donné en vertu de la Loi sur le ministère de la Santé.
Conformément au Rapport sur les plans et les priorités 2013-2014 du Ministère, l'élaboration, la tenue à jour et la mise en place d'un cadre réglementaire pour les produits de santé relèvent de l'activité du Programme sur les produits de santé de Santé Canada, laquelle s'inscrit dans le résultat stratégique 2 : « Les risques et avantages pour la santé associés aux aliments, aux produits, aux substances et aux facteurs environnementaux sont gérés de façon appropriée et communiqués aux Canadiens. »
Le mandat de la Direction générale des produits de santé et des aliments (DGPSA) est d'adopter une approche intégrée à la gestion des risques et des avantages pour la santé liés aux produits de santé et aux aliments. L'activité du Programme sur les produits de santé de la DGPSA est appuyée par quatre sous-programmes :
- Médicaments pharmaceutiques : Ce sous-programme régit les médicaments pharmaceutiques à usage humain et vétérinaire, y compris les médicaments sur ordonnance et en vente libre, les désinfectants et les agents d'assainissement aux propriétés désinfectantes.
- Produits biologiques et radiopharmaceutiques : Ce sous-programme porte sur la réglementation des produits biologiques (produits issus de sources vivantes) destinés à un usage humain. Les produits réglementés comprennent notamment le sang et les produits sanguins, les vaccins antiviraux et bactériens, les produits de thérapie génique, les tissus, les organes et les xénogreffes, qu'ils soient fabriqués au Canada ou à l'étranger.
- Instruments médicaux : Ce sous-programme porte sur la réglementation des instruments médicaux destinés à un usage humain. Les instruments médicaux couvrent un large éventail d'instruments utilisés en santé et en médecine pour le traitement, l'atténuation, le diagnostic ou la prévention de maladies ou d'états physiques anormaux chez les humains.
- Produits de santé naturels : Ce sous-programme fournit le cadre réglementaire nécessaire au développement, à la mise à jour et à la mise en œuvre du programme des produits de santé naturels, qui comprennent les remèdes à base de plantes médicinales, les remèdes homéopathiques, les vitamines, les minéraux, les produits médicinaux traditionnels, les probiotiques, les amino-acides et les acides gras essentiels.
Les principales lois et politiques liées au recouvrement des coûts des produits de santé comprennent la Loi sur l'administration financière, la Loi sur le ministère de la Santé, la Loi sur les frais d'utilisation, ainsi que la Politique sur les autorisations spéciales de dépenser les recettes du Secrétariat du Conseil du Trésor, le Règlement sur les prix à payer à l'égard des drogues et instruments médicaux et le Règlement sur les aliments et drogues.
Les activités réglementées concernant les médicaments et les instruments médicaux à usage humain pour lesquels des frais ont été recueillis sont regroupées en six catégories de frais, comme suit :
- Évaluation des produits pharmaceutiques;
- Licence d'exploitation pour les produits pharmaceutiques;
- Droit de vente de produits pharmaceutiques;
- Évaluation des instruments médicaux;
- Licence d'exploitation pour les instruments médicaux;
- Droit de vente d'instruments médicaux.
Ces activités sont décrites plus en détail à l'annexe C.
La DGPSA est appuyée par d'autres directions générales dans la prestation d'activités d'imposition de frais d'utilisation. Le Bureau des régions et des programmes (BRP) mène des activités de vérification de la conformité et d'application de la loi relativement à la fabrication de produits de santé, à l'emballage/étiquetage, à la distribution, à l'importation, aux essais de produits et aux établissements de gros, principalement grâce aux services d'inspection requis pour la prestation et le renouvellement des licences d'exploitation pour les produits pharmaceutiques (LEPP) et les licences d'exploitation pour les instruments médicaux (LEIM). Il offre également un service de vérification de la conformité. La Direction générale du dirigeant principal des finances (DGDPF) et la Direction générale des services de gestion (DGSG) fournissent pour leur part un soutien fonctionnel (finances, TI et RH).
Les activités d'imposition de frais d'utilisation sont financées grâce à une combinaison de fonds publics (crédits) et de revenus tirés des frais d'utilisation payés par les demandeurs (autorisations de crédit net). Les frais d'utilisation pour les produits de santé pour usage humain ont été introduits au milieu des années 1990. Dès 2010, la proportion de financement public pour les activités réglementaires relatives aux produits de santé à usage humain était passée de 50 % à plus de 75 %.
En 2006-2007, la DGPSA a entrepris de renouveler les frais d'utilisation pour les médicaments et les instruments médicaux à usage humain et a mis au point un modèle d'établissement des coûts par activité afin de déterminer le coût unitaire total des services de réglementation. Un cadre de recouvrement des coûts a été élaboré et des consultations ont eu lieu avec l'industrie, conformément aux exigences prévues dans la Loi sur les frais d'utilisation (LFU), ce qui a mené, en 2010, à une Proposition au Parlement pour les frais d'utilisation et les normes de service pour le Programme des médicaments pour usage humain et des instruments médicaux. Conformément à la Proposition, les frais d'utilisation ont été établis de manière à atteindre un équilibre entre les sources de financement publiques et privées pour chaque catégorie de frais. Les ratios de partage des coûts ont été définis en fonction de l'avantage relatif pour l'industrie par rapport à l'avantage public tiré des activités réglementées.
Le Règlement sur les prix à payer à l'égard des drogues et instruments médicaux révisé a été mis en application en décembre 2011. Voir l'annexe D et l'annexe E pour consulter la grille des frais d'utilisation et des normes de service, ainsi que les résultats de rendement depuis la mise en place du nouveau règlement. Les autorisations de crédit net du Ministère pour recueillir et utiliser les revenus tirés des frais d'utilisation ont été augmentées en 2011.
La Proposition au Parlement soumise en 2010 comportait les revenus prévus et les coûts totaux pour les activités réglementées relatives aux médicaments pour usage humain et instruments médicaux. Le tableau 1 présente une comparaison entre les revenus et les coûts prévus au départ, ainsi que les prévisions révisées et les résultats réels de 2010-2011 à 2013-2014.
| Total des coûts (en millions de dollars) |
Total des revenus (en millions de dollars) |
Ratio du recouvrement des coûts | ||
|---|---|---|---|---|
| Sources d'information : Prévisions initiales : Proposition de Santé Canada au Parlement pour les frais d'utilisation et les normes de service pour le Programme des médicaments pour usage humain et des instruments médicaux (déposée en 2010); Prévisions révisées et chiffres réels : Rapport ministériel sur le rendement et rapports financiers fournis par l'agent principal des finances de la direction générale. |
||||
| 2010-2011 (anciens frais d'utilisation) | Réel | 172,0 $ | 48,9 $ | 28 % |
| 2011-2012 | Prévision initiale | 227,8 $ | 112,4 $ | 49 % |
| Réel | 203,5 $ | 73,8 $ | 36 % | |
| 2012-2013 | Prévision initiale | 244,9 $ | 114,6 $ | 47 % |
| Prévision révisée | 207,6 $ | 83,3 $ | 40 % | |
| Réel | 202,1 $ | 81,8 $ | 41 % | |
| 2013-2014 | Prévision initiale | 262,1 $ | 116,9 $ | 45 % |
| Prévision révisée | 206,1 $ | 88,3 $ | 43 % | |
| Réel | 211,5 $ | 84,2 $ | 41 % | |
2. Objectif de l'audit
L'objectif de l'audit était d'évaluer l'efficacité du cadre de contrôle de gestion en place pour appuyer les activités de recouvrement des coûts relatives aux produits de santé et assurer la conformité avec les lois et les règlements applicables.
3. Portée de l'audit
La portée de l'audit était centrée sur les activités de programme entreprises pendant l'exercice financier 2013-2014. Elle comprenait les activités de recouvrement des coûts pour les frais de médicaments pour usage humain et d'instruments médicaux. L'audit a permis d'examiner la gouvernance, la gestion des risques et les mesures de contrôle en place pour assurer la surveillance des coûts annuels, des revenus tirés des frais imposés, du rendement en regard des normes de services et des répercussions des frais d'utilisation sur la gestion du budget.
La portée ne comprenait pas les produits de santé naturels, puisqu'aucun droit pour le recouvrement des coûts n'est associé à ces produits. Quant aux médicaments à usage vétérinaire, ils n'ont pas été inclus parce qu'ils ne s'inscrivent pas dans la portée du Règlement sur les prix à payer à l'égard des drogues et instruments médicaux.
4. Méthode d'audit
La méthode d'audit comprend un examen du cadre législatif (lois, politiques et règlements) ainsi que de la documentation ministérielle comme les mandats, les comptes-rendus de décisions, les documents de procédures, les méthodologies d'établissement des coûts, les documents financiers et rapports de rendement internes et externes, les entrevues, les observations et les enquêtes. Cette méthode inclut également la vérification et l'analyse des transactions dans les six grandes catégories de frais, telles qu'elles sont énoncées à l'annexe C, pour évaluer l'exactitude de l'information sur les finances et le rendement fournie à la direction.
Les critères de l'audit, énoncés à l'annexe A, sont dérivés des lois, règlements et politiques du CT qui s'appliquent, ainsi que de la publication du Secteur de la vérification interne du Bureau du contrôleur général intitulée Critères de vérification liés au Cadre de responsabilisation de gestion : outil à l'intention des vérificateurs internes (mars 2011).
L'audit a été mené conformément à la Politique sur la vérification interne du gouvernement du Canada. Il a permis d'examiner des données probantes suffisantes, pertinentes, fiables et utiles et d'obtenir assez d'information pour offrir un niveau d'assurance raisonnable à l'appui de la conclusion de l'audit. Ce travail concret a été réalisé à l'administration centrale de Santé Canada.
5. Déclaration de conformité
Selon le jugement professionnel de la dirigeante principale de la vérification, les procédures d'audit et les données probantes recueillies étaient suffisantes et appropriées pour corroborer l'exactitude de la conclusion de l'audit. Les constatations et la conclusion de l'audit sont fondées sur une comparaison des conditions qui existaient à la date de l'audit, par rapport à des critères établis en collaboration avec la direction. En outre, les données probantes ont été recueillies conformément aux Normes relatives à la vérification interne au sein du gouvernement du Canada et aux Normes internationales pour la pratique professionnelle de l'audit interne. L'audit est conforme aux Normes relatives à la vérification interne au sein du gouvernement du Canada, comme le soutiennent les résultats du programme d'amélioration et d'assurance de la qualité.
B - Constatations, recommandations et réponses de la direction
1. Gouvernance
1.1 Structure de gouvernance et surveillance
Critère d'audit : Il y a une structure de gouvernance en place qui permet d'assurer la surveillance du recouvrement des coûts des activités liées aux produits de santé et qui soutient l'obtention de résultats.
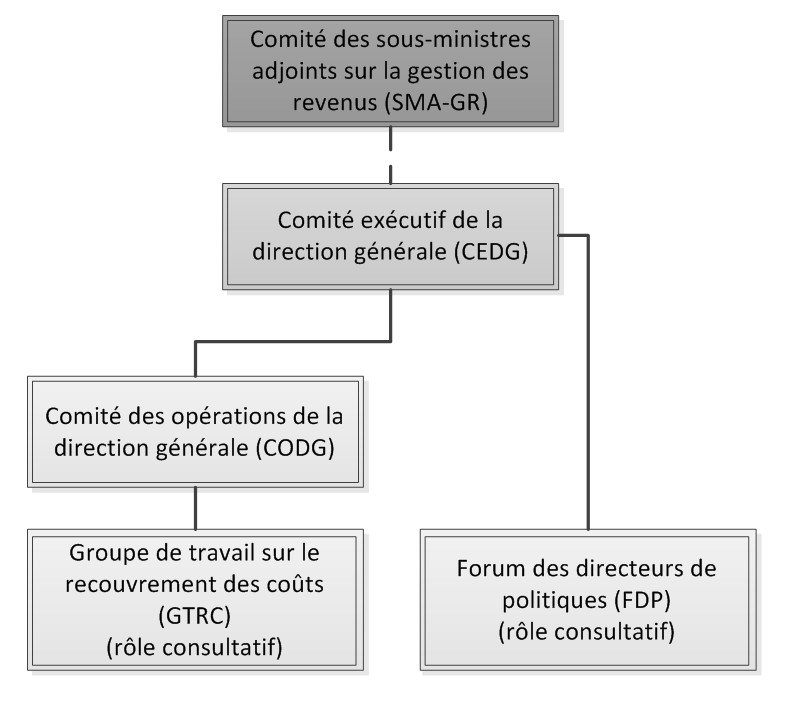
Une structure de gouvernance efficace s'avère essentielle pour veiller à ce que les activités réglementées dont les coûts sont recouvrables soient gérées et surveillées de manière à soutenir la prestation des services réglementaires de la Direction générale des produits de santé et des aliments (DGPSA) pour les médicaments pour usage humain et instruments médicaux, conformément aux lois, aux politiques et aux règlements qui s'appliquent. La prestation de services particuliers pour lesquels des frais d'utilisation sont imposés comporte plusieurs étapes et nécessite la contribution de plusieurs unités fonctionnelles de Santé Canada (le Ministère) (voir l'annexe F). La complexité des processus varie selon le niveau de dépendance entre les intervenants, la nature novatrice, la qualité et la quantité des données fournies avec les demandes et la synchronisation et la complexité de la facturation et de la collecte de revenus.
À l'échelon de l'ensemble des directions générales, le Comité des sous-ministres adjoints sur la gestion des revenus (SMA-GR) est l'organe décisionnel le plus élevé relativement à la gestion des revenus tirés des frais d'utilisation. Le président est le SMA de la DGPSA et les membres comprennent le SMA de la Direction générale des services de gestion (DGSG), le dirigeant principal des finances (DPF), le directeur général principal du Bureau des régions et des programmes (BRP) et le directeur général de la Direction de la gestion des ressources et des opérations (DGRO). Les réunions ont lieu tous les trimestres, ou à la demande du SMA de la DGPSA et la présence des membres varie selon les besoins. Le SMA-GR prend les décisions concernant la répartition annuelle des revenus entre les directions générales; la collecte des revenus et les écarts par rapport aux prévisions font l'objet d'une surveillance pendant l'exercice financier. Même s'il n'existe pas de mandat pour ce comité, les ordres du jour des réunions du SMA-GR, les discussions et les décisions sont documentés.
Au sein de la DGPSA, le Comité exécutif de la direction générale (CEDG) est l'organe décisionnel le plus élevé; il est responsable de la gestion cohérente et stratégique globale des ressources, des politiques et des responsabilités générales de la direction générale pour l'atteinte des objectifs. Les membres se réunissent chaque mois et son mandat consiste à jouer un rôle de leadership, à établir l'orientation quant à la stratégie, aux politiques et aux risques, et à en assurer une surveillance en vue de l'atteinte des résultats. Sa fonction de secrétariat est assumée par la DGRO. Les membres comprennent le SMA de la DGPSA (président) et tous les directeurs généraux (DG) de la DGPSA. Le CEDG reçoit périodiquement les tableaux de bord contenant des renseignements financiers, opérationnels et de rendement sur les médicaments pour usage humain et les instruments médicaux en vue des activités de recouvrement des coûts. L'information sur le rendement financier de la DGPSA est communiquée au CEDG au moyen du processus de production de rapports sur les écarts financiers (REF). Les ordres du jour des réunions, discussions et décisions du CEDG sont officiellement documentés.
Le Comité des opérations de la direction générale (CODG) assure une orientation stratégique et une méthode intégrée pour la gestion et les activités de la direction générale, selon un mandat plus large centré sur la gestion interne, les politiques, procédures et initiatives opérationnelles et les questions de gestion ministérielle. Il est présidé par le directeur général de la DGRO. Les autres membres sont les directeurs de chaque direction au sein de la Direction générale, un représentant de la Direction des ressources humaines et l'agent principal des finances de la direction générale. Dans le cadre de l'audit, le mandat quelques ordres du jour et comptes-rendus de décisions ont été examinés et il a été observé que les activités liées aux programmes font l'objet de discussions périodiques.
Le Forum des directeurs des politiques (FDP) joue un rôle consultatif auprès du CEDG. Le directeur de la Direction des politiques, de la planification et des affaires internationales (DPPAI) préside ce comité. Parmi les membres, on compte un directeur de chaque direction ayant un mandat portant sur les questions stratégiques en matière de programmes, y compris celles touchant le recouvrement des coûts. Les rencontres ont lieu officieusement de façon ponctuelle; des problèmes liés aux politiques sur le recouvrement des coûts font l'objet de discussions et des recommandations sont soumises au CEDG.
À l'échelon opérationnel, le Groupe de travail sur le recouvrement des coûts (GTRC) discute des questions qui touchent les activités d'imposition de frais d'utilisation à l'échelon de la direction et formule des recommandations au CODG et au CEDG. Bien que le GTRC ne prenne pas de décisions, il joue un rôle clé dans l'offre de conseils d'experts à la direction en ce qui a trait aux questions relatives au recouvrement des coûts. Le mandat comprend l'élaboration de données de base pour l'établissement des coûts pour l'ensemble des activités de la direction générale, à l'appui de plusieurs objectifs de la direction générale comme l'examen des frais actuels et la proposition de mises à jour ainsi l'amélioration de l'efficacité relativement à l'administration des frais d'utilisation. Plus précisément, le groupe de travail se penche sur l'élaboration de données de base pour l'établissement des coûts à l'échelon de la direction, lesquelles sont précisément centrées sur les activités de recouvrement des coûts. Le groupe de travail est présidé par un représentant de la DGRO et les membres sont des représentants de l'ensemble des directions de la DGPSA. L'Inspectorat de la Direction générale des produits de santé et des aliments (l'Inspectorat) mène le programme et représente les intérêts du BRP en ce qui a trait à l'établissement des coûts des activités.
Depuis la mise en place d'un règlement sur les frais d'utilisation en 2011, le rôle du GTRC a évolué et est désormais plus centré sur l'offre de soutien aux directions concernant les nouvelles questions de nature réglementaire, l'aide à la création d'outils d'analyse relatifs aux données de base pour l'établissement des coûts et la formulation de recommandations à la haute direction sur les questions stratégiques. Le mandat du GTRC n'a pas été mis à jour depuis mai 2012 et de nombreuses activités ne sont plus en cours (par exemple, le plan du projet de recouvrement des coûts de la direction générale et les budgets base zéro). La fréquence des réunions a aussi été modifiée, passant de bimensuelle à mensuelle. La direction générale pourrait profiter d'une mise à jour du mandat du GTRC afin d'assurer que sa mission, ses livrables et sa structure hiérarchique sont en lien avec les priorités actuelles de la direction générale.
Le graphique 1 illustre la structure de gouvernance avant mars 2015.
Graphique 1 : Structure de gouvernance avant mars 2015
{kind=link}

Le 18 mars 2015, la direction générale a dévoilé son nouveau modèle de gouvernance, qui a pour but de simplifier et d'améliorer la prise de décision, de mieux s'harmoniser avec la structure de gouvernance établie au Ministère et de centrer davantage les activités du CEDG sur la prise de décision stratégique (voir l'annexe G). Ce nouveau modèle de gouvernance sera appuyé par trois sous-comités.
- Le Sous-comité sur l'intégration des politiques, des programmes et de la science (CEDG-IPPS) sera responsable de soutenir les diverses initiatives de la Direction générale tout au long de leur élaboration et d'aligner celles-ci aux autres priorités de la direction générale. Ces initiatives comprennent l'élaboration de politiques, de lois, de règlements, de programmes et de projets internationaux et scientifiques.
- Le Sous-comité sur la transformation, la transparence, les investissements et les finances (CEDG-TTIF) travaillera à accroître l'efficacité et le rendement de la direction générale en abordant les questions financières, y compris la planification et la création de plans d'investissements.
- Le Sous-comité sur la gestion des talents et des ressources humaines (CEDG-GTRH) offre des solutions aux problèmes qui touchent le personnel de la DGPSA.
L'audit a conclu que la structure de gouvernance en place permet d'assurer une surveillance adéquate des activités de recouvrement des coûts liées aux produits de santé et que celle-ci soutient l'obtention de résultats. La DGPSA profiterait d'une mise à jour du mandat du Groupe de travail sur le recouvrement des coûts. Cependant, étant donné que la nouvelle structure de gouvernance a récemment été déployée, aucune recommandation ne sera formulée pour le moment.
1.2 Rôles et responsabilités
Critère d'audit : Les rôles et les responsabilités sont définis et communiqués.
Au moment d'examiner les rôles et les responsabilités dans le cadre de l'audit, on s'attendait à ce que les responsabilités soient clairement établies, communiquées et comprises, et que les processus relatifs aux intrants provenant des diverses directions générales, directions et divisions pour la coordination du travail, de la collecte de données, de l'analyse et de la production de rapports soient en place et efficaces.
La Direction générale des produits de santé et des aliments (DGPSA) assume la responsabilité principale en matière de gestion des risques liés à la santé et des avantages des produits de santé, y compris le développement d'un cadre de gestion pour le recouvrement des coûts des activités réglementés touchant les médicaments pour usage humain et les instruments médicaux.
Les activités réglementées de la DGPSA touchant les médicaments pour usage humain et les instruments médicaux comprennent l'évaluation précommercialisation et la surveillance post-commercialisation de la sécurité, de la qualité et de l'efficacité des médicaments, des vaccins, des instruments médicaux et autres produits thérapeutiques. Les activités de précommercialisation comprennent la délivrance de licences pour les instruments médicaux, de numéros d'identification des médicaments (DIN) et de licences d'exploitation. Les activités post-commercialisation comprennent la surveillance et la supervision des médicaments et des instruments médicaux homologués, ainsi que les activités de vérification de la conformité et d'application de la Loi, afin d'assurer que les parties réglementées satisfont aux exigences réglementaires avant que les licences d'exploitation puissent être renouvelées.
Les activités réglementées concernant les médicaments pour usage humain (produits pharmaceutiques, biologiques et radiopharmaceutiques) et les instruments médicaux pour lesquels des frais ont été recueillis sont regroupées en six catégories de frais, comme suit :
- évaluation des médicaments (EVAL-MED, produits pharmaceutiques et biologiques);
- licence d'exploitation pour les produits pharmaceutiques (LEPP);
- droit de vente de produits pharmaceutiques (DVPP);
- évaluation des instruments médicaux (EVAL-IMED);
- licence d'exploitation pour les instruments médicaux (LEIM);
- droit de vente d'instruments médicaux (DVIM).
Les descriptions de ces activités sont présentées plus en détail à l'annexe C.
Les directions au sein de la direction générale sont responsables de mener à bien les activités associées aux catégories de frais particulières, comme il est décrit à l'annexe F.
La Direction des produits thérapeutiques (DPT) régit les produits pharmaceutiques (EVAL-MED - produits pharmaceutiques) pour usage humain et les instruments médicaux (EVAL-IMED). Avant d'obtenir une autorisation de mise en marché, le fabricant doit présenter une preuve scientifique considérable de la sécurité, de l'efficacité et de la qualité de son produit, conformément à la Loi sur les aliments et drogues et au Règlement sur les aliments et drogues.
La Direction des produits biologiques et des thérapies génétiques (DPBTG) régit les médicaments biologiques (EVAL-MED - produits biologiques) et les produits radiopharmaceutiques pour usage humain au Canada, qu'ils aient été fabriqués au Canada ou ailleurs. Avant d'obtenir une autorisation de mise en marché, le fabricant doit présenter une preuve scientifique considérable de la sécurité, de l'efficacité et de la qualité de son produit, conformément à la Loi sur les aliments et drogues et au Règlement sur les aliments et drogues.
La Direction des produits de santé commercialisés (DPSC) est responsable de coordonner et de mettre en place une méthode cohérente de surveillance post-commercialisation, d'évaluation et d'intervention concernant tous les médicaments et instruments médicaux réglementés qui ont été mis sur le marché. Certains des coûts de ces activités sont couverts par les frais d'utilisation exigés pour le renouvellement annuel des licences de produits (DVPP et DVIM).
L'Inspectorat de la Direction générale des produits de santé et des aliments (l'Inspectorat) gère et exécute un programme national de vérification de la conformité et d'application de la Loi concernant le sang et le sperme de donneurs; les cellules, tissus et organes; les médicaments (pour usage humain et vétérinaire); les instruments médicaux et les produits de santé naturels. Il est aussi responsable de gérer le programme de vérification de la conformité et d'application de la loi à l'échelon national pour l'ensemble des produits s'inscrivant dans le mandat de la DGPSA, à l'exception des produits alimentaires. L'Inspectorat travaille de concert avec le Bureau des régions et des programmes (BRP), lequel procède aux inspections sur place qui sont requises pour le renouvellement des licences d'exploitation pour les produits pharmaceutiques et les instruments médicaux, ainsi qu'aux activités de vérification de la conformité. L'Inspectorat collabore aussi avec le Programme de laboratoire national, qui fournit une expertise scientifique et des résultats d'analyse à l'appui des activités d'intégrité frontalière, des inspections, de la vérification de la conformité et des enquêtes concernant les médicaments, les produits de santé naturels et certains instruments médicaux.
La Direction des politiques, de la planification et des affaires internationales (DPPAI) a pour mandat d'assurer un leadership dans l'élaboration et le développement des programmes politiques et internationaux de la direction générale. Cela comprend l'élaboration de politiques sur des questions horizontales; la modernisation des lois et des règlements; des activités visant à accroître l'influence du Canada en tant qu'autorité mondiale chargée de la réglementation; et l'intégration des politiques scientifiques.
La Direction de la gestion des ressources et des opérations (DGRO) a pour mandat d'améliorer le cadre de gestion de la direction générale, afin de veiller à ce que la DGPSA s'acquitte de ses obligations en matière de gestion et de gérance relativement aux ressources de la direction générale, et d'assurer que tous les gestionnaires de la direction générale disposent des outils dont ils ont besoin pour gérer efficacement les programmes. Plus précisément, la DGRO assure une coordination et un encadrement horizontaux à l'échelle de la Direction générale concernant la gestion cohérente, efficace et efficiente des activités et des ressources pour les programmes des médicaments pour usage humain et des instruments médicaux.
La DGRO a joué un rôle clé dans le soutien des directions tout au long de la mise en œuvre du nouveau règlement sur les frais, de la modernisation de la structure d'activités utilisée dans l'actuel modèle d'établissement des coûts et pendant la mise en place de SAP-PS, un système de suivi du temps qui permet aux employés d'enregistrer leurs heures de travail en regard d'activités prédéterminées. La DGRO continue de soutenir les directions et la direction générale concernant la gestion des nouveaux enjeux en matière de frais d'utilisation. De plus la DGRO recueille auprès des directions des renseignements sur les activités et le rendement, puis en fait l'examen. Cette information est consolidée dans des tableaux de bord et présentée à la haute direction lors des réunions du CODG et du CEDG.
La DGPSA est appuyée par d'autres directions générales dans la prestation d'activités d'imposition de frais d'utilisation. La Direction générale du dirigeant principal des finances (DGDPF) et la Direction générale des services de gestion (DGSG) lui fournissent un soutien fonctionnel (finances, TI et RH).
L'audit a permis d'examiner de la documentation comme des organigrammes, les mandats de comités, les plans d'activités des divisions et les documents d'orientation opérationnelle au sujet de diverses activités d'imposition de frais d'utilisation. Des entrevues réalisées auprès du personnel ont confirmé que les rôles et les responsabilités concernant les activités comme la production de rapports ministériels sur le rendement, l'établissement de rapports sur les écarts financiers et les tableaux de bord ont été définis et communiqués pour ce qui est de la responsabilité, la préparation, la présentation, l'examen et l'approbation.
L'audit a révélé que les rôles et les responsabilités ont été communiqués aux diverses unités opérationnelles, groupes de travail et comités, et que les procédures et lignes directrices visant à faciliter la gestion des activités d'imposition de frais d'utilisation et la prise de décisions étaient en place.
1.3 Information nécessaire à la prise de décisions
Critère d'audit : La haute direction reçoit de l'information complète, opportune et exacte à l'appui du processus de prise de décisions.
Il est essentiel que les organes de gouvernance reçoivent de l'information exacte et opportune pour soutenir la planification, la surveillance, la gestion des risques et la prise de décisions. La prise de décisions dans les activités quotidiennes ainsi que dans la planification à long terme, comme pour le renouvellement des frais d'utilisation, ne peut se faire sans des renseignements complets, opportuns et exacts.
Les renseignements opérationnels sur les charges de travail et les arriérés, le rendement en regard des normes de service et la situation financière sont recueillis et approuvés à l'échelon de la direction. Cette information est ensuite consolidée par la DGRO dans des tableaux de bord aux fins d'examen et d'approbation chaque mois par le sous-ministre adjoint (SMA), et chaque trimestre par le sous-ministre (SM). Les renseignements sur l'état financier et les prévisions sont examinés et approuvés depuis l'échelon du centre de coût jusqu'à l'échelon de la direction générale, pendant le processus d'établissement de rapports sur les écarts financiers (REF). L'agent principal des finances de la direction générale (APFDG) présente la situation financière de la direction générale au SMA afin d'obtenir son approbation, puis ces renseignements sont transmis aux membres du CEDG à titre d'information. L'audit a révélé que l'information opérationnelle contenue dans les tableaux de bord et les rapports financiers était opportune et complète et qu'elle soutenait la prise de décisions.
Les frais d'utilisation actuels sont fondés sur des ratios de partage de coûts privés/publics particuliers pour chaque catégorie de frais qui ont été établis selon le principe de l'atteinte d'un équilibre raisonnable entre le financement public et le financement privé. L'industrie, le Ministère et le Secrétariat du Conseil du Trésor (SCT) se sont mis d'accord sur les ratios à la suite de discussions et de négociations.
Les revenus tirés des frais d'utilisation et les coûts totaux par catégorie de frais sont publiés annuellement dans les tableaux du Rapport ministériel sur le rendement (RMR), conformément aux exigences de la Loi sur les frais d'utilisation (LFU). Un examen des tableaux du RMR a révélé que, depuis la mise en œuvre du nouveau règlement qui comprend une augmentation annuelle de 2 % des frais d'utilisation, les revenus générés depuis 2011 n'ont pas été aussi élevés que prévu et les ratios de recouvrement des coûts établis au départ n'ont pas été atteints (voir le tableau 2 pour de plus amples détails).
| Catégories de frais | Avant la proposition |
Après la proposition | Ratio d'avantages privé-public, par propositionTableau 2 - Note de bas de page * | |||
|---|---|---|---|---|---|---|
| Résultats réels 2010-2012 |
Résultats réels 2011-2012 |
Résultats réels 2012-2013 |
Résultats réels 2013-2014 |
Moyenne de 3 ans par frais d'utilisation |
||
|
Tableau 2 - Notes de bas de page
|
||||||
| Évaluation des médicaments (EVAL-MED) | 29 % | 37 % | 45 % | 46 % | 43 % | 75 % |
| Évaluation des instruments médicaux (EVAL-IMED) | 34 % | 34 % | 39 % | 32 % | 35 % | 75 % |
| Licence d'exploitation pour les produits pharmaceutiques (LEPP) | 39 % | 50 % | 47 % | 47 % | 48 % | 100 % |
| Licence d'exploitation pour les instruments médicaux (LEIM) | 36 % | 81 % | 56 % | 69 % | 69 % | 85 % |
| Droit de vente de produits pharmaceutiques (DVPP) | 16 % | 16 % | 20 % | 20 % | 19 % | 50 % |
| Droit de vente d'instruments médicaux (DVIM) | 36 % | 61 % | 75 % | 57 % | 64 % | 50 % |
La DGPSA a élaboré un processus officiel pour la prévision et l'analyse des revenus au niveau des catégories de frais (voir la section 3.1). Le suivi des heures consacrées aux activités de recouvrement des coûts est saisi dans le système de suivi du temps, jusqu'au niveau de la demande d'homologation dans certains cas; des efforts sont actuellement déployés pour mettre au point des outils d'établissement du coût unitaire (voir la section 3.2).
Dans le cadre de la Proposition au Parlement de 2010, Santé Canada s'est engagé à revoir les frais et les coûts de ses services après trois ans. Ainsi, la DGPSA a publié l'Examen de 2014 du Règlement sur les prix à payer à l'égard des drogues et instruments médicaux. Ce rapport donne un aperçu des activités réalisées au cours des trois dernières années, et fait référence au nombre croissant de cas où les demandes d'homologation deviennent plus complexes, contiennent plus de données sur les essais cliniques et nécessitent un niveau d'effort plus élevé à l'examen. Cependant, aucun renseignement sur l'établissement des coûts n'est fourni pour indiquer les répercussions de ces facteurs de coût. Bien que des efforts soient actuellement déployés pour la création d'outils d'analyse des coûts, il n'existe aucun processus officiel permettant d'analyser et de référencer les coûts annuels au niveau de la catégorie de frais. De tels renseignements s'avèrent essentiels à la direction pour définir une stratégie à long terme et déterminer à quel moment un renouvellement des frais actuels est justifié, le cas échéant.
La haute direction reçoit de l'information complète et opportune à l'appui du processus quotidien de prise décisions. L'analyse et la référenciation des coûts annuels au niveau des catégories de frais permettraient à la direction de déterminer le moment opportun pour le renouvellement des frais d'utilisation.
Recommandation 1
Il est recommandé que le sous-ministre adjoint, Direction générale des produits de santé et des aliments, mette au point un processus officiel d'analyse et de référenciation des coûts annuels au niveau des catégories de frais, afin d'appuyer la direction dans la détermination du moment opportun pour le renouvellement des frais d'utilisation.
Réponse de la direction
La direction souscrit à cette recommandation et établira un processus d'analyse formelle pour appuyer la décision quant à la détermination du moment opportun pour le renouvellement des frais d'utilisation.
Les coûts et les revenus sont présentés annuellement dans le Rapport ministériel sur le rendement. L'analyse des écarts est aussi effectuée annuellement. Dans le résumé de l'étude d'impact de la réglementation relative aux frais d'utilisation, Santé Canada s'est engagé à examiner les frais et les coûts aux trois ans et de proposer, le cas échéant, de nouveaux frais ou encore d'amender les frais existants en fonction de cet examen. Ce premier rapport d'examen du recouvrement des coûts a été publié en octobre 2014; le prochain rapport d'examen est prévu pour 2017-2018, y compris une analyse des coûts par catégorie de frais. Une révision des frais requiert des consultations exhaustives et une présentation au Parlement, et ce processus peut prendre de deux à quatre ans à compléter.
2. Gestion du risque
2.1 Gestion des risques
Critère d'audit : Les risques qui ont une incidence sur le recouvrent des coûts des produits de santé sont définis, évalués, documentés et gérés de façon continue.
Au moment d'examiner les pratiques de gestion des risques, on s'attendait qu'il y ait en place des processus d'identification, d'examen, de communication, d'atténuation et de surveillance des risques relativement aux activités de recouvrement des coûts.
Chaque année, les questions relatives aux risques sont saisies dans le Rapport sur les plans et les priorités, les plans de continuité des activités et les plans opérationnels de la direction générale. La direction générale indique dans son plan opérationnel que les risques font l'objet de discussions au sein de divers comités. Toutefois, de nouveaux risques ont émergé depuis la mise en application du règlement en 2011. Dans de nombreux cas, le montant d'effort requis pour traiter les demandes d'homologation augmente étant donné la complexité accrue de la science qui sous-tend le développement de nouveaux médicaments et instruments médicaux. En outre, les ratios d'avantages privé-public pour les revenus tirés des frais d'utilisation n'ont pas encore été réalisés, et les répercussions sur les revenus tirés des mesures d'atténuation des frais demeurent substantielles. Ces risques créent des pressions financières et opérationnelles pour lesquelles le Ministère doit prendre des mesures afin de pouvoir continuer de respecter ses engagements en matière de réglementation.
L'audit a révélé que les risques liés à la gestion financière sont documentés, communiqués et approuvés par les gestionnaires et directeurs, les DG et le SMA au niveau de la direction générale, au moyen de rapports sur les écarts financiers (REF). Les risques liés à la collecte des revenus font également l'objet de discussions lors des réunions du SMA-GR. Les risques associés aux activités et au rendement en regard des normes de service sont documentés et communiqués à l'aide de tableaux de bord consolidés, approuvés par les DG, et sont remis au SMA chaque mois et au SM chaque trimestre aux fins d'examen et d'approbation.
Les risques associés aux questions opérationnelles, réglementaires et politiques sont définis au moyen d'un processus informel au sein de chaque direction. Ces éléments sont ensuite présentés au DG approprié, au cas par cas, en fonction de l'importance du problème. Les problèmes non résolus par les DG sont acheminés à la DGRO, où ils sont enregistrés sur une liste permanente appelée « Questions en suspens », aux fins d'examen éventuel. L'examen des documents fournis par la DGRO a démontré que la plupart des questions relevées pendant l'audit ont été enregistrées. Cependant, certains documents n'étaient pas datés, les questions n'étaient pas classées en ordre de priorité selon le niveau de risque ni assignées à une division ou à une personne et les mesures potentielles, les autorisations requises et les échéanciers n'étaient pas indiqués. Ce genre de liste constitue un important outil de gestion des risques, permettant de prioriser les mesures requises pour le renouvellement éventuel des frais d'utilisation.
L'audit a révélé que les risques opérationnels et financiers quotidiens liés aux activités de recouvrement des coûts sont bien gérés au moyen de tableaux de bord et de rapports sur les écarts financiers. Toutefois, la DGPSA devrait renforcer la gestion des questions qui nécessitent des solutions à plus long terme en mettant au point et en maintenant un registre permanent des risques.
Recommandation 2
Il est recommandé que le sous-ministre adjoint, Direction générale des produits de santé et des aliments, mette au point un registre permanent des risques pour renforcer la gestion des questions qui nécessitent des solutions à plus long terme.
Réponse de la direction
La direction souscrit à cette recommandation. Elle développera un registre des risques pour traiter des questions relatives au recouvrement des coûts.
La DGPSA mettra au point un registre permanent des risques pour éclairer la planification à plus long terme.
3. Contrôles internes
3.1 Revenus tirés des frais d'utilisation
Critère d'audit : Les systèmes de suivi et de production de rapports fournissent des renseignements complets, exacts et opportuns sur les revenus générés grâce aux frais d'utilisation.
Il est important que des processus et des systèmes soient en place pour soutenir, de façon rapide et exacte, l'enregistrement, le suivi, la collecte et l'établissement de rapports relativement aux revenus tirés des frais d'utilisation, et ce, afin d'assurer une surveillance adéquate de la gestion des revenus.
Les revenus tirés des frais d'utilisation sont générés grâce au traitement des présentations et demandes d'homologation reçues de l'industrie tout au long de l'année. Le Règlement sur les aliments et drogues comprend des dispositions énonçant le montant des frais à facturer et le moment où le paiement est dû, et ce, pour chaque catégorie de frais (voir l'annexe D et l'annexe E). Les frais d'utilisation consistent principalement en un tarif uniforme pour les demandes d'homologation et les présentations s'inscrivant dans une catégorie de frais, à l'exception des frais de licence d'exploitation pour les produits pharmaceutiques qui sont fondés sur le nombre de « composantes de coût » compris dans la demande de licence. La DGPSA prépare des prévisions de revenus annuels pour chaque catégorie de frais en fonction des volumes d'activités antérieurs et de l'information prospective reçue de l'industrie. Une fois les prévisions de revenus approuvées, les revenus prévus sont répartis entre la DGPSA, le BRP, la DGDPF et la DGSG au moyen du système financier SAP en tant que montants annuels prévus au budget. Après que les montants des revenus sont inscrits dans le budget, seuls les fonds provenant des revenus recueillis peuvent être dépensés; ainsi il y a toujours un risque de manque à gagner entre les collectes réelles et les revenus prévus. Le calendrier des paiements n'est pas le même pour l'ensemble des catégories de frais et certains frais sont recueillis dans un exercice financier différent de l'année dans laquelle le travail a été achevé. Dans le cas des demandes d'homologation de médicaments, lorsque les frais dépassent les 10 000 $, une proportion de 75 % des revenus est recueillie avant que la présentation ou la demande d'homologation ait été entièrement traitée. Ainsi, le traitement de certaines demandes d'homologation peut se faire sur plus d'un exercice financier. Afin d'assurer une correspondance adéquate des revenus et des dépenses, la DGPSA reporte à l'exercice suivant la portion des revenus qui n'a pas été gagnée au cours d'un exercice donné.
Les directions offrant des services pour lesquels des frais d'utilisation sont imposés sont responsables de l'exactitude des factures émises. Bien que les demandeurs soumettent le paiement des frais d'utilisation avec leurs demandes, les montants finaux à facturer sont déterminés et saisis dans le système de gestion financière du Ministère (SAP) par le personnel de la division qui s'occupe du tri des demandes d'homologation de médicaments et d'instruments médicaux ou du traitement du renouvellement annuel des demandes de licences de produits pharmaceutiques et d'exploitation. Les factures sont générées dans SAP. Chaque type de frais d'utilisation dans chacune des catégories de frais est associé à un code matières, lequel est relié à un compte de grand livre (GL) prédéterminé dans SAP. Les rapports SAP fournissent de l'information sur les revenus en fonction de la catégorie de frais ou du code matières. Les montants reçus des directions sont acheminés à la DGDPF et sont déposés sur-le-champ.
Les revenus recueillis sont répartis entre la DGPSA, le BRP, la DGDPF et la DGSG selon des proportions préétablies approuvées par le comité du SMA-GR. La DGPSA réattribue ensuite les revenus aux différentes directions et divisions qui mènent des activités d'imposition de frais d'utilisation. L'annexe H présente en détail la répartition des revenus par catégorie de frais et par direction générale pour 2013-2014. La section 3.4 du présent rapport contient d'autres explications concernant le processus de gestion budgétaire.
L'audit comprenait un examen des règlements applicables, des processus de facturation et des mesures de contrôle, ainsi qu'une vérification des transactions de revenus pour l'ensemble des catégories de frais. L'audit a permis de constater que, dans l'ensemble, les systèmes de suivi et d'établissement de rapports offrent des informations complètes et exactes sur les revenus tirés des frais d'utilisation, et ce, en temps opportun. Toutefois, l'audit a constaté les faits suivants.
- La date de renouvellement des licences d'exploitation crée des défis relatifs à la gestion de la trésorerie, puisque ces revenus sont perçus à la fin de l'exercice financier.
- La complexité de la structure tarifaire pour les licences d'exploitation de produits pharmaceutiques comporte un risque possible d'erreurs de facturation.
- Aucun processus n'est en place pour déterminer les conditions d'admissibilité à la remise de frais (c'est-à-dire, une réduction du montant des frais d'utilisation).
Date de renouvellement des licences d'exploitation (LE)
La réglementation actuelle exige que les demandes et le paiement des frais d'utilisation pour le renouvellement annuel des LE soient soumis par les titulaires de licence avant le 1er avril de chaque année, ce qui génère l'entrée de revenus considérables à la fin de l'exercice financier. Le traitement des demandes de renouvellement des LE reçues à la fin de l'exercice financier risque fort de se faire pendant l'exercice suivant. Comme les revenus ne peuvent pas être reportés d'un exercice financier à l'autre, la gestion de la trésorerie devient plus complexe en raison des revenus qui entrent à la fin de l'exercice financier. Il est difficile de prévoir le montant des frais d'utilisation qui sera recueilli avec les renouvellements avant la fin de l'exercice financier et au début du nouvel exercice (1er avril), ce qui réduit la stabilité et la prévisibilité des revenus de la LE servant à financer les activités liées aux LE. Dans le cas des licences d'exploitation, en 2012, cette situation a entraîné l'entrée d'environ 5 millions de dollars en revenus après le 31 mars en liaison avec des licences de l'exercice financier précédent (c'est-à-dire, 2011-2012). Cette situation s'est répétée en 2012-2013 et a eu une incidence encore plus grande, puisque 9 millions de dollars ont été recueillis dans l'exercice financier subséquent, soit 2013-2014.
Depuis 2013-2014, afin de revoir les demandes et de recueillir les revenus connexes dans le même exercice financier, la DGPSA encourage ses clients à soumettre leur demande d'examen annuel des LE avant la mi-février, afin d'accélérer le traitement et l'émission de la facture. Cela a permis à l'Inspectorat de renouveler bon nombre de licences et de recueillir des revenus avant la fin de l'exercice financier. La direction a mentionné que des efforts étaient actuellement déployés pour ajuster la date de renouvellement dans le Règlement sur les aliments et drogues.
Complexité de la structure tarifaire pour les licences d'exploitation de produits pharmaceutiques
L'audit a permis de constater que la facturation des LEPP axée sur les composantes de coût s'avère une méthode manuelle complexe et fastidieuse qui nécessite une bonne compréhension de la structure tarifaire afin de déterminer le total des frais pour chaque demande; le risque d'erreur humaine est donc élevé. L'Inspectorat reconnait que la structure tarifaire est complexe, et qu'une simplification de celle-ci permettrait de réduire la possibilité de risques d'erreurs dans la facturation.
À la section 3.2 du présent rapport, la recommandation 5 porte sur le fait que la structure tarifaire pour les LEPP et la structure des activités du modèle d'établissement des coûts doivent être davantage harmonisées pour appuyer la définition de frais d'utilisation éventuels. Cette initiative donnerait l'occasion à la DGPSA de simplifier la structure tarifaire pour les LEPP et de réduire le risque d'erreurs dans la facturation.
Remise des frais d'utilisation
De nouvelles mesures d'atténuation ont été introduites dans la réglementation des frais révisée, pour les cas où les frais entraîneraient un fardeau déraisonnable pour certains groupes ou payeurs individuels de frais d'utilisation. La méthode du Ministère concernant la réduction des frais est centrée sur l'offre d'un accès moins restrictif aux produits afin d'aider les Canadiens à maintenir et à améliorer leur santé, et de soutenir l'accès aux nouveaux produits. Par exemple, la remise des frais est accessible aux entreprises à faible revenu. Les nouvelles entreprises peuvent demander un report de paiement des frais, et une exemption spéciale est accordée pour les médicaments et instruments médicaux destinés à la vente à des fins humanitaires internationales. Cependant, il est entendu que la réduction des frais a une incidence sur les attentes en matière de revenus. Comme il est illustré dans le tableau 3, les répercussions de la réduction des frais ont augmenté, passant de 14 % des frais d'utilisation totaux en 2011-2012 à 20 % en 2013-2014.
| Remise | 2011-2012 | 2012-2013 | 2013-2014 | |||
|---|---|---|---|---|---|---|
| Montant remis | Pourcentage des revenus | Montant remis | Pourcentage des revenus | Montant remis | Pourcentage des revenus | |
| Source : Examen de 2014 du Règlement sur les prix à payer à l'égard des drogues et instruments médicaux (non audité). | ||||||
| EVAL-MED | 110 809 $ | < 1 % | 411 407 $ | 1 % | 99 445 $ | <1 % |
| EVAL-IMED | 136 853 $ | 2 % | 270 128 $ | 4 % | 210 600 $ | 3 % |
| DVPP | 3 437 969 $ | 27 % | 4 000 554 $ | 25 % | 4 246 334 $ | 25 % |
| DVIM | 2 343 880 $ | 26 % | 2 941 174 $ | 25 % | 3 335 602 $ | 27 % |
| LEPP | 1 420 750 $ | 9 % | 1 615 180 $ | 10 % | 2 173 412 $ | 15 % |
| LEIM | 4 562 381 $ | 36 % | 4 408 768 $ | 36 % | 12 004 643 $ | 53 % |
| Total | 12 012 641 $ | 14 % | 13 647 211 $ | 12 % | 22 070 036 $ | 20 % |
Le règlement actuel permet aux demandeurs de demander une remise des frais (c'est-à-dire, une réduction des frais), en fonction d'un pourcentage du revenu brut ou des ventes de produits. Une telle demande exige des demandeurs qu'ils soumettent de l'information financière à l'appui de la demande de remise des frais, ainsi qu'une attestation confirmant la véracité de l'information fournie. La Ministre peut exiger que le demandeur fournisse de l'information financière auditée. Pendant l'examen du processus de facturation, l'audit a révélé qu'il n'y avait pas d'outil pour guider le personnel au moment de déterminer si l'information financière auditée doit être exigée. Étant donné que la remise des frais a une grande incidence sur les revenus, il serait préférable que la DGPSA mette en place un processus d'évaluation des risques pour déterminer à quel moment l'information financière auditée doit être exigée.
Pour conclure, donc, dans l'ensemble, les systèmes en place fournissent des renseignements complets, exacts et opportuns qui permettent le suivi et l'établissement de rapports sur les revenus tirés des frais d'utilisation. Toutefois, l'audit a permis de constater qu'en raison de la date de renouvellement des licences d'exploitation, la DGPSA éprouve des difficultés quant à la gestion de la trésorerie du fait que les revenus entrent à la fin de l'exercice financier. De plus, l'audit a révélé l'absence d'un processus établi pour déterminer l'admissibilité à la remise des frais. Finalement, l'audit a relevé une occasion de simplifier la structure tarifaire pour les LEPP et de réduire le risque d'erreurs dans la facturation (voir la recommandation 5).
Recommandation 3
Il est recommandé que le sous-ministre adjoint, Direction générale des produits de santé et des aliments, modifie la date de renouvellement des licences d'exploitation dans le Règlement sur les aliments et drogues afin que les services puissent être offerts pendant l'année où les revenus sont recueillis.
Réponse de la direction
La direction souscrit à la recommandation. En déplaçant la date ainsi, elle croit qu'elle pourra améliorer la prévisibilité et la disponibilité des revenus afin de financer les activités relatives aux licences d'exploitation dans l'année pendant laquelle les revenus sont recueillis.
La date actuelle du 1er avril établie dans le Règlement sur les aliments et drogues pour le renouvellement des licences d'exploitation entraîne certaines difficultés quant à la gestion de la trésorerie, du fait qu'une partie importante des revenus entre à la fin de l'exercice financier. La DGPSA proposera une date convenable et entamera des discussions informelles avec les intervenants. Le processus de révision d'un règlement est complexe et se fait selon les priorités du Ministère.
Recommandation 4
Il est recommandé que le sous-ministre adjoint, Direction générale des produits de santé et des aliments, mette au point un processus d'évaluation des risques afin de déterminer à quel moment l'information financière auditée sera exigée des personnes qui demandent un remboursement de frais.
Réponse de la direction
La direction souscrit à la recommandation et convient qu'un processus d'évaluation des risques serait une façon appropriée de déterminer le moment opportun de demander les relevés de ventes audités.
Un nombre important d'entreprises et de produits font la demande et sont admissibles pour une remise des frais en raison de leur faible revenu, ce qui a une incidence appréciable sur les revenus de Santé Canada. La Ministre est autorisée d'exiger un relevé de ventes audité pour appuyer une demande de remise de frais. Une méthode sera développée et un projet pilote à échantillon restreint sera établi. Une analyse des résultats et du projet pilote aidera à déterminer une stratégie et un processus éventuels.
3.2 Établissement du coût des activités
Critère d'audit : Des systèmes de suivi et d'établissement de rapports fournissent de l'information complète, exacte et opportune sur le coût total des activités d'imposition de frais d'utilisation, à l'appui de la prise de décisions.
Les systèmes d'établissement des coûts fournissent à la direction les données brutes dont elle a besoin pour assurer le suivi et l'établissement de rapports sur le coût des activités, à l'appui de la prise de décisions opérationnelles et à long terme. On s'attend à ce que les systèmes soient en place pour enregistrer et communiquer les coûts opérationnels aux fins de la gestion financière en cours d'année, ainsi que pour la planification des activités futures. Il était également prévu que le processus d'établissement des coûts des activités fournirait suffisamment de détails pour définir précisément et analyser les coûts associés à la structure tarifaire des frais d'utilisation conformément à la LFU, afin de fournir à la direction l'information requise pour éclairer la prise de décision stratégique concernant d'éventuelles améliorations aux frais d'utilisation.
L'alinéa 4(1)d) de la LFU exige aussi qu'avant d'établir ou d'augmenter les frais d'utilisation, d'en élargir l'application ou d'en prolonger la durée d'application, l'organisme de réglementation doit expliquer clairement aux clients la façon dont les frais d'utilisation sont établis et en indiquer le coût et la structure tarifaire.
L'information sur l'établissement des coûts est générée par deux systèmes principaux. Premièrement, les dépenses et revenus d'exploitation sont enregistrés et communiqués à l'aide de divers modules du SAP. Le Ministère utilise ce système pour générer des soldes de vérification mensuelles et des rapports de gestion financière. L'établissement de rapports sur l'information financière de la DGPSA se fait par direction générale, la direction, la division et le bureau, et comprend les revenus tirés de frais d'utilisation ainsi que les dépenses issues des activités dont les coûts sont recouvrables et des activités dont les coûts ne sont pas recouvrables. En raison de limites en matière de codage, SAP ne peut pas faire la distinction entre les activités dont les coûts sont recouvrables et celles dont les coûts ne sont pas recouvrables et produire les rapports connexes. Il ne peut donc pas être utilisé seul pour déterminer les coûts liés aux activités d'imposition de frais d'utilisation en vue de déterminer les frais d'utilisation.
Deuxièmement, les employés de la DGPSA, ainsi que ceux du BRP qui travaillent aux activités de programme de l'Inspectorat, inscrivent les heures travaillées dans « SAP-Project Systems, Cross Application Time Sheets » (SAP-PS/CATS). Ce système a été mis en place en 2011 afin de saisir des renseignements exacts au sujet du temps et des efforts consacrés à l'ensemble des activités de la direction générale, afin de comprendre les efforts et les ressources nécessaires à l'exécution des activités. Plus précisément, l'objectif du système SAP-PS/CATS est de fournir des données fiables permettant de déterminer les coûts totaux des activités et d'établir une méthode de calcul des coûts unitaires détaillés à l'appui d'éventuelles initiatives de modernisation des frais d'utilisation et aux fins d'établissement de rapports sur les frais d'utilisation dans le RMR.
La DGRO, en collaboration avec le personnel des directions et des divisions, a mis au point une structure d'activités et des codes connexes qui définissent les catégories d'activités dont les coûts sont recouvrables et celles dont les coûts ne sont pas recouvrables. Cette structure d'activités a été chargée dans SAP-PS/CATS et les codes d'activité sont reliés à des éléments pertinents dans SAP. Pour certaines activités SAP-PS/CATS peut enregistrer les coûts liés au salaire, et ce, jusqu'au niveau de la demande d'homologation. Les coûts liés au salaire représentent environ 85 % des dépenses totales de la DGPSA.
Calcul du coût des activités de vérification de la conformité et d'application de la Loi
Les frais de licences d'exploitation pour les produits pharmaceutiques (LEPP) et les instruments médicaux (LEIM) sont établis en fonction du coût des activités de vérification de la conformité et de l'application de la Loi. L'alinéa 4(1)d) de la LFU exige des ministères qu'ils expliquent clairement aux clients la façon dont les frais d'utilisation sont établis et en indiquent les composantes de coût et de revenus. Bien que le modèle de calcul des coûts aligne clairement les activités et la structure tarifaire pour les autres catégories de frais, l'audit a révélé que la structure d'activités pour la vérification de la conformité et de l'application de la Loi n'est pas clairement harmonisée avec la structure tarifaire établie pour les frais d'utilisation relatifs aux LEPP. Il est à noter que la même observation a été faite au sujet du modèle de calcul des coûts de 2007, lequel était utilisé pour établir les frais des LEPP dans la proposition de 2010.
Des améliorations doivent être apportées à la structure tarifaire pour les LEPP ainsi qu'à la structure d'activité du modèle d'établissement des coûts afin d'assurer qu'ils sont harmonisés et que les coûts utilisés pour établir les frais d'utilisation relatifs aux LEPP sont clairement expliqués dans les prochaines initiatives de renouvellement des frais d'utilisation. Cette initiative pourrait également s'avérer une occasion de simplifier la structure tarifaire pour les LEPP et réduire le risque d'erreurs de facturation, comme il a été souligné à la section 3.1 du présent rapport.
Analyse des données SAP-PS et calcul du coût unitaire
Depuis la mise en place du système SAP-PS en 2011, la DGPSA a réalisé d'importants progrès dans l'amélioration de la conformité de la saisie des heures en encourageant le personnel des directions à enregistrer rapidement les heures qu'ils ont travaillées dans le système SAP-PS.
Depuis 2013-2014, dans le cadre de la phase 3 de la feuille de route sur le recouvrement des coûts de la DGPSA, l'accent a porté davantage sur l'analyse des données SAP-PS à l'échelon de la direction, afin d'élaborer des méthodes de calcul des coûts visant à analyser les fluctuations dans le coût de la main-d'œuvre et d'autres mesures opérationnelles. Cette initiative, coordonnée par l'entremise du GTRC, en est toujours à ses débuts. Comme il a été mentionné précédemment, l'examen de 2014 du Règlement sur les prix à payer à l'égard des drogues et instruments médicaux indique qu'il y a un nombre croissant de cas où les demandes d'homologation sont de plus en plus complexes, contiennent plus de données sur des essais cliniques et s'avèrent plus difficiles à examiner; toutefois, le rapport ne quantifie pas ces phénomènes. Le développement d'outils de calcul des coûts est essentiel pour comprendre les répercussions sur les activités et pour obtenir l'information requise à l'appui des prochaines initiatives de renouvellement des frais d'utilisation. L'audit a révélé que les efforts et les progrès réalisés pour mettre au point des outils de calcul des coûts varient d'une direction à l'autre quant à leur portée et leur profondeur. Il serait avantageux pour la DGPSA de s'assurer que les efforts déployés pour la mise en place d'outils de calcul des coûts sont uniformes à l'échelle des directions.
Avant 2013-2014, les revenus tirés des frais d'utilisation et les coûts connexes dans le RMR étaient déterminés à l'aide de l'information issue du SAP. Depuis 2013-2014, le coût des salaires est déterminé à l'aide des données SAP-PS, alors que les revenus et les coûts d'exploitation non liés au salaire sont obtenus à partir du SAP et répartis de façon semblable aux années précédentes. L'audit a révélé que le processus utilisé pour établir les revenus et les coûts dans le RMR 2013-2014 fournissait des renseignements exacts et opportuns.
Dans l'ensemble, les systèmes de suivi et de production de rapports fournissent de l'information complète, exacte et opportune sur le coût total des activités d'imposition de frais d'utilisation, à l'appui de la prise de décisions. Cependant, des améliorations pourraient être apportées à la structure des activités de vérification de la conformité et d'application de la Loi qui dictent les frais des LEPP, conformément à l'alinéa 4(1)d) de la LFU.
Recommandation 5
Il est recommandé que le sous-ministre adjoint, Direction générale des produits de santé et des aliments, examine la structure tarifaire pour les licences d'exploitation pour les produits pharmaceutiques (LEPP), afin qu'elle soit harmonisée davantage avec le modèle de calcul des coûts connexes, de sorte que les frais d'utilisation relatifs aux LEPP soient clairement identifiés et expliqués.
Réponse de la direction
La direction souscrit à la recommandation. Elle examinera et traitera de la complexité de la structure tarifaire des LEPP, qui comprend plusieurs composantes comme, par exemple, l'activité précise, la catégorie de médicament et la forme posologique.
Les revenus tirés des frais de LEPP viennent appuyer la prestation du programme national de vérification de la conformité et d'application de la Loi, y compris les inspections relatives aux bonnes pratiques de fabrication; il ne s'agit pas, toutefois, d'une rémunération à l'acte. Les frais seront refondus afin de mieux refléter le montant d'effort requis pour les différentes activités liées aux licences. La révision des frais requiert des consultations exhaustives et une présentation au Parlement, et le processus peut prendre de deux à quatre ans à compléter.
3.3 Rendement en regard des normes de service
Critère d'audit : Les résultats du rendement font l'objet d'une surveillance et de rapports en regard des normes de service relatives aux frais d'utilisation.
Il est essentiel de satisfaire aux exigences en matière de rendement pour répondre aux attentes de l'industrie et pour maintenir une source de financement stable grâce à la collecte de frais d'utilisation. Pendant l'audit, on s'attendait à ce que des processus et des mesures de contrôle soient en place afin de mesurer, d'évaluer et de communiquer dans des rapports rapidement et exactement le rendement en regard des normes de service, et d'informer la direction des résultats et des facteurs de risque à l'appui de la planification et de la prise de décision.
Conformément à la LFU, des pénalités financières peuvent être engagées sous forme de réduction de frais, si le Ministère ne respecte pas ses normes de services relativement aux frais d'utilisation à l'intérieur de certains paramètres. Par conséquent, la capacité du Ministère de gérer ses activités et donc, de respecter ces normes de services, conformément à ce qui est énoncé à l'annexe D et l'annexe E, s'avère essentielle.
La LFU exige qu'avant d'établir ou d'augmenter les frais d'utilisation, d'en élargir l'application ou d'en prolonger la durée d'application, l'organisme de réglementation doit établir des normes en regard desquelles le rendement de l'organisme de réglementation peut être évalué. De plus, si, pour un exercice donné, le rendement d'un organisme de réglementation à l'égard de frais d'utilisation est inférieur aux normes de rendement qu'il a établies pour cet exercice dans une proportion dépassant dix pour cent, les frais d'utilisation seront réduits. La LFU exige également que le ministre produise un rapport annuel sur les niveaux de rendement réels qui ont été atteints en regard des normes de service établies.
Les renseignements sur le rendement font l'objet d'un suivi en fonction des catégories de frais, à l'aide de plusieurs systèmes. L'information est enregistrée dans le système approprié par le personnel de la division qui est responsable de ces processus particuliers. Le suivi du rendement et la surveillance des présentations et demandes d'homologation individuelles sont vérifiés par la direction à tous les échelons de la direction générale. Ce processus permet de veiller à ce que les charges de travail soient bien gérées, que les normes de rendement soient satisfaites ou des mesures appropriées soient prise pour corriger toute erreur.
L'audit comprenait des fichiers choisis parmi les diverses catégories de frais et des données sur le rendement qui avaient été enregistrées dans les systèmes de suivi appropriés. L'audit a révélé que dans l'ensemble, les résultats sur le rendement ont fait l'objet d'une surveillance et de rapports en regard des normes de service relatives aux frais d'utilisation. Toutefois, il a été observé que les mesures de contrôle internes doivent être améliorées afin de rehausser le suivi du rendement pour les demandes relatives aux instruments médicaux (IMED). L'audit a aussi constaté que la direction générale aurait intérêt à améliorer les documents sur les LEPP et le LEIM à l'appui du RMR.
Le Bureau des matériels médicaux de la Direction des produits thérapeutiques (DPT) est responsable du traitement des demandes d'homologation pour les instruments médicaux. Comme il est indiqué dans le document intitulé Gestion des demandes d'homologation d'instruments médicaux et d'autorisation d'essais expérimentaux, toutes les demandes d'homologation d'instruments médicaux font l'objet d'un suivi, depuis le moment où elles sont reçues jusqu'à la décision ultime. Par conséquent, lorsqu'une demande pour un instrument médical est reçue, elle fait d'abord l'objet d'un examen préliminaire afin de déterminer si elle est recevable sur le plan administratif et technique avant d'être acheminée à l'étape de l'examen. Une lettre d'acceptation (demandes de classe III et IV) est préparée à la fin du processus d'examen préliminaire, ce qui marque le début de l'étape de l'examen.
Le rendement des demandes d'homologation pour un instrument médical consigné dans le RMR est fondé sur le nombre de jours civils requis pour un premier examen. La décision suivant le premier examen peut donner lieu à l'octroi d'une licence, à l'envoi d'une lettre demandant des renseignements supplémentaires ou à un refus. Le temps requis pour le premier examen est calculé depuis le moment où la lettre d'acceptation est envoyée à la suite de l'examen préliminaire, mais ne comprend pas le temps requis pour l'examen préliminaire en soi. Le rendement relatif au processus d'examen préliminaire fait l'objet d'un suivi et d'un rapport à l'interne seulement.
Pendant l'examen du suivi des dossiers de demandes d'homologation de classe III et IV, l'audit a révélé que les objectifs de l'examen préliminaire des dossiers vérifiés étaient souvent dépassés, alors que les objectifs de rendement pour la première étape de l'examen étaient en grande partie satisfaits. L'audit a permis d'observer que la responsabilité concernant l'examen préliminaire technique et la vérification des demandes de classe III et IV relève de la même division d'évaluation, ce qui est différent des pratiques utilisées au sein de la division de l'examen des produits pharmaceutiques de la DPT. Afin d'assurer une méthode efficace relative aux examens au sein de la DPT et pour renforcer les mesures de suivi et d'établissement de rapports en matière de rendement, il faudrait mettre en place un processus selon lequel les activités d'examen préliminaire et de premier examen des demandes d'homologation d'instruments médicaux de classes III et IV seraient clairement séparées.
Pour conclure, donc, dans l'ensemble, l'information sur le rendement fait l'objet d'une surveillance et de rapports en regard des normes de service relatives aux frais d'utilisation. Cependant, des améliorations sont requises pour renforcer les méthodes de suivi du rendement et d'établissement de rapports connexes concernant les demandes relatives aux instruments médicaux.
Recommandation 6
Il est recommandé que le sous-ministre adjoint, Direction générale des produits de santé et des aliments, établisse un processus selon lequel les activités de l'examen préliminaire et du premier examen des demandes d'homologation pour des instruments médicaux de classe III et IV soient clairement séparées.
Réponse de la direction
La direction souscrit à la recommandation. Afin de réduire les perceptions erronées, la DGPSA convient qu'une séparation claire des responsabilités relatives à l'examen préliminaire et le premier examen des demandes d'homologation pourrait renforcer l'intégrité du processus.
Le but de l'examen préliminaire est de déterminer si la demande est recevable sur le plan administratif et technique. Un projet pilote sur l'examen préliminaire est actuellement en cours afin de déterminer la viabilité de compléter l'examen préliminaire uniquement au sein de la Division des services d'enregistrement des matériaux, plutôt que d'effectuer certains des examens techniques dans d'autres secteurs d'examen.
3.4 Gestion des ressources financières
Critère d'audit : Les ressources financières sont affectées et réaffectées en regard des activités correspondantes d'imposition de frais d'utilisation.
Le financement des activités dont les coûts sont recouvrables provient d'une combinaison de crédits et de revenus obtenus auprès du secteur privé par le paiement de frais d'utilisation des services.
Une fois que les revenus tirés des frais d'utilisation ont été facturés et recueillis, l'APFDG redistribue d'abord les revenus tirés des frais d'utilisation entre les directions générales participantes (DGPSA, BRP, DGPDF et DGSG) en fonction de la proportion selon laquelle chaque direction générale contribue au coût total des activités. Cette répartition est approuvée chaque année (ou au besoin) par le SMA-GR. Ensuite, chacune des directions générales répartit les revenus parmi ses unités administratives, de manière à couvrir les coûts liés aux activités connexes. Voir l'annexe F et l'annexe H pour obtenir d'autres détails sur la structure organisationnelle et la répartition des revenus tirés des frais d'utilisation.
La Loi sur les frais d'utilisation (LFU) exige que soit déposé devant le Parlement un rapport indiquant le montant total des frais d'utilisation recueillis par catégorie de frais, les coûts que les frais d'utilisation ont permis de couvrir, les normes de rendement établies et les niveaux de rendement réels qui ont été atteints. L'audit a révélé que le Ministère a satisfait à cette exigence en publiant cette information dans les tableaux supplémentaires du RMR annuel.
La Politique sur les autorisations spéciales de dépenser les recettes du CT stipule que les ministères devront assurer que les dépenses encourues dans la prestation d'un service sont liées directement au revenu perçu de la vente de ce service. Les revenus doivent donc être consacrés aux utilisations voulues et l'interfinancement n'est pas permis. Le Ministère est autorisé de dépenser les revenus perçus, conformément au règlement sur les frais d'utilisation qui s'applique aux programmes de médicaments pour usage humain et d'instruments médicaux. En 2015, le Ministère a publié un Bulletin d'informations financières, qui offre une orientation relative à la gestion des revenus tirés des programmes de médicaments et d'instruments médicaux.
Le bulletin indique les revenus tirés des programmes de médicaments et d'instruments médicaux devront demeurer dans les sous-programmes listés dans l'Architecture d'alignement des programmes (AAP) : Médicaments (AAP 2.1.1 et 2.1.2) et Matériel médical (AAP 2.1.3), selon la façon dont les frais d'utilisation sont définis et facturés. Les revenus ne seront pas répartis à des programmes pour lesquels les coûts sont récupérés.
L'audit a examiné la documentation supplémentaire pertinente portant sur la répartition des revenus et a conclu que dans l'ensemble, les revenus recueillis ont été affectés à des activités correspondantes d'imposition de frais d'utilisation au sein des sous-programmes.
C - Conclusion
L'audit a permis de conclure que le cadre de contrôle de gestion en place pour appuyer les activités de recouvrement des coûts pour les produits de santé et assurer la conformité avec les lois et règlements nécessite quelques améliorations.
L'audit a révélé que la Direction générale dispose d'une structure de gouvernance qui lui permet de veiller à la surveillance adéquate du recouvrement des coûts liés aux produits de santé à usage humain. Les rôles et les responsabilités sont définis et communiqués, et la haute direction reçoit des renseignements complets et opportuns pour l'appuyer dans la prise de décisions quotidienne. Cependant, il serait bénéfique de fournir au Ministère, sur une base annuelle, de l'information détaillée sur les coûts; la direction pourrait ainsi déterminer le moment du renouvellement des frais d'utilisation. En mars 2015, la direction générale a mis en place une nouvelle structure de gouvernance dans le but de simplifier et d'améliorer le processus de prise de décisions par le Comité exécutif de la direction générale (CEDG).
En ce qui concerne la gestion des risques, les risques opérationnels et financiers quotidiens liés aux activités de recouvrement des coûts sont communiqués au moyen de tableaux de bord et de rapports sur les écarts financiers. La gestion des questions opérationnelles, réglementaires et politiques qui nécessitent des solutions à plus long terme pourrait être renforcée par l'examen et la mise à jour périodiques d'un registre permanent des risques.
Dans l'ensemble, les systèmes de suivi et de production de rapports fournissent des renseignements complets, exacts et opportuns sur les revenus générés grâce aux frais d'utilisation et les activités d'imposition de frais d'utilisation. Les mesures de contrôle internes pourraient être rehaussées davantage par la mise en place d'un processus d'admissibilité à une remise des frais; par la modification de la date de renouvellement de la LE dans la réglementation, afin qu'elle soit harmonisée avec le cycle opérationnel; et par la modification de la structure des frais d'utilisation exigés pour l'obtention d'une LEPP, afin qu'ils correspondent de plus près au modèle de coût connexe, de manière à ce que les frais d'utilisation liés exigés pour la LEPP puissent être clairement identifiés et expliqués dans les prochaines initiatives de renouvellement des frais d'utilisation.
Dans l'ensemble, l'information sur le rendement fait l'objet d'une surveillance et de rapports en regard des normes relatives aux frais d'utilisation des services. Les responsabilités concernant le tri et le premier examen des demandes d'homologation d'instruments médicaux de classes III et IV devraient être déléguées à des tiers indépendants. En ce qui concerne la gestion des ressources financières, les revenus perçus ont été répartis aux sous-programmes des médicaments et des instruments médicaux en fonction des activités correspondantes relatives aux frais d'utilisation.
Les possibilités d'amélioration mises en évidence renforceront collectivement le cadre de contrôle de la gestion, et ce, afin de mieux appuyer les activités de recouvrement des coûts pour les produits de santé au sein du Ministère.
Annexe A - Champs d'enquête et critères
| Titre du critère | Critère d'audit | |
|---|---|---|
|
Annexe A - Notes de bas de page
|
||
| Champ d'enquête 1 : Gouvernance | ||
| 1.1 | Structure de gouvernance et surveillanceAnnexe A - Note de bas de page 1 | Il y a une structure de gouvernance en place qui permet d'assurer la surveillance du recouvrement des coûts des activités liées aux produits de santé et qui soutient l'obtention de résultats. |
| 1.2 | Rôles et responsabilitésAnnexe A - Note de bas de page 1 | Les rôles et les responsabilités sont définis et communiqués. |
| 1.3 | Information nécessaire à la prise de décisionsAnnexe A - Note de bas de page 1 | La haute direction reçoit de l'information complète, opportune et exacte à l'appui du processus de prise de décisions. |
| Champ d'enquête 2 : Gestion du risque | ||
| 2.1 | Gestion des risquesAnnexe A - Note de bas de page 1 | Les risques qui ont une incidence sur le recouvrent des coûts des produits de santé sont définis, évalués, documentés et gérés de façon continue. |
| Champ d'enquête 3 : Contrôles internes | ||
| 3.1 | Revenus tirés des frais d'utilisationAnnexe A - Note de bas de page 2,Annexe A - Note de bas de page 3,Annexe A - Note de bas de page 4 | Les systèmes de suivi et de production de rapports fournissent des renseignements complets, exacts et opportuns sur les revenus générés grâce aux frais d'utilisation. |
| 3.2 | Calcul du coût des activitésAnnexe A - Note de bas de page 2,Annexe A - Note de bas de page 3,Annexe A - Note de bas de page 4 | Des systèmes de suivi et d'établissement de rapports fournissent de l'information complète, exacte et opportune sur le coût total des activités d'imposition de frais d'utilisation, à l'appui de la prise de décisions. |
| 3.3 | Rendement en regard des normes de serviceAnnexe A - Note de bas de page 3,Annexe A - Note de bas de page 4 | Les résultats du rendement font l'objet d'une surveillance et de rapports en regard des normes de service relatives aux frais d'utilisation. |
| 3.4 | Gestion des ressources financièresAnnexe A - Note de bas de page 5 | Les ressources financières sont affectées et réaffectées en regard des activités correspondantes d'imposition de frais d'utilisation. |
Annexe B - Grille d'évaluation
| Critère | Note | Conclusion | Nº de la rec. |
|---|---|---|---|
| Gouvernance | |||
| 1.1 Structure de gouvernance et surveillance | Nécessite des améliorations mineures | Une structure de gouvernance est en place pour assurer une surveillance et pour appuyer l'atteinte des résultats. La direction générale profiterait d'une mise à jour du mandat du Groupe de travail sur le recouvrement des coûts. | |
| 1.2 Rôles et responsabilités | Satisfaisant | Les rôles et les responsabilités sont définis et communiqués | |
| 1.3 Information nécessaire à la prise de décisions | Nécessite des améliorations modérées | La direction reçoit de l'information complète et opportune à l'appui du processus quotidien de prise décisions. Le Ministère devrait recevoir de l'information sur les revenus et les coûts annuels afin de déterminer le moment opportun pour le renouvellement des frais d'utilisation. | 1 |
| Gestion du risque | |||
| 2.1 Gestion des risques | Nécessite des améliorations mineures | Les risques opérationnels et financiers associés au fonctionnement quotidien sont bien gérés. Un registre permanent des risques devrait être créé afin de renforcer la gestion des questions qui nécessitent des solutions à plus long terme. | 2 |
| Critère | Note | Conclusion | Nº de la rec. | |||||
|---|---|---|---|---|---|---|---|---|
| Contrôles internes (notation selon les catégories de frais pour les critères 3.1, 3.2 et 3.3) | ||||||||
| (voir l'annexe C pour une description des catégories de frais) | EVAL-MED | LEPP | DVPP | EVAL-IMED | LEIM | DVIM | EVAL-MED : Évaluation des médicaments LEPP : Licence d'exploitation pour les produits pharmaceutiques DVPP : Droit de vente de produits pharmaceutiques EVAL-IMED : Évaluation des instruments médicaux LEIM : Licence d'exploitation pour les instruments médicaux DVIM : Droit de vente d'instruments médicaux |
|
| 3.1 Revenus tirés des frais d'utilisations | Nécessite des améliorations mineures | Nécessite des améliorations modérées | Nécessite des améliorations mineures | Nécessite des améliorations mineures | Nécessite des améliorations modérées | Nécessite des améliorations mineures | Dans l'ensemble, les systèmes de suivi et de production de rapports fournissent des renseignements complets, exacts et opportuns sur les revenus générés grâce aux frais d'utilisation. Cependant, la date de renouvellement pour les EVAL-MED et les EVAL-IMED devrait être modifiée dans la réglementation afin qu'elle soit harmonisée avec le cycle opérationnel. De plus, un processus d'évaluation des risques devrait être développé et mis en place afin de pouvoir déterminer l'admissibilité pour une remise des frais d'utilisation pour toutes les catégories de frais. | 3, 4 |
| Il existe une occasion de simplifier la structure tarifaire pour les LEPP et de réduire le risque d'erreurs dans la facturation. | Voir rec. 5 | |||||||
| 3.2 Calcul du coût des activités | Satisfaisant | Nécessite des améliorations modérées | Satisfaisant | Satisfaisant | Satisfaisant | Satisfaisant | Dans l'ensemble, les systèmes de suivi et de production de rapports fournissent des renseignements complets, exacts et opportuns sur le coût total des activités d'imposition de frais d'utilisation. Cependant, des améliorations doivent être apportées à la structure tarifaire pour les LEPP ainsi qu'à la structure d'activité du modèle d'établissement des coûts afin d'assurer qu'ils sont harmonisés et que les coûts utilisés pour établir les frais d'utilisation relatifs aux LEPP sont clairement identifiés et expliqués dans les prochaines initiatives de renouvellement des frais d'utilisation. | 5 |
| 3.3 Rendement en regard des normes de service | Satisfaisant | Nécessite des améliorations mineures | Satisfaisant | Nécessite des améliorations modérées | Nécessite des améliorations mineures | Satisfaisant | Dans l'ensemble, l'information sur le rendement fait l'objet d'une surveillance et de rapports en regard des normes de service relatives aux frais d'utilisation. Cependant, des améliorations sont requises pour renforcer les méthodes de suivi du rendement et d'exploitation de rapports connexes concernant les demandes relatives aux instruments médicaux. La direction générale aurait intérêt à améliorer les documents sur les LEPP et le LEIM à l'appui du RMR. | 6 |
| 3.4 Gestion des ressources financières | Satisfaisant | Les revenus perçus ont été répartis aux sous-programmes des médicaments et des instruments médicaux en fonction des activités correspondantes relatives aux frais d'utilisation. | ||||||
Annexe C - Frais d'utilisation et activités connexes
Évaluation des médicaments (EVAL-MED) : Ces activités concernent l'évaluation de demandes d'homologation pour des produits pharmaceutiques et biologiques. Des frais sont exigés pour l'évaluation de la documentation qu'un fabricant a soumise pour démontrer la sécurité, l'efficacité et la qualité d'un produit dans des conditions particulières d'emploi, avant sa mise en marché au Canada.
Licence d'exploitation pour les produits pharmaceutiques (LEPP) : Ces activités sont directement liées à l'un des principaux outils de vérification de la conformité et de l'application de la loi de Santé Canada. Le respect des exigences réglementaires est évalué au moyen d'inspections, lesquelles sont à la base de l'octroi de la licence d'exploitation. Une LEPP est requise pour qu'un établissement qui propose de mener ou qui mène une activité réglementée soit autorisé à le faire. Des frais sont exigés pour la licence initiale, les examens annuels de la licence, la modification de la licence et le rétablissement de la licence.
Droit de vente de produits pharmaceutiques (DVPP) : Ces frais annuels permettent à un fabricant de continuer à vendre un produit autorisé ou homologué au Canada. Santé Canada assure la surveillance d'un grand nombre de médicaments vendus sur le marché canadien au moyen d'activités de surveillance post-commercialisation, de communication avec les intervenants, d'élaboration de politiques et de technologies, de surveillance de la gestion de la qualité, d'essais de produits et d'analyse en laboratoire ainsi que de vérification de la conformité et d'application de la Loi.
Évaluation des instruments médicaux (EVAL-IMED) : Ces activités concernent l'évaluation médicale/scientifique des demandes de licences pour instruments médicaux et les demandes de modifications connexes. Des frais sont imposés pour l'examen et l'évaluation de la documentation scientifique soumise pour démontrer la sécurité et l'efficacité des instruments avant leur mise en marché au Canada.
Licence d'exploitation pour les instruments médicaux (LEIM) : Ces frais d'utilisation soutiennent l'autorisation accordée aux établissements qui proposent de s'engager ou qui s'engagent dans l'importation ou la vente d'instruments médicaux et aux fabricants d'instruments médicaux présentant le plus faible risque possible. Une licence d'exploitation pour les instruments médicaux autorise la partie licenciée à mener une activité contrôlée au Canada dans un établissement divulgué. Des frais sont exigés pour la licence initiale, l'examen annuel de la licence et le rétablissement de la licence.
Droit de vente d'instruments médicaux (DVIM) : Il s'agit de frais annuels pour l'autorisation de tenir et de vendre des instruments médicaux au Canada, à l'exception de ceux classés comme présentant le plus faible risque. Les activités soutenues à l'aide de ces frais comprennent, sans toutefois s'y limiter : la surveillance post-commercialisation, la communication avec les intervenants, l'élaboration de politiques et de technologies et la surveillance de la gestion de la qualité.
(Source : Proposition de 2010 au Parlement)
Annexe D - Normes de service, frais et résultats de rendement relativement à l'évaluation de médicaments
Source d'information : Normes de service : Gazette du Canada, vol. 148, nº 11 - 15 mars 2014; Frais de service : Rapports ministériels sur le rendement de Santé Canada pour les exercices financiers de 2011-2012 à 2013-2014.
Évaluation des médicaments :
La catégorie « Évaluation des médicaments » représente les activités d'autorisation de Santé Canada qui permettent à un fabricant de vendre son produit au Canada. Il existe de plusieurs types et catégories de produits différents et de nombreux changements aux produits qui nécessitent une autorisation de Santé Canada. Par exemple : médicaments biologiques ou médicaments chimiques (produits pharmaceutiques); médicaments de marque ou médicaments génériques; nouveaux médicaments ou anciens médicaments; ajout de nouvelles indications ou modification d'un site de fabrication. La taille et la complexité de ces différentes évaluations de médicaments sont très diversifiées et donc, des normes de rendement différentes sont requises pour représenter cette diversité. Le rapport de rendement complexe ci-dessous représente les divers types de produits et d'évaluations de médicaments qui sont soumis par différents sous-secteurs de l'industrie pharmaceutique.
Rendement :
Délai moyen en jours civils pour rendre une première décision d'examen (examen 1).
La Loi sur les frais d'utilisation exige que, si le rendement est inférieur aux normes de rendement établies dans une proportion dépassant dix pour cent, ces frais d'utilisation soient réduits (jusqu'à concurrence de 50 % des frais d'utilisation). Les frais d'utilisation réduits s'appliquent à compter du jour où le rapport annuel pour l'exercice financier est déposé, jusqu'au jour où le rapport annuel suivant est déposé.
| Frais d'utilisation (catégories de frais) |
Type et catégorie de frais | Frais avec pénalités, en date du 6 novembre 2013 |
Frais en date du 1er avril 2014 |
Nouvelles normes de service pour les frais d'utilisation, en vigueur le 1er avril 2011 (jours) |
Résultats de rendement (jours) |
||
|---|---|---|---|---|---|---|---|
| 2011-2012 | 2012-2013 | 2013-2014 | |||||
|
Annexe D - Notes de bas de page
|
|||||||
| Frais d'évaluation des médicaments (EVAL-MED) | Produits pharmaceutiques | ||||||
| Nouvelle substance active | 322 056 $ | ||||||
|
Nouvelle substance active : Présentation de nouveau médicament (RNA : PNM)
|
300 | 220 | 256 | 268 | |||
| Données cliniques ou non cliniques et données sur la composition chimique et la fabrication | 163 120 $ | ||||||
|
CLIN C&F : PNM (Présentation de nouveau médicament)
|
300 | 236 | 302Annexe D - Note de bas de page * | 291 | |||
|
CLIN C&F : SPNM (Supplément à une présentation de nouveau médicament)
|
300 | 235 | 271 | 293 | |||
| Données cliniques ou non cliniques seulement | 76 132 $ | ||||||
|
CLIN : PNM (Présentation de nouveau médicament)
|
300 | - | - | 297 | |||
|
CLIN : SPNM (Supplément à une présentation de nouveau médicament)
|
300 | 226 | 266 | 276 | |||
|
CLIN : DIN A (Numéro d'identification du médicament)
|
210 | 188 | 160 | 205 | |||
| Études comparatives des données sur la composition chimique et la fabrication | 46 016 $ | ||||||
|
COMP C&F : PNM (Présentation de nouveau médicament)
|
180 | 175 | 154 | - | |||
|
COMP C&F : PAMN (Présentation abrégée d'un médicament nouveau)
|
23 910 $ | 24 388 $ | 180 | 172 | 264Annexe D - Note de bas de page * | 196Annexe D - Note de bas de page * | |
|
COMP C&F : SPAMN (Supplément à une présentation abrégée de médicament nouveau)
|
22 557 $ | 23 008 $ | 180 | 177 | 340Annexe D - Note de bas de page * | 191Annexe D - Note de bas de page * | |
|
COMP C&F : SPNM (Supplément à une présentation de nouveau médicament)
|
180 | 159 | 175 | 174 | |||
|
COMP C&F : DIN A (Numéro d'identification du médicament)
|
210 | - | 210 | 193 | |||
| Données sur la composition chimique et la fabrication seulement | 21 756 $ | ||||||
|
C&F : PNM (Présentation de nouveau médicament)
|
180 | - | 110 | 178 | |||
|
C&F : PAMN (Présentation abrégée d'un médicament nouveau)
|
15 144 $ | 15 447 $ | 180 | 121 | 233Annexe D - Note de bas de page * | 288Annexe D - Note de bas de page * | |
|
C&F : SPAMN (Supplément à une présentation abrégée de médicament nouveau)
|
180 | 132 | 198Annexe D - Note de bas de page * | 149 | |||
|
C&F : SPNM (Supplément à une présentation de nouveau médicament)
|
180 | 115 | 150 | 157 | |||
|
C&F : DIN A (Numéro d'identification du médicament)
|
210 | 201 | 117 | 177 | |||
| Données publiées | 18 041 $ | ||||||
|
DONNÉES PUBLIÉES : SPNM (Supplément à une présentation de nouveau médicament)
|
300 | 243 | 213 | 262 | |||
|
DONNÉES PUBLIÉES : DIN A (Numéro d'identification du médicament)
|
210 | - | 200 | 159 | |||
| Transfert de statut - de médicament sur ordonnance à médicament en vente libre | 43 808 $ | ||||||
|
Transfert - ordonnance à vente libre SPNM (Supplément à une présentation de nouveau médicament)
|
180 | - | 167 | 172 | |||
| Désinfectants | 4 055 $ | ||||||
|
DÉSINFECTANT : PNM-D (Présentation de nouveau médicament-Désinfectant)
|
300 | - | 266 | 327Annexe D - Note de bas de page * | |||
|
DÉSINFECTANT : SPNM-D (Supplément à une présentation de nouveau médicament-Désinfectant)
|
300 | - | - | 290 | |||
|
DÉSINFECTANT : DIN D 180 (Numéro d'identification du médicament)
|
180 | 7 | - | - | |||
|
DÉSINFECTANT : DIN D 210 (Numéro d'identification du médicament)
|
210 | 181 | 194 | 210 | |||
| Étiquetage seulement | 2 931 $ | ||||||
|
ÉTIQUETAGE SEULEMENT : PNM (Présentation de nouveau médicament)
|
60 | 47 | 56 | 50 | |||
|
ÉTIQUETAGE SEULEMENT : PAMN (Présentation abrégée d'un médicament nouveau)
|
60 | - | 58 | 48 | |||
|
ÉTIQUETAGE SEULEMENT : SPAMN (Supplément à une présentation abrégée de médicament nouveau)
|
60 | 27 | 32 | 29 | |||
|
ÉTIQUETAGE SEULEMENT : SPNM (Supplément à une présentation de nouveau médicament)
|
60 | 46 | 54 | 52 | |||
|
ÉTIQUETAGE SEULEMENT : DIN A
|
180 | 103 | 127 | 127 | |||
| Demandes de numéro d'identification de médicament - Norme d'étiquetage | 1 625 $ | ||||||
|
NORME D'ÉTIQUETAGE : DIN A (Numéro d'identification du médicament)
|
45 | 32 | 32 | 37 | |||
|
NORME D'ÉTIQUETAGE : DIN D (Numéro d'identification du médicament)
|
45 | 22 | 35 | 42 | |||
|
NORME D'ÉTIQUETAGE : DIN F (Numéro d'identification du médicament)
|
45 | 32 | 34 | 34 | |||
| Demande administrative | 303 $ | ||||||
|
ADMINISTRATIVE : PNM (Présentation de nouveau médicament)
|
45 | 31 | 22 | 29 | |||
|
ADMINISTRATIVE : PAMN (Présentation abrégée d'un médicament nouveau)
|
45 | 30 | 21 | 28 | |||
|
ADMINISTRATIVE : SPAMN (Supplément à une présentation abrégée de médicament nouveau)
|
45 | 30 | 24 | 30 | |||
|
ADMINISTRATIVE : SPNM (Supplément à une présentation de nouveau médicament)
|
45 | 33 | 35 | 31 | |||
|
ADMINISTRATIVE : DIN A (Numéro d'identification du médicament)
|
45 | 16 | 21 | 20 | |||
|
ADMINISTRATIVE : DIN D (Numéro d'identification du médicament)
|
45 | 17 | 32 | 32 | |||
| Produits biologiques | |||||||
| Nouvelle substance active | 322 056 $ | ||||||
|
RNA PNM (Présentation de nouveau médicament)
|
300 | 238 | 230 | 239 | |||
| Données cliniques ou non cliniques et données sur la composition chimique et la fabrication | 163 120 $ | ||||||
|
CLIN C&F : PNM (Présentation de nouveau médicament)
|
300 | 244 | 275 | 280 | |||
|
CLIN C&F : SPNM (Supplément à une présentation de nouveau médicament)
|
300 | 291 | 296 | 264 | |||
|
CLIN C&F : DIN B
|
210 | - | - | 209 | |||
| Données cliniques ou non cliniques seulement | 76 132 $ | ||||||
|
CLIN : SPNM (Supplément à une présentation de nouveau médicament)
|
300 | 233 | 272 | 286 | |||
| Données sur la composition chimique et la fabrication seulement | 21 756 $ | ||||||
|
C&F : PAMN (Présentation abrégée d'un médicament nouveau)
|
180 | - | 180 | 180 | |||
|
C&F : SPNM (Supplément à une présentation de nouveau médicament)
|
180 | 146 | 168 | 168 | |||
|
C&F : DIN B (Numéro d'identification du médicament)
|
210 | 153 | 192 | 206 | |||
| Études comparatives des données sur la composition chimique et la fabrication | 46 016 $ | ||||||
|
COMP C&F : SPNM (Supplément à une présentation de nouveau médicament)
|
180 | 179 | 177 | 179 | |||
| Étiquetage seulement | 2 931 $ | ||||||
|
ÉTIQUETAGE SEULEMENT : SPNM (Supplément à une présentation de nouveau médicament)
|
60 | 33 | 55 | 50 | |||
| Données publiées | 18 041 $ | ||||||
|
DONNÉES PUBLIÉES : SPNM
|
300 | 177 | 140 | 291 | |||
| Demande administrative | 303 $ | ||||||
|
ADMINISTRATIVE : PNM (Présentation de nouveau médicament)
|
45 | 38 | 42 | 28 | |||
|
ADMINISTRATIVE : DIN B (Numéro d'identification du médicament)
|
45 | 36 | - | - | |||
| Remise (frais de traitement applicable à tous les types de frais) | 532 $ | Par catégorie/type de frais | s.o. | s.o. | s.o. | ||
Annexe E - Normes de service, frais et résultats de rendement concernant les évaluations d'instruments médicaux, les licences d'exploitation et le droit de vente
Source d'information : Normes de service : Gazette du Canada, vol. 148, nº 11 - 15 mars 2014; Frais de service : Rapports ministériels sur le rendement de Santé Canada pour les exercices financiers de 2011-2012 à 2013-2014.
Évaluation des instruments médicaux
Cette catégorie est plus complexe. En ce qui concerne l'évaluation des instruments médicaux, il existe différents frais et différentes normes de rendement, selon la catégorie d'instrument médical. Les frais augmentent, tout comme le temps nécessaire à l'évaluation d'un produit, selon la complexité du produit et le niveau de risque que peut poser ce produit. Par conséquent, l'évaluation d'un instrument médical de classe II est associée à des frais d'utilisation moins élevés et à une norme de rendement moins importante qu'un instrument de classe IV, qui est définie comme présentant un risque plus élevé.
Rendement :
Délai moyen en jours civils pour rendre une première décision d'examen (examen 1).
La Loi sur les frais d'utilisation exige que, si le rendement est inférieur aux normes de rendement établies dans une proportion dépassant dix pour cent, ces frais d'utilisation soient réduits (jusqu'à concurrence de 50 % des frais d'utilisation). Les frais d'utilisation réduits s'appliquent à compter du jour où le rapport annuel pour l'exercice financier est déposé, jusqu'au jour où le rapport annuel suivant est déposé.
| Frais d'utilisation (catégories de frais) |
Type et catégorie de frais | Frais en date du 1er avril 2014 |
Nouvelles normes de service pour les frais d'utilisation, en vigueur le 1er avril 2011 (jours) |
Résultats de rendement (jours) |
||
|---|---|---|---|---|---|---|
| 2011-2012 | 2012-2013 | 2013-2014 | ||||
|
Annexe E - Tableau 1 - Notes de bas de page
|
||||||
| Évaluation des instruments médicaux (EVAL-IMED) | Classe II | |||||
|
Classe II Nouveau
|
373 $ | 15 | 10.49 | 12.99 | 11 | |
| Classe III | ||||||
|
Classe III Nouveau
|
5 361 $ | 60 | 45.02 | 51.46 | 53 | |
|
Classe III Près du patient
|
9 127 $ | 60 | 40.86 | 50.42 | 49 | |
|
Classe III Modification à la fabrication
|
1 349 $ | 60 | 25.92 | 37.8 | 44 | |
|
Classe III Modification importante
|
5 021 $ | 60 | 43.25 | 52.14 | 53 | |
| Classe IV | ||||||
|
Classe IV Nouveau
|
12 470 $ | 75 | 67.33 | 68.09 | 78Annexe E - Tableau 1 - Note de bas de page * | |
|
Classe IV Tissu animal-humain
|
11 633 $ | 75 | 70 | 67 | 62 | |
|
Classe IV Modification à la fabrication
|
1 349 $ | 75 | 32.72 | 43.1 | 59 | |
|
Classe IV Modification importante
|
5 721 $ | 75 | 52.14 | 63.64 | 67 | |
| Remboursement (frais de traitement applicable à tous les types de frais) | ||||||
|
Remise
|
55 $ | Par catégorie/type de frais | s.o. | s.o. | s.o. | |
Licence d'exploitation et droit de vente :
Ces frais sont imposés pour les instruments médicaux ainsi que pour les médicaments. La licence d'exploitation permet à une industrie de fabriquer, de distribuer, d'importer, de vendre en gros et de tester des produits réglementés au Canada. Le droit de vente permet à l'industrie de vendre officiellement un produit réglementé au Canada. Ces normes de rendement sont claires. Il n'y a qu'un seul frais et une seule norme de rendement pour chaque service et catégorie de frais.
| Frais d'utilisation (catégories de frais) |
Type et catégorie de frais |
Frais en date du 1er avril 2014 |
Nouvelles normes de service pour les frais d'utilisation en vigueur le 1er avril 2011 |
Résultats de rendement (jours) | ||
|---|---|---|---|---|---|---|
| 2011-2012 | 2012-2013 | 2013-2014 | ||||
| Licence d'exploitation | ||||||
| Licence d'exploitation pour les produits pharmaceutiques (LEPP) |
Plusieurs frais peuvent s'appliquer selon le type et la taille du fabricant, de l'importateur ou du distributeur. Toutefois, la norme de service à respecter pour l'octroi d'une licence d'exploitation pour les produits pharmaceutiques est la même, peu importe la quantité de frais qui s'appliquent. | 250 jours civils pour octroyer/renouveler une licence | Satisfait : Nombre moyen de jours : 121 | Satisfait : Nombre moyen de jours : 183 | Satisfait : Nombre moyen de jours : 153 | |
| Catégorie de frais | Type de frais | Frais | ||||
| Fabrication de médicaments -- Annexe 2 | Frais de base | 16 397 $ | ||||
| Chaque catégorie supplémentaire | 4 108 $ | |||||
| Classes de forme posologique | ||||||
|
2 classes
|
8 204 $ | |||||
|
3 classes
|
16 397 $ | |||||
|
4 classes
|
20 505 $ | |||||
|
5 classes
|
24 600 $ | |||||
|
6 classes
|
28 696 $ | |||||
|
Chaque classe supplémentaire
|
1 646 $ | |||||
| Formes posologiques stériles | 8 204 $ | |||||
| Emballage/étiquetage de médicaments -- Annexe 3 | Frais de base | 10 963 $ | ||||
| Chaque catégorie supplémentaire | 2 739 $ | |||||
| Classes de forme posologique | ||||||
|
2 classes
|
5 467 $ | |||||
|
3 classes ou plus
|
8 204 $ | |||||
| Importation et distribution de médicaments -- Annexe 4 | Frais de base | 6 836 $ | ||||
| Chaque catégorie supplémentaire | 1 710 $ | |||||
| Classes de forme posologique | ||||||
|
2 classes
|
3 419 $ | |||||
|
3 classes ou plus
|
6 836 $ | |||||
| Chaque fabricant | 1 646 $ | |||||
| Chaque classe de forme posologique supplémentaire pour chaque fabricant | 829 $ | |||||
| Distribution ou vente en gros | Frais de distribution ou de vente en gros | 4 108 $ | ||||
| Tests | Frais de test | 2 739 $ | ||||
| Analyse de médicament -- Annexe 5 | Vaccins | 27 327 $ | ||||
| Médicaments, non inclus aux points 1, 6 et 9 de la présente annexe, et qui figurent à l'annexe D de la Loi sur les aliments et drogues, 25 | 10 932 $ | |||||
| Médicaments pour usage humain qui sont des médicaments sur ordonnance, des médicaments contrôlés ou des narcotiques | 8 204 $ | |||||
| Médicaments pour usage humain qui ne sont pas inclus dans aucun autre point, pour lesquels un numéro d'identification a été assigné | 4 108 $ | |||||
| Licence de distributeur | Frais d'examen des demandes de licence de distributeur | Licence de distributeur Norme de service = 250 jours pour le renouvellement |
4 788 $ | |||
| Licence d'exploitation pour les instruments médicaux (LEIM) | Licence d'exploitation pour les instruments médicaux | 7 641 $ | 120 jours civils pour octroyer/renouveler une licence | Satisfait : Moyenne de 77 jours | Satisfait : Moyenne de 101 jours | Satisfait : Moyenne de 90 jours |
| Droit de vente | ||||||
| Droit de vente de produits pharmaceutiques (DVPP) | Droit de vente de produits pharmaceutiques | 1 084 $ | 120 jours civils pour mettre à jour la base de données des produits pharmaceutiques à la suite de l'avis | Satisfait : Nombre moyen de jours : 9,1 | Satisfait : Nombre moyen de jours = 7,1 | Satisfait : 100 % dans les 120 jours |
| Droit de vente d'instruments médicaux (DVIM) * Classe II, III ou IV, instruments médicaux seulement |
Frais - revenus bruts annuels de la vente d'instruments médicaux inférieurs à 20 000 $ | 55 $ | 20 jours civils à compter de la date de réception de l'avis annuel de mise à jour de la base de données des licences actives pour des instruments médicaux (MDALL) | Satisfait : 99,8% dans les 20 jours civils | Satisfait : 99,9% dans les 20 jours civils | Satisfait : 99,9% dans les 20 jours civils |
| Frais - tous les autres cas | 351 $ | |||||
Annexe F - Organigramme des activités de recouvrement des coûts
{kind=link}

Annexe G - Structure de gouvernance
{kind=link}

Annexe H - Répartition des revenus tirés des frais d'utilisation pour 2013-2014, par direction générale et catégorie de frais (en milliers de dollars)
| Catégorie de frais | DGPSA | BRP | DGSG | DGDPF | Total | Régime d'avantages sociaux des employés | Somme totale |
|---|---|---|---|---|---|---|---|
| Source : Données SAP fournies par l'agent principal des finances de la direction générale (non auditées) | |||||||
| Évaluation des médicaments pharmaceutiques | 24 334 | 76 | 1 805 | 337 | 26 552 | 3 182 | 29 734 |
| Évaluation des médicaments biologiques | 6 537 | 20 | 485 | 91 | 7 133 | 855 | 7 988 |
| Évaluation d'instruments médicaux | 5 150 | 16 | 380 | 71 | 5 617 | 669 | 6 286 |
| Licences d'exploitation pour les produits pharmaceutiques | 4 523 | 4 972 | 702 | 131 | 10 328 | 1 238 | 11 566 |
| Licences d'exploitation pour les instruments médicaux | 3 504 | 3 898 | 551 | 103 | 8 058 | 971 | 9 027 |
| Droit de vente de drogues | 7 035 | 2 165 | 680 | 127 | 10 007 | 1 199 | 11 206 |
| Droit de vente d'instruments médicaux | 5 282 | 1 624 | 510 | 95 | 7 511 | 899 | 8 410 |
| Total | 56 365 | 12 771 | 5 113 | 955 | 75 204 | 9 013 | 84 217 |
Détails de la page
- Date de modification :