
Évaluation du programme des médicaments à usage humain de 1999-2000 à 2011-2012
Préparé par la
Direction de l'évaluation
Santé Canada et l'Agence de la santé publique du Canada
Mai 2014
Table des matières
- Résumé
- Réponse et plan d'action de la direction
- 1.0 Introduction
- 2.0 Profil du Programme des médicaments à usage humain
- 3.0 Méthode d'évaluation
- 4.0 Résultats - Pertinence
- 5.0 Résultats - Gouvernance et mise en œuvre
- 6.0 Résultats - Résultats atteints
- 6.1 Sensibilisation et compréhension des intervenants
- 6.2 Sensibilisation et compréhension de l'industrie
- 6.3 Sécurité et efficacité
- 6.4 Conformité de l'industrie
- 6.5 Adoption de comportements sûrs
- 6.6 Utilisation de données scientifiques et d'analyses risques-avantages
- 6.7 Intervention réglementaire rapide à l'égard des risques
- 6.8 Harmonisation internationale
- 6.9 Résultats à long terme
- 6.10 Conséquences inattendues
- 7.0 Constatations - Efficacité et économie
- 8.0 Conclusions et recommandations
- Annexe A - Grille d'évaluation
- Annexe B - Liste des ouvrages de référence
- Annexe C - Tableaux de données supplémentaires
Résumé
Le Programme des médicaments à usage humain (PMUH) est géré par la Direction générale des produits de santé et des aliments (DGPSA) de Santé Canada. Il inclut les médicaments à usage humain (aussi appelés médicaments pharmaceutiques), soit les médicaments sur ordonnance et les médicaments en vente libre destinés à l'usage humain, mais exclut les médicaments biologiques, les vaccins, le sang et les produits sanguins, les cellules, les tissus et les organes, lesquels relèvent de la compétence du Programme des produits biologiques de la DGPSA.
Les principaux partenaires participant à la mise en œuvre du PMUH sont la Direction des produits thérapeutiques (DPT), la Direction des produits de santé commercialisés (DPSC), l'Inspectorat de la DGPSA (l'Inspectorat), le Bureau des régions et des programmes (BRP), la Direction des politiques, de la planification et des affaires internationales (DPPAI), et la Direction de la gestion des ressources et des opérations (DGRO). Le programme comprend les activités suivantes : la création et la tenue à jour d'un cadre de réglementation, l'interaction et la communication avec les partenaires et les intervenants, la réalisation d'évaluations des avantages et des risques, le contrôle et la surveillance post-commercialisation, et la conformité et l'application du cadre de réglementation.
L'évaluation du PMUH fait partie du plan quinquennal d'évaluation de Santé Canada. Selon la Politique sur l'évaluation du Conseil du Trésor (SCT, 2009), elle porte sur la pertinence et le rendement (efficacité, efficience et économie) des activités de Santé Canada dans le cadre du programme. L'évaluation couvre la période allant de 1999 à la fin de l'année civile 2012, plus précisément les cinq dernières années, même si le rapport rend compte des activités du programme jusqu'à la fin de juin 2013. Les résultats de l'évaluation éclaireront la mise en œuvre des activités en cours et à venir.
Une société d'experts-conseils indépendante en matière d'évaluation a réalisé l'évaluation pour le compte de Santé Canada. Elle s'est inspirée de plusieurs sources de données, dont la revue de la littérature, l'examen des documents, l'examen des données de gestion, les études de cas, les sondages menés auprès de fabricants et d'autres intervenants, et les entrevues avec des informateurs clés externes. Tout au long du processus, des représentants du programme ont été consultés au moyen de conférences téléphoniques et de courriels en vue d'obtenir des renseignements supplémentaires et de clarifier des questions. Le présent rapport dévoile les résultats de l'évaluation, tire des conclusions et formule des recommandations.
Résultats
Pertinence
L'évaluation a confirmé un besoin continu de surveillance gouvernementale en ce qui a trait aux médicaments à usage humain en vue de protéger la santé et la sécurité des Canadiens. Comme ces produits sont de plus en plus populaires en raison de l'accroissement et du vieillissement de la population, ainsi que de la publicité faite par les représentants de l'industrie, un nombre accru de Canadiens seront exposés à leurs risques et à leurs avantages. De plus, certaines tendances, comme l'émergence des produits mixtes et la mondialisation de la chaîne d'approvisionnement, créent des incertitudes qui accentuent la nécessité d'une intervention gouvernementale afin de protéger la santé et la sécurité des Canadiens. En outre, ce rôle cadre bien avec les rôles et les responsabilités du gouvernement fédéral et de Santé Canada décrits dans les lois et les règlements fédéraux, et est en harmonie avec le résultat stratégique de Santé Canada qui consiste à informer et à protéger les Canadiens des risques pour la santé que présentent les aliments, les produits, les substances et les environnements.
Les activités du PMUH correspondent bien aux priorités fédérales portant sur le renforcement de la sécurité du consommateur. Au cours de la dernière décennie, le gouvernement fédéral a consacré d'importantes ressources au développement d'initiatives visant l'amélioration de l'innocuité des produits de santé, notamment les médicaments à usage humain, grâce à la modernisation du cadre de réglementation de ces produits. Les principes clés de cette modernisation incluent l'adoption d'une approche axée sur le cycle de vie des produits, laquelle consiste à évaluer les risques et les avantages des produits thérapeutiques tout au long de leur cycle de vie; la mise en place de mesures réglementaires proportionnelles aux risques, et l'amélioration de la transparence et de l'ouverture du système de réglementation. Le gouvernement fédéral a récemment confirmé son engagement à l'égard de la viabilité à long terme du PMUH en ajoutant les médicaments à usage humain aux dernières mises à jour relatives à son cadre de recouvrement des coûts et aux frais d'utilisation.
Rendement - Mise en œuvre du programme
Au cours de la période visée par l'évaluation, Santé Canada a fait des progrès en mettant en œuvre les activités prévues et, par le fait même, en répondant à de nombreux problèmes et enjeux nouveaux. Cependant, plusieurs de ces problèmes et enjeux ne sont toujours pas résolus.
Essais cliniques
Santé Canada a renforcé le cadre de réglementation des essais cliniques en concrétisant des approches axées sur les risques en vue de contrôler les rapports sur les réactions indésirables survenues au cours d'un essai clinique, en lançant un programme d'inspection des sites d'essai clinique et en exigeant l'établissement de nouvelles normes facultatives pour les comités d'éthique de la recherche. En mai 2013, Santé Canada a lancé une nouvelle base de données publique sur les essais cliniques de médicaments qui avaient été autorisés par le ministère. Même si elle est obligatoire pour les promoteurs, la base de données contient moins de renseignements que les registres obligatoires depuis 1997 aux États-Unis et 2004 dans l'Union européenne. De plus, contrairement aux États-Unis (et bientôt à l'UE), Santé Canada n'oblige pas les promoteurs à publier les résultats des essais cliniques. Une augmentation de la quantité de renseignements disponibles sur les essais cliniques, y compris les résultats de ces essais, cadrerait avec l'engagement de Santé Canada visant à accroître la transparence et l'ouverture dans le cadre de la modernisation de la réglementation, et permettrait au ministère d'harmoniser davantage son approche avec celle de ses principaux homologues internationaux.
Recommandation 1:
Conformément aux tendances internationales et à son engagement visant à accroître la transparence et l'ouverture dans le cadre de la modernisation de la réglementation, Santé Canada devrait publier davantage de renseignements sur les essais cliniques, y compris les résultats de ces essais.
Examen des présentations et autorisation de mise sur le marché
Santé Canada a franchi une étape importante avec l'entrée en vigueur du Règlement sur les prix à payer à l'égard des drogues et instruments médicaux en avril 2011. Le règlement actualise les frais d'utilisation pour différents services de réglementation, y compris l'examen des présentations et les licences d'établissement de produits pharmaceutiques, dans le but de rétablir le ratio de partage des coûts de 50 % en vigueur lors de l'adoption des frais d'utilisation au milieu des années 1990. On s'attend à ce que l'augmentation des revenus résultant de la mise à jour des frais d'utilisation favorise la stabilité de la plateforme financière, ce qui permettra à Santé Canada d'offrir plus de services de réglementation.
En plus du nouveau cadre de recouvrement des coûts, Santé Canada a également lancé de nombreuses initiatives visant l'amélioration de la qualité et de l'efficience du processus d'examen des présentations et d'autorisation de mise sur le marché. Par exemple : la mise en œuvre d'une approche de gestion de projet pour l'examen des présentations; l'élaboration de Bonnes pratiques de lignes directrices et de Bonnes pratiques d'examen en vue d'accroître l'uniformité de ces processus; la collaboration avec la Food and Drug Administration (FDA) par l'intermédiaire du Conseil de coopération en matière de réglementation (CCR) en vue d'élaborer le Portail commun de demandes électroniques, lequel permettra aux promoteurs de présenter des demandes simultanément à Santé Canada et à la FDA; le renforcement de la capacité d'examen scientifique; et la prise de mesures dans le but d'augmenter le nombre d'examens étrangers effectués relativement aux demandes canadiennes.
L'incidence de ces initiatives sur l'efficience et la qualité du processus d'examen donne matière à de plus amples analyses. Au cours de la période visée par l'évaluation, les délais relatifs à l'examen des présentations variaient et la cible de rendement de Santé Canada n'était généralement pas atteinte. Toutefois, depuis la mise en œuvre du nouveau cadre de recouvrement des coûts en avril 2011, la DPT a atteint la majorité de ses cibles de rendement en matière de recouvrement des coûts, à l'exception de ceux relatifs aux médicaments génériques, et le rendement des examens des présentations est demeuré assez stable.
Surveillance post-commercialisation
Depuis toujours, les rapports sur les réactions indésirables sont les piliers sur lesquels s'appuie l'approche de Santé Canada en matière de surveillance post-commercialisation. En règle générale, les rapports obligatoires sur les réactions indésirables par les représentants de l'industrie ne posent pas problème. En vue de traiter les enjeux de longue date relatifs à la sous-déclaration des professionnels de la santé, Santé Canada a exigé la création de nouvelles normes nationales concernant les rapports sur les réactions indésirables à l'intention des établissements de santé et des hôpitaux agréés. Ces normes ont été publiées en janvier 2013. À ce moment, Santé Canada a mentionné qu'il envisageait de surveiller l'incidence de ces nouvelles normes avant de décider de procéder à des modifications réglementaires. Les rapports obligatoires sur les réactions indésirables pour les établissements de santé avaient déjà fait l'objet du projet de loi C-51 en 2008, soit une mesure législative d'envergure visant à aborder les différentes lacunes du cadre de réglementation actuel relativement aux produits de santé. Le projet de loi C-51 est mort au Feuilleton et n'est jamais devenu une loi.
Santé Canada procède actuellement à l'élaboration de systèmes d'information en vue d'accepter les rapports sur les réactions indésirables par voie électronique, de les contrôler et de les analyser dans le but de détecter les signaux d'innocuité. Plus particulièrement, Santé Canada met en œuvre un système électronique pour la présentation de rapports sur les réactions indésirables destiné aux représentants de l'industrie, ainsi que des stratégies visant à contrôler systématiquement ces rapports au moyen d'une surveillance ciblée et de l'exploration des données. Malgré les délais causés par des difficultés techniques, on s'attend à ce que ces initiatives renforcent la capacité du ministère à analyser les rapports sur les réactions indésirables à l'échelle nationale en vue de détecter les signaux d'innocuité. On espère également que la présentation électronique de rapports sur les réactions indésirables et l'exploration des données permettront à Santé Canada d'analyser les rapports étrangers, lesquels représentent plus de 90 % des tous les rapports reçus par la Direction générale. En attendant que le ministère mette ces systèmes d'information et ces fonctions en place, les initiatives (comme les normes nationales concernant les rapports sur les réactions indésirables) visant à accroître le nombre de rapports reçus sont peut-être prématurées.
Au cours des dernières années, comme d'autres organismes de réglementation partout dans le monde, Santé Canada a reconnu les limites des rapports spontanés sur les réactions indésirables pour détecter les signaux d'innocuité et a cherché à renforcer la surveillance post-commercialisation de nombreuses façons. En plus des rapports sur les réactions indésirables, Santé Canada surveille désormais d'autres sources d'information visant à détecter les signaux d'innocuité, et a normalisé le processus de détection des signaux et d'évaluation pour l'ensemble de ses gammes de produits. Toutefois, le ministère ne dispose pas d'une approche normalisée et centralisée lui permettant d'effectuer un suivi systématique de ses activités liées aux signaux et de sa réponse aux mesures recommandées découlant des évaluations de signaux réalisées. À l'heure actuelle, cette information n'est pas mise à jour régulièrement et est très dispersée au sein de la DGPSA. Étant donné les risques pour la santé et la sécurité des Canadiens, une approche globale, uniforme et centralisée quant à la gestion de l'information des activités de surveillance post-commercialisation de Santé Canada semble justifiée.
Recommandation 2:
Santé Canada devrait améliorer ses systèmes d'information pour le contrôle et la surveillance post-commercialisation. Cette amélioration devrait inclure les points suivants :
- Mise en œuvre intégrale des systèmes d'information en vue d'appuyer la présentation électronique de rapports sur les réactions indésirables survenues au Canada et à l'étranger, ainsi que le contrôle et l'analyse systématiques de ces présentations au moyen d'une surveillance ciblée et de l'exploration des données;
- Élaboration et mise en œuvre d'une approche approfondie et centralisée en matière de gestion de l'information en ce qui a trait aux activités de surveillance post-commercialisation. Plus particulièrement, il faut mettre en place un mécanisme centralisé afin d'effectuer le suivi des activités de détection des signaux d'innocuité, notamment la réponse de Santé Canada aux mesures recommandées découlant des évaluations de signaux réalisées
Malgré l'annonce faite à cet égard dans le cadre du Plan d'action pour assurer la sécurité des produits alimentaires et de consommation (PASPAC), Santé Canada n'a toujours pas concrétisé un seul des éléments des approches renforcées en matière de surveillance post-commercialisation en place ailleurs, à savoir l'autorité nécessaire afin d'obliger les fabricants à fournir des plans de gestion des risques et des rapports réguliers concernant l'innocuité. Les représentants de l'industrie peuvent toutefois présenter ces documents de façon volontaire en réponse aux demandes de Santé Canada. Même si le ministère reconnaît que l'absence de ces autorités représente une limite de son approche, on ne sait pas vraiment s'il a l'intention de les obtenir dans le futur. Un autre domaine d'incertitude est la mesure dans laquelle Santé Canada contrôle la conformité des fabricants aux conditions imposées dans le cadre des avis de conformité avec conditions.
Recommandation 3:
Santé Canada devrait évaluer s'il est toujours pertinent de poursuivre la mise en place d'organismes de réglementation post-commercialisation comme il s'y est engagé dans le cadre du PASPAC, à savoir accorder l'autorité nécessaire afin d'obliger les fabricants à fournir des plans de gestion des risques et des rapports réguliers concernant l'innocuité.
Conformité et application
En ce qui concerne la conformité et l'application, Santé Canada a renforcé les inspections des essais cliniques en élaborant un processus de sélection des sites axé sur les risques; a consolidé la surveillance en ce qui a trait aux produits importés par l'intermédiaire du Programme national de l'intégrité frontalière; a changé le nom du Programme d'inspection de la conformité aux exigences de déclaration après commercialisation (CEDAC), qui se nomme maintenant le programme d'inspection des Bonnes pratiques de pharmacovigilance (BPV), afin de respecter les exigences internationales; a élargi les exigences relatives aux Bonnes pratiques de fabrication (BPF) et à la licence d'établissement de produits pharmaceutiques (LEPP) en vue d'englober les ingrédients pharmaceutiques actifs (IPA); et a adopté une approche axée sur les risques relativement aux inspections des BPF des établissements pharmaceutiques canadiens.
Les événements récents, comme le ralentissement de la production d'un fabricant de produits génériques à la suite des inspections de la FDA, lesquelles ont révélé plusieurs non-conformités, ont porté l'attention du public sur le programme d'inspection des BPF de Santé Canada, en plus de soulever des questions à cet égard. Même si certains informateurs clés externes se sont montrés préoccupés par les interprétations divergentes quant aux normes et à la qualité des inspections, Santé Canada a mentionné que celles-ci sont probablement attribuables aux différences relatives aux approches des deux organismes de réglementation en matière d'inspection, et à la portée de ces inspections.
Un autre problème concerne peut-être la surveillance qu'exerce Santé Canada sur les établissements étrangers qui fabriquent des médicaments qui seront importés au Canada. Même si le Canada a signé des accords de reconnaissance mutuelle (ARM) avec plusieurs pays et qu'il reconnaît ainsi l'équivalence de leurs programmes de conformité aux BPF, la majorité des produits pharmaceutiques et des IPA importés au Canada proviennent de pays avec lesquels le Canada n'a pas conclu d'accord. Dans le cas des IPA, l'importation représente 85 % du marché canadien, et la Chine et l'Inde sont les principaux fournisseurs. De plus, même si les établissements canadiens sont inspectés régulièrement conformément aux critères axés sur les risques, Santé Canada n'effectue pas souvent d'inspections dans les établissements étrangers. On s'attend donc à ce que l'élargissement récent des exigences relatives aux BPF et à la LEPP en vue d'englober les IPA réduise le risque que des produits de santé de mauvaise qualité ou de contrefaçon se retrouvent sur le marché canadien.
Afin de surmonter les difficultés associées aux inspections des BPF, Santé Canada et la FDA des États-Unis ont lancé une initiative par l'intermédiaire du CCR dont le but est d'accroître, dans chaque pays, le recours aux rapports d'inspection des BPF préparés par l'autre pays. À l'heure actuelle, l'initiative s'applique aux sites canadiens et américains, bien qu'il soit possible qu'elle s'applique à d'autres pays dans le futur. En outre, Santé Canada est membre du Pharmaceutical Inspection Cooperation Scheme (PIC/S), un organisme composé de 43 autorités internationales participantes qui se sont réunies en vue d'harmoniser les approches et les exigences en matière d'inspection des BPF partout dans le monde. Santé Canada examine les rapports d'inspection du PIC/S afin d'approuver les sites étrangers pour les importateurs canadiens.
Recommandation 4:
Santé Canada devrait examiner les façons d'améliorer sa surveillance des établissements producteurs à l'étranger.
Même si l'un des principes clés de l'initiative du CCR porte sur la normalisation et le partage des rapports d'inspection des BPF, Santé Canada et la FDA optent pour des approches d'établissement de rapport différentes. En effet, la FDA établit des rapports en fonction de la catégorie des produits, alors que Santé Canada rassemble tous les rapports sur les BPF, peu importe le type de produit concerné. Il est possible que l'atteinte des objectifs de l'initiative nécessite une approche commune en matière d'établissement de rapports de conformité.
Enfin, Santé Canada a inclus de nouvelles mesures de gestion et d'application dans le cadre du projet de loi C-51. Ces mesures comprenaient le pouvoir d'ordonner le rappel de produits thérapeutiques et celui d'imposer des amendes plus lourdes en cas de non-conformité. Comme il a été mentionné précédemment, le projet de loi C-51 n'est pas devenu une loi. Lors d'entrevues, certains informateurs clés externes se sont montrés préoccupés par ce qu'ils considèrent comme le peu d'options dont dispose Santé Canada en matière d'application.
Recommandation 5:
Santé Canada devrait se pencher sur la question à savoir s'il est encore justifié d'obtenir le pouvoir de prendre des mesures de conformité et d'application comme il s'y est engagé dans le cadre du PASPAC, à savoir le pouvoir d'ordonner le rappel de produits thérapeutiques et celui d'imposer des amendes plus lourdes en cas de non-conformité.
Communications et mobilisation des intervenants
Santé Canada a entrepris de nombreuses initiatives en vue d'améliorer les communications et la mobilisation des intervenants. Par exemple, depuis 2005, Santé Canada fournit au public des renseignements sur les décisions relatives aux examens par l'intermédiaire du Sommaire des motifs de décision (SMD). En 2012, en partie en vue d'aborder les préoccupations exprimées par le Bureau du vérificateur général (BVG) selon lesquelles le ministère ne divulguait pas de renseignements liés aux avis de conformité avec conditions (AC-C), aux médicaments rejetés et aux médicaments retirés du processus d'examen, Santé Canada a mis en place le tableau des activités postautorisation (TAPA). Les TAPA fournissent des renseignements à jour sur les produits approuvés. Ils incluent un résumé des activités portant sur l'utilisation sécuritaire et efficace du produit, comme les renseignements liés aux présentations en vue d'une nouvelle utilisation du produit (peu importe si la décision de Santé Canada était positive ou négative), les présentations soumises dans le but de respecter les conditions (pour les produits approuvés en vertu des lignes directrices de l'AC-C), et les décisions réglementaires, comme l'annulation du numéro d'identification du médicament (DIN). Santé Canada ne publie pas de SMD en cas de décisions négatives et ne divulgue pas les raisons de ces décisions, contrairement à la FDA et à l'Agence européenne des médicaments (EMA).
Afin d'accroître la qualité et la disponibilité d'étiquettes de produits pharmaceutiques faciles à comprendre, Santé Canada a amélioré la monographie des produits, a créé, par l'intermédiaire du CCR, une monographie commune avec la FDA pour les médicaments en vente libre, et a apporté des modifications réglementaires dans le cadre de l'initiative d'étiquetage en langage clair. À l'heure actuelle, Santé Canada n'a pas l'autorité nécessaire pour exiger aux fabricants de modifier l'étiquetage d'un produit lorsque ce dernier a reçu un avis de conformité. En revanche, la FDA peut exiger que l'étiquetage des produits soit mis à jour en fonction des nouveaux renseignements disponibles sur l'innocuité d'un médicament, y compris le pouvoir d'exiger un encadré à cet effet. La nouvelle législation de l'UE en matière de pharmacovigilance ordonne qu'un triangle noir inversé soit placé sur le dépliant des médicaments faisant l'objet d'une surveillance post-commercialisation accrue.
Au cours de la période visée par l'évaluation, Santé Canada a publié des renseignements sur les risques et l'innocuité des médicaments sur la page « Avis, mises en garde et retraits » du site Web de MedEffet, et au moyen d'autres mécanismes de diffusion variés Au début de l'année 2013, il a lancé la Base de données sur les rappels et les avis de sécurité, laquelle comprend une fonction de recherche avancée et un nouveau format aux fins de communication des risques. Le ministère a mentionné qu'il procède actuellement à l'examen de ses cibles de rendement actuelles concernant la préparation et la diffusion de documents de communication des risques, et qu'il se penche sur des domaines dans lesquels il serait possible d'apporter des améliorations axées davantage sur la communication des risques. En outre, Santé Canada a récemment entrepris l'évaluation de sa communication des risques relativement aux produits de santé, y compris les médicaments à usage humain, en donnant suite à des plans de longue date visant à évaluer l'efficacité de ses produits de communication des risques.
Santé Canada favorise la mobilisation des intervenants de multiples façons, comme la tenue de consultations publiques sur les lignes directrices, les politiques et les modifications réglementaires proposées, ainsi que la mise en place de comités consultatifs en vue d'éclairer l'élaboration de politiques et de règlements. Le ministère consulte également les représentants de l'industrie au moyen de réunions préalables au dépôt des présentations et de programmes des réunions bilatérales (PRB). Même si ces possibilités particulières ne sont pas offertes aux professionnels de la santé et aux consommateurs/patients, Santé Canada a indiqué qu'il rencontre toutefois les associations pharmaceutiques, les associations médicales et les associations des hôpitaux qui représentent ces professionnels. Certains informateurs clés externes se sont dits préoccupés par le fait que le processus de consultation et de mobilisation puisse favoriser les représentants de l'industrie plutôt que les autres intervenants.
Rendement - Résultats atteints
Au cours de la période visée par l'évaluation, Santé Canada a participé à de nombreuses activités qui devraient favoriser l'atteinte des résultats du PMUH. Toutefois, pour différentes raisons, les données appuyant les conclusions définitives quant aux résultats atteints sont relativement limitées.
Résultats immédiats
À très court terme, on s'attend à ce que les activités de Santé Canada accroissent la sensibilisation et la compréhension des intervenants externes quant aux risques et aux avantages des médicaments à usage humain. Les sondages menés entre 2003 et 2007 ont révélé des façons d'accroître la sensibilisation des consommateurs et des professionnels de la santé en ce qui a trait à l'information sur l'innocuité des médicaments disponible auprès de Santé Canada, mais aucun renseignement précis n'était disponible sur les médicaments à usage humain. Le ministère procède actuellement à l'évaluation de l'efficacité de ses communications des risques en ce qui concerne les produits thérapeutiques.
À très court terme, on s'attend à ce que les activités de Santé Canada accroissent la sensibilisation et la compréhension des représentants de l'industrie quant aux activités de réglementation du ministère relativement aux médicaments à usage humain. Les données disponibles, bien qu'elles soient limitées, indiquent certains aspects susceptibles d'amélioration. Par exemple, même si les répondants au sondage mené auprès des représentants de l'industrie croient que leur entreprise connaît bien les exigences de Santé Canada en ce qui concerne les demandes d'approbation de mise en marché, les informateurs clés de l'industrie ont affirmé que des précisions seraient nécessaires en ce qui a trait à la classification des nouveaux produits de santé et à l'utilisation que fait Santé Canada des lignes directrices et des examens étrangers dans le cadre du processus d'examen. À court terme, les activités de Santé Canada visent également à augmenter l'innocuité et l'efficacité des médicaments à usage humain. Des processus préalables à la mise en marché et après la mise en marché sont en place en vue de s'assurer que les médicaments à usage humain sont sécuritaires et efficaces; toutefois, aucune donnée ne confirme des améliorations dans ces domaines. Il serait possible de prétendre que l'autorité de Santé Canada consiste uniquement à veiller à ce que les produits disponibles sur le marché canadien soient sécuritaires et efficaces.
Enfin, à court terme, on s'attend à ce que les activités de Santé Canada favorisent la conformité des représentants de l'industrie aux exigences réglementaires. Les données disponibles indiquent que les non-conformités graves sont assez rares. Au cours de la période visée par l'évaluation, Santé Canada n'a pas produit de rapports de façon régulière ou systématique sur la nature, la gravité, la fréquence ou la prévalence des cas de non-conformité liés aux médicaments à usage humain; il a plutôt axé ses rapports sur la mesure des activités et des extrants. De plus, la majorité des données de conformité de Santé Canada, y compris celles sur les BPF, les Bonnes pratiques cliniques (BPC) et les BPV, sont regroupées au sein de différentes catégories de produits, englobant les médicaments à usage humain et biologiques (ainsi que les médicaments vétérinaires dans le cas des BPF).
Ceci étant dit, l'Inspectorat a récemment rédigé un rapport sommaire des inspections annuelles, lequel sera publié sur le site Web de Santé Canada. Le rapport 2012-2013 fournit une description des activités et des extrants de l'Inspectorat, décrit le taux de conformité global de l'industrie et dresse une liste des observations communes citées dans les établissements non conformes. Le rapport ne contient aucun renseignement sur les mesures prises par l'Inspectorat en réponse aux cas de non-conformité. Il ne décompose pas non plus l'information portant sur la conformité des BPF, des BPC et des BPV par catégorie de produits.
Recommandation 6:
Santé Canada devrait continuer de miser sur son approche actuelle en matière de rapports sur la conformité en se concentrant davantage sur les résultats des activités de conformité et d'application, et en renforçant sa capacité à produire des rapports sur la conformité par catégorie de produits.
Résultats intermédiaires
À moyen terme, on s'attend à ce que les activités de Santé Canada encouragent les intervenants à adopter des comportements sécuritaires en ce qui a trait à l'utilisation de médicaments à usage humain. Même si la littérature confirme que des pratiques dangereuses, comme l'abus de produits pharmaceutiques sur ordonnance, sont utilisées au Canada, on ne connaît pas l'incidence que peuvent avoir les activités du ministère à cet égard. L'évaluation continue de Santé Canada quant à l'efficacité de ses communications des risques peut indiquer l'influence qu'ont les activités du ministère sur le comportement des intervenants. Dans ce contexte, il est important de noter que la pratique de la médecine, laquelle est régie par les provinces et les territoires, influe également sur le comportement des intervenants.
Les activités de Santé Canada devraient également favoriser l'utilisation des données scientifiques et de l'analyse des avantages et des risques en vue d'éclairer la prise de décisions relative aux médicaments à usage humain. L'utilisation des données scientifiques et de l'analyse des avantages et des risques a été officiellement intégrée au cadre décisionnel de Santé Canada pour la détermination, l'évaluation et la gestion des risques pour la santé, ainsi qu'à différents processus préalables à la mise en marché et après la mise en marché. Le ministère a créé de nombreux groupes consultatifs d'experts/de scientifiques afin d'orienter l'élaboration de politiques et de règlements, et a concrétisé une partie de leurs recommandations.
À moyen terme, Santé Canada espère réagir rapidement aux risques liés aux médicaments à usage humain. En admettant que l'élaboration de politiques et de règlements soit souvent un processus très long, l'évaluation a permis de relever des exemples dans lesquels la mise en œuvre de la réponse de Santé Canada a pris plus de temps que prévu. Par exemple, plus d'une décennie s'est écoulée entre le moment où Santé Canada a annoncé son intention de mettre en place des exigences relatives aux BPF et à la LEPP en vue d'englober les IPA et la modification réglementaire. Ou, le ministère s'est engagé à accroître la transparence des renseignements sur les essais cliniques dès 2007, mais n'a mis en place une base de données publique à cet effet qu'en mai 2013.
L'analyse des données relatives aux signaux de Santé Canada a permis de révéler que de nombreux secteurs risquent de connaître des retards. Ces données indiquent que même si le ministère se fie à de nombreuses sources pour détecter les signaux d'innocuité liés aux médicaments à usage humain, y compris les données des promoteurs, les deux principales sources demeurent les ouvrages scientifiques et les autres organismes de réglementation. Comme il peut être très long avant que les résultats des études scientifiques soient publiés et que les autres organismes de réglementation évaluent l'innocuité, il est possible que Santé Canada ait de la difficulté à réagir rapidement. Les efforts du ministère en vue de promouvoir la présentation de rapports et d'analyses sur les réactions indésirables peuvent contribuer à la résolution de ce problème, qui dépendra alors moins des études externes.
La rapidité du processus d'évaluation des signaux s'est améliorée depuis la mise en place des normes de service relatives à l'évaluation des signaux. En 2011-2012, 93 % des évaluations des signaux après la mise en marché concernant les produits pharmaceutiques, les instruments médicaux, les produits biologiques et les produits de santé naturels ont été réalisées à l'intérieur du délai de 130 jours prescrit par la norme de service. Toutefois, l'évaluation des signaux représente seulement un aspect de la réponse globale de Santé Canada en matière de signaux d'innocuité. L'évaluation a révélé que les retards peuvent survenir à d'autres moments pendant le processus. Notamment, une période de temps considérable peut s'écouler entre le moment où les signaux sont détectés et celui où on les évalue, de même qu'entre l'approbation de l'évaluation du signal et la communication du risque.
Ce résultat cadre avec le rapport 2011 du BVG, lequel indique que l'évaluation et la réponse du ministère à l'égard des signaux d'innocuité potentiels n'ont pas lieu en temps opportun. Même si la majorité des évaluations de l'innocuité étudiées par le BVG dans le cadre de sa vérification ont été réalisées dans les délais prescrits, l'approche employée par le ministère pour mesurer son rendement n'a pas tenu compte du temps écoulé avant que le problème d'innocuité potentiel ne soit évalué; du temps d'attente avant le début de l'évaluation; du temps nécessaire pour obtenir des renseignements supplémentaires auprès des parties externes (comme les fabricants); et du nombre total de jours civils, plutôt que de jours ouvrables, requis pour effectuer l'évaluation. Le BVG a constaté que lorsque ces facteurs ont été pris en considération, il a fallu au moins une année à Santé Canada pour réaliser 34 de ses 54 évaluations, et même beaucoup plus de temps dans certains cas - deux ou trois ans.
Les représentants du programme ont signalé que la préparation des documents de communication des risques nécessite habituellement de nombreuses négociations avec le détenteur d'une autorisation de mise sur le marché, et que la diffusion du document peut être retardée jusqu'à ce que les changements appropriés aient été apportés à l'étiquette du produit. En raison de ces facteurs, le temps écoulé entre le moment où le signal est détecté et la diffusion du document de communication du risque peut être très long. En ce qui concerne 14 cas pour lesquels des données complètes étaient disponibles (portant sur des évaluations des signaux réalisées entre 2010 et 2012), le temps total écoulé entre la détection du signal et la publication du document de communication du risque variait entre 232 jours et 1 481 jours (4 ans), avec une médiane de 1,4 an. Comme il a été mentionné, Santé Canada procède à l'examen de ses normes de rendement actuelles relativement à la préparation et à la diffusion de documents de communication des risques.
Recommandation 7:
Santé Canada devrait prendre des mesures pour augmenter la rapidité avec laquelle il donne suite aux signaux relatifs à l'innocuité (de la détection de ces signaux à la diffusion d'une communication des risques).
À moyen terme, Santé Canada espère accroître l'uniformité des exigences réglementaires concernant les médicaments à usage humain à l'échelle internationale. Parmi de nombreux autres engagements internationaux, Santé Canada est un observateur officiel de la Conférence internationale sur l'harmonisation (CIH) des exigences techniques pour l'enregistrement des médicaments à usage humain, en plus d'y participer; a élaboré des ARM quant à la conformité aux BPF avec plusieurs autres pays; a conclu des ententes sur l'échange de renseignements avec la FDA, la Therapeutic Goods Administration (TGA) de l'Australie et l'Union européenne; et participe, avec la FDA, au CCR, dont le principal objectif consiste à améliorer l'uniformité des approches réglementaires des deux pays. Comme il a été mentionné, Santé Canada est aussi membre du PIC/S. D'importants informateurs internationaux perçoivent Santé Canada comme un participant constructif dans les engagements bilatéraux et multilatéraux.
Résultats à long terme
À long terme, les activités de Santé Canada contribueront probablement à la réduction des risques pour la santé et des événements indésirables liés à l'utilisation des médicaments à usage humain. D'après les données d'enquête disponibles, le taux de confiance des consommateurs en ce qui concerne l'innocuité des médicaments et la sécurité du système de réglementation en place est élevé, même si aucun renseignement précis sur les médicaments à usage humain n'est disponible.
En définitive, Santé Canada espère offrir un système de réglementation viable, rentable, adapté et fondé sur la science en ce qui a trait aux médicaments à usage humain au Canada. L'évaluation a révélé que le ministère a recours aux données scientifiques et consulte des intervenants pour l'élaboration de politiques et de règlements, et que les récentes mises à jour concernant les frais d'utilisation relatifs aux médicaments à usage humain appuieront la viabilité du système de réglementation de ces produits.
Rendement - efficience et économie
Au cours de la période visée par l'évaluation, les changements apportés à l'approche de la DGPSA en ce qui concerne l'établissement de rapports financiers ont compliqué l'harmonisation des dépenses et des budgets relatifs au PMUH permettant la comparaison et l'analyse ultérieures de ces renseignements. La DGPSA a récemment réorganisé la façon dont elle communique l'information financière afin de respecter les exigences du Conseil du Trésor; elle devrait ainsi accroître l'exactitude de ces renseignements et faciliter les prochaines analyses.
Au sein de la DPT, même si aucun rapport financier axé sur les activités n'a été rédigé depuis 2008-2009, le codage a été mis à jour en 2012-2013, de sorte que les renseignements sur les activités fonctionnelles seront disponibles ultérieurement. Les rapports axés sur les activités permettent d'appuyer la comptabilité par activités, laquelle est essentielle à l'analyse de l'efficience des activités de programme réalisées. La DGPSA a entrepris un exercice de comptabilité par activités en 2007 en vue de soutenir la proposition visant à mettre à jour les frais d'utilisation. Elle a ensuite utilisé les résultats de cette analyse pour calculer les coûts unitaires de différentes activités de réglementation, y compris l'examen des présentations. Les données disponibles indiquent que la rapidité à laquelle sont effectués les examens des présentations de la majorité des catégories de médicaments à usage humain s'est améliorée grâce au nouveau cadre de recouvrement des coûts. Toutefois, sans analyser les coûts unitaires de l'examen des présentations et d'autres activités de recouvrement des coûts, on ne sait pas si la rapidité accrue a permis la réalisation de gains d'efficacité. En plus de permettre l'évaluation de la portée des gains d'efficacité réalisés grâce au nouveau cadre, l'analyse permettra également au PMUH de déterminer les ajustements nécessaires. À cette fin, la DGPSA examine donc les coûts, les frais et le rendement associés au PMUH conformément à son engagement dans le cadre de la proposition de frais d'utilisation et du Résumé de l'étude d'impact de la réglementation liés au Règlement sur les prix à payer à l'égard des drogues et instruments médicaux.
| Recommandation | Réponse | Activités principales | Livrables | Direction responsable | Échéance |
|---|---|---|---|---|---|
1. Conformément aux tendances internationales et à son engagement visant à accroître la transparence et l'ouverture dans le cadre de la modernisation de la réglementation, Santé Canada devrait publier davantage de renseignements sur les essais cliniques, y compris les résultats de ces essais. |
Acceptée |
Le Ministère a pris de nombreuses initiatives en vue de respecter son engagement visant à accroître l'accessibilité des renseignements administratifs sur les essais cliniques afin que les patients prennent des décisions éclairées. En 2007, Santé Canada a publié un avis aux promoteurs pour les inciter à inscrire volontairement des essais cliniques dans des registres publics reconnus par l'Organisation mondiale de la Santé (OMS). Cet avis a été mis à jour en 2012 pour laisser savoir que Santé Canada continue d'appuyer la transparence dans les essais cliniques par l'enregistrement des protocoles des essais. En novembre 2011, Santé Canada a également ajouté dans son attestation de non-objection (autorisation délivrée aux promoteurs d'essais cliniques) un rappel sur l'enregistrement de toutes les phases des essais cliniques dans les registres publics. En mai 2013, Santé Canada a publié la version révisée du document d'orientation Considérations relatives à l'inclusion des femmes dans les essais cliniques et à l'analyse de données selon le sexe et l'a mis à la disposition des intervenants participant au développement de médicaments et à la recherche sur les médicaments. Depuis la fin de cette évaluation, soit en mai 2013, le Ministère a lancé une base de données regroupant les essais cliniques de médicaments autorisés. Cette base de données est une source centrale de renseignements de haut niveau (numéro de protocole, titre du protocole, nom du médicament, condition médicale, population à l'étude, date d'autorisation, nom du promoteur, numéro de contrôle de Santé Canada, dates de début et de fin de l'essai clinique, le cas échéant) sur les essais cliniques de phase I, II et III sur des patients. Entre le 1er avril et le 22 octobre 2013, le Ministère a publié 328 essais cliniques. Cette base de données contribue à combler une lacune en matière d'information relevée dans une enquête sur les registres internationaux, qui a révélé que seulement 24 % des essais cliniques autorisés par Santé Canada étaient inscrits dans un registre public et que seulement environ 50 % des essais cliniques sur des patients étaient enregistrés. De plus, Santé Canada a publié une Ligne directrice à l'intention des promoteurs d'essais cliniques : Demandes d'essais cliniques. Santé Canada a également travaillé avec l'Office des normes générales du Canada (ONGC) pour créer une nouvelle norme volontaire pour les comités d'éthique de la recherche afin de les inciter à publier leurs procédures opérationnelles normalisées. Cette norme est accessible dans le site Web de Travaux publics et Services gouvernementaux Canada, à l'adresse http://www.tpsgc-pwgsc.gc.ca/ongc-cgsb/publications/nouvelles-news/nncvcb-ncsreo-fra.htm. |
La DPT dressera un plan présentant les améliorations pouvant être apportées à la base de données. |
DPT |
Juin 2014 |
Au sujet des résultats des essais cliniques, le Règlement sur les aliments et drogues n'oblige pas, à l'heure actuelle, les promoteurs à fournir les résultats de toutes les études cliniques réalisées tout au long du processus de développement d'une drogue, à moins qu'il n'y ait des problèmes qui doivent être signalés. De plus, les promoteurs qui demandent une autorisation de mise en marché peuvent être tenus de présenter des résultats plus poussés relativement à leurs essais cliniques si SC juge que l'information figurant sur la présentation de la drogue est insatisfaisante ou si les évaluateurs sont au fait d'autres études qui pourraient influencer leur prise de décision. Dans certaines circonstances, des données supplémentaires seront demandées pour confirmer l'innocuité, l'efficacité et la qualité de la drogue. Actuellement, il n'existe aucune norme internationale régissant la publication des résultats (c.-à-d. le contenu et la présentation). Nous suivrons de près l'évolution des discussions sur la divulgation des résultats liés aux études sur la scène internationale et orienterons nos travaux politiques et réglementaires futurs en conséquence. |
|||||
2. Santé Canada devrait améliorer ses systèmes d'information pour le contrôle et la surveillance post-commercialisation. Cette amélioration devrait inclure les points suivants :
|
Acceptée |
Avec l'aide du Groupe d'experts de Canada Vigilance, Santé Canada instaure la déclaration électronique des réactions indésirables pour l'industrie et les promoteurs d'essais cliniques. Les déclarations, qui comprendront des renseignements sur les réactions indésirables après la mise en marché et dans le cadre d'essais cliniques conformément aux normes internationales et aux normes de Santé Canada, permettront d'améliorer la capacité du Ministère à évaluer activement les tendances relatives aux réactions indésirables aux produits pharmaceutiques et aux médicaments à usage humain tant à l'échelle nationale qu'internationale. Il s'agira de l'un des outils qui contribueront à la surveillance active des questions relatives à l'innocuité après la mise en marché. Le Ministère continue d'améliorer ses stratégies de surveillance des déclarations de réactions indésirables grâce à une surveillance ciblée et à des techniques améliorées d'exploration de données (analyse statistique). Dans ce contexte, la surveillance ciblée vise à orienter les efforts sur les secteurs considérés comme à risque élevé d'après les problèmes éventuels d'innocuité relevés grâce à l'évaluation des signaux et aux déclarations de réactions indésirables reçues.
|
Afin d'assurer la solidité des systèmes de surveillance après la mise en marché et de surveillance de l'information, la DGPSA élaborera une proposition sur les besoins liés aux éléments suivants : |
DPSC |
|
|
DPSC |
Février 2014 |
|||
La surveillance ciblée et les initiatives d'exploration de données sont relativement récentes et encore en évolution. Le Ministère élabore un plan d'amélioration continue de ces stratégies. Il s'agit d'un élément important dans la transition d'une approche de surveillance passive à une approche de surveillance active. |
Les besoins pour de nouveaux outils seront présentés au Ministère, qui les examinera. |
DPSC |
Décembre 2014 |
||
3. Santé Canada devrait se pencher sur la question de savoir s'il est encore justifié d'obtenir les pouvoirs de réglementation après la mise sur le marché, conformément à son engagement dans le Plan d'action pour assurer la sécurité des produits alimentaires et de consommation (PASPAC), à savoir le pouvoir de contraindre les fabricants à présenter des plans de gestion des risques et des rapports périodiques de pharmacovigilance. |
Acceptée |
En 2008, le gouvernement a présenté le projet de loi C-51, qui comprenait le pouvoir d'établir des modalités relatives à une licence de vente d'un médicament. Ces dispositions auraient appuyé les dispositions obligatoires des plans de gestion des risques (PGR) et des Rapports périodiques de pharmacovigilance (RPPV), mais le projet de loi est mort au feuilleton en raison du déclenchement des élections en 2008. En 2009 Santé Canada a adopté le thème E2E de l'International Conference on Harmonisation (ICH) : Planification de la pharmacovigilance et a instauré des processus visant à faciliter la présentation volontaire de plans de gestion des risques. À ce moment, le Ministère était aussi déterminé à élaborer et à appliquer un cadre de pharmacovigilance par la modernisation de la réglementation qui aurait appuyé la mise en œuvre complète de la planification en matière de gestion des risques. En 2011, Santé Canada a modifié le Règlement sur les aliments et drogues (RAD) afin d'obliger les fabricants à préparer un rapport de synthèse annuel regroupant tous les renseignements relatifs aux réactions indésirables aux drogues et aux réactions indésirables graves aux drogues et à informer le ministre de tout changement sur le plan de l'innocuité relevé dans ce rapport. Le ministre peut demander une copie du rapport de synthèse annuel et demander à ce qu'il soit présenté conformément aux normes de l'ICH relatives aux RPPV. Exception faite de ces exigences, Santé Canada reçoit uniquement les RPPV qui sont présentés sur une base volontaire. Il faut de nouveaux pouvoirs pour obliger les fabricants à présenter des RPPV plus régulièrement s'ils veulent obtenir une autorisation de mise sur le marché. Dans le discours du Trône d'octobre 2013, le gouvernement s'est engagé à déposer un nouveau projet de loi sur la sécurité des patients afin de contribuer à déterminer les médicaments potentiellement dangereux. Le 6 décembre 2013, la ministre de la Santé a déposé un projet de loi au Parlement prévoyant de nouveaux pouvoirs de réglementation visant à imposer des modalités relatives aux produits autorisés, à titre d'exigence après la mise en marché, comme les plans de gestion des risques. Si ces modalités ne sont pas respectées, de nouvelles amendes et pénalités seront imposées pour les produits thérapeutiques. |
Présentation au Parlement d'un projet de loi sur la sécurité des patients permettant entre autres aux responsables de la réglementation d'ordonner la déclaration de données sur l'innocuité après la mise en marché, comme des plans de gestion des risques et des Rapports périodiques de pharmacovigilance. |
DPPAI |
Décembre 2013 (terminée) |
4. Santé Canada devrait examiner les façons d'améliorer sa surveillance des établissements de fabrication à l'étranger. |
Acceptée |
Étant donné la nature mondiale de l'industrie de la fabrication de médicaments et qu'aucun pays ne peut inspecter tous les établissements étrangers de fabrication, les pays doivent travailler ensemble pour protéger leurs citoyens et s'assurer de l'innocuité des médicaments vendus dans leur pays peu import où ils sont fabriqués et utilisés. Santé Canada tire profit des inspections réalisées par des partenaires réglementaires de confiance dans des établissements à l'étranger qui produisent des médicaments vendus au Canada. Ils comptent sur nos inspections des établissements canadiens qui font des affaires à l'étranger. Toutefois, lorsque l'information ou les résultats d'inspections issus de partenariats officiels au terme d'un accord de reconnaissance mutuelle (ARM), des États membres de la Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme ou d'autres sources d'information ne peuvent être utilisés, Santé Canada utilise un modèle axé sur les risques liés à l'établissement et au produit lorsqu'il choisit des établissements pour réaliser des inspections à l'étranger. |
|
IDGPSA |
|
Santé Canada réalise également des inspections de bonnes pratiques de fabrication dans des établissements étrangers si d'importantes questions sont soulevées quant aux pratiques de fabrication, et ce, même si l'établissement a été inspecté par un partenaire de confiance. Aucun produit pharmaceutique ne peut être importé au Canada d'un établissement étranger à moins que Santé Canada ne dispose de données probantes satisfaisantes témoignant du fait que les normes canadiennes relatives aux bonnes pratiques de fabrication sont respectées. Les établissements étrangers approuvés desquels des médicaments peuvent être importés sont identifiés sur la licence d'établissement d'un importateur canadien. |
2 Dans le cadre de l'engagement du CCMR, analyser les inspections d'observation de phase I et préparer un rapport sur l'issue des inspections d'observation de la FDA et de Santé Canada. |
IDGPSA |
|
||
Santé Canada a présenté une modification du Règlement sur les aliments et drogues visant à accroître la surveillance des fabricants et des importateurs d'ingrédients pharmaceutiques actifs (IPA) dans la chaîne d'approvisionnement en médicaments. Ce règlement est entré en vigueur le 8 novembre 2013. Il améliorera la capacité de Santé Canada à remonter aux établissements étrangers s'il y a des problèmes liés aux bonnes pratiques de fabrication. Un programme d'inspection des ingrédients pharmaceutiques actifs a été lancé en novembre 2013. En ce qui concerne les fabricants étrangers se trouvant dans les pays signataires d'un ARM, Santé Canada a entamé des discussions dans le but d'élargir la portée de cet accord et d'y intégrer les ingrédients pharmaceutiques actifs. Santé Canada pourrait ainsi échanger des certificats de conformité pour les inspections des BPF aux établissements d'ingrédients pharmaceutiques actifs qui ont été réalisées par des organismes de réglementation de la santé de pays signataires de l'ARM. |
|
IDGPSA |
|
||
Santé Canada travaille avec la Food and Drug Administration (FDA) des États-Unis dans le cadre de l'initiative du Conseil de coopération en matière de réglementation (CCMR) afin d'éviter la réalisation d'inspections en double et d'accroître la collaboration concernant la conformité et l'application. Cette initiative a renforcé la relation avec la FDA et permet d'échanger des renseignements sur la conformité et l'application dans des établissements d'intérêt commun (à l'échelle nationale et internationale). Santé Canada travaille avec la Therapeutics Goods Administration (TGA) de l'Australie dans le cadre de l'initiative de coopération en matière de réglementation (ICMR) afin de profiter de l'échange de renseignements sur des établissements étrangers d'intérêt commun. La phase I de ce projet est terminée depuis peu tandis qu'il y a eu des discussions sur la phase II, qui est en cours d'achèvement. |
|||||
5. Santé Canada devrait examiner s'il est encore justifié d'obtenir le pouvoir de prendre des mesures de conformité et d'application comme il s'y est engagé dans le cadre du PASPAC, à savoir le pouvoir d'ordonner le rappel de produits thérapeutiques et celui d'imposer des amendes plus lourdes en cas de non-conformité. |
Acceptée |
Depuis son engagement aux termes du PASPAC, Santé Canada a entrepris diverses activités appuyant la poursuite des mesures de conformité et d'application, à savoir le pouvoir d'ordonner des rappels. Au printemps 2008, le gouvernement a présenté le projet de loi C-51, qui comprenait un nouveau pouvoir d'ordonner des rappels. L'engagement des intervenants à l'égard du projet de loi a appuyé la nécessité d'octroyer ce nouveau pouvoir. Le projet de loi est mort au feuilleton en raison du déclenchement des élections en 2008. En janvier 2011, Santé Canada a organisé une série de discussions techniques avec des intervenants sur la modernisation de la réglementation régissant les produits thérapeutiques (médicaments et matériels médicaux) afin de mettre à l'essai et de confirmer les activités proposées qui sont liées à la réglementation d'un produit tout au long de son cycle de vie. La troisième et dernière séance portait sur les activités postérieures à l'autorisation, y compris les rappels obligatoires. En avril 2013, le gouvernement a demandé la réalisation d'un examen indépendant à la suite du rappel d'Alysena 28. Le rapport exigeait le renforcement du pouvoir d'intervention de Santé Canada lorsque des médicaments présentent un danger. Le gouvernement a accepté les recommandations formulées dans le rapport et s'est engagé à examiner la possibilité d'élargir les pouvoirs en matière de rappels. Dans le discours du Trône d'Octobre 2013, le gouvernement s'est engagé à introduire une nouvelle législation pour des patients pour aider à identifier les médicaments potentiellement dangereux et assurer le retrait rapide des médicaments dangereux. Le 6 décembre la ministre de la Santé a déposé un projet de loi au Parlement prévoyant un pouvoir permettant au ministre d'ordonner à une personne vendant un produit thérapeutique de rappeler ce produit ou de l'envoyer à un endroit déterminé par le ministre si celui-ci estime que le produit présente un risque grave ou imminent pour la santé. Le projet de loi prévoyait également des amendes et des pénalités accrues pour mieux représenter la gravité de l'infraction. La pénalité maximale est passée de 5 0000 $ à 50 000 0 $ ou à une peine d'emprisonnement de deux ans. |
Dépôt au Parlement d'une loi sur la sécurité des patients qui prévoit un pouvoir d'ordonner des rappels obligatoires pour des produits thérapeutiques et un pouvoir d'imposer des amendes accrues en cas de non-conformité. |
DPPAI |
Décembre 2013 |
6. Santé Canada devrait continuer de miser sur son approche actuelle en matière de rapports sur la conformité en se concentrant davantage sur les résultats des activités de conformité et d'application, et en renforçant sa capacité à produire des rapports sur la conformité par catégorie de produits. |
Acceptée |
Santé Canada utilise actuellement la cote de conformité globale (c.-à-d. pourcentage de conformité) de l'industrie comme indicateur de rendement fondé sur les résultats pour mesurer et évaluer les résultats de l'Inspectorat (c.-à-d. objectifs en matière d'inspection, nombre d'incidents ouverts ou fermés, etc.) dans l'atteinte des objectifs du programme. Cette mesure du rendement est déclarée chaque mois et intégrée à la planification de Santé Canada. Elle est aussi utilisée dans le Rapport ministériel sur le rendement (RMR), le Rapport sur les plans et les priorités (RPP) et le cadre de mesure du rendement (CMR). Depuis la période visée par cette évaluation, Santé Canada a également élaboré un rapport de synthèse annuel sur les inspections publié à l'externe. Ce rapport décrit en détail la cote de conformité globale de l'industrie par programme et énumère les observations fréquentes relevées dans les établissements non conformes. Il donne également des exemples précis d'observations se rapportant à la Loi sur les aliments et drogues et au Règlement sur les aliments et drogues, et il établit des liens entre les inspections réalisées par activité (c.-à-d. fabricant, importateur, distributeur, etc.), les observations notées et leur catégorie de risque. Ce rapport est publié pour obtenir de nombreux résultats prévus dans le CMR, y compris l'amélioration de la connaissance et de la compréhension du cadre de réglementation de Santé Canada au sein de l'industrie ainsi que la conformité accrue de l'industrie aux exigences réglementaires de Santé Canada. |
Rapports de synthèse annuels sur les inspections prêts à être publiés. |
Inspectorat |
Mars 2014 |
7. Santé Canada devrait prendre des mesures pour augmenter la rapidité avec laquelle il donne suite aux signaux relatifs à l'innocuité (de la détection de ces signaux à la diffusion d'une communication des risques). |
Acceptée |
La norme de service pour l'achèvement des évaluations des signaux est de 130 jours et l'objectif est de 90 %. Santé Canada atteint constamment cet objectif. En fait, le temps moyen pour terminer une évaluation des signaux est de 90 jours. Le Ministère évalue également l'efficacité des produits de communication sur les risques des produits de santé. |
Le Ministère élabore une stratégie visant à passer d'un modèle de surveillance passive à un modèle de surveillance active pour la gestion des risques liés aux produits de santé. Cette stratégie devrait permettre d'effectuer rapidement des examens de la sécurité et des interventions dynamiques, et de publier des examens sur la sécurité. |
DPSC |
Juin 2014 |
1.0 Introduction
Le Programme des médicaments à usage humain (PMUH) est géré par la Direction générale des produits de santé et des aliments (DGPSA) de Santé Canada. Les principaux partenaires participant à la mise en œuvre du PMUH sont la Direction des produits thérapeutiques (DPT), la Direction des produits de santé commercialisés (DPSC), l'Inspectorat de la Direction générale des produits de santé et des aliments (l'Inspectorat), le Bureau des régions et des programmes (BRP), la Direction des politiques, de la planification et des affaires internationales (DPPAI), et la Direction de la gestion des ressources et des opérations (DGRO). Le programme comprend les activités suivantes : la création et la tenue à jour d'un cadre de réglementation, l'interaction et la communication avec les partenaires et les intervenants, la réalisation d'évaluations des avantages et des risques, la surveillance post-commercialisation, et la conformité et l'application du cadre de réglementation.
L'évaluation du PMUH fait partie du plan quinquennal d'évaluation de Santé Canada. Selon la Politique sur l'évaluation du Conseil du Trésor (SCT, 2009), elle porte sur la pertinence et le rendement (efficacité, efficience et économie) des activités de Santé Canada dans le cadre du programme. L'évaluation couvre la période allant de 1999 à la fin de l'année civile 2012, plus précisément les cinq dernières années, même si le rapport rend compte des activités du programme jusqu'à la fin de juin 2013. Les résultats de l'évaluation éclaireront la mise en œuvre des activités en cours et à venir.
Une société d'experts-conseils indépendante en matière d'évaluation a réalisé l'évaluation pour le compte de Santé Canada. Elle s'est inspirée de plusieurs sources de données, dont la revue de la littérature, l'examen des documents, l'examen des données de gestion, les études de cas, les sondages menés auprès de fabricants et d'autres intervenants, et les entrevues avec des informateurs clés. Le présent rapport dévoile les résultats de l'évaluation, tire des conclusions et formule des recommandations.
1.1 Structure du rapport
Le rapport est divisé en plusieurs sections. La section 2 présente le profil du PMUH, et la section 3 décrit la méthode d'évaluation. Les sections 4 à 7 dévoilent les résultats de l'évaluation en ce qui a trait à la pertinence, à la gouvernance et à la mise en œuvre, à l'atteinte des résultats, et à l'efficience et à l'économie. La section 8 présente la conclusion et les recommandations. Trois annexes accompagnent le rapport principal. L'annexe A comprend la matrice d'évaluation, l'annexe B dresse une liste de références et l'annexe, l'annexe C inclut des tableaux de données supplémentaires.
2.0 Profil du Programme des médicaments à usage humain
Le PMUH mène ses activités en vertu de la Loi sur les aliments et drogues. Selon cette loi, « sont compris parmi les drogues les substances ou mélanges de substances fabriqués, vendus ou présentés comme pouvant servir :
- au diagnostic, au traitement, à l'atténuation ou à la prévention d'une maladie, d'un désordre, d'un état physique anormal ou de leurs symptômes, chez l'être humain ou les animaux;
- à la restauration, à la correction ou à la modification des fonctions organiques chez l'être humain ou les animaux;
- à la désinfection des locaux où des aliments sont gardés. (GC, 2008b, art. 2).
Le PMUH inclut les médicaments à usage humain (aussi appelés médicaments pharmaceutiques), soit les médicaments sur ordonnance et les médicaments en vente libre destinés à l'usage humain, mais exclut les médicaments biologiques, les vaccins, le sang et les produits sanguins, les cellules, les tissus et les organes, lesquels relèvent de la compétence du Programme des produits biologiques de la DGPSA. L'objectif ultime du PMUH consiste à donner accès à des médicaments à usage humain sécuritaires et efficaces et à fournir de l'information sur des choix santé afin d'aider les Canadiens à maintenir et à améliorer leur état de santé (Santé Canada, 2012).
2.1 Rôles et responsabilités des partenaires du programme
Les principaux partenaires du PMUH sont la DPT, la DPSC, l'Inspectorat et le BRP. On compte parmi les autres partenaires participant à la mise en œuvre et à la gestion du programme la DPPAI et la DGRO. Les rôles et les responsabilités de chacun de ces partenaires sont décrits brièvement ci-dessous.
2.1.1 Direction des produits thérapeutiques
La DPT est l'autorité qui réglemente les produits pharmaceutiques destinés à un usage humain en vertu de la Loi sur les aliments et drogues et du Règlement sur les aliments et drogues. Plus particulièrement, elle assume les responsabilités suivantes :
- Élaborer des règlements, des politiques, des lignes directrices et des procédures opérationnelles normalisées (PON), et tenir et mettre à jour les documents actuels;
- Effectuer un examen préalable à la mise en marché concernant les présentations de produits afin de déterminer l'innocuité, l'efficacité et la qualité de ces produits, et la pertinence de leur étiquette, et autoriser leur vente au Canada;
- Examiner les demandes d'essais cliniques, assurer une conception appropriée, veiller à ce que les participants ne courent aucun risque inacceptable et surveiller les réactions indésirables pendant les essais cliniques;
- Évaluer les événements indésirables après la mise en marché, les plaintes et les rapports de problème (y compris les évaluations des risques pour la santé) en vue d'appuyer les activités post-commercialisation de la DPSC et de l'Inspectorat;
- Passer en revue les renseignements que les fabricants de produits fournissent aux professionnels de la santé et aux consommateurs au sujet de leurs produits (p. ex., les monographies et les prospectus);
- Fournir des renseignements scientifiques au public canadien sur ces produits.
Il incombe aussi à la DPT de permettre aux médecins d'accéder aux médicaments à usage humain non commercialisés et aux produits biologiques par l'intermédiaire du Programme d'accès spécial lorsque les traitements classiques se sont révélés inefficaces, ne conviennent pas ou ne sont pas disponibles.
2.1.2 Direction des produits de santé commercialisés
La DPSC « veille à ce que les programmes de la DGPSA procèdent de façon uniforme pour la surveillance post-approbation, l'évaluation des signaux et des tendances concernant l'innocuité et la communication des risques à l'égard de tous les types de produits de santé réglementés qui sont commercialisés » (Santé Canada, 2012g). Les responsabilités de la DPSC relativement aux médicaments à usage humain sont les suivantes :
- Gérer le Programme Canada Vigilance, soit le programme de surveillance après la mise en marché de Santé Canada qui recueille et évalue les rapports sur les réactions indésirables présumées associées aux produits commercialisés au Canada;
- Surveiller les renseignements sur les réactions indésirables et les incidents liés aux médicaments recueillis par l'intermédiaire du Programme Canada Vigilance;
- Effectuer des évaluations approfondies de l'innocuité, comme des évaluations des signaux, des évaluations des risques et des avantages, et des examens des plans de gestion des risques (PGR) concernant les produits de santé commercialisés, ainsi que des examens des PGR en ce qui a trait à certains produits de santé présentés aux fins d'approbation;
- Coordonner les activités de réglementation concernant la publicité;
- Communiquer des renseignements aux professionnels de la santé et au public au sujet des risques associés aux produits;
- Élaborer des politiques et des stratégies aux fins de surveillance post-commercialisation.
2.1.3 Inspectorat et BRP
L'Inspectorat a pour rôle principal « d'exécuter un programme national de conformité et d'application de la loi pour tous les produits visés par le mandat de la Direction générale, à l'exception des produits alimentaires qui relèvent de l'Agence canadienne d'inspection des aliments » (Santé Canada, 2011b). Les responsabilités de l'Inspectorat relativement aux médicaments à usage humain sont les suivantes :
- Élaborer une politique portant sur la conformité et l'application de la réglementation;
- Décerner des licences d'établissement de produits conformes aux exigences réglementaires et modifier, suspendre ou refuser toute licence d'établissement de produits pharmaceutiques (LEPP) en cas de non-conformité;
- Effectuer des inspections afin d'assurer la conformité des fabricants aux exigences réglementaires relatives aux Bonnes pratiques cliniques (BPC), aux Bonnes pratiques de fabrication (BPF) et aux Bonnes pratiques de pharmacovigilance (BPV)Note de bas de page 2;
- Vérifier la conformité en réponse aux plaintes particulières ou aux risques relevés en ce qui concerne les médicaments à usage humain et les essais cliniques;
- Fournir des services d'analyses chimiques, physiques et microbiologiques par l'intermédiaire du programme de laboratoire en vue d'appuyer les activités d'inspection et d'enquête;
- Réaliser des activités de promotion de la conformité avec les intervenants de l'industrie.
En ce qui concerne les substances contrôlées, l'Inspectorat est également responsable des inspections d'innocuité afin d'assurer la conformité des fabricants aux exigences réglementaires liées aux substances et aux drogues contrôlées, et de la surveillance de la destruction des substances assujetties à la Loi réglementant certaines drogues et autres substances (LRDS).
Le BRP est l'organisme opérationnel de Santé Canada dans les régions. Il est responsable des activités de promotion de la conformité, ainsi que de l'application des lois et des règlements au moyen d'inspections, d'enquêtes, d'actions en justice et d'évaluations de la conformité aux normes touchant la fabrication, l'emballage et l'étiquetage, l'analyse, l'importation, la distribution et le commerce de gros des produits de santé destinés aux consommateurs.
2.1.4 Autres partenaires
Deux autres partenaires de Santé Canada ont des responsabilités dans le cadre du PMUH.
- La DPPAI assure le leadership et le soutien à la planification stratégique, ainsi qu'à l'élaboration et à la planification de politiques sur des questions horizontales d'importance stratégique. Elle exerce également un leadership dans les affaires internationales.
- La DGRO est responsable de l'attribution des ressources, y compris la politique de recouvrement des coûts et sa mise en œuvre, ainsi que de la synthèse des renseignements sur la mesure du rendement en vue de produire des rapports sur le rendement du ministère et de la Direction générale. Ces responsabilités sont passées de la DPPAI à la DGRO en juillet 2011.
2.2 Logique et activités du programme
Le PMUH comprend cinq activités principales offertes par les partenaires du programme :
- Élaboration et mise en place du cadre de réglementation;
- Communication et mobilisation des intervenants;
- Évaluations des avantages et des risques associés au processus d'autorisation d'essai clinique et d'autorisation de mise sur le marché;
- Contrôle et surveillance post-commercialisation;
- Conformité et application du cadre de réglementation.
Ces activités correspondent aux résultats particuliers à court, à moyen et à long terme. À très court terme, on s'attend à ce que les activités du programme accroissent la sensibilisation et la compréhension des intervenants externes quant aux risques et aux avantages des médicaments à usage humain. Elles devraient également accroître la sensibilisation et la compréhension des représentants de l'industrie quant aux activités de réglementation de Santé Canada relativement à ces médicaments, augmenter l'innocuité et l'efficacité de ces produits, et favoriser la conformité des représentants de l'industrie aux exigences réglementaires pertinentes.
L'atteinte de ces résultats immédiats devrait entraîner des résultats intermédiaires, dont l'adoption de comportements sécuritaires de la part des intervenants externes en ce qui a trait aux médicaments à usage humain; l'utilisation accrue de données scientifiques et d'analyses des avantages et des risques par Santé Canada en vue d'éclairer la prise de décisions; l'intervention réglementaire rapide concernant les risques cernés; la diminution de l'exposition aux risques pour la santé associés à l'utilisation de ces produits; l'harmonisation du cadre de réglementation du Canada concernant les approches utilisées à l'échelle internationale.
À long terme, Santé Canada souhaite réduire les événements indésirables associés à l'utilisation de médicaments à usage humain; améliorer la confiance du public par rapport à ces médicaments; et établir un système de réglementation viable, rentable, adapté et fondé sur la science en ce qui a trait à ces produits au Canada.
Un modèle logique illustrant les liens entre les activités, les extrants et les résultats attendus du PMUH est décrit ci-dessous (Section 2.3).
2.3 Description du modèle logiqueNote de bas de page 3
Le Programme des médicaments à usage humain utilise différentes ressources (intrants) pour mener à bien ses activités, produire des extrants et atteindre ses résultats, notamment le financement, les ressources humaines, les installations, l'infrastructure, les lois, les règlements, les politiques et les priorités, la science et la technologie et les données de recherche.
Le Programme des médicaments à usage humain se divise en cinq principales activités assurées par ses partenaires, soient :
- l'élaboration, la mise en œuvre et la tenue à jour du cadre de réglementation;
- les communications avec les partenaires et les intervenants;
- l'évaluation des risques et des avantages associés aux processus d'autorisation d'essai clinique et d'autorisation de mise sur le marché;
- la surveillance et le suivi des activités après commercialisation;
- la conformité et l'application du cadre de réglementation.
Ces activités ciblent différents groupes.
- L'élaboration, la mise en œuvre et la tenue à jour du cadre de réglementation visent :
- l'industrie, les médias, les consommateurs, les gouvernements et les organismes internationaux, les praticiens de la santé, le milieu universitaire, les gouvernements fédéral, provinciaux et territoriaux, et les associations industrielles et professionnelles.
- Les communications avec les partenaires et les intervenants visent :
- l'industrie, les médias, les consommateurs, les gouvernements et les organismes internationaux, les praticiens de la santé, le milieu universitaire, les gouvernements fédéral, provinciaux et territoriaux, et les associations industrielles et professionnelles.
- L'évaluation des risques et des avantages associés aux processus d'autorisation d'essai clinique et d'autorisation de mise sur le marché vise :
- l'industrie, les organismes internationaux, le milieu universitaire, les professionnels de la santé et les gouvernements fédéral, provinciaux et territoriaux.
- La surveillance et le suivi des activités après commercialisation visent :
- les responsables de la réglementation de Santé Canada et de l'étranger, le milieu universitaire et les organisations scientifiques et de recherche.
- La conformité et l'application du cadre de réglementation visent
- l'industrie, Santé Canada, les intervenants et les partenaires fédéraux, provinciaux, territoriaux et internationaux du domaine de la réglementation, et les consommateurs.
Chaque activité permet au Programme de générer différents produits et services.
- L'élaboration, la mise en œuvre et la tenue à jour du cadre de réglementation donnent lieu à :
- des lois et des règlements, des politiques, des lignes directrices, des accords de reconnaissance mutuelle et des protocoles d'entente.
- Les communications avec les partenaires et les intervenants donnent lieu à :
- des sources et des systèmes de données, des sites Web, des données de recherche, du matériel et des activités de sensibilisation du public et des intervenants, des publications, des mises en garde, des avis (y compris des lettres à l'intention des professionnels de la santé), de la correspondance et des demandes de renseignements du public.
- L'évaluation des risques et des avantages associés aux processus d'autorisation d'essai clinique et d'autorisation de mise sur le marché donne lieu à :
- des évaluations des risques et des avantages (initiales et continues, y compris des évaluations des risques pour la santé), des autorisations de mise sur le marché, des licences d'établissement, des autorisations d'essai clinique et des décisions prises en vertu du Programme d'accès spécial.
- La surveillance et le suivi des activités après commercialisation donnent lieu au :
- Programme Canada Vigilance et à des données, des analyses, des preuves, des rapports, des recommandations réglementaires, l'identification et la caractérisation des signaux, des études épidémiologiques et des plans de gestion et d'atténuation des risques.
- La conformité et l'application du cadre de réglementation donnent lieu à
- des rapports de conformité et d'application de la loi, des mesures de conformité et d'application de la loi, dont des poursuites en justice, des suspensions de licences, des saisies, des ordres d'arrêt de vente et des perquisitions.
Ces activités correspondent à des résultats particuliers à court, à moyen et à long terme. À court terme, on s'attend à ce que les activités du Programme accroissent la sensibilisation et la compréhension des intervenants externes quant aux risques et aux avantages des médicaments à usage humain. Elles devraient également accroître la sensibilisation et la compréhension des représentants de l'industrie quant aux activités de réglementation de Santé Canada relativement à ces médicaments, augmenter l'innocuité et l'efficacité de ces produits, et favoriser la conformité des représentants de l'industrie aux exigences réglementaires pertinentes.
L'atteinte de ces résultats immédiats devrait entraîner des résultats intermédiaires, dont l'adoption de comportements sécuritaires de la part des intervenants externes en ce qui a trait aux médicaments à usage humain; l'utilisation accrue de données scientifiques et d'analyses des avantages et des risques par Santé Canada en vue d'éclairer la prise de décisions; l'intervention réglementaire rapide à l'égard des risques cernés; la diminution de l'exposition aux risques pour la santé associés à l'utilisation des produits à usage humain; l'harmonisation du cadre de réglementation du Canada avec les approches adoptées à l'échelle internationale.
À long terme, Santé Canada souhaite réduire les événements indésirables associés à l'utilisation de médicaments à usage humain; améliorer la confiance du public par rapport à ces médicaments et au système de réglementation connexe, en plus d'améliorer ce dernier de sorte qu'il soit viable, rentable, adapté et fondé sur la science.
L'objectif ultime du Programme est d'aider à offrir aux Canadiens des médicaments à usage humain sûrs et efficaces ainsi que de l'information qui permette de faire des choix sains.
2.4 Ressources
Le PMUH est financé par Santé Canada, ainsi que par l'industrie au moyen du recouvrement des coûts. En 2011-2012, le total des dépenses relatives au PMUH s'élevait à 99,8 millions de dollars. Les recettes tirées du recouvrement des coûts étaient de 38,7 millions de dollars.
2.5 Contexte du programme
Le contexte de gestion et de mise en œuvre du PMUH, comme tous les programmes de réglementation fédéraux, dépend de plusieurs politiques, directives et mesures législatives très importantes. Les règlements sont l'un des nombreux instruments d'intervention gouvernementale utilisés pour atteindre des résultats stratégiques. La Directive du Cabinet sur la gestion de la réglementation de 2012 exige que les programmes de réglementation démontrent l'efficience et l'efficacité en s'assurant que les avantages de la réglementation justifient les coûts et en présentant des résultats tangibles aux Canadiens (GC, 2012a). La directive entraîne le besoin de consulter les organismes concernés à toutes les étapes du processus de réglementation; de sélectionner la combinaison appropriée d'instruments d'intervention gouvernementale, y compris les mesures réglementaires et non réglementaires; d'imposer aux entreprises et aux Canadiens le plus petit coût possible nécessaire à l'atteinte des objectifs stratégiques; de tirer parti des possibilités de coordination et de collaboration avec les gouvernements provinciaux et territoriaux, et à l'échelle internationale, en limitant, par exemple, le nombre d'exigences réglementaires propres au Canada et en réduisant au minimum les différences entre les principaux partenaires commerciaux (c.-à-d. les États-Unis); et d'évaluer le rendement des programmes de réglementation et de présenter un rapport aux Canadiens à cet effet en temps opportun.
En mai 2012, la DGPSA a publié la Feuille de route de la réglementation pour les produits de santé et aliments (Santé Canada, 2012j). Celle-ci prévoit un système de réglementation efficient et transparent qui protège et améliore la sécurité des consommateurs, qui allège le fardeau de la réglementation pour les petites entreprises, qui appuie l'innovation scientifique et qui permettra aux Canadiens d'avoir accès au plus large éventail possible d'options et d'avantages sur le plan de la santé. Ces objectifs sont conformes à la Directive du Cabinet sur la gestion de la réglementation, laquelle exige que tous les ministères fédéraux examinent les répercussions sur les petites entreprises de l'adoption de nouveaux règlements ou de la modification de règlements existants, tout en assurant la protection et la promotion de l'intérêt public en matière de santé et de sécurité. Ces efforts s'inspirent du Plan d'action pour la réduction du fardeau administratif du gouvernement du Canada, dont le but est de réduire le fardeau administratif des entreprises.
Les programmes réglementaires qui imposent des frais d'utilisation, comme le PMUH, sont assujettis à la Loi sur les frais d'utilisation (GC, 2004).La loi exige que les organismes de réglementation consultent les clients et les bénéficiaires des services et établissent des normes de services comparables à celles établies par d'autres pays avant de modifier les frais ou d'en créer de nouveaux.
La Politique de communication du gouvernement du Canada oblige les institutions fédérales, entre autres, à fournir au public des renseignements sur ses politiques, programmes, services et initiatives qui sont « opportuns, exacts, clairs, objectifs et complets », et ce, dans toutes les régions du Canada et sur de nombreux supports, et de consulter le public au moment d'établir des priorités et de planifier des programmes et des services (GC, 2012b).
3.0 Evaluation methodology
La présente section décrit la méthode d'évaluation.
3.1 Structure de l'évaluation et méthodes utilisées
La conception de l'évaluation se fonde sur les résultats d'une évaluation de l'évaluabilité, qui constituait la première étape des évaluations. Dans le cadre de cette évaluation de l'évaluabilité, on a procédé à un examen préliminaire ainsi qu'à l'analyse des documents et des données de gestion accessibles.
La conception de l'évaluation et la matrice d'évaluation (annexe A) sont fondées sur cette évaluation de l'évaluabilité. La matrice d'évaluation aborde dix questions clés et de nombreuses sous-questions liées à la pertinence et au rendement du programme (efficacité, efficience et économie), conformément à la Politique sur l'évaluation du Conseil du Trésor.
Plusieurs méthodes de collecte de données ont été utilisées aux fins de l'évaluation. Certaines de ces méthodes, en particulier les sondages menés auprès des intervenants, les entrevues avec les informateurs clés, les études de cas et l'examen des données de gestion, ont été conçues pour obtenir des renseignements en vue d'appuyer l'une des deux évaluations parallèles, ou les deux, d'autres programmes de réglementation de la DGPSA, notamment le Programme des produits biologiques et le Programme des matériels médicaux.
Revue de la littérature. La revue de la littérature a contribué à répondre aux questions d'évaluation se rapportant à la pertinence du programme, aux résultats à long terme et aux autres méthodes. On a également tenu compte des évaluations par les pairs (c.-à-d. en milieu scientifique et universitaire) et de la littérature grise. La documentation pertinente a été trouvée au moyen de recherches en ligne.
Examen des documents.L'examen des documents a contribué à répondre à toutes les questions d'évaluation, dans la mesure où des documents à l'appui étaient accessibles. L'examen a porté sur des documents gouvernementaux, principalement produits par Santé Canada, qui se rapportaient à la planification et à la gestion du PMUH, ainsi qu'aux activités continues. Plusieurs centaines de documents ont été examinés dans le cadre des évaluations.
Examen des données de gestion. L'examen des données de gestion a contribué à répondre aux questions d'évaluation se rapportant aux résultats du programme. Il a tenu compte des données produites par la DPT, la DPSC et l'Inspectorat. Bien qu'ils soient différents, en théorie, l'examen des documents et l'examen des données de gestion constituaient, en pratique, deux aspects de la même tâche, car la majorité des données de gestion étaient incluses dans la documentation relative au programme.
Afin d'appuyer la revue de la littérature, l'examen des documents et l'examen des données de gestion, une bibliothèque comptant environ 3 500 documents individuels, sources de données de gestion et ouvrages a été créée, y compris environ 1 650 documents; 980 pages Web; 570 articles de journaux et différents types de sources, lesquels ont été examinés au cours de l'évaluation.
Études de cas.Deux études de cas ont été réalisées en vue d'appuyer l'évaluation du PMUH. Une des deux études portait sur l'approche de Santé Canada relativement aux essais cliniques. Même si l'étude de cas sur les essais cliniques a été menée dans le contexte de l'évaluation du PMUH, comme les produits biologiques et les médicaments à usage humain font l'objet du même cadre de réglementation, elle a également été utilisée pour orienter l'évaluation du Programme des produits biologiques.
L'autre étude de cas se penchait sur la détection des signaux, l'évaluation des signaux et les activités de gestion des risques de Santé Canada relativement aux médicaments à usage humain et aux produits biologiques. Comme pour l'étude de cas sur les essais cliniques, cette étude a aussi été utilisée aux fins des deux évaluations.
Sondage auprès des représentants de l'industrie. Le sondage bilingue réalisé en ligne auprès de représentants de l'industrie était axé sur les questions d'évaluation se rapportant aux résultats. Dans le cadre de l'évaluation, compte tenu des recommandations et des directives de Travaux publics et Services gouvernementaux Canada en matière de sondages et de recherche sur l'opinion publique, le sondage devait porter uniquement sur les personnes qui avaient eu affaire à Santé Canada pour des raisons liées au PMUH. Au départ, le sondage était supposé porter sur tous les fabricants autorisés à fabriquer des médicaments à usage humain au Canada (soit l'approche choisie pour l'évaluation du Programme des matériels médicaux). Toutefois, l'échantillon a plutôt été tiré par Santé Canada à partir de la base de données du Système de gestion de l'information sur les intervenants (SGII) du ministère et n'est probablement pas aléatoire ni représentatif de la population de fabricants dont les activités ont lieu au Canada.
Une fois les entrées sans adresse électronique et les doublons supprimés, l'échantillon final du sondage auprès des représentants de l'industrie des médicaments à usage humain portait sur 380 fabricants. Ce nombre comprenait uniquement les fabricants qui produisaient exclusivement des médicaments à usage humain et excluait les fabricants de produits biologiques.
Le sondage auprès des représentants de l'industrie des médicaments à usage humain a été réalisé auprès de 24 répondants, soit un pourcentage de réponses de 6,6 %Note de bas de page 4. Bien qu'un taux de réponse de 10 % ne soit pas inhabituel pour des sondages en ligne auprès des représentants de l'industrie, ce taux est moins élevé que prévu; ce résultat peut être attribuable au fait que Santé Canada n'a pas envoyé d'avis aux répondants susceptibles de participer au sondage. L'invitation au sondage a plutôt été diffusée par l'évaluateur.
Sondage auprès d'autres intervenants.Un sondage bilingue en ligne a également été réalisé pour rejoindre les utilisateurs de médicaments à usage humain, y compris les professionnels de la santé et les patients/consommateurs. Ce sondage avait pour but de recueillir des renseignements sur les utilisateurs de produits biologiques et d'instruments médicaux en vue d'appuyer les évaluations parallèles sur les activités réglementaires de Santé Canada dans ces domaines. Plus particulièrement, le sondage visait à obtenir de l'information sur les répercussions des activités de communication et de consultation du ministère relativement à la sensibilisation et à la compréhension des intervenants quant aux risques associés aux produits thérapeutiques, ainsi que des données sur l'incidence de ces activités en ce qui a trait à l'utilisation que font les intervenants de ces produits.
Dans le cadre de l'évaluation, comme dans le cas du sondage auprès des représentants de l'industrie, le sondage devait porter uniquement sur les personnes qui avaient eu affaire à Santé Canada. On a donc exigé à ce que la liste de diffusion électronique du programme MedEffet de Santé Canada, laquelle comprend plus de 20 000 abonnés, soit utilisée comme échantillon. Cependant, eu égard aux préoccupations relatives à la protection des renseignements personnels, à la possibilité que l'utilisation de la liste de diffusion aux fins de sondages entraîne le désabonnement de certains abonnés à MedEffet et au fait que cette liste inclut des personnes qui ne font pas partie du groupe ciblé par le sondage (comme les employés de Santé Canada, et les professionnels de la santé et les consommateurs vivant à l'extérieur du Canada), la liste de diffusion n'a pas été fournie.
En appui au sondage, Santé Canada a présenté une liste d'intervenants trouvés dans la base de données de son SGII. Une fois épuré, l'échantillon final comptait 651 répondants susceptibles de participer au sondage.
Le questionnaire comprenait des modules distincts pour les instruments médicaux, les médicaments à usage humain et les produits biologiques. Un total de 16 intervenants ont répondu au sondage, soit un pourcentage de réponses de 2,6 %Note de bas de page 5. De ce nombre, 10 (1,6 % des répondants) ont répondu au module sur les médicaments à usage humain. Bien que ce faible taux de participation demeure inexpliqué, il est peut-être attribuable au manque de communication de la part de Santé Canada au début du processus.
Entrevues avec des informateurs clés externes. Les entrevues réalisées avec des informateurs clés étaient axées sur les questions d'évaluation se rapportant à la mise en œuvre et à l'efficacité du programme Elles portaient sur les instruments médicaux, les médicaments à usage humain et les produits biologiques en vue d'appuyer les trois évaluations sans fatiguer les informateurs clés avec de multiples demandes d'entrevue. Les répondants étaient des représentants de l'industrie, des chercheurs et des scientifiques, des patients et des organismes de défense des consommateurs, des fournisseurs de soins de santé, des associations professionnelles, des informateurs clés internationaux et d'autres intervenants, entre autres.
Au total, 93 informateurs clés susceptibles de vouloir passer une entrevue ont été répertoriés, et 53 personnes ont donné une entrevue ou ont répondu par écrit aux questions d'entrevue. De ce nombre, 42 ont formulé des commentaires sur les médicaments à usage humain. Les entrevues se sont déroulées par téléphone, dans la langue officielle préférée des intervenants. Elles ont été enregistrées aux fins d'exactitude; les notes ont ensuite été envoyées aux informateurs clés aux fins d'examen et d'approbation. Les intervenants ont été assurés de l'anonymat de leurs réponses.
Pour la préparation du rapport final, des données provenant de toutes les sources ont été intégrées ou triangulées en vue d'aboutir aux conclusions générales de l'évaluation. La triangulation désigne un processus comparant les réponses aux questions de recherche obtenues par différentes méthodes de collecte de données. Lorsque des méthodes différentes donnent des résultats semblables, la validité de ces conclusions est considérée comme supérieure, ce qui justifie une confiance accrue envers les résultats.
3.2 Limites des méthodes et stratégies d'atténuation
Il importe de souligner plusieurs limites importantes relatives aux méthodes. D'abord, étant donné la grande portée et la complexité du sujet, l'examen des documents et la revue de la littérature devaient respecter les échéanciers et les ressources disponibles. L'examen des documents était de plus limité aux documents fournis aux fins de l'évaluation par Santé Canada ou à ceux accessibles au public. Bien qu'on ait déployé tous les efforts nécessaires pour obtenir les documents pertinents auprès du ministère au fur et à mesure que la recherche avançait, il a été impossible de déterminer tous ceux qui auraient pu être utiles. Il incombe donc aux partenaires du programme d'assumer une partie de la responsabilité qui consiste à relever les documents pertinents.
Le sondage auprès des intervenants était limité en raison du processus de développement de l'échantillon. Plus particulièrement, l'échantillon de l'enquête était petit, surtout par rapport aux populations réelles de fournisseurs de soins de santé et de patients/consommateurs. De plus, pour des raisons qui demeurent en partie inconnues, le sondage a obtenu un taux de réponse très faible (2,6 %), et seulement dix intervenants ont répondu aux questions portant sur les médicaments à usage humain. Même s'il est possible que la liste de MedEffet n'ait pas permis d'obtenir un taux de réponse plus élevé, elle aurait au moins augmenté le pourcentage de réponses à un niveau digne de mention. Ainsi, elle aurait fourni des données en appui aux questions portant sur l'incidence de la communication des risques de Santé Canada quant à la sensibilisation, à la compréhension et au comportement des intervenants. Sans données d'enquête, très peu de renseignements soutiennent les conclusions formulées par rapport à ces questions. Ceci étant dit, même si on avait utilisé la liste de MedEffet, les commentaires et les renseignements recueillis n'auraient pas été représentatifs de la population générale d'intervenants au Canada. Cette information aurait plutôt reflété un groupe d'intervenants qui reçoit souvent des communications de Santé Canada.
Comme le sondage mené auprès des intervenants, le sondage mené auprès des représentants de l'industrie était également limité en raison d'un échantillon petit et non représentatif, combiné à un faible taux de réponse. Par conséquent, les résultats de l'enquête sont utilisés avec parcimonie tout au long du présent rapport. Les résultats mentionnés, le cas échéant, sont considérés comme de l'information qualitative.
En ce qui concerne les entrevues réalisées avec des informateurs clés externes, environ la moitié de ceux ayant été répertoriés n'ont finalement pas passé d'entrevue. On ne sait pas si les résultats auraient été très différents si ces personnes avaient accepté de donner une entrevue. Toutefois, il est important de noter que plusieurs des personnes qui ont choisi de ne pas participer ont mentionné ne pas connaître assez bien le programme pour fournir des réponses pertinentes. Comme toutes les données portant sur les informateurs clés, les conclusions tirées des entrevues ne doivent pas être interprétées comme si elles représentaient l'opinion de l'ensemble des intervenants.
Même si des entrevues avec les représentants de Santé Canada étaient prévues, elles n'ont pas eu lieu. Les entrevues étaient la dernière activité de la collecte des données et avaient pour but de permettre au programme de répondre aux résultats de l'évaluation préliminaire et d'aborder les lacunes des renseignements. En raison de contraintes de temps, on a procédé à la production du rapport final sans réaliser les entrevues. Cependant, tout au long du processus, des employés du programme ont été consultés au moyen de conférences téléphoniques et de courriels en vue d'obtenir des renseignements supplémentaires et de clarifier des questions.
4.0 Résultats - Pertinence
La présente section du rapport révèle les résultats de l'évaluation sur la pertinence.
4.1 Besoin continu
L'évaluation a confirmé un besoin continu de surveillance gouvernementale en ce qui a trait aux médicaments à usage humain en vue de protéger la santé et la sécurité des Canadiens. L'utilisation de tels produits prend de l'ampleur en raison de changements démographiques, comme l'accroissement et le vieillissement de la population, et de la publicité faite par les représentants de l'industrie. Par conséquent, un nombre accru de Canadiens seront exposés aux risques et aux avantages de ces produits, d'où la nécessité de les gérer au moyen d'une surveillance continue de la part Santé Canada. De plus, certaines tendances, comme l'émergence des produits mixtes et la mondialisation de la chaîne d'approvisionnement, créent des incertitudes qui accentuent la nécessité d'une intervention gouvernementale afin de protéger la santé et la sécurité des Canadiens.
Les médicaments pharmaceutiques et les produits biologiques contribuent grandement à la santé des Canadiens et sont un élément important du système de soins de santé du Canada.Note de bas de page 6Les dépenses au titre des médicaments augmentent de façon constante depuis les années 1970, atteignant 32,0 milliards de dollars (16 % du total des dépenses du système) en 2010 (ICIS, 2011, p. 22; Santé Canada, 2011a). Environ la moitié des Canadiens prennent au moins un médicament sur ordonnance (Conseil canadien de la santé, 2010, p. 17), comme le font 76 % des aînés vivant en résidence privée et pratiquement tous les aînés résidant dans des établissements de santé (Ramage-Morin, 2009). De plus, l'utilisation de ces produits a augmenté de 77,6 % de 1999 à 2009, passant de 272 à 483 millions de prescriptions par année, respectivement (Conseil canadien de la santé, 2010, p. 18). L'utilisation accrue de médicaments par personne, attribuable, en partie, à la publicité faite par les représentants de l'industrie et à l'accroissement et au vieillissement de la population, semble indiquer que de plus en plus de Canadiens sont et seront exposés aux risques et aux avantages associés aux médicaments approuvés par Santé Canada (Lemmens & Bouchard, 2007, p. 312).
Différents risques pour la santé et la sécurité sont associés à l'utilisation des médicaments à usage humain, d'où la nécessité de les gérer au moyen d'un certain degré de surveillance réglementaire.
- Risques attribuables à une compréhension limitée des effets d'un produit sur la santé. Malgré les processus préalables à la mise en marché établis au Canada et dans d'autres pays depuis les années 1960, les préoccupations en matière d'innocuité entraînent périodiquement le retrait du marché de produits approuvés. Depuis 2004, au Canada, 28 médicaments de marque déposée et génériques (concernant 22 ingrédients actifs) ont été inactivés par le fabricant pour des raisons de sécurité (Santé Canada, 2013b).Note de bas de page 7 Although it is difficult to identify estimates in the literature of how many Canadians have been affected by exposure to unsafe drugs, the numbers are likely to be significant. For instance, Vioxx may have been associated with as many as 4,000 to 7,000 deaths in Canada before being withdrawn from the market in late 2004 (Abraham, 2005).
- Risques attribuables à un emploi non conforme à l'étiquetage. Le principal problème lié à l'emploi non conforme à l'étiquetage d'un médicament est l'absence de données cliniques démontrant l'efficacité et l'innocuité du médicament prescrit à des fins autres que celles à l'origine de son approbation. La prescription non conforme à l'étiquetage semble courante dans les soins de santé canadiens. Une étude récente estime que 11 % des médicaments prescrits dans la pratique le sont à des fins non conformes; dans 79 % de ces cas, aucune donnée scientifique solide ne confirmait l'efficacité des produits en question en ce qui a trait à leur utilisation prévue (Eguale et coll., 2012). Même si son autorité réglementaire est limitée quant à l'utilisation des médicaments, Santé Canada peut néanmoins influer indirectement sur l'utilisation par d'autres moyens, comme la préparation et la diffusion de produits de connaissance. En vertu de la Loi sur les aliments et drogues, il peut également poursuivre les entreprises qui encouragent l'emploi non conforme à l'étiquetage.
- Risques attribuables à un abus ou à un mauvais usage de médicaments sur ordonnance. L'abus ou le mauvais usage de médicaments sur ordonnance est une forme d'emploi non conforme à l'étiquetage qui présente des défis uniques, car ces médicaments sont souvent importants pour le traitement de la douleur chronique. Les types de médicaments sur ordonnance qui risquent le plus de faire l'objet d'un mauvais usage sont les analgésiques opioïdes (p. ex., fentanyl, Percodan, Demerol et OxyContin), les stimulants (p. ex., Ritalin, Concerta, Adderall et Dexedrine), et les tranquillisants et les sédatifs (p. ex., benzodiazépine, Valium, Ativan et Xanax) (CCLT, 2012). Récemment, le Canada est devenu le deuxième consommateur mondial par personne d'opioïdes sur ordonnance, soit une augmentation en corrélation avec la hausse des dépendances, des surdoses et des décès liés aux opioïdes (CCLT, 2012). Une étude a révélé que les décès liés aux opioïdes en Ontario ont doublé, passant de 13,7 par million en 1991 à 27,2 par million en 2004, et qu'une grande partie de cette augmentation était associée à l'ajout de l'oxycodone à action prolongée (c.-à-d. l'OxyContin) au formulaire de médicaments de l'Ontario (Dhalla et coll., 2009). L'OxyContin était le huitième médicament le plus vendu au Canada en 2011 (voir Tableau 1).
- Risques survenant pendant la fabrication et la distribution. Les préoccupations concernant la tendance vers la mondialisation de la chaîne d'approvisionnement des ingrédients actifs et des produits finis sont liées aux possibilités perçues de contrefaçon, de falsification, d'adultération, de contamination et d'autres risques qui pourraient nuire aux utilisateurs finaux des produits (Woo, Wolfgang et Batista, 2008, p. 494). À l'heure actuelle, une grande proportion des produits de santé finis vendus au Canada sont maintenant importés; dans le cas des ingrédients pharmaceutiques actifs (IPA), Santé Canada mentionne que l'importation représente 85 % du marché, et la Chine et l'Inde sont les principaux fournisseurs (Santé Canada, 2012c). Toutefois, il serait faux de penser que la qualité pose problème seulement dans les installations de fabrication étrangères. En plus des dommages directement causés par des ingrédients actifs contaminés ou de qualité inférieure aux normes, ou même par des produits chimiques toxiques, les médicaments de contrefaçon ou de mauvaise qualité n'auront peut-être pas les effets thérapeutiques nécessaires et produiront des effets nuisibles ou un malaise chez le patient. De plus, les ingrédients ou les produits de santé fabriqués de qualité inférieure pourraient entraîner une résistance aux médicaments ou une pénurie s'ils font l'objet d'un rappel (Santé Canada, 2012c). Dans un exemple très médiatisé, des centaines d'événements indésirables et de décès potentiels découlant de réactions de type allergique et d'hypotension chez des patients américains subissant une dialyse ont finalement été associés à l'adultération, en Chine, de l'héparine, un anticoagulant, contaminée par du sulfate de chondroïtine sursulfaté, un matériel synthétique doté de caractéristiques semblables à celles de l'héparine, mais cent fois moins dispendieux (Pew Health Group, 2011, p. 16-20).Note de bas de page 8
- Risques attribuables à la préparation de médicaments en pharmacie. Santé Canada définit la préparation en pharmacie comme suit : La combinaison ou le mélange de deux ingrédients ou plus (dont au moins un ingrédient est une drogue ou une composante pharmacologique active) pour créer un produit final sous forme dosifiée appropriée (Inspectorat, 2009b, p. 7).Note de bas de page 9 Les produits préparés en pharmacie présentent un risque pour la sécurité des patients. Récemment, on a beaucoup parlé du problème de la préparation en pharmacie en raison d'une éclosion de méningite fongique aux États-Unis chez des patients recevant des injections de stéroïdes contaminés. En date du 25 octobre 2012, 328 patients avaient été infectés dans 18 états. L'éclosion s'est soldée par 24 décès (CDC, 2012).
- Risques attribuables aux pénuries de médicaments. Depuis plusieurs années, les pénuries de médicaments au Canada représentent un nouvel enjeu (Duffin, 2012; CPS, 2012; Labrie, 2012). Par exemple, des pénuries de médicaments ont été causées par les récents ralentissements de la production d'un fabricant important de médicaments génériques et, plus particulièrement, par les événements survenus dans une installation de fabrication du Québec qui produit environ la moitié des médicaments génériques injectables du Canada (CPS, 2012, p. 3). Les pénuries de médicaments peuvent accroître le recours à des médicaments de substitution inappropriés, retarder les actes médicaux et augmenter la probabilité d'erreurs d'administration d'un médicament.
Selon cet aperçu des risques pour la santé et la sécurité liés aux médicaments à usage humain, un rôle permanent du gouvernement semble justifié afin de protéger la santé et la sécurité des humains. Dans ce contexte, il est important de noter les dernières tendances de l'industrie qui influent sur les organismes de réglementation. Par exemple, pendant un certain temps, l'industrie pharmaceutique a fait face à la « chute des brevets », laquelle fait référence à la perte d'une protection conférée par un brevet pour plusieurs de ces meilleurs vendeurs. Elle a réagi à cette situation de deux façons. D'abord, elle s'est concentrée sur la prolongation de la protection conférée par un brevet pour les produits existants, en plus de développer de nouveaux médicaments pharmaceutiques semblables aux médicaments déjà sur le marché (Ferguson et Lybecker, 2012). Ensuite, elle s'est orientée vers le développement de produits à coût élevé pour les troubles médicaux moins fréquents, notamment les produits biologiques. Même si des produits biologiques sont utilisés au Canada depuis de nombreuses années, ceux produits à l'aide de la biotechnologie ont rapidement été acceptés sur le marché canadien. Comme l'indique Tableau 1, certains de ces produits biologiques (Remicade, Enbrel, Humira et Lucentis) font actuellement partie des médicaments les plus vendus au pays.
| Rang | Nom du produit | Sous-classe thérapeutique | Ventes totales (en millions de dollars) | Croissance en 2010 (%) | Entreprise |
|---|---|---|---|---|---|
| 1 | Crestor | Hypocholestérolémiant | 742,20 | 14,2 | AstraZeneca |
| 2 | Remicade | Agent antiarthritique | 504,80 | 16,3 | Schering |
| 3 | Plavix | Antiplaquettaire | 306,70 | 4,4 | BMS |
| 4 | Humira | Agent antiarthritique | 301,90 | 22,4 | Nycomed |
| 5 | Enbrel | Agent antiarthritique | 292,70 | 7,1 | Amgen |
| 6 | Nexium | Agent antiacide | 306,70 | 4,4 | AstraZeneca |
| 7 | Lucentis | Préparation ophtalmique | 238,00 | 33,8 | Novartis |
| 8 | OxyContin | Analgésique | 234,00 | 2,1 | Purdue |
| 9 | Lyrica | Troubles convulsifs | 206,00 | 14,0 | Pfizer |
| 10 | Advair | Traitement contre l'asthme | 202,90 | 2,9 | Abbott |
Source: (Industrie Canada, 2012) |
|||||
En outre, d'autres catégories de médicaments biologiques commencent à faire leur apparition. On parle, par exemple, des produits biologiques de seconde génération, également appelés « biobetters » ou « biosuperiors », qui sont des produits dont la structure ou la fonction a été modifiée en vue d'atteindre un rendement clinique accru ou différent; des succédanés biologiques ou des produits biologiques « non innovants », lesquels ont été développés indépendamment et n'ont peut-être pas été comparés directement aux produits fabriqués sous licence; et des produits biologiques ultérieurs (PBU), également appelés « biosimilaires », lesquels diffèrent des produits biologiques non innovants, car ils ont été officiellement comparés à un produit actuellement fabriqué sous licence (Weise et coll., 2011, p. 691). Comme la protection conférée par un brevet de nombreux médicaments tire à sa fin, les organismes de réglementation se heurtent maintenant à un nouvel enjeu important qui porte sur la gestion de l'approbation et du remboursement de ces catégories de médicaments biologiques.
L'émergence des produits mixtes pose elle aussi des défis aux organismes de réglementation. Ces produits mixtes sont, par exemple, les endoprothèses à élution de médicaments et les « pilules intelligentes » dotées d'un instrument médical qui contrôle la libération de l'ingrédient actif. Du point de vue de la réglementation, il est alors plus difficile de déterminer les mesures législatives qui régissent le produit en question ou les organismes de réglementation qui détiennent l'autorité en matière d'essais, d'approbation et d'utilisation.
Enfin, la mondialisation de la chaîne d'approvisionnement des produits thérapeutiques présente des défis pour les organismes de réglementation nationaux en ce qui a trait à la gestion des risques à différentes étapes du processus de fabrication et du cycle de vie du produit, et crée des pressions pour une meilleure harmonisation des approches réglementaires. Bref, le nombre de Canadiens possiblement touchés, les risques associés à l'utilisation de médicaments à usage humain, et les tendances et les développements actuels de l'industrie confirment qu'une surveillance gouvernementale continue de ces produits est nécessaire afin de protéger la santé et la sécurité des Canadiens.
4.2 Concordance avec les priorités fédérales
L'évaluation a révélé que le PMUH est en harmonie avec les priorités du gouvernement du Canada. Au cours de la dernière décennie, le gouvernement fédéral a consacré d'importantes ressources au développement d'initiatives visant l'amélioration de l'innocuité des produits de santé en général grâce à la modernisation du cadre de réglementation de ces produits. Le but du renouvellement est de fournir aux Canadiens un accès rapide et continu à des produits de santé sécuritaires et efficaces malgré les nombreux problèmes d'envergure auxquels fait face le système de réglementation actuel. Selon Santé Canada, ces défis englobent des outils de réglementation désuets; l'incapacité actuelle du système à envisager des produits durant tout leur « cycle de vie », de la découverte jusqu'à l'examen des avantages et des risques concrets; l'incidence des changements technologiques et scientifiques, la mondialisation du marché, et la connaissance et la mobilisation accrues du public; et le manque de ressources en vue d'assurer l'efficacité et la viabilité à long terme (DGPSA, 2007a).
Les plans de la Direction générale concernant le renouvellement de la réglementation ont d'abord été énoncés dans le Plan de renouveau de 2007 (DGPSA, 2007a). Voici certains des objectifs généraux exprimés dans ce document :
- Adopter une approche réglementaire axée sur le « cycle de vie des produits » ou l'« homologation progressive », c.-à-d. une approche régissant les produits de santé qui englobe toutes les étapes de développement des produits, et sur laquelle s'appuie l'évaluation de la qualité, des incertitudes, des bienfaits et des effets nuisibles relatifs aux produits aux étapes précédant et suivant la mise en marché;
- Mettre en œuvre des mesures réglementaires proportionnelles aux risques;
- Renforcer la surveillance post-commercialisation, la conformité et l'application;
- Apprendre des homologues internationaux et collaborer avec eux;
- Accroître la transparence et l'ouverture;
- Appuyer la réglementation sur des données scientifiques;
- Assurer la viabilité du cadre de réglementation
Le gouvernement fédéral a entrepris plusieurs initiatives qui sont clairement en harmonie avec les objectifs du renouvellement de la réglementation. À compter de 2003-2004, il a fourni 190 millions de dollars sur cinq ans dans le cadre de la Stratégie d'accès aux produits thérapeutiques (SAPT) « pour accélérer les méthodes de réglementation de Santé Canada en ce qui touche les médicaments [à] usage humain » (ministère des Finances, 2003). L'un des principaux objectifs du SAPT consistait à accroître l'efficacité, la rapidité et la transparence du processus de prise de décisions réglementaires avant la mise en marché.
À compter de 2006, le gouvernement fédéral a alloué 172,5 millions de dollars sur cinq ans à l'Initiative d'innocuité des produits thérapeutiques (IIPT). L'IIPT portait essentiellement sur les processus après la mise en marché relatifs à l'innocuité des produits biologiques.
Plus récemment, le budget de 2008 a consacré 489,4 millions de dollars dans le cadre du Plan d'action pour assurer la sécurité des produits alimentaires et de consommation (PASPAC) en vue de s'assurer que les produits alimentaires et de consommation sont sécuritaires et qu'ils contribuent à la santé des Canadiens (Santé Canada, 2008a). Le PASPAC est une initiative horizontale touchant Santé Canada, l'Agence de la Santé publique du Canada (ASPC), l'Agence canadienne d'inspection des aliments (ACIA) et les Instituts de recherche en santé du Canada (IRSC) dont l'objectif général consiste à moderniser les pratiques et les règlements concernant la sécurité des aliments et des produits de santé et de consommation au Canada. À long terme, on s'attend à ce que les activités du PASPAC permettent d'améliorer l'innocuité des produits de santé sur le marché.
En 2008, le gouvernement du Canada a mis en place une législation exhaustive (projet de loi C-51) contenant plusieurs des éléments de la modernisation de la réglementation, notamment :
- Intégrer un cadre d'homologation progressive selon lequel « l'évaluation des avantages et des risques doit se fonder sur une preuve scientifique et objective » et qui prévoit « l'évaluation continue de l'information sur un produit thérapeutique pendant son cycle de vie »;
- Obliger les établissements de santé appartenant à une catégorie réglementaire de fournir des rapports sur les réactions indésirables;
- Ajouter de nouvelles mesures d'administration et d'application, notamment le rappel obligatoire de produits thérapeutiques et le pouvoir fédéral de retirer les stocks de médicaments restants suivant le retrait de ces produits du marché en raison de problèmes d'innocuité;
- Mettre en place un cadre modernisé relatif aux sanctions et aux amendes pécuniaires en augmentant considérablement les amendes en cas de non-conformité;
- Accroître les pouvoirs du ministre lui permettant ainsi d'ordonner aux titulaires d'autorisation d'essai clinique, d'autorisation de mise en marché ou de licence d'établissement de compiler des renseignements, d'effectuer des essais ou des études ou de surveiller l'expérimentation des effets d'un produit thérapeutique sur la santé ou la sécurité, et de communiquer ces renseignements ou ces résultats au ministre (GC, 2008a).
En raison de la dissolution du Parlement le 7 septembre 2008, le projet de loi C-51 n'est pas devenu une loi. Depuis ce temps, le gouvernement fédéral aborde la modernisation de la réglementation à l'aide d'une approche progressive. Il exprime ses plus récentes idées sur la modernisation dans la Feuille de route de la réglementation pour les produits de santé et aliments, publiée en mai 2012 (Santé Canada, 2012j). Celle-ci prévoit une approche progressive à l'égard de la modernisation, et la mise en œuvre intégrale de toutes les phases devrait prendre au moins cinq ans. La phase I consiste à élaborer des modifications réglementaires visant l'abrogation de l'annexe F (médicaments sur ordonnance) du Règlement sur les aliments et drogues et de la remplacer par une Liste des drogues sur ordonnance; à créer un cadre de réglementation pour les médicaments orphelins (c.-à-d. les médicaments développés spécifiquement pour soigner des maladies rares); et mettre en œuvre l'initiative d'étiquetage en langage clair (DGPSA, 2012d).
Le gouvernement fédéral a annoncé son engagement à l'égard de la modernisation de la réglementation des produits de santé en révisant de son cadre de recouvrement des coûts actuel. L'Initiative de recouvrement des coûts (IRC) de 2007 était, en partie, une réponse aux préoccupations soulevées par le Bureau du vérificateur général du Canada (BVG) en 2004 et en 2006 quant à la capacité de Santé Canada à satisfaire ses exigences réglementaires étant donné ses niveaux de ressources.Note de bas de page 10L'IRC devait contribuer à la stabilité de la plateforme financière des services de réglementation en faisant passer les recettes annuelles de Santé Canada de 47 millions de dollars à 112,4 millions de dollars, lesquelles restaureraient le ratio initial de partage des coûts d'environ 50 %Note de bas de page 11en vigueur lorsque les frais ont été mis en place (Santé Canada, 2010c). Une étape importante de l'IRC de 2007 a été franchie grâce à la prise d'effet du Règlement sur les prix à payer à l'égard des drogues et instruments médicaux le 1er avril 2011 (GC, 2011).
Les principales initiatives gouvernementales décrites ci-dessus montrent clairement l'harmonisation du PMUH à la DGPSA et aux priorités fédérales, surtout en ce qui concerne la modernisation de la réglementation des produits de santé. De plus, les activités du PMUH cadrent avec l'architecture d'harmonisation de programmes (AHP) actuelle de Santé Canada et, plus particulièrement, avec le résultat stratégique visé ayant pour but de s'assurer que les Canadiens sont informés et protégés des risques pour la santé que présentent les aliments, les produits, les substances et les environnements, que les produits qu'ils utilisent présentent le moins de risque possible et que les dangers pour la santé sont traités adéquatement (Santé Canada, 2012b).
4.3 Concordance avec les rôles et les responsabilités du gouvernement fédéral
Le PMUH est en harmonie avec les rôles et les responsabilités du gouvernement fédéral, dont certains sont définis dans la législation. Le mandat de Santé Canada est défini dans la Loi sur le ministère de la Santé, selon laquelle les tâches du ministre incluent, entre autres, le maintien de la santé et du bien-être des Canadiens, la protection de la population contre la propagation de la maladie et les risques pour la santé, les enquêtes et les recherches sur la santé publique, l'établissement et le contrôle des normes de sécurité des produits de consommation ainsi que de l'information relative à la sécurité dont ceux-ci doivent être accompagnés, et la collecte, l'analyse, l'interprétation, la publication et la diffusion de l'information sur la santé publique (GC, 2006, par. 4(2)). De façon plus générale, la compétence du ministre couvre toutes les questions liées à la santé des Canadiens qui n'ont pas été confiées à un autre organisme par le gouvernement.
La Loi sur les aliments et drogues et le Règlement sur les aliments et drogues définissent les paramètres d'une drogue et offrent un soutien législatif à Santé Canada dans son rôle visant à régir l'utilisation des médicaments à usage humain et à prendre des mesures en vue de faire respecter ces dispositions. La loi restreint les raisons pour lesquelles un aliment ou un produit de santé général peut être annoncé ou vendu (GC, 2008b, art. 3). Les articles 8 à 11 de la loi, lesquels font directement référence aux drogues, interdisent la vente de drogues falsifiées ou non hygiéniques, l'utilisation d'une étiquette inexacte, la fausse représentation de substances de manière qu'elles puissent être confondues avec des drogues et la fabrication de drogues dans des conditions non hygiéniques. Le règlement fournit des détails sur les exigences relatives à l'innocuité et à l'efficacité des drogues conçues, fabriquées ou distribuées au Canada, les normes concernant l'étiquetage et la publicité, les exigences quant aux licences d'établissement, les BPF, les essais cliniques et les déclarations des effets ou des événements indésirables. Il précise également les mesures correctives à prendre en cas de doute quant au respect de ces exigences (GC, 2012c).
Santé Canada participe aussi à la réglementation des drogues contrôlées en vertu de la Loi réglementant certaines drogues et autres substances (LRCDAS) et du Règlement sur les stupéfiants. Les responsabilités de l'Inspectorat incluent la réalisation d'inspections d'innocuité en vue de déterminer si l'entreposage des drogues contrôlées est approprié avant de délivrer une licence à un distributeur autorisé et la simplification de la destruction des substances contrôlées (Inspectorat, 2012d). D'autres responsabilités sont assumées par différents bureaux de la Direction générale de la santé environnementale et de la sécurité des consommateurs (DGSESC), notamment le Bureau des substances contrôlées (BSC) et le Bureau de la recherche et de la surveillance (BRS). Le rôle du BSC consiste à délivrer des licences aux distributeurs de substances contrôlées et à élaborer des lois, des règlements, des politiques et des mesures en appui au contrôle des drogues illicites et d'autres substances (DGSESC, 2009). Le BRS, quant à lui, a pour mission de surveiller les tendances relatives à l'utilisation de drogues illicites au Canada et d'examiner les problèmes et les nouvelles tendances concernant l'abus de drogues à l'intérieur et à l'extérieur du Canada (DGSESC, 2009).
5.0 Résultats - Gouvernance et mise en œuvre
La présente section du rapport révèle les résultats de l'évaluation sur la gouvernance et la mise en œuvre du programme.
5.1 Gouvernance du programme
Le PMUH inclut toutes les activités entreprises par la DPT relativement aux médicaments à usage humain, ainsi que toutes les activités pertinentes réalisées par la DPSC, l'Inspectorat/le BRP, la DPPAI et la DGRO. Les représentants de Santé Canada ont signalé que le Comité de coordination des directeurs généraux a été créé en 2010 en vue de gérer le PMUH.Note de bas de page 12
En 2012-2013, la DGPSA a mis en œuvre une nouvelle structure de gouvernance. Dans le cadre de cette structure, les médicaments pharmaceutiques, les produits biologiques, les instruments médicaux, les produits de santé naturels, la salubrité des aliments et la nutrition, et la politique et la promotion de la nutrition sont considérés comme des programmes distincts, et chacun d'entre eux est gouverné par un sous-comité du comité exécutif du programme. Le Programme des médicaments pharmaceutiques (PMP) englobe les médicaments vétérinaires et les médicaments à usage humain. La DGPSA est composée de différents comités et groupes de travail qui participent à la gestion des aspects variés des activités du PMUH.
Dans le cadre de la structure de gouvernance précédente, selon des renseignements fournis par la Direction générale, la planification était effectuée au niveau de la direction et les décisions étaient présentées aux hauts fonctionnaires en fonction des programmes. Dans le nouveau modèle, le but est que les responsables des programmes dirigent la planification et la production de rapports et que les directions offrent les activités fonctionnelles nécessaires en vue de présenter les résultats du programme. La nouvelle approche devrait accroître l'harmonisation du travail effectué par l'ensemble des directions aux stratégies, aux résultats et aux priorités des programmes.
5.2 Collaboration avec les partenaires externes
Les partenaires du PMUH collaborent avec plusieurs autres ministères et organismes fédéraux, ainsi qu'avec d'autres partenaires externes, dans le but d'offrir des activités réglementaires relatives aux médicaments à usage humain. Dans le cadre du PMUH, l'Inspectorat collabore avec l'Agence des services frontaliers du Canada (ASFC) afin d'assurer l'administration uniforme de la Loi sur les aliments et drogues et du Règlement sur les aliments et drogues par l'intermédiaire du Programme de l'intégrité des frontières, dont l'objectif est de renforcer la capacité de Santé Canada à prendre et à appuyer des décisions relatives à l'admissibilité des produits de santé à la frontière.
Dans le cadre du PMUH, l'Inspectorat collabore également avec le BSC de la DGSESC de Santé Canada. Le rôle du BSC consiste à délivrer des licences aux fabricants et aux distributeurs de substances contrôlées, à autoriser la destruction de drogues illicites, à gérer les processus d'exemption permettant à des personnes ayant des raisons scientifiques ou de santé d'accéder aux substances contrôlées, et à travailler avec d'autres groupes afin de faire respecter la conformité (DGSESC, 2009). Les responsabilités de l'Inspectorat incluent la réalisation d'inspections d'innocuité en vue de déterminer si l'entreposage des drogues contrôlées est approprié avant de délivrer une licence à un distributeur autorisé et la simplification de la destruction des substances contrôlées.
La DPT et l'Inspectorat collaborent aussi avec les représentants de l'industrie et d'autres intervenants dans le cadre du Programme des réunions bilatérales (PRB). Par l'intermédiaire de ce programme, les directions organisent, d'une à quatre fois par année, des réunions bilatérales avec différentes organisations nationales. Ces réunions ont lieu dans le but de discuter des problèmes de réglementation, d'échanger des renseignements, et de faire part de son expertise et de ses responsabilités. Enfin, différents groupes et comités consultatifs d'experts et de chercheurs ont participé à l'orientation des activités portant sur les médicaments à usage humain tout au long de la période visée par l'évaluation.
5.3 Mesure du rendement
De nombreuses stratégies de mesure du rendement (SMR) concernent directement le PMUH. On parle, par exemple, des cadres de l'IIPT, du PASPAC et de l'IRC, ainsi que du modèle logique de la DGPSA et de sa SMR. Les données sur le rendement des examens des présentations, soit les mesures sur le volume de demandes reçues, les délais d'examen, la charge de travail et les arriérés, sont, depuis toujours, les principaux éléments des rapports sur le rendement du PMUH. Les rapports annuels détaillés sur le rendement des examens des présentations sont utilisés pour éclairer les gestionnaires et les autres décideurs, et sont aussi transmis aux représentants de l'industrie. En outre, les rapports mensuels du tableau de bord ont été mis en œuvre avec l'IRC et sont employés pour orienter les décisions sur les priorités et l'attribution de ressources. Chaque année, Santé Canada présente également des rapports sur les normes de rendement établies relativement au PMUH dans le Rapport ministériel sur le rendement, lequel est déposé au Parlement.
5.4 Mise en œuvre du programme
Au cours de la période visée par l'évaluation, le PMUH a fait des progrès en mettant en œuvre les activités prévues et, par le fait même, en répondant à de nombreux problèmes et enjeux nouveaux. L'analyse ci-dessous fournit un aperçu, en guise d'introduction, du processus de réglementation des médicaments, puis décrit les activités préalables à la mise en marché, le contrôle et la surveillance post-commercialisation, la conformité et l'application, et les communications et les activités de mobilisation des intervenants au cours de la période visée par l'évaluation.Note de bas de page 13
5.4.1 Processus de réglementation des médicaments
The current system of drug regulation in Canada focuses mostly on pre-market activities (TPD, 2010b). It is a "point-in-time" system, which is characterized by discrete regulatory interventions at specific, defined points, and in which drugs are evaluated for their risks and benefits primarily at the pre-market stage. Figure 1 illustrates the process, showing the order of the components and the areas where Health Canada has regulatory authority. The regulatory process depicted below applies to all drugs, as defined in the Food and Drugs Act.
Figure 1: Aperçu du processus de réglementation des médicaments (DPT, 2007)
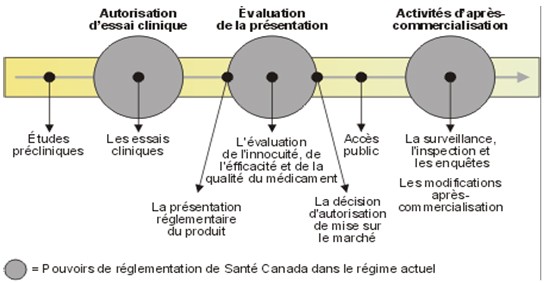
Équivalent textuel
L'ensemble du processus de réglementation peut se diviser en différentes étapes, soient :
- Les études précliniques
- L'autorisation d'essai clinique, dont font partie :
- Les essais cliniques
- La demande d'essai clinique ou une modification à la demande d'essai clinique (titre 5)
- Les essais cliniques
- Le Programme d'accès spécial
- L'évaluation de la demande, qui inclut :
- La présentation du produit conformément à la réglementation
- L'évaluation de l'innocuité, de l'efficacité et de la qualité du médicament
- La demande d'identification numérique du médicament (titre 1)
- La présentation de drogue nouvelle (titres 3, 4 et 8)
- La décision d'autorisation de mise sur le marché
- L'accès au public
- Les activités après commercialisation, qui incluent :
- La surveillance, l'inspection et les enquêtes
- La déclaration des effets indésirables
- Les modifications après commercialisation
- Le changement d'identification numérique de drogue
- Les modifications d'une nouvelle drogue
- La surveillance, l'inspection et les enquêtes
Santé Canada dispose de pouvoirs de réglementation pendant les étapes suivantes du processus :
- L'autorisation d'essai clinique
- L'évaluation de la présentation
- Les activités après commercialisation
Les sections 5.4.2 à 5.4.5 décrivent les activités du PMUH au cours de la période visée par l'évaluation en lien avec les quatre principales étapes du processus de réglementation : les essais cliniques, l'examen des présentations et l'autorisation de mise sur le marché, le contrôle et la surveillance post-commercialisation, et la conformité et l'application. Une description des activités du PMUH en matière de communication et de mobilisation des intervenants suit.
5.4.2 Essais cliniques
L'autorisation d'essai clinique est la première étape du processus de délivrance de licences et la première étape pour laquelle Santé Canada fait autorité en matière de réglementation. Pour démarrer le processus, les promoteurs présentent une demande d'essai clinique (DEC). Les DEC sont nécessaires afin d'obtenir l'autorisation d'effectuer des essais cliniques sur les nouveaux médicaments au Canada ou les drogues commercialisées dont l'usage proposé diffère de l'utilisation à l'origine de l'approbation.
À la réception de la DEC ou de la DEC-M d'un promoteur, Santé Canada entame l'examen de l'innocuité, de l'efficacité et de la qualité. Il vérifie l'intégralité de la demande et détermine si elle est acceptable d'un point de vue scientifique et réglementaire. Ce processus est réalisé dans un délai de 30 jours civils. Si le ministère trouve une lacune, il enverra, avant la date de défaut, une lettre ou un avis précis au promoteur, selon le contexte de la lacune.
Si Santé Canada ne trouve aucune lacune relative à la DEC ou à la DEC-M, il enverra une lettre de non-objection (LNO); le promoteur pourra alors procéder à l'essai clinique. Si le ministère omet de réaliser l'examen à l'intérieur du délai de 30 jours ciblé, le promoteur a aussi le droit d'effectuer l'essai clinique. Santé Canada a le pouvoir de suspendre ou d'annuler l'autorisation d'essai clinique s'il reçoit des renseignements portant à croire que le rapport risques-avantages de l'essai est inacceptable.
En 2010, Santé Canada a reçu 1 191 DEC relatives à des médicaments à usage humain, et 1 162 ont été approuvées. Depuis 2004, le nombre de DEC reçues ou approuvées relativement à des médicaments à usage humain a diminué de 30 %. En revanche, Santé Canada a reçu 272 DEC relatives à des produits biologiques en 2010, et en a approuvé 263, soit une augmentation de 5 % des DEC reçues et de 15 % des demandes approuvées. Ces tendances cadrent avec les changements de l'industrie décrits à la section 4.1 concernant le développement des produits biologiques. Ceci étant dit, Santé Canada a mentionné que cette tendance à la baisse en ce qui a trait aux DEC relatives aux médicaments à usage humain s'est stabilisée aux alentours de 2010; récemment (en juin 2013), le nombre de DEC a augmenté.Note de bas de page 14Veuillez consulter l'annexe C pour obtenir des renseignements détaillés.
Renforcer le cadre de réglementation des essais cliniques
Le cadre actuel des essais cliniques sur les médicaments nécessitant la participation de sujets humains est entré en vigueur le 1er septembre 2001. La réglementation a mis en place une exigence prévoyant que le protocole d'essai clinique soit approuvé par un comité d'éthique de la recherche (CER) pour chaque site d'essai clinique et que l'essai clinique soit effectué conformément aux BPC, ainsi que des exigences concernant la tenue de dossiers et les rapports sur les réactions indésirables. Un programme d'inspection a été lancé afin de s'assurer que les essais cliniques sont en harmonie avec la nouvelle réglementation.Note de bas de page 15
Depuis l'adoption de ces règlements, Santé Canada a renforcé le cadre de réglementation des essais cliniques en élaborant et en mettant à jour des documents d'orientation destinés aux promoteurs, y compris, la mise à jour récente des lignes directrices générales et l'ajout de lignes directrices sur l'inclusion des femmes dans les essais cliniques; la mise en œuvre d'approches axées sur les risques en vue de contrôler et d'évaluer les rapports sur les réactions indésirables survenus au cours d'un essai clinique, et de sélectionner les inspections de sites d'essai clinique, en réponse aux recommandations de 2011 du BVG; et la requête formulée auprès de l'Office des normes générales du Canada afin que ce dernier établisse de nouvelles normes facultatives pour les CER, lesquelles ont été officiellement approuvées par le Conseil canadien des normes en mai 2013 et qui peuvent maintenant être achetées par le public sur le site Web de Travaux publics et Services gouvernementaux Canada.
Le 29 mai 2013, Santé Canada a lancé une nouvelle base de données publique sur les essais cliniques de médicaments qui avaient été autorisés par le ministère. La base de données est obligatoire pour les représentants de l'industrie et contient des renseignements précis sur les phases I, II et III des essais cliniques chez les patients, y compris le numéro et le titre du protocole, le nom du médicament, le trouble médical, la population à l'étude, la date de la LNO, le nom du promoteur, la date de début et de fin de l'étude, et l'état d'avancement de l'essai. La base de données comprend des essais qui ont été autorisés par Santé Canada depuis le 1er avril 2013 (Santé Canada, 2013f).
La mise en œuvre de la base de données sur les essais cliniques de médicaments a permis à Santé Canada d'aborder son engagement de longue date visant à accroître la transparence des renseignements sur les essais cliniques.Note de bas de page 16Toutefois, comme le mentionne le ministère, la base de données n'est pas un registre des essais et ne contient pas toute l'information sur chaque essai clinique (Santé Canada, 2013f). En revanche, les États-Unis et l'UE ont mis en place des registres des essais cliniques obligatoires et ont pris les mesures nécessaires pour exiger la divulgation des résultats des essais cliniques.
Aux États-Unis, les registres sur les essais cliniques sont obligatoires depuis la mise en place du site ClinicalTrials.gov à la suite de l'adoption de la Food and Drug Administration Modernization Act de 1997, et l'obligation de présenter les résultats des essais cliniques au plus tard 12 mois après l'achèvement de l'essai a vu le jour en 2007 à la suite de l'adoption de la Food and Drug Administration Amendments Act (NIH, 2012; gouvernement des États-Unis, 2007). Depuis 2004, dans l'UE, les essais cliniques doivent aussi être consignés dans la base de données EudraCT. Depuis 2011, par l'intermédiaire du EU Clinical Trials Register, le public peut accéder à des renseignements descriptifs sur les phases I à IV des essais cliniques chez les adultes et des essais cliniques pédiatriques dont le site d'essai se situe dans l'UE/Espace économique européen (EMEA, 2012). On s'attend à ce que la publication des résultats fasse partie de la mise à jour d'une directive sur les essais cliniques qui devrait voir le jour en 2016.
Il est important de divulguer les résultats des essais cliniques étant donné les preuves tirées de la littérature confirmant que la publication de résultats sélectifs ou faussés sur les essais cliniques, c.-à-d. déclarer uniquement les résultats positifs et omettre les résultats négatifs, est assez courante. En outre, comme dans le cas du Vioxx, les rapports sélectifs peuvent entraîner des conséquences graves pour la sécurité des patients. Une augmentation de la quantité de renseignements disponibles sur les essais cliniques, y compris les résultats de ces essais, cadrerait avec l'engagement de Santé Canada visant à accroître la transparence et l'ouverture dans le cadre de la modernisation de la réglementation, et permettrait au ministère d'harmoniser davantage son approche avec celle de ses principaux homologues internationaux.
Un autre domaine présentant des divergences en matière de réglementation porte sur les essais cliniques pédiatriques. Contrairement à Santé Canada, les États-Unis et l'UE ont le pouvoir d'obliger les fabricants à effectuer des essais pédiatriques. Les représentants de Santé Canada ont mentionné que le ministère exige des essais pédiatriques avant d'autoriser la mise en marché si le fabricant à l'intention d'obtenir une indication pédiatrique; sinon, l'approbation ne sera pas accordée. Le ministère a pris des mesures pour encourager les promoteurs à réaliser des études pédiatriques.Note de bas de page 17 Aux États-Unis, la Pediatric Research Equity Act de 2003 a accordé à la FDA le pouvoir d'obliger les fabricants à effectuer des études pédiatriques sur les nouveaux médicaments qui seront probablement utilisés chez de nombreux enfants, ou qui devraient offrir des avantages considérables aux enfants comparativement aux traitements existants (IOM, 2008). Une loi semblable a été adoptée aux États-Unis en 2007, dont une exigence voulant que toutes les demandes d'autorisation de mise sur le marché relatives aux nouveaux médicaments incluent les résultats des toutes les études réalisées dans le cadre d'un plan d'investigation pédiatrique obligatoire, lequel vise à assurer que les données nécessaires sont recueillies « afin de déterminer les conditions d'autorisation d'un médicament destiné à la population pédiatrique » (MHRA, 2009).
L'absence de règlements semblables au Canada a incité le Comité permanent à recommander, dans son rapport 2012, que Santé Canada adopte des règlements visant à exiger à ce que les essais cliniques soient conçus de façon à faire appel à la même « population que celle qui risque vraisemblablement de faire usage d'un médicament », une fois que sa mise en marché au Canada aura été approuvée, et qu'il modifie le processus d'approbation des médicaments de sorte que la mise en marché ne sera approuvée que si les preuves cliniques démontrant l'innocuité et l'efficacité du produit comprennent des données sur tous les groupes de population qui risquent vraisemblablement de faire usage de ce médicament, une fois que sa mise en marché au Canada aura été approuvée (CPASST, 2012, p 35). En réponse à la recommandation du Comité, Santé Canada a mentionné que même si, à l'heure actuelle, le ministère n'oblige pas la conception d'essais cliniques, il a adopté les lignes directrices de la CIH, dont le but est d'encourager et d'appuyer le développement de médicaments aux fins d'une utilisation dans des populations spéciales, comme en gériatrie et en pédiatrie; a élaboré un additif canadien aux lignes directrices de la CIH pour le développement de médicaments dans les populations pédiatriques; et a collaboré avec les Instituts de recherche en santé du Canada et de nombreux groupes d'intervenants en vue de préparer Les pratiques exemplaires dans la recherche en santé avec des enfants et des adolescents.
Les énoncés de politique décrits dans les lignes directrices de Santé Canada récemment publiées sur l'inclusion des femmes dans les essais cliniques semblent cadrer avec les recommandations du Comité, en plus de reconnaître qu'il faut inclure dans les essais cliniques des sujets représentatifs de la population ou des populations chez lesquelles l'utilisation du produit est prévue (Santé Canada, 2012e).
5.4.3 Examen des présentations et autorisation de mise sur le marché
La deuxième étape du processus de délivrance de licences est la présentation réglementaire. Après avoir effectué les essais cliniques, le promoteur peut essayer d'obtenir une autorisation de mise sur le marché du médicament en présentant une demande auprès de Santé Canada. Le rôle de Santé Canada à l'étape de l'examen préalable à la mise en marché consiste à évaluer l'innocuité, la qualité et l'efficacité des médicaments à usage humain en vue de bien démontrer que leurs avantages l'emportent sur les risques potentiels. Le processus d'approbation préalable à la mise en marché s'applique à toutes les drogues, telles qu'elles sont définies dans la Loi sur les aliments et drogues.
Principaux types de présentations de drogue
- Demande de DIN (titre 1)
- Présentation de drogue nouvelle (PDN) (titre 8)
- Demande de PDN prioritaire
- Demande d'avis de conformité avec conditions (AC-C)
- Présentation abrégée de drogue nouvelle (PADN) (médicaments génériques)
- Supplément à une présentation de drogue nouvelle (SPDN)
- Supplément à une présentation abrégée de drogue nouvelle (SPADN)
- Demande de drogue nouvelle pour usage exceptionnel (DNUE)
Source : Processus et types de présentations
Il existe plusieurs types de présentations de drogue. Le processus relatif à une demande de DIN s'applique aux drogues relevant du « titre 1 ». Note de bas de page 18Une drogue relevant du titre 1 est habituellement une drogue qui se trouve sur le marché depuis de nombreuses années et dont l'innocuité et l'efficacité sont bien établies. Ces médicaments sont souvent disponibles en vente libre (MSO) (c.-à-d. aucune ordonnance n'est requise). Les demandes retenues reçoivent un numéro d'identification du médicament (DIN).
Le processus relatif à la présentation de drogue nouvelle (PDN) s'applique aux drogues relevant du « titre 8 ». Les drogues relevant du titre 8 sont les nouvelles drogues, ou celles qui n'ont pas été sur le marché longtemps. La PDN est semblable à la demande de DIN, malgré certaines différences importantes, comme des exigences réglementaires beaucoup plus rigoureuses. Le processus relatif à la PDN entraîne un avis de conformité (AC), lequel autorise le médicament à être vendu sur le marché, ainsi que l'attribution d'un DIN.
Pour entamer le processus relatif à la PDN, le promoteur peut présenter une PDN habituelle (tel que mentionné ci-dessus), faire une demande de PDN prioritaire ou demander un avis de conformité avec conditions (AC-C). Il est possible de formuler une demande de statut prioritaire lorsqu'un nombre suffisant de données portent à croire que la drogue proposée sera utile dans le cas d'une maladie ou d'un trouble débilitant ou mortel, ou pour lequel il n'existe aucun autre traitement commercialisé au Canada, ou lorsque le profil risques-avantages présente une amélioration par rapport aux traitements actuellement commercialisés au pays. Les délais relatifs à la présélection et à l'examen sont plus courts dans le cas d'une PDN prioritaire que dans le cas d'une PDN habituelle.
Comme pour un statut d'examen prioritaire, un AC-C peut permettre de fournir un accès anticipé à un « médicament pouvant sauver des vies ». Toutefois, l'AC-C diffère de l'examen prioritaire en ce sens qu'il met en place des conditions selon lesquelles le fabricant doit mener des études approfondies visant à confirmer les avantages du médicament. Ces études de surveillance post-commercialisation ont pour but de surveiller l'innocuité et l'efficacité du produit.
En revanche, le promoteur peut soumettre une présentation abrégée de drogue nouvelle (PADN) lorsque le médicament proposé est jugé comme équivalent et bioéquivalent, sur le plan pharmaceutique, à un médicament existant sur le marché. Le cas échéant, la présentation doit fournir l'information permettant de comparer le rendement du produit « générique » à celui du produit de marque. On fournit un supplément à une présentation de drogue nouvelle (SPDN) ou un supplément à une présentation abrégée de drogue nouvelle (SPADN) lorsque des changements importants, comme le dosage, le processus de fabrication ou l'étiquetage, sont apportés aux nouveaux médicaments qui ont déjà reçu un AC.
Enfin, depuis 2011, les promoteurs peuvent présenter une demande de drogue nouvelle pour usage exceptionnel (DNUE). Il s'agit d'un mécanisme d'autorisation des nouveaux médicaments utilisés dans des circonstances exceptionnelles permettant aux promoteurs de se servir des résultats d'études chez les animaux conjointement à ceux de quelques études sur l'innocuité et l'efficacité pour les humains à l'appui de leur présentation de drogue (Santé Canada, 2013a). Les DNUE sont destinées à un usage exceptionnel en réponse à une exposition à une substance chimique, biologique, radiologique ou nucléaire nécessitant une intervention en vue de traiter, d'atténuer ou de prévenir une maladie ou un trouble grave ou mortel découlant de cette exposition, ou à des fins d'utilisation préventive chez les personnes qui risquent d'être exposés à de telles substances.
Examen et autorisation de mise sur le marché
Une fois présentés, les processus de présélection et d'examen de l'innocuité, de l'efficacité et de la qualité peuvent être mis en œuvre. Comparativement aux drogues relevant du titre 1, dont l'innocuité et l'efficacité sont bien connues, le processus d'examen d'une PDN requiert une évaluation clinique approfondie des études réalisées sur des animaux et sur des humains. En cas de lacunes ou de non-conformités, l'entreprise peut fournir des réponses aux fins d'examen avant qu'une décision sans appel ne soit prise. Par conséquent, des cycles d'examen multiples peuvent être nécessaires.
Après avoir achevé avec succès le processus de présélection et d'examen de l'innocuité, de l'efficacité et de la qualité, le médicament obtient un DIN et un AC. L'AC atteste que le médicament est conforme à la Loi sur les aliments et drogues et au Règlement sur les aliments et drogues, et autorise sa mise en marché. Par contre, le médicament reçoit un AC-C, lequel autorise la mise sur le marché d'un produit de qualité supérieure dont l'innocuité est acceptable, pourvu que les fabricants continuent de respecter les conditions connexes (DGPSA, 2011b). Le but de l'AC-C est de fournir l'accès à une mesure de prévention, à un traitement ou à un diagnostic nouveau ou grandement amélioré relativement à une maladie ou à un trouble grave pour lequel il n'existe aucun médicament ni aucun médicament de qualité inférieure, tout en continuant de surveiller les risques qu'il présente et d'évaluer son profil général risques-avantages.
Changements post-commercialisation
Il incombe aux promoteurs d'informer Santé Canada de toute modification apportée à un médicament ayant déjà obtenu une autorisation de mise sur le marché. Le processus suivi dépendra de la nature du changement et de son incidence probable ou potentielle sur l'innocuité, la qualité et l'efficacité du produit. Même si aucun changement n'est apporté à un médicament, le promoteur doit malgré tout présenter des avis annuels de la situation à Santé Canada (Santé Canada, 2011c).
Programme d'accès spécial
Enfin, dans le cadre du Programme d'accès spécial (PAS), les professionnels de la santé peuvent obtenir un accès aux médicaments qui n'ont pas encore reçu l'autorisation de mise sur le marché au Canada. Le PAS est destiné aux « maladies graves ou mortelles, lorsque les traitements classiques se sont révélés inefficaces, ne conviennent pas ou ne sont pas disponibles, sous la forme de produits commercialisés ou dans le cadre d'essais cliniques » (DGPSA, 2008). Il peut également permettre de clarifier les circonstances dans lesquelles une autorisation est accordée ou refusée en cas d'éclosions de maladies transmissibles; d'offrir un examen approfondi des autorisations si l'on soupçonne le contournement des exigences relatives à un essai clinique ou à une PDN; d'autoriser la distribution en bloc de produits dans le cas d'urgences de santé publique; et d'établir un cadre éthique permettant l'accès aux nouveaux traitements pour des raisons humanitaires en dehors de la recherche clinique (DGPSA, 2007a).
Dans le cadre du renouvellement de la réglementation, Santé Canada a planifié l'examen et la modernisation du cadre de réglementation du PAS, établi en 1966. Le statut de cette initiative demeure inconnu.
Amélioration du processus d'examen des présentations et d'autorisation de mise sur le marché
L'amélioration de la qualité et de l'efficacité du processus d'examen des présentations et d'autorisation de mise sur le marché relatif aux produits thérapeutiques, y compris les médicaments à usage humain, a été une priorité de la DGPSA tout au long de la période visée par l'évaluation. Faisant face à une augmentation des coûts et à des arriérés considérables relativement au processus d'examen, la DGPSA a entrepris différentes initiatives visant à réduire les coûts et à accroître l'efficacité et la qualité de l'examen des présentations. Ces initiatives sont décrites brièvement ci-dessous.
- Normes de service pour les activités de recouvrement des coûts. Afin de se conformer aux exigences de la Loi sur les frais d'utilisation, Santé Canada a établi des normes de service pour les activités de recouvrement des coûts, y compris l'examen des présentations (GC, 2004, par. 4(1)). Les normes de service relatives à l'examen des présentations varient en fonction du type et de la catégorie de la présentation; pour la majorité des PDN et des PADN, la norme est de 300 jours et de 180 jours, respectivement (DGPSA, 2011c).
- Nouveau cadre de recouvrement des coûts. En avril 2011, en réponse aux préoccupations du BVG portant sur la capacité de la DGPSA à offrir des services de réglementation étant donné la disponibilité de ses ressources, un nouveau cadre de recouvrement des coûts relatifs aux médicaments et aux instruments médicaux a été mis en œuvre avec l'entrée en vigueur du Règlement sur les prix à payer à l'égard des drogues et instruments médicaux (GC, 2011; DGPSA, 2011a). Au même moment, la DGPSA a ajusté sa cible de rendement concernant l'examen des présentations. Avant la mise en œuvre du nouveau cadre, la cible de rendement de la DGPSA consistait à achever 90 % de toutes les décisions initiales pour chaque catégorie de présentation dans les délais prescrits. Même si la DGPSA se doit toujours d'atteindre cette cible de rendement, une nouvelle mesure a été établie avec la mise en œuvre du nouveau cadre de recouvrement des coûts : veiller à ce que la moyenne des présentations respecte les délais d'examen prévus en ce qui concerne l'examen initial de leur catégorie de présentation.Note de bas de page 19En théorie, le traitement des présentations selon les cibles de rendement établies devrait faciliter l'accès rapide à des produits thérapeutiques efficaces, augmentant ainsi le nombre d'options de traitement possibles sans mettre les utilisateurs finaux en danger.
- Approche de gestion de projet pour l'examen des présentations. Santé Canada a mis en place une approche de gestion de projet pour l'examen des présentations. Celle-ci a nécessité la création de nouveaux postes de « gestionnaire de projets réglementaires » au sein de la DPT en vue de coordonner et d'orienter chaque présentation au moyen du processus, l'organisation de forums sur la charge de travail dans le but de fournir un examen mensuel de la charge de travail avec les directeurs et les chefs de division, et l'établissement de rapports sur la charge de travail.
- Bonnes pratiques de lignes directrices (BPLD) et Bonnes pratiques d'examen (BPE). La DPT a créé les BPLD et les BPE en vue d'accroître l'uniformité relative à l'élaboration de lignes directrices et aux examens des présentations, respectivement.
- Présentations électroniques. Santé Canada a adopté le format electronic Common Technical Document (eCTD) établi par la Conférence internationale sur l'harmonisation (CIH) des exigences techniques pour l'enregistrement des médicaments à usage humain aux fins d'examen électronique. En 2011, malgré une mise en œuvre des présentations électroniques plus lente que prévu, Santé Canada a augmenté le nombre de types de présentations acceptées en format de dépôt uniquement électronique eCTD, et le nombre de présentations électroniques s'est accru. Récemment, par l'intermédiaire du CCR, Santé Canada et la FDA des États-Unis ont mis en œuvre un Portail commun de demandes électroniques à l'aide du portail actuel des États-Unis, pour permettre aux « clients de l'industrie de soumettre facilement de gros documents électroniques à Santé Canada et à la FDA des États-Unis » (GC, sans date). Grâce au Portail commun de demandes électroniques, les présentations seront transmises à Santé Canada par l'intermédiaire de l'infrastructure de la technologie de l'information de la FDA. Un projet pilote de trois mois sur le portail est en cours.Note de bas de page 20
- Capacité d'examen accrue. En 2007, la DPT a lancé le Répertoire des experts scientifiques en vue de faciliter l'accès à des experts externes pour l'examen des présentations de drogue et de participer aux comités et aux groupes consultatifs de scientifiques. Santé Canada a signalé que le répertoire compte un total de 564 experts dont les services peuvent être sollicités pour effectuer certains aspects de l'examen des présentations. Dans le cas des produits pharmaceutiques, ces contrats portent principalement sur des examens précliniques, bien qu'ils puissent aussi concerner d'autres aspects de l'examen, comme l'examen de l'étiquette, l'examen de la composition chimique et du processus de fabrication, ou l'examen de la bioéquivalence, entre autres. Selon Santé Canada, les coûts associés aux contrats externes ont augmenté, passant d'environ 350 000 dollars en 2006-2007 à une prévision de 1 million de dollars en 2013-2014.
- Réunions préalables au dépôt des présentations. Même si les réunions préalables au dépôt des présentations avec les promoteurs étaient bien établies au début des années 1990, le Plan de renouveau et le PASPAC ont permis de trouver des façons d'améliorer ce processus. L'un des principaux objectifs des réunions préalables au dépôt des présentations, lesquelles sont à la discrétion des promoteurs, consiste à fournir des « conseils scientifiques et réglementaires aux promoteurs aux étapes préliminaires de la mise au point d'un produit » (DGPSA, 2007a, p. 22-23). En 2010-2011, les lignes directrices destinées aux représentants de l'industrie quant aux réunions préalables au dépôt des présentations ont été mises à la disposition du public. Les données fournies par Santé Canada révèlent qu'entre 2002 et le 23 novembre 2012, la DPT a tenu 1 279 réunions préalables au dépôt des présentations, parmi lesquelles 13 % étaient des réunions préalables à des DEC et 59 % étaient des réunions préalables à des PDN (DGPSA, 2012a). Les informateurs clés de l'industrie ont trouvé des façons d'améliorer le processus de réunions préalables au dépôt des présentations en abordant la longue attente avant les réunions et en veillant à ce que l'évaluateur principal soit présent.
- Utilisation des examens étrangers. En octobre 2011, dans le cadre de plans généraux visant l'amélioration de l'efficacité des examens au moyen d'une utilisation accrue des examens étrangers, Santé Canada a lancé le Projet pilote sur l'utilisation des examens étrangers, qui devait se poursuivre jusqu'en mars 2013. Les produits abordés par le projet pilote sont les PDN, les PADN, les SPDN, les SPADN, les AC et les plans de gestion des risques (PGR). Le nombre de présentations de médicaments à usage humain dans le cadre du projet pilote demeure inconnu. Dans une mise à jour de mars 2013 présentée au BVG, Santé Canada a signalé que les « résultats du projet pilote [étaient] encourageants » (Santé Canada, 2013l), alors qu'un document interne datant du milieu de l'exercice 2012-2013 a révélé que le projet pilote « avait peu de succès en raison du manque de participation et de renseignements de la part des sources externes » (DGPSA, 2012c). Les informateurs clés externes ont encouragé Santé Canada à accepter et à utiliser plus de renseignements et d'approbations provenant de sources externes afin d'améliorer l'efficacité du processus d'examen.Note de bas de page 21
On s'attend à ce que ces initiatives améliorent l'efficacité et la qualité du processus d'examen des présentations de médicaments à usage humain. Les données disponibles sur le rendement des examens des présentations, lesquels couvrent la période allant du début de l'année civile 2004 à la fin de l'année civile 2012 (voir Tableau 2) dévoilent les renseignements suivants :
- Pour chaque type de présentation, la rapidité des examens a varié au fil des ans, et souvent, la cible de 90 % n'a pas été atteinte.Note de bas de page 22Ceci étant dit, les plus récentes données disponibles, pour 2012, indiquent une amélioration par rapport à l'année précédente quant à la proportion des cycles d'examen respectant la norme de service pour tous les types de présentation. En ce qui concerne les PDN, la proportion des cycles d'examen atteignant la norme de service s'améliore depuis 2009, alors que 71 % des cycles d'examen respectaient la norme. En 2012, cette proportion avait atteint 97 %.
- L'examen rapide des PADN (présentations de produits génériques) et des SPADN a été plus difficile dans les dernières années. La charge de travail relative à l'examen des PDN a continué d'augmenter, passant de 81 examens en 2007 à 282 en 2011. En 2011, 11 % des cycles d'examen des PADN étaient achevés dans les délais, le temps médian d'approbation avait atteint 645 jours civils et 61 % des PADN présentaient un arriéré, soit la proportion la plus élevée depuis 2004. Sur une note positive, 2012 a connu des améliorations, dont une augmentation de 125 % du nombre d'examens réalisés (320 en 2012 comparativement à 140 en 2011), ce qui a permis de réduire la charge de travail relative aux examens des PADN de fin d'année à 222 examens, et un arriéré de 45 % (diminution de 16 % par rapport à 2011). En 2012, 18 % des cycles d'examen des PDN étaient achevés dans les délais, et le temps médian d'approbation avait diminué à 543 jours.
- Entre 2004 et 2012, le temps médian d'approbation avait diminué de 40 % pour les PDN et de 23 % pour les SPDN. Dans la même période, le délai d'approbation médian a augmenté de 48 % pour les PADN et de 23 % pour les SPADN. En 2012, le temps médian d'approbation pour les PDN était de 349 jours civils, comparativement à 543 jours civils pour les PADN.
Le rendement récent de Santé Canada quant à l'examen des présentations de produits génériques semble être attribuable, du moins en partie, à l'augmentation du nombre de ces présentations. Les informateurs clés de l'industrie ont attribué ce rendement non seulement aux volumes croissants, mais aussi à la décision du ministère de limiter les ressources consacrées à ces examens. Il a été impossible d'accéder aux renseignements concernant les ressources humaines affectées à l'examen des médicaments génériques et à d'autres présentations, et on ne pouvait pas analyser le lien entre le nombre de présentations, les niveaux de ressources et la rapidité des examens.
Dans l'ensemble, le rendement des examens des présentations de médicaments à usage humain s'est amélioré avec le nouveau cadre de recouvrement des coûts, et ce, pour toutes les catégories de présentation. Toutefois, à l'exception des PADN, les améliorations relatives au rendement des examens étaient déjà en cours pour toutes les catégories de présentation avant la mise en œuvre du nouveau cadre et sont peut-être attribuables aux autres initiatives de Santé Canada visant à accroître l'efficacité et la qualité du processus d'examen décrit ci-dessus.
Une analyse détaillée des données sur les examens des présentations, tenant compte de facteurs comme les changements relatifs aux niveaux de ressources et le nombre de présentations au fil du temps, fournirait des informations supplémentaires sur le rendement des examens de Santé Canada, y compris la portée des gains d'efficacité réalisés dans le cadre du nouveau système. Par exemple, il serait profitable d'étudier le rendement annuel des examens compte tenu du ratio d'équivalents temps plein (ETP) par rapport au nombre de demandes annuelles formulées pour chaque catégorie de présentation. Il serait tout aussi profitable de se pencher sur les coûts unitaires des examens pour chaque catégorie de présentation (veuillez consulter la section 7.0 pour une analyse détaillée).
Enfin, dans le contexte de l'examen des présentations, il est important de noter, en passant, que certains informateurs clés externes se sont dits préoccupés par l'efficacité de l'approche de Santé Canada visant à régir les produits mixtes. Par exemple, ces derniers ont mentionné que le cadre actuel de réglementation a empêché Santé Canada de fournir un aperçu approprié concernant les cigarettes électroniques, qui peuvent être considérées à la fois comme une drogue et un instrument médical. Dans un autre exemple, la réglementation des trousses contenant plusieurs catégories de produits est considérée comme très dispendieuse, particulièrement lorsque les articles de la trousse ne sont pas tous produits par le même fabricant. Les produits mixtes ont fait l'objet d'une analyse approfondie dans l'évaluation du Programme des matériels médicaux. Cette analyse n'est pas répétée ici.
| Présentations | 2004 | 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2011 | 2012 |
|---|---|---|---|---|---|---|---|---|---|
| Présentation de drogue nouvelle (PDN) | |||||||||
| Nombre de présentations reçues | 47 | 51 | 53 | 51 | 53 | 64 | 54 | 53 | 69 |
| Nombre de présentations dans la charge de travail (à la fin de l'année) | 45 | 47 | 46 | 38 | 51 | 49 | 54 | 42 | 45 |
| Pourcentage de l'arriéré | 18 % | 4 % | 0 % | 0 % | 12 % | 8 % | 15 % | 0 % | 0 % |
| Nombre de cycles d'examen achevés | 84 | 68 | 74 | 77 | 58 | 75 | 67 | 86 | 65 |
| Pourcentage de cycles d'examen achevés dans les délais | 17 % | 62 % | 84 % | 92 % | 88 % | 71 % | 73 % | 80 % | 97 % |
| Nombre d'approbations | 40 | 32 | 40 | 40 | 27 | 34 | 35 | 61 | 39 |
| Temps médian d'approbation (jours civils) | 581 | 430 | 397 | 384 | 345 | 428 | 433 | 375 | 349 |
| Supplément à une présentation de drogue nouvelle (SPDN) | |||||||||
| Nombre de présentations reçues | 114 | 133 | 186 | 154 | 154 | 132 | 101 | 111 | 162 |
| Nombre de présentations dans la charge de travail (à la fin de l'année) | 95 | 91 | 122 | 116 | 104 | 92 | 76 | 88 | 95 |
| Pourcentage de l'arriéré | 8 % | 1 % | 0 % | 1 % | 2 % | 7 % | 4 % | 2 % | 1 % |
| Nombre de cycles d'examen achevés | 163 | 146 | 160 | 195 | 175 | 158 | 138 | 110 | 160 |
| Pourcentage de cycles d'examen achevés dans les délais | 38 % | 73 % | 92 % | 97 % | 92 % | 88 % | 79 % | 94 % | 95 % |
| Nombre d'approbations | 103 | 93 | 123 | 158 | 139 | 121 | 103 | 76 | 131 |
| Temps médian d'approbation (jours civils) | 369 | 344 | 337 | 323 | 322 | 325 | 338 | 336 | 285 |
| Présentation abrégée de drogue nouvelle (PADN) | |||||||||
| Nombre de présentations reçues | 117 | 137 | 124 | 142 | 161 | 179 | 185 | 208 | 215 |
| Nombre de présentations dans la charge de travail (à la fin de l'année) | 71 | 77 | 92 | 81 | 97 | 153 | 212 | 282 | 222 |
| Pourcentage de l'arriéré | 32 % | 0 % | 3 % | 6 % | 5 % | 35 % | 44 % | 61 % | 45 % |
| Nombre de cycles d'examen achevés | 111 | 165 | 195 | 222 | 204 | 168 | 164 | 140 | 320 |
| Pourcentage de cycles d'examen achevés dans les délais | 19 % | 64 % | 90 % | 92 % | 90 % | 45 % | 24 % | 11 % | 18 % |
| Nombre d'approbations | 82 | 77 | 81 | 121 | 122 | 96 | 107 | 86 | 225 |
| Temps médian d'approbation (jours civils) | 368 | 378 | 525 | 532 | 453 | 467 | 536 | 645 | 543 |
| Supplément à une présentation abrégée de drogue nouvelle (SPADN) | |||||||||
| Nombre de présentations reçues | 8 | 7 | 26 | 12 | 19 | 32 | 28 | 28 | 60 |
| Nombre de présentations dans la charge de travail (à la fin de l'année) | 5 | 12 | 10 | 4 | 8 | 20 | 36 | 36 | 33 |
| Pourcentage de l'arriéré | 60 % | 0 % | 0 % | 0 % | 0 % | 35 % | 64 % | 67 % | 30 % |
| Nombre de cycles d'examen achevés | 15 | 14 | 25 | 21 | 13 | 19 | 26 | 33 | 62 |
| Pourcentage de cycles d'examen achevés dans les délais | 33 % | 57 % | 92 % | 95 % | 92 % | 68 % | 12 % | 27 % | 37 % |
| Nombre d'approbations | 11 | 10 | 17 | 20 | 10 | 12 | 18 | 23 | 51 |
| Temps médian d'approbation (jours civils) | 328 | 382 | 323 | 305 | 245 | 220 | 414 | 507 | 402 |
Définitions : Présentations reçues : Le nombre de présentations reçues pendant un exercice financier en employant la date de dépôt (date à laquelle la présentation est considérée comme complète du point de vue administratif par Santé Canada. Charge de travail : Le nombre de présentations en examen à la fin de l'année. Ce nombre inclut les présentations subissant un examen initial ainsi que les présentations pour lesquelles un autre cycle d'examen a été requis afin d'examiner les réponses de l'entreprise aux questions soulevées par Santé Canada. Arriéré : La portion de la charge de travail qui a dépassé les délais prescrits. Approbations : L'avis de conformité (AC) délivré ou délivrable. Un AC d'une présentation est dit délivrable lorsqu'il est temporairement suspendu, attend l'autorisation de mise en marché, ou résulte de la réglementation sur les brevets ou encore du retrait du produit de l'annexe F (il passe de médicament sur ordonnance à médicament en vente libre). Temps d'approbation : Le nombre de jours civils entre la date de dépôt d'une présentation et la date de son approbation; inclut le temps de l'entreprise, le cas échéant. Cycle d'examen achevé : Comptabilisé lorsqu'un examen scientifique approfondi aboutit à une décision d'approbation ou de non-approbation. Une décision est jugée prise dans les délais lorsque les jours nécessaires à l'achèvement de l'examen respectent la norme de rendement. |
|||||||||
5.4.4 Contrôle et surveillance post-commercialisation
À la suite de l'autorisation de mise sur le marché, il incombe à Santé Canada d'effectuer une surveillance post-commercialisation, aussi appelée la pharmacovigilance, afin de s'assurer que l'équilibre entre les avantages et les risques du produit demeure favorable. Santé Canada définit la pharmacovigilance comme « la science et les activités liées à la détection, à l'évaluation, à la compréhension et à la prévention des effets indésirables des médicaments ou de tout autre problème relié aux médicaments » (DPSC, 2009).
Depuis toujours, les rapports sur les réactions indésirables sont les piliers sur lesquels s'appuie l'approche de Santé Canada (et d'autres organismes de réglementation) en matière de pharmacovigilance. Toutefois, au cours de la dernière décennie, de plus en plus d'organismes de réglementation ont reconnu le besoin d'augmenter la surveillance post-commercialisation afin de mieux protéger la santé et la sécurité des patients. Santé Canada a admis les nombreuses limites de son approche actuelle à cet égard, notamment les points suivants :
- Recours à la pharmacovigilance de routine (c.-à-d. des rapports spontanés sur les réactions indésirables) pour détecter les signaux d'innocuité qui sont difficiles à percevoir à l'aide des données d'une étude de casNote de bas de page 23
- Manque de suivi et d'évaluation des risques dans les populations qui ne sont pas étudiées dans les essais cliniques;
- Défaut d'aborder les problèmes liés à l'emploi non conforme à l'étiquetage;
- Défaut de relever les risques potentiels dans le contexte canadien et d'en faire le suivi;
- Manque d'autorité pour ordonner la tenue d'études avant la mise en marché ou d'autres stratégies de gestion des risques;
- Absence d'une évaluation systématique de la pratique de gestion des risques en vue de déterminer l'efficacité (DGPSA, 2009, 2012g).
Dans le cadre de son initiative générale de modernisation de la réglementation et de son orientation souhaitée vers une approche de réglementation fondée sur le cycle de vie d'un produit, Santé Canada a pris les mesures nécessaires en vue d'aborder ces limites et de renforcer son approche en matière de contrôle et de surveillance post-commercialisation.
Accroître le nombre de rapports et d'analyses sur les réactions indésirables
Conformément au Règlement sur les aliments et drogues, les fabricants sont obligés de présenter des « rapports sur les réactions indésirables graves aux drogues ». De plus, les professionnels de la santé, les établissements de santé et les membres du public peuvent volontairement fournir des rapports sur les réactions indésirables. Les points ci-dessous fournissent des renseignements détaillés concernant les rapports sur les réactions indésirables.
Rapports sur les réactions indésirables aux médicaments à usage humain au Canada
- Fabricants : Conformément au Règlement sur les aliments et drogues, les fabricants sont obligés de présenter des « rapports sur les réactions indésirables graves aux drogues ». Les fabricants doivent fournir tous les renseignements liés à l'incident dans les quinze jours après avoir été informés d'une réaction indésirable grave, ou après en avoir pris connaissance, que cela se produise à l'intérieur ou à l'extérieur du Canada. Les rapports dûment remplis peuvent être envoyés à Santé Canada par la poste ou par télécopieur. Depuis 2011, les fabricants sont également tenus de préparer des rapports sommaires annuels de toutes les réactions indésirables survenues au cours de l'année précédente en plus de leurs analyses de ces incidents, et de présenter ces rapports au ministère de la Santé, sur demande.
- Professionnels de la santé, établissements de santé et membres du public : Les rapports sur les réactions indésirables sont facultatifs pour ces intervenants. Des rapports officiels sont à la disposition des consommateurs et les professionnels de la santé; ils peuvent être envoyés par la poste ou par télécopieur, ou remplis en ligne. Il est aussi possible de remplir les rapports par téléphone.
Tous les rapports sur les réactions indésirables sont présentés au Programme Canada Vigilance, qui, en 2007, a remplacé le Programme canadien de surveillance des effets indésirables des médicaments. Depuis ce temps, les rapports sur les réactions indésirables ont augmenté de façon considérable.
La Figure 2 montre la croissance du nombre de rapports sur les réactions indésirables survenues au Canada en lien avec les médicaments pharmaceutiques reçus par Santé Canada entre 1999 et 2011; les produits biologiques sont inclus aux fins de comparaison.Note de bas de page 24Au total, 147 667 rapports sur les réactions indésirables ont été reçus concernant les produits pharmaceutiques. Le total annuel de rapports portant sur des produits pharmaceutiques a augmenté de 456 % au cours de cette période, passant de 4 870 rapports en 1999 à 27 077 en 2011. Une grande partie de cette augmentation a eu lieu depuis 2007.
Les rapports sur les réactions indésirables survenues au Canada représentent seulement une fraction du nombre total de rapports sur les réactions indésirables reçus par Santé Canada, car les rapports étrangers, qui comptent pour plus de 90 % de tous les rapports reçus, ne sont pas consignés dans la base de données de Canada Vigilance.
Figure 2: Rapports sur les réactions indésirables survenues au Canada : les produits pharmaceutiques par rapport aux produits biologiques
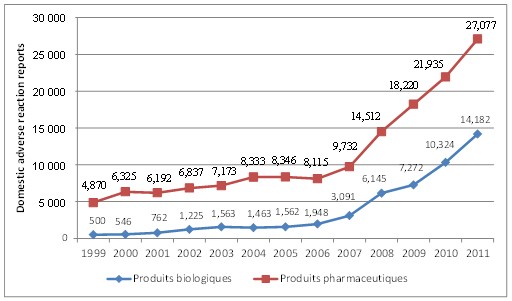
Équivalent textuel
La figure 2 montre la croissance du nombre de déclarations d'effets indésirables associés à des médicaments pharmaceutiques au Canada qui ont été présentées à Santé Canada entre 1999 et 2011; les produits biologiques sont inclus aux fins de comparaison. Au total, 147 667 déclarations d'effets indésirables mettant en cause des produits pharmaceutiques ont été présentées. Le nombre annuel de déclarations d'effets indésirables associés à ces produits a augmenté de 456 % au cours de cette période, passant de 4 870 déclarations en 1999 à 27 077 déclarations en 2011. Une grande partie de cette augmentation a eu lieu depuis 2007.
Description du modèle logique (annexe A, et non B)
Le Programme des médicaments à usage humain utilise différentes ressources (intrants) pour mener à bien ses activités, produire des extrants et atteindre ses résultats, notamment le financement, les ressources humaines, les installations, l'infrastructure, les lois, les règlements, les politiques et les priorités, la science et la technologie et les données de recherche.
Le Programme des médicaments à usage humain se divise en cinq principales activités assurées par ses partenaires, soient :
-
- l'élaboration, la mise en œuvre et la tenue à jour du cadre de réglementation;
- les communications avec les partenaires et les intervenants;
- l'évaluation des risques et des avantages associés aux processus d'autorisation d'essai clinique et d'autorisation de mise sur le marché;
- la surveillance et le suivi des activités après commercialisation;
- la conformité et l'application du cadre de réglementation.
Ces activités ciblent différents groupes:
- L'élaboration, la mise en œuvre et la tenue à jour du cadre de réglementation visent
- l'industrie, les médias, les consommateurs, les gouvernements et les organismes internationaux, les praticiens de la santé, le milieu universitaire, les gouvernements fédéral, provinciaux et territoriaux, et les associations industrielles et professionnelles
- Les communications avec les partenaires et les intervenants visent
- l'industrie, les médias, les consommateurs, les gouvernements et les organismes internationaux, les praticiens de la santé, le milieu universitaire, les gouvernements fédéral, provinciaux et territoriaux, et les associations industrielles et professionnelles
- L'évaluation des risques et des avantages associés aux processus d'autorisation d'essai clinique et d'autorisation de mise sur le marché vise
- l'industrie, les organismes internationaux, le milieu universitaire, les professionnels de la santé et les gouvernements fédéral, provinciaux et territoriaux
- La surveillance et le suivi des activités après commercialisation visent
- les responsables de la réglementation de Santé Canada et de l'étranger, le milieu universitaire et les organisations scientifiques et de recherche
- La conformité et l'application du cadre de réglementation visent
- l'industrie, Santé Canada, les intervenants et les partenaires fédéraux, provinciaux, territoriaux et internationaux du domaine de la réglementation, et les consommateurs
Chaque activité permet au Programme de générer différents produits et services :
- L'élaboration, la mise en œuvre et la tenue à jour du cadre de réglementation donnent lieu à
- des lois et des règlements, des politiques, des lignes directrices, des accords de reconnaissance mutuelle et des protocoles d'entente
- Les communications avec les partenaires et les intervenants donnent lieu à
- des sources et des systèmes de données, des sites Web, des données de recherche, du matériel et des activités de sensibilisation du public et des intervenants, des publications, des mises en garde, des avis (y compris des lettres à l'intention des professionnels de la santé), de la correspondance et des demandes de renseignements du public
- L'évaluation des risques et des avantages associés aux processus d'autorisation d'essai clinique et d'autorisation de mise sur le marché donne lieu à
- des évaluations des risques et des avantages (initiales et continues, y compris des évaluations des risques pour la santé), des autorisations de mise sur le marché, des licences d'établissement, des autorisations d'essai clinique et des décisions prises en vertu du Programme d'accès spécial
- La surveillance et le suivi des activités après commercialisation donnent lieu au
- Programme Canada Vigilance et à des données, des analyses, des preuves, des rapports, des recommandations réglementaires, l'identification et la caractérisation des signaux, des études épidémiologiques et des plans de gestion et d'atténuation des risques
- La conformité et l'application du cadre de réglementation donnent lieu à
- des rapports de conformité et d'application de la loi, des mesures de conformité et d'application de la loi, dont des poursuites en justice, des suspensions de licences, des saisies, des ordres d'arrêt de vente et des perquisitions.
Ces activités correspondent à des résultats particuliers à court, à moyen et à long terme. À court terme, on s'attend à ce que les activités du Programme accroissent la sensibilisation et la compréhension des intervenants externes quant aux risques et aux avantages des médicaments à usage humain. Elles devraient également accroître la sensibilisation et la compréhension des représentants de l'industrie quant aux activités de réglementation de Santé Canada relativement à ces médicaments, augmenter l'innocuité et l'efficacité de ces produits, et favoriser la conformité des représentants de l'industrie aux exigences réglementaires pertinentes.
L'atteinte de ces résultats immédiats devrait entraîner des résultats intermédiaires, dont l'adoption de comportements sécuritaires de la part des intervenants externes en ce qui a trait aux médicaments à usage humain; l'utilisation accrue de données scientifiques et d'analyses des avantages et des risques par Santé Canada en vue d'éclairer la prise de décisions; l'intervention réglementaire rapide à l'égard des risques cernés; la diminution de l'exposition aux risques pour la santé associés à l'utilisation des produits à usage humain; l'harmonisation du cadre de réglementation du Canada avec les approches adoptées à l'échelle internationale.
À long terme, Santé Canada souhaite réduire les événements indésirables associés à l'utilisation de médicaments à usage humain; améliorer la confiance du public par rapport à ces médicaments et au système de réglementation connexe, en plus d'améliorer ce dernier de sorte qu'il soit viable, rentable, adapté et fondé sur la science.
L'objectif ultime du Programme est d'aider à offrir aux Canadiens des médicaments à usage humain sûrs et efficaces ainsi que de l'information qui permette de faire des choix sains.
Santé Canada entreprend plusieurs initiatives en vue de promouvoir la présentation de rapports et d'analyses sur les réactions indésirables.
Lutter contre la sous-déclaration des professionnels de la santé
En vue d'aborder le problème de longue date concernant la sous-déclaration des réactions indésirables aux drogues par les professionnels de la santé, les coordonnateurs régionaux du Programme Canada Vigilance organisent des activités de sensibilisation et d'éducation dans le but de fournir des renseignements sur la façon et l'importance de présenter des rapports sur les réactions indésirables.Note de bas de page 25En outre, Santé Canada avait l'intention de mettre en place des rapports obligatoires sur les réactions indésirables graves aux drogues pour les établissements de santé dans le cadre du PASPAC. Une disposition obligeant ces établissements à présenter des rapports a été ajoutée au projet de loi C-51 en 2008, mais celui-ci n'est jamais devenu une loi. Depuis ce temps, Santé Canada a rédigé des lignes directrices en vue d'aider les professionnels de la santé à déclarer les événements indésirables (DPSC, 2011a); a procédé, en 2011, à l'essai d'un système de déclaration électronique permettant aux professionnels de la santé de présenter des rapports sur les réactions indésirables par l'intermédiaire du site Web de MedEffet (DPSC, 2011a); et a eu recours aux services d'Agrément Canada, l'organisme responsable d'agréer les hôpitaux canadiens et d'autres établissements de santé, pour établir des normes nationales relatives aux rapports sur les réactions indésirables dans le cadre du système d'accréditation actuel.
En janvier 2013, Agrément Canada a publié des lignes directrices concernant les rapports sur les réactions indésirables dans ses plus récentes normes sur la gestion des médicaments à l'intention des établissements de santé. En vue d'appuyer les nouvelles lignes directrices, Santé Canada envisage d'entreprendre des activités de marketing destinées aux professionnels de la santé dans le but de fournir des renseignements sur la façon et l'importance de présenter des rapports sur les réactions indésirables, et le moyen de rester au fait des nouvelles informations en matière d'innocuité (Santé Canada, 2013i). Les activités d'orientation et de marketing permettront peut-être d'aborder l'une des principales raisons soulevées par les informateurs clés externes de la sous-déclaration des professionnels de la santé, notamment le manque de clarté concernant les exigences à cet égard. Les intervenants ont relevé de nombreux exemples de problèmes auxquels ils ont dû faire face au moment de décider de présenter, ou non, un rapport sur un événement observé. Par exemple, à qui revient la responsabilité de présenter le rapport dans les cas où le patient est suivi par plusieurs médecins; faut-il signaler une réaction prévue ou attribuable, en totalité ou en partie, à l'usage inapproprié d'un produit; quel doit être le degré de gravité de l'événement pour qu'il mérite un rapport; et comment, ou faut-il, déclarer des événements indésirables liés à des produits présentant un faible risque. D'autres raisons de la sous-déclaration, selon les informateurs clés externes, sont les contraintes de temps, la non-convivialité du processus de présentation de rapports, les préoccupations relatives aux conséquences négatives potentielles et le manque de rétroaction de Santé Canada à savoir si les renseignements fournis seront utilisés, et de quelle façon.
Les recommandations formulées par les informateurs clés externes quant à l'augmentation des rapports volontaires visaient à intensifier les efforts de marketing, à inciter les associations de professionnels et de l'industrie à encourager leurs membres à présenter des rapports, à simplifier le processus de présentation de rapports, à fournir une rétroaction aux auteurs des rapports, à examiner les approches employées dans d'autres pays, et à rembourser aux praticiens les coûts assumés pour avoir présenté un rapport et mis en œuvre les rapports obligatoires. En ce qui concerne le dernier point, lors de la publication des nouvelles normes, Santé Canada a manifesté son intention de surveiller l'incidence de ces nouvelles normes avant de décider de procéder à des modifications réglementaires (Toronto Star, 2013a). Il est important de mentionner que, pour le moment, les établissements et les fournisseurs de soins de santé des États-Unis et de l'Australie ne sont pas obligés de présenter des rapports sur les réactions indésirables (FDA, 2012a; TGA, 2012). Au sein de l'UE, les exigences relatives à la présentation de rapports à l'intention des professionnels de la santé varient; certains états (comme le Danemark, la France, l'Autriche et l'Italie) obligent les professionnels de la santé à présenter des rapports sur les réactions indésirables graves aux drogues, alors que d'autres (dont le Royaume-Uni, l'Irlande, les Pays-Bas et la Roumanie) comptent sur les rapports volontaires des professionnels de la santé (Srba, Descikova et Vlcek, 2012, p. 1061).
Comparativement aux rapports volontaires, les rapports obligatoires sur les réactions indésirables par les représentants de l'industrie ne semblent pas, dans la majorité des cas, poser problème; dans l'ensemble, les informateurs clés externes et les répondants au sondage mené auprès des représentants de l'industrie reconnaissent que Santé Canada a clairement défini les exigences concernant les rapports obligatoires et que les représentants de l'industrie comprennent et respectent ces exigences.
Mettre en œuvre la présentation électronique de rapports sur les réactions indésirables
Afin de promouvoir la présentation électronique de rapports et d'analyses sur les réactions indésirables, y compris l'analyse de rapports étrangers, Santé Canada a entamé la mise en œuvre de la présentation électronique de rapports sur les réactions indésirables destinée aux représentants de l'industrie. La mise en œuvre a pris plus de temps que prévu en raison d'exigences techniques (Santé Canada, 2013l). Le 1er mai 2013, les représentants du programme ont signalé qu'un détenteur d'une autorisation de mise sur le marché (DAMM) s'était inscrit à la présentation électronique de rapports, et que d'autres DAMM allaient le faire au cours des prochaines années.La présentation électronique est obligatoire pour les représentants de l'industrie aux États-Unis depuis 2000 (FDA, 2012b). Elle est également possible dans l'UE.
Accroître le nombre d'analyses des rapports sur les réactions indésirables
Santé Canada examine chaque rapport sur les réactions indésirables survenues au Canada présenté à Canada Vigilance et a pour objectif de réaliser 95 % de ces examens en respectant la norme de service établie de 15 jours pour l'examen des rapports initiaux prioritaires (c.-à-d. ceux impliquant des décès ou concernant des situations mortelles).Note de bas de page 26
En outre, Santé Canada a entamé l'élaboration et la mise en œuvre de stratégies en vue de contrôler systématiquement les rapports sur les réactions indésirables, y compris la surveillance ciblée et de l'exploration des données, afin de détecter les signaux d'innocuité potentiels. La mise en œuvre de la base de données de Canada Vigilance visait à améliorer la capacité de Santé Canada à surveiller et à analyser les données sur les réactions indésirables aux drogues à l'aide d'un nouveau logiciel analytique appelé agSignals, anciennement SafetyMart, qui a d'abord été lancé en mars 2008 comme composant de la nouvelle base de données (DPSC, 2010). En raison d'un retard considérable dans la mise en œuvre du logiciel agSignals, on s'attend maintenant à ce que l'exploration des données coïncide avec la mise en place de la présentation électronique. Depuis 2010, la surveillance ciblée pour les événements médicaux ciblés en fonction de produits de santé, les événements médicaux désignés et les nouvelles substances actives a été mise en œuvre. La surveillance des populations spéciales devrait commencer en 2013. Les plans concernant la surveillance ciblée des cas où l'on observe une issue fatale ont été remplacés par une nouvelle directive de travail axée sur les risques. Celle-ci s'applique à l'évaluation des rapports sur les réactions indésirables soumis par voie électronique à la base de données de Canada Vigilance.
Comme la surveillance ciblée et l'exploration des données sont des initiatives relativement récentes et encore en élaboration, on ne sait pas si Santé Canada a beaucoup abordé l'analyse des rapports sur les réactions indésirables au cours de la période visée par l'évaluation. Dans un cas comme dans l'autre, en attendant que Santé Canada mette les systèmes d'information en place pour recevoir les rapports par voie électronique et surveiller et analyser systématiquement ces rapports afin de détecter les signaux d'innocuité potentiels, les initiatives visant à accroître le nombre de rapports reçus sont peut-être prématurées.
Un sous-ensemble des données tirées des rapports sur les réactions indésirables est accessible au public par l'intermédiaire de la base de données en ligne de Canada Vigilance, laquelle est mise à jour tous les trimestres. Santé Canada a relevé de nombreuses mises en garde entourant l'utilisation de ces données (Santé Canada, 2012f). Dans une étude réalisée en 2008 par le Comité permanent de la santé, on a critiqué l'accès limité à la base de données et le manque de transparence concernant l'analyse fondée sur les rapports de la base de données (CPS, 2008).Note de bas de page 27
Accroître la détection et l'évaluation des signaux
En plus de promouvoir la présentation de rapports et d'analyses sur les réactions indésirables, Santé Canada a également cherché, dans les dernières années, à stimuler la détection et l'évaluation des signaux d'innocuité en augmentant le nombre de sources d'information qu'il surveille dans le but de détecter les signaux potentiels et en développant des PON afin d'orienter le processus. Selon Santé Canada, un signal d'innocuité est une « nouvelle association potentiellement causale ou un nouvel aspect d'une association connue entre une intervention et un événement ou un ensemble d'événements reliés, indésirables ou bénéfiques, dont la probabilité est estimée assez élevée pour justifier des mesures de vérification supplémentaires » (DPSC, 2012e).
Les sources surveillées en vue de détecter les signaux d'innocuité potentiels sont les ouvrages scientifiques, les communications des médias, les communications publiques provenant d'autres organismes de réglementation, les renseignements sur l'innocuité obtenus auprès des DAMM (comme les rapports sur les réactions indésirables et les rapports périodiques de pharmacovigilance), l'information sur les réactions indésirables volontairement soumise au Programme Canada Vigilance ou d'autres bases de données sur les réactions indésirables, et les données fournies par les bureaux de Santé Canada responsables de la mise en marché (DPSC, 2013). Au sein de la DPSC, trois bureaux ont la tâche de surveiller ces sources afin de détecter les signaux potentiels, dont deux ont des responsabilités relatives aux médicaments à usage humain, comme l'indique Tableau 3. Le Bureau des produits pharmaceutiques et des instruments médicaux commercialisés surveille les rapports sur les réactions indésirables, alors que le Bureau des initiatives stratégiques et de la planification s'occupe de contrôler toutes les autres sources d'information afin de détecter des signaux d'innocuité potentiels liés aux médicaments à usage humain (et aux instruments médicaux).
| Bureau | Responsabilité |
|---|---|
| Bureau des produits pharmaceutiques et des instruments médicaux commercialisés | Surveille les rapports spontanés sur les réactions indésirables présentés au Programme Canada Vigilance à l'aide de l'analyse des études de cas et de série de cas, de la surveillance ciblée et de l'exploration des données. |
| Bureau des initiatives stratégiques et de la planification | Quatre groupes de travail spécialisés au sein de ce bureau sont responsables de détecter les signaux pharmaceutiques provenant d'organismes de réglementation étrangers, de DAMM, de la littérature médicale et scientifique, et des renseignements sur l'innocuité obtenus auprès de la DPT, respectivement. |
| Bureau des produits biologiques, biotechnologiques et de santé naturels commercialisés | Le groupe de travail sur la coordination et la détection des signaux de ce bureau a la responsabilité de détecter les signaux potentiels liés aux produits biologiques en examinant toutes les sources d'information. |
Lorsqu'un signal potentiel détecté, celui-ci fait l'objet d'une évaluation préliminaire et est ensuite présenté au comité de priorisation des signaux aux fins de discussion. Le comité est responsable de la classification des signaux potentiels selon l'une des trois façons suivantes :
- Un signal détecté : si des renseignements portent à croire qu'un lien existe entre le produit et l'événement, et que la gestion des risques est possible;
- Un problème potentiel relativement à un produit en attente de renseignements supplémentaires : s'il n'y a pas assez d'information pour appuyer le lien en ce moment, mais que d'autres renseignements pourraient être disponibles dans un délai de 18 mois et permettre la gestion des risques;
- Un signal rejeté : si les renseignements qui appuient le lien entre le produit et l'événement ne sont pas disponibles en ce moment et ont peu de chances de l'être bientôt, ou si la gestion des risques est impossible peu importe les résultats de l'enquête (DPSC, 2012e).
Les signaux détectés sont confiés aux évaluateurs qui procèdent alors à une évaluation critique exhaustive des données disponibles, analysent les options de gestion des risques possibles et recommandent des mesures à prendre (DPSC, 2012f). On peut, par exemple, recommander de mettre en place une surveillance normalisée ou accrue; de demander des renseignements supplémentaires au DAMM; d'exiger que le DAMM effectue une évaluation des risques et des avantages, développe un plan de pharmacovigilance ou élabore un plan de gestion des risques; de proposer des modifications à l'étiquetage du produit; de conseiller la communication du risque; de demander un examen du Réseau sur l'innocuité et l'efficacité des médicaments (RIEM); et de suggérer une analyse détaillée des
Au cours des dernières années, de nombreuses PON ont été créées en vue de soutenir la détection, la priorisation et l'évaluation des signaux, et d'autres sont en élaboration.
En s'appuyant sur l'analyse des données de gestion fournies par Santé Canada, il est possible de formuler les observations suivantes à propos des activités de détection et d'évaluation de la DPSC :
- Entre 2006 et 2012, parmi les 1 980 signaux détectés liés aux médicaments à usage humains, environ 81 % ont été rejetés et 7 % ont été évalués en priorité. En revanche, environ 97 % des 6 505 signaux détectés liés aux produits biologiques ont été rejetés, et moins de 1 % a été évalué en priorité.Note de bas de page 28Au cours des dernières années, même si la DPSC a développé une approche normalisée pour la détection et l'analyse des signaux, les écarts entre le nombre de signaux détectés concernant les produits pharmaceutiques et les produits biologiques, ainsi que les écarts entre ce nombre et la proportion de signaux rejetés, peuvent sous-entendre que la DPSC a néanmoins utilisé des processus différents ou appliqué des critères distincts pour la détection des signaux liés aux produits pharmaceutiques et aux produits biologiques.
- Un total de 272 évaluations des signaux liés aux médicaments à usage humain ont été réalisées entre 2003 et 2012. Les sources des signaux d'innocuité les plus souvent consignées étaient un organisme de réglementation international (26 %), dans la majorité des cas la FDA (14 %); des ouvrages scientifiques (26 %); et la base de données de Canada Vigilance (8,5 %).
- Les recommandations les plus souvent formulées à la suite des évaluations des signaux proposaient de modifier l'étiquetage du produit (54 %); de mettre en place une surveillance normalisée (36 %); et d'exiger la communication du risque (24 %).
Veuillez consulter l'annexe C pour obtenir des renseignements détaillés.
Différentes limites des données ont compliqué l'analyse ci-dessus. Par exemple, le suivi des activités de détection et d'évaluation est effectué au moyen d'une série de feuilles de calcul Excel tenues à jour par différents groupes au sein de la DPSC, plutôt que par l'intermédiaire d'une base de données intégrée ou centralisée à la disposition de toutes les parties concernées. Il n'y a aucune façon de relier facilement les enregistrements contenus sur ces feuilles de calcul, car des identifiants uniques sont attribués aux évaluations des signaux, mais pas aux activités de détection des signaux; par conséquent, le lien entre la détection des signaux et les enregistrements des évaluations doit être fait manuellement. De plus, on constate de nombreuses incohérences dans les façons employées pour consigner les données, et des données sont manquantes, ce qui a une incidence sur la fiabilité et la validité des données. L'utilisation généralisée de zones de texte illimitées pour la saisie de données oblige le recours à un codage chronophage afin de permettre l'analyse ou l'évaluation quantitative.
En outre, aucun outil ou mécanisme centralisé n'a été mis en place afin de faire le suivi systématique de la réponse de Santé Canada aux mesures recommandées résultant des évaluations des signaux effectuées. En 2011, le BVG a conseillé la mise en œuvre d'un système semblable, ce à quoi Santé Canada a répondu que des PON relatives à de nouveaux systèmes de suivi normalisés étaient en élaboration, et que celles-ci seraient réalisées avant la fin de mars 2013 (Santé Canada, 2011d, p. 27). En mars 2013, Santé Canada a signalé que l'outil de suivi des recommandations en matière d'innocuité concernant tous les produits pharmaceutiques avait été achevé et que des procédures opérationnelles avaient été mises en place (Santé Canada, 2013l). L'outil est propre à la DPT et se limite aux mesures qui relèvent de son domaine de responsabilité (c.-à-d. les modifications apportées à l'étiquetage d'un produit). On ne sait pas si un système semblable a été mis en œuvre en vue d'assurer le suivi de la réponse de Santé Canada aux mesures recommandées qui relèvent du domaine de compétence d'autres directions ou de celles portant sur les signaux liés à des produits biologiques.
Mettre en œuvre la gestion des risques et la planification de la pharmacovigilance
La planification de la gestion des risques consiste à détailler les risques potentiels que présente un produit de santé et les mesures à prendre en vue de réduire la possibilité de dommages (Santé Canada, 2009b), alors que la planification de la pharmacovigilance fait référence à l'élaboration de la pré-autorisation d'un processus de surveillance dans le but de recueillir des renseignements de façon continue une fois qu'un médicament est mis sur le marché. Dans le cadre du PASPAC, Santé Canada avait l'intention de mettre en œuvre une approche structurée, exhaustive et systématique relativement à la planification de la pharmacovigilance et, pour compléter les plans de pharmacovigilance (PPV), de créer un nouveau programme demandant aux représentants de l'industrie de fournir volontairement des PGR. Santé Canada souhaitait éventuellement proposer des modifications législatives et réglementaires visant à obliger les fabricants à présenter des PGR et des PPV régulièrement dans le cadre du processus préalable à la mise en marché.
Exigences relatives aux PGR au Canada
Composantes obligatoires :
- La description du profil d'innocuité, qui est un résumé des informations importantes sur l'innocuité du produit, y compris les écarts des savoirs;
- Le plan de pharmacovigilance, qui inclut les problèmes d'innocuité connus et potentiels et les méthodes employées par le fabricant pour examiner les nouveaux renseignements sur l'innocuité;
- Un plan de minimisation des risques, qui propose des façons de réduire au minimum les risques connus et potentiels concernant l'innocuité.
Présentation d'un PGR :
- Comme élément d'une demande de licence de mise en marché;
- À la demande de Santé Canada dans tous les cas où le ministère la juge pertinente pour la prise de décisions sur les risques et les avantages, comme une nouvelle substance active, une modification de l'indication ou l'ajout d'un produit dans une catégorie qui présentait déjà un risque en matière d'innocuité
Comme ces plans ont été annoncés, la planification de la pharmacovigilance a été incluse dans le concept général de la planification de la gestion des risques. En 2009, Santé Canada a adopté le thème E2E de la CIH sur la planification de la pharmacovigilance et a mis en place des exigences provisoires concernant les PGR fondées sur les lignes directrices de l'Agence européenne des médicaments (EMA).
Les données fournies par Santé Canada indiquent que, de façon générale, le nombre de PGR augmente depuis 2005 pour les médicaments à usage humain et les produits biologiques, passant d'un total de 2 en 2005 à 75 en 2012. En 2012, 49 PGR étaient associés à la DPT. On ne sait pas si ces données représentent le nombre de PGR demandés, reçus ou examinés par Santé Canada. Veuillez consulter l'annexe C pour obtenir d'autres renseignements.
Santé Canada a récemment annoncé qu'il exigerait des PGR pour les versions génériques de l'oxycodone qu'il a approuvées en novembre 2012. On ne sait pas si on demande régulièrement des PGR pour tout autre type de médicament pharmaceutique.Note de bas de page 29À l'heure actuelle, Santé Canada n'a pas l'autorité réglementaire pour obliger la présentation de PGR, et l'état d'avancement de ses plans visant à proposer des modifications qui lui accorderaient une telle autorité reste inconnu.
L'EMA et la FDA ont toutes les deux l'autorisation légale d'obliger des présentations et d'imposer des amendes en cas de non-conformité (DPSC, 2009). Depuis 2007, la FDA a l'autorité d'obliger les promoteurs à soumettre une stratégie d'évaluation et d'atténuation des risques (SEAR) afin de s'assurer qu'un médicament présente plus d'avantages que de risques (gouvernement des États-Unis, 2007), et ce, pour les nouveaux médicaments et les médicaments déjà sur le marché, selon des critères précis. Dans l'UE, depuis l'adoption de la nouvelle législation en matière de pharmacovigilance en juillet 2012, les PGR sont obligatoires pour toutes les demandes d'autorisation de mise sur le marché; ils sont aussi habituellement requis lorsqu'une modification importante est apportée à l'autorisation, comme le dosage, la voie d'administration ou l'indication. Un PGR peut également être exigé par un organisme de réglementation chaque fois que l'on craint qu'un risque nuise à l'équilibre entre les avantages et les risques d'un médicament, ou présenté volontairement par le fabricant relativement aux nouveaux problèmes d'innocuité relevés. La législation de l'UE demande également à ce qu'un résumé du PGR soit rendu public (EMEA, 2013a; DPSC, 2009).
Accroître la présentation de rapports d'innocuité après la mise en marché par les fabricants
Le Rapport périodique de pharmacovigilance (RPPV) est un rapport normalisé à l'échelle internationale qu'un fabricant présente à un organisme de réglementation quant à l'innocuité mondiale d'un produit de santé commercialisé (DPSC, 2012d). Au Canada, on s'attend à ce que les RPPV couvrent l'information par intervalles de six mois. Ils peuvent être présentés volontairement par les fabricants, ou Santé Canada peut en exiger un si un problème d'innocuité a été relevé (DPSC, 2012d). La présentation d'un RPPV peut également être requise à la suite d'un AC-C ou d'un autre engagement suivant l'autorisation de mise sur le marché, ou être négociée pendant le processus d'autorisation. Sinon, les RPPV ne sont pas obligatoires en vertu de la réglementation actuelle.
Contrairement aux PGR, dont le nombre augmente tranquillement, il n'y a aucune tendance précise concernant le nombre de RPPV. Les données fournies par Santé Canada montrent que depuis 2010, le nombre de RPPV a diminué, passant d'un total de 72 à 28 en 2012. Les RPPV liés à la DPT ont chuté de 48 à 19 au cours de cette période. Comme pour les PGR, on ne sait pas si ces données représentent le nombre de RPPV demandés, reçus ou examinés par Santé Canada. Veuillez consulter l'annexe C pour obtenir d'autres renseignements.
Dans le cadre du PASPAC, Santé Canada a planifié renforcer son programme actuel de collecte et d'examen des RPPV et a proposé des modifications à la Loi sur les aliments et drogues en vue d'obtenir l'autorité législative pour obliger les fabricants à présenter des RPPV. Le ministère a mis en place deux niveaux d'examen des RPPV et a établi des normes de rendement en matière d'examen. Récemment, il a annoncé qu'il progressait vers la mise en application de la directive E2C (R2) de la CIH et du Rapport périodique d'évaluation des avantages et des risques (RPEAR) « afin de s'aligner sur les meilleures pratiques internationales et de réduire le fardeau de l'industrie en [permettant aux fabricants] de présenter soit [un RPPV ou un RPEAR] pour satisfaire aux exigences réglementaires applicables au Canada » (Santé Canada, 2013j).
Les « exigences réglementaires applicables » en question sont celles de l'article C.01.018 du Règlement sur les aliments et drogues qui, depuis 2011, oblige les DAMM à analyser les données portant sur les réactions indésirables aux drogues et à préparer un rapport de synthèse annuel, lequel doit être fourni à la demande de Santé Canada. Par conséquent, le RPEAR peut maintenant être présenté à titre de rapport de synthèse annuel.
Dans l'UE, la présentation de RPPV est obligatoire depuis 2001 pour tous les produits autorisés (directive du Conseil, 2001, p. 96), bien que la nouvelle législation en matière de pharmacovigilance abandonne l'obligation de présenter des RPPV régulièrement pour des produits médicaux génériques et des produits médicaux bien établis (EMEA, 2013d, p. 7). Dans le même ordre d'idées, depuis au moins 1996, la FDA a le pouvoir d'obliger les demandeurs à fournir des rapports réguliers concernant l'innocuité après la mise en marché pour chaque demande approuvée, et ce, tous les trimestres au cours des trois premières années suivant la date d'approbation des États-Unis, et tous les ans par la suite (gouvernement des États-Unis, 1996, par. 314.80(2)).Note de bas de page 30
En plus d'exiger la présentation de rapports périodiques concernant l'innocuité, l'EMA et la FDA, contrairement à Santé Canada, bénéficient de l'autorité législative leur permettant d'obliger les DAMM à préparer et à fournir des études d'innocuité après la mise en marché (EMEA, 2013b; FDA, 2011a).
Mettre en œuvre des normes de service pour les activités suivant la mise en marché
Dans un effort visant la mise en place d'une approche normalisée en matière de surveillance post-commercialisation, la DPSC a établi des cibles de rendement pour certaines de ses activités de surveillance post-commercialisation. En ce qui concerne les rapports sur les réactions indésirables, la DPSC s'est donnée comme objectif de respecter la norme dans 95 % des cas; pour ce qui est des cibles liées à d'autres activités suivant la mise en marché, elle souhaite être en mesure de respecter la norme dans 90 % des cas. Les cibles sont actuellement en élaboration aux fins d'évaluations de la causalité, et de rédaction et de diffusion du contenu des communications des risques (DPSC, 2012c). Veuillez consulter l'annexe C pour obtenir d'autres renseignements.
En avril 2007, la DPSC a adopté des normes de rendement non officielles concernant les évaluations des signaux fondées sur le nombre de jours ouvrables entre l'affectation de l'évaluation du signal à un évaluateur et son approbation. La norme de service variait selon le niveau de priorité attribué au signal : 200 jours ouvrables pour les évaluations peu prioritaires; 130 jours ouvrables pour les évaluations moyennement prioritaires; et 80 jours ouvrables pour les évaluations hautement prioritaires. Ces normes de service sont devenues officielles en novembre 2011, mais ont été ajustées en avril 2012 en vue d'établir un délai unique de 130 jours. Les représentants du programme ont affirmé que ces changements ont été apportés à la suite de la décision de la DPSC prise en janvier 2012 d'arrêter d'attribuer des niveaux de priorité aux signaux; les raisons de cette décision sont toutefois nébuleuses. Une analyse de la rapidité des activités de Santé Canada suivant la mise en marché se trouve à la section 6.7.
Accroître le nombre de partenariats et l'utilisation des bases de données existantes
Enfin, Santé Canada privilégie une surveillance post-commercialisation active en s'associant à des organisations externes et en utilisant les bases de données existantes. Par exemple, dans le cadre du PASPAC, le ministère a entrepris le développement du Réseau sur l'innocuité et l'efficacité des médicaments (RIEM). Le RIEM est un réseau virtuel reliant des centres d'excellence partout au pays dirigé par quelques principaux centres dotés d'un leadership distinctif dans le domaine des compétences et des méthodologies de recherche (Risk Sciences International, 2012). On s'attend à ce que le RIEM améliore la capacité du Canada à s'occuper des problèmes urgents en matière de recherche dès leur apparition et à coordonner à l'échelle nationale la surveillance post-commercialisation dans le but ultime d'éclairer la prise de décisions réglementaires.
La DPSC se penche également sur la possibilité d'utiliser les bases de données existantes et les dossiers électroniques de santé (p. ex., bases de données provinciales, Institut canadien d'information sur la santé, Statistique Canada) comme sources potentielles pour réaliser des études en appui à la surveillance et à la détection des signaux (DPSC, 2011c). Depuis 1996, la DPSC collabore avec la Société canadienne de pédiatrie au Programme canadien de surveillance pédiatrique (PCSP), qui recueille, tous les mois, des données auprès des pédiatres et des pédiatres avec surspécialité, afin de surveiller les maladies rares chez les enfants canadiens (Société canadienne de pédiatrie, sans date). Le PCSP est passé de l'étude de trois maladies, au cours de l'année pilote, à celle de 50 pathologies à l'heure actuelle (Société canadienne de pédiatrie, sans date).
Le système-sentinelle de la FDA des États-Unis est l'un des modèles de surveillance post-commercialisation active s'appuyant sur les bases de données existantes et les dossiers électroniques de santé. Lancé en 2008, le système-sentinelle a été revendiqué par des modifications législatives apportées à la Food and Drug Administration Amendments Act (art. 905), qui exigeaient une analyse et une surveillance post-commercialisation active de l'innocuité (FDA, 2008). Le système est actuellement opérationnel à titre de projet pilote appelé « Mini-Sentinel » et concerne plus de 20 partenaires. Les éléments clés du projet portent, entre autres, sur la surveillance active de l'activité suivant la mise en marché au moyen d'une évaluation régulière des données de santé électroniques recueillies en réponse aux préoccupations de la FDA; l'évaluation des changements apportés à l'utilisation des produits médicaux en réponse à la mesure réglementaire de la FDA; l'utilisation d'un réseau de données distribuées dans lequel les partenaires de données gardent le contrôle des données, mais utilisent des programmes informatiques normalisés au sein de leurs institutions et échangent des résultats cumulatifs; et le contrôle de la qualité (p. ex., tester et améliorer les méthodes statistiques) (Mini-Sentinel, sans date).
Plusieurs informateurs clés externes ont encouragé Santé Canada à coordonner la collecte de données liées à la pharmacovigilance provenant d'autres ministères fédéraux, ainsi que des gouvernements provinciaux et d'autres organismes, comme les centres antipoison. Ils ont également favorisé l'échange des données obtenues après la mise en marché avec les organismes de réglementation des autres provinces dans le but de relever rapidement les problèmes d'innocuité, car une population nombreuse sera surveillée. Ainsi, les organismes de réglementation pourront répondre promptement aux risques liés à l'innocuité et à la santé.
5.4.5 Conformité et application
Même si on pense souvent que la conformité et l'application sont les dernières étapes du processus de réglementation des médicaments, Santé Canada a la responsabilité de surveiller la conformité et de faire respecter le cadre de réglementation des médicaments à usage humain aux étapes précédant et suivant la mise en marché. Pour ce faire, l'Inspectorat effectue des inspections de la surveillance afin d'assurer la conformité des fabricants aux exigences réglementaires relatives aux BPC, aux BPF et aux BPV, et de veiller à ce que les substances contrôlées respectent elles aussi les exigences réglementaires. Santé Canada réalise également des vérifications de la conformité en réponse aux plaintes particulières ou aux risques relevés. En cas de non-conformité, l'Inspectorat précisera ce qui doit être fait pour atteindre l'objectif de conformité et prendra peut-être des mesures d'application de la loi si la partie réglementée ne procède pas volontairement à des mesures de conformité (veuillez consulter la liste ci-dessous pour un sommaire des mesures volontaires et d'application disponibles).
Mesures de conformité et d'application disponibles
Mesures volontaires
- Consentement à la confiscation
- Rétention du produit
- Élimination du produit
- Arrêt de la vente
- Rappel
Mesures réglementaires/d'application
- Activités douanières
- Injonction
- Poursuite judiciaire
- Confiscation après saisie
- Mise en garde ou avis au public
- Lettres destinées au secteur et aux parties réglementées
- Arrêt réglementaire de la vente
- Perquisition et saisie
- Saisie et rétention
- Suspension ou annulation de l'autorisation de commercialisation ou de la licence de produit
- Refus, suspension ou modification de la licence d'établissement
- Lettre de mise en garde
Le renforcement des outils et des approches en matière de conformité dans le but de stimuler la conformité de l'industrie au cadre de réglementation relatif aux médicaments à usage humain (pharmaceutiques et biologiques) était l'un des principaux objectifs de Santé Canada au cours de la période visée par l'évaluation. Les activités réalisées dans ce domaine sont la mise en œuvre de nouveaux pouvoirs et outils en matière de conformité et d'application, le renforcement des inspections des essais cliniques et l'adoption d'une approche axée sur les risques relativement aux inspections des BPF, entre autres. Les améliorations prévues aux pouvoirs de Santé Canada en matière de conformité et d'application ne se sont pas concrétisées, car les modifications législatives n'ont pas eu lieu.
Mettre en œuvre de nouveaux pouvoirs et outils en matière de conformité et d'application
Le Plan de renouveau et le PASPAC décrivent l'intention de Santé Canada de chercher à obtenir de nouvelles autorités réglementaires et législatives en matière de conformité et d'application, comme le pouvoir d'ordonner des mesures correctives et la modernisation du cadre actuel concernant les sanctions et les amendes (DGPSA, 2007a). Le projet de loi C-51 incluait de nouvelles mesures d'administration et d'application, notamment le rappel obligatoire de produits thérapeutiques et le pouvoir fédéral de retirer les stocks de médicaments restants suivant le retrait de ces produits du marché en raison de problèmes d'innocuité; Il englobait également un cadre modernisé relatif aux sanctions et aux amendes pécuniaires en augmentant considérablement les amendes en cas de non-conformité. Comme il a été mentionné précédemment, le projet de loi C-51 n'est pas devenu une loi.
Santé Canada ne bénéficie pas encore de ces pouvoirs, et on ne sait pas s'il a toujours l'intention d'essayer de les obtenir. On ne sait pas non plus si les plans visant à développer des outils non législatifs en cas de non-conformité (les documents de l'Inspectorat indiquent un « programme de contraventions » en cas de non-conformité, par exemple) se sont concrétisés. Dans les entrevues, certains informateurs clés se sont montrés préoccupés par ce qu'ils considèrent comme le peu d'options dont dispose Santé Canada en matière d'application, et ont mentionné que l'adoption d'une approche axée sur les risques relativement à la conformité et à l'application signifie que le ministère sera peut-être moins susceptible de répondre aux problèmes qui, selon lui, ne présentent qu'un faible risque pour les consommateurs. En outre, même si les approches axées sur les risques en matière de conformité et d'application sont appropriées dans un contexte de ressources limitées, elles ne permettent pas nécessairement de bien comprendre la conformité au sein de l'industrie réglementée et n'offrent pas toujours les outils nécessaires pour déterminer les nouveaux risques (Sparrow, 2000, p. 290).
Renforcer les inspections des essais cliniques
En 2012, Santé Canada a lancé le programme d'inspection des essais cliniques visant deux objectifs : protéger la sécurité des participants aux essais et vérifier la qualité des données des essais cliniques. Chaque année, dans le cadre de ce programme, jusqu'à 2 % de tous les essais cliniques canadiens (y compris les médicaments biologiques et à usage humain) sont sélectionnés aux fins d'inspection. Santé Canada estime qu'environ 4 000 essais cliniques sont effectués dans une année au Canada; étant donné la cible de 2 %, cela représente environ 80 inspections par année. La sélection des sites s'appuie sur des critères de risque, notamment le nombre d'essais cliniques réalisés sur le site; le nombre de sujets concernés; le nombre de réactions indésirables graves et imprévues aux drogues sur le site; et les observations formulées lors des inspections précédentes. Deux évaluations sont possibles : conforme ou non conforme.
Au cours des dernières années, le BVG et le Comité permanent ont relevé des lacunes dans l'approche de Santé Canada en matière d'inspections des essais cliniques. Par exemple, le ministère a omis de recueillir régulièrement tous les renseignements nécessaires en vue d'évaluer les critères de risque concernant la sélection des sites, de respecter la cible de 2 % relative aux inspections et de fixer des échéances pour informer les promoteurs en cas de non-conformité (BVG, 2011; CPASST, 2012). En réponse à ces affirmations, Santé Canada s'est engagé à publier des rapports annuels sur ses inspections des essais cliniques, a révisé ses procédures d'inspection afin d'inclure des échéances aux principales étapes du processus d'inspection, ce qui comprend les avis de non-conformité et l'examen des mesures correctives proposées, et a élaboré un processus axé sur les risques pour sélectionner les sites d'essai clinique à inspecter. Parmi les critères axés sur les risques utilisés pour choisir les sites à inspecter, on retrouve, entre autres, la phase dans le processus de développement de médicament, la complexité de la conception de l'essai, notamment sa population, le niveau de risques pour les Canadiens, les nouvelles thérapies/formes posologiques, les rapports importants ou fréquents sur les événements indésirables, les avis des promoteurs concernant les déviations du protocole, et les nouveaux thèmes ou nouvelles tendances (Inspectorat, 2013). Selon Santé Canada, on peut également avoir recours à d'autres éléments, comme les meilleures pratiques internationales.
Santé Canada a mentionné qu'une formation sur le nouveau processus a été donnée à l'équipe des BPC de l'Inspectorat en mai 2013 et aux évaluateurs de la DPT en octobre 2013. Le nouveau processus a été mis en œuvre le 1er juin 2013 au moyen d'une approche progressive utilisant l'ancien et le nouveau système de sélection. Une fois bien mis en œuvre et documenté, le processus axé sur les risques relatifs à la sélection des sites permettra peut-être d'aborder les préoccupations exprimées lors des entrevues concernant le manque de transparence en ce qui a trait à la façon de sélectionner les sites d'essai clinique aux fins d'inspection.
Quelques informateurs clés externes se sont dits préoccupés par le fait que le programme d'inspection met l'accent sur des facteurs qui, selon eux, n'ont rien à voir avec la sécurité des participants aux essais ou la qualité de la recherche, ou qui font déjà l'objet d'une surveillance par d'autres sources.
Augmenter le nombre de licences d'établissement de produits pharmaceutiques et d'inspections des BPF
Les établissements qui fabriquent, emballent/étiquettent, analysent, importent, distribuent et vendent en gros des médicaments doivent obtenir une licence d'établissement de produits pharmaceutiques (LEPP) en vue d'exercer leurs activités légalement. Afin de recevoir une LEPP, les établissements doivent subir une évaluation vérifiant la conformité aux titres 2 à 4 de l'article C du Règlement sur les aliments et drogues, lesquels font référence aux Bonnes pratiques de fabrication (BPF). Cela inclut des exigences sur les locaux, l'équipement, le personnel, l'hygiène, l'analyse des matières premières, le contrôle de la fabrication et de la qualité, l'analyse du matériel d'emballage et du produit fini, la tenue de dossiers, la conservation des échantillons, la manipulation des produits stériles et la stabilité des produits (GC, 2012c). Dans les établissements nationaux, les inspections des BPF ont lieu tous les 24 mois pour les fabricants, les emballeurs/étiqueteurs et les laboratoires d'essai, et tous les 36 mois pour les importateurs, les distributeurs et les grossistes. Les nouveaux établissements qui font une demande de LEPP doivent être prêts à subir une inspection lorsqu'ils présentent leur demande, car l'Inspectorat tente d'effectuer l'inspection initiale dans les trois mois suivant la réception de la demande. Une inspection régulière est réalisée dans un délai de 12 mois suivant l'inspection initiale.
Les établissements nationaux se voient attribuer soit une cote conforme (C) ou non conforme (NC) selon les risques ayant donné lieu à des observations notées lors d'une inspection (Inspectorat, 2012f). Il existe trois niveaux de risque, le risque 1 étant le plus élevé et le risque 3, le plus faible. Si une ou plusieurs observations comportent des risques de classe 1, le rapport global de l'inspection pourra indiquer que l'établissement est NC.
Jusqu'à tout récemment, les LEPP étaient requises relativement aux médicaments sous forme posologique et aux médicaments sous forme de produits intermédiaires en vrac visés à l'annexe C (produits radiopharmaceutiques) et à l'annexe D (produits biologiques) (Inspectorat, 2012e). En mai 2013, Santé Canada a donné suite à un engagement de longue date visant à élargir les exigences relatives aux BPF et aux LEPP en vue d'englober les IPA (Santé Canada, 2013e). La nouvelle réglementation, qui entrera en vigueur en novembre 2013, harmonise les exigences de Santé Canada avec celles de ses homologues aux États-Unis, dans l'UE, en Australie et au Japon, où des exigences relatives aux BPF des IPA sont en place depuis environ dix ans (CIH, 2013).Note de bas de page 31
Conformité des sites nationaux aux BPF
Au cours de la période visée par l'évaluation, Santé Canada a revu son programme d'inspection des BPF nationales dans le but de réaliser des gains d'efficacité qui permettraient d'atteindre la stabilité et d'éviter les arriérés, lesquels ont posé d'importants problèmes pendant plusieurs années (Inspectorat, 2007a). Les recommandations formulées à partir de l'examen portaient sur la direction d'inspections axées sur les risques, d'inspections d'établissements ayant plusieurs sites et d'« inspections d'appoint ciblées » (Inspectorat, 2012b, p. 6). Comme l'examen était terminé, Santé Canada a éliminé les inspections de renouvellement qui avaient normalement lieu lorsque la licence annuelle d'un établissement expirait. Ce changement devrait permettre aux inspecteurs de se concentrer davantage sur les établissements de produits pharmaceutiques à risque élevé (BVG, 2011, p. 29). Les autres mesures prises en vue de concrétiser les recommandations formulées à partir de l'examen demeurent inconnues. Le passage aux inspections axées sur les risques est conforme à l'approche adoptée par la FDA.Note de bas de page 32
Des événements récents ont attiré l'attention sur le programme d'inspection des BPF de Santé Canada.
- En 2011, la FDA a envoyé une lettre de mise en garde à l'usine du Québec de Sandoz Canada après que trois inspections effectuées cette même année ont révélé que l'entreprise ne respectait pas les procédures appropriées « visant à prévenir la contamination microbiologique des produits pharmaceutiques soi-disant stériles », et en raison de pratiques incomplètes ou inexactes en matière de documentation et d'enquête (FDA, 2011b; Globe and Mail, 2012a). Les répercussions ont été considérables. Sandoz a réduit la production de certains médicaments (principalement des analgésiques, des antibiotiques et des anesthésiques) dans le but de perfectionner les opérations, ce qui a entraîné des pénuries de médicaments et l'annulation d'opérations chirurgicales non urgentes (CBC News, 2012). Finalement, Santé Canada a annoncé des plans visant à trouver rapidement de nouvelles sources pour certains médicaments en réponse aux pénuries (CBC News, 2012).
- En février 2013, la FDA a envoyé une lettre de mise en garde à Apotex inc. après que son inspection a révélé des « violations considérables » des BPF actuelles et a menacé d'imposer une interdiction d'importation sur les produits fabriqués par Apotex dans deux installations de la région de Toronto (FDA, 2013f). Les inspecteurs canadiens n'avaient pas inspecté les installations en question depuis 2011 (Globe and Mail, 2013b). La FDA avait déjà imposé une interdiction d'importation de deux ans sur les produits d'Apotex en 2009 après que des inspections ont révélé des lacunes importantes relatives aux BPF actuelles (Globe and Mail, 2013b).
Lors d'entrevues, certains informateurs clés externes se sont montrés préoccupés par la formation donnée aux inspecteurs et par leurs qualifications, mentionnant qu'ils ont observé une interprétation irrégulière des normes et des inspections de qualité variable. Selon Santé Canada, les raisons expliquant les divergences entre les résultats du ministère et ceux de la FDA sont complexes, et sont probablement attribuables à des différences dans les approches employées par les deux organismes de réglementation pour effectuer des inspections, et à la portée de ces inspections.
Conformité des sites étrangers aux BPF
Les sites étrangers qui fabriquent des médicaments destinés à l'importation au Canada par des fabricants canadiens sont, comme les sites nationaux, assujettis à des licences d'établissement et à des exigences relatives aux BPF, mais ils sont rarement inspectés par Santé Canada. L'Inspectorat évalue plutôt la demande initiale de LEPP pour un site étranger, ainsi que la conformité du site étranger, en s'appuyant sur les données fournies par le promoteur. Les données à présenter dépendent de l'emplacement du site, à savoir s'il est situé dans un pays avec qui Santé Canada a conclu un accord de reconnaissance mutuelle (ARM), soit une entente entre le ministère et un autre pays quant à l'équivalence de leurs programmes de conformité des BPF respectifs (Santé Canada, 2009a). À l'heure actuelle, le Canada a signé des ARM avec l'UE, la Suisse, l'Islande, le Liechtenstein, la Norvège et l'Australie (Inspectorat, 2012i). Toutefois, Santé Canada estime que 76 % des produits pharmaceutiques importés au Canada proviennent de pays avec qui le ministère n'a pas conclu d'ARM; au cours des cinq dernières années, le ministère a inspecté 35 sites étrangers (Cassels, 2012). Dans le cas des IPA, Santé Canada indique que l'importation représente 85 % du marché canadien, et la Chine et l'Inde sont les principaux fournisseurs (Santé Canada, 2012c).
Certains informateurs clés externes se sont montrés préoccupés par le manque d'inspections des BPF des installations étrangères dans des pays avec qui le Canada n'a pas signé d'ARM, s'inquiétant de la possibilité que les Canadiens soient exposés à des produits de santé de mauvaise qualité ou de contrefaçon. Par exemple, en 2013, Santé Canada a averti les femmes canadiennes que deux contraceptifs oraux génériques avaient fait l'objet d'un rappel après qu'on a découvert des emballages dans lesquels il y avait un comprimé placebo à l'endroit où devait se trouver un comprimé actif dans des emballages; un troisième rappel a récemment été annoncé, car le fabricant « n'a pas été en mesure d'écarter la possibilité » que la pilule soit touchée par le même problème (Globe and Mail, 2013a). Deux des pilules retirées du marché avaient été fabriquées en Inde (Toronto Star, 2013b); l'autre, en Espagne par Apotex inc. L'élargissement des exigences relatives aux BPF et à la LEPP en vue d'englober les IPA devrait permettre d'aborder la possibilité que des produits de santé de mauvaise qualité ou de contrefaçon se retrouvent sur le marché canadien.
Les informateurs clés de l'industrie, quant à eux, ont affirmé que l'approche de Santé Canada à l'égard des inspections des sites étrangers impose un fardeau excessif sur les intervenants qui importent des produits de santé (p. ex., la faible fréquence des inspections oblige les importateurs à assumer la responsabilité de la conformité des établissements dans d'autres pays). Des représentants de l'industrie ont mentionné que le processus relatif aux inspections des BPF des sites étrangers serait plus simple si Santé Canada acceptait que les inspections soient effectuées par des organismes de réglementation étrangers ou des tiers vérificateurs étrangers approuvés.
Aux États-Unis, la FDA a adopté une approche de plus en plus agressive relativement à la surveillance des installations étrangères à la suite de l'incident de l'héparine falsifiée (Grosh et coll., 2013, p. 30-36). Malgré ces efforts, la littérature porte à croire que la FDA n'a pas les ressources nécessaires pour effectuer un nombre suffisant d'inspections étrangères (Liu, Zhang et Linhardt, 2009).
Une initiative récemment lancée entre le Canada et les États-Unis par l'intermédiaire du Conseil de coopération en matière de réglementation (CCR) vise à aborder les enjeux liés aux inspections des BPF en augmentant, dans chaque pays, le recours aux rapports d'inspection des BPF des installations de fabrication de médicaments préparés par l'autre pays, plutôt que de redoubler d'efforts inutilement (groupe de travail du CCR du secteur des produits de soins personnels et des produits pharmaceutiques, 2012a). Les principales activités comprises dans le plan de travail sont les évaluations et les échanges habituels de rapports d'inspection entre les pays et l'engagement régulier des intervenants au moyen de publications Web et de réunions trimestrielles. En définitive, les pays ont l'intention d'élaborer un cadre en vue d'assurer le recours aux rapports d'inspection de chacun. Ce cadre pourrait inclure une base de données commune des BPF afin d'uniformiser l'échange de rapports d'inspection; l'échange systématique de rapports d'inspection et des mesures/changements réglementaires importants; des inspections conjointes d'établissements sélectionnés; et la mise en place d'un forum pour discuter des modifications réglementaires de l'un ou l'autre des pays, et de s'y ajuster.
Malgré le fait que le plan de travail sur les BPF mette l'accent sur la normalisation et l'échange des rapports d'inspection, Santé Canada et la FDA optent actuellement pour des approches d'établissement de rapport différentes. En effet, la FDA établit des rapports en fonction du centre responsable de la réglementation de différentes catégories de produits - déclarant les données d'inspection séparément pour le Center for Veterinary Medicine (médicaments vétérinaires), le Center for Drug Evaluation and Research (médicaments à usage humain) et le Center for Biologics Evaluation and Research (produits biologiques) (FDA, 2013d) - alors que Santé Canada rassemble tous les rapports sur les BPF, peu importe la gamme de produits. Santé Canada a mentionné qu'il a choisi cette approche, car elle s'appuie sur les installations, et qu'à l'heure actuelle, il n'est pas possible d'effectuer le suivi des inspections par gamme de produits. Il est toutefois possible que l'atteinte des objectifs du plan de travail sur les BPF nécessite une approche commune en matière d'établissement de rapports de conformité.
Finalement, Santé Canada est membre du Pharmaceutical Inspection Cooperation Scheme (PIC/S), un organisme composé de 43 autorités internationales participantes qui se sont réunies en vue d'harmoniser les approches et les exigences en matière d'inspection des BPF partout dans le monde. Santé Canada examine les rapports d'inspection du PIC/S afin d'approuver les sites étrangers pour les importateurs canadiens.
Autres initiatives visant à renforcer la conformité et l'application
Finalement, Santé Canada a entrepris plusieurs autres initiatives en vue de renforcer la conformité au cadre de réglementation relatif aux médicaments à usage humain, et son l'application.
- Effectuer des inspections des rapports après la mise en marché. En 2004, Santé Canada a lancé le programme de conformité aux exigences de déclaration après commercialisation (CEDAC) dans le but de s'assurer « que les fabricants sont conformes aux exigences réglementaires relatives à la réception, à l'analyse et à la présentation d'information sur l'innocuité des médicaments à Santé Canada » (Inspectorat, 2012a, p. 2). En 2013, le programme est devenu le programme d'inspection des Bonnes pratiques de pharmacovigilance (BPV); les représentants de Santé Canada ont signalé que ce changement permet de s'harmoniser davantage avec les organismes de réglementation internationaux. En février 2013, une approche axée sur les risques a été adoptée pour la planification des inspections, ainsi qu'une nouvelle ligne directrice destinée aux représentants de l'industrie.
- Renforcer la surveillance des produits importés. Santé Canada a renforcé la surveillance réglementaire des produits importés en créant le Programme national de l'intégrité frontalière en collaboration avec l'ACIA, l'ASFC et la FDA. L'objectif de ce programme consiste à renforcer la capacité de Santé Canada de prendre et d'appuyer des décisions concernant l'admissibilité à la frontière en rapport avec les produits de santé. Le programme s'appuie, entre autres, sur le pouvoir d'inspecter et de prélever des échantillons de produits de santé importés, de demander à l'ASFC de cibler et de retenir certains envois afin d'en déterminer l'admissibilité, et de prendre des mesures de conformité et d'application; le recours à des partenariats et aux meilleures pratiques; les activités de sensibilisation et d'éducation; et l'utilisation des technologies de l'information de manière efficace en participant à l'initiative du guichet unique menée par l'ASFC, dont le but est de créer une approche automatisée axée sur les risques pour relever les marchandises à haut risque et accélérer la circulation des marchandises à faible risque (Inspectorat, 2010a).
- Élaborer une politique sur les produits de santé de contrefaçon. En 2010, dans une initiative connexe, Santé Canada a publié une politique sur les produits de santé de contrefaçon, dont l'objectif principal consiste à « gérer le risque pour les Canadiennes et les Canadiens et de faire enlever les produits contrefaits du marché en appliquant le niveau d'intervention le plus approprié et en avisant les parties à risque » (Inspectorat, 2010c). Selon la documentation de l'Inspectorat, une stratégie anti-contrefaçon et un plan de mise en œuvre progressive ont été élaborés, un groupe de travail sur la contrefaçon a été créé; et l'Inspectorat et la Gendarmerie royale du Canada ont développé un protocole d'entente (PE) (Inspectorat, 2009a, p. 24). Toutefois, l'évaluation n'a pas permis de trouver d'autres renseignements sur le plan de mise en œuvre progressive, le groupe de travail sur la contrefaçon ou le PE.
5.4.6 Communications et mobilisation des intervenants
Les sections précédentes décrivaient les activités de Santé Canada en lien avec les quatre principales étapes du processus de réglementation : les essais cliniques, l'examen des présentations et l'autorisation de mise sur le marché, le contrôle et la surveillance post-commercialisation, et la conformité et l'application. Santé Canada utilise également ce processus pour communiquer avec les intervenants, comme les consommateurs/patients, les fournisseurs de soins et les représentants de l'industrie. Au cours de la période visée par l'évaluation, Santé Canada a cherché à atteindre deux principaux objectifs en lien avec les communications et la mobilisation des intervenants : offrir aux Canadiens plus d'information sur les produits de santé, notamment des renseignements opportuns et accessibles, et accroître l'ouverture des systèmes de réglementation à l'avis et à la participation des consommateurs et des autres intervenants. Les activités réalisées dans ces domaines sont décrites ci-dessous.
Accroître la transparence du processus d'examen des médicaments à usage humain
Conformément à son objectif visant à fournir aux Canadiens plus d'information sur les produits de santé, Santé Canada a lancé, en 2005, l'initiative du Sommaire des motifs de décision (SMD) en vue d'accroître la transparence du processus d'examen des médicaments à usage humain. Les SMD offrent aux professionnels de la santé, aux consommateurs et aux patients les analyses scientifiques axées sur les avantages et les risques utilisées dans le processus d'autorisation de mise sur le marché. Entre 2005 et septembre 2012, un total de 131 SMD ont été publiés sur les médicaments à usage humain.
Une évaluation de la phase I de l'initiative, ainsi que du rapport 2011 du BVG sur la réglementation des produits pharmaceutiques, a entraîné plusieurs changements à la phase II, notamment la réduction du délai de publication du SMD visé, la limitation de la capacité des DAMM à exiger des changements et à en appeler du SMD, la mise en place d'un format de questions et réponses (Q et R) et d'autres renseignements sur l'analyse des avantages et des risques de Santé Canada, et la publication d'un tableau des activités postautorisation (TAPA) relativement aux produits admissibles (DPT, 2012c). La phase II du projet de SMD s'applique aux nouveaux médicaments composés de nouvelles substances actives, ainsi qu'aux PBU, autorisés après le 1er septembre 2012.
Le TAPA a été lancé, en partie, en vue d'aborder les préoccupations du BVG concernant le fait que Santé Canada omettait de divulguer des renseignements liés aux AC-C, aux médicaments rejetés et aux médicaments retirés du processus d'examen (BVG, 2011, p. 18). Les TAPA fournissent des renseignements à jour sur les produits approuvés. Ils incluent un résumé des activités portant sur l'utilisation sécuritaire et efficace du produit, comme les renseignements liés aux présentations en vue d'une nouvelle utilisation du produit (peu importe si la décision de Santé Canada était positive ou négative), les présentations soumises dans le but de respecter les conditions (pour les produits approuvés en vertu des lignes directrices de l'AC-C), et les décisions réglementaires, comme l'annulation du DIN.
À ce jour, Santé Canada n'a pas publié de SMD concernant des décisions négatives - même s'il a indiqué qu'il envisagera peut-être cette possibilité (DPT, 2012c, p. 1) - et ne fournit pas de mises à jour au public quant aux progrès réalisés par les fabricants en vue de se conformer aux conditions post-commercialisation. La FDA présente, quant à elle, des mises à jour des progrès réalisés par les fabricants en vue de se conformer aux conditions post-commercialisation, comme des améliorations relativement aux études post-approbation. De plus, le site Web Drugs@FDA de la FDA fournit une base de données interrogeable contenant de la documentation relative aux marques approuvées, aux médicaments à usage humain génériques en vente libre et sur ordonnance, et aux produits thérapeutiques biologiques, soit l'ensemble de la trousse d'approbation des médicaments (y compris les rapports d'examen complets de la FDA, ainsi que les lettres d'approbation, mais pas les présentations en soi), et toutes les versions des étiquettes des produits approuvés depuis 1998. Toutefois, il ne semble pas avoir de sommaire semblable au SMD (FDA, 2013a).
Dans l'UE, l'EMA publie un rapport européen public d'évaluation (EPAR) pour tout médicament ayant reçu une autorisation centrale de mise sur le marché par la Commission européenne. Les renseignements disponibles incluent un sommaire en format Q et R et le dépliant de l'emballage. Des informations sont également fournies sur les médicaments dont l'autorisation de mise sur le marché a été refusée, ou qui ont été suspendus ou retirés du marché après avoir été approuvés (EMEA, 2013e).
Selon certains informateurs clés, Santé Canada communique très peu de renseignements au public sur le processus d'examen préalable à la mise en marché. Ils ont mentionné que Santé Canada ne fournit aucune donnée sur les médicaments en cours d'examen, offre très peu d'information sur la façon dont l'examen a été mené et les conclusions des évaluateurs à propos des produits en question, et ne présente aucun renseignement sur les réunions avec les représentants de l'industrie quant aux produits en voie de commercialisation. De plus, dans le cas des AC-C, Santé Canada ne communique aucune information sur les mesures prises par les entreprises en vue de satisfaire les conditions qui leur ont été imposées lorsqu'ils ont été autorisés à vendre leurs médicaments au Canada. On a laissé entendre que si les problèmes liés à un médicament sont assez graves pour qu'il vaille la peine d'imposer des conditions à l'approbation, Santé Canada devrait s'assurer qu'elles sont respectées, en plus d'informer le public quant à la conformité des fabricants à leur égard. L'évaluation n'a pas permis de déterminer l'ampleur actuelle de la surveillance de Santé Canada quant à la conformité des promoteurs aux conditions imposées dans les AC-C.
Améliorer l'étiquetage des produits pharmaceutiques
Santé Canada a entrepris plusieurs initiatives en vue d'accroître la qualité et la disponibilité d'étiquettes de produits pharmaceutiques faciles à comprendre. L'étiquetage des produits pharmaceutiques fait référence à la monographie du produit, au matériel inclus dans l'emballage et à la documentation fournie au consommateur au moment de l'achat. Cela comprend l'étiquette de l'emballage, les dépliants, les fiches techniques et tout autre document contenant de l'information précise sur le produit pharmaceutique (DPT, 2011). Il est important d'avoir accès à des renseignements clairs et exacts sur les médicaments, car ceux-ci permettent aux professionnels de la santé et aux consommateurs de prendre des décisions éclairées en matière de pharmacothérapie (DPT, 2011). Voici certaines des initiatives mises en œuvre dans ce domaine :
- Apporter des améliorations à la monographie du produit. La monographie du produit est un document scientifique factuel qui inclut de l'information sur les propriétés d'un médicament, les conditions d'utilisation et tout autre renseignement pouvant être requis pour une utilisation sûre, efficace et optimale. L'examen de la monographie fait partie du processus d'examen des présentations de drogue, et aucun AC n'est attribué si elle n'est pas disponible. La monographie du produit comprend trois sections, chacune visant un public précis : professionnels de la santé, scientifiques et (depuis 2004) consommateurs (DPT, 2008b). En 2008, dans le cadre du PASPAC, Santé Canada a commencé à publier des monographies de produit, y compris la partie destinée aux consommateurs, sur son site Web par l'intermédiaire de la Base de données sur les produits pharmaceutiques (BDPP).Note de bas de page 33Depuis ce temps, Santé Canada a également revu le modèle de monographie de produit afin d'y inclure un nouvel « encadré » pour les renseignements sur le traitement des surdoses (DPT, 2010c).
- Entreprendre l'élaboration de monographies de produit communes avec la FDA en ce qui concerne les MSO. Dans le cadre du CCR, Santé Canada et la FDA ont entrepris un projet en vue de développer et d'adopter des éléments de monographies harmonisés (notamment, les indications, les mises en garde, les conditions d'utilisation et toutes autres informations) pour les produits à faible risque. Toutefois, il est important de noter que les objectifs de cette initiative ne sont pas nécessairement d'améliorer l'étiquetage des produits, mais plutôt « de rationaliser les coûts pour les fabricants et les distributeurs et d'améliorer l'accès de ces produits thérapeutiques aux consommateurs de part et d'autre de la frontière canado-américaine » (groupe de travail du CCR du secteur des produits de soins personnels et des produits pharmaceutiques du, 2012b, p. 2).
- Modifier la réglementation afin d'obliger les fabricants à dresser une liste des ingrédients non médicinaux (INM) sur les étiquettes des médicaments en vente libre. Santé Canada a modifié la réglementation afin d'obliger les fabricants à dresser une liste des INM sur les étiquettes des médicaments en vente libre, ce qui devrait permettre d'informer les consommateurs qui ont des antécédents de réactions indésirables à certains INM, de réduire les réactions indésirables et, éventuellement, de diminuer les coûts du système de soins de santé. Les changements proposés sont devenus officiels en mai 2010 et sont entrés en vigueur en 2012 (GC, 2010).
- Définir des exigences en matière d'étiquetage concernant certains médicaments particuliers. Santé Canada a adopté des exigences en matière d'étiquetage concernant certains médicaments pédiatriques en vente libre contre la toux et le rhume administrés par voie orale (DPT, 2008c); a élaboré une ligne directrice portant sur l'étiquetage de l'acétaminophène (DGPSA, 2009); et a tenu des consultations sur la conception d'une ligne directrice sur l'étiquetage de l'acide acétylsalicylique (DPT, 2012a, 2012b).
- Apporter des modifications réglementaires concernant l'étiquetage, l'emballage et les marques nominatives des médicaments à usage humain. En juin 2013, dans le cadre de l'initiative d'étiquetage en langage clair - un élément de la phase I des plans de Santé Canada quant à la modernisation de la réglementation -, les modifications proposées aux exigences en matière d'étiquetage conformément au Règlement sur les aliments et drogues ont été publiées dans la Gazette du Canada. La réglementation proposée mettrait en place une exigence générale portant sur l'utilisation d'un langage clair sur les étiquettes des médicaments, et d'un format de présentation qui ne gêne pas la compréhension. Il adopterait également des exigences préalables à la commercialisation, notamment l'inscription des coordonnées de l'entreprise pour que les consommateurs puissent signaler tout problème concernant le produit; un tableau normalisé sur l'étiquette des médicaments en vente libre; la présentation de maquettes d'étiquettes et d'emballages dans le cadre du processus de présentations préalables à la mise en marché; et la codification de la politique actuelle sur les produits de santé à présentation et à consonance semblables (PSPCS)Note de bas de page 34(GC, 2013).).
Des exigences semblables concernant l'étiquetage en langage clair ont été adoptées ailleurs. Par exemple, l'UE a mis en place des exigences portant sur l'étiquetage normalisé et facile à lire il y a plus de dix ans. La Directive 2001/83/EC indique que l'étiquetage de tous les produits médicinaux (y compris les médicaments en vente libre) doit être « facile à lire et à comprendre, et indélébile », respecter le format obligatoire de l'EMEA aux fins d'uniformité, et fournir des maquettes de couleur aux fins d'examen (EMEA, 2009). Aux États-Unis, la réglementation concernant le tableau « Drug Facts » de 1999 sur les MSO exige que les fabricants apposent des étiquettes normalisées et faciles à lire sur la majorité des MSO d'ici 2002 (FDA, 2009). En 2006, l'agence a mis en place les exigences de la Physician Label Rule (PLR) relativement aux renseignements d'ordonnance (c.-à-d. les dépliants des emballages), lesquelles avaient pour but d'accroître l'utilisation sécuritaire et efficace des médicaments sur ordonnance en fournissant aux professionnels de la santé des renseignements clairs et précis, et plus faciles à obtenir, à lire et à utiliser (FDA, 2013e). La réglementation concernant les exigences de la PLR exige que les renseignements d'ordonnance contiennent des éléments d'information précis présentés dans un format de synthèse de type « faits saillants »Note de bas de page 35
La réglementation proposée permettrait d'harmoniser Santé Canada à ses homologues internationaux exigeant des étiquettes de produits faciles à comprendre. Toutefois, à l'heure actuelle, Santé Canada n'a pas l'autorité nécessaire pour exiger à un fabricant de modifier l'étiquette ou la monographie d'un produit lorsque ce dernier a reçu un AC (DPT, 2011).Note de bas de page 36 En revanche, la FDA peut exiger que l'étiquetage des produits soit mis à jour en fonction des nouveaux renseignements disponibles sur l'innocuité d'un médicament (gouvernement des États-Unis, 2012b). Par exemple, la FDA a le pouvoir d'exiger qu'un encadré soit ajouté au sommaire des renseignements d'ordonnance (faits saillants) et à la brochure complète de ces renseignements; l'encadré contient une mise en garde en caractères gras indiquant que le médicament peut causer des blessures graves ou la mort et fournit une courte explication du risque avant de renvoyer les lecteurs à la section appropriée de la brochure pour de plus amples renseignements (gouvernement des États-Unis, 2012a). Par conséquent, la FDA utilise l'encadré de l'étiquette pour communiquer aux médecins des renseignements sur l'innocuité et les risques (FDA, 2010).
Dans le même ordre d'idées, la nouvelle législation en matière de pharmacovigilance de l'UE ordonne que le dépliant des médicaments faisant l'objet d'une surveillance post-commercialisation accrue affiche un triangle noir inversé, ainsi que des énoncés indiquant que le produit est sous surveillance et qu'on encourage les patients à signaler les réactions indésirables (EMEA, 2013c). Le symbole et les énoncés sont inclus sur les produits biologiques, biosimilaires et pharmaceutiques contenant de nouvelles substances actives autorisées depuis le 1er janvier 2011, ainsi que sur les produits qui font actuellement l'objet d'études d'innocuité après la mise en marché ou les produits assujettis à des conditions ou à des restrictions en matière d'innocuité et d'efficacité, conformément au plan de gestion des risques (Parlement européen, 2010a, 2010b).
Accroître les communications des risques et l'innocuité après la mise en marché
Depuis 2005, le site Web de MedEffet est le principal mécanisme employé par Santé Canada pour communiquer l'information sur l'innocuité et les risques après la mise en marché aux professionnels de la santé et au public. L'Initiative MedEffet Canada a été créée dans le but d'améliorer l'accès aux renseignements sur l'innocuité et les réactions indésirables des produits de santé commercialisés, tout en offrant un guichet unique pour les activités de surveillance post-commercialisation. Le guichet unique a pour but d'offrir au public et aux professionnels de la santé des renseignements en ligne centralisés et faciles à trouver en vue d'accroître la sensibilisation quant à l'importance de signaler les événements indésirables liés à des produits de santé, tout en facilitant la façon de déclarer ces événements pour les professionnels de la santé et les membres du public.
Jusqu'à tout récemment, Santé Canada publiait des renseignements sur les risques et l'innocuité des médicaments sur la page « Avis, mises en garde et retraits » du site Web de MedEffet. D'autres méthodes de distribution ont été utilisées, et le sont encore, y compris les envois postaux et les publications imprimées, la diffusion aux associations professionnelles concernées en vue d'encourager l'affichage sur leurs sites Web et la publication dans leurs revues et leurs bulletins, et la diffusion aux organismes d'attribution de licences, aux ministères provinciaux et aux organismes de réglementation étrangers. De plus, la liste de diffusion électronique gratuite du programme MedEffet envoie les avis, les mises en garde et les rappels, le Bulletin canadien des effets indésirables trimestriel et les mises à jour du contenu de MedEffet directement à environ 20 000 abonnés. Afin de communiquer les risques concernant l'innocuité d'un produit, Santé Canada a recours à différents documents de communication des risques ciblant le public et les professionnels de la santé, et conçus pour des situations présentant un risque faible, moyen ou élevé.
Les analyses des communications des risques liées aux médicaments à usage humain affichées sur le site de MedEffet montrent qu'entre 2005 et le 23 août 2012 (DPSC, 2012b), 419 communications des risques liées aux médicaments à usage humain ont été affichées sur le site de MedEffet; plus de la moitié (51 %) était destinée au public et le tiers (34 %) ciblait les professionnels de la santé. L'analyse a également révélé certaines limites associées à la communication des risques. Par exemple, les communications concernant les rappels de produits n'indiquaient pas toujours clairement si le rappel du produit était communiqué. De plus, jusqu'à tout récemment, les renseignements sur les rappels de produits étaient accessibles à deux endroits sur le site Web de Santé Canada, soit sur la page « Avis, mises en garde et retraits » du site Web de MedEffet et sur les Listes de retrait de marché des médicaments. Cette situation risquait de prêter à confusion et d'entraîner des irrégularités dans les renseignements affichés sur les deux sites.Note de bas de page 37
En février 2013, Santé Canada a lancé la Base de données sur les rappels et les avis de sécurité, soit un système révisé dont le but est de diffuser les communications des risques aux professionnels de la santé et au public.Note de bas de page 38La nouvelle base de données semble être une amélioration de la liste « Avis, mises en garde et retraits » du site Web de MedEffet en plusieurs points, y compris la disponibilité d'une fonction de recherche avancée et d'un nouveau format aux fins de communication des risques. Toutes les communications des risques sont maintenant classées dans l'une des deux principales catégories - avis et retraits -, ce qui devrait permettre d'éliminer toute incertitude à savoir si le rappel d'un produit est communiqué.Note de bas de page 39Encore plus récemment, au milieu de mars 2013, Santé Canada a supprimé les Listes de retrait de marché des médicaments en ligne et consolidé les renseignements sur les retraits de produits dans la Base de données sur les rappels et les avis de sécurité. Ce changement a permis d'écarter la possibilité d'irrégularités, quoiqu'il n'y ait plus de liste exhaustive des retraits concernant tous les types de risques pour la santé disponible sur le site Web de Santé Canada.
Afin d'améliorer davantage le processus de communication des risques, la DPSC a mentionné qu'elle procède actuellement à l'examen de ses cibles de rendement existantes relativement à la préparation et à la diffusion de documents de communication des risques, lesquels font partie de ses lignes directrices destinées aux représentants de l'industrie quant à la communication des risques. La ligne directrice indique que les communications destinées aux professionnels de la santé « doivent être rédigées et diffusées dans les douze jours ouvrables suivant la date à laquelle la lettre de demande a été reçue » et que la communication au public connexe « doit être diffusée trois (3) jours ouvrables après la diffusion » de la communication aux professionnels de la santé (Santé Canada, 2010b, p. 7). La lettre de demande est la lettre envoyée au DAMM par Santé Canada décrivant la nature du problème d'innocuité et demandant au DAMM de rédiger une communication des risques à l'intention des professionnels de la santé et du public. La DPSC a affirmé que, à l'heure actuelle, il n'y a pas de délai précis concernant l'envoi de la lettre de demande; l'envoi dépend de la nature du risque.
Elle a aussi mentionné que Santé Canada se penche sur des domaines dans lesquels il serait possible d'apporter des améliorations axées davantage sur la communication des risques. Enfin, la DGPSA procède à l'élaboration d'une ligne directrice interne sur le processus de communication des risques pour les produits de santé à usage humain, laquelle décrit le ou les processus à suivre et les responsabilités des parties concernées, y compris la Direction des sciences (c.-à-d. la DPT dans le cas des médicaments pharmaceutiques), l'Inspectorat, la DGPSA, la Direction générale des communications et des affaires publiques (DGCAP), le DAMM et le sous-ministre adjoint (SMA) (Santé Canada, 2013g).
Ouvrir le système de réglementation à l'avis et à la participation des intervenants
Conformément au processus établi concernant le développement de la réglementation au niveau fédéral, Santé Canada organise des consultations publiques sur les modifications réglementaires proposées au Règlement sur les aliments et drogues et à d'autres règlements connexes. Le ministère a également entrepris plusieurs initiatives en vue d'accroître l'ouverture des systèmes de réglementation à l'avis et à la participation des intervenants. En voici des exemples.
- La création, en 2005, d'un Cadre de participation du public à la suite de consultations internes et externes avec les représentants de l'industrie, les groupes de patients et de consommateurs, ainsi que le milieu universitaire et les professionnels de la santé. Le cadre vise à orienter les activités de mobilisation du public dans l'ensemble des responsabilités de la Direction générale (DGPSA, 2005b, p. 1).
- L'élaboration, en 2007, d'une politique sur l'avis du public dans le cadre du processus d'examen des produits réglementés qui vise à corriger les lacunes de l'ancien cadre de réglementation, selon lequel les fabricants avaient la responsabilité de fournir tous les renseignements sur un produit présenté aux fins d'approbation, et qui n'a pas clairement permis l'analyse des renseignements donnés par les consommateurs, les groupes de patients, les médecins et d'autres intervenants dans le processus de prise de décision. La politique décrit les processus à utiliser lorsque la Direction générale « relève une situation pour laquelle la prise de décision concernant un produit réglementé bénéficierait de l'avis du public » (DGPSA, 2007b, p. 1). Elle stipule également que la Direction générale présenterait des rapports publics (par l'intermédiaire du site Web de Santé Canada) sur les avis reçus du public. Ces rapports énonceraient les objectifs visés et les méthodes employées dans le cadre de la collecte des commentaires du public et fourniraient un aperçu des avis reçus. Enfin, la politique fournit une définition des informations commerciales confidentielles et décrit la façon dont la DGPSA traite ces renseignements.
- La création de différents groupes et comités consultatifs d'experts/de scientifiques afin d'orienter l'élaboration de politiques et de règlements et de fournir des recommandations concernant des problèmes scientifiques particuliers. On parle, par exemple, du Comité consultatif d'experts sur la vigilance des produits de santé, qui offre une expertise externe à la DGPSA concernant la surveillance post-commercialisation, les communications des risques et la surveillance de la réglementation de la publicité (DPSC, 2008); le Comité consultatif d'experts sur les initiatives pédiatriques, qui permet d'« obtenir l'avis d'experts et l'apport du public en ce qui a trait à la mise au point et à l'homologation des produits de santé commercialisés destinés aux enfants et aux femmes enceintes ou qui allaitent » (Santé Canada, 2012h).
- L'organisation de consultations avec le public, les intervenants et les représentants de l'industrie sur l'élaboration des lignes directrices et des politiques proposées (en plus des consultations sur les modifications réglementaires proposées).
- Les réunions régulières avec les représentants de l'industrie et d'autres intervenants dans le cadre du Programme des réunions bilatérales (PRB). Grâce à ces réunions, la DPT rencontre régulièrement les représentants de l'industrie et les autres associations d'intervenants; ces réunions font souvent appel à la participation d'autres directions de la DGPSA et permettent aux intervenants de discuter des problèmes de réglementation relatifs à des intérêts communs, d'échanger des renseignements et de faire part de leur expertise. Les associations de l'industrie concernées par le PRB de la DPT sont les suivantes : l'Association canadienne des chaînes de pharmacies, L'Association canadienne de produits de consommation spécialisés, l'Association canadienne des cosmétiques, produits de toilette et parfums, Produits de santé consommateurs du Canada, l'Association canadienne du médicament générique, Les compagnies de recherche pharmaceutique du Canada, l'Association des pharmaciens du Canada, l'Association de ventes directes du Canada, le Groupement provincial de l'industrie du médicament et l'Association nationale des organismes de réglementation de la pharmacie. Les sujets abordés sont le rendement de l'examen des présentations, les activités de conformité, le recouvrement des coûts, les modifications réglementaires proposées et l'élaboration de nouvelles lignes directrices.
- L'affichage de renseignements destinés aux représentants de l'industrie sur le processus de demande et d'examen relatif aux médicaments à usage humain, ainsi que les politiques et lignes directrices connexes.
- L'organisation de réunions préalables au dépôt des présentations avec les promoteurs avant qu'ils présentent une PDN ou une DEC.
- La réalisation d'activités de conformité, d'éducation et de promotion, comme des tournées et des présentations, en vue d'informer les représentants de l'industrie au sujet de leurs obligations.
Le bureau de l'ombudsman public, créé dans le cadre de l'IIPT en vue de répondre aux plaintes du public et des intervenants sur des enjeux réglementaires (Santé Canada, 2006, p. 9), ne semble plus exister.
Certains informateurs clés externes croient que la consultation et la mobilisation des intervenants puissent favoriser les représentants de l'industrie plutôt que les consommateurs, les patients et les professionnels de la santé. On a noté que les représentants de l'industrie seront plus susceptibles d'être informés des possibilités de consultation en raison de leur intérêt prononcé quant à la façon dont leurs produits sont touchés par les modifications réglementaires ou politiques, et peuvent disposer de plus de ressources que les autres intervenants, particulièrement lorsque des déplacements sont nécessaires.Note de bas de page 40L'évaluation n'a pas permis de déterminer si les représentants de l'industrie participent plus souvent aux consultations publiques que les autres groupes d'intervenants. De plus, les représentants de l'industrie ont la possibilité de consulter Santé Canada par l'intermédiaire du PRB et des réunions préalables au dépôt des présentations et à celles sur les produits en voie de commercialisation, qui ne sont pas accessibles aux consommateurs, aux patients ni aux professionnels de la santé.
6.0 Résultats - Résultats atteints
La présente section expose les constatations tirées des réponses obtenues aux questions de l'évaluation liées aux résultats. Bien que Santé Canada ait participé à de nombreuses activités qui devraient, en théorie, contribuer à l'obtention des résultats escomptés, les données appuyant une conclusion définitive sur la mesure dans laquelle les résultats attendus ont été atteints sont relativement limitées. Pour cette raison, les conclusions de l'évaluation portant sur les résultats du programme seront considérées comme une base de référence.
6.1 Sensibilisation et compréhension des intervenants
À court terme, les activités de Santé Canada, en particulier ses activités de communication et visant à favoriser la participation des intervenants, devraient entraîner un accroissement de la sensibilisation et de la compréhension des intervenants ne faisant pas partie de l'industrie (c'est-à-dire les consommateurs et les professionnels de la santé) à l'égard des risques et des avantages associés aux médicaments pour usage humain. Une série d'études.Note de bas de page 41parrainées depuis 2003 par Santé Canada fournit à ce titre des renseignements pertinents. Dans l'ensemble, ces études proposent des moyens d'améliorer la sensibilisation des consommateurs et des professionnels de la santé à propos de l'information sur la sécurité des médicaments accessible auprès de Santé Canada, bien que les résultats ne soient pas subdivisés par type de produits (médicaments pour usage humain ou médicaments biologiques). Les études ont par exemple permis de constater les éléments suivants :
- En 2003 et en 2006, environ le tiers des consommateurs savaient que de l'information sur l'innocuité des médicaments est accessible depuis le site Web de Santé Canada. Toutefois, seulement 10 % d'entre eux approximativement avaient consulté cette information au cours des six mois précédents. Inversement, près des deux tiers étaient au courant des avis et des avertissements publiés dans les médias (Centre de recherche Décima, 2003, 2006). Très peu (1 %) étaient par ailleurs abonnés à l'Avis électronique MedEffect (Centre de recherche Décima, 2006).
- En 2003, un peu plus de la moitié des professionnels de la santé connaissaient bien les Lettres aux professionnels de la santé (LPS) diffusés par les fabricants ainsi que le Bulletin canadien des effets indésirables, mais ils étaient moins nombreux à connaître les LPS diffusée par Santé Canada, les avis en ligne de Santé Canada sur la sécurité des médicaments et la liste d'envoi par courrier électronique de Santé Canada (Centre de recherche Décima, 2003). En 2007, 12 % des professionnels de la santé ont indiqué que Santé Canada et MedEffect étaient pour eux des sources d'information nouvelle sur l'innocuité des médicaments (Environics Research Group, 2007). Une étude qualitative réalisée la même année a observé qu'un très petit nombre de professionnels de la santé étaient au courant de l'existence du site Web MedEffect, et qu'un seul des répondants était abonné à l'avis électronique de ce site (The Antima Group et TNS Canada, 2007).
- En 2003 et en 2006, environ la moitié des professionnels de la santé ont affirmé que le processus de déclaration d'un effet indésirable leur était familier. Les pharmaciens et, à un degré moindre, les médecins avaient tendance à bien connaître le processus, tandis que les infirmières, les naturopathes et les dentistes avaient tendance à mal le connaître (Centre de recherche Décima, 2003; Environics Research Group, 2007). Dans le même ordre d'idées, l'étude qualitative de 2007 a permis de constater que les professionnels de la santé ne savent pas comment ni pourquoi une déclaration devrait être effectuée, ni à qui revient la tâche de déclarer un effet indésirable auprès de Santé Canada (The Antima Group & TNS Canada, 2007).
Voir l'annexe C pour consulter un résumé plus détaillé des résultats de ces sondages.
Depuis la réalisation de ces études, Santé Canada n'a mené aucune autre recherche sur l'efficacité de ses communications sur les risques ou de l'information transmise aux intervenants. Le sondage auprès des intervenants visait à combler cette lacune, mais en raison de contraintes méthodologiques et d'un faible taux de réponse, ses résultats ne sont pas fiables et n'ont pas été incorporés au présent rapport. Par conséquent, les données fournies se limitent à l'information obtenue des informateurs clés externes, qui n'avaient en général pas d'opinion très tranchée sur la communication des risques par Santé Canada ou sur les autres renseignements transmis par le Ministère aux intervenants. Ils ont néanmoins cerné des problèmes et proposé des améliorations, résumés ci-dessous.
Perception des intervenants clés externes à l'égard des communications et de l'information de Santé Canada
- Il est possible que les communications sur les risques soient trop techniques pour être comprises de tout le public.
- Les communications sur les risques devraient fournir une information plus détaillée aux patients et aux professionnels de la santé.
- L'information sur les risques et l'innocuité devrait être publiée plus rapidement.
- Il est difficile de naviguer dans le site Web de Santé Canada et d'y faire des recherches. Il serait possible d'améliorer sa conception en créant des pages distinctes pour les consommateurs et les professionnels de la santé ou en améliorant les liens qui figurent dans le site.
- L'affichage dans le site Web, bien que nécessaire, n'est pas un moyen de communication suffisant avec les intervenants. Santé Canada devrait intégrer à ses activités de diffusion d'information une utilisation accrue des médias sociaux et conventionnels.
- Santé Canada devrait fournir au public davantage d'information sur les examens préalables à la mise en marché et l'analyse des déclarations d'effets indésirables.
Santé Canada a récemment entrepris de s'acquitter de son engagement de longue date (datant au moins de 2007) d'effectuer une évaluation de l'efficacité de ses activités de communication des risques associés aux produits de santé. Le projet Évaluation de l'efficacité de la communication des risques évaluera les LPS produites ou endossées par Santé Canada, les avis aux hôpitaux et les communications au public au cours de la période d'un an s'étendant du 1er septembre 2011 au 1er septembre 2012 (Santé Canada, 2013d). L'échéance prévue de cette évaluation est décembre 2013.Note de bas de page 42
6.2 Sensibilisation et compréhension de l'industrie
À court terme, les activités de Santé Canada devraient entraîner un accroissement de la sensibilisation et de la compréhension des intervenants de l'industrie à l'égard des cadres réglementaires associés aux médicaments pour usage humain. Comme il a été mentionné précédemment dans le présent rapport, l'industrie a la possibilité d'exprimer ses commentaires et ses propositions sur les projets de règlements et de politiques grâce au processus de consultation en ligne de Santé Canada et au processus de publication des projets réglementaires dans la Gazette du Canada, en plus de pouvoir consulter Santé Canada par l'entremise du PRB ainsi que des réunions préalables à la présentation et sur les produits en voie de commercialisation.
En l'absence de données de référence, les données disponibles ne permettent pas de tirer des conclusions sur la mesure dans laquelle la sensibilisation et la compréhension des intervenants ont pu s'accroître, mais elles révèlent néanmoins l'existence possible de lacunes ou d'éléments à améliorer.
- Exigences relatives au processus de présentation. La plupart des répondants au sondage mené auprès d'intervenants de l'industrie des médicaments pour usage humain ont indiqué que leur entreprise possédait une compréhension approfondie des exigences de Santé Canada relatives au processus de présentation d'un médicament en vue de sa mise en marché. Les données qualitatives issues des entrevues menées avec des intervenants de l'industrie laissent cependant entendre que ces directives, dans certains domaines, nécessitent d'être clarifiées. Plus particulièrement, les informateurs clés de l'industrie ont signalé le besoin de directives ou d'une information plus claires sur les produits mixtes, la classification des nouveaux produits de santé et l'utilisation par Santé Canada de directives et d'examens étrangers dans le cadre du processus d'examen des médicaments.
- Exigences relatives aux déclarations d'effets indésirables. Une majorité des répondants au sondage mené auprès d'intervenants de l'industrie des médicaments pour usage humain sont d'avis que Santé Canada expose clairement les directives et exigences relatives à la déclaration des effets indésirables et a défini les effets qui doivent être signalés. Ils conviennent également que le formulaire de déclaration obligatoire est clair et compréhensible. La plupart ont aussi indiqué que leur entreprise était en mesure de fournir les renseignements requis et de remplir le formulaire de déclaration dans les délais impartis.
- Autres exigences. La plupart des répondants au sondage mené auprès d'intervenants de l'industrie des médicaments pour usage humain ont affirmé que leur entreprise possède une compréhension approfondie des exigences de Santé Canada relativement aux BPF, à l'agrément des établissements, aux activités en lien avec la conformité réglementaire et aux mesures d'application connexes. Une minorité des répondants ont qualifié d'approfondie leur connaissance des exigences liées aux BPC.
Les intervenants clés de l'industrie et les répondants au sondage étaient généralement satisfaits de l'information fournie par Santé Canada. Un petit nombre d'informateurs clés externes ont recommandé que Santé Canada s'appuie moins sur les associations industrielles pour la diffusion de l'information sur les politiques et règlements aux fabricants, car cette façon de faire tend à exclure les entreprises qui ne font partie d'aucune association.
6.3 Sécurité et efficacité
À court terme, les activités de Santé Canada devraient entraîner un accroissement de la sécurité et de l'efficacité des médicaments pour usage humain sur le marché canadien. De façon générale, Santé Canada a mis en place des procédures et des processus d'autorisation visant à contribuer à la sécurité et à l'efficacité des médicaments pour usage humain sur le marché, mais subsistent certaines lacunes, dont la résolution permettra une meilleure atteinte de cet objectif.
Étape préalable à la commercialisation
À l'étape qui précède la commercialisation, les exigences en matière d'information liées aux examens préalables à la mise en marché sont complexes et détaillées. De façon générale, les intervenants clés externes reconnaissent que l'information examinée par Santé Canada dans le cadre des examens préalables à la mise en marché est d'un type et d'une qualité appropriés, bien que certains aimeraient que Santé Canada incorpore dans le processus d'examen plus de données étrangères, y compris des approbations d'autres pays. Les répondants au sondage mené auprès d'intervenants de l'industrie des médicaments pour usage humain ont affirmé que les examens préalables à la mise en marché de Santé Canada sont rigoureux et que les procédures en place sont adéquates pour assurer la qualité et l'efficacité des produits de santé, de même qu'un niveau de sécurité suffisant.
En revanche, dans la mesure où nombre de produits ne font l'objet d'aucun essai sur la population pour laquelle ils sont destinés, les données de présentation d'un médicament ne reflètent pas toujours le profil complet d'efficacité ou d'innocuité du produit, la population pédiatrique constituant un exemple typique à ce propos. De plus, certaines études canadiennes et américaines ont observé un lien, bien qu'il ne soit pas causal, entre la rapidité du processus d'approbation et les problèmes d'innocuité après la commercialisation.Note de bas de page 43L'importante question empirique que doivent par conséquent se poser les organismes de réglementation est celle de savoir si des délais plus courts d'approbation entraînent l'arrivée d'un plus grand nombre de produits non sécuritaires sur le marché.
De façon plus générale, l'existence d'un processus d'examen rigoureux ne veut pas nécessairement dire que, avec le temps, des médicaments plus efficaces et plus sécuritaires sont mis en marché au Canada, ou qu'il y a amélioration globale de l'innocuité et de l'efficacité des médicaments sur le marché. En fait, les présentations de médicaments pour usage humain génériques ont représenté, en moyenne, 43 % de toutes les présentations de médicaments pour usage humain entre 2004 et 2012, et leur proportion a augmenté au cours des dernières années pour atteindre 52 % en 2011, avant de redescendre à 42 % en 2012. Or, par définition, les médicaments génériques ne sont pas plus efficaces ni plus sécuritaires que ceux qui sont déjà sur le marché.
Étape après la mise en marché
Santé Canada a reconnu, de concert avec d'autres organismes de réglementation, la nécessité d'accroître la surveillance après la mise en marché afin de mieux protéger la santé et la sécurité des patients. Il y a néanmoins eu des progrès dans certains domaines, comme il a été mentionné à la section 5.4.4 , et les données disponibles donnent à penser que les rappels de type I, déclenchés lorsqu'il existe une probabilité raisonnable que l'utilisation d'un produit ou l'exposition à celui-ci entraînent la mort ou des effets indésirables graves sur la santé, sont relativement rares. Entre 2005 et août 2012, 10 % des avis de rappel concernant des médicaments pour usage humain étaient de type I. Consulter l'annexe C pour obtenir de plus amples détails à ce chapitre.
Cela dit, Santé Canada n'a pas mis en œuvre certains éléments de renforcement de la surveillance après la commercialisation qu'on trouve ailleurs, à savoir le pouvoir d'obliger l'élaboration d'un PGR et la réalisation d'études post-commercialisation, et a récemment reconnu que l'absence d'un tel pouvoir constitue une limitation de son approche actuelle. Santé Canada a également admis que son approche comporte d'autres limitations, notamment l'absence de mesures concernant le problème des utilisations hors indication et le manque de suivi ou d'évaluation des risques touchant les groupes de la population qui n'ont fait l'objet d'aucune étude lors des essais cliniques.
En matière de conformité et d'application de la réglementation, Santé Canada ne possède pas actuellement le pouvoir d'obliger l'industrie à déclencher des rappels, bien que le projet de loi C-51 ait comporté une disposition à ce titre. Dans le même ordre d'idées, Santé Canada ne dispose pas de l'autorité législative nécessaire pour renforcer les sanctions imposées en cas de non-conformité, et certains informateurs clés externes se sont dits préoccupés par l'éventail à leur avis plutôt limité des possibilités qui s'offrent à Santé Canada pour régler les problèmes de non-conformité. Les autres sujets de préoccupation évoqués par les informateurs clés externes touchent la mesure dans laquelle Santé Canada s'assure du respect des ACC et surveille les installations de fabrication à l'étranger.
La majorité des répondants au sondage auprès d'intervenants de l'industrie des médicaments pour usage humain ont admis que les activités de contrôle et de surveillance après la commercialisation ainsi que les activités relatives à la conformité et à l'application de la réglementation de Santé Canada permettent efficacement d'assurer un niveau adéquat de qualité et d'innocuité des médicaments pour usage humain au Canada.
Enfin, il est important de signaler la possibilité d'une certaine évolution de la façon dont les organismes de réglementation définissent la sécurité des produits. La décision récente de la FDA de ne pas approuver les versions génériques de l'OxyContin parce qu'elles posent le risque d'un [traduction] « accroissement éventuel de certains types de mésusage du médicament » (FDA, 2013b) laisse entrevoir une conception de la sécurité tenant compte de la façon dont le médicament est consommé après sa mise en marché. Une telle conception de la sécurité des produits est possiblement plus en phase avec une approche réglementaire axée sur le cycle de vie des produits (approche mettant l'accent sur la consommation réelle d'un médicament et l'information sur la sécurité) qu'avec la conception traditionnelle de la sécurité, qui s'appuie sur l'hypothèse que le médicament sera « utilisé de la façon indiquée ».Note de bas de page 44
6.4 Conformité de l'industrie
À court terme, les activités de Santé Canada devraient entraîner une conformité accrue de l'industrie aux exigences réglementaires liées aux médicaments pour usage humain. Bien que de façon générale l'information disponible donne à penser que le niveau de conformité au sein de l'industrie est élevé, diverses contraintes touchant les données rendent difficile l'établissement d'un portrait plus détaillé de la situation, notamment des incohérences dans les déclarations qui se sont succédé dans le temps, l'impossibilité d'extrapoler le taux de conformité global de l'industrie réglementée, des regroupements de déclarations recoupant plusieurs catégories de produits et le peu de données accessibles sur la nature, la gravité, la fréquence et la prévalence des non-conformités.
Sont présentés ci-dessous les renseignements disponibles sur la conformité de l'industrie :
- Conformité des essais cliniques. Le taux de conformité des établissements d'essais cliniques inspectés est élevé. Selon le sommaire des rapports des inspections menées entre avril 2004 et mars 2011, 92 % des lieux inspectés ont été jugés conformes (Inspectorat, 2012g). La majorité des observations relevées lors de ces inspections portent sur des lacunes constatées dans les systèmes de qualité et les procédures (principalement les BPC et les registres). De ces observations, 54 % sont considérées d'ordre mineur, 39 % d'ordre majeur et 1 % d'ordre critique. Dans la mesure où seulement 2 % des établissements ont fait l'objet d'une inspection, ce taux de conformité ne peut pas être généralisé à l'ensemble de l'industrie des essais cliniques. De plus, étant donné que les données d'inspection sur les BPC et la conformité ne sont pas subdivisées par type de produits (médicaments pour usage humain ou médicaments biologiques), il est impossible d'analyser séparément le taux de conformité aux BPC de chacun de ces deux secteurs.
- Conformité aux BPF. Le taux de conformité aux BPF des établissements pharmaceutiques canadiens est élevé. Entre 2005-2006 et 2010-2011, le taux de conformité aux BPF au sein des établissements inspectés oscillait entre 94 % et 98 % (Inspectorat, 2007b, 2010b, 2012h).Note de bas de page 45Toutefois, comme dans le cas des essais cliniques, les données d'inspection sur les BPF ne sont pas subdivisées par catégorie de produits et comprennent les données canadiennes touchant les médicaments pour usage humain, les médicaments biologiques et les médicaments vétérinaires. Par conséquent, il est impossible d'analyser séparément le taux de conformité aux BPF du secteur de l'industrie pharmaceutique associé aux médicaments pour usage humain.
- Conformité aux exigences de déclaration après commercialisation. Entre 2004-2005 et 2009-2010, le programme d'inspection de la CEDAC a permis de constater un taux de conformité aux exigences de déclaration après la commercialisation de 100 % au sein des établissements inspectés des secteurs des médicaments pour usage humain et des médicaments biologiques. Toutefois, comme dans le cas des BPF et des BPC, ces données ne sont pas subdivisées par type de produits. Il est donc difficile de savoir s'il est possible d'extrapoler à partir de ce taux de conformité un taux général pour l'ensemble des établissements.
- Conformité des produits importés. Les données du Programme national de l'intégrité frontalière indiquent ce qui suit :
- Au total, 41 669 envois de médicaments figurant à l'annexe F médicaments sur ordonnance ont été inspectés entre 2010-2011 et 2011-2012. Dans l'ensemble, 94 % de ces envois ont été refusés à la frontière pendant les deux années visées, et 24 % (de tous les envois) l'ont été en raison d'un soupçon qu'ils puissent contenir des produits contrefaits. Cette proportion peut sembler considérable, mais il est possible de présumer que les envois inspectés ont été ciblés en fonction de critères de risque. Par conséquent, le taux de non-conformité relevé ne reflète pas nécessairement l'ensemble des envois de médicaments figurant à l'annexe F du Règlement sur les aliments et drogues.
- Au total, 3 812 envois de médicaments en vente libre (ou hors annexe) ont été inspectés entre 2010-2011 et 2011-2012. Du nombre, 13 % ont été refusés pour des raisons de non-conformité, mais aucun sous le prétexte d'un soupçon de contrefaçon.
Voir l'annexe C pour obtenir de plus amples renseignements sur la conformité de l'industrie.
Les données disponibles semblent laisser croire que les cas graves de non-conformité sont relativement peu communs. Cependant, au cours de la période visée par l'évaluation, Santé Canada n'a pas produit de rapports de façon régulière ou systématique sur la nature, la gravité, la fréquence ou la prévalence des cas de non-conformité liés aux médicaments pour usage humain, ayant plutôt axé ses rapports sur la mesure des activités et des extrants. De plus, la majeure partie des rapports de Santé Canada sur la conformité, y compris les données sur le respect des BPF, des BPC et des BPV, recoupent plusieurs catégories de produits, sans distinguer les médicaments pharmaceutiques des médicaments biologiques.
Cela dit, l'Inspectorat a récemment élaboré un rapport sommaire annuel des inspections qui sera publié sur le site Web de Santé Canada. L'édition 2012-2013 de ce rapport comprend une description des activités et des extrants de l'Inspectorat, présente le taux de conformité global de l'industrie, expose les niveaux de risque associés aux observations relevées lors des inspections et donne des exemples des observations les plus communes mentionnées dans les rapports d'inspection des établissements non conformes (Santé Canada, 2013h). D'après ce rapport, dans le cadre de l'ensemble de ses programmes d'inspection (programmes touchant les médicaments pour usage humain, les instruments médicaux, les produits de santé naturels, le sang, le sperme, les cellules, les tissus et les organes), Santé Canada a mené 1 387 inspections en 2012-2013, a relevé des centaines d'observations nécessitant des mesures correctives et a remis 33 avis de non-conformité. Le rapport ne comporte aucune information sur les mesures prises par l'Inspectorat concernant les cas de non-conformité et les données qu'il contient sur la conformité aux BPF, aux BPC et aux BPV n'opèrent aucune distinction entre les médicaments pharmaceutiques et les médicaments biologiques.
6.5 Adoption de comportements sûrs
À moyen terme, les activités de Santé Canada devraient inciter les intervenants externes à adopter des comportements sûrs en ce qui concerne les médicaments pour usage humain. La mesure dans laquelle cet objectif a été atteint n'a pas pu être déterminée dans le cadre de cette évaluation.
À ce jour, Santé Canada n'a pas évalué les répercussions de ses activités visant à influencer le comportement des intervenants, mais prévoit effectuer une évaluation de l'efficacité de ses activités de communication des risques au cours de l'année 2013-2014. Le sondage mené auprès des intervenants avait pour objectif de recueillir des données qui permettaient de tirer des conclusions sur cette question, mais cet objectif n'a pas pu être atteint dans la pratique en raison du faible taux de réponse au sondage.
La revue de la littérature a permis de relever certains faits indiquant l'existence de comportements ou de pratiques non sécuritaires associés à l'utilisation des médicaments pharmaceutiques. Une étude récente estime que 11 % des médicaments prescrits dans le cadre des soins de première ligne le sont en vue d'un usage autre que celui qui est prévu et que dans 79 % des cas il n'y a pas de preuves scientifiques solides de l'efficacité du produit relativement à cet usage (Eguale et coll., 2012). L'abus de médicaments d'ordonnance est une forme d'usage à des fins autres que celles approuvées pouvant provoquer une dépendance, une surdose ou la mort.
Bien que n'ayant pas actuellement l'autorité de réglementer l'utilisation des médicaments, Santé Canada peut néanmoins exercer une influence indirecte sur celle-ci au moyen d'autres voies, comme l'étiquetage des produits de même que la préparation et la diffusion de publications et de documents sur les risques. De plus, la DGPSA a la responsabilité de l'examen des produits ainsi que le pouvoir de refuser l'autorisation de mise en marché des produits que ne sont pas sécuritaires.
6.6 Utilisation de données scientifiques et d'analyses risques-avantages
À moyen terme, le PMUH prévoit une hausse de l'utilisation de données scientifiques et d'analyses risques-avantages par Santé Canada afin d'éclairer sa prise de décisions. Santé Canada a intégré l'utilisation de données scientifiques et d'analyses risques-avantages à ses activités et ses processus décisionnels, mais il n'a pas été possible de déterminer si cette utilisation a augmenté au cours de la période visée par l'évaluation.
L'utilisation de données scientifiques et d'analyses risques-avantages fait officiellement partie du processus décisionnel de Santé Canada. Le Cadre décisionnel de Santé Canada pour la détermination, l'évaluation et la gestion des risques pour la santé propose en effet une démarche de prise de décisions au sein de laquelle l'analyse et la gestion des risques occupent une place centrale (Santé Canada, 2000). Ce cadre est utilisé pour orienter le processus décisionnel du PMUH, mais il n'a pas possible d'évaluer avec quelle régularité il est appliqué dans la pratique.
L'utilisation de données scientifiques et d'analyses risques-avantagesNote de bas de page 46(ou d'analyses axées sur les risques) fait aussi partie des mécanismes de Santé Canada préalables et consécutifs à la commercialisation. Sont présentés ci-dessous certains exemples à ce chapitre (la liste donnée ne vise pas à l'exhaustivité).
- Les exigences en matière d'information liées aux présentations de médicaments pour usage humain sont complexes et détaillées.
- Les responsables du PMHU ont récemment élaboré et mis en œuvre une approche normalisée des activités après la commercialisation pour l'ensemble des catégories de produits réglementées par la DGPSA. Les processus normalisés mis en place touchent la détection des signaux ainsi que l'évaluation et la gestion des risques, en plus d'intégrer une prise en compte des données scientifiques et des analyses risques-avantages.
- Le Guide d'évaluation du risque par rapport à la conformité et à l'application de la loi de l'Inspectorat, élaboré récemment, propose une méthode structurée d'évaluation et de gestion des risques en santé se déployant par l'entremise des activités liées à la conformité et à l'application de la réglementation (Inspectorat, 2012c).
- Les programmes d'inspection de l'Inspectorat, y compris ceux visant les BPF et les BPC, sont axés sur les riches et ont pour objectif de permettre à l'Inspectorat de concentrer ses ressources dans les secteurs à haut risque.
En plus de tenir compte des données scientifiques et d'intégrer à ses processus des analyses risques-avantages (axés sur les risques), Santé Canada a constitué un certain nombre de comités et de groupes d'experts ayant pour rôle d'orienter l'élaboration des politiques et règlements dans divers domaines. Toutes les recommandations de tous les groupes consultatifs n'ont pas été mises en œuvre, mais certaines ont été utilisées pour guider le travail de préparation des politiques et règlements. Quelques exemples des comités constitués sont brièvement présentés ci-dessous. Pour consulter la liste complète, voir l'annexe C.
- Le Comité consultatif d'experts sur la vigilance des produits de santé (CCE-VPS) a été mis sur pied en 2006 afin de fournir à la DGPSA, sur une base continue, des conseils stratégiques d'experts sur les politiques en lien avec la sécurité et l'efficacité des produits de santé pour usage humain sur le marché. Depuis sa création en 2006, le Comité a formulé pour la DGPSA des avis sur la communication des risques, sur les déclarations de consommateurs auprès de Canada Vigilance, sur la détection et de l'évaluation des signaux, sur les analyses risques-avantages, sur le suivi des effets indésirables liés à des usages autres que ceux approuvés et sur l'augmentation de la surveillance après la commercialisation au moyen d'une collaboration avec les systèmes provinciaux et les chercheurs associés au RIEM. Les responsables du PMUH ont utilisé les avis du Comité dans le cadre de la préparation du document Recommandations pour l'usage approprié des produits contre la toux et le rhume chez les enfants.
- Le Comité consultatif d'experts sur les initiatives pédiatriques (CCEIP) a été formé en 2009 afin de fournir à la DGPSA des avis d'experts et un apport du public en ce qui a trait à la mise au point, à l'homologation et à la surveillance après la commercialisation des produits de santé destinés aux enfants et aux femmes enceintes ou qui allaitent. L'examen réalisé par le CCEIP de l'utilisation de médicaments à des fins autres que celles prévues au sein de la population pédiatrique a motivé l'étude « Prendre garde aux lacunes », entreprise par le Bureau des initiatives pédiatriques afin d'aborder les questions relatives à l'utilisation pour les enfants de médicaments à des fins autres que celles prévues.
- Le Comité consultatif scientifique sur les thérapies respiratoires et le traitement des allergies (CCS-TRTA) a mis au point deux projets de document sur les exigences de présentation des produits de commercialisation subséquente en lien avec les corticostéroïdes et les produits administrés par voie nasale servant au traitement de l'asthme et de la rhinite allergique.
- Plusieurs comités ont fourni des avis à Santé Canada à propos de produits précis et leurs conseils ont contribué à éclairer le processus décisionnel du Ministère concernant ces produits. Par exemple, en fonction des recommandations du Comité sur le nouveau médicament Adderall XR, Santé Canada a suspendu l'avis de conformité de ce produit.Note de bas de page 47
Divers autres comités ont été mis sur pied, assez récemment dans le cas de certains, afin d'aborder les questions liées aux traitements métaboliques et endocriniens, aux traitements contre le cancer, aux sciences pharmaceutiques, à la pharmacologie clinique, à la bioéquivalence spécifique au sexe pour les médicaments et aux médicaments en vente libre. Ces comités ont été actifs à divers degrés et plusieurs d'entre eux, travaillant notamment sur les techniques de reproduction humaine, les formes pharmaceutiques à libération modifiée, l'abus d'analgésiques opioïdes et le PAS, ont terminé leur mandat, alors que deux, fournissant des conseils sur les traitements musculo-squelettiques et sur les traitements neurologiques, ont été dissous.
Du point de vue de l'évaluation, il est difficile de déterminer clairement comment une augmentation de l'utilisation de données scientifiques et d'analyses risques-avantages (axées sur les risques) peut être mesurée ou réalisée de façon significative. Il est possiblement plus raisonnable de s'attendre à ce que Santé Canada utilise de façon constante des données scientifiques et des analyses risques-avantages (axées sur les risques) pour éclairer ses prises de décisions.
6.7 Intervention réglementaire rapide à l'égard des risques
À moyen terme, les activités du PMHU devraient entraîner une intervention réglementaire rapide à l'égard des risques établis. Il est toutefois complexe de tirer une conclusion générale à propos de ce résultat, car il existe diverses façons de le mesurer et il est difficile de déterminer le moment à partir duquel on peut considérer qu'un risque a initialement été décelé par Santé Canada.
Rapidité avec laquelle Santé Canada élabore des interventions réglementaires ou en matière de politique
Une façon de mesurer ce résultat est d'examiner la rapidité avec laquelle Santé Canada élabore une intervention réglementaire ou en matière de politique après la détermination d'un risque. Cependant, il est généralement difficile de savoir à quel moment un risque donné a initialement été établi par Santé Canada. De plus, même s'il est bien connu que l'élaboration de mesures réglementaires (ou de politiques) est un processus habituellement long, il n'existe pas de norme objective à partir de laquelle la rapidité d'intervention de Santé Canada peut être évaluée.Note de bas de page 48
Tout en tenant compte de ces restrictions, l'évaluation a permis de constater plusieurs cas où l'intervention de Santé Canada après la reconnaissance d'un risque a nécessité un temps de mise en œuvre plus long que prévu. Un exemple à ce titre est la mise au point d'un cadre réglementaire entourant les IPA. Comme il a été mentionné à la section 4.1, environ 85 % des IPA vendus au Canada sont importés, principalement de Chine et d'Inde, ce qui soulève des préoccupations quant à la possibilité que des ingrédients contaminés ou ne répondant pas aux normes aient des conséquences néfastes sur la santé de patients. Santé Canada a annoncé en 2002 vouloir adopter la directive Q7 de la CIH et souhaiter entreprendre l'élaboration d'un cadre réglementaire pour les IPA. Il a toutefois fallu attendre mai 2013 avant que soit publiée dans la partie I de la Gazette du Canada une modification réglementaire exigeant que les établissements qui produisent des IPA possèdent une LEPPP et respectent les BPF (Santé Canada, 2013e). Selon les explications des représentants de Santé Canada, cet écart de dix ans entre l'annonce de Santé Canada et la modification réglementaire est dû à la complexité du règlement et à l'évolution dans le temps des priorités de la DGPSA en ce qui concerne l'élaboration des règlements. Des règlements similaires ont été adoptés ailleurs dans le monde depuis une décennie, notamment aux États-Unis, dans l'Union européenne et au Japon.
La réponse de Santé Canada aux recommandations demandant une divulgation accrue des données des essais cliniques est un autre exemple. Santé Canada a promis d'augmenter l'accès du public aux données des essais cliniques dans son Plan de renouveau de 2007 et a annoncé en 2013 la création d'une base de données publique sur les essais cliniques. Santé Canada a toutefois admis que la base de données contient moins de renseignements que les registres obligatoires depuis 1997 aux États-Unis et depuis 2004 dans l'Union européenne. En plus de ces registres, la divulgation des résultats des essais est obligatoire aux États-Unis depuis 2007, et l'Union européenne travaille actuellement à la mise en œuvre de plans similaires.
Les retards dans les interventions de Santé Canada sont possiblement attribuables à la complexité des règlements et à l'évolution des priorités dans le temps, bien que d'autres facteurs puissent également entrer en ligne de compte. À ce titre, il est important de signaler que dans certains cas, malgré la détermination de risques et l'élaboration d'interventions réglementaires répondant à ceux-ci, Santé Canada n'a pas été en mesure de procéder à la mise en œuvre complète des initiatives prévues, faute de l'adoption d'une loi habilitante. Plusieurs initiatives prévues ont en effet été touchées par le fait que le projet de loi C-51 n'a jamais été adopté, notamment la mise en œuvre d'un cadre d'homologation progressive, l'instauration de déclarations des effets indésirables obligatoires dans les établissements de soins de santé et la mise en place de mesures administratives et d'application de la réglementation nouvelles, dont le pouvoir d'ordonner des rappels, entre autres choses. Dans d'autres cas, il n'a pas été possible de déterminer dans le cadre de l'évaluation pourquoi des modifications réglementaires prévues n'ont pas été mises en œuvre, par exemple l'instauration d'une autorité réglementaire ayant le pouvoir d'obliger les fabricants à élaborer des PGR et à produire des RPPV, projet qui avait pourtant été annoncé dans le cadre du PASPAC.
Rapidité d'intervention à l'égard des problèmes d'innocuité après la commercialisation
Une autre façon de mesurer la rapidité avec laquelle Santé Canada intervient après l'établissement d'un risque est d'examiner sa rapidité d'intervention à l'égard des problèmes d'innocuité après la commercialisation. Le RMR pour 2011-2012 de Santé Canada indique que, durant l'exercice, 93 % des évaluations de signaux après la commercialisation touchant des produits pharmaceutiques, des instruments médicaux, des médicaments biologiques et des produits de santé naturels ont été réalisées dans le respect des normes de service établies (Santé Canada, 2012a). Le rapport ne fournit toutefois pas l'analyse des données par catégorie de produits.
Le BVG a effectué dans son rapport de 2011 un examen de la rapidité d'intervention de Santé Canada à l'égard des signaux indiquant un problème possible d'innocuité. Tout en constatant que 34 des 54 évaluations de signaux examinées ont été réalisées dans les délais prévus, le BVG a également relevé que la méthode utilisée par Santé Canada pour mesurer son rendement ne tenait pas compte du délai précédant l'évaluation d'un problème d'innocuité potentiel, de la période pendant laquelle une évaluation peut être suspendue, du temps qu'il faut pour obtenir des renseignements supplémentaires auprès de tierces parties extérieures (comme le fabricant) et du nombre total de jours civils, plutôt que de jours ouvrables, consacrés à la réalisation de l'évaluation (BVG, 2011, p. 24). Ainsi, en tenant compte de ces facteurs, le BVG a pu établir que Santé Canada a pris au moins une année pour effectuer 34 des 54 évaluations examinées, et beaucoup plus de temps dans certains cas - deux ou trois ans (BVG, 2011, p. 24), ce qui l'a amené a conclure que Santé Canada « n'effectue pas l'évaluation des problèmes d'innocuité potentiels, ni n'y donne suite, en temps opportun » (BVG, 2011, p. 22).
Pour jeter un éclairage supplémentaire sur le temps d'intervention de Santé Canada à l'égard des problèmes d'innocuité après la commercialisation de médicaments pour usage humain (et de médicaments biologiques), les auteurs de l'évaluation ont entrepris dans le cadre de l'étude de cas relative à la surveillance après la commercialisation une analyse détaillée, au moyen des données administratives du PMUH, de la réaction du Ministère à l'égard des signaux indiquant un problème éventuel d'innocuité. Les données détaillées sont présentées à l'annexe C. L'analyse a permis de constater les éléments suivants :
- À l'étape de la détection d'un signal, comme il a déjà été indiqué, Santé Canada s'appuie de façon importante sur la littérature scientifique et sur les activités menées par les autres organismes de réglementation pour détecter les signaux liés à l'innocuité des médicaments pour usage humain. Or, dans la mesure où la publication des résultats d'études scientifiques et où la réalisation d'évaluations et la communication des risques par les autres organismes de réglementation peuvent prendre un temps considérable, il est possible que ces facteurs aient une incidence sur la rapidité des activités de détection de Santé Canada.
- Dans le cas des médicaments pour usage humain, une longue période peut s'écouler entre le moment où un signal est reçu et celui où une évaluation est prévue pour l'examiner. S'écoulent en moyenne 65 jours avant que soit planifié l'évaluation d'un signal lié à un médicament pour usage humain, et dans 16 % des cas ce délai peu prendre une année. En comparaison, le délai moyen de planification d'une évaluation pour les signaux liés aux médicaments biologiques est de 14 jours.
- La rapidité du processus d'évaluation des signaux liés aux médicaments pour usage humain s'est améliorée depuis l'instauration en avril 2007 d'une norme de rendement (non officielle) pour les signaux dont la priorité est élevée, moyenne et basse. Un peu plus de la moitié (51 %) des évaluations de signaux liés à des médicaments pour usage humain réalisées après avril 2007 ont été achevées dans un délai de 130 jours ouvrables. Le temps de traitement médian des évaluations de signaux effectuées durant cette période est de 128 jours ouvrables.
- En ce qui concerne les médicaments biologiques, 92 % des évaluations de signaux prévues entre novembre 2008 et octobre 2012 ont été réalisées dans un délai de 130 jours ouvrables. Note de bas de page 49Le temps de traitement médian des évaluations de signaux effectuées durant cette période est de 46 jours.
La raison de ces écarts dans la rapidité d'exécution des évaluations de signaux liés à des médicaments pour usage humain et à des médicaments biologiques est inconnue, mais il est possible qu'elle soit rattachée au nombre des évaluations par rapport au niveau des ressources disponibles ou à la complexité relative des dossiers. La présente évaluation n'a pas pu examiner la relation entre le niveau des ressources et la rapidité des évaluations, car il n'a pas été possible d'avoir accès aux données sur le nombre d'ETP affectés à cette activité.
Bien qu'il soit important pour Santé Canada d'effectuer les évaluations de signaux rapidement, il lui importe tout autant, du point de vue de la sécurité, d'exécuter promptement les mesures recommandées consécutivement aux évaluations réalisées. Les auteurs de la présente évaluation ont donc entrepris d'examiner le sous-ensemble des évaluations de signaux liés à des médicaments pour usage humain qui ont été menées en 2010, en 2011 et en 2012 et ont donné lieu à la recommandation d'une communication des risques.Note de bas de page 50Cet examen a permis de constater un délai médian entre l'approbation de l'évaluation d'un signal et l'affichage de la communication des risques de 200 jours pour les signaux de faible priorité et de 176 jours pour les signaux de priorité moyenne. Dans le cas de l'unique évaluation d'un signal de priorité élevée réalisée, un délai de 203 jours s'est écoulé entre l'approbation et l'affichage de la communication des risques.
En raison de la taille réduite de l'échantillon, il est difficile de tirer des conclusions de ces données. De plus, comme il a déjà été mentionné, les normes de rendement en place au sein de Santé Canada relativement à l'élaboration du contenu et à la diffusion des documents de communication des risques produits par l'industrie ne tiennent pas compte du délai entre l'approbation par Santé Canada de l'évaluation d'un signal et la présentation à l'industrie d'une demande de préparation d'un document de communication des risques.
Santé Canada a indiqué que ces normes de rendement sont en cours de révision. Les représentants du programme ont quant à eux expliqué que la préparation d'un document de communication des risques nécessite un temps considérable de négociation entre Santé Canada et le détenteur de l'AMM, et que l'ébauche et l'affichage du document peuvent devoir être reportés jusqu'à ce que les modifications appropriées aient été apportées à l'étiquette du produit. L'ébauche d'une ligne directrice de la DGPSA sur la communication des risques vise à intégrer cette négociation dans le processus (Santé Canada, 2013g). Il est toutefois difficile de savoir ce qui se produit lorsqu'une négociation échoue ou se prolonge indéfiniment, et si Santé Canada peut, dans certaines circonstances, diffuser un document de communication des risques sans que le détenteur de l'AMM ait au préalable modifié l'étiquette du produit visé ou sans qu'une entente ait pu être conclue avec celui-ci concernant le contenu du document de communication.
Enfin, on a tenté d'analyser dans le cadre de l'évaluation la durée totale du délai s'écoulant entre la détection initiale par Santé Canada d'un signal indiquant un problème possible d'innocuité et le résultat éventuel ou la mesure prise par le Ministère. Les données complètes n'étaient cependant disponibles que pour un petit nombre des évaluations de signaux réalisées (n=14).Note de bas de page 51Dans les 14 cas pour lesquels les données étaient accessibles, la détection initiale du signal a eu lieu entre 2007 et 2011, alors que l'évaluation a été effectuée entre 2010 et 2012. L'analyse de ces dossiers a permis de constater que le nombre total de jours civils qui se sont écoulés entre la détection d'un signal et la communication des risques se situait entre 232 dans le cas du Pradax et 1 481 (quatre ans) dans le cas de l'Avandia, pour une moyenne de 515 jours (1,4 an). Ces deux signaux avaient été classés comme étant de faible priorité. En l'absence d'un échantillon d'analyse plus important, il est impossible de formuler des conclusions à partir de ces données.
Bien qu'il soit important pour Santé Canada d'établir et de respecter des normes de rendement propres à ses activités après la commercialisation, il importe tout autant de comprendre la rapidité d'intervention globale du Ministère à l'égard des risques liés à l'innocuité, de leur détection initiale à la prise des mesures destinées à les atténuer. Une comparaison de la vitesse d'intervention de Santé Canada avec celle d'organismes homologues ailleurs dans le monde permettrait de produire une analyse plus objective à ce chapitre.Note de bas de page 52
Actuellement, les données requises pour faire une analyse détaillée des activités après commercialisation de Santé Canada sont grandement dispersées et ne sont pas tenues de façon constante. Or, comme Santé Canada a exprimé l'intention d'accroître ses activités de surveillance après la mise en marché, il semble justifier de préconiser une méthode de collecte des données et de gestion de l'information sur les activités après la commercialisation plus exhaustive, plus uniforme et plus centralisée. La mise sur pied d'une base de données serait à ce titre une solution idéale, mais il est également possible d'améliorer le système actuel fonctionnant sur Excel au moyen d'ajustements relativement simples et peu coûteux. Parmi ces ajustements, on peut citer entre autres exemples une normalisation de la saisie des données à l'aide de codes prédéterminés, l'attribution d'identificateurs uniques aux dossiers dès l'étape de la détection des signaux et l'utilisation de ces identificateurs à toutes les étapes du processus de traitement des signaux afin qu'il soit possible de relier entre eux les éléments des dossiers qui sont dispersés dans différents chiffriers.
6.8 Harmonisation internationale
À moyen terme, les activités du PMUH devraient entraîner une meilleure harmonisation internationale des exigences réglementaires concernant les médicaments pour usage humain. Selon la DPT, l'harmonisation « fait référence à l'élaboration, à l'adoption et à la mise en œuvre de normes techniques internationales relativement à la mise au point, à l'homologation et au contrôle des produits pharmaceutiques et des matériels médicaux », de même qu'à une « convergence des processus et pratiques réglementaires » (DPT, 2004, p. 9).
Dans ce domaine, Santé Canada a concentré ses efforts sur l'établissement et le renforcement d'activités de coopération et de travail partagé avec ses principaux homologues internationaux, sur une participation active à l'harmonisation des normes internationales et exigences techniques et aux initiatives de convergence en matière de réglementation, et sur la création de stratégies avec les pays dont les systèmes de réglementation sont en cours d'élaboration de manière à contribuer au renforcement des capacités de ceux-ci. L'évaluation a permis de recueillir des données démontrant que des efforts ont été déployés dans le cadre du PMUH afin d'accroître l'harmonisation internationale et que des progrès dans certains secteurs ont été réalisés à cet égard.
- Santé Canada est un observateur et un participant actif de la CIH, qui a été créée par les organismes de réglementation et les industries pharmaceutiques d'Europe, du Japon et des États-Unis dans le but d'harmoniser les exigences techniques et d'assurer la sécurité, la qualité et l'efficacité des produits pharmaceutiques pour usage humain (DGPSA, 2012b, p. 9). Dans l'exercice de ce rôle, Santé Canada participe à l'élaboration et à la révision des directives et des normes de la CIH, en plus d'être membre de divers comités et groupes de travail et de présider un groupe d'étude spécial dont le mandat est d'examiner les moyens de mettre au point pour la CIH un plan stratégique en matière de formation. Santé Canada prend également part à l'élaboration d'une norme de présentation électronique (eCTD) (DGPSA, 2012b, p. 9) et a adopté certaines des normes de la CHI, y compris le Common Technical Document (CTD) (Santé Canada, 2003) et diverses autres normes présentées précédemment dans le présent rapport. Il est toutefois difficile de déterminer combien exactement de normes et de directives de la CHI ont été adoptées par Santé Canada jusqu'à ce jour.
- La DGPSA a conclu avec l'Union européenne, la Suisse, l'Islande, le Liechtenstein, la Norvège et l'Australie un ARM concernant la conformité aux BPF.
- Santé Canada est membre du Pharmaceutical Inspection Cooperation Scheme (PIC/S), participe à une mise en commun des rapports d'inspection et prend parts à diverses activités en matière d'inspection.
- La DGPSA a conclu des ententes de mise en commun de l'information avec la FDA des États-Unis, la Therapeutic Goods Administration (TGA) d'Australie et l'Union européenne.
- Santé Canada et la FDA participent au CCR, dont l'objectif général est de mieux harmoniser les approches réglementaires du Canada et des États-Unis. Des initiatives en lien avec l'élaboration de présentations électroniques communes et de monographies communes sur les médicaments en vente libre et les BPF sont actuellement en cours.
- La DGPSA et la TGA ont mis sur pied une initiative améliorée de travail partagé dans les secteurs des médicaments génériques, des évaluations de la conformité administrative aux BPF des établissements et des monographies sur les médicaments en vente libre (DGPSA, 2012b, p. 36).
- La DGPSA participe également à une initiative d'intégration d'experts avec l'EMEA dans le cadre de laquelle un membre de son personnel a été intégré à l'agence européenne afin de travailler sur des projets conjoints entre les deux organismes. Les activités proposées comprennent une participation à titre d'observateur aux réunions du comité consultatif pour l'évaluation des risques en matière de pharmacovigilance, la mise en œuvre de stratégies contribuant à faire évoluer les directives de la CHI concernant la pharmacovigilance, l'harmonisation des méthodes et des normes pour l'évaluation des analyses risques-avantages, et l'élaboration de stratégies efficaces d'atténuation et de communication des risques. La DGPSA assiste également aux réunions de plusieurs autres groupes de travail de l'EMEA en lien avec la pharmacovigilance, la qualité et l'inspection des BPC (DGPSA, 2012b, p. 33-35).
- Santé Canada participe à plusieurs initiatives de l'Organisation mondiale de la Santé (OMS). Le Ministère contribue notamment au Programme de préqualification des médicaments et au Programme international de pharmacovigilance de l'OMS, en plus d'être membre du Conseil des organisations internationales des sciences médicales.
- Les autres forums internationaux auxquels participe Santé Canada comprend celui de la Coopération économique de la zone Asie-Pacifique, le Consortium des chefs d'agence, le Forum international des laboratoires sur les médicaments contrefaits, le Groupe international des organismes de réglementation des médicaments génériques, le Réseau des laboratoires officiels de contrôle des médicaments, l'Organisation panaméricaine de la santé, le Réseau panaméricain pour la coopération et l'harmonisation des réglementations pharmaceutiques, et plusieurs autres.
Tous les informateurs clés internationaux jugent que Santé Canada est un acteur positif des efforts bilatéraux et multilatéraux auxquels prend part le Ministère, qu'ils décrivent comme ayant le même esprit qu'eux et comme étant constant, fiable, stratégique, créatif, bien informé, enclin à la coopération et constructif. Plusieurs ont relevé que Santé Canada, à titre de participant aux forums internationaux, est perçu comme étant plus neutre et plus consciencieux que certains autres acteurs d'importance. Les informateurs clés internationaux ont tout de même signalé que Santé Canada, de la même façon que les autres organismes de réglementation, doit surmonter divers obstacles pour accroître son harmonisation et sa collaboration internationale, notamment des ressources limitées pour contribuer aux activités internationales, des facteurs politiques ou économiques ayant la priorité, des contraintes législatives empêchant l'harmonisation (p. ex., l'absence de fondement législatif pour les processus harmonisés), une législation nationale toujours plus détaillée et normative, et des différences de nature philosophique (différences perçues principalement comme existant entre l'approche des États-Unis et le reste du monde).
Enfin, il est important d'indiquer que le principal forum auquel Santé Canada participe afin d'accroître l'accès aux médicaments dans les pays en développement consiste en le Régime canadien d'accès aux médicaments, qui a été adopté dans le cadre d'une loi en 2004. Selon ce régime, les pays admissibles peuvent importer du Canada des versions génériques et moins chères de médicaments et d'instruments médicaux brevetés (Santé Canada, 2008c). En l'espace de huit années, le régime a permis d'acheminer deux lots d'un médicament générique vers un pays (Globe and Mail, 2012b, p. 1). En novembre 2012, un projet de loi d'initiative parlementaire visant à éliminer certains des obstacles qui empêchent les entreprises canadiennes de participer au régime a été défait à la Chambre des communes.
6.9 Résultats à long terme
À long terme, les activités du PMUH devraient contribuer à réduire les risques pour la santé et les événements indésirables associés à la consommation des médicaments pour usage humain, et accroître la confiance du public dans ces produits et dans les systèmes de réglementation connexes. Les activités du PMUH ont sans doute un effet sur ces deux résultats, quoique plusieurs autres facteurs puissent également entrer en ligne de compte.
Les activités du PMUH, notamment l'approbation de médicaments pour usage humain sécuritaires dont les avantages dépassent les risques, la surveillance et le contrôle après la mise en marché et le travail de vérification de la conformité et d'application des cadres réglementaires, devraient contribuer à réduire les risques pour la santé et les événements indésirables associés à la consommation des médicaments pour usage humain. Par exemple, retirer du marché les produits non sécuritaires au moyen de rappels et ne pas autoriser l'entrée au Canada de produits non conformes sont des mesures susceptibles d'éviter des effets néfastes sur la santé humaine qui pourraient se produire si elles n'étaient pas prises.
Bien qu'il ne fasse pas de doute que ces activités et les autres activités du PMUH ont contribué à réduire les risques pour la santé et les événements indésirables associés à la consommation des médicaments pour usage humain, il demeure difficile de trouver des données concrètes démontrant ces résultats. Les indicateurs disponibles peuvent en effet prêter à diverses interprétations. Par exemple, le nombre des déclarations d'effets indésirables transmises dans le système Canada Vigilance a connu dans le temps une croissance constante. Toutefois, cette croissance est possiblement attribuable à une augmentation de la quantité et de la diversité des produits sur le marché ainsi qu'à une plus grande reconnaissance du besoin ou de l'obligation de déclarer les effets indésirables, et n'est de ce fait pas nécessairement liée à une simple augmentation du nombre des produits non sécuritaires. En réalité, la proportion des déclarations d'effets indésirables classés comme étant « graves » -- c'est-à-dire qui ont été associés à une anomalie congénitale, à la mort, à une incapacité, à une hospitalisation ou à un autre trouble médical important, ou encore qui constituent un danger de mort (Santé Canada, 2012d) -- est demeurée stable depuis 2001, représentant un peu plus des deux tiers de toutes les déclarations transmises chaque année (DPSC, 2012a).
En ce qui a trait à la confiance du public, les données de sondage indiquent que les consommateurs ont un niveau de confiance élevé dans la sécurité des médicaments et dans le système de réglementation. En 2003 et en 2006, 84 % et 86 %, respectivement, des consommateurs avaient confiance dans la sécurité des médicaments d'ordonnance, et 75 % d'entre eux, pour les deux années, avaient confiance dans la sécurité des médicaments en vente libre (Centre de recherche Décima, 2003, 2006). Le pourcentage des consommateurs ayant confiance a cependant légèrement diminué en 2010, alors que 65 % des Canadiens estimaient que les médicaments approuvés par Santé Canada sont sécuritaires (Nanos Research, 2010).
La majorité des consommateurs interrogés ont dit avoir confiance dans le système de réglementation. En 2003, 85 % d'entre eux ont affirmé ne pas douter de l'efficacité des systèmes et des mesures de protection mis en place pour garantir la sécurité des médicaments d'ordonnance vendus au Canada (Centre de recherche Décima, 2003). En 2006, environ 78 % des répondants ont dit avoir confiance dans la façon dont le gouvernement fédéral surveille et encadre la sécurité et l'efficacité des médicaments (Centre de recherche Décima, 2006).
En définitive, Santé Canada espère offrir un système viable, rentable, adapté aux besoins et fondé sur la science pour la réglementation des médicaments pour usage humain au Canada. L'évaluation a permis de constater que Santé Canada utilise des données scientifiques et consulte les intervenants dans l'élaboration des politiques et des règlements. Également, la récente mise à jour des frais d'utilisation pour les médicaments pour usage humain permettra de soutenir la viabilité du système de réglementation associé à ces produits.
6.10 Conséquences inattendues
Les informateurs clés externes ont signalé un certain nombre de conséquences inattendues découlant de l'approche de Santé Canada en matière de réglementation des médicaments pour usage humain. La plupart de ces conséquences étaient liées à un accroissement du fardeau et des coûts associés au respect des exigences réglementaires, et des informateurs ont également laissé entendre que dans certains cas elles pouvaient finir par avoir un effet négatif sur la recherche, sur le développement de produits ou la croissance de certains domaines médicaux, et sur l'accès aux produits de santé. Les informateurs ont cité les exemples suivants :
- La réglementation encadrant les essais cliniques pourrait entraîner une réduction de la recherche en milieu académique par rapport à la recherche réalisée par l'industrie, car seules les grandes sociétés pharmaceutiques et de biotechnologies sont susceptibles de posséder les ressources nécessaires pour respecter la réglementation en place.Note de bas de page 53
- Bien qu'il y ait concurrence sur le marché entre les médicaments en vente libre et les produits de santé naturels, les premiers font l'objet d'une réglementation plus stricte, ce qui procure un avantage concurrentiel aux seconds.
- Dans la mesure où les médicaments existants pour lesquels sont mises au point de nouvelles indications ne sont pas toujours admissibles à la protection des données au Canada, il arrive que certains de ces produits ne soient pas commercialisés au pays.
Il n'a pas été possible d'évaluer dans le cadre de l'évaluation la mesure dans laquelle ces conséquences se sont réellement produites.
7.0 Constatations - Efficacité et économie
Globalement, l'information sur les ressources humaines et financières est insuffisante pour produire une analyse de l'efficacité et de l'économie du programme. L'évaluation n'a par conséquent pas permis de déterminer la mesure dans laquelle les ressources du programme ont été utilisées comme prévu ni d'établir si les extrants du programme ont été produits de façon efficiente ou si les résultats attendus ont été produits de façon économique.
En ce qui concerne l'exercice 2007-2008 et les exercices antérieurs, les rapports financiers ont été préparés en fonction de l'ancienne AHP de Santé Canada, dans le cadre de laquelle les activités liées aux médicaments pour usage humain étaient rattachées à l'activité de programme associée aux produits alimentaires et de santé. Cette activité de programme comportait les sous-activités suivantes :
- l'évaluation réglementaire avant la mise sur le marché et l'amélioration du processus;
- l'information, l'éducation et la sensibilisation concernant les produits de santé, les aliments et la nutrition;
- la surveillance de l'innocuité et de l'efficacité thérapeutique, et la gestion du risque;
- la transparence et la responsabilité à l'égard du public, et les relations avec les intervenants.
Les rapports financiers étaient donc liés à ces quatre sous-activités de programme ainsi qu'aux sous-sous-activités représentant, à un niveau de détail plus fin, les tâches fonctionnelles exécutées par le personnel de Santé Canada. Il est toutefois impossible de rattacher cette information à ce qu'est devenue l'AHP à partir de 2008-2009.
L'AHP de Santé Canada a fait l'objet d'une restructuration en 2007-2008. Dans le cadre cette nouvelle version de l'AHP, l'activité de programme pertinente pour les médicaments pour usage humain recouvre désormais la catégorie des produits de santé, et ses sous-activités de programme se répartissent de la façon suivante :
- les médicaments à usage humain;
- les produits biologiques et radiopharmaceutiques;
- les instruments médicaux;
- les médicaments vétérinaires
Une nouvelle restructuration de l'AHP réalisée en 2012 a conduit à la création d'un Programme des médicaments élargi combinant le Programme des médicaments pour usage humain et le Programme des médicaments vétérinaires.
De 2008-2009 à 2011-2012, les rapports financiers étaient liés aux quatre groupes de sous-activités de programme énumérés ci-dessus. Le tableau 4 présente les données disponibles sur les dépenses du PMUH de 2009-2010 à 2011-2012, avec et sans revenus. Il est important de signaler que ces données sur les dépenses ne comprennent pas les coûts indirects, dont le codage est associé à l'échelon du programme (Produits de santé) et non à l'échelon du sous-programme. Par conséquent, l'information financière disponible ne permet pas de dresser un portrait exact des dépenses globales du programme.
| Direction | 2009-2010 | 2010-2011 | 2011-2012 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sans revenus | Avec revenus | Revenus | Sans revenus | Avec revenus | Revenus | Sans revenus | Avec revenus | Revenus | |
| Produits thérapeutiques | 44 791 057 | 22 990 326 | -21 800 731 | 42 944 109 | 21 697 816 | -21 247 248 | 49 412 876 | 29 126 690 | -20 225 641 |
| Inspectorat | 3 609 648 | -336 913 | -4 027 954 | 4 225 428 | 1 099 902 | -3 125 526 | 7 861 380 | 3 096 316 | -4 765 064 |
| Produits de santé commercialisés | 13 730 333 | 13 730 333 | 0 | 13 623 268 | 13 617 856 | 0 | 15 634 943 | 13 288 412 | -2 351 741 |
| Médicaments vétérinaires | 0 | 0 | 0 | 0 | 0 | 0 | 1 979 897 | 1 341 185 | -638 687 |
| Autres coûts de la DGPSA | 5 990 811 | 5 990 811 | 0 | 4 584 730 | 4 584 730 | 0 | 7 825 883 | 4 786 885 | -3 038 998 |
| DGPSA - Régions | 2 146 947 | 2 146 947 | 0 | 815 712 | 815 712 | 0 | 0 | 0 | 0 |
| Dépenses totales du PMUH - DGPSA | 70 268 796 | 44 521 504 | -25 828 685 | 66 193 247 | 41 816 016 | -24 372 774 | 82 714 979 | 51 639 488 | -31 020 131 |
| BRP | 12 135 809 | 12 285 455 | 17 098 529 | 9 412 834 | -7 685 695 | ||||
| Dépenses totales du PMUH - DGPSA et BRP | 82 404 605 | 78 478 702 | 99 813 508 | 61 052 322 | -38 705 826 | ||||
Remarques :
Sources : DGPSA et BRP. |
|||||||||
Dans le même ordre d'idées, l'information disponible sur les budgets du PMUH demeure limitée. Par exemple, en 2010-2011 et en 2011-2012 les rapports financiers à l'échelon des sous-activités de programme ne comprenaient pas les montants des budgets. Ces derniers ne figuraient que dans les rapports produits à l'échelon du programme (Produits de santé). Cette situation, combinée à certaines difficultés dans la compréhension des dépenses globales du PMUH, a fait en sorte qu'il a été impossible de comparer dans le temps les montants des budgets avec les dépenses réelles.
- Les représentants de Santé Canada ont indiqué que la DGPSA a récemment mis en œuvre plusieurs améliorations de sa méthode de communication de l'information financière.
- Bien que les dépenses engagées par les organismes qui chapeautent le programme, comme le SMA ou la DPPAI, n'aient pas dans le passé été codées de façon constante de manière à ce qu'elles soient associées au PMUH, depuis 2011-2012 des procédures ont été mises en place afin de veiller à ce que toutes les dépenses soient rattachées par code à la sous-activité appropriée (médicaments pharmaceutiques, instruments médicaux, etc.) plutôt qu'au programme général (Produits de santé).
- À partir de 2013-2014, les budgets nationaux ont été revus afin de s'assurer que les affectations appropriées se font à l'échelon des sous-activités.
- À compter de 2014-2015, le SCT exigera que tous les rapports sur la gestion des dépenses du gouvernement renvoient l'échelon des sous-activités ou à un échelon inférieur, selon l'AHP approuvée. Les représentants de Santé Canada ont indiqué que la DGPSA est en très bonne voie de pouvoir établir sa planification, ses budgets et ses rapports à partir du nouvel échelon requis.
En plus des contraintes, présentées ci-dessus, que pose l'ancienne méthode utilisée par la DGPSA pour produire des rapports sur les budgets et les dépenses du PMUH, les données disponibles comportent plusieurs autres lacunes qui les rendent difficilement utilisables dans une analyse de l'efficacité et de l'économie. Par exemple, bien qu'il y ait de l'information sur le nombre d'ETP affectés aux différentes directions de la DGPSA ou des autres entités liées au PMUH, le nombre d'ETP affectés aux diverses activités de programme est quant à lui inconnu.Note de bas de page 54
Une autre difficulté, du point de vue de l'évaluation de l'efficacité et de l'économie, réside dans le fait que depuis 2008-2009 il n'y a plus de rapports qui sont produits en fonction des activités fonctionnelles, comme c'était le cas dans le cadre de l'ancienne AHP. Les activités fonctionnelles, aussi appelées domaines fonctionnels, comprennent les examens préliminaires, les évaluations de produits, les présentations de nouveaux produits, les activités de contrôle et de surveillance, l'éducation et la sensibilisation. De tels rapports axés sur des tâches ou des activités sont essentiels pour analyser l'efficacité et l'économie, car ils tiennent compte du temps pris par le personnel du programme pour exécuter différentes tâches ou activités et pour produire divers extrants. Cette information est importante pour évaluer :
- l'efficacité de l'attribution des ressources, qui est exprimée par la relation entre les ressources et les résultats;
- l'efficience opérationnelle, qui est exprimée par la relation entre les ressources et les extrants;
- l'économie, qui est exprimée par l'optimisation (y compris la minimisation) de l'utilisation des ressources.
Au sein de la DPT, le codage a été mis à jour à partir de 2012-2013 de manière à ce que l'information sur les activités fonctionnelles soit disponible dans l'avenir. Dans un rapport d'étape présenté récemment au Conseil du Trésor sur la gestion financière et le rendement des programmes relatifs aux instruments médicaux et aux médicaments pour usage humain (y compris les médicaments biologiques) dans le cadre du nouveau régime de recouvrement des coûts, Santé Canada a indiqué avoir mis en place des mesures de contrôle de la gestion financière, notamment un système amélioré de suivi du temps, destinées à valider les données sur le calcul des coûts (Santé Canada, 2013k). Il est donc possible qu'on ait recommencé à produire des rapports en fonction des activités fonctionnelles. En outre, Santé Canada a affirmé avoir instauré diverses autres mesures visant à soutenir une prestation efficace des services de réglementation, notamment les suivantes :
- une rationalisation des processus d'examen;
- la mise en place de systèmes, comme des PON, des lignes directrices et de la formation, permettant d'accroître l'efficacité des examens et pouvant être appliqués de façon constante pour améliorer la planification des présentations;
- l'utilisation de rapports d'examen étrangers et la collaboration avec d'autres organismes de réglementation;
- le renforcement des capacités scientifiques par l'embauche et le maintien en poste d'employés qualifiés;
- l'amélioration des systèmes de technologie de l'information soutenant le processus d'examen des présentations.
Dans le même rapport, Santé Canada a indiqué que grâce à l'augmentation des revenus découlant de la mise à jour des frais d'utilisation les activités ont bénéficié de meilleures ressources et « les cibles de recouvrement de coûts pour les premières décisions ont respecté la totalité des normes de rendement dans tous les secteurs de programme ». La même information figure également dans le RMR de 2011-2012.
Comme il a été mentionné à la section 5.4.3, l'application du nouveau cadre de recouvrement des coûts a donné lieu à une amélioration du rendement global des examens de présentations de médicaments pour usage humain dans toutes les catégories de présentations. Toutefois, à l'exception des PADN, les améliorations observées du rendement des examens ont commencé à apparaître dans toutes les catégories de présentations avant la mise en œuvre du nouveau cadre et pourraient par conséquent être attribuables aux diverses initiatives entreprises par Santé Canada au cours des dernières années pour améliorer son processus d'examen.
Il est important de signaler qu'une amélioration de la rapidité d'exécution ne s'accompagne pas nécessairement d'un accroissement de l'efficacité des examens des présentations, c'est-à-dire d'une production à coût moindre des extrants du programme, à savoir dans le cas présent les examens achevés. Une analyse plus détaillée des coûts permettrait de mieux saisir l'étendue des gains en efficacité éventuellement attribuables au nouveau régime de recouvrement des coûts, comme l'analyse réalisée en 2007 pour appuyer la proposition de mise à jour des frais d'utilisation, dans le cadre de laquelle a été calculé le coût unitaire des diverses activités de programme sujettes au recouvrement des coûts (Santé Canada et Spearhead Management Canada Limited, 2007).
Santé Canada a entrepris un examen des coûts, des frais et du rendement du PMHU en fonction de son engagement concernant la Proposition relative aux frais d'utilisation et le Résumé de l'étude d'impact de la réglementation associés au Règlement sur les prix à payer à l'égard des drogues et instruments médicaux. Les résultats préliminaires sont attendus pour 2014.
8.0 Conclusions et recommandations
Pertinence
L'évaluation a confirmé un besoin continu de surveillance gouvernementale en ce qui a trait aux médicaments à usage humain en vue de protéger la santé et la sécurité des Canadiens. Comme ces produits sont de plus en plus populaires en raison de l'accroissement et du vieillissement de la population, ainsi que de la publicité faite par les représentants de l'industrie, un nombre accru de Canadiens seront exposés à leurs risques et à leurs avantages. De plus, certaines tendances, comme l'émergence des produits mixtes et la mondialisation de la chaîne d'approvisionnement, créent des incertitudes qui accentuent la nécessité d'une intervention gouvernementale afin de protéger la santé et la sécurité des Canadiens. En outre, ce rôle cadre bien avec les rôles et les responsabilités du gouvernement fédéral et de Santé Canada décrits dans les lois et les règlements fédéraux, et est en harmonie avec le résultat stratégique de Santé Canada qui consiste à informer et à protéger les Canadiens des risques pour la santé que présentent les aliments, les produits, les substances et les environnements.
Les activités du PMUH correspondent bien aux priorités fédérales portant sur le renforcement de la sécurité du consommateur. Au cours de la dernière décennie, le gouvernement fédéral a consacré d'importantes ressources au développement d'initiatives visant l'amélioration de l'innocuité des produits de santé, notamment les médicaments à usage humain, grâce à la modernisation du cadre de réglementation de ces produits. Les principes clés de cette modernisation incluent l'adoption d'une approche axée sur le cycle de vie des produits, laquelle consiste à évaluer les risques et les avantages des produits thérapeutiques tout au long de leur cycle de vie; la mise en place de mesures réglementaires proportionnelles aux risques, et l'amélioration de la transparence et de l'ouverture du système de réglementation. Le gouvernement fédéral a récemment confirmé son engagement à l'égard de la viabilité à long terme du PMUH en ajoutant les médicaments à usage humain aux dernières mises à jour relatives à son cadre de recouvrement des coûts et aux frais d'utilisation.
Rendement - Mise en œuvre du programme
Au cours de la période visée par l'évaluation, Santé Canada a fait des progrès en mettant en œuvre les activités prévues et, par le fait même, en répondant à de nombreux problèmes et enjeux nouveaux. Cependant, plusieurs de ces problèmes et enjeux ne sont toujours pas résolus.
Essais cliniques
Santé Canada a renforcé le cadre de réglementation des essais cliniques en concrétisant des approches axées sur les risques en vue de contrôler les rapports sur les réactions indésirables survenues au cours d'un essai clinique, en lançant un programme d'inspection des sites d'essai clinique et en exigeant l'établissement de nouvelles normes facultatives pour les comités d'éthique de la recherche. En mai 2013, Santé Canada a lancé une nouvelle base de données publique sur les essais cliniques de médicaments qui avaient été autorisés par le ministère. Même si elle est obligatoire pour les promoteurs, la base de données contient moins de renseignements que les registres obligatoires depuis 1997 aux États-Unis et 2004 dans l'Union européenne. De plus, contrairement aux États-Unis (et bientôt à l'UE), Santé Canada n'oblige pas les promoteurs à publier les résultats des essais cliniques. Une augmentation de la quantité de renseignements disponibles sur les essais cliniques, y compris les résultats de ces essais, cadrerait avec l'engagement de Santé Canada visant à accroître la transparence et l'ouverture dans le cadre de la modernisation de la réglementation, et permettrait au ministère d'harmoniser davantage son approche avec celle de ses principaux homologues internationaux.
Recommandation 1:
Conformément aux tendances internationales et à son engagement visant accroître la transparence et l'ouverture dans le cadre de la modernisation de la réglementation, Santé Canada devrait publier davantage de renseignements sur les essais cliniques, y compris les résultats de ces essais.
Examen des présentations et autorisation de mise sur le marché
Santé Canada a franchi une étape importante avec l'entrée en vigueur du Règlement sur les prix à payer à l'égard des drogues et instruments médicaux en avril 2011. Le règlement actualise les frais d'utilisation pour différents services de réglementation, y compris l'examen des présentations et les licences d'établissement de produits pharmaceutiques, dans le but de rétablir le ratio de partage des coûts de 50 % en vigueur lors de l'adoption des frais d'utilisation au milieu des années 1990. On s'attend à ce que l'augmentation des revenus résultant de la mise à jour des frais d'utilisation favorise la stabilité de la plateforme financière, ce qui permettra à Santé Canada d'offrir plus de services de réglementation.
En plus du nouveau cadre de recouvrement des coûts, Santé Canada a également lancé de nombreuses initiatives visant l'amélioration de la qualité et de l'efficience du processus d'examen des présentations et d'autorisation de mise sur le marché. Par exemple : la mise en œuvre d'une approche de gestion de projet pour l'examen des présentations; l'élaboration de Bonnes pratiques de lignes directrices et de Bonnes pratiques d'examen en vue d'accroître l'uniformité de ces processus; la collaboration avec la Food and Drug Administration (FDA) par l'intermédiaire du Conseil de coopération en matière de réglementation (CCR) en vue d'élaborer le Portail commun de demandes électroniques, lequel permettra aux promoteurs de présenter des demandes simultanément à Santé Canada et à la FDA; le renforcement de la capacité d'examen scientifique; et la prise de mesures dans le but d'augmenter le nombre d'examens étrangers effectués relativement aux demandes canadiennes.
L'incidence de ces initiatives sur l'efficience et la qualité du processus d'examen donne matière à de plus amples analyses. Au cours de la période visée par l'évaluation, les délais relatifs à l'examen des présentations variaient et la cible de rendement de Santé Canada n'était généralement pas atteinte. Toutefois, depuis la mise en œuvre du nouveau cadre de recouvrement des coûts en avril 2011, la DPT a atteint la majorité de ses cibles de rendement en matière de recouvrement des coûts, à l'exception de ceux relatifs aux médicaments génériques, et le rendement des examens des présentations est demeuré assez stable.
Surveillance post-commercialisation
Depuis toujours, les rapports sur les réactions indésirables sont les piliers sur lesquels s'appuie l'approche de Santé Canada en matière de surveillance post-commercialisation. En règle générale, les rapports obligatoires sur les réactions indésirables par les représentants de l'industrie ne posent pas problème. En vue de traiter les enjeux de longue date relatifs à la sous-déclaration des professionnels de la santé, Santé Canada a exigé la création de nouvelles normes nationales concernant les rapports sur les réactions indésirables à l'intention des établissements de santé et des hôpitaux agréés. Ces normes ont été publiées en janvier 2013. À ce moment, Santé Canada a mentionné qu'il envisageait de surveiller l'incidence de ces nouvelles normes avant de décider de procéder à des modifications réglementaires. Les rapports obligatoires sur les réactions indésirables pour les établissements de santé avaient déjà fait l'objet du projet de loi C-51 en 2008, soit une mesure législative d'envergure visant à aborder les différentes lacunes du cadre de réglementation actuel relativement aux produits de santé. Le projet de loi C-51 est mort au Feuilleton et n'est jamais devenu une loi.
Santé Canada procède actuellement à l'élaboration de systèmes d'information en vue d'accepter les rapports sur les réactions indésirables par voie électronique, de les contrôler et de les analyser dans le but de détecter les signaux d'innocuité. Plus particulièrement, Santé Canada met en œuvre un système électronique pour la présentation de rapports sur les réactions indésirables destiné aux représentants de l'industrie, ainsi que des stratégies visant à contrôler systématiquement ces rapports au moyen d'une surveillance ciblée et de l'exploration des données. Malgré les délais causés par des difficultés techniques, on s'attend à ce que ces initiatives renforcent la capacité du ministère à analyser les rapports sur les réactions indésirables à l'échelle nationale en vue de détecter les signaux d'innocuité. On espère également que la présentation électronique de rapports sur les réactions indésirables et l'exploration des données permettront à Santé Canada d'analyser les rapports étrangers, lesquels représentent plus de 90 % des tous les rapports reçus par la Direction générale. En attendant que le ministère mette ces systèmes d'information et ces fonctions en place, les initiatives (comme les normes nationales concernant les rapports sur les réactions indésirables) visant à accroître le nombre de rapports reçus sont peut-être prématurées.
Au cours des dernières années, comme d'autres organismes de réglementation partout dans le monde, Santé Canada a reconnu les limites des rapports spontanés sur les réactions indésirables pour détecter les signaux d'innocuité et a cherché à renforcer la surveillance post-commercialisation de nombreuses façons. En plus des rapports sur les réactions indésirables, Santé Canada surveille désormais d'autres sources d'information visant à détecter les signaux d'innocuité, et a normalisé le processus de détection des signaux et d'évaluation pour l'ensemble de ses gammes de produits. Toutefois, le ministère ne dispose pas d'une approche normalisée et centralisée lui permettant d'effectuer un suivi systématique de ses activités liées aux signaux et de sa réponse aux mesures recommandées découlant des évaluations de signaux réalisées. À l'heure actuelle, cette information n'est pas mise à jour régulièrement et est très dispersée au sein de la DGPSA. Étant donné les risques pour la santé et la sécurité des Canadiens, une approche globale, uniforme et centralisée quant à la gestion de l'information des activités de surveillance post-commercialisation de Santé Canada semble justifiée.
Recommandation 2:
Santé Canada devrait améliorer ses systèmes d'information pour le contrôle et la surveillance post-commercialisation. Cette amélioration devrait inclure les points suivants :
- Mise en œuvre intégrale des systèmes d'information en vue d'appuyer la présentation électronique de rapports sur les réactions indésirables survenues au Canada et à l'étranger, ainsi que le contrôle et l'analyse systématiques de ces présentations au moyen d'une surveillance ciblée et de l'exploration des données;
- Élaboration et mise en œuvre d'une approche approfondie et centralisée en matière de gestion de l'information en ce qui a trait aux activités de surveillance post-commercialisation. Plus particulièrement, il faut mettre en place un mécanisme centralisé afin d'effectuer le suivi des activités de détection des signaux d'innocuité, notamment la réponse de Santé Canada aux mesures recommandées découlant des évaluations de signaux réalisées.
Malgré l'annonce faite à cet égard dans le cadre du Plan d'action pour assurer la sécurité des produits alimentaires et de consommation (PASPAC), Santé Canada n'a toujours pas concrétisé un seul des éléments des approches renforcées en matière de surveillance post-commercialisation en place ailleurs, à savoir l'autorité nécessaire afin d'obliger les fabricants à fournir des plans de gestion des risques et des rapports réguliers concernant l'innocuité. Les représentants de l'industrie peuvent toutefois présenter ces documents de façon volontaire en réponse aux demandes de Santé Canada. Même si le ministère reconnaît que l'absence de ces autorités représente une limite de son approche, on ne sait pas vraiment s'il a l'intention de les obtenir dans le futur. Un autre domaine d'incertitude est la mesure dans laquelle Santé Canada contrôle la conformité des fabricants aux conditions imposées dans le cadre des avis de conformité avec conditions.
Recommandation 3:
Santé Canada devrait évaluer s'il est toujours pertinent de poursuivre la mise en place d'organismes de réglementation post-commercialisation comme il s'y est engagé dans le cadre du PASPAC, à savoir accorder l'autorité nécessaire afin d'obliger les fabricants à fournir des plans de gestion des risques et des rapports réguliers concernant l'innocuité.
Conformité et application
En ce qui concerne la conformité et l'application, Santé Canada a renforcé les inspections des essais cliniques en élaborant un processus de sélection des sites axé sur les risques; a consolidé la surveillance en ce qui a trait aux produits importés par l'intermédiaire du Programme national de l'intégrité frontalière; a changé le nom du Programme d'inspection de la conformité aux exigences de déclaration après commercialisation (CEDAC), qui se nomme maintenant le programme d'inspection des Bonnes pratiques de pharmacovigilance (BPV), afin de respecter les exigences internationales; a élargi les exigences relatives aux Bonnes pratiques de fabrication (BPF) et à la licence d'établissement de produits pharmaceutiques (LEPP) en vue d'englober les ingrédients pharmaceutiques actifs (IPA); et a adopté une approche axée sur les risques relativement aux inspections des BPF des établissements pharmaceutiques canadiens.
Les événements récents, comme le ralentissement de la production d'un fabricant de produits génériques à la suite des inspections de la FDA, lesquelles ont révélé plusieurs non-conformités, ont porté l'attention du public sur le programme d'inspection des BPF de Santé Canada, en plus de soulever des questions à cet égard. Même si certains informateurs clés externes se sont montrés préoccupés par les interprétations divergentes quant aux normes et à la qualité des inspections, Santé Canada a mentionné que celles-ci sont probablement attribuables aux différences relatives aux approches des deux organismes de réglementation en matière d'inspection, et à la portée de ces inspections.
Un autre problème concerne peut-être la surveillance qu'exerce Santé Canada sur les établissements étrangers qui fabriquent des médicaments qui seront importés au Canada. Même si le Canada a signé des accords de reconnaissance mutuelle (ARM) avec plusieurs pays et qu'il reconnaît ainsi l'équivalence de leurs programmes de conformité aux BPF, la majorité des produits pharmaceutiques et des IPA importés au Canada proviennent de pays avec lesquels le Canada n'a pas conclu d'accord. Dans le cas des IPA, l'importation représente 85 % du marché canadien, et la Chine et l'Inde sont les principaux fournisseurs. De plus, même si les établissements canadiens sont inspectés régulièrement conformément aux critères axés sur les risques, Santé Canada n'effectue pas souvent d'inspections dans les établissements étrangers. On s'attend donc à ce que l'élargissement récent des exigences relatives aux BPF et à la LEPP en vue d'englober les IPA réduise le risque que des produits de santé de mauvaise qualité ou de contrefaçon se retrouvent sur le marché canadien.
Afin de surmonter les difficultés associées aux inspections des BPF, Santé Canada et la FDA des États-Unis ont lancé une initiative par l'intermédiaire du CCR dont le but est d'accroître, dans chaque pays, le recours aux rapports d'inspection des BPF préparés par l'autre pays. À l'heure actuelle, l'initiative s'applique aux sites canadiens et américains, bien qu'il soit possible qu'elle s'applique à d'autres pays dans le futur. En outre, Santé Canada est membre du Pharmaceutical Inspection Cooperation Scheme (PIC/S), un organisme composé de 43 autorités internationales participantes qui se sont réunies en vue d'harmoniser les approches et les exigences en matière d'inspection des BPF partout dans le monde. Santé Canada examine les rapports d'inspection du PIC/S afin d'approuver les sites étrangers pour les importateurs canadiens.
Recommandation 4:
Santé Canada devrait examiner les façons d'améliorer sa surveillance des établissements producteurs à l'étranger.
Même si l'un des principes clés de l'initiative du CCR porte sur la normalisation et le partage des rapports d'inspection des BPF, Santé Canada et la FDA optent pour des approches d'établissement de rapport différentes. En effet, la FDA établit des rapports en fonction de la catégorie des produits, alors que Santé Canada rassemble tous les rapports sur les BPF, peu importe le type de produit concerné. Il est possible que l'atteinte des objectifs de l'initiative nécessite une approche commune en matière d'établissement de rapports de conformité.
Enfin, Santé Canada a inclus de nouvelles mesures de gestion et d'application dans le cadre du projet de loi C-51. Ces mesures comprenaient le pouvoir d'ordonner le rappel de produits thérapeutiques et celui d'imposer des amendes plus lourdes en cas de non-conformité. Comme il a été mentionné précédemment, le projet de loi C-51 n'est pas devenu une loi. Lors d'entrevues, certains informateurs clés externes se sont montrés préoccupés par ce qu'ils considèrent comme le peu d'options dont dispose Santé Canada en matière d'application.
Recommandation 5:
Santé Canada devrait se pencher sur la question à savoir s'il est encore justifié d'obtenir le pouvoir de prendre des mesures de conformité et d'application comme il s'y est engagé dans le cadre du PASPAC, à savoir le pouvoir d'ordonner le rappel de produits thérapeutiques et celui d'imposer des amendes plus lourdes en cas de non-conformité.
Communications et mobilisation des intervenants
Santé Canada a entrepris de nombreuses initiatives en vue d'améliorer les communications et la mobilisation des intervenants. Par exemple, depuis 2005, Santé Canada fournit au public des renseignements sur les décisions relatives aux examens par l'intermédiaire du Sommaire des motifs de décision (SMD). En 2012, en partie en vue d'aborder les préoccupations exprimées par le Bureau du vérificateur général (BVG) selon lesquelles le ministère ne divulguait pas de renseignements liés aux avis de conformité avec conditions (AC-C), aux médicaments rejetés et aux médicaments retirés du processus d'examen, Santé Canada a mis en place le tableau des activités postautorisation (TAPA). Les TAPA fournissent des renseignements à jour sur les produits approuvés. Ils incluent un résumé des activités portant sur l'utilisation sécuritaire et efficace du produit, comme les renseignements liés aux présentations en vue d'une nouvelle utilisation du produit (peu importe si la décision de Santé Canada était positive ou négative), les présentations soumises dans le but de respecter les conditions (pour les produits approuvés en vertu des lignes directrices de l'AC-C), et les décisions réglementaires, comme l'annulation du numéro d'identification du médicament (DIN). Santé Canada ne publie pas de SMD en cas de décisions négatives et ne divulgue pas les raisons de ces décisions, contrairement à la FDA et à l'Agence européenne des médicaments (EMA).
Afin d'accroître la qualité et la disponibilité d'étiquettes de produits pharmaceutiques faciles à comprendre, Santé Canada a amélioré la monographie des produits, a créé, par l'intermédiaire du CCR, une monographie commune avec la FDA pour les médicaments en vente libre, et a apporté des modifications réglementaires dans le cadre de l'initiative d'étiquetage en langage clair. À l'heure actuelle, Santé Canada n'a pas l'autorité nécessaire pour exiger aux fabricants de modifier l'étiquetage d'un produit lorsque ce dernier a reçu un avis de conformité. En revanche, la FDA peut exiger que l'étiquetage des produits soit mis à jour en fonction des nouveaux renseignements disponibles sur l'innocuité d'un médicament, y compris le pouvoir d'exiger un encadré à cet effet. La nouvelle législation de l'UE en matière de pharmacovigilance ordonne qu'un triangle noir inversé soit placé sur le dépliant des médicaments faisant l'objet d'une surveillance post-commercialisation accrue.
Au cours de la période visée par l'évaluation, Santé Canada a publié des renseignements sur les risques et l'innocuité des médicaments sur la page « Avis, mises en garde et retraits » du site Web de MedEffet, et au moyen d'autres mécanismes de diffusion variés Au début de l'année 2013, il a lancé la Base de données sur les rappels et les avis de sécurité, laquelle comprend une fonction de recherche avancée et un nouveau format aux fins de communication des risques. Le ministère a mentionné qu'il procède actuellement à l'examen de ses cibles de rendement actuelles concernant la préparation et la diffusion de documents de communication des risques, et qu'il se penche sur des domaines dans lesquels il serait possible d'apporter des améliorations axées davantage sur la communication des risques. En outre, Santé Canada a récemment entrepris l'évaluation de sa communication des risques relativement aux produits de santé, y compris les médicaments à usage humain, en donnant suite à des plans de longue date visant à évaluer l'efficacité de ses produits de communication des risques.
Santé Canada favorise la mobilisation des intervenants de multiples façons, comme la tenue de consultations publiques sur les lignes directrices, les politiques et les modifications réglementaires proposées, ainsi que la mise en place de comités consultatifs en vue d'éclairer l'élaboration de politiques et de règlements. Le ministère consulte également les représentants de l'industrie au moyen de réunions préalables au dépôt des présentations et de programmes des réunions bilatérales (PRB). Même si ces possibilités particulières ne sont pas offertes aux professionnels de la santé et aux consommateurs/patients, Santé Canada a indiqué qu'il rencontre toutefois les associations pharmaceutiques, les associations médicales et les associations des hôpitaux qui représentent ces professionnels. Certains informateurs clés externes se sont dits préoccupés par le fait que le processus de consultation et de mobilisation puisse favoriser les représentants de l'industrie plutôt que les autres intervenants.
Rendement -- Résultats atteints
Au cours de la période visée par l'évaluation, Santé Canada a participé à de nombreuses activités qui devraient favoriser l'atteinte des résultats du PMUH. Toutefois, pour différentes raisons, les données appuyant les conclusions définitives quant aux résultats atteints sont relativement limitées.
Résultats immédiats
À très court terme, on s'attend à ce que les activités de Santé Canada accroissent la sensibilisation et la compréhension des intervenants externes quant aux risques et aux avantages des médicaments à usage humain. Les sondages menés entre 2003 et 2007 ont révélé des façons d'accroître la sensibilisation des consommateurs et des professionnels de la santé en ce qui a trait à l'information sur l'innocuité des médicaments disponible auprès de Santé Canada, mais aucun renseignement précis n'était disponible sur les médicaments à usage humain. Le ministère procède actuellement à l'évaluation de l'efficacité de ses communications des risques en ce qui concerne les produits thérapeutiques.
À très court terme, on s'attend à ce que les activités de Santé Canada accroissent la sensibilisation et la compréhension des représentants de l'industrie quant aux activités de réglementation du ministère relativement aux médicaments à usage humain. Les données disponibles, bien qu'elles soient limitées, indiquent certains aspects susceptibles d'amélioration. Par exemple, même si les répondants au sondage mené auprès des représentants de l'industrie croient que leur entreprise connaît bien les exigences de Santé Canada en ce qui concerne les demandes d'approbation de mise en marché, les informateurs clés de l'industrie ont affirmé que des précisions seraient nécessaires en ce qui a trait à la classification des nouveaux produits de santé et à l'utilisation que fait Santé Canada des lignes directrices et des examens étrangers dans le cadre du processus d'examen.
À court terme, les activités de Santé Canada visent également à augmenter l'innocuité et l'efficacité des médicaments à usage humain. Des processus préalables à la mise en marché et après la mise en marché sont en place en vue de s'assurer que les médicaments à usage humain sont sécuritaires et efficaces; toutefois, aucune donnée ne confirme des améliorations dans ces domaines. Il serait possible de prétendre que l'autorité de Santé Canada consiste uniquement à veiller à ce que les produits disponibles sur le marché canadien soient sécuritaires et efficaces.
Enfin, à court terme, on s'attend à ce que les activités de Santé Canada favorisent la conformité des représentants de l'industrie aux exigences réglementaires. Les données disponibles indiquent que les non-conformités graves sont assez rares. Au cours de la période visée par l'évaluation, Santé Canada n'a pas produit de rapports de façon régulière ou systématique sur la nature, la gravité, la fréquence ou la prévalence des cas de non-conformité liés aux médicaments à usage humain; il a plutôt axé ses rapports sur la mesure des activités et des extrants. De plus, la majorité des données de conformité de Santé Canada, y compris celles sur les BPF, les Bonnes pratiques cliniques (BPC) et les BPV, sont regroupées au sein de différentes catégories de produits, englobant les médicaments à usage humain et biologiques (ainsi que les médicaments vétérinaires dans le cas des BPF).
Ceci étant dit, l'Inspectorat a récemment rédigé un rapport sommaire des inspections annuelles, lequel sera publié sur le site Web de Santé Canada. Le rapport 2012-2013 fournit une description des activités et des extrants de l'Inspectorat, décrit le taux de conformité global de l'industrie et dresse une liste des observations communes citées dans les établissements non conformes. Le rapport ne contient aucun renseignement sur les mesures prises par l'Inspectorat en réponse aux cas de non-conformité. Il ne décompose pas non plus l'information portant sur la conformité des BPF, des BPC et des BPV par catégorie de produits.
Recommandation 6:
Santé Canada devrait continuer de miser sur son approche actuelle en matière de rapports sur la conformité en se concentrant davantage sur les résultats des activités de conformité et d'application, et en renforçant sa capacité à produire des rapports sur la conformité par catégorie de produits.
Résultats intermédiaires
À moyen terme, on s'attend à ce que les activités de Santé Canada encouragent les intervenants à adopter des comportements sécuritaires en ce qui a trait à l'utilisation de médicaments à usage humain. Même si la littérature confirme que des pratiques dangereuses, comme l'abus de produits pharmaceutiques sur ordonnance, sont utilisées au Canada, on ne connaît pas l'incidence que peuvent avoir les activités du ministère à cet égard. L'évaluation continue de Santé Canada quant à l'efficacité de ses communications des risques peut indiquer l'influence qu'ont les activités du ministère sur le comportement des intervenants. Dans ce contexte, il est important de noter que la pratique de la médecine, laquelle est régie par les provinces et les territoires, influe également sur le comportement des intervenants.
Les activités de Santé Canada devraient également favoriser l'utilisation des données scientifiques et de l'analyse des avantages et des risques en vue d'éclairer la prise de décisions relative aux médicaments à usage humain. L'utilisation des données scientifiques et de l'analyse des avantages et des risques a été officiellement intégrée au cadre décisionnel de Santé Canada pour la détermination, l'évaluation et la gestion des risques pour la santé, ainsi qu'à différents processus préalables à la mise en marché et après la mise en marché. Le ministère a créé de nombreux groupes consultatifs d'experts/de scientifiques afin d'orienter l'élaboration de politiques et de règlements, et a concrétisé une partie de leurs recommandations.
À moyen terme, Santé Canada espère réagir rapidement aux risques liés aux médicaments à usage humain. En admettant que l'élaboration de politiques et de règlements soit souvent un processus très long, l'évaluation a permis de relever des exemples dans lesquels la mise en œuvre de la réponse de Santé Canada a pris plus de temps que prévu. Par exemple, plus d'une décennie s'est écoulée entre le moment où Santé Canada a annoncé son intention de mettre en place des exigences relatives aux BPF et à la LEPP en vue d'englober les IPA et la modification réglementaire. Ou, le ministère s'est engagé à accroître la transparence des renseignements sur les essais cliniques dès 2007, mais n'a mis en place une base de données publique à cet effet qu'en mai 2013.
L'analyse des données relatives aux signaux de Santé Canada a permis de révéler que de nombreux secteurs risquent de connaître des retards. Ces données indiquent que même si le ministère se fie à de nombreuses sources pour détecter les signaux d'innocuité liés aux médicaments à usage humain, y compris les données des promoteurs, les deux principales sources demeurent les ouvrages scientifiques et les autres organismes de réglementation. Comme il peut être très long avant que les résultats des études scientifiques soient publiés et que les autres organismes de réglementation évaluent l'innocuité, il est possible que Santé Canada ait de la difficulté à réagir rapidement. Les efforts du ministère en vue de promouvoir la présentation de rapports et d'analyses sur les réactions indésirables peuvent contribuer à la résolution de ce problème, qui dépendra alors moins des études externes.
La rapidité du processus d'évaluation des signaux s'est améliorée depuis la mise en place des normes de service relatives à l'évaluation des signaux. En 2011-2012, 93 % des évaluations des signaux après la mise en marché concernant les produits pharmaceutiques, les instruments médicaux, les produits biologiques et les produits de santé naturels ont été réalisées à l'intérieur du délai de 130 jours prescrit par la norme de service. Toutefois, l'évaluation des signaux représente seulement un aspect de la réponse globale de Santé Canada en matière de signaux d'innocuité. L'évaluation a révélé que les retards peuvent survenir à d'autres moments pendant le processus. Notamment, une période de temps considérable peut s'écouler entre le moment où les signaux sont détectés et celui où on les évalue, de même qu'entre l'approbation de l'évaluation du signal et la communication du risque.
Ce résultat cadre avec le rapport 2011 du BVG, lequel indique que l'évaluation et la réponse du ministère à l'égard des signaux d'innocuité potentiels n'ont pas lieu en temps opportun. Même si la majorité des évaluations de l'innocuité étudiées par le BVG dans le cadre de sa vérification ont été réalisées dans les délais prescrits, l'approche employée par le ministère pour mesurer son rendement n'a pas tenu compte du temps écoulé avant que le problème d'innocuité potentiel ne soit évalué; du temps d'attente avant le début de l'évaluation; du temps nécessaire pour obtenir des renseignements supplémentaires auprès des parties externes (comme les fabricants); et du nombre total de jours civils, plutôt que de jours ouvrables, requis pour effectuer l'évaluation. Le BVG a constaté que lorsque ces facteurs ont été pris en considération, il a fallu au moins une année à Santé Canada pour réaliser 34 de ses 54 évaluations, et même beaucoup plus de temps dans certains cas - deux ou trois ans.
Les représentants du programme ont signalé que la préparation des documents de communication des risques nécessite habituellement de nombreuses négociations avec le détenteur d'une autorisation de mise sur le marché, et que la diffusion du document peut être retardée jusqu'à ce que les changements appropriés aient été apportés à l'étiquette du produit. En raison de ces facteurs, le temps écoulé entre le moment où le signal est détecté et la diffusion du document de communication du risque peut être très long. En ce qui concerne 14 cas pour lesquels des données complètes étaient disponibles (portant sur des évaluations des signaux réalisées entre 2010 et 2012), le temps total écoulé entre la détection du signal et la publication du document de communication du risque variait entre 232 jours et 1 481 jours (4 ans), avec une médiane de 1,4 an. Comme il a été mentionné, Santé Canada procède à l'examen de ses normes de rendement actuelles relativement à la préparation et à la diffusion de documents de communication des risques.
Recommandation 7:
Santé Canada devrait prendre des mesures pour augmenter la rapidité avec laquelle il donne suite aux signaux relatifs à l'innocuité (de la détection de ces signaux à la diffusion d'une communication des risques).
À moyen terme, Santé Canada espère accroître l'uniformité des exigences réglementaires concernant les médicaments à usage humain à l'échelle internationale. Parmi de nombreux autres engagements internationaux, Santé Canada est un observateur officiel de la Conférence internationale sur l'harmonisation (CIH) des exigences techniques pour l'enregistrement des médicaments à usage humain, en plus d'y participer; a élaboré des ARM quant à la conformité aux BPF avec plusieurs autres pays; a conclu des ententes sur l'échange de renseignements avec la FDA, la Therapeutic Goods Administration (TGA) de l'Australie et l'Union européenne; et participe, avec la FDA, au CCR, dont le principal objectif consiste à améliorer l'uniformité des approches réglementaires des deux pays. Comme il a été mentionné, Santé Canada est aussi membre du PIC/S. D'importants informateurs internationaux perçoivent Santé Canada comme un participant constructif dans les engagements bilatéraux et multilatéraux.
Résultats à long terme
À long terme, les activités de Santé Canada contribueront probablement à la réduction des risques pour la santé et des événements indésirables liés à l'utilisation des médicaments à usage humain. D'après les données d'enquête disponibles, le taux de confiance des consommateurs en ce qui concerne l'innocuité des médicaments et la sécurité du système de réglementation en place est élevé, même si aucun renseignement précis sur les médicaments à usage humain n'est disponible.
En définitive, Santé Canada espère offrir un système de réglementation viable, rentable, adapté et fondé sur la science en ce qui a trait aux médicaments à usage humain au Canada. L'évaluation a révélé que le ministère a recours aux données scientifiques et consulte des intervenants pour l'élaboration de politiques et de règlements, et que les récentes mises à jour concernant les frais d'utilisation relatifs aux médicaments à usage humain appuieront la viabilité du système de réglementation de ces produits.
Rendement - efficience et économie
Au cours de la période visée par l'évaluation, les changements apportés à l'approche de la DGPSA en ce qui concerne l'établissement de rapports financiers ont compliqué l'harmonisation des dépenses et des budgets relatifs au PMUH permettant la comparaison et l'analyse ultérieures de ces renseignements. La DGPSA a récemment réorganisé la façon dont elle communique l'information financière afin de respecter les exigences du Conseil du Trésor; elle devrait ainsi accroître l'exactitude de ces renseignements et faciliter les prochaines analyses.
Au sein de la DPT, même si aucun rapport financier axé sur les activités n'a été rédigé depuis 2008-2009, le codage a été mis à jour en 2012-2013, de sorte que les renseignements sur les activités fonctionnelles seront disponibles ultérieurement. Les rapports axés sur les activités permettent d'appuyer la comptabilité par activités, laquelle est essentielle à l'analyse de l'efficience des activités de programme réalisées. La DGPSA a entrepris un exercice de comptabilité par activités en 2007 en vue de soutenir la proposition visant à mettre à jour les frais d'utilisation. Elle a ensuite utilisé les résultats de cette analyse pour calculer les coûts unitaires de différentes activités de réglementation, y compris l'examen des présentations. Les données disponibles indiquent que la rapidité à laquelle sont effectués les examens des présentations de la majorité des catégories de médicaments à usage humain s'est améliorée grâce au nouveau cadre de recouvrement des coûts. Toutefois, sans analyser les coûts unitaires de l'examen des présentations et d'autres activités de recouvrement des coûts, on ne sait pas si la rapidité accrue a permis la réalisation de gains d'efficacité. En plus de permettre l'évaluation de la portée des gains d'efficacité réalisés grâce au nouveau cadre, l'analyse permettra également au PMUH de déterminer les ajustements nécessaires. À cette fin, la DGPSA examine donc les coûts, les frais et le rendement associés au PMUH conformément à son engagement dans le cadre de la proposition de frais d'utilisation et du Résumé de l'étude d'impact de la réglementation liés au Règlement sur les prix à payer à l'égard des drogues et instruments médicaux.
Annexe A - Grille d'évaluation
| Enjeux et questions d'évaluation | Indicateurs | Sources possibles de données | |
|---|---|---|---|
| Section 1 : Pertinence | |||
| Enjeu 1 : Nécessité continue du programme | |||
| 1. Existe-t-il toujours un besoin pour le PMUH? |
|
Examen de la documentation :
|
|
|
Examen de la documentation :
Revue de la littérature |
||
|
Entrevues auprès d'informateurs clés (internes et externes). | ||
|
Examen de la documentation. |
||
| Enjeu 2 : Respect des priorités du gouvernement | |||
| 2.Le PMUH concorde-t-il avec les priorités du gouvernement du Canada? |
|
Examen de la documentation :
|
|
|
Examen de la documentation :
|
||
| Enjeu 3 : Conformité avec les rôles et les responsabilités du gouvernement fédéral | |||
| 3. Le PMUH cadre-t-il avec les rôles et les responsabilités du gouvernement fédéral? |
|
Examen de la documentation :
|
|
|
Examen de la documentation :
|
||
| Section 2 : rendement (efficacité, efficience et économie) | |||
| Enjeu 4 : Obtention des résultats escomptés | |||
| 4. La structure de gouvernance du PMUH est-elle susceptible de favoriser l'atteinte des résultats escomptés? | |||
| a) Y a-t-il une structure de gouvernance établie permettant de coordonner la prestation du PMUH? |
|
Examen de la documentation :
Entrevues auprès d'informateurs clés (internes et externes, c.-à-d. d'autres ministères fédéraux) |
|
Portée de la collaboration entre les partenaires internes et interministériels, démontrée par les éléments suivants :
|
Examen de la documentation :
Entrevues auprès d'informateurs clés (internes et externes, c.-à-d. d'autres ministères fédéraux) |
||
|
Examen de la documentation :
Entrevues auprès d'informateurs clés (internes et externes). |
||
| b) Un cadre de mesure du rendement a-t-il été conçu et mis en place? |
|
Examen de la documentation :
|
|
|
Examen de la documentation :
Entrevues auprès d'informateurs clés (internes) |
||
| c) Le cadre de mesure du rendement est-il utilisé pour appuyer les processus décisionnels? |
|
Examen de la documentation :
Entrevues auprès d'informateurs clés (internes) |
|
| 5. Dans quelle mesure le PMUH a-t-il été mis en œuvre comme prévu? | |||
| a) Les responsables du PMUH ont-ils donné suite aux défis, aux problèmes émergents et à l'évolution des priorités de façon efficace? | Mesure dans laquelle les responsables du programme ont donné suite aux défis, aux problèmes émergents et à l'évolution des priorités de façon efficace, en ce qui concerne, par exemple :
|
Examen de la documentation (tous les documents) |
|
b) Les activités ont-elles été mises en œuvre comme prévu? c) Les activités ont-elles produit les résultats escomptés? |
|
Examen de la documentation - planification :
|
Examen de la documentation - activités réelles :
|
| d) Les exigences ou engagements à l'égard des organismes centraux (Bureau du vérificateur général, Directive du Cabinet sur la rationalisation de la réglementation, Politique sur la consultation du public, Politique en matière d'analyse comparative entre les sexes) ont-ils été respectés? |
|
Examen de la documentation (tous les documents) |
|
| 6) Dans quelle mesure les résultats escomptés ont-ils été atteints? | |||
| Résultats immédiats | |||
| a. Dans quelle mesure les intervenants externes connaissent-ils et comprennent-ils davantage les risques et les avantages liés aux médicaments pour usage humain? | Fréquence et nature des communications de Santé Canada avec les intervenants externes, par type et groupe cible, y compris :
|
Examen de la documentation et des données administratives, y compris les données accessibles au public depuis le site Web de Santé Canada | |
|
Examen de la documentation | ||
|
Examen de la documentation | ||
|
Examen de la documentation |
||
|
Examen des documents et des données administratives | ||
|
Sondage mené auprès des fournisseurs de soins de santé |
||
Évaluation par les intervenants de la qualité de l'information et des communications de Santé Canada, en ce qui concerne :
|
Sondage mené auprès des fournisseurs de soins de santé |
||
|
Sondage mené auprès des fournisseurs de soins de santé |
||
|
Sondage mené auprès des fournisseurs de soins de santé |
||
| b. Dans quelle mesure l'industrie connaît-elle et comprend-elle davantage le cadre de réglementation des médicaments pour usage humain de Santé Canada? | Fréquence et nature des communications, rencontres et consultations de Santé Canada avec les intervenants de l'industrie concernant le cadre de réglementation des médicaments pour usage humain, y compris :
|
Examen des documents et des données administratives | |
|
Examen de la documentation : | ||
|
Examen de la documentation : | ||
|
Sondage auprès de l'industrie |
||
Évaluation par l'industrie de la qualité de l'information et des communications de Santé Canada, en ce qui concerne :
|
Sondage auprès de l'industrie |
||
|
Sondage auprès de l'industrie |
||
|
Sondage auprès de l'industrie Entrevues auprès d'informateurs clés (externes) |
||
| c. Dans quelle mesure les médicaments pour usage humain sont-ils plus sécuritaires et efficaces? |
|
Examen de la documentation |
|
|
Examen de la documentation |
||
Étendue et nature des initiatives, outils, approches et activités préalables à la commercialisation visant à accroître la sécurité et l'efficacité, par exemple :
|
Examen des documents et des données administratives |
||
Étendue et nature des initiatives, outils, approches et activités après la commercialisation visant à accroître la sécurité et l'efficacité, par exemple :
|
Examen des documents et des données administratives |
||
|
Examen des documents et des données administratives, surtout :
|
||
|
Examen des documents et des données administratives :
|
||
|
Examen des documents et des données administratives | ||
|
Examen des documents et des données administratives |
||
|
Examen des documents et des données administratives |
||
|
Examen des données administratives |
||
|
Sondage mené auprès de l'industrie, des fournisseurs de soins de santé et des clients/patients |
||
|
Revue de la littérature |
||
| d. Dans quelle mesure l'industrie respecte-t-elle davantage les exigences réglementaires de Santé Canada relatives aux médicaments pour usage humain? |
|
Examen de la documentation | |
|
Examen de la documentation |
||
|
Examen de la documentation |
||
|
Examen des documents et des données administratives
|
||
(Conformité après la commercialisation) Tendances relatives au nombre des inspections, des vérifications de la conformité et des autres activités de contrôle de la conformité et d'application de la réglementation en lien avec :
|
Examen des documents et des données administratives
|
||
|
Examen des documents et des données administratives
|
||
(Conformité post-commercialisation) Tendances relatives au nombre et aux résultats des mesures de respect et d'application de la réglementation, y compris :
|
Examen des documents et des données administratives
|
||
|
Examen des documents et des données administratives
|
||
| Résultats intermédiaires | |||
| e. Dans quelle mesure les intervenants externes adoptent-ils des comportements sécuritaires au chapitre des médicaments pour usage humain? |
|
Entrevues auprès d'informateurs clés (informateurs externes, en particulier des établissements/professionnels en santé et des groupes de clients/patients) |
|
Nombres des intervenants qui ont indiqué avoir modifié leur comportement en raison de documents de communication des risques produits par Santé Canada, par exemple :
|
Sondage mené auprès des fournisseurs de soins de santé |
||
|
Revue de la littérature |
||
| f. Dans quelle mesure y a-t-il une utilisation accrue des données scientifiques et des analyses risques-avantages de Santé Canada dans les processus décisionnels? |
|
Examen de la documentation |
|
Étendue et nature des activités post-commercialisation de Santé Canada pour accroître l'utilisation de données scientifiques et d'analyses axées sur les risques dans les processus décisionnels, par exemple :
|
Examen des documents et des données administratives |
||
Nombre de signaux indiquant un possible problème d'innocuité captés grâce à des activités de détection des signaux post-commercialisation, y compris :
|
Examen des documents et des données administratives |
||
Mesure dans laquelle l'information réunie dans le cadre des activités de détection de signaux post-commercialisation sert à orienter les prises de décision :
|
Examen des documents et des données administratives |
||
|
Examen de la documentation
|
||
|
Examen de la documentation |
||
|
Entrevues/consultations auprès d'informateurs clés (internes et externes) |
||
| g. Dans quelle mesure le système réglementaire intervient-il rapidement à l'égard des risques cernés? |
|
Examen de la documentation | |
|
Examen des documents (si l'information est disponible) |
||
|
Examen des documents et des données administratives
Étude de cas (post-commercialisation) |
||
|
Entrevues auprès d'informateurs clés (internes et externes) |
||
| h. Dans quelle mesure le cadre de réglementation des médicaments pour usage humain du Canada est-il en phase avec les approches internationales? |
|
Revue de la littérature
Entrevues auprès d'informateurs clés (internes et externes) |
|
|
Entrevues auprès d'informateurs clés (internes) | ||
|
Examen de la documentation |
||
|
Examen de la documentation | ||
|
Examen de la documentation |
||
Mesure dans laquelle Santé Canada est reconnu comme un organisme de réglementation et une autorité scientifique responsables en matière de médicaments pour usage humain (aux échelles nationale et internationale), comme en témoignent :
|
Examen de la documentation |
||
| i. Dans quelle mesure l'exposition aux risques cernés liés à l'utilisation de médicaments pour usage humain est-elle réduite? |
|
Examen des documents et des données administratives
|
|
|
Examen des documents et des données administratives
|
||
|
Examen des documents et des données administratives
|
||
|
Examen des documents et des données administratives
|
||
|
Entrevues auprès d'informateurs clés (internes et externes) |
||
| Résultats à long terme | |||
| j. Dans quelle mesure les effets indésirables associés à l'utilisation de médicaments pour usage humain ont-ils diminué? |
|
Examen des documents et des données administratives |
|
|
Examen des documents et des données administratives (selon la disponibilité) | ||
| k. Dans quelle mesure la confiance du public à l'égard des médicaments pour usage humain et du système réglementaire connexe a-t-elle augmenté? |
|
Examen de la documentation :
Entrevues auprès d'informateurs clés (externes) |
|
|
Examen de la documentation | ||
| l. Dans quelle mesure le système de réglementation des médicaments pour usage humain du Canada est-il viable, rentable, adapté aux besoins et fondé sur la science? |
|
Toutes les sources de données |
|
| 7. Le PMUH a-t-il entraîné des conséquences inattendues, de nature positive ou négative? |
|
Entrevues ou consultations auprès des informateurs clés (internes et externes) |
|
| Enjeu 5 : Efficience et économie | |||
| 8. Les ressources du PMUH ont-elles été utilisées selon ce qui était prévu? Qu'est-ce qui explique les dépassements ou les dépenses plus faibles que prévu? |
|
Examen des données administratives, par exemple :
Entrevues auprès d'informateurs clés (internes) |
|
| 9. Existe-t-il des moyens d'obtenir les extrants du PMUH à coût moindre? |
|
Entrevues auprès d'informateurs clés (internes) |
|
| 10. Peut-on obtenir autrement des résultats semblables à moindre coût? |
|
Revue de la littérature |
|
Annexe B - Liste des ouvrages de référence
Abraham, C. (2005). Vioxx took deadly toll: study. The Globe and Mail. Tiré de http://www.theglobeandmail.com/life/vioxx-took-deadly-toll-study/article1113848/.
Société canadienne de pédiatrie. (s.d.). Programme canadien de surveillance pédiatrique. Consulté le 4 octobre 2012 sur http://www.pcsp.cps.ca/.
Cassels, A. (2012). Most of our prescription drugs are manufactured overseas -- but are they safe? Journal de l'Association médicale canadienne, 184(14), 1648.
CBC News. (2012). Sandoz drug shortage prompts Ottawa fast-track efforts. Consulté le 10 septembre 2013 au http://www.cbc.ca/news/health/story/2012/03/06/sandoz-drug-supply-concerns.html.
CCLT. (2012). Mauvais usage de médicaments sur ordonnance. Consulté le 29 octobre 2012 au http://www.ccsa.ca/fra/priorities/prescription-drug-misuse/pages/default.aspx.
CDC. (2012). Multistate Fungal Meningitis Outbreak Investigation. Website of the Centers for Disease Control and Prevention. Consulté le 25 octobre 2012 au http://www.cdc.gov/HAI/outbreaks/meningitis.html.
ICIS. (2011). Tendances des dépenses nationales de santé, 1975 à 2011. Tiré de https://secure.cihi.ca/estore/productFamily.htm?locale=fr&pf=PFC1671.
Collier, R. (2013). Propecia lawsuits: the lasting effects of delayed drug warnings. Journal de l'Association médicale canadienne, 185(10), E455-E456. doi:10.1503/cmaj.109-4488
Parlement européen et le Conseil de l'Union européenne. (2001). Directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain, Journal officiel des Communautés européennes, 311. Tiré de http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2001:311:0067:0128:fr:PDF.
Centre de recherche Décima. (2003). Sondage sur l'opinion du public quant aux enjeux principaux liés à la surveillance des produits de santé commercialisés au Canada.
Centre de recherche Décima. (2006). Rapport final - Sondage d'opinion publique sur les principaux enjeux relatifs à la surveillance des produits de santé commercialisés au Canada (2006).
Ministère des Finances. (2003). Le plan budgétaire de 2003. Consulté le 21 août 2012 sur http://fin.gc.ca/budget03/pdf/bp2003f.pdf.
Dhalla, I., Mamdani, M. M., Sivilotti, M. L. A., Kopp, A., Qureshi, O., & Juurlink, D. N. (2009). Prescribing of opioid analgesics and related mortality before and after the introduction of long-acting oxycodone. Journal de l'Association médicale canadienne, 181(12), 891-896. doi:10.1503/cmaj.090784
Duffin, J. (2012). Home: What is the drug shortage? Canadian Drug Shortage - A site for information about the situation in Canada. Consulté le 25 octobre 2012 sur http://www.canadadrugshortage.com/.
Eguale, T., Buckeridge, D. L., Winslade, N. E., Benedetti, A., Hanley, J. A., & Tamblyn, R. (2012). Drug, Patient, and Physician Characteristics Associated With Off-label Prescribing in Primary Care. Archives of Internal Medicine.
EMEA. (2009). Guideline on the Readability of the Labelling and Package Leaflet of Medicinal Products for Human Use. Consulté le 8 juillet 2013 sur http://ec.europa.eu/health/files/eudralex/vol-2/c/2009_01_12_readability_guideline_final_en.pdf.
EMEA. (2012). EU Clinical Trials Register - About Page | More information on EU-CTR search and website. Consulté le 26 février 2013 sur https://www.clinicaltrialsregister.eu/about.html#whatsnew.
EMEA. (2013a). Agence européenne des médicaments - Pharmacovigilance - Risk-management plans. Consulté le 4 juillet 2013 sur http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/
document_listing_000360.jsp&mid=WC0b01ac058067a113.
EMEA. (2013b). Agence européenne des médicaments - Post-authorisation - Human post-authorisation Q&A: Introduction. Consulté le 5 juillet 2013 sur http://www.emea.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content
_000166.jsp&mid=WC0b01ac0580023399.
EMEA. (2013c). European Medicines Agency updates product-information template to label medicines subject to additional monitoring and encourage adverse-reaction reporting. Consulté le 9 juillet 2013 sur http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2013/03/
news_detail_001740.jsp&mid=WC0b01ac058004d5c1.
EMEA. (2013d). Guideline on good pharmacovigilance practices (GVP). Consulté le 4 juillet 2013 sur http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/
WC500129136.pdf.
EMEA. (2013e). Human medicines - European public assessment reports. Consulté le 9 septembre 2013 sur http://www.ema.europa.eu/ema/index.jsp?searchType=name&startLetter=O&
taxonomyPath=&keyword=opioid&alreadyLoaded=true&curl=pages%2Fmedicines%2
Flanding%2Fepar_search.jsp&status=Authorised&status=Withdrawn&status=Suspended
&status=Refused&mid=WC0b01ac058001d125&searchGenericType=generics&treeNumber
=&searchTab=&pageNo=2.
Environics Research Group. (2007). Sondage des professionnels de la santé - Déclaration d'effets indésirables. Consulté le 11 octobre 2012 sur http://epe.lac-bac.gc.ca/100/200/301/pwgsc-tpsgc/por-ef/health/2007/432-06/sommaire.pdf.
Parlement européen. (2010a). Directive 2010/84/UE du Parlement européen et du Conseil du 15 décembre 2010, Journal officiel de l'Union européenne, 348. Tiré de http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:348:0074:0099:FR:PDF.
Parlement européen. (2010b). Règlement (UE) nº 1235/2010 du Parlement européen et du Conseil du 15 décembre 2010, Journal officiel de l'Union européenne, 348. Tiré de http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:348:0001:0016:FR:PDF.
FDA. (2002). Compliance Program Guidance Manual Program: Drug Manufacturing Inspections. Consulté le 5 juillet 2013 sur http://www.fda.gov/downloads/ICECI/ComplianceManuals/
ComplianceProgramManual/UCM125404.pdf.
FDA. (2008). Sentinel Initiative - Transforming How We Monitor Product Safety > The Sentinel Initiative: A National Strategy for Monitoring Medical Product Safety. Consulté le 5 juillet 2013 sur http://www.fda.gov/Safety/FDAsSentinelInitiative/ucm089474.htm.
FDA. (2009). Information for Consumers (Drugs) > OTC Drug Facts Label. Consulté le 8 juillet 2013 sur http://www.fda.gov/Drugs/ResourcesForYou/Consumers/ucm143551.htm.
FDA. (2010). Boxed warnings and other FDA communication tools. American Family Physician, 81(3). Tiré de http://www.fda.gov/downloads/Safety/MedWatch/UCM201430.pdf.
FDA. (2011a). Guidance for Industry Postmarketing Studies and Clinical Trials -- Implementation of Section 505(o)(3) of the Federal Food, Drug, and Cosmetic Act. Consulté le 5 juillet 2013 sur http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/
Guidances/UCM172001.pdf.
FDA. (2011b). Warning Letter. Novartis International AG 11/18/11. Consulté le 10 septembre 2013 sur http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm281843.htm.
FDA. (2012a). FDA Adverse Event Reporting System (FAERS) (formerly AERS). Consulté le 2 mai 2013 sur http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/
AdverseDrugEffects/default.htm.
FDA. (2012b). FDA Adverse Events Reporting System (FAERS) Electronic Submissions. Consulté le 2 mai 2013 sur http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/
AdverseDrugEffects/ucm115894.htm.
FDA. (2012c). Risk Evaluation and Mitigation Strategy (REMS) for Extended-Release and Long-Acting Opioids. Consulté le 16 novembre 2012 sur http://www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm163647.htm.
FDA. (2013a). Drugs@FDA: FDA Approved Drug Products. Consulté le 9 septembre 2013 sur http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm.
FDA. (2013b). FDA News Release. FDA approves abuse-deterrent labeling for reformulated OxyContin. Consulté le 9 septembre 2013 sur http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm348252.htm.
FDA. (2013c). Guidance for Industry Providing Postmarket Periodic Safety Reports in the ICH E2C(R2) Format (Periodic Benefit-Risk Evaluation Report). Consulté le 4 juillet 2013 sur http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/
Guidances/UCM346564.pdf.
FDA. (2013d). Inspections, Compliance, Enforcement, and Criminal Investigations. Consulté le 9 septembre 2013 http://www.fda.gov/iceci/enforcementactions/ucm222557.htm.
FDA. (2013e). Laws, Acts, and Rules > PLR Requirements for Prescribing Information. Consulté le 8 juillet 2013 sur http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/LawsActsandRules
/ucm084159.htm.
FDA. (2013f). Warning Letter. Apotex Inc. February 21, 2013. Consulté le 7 octobre 2013 sur http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2013/ucm344476.htm.
Ferguson, B., & Lybecker, K. (2012). Economics of intellectual property protection in the pharmaceutical sector: Pills, patients, & profits II. Tiré de http://www.macdonaldlaurier.ca/files/pdf/Economics-of-Intellectual-Property-Protection-in-the-Pharmaceutical-Sector-January-2012.pdf.
Globe and Mail. (2012a). Sandoz Canada's production slows to a crawl after harsh criticism from U.S. regulators. Consulté le 10 septembre 2013 sur http://m.theglobeandmail.com/globe-investor/sandoz-canadas-production-slows-to-a-crawl-after-harsh-criticism-from-us-regulators/article2343341/?service=mobile.
Globe and Mail. (2012b, November 29). Tories block bid to make cheaper medicines for poor nations.
Globe and Mail. (2013a). Health Canada announces third birth-control recall this year. Consulté le 10 septembre 2013 sur http://www.theglobeandmail.com/life/health-and-fitness/health/health-canada-announces-another-birth-control-recall-of-esme-28/article14120836/.
Globe and Mail. (2013b, April 26). U.S. regulator warns Canadian drug maker Apotex about quality control. Consulté le 7 octobre 2013 sur http://www.theglobeandmail.com/life/health-and-fitness/health/us-regulator-warns-canadian-drug-maker-apotex-about-quality-control/article11560463/.
GdC. (2004). Loi sur les frais d'utilisation (L.C. 2004, ch. 6).
GdC. (2006). Loi sur le ministère de la Santé (L.C. 1996, ch. 8).
GdC. (2008a). Projet de loi C-51 : Loi modifiant la Loi sur les aliments et drogues
et modifiant d'autres lois en conséquence.
GdC. (2008b). Loi sur les aliments et drogues (L.R.C., 1985, ch. F-27).
GdC. (2010). Règlement modifiant le Règlement sur les aliments et drogues (743 - Ingrédients non médicinaux) (Partie II), Gazette du Canada, Partie II, vol. 144, nº 11. Tiré de http://gazette.gc.ca/rp-pr/p2/2010/2010-05-26/html/sor-dors105-fra.html.
GdC. (2011). Règlement sur les prix à payer à l'égard des drogues et instruments médicaux, Gazette du Canada, Partie II, vol. 145, nº 8. Tiré de http://www.gazette.gc.ca/rp-pr/p2/2011/2011-04-13/html/sor-dors79-fra.html.
GdC. (2012a). Directive du Cabinet sur la gestion de la réglementation. Consulté le 9 septembre 2013 sur http://www.tbs-sct.gc.ca/rtrap-parfa/cdrm-dcgr/cdrm-dcgrtb-fra.asp.
GdC. (2012b). Politique de communication du gouvernement du Canada. Consulté le 9 septembre 2013 sur http://www.tbs-sct.gc.ca/pol/doc-fra.aspx?id=12316§ion=text.
GdC. (2012c). Règlement sur les aliments et drogue (C.R.C., ch. 870).
GdC. (2013). Canada Gazette - Règlement modifiant le Règlement sur les aliments et drogues (étiquetage, emballage et marques nominatives des drogues pour usage humain), Gazette du Canada, Partie I, vol. 147, nº 25. Tiré de http://www.gazette.gc.ca/rp-pr/p1/2013/2013-06-22/html/reg2-fra.html#reg.
GdC. (s.d.). Plan de travail sur le portail commun de demandes électroniques. Consulté le 3 juin 2013 sur http://actionplan.gc.ca/fr/page/rcc-ccr/plan-de-travail-portail-commun-de-demandes.
Grosh, L., Sharpe, J., Bergman, N., Bigge, D., Blanck, J., Cate, A., & Thornton, N. (2013). In the Arbitration Under Chapter Eleven of the North American Free Trade Agreement and the ICSID Arbitration (Additional Facility) Rules Between Apotex Holdings Inc. and Apotex Inc. (Claimants/Investors) and the United States of America (Respondent/Party) Case No. ARB(AF)/12/1. Consulté le 16 septembre 2013 sur http://italaw.com/sites/default/files/case-documents/italaw1242.pdf.
Santé Canada. (2000). Cadre décisionnel de Santé Canada pour la détermination, l'évaluation et la gestion des risques pour la santé.
Santé Canada. (2003). Avis - Common Technical Document - Sujet M4 de l'ICH. Consulté le 18 octobre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/ctd_m4_notice-fra.pdf.
Santé Canada. (2006). Results-based Management and Accountability Framework - Therapeutic Products Safety Initiative submission.
Santé Canada. (2008a). Cadre de gestion et de responsabilisation axé sur les résultats (CGRR) du PASPAC.
Santé Canada. (2008b). Description des documents actuels de communication des risques concernant les produits de santé commercialisés destinés aux humains - Lignes directrices. Tiré de http://www.hc-sc.gc.ca/dhp-mps/pubs/medeff/_guide/2008-risk-risques_comm_guid-dir/index-fra.php.
Santé Canada. (2008c). Régime canadien d'accès aux médicaments - Introduction. Page d'accueil. Consulté le 28 août 2012 sur http://www.camr-rcam.gc.ca/intro/index-fra.php.
Santé Canada. (2009a). Guide sur les preuves de conformité aux BPF des médicaments provenant de sites étrangers (GUI-0080). Consulté le 24 juillet 2013 sur http://www.hc-sc.gc.ca/dhp-mps/compli-conform/gmp-bpf/docs/gui-0080-fra.php.
Santé Canada. (2009b). Avis concernant la mise en œuvre de la planification de gestion des risques, y compris l'adoption des lignes directrices « Planification de la pharmacovigilance » - Thème E2E de l'ICH.
Santé Canada. (2009c). The Therapeutic Access Strategy (TAS) Formative Evaluation (Final Report).
Santé Canada. (2010a). Ébauche de la ligne directrice l'étiquetage des médicaments à usage humain - 2010 Health Canada Consultation Document. Consulté le 9 juillet 2013 sur http://www.hc-sc.gc.ca/dhp-mps/consultation/drug-medic/draft_ebauche_label_guide-eng.php#a31 lien périmé; visible en français à http://web.archive.org/web/20131224205218/http://www.hc-sc.gc.ca/dhp-mps/consultation/drug-medic/draft_ebauche_label_guide-fra.php mais on ne peut ouvrir le fichier PDF et en anglais à http://web.archive.org/web/20131220182155/http://www.hc-sc.gc.ca/dhp-mps/consultation/drug-medic/draft_ebauche_label_guide-eng.php#a31. « Health Canada Consultation Document » ne figure pas sur la page Web.
Santé Canada. (2010b). Document d'orientation à l'intention de l'industrie - Diffusion par les détenteurs d'une autorisation de mise sur le marché de communications aux professionnels de la santé et de communications au public. Consulté le 27 novembre 2013 sur http://www.hc-sc.gc.ca/dhp-mps/pubs/medeff/_guide/2010-guid-dir_indust_hppc-cpsp/index-fra.php.
Santé Canada. (2010c). Proposition de Santé Canada soumise au Parlement au sujet des frais d'utilisation et des normes de service pour les programmes des médicaments pour usage humain et des matériels médicaux. Tiré de http://www.hc-sc.gc.ca/dhp-mps/finance/costs-couts/fee-propo-frais-fra.php.
Santé Canada. (2011a). Le système de soins de santé du Canada, site Web de Santé Canada. Consulté le 5 octobre 2012 sur http://www.hc-sc.gc.ca/hcs-sss/pubs/system-regime/2011-hcs-sss/index-fra.php.
Santé Canada. (2011b). Bureau du Directeur général - Inspectorat de la Direction générale des produits de santé et des aliments - Santé Canada. Description organisationnelle. Consulté le 20 août 2012 sur http://www.hc-sc.gc.ca/ahc-asc/branch-dirgen/hpfb-dgpsa/hpfi-ipsa/dgo-bdg-fra.php.
Santé Canada. (2011c). Ébauche - Ligne directrice - Changements survenus après l'avis de conformité (AC) : document-cadre. Consulté le 3 juillet 2013 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/postnoc_change_apresac/noc_pn_framework_ac_sa_cadre-fra.php.
Santé Canada. (2011d). Health Canada's Management Response and Action Plan (MRAP) - Audit of Chapter 4, Regulating Pharmaceutical Drugs.
Santé Canada. (2012a). 2011-12 - Rapport ministériel sur le rendement. Consulté le 9 septembre 2013 sur http://www.hc-sc.gc.ca/ahc-asc/performance/estim-previs/dpr-rmr/2011-2012/report-rapport-fra.php#a221.
Santé Canada. (2012b). 2011-12 - Rapport ministériel sur le rendement. Consulté le 3 juillet 2013sur http://www.hc-sc.gc.ca/ahc-asc/performance/estim-previs/dpr-rmr/2011-2012/report-rapport-fra.php.
Santé Canada. (2012c). Analyse coûts-avantages : Règlement modifiant le Règlement sur les aliments et drogues - Bonnes pratiques de fabrication applicables aux ingrédients actifs. Consulté le 30 octobre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/compli-conform/info-prod/drugs-drogues/actingred-cba-aca-fra.php.
Santé Canada. (2012d). Glossaire des champs de la base de données en ligne des effets indésirables de Canada Vigilance. Terminologie. Consulté le 22 octobre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/medeff/databasdon/glossary_definition-fra.php.
Santé Canada. (2012e). Document d'orientation de Santé Canada - Considérations relatives à l'inclusion des femmes dans les essais cliniques et à l'analyse des données selon le sexe. Consulté le 26 février 2013 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/consultation/biolog/draft_iwct_ebauche_ifec/draft_ebauche_iwct_ifec-fra.pdf.
Santé Canada. (2012f). Interprétation des renseignements sur les effets indésirables présumés. Consulté le 9 septembre 2013 sur http://www.hc-sc.gc.ca/dhp-mps/medeff/databasdon/interpretation-fra.php.
Santé Canada. (2012g). Direction des produits de santé commercialisés. Description organisationnelle. Consulté le 20 août 2012 sur http://www.hc-sc.gc.ca/ahc-asc/branch-dirgen/hpfb-dgpsa/mhpd-dpsc/index-fra.php
Santé Canada. (2012h). Comité consultatif d'experts sur les initiatives pédiatriques (CCEIP) - Santé Canada. Liste des ressources. Consulté le 4 octobre 2012 sur http://www.hc-sc.gc.ca/ahc-asc/branch-dirgen/hpfb-dgpsa/opi-bip/peac-cceip/index-fra.php.
Santé Canada. (2012i). Performance Measurement and Evaluation Plan - EERC One-year Study.
Santé Canada. (2012j). Feuille de route de la réglementation pour les produits de santé et aliments. Tiré de http://www.hc-sc.gc.ca/ahc-asc/alt_formats/pdf/activit/mod/roadmap-feuillederoute-fra.pdf.
Santé Canada. (2012k). Déclaration sur l'autorisation des versions génériques d'OxyContin. Consulté le 6 décembre 2012 sur http://www.hc-sc.gc.ca/ahc-asc/media/ftr-ati/_2012/2012_176-fra.php.
Santé Canada. (2012l). Summative evaluation framework: Human Drugs Activities.
Santé Canada. (2012m). Use of international paediatric information by Health Canada.
Santé Canada. (2013a). Ébauche - Lignes directrices à l'intention de l'industrie - Exigences en matière de présentation de renseignements relatifs aux drogues nouvelles pour usage exceptionnel (DNUE).
Santé Canada. (2013b). Drugs removed from the market for safety reasons, 2004-2012, data extract.
Santé Canada. (2013c). E-Communications: Interim Web Publishing Criteria and Process.
Santé Canada. (2013d). Evaluating the Effectiveness of (Health Product) Risk Communications (EERC) in the Health Products and Food Branch: Project Charter.
Santé Canada. (2013e). Le gouvernement Harper renforce l'innocuité des médicaments à l'aide de nouvelles normes - Communiqué de presse de Santé Canada, le 8 mai 2013. Consulté le 5 juillet 2013 sur http://www.hc-sc.gc.ca/ahc-asc/media/nr-cp/_2013/2013-58-fra.php.
Santé Canada. (2013f). Base de données sur les essais cliniques de Santé Canada. Consulté le 3 juin 2013 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/databasdonclin/index-fra.php.
Santé Canada. (2013g). HPFB Risk Communication Process for Human Health Products (draft).
Santé Canada. (2013h). Inspectorate Program: Inspectorate Annual Report 2012-2013.
Santé Canada. (2013i). Parution aujourd'hui d'une nouvelle norme hospitalière sur la déclaration des effets indésirables des médicaments - Des activités de marketing viendront appuyer la norme pour accroître le signalement chez les professionnels de la santé. Consulté le 21 mars 2013 sur http://www.hc-sc.gc.ca/ahc-asc/media/nr-cp/_2013/2013-01-fra.php.
Santé Canada. (2013j). Avis - L'adoption de la directive de l'International Conference on Harmonisation (ICH) intitulée: rapport périodique d'évaluation des avantages et des risques - ICH EC2 (R2), à partir du 1er mars 2013. Consulté le 4 juillet 2013 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/ich/efficac/e2cr2_notice_avis-fra.php.
Santé Canada. (2013k). Status Report on the Financial Management and Performance of the Human Drugs and Medical Devices Programs.
Santé Canada. (2013l). Mise à jour des réponses aux recommandations formulées par le BVG dans son rapport de l'automne 2011 concernant la réglementation des médicaments pharmaceutiques - Plan d'action de Santé Canada au 28ième mars 2013.
Santé Canada, & Spearhead Management Canada Limited. (2007). Costing model report - final deliverable.
Conseil canadien du Canada. (2010). Décisions, décisions : les médecins de famille en tant que gardes de l'accès aux médicaments d'ordonnance et à l'imagerie diagnostique au Canada. Tiré de http://www.healthcouncilcanada.ca/rpt_det.php?id=154.
DGSESC. (2009). Programme de la stratégie antidrogue et des substances contrôlées - Direction générale de la santé environnementale et de la sécurité des consommateurs. Consulté le 24 octobre 2012 sur http://www.hc-sc.gc.ca/ahc-asc/branch-dirgen/hecs-dgsesc/dscsp-psasc/index-fra.php.
Comité permanent de la santé du Parlement. (2008). La surveillance post-commercialisation des produits pharmaceutiques - Rapport du Comité permanent de la santé. Tiré de http://publications.gc.ca/collections/collection_2008/parl/XC62-392-1-1-04F.pdf.
Comité permanent de la santé du Parlement. (2012). L'approvisionnement en médicaments au Canada : une réponse multilatérale - Rapport du Comité permanent de la santé. Tiré de http://www.parl.gc.ca/content/hoc/Committee/411/HESA/Reports/RP5640047/
hesarp09/hesarp09-f.pdf.
DGPSA. (2005a). Ébauche de ligne directrice à l'intention de l'industrie - Évaluation de noms de produits de santé commercialisés : noms des produits de santé à présentation et à consonance semblables (PSPCS).
DGPSA. (2005b). Direction générale des produits de santé et des aliments - Cadre de participation du public.
DGPSA. (2007a). Plan de renouveau II.
DGPSA. (2007b). Examen des produits réglementés : Politique sur la participation du public.
DGPSA. (2008). Ligne directrice à l'intention de l'industrie et des praticiens - Programme d'accès spécial : médicaments.
DGPSA. (2009). Ligne directrice - Norme d'étiquetage pour l'acétaminophène. Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/label_stand_guide_ld-fra.pdf,
DGPSA. (2011a). Cost recovery: Project summary.
DGPSA. (2011b). Ligne directrice - Avis de conformité avec conditions (AC-C).
DGPSA. (2011c). Ligne directrice de l'industrie : gestion des présentations de drogues.
DGPSA. (2012a). DSTS Premeeting submission types 2002-2012.
DGPSA. (2012b). HPFB international activities: Inventory of significant international activities in HPFB (ébauche, mise à jour avril 2012).
DGPSA. (2012c). HPFB mid-year report 2012-13: Summary of Successes and Challenges.
DGPSA. (2012d). Pharmaceutical Drugs Program: 2012-13 mid-year review.
ICH. (2013). Quality Guidelines: ICH. Consulté le 5 juillet 2013 sur http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html.
Industrie Canada. (2012). Profil de l'industrie pharmaceutique - Industries canadiennes - Sciences de la vie. Consulté le 23 mai 2013 sur http://www.ic.gc.ca/eic/site/lsg-pdsv.nsf/fra/h_hn01703.html.
Inspectorat. (2002). Avis d'intention publié dans la Gazette du Canada, Partie I, le 7 décembre 2002 et Ligne directrice sur les Bonnes pratiques de fabrication applicables aux ingrédients pharmaceutiques actifs (ICH thème Q7A). Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/compli-conform/legislation/gazette1-q7a_tc-tm-fra.php.
Inspectorat. (2007a). A2.2 Human Drugs - GMP Inspections operational planning (2007-08).
Inspectorat. (2007b). Inspectorate performance report (2006-07, year-end).
Inspectorat. (2009a). Plan stratégique de l'Inspectorat pour 2009-2012.
Inspectorat. (2009b). Politique sur la fabrication et la préparation en pharmacie de produits pharmaceutiques au Canada. Tiré de http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/compli-conform/pol_0051-fra.pdf.
Inspectorat. (2010a). L'Approche de l'intégrité frontalière (POL-0059). Consulté le 20 juillet 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/compli-conform/import-export/pol_0059_BI_approach-fra.pdf.
Inspectorat. (2010b). Inspectorate performance report (2009-10).
Inspectorat. (2010c). Politique sur les produits de santé de contrefaçon (POL-0048) [Santé Canada, 2010]. Consulté le 13 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/compli-conform/activit/pol_0048_counterfeit-contrefacon-fra.php.
Inspectorat. (2012a). A2.10 PMRC - Drug Inspections operational planning (2011-12) (v2).
Inspectorat. (2012b). A2.2 Human Drugs - GMP Inspections operational planning (2011-12) ("Clean").
Inspectorat. (2012c). Compliance and Enforcement Risk Evaluation Guide: An approach to decision-making.
Inspectorat. (2012d). Documents d'orientation, directives et politiques. Consulté le 21 novembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/compli-conform/info-prod/drugs-drogues/docs-fra.php.
Inspectorat. (2012e). Document d'orientation sur les licences d'établissement et le prix à payer pour les licences d'établissement (GUI-0002). Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/compli-conform/licences/directives/gui-002-fra.pdf.
Inspectorat. (2012f). Classification des observations liées aux Bonnes pratiques de fabrication (BPF) en fonction du risque (GUI-0023). Consulté le 21 novembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/compli-conform/gmp-bpf/docs/gui-0023-fra.pdf.
Inspectorat. (2012g). Rapport sommaire des inspections d'essais cliniques réalisées d'avril 2004 à mars 2011. Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/compli-conform/clini-pract-prat/report-rapport/2004-2011-fra.pdf.
Inspectorat. (2012h). Rapport sommaire sur le Programme d'inspection des Bonnes pratiques de fabrication (BPF) des drogues du 1er avril 2006 au 31 mars 2011. Consulté le 26 novembre 2013 sur http://www.hc-sc.gc.ca/dhp-mps/compli-conform/gmp-bpf/2011-03-31-report-rapport-fra.php.
Inspectorat. (2012i). Mises à jour - Accords de reconnaissance mutuelle. Consulté le 21 août 2013 sur http://www.hc-sc.gc.ca/dhp-mps/compli-conform/int/mra-arm/update-miseajour/index-fra.php.
Inspectorat. (2013). Procedure or Work Instruction: Clinical Trial Site Selection for Inspection: SOP- 0548.
IOM. (2008). Forum on Drug Discovery, Development, and Translation: Addressing the Barriers to Pediatric Drug Development: Workshop Summary - Chapter 2: Regulatory Framework. Consulté le 26 février 2013 sur http://www.ncbi.nlm.nih.gov/books/NBK3997/.
Labrie, Y. (2012). Comment éviter les pénuries de médicaments? Retrieved from http://www.iedm.org/files/note0912_fr.pdf.
Lemmens, T., & Bouchard, R. (2007). Regulation of pharmaceuticals in Canada. In J. Downie, T. Caulfield, & C. Flood (Eds.), Canadian Health Law and Policy (3rd ed., p. 311-365).
Lexchin, J. (2012). New Drugs and Safety: What Happened to New Active Substances Approved in Canada Between 1995 and 2010? Archives of Internal Medicine, 1. doi:10.1001/archinternmed.2012.4444
Liu, H., Zhang, Z., & Linhardt, R. J. (2009). Lessons learned from the contamination of heparin. Natural Product Reports, 26(3), 313. doi:10.1039/b819896a
DPSC. (2008). Direction des produits de santé commercialisés - Rétrospective : les cinq premières années | 2002-2007 (rapport d'étape).
DPSC. (2009). Towards Comprehensive Risk Management Planning: Health Canada's Proposed Approach. Consulté le 3 juillet 2013 sur http://www.mclaughlincentre.ca/events/Parmaco2/Nashwa%20Irfan_UofO%20
workshop%20May%2028-29%202009.pdf.
DPSC. (2010). Canada Vigilance_Signal Detection Business Transformation: Project overview - Office of the Auditor General.
DPSC. (2011a). La déclaration des effets indésirables et les renseignements concernant l'innocuité des produits de santé - Guide à l'intention des professionnels de la santé. Guide. Consulté le 18 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/pubs/medeff/_fs-if/2011-ar-ei-guide-prof/index-fra.php.
DPSC. (2011b). Procédure - La divulgation au public de renseignements tirés des déclarations d'effets indésirables et d'incidents concernant des matériels médicaux [Santé Canada, 2011]. Guide. Consulté le 14 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/pubs/medeff/_guide/2011-procedure/index-fra.php.
DPSC. (2011c). Strengthening HPFB's Health Product Vigilance System into the Future - DRAFT.
DPSC. (2012a). Base de données des effets indésirables - Base de données en ligne des effets indésirables de Canada Vigilance [2012]. Consulté le 27 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/medeff/databasdon/index-fra.php.
DPSC. (2012b). MedEffet Canada - Avis, mises en garde et retraits. Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/medeff/advisories-avis/index-fra.php.
DPSC. (2012c). MHPD performance standards.
DPSC. (2012d). Standard Operating Procedure: Periodic Safety Update Report (PSUR) Screening-(Level I review).
DPSC. (2012e). Standard Operating Procedure: Prioritization and management of potential signal files in MHPD.
DPSC. (2012f). Standard Operating Procedure: Signal Assessment.
DPSC. (2012g). The Past and Future of Pharmacovigilance Activities at the Marketed Health Products Directorate, Symposium international sur la pharmacovigilance de la Direction générale des produits de santé et des aliments.
DPSC. (2013). Standard Operating Procedure: MHPD Signal Detection (MBBNHPB).
MHRA. (2009). Summary of Regulation on Me Dicines for Paediatric Use. Consulté le 26 février 2013 sur http://www.mhra.gov.uk/home/groups/pl-a/documents/websiteresources/con2025602.pdf.
Mini-Sentinel. (n.d). Background | About Mini-Sentinel | Mini-Sentinel. Consulté le 5 juillet 2013 sur http://mini-sentinel.org/about_us/.
Nanos Research. (2010). Results of drug safety survey of Canadians.
NIH. (2012). FDAAA 801 Requirements - ClinicalTrials.gov. Consulté le 26 février 2013 sur http://clinicaltrials.gov/ct2/manage-recs/fdaaa.
BVG. (2006). Rapport de la vérificatrice générale du Canada à la Chambre des communes, Chapitre 8 - L'affectation des fonds aux programmes de réglementation - Santé Canada.
BVG. (2011). Rapport de la vérificatrice générale du Canada à la Chambre des communes, Chapitre 4 - La réglementation des médicaments - Santé Canada.
Olson, M. (2002). Pharmaceutical Policy Change and the Safety of New Drugs. Journal of Law and Economics, 45(S2), 615-642.
Pew Health Group. (2011). After Heparin: Protecting Consumers from the Risks of Substandard and Counterfeit Drugs. Tiré de http://www.pewtrusts.org/uploadedFiles/wwwpewtrustsorg/Reports/Health/
Pew_Heparin_Final_HR.pdf.
Ramage-Morin, P. (2009). « Consommation de médicaments chez les Canadiens âgés », Rapports sur la santé, nº 82-003-X au catalogue, Statistique Canada. Tiré de http://www.statcan.gc.ca/pub/82-003-x/2009001/article/10801-fra.pdf.
Groupe de travail sur les produits de soins personnels et produits pharmaceutiques. (2012a). Plan de travail sur les bonnes pratiques de fabrication. Consulté le 24 juillet 2013 sur http://actionplan.gc.ca/grfx/BAP-RCC/GMP_Reformat_french.pdf.
Groupe de travail sur les produits de soins personnels et produits pharmaceutiques. (2012b). Plan de travail sur l'approbation de produits thérapeutiques en vente libre et de licences. Consulté le 25 juillet 2013 sur http://actionplan.gc.ca/grfx/BAP-RCC/Common_Monograph_OTC_french.pdf.
Risk Sciences International. (2012). Ensuring drug safety and effectiveness through pharmacovigilance. Tiré de http://www.risksciencesint.com/app/wa/mediaEntry?mediaEntryId=11780
Sparrow, M. (2000). The Regulatory Craft: Controlling Risks, Solving Problems, and Managing Compliance. Washington, D.C.: Brookings Institution Press.
Srba, J., Descikova, V., & Vlcek, J. (2012). Adverse drug reactions: Analysis of spontaneous reporting system in Europe in 2007-2009. European Journal of Clinical Pharmacology, 68(7), 1057-1063. doi:10.1007/s00228-012-1219-4
CSPASST. (2012). L'infrastructure des essais cliniques au Canada : ordonnance pour améliorer l'accès aux nouveaux médicaments. Consulté le 26 novembre 2012 sur http://www.parl.gc.ca/Content/SEN/Committee/411/soci/rep/rep14nov12-f.pdf.
SCT. (2009). Politique sur l'évaluation. Tiré de http://www.tbs-sct.gc.ca/pol/doc-fra.aspx?section=text&id=15024.
TGA. (2012). Therapeutic product vigilance. Consulté le 3 juillet 2013 sur http://www.tga.gov.au/about/tga-therapeutic-product-vigilance.htm
Le Groupe Antima & TNS Canada. (2007). Underutilization of the Adverse Reaction Reporting System. Consulté 11 octobre 2012 sur http://epe.lac-bac.gc.ca/100/200/301/pwgsc-tpsgc/por-ef/health/2007/385-06/sommaire.pdf
Toronto Star. (2013a). Health Canada considers mandatory reporting of adverse drug reactions. Consulté le 2 mai 2013 sur http://www.thestar.com/news/canada/2013/04/16/conservative_government_aims_
to_improve_safety_of_food_childrens_products_and_prescription_drugs.html.
Toronto Star. (2013b, September 6). Recalls of faulty birth control pills alarm Canadian doctors. Consulté le 7 octobre 2013 sur http://www.thestar.com/news/canada/2013/09/06/recalls_of_faulty_birth_control_pills_
alarm_canadian_doctors.html.
DPT. (2004). DPT - Rapport d'étape sur la transformation opérationnelle (2003-04).
DPT. (2007). Démarche d'homologation des médicaments.
DPT. (2008a). Rapport annuel de rendement sur les présentations de drogues - DPT - 2008.
DPT. (2008b). Foire aux questions - Monographies de produit affichées sur le site Web de Santé Canada. Consulté le 4 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/pm_qa_mp_qr-fra.pdf.
DPT. (2008c). Avis aux détenteurs d'une autorisation de mise en marché - Décision de Santé Canada concernant l'étiquetage de certains médicaments pédiatriques (0 à moins de 12 ans) pour la toux et le rhume administrés par voie orale en vente libre au Canada. Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/notice_avis_decision_pedlscc_pednecr-eng.pdf. (Lien périmé; seule la version finale est disponible sur le Web; elle fait mention de l'ancien titre : http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/pedlscc_pednecr-fra.pdf.)
DPT. (2010a). Rapport annuel de rendement sur les présentations de drogues - DPT - 2010. Consulté le 31 août 2012 sur http://www.jptcp.com/files/tpd_dpt_annual_annuel_10_eng.pdf. (Hyperlien non trouvé pour la version française).
DPT. (2010b). Présentation du régime actuel (Règlement sur les médicaments au Canada).
DPT. (2010c). Modifications aux modèles de monographie de produit (Avis de révisions). Consulté le 4 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/monograph/pmappe_mpanne-fra.pdf.
DPT. (2011). Product information (labelling).
DPT. (2012a). Avis - Ébauche de la ligne directrice - Norme d'étiquetage pour l'acide
acétylsalicylique. Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/consultation/drug-medic/consult_asa_gd_draft_aas_ld_ebauche-eng.php. (Lien périmé; visible à http://web.archive.org/web/20131002222124/http://www.hc-sc.gc.ca/dhp-mps/consultation/drug-medic/asa_gd_draft_aas_ld_ebauche-fra.php.)
DPT. (2012b). L'ébauche de la ligne directrice - Norme d'étiquetage de l'acide acétylsalicylique - Avis de consultation 2012 de Santé Canada Consultation Notice. Consulté le 12 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/consultation/drug medic/consult_asa_gd_draft_aas_ld_ebauche-eng.php. (Lien périmé; visible à http://web.archive.org/web/20131002222124/http://www.hc-sc.gc.ca/dhp-mps/consultation/drug-medic/asa_gd_draft_aas_ld_ebauche-fra.php.)
DPT. (2012c). Foire aux questions - Phase II du projet de sommaires des motifs de décision (SMD).
DPT & BGTD. (2009). Résultats de l'évaluation de la phase I de l'Initiative du sommaire des motifs de décision.
Gouvernement des États-Unis. (1996). 21 CFR 314.80 Postmarketing reporting of adverse drug experiences. Consulté le 5 juillet 2013 sur http://www.gpo.gov/fdsys/pkg/CFR-1996-title21-vol5/pdf/CFR-1996-title21-vol5-chap-id2.pdf.
Gouvernement des États-Unis. Public Law 110-85: An Act to Amend the Federal Food, Drug and Cosmetic Act to revise and extend the user-fee programs for prescription drugs and for medical devices, to enhance postmarket authorities of the FDA with respect to the safety of drugs and for other purposes. , Pub. L. No. 110-85 (2007). Tiré de http://www.gpo.gov/fdsys/pkg/PLAW-110publ85/pdf/PLAW-110publ85.pdf#page=85.
Gouvernement des États-Unis. (2012a). CFR - Code of Federal Regulations Title 21 - 201.57 - Subpart B--Labeling Requirements for Prescription Drugs and/or Insulin. Consulté le 9 juillet 2013 sur http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm.
Gouvernement des États-Unis. (2012b). CFR - Code of Federal Regulations Title 21 - Sec. 201.56 Requirements on content and format of labeling for human prescription drug and biological products. Consulté le 8 juillet 2013 sur http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=201.56&utm
_campaign=Google2&utm_source=fdaSearch&utm_medium=website&utm_term=21%20
CFR%20201.56&utm_content=1.
Weise, M., Bielsky, M., De Smet, K., Ehmann, F., Ekman, N., Narayanan, G., ... Schneider, C. (2011). Biosimilars - why terminology matters. Nature Biotechnology, 29(8), 690-693.
Woo, J., Wolfgang, S., & Batista, H. (2008). The effect of globalization of drug manufacturing, production, and sourcing and challenges for American drug safety. Clinical Pharmacology & Therapeutics, 83(3), 494-497.
Annexe C - Tableaux de données supplémentaires
| Année | Médicaments pour usage humain | Médicaments biologiques | ||
|---|---|---|---|---|
| Demandes reçues | Demandes approuvées (LNO) | Demandes reçues | Demandes approuvées (LNO) | |
| 2004 | 1730 | 1677 | 258 | 229 |
| 2005 | 1732 | 1658 | 239 | 210 |
| 2006 | 1685 | 1621 | 272 | 245 |
| 2007 | 1724 | 1633 | 278 | 253 |
| 2008 | 1613 | 1579 | 267 | 232 |
| 2009 | 1400 | 1341 | 266 | 247 |
| 2010 | 1191 | 1162 | 272 | 263 |
| % change, 2004-2010 | -31% | -31% | 5% | 15% |
Source: (BGTD, 2010; TPD, 2008a, 2008b, 2010a) |
||||
| Nombre | % | % combiné | |
|---|---|---|---|
| Signaux rejetés | |||
| Signaux rejetés au premier examen préliminaire | 1 209 | 61,1% | 80,5% |
| Signaux rejetés au deuxième examen préliminaire | 304 | 15,4% | |
| Signaux rejetés au moment de la rencontre de coordination | 32 | 1,6% | |
| Signaux rejetés au moment de l'établissement des signaux prioritaires ou lors du troisième examen préliminaire | 49 | 2,5% | |
| Total des signaux rejetés | 1 594 | ||
| Autres résultats | |||
| Communication avec la DPT/participation de celle-ci | 77 | 3,9% | 12,1% |
| RPPV | 35 | 1,8% | |
| Communication avec le Bureau de la gestion des risques et de la science (BGRS)/participation de celui-ci | 4 | 0,2% | |
| Communication avec le BPPIMC/participation de celui-ci | 4 | 0,2% | |
| Autre | 35 | 1,8% | |
| Nul | 84 | 4,2% | |
| Total - autres | 239 | ||
| Total - autres | |||
| Vert | 69 | 3,5% | 7,4% |
| Jaune | 59 | 3,0% | |
| Rouge | 2 | 0,1% | |
| Signaux jugés prioritaires | 17 | 0,9% | |
| Total des signaux jugés prioritaires | 147 | ||
| Total | 1 980 | ||
Sources: fichiers intitulés MAHSI WG tracking sheet.xls, PPSD WG tracking sheet.xls et Signal Detection Tracking Sheet.xls |
|||
| Nombre | % | % combiné | |
|---|---|---|---|
| Signaux rejetés | |||
| Signaux rejetés avant l'examen préliminaire | 6 288 | 96,7% | 96,9% |
| Signaux rejetés avant l'établissement des priorités | 1 | 0,0% | |
| Signaux rejetés par le comité d'établissement des priorités | 15 | 0,2% | |
| Total des signaux rejetés | 6 ,304 | ||
| Autres mesures | |||
| Surveillance continue/surveillance lors du prochain RPPV/aucune évaluation supplémentaire | 133 | 2,0% | 2,6% |
| Demandes ponctuelles/analyse des écarts | 10 | 0,2% | |
| RPPV | 4 | 0,1% | |
| Autre (S.O., nul, acheminement vers le BIIEPSC, la DPBTG, etc.) | 19 | 0,3% | |
| Total - autres | 166 | ||
| Signaux prioritaires aux fins d'une évaluation | |||
| Priorité faible | 18 | 0,3% | 0,5% |
| Priorité moyenne | 10 | 0,2% | |
| Priorité élevée | - | - | |
| Signaux jugés prioritaires | 7 | 0,1% | |
| Total des signaux jugés prioritaires | 35 | ||
| Total | 6 505 | ||
Source: MBB_SIC_WG_ tracking_2012-07-09.xls - worksheets "Prelim. Assessment Completed" and "No Action" |
|||
| Sources | Nombre d'évaluations | % du total | % combiné |
|---|---|---|---|
| Organismes de réglementation internationaux | |||
| États-Unis (FDA) | 39 | 14,3% | 26,1% |
| EMEA | 15 | 5,5% | |
| France (Affsaps) | 6 | 2,2% | |
| Italie (SIFA) | 5 | 1,8% | |
| R.-U. (MHRA) | 4 | 1,5% | |
| Nouvelle-Zélande (Medsafe) | 3 | 1,1% | |
| OMS | 2 | 0,7% | |
| Australie (TGA) | 1 | 0,4% | |
| Santé Canada (mécanismes) | |||
| Base de données de Canada Vigilance | 23 | 8,5% | 16,5% |
| RPPV | 15 | 5,5% | |
| Évaluation de signaux | 7 | 2,6% | |
| Santé Canada (organismes) | |||
| DPT | 19 | 7,0% | 8,5% |
| DPSC | 3 | 1,1% | |
| Inspectorat | 1 | 0,4% | |
| Autres sources | |||
| Littérature scientifique | 70 | 25,7% | - |
| Détenteurs d'AMM | 18 | 6,6% | - |
| RCEOEM | 2 | 0,7% | - |
| RIEM | 1 | 0,4% | - |
| Autre | 4 | 1,5% | - |
| Sources non préciséesd | 40 | 14,7% | - |
| Nombre total des évaluations de signaux | 272 | - | - |
Source: Suivi des activités d'évaluation des signaux du BPPIMC (2006-2012) (fichier Excel reçu en février 2013). Nom du fichier : Copy of 2013-02 MPMDB Activity_Signal Tracking.xls Remarque : les totaux des colonnes ne correspondent au total des sources, car les évaluations de signaux peuvent reposer sur plusieurs sources. *Le pourcentage du total et le pourcentage combiné peuvent ne pas être égaux parce que ce dernier ne vise qu'à déterminer si oui ou non les évaluations de signaux ont utilisé comme source un organisme de réglementation internationale, un mécanisme de Santé Canada ou une direction de Sanaté Canada. Les écarts entre les pourcentages sont liés au fait que les sources de certaines évaluations de signaux comprennent plusieurs organismes de réglementation internationaux ou directions de Sanaté Canada. |
|||
| Recommandation | Nombre | Pourcentage |
|---|---|---|
| Effectuer des modifications à l'étiquette d'un produit | 148 | 54% |
| Effectuer une surveillance standard | 99 | 36% |
| Demander la diffusion d'un document de communication des risques | 66 | 24% |
| Demander des renseignements supplémentaires sur l'innocuité | 28 | 10% |
| Réacheminer vers le BIIEPSC pour réévaluation | 24 | 9% |
| Demander un examen par le RIEM | 17 | 6% |
| Demander la présentation d'un plan de pharmacovigilance (PPV) ou de gestion des risques (PGR) | 11 | 4% |
| Accroître la surveillance | 7 | 3% |
| Demander une analyse risques-avantages au détenteur de l'AMM | 5 | 2% |
| Demander de l'information auprès d'un autre organisme de réglementation | 4 | 1% |
| Effectuer un résumé analytique de la question | 3 | 1% |
| Procéder à une intervention réglementaire s'appuyant sur les articles C.01.013, C.01.014 et C.08.006 de la Loi sur les aliments et drogues et du Règlement sur les aliments et drogues | 3 | 1% |
| Mettre en commun l'information avec les autres directions | 2 | 1% |
| Autre | 5 | 2% |
| Clore le dossier | 6 | 2% |
| Total | 272 | |
Source: fichier intitulé Copy of 2013-02 MPMDB Activity_Signal Tracking.xls Remarque : chaque évaluation de signal peut donner lieu à plus qu'une recommandation. Par conséquent, le total des colonnes ne correspondra pas au total des évaluations. |
||
| 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2011 | 2012 | Total | |
|---|---|---|---|---|---|---|---|---|---|
| DPT | 2 | 3 | 11 | 27 | 13 | 25 | 31 | 49 | 161 |
| DPBTG | 0 | 5 | 4 | 13 | 24 | 13 | 13 | 26 | 98 |
| DPSC | 0 | 0 | 1 | 0 | 3 | 2 | 3 | 0 | 9 |
| DPPR | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Total | 2 | 8 | 16 | 41 | 40 | 40 | 47 | 75 | 269 |
Source: Santé Canada Remarques : la DPPR (Division des politiques sur les présentations et renseignements) est une division de la DPT. Les PGR associés à la DPPR concernent les médicaments pour usage humain. Il est difficile de départager le nombre de PGR associés à la DPSC portant sur des médicaments pour usage humain d'une part et sur des médicaments biologiques d'autre part. Il n'est pas clair si ces données correspondent au nombre de PGR demandés, reçus ou examinés par Santé Canada. |
|||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2011 | 2012 | Total | |
|---|---|---|---|---|---|---|---|---|---|
| DPT | 51 | 24 | 21 | 8 | 14 | 48 | 29 | 19 | 214 |
| DPBTG | 3 | 6 | 13 | 5 | 17 | 23 | 7 | 8 | 82 |
| DPSC | 0 | 2 | 4 | 7 | 3 | 0 | 3 | 1 | 20 |
| DPPR | 18 | 33 | 8 | 4 | 2 | 1 | 0 | 0 | 66 |
| Total | 72 | 65 | 46 | 24 | 36 | 72 | 39 | 28 | 382 |
Source: Santé Canada Remarques : la DPPR (Division des politiques sur les présentations et renseignements) est une division de la DPT. Les RPPV associés à la DPPR concernent les médicaments pour usage humain. Il est difficile de départager le nombre de RPPV associés à la DPSC portant sur des médicaments pour usage humain d'une part et sur des médicaments biologiques d'autre part. Il n'est pas clair si ces données correspondent au nombre de RPPV demandés, reçus ou examinés par Santé Canada. |
|||||||||
| Activités post-commercialisation | Nombre de jours ciblé pour la réalisation |
|---|---|
| Déclarations d'effets indésirables | |
| Déclarations initiales prioritaires (effets ayant causé la mort ou pouvant causer la mort) | 15 |
| Déclarations initiales | 42 |
| Toutes les déclarations initiales et suivantes (normales et prioritaires) | 84 |
| Autres activités post-commercialisation | |
| Évaluation des signaux | 130 |
| Examen des PGR | 90 |
| Examen préliminaire des RPPV (niveau I) | 30 |
| Examen des RPPV (niveau II) | 90 |
| Examens de conformité ponctuels | 60 |
| Examen des enjeux liés à la publicité | 15 |
Source : Santé Canada Remarques : la DPPR (Division des politiques sur les présentations et renseignements) est une division de la DPT. Les RPPV associés à la DPPR concernent les médicaments pour usage humain. Il est difficile de départager le nombre de RPPV associés à la DPSC portant sur des médicaments pour usage humain d'une part et sur des médicaments biologiques d'autre part. Il n'est pas clair si ces données correspondent au nombre de RPPV demandés, reçus ou examinés par Santé Canada. |
|
| Sondage | Cible | Constatations pertinentes |
|---|---|---|
Sondage sur l'opinion du public quant aux enjeux principaux liés à la surveillance des produits de santé commercialisés au Canada (Centre de recherche Décima, 2003) |
Public |
Perception de l'innocuité des médicaments
Source d'information nouvelle sur l'innocuité des médicamentsTableau 8 note de bas de page 1
|
Professionnels de la santé |
Bonne connaissance des sources d'information sur l'innocuité des médicaments Lettres aux professionnels de la santé (LPS) produites par les fabricants
LPS produites par Santé Canada
Bulletin canadien des effets indésirables
Avis en ligne de Santé Canada sur l'innocuité des médicaments
Liste d'envoi électronique de Santé Canada
Effets indésirables
|
|
Sondage sur l'opinion du public quant aux enjeux principaux liés à la surveillance des produits de santé commercialisés au Canada (2006), rapport final (Suivi du sondage de 2003) (Centre de recherche Décima, 2006) |
Public |
Perception de l'innocuité des médicaments
Information nouvelle sur l'innocuité d'un médicament
|
Sondage des professionnels de la santé - Déclaration des effets indésirables (Suivi du sondage de 2003) (Seul le résumé des résultats touchant les professionnels occupant des postes de direction est accessible en ligne) (Environics Research Group, 2007) |
Professionnels de la santé |
Sources d'information nouvelle sur les médicaments
Effets indésirables
|
Sous utilisation de système de déclaration des effets indésirables (Seul le résumé des résultats touchant les professionnels occupant des postes de direction est accessible en ligne) (The Antima Group et TNS Canada, 2007) |
Professionnels de la santé |
Sources d'information nouvelle sur les médicaments
|
l'innocuité des médicaments menés auprès des Canadiens Nanos, années diverses (Nanos Research, 2010) |
Public |
|
Sensibilisation des Canadiens à l'égard des questions de santé et d'innocuité liées aux produits de consommation (Phoenix Strategic Perspectives Inc., 2011) |
Public Sondage Groupes de discussion (x8) |
|
Table 8 footnotes
|
||
| 2004 | 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2011 | 2012 | Total/ moyenne |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Total des présentations | 286 | 328 | 389 | 359 | 387 | 407 | 368 | 400 | 506 | 3 430 |
| % des PNM par rapport au total | 16,4% | 15,5% | 13,6% | 14,2% | 13,7% | 15,7% | 14,7% | 13,3% | 13,6% | 14,5% |
| % des PADM par rapport au total | 40,9% | 41,8% | 31,9% | 39,6% | 41,6% | 44,0% | 50,3% | 52,0% | 42,4% | 42,6% |
Sources: DPT, 2008a, 2010a. Données de 2011 et 2012 fournies par Santé Canada. Total et pourcentages de 2012 s'appuyant sur un nombre de PANM établi à 215. |
||||||||||
| Exercice | Arriéré de départ | Objectif annuel (nombre d'inspections) | Inspections* | Degré d'atteinte de l'objectif annuel | Non conforme | Conforme | Arriéré reporté |
|---|---|---|---|---|---|---|---|
| 2005-06 | N/A | 567 | 420 | 74% | 21 | 95% | 165 |
| 2006-07 | 165 | 543 | 356 | 66% | 6 | 98% | 187 |
| 2007-08 | 165 | 484 | 446 | 92% | 9 | 98% | 170 |
| 2008-09 | 210 | 689 | 440 | 64% | 8 | 98% | 57 |
| 2009-10 | 117 | 475 | 406 | 86% | 15 | 96% | 78 |
Source: Inspectorat, 2007, 2010b |
|||||||
| Exercice | Objectif | Inspections terminées | Degré d'atteinte de l'objectif annuel | Non conforme | Conforme |
|---|---|---|---|---|---|
| 2004-05 | - | 25 | - | 0 | 100% |
| 2005-06 | 243 | 75 | 31% | 0 | 100% |
| 2006-07 | 256 | 120 | 47% | 0 | 100% |
| 2007-08 | 242 | 153 | 63% | 0 | 100% |
| 2008-09 | 94 | 76 | 81% | 0 | 100% |
| 2009-10 | 64 | 72 | 113% | 0 | 100% |
Source: Inspectorat, 2004, 2006, 2007, 2010b |
|||||
| Exercice | Envois personnels | Envois commerciaux | Total | ||||
|---|---|---|---|---|---|---|---|
| Refus | Mainlevées | Refus | Mainlevées | ||||
| Contrefaçon (soupçon) | Autres raisons | Contrefaçon (soupçon) | Autres raisons | ||||
| Produits biologiques (annexe C) | |||||||
| 2010-2011 | 2 | 2 | |||||
| 2011-2012 | 17 | 8 | 25 | ||||
| Produits biologiques (annexe D) | |||||||
| 2010-2011 | 1 | 9 | 1 | 4 | 15 | ||
| 2011-2012 | 17 | 14 | 15 | 46 | |||
| Médicaments sur ordonnance (annexe F) | |||||||
| 2010-2011 | 3 789 | 20 577 | 935 | 86 | 585 | 201 | 26 173 |
| 2011-2012 | 6 007 | 7 202 | 1 011 | 244 | 872 | 160 | 15 496 |
| Médicaments en vente libre (aucune annexe) | |||||||
| 2010-2011 | 10 | 1 178 | 150 | 66 | 1 404 | ||
| 2011-2012 | 3 | 1 945 | 347 | 113 | 2 408 | ||
Source: Inspectorat, 2010a, 2011, 2012a |
|||||||
| Nom | Description/mandat | Activités consignées |
|---|---|---|
| Comité consultatif d'experts sur la vigilance des produits de santé (CCE-VPS) | Le CCE-VPS a pour mandat de « fournir, sur une base permanente, à la DGPSA des avis généraux d'experts de l'extérieur au sujet des politiques et des stratégies concernant l'innocuité et l'efficacité thérapeutique des produits de santé commercialisés à l'usage des humains. Il est aussi chargé d'établir un mécanisme favorisant la participation du public et permettant aux citoyens de faire connaître leurs points de vue aux experts, qui pourront par la suite les examiner et les intégrer dans les avis qu'ils seront appelés à fournir » (DPSC,2006). | Selon l'information consignée, le Comité s'est réuni neuf fois ente 2007 et 2009. Le Comité a tenu des discussions et formulé des conseils à propos de 47 sujets proposés. Ses avis ont été utiles dans le cadre de l'élaboration du document Recommandations pour l'usage approprié des produits contre la toux et le rhume chez les enfants (DPSC, 2008). Le Comité a élaboré un plan de participation du public. Des 450 intervenants invités à fournir des commentaires et suggestions sur trois questions, 58 l'ont fait avant la réunion et 16 pendant la réunion. |
| Comité consultatif d'experts sur les initiatives pédiatriques | Le Comité, formé en 2009, a pour objectif de permettre à Santé Canada « d'obtenir l'avis d'experts et l'apport du public en ce qui a trait à la mise au point et à l'homologation des produits de santé commercialisés -- produits pharmaceutiques, matériels médicaux et produits biologiques, y compris les vaccins et les produits de santé naturels -- destinés aux enfants et aux femmes enceintes ou qui allaitent » (Santé Canada, 2012b). Le Comité présente aussi à Santé Canada son point de vue sur les normes de nutrition et de salubrité des aliments. Il est composé de 15 experts, notamment des spécialistes en pédiatrie, des professeurs d'université, des pharmaciens, des chercheurs ainsi que des représentants de l'industrie, de groupes de patients, d'organismes sans but lucratif et de parents (Santé Canada, 2012b). | En mai 2010, le Comité a discuté de l'utilisation de médicaments à des fins autres que les usages indiqués sur l'étiquette au sein de la population pédiatrique. Les documents donnent à comprendre que les discussions ont motivé l'étude « Prendre garde aux lacunes », entreprise par le Bureau des initiatives pédiatriques afin d'aborder les questions relatives à l'utilisation pour les enfants de médicaments à des fins autres que celles prévues (DPSC, 2011). Le Comité a parrainé une initiative visant à « réaliser une évaluation fondée sur des preuves et faisant autorité sur la situation au Canada et à l'étranger concernant les produits thérapeutiques destinés aux enfants » (gouvernement du Canada, 2012, p. 16:10) |
| Comité sur le nouveau médicament Adderall XR | Le Comité a été formé à la demande de Santé Canada et de la société pharmaceutique Shire BioChem Inc., fabricant du médicament Adderall XR, afin de formuler des recommandations concernant la décision de suspendre la formulation du médicament en question (Santé Canada, 2005). | Le Comité a produit un rapport sur le nouveau médicament Adderall XR formulant des recommandations appuyant la décision de suspendre l'avis de conformité du Adderall XR. Les recommandations du Comité ont par la suite été adoptées par Santé Canada (l'avis de conformité a été suspendu en février 2005, puis a été réinstauré par la suite). |
| Comité consultatif scientifique sur l'abus d'analgésiques opioïdes (CCS-AAO) | Le CCS-AAO « agit à titre de forum pour recueillir les commentaires et de groupe de rétroaction pour la direction et les scientifiques de Santé Canada. » On le consulte pour obtenir des avis sur les enjeux touchant l'innocuité des nouveaux analgésiques opioïdes d'ordonnance et leur consommation abusive, des propositions concernant la nature des renseignements que devraient comprendre les monographies de ce type de produits pharmaceutiques, et des conseils sur les stratégies de gestion des risques à adopter (DPT, 2011f). | Selon l'information consignée, le Comité s'est réuni une fois, le 29 mars 2011. Les observations formulées à la fin de la réunion indiquent que les participants estimaient avoir eu des discussions productives et qu'il n'était pas nécessaire de tenir une seconde réunion (DPT, 2011e). |
| Comité consultatif scientifique sur les médicaments en vente libre (CCS-MVL) | Le CCS-MVL « agit à titre de forum pour recueillir les commentaires et de groupe de rétroaction pour la direction et les scientifiques de Santé Canada. » Le Comité fournit des conseils scientifiques, techniques et médicaux à la DPT afin de l'aider dans l'élaboration de la réglementation touchant les médicaments en vente libre (DPT, 2011c). | Selon l'information consignée, le Comité s'est réuni une fois, le 24 novembre 2012 (DPT, 2012a). |
| Comité consultatif scientifique sur les thérapies oncologiques (CCS-TO) | Le CCS-TO « fournit à Santé Canada, en temps opportun, des conseils scientifiques, techniques et médicaux relatifs à la réglementation des thérapies oncologiques. La participation des scientifiques, des médecins et des communautés d'intérêts au processus d'examen réglementaire devrait accroître la transparence et permettre d'obtenir de façon proactive des conseils externes facilitant ainsi le processus d'examen des médicaments pour le cancer. Le Comité émet des conseils et des recommandations à Santé Canada, mais la prise de décisions incombe au Ministère » (DPT, 2010b). | Selon l'information consignée, le Comité s'est réuni quatre fois : le 13 septembre 2007, le 26 février 2009, le 15 avril 2010 et le 6 décembre 2011. Lors de ces réunions, les membres du Comité ont apporté des réponses et formulé des recommandations concernant six sujets qui leur avaient été soumis (DPT, 2011b). |
| Comité consultatif scientifique sur les sciences pharmaceutiques et la pharmacologie clinique (CCS-SPPC) | Le CCS-SPPC fournit « à Santé Canada, en temps opportun, des conseils scientifiques, techniques et cliniques sur des questions reliées aux sciences pharmaceutiques et à la pharmacologie clinique touchant les exigences relatives aux données pour de nouvelles formulations de produits pharmaceutiques. Par exemple, ces exigences pourraient porter sur l'équivalence pharmaceutique des médicaments génériques, les nouveaux sels, les formulations complexes, etc. » (DPT, 2011d). | L'unique activité consignée de ce comité est un appel de candidature et l'annonce d'une réunion (DPT, 2011a, 2012c). Le mandat n'a jamais été finalisé. |
| Comité consultatif scientifique sur les thérapies respiratoires et le traitement des allergies (CCS-TRTA) | Le CCS-TRTA « donne à Santé Canada, de façon continue et en temps opportun, des conseils scientifiques, techniques, médicaux et cliniques pour l'évaluation de l'innocuité et de l'efficacité des médicaments utilisés dans le traitement des maladies respiratoires et des allergies, et en matière de politiques » (DPT, 2010c). | Selon l'information consignée, le Comité s'est réuni huit fois depuis 2006 (DPT, 2012b). Le Comité a élaboré les ébauches de deux documents qui n'ont pas encore été finalisés : Exigences relatives à la présentation de corticostéroïdes en inhalation de commercialisation subséquente utilisés dans le traitement de l'asthme et Exigences relatives à la présentation de stéroïdes nasaux de commercialisation subséquente utilisés dans le traitement de la rhinite allergique (DPT, 2012b). |
| Groupe consultatif scientifique sur les normes de bioéquivalence pour les médicaments spécifique au sexe (GCS-MSS) | Le GCS-MSS « transmet des avis à Santé Canada sur les exigences en matière d'études de bioéquivalence à l'égard des produits pharmaceutiques destinés à être administrés exclusivement aux femmes ou exclusivement aux hommes. Revêtent un intérêt tout particulier les médicaments au sujet desquels des craintes ont été soulevées en lien avec les études de bioéquivalence actuellement exigées qui ne seraient pas adéquates, par exemple, les médicaments contenant du succinate de doxylamine et du chlorhydrate de pyridoxine pour le traitement des nausées durant la grossesse » (Santé Canada, 2011c). | Selon l'information consignée, le Groupe s'est réuni une fois, le 2 juin 2011 (Santé Canada, 2011d). |
| Groupe consultatif scientifique sur les formes pharmaceutiques à libération modifiée (GCS-FPLM) | Le GCS-FPLM « fournit à Santé Canada des conseils relatifs aux normes de bioéquivalence qui s'appliquent aux FPLM des médicaments pharmaceutiques. Il est intéressant de s'attarder aux médicaments pour lesquels des inquiétudes ont été soulevées voulant que les normes en vigueur ne conviennent peut-être pas, comme pour le méthylphénidate et la nifédipine » (DPT, 2010d). | Selon l'information consignée, le Groupe s'est réuni une fois, le 11 juin 2010. Lors de cette réunion, les membres du Groupe ont formulé des recommandations concernant deux sujets qui leur avaient été soumis par Santé Canada (DPT, 2010e). Le mandat n'a jamais été finalisé (DPT, 2010d). |
| Groupe consultatif d'experts sur le Programme d'accès spécial (GCE-PAS) | Le GCE-PAS « agit à titre de forum pour recueillir les commentaires et de groupe de rétroaction pour la direction et les scientifiques de [la DGPSA]. Les membres du groupe [...] se réunissent pour discuter des moyens d'améliorer le cadre réglementaire qui régira le fonctionnement du Programme d'accès spécial » (Santé Canada, 2007). | Les documents fournis à PRA Inc. ne comportaient aucune information sur les activités de ce groupe consultatif. Selon le site Web de Santé Canada, son mandat a été réalisé (Santé Canada, 2012c). |
| Comité consultatif scientifique sur les traitements liés à la procréation (CCS-TP) | Le CCS-TP « fournit à Santé Canada, en temps opportun, des avis scientifiques, techniques et médicaux concernant l'évaluation des données sur l'innocuité et l'efficacité des traitements liés à la procréation, qui sont soumises dans le cadre du processus d'examen des présentations de médicaments ou d'instruments médicaux ou des activités de surveillance post-commercialisation. » Le Comité devrait « accroître la transparence du processus et permettre une contribution externe proactive » (DPT, 2003). | Les documents fournis à PRA Inc. ne comportaient aucune information sur les activités de ce comité. Selon le site Web de Santé Canada, son mandat a été réalisé (Santé Canada, 2012a). |
| Comité consultatif scientifique sur les traitements métaboliques et endocriniens (CCS-TME) | Le CCS-TME fournit à Santé Canada « des conseils liés à l'évaluation des données sur l'efficacité et l'innocuité des médicaments servant aux traitements métaboliques, qui sont soumises dans le cadre du processus d'examen des présentations de médicaments ou d'instruments médicaux ou des activités de surveillance post-commercialisation. La participation [du Comité] au processus d'examen réglementaire devrait accroître la transparence et permettre d'obtenir de façon proactive des conseils externes facilitant ainsi le processus d'examen des médicaments et des instruments médicaux » (Santé Canada, 2006). | Selon l'information consignée, le Comité s'est réuni une fois, le 15 novembre 2007(Santé Canada, 2006). |
| Comité consultatif scientifique sur les traitements musculosquelettiques (CCS-TMS) | Aucune information n'est disponible. | Le Comité a été dissous (Santé Canada, 2011a). |
| Comité consultatif scientifique sur les thérapies neurologiques (CCS-TN) | Aucune information n'est disponible. | Le Comité a été dissous (Santé Canada, 2011a). |
| Médicaments humains 2003-2012 |
Médicaments biologiques 2005-2012 |
|
|---|---|---|
| Nombre d'évaluations de signaux réalisées | 272 | 113 |
| Nombre médian de jours ouvrables entre la réception d'un signal et son évaluation | 65 jours | 14 jours |
| Nombre moyen de jours ouvrables entre la réception d'un signal et son évaluation | 157,7 jours | 70 jours |
| Évaluations réalisées dans un délai de 130 jours ouvrables, avant avril 2007 | 10% | N/A |
| Évaluations réalisées dans un délai de 130 jours ouvrables, après avril 2007 | 51 % | 92 % |
| Évaluations de faible priorité réalisées à l'intérieur du délai cible de 200 jours ouvrables, après avril 2007 | 68% | 97 % |
| Évaluations de priorité moyenne réalisées à l'intérieur du délai cible de 130 jours ouvrables, après avril 2007 | 44% | 99 % |
| Évaluations de priorité élevée réalisées à l'intérieur du délai cible de 80 jours ouvrables, après avril 2007 | 75 % | 100 % |
| Temps de traitement médian en jours ouvrables, avant avril 2007 | 348 jours | S.O. |
| Temps de traitement médian en jours ouvrables, après avril 2007 | 128 jours | 46 jours |
| Temps de traitement médian en jours ouvrables, avant avril 2007 | 481 jours | S.O. |
| Temps de traitement médian en jours ouvrables, après avril 2007 | 161 jours | 70 jours |
| Niveau de priorité | Période d'activité | Jours civils | Nombre total de cas pour ce niveau de priorité et cette période d'activité | ||
|---|---|---|---|---|---|
| Min.* | Max.* | Moy.* | |||
| Vert (faible) (n=15) |
De la détection à la réception | 1 | 91 | 25 | 10 |
| De la réception à l'attribution | 1 | 1 310 | 199 | 15 | |
| De l'attribution à l'approbation | 51 | 538 | 238 | 15 | |
| De l'approbation à l'affichage | 115 | 382 | 200 | 10 | |
| Jaune (moyen) (n=19) |
De la détection à la réception | 0 | 96 | 21 | 13 |
| De la réception à l'attribution | 0 | 223 | 31 | 19 | |
| De l'attribution à l'approbation | 54 | 835 | 199 | 19 | |
| De l'approbation à l'affichage | 32 | 507 | 176 | 17 | |
| Rouge (élevé) (n=1) |
De la détection à la réception | 37 | 37 | - | 1 |
| De la réception à l'attribution | 6 | 6 | - | 1 | |
| De l'attribution à l'approbation | 106 | 106 | - | 1 | |
| De l'approbation à l'affichage | 203 | 203 | - | 1 | |
| Évaluations prioritaires (aucun niveau de priorité attribué) (n=3) |
De la détection à la réception | 42 | 42 | - | 1 |
| De la réception à l'attribution | 0 | 12 | 6 | 2 | |
| De l'attribution à l'approbation | 94 | 276 | 185 | 2 | |
| De l'approbation à l'affichage | 98 | 98 | - | 1 | |
*Les valeurs négatives sont exclues. |
|||||
| Activité/forum | Description |
|---|---|
| American Association of Tissue Banks (AATB) | Ce groupe publie des normes et délivre des agréments aux banques de tissus à l'échelle internationale, interagit avec les organismes de réglementation, organise des conférences éducatives et donne de la formation. La DPBTG est membre de son comité responsable de l'élaboration des normes et a notamment participé à la préparation de la 13e édition du guide des normes de l'AATB Standards for Tissue Banking, publiée en mars 2012. |
| Forum de Coopération économique de la zone Asie-Pacifique (APEC) par l'entremise de son comité directeur sur l'harmonisation réglementaire (RHSC) | La DPT occupe la présidence du RHSC, dont le mandat est de promouvoir l'harmonisation et de cerner les projets de grande valeur en partenariat avec l'APEC Harmonization Center ainsi qu'avec d'autres initiatives d'harmonisation, des organismes de formation et des acteurs internationaux comme l'OMS. En 2011, les ministres de l'APEC ont approuvé un cadre stratégique visant une convergence des réglementations sur les produits médicaux pour 2020. Santé Canada préside actuellement le groupe de travail sur les sciences de la vie de l'APEC. |
| Groupe de travail de l'APEC sur l'inspection des BPC | L'Inspectorat est membre de ce groupe, qui œuvre à promouvoir l'harmonisation des BPC et offre des possibilités de réseautage et de mise en commun des meilleures pratiques avec des collègues d'autres pays. Le groupe apporte également du soutien pour l'élaboration de cadres réglementaires dans les pays où les systèmes réglementaires sont moins avancés. |
| Harmonisation de l'homologation des médicaments en Afrique (HHMA) | L'HHMA aide les pays africains à harmoniser leur processus d'homologation des médicaments et à accélérer l'homologation des médicaments prioritaires et essentiels. Les partenaires de l'HHMA prêtent main-forte aux communautés économiques régionales d'Afrique dans l'élaboration de propositions de projets d'harmonisation de l'homologation des médicaments. La réunion inaugurale des intervenants a eu lieu le 28 mars 2012, en Tanzanie. |
| Initiative canadienne de vaccin contre le VIH (ICVV) | Il s'agit d'une initiative de cinq ans qui vise la collaboration entre le gouvernement du Canada et la Bill & Melinda Gates Foundation. Dans le cadre de cette initiative, la DPBTG offre de la formation aux nouvelles autorités réglementaires nationales (ARN) des pays à revenu faible et moyen afin de renforcer les cadres réglementaires régissant les essais cliniques et les autorisations de mise en marché des vaccins. Les initiatives ci-après sont soutenues par l'entremise de l'ICVV. |
| a. African Vaccines Regulatory Forum | La DPBTG participe aux activités de ce groupe, qui offre aux organismes de réglementation des avis d'experts sur l'évaluation des vaccins. Ce forum aide à la prise de décisions en ce qui concerne l'autorisation des essais cliniques, l'évaluation des dossiers d'homologation et les autres enjeux liés à l'évaluation des vaccins. |
| b. Programme de mentorat sur le renforcement des capacités réglementaires de l'ICVV | Ce programme de mentorat vise à offrir des occasions de formation individualisée aux ARN afin de renforcer les capacités réglementaires relatives aux médicaments biologiques et aux vaccins. Dans le cadre de ce programme, la DPBTG a lancé en 2012 un programme de mentorat avec le Malawi. |
| c. Réseau des autorités de réglementation des vaccins des pays en développement | La DPBTG participe aux activités de ce réseau, qui vise à favoriser le renforcement des capacités réglementaires des ARN par l'échange d'expertise et d'information concernant l'évaluation des propositions d'essais cliniques et les données sur les essais cliniques. |
| d. Second International Congress on Pharmacology of Vaccines | La DPBTG a participé à ce congrès (VacciPharma 2012) avec les pays de l'Organisation panaméricaine de la santé afin de discuter de l'utilisation des vaccins prophylactiques contre le VIH. |
| Consortium des chefs d'agence | Le Consortium des chefs d'agence comprend également la TGA, Swissmedic et la Health Sciences Authority de Singapour. Son objectif est d'augmenter le niveau d'expertise et de connaissance et de cerner les possibilités de partage du travail. Le groupe a élaboré un plan d'action touchant les médicaments génériques et a créé un processus de même qu'un modèle normalisés permettant la mise en commun des demandes en attente et l'établissement des demandes communes. Des efforts sont actuellement déployés pour que les organismes partenaires travaillent en collaboration pour la revue des RPPV. |
| Forum international des laboratoires sur les médicaments contrefaits (ILFCM) | Santé Canada participe aux activités du ILFCM, qui est un forum non officiel de mise en commun de l'information où les experts peuvent partager leurs expériences à propos d'enjeux opérationnels importants comme l'inspection et l'analyse des produits contrefaits. Les membres du forum se réunissent semestriellement. |
| Groupe international des organismes de réglementation des médicaments génériques (IGDRG) | Santé Canada participe aux activités du IGDRG, qui est un forum destiné aux organismes de réglementation et visant à examiner les possibilités de collaboration dans le domaine des médicaments génériques. Les possibilités de collaboration comprennent le travail partagé et l'accroissement de l'harmonisation réglementaire. Les membres du groupe se sont réunis physiquement au moins deux fois et, durant ces rencontres, s'est dégagé un consensus quant aux buts, aux objectifs et aux axes de collaboration prioritaires. |
| Forum international sur la réglementation | Ce forum annuel vise à contribuer au développement des réglementations en Afrique et en Asie et à la collaboration entre les Amériques. Le quatrième Forum international sur la réglementation annuel s'est tenu du 24 au 28 septembre 2012 à Ottawa et a accueilli environ 100 participants de plus de 40 pays. |
| Réseau des laboratoires officiels de contrôle des médicaments (OMCL) | Les laboratoires de l'Inspectorat font partie de ce réseau, dont les réunions permettent à ses membres d'échanger sur les meilleures pratiques et sur des questions d'intérêt pour les gestionnaires de laboratoires de contrôle des médicaments en Europe, en Australie et au Canada. |
| Organisation panaméricaine de la santé (OPS) | L'OPS a demandé à ce que la DGPSA fasse l'objet d'une évaluation de sa réglementation par l'OMS et l'OPS dans le cadre de la résolution du conseil directeur de l'OPS sur les ARN. Cet exercice permettra de faire en sorte que Santé Canada puisse servir de modèle pour les autres ARN de l'hémisphère et améliorer ses propres programmes de réglementation (le programme de la DPBGT touchant les vaccins a déjà été évalué favorablement). |
| Coopération du Réseau panaméricain d'harmonisation de la réglementation pharmaceutique (PANDRH) | Ce réseau appuie le processus d'harmonisation de la réglementation pharmaceutique dans les Amériques. La DPT est membre du comité directeur et la DPBTG participe aux activités de deux des groupes de travail du PANDRH (sur les vaccins et les produits biologiques), lesquels ont produit des lignes directrices internationales sur ces sujets. |
| Pharmaceutical Inspection Co-operation Scheme (PIC/S) et collaboration spéciale PIC/S-accords de reconnaissance mutuelle (ARM) | Le PIC/S vise plusieurs objectifs, notamment :
La DGPSA fait partie de divers groupes de travail du PIC/S. Également, Santé Canada participe actuellement à un projet conjoint spécial destiné à évaluer la faisabilité d'une collaboration plus étroite avec le PIC/S dans le cadre d'un programme d'évaluation conjoint visant les pays qui souhaitent joindre le programme des ARM et du PIC/S. Certains représentants du PIC/S travaillent, au cours du présent exercice, de concert avec un agent ARM et l'autorité VMD sur un projet pilote d'évaluation de cette collaboration éventuelle. L'équipe d'évaluateurs est composée de la République tchèque, qui est à la tête, ainsi que de la Suisse et de Santé Canada. |
| Organisation internationale de normalisation | La DGPSA a participé à l'élaboration de la norme ISO - TC215, dont l'objectif est de permettre un partage libre et ouvert des données internationales sur la sécurité, de favoriser l'interfonctionnement entre des systèmes indépendants et de rendre les données et l'information en santé compatibles et uniformes. La DPSC fait partie du groupe de travail de travail 6 de la norme TC215 sur l'informatique en santé, dont les travaux portent sur le domaine pharmaceutique et des médicaments. |
| Téléconférence sur la pharmacovigilance | La DGPSA participe à une téléconférence sur la pharmacovigilance amorcée en 1998 et comprenant la DPSC (DGPSA), le Centre for Drug Evaluation and Research (FDA), l'Adverse Drug Reaction Unit (TGA), Medsafe (Nouvelle-Zélande) et la Health Sciences Authority (Singapour). Cette téléconférence est un forum qui permet les échanges et la discussion sur la surveillance des produits de santé et favorise la coordination des efforts de pharmacovigilance entre les différentes autorités réglementaires en faisant en sorte que chacune soit sensibilisée aux nouveaux enjeux en matière de sécurité auxquels les autres doivent faire face. Les réunions ont lieu tous les deux mois et la présidence est tournante. |
| Coopération avec l'OMS pour la normalisation biologique et l'évaluation des médicaments biologiques | La DPBTG a collaboré avec l'OMS pour la normalisation biologique et l'évaluation des médicaments biologiques, notamment en travaillant sur des domaines clés comme les vaccins et les vaccins contre le VIH/SIDA. Les activités menées en collaboration comprennent : offrir des conseils et de l'aide technique pour l'élaboration de normes réglementaires touchant les produits biologiques, apporter de l'aide concernant le programme de préqualification de l'OMS et mettre en œuvre les directives de l'OMS dans les pratiques réglementaires. |
| Programme international de pharmacovigilance de l'OMS | Le programme vise à accroître la collaboration entre les organismes de réglementation au moyen de la discussion et d'une mise en commun de l'information. Les forums permettent à Santé Canada de partager son expertise en pharmacovigilance afin d'aider les autres pays dans la mise sur pied de programmes de pharmacovigilance. Dans le cadre de sa participation aux forums, Santé Canada téléverse ses déclarations d'effets indésirables dans la base de données de l'OMS Vigibase. |
| Conseil international des organisations des sciences médicales (CIOMS) de l'OMS | Le CIOMS vise à améliorer les méthodes et la normalisation dans le domaine des sciences biomédicales (c'est-à-dire, minimiser les risques). Les rapports produits par le CIOMS sont devenus la référence de base de nombreux documents d'orientation internationaux en lien avec la surveillance et l'examen des produits de santé. La DPSC est membre de plusieurs des groupes de travail du CIOMS. |
| Programme de préqualification des médicaments de l'OMS | Ce programme évalue les demandes de médicaments présentées à l'OMS après une déclaration d'intérêt de l'organisme. Les évaluations permettent de garantir la qualité des médicaments achetés par les organismes de financement de l'ONU et destinés à être acheminés dans les pays admissibles, et contribuent à l'atteinte de l'objectif onusien de lutter contre la propagation des maladies dans les pays où l'accès à des médicaments de qualité est restreint. Jusqu'à six séances d'examen sont menées chaque année. |
| Groupe spécial international chargé de la lutte contre les contrefaçons de produits médicaux (IMPACT) | IMPACT travaille à trouver les mesures appropriées pour lutter contre l'accessibilité à des produits médicaux défectueux. |
| Conférence sur l'application proactive des BPC d'ExL Pharma | Cette conférence de trois jours se tient à Arlington en Virginie et constitue une occasion d'éduquer les intervenants internationaux à propos des exigences réglementaires canadiennes concernant les essais cliniques, ainsi que de faire du réseautage avec des organismes de réglementation d'autres pays. |
Données sur les rappels
Pour obtenir les données sur les produits retirés du marché, PRA Inc. a compilé l'information sur les rappels figurant dans la liste des retraits de médicament en ligne. Les données analysées s'étendent jusqu'au 23 août 2012, inclusivement. Un tableau énumérant tous les rappels cernés de cette façon a été acheminé à la DPT et à la DPBTG afin qu'elles identifient, respectivement, les rappels touchant des médicaments pour usage humain et ceux touchant des médicaments biologiques. Un total de 181 retraits ont été retranchés de la liste initialement établie pour les raisons suivantes : 106 rappels visaient des produits que la DPT n'a pas pu identifier, généralement parce que la liste ne comportait pas de numéro d'autorisation; 40 visaient de produits de santé naturels contenant des ingrédients pharmaceutiques; 35 visaient des produits pour lesquels la DPT n'a pas pu déterminer de façon certaine s'il s'agissait de produits pharmaceutiques ou de santé naturels. Dans l'ensemble, 666 avis de rappel touchant des médicaments pour usage humain et des médicaments biologiques ont été analysés. Étant donné les étapes nécessaires pour sa réalisation, l'analyse n'a pas été mise à jour et ne comporte pas toutes les données de l'année 2012.
| Année* | Type I | Type II | Type III | Autre/non précisé** | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Biol. | Pharm. | Total | Biol. | Pharm. | Total | Biol. | Pharm. | Total | Biol. | Pharm. | Total | Biol. | Pharm. | |
| 2005 | 11 | 0 | 11 | 12 | 0 | 12 | 33 | 2 | 31 | 7 | 0 | 7 | 63 | 2 | 61 |
| 2006 | 11 | 1 | 10 | 53 | 0 | 53 | 17 | 0 | 17 | 4 | 0 | 4 | 85 | 1 | 84 |
| 2007 | 4 | 0 | 4 | 10 | 3 | 7 | 15 | 0 | 15 | 3 | 0 | 3 | 32 | 3 | 29 |
| 2008 | 13 | 5 | 8 | 37 | 1 | 36 | 20 | 0 | 20 | 1 | 0 | 1 | 71 | 6 | 65 |
| 2009 | 5 | 0 | 5 | 57 | 1 | 56 | 29 | 1 | 28 | 0 | 0 | 0 | 91 | 2 | 89 |
| 2010 | 7 | 0 | 7 | 63 | 6 | 57 | 34 | 2 | 32 | 6 | 0 | 6 | 110 | 8 | 102 |
| 2011 | 17 | 0 | 17 | 71 | 4 | 67 | 52 | 2 | 50 | 0 | 0 | 0 | 140 | 6 | 134 |
| 2012 | 3 | 0 | 3 | 51 | 3 | 48 | 19 | 0 | 19 | 0 | 0 | 0 | 73 | 3 | 70 |
| Unspecified | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Total | 71 | 6 | 65 | 355 | 18 | 337 | 219 | 7 | 212 | 21 | 0 | 21 | 666 | 31 | 635 |
Source: Inspectorate, 2012b. Les données de 2012 étaient à jour en date du 23 août 2012. Remarque : Les données du tableau ne comprennent pas les cas de rappel suivants : 106 rappels visaient des produits que la DPT n'a pas pu identifier, généralement parce que la liste ne comportait pas de numéro d'autorisation; 40 visaient de produits de santé naturels contenant des ingrédients pharmaceutiques; 35 visaient des produits pour lesquels la DPT n'a pas pu déterminer de façon certaine s'il s'agissait de produits pharmaceutiques ou de santé naturels. *Un rappel fait partie d'une année donnée si la date de commencement de ce rappel est incluse dans cette année. **Ces colonnes comprennent les cas qui ont été classés par l'Inspectorat dans les catégories suivantes : S.O., risque inacceptable pour la santé, retrait du marché ordonné par Santé Canada ou nul. |
|||||||||||||||
| Année | Effets indésirables mineurs | Effets indésirables graves | Total | ||
|---|---|---|---|---|---|
| Nombre | Pourcentage | Nombre | Pourcentage | ||
| 1999 | 2 628 | 47,0% | 2 960 | 53,0% | 5 588 |
| 2000 | 3 251 | 45,7% | 3 862 | 54,3% | 7 113 |
| 2001 | 1 976 | 27,2% | 5 292 | 72,8% | 7 268 |
| 2002 | 2 561 | 30,5% | 5 828 | 69,5% | 8 389 |
| 2003 | 2 605 | 29,0% | 6 392 | 71,0% | 8 997 |
| 2004 | 3 184 | 31,6% | 6 904 | 68,4% | 10 088 |
| 2005 | 3 134 | 30,5% | 7 147 | 69,5% | 10 281 |
| 2006 | 3 454 | 33,0% | 7 007 | 67,0% | 10 461 |
| 2007 | 4 169 | 33,6% | 8 227 | 66,4% | 12 396 |
| 2008 | 4 918 | 31,6% | 10 646 | 68,4% | 15 564 |
| 2009 | 5 210 | 28,0% | 13 414 | 72,0% | 18 624 |
| 2010 | 6 604 | 29,7% | 15 631 | 70,3% | 22 235 |
| 2011 | 8 064 | 28,1% | 20 595 | 71,9% | 28 659 |
| 2012 | 2 286 | 26,6% | 6 313 | 73,4% | 8 599 |
| Ne sais pas | 54 044 | 31,0% | 120 218 | 69,0% | 174 262 |
Source: (MHPD, 2012a) |
|||||
| Marque déposée | Ingrédient actif | Annexe |
|---|---|---|
| Anzemet/Anzemet (injection) | Mésylate de dolasétron | F |
| Apo-Phenylbutazone | Phénylbutazone | F |
| Apo-Thioridazine/Dom Thioridazine/Novo-Rizadine/PMS-Thioridazine/Thioridazine | Chlorhydrate de thioridazine | F |
| Apo-Sibutramine/Meridia | Chlorhydrate de sibutramine | F |
| BCI Pravastatin/Riva-Pravastatin | Pravastatine sodique | F |
| Bextra | Valdécoxib | F |
| Climacteron (injection) | benzoate d'oestradiol, diénanthate d'oestradiol, énanthate de testostérone | G (LRCDAS IV) |
| Darvon-N | Dextropropoxyphène | Stupéfiant (LRCDAS I) |
| Depakene | Acide valproïque | F |
| Fluotic | Fluorure de sodium | F |
| Fucidin | Fusidate de sodium | F |
| Levo-T | Lévothyroxine sodique | F |
| Palladone XL | Chlorhydrate d'hydromorphone | Stupéfiant (LRCDAS I) |
| Permax/Shire-Pergolide | Mésylate de pergolide | F |
| Prexige | Lumiracoxib | F |
| Raptiva | Efalizumab | D, F |
| Thelin | Sitaxsetan sodique | F |
| Vioxx | Rofécoxib | F |
| Xigris | Drotrécogine alfa | D, F |
| Zelnorm | Maléate de tégasérod | F |
Source: Santé Canada (2013) |
||
Liste des ouvrages de référence pour l'annexe C - Tableaux de données supplémentaires
DPBTG. (2010). Drug submission performance annual report (2010).
Decima Research. (2003). Public opinion survey on key issues pertaining to post-market surveillance of marketed health products in Canada.
Decima Research. (2006). Final report: 2006 General public opinion survey on key issues pertaining to post-market surveillance of marketed health products in Canada.
Environics Research Group. (2007). Adverse Reaction Reporting - Survey with Health Professionals. Consulté le 11 octobre 2012 sur http://epe.lac-bac.gc.ca/100/200/301/pwgsc-tpsgc/por-ef/health/2007/432-06/summary.pdf
FFAMC. (2012). Proceedings: SSCSAST, Issue No. 16 (May 9-10, 2012).
Santé Canada. (2005). Report of the "Adderall XR New Drug Committee." Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/ndca_rep_cnma_rap_2005-08-25-eng.pdf (lien périmé).
Santé Canada. (2006). Metabolic and Endocrine Therapies TOR. Consulté le 4 avril 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/sacmet_tor_ccstme_att-eng.pdf (lien périmé).
Santé Canada. (2007). Expert Advisory Panel on the Special Access Programme: Terms of Reference.
Santé Canada. (2011a). Complété ou actif - Organismes consultatifs scientifiques (2011). Consulté le 19 mars 2012 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/activit/sci-com/sab_ocs_comm_list-fra.php.
Santé Canada. (2011b). Performance measurement and evaluation plan for the proposed fees in respect of Human Drugs and Medical Devices Regulations (version 8).
Santé Canada. (2011c). Groupe consultatif scientifique sur les normes de bioéquivalence pour les médicaments spécifiques au sexe - Attributions. Consulté le 4 octobre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-consult/gender-sexe/sapgsdp_tor_gcsmss_att-fra.pdf.
Santé Canada. (2011d). Groupe consultatif scientifique sur les normes de bioéquivalence pour les médicaments spécifiques au sexe (GCS-MSS) : Compte rendu des délibérations 2011-06-02. Consulté le 28 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-consult/gender-sexe/sapgsdp-rop_gcsmss-crd_2011-06-02_ati-fra.pdf.
Santé Canada. (2012a). Complété ou actif - Organismes consultatifs scientifiques (2012). Consulté le 18 octobre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/activit/sci-com/sab_ocs_comm_list-fra.php.
Santé Canada. (2012b). Comité consultatif d'experts sur les initiatives pédiatriques (CCEIP) - Santé Canada. Liste des ressources. Consulté le 4 octobre 2012 sur http://www.hc-sc.gc.ca/ahc-asc/branch-dirgen/hpfb-dgpsa/opi-bip/peac-cceip/index-fra.php.
Santé Canada. (2012c). Groupes consultatifs d'experts/scientifiques - Médicaments et produits de santé - Santé Canada. Consulté le 18 octobre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/activit/sci-consult/sab_ocs_pan_gr_list-fra.php.
Santé Canada. (2013). Drugs removed from the market for safety reasons, 2004-2012, data extract.
Inspectorat. (2004). A2.10 PMRC - Drug Inspections performance report (2003-04, year-end).
Inspectorat. (2006). A2.10 PMRC - Drug Inspections performance report (2005-06, year-end) (final, v2).
Inspectorat. (2007). Inspectorate performance report (2006-07, year-end).
Inspectorat. (2010a). B1.5 Regional Border Integrity Unit performance report (2009-10) (feuille de calcul Excel).
Inspectorat. (2010b). Inspectorate performance report (2009-10).
Inspectorat. (2011). B1.5 Regional Border Integrity Unit performance report (2010-11) (feuille de calcul Excel).
Inspectorat. (2012a). B1.5 Regional Border Integrity Unit performance report (2011-2012) (feuille de calcul Excel).
Inspectorat. (2012b). Liste de retrait de marché des médicaments et des instruments (août 2012). Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/compli-conform/recall-retrait/_list/index-eng.php (lien périmé).
DPSC. (2006). Terms of reference of the Expert Advisory Committee on the Vigilance of Health Products (EAC-VHP)
DPSC. (2008). Rapport sommaire de réunion - Comité consultatif d'experts sur la vigilance des produits de santé (CCE-VPS) - les 21 et 22 février 2008.
DPSC. (2011). Rapport sommaire de réunion - Comité consultatif d'experts sur la vigilance des produits de santé (CCE-VPS) - 31 mai, 1er et 2 juin 2011.
DPSC. (2012a). Base de données des effets indésirables : Base de données en ligne des effets indésirables de Canada Vigilance [2012]. Consulté le 27 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/medeff/databasdon/index-fra.php.
DPSC. (2012b). Normes de rendement de la DPSC.
Nanos Research. (2010). Results of drug safety survey of Canadians.
Phoenix Strategic Perspectives Inc. (2011). Canadians' Awareness of Health and Safety Issues Related to Consumer Products (HC POR 10-09).
Le Groupe Antima et TNS Canada. (2007). La sous-utilisation du système de déclaration des réactions indésirables. Consulté le 11 octobre 2012 sur http://epe.lac-bac.gc.ca/100/200/301/pwgsc-tpsgc/por-ef/health/2007/385-06/sommaire.pdf.
DPT. (2003). Comité consultatif scientifique sur les traitements liés à la procréation (CCS-TP) Attributions.
DPT. (2008a). Rapport annuel de rendement sur les présentations de médicaments - DPT - 2008.
DPT. (2008b). Rapport annuel de rendement sur les présentations de médicaments (2008).
DPT. DSPC. (2010a). Rapport annuel de rendement sur les présentations de médicaments - DPT - 2010. Consulté le 31 août 2012 sur http://www.jptcp.com/files/tpd_dpt_annual_annuel_10_eng.pdf (lien inexistant en français).
DPT. (2010b). Comité consultatif scientifique sur les thérapies oncologiques : Attributions. Consulté le 26 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-com/onco/sacot_tor_ccsto_att-fra.pdf.
DPT. (2010c). Comité consultatif scientifique sur les thérapies respiratoires et le traitement des allergies (CCS-TRTA). Consulté le 28 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-com/resp_allerg/sacrat_tor_ccstrta_att-eng.pdf (lien périmé).
DPT. (2010d). Groupe consultatif scientifique sur les exigences en matière de bioéquivalence pour les formes pharmaceutiques à libération modifiée (GCS-FPLM). Consulté le 28 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-consult/mrdf-fplm/mrdf_tor_dr_fplm_att_eb-fra.pdf.
DPT. (2010e). Groupe consultatif scientifique sur les exigences en matière de bioéquivalence pour les formes pharmaceutiques à libération modifiée (GCS-FPLM): Compte rendu des délibérations 2010-06-11. Consulté le 28 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-consult/mrdf-fplm/sapmrdf_rop_gscfplm_crd_2010-06-11-eng.pdf (lien périmé).
DPT. (2011a). Avis : Appel de candidatures - Comité consultatif scientifique sur les sciences pharmaceutiques et la pharmacologie clinique (CCS-SPPC). Consulté le 27 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-com/pharma/sacpscp_ccssppc_inv-eng.pdf.
DPT. (2011b). Oncothérapies - Comités consultatifs d'experts scientifiques. Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/activit/sci-com/onco/index-fra.php.
DPT. (2011c). Comité consultatif scientifique sur les médicaments en vente libre (CCS-MVL) : Attributions. Consulté le 26 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-com/nonpres/sacnpd_tor_ccsmvl_att-fra.pdf.
DPT. (2011d). Comité consultatif scientifique sur les sciences pharmaceutiques et la pharmacologie clinique (CCS-SPPC) : Attributions provisoires. Consulté le 27 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-com/pharma/sacpscp_tor_ccssppc_att-fra.pdf.
DPT. (2011e). Groupe consultatif scientifique sur l'abus d'analgésiques opioïdes (GCS-AAO) : Compte rendu des délibérations 2011-03-29. Consulté le 26 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-consult/opioid/sapoaa_rop_gcsaao_crd_2011-03-29-eng.pdf (lien périmé).
DPT. (2011f). Groupe consultatif scientifique sur l'abus d'analgésiques opioïdes (GCS-AAO) : Attributions. Consulté le 26 septembre 26 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-consult/opioid/sapoaa_tor_gcsaao_att-eng.pdf (lien périmé).
DPT. (2012a). Médicaments en vente libre - Comités consultatifs d'experts/scientifiques. Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/activit/sci-com/nonpres/index-fra.php.
DPT. (2012b). Thérapies respiratoires et le traitement des allergies - Comités consultatifs d'experts/scientifiques. Consulté le 21 août 2012 sur http://www.hc-sc.gc.ca/dhp-mps/prodpharma/activit/sci-com/resp_allerg/index-fra.php.
DPT. (2012c). Comité consultatif scientifique sur les sciences pharmaceutiques et la pharmacologie clinique (CCS-SPPC) Annonce de la réunion et invitation à participer - 2012-06-26. Consulté le 27 septembre 2012 sur http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/activit/sci-com/pharma/sacpscp_ccssppc_anno-eng.pdf (lien périmé).
Liste des acronymes
- SMA
- Sous-ministre adjoint
- PADN
- Présentation abrégée de drogue nouvelle
- IPA
- Ingrédient pharmaceutique actif
- DPBTG
- Direction des produits biologiques et des thérapies génétiques
- PRB
- Programme des réunions bilatérales
- ASFC
- Agence des services frontaliers du Canada
- CCLT
- Centre canadien de lutte contre les toxicomanies
- CDC
- Centres pour le contrôle et la prévention des maladies
- LRCDAS
- Loi réglementant certaines drogues et autres substances
- ACIA
- Agence canadienne d'inspection des aliments
- BPFa
- Bonnes pratiques de fabrication actuelles
- ICIS
- Institut canadien d'information sur la santé
- IRSC
- Instituts de recherche en santé du Canada
- DGCAP
- Direction générale des communications et des affaires publiques
- PCSP
- Programme canadien de surveillance pédiatrique
- IRC
- Initiative de recouvrement des coûts
- DEC
- Demande d'essai clinique
- DEC-M
- Modification à une demande d'essai clinique
- CTD
- Common Technical Document
- CTO
- Cellules, tissus et organes
- LEPP
- Licence d'établissement de produits pharmaceutiques
- LPS
- Lettre adressée aux professionnels de la santé
- DIN
- Numéro d'identification du médicament
- BDPP
- Base de données sur les produits pharmaceutiques
- RMR
- Rapport ministériel sur le rendement
- RIEM
- Réseau sur l'innocuité et l'efficacité des médicaments
- CCE
- Comité consultatif d'experts
- GCE
- Groupe consultatif d'experts
- RASE
- Régime d'avantages sociaux des employés
- eCTD
- electronic Common Technical Document
- EMA/EMEA
- Agence européenne des médicaments
- EPAR
- Rapport européen public d'évaluation
- LP/AP
- Libération prolongée/action prolongée
- EU
- Union européenne
- DNUE
- Drogue nouvelle pour usage exceptionnel
- PASPAC
- Plan d'action pour assurer la sécurité des produits alimentaires et de consommation
- FDA
- Food and Drug Administration (États-Unis)
- ETP
- Équivalent temps plein
- BPC
- Bonnes pratiques cliniques
- BPLD
- Bonnes pratiques de lignes directrices
- BPF
- Bonnes pratiques de fabrication
- GC
- Gouvernement du Canada
- BPE
- Bonnes pratiques d'examen
- BPV
- Bonnes pratiques de pharmacovigilance
- PMUH
- Programme des médicaments à usage humain
- DGSESC
- Direction générale de la santé environnementale et de la sécurité des consommateurs
- CPS
- Comité permanent de la santé
- DGPSA
- Direction générale des produits de santé et des aliments
- CIH
- Conférence internationale sur l'harmonisation des exigences techniques pour l'enregistrement des médicaments à usage humain
- Institute of Medicine
- IOM
- Institute of Medicine
- PSPCS
- Produits de santé à présentation et à consonance semblables
- DAMM
- Détenteur d'une autorisation de mise sur le marché
- DPSC
- Direction des produits de santé commercialisés
- MHRA
- Medicines and Healthcare Products Regulatory Agency
- PE
- Protocole d'entente
- ARM
- Accord de reconnaissance mutuelle
- NSA
- Nouvelle substance active
- NC
- Non conforme
- PDN
- Présentation de drogue nouvelle
- NIH
- National Institutes of Health (États-Unis)
- INM
- Ingrédient non médicinal
- AC
- Avis de conformité
- AC-C
- Avis de conformité avec conditions
- LNO
- Lettre de non-objection
- BVG
- Bureau du vérificateur général du Canada
- BSC
- Bureau des substances contrôlées
- BRS
- Médicament en vente libre
- MSO
- Program Activity Architecture
- AHP
- Architecture d'harmonisation de programmes
- TAPA
- Tableau des activités postautorisation
- RPEAR
- Rapport périodique d'évaluation des avantages et des risques
- PMP
- Programme des médicaments pharmaceutiques
- CCEIP
- Comité consultatif d'experts sur les initiatives pédiatriques
- ASPC
- Agence de la santé publique du Canada
- PIC/S
- Pharmaceutical Inspection Cooperation Scheme
- PLR
- Physician Label Rule (Règle des médecins concernant l'étiquetage)
- CEDAC
- Conformité aux exigences de déclaration après commercialisation
- SMR
- Stratégie de mesure du rendement
- DPPAI
- Direction des politiques, de la planification et des affaires internationales
- RPPV
- Rapport périodique de pharmacovigilance
- PPV
- Plans de pharmacovigilance
- Q et R
- Questions et réponses
- BRP
- Bureau des régions et des programmes
- CCR
- Conseil de coopération en matière de réglementation
- CER
- Comité d'éthique de la recherche
- SEAR
- Stratégie d'évaluation et d'atténuation des risques
- DGRO
- Direction de la gestion des ressources et des opérations
- PGR
- Plan de gestion des risques
- CCS
- Comité consultatif scientifique
- SPADN
- Supplément à une présentation abrégée de drogue nouvelle
- PAS
- Programme d'accès spécial
- SMD
- Sommaire des motifs de décision
- PBU
- Produits biologiques ultérieurs
- SGII
- Système de gestion des informations des intervenants
- SPDN
- Supplément à une présentation de drogue nouvelle
- PON
- Procédure opérationnelle normalisée
- CPASST
- Comité permanent des affaires sociales, des sciences et de la technologie
- SAPT
- Stratégie d'accès aux produits thérapeutiques
- TGA
- Therapeutic Goods Administration
- DPT
- Direction des produits thérapeutiques
- IIPT
- Initiative d'innocuité des produits thérapeutiques
- OMS
- Organisation mondiale de la Santé
Notes de bas de page
- Notes de bas de page 1
-
La RPAD a été élaborée par des organismes participants [c.-à-d. la Direction des produits thérapeutiques (DPT), la Direction des produits de santé commercialisés (DPSC), la Direction des produits biologiques et des thérapies génétiques (DPBTG), l'Inspectorat de la DGPSA (Inspectorat), la Direction des politiques, de la planification et des affaires internationales (DPPAI) de la Direction générale des produits de santé et des aliments (DGPSA); le Bureau des régions et des programmes (BRP)] en réponse aux recommandations formulées dans l'évaluation du Programme des matériels médicaux. La responsabilité de rendre des comptes sur les activités principales relève entièrement des directeurs généraux
- Notes de bas de page 2
-
Il est important de noter que les programmes d'inspection des BPC, des BPF et des BPV englobent les médicaments pharmaceutiques et les médicaments biologiques
- Notes de bas de page 3
-
Calculé à partir de 365 adresses électroniques valides.
- Notes de bas de page 4
-
Calculé à partir de 622 adresses électroniques valides.
- Notes de bas de page 5
-
La littérature ne fait pas toujours la distinction entre les médicaments pharmaceutiques et les médicaments biologiques. Par conséquent, le reste de cette étude décrit les tendances et les problèmes généraux, et différencie les médicaments pharmaceutiques des médicaments biologiques lorsque cela est possible.
- Notes de bas de page 6
-
De ce nombre, deux étaient des produits biologiques (annexes D et F), deux étaient des stupéfiants (LRCDAS I), un faisait partie de l'annexe G (LRCDAS IV) et les autres étaient médicaments sur ordonnance (annexe F).
- Notes de bas de page 7
-
L'héparine est actuellement régie comme un produit biologique, mais à ce moment-là, était régie comme un produit pharmaceutique.
- Notes de bas de page 8
-
De plus, la préparation en pharmacie « peut nécessiter des substances chimiques brutes ou l'altération des caractéristiques physiques ou de la puissance des produits disponibles sur le marché », et « peut comprendre la reformulation pour permettre la délivrance d'un nouveau médicament », mais « ne comprend pas le mélange, la reconstitution ou d'autres manipulations réalisées conformément aux directives d'utilisation indiquées sur l'étiquette des médicaments homologués » (Inspectorat, 2009b, p. 7). Au Canada, la fabrication de médicaments relève de la compétence fédérale, alors que la préparation de médicaments en pharmacie relève principalement de la compétence provinciale ou territoriale; les distinctions entre la préparation en pharmacie et la fabrication sont établies au cas par cas (Inspectorat, 2009b).
- Notes de bas de page 9
-
Plus particulièrement, en 2006, le BVG a recommandé l'élaboration de renseignements de base sur la mesure du rendement, l'établissement de frais d'utilisation fondés sur des normes de service claires et mesurables, et l'examen du financement de base afin de déterminer s'il est suffisant (BVG, 2006).
- Notes de bas de page 10
-
Un ratio de partage des coûts de 50 % signifie que 50 % des coûts du programme sont couverts par les frais d'utilisation.
- Notes de bas de page 11
-
Veuillez noter que Santé Canada n'établit pas son autorité uniquement en fonction de la législation; en fait, une bonne partie de son rôle découle de l'exercice du pouvoir fédéral de dépenser.
- Notes de bas de page 12
-
Avant 2009, aucune donnée ne confirme l'organisation de réunions ordinaires avec les cadres supérieurs des directions partenaires dans le but de donner une orientation stratégique générale au PMUH. Cela est peut-être attribuable au fait que les activités de Santé Canada relativement aux médicaments à usage humain n'ont jamais été conçues en tant que « programme ». Cette désignation est relativement récente et semble être apparue, en partie, en réponse au besoin d'évaluation et à la réorganisation de l'AHP de Santé Canada en 2007-2008.
- Notes de bas de page 13
-
Afin de simplifier la présentation de l'information, l'organigramme ne cadre pas précisément avec le modèle logique du programme. Néanmoins, toutes les activités pertinentes du programme sont abordées.
- Notes de bas de page 14
-
Selon Santé Canada, peu de nouvelles molécules pharmaceutiques ont connu du succès entre 2005 et 2011, ce qui a accentué la diminution des essais cliniques sur des produits pharmaceutiques et l'augmentation simultanée des DEC relatives à des produits biologiques. Le ministère a également fait mention d'une augmentation considérable des DEC-M au cours de la même période.
- Notes de bas de page 15
-
Le programme d'inspection des essais cliniques est décrit en détail à la section 5.4.5.
- Notes de bas de page 16
-
Santé Canada s'est engagé à accroître l'accès du public aux renseignements sur les essais cliniques dans le Plan de renouveau de 2007 et, depuis ce temps, encourage les promoteurs à consigner les essais cliniques de leurs produits thérapeutiques dans un délai de 21 jours suivant le début de l'essai dans l'un des trois registres du réseau de l'Organisation mondiale de la Santé (OMS). Au cours des dernières années, le BVG et le Comité permanent des affaires sociales, des sciences et de la technologie (CPASST) ont critiqué le ministère pour ne pas avoir mis davantage de renseignements sur les essais cliniques à la disposition des Canadiens, et ce, malgré son engagement (BVG, 2011; CPASST, 2012).
- Notes de bas de page 17
-
Santé Canada a pris certaines mesures en vue d'encourager les promoteurs à effectuer des études pédiatriques. En 2006, des mesures incitatives en matière de réglementation sur les études pédiatriques sont entrées en vigueur, offrant ainsi une prolongation de la période de protection des données de six mois aux promoteurs fournissant des renseignements sur des essais cliniques pédiatriques dans le cadre d'une présentation de drogue novatrice (DGPSA, 2007a, p. 27). De plus, Santé Canada a récemment publié un document de travail sur l'utilisation des renseignements pédiatriques internationaux, lequel décrivait trois approches possibles pour aborder l'absence d'une politique ou d'un mécanisme de réglementation au sein du ministère visant à encourager ou à obliger les promoteurs de médicaments à présenter de nouvelles données sur des essais cliniques pédiatriques recueillies à l'échelle internationale (Santé Canada, 2012m).
- Notes de bas de page 18
-
Le DIN est un code numérique de huit chiffres qui indique la marque nominative, le fabricant, les ingrédients, le dosage, la forme posologique et la voie d'administration d'un produit pharmaceutique. À l'exception des produits radiopharmaceutiques, tout produit pharmaceutique commercialisé au Canada (en vertu de la Loi sur les aliments et drogues et du Règlement sur les aliments et drogues) a un DIN.
- Notes de bas de page 19
-
Conformément à la Loi sur les frais d'utilisation, « si, pour un exercice donné, le rendement d'un organisme de réglementation à l'égard de frais d'utilisation est inférieur aux normes de rendement qu'il a établies pour cet exercice dans une proportion dépassant dix pour cent, ces frais d'utilisation sont réduits d'un pourcentage – d'au plus cinquante pour cent – équivalent à l'insuffisance du rendement » (GC, 2004, par. 5(1)). La sanction s'applique lorsque le rendement moyen est supérieur à 110 % du délai prescrit.
- Notes de bas de page 20
-
L'incidence de cette initiative sur l'infrastructure de présentations électroniques actuelle de Santé Canada, en cours depuis plusieurs années, demeure inconnue.
- Notes de bas de page 21
-
Le projet portant sur l'utilisation des examens étrangers est l'un des aspects d'un projet d'envergure sur l'utilisation des renseignements réglementaires étrangers qui inclut également un comité de réglementation scientifique étranger, des avis scientifiques, des examens parallèles et conjoints, et des groupes de discussion auxquels différents organismes de réglementation prennent part (DGPSA, 2012b). Un comité de réglementation scientifique étranger en est à la phase pilote, et Santé Canada s'engage également dans des discussions et des avis scientifiques en temps réel avec d'autres organismes de réglementation (FDA, EMA) au moyen de « groupes » d'oncologie, de pédiatrie et sanguins.
- Notes de bas de page 22
-
Des données sur le pourcentage de cycles d'examen achevés dans les délaissont présentées. Cet indicateur a été sélectionné en collaboration avec Santé Canada et comprend toutes les catégories de présentation, à l'exception de l'étiquetage, compris dans ces types de présentation, ainsi que tous les cycles d'examen. Ce n'est pas la seule façon d'analyser le rendement des examens. Le rapport 2011 du BVG a seulement tenu compte de l'examen 1, comme le font les Rapport ministériel sur le rendement (RMR) de Santé Canada.
- Notes de bas de page 23
-
On parle, par exemple, des interactions médicamenteuses et des réactions indésirables avec une longue période de latence ou une incidence élevée au sein de la population.
- Notes de bas de page 24
-
Ces données ont été extraites de la Base de données de Canada Vigilance par Santé Canada.
- Notes de bas de page 25
-
Les activités de sensibilisation et d'éducation ciblent les consommateurs et les professionnels de la santé.
- Notes de bas de page 26
-
Une analyse détaillée des normes de service et des cibles de rendement de Santé Canada concernant les activités suivant la mise en marché se trouve plus loin dans cette section.
- Notes de bas de page 27
-
En 2011, Santé Canada a publié sa procédure pour la diffusion publique de renseignements obtenus des rapports sur les réactions indésirables (DPSC, 2011b). La procédure précise que les renseignements personnels et confidentiels de tiers sont protégés et que les demandes concernant des renseignements non disponibles sur le site Web ou dans un format récapitulatif standard, ainsi que des renseignements tirés des rapports sur les réactions indésirables survenues au cours d'un essai clinique, doivent être formulées auprès de la Division de l'accès à l'information et de la protection des renseignements personnels de Santé Canada.
- Notes de bas de page 28
-
L'analyse portant sur les médicaments à usage humain couvrait la période de 2006 à 2012. Celle sur les produits biologiques s'échelonnait de 2008 à 2012.
- Notes de bas de page 29
-
La DPBTG exige actuellement la présentation de PGR dans le cadre de demandes de produits biologiques ultérieurs (PBU).
- Notes de bas de page 30
-
La FDA des États-Unis a récemment annoncé qu'elle accepterait les rapports réguliers concernant l'innocuité dans le format des RPEAR (FDA, 2013c).
- Notes de bas de page 31
-
Santé Canada a d'abord publié son avis d'intention d'adopter la directive Q7 de la CIH et procédé à l'élaboration d'un cadre de réglementation relatif aux IPA en 2002 (Inspectorat, 2002). Les représentants du ministère ont expliqué que le délai de dix ans entre l'annonce de l'intention et la modification réglementaire est attribuable à la complexité du règlement et aux changements apportés aux priorités de la DGPSA en ce qui a trait à l'élaboration de règlements au fil du temps.
- Notes de bas de page 32
-
La FDA fait référence aux BPF sous l'appellation Bonnes pratiques de fabrication actuelles (BPFa). Comme au Canada, les inspections de sites de fabrication ont lieu tous les deux ans, mais puisque la FDA « ne dispose pas des ressources nécessaires pour vérifier tous les aspects d'une BPFa à chaque inspection », l'exhaustivité de l'inspection dépend de l'historique de conformité de l'établissement, de la technologie utilisée et des caractéristiques des produits fabriqués (FDA, 2002). Des inspections abrégées peut être réalisées dans les établissements où l'historique de conformité aux BPFa est satisfaisant, alors qu'on optera plutôt pour une inspection intégrale lorsqu'on ne connaît pas ou très peu l'historique de conformité, comme dans le cas des nouvelles entreprises ou des entreprises où l'on doute de la conformité, selon leur historique de conformité/récidive (FDA, 2002).
- Notes de bas de page 33
-
Selon les renseignements fournis par Santé Canada, les monographies de produit sont habituellement ajoutées à la BDPP 24 heures après l'attribution de l'AC. Toutefois, le public peut seulement les voir lorsque l'entreprise avise Santé Canada qu'elle a l'intention de commercialiser le produit au Canada. Une entreprise dispose de 30 jours suivant le début de la vente du produit pour informer le ministère. La monographie du produit est rendue visible au public lorsque Santé Canada reçoit l'avis. Cela se produit habituellement de 24 à 48 heures suivant la réception de l'avis.
- Notes de bas de page 34
-
Les noms des produits de santé à présentation et à consonance semblables (PSPCS) désignent les « noms de différents produits de santé qui ont une orthographe ou une prononciation similaire (c.-à-d. que la façon de les écrire ou de les prononcer est semblable) » (DGPSA, 2005a, p. 1). Ces similitudes peuvent constituer un risque pour la santé en entraînant des erreurs lors de la prescription, de la dispensation ou de l'administration d'un produit. La modification réglementaire proposée obligerait les fabricants à soumettre une évaluation de la marque nominative du médicament, montrant qu'il est peu probable que ce produit soit confondu avec un autre médicament (GC, 2013).
- Notes de bas de page 35
-
Ces renseignements incluent le nom du produit, l'encadré, les récents changements importants, les indications et l'utilisation, le dosage et l'administration, les contre-indications, les mises en garde et les avertissements, les réactions indésirables, les interactions médicamenteuses et l'utilisation dans des populations particulières.
- Notes de bas de page 36
-
Le sous-alinéa C.01.004(c)(iii) du Règlement sur les aliments et drogues exige que les étiquettes intérieure et extérieure indiquent « le mode d'emploi approprié de la drogue » (Santé Canada, 2010a), ce qui inclut toute mise en garde ou précaution explicitement requise par le règlement. Dans certains cas, une mise en garde ou une précaution spécifique sera nécessaire pour un produit particulier ou une catégorie de produits, mais la mise en garde en soi ne peut pas être un énoncé réglementé. Les mises en garde sont plutôt considérées comme une expression signifiant le mode d'emploi approprié fondé sur des raisons médicales ou pharmacologiques (Santé Canada, 2010a).
- Notes de bas de page 37
-
Jusqu'à tout récemment, tous les rappels de médicaments, peu importe le type, se retrouvaient dans les Listes de retrait de marché des médicaments, et certains aboutissaient dans une communication des risques affichée sur le site de MedEffet. La ligne directrice de Santé Canada stipule que « [l]es avis des entreprises peuvent être affichés sur MedEffet Canada quand Santé Canada juge qu'il est important de mettre les renseignements détaillés sur l'innocuité à la disposition de quiconque pourrait avoir besoin de les connaître » (Santé Canada, 2008b, p. 29). Les tentatives entreprises dans le cadre de l'évaluation en vue de quantifier les rappels de type I dans les Listes de retrait de marché des médicaments ayant abouti à une communication sur le site de MedEffet ont été abandonnées en raison de problèmes méthodologiques.
- Notes de bas de page 38
-
Ces initiatives sont mises en œuvre en réponse à une règle de la Cour fédérale voulant que tous les sites du gouvernement fédéral respectent des normes internationales en matière d'accessibilité, et à une directive du Conseil du Trésor visant à réduire de moitié le nombre de pages du site Web d'ici le 31 juillet 2013 (Santé Canada, 2013c).
- Notes de bas de page 39
-
Ceci étant dit, un examen des affichages indique que certaines communications décrivent des retraits et emploient le mot « retrait » dans leur titre, mais sont toutefois classées comme des « avis » et non des « retraits ». L'évaluation n'a pas permis de déterminer la raison ou le critère utilisé par Santé Canada pour classer les communications des risques en tant que « retraits » ou « avis ».
- Notes de bas de page 40
-
Santé Canada ne paie pas les frais de déplacement des participants.
- Notes de bas de page 41
-
Portant sur la perception du public et des professionnels de la santé à propos de l'innocuité des médicaments en général et plus particulièrement de l'information sur la question produite par Santé Canada, ces études visaient à évaluer les pratiques de recherche d'information des répondants, leur connaissance des sources d'information existantes et leur degré de satisfaction à l'égard de ces sources.
- Notes de bas de page 42
-
Selon le plan de mesure du rendement et d'évaluation du projet, les méthodes d'évaluation utilisées comprendront une analyse de la rapidité et du degré de préparation des communications de Santé Canada sur les risques, une analyse des sites Web, une comparaison visant à déterminer à quel point l'approche de Santé Canada (rapidité et transmission du message) est cohérente avec celle des autres organismes de réglementation, un sondage instantané mené auprès des utilisateurs du site Web MedEffect, des analyses des tendances en matière de prescription et de déclaration des effets indésirables, la tenue de groupes de discussion formés d'usagers afin d'examiner les attitudes, les opinions et le niveau de satisfaction, et un sondage mené auprès de la population canadienne sur des questions similaires (Santé Canada, 2012i).
- Notes de bas de page 43
-
Aux États-Unis, Olson (2002) a observé qu'une diminution d'un mois de la durée du processus d'approbation d'un médicament par la FDA est associée à une augmentation de 1 % des déclarations escomptées d'effets indésirables ayant provoqué une hospitalisation et de 2 % des déclarations escomptées d'effets indésirables ayant provoqué la mort (p. 27). Au Canada, Lexchin (2012) a constaté que 23,7 % (84 sur 434) des nouvelles substances actives (NAS) approuvées par Santé Canada entre le 1er janvier 1995 et le 31 décembre 2010 ont présenté des problèmes d'innocuité et ont été retirées du marché ou fait l'objet d'un avertissement sérieux concernant leur innocuité. Ces NAS comprenaient 68 produits visés par un avertissement sérieux concernant l'innocuité, 9 visés par un tel avertissement et ensuite retirés du marché et 7 retirés du marché sans avertissement préalable. L'étude estime que les NSA ayant fait l'objet d'un examen normal présentaient une probabilité de 19,8 % de donner lieu à un problème d'innocuité, par rapport à une probabilité de 34,2 % pour les NAS ayant fait l'objet d'un examen prioritaire. En ce qui concerne les NAS qui n'offraient pas d'amélioration thérapeutique importante (n=81), la probabilité était de 36,0 % (Lexchin, 2012, p. E1).
- Notes de bas de page 44
-
La FDA a en outre indiqué que la mise au point de formulations destinées à décourager la consommation abusive est une de ses priorités en matière de santé publique et a approuvé, en juillet 2012, une SEAR pour la catégorie de médicaments comprenant tous les opioïdes à libération prolongée et à effet prolongé dont le but précis est de réduire [traduction] « les conséquences indésirables découlant d'un mésusage, d'une consommation abusive et d'une prescription inappropriée » des analgésiques opioïdes (FDA, 2012c). En contraste avec la FDA, Santé Canada a autorisé en novembre 2012 six fabricants à produire des versions génériques de l'oxycodone pour le marché canadien, indiquant que ces « produits seront […] sûrs et efficaces s'ils sont utilisés aux fins recommandées ». (Santé Canada, 2012k). Santé Canada a également exigé des fabricants qu'ils fournissent un PGR exposant les stratégies qu'ils proposent pour surveiller les risques connus et éventuels, pour intervenir en cas de besoin et pour éduquer le public et les professionnels de la santé à propos de ces risques, en plus d'annoncer des conditions d'homologation supplémentaires visant spécifiquement les produits à libération contrôlée contenant de l'oxycodone.
- Notes de bas de page 45
-
Comme il a été mentionné précédemment, une cote « Conforme » ne signifie pas qu'aucun écart relativement aux exigences des BPF n'a été observé. Dans les faits, les entreprises jugées « conformes » font généralement l'objet d'observations pour lesquelles elles doivent mettre en œuvre des mesures correctives.
- Notes de bas de page 46
-
Bien qu'il soit question dans le cadre d'évaluation élaboré par Santé Canada d'« analyses risques-avantages », de nombreux documents du Ministère parlent d'analyses des risques, d'analyses axées sur les riches et d'approches axées sur les risques. Ces concepts sont tous distincts, mais sont traités comme ayant globalement le même sens dans le cadre du présent rapport.
- Notes de bas de page 47
-
L'avis de conformité a été par la suite rétabli.
- Notes de bas de page 48
-
Le processus d'élaboration réglementaire comporte trois principales étapes au Canada : la recherche préliminaire et la décision de réglementer; la publication d'un projet de règlement; l'approbation, la publication et l'enregistrement du règlement proposé. Tandis que les étapes deux et trois combinées nécessitent généralement de 6 à 24 mois, aucune durée n'est associée à l'étape de la recherche préliminaire.
- Notes de bas de page 49
-
Il n'a pas été possible de comparer les données avant et après avril 2007 pour les médicaments biologiques, car toutes les évaluations consignées de signaux indiquant la possibilité d'un problème d'innocuité concernant un médicament biologique se sont vu attribuer une date ultérieure à avril 2007, les dates attribuées se situant plus particulièrement en novembre 2008 et octobre 2012.
- Notes de bas de page 50
-
Dans le cadre de l'évaluation, ont été examinées toutes les évaluations de signaux indiquant la possibilité d'un problème d'innocuité lié à un médicament pour usage humain réalisées en 2010, en 2011 et en 2012, et ayant mené à la recommandation d'une communication des risques (n=38). L'évaluation devait initialement déterminer par analyse si toutes les recommandations qui ont découlé de l'ensemble des évaluations de signaux exécutées ont été mises en œuvre par Santé Canada, et le délai requis pour chacune. Cependant, les données pour réaliser l'analyse n'étaient pas disponibles, car pour y avoir accès il aurait fallu que les ressources internes de Santé Canada les compilent depuis de nombreuses sources. L'analyse effectuée dans le cadre de l'évaluation s'est donc limitée aux évaluations de signaux qui ont été réalisées en 2010, en 2011 et en 2012 et qui ont donné lieu à la recommandation d'une communication des risques, puisque les données nécessaires pour une telle analyse pouvaient être compilées depuis des sources publiques (p. ex., le site Web MedEffect). Une analyse similaire n'a pas pu être entreprise pour les médicaments biologiques en raison du trop petit nombre d'évaluations de signaux ayant motivé la recommandation d'une communication des risques.
- Notes de bas de page 51
-
Il a été possible de constituer dans le cadre de l'évaluation un « dossier complet » pour seulement 14 des 38 évaluations de signaux liés à un médicament pour usage humain qui ont donné lieu à une communication des risques, c'est-à-dire qu'il n'a été possible d'associer avec certitude une détection de signal avec une évaluation de signal et l'affichage d'un document de communication d'un risque que pour 14 des évaluations effectuées. Une analyse similaire n'a pas pu être réalisée pour les médicaments biologiques en raison du trop petit nombre de cas.
- Notes de bas de page 52
-
Dans le cas d'un exemple bien connu, Santé Canada a mis à jour les étiquettes du Propecia et du Proscar afin d'y ajouter l'avertissement que les problèmes de dysfonction sexuelle liés à ces produits peuvent se poursuivre après l'arrêt du traitement trois ans après les organismes réglementaires de la Suède, de l'Italie et du Royaume-Uni (Collier, 2013).
- Notes de bas de page 53
-
Santé Canada a indiqué que ses directives actuelles concernant les demandes d'essais cliniques visent tout particulièrement les universités et ont pour objectif de mieux répondre aux besoins des recherches réalisées à plus petite échelle.
- Notes de bas de page 54
-
Selon les données fournies par la DGPSA, le nombre d'ETP affectés au PMUH au sein de la DGPSA a augmenté de 576,53 en 2009-2010 à 712,91 en 2011-2012.
Détails de la page
- Date de modification :