
Ligne directrice : diffusion publique des renseignements cliniques
Avant-propos
Les lignes directrices sont des documents destinés à guider l'industrie et les professionnels de la santé sur la façon de se conformer aux lois et aux règlements qui régissent leurs activités. Elles servent également de guide au personnel pour les aider à déterminer comment réaliser le mandat et les objectifs de Santé Canada d'une manière équitable, uniforme et efficace.
Les lignes directrices sont des outils administratifs qui n'ont pas force de loi, ce qui permet une certaine souplesse dans leur mise en œuvre. D'autres approches, outre celles décrites dans le présent document pour la mise en œuvre des principes, considérations et exigences qui y sont énoncés, pourraient être acceptables, à condition qu'elles respectent les lois pertinentes. Ces autres approches devraient être examinées préalablement en consultation avec le programme concerné pour s'assurer qu'elles respectent les exigences des lois et des règlements applicables.
Corollairement à ce qui précède, il importe également de mentionner que Santé Canada se réserve le droit de demander des renseignements ou du matériel supplémentaires, ou de définir des conditions dont il n'est pas explicitement question dans la ligne directrice. Santé Canada s'engage à justifier de telles demandes et à documenter clairement ses décisions.
| Version | Date |
|---|---|
| 1.0 | 12 mars 2019 |
Table des matières
- 1. Introduction
- 2. Portée et application
- 3. Lignes directrices générales relatives à la mise en œuvre
- 3.1 Considérations concernant les analyses intermédiaires
- 3.2 Dossiers des patients (listes individuelles de patients et formulaires d'exposé de cas)
- 3.3 Calendrier de mise en œuvre pour la divulgation proactive des renseignements cliniques contenus dans les présentations de drogues et les demandes d'homologation d'instruments médicaux
- 3.4 Publication sur demande de renseignements cliniques provenant d'anciennes présentations de drogues
- 4. Procédures générales relatives aux renseignements cliniques dans les présentations de drogues
- 4.1 Réunion de lancement du processus de diffusion publique des renseignements cliniques
- 4.2 Début du processus de diffusion publique des renseignements cliniques par Santé Canada
- 4.3 Comment soumettre une demande de renseignements cliniques pour des présentations antérieures
- 4.4 Présentation des documents annotés, incluant les caviardages et anonymisations proposés pour les RCC
- 4.5 Examen des documents annotés par Santé Canada
- 4.6 Version finale des documents
- 4.7 Publication de la version finale des documents
- 5. Exigences relatives au caviardage des renseignements commerciaux confidentiels
- 6. Anonymisation des renseignements personnels
- 7. Coordonnées
- 8. Références
- 9. Annexes
- Annexe A : Structure et contenu du CTD/eCTD de l'ICH M2.7 M2.5 et M5
- Annexe B : Structure et contenu du CTD/eCTD de l'ICH Module 5.3 Rapports d'études cliniques
- Annexe C : Diagramme du processus
- Annexe D : Convention d'appellation des noms de documents pour les présentations par le PCDE
- Annexe E : Feuille de contrôle des caviardages proposés
- Annexe F : Modèle de rapport d'anonymisation
- Annexe G : Lettre de certification et tableau des renseignements précédemment caviardés
- Annexe H : Conditions d'utilisation
1. Introduction
1.1 Objectif stratégique
L'objectif de Santé Canada est de permettre au public d'avoir accès, à des fins non commerciales, aux renseignements cliniques anonymisés des présentations de drogues et des demandes d'homologation d'instruments médicaux, une fois le processus d'examen réglementaire terminé, tout en respectant les dispositions de la Loi sur la protection des renseignements personnels.
Le Canada reçoit des renseignements cliniques qui servent à évaluer l'innocuité et l'efficacité des médicaments et instruments médicaux qui sont soumis au processus d'approbation pour la vente au Canada. L'accès du public à ces renseignements cliniques, en plus d'aider les Canadiens à prendre des décisions éclairées sur leur santé, peut favoriser la conduite de nouvelles analyses de données indépendantes et l'examen de nouvelles questions de recherche.
Ce document vise à aider le public, l'industrie, les professionnels de la santé et d'autres intervenants à mieux comprendre la mise en œuvre de l'initiative sur la diffusion publique des renseignements cliniques (DPRC) de Santé Canada, notamment les procédures relatives à la préparation des renseignements à diffuser, les catégories de renseignements qui demeurent assujettis à la définition de renseignements commerciaux confidentiels (RCC) et qui pourraient faire l'objet d'un caviardage, ainsi que la protection des renseignements personnels.
1.2 Terminologie et définitions
- Anonymisation:
- Processus par lequel des renseignements personnels sont modifiés, en supprimant des identificateurs directs et tout autre code connexe qui permettraient d'établir un lien avec des renseignements d'identification et en veillant à ce que les identificateurs indirects restant n'offrent pas de fortes possibilités de ré-identifier une personne.
- Caviardage:
- Technique d'anonymisation qui consiste à supprimer des renseignements personnels en recouvrant le texte ou l'image d'un encadré opaque.
- Compensation:
- Technique d'anonymisation qui consiste à remplacer des données numériques par l'addition ou la soustraction d'une quantité fixe.
- CTD:
- Common Technical Document Format de présentation de l'International Conference on Harmonisation (ICH).
- Demande de changement (Rx-switch):
- Présentation visant à modifier le statut d'un ingrédient médicinal autorisé, de « sur ordonnance » à « sans ordonnance »
- eCTD :
- Electronic Common Technical Document Format de présentation électronique de la Conférence internationale sur l'harmonisation des exigences techniques relatives à l'homologation des produits pharmaceutiques à usage humain (ICH).
- Fabricant:
- Pour les médicaments, la définition est la même que celle décrite par l'article A.01.010 du RAD.
- Pour les instruments médicaux, la définition est la même que celle décrite dans le RIM
- Fins non commerciales:
- Renseignements qui ne seront pas utilisés pour appuyer une demande d'autorisation de mise en marché où que ce soit dans le monde, ou qui ne seront pas vendus ou échangés avec une autre personne.
- Généralisation:
- Technique d'anonymisation qui consiste à utiliser une nouvelle catégorisation à l'intérieur d'un intervalle donné, dans le but d'augmenter le nombre de personnes « semblables ».
- ICH:
- International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (Conférence internationale sur l'harmonisation des exigences techniques relatives à l'homologation des produits pharmaceutiques à usage humain).
- Instrument médical:
- S'entend d'un instrument au sens du Règlement sur les instruments médicaux (RIM).
- Pour plus de renseignements sur la classification des instruments médicaux, veuillez consulter la ligne directrice : Orientation pour le système de classification fondé sur le risque des instruments diagnostiques in vitro (IDIV) et la ligne directrice : Orientation sur le système de classification fondé sur le risque des instruments autres que les instruments diagnostiques in vitro (IDIV).
- LAD:
- Loi sur les aliments et drogues
- Population de référence:
- Groupe de personnes utilisé pour déterminer le risque de ré-identification.
- Pseudonymisation:
- Technique d'anonymisation selon laquelle les informations personnelles (p. ex. le numéro d'identification du sujet) sont recodées afin de dissocier l'information du patient.
- RAD:
- Règlement sur les aliments et drogues
- Randomisation:
- Technique d'anonymisation qui consiste à effectuer de petites modifications de façon aléatoire sur des variables pour réduire la possibilité que ces données puissent identifier une personne.
- Rapport d'études cliniques:
- S'entend d'un rapport complet intégré d'une étude individuelle menée auprès de patients sur tout agent thérapeutique, prophylactique ou diagnostique (fait référence ici à une drogue - médicament ou traitement ou un instrument médical), dans lequel les descriptions cliniques et statistiques ainsi que les présentations et les analyses sont intégrées dans un seul rapport. Le rapport incorpore des tableaux et des figures dans le corps du texte principal ou à la fin du texte et inclus des annexes contenant le protocole, des exemples de rapports de cas, des informations relatives aux investigateurs, des informations relatives aux médicaments à tester / produits expérimentaux, y compris les contrôles actifs / comparateurs, la documentation statistique technique, des publications connexes, les listes de données de patients et les détails statistiques techniques tels que dérivations, calculs, analyses et résultats informatiques.
- RCC :
- Renseignements commerciaux confidentiels au sens de l'article 2 de la Loi sur les aliments et drogues :
- « Renseignements commerciaux confidentiels » sous réserve des règlements, renseignements commerciaux qui se rapportent à l'entreprise d'une personne ou à ses activités et, à la fois :
-
- qui ne sont pas accessibles au public;
- à l'égard desquels la personne a pris des mesures raisonnables dans les circonstances pour qu'ils demeurent inaccessibles au public;
- qui ont une valeur économique réelle ou potentielle pour la personne ou ses concurrents parce qu'ils ne sont pas accessibles au public et que leur divulgation entraînerait une perte financière importante pour elle ou un gain financier important pour ses concurrents.
- Renseignements cliniques:
- S'entend des renseignements relatifs à un essai clinique, selon le sens du paragraphe C.08.009.1(1) du RAD, ou des renseignements relatifs à des études cliniques ou essais expérimentaux au sens de l'article 43.11 du RIM. Pour plus de clarté, cela comprend les aperçus, résumés et rapports d'études cliniques sur des médicaments, ainsi que les résumés et renseignements détaillés de toutes les études cliniques et essais expérimentaux fournissant des données probantes sur l'innocuité et l'efficacité d'instruments médicaux.
- Renseignements qui permettent l'identification directe:
- Renseignements d'identification qui peuvent être reproduits, qui sont reconnaissables et qui peuvent être connus.
- Renseignements qui permettent l'identification indirecte:
- Renseignements qui peuvent aider à identifier une personne à partir d'une combinaison d'identificateurs indirects.
- Renseignements personnels:
- Renseignements au sens de l'article 3 de la Loi sur la protection des renseignements personnels.
- Resynthèse:
- Technique appliquée après la généralisation des données, qui consiste à convertir une plage de données en un point de données précis à l'intérieur de la plage généralisée initiale.
- RIM:
- Règlement sur les instruments médicaux
- RLP:
- Réunion de lancement du processus
- Tm-IMDRF:
- Table des matières de l'International Medical Device Regulators Forum (en anglais)
1.3 Harmonisation internationale
Santé Canada reconnaît l'importance d'une harmonisation internationale en matière d'échange de l'information clinique, notamment en ce qui concerne la publication d'information clinique rendue disponible ailleurs. Par la mise en œuvre de son initiative sur la diffusion publique des renseignements cliniques, Santé Canada cherche à harmoniser ses pratiques avec les pratiques exemplaires internationales lorsque celles-ci sont conformes aux obligations légales du Canada et qu'elles contribuent à la réalisation des objectifs de la politique canadienne. La collaboration avec les programmes de publication d'information clinique de nos partenaires en réglementation aidera à réduire le fardeau administratif et à promouvoir une uniformité en matière de protection des renseignements personnels.
2. Portée et application
2.1 Portée générale et application
Le présent document s'applique aux modifications apportées au Règlement sur les aliments et drogues et au Règlement sur les instruments médicaux, qui sont entrées en vigueur le 28 février 2019. Ces modifications réglementaires précisent les renseignements cliniques dans les présentations de drogues et les demandes d'homologation d'instruments médicaux qui cessent d'être des renseignements commerciaux confidentiels lorsque la décision réglementaire finale a été rendue, et elles autorisent Santé Canada à diffuser publiquement ces renseignements. Le présent document décrit la portée des renseignements cliniques pouvant faire l'objet d'une diffusion publique, ainsi que les procédures à suivre pour la suppression des renseignements qui demeurent des RCC et la protection des renseignements personnels avant la diffusion publique.
Les modifications réglementaires s'appliquent aux renseignements cliniques qui ont déjà été soumis à Santé Canada ainsi qu'à ceux qui le seront dans l'avenir.
La diffusion proactive de cette information sera échelonnée sur une période de quatre ans tel que décrit dans la section 3.3. de la présente ligne directrice
Santé Canada prévoit publié de manière proactive l'information contenue dans les présentations de drogues et les demandes d'homologation d'instruments médicaux, pour lesquelles la décision réglementaire finale sera rendue après l'entrée en vigueur de la réglementation (nouvelles présentations). La présente ligne directrice décrit le processus à suivre pour présenter une demande en vue d'obtenir des renseignements contenus dans des présentations antérieures ainsi que le calendrier prévu pour la publication proactive des nouvelles présentations.
Ce document ne s'applique pas au pouvoir de divulgation des RCC aux termes de l'alinéa 21.1(3)(c) de la Loi sur les aliments et drogues, lequel permet au Ministre de communiquer des RCC à certaines personnes aux fins de la protection ou de la promotion de la santé humaine ou de la sécurité du public. Les renseignements visés par ce pouvoir sont précisés dans le document Ligne directrice - Communication de renseignements commerciaux confidentiels aux termes de l'alinéa 21.1(3)(c) de la Loi sur les aliments et drogues.
2.2 Renseignements cliniques dans les présentations de drogues
L'article C.08.009.2 du RAD décrit les circonstances dans lesquelles les renseignements cliniques inclus dans les présentations de drogues cessent d'être des RCC. Il s'agit de :
- la délivrance d'un avis de conformité (AC);
- la délivrance d'un avis de non-conformité - retrait (ANC-R);
- la délivrance d'un avis de déficience - retrait (ADI-R)
En vertu de l'article C.08.009.3 du RAD, le ministre est habilité à communiquer, sans avis ni consentement, des renseignements cliniques qui cessent d'être des RCC dans les circonstances précitées.
Les renseignements cliniques sur les médicaments sont présentés à Santé Canada en vertu de l'article 8 du RAD, conformément à la structure internationalement harmonisée de l'eCTD (electronic Common Technical Document (eCTD). Santé Canada communiquera les renseignements cliniques contenus dans les modules 2.5 (aperçus d'études cliniques), 2.7 (résumés d'études cliniques) et 5.3 (rapports d'études cliniques) de l'eCTD. Voir l'annexe A pour de plus amples détails. Santé Canada communiquera également les renseignements cliniques présentés dans les annexes suivantes jointes aux rapports d'études cliniques : 16.1.1 (protocole et modification du protocole), 16.1.2 (formulaires types d'exposé de cas) et 16.1.9 (plan d'analyse statistique). Voir l'annexe B pour de plus amples détails.
Les types suivants de présentation de drogue peuvent contenir des renseignements cliniques :
- Présentation de drogue nouvelle (PDN)
- Supplément à une présentation de drogue nouvelle (SPDN)
- Présentation abrégée de drogue nouvelle (PADN)
- Supplément à une présentation abrégée de drogue nouvelle (SPADN)
- Présentation de drogue nouvelle pour usage exceptionnel (PDNUE)
- Supplément à une présentation de drogue nouvelle pour usage exceptionnel (SPDNUE)
Quel que soit leur emplacement dans une présentation de nouvelle drogue, les renseignements sur la chimie, la fabrication et autres renseignements non cliniques continueront d'être des renseignements commerciaux confidentiels au sens de la LAD.
Santé Canada a l'intention de publier de manière proactive les informations cliniques dans les présentations de drogue nouvelle à partir de 2019. La publication proactive sera introduite progressivement par type de présentation, comme décrit à la section 3.3 de ces directives. Les informations contenues dans les présentations de drogues qui ne sont pas sujettes à une diffusion proactive sont disponibles sur demande.
2.3 Renseignements cliniques dans les demandes d'homologation d'instruments médicaux
Le paragraphe 43.12(1) du RIM décrit les circonstances dans lesquelles les renseignements cliniques contenus dans les demandes d'homologation d'instruments médicaux cessent d'être des RCC. Il s'agit de :
- la délivrance d'une homologation pour un instrument médical;
- la délivrance d'une homologation modifiée pour un instrument médical,
- la délivrance d'une lettre de refus.
En vertu de l'article 43.13 du RIM, le ou la ministre est habilité à communiquer, sans avis ni consentement, des renseignements cliniques qui cessent d'être des RCC dans les circonstances précitées.
Pour les instruments médicaux de classe III et IV, les renseignements cliniques sont actuellement présentés en vertu des paragraphes 32(3) et (4) du RIM. Les instruments médicaux de classe 1 et 2 sont hors de la portée de ce document puisque les instruments de classe 1 n'ont pas besoin d'obtenir une licence médicale et les fabricants d'instruments médicaux de classe 2 ne sont généralement pas tenus de présenter des renseignements cliniques à l'appui d'une demande de licence sauf dans certaines circonstances.
Les types suivants de demandes d'homologation d'instruments médicaux peuvent contenir des renseignements cliniques :
- Demande d'homologation d'un instrument médical de classe III
- Modification d'une demande d'homologation d'un instrument médical de classe III
- Demande d'homologation d'un instrument médical de classe IV
- Modification d'une demande d'homologation d'un instrument médical de classe IV
Quel que soit leur emplacement dans une demande d'homologation d'instrument médical, les renseignements sur la fabrication et autres renseignements non cliniques continueront d'être des renseignements commerciaux confidentiels au sens de la LAD.
Santé Canada prévoit publier de manière proactive les renseignements cliniques qui seront présentés dans les nouvelles demandes d'homologation d'instruments médicaux à compter de 2021. La diffusion proactive sera échelonnée selon le type d'application, tel que décrit dans la section 3.3 de la présente ligne directrice.
2.4 Sommaire
Le tableau ci-dessous résume comment le règlement s'applique aux présentations de drogues et aux demandes d'homologations d'instruments médicaux, et comment Santé Canada a l'intention de mettre en œuvre la divulgation publique des informations cliniques qu'elles contiennent.
| Entrée en vigueur de la réglementation | Implantation | |||
|---|---|---|---|---|
| Présentations de drogues | Demandes d'homologation d'instruments médicaux | Présentations de drogues | Demandes d'homologation d'instruments médicaux | |
| Nouvelles demandes d'homologation/ applications | ✔ | ✔ | Début de la diffusion proactive publication en 2019* | Début de la diffusion proactive publication en 2021* |
| Demandes d'homologation antérieures / applications | ✔ | ✔ | Disponible sur demande à partir du 20 mars 2019 | Disponible sur demande à partir du 20 mars, 2019 |
| * voir le calendrier de la mise en œuvre progressive pour la publication proactive dans la section 3.3 de la présente ligne directrice. | ||||
3. Lignes directrices générales relatives à la mise en œuvre
3.1 Considérations concernant les analyses intermédiaires
Une analyse intermédiaire est une analyse qui répond à la définition de la ligne directrice ICH E9 : Principes statistiques pour les essais cliniques et qui a pour but de comparer l'innocuité ou l'efficacité entre les groupes de traitement, à tout moment avant la fin d'un essai.
La divulgation prématurée d'information clinique, avant la fin d'un essai clinique, peut toutefois compromettre la fiabilité des données de l'essai en influant sur le recrutement des patients, en introduisant un biais dans la collecte de données et les analyses, ou en réduisant la confiance envers les conclusions de l'étude. La communication des analyses intermédiaires sera donc soupesée en regard de la nécessité de préserver l'intégrité scientifique des essais cliniques.
La décision de communiquer des renseignements cliniques provenant d'analyses intermédiaires sera prise au cas par cas, en tenant compte des considérations suivantes :
| Situation relative à l'analyse intermédiaire : | Avancement de l'étude clinique | Diffusion |
|---|---|---|
| Analyse intermédiaire ayant établi la supériorité évidente du traitement dans les conditions d'utilisation, utilisée pour mettre fin plus tôt à l'essai | Aucune étude clinique en cours | Les résultats de l'analyse intermédiaire peuvent être communiqués. |
| Analyse intermédiaire d'une étude clinique qui est terminée ou qui a été interrompue | Aucune étude clinique en cours | Les résultats de l'analyse intermédiaire peuvent être communiqués. |
| Analyse intermédiaire d'un essai en cours qui, si elle était communiquée, pourrait avoir une incidence sur l'intégrité des résultats de l'étude et diminuer la confiance à l'égard des conclusions tirées | Étude clinique en cours | Les résultats de l'analyse intermédiaire ne peuvent pas être communiqués. |
3.2 Dossiers des patients (listes individuelles de patients et formulaires d'exposé de cas)
Les formulaires de rapport d'études cliniques (p. ex. ICH E3 16.3) sont des documents destinés à la saisie de renseignements sur chacun des participants de l'étude, comme l'exige le protocole d'étude clinique. Les listes individuelles de patients (p. ex. ICH E3 16.2) incluent des données démographiques, des données sur l'efficacité de réponse individuelle, ainsi que des listes de mesures individuelles obtenues en laboratoire pour chaque patient. Comme de multiples dossiers sont présentés pour chaque participant aux essais, ces renseignements représentent une forte proportion des données contenues dans une présentation de drogue type.
Puisque les dossiers de chaque patient contiennent une grande quantité de renseignements personnels structurés et non structurés, d'importantes modifications sont nécessaires pour l'anonymisation de ces renseignements. L'application des méthodes actuelles d'anonymisation exige toutefois des ressources considérables et réduit considérablement la valeur des renseignements aux fins de recherche. Par conséquent, les dossiers individuels des patients ne seront pas diffusés publiquement avec les autres renseignements cliniques.
Les chercheurs qui aimeraient obtenir, à des fins de recherches ou d'analyses statistiques, les dossiers individuels de patients liés à des renseignements sur des produits thérapeutiques peuvent présenter une demande en ce sens en vertu des dispositions de la Loi sur la protection des renseignements personnels qui s'appliquent.
Les événements indésirables et autres résultats importants au niveau des participants sont rapportés dans la partie principale du rapport d'études cliniques. La section 6 de ce document définit la marche à suivre afin de pouvoir anonymiser des renseignements personnels contenus dans les rapports d'études cliniques avant la diffusion publique de ceux-ci.
3.3 Calendrier de mise en œuvre pour la divulgation proactive des renseignements cliniques contenus dans les présentations de drogues et les demandes d'homologation d'instruments médicaux
Santé Canada prévoit procéder à une diffusion proactive progressive des renseignements cliniques contenus dans les présentations de drogues et les demandes d'homologation d'instruments médicaux, pour lesquelles la décision réglementaire finale sera rendue après l'entrée en vigueur du présent règlement (nouvelles présentations). La publication proactive de ces renseignements devrait se faire conformément au calendrier ci-après :
| Étape | Mise en œuvre progressive proposée | Portée des types de demandes |
|---|---|---|
| 1 | Année 1 | PDN-SAN, SPDN-C et demandes de changement |
| 2 | Année 2 | Toutes les PDN + SPDN-C et demandes de changement |
| 3 | Année 3 | Toutes les PDN, toutes les SPDN et instruments médicaux de classe IV |
| 4 | Année 4 | Toutes les PDN, SPDN, PADN, SPADN et instruments médicaux de classe III et IV |
Étape 1 de la mise en œuvre
Dans la première phase de mise en œuvre de la divulgation publique proactive, Santé Canada souhaite publier les renseignements cliniques figurant dans les présentations de drogues suivantes :
- substance active nouvelle (PDN-SAN), à savoir les présentations portant sur des médicaments qui ne sont pas des variations d'ingrédients médicinaux préalablement approuvés au Canada, c.-à-d. les drogues « innovantes »;
- suppléments à une présentation de drogue nouvelle contenant des essais de confirmation (SPDN-C) après la délivrance d'un avis de conformité avec conditions conformément à la lettre d'engagement;
- demande de changement visant à modifier le statut d'un ingrédient médicinal autorisé, de « sur ordonnance » à « sans ordonnance » (changement partiel ou complet).
Étape 2 de la mise en œuvre
À l'étape 2, Santé Canada prévoit ajouter la publication proactive des renseignements cliniques contenus dans toutes les présentations de drogues nouvelles (PDN-SAN et substances non classées comme substances actives nouvelles).
Étape 3 de la mise en œuvre
À cette étape, Santé Canada procédera à la publication proactive des renseignements cliniques contenus dans tous les suppléments à une présentation de drogue nouvelle (SPDN), p. ex. les présentations portant sur de nouvelles indications pour un produit commercialisé, ainsi que des renseignements cliniques dans les demandes d'homologation d'instruments médicaux de classe IV.
La mise en œuvre de l'étape 3 devrait se faire au moment de l'adoption de la structure Tm-IMDRF pour les demandes d'homologation d'instruments médicaux, afin de favoriser la publication efficace des renseignements cliniques sur les instruments médicaux de classe III et IV.
Étape 4 de la mise en œuvre
Les présentations abrégées de drogue nouvelle (PADN), c.-à-d. les médicaments génériques, et les demandes d'homologation pour des instruments médicaux de classe III, seront incluses à l'étape 4.
3.4 Publication sur demande de renseignements cliniques provenant d'anciennes présentations de drogues et demandes d'homologation d'instruments médicaux
Des demandes peuvent être présentées par l'entremise du portail de Santé Canada sur les renseignements cliniques, en vue d'obtenir des renseignements cliniques contenus dans d'anciennes présentations de drogues et demandes d'homologation d'instruments médicaux (pour lesquelles la décision réglementaire finale a été rendue avant l'entrée en vigueur du règlement).
Les informations cliniques provenant de présentations de drogues et de demandes d'homologation d'instruments médicaux (qui ont fait l'objet d'une décision finale avant l'entrée en vigueur de la réglementation le 20 mars 2019) peuvent être demandées via le portail d'information clinique de Santé Canada. Les informations contenues dans les soumissions et les applications ayant reçu une décision finale après cette date mais qui ne sont pas encore sujet à la publication proactive sont également disponibles sur demande.
L'information contenue dans les types de présentations et demandes d'homologation suivantes est disponible sur demande :
- Présentation de drogue nouvelle (PDN)
- Supplément à une présentation de drogue nouvelle (SPDN)
- Présentation abrégée de drogue nouvelle (PADN)
- Supplément à une présentation abrégée de drogue nouvelle (PSADN)
- Présentation de drogue nouvelle pour usage exceptionnel (PDNUE)
- Supplément présentation de drogue nouvelle pour usage exceptionnel (SPDNUE)
- Demande d'homologation d'instrument médical de classe III
- Demande d'homologation d'instrument médical de classe III
- Demande d'homologation d'instrument médical de classe IV
- Modification d'une demande d'homologation d'instrument médical de classe IV
Les demandes de renseignements cliniques pour une présentation antérieure doivent être soumises à l'aide du formulaire de demande en ligne sur le portail de renseignements cliniques de Santé Canada, ainsi qu'il est décrit à la section 4.3.
4. Procédures relatives aux renseignements cliniques dans les présentations de drogues et les demandes d'homologation d'instruments médicaux
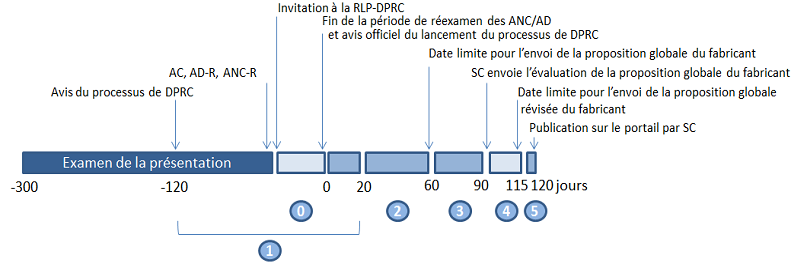
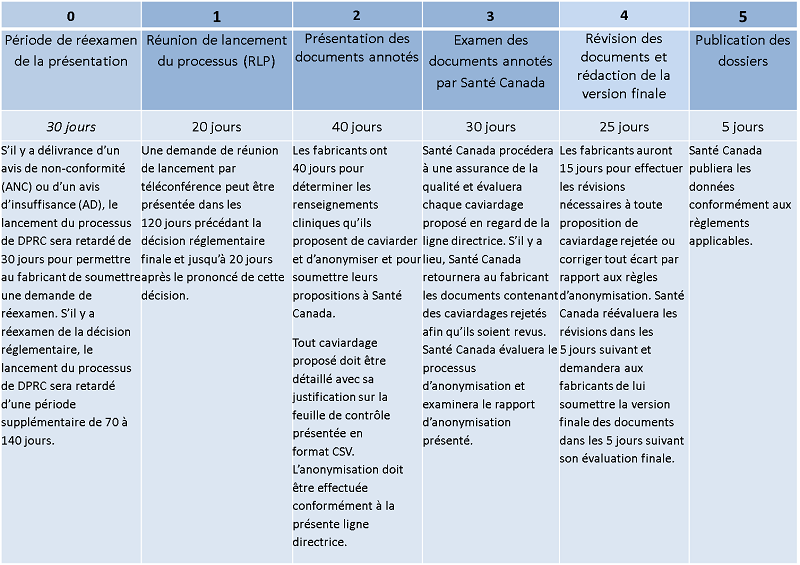
La publication de renseignements cliniques dans le cadre de l'initiative sur la diffusion publique des renseignements cliniques se fera en cinq phases distinctes - le début du processus, la soumission des documents, examen, la mise au point finale et la publication.
L'objectif de Santé Canada est de télécharger un dossier final de renseignements cliniques anonymisés et caviardés sur le portail de renseignements cliniques de Santé Canada dans les 120 jours civils suivant le début du processus. Si une demande concerne des renseignements cliniques qui ne sont actuellement pas disponibles en format électronique, un délai additionnel pourrait être nécessaire pour la numérisation des documents papier.
4.1 Réunion de lancement du processus de diffusion publique des renseignements cliniques
Avant de procéder à la publication de renseignements cliniques, un promoteur peut demander de participer à une réunion individuelle de lancement du processus.
Cette réunion a pour but de permettre au promoteur de consulter Santé Canada afin de savoir quels sont les documents cliniques visés par l'initiative de diffusion publique des renseignements cliniques et d'obtenir des précisions sur les exigences et le processus de Santé Canada.
Les promoteurs qui ne sont pas familier avec cette initiative sont encouragés à demander la tenue d'une telle réunion avant le début du processus. Une demande de réunion peut être présentée dans les 120 jours civils précédant une décision réglementaire finale, et jusqu'à 20 jours civils après le prononcé de cette décision.
Les demandes de réunion doivent être envoyées à l'adresse suivante : hc.clinicaldata-donneescliniques.sc@canada.ca
Il est recommandé que le promoteur propose plusieurs dates et heures pour la tenue de la réunion à l'intérieur de la période d'admissibilité. Les renseignements suivants doivent aussi être joints à la demande de réunion :
- Un ordre du jour ou un bref plan d'ensemble de la réunion;
- Une liste des questions, le cas échéant, devant être examinées durant la réunion par l'équipe chargée de l'initiative sur la diffusion publique des renseignements cliniques.
Santé Canada enverra un accusé de réception de la demande et confirmera la date de la réunion par courriel.
Veuillez noter que ces réunions se dérouleront uniquement par téléconférence.
4.2 Début du processus de diffusion publique des renseignements cliniques par Santé Canada
Décisions réglementaires favorables
La publication des renseignements cliniques débute dès que la décision réglementaire est rendue au sujet de la présentation de drogue ou demande d'homologation d'instrument médical; bien que le processus débute par l'envoi d'un avis par courriel au fabricant, le fabricant peut commencer à préparer les renseignements cliniques destinés à être publiés avant de recevoir l'avis de Santé Canada.
L'avis par courriel précise la présentation de drogue ou la demande d'homologation visée et énumère la liste des documents que Santé Canada rendra publics.
Les annexes A et B précisent quelles sections d'une présentation de drogue sont éligibles pour diffusion, selon le format eCTD. Puisque la structure des demandes d'homologation d'instruments médicaux varie présentement, Santé Canada déterminera les sections éligibles pour la diffusion publique dans une approche cas par cas
De futures lignes directrices seront élaborées afin de s'aligner sur les initiatives de transparence d'autres organismes de réglementation et aussi sur un nouveau format harmonisé internationalement qui est présentement en développement (IMDRF ToC).
Sur cet avis, Santé Canada demande au fabricant de lui soumettre les documents anonymisés avec les caviardages proposés dans un délai de 60 jours civils. La fiche de contrôle des caviardages proposés (en format .CSV; voir le modèle à l'annexe E) et le rapport d'anonymisation (voir le modèle à l'annexe F) doivent aussi être présentés, ainsi qu'il est indiqué ci-après.
Décisions réglementaires défavorables
Si une présentation de drogue est jugée non conforme au RAD, Santé Canada entreprendra la publication dans un délai de 31 jours civils après la date de l'avis, à moins que le fabricant ne lui fasse parvenir une lettre d'intention de réexamen.
Si le fabricant soumet une lettre d'intention de réexamen, Santé Canada entreprendra la publication à la fin du processus de réexamen, comme le prévoit la ligne directrice « Révision des décisions sur les présentations de drogue pour usage humain ». Pour le processus de révision des décisions, prévoir un délai de 70 à 140 jours civils, dépendamment si la demande doit faire l'objet d'un réexamen interne ou externe.
Dans l'éventualité où une demande d'homologation pour un instrument médical est jugée non conforme au RIM, et que le fabricant soumet une lettre d'intention de réexamen, Santé Canada entamera le processus de publication dès qu'une décision est rendue par rapport à la procédure d'appel, conformément à la politique « Gestion des demandes d'homologation d'instruments médicaux et d'autorisation d'essais expérimentaux ».
Dans l'éventualité d'un appel au premier palier, si le fabricant ne soumet pas la documentation nécessaire pour supporter le processus d'appel, Santé Canada entamera le processus de publication 21 jours suivant la réception de la lettre d'intention d'appel au Directeur du Bureau.
Dans l'éventualité d'un appel au deuxième palier, Santé Canada entamera le processus de publication après avoir une fois informé le fabricant de la décision de la Direction concernant le processus d'appel.
Les fabricants seront avisés par courriel lorsque Santé Canada commencera la publication de l'information clinique relative à une décision négative. L'avis comportera les mêmes informations et procédures que dans le cas de décisions positives (voir ci-dessus).
4.3 Comment soumettre une demande de renseignements cliniques pour des présentations de drogues antérieures et demandes d'homologation d'instruments médicaux
Santé Canada prévoit publier les renseignements cliniques de présentations de drogues antérieures ou de demandes d'homologation d'instruments médicaux sur réception d'une demande du public via le portail, dans les limites de sa compétence administrative. Les demandes portant sur de multiples présentations de drogue seront traitées dans l'ordre, en fonction de leur priorité.
Les membres du public peuvent demander des renseignements cliniques contenus dans des présentations de antérieures et demandes d'homologation, par l'entremise du portail de renseignements cliniques de Santé Canada; ils doivent pour ce faire présenter une formulaire de demande électronique sur lequel figure le nom du produit, le nom du fabricant, l'indication connexe, les renseignements demandés (p. ex. rapport d'étude clinique, aperçu d'étude clinique, résumé d'étude clinique), les motifs de la demande ainsi que les coordonnées de l'auteur de la demande.
Dans la mesure du possible, l'auteur de la demande doit fournir à Santé Canada le numéro de contrôle de la présentation ou le numéro de demande d'homologation. Ces renseignements supplémentaires figurent dans les documents Sommaire des motifs de décision et Sommaire des décisions réglementaires de Santé Canada associés à la présentation de drogue ou à la demande d'homologation de l'instrument médical.
Santé Canada confirmera la réception des demandes par l'envoi d'un avis direct à chaque demandeur. Santé Canada mettra aussi régulièrement à jour la liste des demandes sur son portail Web.
Établissement de l'ordre de priorité des demandes
Si les demandes de renseignements cliniques dépassent la compétence administrative de Santé Canada, elles seront traitées par ordre de priorité. L'ordre de priorité des demandes sera établi en fonction de paramètres d'identification des produits et de renseignements ayant une grande incidence sur le système de santé. Parmi les facteurs qui seront pris en compte, mentionnons les médicaments et les instruments médicaux qui font l'objet de demandes continues de la part d'organisations de santé, les produits qui sont largement utilisés et les produits qui présentent un grand intérêt public. Santé Canada informera les demandeurs lorsque le traitement de leur demande débutera et mettra régulièrement à jour son portail Web pour indiquer quelles demandes sont actuellement en cours de traitement.
4.4 Présentation des documents annotés, incluant les caviardages et anonymisations proposés pour les RCC
Santé Canada a défini les circonstances restreintes et précises, prévues par le règlement, dans lesquelles les renseignements cliniques contenus dans une présentation de drogue ou une demande d'homologation d'instrument médical peuvent conserver une valeur commerciale après le prononcé de la décision réglementaire finale. Les catégories précises de renseignements dont Santé Canada envisagera le caviardage (avec justification à l'appui) dans les présentations de drogues sont décrites à la section 5 de la présente ligne directrice.
De futures lignes directrices spécifieront ces catégories pour demandes d'homologation d'instruments médicaux sur la base du format normalisé de la table des matières IMDRF, actuellement en cours de développement. Entre temps, Dans l'intervalle, les fabricants d'instruments médicaux devraient se reporter à la section 5 pour savoir comment proposer caviardages conformes aux exceptions spécifiées dans le paragraphe 43.12 (2) du Règlement sur les instruments médicaux.
Pour justifier certains caviardages, le fabricant pourrait devoir présenter des renseignements extraits de ses plans internes d'entreprise (p. ex. développement futur de nouvelles indications basées sur des résultats secondaires). Santé Canada demande donc au fabricant de lui soumettre une version annotée de tous les renseignements cliniques assujettis à la publication, dans laquelle tous les caviardages proposés seront indiqués en surbrillance. Tout texte que le fabricant propose de caviarder doit rester lisible, et tous les caviardages proposés doivent être accompagnés d'une justification détaillée et précise et être inscrits sur la « feuille de contrôle des caviardages proposés » présentée en format .CSV. Veuillez-vous reporter à l'annexe E pour un modèle de la feuille de contrôle.
Les documents annotés dans les présentations de drogues et les demandes d'homologation d'instruments médicaux doivent être anonymisés conformément à la procédure décrite à la section 5 de la présente ligne directrice, et le processus d'anonymisation des données doit être décrit en détail dans un rapport d'anonymisation distinct (voir l'annexe F pour un modèle de rapport d'anonymisation).
Lorsque le fabricant a terminé de préparer les documents précités, il doit les transmettre à Santé Canada au moyen du portail commun de demandes électroniques (PCDE). Les documents envoyés au moyen du PCDE doivent respecter la convention d'appellation définie à l'annexe D.
Santé Canada peut renvoyer un document au fabricant une seule fois pour lui demander de lui fournir des justifications supplémentaires. La décision finale quant aux renseignements cliniques qui seront diffusés publiquement appartient à Santé Canada.
Utilisation de documents préalablement caviardés
Sous réserve d'une certification appropriée, le fabricant peut soumettre à Santé Canada des documents finaux caviardés pour une présentation de drogue qui ont déjà été acceptés par l'Agence européenne des médicaments (EMA). Si le fabricant reçoit un avis de Santé Canada l'informant qu'il doit préparer des documents annotés pour diffusion publique, et que ces renseignements cliniques ont déjà été publiés par l'EMA en vertu de la politique sur la publication d'information clinique pour les produits médicinaux destinés à l'usage humain (politique de l'EMA 0070), le fabricant peut choisir de soumettre les mêmes documents que ceux soumis à l'EMA.
Comme il est indiqué ci-dessus, les fabricants doivent soumettre la version finale de leurs documents caviardés au moyen du PCDE.
Santé Canada demande aux fabricants de soumettre leur certification à l'aide du modèle présenté à l'annexe G. Ce formulaire atteste que les renseignements cliniques visés par l'initiative de diffusion publique des renseignements cliniques de Santé Canada sont identiques à ceux publiés en vertu de la politique 0070.
Dans les cas où une seulement partie des renseignements demandés par Santé Canada à des fins de diffusion publique a été caviardée avant d'être présentée à l'EMA, les fabricants peuvent présenter les mêmes renseignements avec la certification et ne caviarder que les éléments qui ne l'ont pas été et qui doivent être présentés à Santé Canada.
4.5 Examen des documents annotés par Santé Canada
Santé Canada examinera les justifications fournies par le fabricant pour chaque caviardage proposé dans les documents annotés. Tous les caviardages proposés seront évalués par rapport aux exceptions autorisées par la réglementation (voir la section 5). Après cet examen, les caviardages proposés seront acceptés ou refusés avant la diffusion publique finale des renseignements cliniques.
Les caviardages proposés peuvent être rejetés pour les raisons suivantes :
- lorsque le fabricant est incapable de démontrer que l'information n'a pas servi à étayer les conditions d'utilisation ou l'indication de la drogue ou de l'instrument médical, comme il est énoncé dans la présentation ou la demande d'homologation;
- lorsque le fabricant ne justifie pas adéquatement que l'information proposée décrit un test, une méthode ou une épreuve qui est utilisé exclusivement par le fabricant;
- lorsque le fabricant ne démontre pas que l'information proposée n'est pas visée par le règlement;
- lorsque le caviardage proposé concerne de l'information qui est déjà du domaine public.
Santé Canada informera le fabricant de tout caviardage proposé que le Ministère rejette. Le fabricant aura une seconde chance de mieux justifier un caviardage à la suite de l'examen par Santé Canada.
Ainsi qu'il est indiqué à la section 6, Santé Canada demandera au fabricant d'anonymiser les renseignements cliniques à l'aide d'un processus d'anonymisation basé sur le risque. Santé Canada rejettera toute modification de données qui n'est pas accompagnée d'une justification adéquate. La décision finale concernant la diffusion publique des renseignements incombe à Santé Canada.
4.6 Version finale des documents
À la suite de l'examen mené par Santé Canada, le fabricant doit soumettre une version finale des documents, préparée conformément aux instructions suivantes :
- Tous les caviardages proposés qui sont acceptés doivent être convertis en texte non lisible; le texte caviardé ne doit pas être lisible par machine et aucune recherche ne doit pouvoir y être effectuée. Les caviardages doivent être clairement visibles (généralement en utilisant une trace opaque) et devraient être identifiés de manière à faire la distinction entre des RCC et les informations personnelles. D'une autre part, les spécifications de l'EMA concernant le caviardage sont autorisées.
- Une version finale de toutes les modifications de données proposées aux fins de l'anonymisation doit être présentée, et un rapport d'anonymisation révisé, excluant tout renseignement personnel, doit être joint à la version finale des documents cliniques anonymisés (voir la section 5 pour de plus amples renseignements sur les exigences relatives au rapport d'anonymisation).
- La version finale des documents doit être nommée conformément à la convention d'appellation de noms définies à l'annexe D et transmise à Santé Canada au moyen du PCDE.
4.7 Publication de la version finale des documents
La version finale des documents sera accessible au public à des fins non commerciales sur le portail des renseignements cliniques de Santé Canada. Toutes les pages du document final porteront un filigrane (non lisible par machine) visant à informer les utilisateurs des conditions d'utilisation qui indiquent que les renseignements sont divulgués par Santé Canada à des fins non commerciales. Santé Canada s'efforcera de publier les renseignements cliniques dans les 120 jours civils suivant le début du processus.
5. Exigences relatives au processus de caviardage des renseignements commerciaux confidentiels
Deux catégories de renseignements cliniques continueront de répondre à la définition de RCC selon la LAD. Sous réserve d'une justification valable du fabricant, Santé ne publiera pas les éléments suivants :
- Renseignements cliniques que le fabricant n'a pas utilisés dans la présentation ou le supplément pour étayer les conditions d'utilisation qu'il propose pour la nouvelle drogue ou l'indication pour laquelle la drogue est recommandée;
- Renseignements cliniques qui décrivent les tests, méthodes et essais utilisés exclusivement par le fabricant.
1) En vertu de l'alinéa C.08.009.2(2)(a) du RAD, les renseignements cliniques fournis par le fabricant, qui n'ont pas servi à appuyer les conditions d'utilisation recommandées ou le but pour lequel le médicament est recommandé, restent des renseignements commerciaux confidentiels à la suite de la décision réglementaire finale de Santé Canada.
Conformément à l'alinéa 43.12(2)(a) du RIM, les renseignements cliniques fournis par le fabricant, qui n'ont pas servi à appuyer les caractéristiques de l'appareil qui lui permettent d'être utilisé pour les conditions médicales, buts et utilisations pour lesquels il est fabriqué, vendu ou présenté restent des renseignements commerciaux confidentiels à la suite de la décision réglementaire finale de Santé Canada.
Par exemple, un fabricant peut utiliser des indicateurs de résultats secondaires ou exploratoires décrits dans les renseignements cliniques pour appuyer de futurs essais visant à faire approuver une nouvelle indication. La divulgation de cette information pourrait fournir à un concurrent de l'information sur les utilisations futures d'une drogue.
Il est possible que les rapports, aperçus et résumés d'études cliniques sur un produit pour lequel le fabricant cherche à obtenir une autorisation de mise en marché contiennent de l'information ou des données sur des résultats secondaires ou exploratoires qui ne servent pas à appuyer les conditions d'utilisation recommandées, ou le but du produit, décrits dans la présentation ou dans la demande d'homologation. Dans les rares cas où ces renseignements peuvent faire partie d'un programme de développement en cours sur de nouvelles allégations, Santé Canada protégera ces renseignements et ne les diffusera pas publiquement si une justification adéquate est présentée.
2) En vertu de l'alinéa C.08.009.2(2)(b) du RAD, et de l'alinéa 43.12(2)(b) du RIM les renseignements cliniques sur les tests, méthodes et essais qui sont utilisés exclusivement par le fabricant continuent d'être des renseignements commerciaux confidentiels après que Santé Canada a rendu sa décision réglementaire finale.
Par exemple, un fabricant peut apporter de nouvelles modifications à un essai biologique qui est ensuite utilisé pour recueillir des données cliniques. Dans certains cas, ces modifications peuvent nécessiter beaucoup d'efforts et d'investissements de la part du fabricant et être utilisées pour d'autres études en cours ou dans le cadre d'utilisations courantes. Ces modifications peuvent être considérées comme étant utilisées exclusivement par le fabricant.
Il est possible que les rapports, aperçus et résumés d'études cliniques incluent des détails, des spécifications et des renseignements de validation sur des essais ou des méthodes d'analyse qui ont été élaborés exclusivement par le promoteur ou un autre tiers et qui sont utilisés exclusivement par le promoteur de la présentation. Si ces détails méthodologiques n'ont pas été rendus publics, Santé Canada protégera ces renseignements et ne les diffusera pas publiquement si une justification adéquate est présentée.
Quel que soit leur emplacement dans une présentation de drogue, les renseignements sur la chimie, la fabrication et autres informations non cliniques resteront soumises à la définition des renseignements commerciaux confidentiels de la LDA.
6. Anonymisation des renseignements personnels
6.1 Principes régissant la protection des renseignements personnels
La Loi sur la protection des renseignements personnels fédérale définit les « renseignements personnels » comme des renseignements concernant un individu identifiable, quels que soient leur forme et leur support, puis elle présente une liste non exhaustive d'exemples précis de ce type de renseignements. Les renseignements cliniques contiennent des renseignements qui satisfont à cette définition de renseignements personnels.
La Cour fédérale a adopté le critère des « fortes possibilités » pour déterminer à quel moment des renseignements sont des renseignements concernant un individu identifiable (Gordon c. Canada (Santé), 2008 CF 258). Les renseignements cliniques doivent être adéquatement anonymisés avant d'être rendus publics, pour éviter les fortes possibilités qu'un patient participant à un essai clinique puisse être identifié; pour ce faire, un processus d'anonymisation objectif, systématique et documenté doit être utilisé.
Afin de maximiser la diffusion de renseignements utiles sur le plan analytique et de préserver au maximum l'utilité des renseignements cliniques publiés, l'anonymisation des données cliniques doit être effectuée selon les principes suivants :
- 1- Toute transformation des données doit être réalisée dans le seul but d'éviter la divulgation de renseignements personnels;
- 2- Toute transformation des données doit être accompagnée d'une rigoureuse justification et être appliquée uniquement aux variables qui risquent de permettre la ré-identification, et non à de larges sections de renseignements cliniques; et
- 3- La transformation des données favoriserait des méthodes qui permettent de conserver la valeur analytique, p. ex. la généralisation, la répartition aléatoire et la compensation, plutôt que le caviardage.
6.2 Processus d'anonymisation
Il existe actuellement de nombreux cadres d'anonymisation qui sont accessibles au public. Santé Canada recommande de suivre un processus en trois étapes adapté du guide 2016 Information and Privacy Commissioner of Ontario De-identification Guidelines. Le processus d'anonymisation doit généralement suivre les trois étapes suivantes :
- Étape 1 : Identifier et classer les renseignements
- Étape 2 : Mesurer le risque de ré-identification
- Étape 3 : Anonymiser les données
L'adoption d'un processus d'anonymisation qui suit ces étapes permet de réduire de manière fiable le risque de divulgation de renseignements personnels. Un processus d'anonymisation qui comporte une mesure objective du risque et prévoit la sélection d'une population de référence adéquate offre aussi l'avantage de maximiser l'utilité des données, tout en s'ajustant au caractère confidentiel variable de certaines populations à l'étude.
Étape 1 : Identifier et classer les renseignements
Les renseignements qui permettent l'identification directe et indirecte doivent être classés avant d'entamer le processus d'anonymisation des renseignements cliniques.
Les renseignements qui permettent l'identification directe sont généralement décrits comme des renseignements qui répondent aux critères suivants :
- A - Peuvent être reproduits, au sens où les renseignements sont peu susceptibles de varier fréquemment au fil du temps;
- B - Sont reconnaissables, au sens où certains patients peuvent présenter des valeurs reconnaissables distinctes;
- C - Peuvent être connus, au sens où une personne connaît les renseignements associés à un individu donné.
Les renseignements qui permettent l'identification directe peuvent être des identificateurs uniques (p. ex. le numéro d'assurance sociale du patient) ou non uniques (p. ex. nom du patient). Certains renseignements classés parmi les renseignements permettant l'identification directe ne sont pas nécessaires pour comprendre les données cliniques (p. ex. les initiales du patient), alors que d'autres peuvent l'être (p. ex. le numéro d'identification du sujet). La divulgation de renseignements permettant l'identification directe présente de fortes possibilités de ré-identification d'une personne.
Les renseignements qui permettent une identification indirecte sont d'autres variables d'identification qui satisfont à la définition de « renseignements personnels » au sens de la Loi sur la protection des renseignements personnels du Canada. Pour qu'un renseignement permettant l'identification indirecte exige l'anonymisation, sa divulgation doit présenter de fortes possibilités de ré-identification d'une personne lorsque ce renseignement est combiné avec d'autres renseignements disponibles (p. ex. les données démographiques). Les renseignements qui permettent l'identification indirecte peuvent être nécessaires pour comprendre l'information clinique; leur anonymisation doit donc être soigneusement justifiée, en conformité avec le deuxième principe d'anonymisation.
Étape 2 : Mesurer le risque de ré-identification
Après avoir classé les renseignements, il faut mesurer le risque qui y est associé. Cette mesure du risque permettra de justifier toute transformation de données pouvant en découler. Les renseignements qui, seuls ou combinés avec d'autres renseignements, ne présentent pas de fortes possibilités d'identifier une personne ne sont pas considérés comme des renseignements personnels et n'ont pas à être modifiés.
Le risque global de ré-identification associé à la divulgation de données cliniques correspond au produit du risque inhérent aux données et du risque associé au contexte de la diffusion. Le calcul du risque de ré-identification associé à la diffusion publique de renseignements cliniques doit tenir compte de ce contexte; dans le cadre d'une diffusion publique, le risque contextuel ne peut pas être réduit, de sorte que le risque global de ré-identification équivaut au risque inhérent aux données (contrairement à la diffusion de renseignements à un petit groupe choisi de personnes, qui pourrait constituer un contexte de risque plus faible et donc être associée à un plus faible risque de ré-identification).
Mesure du risque de ré-identification associée aux renseignements permettant l'identification directe
Il existe de fortes possibilités qu'un participant à un essai puisse être identifié à partir des renseignements permettant l'identification directe, et le risque de ré-identification doit être considéré comme étant de 100 % (risque = 1,0); ces renseignements nécessitent donc invariablement une anonymisation pour réduire suffisamment le risque de ré-identification des participants à l'essai.
Mesure du risque de ré-identification à partir des renseignements qui permettent l'identification indirecte
Le risque de ré-identification à partir de renseignements associés au patient qui permettent l'identification indirecte doit être calculé en fonction du patient. Une méthode simple de calculer ce risque est de mesurer la taille de l'échantillon.
La taille de l'échantillon correspond au nombre de patients dont les renseignements permettant l'identification indirecte ont la même valeur. Un seuil de risque de 0,09 équivaut à un échantillon cible de 11 patients. Après avoir déterminé les renseignements permettant l'identification indirecte qui doivent être anonymisés, les données des patients correspondants doivent être anonymisées afin d'obtenir une taille d'échantillon de 11.
Population de référence pour les renseignements permettant l'identification indirecte
La taille globale de l'échantillon de patients, ainsi que le degré d'anonymisation (transformation des données) nécessaire pour réduire le risque de ré-identification des patients, varient en fonction de la population de référence choisie. La population de référence peut être définie à partir des patients participant à l'essai en question (population la plus faible) ou encore à partir de l'ensemble des patients participant à des essais semblables menés par un promoteur en particulier, de l'ensemble des patients inscrits à des essais semblables (p. ex. par maladie ou catégorie d'intervention thérapeutique) ou de l'ensemble des patients à l'intérieur d'une zone géographique donnée (population la plus grande).Lorsque la population de référence appropriée est une population autre que celle établie à partir des participants à l'essai clinique en question, une extrapolation de la population à l'essai clinique peut être appliquée pour obtenir une estimation de la taille de la population de référence.
Conformément au premier et deuxième principe directeur, le risque de ré-identification doit être analysé non seulement en fonction du nombre de personnes dans une étude, mais aussi en fonction d'un nombre reflétant le risque en situation réelle.
Seuil de risque
Santé Canada encourage l'adoption d'un seuil de risque de ré-identification de 9 % (risque = 0,09). Cette valeur est conforme au seuil de risque mentionné dans la ligne directrice externe sur la politique 0070 de l'EMA, ainsi qu'à d'autres seuils de risque associés à la divulgation de données publiques. Bien qu'une méthode qualitative puisse être utilisée pour mesurer le risque, la méthode quantitative offre l'avantage d'être basée sur des mesures empiriques et donc d'être plus précise, objective et de fournir généralement des données plus utiles. Lorsque le risque de ré-identification est mesuré par une méthode qualitative, il ne devrait plus exister de fortes possibilités que les renseignements cliniques permettent l'identification d'une personne après le processus d'anonymisation.
Étape 3 : Anonymiser les données
Utilité des données
La méthode utilisée pour l'anonymisation des renseignements cliniques peut réduire l'utilité de ces données. Les données qui sont préservées sont les plus utiles. Par conséquent, il est conseillé de ne pas transformer (anonymiser) les renseignements qui ne contribuent pas au risque de ré-identification et d'adopter des méthodes qui ont le plus faible impact possible sur l'utilité des données.
Anonymisation des renseignements permettant l'identification directe
Les renseignements permettant l'identification directe peuvent être anonymisés par caviardage, pseudonymisation ou répartition aléatoire. Ces renseignements peuvent être caviardés si la suppression de la variable ne nuit pas à la compréhension des renseignements cliniques. Lorsqu'un renseignement permettant l'identification directe est essentiel à la compréhension des données cliniques, d'autres méthodes d'anonymisation doivent être utilisées. Dans le cas, par exemple, du numéro d'identification du sujet, la méthode de pseudonymisation permet d'établir un lien avec les données des participants à l'essai clinique dans l'ensemble des dossiers de l'étude.
Voici des exemples de renseignements permettant l'identification directe qui peuvent être caviardés :
- Nom
- Initiales
- Signature
- Titre de l'emploi/poste
- Adresse
- Numéro de télécopieur
- Adresse courriel
- Numéro d'assurance-maladie du bénéficiaire
- Numéro de lot/de série
- Numéro de téléphone
Anonymisation des renseignements permettant l'identification indirecte
Santé Canada encourage la généralisation des renseignements permettant l'identification indirecte. Ces renseignements peuvent inclure les suivants : ville, état ou province, code postal, données démographiques (race, sexe, etc.), antécédents médicaux, événements indésirables graves, dates, taille, poids et indice de masse corporelle (IMC).
Dans certaines circonstances, les renseignements devraient être resynthétisés après la généralisation pour éviter de donner l'impression d'une anonymisation. La resynthèse subséquente devrait réduire encore plus le risque parce qu'elle empêche d'identifier les renseignements anonymisés.
Documentation du processus d'anonymisation et gouvernance
Le processus d'anonymisation doit être bien documenté afin d'établir la piste d'audit nécessaire. Santé Canada demande aux fabricants de soumettre un rapport d'anonymisation dûment rempli (modèle fourni à l'annexe F) avec tous les renseignements cliniques anonymisés.
7. Coordonnées
Adresse postale :
Division de la science de l’information et de l’ouverture
Direction de la gestion des ressources et des opérations
Direction générale des produits de santé et des aliments
Santé Canada
Immeuble Graham Spry
250, avenue Lanark
Ottawa (Ontario)
K1A 0K9
Téléphone :
613-960-4687
Courriel :
hc.clinicaldata-donneescliniques.sc@canada.ca
8. Références
Portail des données cliniques de Santé Canada
Loi sur les aliments et drogues
Règlement sur les aliments et drogues
Règlement sur les instruments médicaux
9. Annexes
| Section | RCC | Diffusion proactive |
|---|---|---|
| 2.5 Aperçu clinique | Pas RCC | Oui |
| 2.5.1 Justification de l'élaboration du produit | Pas RCC | Oui |
| 2.5.2 Aperçu des données biopharmaceutiques | Pas RCC | Oui |
| 2.5.3 Aperçu de la pharmacologie clinique | Pas RCC | Oui |
| 2.5.4 Aperçu de l'efficacité | Pas RCC | Oui |
| 2.5.5 Aperçu de l'innocuité | Pas RCC | Oui |
| 2.5.6 Conclusions sur les risques et les avantages | Pas RCC | Oui |
| 2.5.7 Références bibliographiques | Pas RCC | Oui |
| 2.7.1 Résumé de l'évaluation biopharmaceutique et des méthodes d'analyse connexes | Pas RCC | Oui |
| 2.7.1.1 Contexte et aperçu | Pas RCC | Oui |
| 2.7.1.2 Résumé des résultats d'études individuelles | Pas RCC | Oui |
| 2.7.1.3 Comparaison et analyse des résultats pour l'ensemble des études | Pas RCC | Oui |
| 2.7.1.4 Annexe | Pas RCC | Oui |
| 2.7.2 Résumé des études de pharmacologie clinique | Pas RCC | Oui |
| 2.7.2.1 Contexte et aperçu | Pas RCC | Oui |
| 2.7.2.2 Résumé des résultats d'études individuelles | Pas RCC | Oui |
| 2.7.2.3 Comparaison et analyse des résultats pour l'ensemble des études | Pas RCC | Oui |
| 2.7.2.4 Études spéciales | Pas RCC | Oui |
| 2.7.2.5 Annexe | Pas RCC | Oui |
| 2.7.3 Résumé de l'efficacité clinique | Pas RCC | Oui |
| 2.7.3.1 Contexte et aperçu de l'efficacité clinique | Pas RCC | Oui |
| 2.7.3.2 Résumé des résultats d'études individuelles | Pas RCC | Oui |
| 2.7.3.3 Comparaison et analyse des résultats pour l'ensemble des études | Pas RCC | Oui |
| 2.7.3.4 Analyse des données cliniques pertinentes pour les recommandations en matière de posologie | Pas RCC | Oui |
| 2.7.3.5 Persistance de l'efficacité ou de la tolérabilité | Pas RCC | Oui |
| 2.7.3.6 Annexe | Pas RCC | Oui |
| 2.7.4 Résumé de l'innocuité clinique | Pas RCC | Oui |
| 2.7.4.1 Exposition au médicament | Pas RCC | Oui |
| 2.7.4.1.1 Plan d'évaluation globale de l'innocuité et description des études sur l'innocuité | Pas RCC | Oui |
| 2.7.4.1.2 Ampleur de l'exposition globale | Pas RCC | Oui |
| 2.7.4.1.3 Caractéristiques démographiques et autres de la population à l'étude | Pas RCC | Oui |
| 2.7.4.2 Événements indésirables | Pas RCC | Oui |
| 2.7.4.2.1 Analyse des événements indésirables | Pas RCC | Oui |
| 2.7.4.2.2 Descriptions | Pas RCC | Oui |
| 2.7.4.3 Évaluations cliniques en laboratoire | Pas RCC | Oui |
| 2.7.4.4 Signes vitaux, signes physiques et autres observations liées à l'innocuité | Pas RCC | Oui |
| 2.7.4.5 Innocuité dans des groupes et situations particulières | Pas RCC | Oui |
| 2.7.4.5.1 Facteurs intrinsèques | Pas RCC | Oui |
| 2.7.4.5.2 Facteurs extrinsèques | Pas RCC | Oui |
| 2.7.4.5.3 Interactions médicamenteuses | Pas RCC | Oui |
| 2.7.4.5.4 Utilisation pendant la grossesse et l'allaitement | Pas RCC | Oui |
| 2.7.4.5.5 Surdose | Pas RCC | Oui |
| 2.7.4.5.6 Abus de médicament | Pas RCC | Oui |
| 2.7.4.5.7 Sevrage et effet rebond | Pas RCC | Oui |
| 2.7.4.5.8 Effets sur la capacité de conduire un véhicule ou de la machinerie ou altérations des facultés mentales | Pas RCC | Oui |
| 2.7.4.6 Données post-commercialisation | Pas RCC | Oui |
| 2.7.4.7 Annexe | Pas RCC | Oui |
| 2.7.5 Références bibliographiques | Pas RCC | Non |
| 2.7.6 Synopsis d'études individuelles | Pas RCC | Non |
| 5.1 Table des matières du module | Pas RCC | Non |
| 5.2 Liste de toutes les études cliniques sous forme de tableau | Pas RCC | Non |
| 5.3.1.1 Rapports d'études de biodisponibilité | Pas RCC | Oui |
| 5.3.1.2 Rapports d'études de biodisponibilité et de bioéquivalence comparatives | Pas RCC | Oui |
| 5.3.1.3 Rapports d'études de corrélation « in vitro-in vivo » | Pas RCC | Non |
| 5.3.1.4 Rapports sur les méthodes bioanalytiques et analytiques utilisées dans les études chez les humains | Pas RCC | Non |
| 5.3.2.1 Rapports d'études sur la liaison à des protéines plasmatiques | Pas RCC | Oui |
| 5.3.2.2 Rapports d'études sur le métabolisme hépatique et les interactions médicamenteuses | Pas RCC | Oui |
| 5.3.2.3 Rapports d'études utilisant d'autres matériaux biologiques humains | Pas RCC | Oui |
| 5.3.3.1 Rapports d'études sur la pharmacocinétique et la tolérabilité initiale chez des sujets en santé | Pas RCC | Oui |
| 5.3.3.2 Rapports d'études sur la pharmacocinétique et la tolérabilité initiale chez des patients | Pas RCC | Oui |
| 5.3.3.3 Rapports d'études pharmacocinétiques évaluant les effets des facteurs intrinsèques | Pas RCC | Oui |
| 5.3.3.4 Rapports d'études pharmacocinétiques évaluant les effets des facteurs extrinsèques | Pas RCC | Oui |
| 5.3.3.5 Rapports d'études pharmacocinétiques basées sur la population | Pas RCC | Oui |
| 5.3.4.1 Rapports d'études pharmacodynamiques et pharmacodynamiques/pharmacocinétiques chez des sujets en santé | Pas RCC | Oui |
| 5.3.4.2 Rapports d'études pharmacodynamiques et pharmacodynamiques/pharmacocinétiques chez des patients | Pas RCC | Oui |
| 5.3.5.1 Rapports d'études cliniques contrôlées pertinentes pour l'évaluation de l'indication proposée | Pas RCC | Oui |
| 5.3.5.2 Rapports d'études cliniques non contrôlées | Pas RCC | Oui |
| 5.3.5.3 Rapports d'analyses de données provenant de plus d'une étude | Pas RCC | Oui |
| 5.3.5.4 Autres rapports d'études | Pas RCC | Oui |
| 5.3.6 Rapports sur des expériences post-commercialisation | Pas RCC | Non |
| 5.3.7 Formulaires d'exposés de cas et listes individuelles de patients (le cas échéant) | Pas RCC | Non |
| 5.4 Références bibliographiques | Pas RCC | Non |
| *Pour une description des éléments de la présentation, veuillez consulter la ligne directrice ICH M4E (R2) | ||
| Section | Description | RCC | Diffusion publique proactive |
|---|---|---|---|
| 1 | Page titre | Pas RCC | Oui |
| 2 | Synopsis | Pas RCC | Oui |
| 3 | Table des matières de chaque rapport d'étude clinique | Pas RCC | Oui |
| 4 | Liste des abréviations et définitions | Pas RCC | Oui |
| 5 | Éthique | Pas RCC | Oui |
| 5.1 | Comité d'éthique indépendant (CEI) ou conseil d'examen de l'établissement (CEE) | Pas RCC | Oui |
| 5.2 | Conduite éthique de l'étude | Pas RCC | Oui |
| 5.3 | Information et consentement des patients | Pas RCC | Oui |
| 6 | Chercheurs et structure administrative de l'étude | Pas RCC | Oui |
| 7 | Introduction | Pas RCC | Oui |
| 8 | Objectifs de l'étude | Pas RCC | Oui |
| 9 | Plan de l'étude | Pas RCC | Oui |
| 9.1 | Conception et plan d'ensemble de l'étude - Description | Pas RCC | Oui |
| 9.2 | Discussion du plan de l'étude, notamment du choix des groupes témoins | Pas RCC | Oui |
| 9.3 | Sélection de la population à l'étude | ||
| 9.3.1 | Critères d'inclusion | Pas RCC | Oui |
| 9.3.2 | Critères d'exclusion | Pas RCC | Oui |
| 9.3.3 | Patients retirés du traitement ou de l'évaluation | Pas RCC | Oui |
| 9.4 | Traitements | ||
| 9.4.1 | Traitements administrés | Pas RCC | Oui |
| 9.4.2 | Nom du ou des produits à l'étude | Pas RCC | Oui |
| 9.4.3 | Méthode de répartition des patients entre les groupes de traitement | Pas RCC | Oui |
| 9.4.4 | Sélection des doses à l'étude | Pas RCC | Oui |
| 9.4.5 | Sélection et chronologie de la dose pour chaque patient | Pas RCC | Oui |
| 9.4.6 | Insu | Pas RCC | Oui |
| 9.4.7 | Traitement antérieur et concomitant | Pas RCC | Oui |
| 9.4.8 | Respect du traitement | Pas RCC | Oui |
| 9.5 | Variables d'efficacité et d'innocuité | ||
| 9.5.1 | Mesures d'efficacité et d'innocuité et diagramme | Pas RCC | Oui |
| 9.5.2 | Pertinence des mesures | Pas RCC | Oui |
| 9.5.3 | Principale(s) variable(s) d'efficacité | Pas RCC | Oui |
| 9.5.4 | Mesures de la concentration de médicaments | Pas RCC | Oui |
| 9.6 | Assurance de la qualité des données | Pas RCC | Oui |
| 9.7 | Méthodes statistiques prévues dans le protocole et détermination de la taille de l'échantillon | ||
| 9.7.1 | Plans statistiques et analytiques | Pas RCC | Oui |
| 9.7.2 | Détermination de la taille de l'échantillon | Pas RCC | Oui |
| 9.8 | Modification du déroulement de l'étude ou des analyses prévues | Pas RCC | Oui |
| 10 | Patients à l'étude | ||
| 10.1 | Retrait de patients | Pas RCC | Oui |
| 10.2 | Écarts par rapport au protocole | Pas RCC | Oui |
| 11 | Évaluation de l'efficacité | ||
| 11.1 | Ensembles de données analysés | Pas RCC | Oui |
| 11.2 | Caractéristiques démographiques et autres caractéristiques de base | Pas RCC | Oui |
| 11.3 | Mesures du respect du traitement | Pas RCC | Oui |
| 11.4 | Résultats sur l'efficacité et mise en tableaux des données individuelles des patients | Pas RCC | Oui |
| 11.4.1 | Analyses de l'efficacité | Pas RCC | Oui |
| 11.4.2 | Questions statistiques et analytiques | Pas RCC | Oui |
| 11.4.2.1 | Ajustements en fonction du choix de covariables | Pas RCC | Oui |
| 11.4.2.2 | Traitement des abandons et des données manquantes | Pas RCC | Oui |
| 11.4.2.3 | Analyses intermédiaires et surveillance des données | Pas RCC | Oui |
| 11.4.2.4 | Études multicentriques | Pas RCC | Oui |
| 11.4.2.5 | Comparaisons multiples et multiplicité | Pas RCC | Oui |
| 11.4.2.6 | Utilisation d'un « sous-ensemble de patients pour l'évaluation de l'efficacité » | Pas RCC | Oui |
| 11.4.2.7 | Études produit actif-témoin visant à démontrer l'équivalence | Pas RCC | Oui |
| 11.4.2.8 | Examen des sous-groupes | Pas RCC | Oui |
| 11.4.3 | Mise en tableaux des données individuelles sur la réponse | Pas RCC | Oui |
| 11.4.4 | Dose du médicament, concentration du médicament et relations avec la réponse | Pas RCC | Oui |
| 11.4.5 | Interactions médicament-médicament et médicament-maladie | Pas RCC | Oui |
| 11.4.6 | Affichages par patient | Pas RCC | Oui |
| 11.4.7 | Conclusions sur l'efficacité | Pas RCC | Oui |
| 12 | Évaluation de l'innocuité | ||
| 12.1 | Ampleur de l'exposition | Pas RCC | Oui |
| 12.2 | Événements indésirables (EI) | Pas RCC | Oui |
| 12.2.1 | Bref résumé des événements indésirables | Pas RCC | Oui |
| 12.2.2 | Affichage des événements indésirables | Pas RCC | Oui |
| 12.2.3 | Analyse des événements indésirables | Pas RCC | Oui |
| 12.2.4 | Liste des événements indésirables par patient | Pas RCC | Oui |
| 12.3 | Décès, autres événements indésirables graves et autres événements indésirables importants | Pas RCC | Oui |
| 12.3.1 | Liste des décès et des autres événements indésirables graves et événements indésirables importants | Pas RCC | Oui |
| 12.3.1.1 | Décès | Pas RCC | Oui |
| 12.3.1.2 | Autres événements indésirables graves | Pas RCC | Oui |
| 12.3.1.3 | Autres événements indésirables importants | Pas RCC | Oui |
| 12.3.2 | Description des décès, des autres événements indésirables graves et de certains autres événements indésirables importants | Pas RCC | Oui |
| 12.3.3 | Analyse et discussion sur les décès, autres événements indésirables graves et autres événements indésirables importants | Pas RCC | Oui |
| 12.4 | Évaluation clinique en laboratoire | Pas RCC | Oui |
| 12.4.1 | Liste des mesures de laboratoire individuelles par patient (16.2.8) et de chaque valeur de laboratoire anormale (14.3.4) | Pas RCC | Oui |
| 12.4.2 | Évaluation de chaque paramètre de laboratoire | Pas RCC | Oui |
| 12.4.2.1 | Valeurs de laboratoire au fil du temps | Pas RCC | Oui |
| 12.4.2.2 | Modifications individuelles par patient | Pas RCC | Oui |
| 12.4.2.3 | Anomalies individuelles cliniquement importantes | Pas RCC | Oui |
| 12.5 | Signes vitaux, signes physiques et autres observations liées à l'innocuité | Pas RCC | Oui |
| 12.6 | Conclusions sur l'innocuité | Pas RCC | Oui |
| 13 | Discussion et conclusions générales | Pas RCC | Oui |
| 14 | Tableaux, figures et graphiques mentionnés, mais ne figurant pas dans le texte | ||
| 14.1 | Données démographiques | Pas RCC | Oui |
| 14.2 | Résumé des données sur l'efficacité -figures et tableaux | Pas RCC | Oui |
| 14.3 | Résumé des données sur l'innocuité - figures et tableaux | Pas RCC | Oui |
| 14.3.1 | Affichage des événements indésirables | Pas RCC | Oui |
| 14.3.2 | Listes des décès et des autres événements indésirables graves et importants | Pas RCC | Oui |
| 14.3.3 | Description des décès, d'autres événements indésirables graves et de certains autres événements indésirables importants | Pas RCC | Oui |
| 14.3.4 | Liste des valeurs de laboratoire anormales (par patient) | Pas RCC | Oui |
| 15 | Liste de références | Pas RCC | Oui |
| 16 | Annexes | Pas RCC | Oui |
| 16.1 | Renseignements sur l'étude | Pas RCC | Oui |
| 16.1.1 | Protocole et modifications du protocole | Pas RCC | Oui |
| 16.1.2 | Formulaire type d'exposé de cas (une page seulement) | Pas RCC | Oui |
| 16.1.3 | Liste des CEI ou des CEE (avec le nom du président du comité si l'organisme de réglementation l'exige) - renseignements écrits représentatifs destinés aux patients et formulaire type de consentement | Pas RCC | Non |
| 16.1.4 | Liste et description des chercheurs et autres principaux participants à l'étude, notamment brefs curriculum vitæ (une page) ou résumés équivalents de la formation et de l'expérience pertinentes aux fins de la réalisation de l'étude clinique | Pas RCC | Non |
| 16.1.5 | Signature du ou des chercheurs principaux, du ou des chercheurs coordonnateurs ou du médecin responsable nommé par le promoteur, selon les exigences de l'organisme de réglementation | Pas RCC | Non |
| 16.1.6 | Liste des patients recevant les médicaments/produits expérimentaux à l'étude provenant de lots spécifiques, lorsque plus d'un lot a été utilisé | Pas RCC | Non |
| 16.1.7 | Schéma et codes de randomisation (identification des patients et traitement attribué) | Pas RCC | Non |
| 16.1.8 | Certificats de vérification (le cas échéant) | Pas RCC | Non |
| 16.1.9 | Documentation des méthodes statistiques | Pas RCC | Oui |
| 16.1.10 | Documentation des méthodes de normalisation et des procédures d'assurance qualité interlaboratoires, le cas échéant | Pas RCC | Non |
| 16.1.11 | Publications fondées sur l'étude | Pas RCC | Non |
| 16.1.12 | Publications importantes citées dans le rapport | Pas RCC | Non |
| 16.2 | Liste des données sur les patients | ||
| 16.2.1 | Patients retirés | Pas RCC | Non |
| 16.2.2 | Écarts par rapport au protocole | Pas RCC | Non |
| 16.2.3 | Patients exclus de l'analyse de l'efficacité | Pas RCC | Non |
| 16.2.4 | Données démographiques | Pas RCC | Non |
| 16.2.5 | Données sur le respect du traitement ou sur la concentration du médicament (le cas échéant) | Pas RCC | Non |
| 16.2.6 | Données individuelles sur l'efficacité | Pas RCC | Non |
| 16.2.7 | Listes des événements indésirables (par patient) | Pas RCC | Non |
| 16.2.8 | Liste des mesures de laboratoire individuelles par patient, selon les exigences des organismes de réglementation | Pas RCC | Non |
| 16.3 | Formulaires d'exposé de cas (FEC) | ||
| 16.3.1 | FEC sur les décès, les autres événements indésirables graves et les retraits dus à des EI | Pas RCC | Non |
| 16.3.2 | Autres formulaires d'exposé de cas présentés | Pas RCC | Non |
| 16.4 | Listes de données individuelles sur les patients (listes d'archives américaines) | Pas RCC | Non |
Annexe C : Diagramme du processus
Annexe D : Convention d'appellation des noms de documents pour les présentations par le PCDE
Les caviardages, ainsi que les tableaux de justification des caviardages et les rapports d'anonymisation accompagnant les renseignements cliniques anonymisés, doivent être soumis à l'aide du PCDE, en utilisant les mêmes titres d'extension de nœud que ceux figurant dans la présentation parente.
Les fichiers soumis doivent être en format PDF.
La convention d'appellation des noms pour tous les documents auxquels des caviardages sont proposés doit être conforme à la convention utilisée dans la présentation initiale; seul le suffixe « -pr » est ajouté. On présume que la convention d'appellation de noms initiale était conforme à la Ligne directrice : Préparation des activités de réglementation des drogues en format Electronic Common Technical Document de Santé Canada.
« -pr » = caviardage proposé
p. ex. clinical-overview-pr.pdf, summary-clin-safety-pr.pdf, study-XXXXXX-pr.pdf
Une fois la version caviardée finale établie, Santé Canada appliquera le suffixe « -red » aux documents anonymisés finaux, avant leur publication.
« -red » = caviardage final
p. ex. clinical-overview-red.pdf, summary-clin-safety-red.pdf, study-XXXXXX-red.pdf
| Nom du document | Numéro(s) de page | Texte proposé à caviarder | Exceptions admissibles selon le règlement | N'est pas un renseignement clinique | Justification détaillée du caviardage proposé | Réponse de Santé Canada au caviardage proposé | Justification de Santé Canada |
|---|---|---|---|---|---|---|---|
| p.ex : CSR-1.pdf | p.ex : 122 | Copier et coller le texte proposé à être caviardé ici. p.ex. : des modifications spécifiques à un essai biologique qui se qualifie pour le caviardage. | Exemples d'exceptions : C.08.009.2(2)a) ou C.08.009.2(2)b) | p. ex. renseignements sur la chimie, la fabrication | p.ex : le texte proposé constitue une modification brevetée de l'essai biologique mis au point par la compagnie A en collaboration avec le fabricant. Cette information, si publiée, portera atteinte aux intérêts commerciaux du fabricant car … | Rejeté / Partiellement accepté/accepté |
p.ex. : Le caviardage a été rejeté puisque le texte proposé constitue de l'information disponible dans le domaine publique |
Annexe F : Modèle de rapport d'anonymisation
1. Méthode d'anonymisation
- Décrire la méthode, le seuil de risque utilisé et la justification de la méthode choisie.
2. Identification des variables (identificateurs directs et indirects)
- Classer les variables considérées comme des renseignements personnels selon le type de ré-identification qu'elles permettent (identification directe ou indirecte).
- Énoncer et justifier les raisons pour lesquelles les données sont considérées comme des renseignements personnels.
3. Mesure du risque de divulgation
- Indiquer et justifier la population de référence utilisée et fournir un résumé de l'ensemble de données utilisé pour l'évaluation du risque.
- Décrire l'analyse du risque en regard des hypothèses formulées relativement aux situations plausibles de ré-identification.
- Évaluer le risque réel de ré-identification associé à chaque sujet de l'essai.
- Discuter de la manière dont les données ont été modifiées pour réduire le risque de ré-identification.
- Fournir des données attestant que le risque de ré-identification après le processus d'anonymisation est inférieur au seuil prescrit.
4. Considérations liées à l'utilité des données
- Indiquer les efforts qui ont été faits pour maximiser l'utilité des renseignements anonymisés.
5. Écarts
- Tout écart par rapport aux procédures d'anonymisation proposées doit être justifié et parfaitement documenté.
6. Conclusions
- Déclaration attestant que les critères d'anonymisation ont été respectés conformément aux exigences de Santé Canada.
- Je, [le fabricant], certifie que le rapport d'anonymisation a été préparé conformément à la ligne directrice de Santé Canada.
Annexe G : Lettre de certification et tableau des renseignements précédemment caviardés
Formulaire de certification
Nom du produit pharmaceutique :
Compagnie :
Je certifie que les renseignements et données mentionnés ci-après et contenus dans cette présentation sont complets et exacts et qu'ils représentent fidèlement les renseignements caviardés et anonymisés, ainsi que le matériel soumis à l'Agence européenne des médicaments (EMA) en vertu de la politique 0070 et la présentation au Canada à laquelle ils renvoient.
Numéro de contrôle de la présentation de Santé Canada :
Procédure de l'EMA :
- Module 2 :
- Section 2.5 (pages xxx à xxx)
- Section 2.7 (pages xxx à xxx)
- Module 5
- Nom de l'étude clinique 1 (pages xxx à xxx)
- Nom de l'étude clinique 2 (pages xxx à xxx)
Je certifie que les renseignements ci-après n'ont pas été caviardés ni anonymisés et qu'ils sont exclusifs à la présentation au Canada :
- Module 2 :
- Section 2.5 (pages xxx à xxx)
- Section 2.7 (pages xxx à xxx)
- Module 5
- Nom de l'étude clinique 1 (pages xxx à xxx)
- Nom de l'étude clinique 2 (pages xxx à xxx)
OU Sans objet
Signature du responsable de la compagnie certifiant l'exactitude de ce document.
Il est interdit à toute personne de faire sciemment une déclaration fausse ou trompeuse au ministre ou de lui fournir sciemment des renseignements faux ou trompeurs relativement à toute question visée par la loi à l'égard des produits thérapeutiques (Loi sur les aliments et drogues, article 21.6).
Annexe H : Conditions d'utilisation
Ces conditions d'utilisation régissent l'accès aux renseignements cliniques publiés par Santé Canada à des fins non commerciales et l'utilisation de ces renseignements. En cliquant sur le bouton « J'accepte » et en acceptant ces conditions d'utilisation, et après avoir obtenu l'accès aux renseignements cliniques, vous et l'organisation au nom de laquelle vous accédez aux renseignements cliniques, le cas échéant, acceptez expressément d'être liés par ces conditions d'utilisation.
IL EST IMPORTANT DE LIRE LES CONDITIONS D'UTILISATION ATTENTIVEMENT.
1. Définitions
« Fabricant » s'entend de la personne ou de l'entreprise à qui une identification numérique de drogue (DIN) ou une homologation d'instrument médical a été délivrée pour un produit auquel les renseignements cliniques se rapportent.
« Ré-identification » pratique consistant à faire correspondre des données anonymes avec d'autres renseignements dans le but de découvrir l'identité de la personne à qui ces données se rapportent.
« Renseignements cliniques » s'entend des renseignements obtenus sur ce site.
« Vous » signifie vous personnellement et, le cas échéant, si vous accédez et utilisez les renseignements au nom de votre employeur, cet employeur et ses sociétés affiliées.
2. Représentations et garanties
Vous représentez et garantissez que :
a. si vous accédez à des renseignements cliniques pour le compte de votre employeur, vous avez l'autorisation légale de lier votre employeur.
b. vous ne désirez accéder aux renseignements cliniques qu'à des fins non commerciales.
3. Utilisation de renseignements cliniques
3.1 Vous reconnaissez que les renseignements cliniques peuvent être protégés par le droit d'auteur ou d'autres droits de propriété intellectuelle du fabricant ou d'un tiers. La permission accordée par le biais de ce site ne confère aucun droit de propriété intellectuelle ni autre droit commercial relatif aux renseignements cliniques autres que ceux énoncés expressément dans la présente.
3.2 Vous êtes autorisé par la présente à télécharger, à sauvegarder, à imprimer, à partager ou à utiliser, à reproduire ou à communiquer des renseignements cliniques à des fins non commerciales, ce qui, sans limiter ce qui précède, exclut expressément l'utilisation de ces renseignements en appui à une demande d'autorisation de mise en marché partout dans le monde ou la vente ou l'échange de renseignements à une autre personne.
3.3 Si vous constatez une divulgation par inadvertance de renseignements personnels dans les renseignements cliniques obtenus par le biais de ce site, veuillez le signaler immédiatement à Santé Canada. Les renseignements cliniques ne peuvent pas être utilisés pour identifier des participants à des essais cliniques ou d'autres personnes par le biais d'une ré-identification ni pour publier des renseignements personnels résultant d'une ré-identification. Si vous savez que des renseignements qui, combinés à des renseignements cliniques, pourraient vraisemblablement conduire à une ré-identification sont disponibles, veuillez en aviser immédiatement à Santé Canada.
3.4 Vous acceptez de ne fournir aucune copie des renseignements cliniques à une autre entité ou à une autre personne sans engagement envers Santé Canada à l'effet que l'autre entité ou personne utilisera ces renseignements uniquement à des fins non commerciales et conformément aux présentes conditions d'utilisation.
3.5 Lorsque vous reproduisez des renseignements cliniques, vous acceptez de ne pas donner une fausse représentation de la source des renseignements cliniques et de reconnaître que la source des renseignements est le fabricant, de même que de ne pas utiliser ces renseignements de telle sorte à laisser entendre que le fabricant approuve votre utilisation des renseignements cliniques à toute autre fin que des fins non commerciales.
3.6 Vous acceptez d'informer Santé Canada de toute utilisation non autorisée de renseignements cliniques.
3.7 Santé Canada se réserve le droit de révoquer l'accès ou toute permission accordée par la présente à toute personne qui enfreint les présentes conditions d'utilisation ou qui fournit des renseignements faux ou trompeurs en lien avec toute demande de renseignements cliniques.
4. Modifications
4.1 Santé Canada se réserve le droit de modifier ces conditions d'utilisation à tout moment, et ce, sans préavis. De telles modifications entreront en vigueur au moment même de l'avis de modifications ou à toute autre date spécifiée dans l'avis.
4.2 Votre acceptation des conditions d'utilisation modifiées indiquera votre accord avec les modifications qui s'appliqueront à votre utilisation, après la date d'acceptation, des renseignements cliniques précédemment consultés, téléchargés, sauvegardés ou imprimés par vous.
5. Limitation de la responsabilité et de l'indemnisation
5.1 Santé Canada décline toute responsabilité quant à votre respect des présentes conditions d'utilisation ou à toute autre façon de faire découlant de vos actes, de vos omissions ou de votre conduite dans l'utilisation des renseignements cliniques.
5.2 Vous vous engagez à indemniser et à mettre hors de cause Santé Canada, Sa Majesté la Reine du chef du Canada, ses ayants droit et successeurs, agents, employés ou mandataires de toute réclamation, de toute action, de tout dommage, de toute perte, de toute dépense et de tout coût, y compris les honoraires d'avocat raisonnables, résultant de toute violation de ces conditions d'utilisation ou de toute instance découlant de quelque manière que ce soit de vos actes, de vos omissions ou de votre conduite en ce qui a trait à l'utilisation des renseignements cliniques.
6. Avis de non-responsabilité
Sans préjudice de toute obligation du fabricant en vertu des lois du Canada, les renseignements cliniques sont fournis « TELS QUELS » et « TELS QUE DISPONIBLES ». En accédant aux renseignements cliniques et en les utilisant, vous acceptez que cet accès et cette utilisation soient entièrement à vos risques. Santé Canada exclut toutes les représentations, garanties, obligations et responsabilités relatives aux renseignements cliniques qui vous sont accessibles dans toute la mesure permise par la loi. Ni Santé Canada ni le fabricant ne sont responsables des erreurs ou des omissions dans les renseignements cliniques auxquels vous avez accès; ils ne sauraient être tenus responsables de toute perte ou tout dommage de quelque nature que ce soit causé par leur utilisation.
7. Dissociabilité
Si une disposition des présentes conditions d'utilisation est déclarée invalide, illégale ou inapplicable par un arbitre ou un tribunal compétent, cette disposition sera séparée des présentes conditions d'utilisation et toutes les autres conditions resteront en vigueur.
8. Lois et compétences applicables
Toutes les questions relatives à votre accès aux renseignements cliniques ou à leur utilisation sont régies par les lois de la province de l'Ontario, à l'exception des règles de droit international privé et des lois du Canada qui s'y appliquent. En ce qui concerne toute action ou poursuite découlant de ces conditions d'utilisation ou de leur objet ou formation (y compris les litiges ou réclamations non contractuels), ou en relation avec celles-ci, l'exclusivité de compétence et de lieu ira aux tribunaux compétents situés dans la province de l'Ontario. Toutefois, si vous ou une autre partie au litige résidez à l'extérieur du Canada, Santé Canada peut, à sa seule discrétion, choisir les tribunaux situés dans la juridiction où vous ou une autre partie réside.