
Ligne directrice : Conduite et analyse des études de biodisponibilité comparatives
Date d’adoption: 2012/02/08
Date de modification : 2023/01/30
Date d’entrée en vigueur: 2018/07/01 (pour les soumissions qui seront déposées à partir du 1er septembre 2018)
Veuillez vous référer à l’Avis aux intervenants ‐ Mise en œuvre de la ligne directrice ICH M13A : Bioéquivalence des formes orales solides à libération immédiate pour obtenir des informations mises à jour concernant l’application de cette ligne directrice.
Registre des révisions du document
Date :
Changement : l’ajout d’une phrase à la fin du troisième paragraphe
« Le choix d’un plan équilibre pour l’analyse de la variance (plan de Williams) ou d’un plan en bloc incomplet séparé devra être justifie. »
Emplacement (section, paragraphe) : Section 2.3.1
Nature du changement et justification : Clarification des exigences statistiques.
Changement : l’ajout de la section 2.3.3 Études pharmacodynamiques
Emplacement (section, paragraphe) : Section 2.3.3
Nature du changement et justification : certains renseignements transférés de la ligne directrice de norme en matière d’étude biodisponibilité comparative puisque l’information était plus pertinent pour la conception de l’étude que de normes.
Changement : modification du deuxième paragraphe
Emplacement (section, paragraphe) : Section 2.3.5
Nature du changement et justification : rédaction des précisions sur la langue.
Changement : la suppression de phrases concernant l’échantillonnage de l’urine.
Emplacement (section, paragraphe) : Section 2.4.6
Nature du changement et justification : Les données sur l’urine ne sont plus utilisées pour l’évaluation des études de biodisponibilité comparatives donc l’information n’est plus pertinent.
Changement : La suppression de deux phrases dans le premier paragraphe concernant l’urine comme un échantillon biologique.
Emplacement (section, paragraphe) : Section 2.4.7
Nature du changement et justification : Les données sur les échantillons d’urine ne sont plus utilisées pour l’évaluation des études de biodisponibilité comparatives donc l’information n’est plus pertinent.
Changement : La suppression du paragraphe concernant des échantillons d’urine et la production de rapports.
Emplacement (section, paragraphe) : Section 2.4.7
Nature du changement et justification : Les données sur les échantillons d’urine ne sont plus utilisées pour l’évaluation des études de biodisponibilité comparatives donc l’information n’est plus pertinent.
Changement : modification de la phrase suivante :
« Il arrive parfois que la concentration du médicament dans un liquide autre que le sang ou l’urine puisse être mieux corrélée avec l’effet. »
Emplacement (section, paragraphe) : Section 2.4.7
Nature du changement et justification : Les données sur les échantillons d’urine ne sont plus utilisées pour l’évaluation des études de biodisponibilité comparatives donc l’information n’est plus pertinent.
Changement : La suppression du paragraphe concernant l’utilisation des concentrations urinaires d’un métabolite jugée non acceptable pour l’évaluation de la bioéquivalence.
Emplacement (section, paragraphe) : Section 2.6.1
Nature du changement et justification : Les données sur les échantillons d’urine ne sont plus utilisées pour l’évaluation des études de biodisponibilité comparatives donc l’information n’est plus pertinent.
Changement : La modification de la phrase suivante :
« Les méthodes bioanalytiques employées pour doser le médicament, ou le métabolite, dans le plasma, le sang total ou le sérum ou l’urine devraient convenir aux fins prévues. »
Emplacement (section, paragraphe) : Section 2.6.2
Nature du changement et justification : Les données sur les échantillons d’urine ne sont plus utilisées pour l’évaluation des études de biodisponibilité comparatives donc l’information n’est plus pertinent.
Changement : L’ajout du paragraphe suivant :
« Pour en savoir davantage sur les tests de stabilité à réaliser durant la validation de la méthode bioanalytique, veuillez-vous reporter à l’Avis : Clarification des procédures de validation des méthodes bioanalytiques (8 octobre 2015, addenda du 9 mars 2016). Ce document est disponible sur le site Web de Sante Canada. »
Emplacement (section, paragraphe) : Section 2.6.2
Nature du changement et justification : Mettre à jour la référence à l’Agence européenne des médicaments des directives.
Changement : La suppression des paramètres pharmacocinétiques l) et m) ainsi que le paragraphe ci-dessous :
« Lorsque la biodisponibilité comparative s’appuie sur des données urinaires, les paramètres suivants devraient être signalés. »
Emplacement (section, paragraphe) : Section 2.7.2
Nature du changement et justification : Les données sur les échantillons d’urine ne sont plus utilisées pour l’évaluation des études de biodisponibilité comparatives donc l’information n’est plus pertinent.
Changement : L’ajout de l’information ci-dessous après le paramètre pharmacocinétique k)
Lorsqu’on a démontré qu’un profil multiple de la concentration plasmatique d’un produit à libération modifiée faisait partie intégrante de l’effet thérapeutique, il convient de consigner les paramètres suivants :
1. La surface qui se trouve sous la courbe de la concentration en fonction du temps suivant un intervalle de temps restreint après l’administration du médicament (SSC partielle).
Emplacement (section, paragraphe) : Section 2.7.2
Nature du changement et justification : L’introduction du paramètre SSC partielle pour les formes pharmaceutiques à libération modifiée.
Changement : L’ajout du terme SSC partielle et sa définition dans le tableau A1-D
SSC partielle Surface partielle qui se trouve sous la courbe suivant un intervalle de temps restreint après
l’administration du médicament.
Emplacement (section, paragraphe) : Section A1.3
Nature du changement et justification : L’introduction du paramètre SSC partielle pour les formes pharmaceutiques à libération modifiée.
Changement : La suppression des termes suivants :
Ae0-T - Quantité cumulative de médicament excrétée dans l’urine, mesurée au moment du dernier échantillonnage.
Rmax - Taux maximal d’excrétion urinaire du médicament.
Emplacement (section, paragraphe) : Annexe 2
Nature du changement et justification : Les données sur les échantillons d’urine ne sont plus utilisées pour l’évaluation des études de biodisponibilité comparatives donc l’information n’est plus pertinent.
Changement : Révision mineure à la définition de la forme posologique pharmaceutique à libération modifiée pour inclure des formulations des produits pharmaceutiques multiphasiques.
- faire en sorte qu’après l’administration d’une dose unique, les multiples pics et creux des courbes concentration sérique en fonction du temps soient similaires à ceux que l’on obtient après des doses répétées de la forme conventionnelle les courbes de concentration en fonction du temps présentent de multiples pics et creux (c’est-à-dire, forme pharmaceutique multiphasique a libération modifiée).
Emplacement (section, paragraphe) : Annexe 2
Nature du changement et justification : Les produits pharmaceutiques à libération modifiée multiphasique sont maintenant abordés dans le présent document.
Changement : L’ajout de la définition de la SSC partielle
Emplacement (section, paragraphe) : Annexe 2
Nature du changement et justification : La définition de nouveau critère (SSC partielle) introduit pour les produits pharmaceutiques à libération modifiée multiphasique est maintenant abordé dans le présent document.
Date : Le 30 janvier 2023
Changement : Mise à jour de la référence aux lignes directrices sur la validation des méthodes bioanalytiques
Emplacement (section, paragraphe) : Section 2.6, 3e paragraphe et section 2.6.2, deuxième paragraphe
Nature du changement et justification : Lignes directrices de l'EMA sur la validation des méthodes bioanalytiques et avis connexe de Santé Canada remplacés par les lignes directrices ICH M10
Avant-propos
Les lignes directrices sont destinées à guider l'industrie et les professionnels de la santé sur la façon de se conformer aux lois et aux règlements en vigueur. Les lignes directrices fournissent également aux membres du personnel des renseignements concernant la façon de mettre en oeuvre le mandat et les objectifs de Santé Canada de manière juste, uniforme et efficace.
Les lignes directrices sont des outils administratifs n'ayant pas force de loi, ce qui permet une certaine souplesse d'approche. Les principes et les pratiques énoncés dans le présent document pourraient être remplacés par d'autres approches, à condition que celles-ci s'appuient sur une justification adéquate. Il faut tout d'abord discuter des autres approches avec les représentants du programme concerné pour s'assurer qu'elles respectent les exigences des lois et des règlements applicables.
Dans la foulée de ce qui précède, il importe également de mentionner que Santé Canada se réserve le droit de demander des renseignements ou du matériel supplémentaires, ou de définir des conditions dont il n'est pas explicitement question dans la ligne directrice, afin que le Ministère puisse être en mesure d'évaluer adéquatement l'innocuité, l'efficacité ou la qualité d'un produit thérapeutique donné. Santé Canada s'engage à justifier de telles demandes et à documenter clairement ses décisions.
Le présent document devrait être lu en parallèle avec l'avis d'accompagnement et les sections pertinentes des autres lignes directrices qui s'appliquent.
Table des matières
- 1 Introduction
- 1.1 Objectifs de la politique
- 1.2 Énoncés de politique
- 1.3 Portée et application
- 1.4 Contexte
- 2 Conseils pour la mise en œuvre
- 2.1 Planification d'une étude de biodisponibilité comparative
- 2.2 Sélection des sujets d'une étude
- 2.2.1 Choix des sujets
- 2.2.2 Critères d'inclusion/exclusion
- 2.3 Plan de l'étude
- 2.3.1 Plan parallèle ou plan croisé
- 2.3.1.1 Nombre de sujets
- 2.3.2 Autres plans d'étude possibles
- 2.3.2.1 Plans séquentiels par groupes
- 2.3.2.2 Plans adaptatifs
- 2.3.3 Prise en compte des abandons et des retraits
- 2.3.4 Prise en compte des valeurs aberrantes
- 2.3.1 Plan parallèle ou plan croisé
- 2.4 Conduite de l'étude
- 2.4.1 Normalisation
- 2.4.2 Étude en insu
- 2.4.3 Administration d'aliments et de liquides
- 2.4.3.1 Étude chez des sujets à jeun
- 2.4.3.2 Étude chez des sujets non à jeun
- 2.4.4 Posture et activité physique
- 2.4.5 Intervalle entre les doses
- 2.4.6 Temps d'échantillonnage
- 2.4.7 Prélèvement des échantillons
- 2.4.8 Traitement des échantillons
- 2.4.9 Détermination des incidents thérapeutiques
- 2.5 Produit à l'essai et produit de référence
- 2.5.1 Propriétés chimiques
- 2.5.2 Posologie et teneur
- 2.5.3 Choix du produit de référence
- 2.6 Méthodes bioanalytiques
- 2.7 Analyse des données
- 2.7.1 Présentation des données
- 2.7.2 Paramètres pharmacocinétiques
- 2.7.3 Collecte des données
- 2.7.4 Analyse statistique
- 2.7.4.1 Analyse des valeurs aberrantes
- 2.7.4.2 Ajustement du modèle
- 2.7.4.3 Analyse des effets fixes
- 2.7.4.4 Estimation des effets aléatoires
- 2.7.4.5 Analyse des données
- Annexe 1 : Analyse type pour une étude de biodisponibilité comparative
- Annexe 2 : Glossaire
1 Introduction
1.1 Objectifs de la politique
Fournir aux promoteurs qui soumettent une présentation de drogue nouvelle les renseignements nécessaires pour se conformer à l'alinéa C.08.002(2)h), au sous-alinéa C.08.002.1(2)c)(ii) et au paragraphe C.08.003(3) du Règlement sur les aliments et drogues (le Règlement) en ce qui concerne les études de biodisponibilité comparatives utilisées pour établir l'innocuité et l'efficacité d'un médicament.
1.2 Énoncés de politique
Les études de biodisponibilité comparatives devraient être menées conformément aux pratiques cliniques généralement acceptées qui visent à garantir la protection des droits, la sécurité et le bien-être des sujets, et aux bonnes pratiques cliniques définies au titre 5 du Règlement et décrites dans la Ligne directrice de l'International Conference on Harmonisation (ICH) E6 : Les bonnes pratiques cliniques. Les bonnes pratiques de fabrication, telles qu'elles sont énoncées à la partie C, titre 2, du Règlement, devraient être respectées lorsqu'elles s'appliquent.
Les recommandations formulées dans le présent document concernant le plan d'étude, la conduite de l'étude, la validation des méthodes bioanalytiques et l'analyse statistique des données devraient être suivies afin de s'assurer de la conformité au Règlement.
1.3 Portée et application
La présente ligne directrice est destinée à être appliquée à toutes les études de biodisponibilité comparatives qui fournissent des preuves essentielles de l'innocuité et de l'efficacité d'un produit, à l'exception des produits biologiques ultérieurs. Voici des exemples de cas où elle s'applique :
- études de biodisponibilité comparatives pour confirmer la bioéquivalence de produits génériques du produit de référence canadien;
- études de transition lorsque la forme pharmaceutique à commercialiser diffère de celle utilisée lors des essais cliniques pivots;
- études visant à appuyer des changements majeurs après une approbation ou des extensions de gamme;
- études d'innocuité de médicaments non systémiques, lorsque les concentrations systémiques du médicament peuvent être mesurées en vue de l'évaluation de l'innocuité de produits contenant un médicament conçu pour agir localement, par exemple un produit administré au moyen d'un aérosol-doseur;
- études de biodisponibilité comparatives à l'appui d'une demande d'identification numérique de drogue (DIN).
Bien que la ligne directrice vise les formes pharmaceutiques orales solides, qu'elles soient à libération immédiate ou à libération modifiée, les principes et les normes qui y sont décrits pourraient aussi s'appliquer, s'il y a lieu, à d'autres formes pharmaceutiques orales ou à des formes non injectables, tels les timbres transdermiques, les suppositoires et autres, qui sont destinées à libérer le médicament dans la circulation générale.
Le présent document devrait être lu en parallèle avec la ligne directrice connexe de Santé Canada intitulée Normes en matière d'études de biodisponibilité comparatives : Formes pharmaceutiques de médicaments à effets systémiques.
1.4 Contexte
La biodisponibilité est un attribut important des formes pharmaceutiques de médicaments utilisées pour leurs effets systémiques. Elle est définie comme la vitesse et le degré de passage d'un médicament dans la circulation générale.
La biodisponibilité est le plus souvent évaluée par des dosages en série du médicament dans le sang. Ces dosages permettent de produire une courbe de concentration du médicament dans le plasma, le sérum ou le sang total en fonction du temps, à partir de laquelle il est possible de calculer plusieurs paramètres pharmacocinétiques importants, notamment la surface sous la courbe (SSC), la concentration maximale observée (Cmax) et le temps pour atteindre la Cmax (tmax). La SSC représente un substitut de la quantité de médicament absorbée dans la circulation générale. Tant le tmax que la Cmax sont des fonctions complexes qui déterminent le point temporel où le taux d'absorption et le taux d'excrétion sont égaux. Même si ces paramètres souffrent d'un manque de robustesse, la Cmax est généralement considérée comme une donnée raisonnable pour définir la vitesse d'absorption. Pour de nombreux médicaments, ensemble, la SSC et la Cmax peuvent caractériser la courbe de concentration en fonction du temps à des fins de comparaison.
Une comparaison des valeurs de la SSC après une administration par voie orale versus une administration intraveineuse d'une dose équivalente du même ingrédient actif permet d'estimer la biodisponibilité absolue de la plupart des médicaments. La comparaison des courbes de concentration plasmatique du médicament en fonction du temps entre le produit à l'essai et le produit de référence renfermant la même quantité du ou des mêmes ingrédients actifs permet d'estimer la biodisponibilité relative.
Le produit à l'essai et le produit de référence sont bioéquivalents si leur forme pharmaceutique est comparable, s'ils contiennent une quantité identique du même ou des mêmes ingrédients médicinaux et si les courbes du médicament sont similaires. Le degré de similitude des courbes nécessaire pour établir la bioéquivalence est déterminé au moyen d'une évaluation statistique appropriée et en respectant les normes établies pour le médicament et les formes pharmaceutiques comparés (voir la ligne directrice de Santé Canada intitulée Normes en matière d'études de biodisponibilité comparatives : Formes pharmaceutiques de médicaments à effets systémiques).
Le terme « bioéquivalence » laisse supposer qu'on peut s'attendre à ce que le produit à l'essai ait les mêmes effets thérapeutiques et le même profil d'innocuité que le produit de référence lorsqu'il est administré à des patients dans les conditions précisées sur l'étiquette.
S'il est impossible d'effectuer une étude de biodisponibilité à l'aide d'une méthode adéquate, on peut recourir à une autre approche, notamment une étude de pharmacodynamie. Dans certains cas, il pourrait être nécessaire d'établir l'équivalence au moyen d'essais cliniques comportant des critères de jugement thérapeutiques.
2 Conseils pour la mise en œuvre
L'acceptabilité des données obtenues au moyen des études de biodisponibilité comparatives sera évaluée conformément aux principes énoncés au titre 5 du Règlement et dans la Ligne directrice de l'ICH E6 : Les bonnes pratiques cliniques. Ces documents aideront les promoteurs à comprendre les exigences à remplir en vue des présentations à Santé Canada, conformément au Règlement, et ce, même si les études, ou une partie des études, sont réalisées dans d'autres pays.
2.1 Planification d'une étude de biodisponibilité comparative
Les objectifs de l'étude devraient être définis clairement dans le protocole.
Il faudrait indiquer quelles normes en matière de bioéquivalence seront appliquées et en donner les raisons. Il faudrait aussi fournir une justification scientifique de tout écart par rapport aux indications du présent document (par exemple la substance au moyen de laquelle la bioéquivalence sera évaluée, ou les écarts par rapport à un repas à teneur élevée en matières grasses et en calories dans les études menées chez des sujets non à jeun). Les promoteurs sont invités à consulter un représentant de Santé Canada avant l'étude si les écarts sont majeurs.
Parmi les sujets abordés dans le Règlement et dans la Ligne directrice de l'ICH E6 : Les bonnes pratiques cliniques, et par conséquent non repris en détail ici, figurent le comité d'examen de l'établissement, les chercheurs ainsi que les installations pour les études cliniques, les études bioanalytiques et les travaux de laboratoire.
2.2 Sélection des sujets d'une étude
En général, il faudrait choisir les sujets de l'étude de façon à réduire 1) le risque pour les sujets et 2) la variabilité intra- et inter-sujets non attribuable au médicament lui-même.
2.2.1 Choix des sujets
Afin que la variabilité soit réduite, les études de biodisponibilité comparatives sont généralement menées auprès de volontaires normaux en bonne santé (hommes et/ou femmes). Il est généralement admis que les conclusions sur la biodisponibilité comparative tirées d'études menées auprès de volontaires en bonne santé peuvent s'appliquer dans la population de patients. Il est plus difficile de réaliser des études de biodisponibilité comparatives de type croisé chez des patients, en partie à cause de la possibilité d'évolution de leur maladie. Dans certains cas, il pourrait s'avérer nécessaire d'effectuer les études chez des patients qui reçoivent déjà le médicament (par exemple, lorsque le profil d'innocuité du médicament empêche son administration à des volontaires en bonne santé). La variabilité des états pathologiques des patients de l'étude sera un élément important à considérer pour décider de la taille de la cohorte qui sera nécessaire pour satisfaire aux normes.
2.2.2 Critères d'inclusion/exclusion
Les attributs suivants devraient être pris en considération pour réduire les variations pharmacocinétiques non liées à des différences entre les produits et pour prévenir les préjudices indus aux sujets de l'étude.
- Âge
L'âge des sujets devrait être compris entre l'âge de la majorité légale et l'âge d'apparition des modifications des fonctions organiques associées au vieillissement. En règle générale, cette fourchette se situe entre 18 et 55 ans inclusivement. - Taille et poids
L'indice de masse corporelle des sujets devrait se situer entre 18,5 et 30 kg/m². - Santé
L'état de santé des volontaires devrait être établi par le médecin responsable à l'aide d'un examen médical comportant une revue des antécédents médicaux et des résultats du bilan hépatique, rénal et hématologique courant. Les résultats de laboratoire aberrants devraient être vérifiés de nouveau et un résumé devrait accompagner l'opinion du médecin concernant les répercussions possibles de ces résultats sur les conclusions de l'étude.
Un test de détection d'alcool et de médicaments dont les sujets pourraient abuser devrait être effectué à chaque période avant l'administration du médicament. - Sécurité
Les documents relatifs à l'étude devraient comprendre un électrocardiogramme (ECG) s'il est reconnu que le médicament peut causer des modifications de l'ECG.
Les sujets atteints d'une maladie grave affectant l'organisme entier ou présentant un état pathologique instable qui pourrait rendre difficile le respect du protocole devraient être exclus.
Les chercheurs devraient s'assurer que les femmes volontaires ne sont ni enceintes, ni allaitantes, ni susceptibles de tomber enceintes au cours de l'étude. L'absence de grossesse devrait être confirmée par un test de grossesse urinaire ou sérique à chaque période avant l'administration du médicament.
2.3 Plan de l'étude
2.3.1 Plan parallèle ou plan croisé
Le plan d’étude standard est le plan croisé à deux périodes, dans lequel le produit à l’essai et le produit de référence sont administrés à chacun des sujets. L’avantage du plan croisé est que l’erreur intra-sujet est utilisée dans la construction des intervalles de confiance pour comparer les différences de moyenne; l’erreur intra-sujet est toujours moindre que l’erreur inter-sujet utilisée dans le plan parallèle.
On peut aussi avoir recours à un plan croisé répété, dans lequel les formes pharmaceutiques sont mises à l’essai plus d’une fois chez les mêmes sujets. Le principal avantage de ce plan est que le nombre de sujets requis est plus petit, mais les sujets doivent être présents pendant plus de périodes.
Lorsqu’il y a plus de deux formes à l’essai ou que les formes sont étudiées dans des conditions différentes, un plan d’ordre supérieur (plus de périodes et de séquences) devrait être envisagé. Comme le terme « erreur intra-sujet » de ces plans a plus de degrés de liberté, les échantillons plus petits sont souvent adéquats. Le choix d’un plan équilibré pour l’analyse de la variance (plan de Williams) ou d’un plan en bloc incomplet séparé devra être justifié.
Un plan croisé exempt de période sans médicament entre les formes peut être utilisé pour les études menées auprès de patients pour lesquels l’interruption du traitement pendant une période de sevrage thérapeutique poserait un problème d’éthique. Au lieu d’avoir recours à une période de sevrage sans médicament, on administre le médicament à l’étude assez longtemps avant l’échantillonnage pour permettre l’élimination de la forme administrée auparavant.
Un plan parallèle pourrait aussi s’avérer utile lorsqu’on étudie un médicament dont la demi-vie d’élimination est très longue ou la forme retard. Le terme d’erreur utilisé est la variance inter-sujet.
2.3.1.1 Nombre de sujets
Le nombre de sujets à recruter dans une étude de biodisponibilité comparative devrait être estimé en tenant compte des objectifs de l'étude, du plan d'étude, des produits pharmaceutiques à comparer et des conditions dans lesquelles sera réalisée l'étude. Le médicament et le produit pharmaceutique déterminent la norme particulière à respecter. Une revue de littérature exhaustive devrait être réalisée pour comprendre le médicament et le produit pharmaceutique. Le nombre de sujets à recruter est déterminé par la norme, la différence moyenne prévue entre la forme à l'essai et la forme de référence et la variance intra-sujet prévue de tous les paramètres énumérés dans la norme, ainsi que la puissance. Tous les calculs doivent viser le maintien d'une erreur de type I globale de 5 %. Le nombre minimum de sujets est de 12, mais un plus grand nombre de sujets est généralement requis.
2.3.2 Autres plans d'étude possibles
Lorsque les estimations proposées de la variance intra-sujet dans la littérature souffrent d'une grande incertitude, il est possible de recueillir les données par stades en s'appuyant sur la variance intra-sujet observée au premier stade. Les deux stratégies de collecte des données par stades sont le plan séquentiel par groupes et le plan adaptatif. Dans ces deux types de plans, l'erreur de type I globale devrait être maintenue à 5 % et l'algorithme devrait être défini à priori dans le protocole. Ces stratégies peuvent être utilisées avec un plan croisé ou un plan parallèle.
2.3.2.1 Plans séquentiels par groupes
La collecte de données dans un plan séquentiel par groupes repose sur des échantillons de taille fixe (Ni) à chaque stade i. Il est recommandé de n'utiliser que deux stades étant donné que ces essais sont très petits comparativement aux études d'issues cliniques. La taille de l'échantillon N1 du premier stade est généralement déterminée par l'estimation la plus probable de la variance intrasujet à laquelle on ajoute quelques sujets pour contrer les abandons. Le nombre de sujets supplémentaires requis pour l'échantillon N2 du deuxième stade est habituellement calculé en fonction du pire scénario à l'aide d'une estimation plus grande de la variance intra-sujet, de sorte que N1 plus N2 égale la taille estimée de l'échantillon pour la plus grande variance intra-sujet. Habituellement, la stratégie avec ce plan consiste à accepter la bioéquivalence au premier stade et à ne passer au deuxième stade que lorsque la variance intra-sujet au premier stade est très grande. Il est recommandé de toujours utiliser la même valeur alpha aux deux stades, conformément à la méthode établie par Pocock (Pocock SJ. Group sequential methods in the design and analysis of clinical trials. Biometrika 1977; 64(2): 191-9), qui donne une valeur alpha de 0,0294 dans ce cas. Avec cette méthode, un effet de stade n'est pas nécessaire dans le modèle.
2.3.2.2 Plans adaptatifs
Lorsqu'on ne dispose que de très peu de données sur la variance intra-sujet, on peut faire appel à une approche similaire au plan séquentiel , soit le plan adaptatif, dans lequel la taille de l'échantillon du deuxième stade est basée sur la variance intra-sujet estimée au premier stade. Il est recommandé d'utiliser la méthode C décrite par Potvin et al. (Potvin D et al. Sequential design approaches for bioequivalence studies with crossover designs. Pharmaceut. Statist. 2008; 7: 245-62).
2.3.3 Études pharmacodynamiques
Dans les cas où des effets pharmacocinétiques ne pourraient pas être mesurés avec fiabilité, il peut être acceptable d’établir une bioéquivalence ou une comparabilité in vivo en menant des études pharmacodynamiques. Le recours à une étude pharmacodynamique et le plan de l’étude devraient être justifiés.
Le plan d’une étude pharmacodynamique devrait prendre en considération la pathologie sous-jacente et les antécédents naturels de l’état traité. Si les conditions de base ne sont pas reproductibles, il peut être nécessaire de recourir à un plan parallèle plutôt qu’à un plan croisé, ce dernier étant habituellement préféré. Les sujets ne répondant pas au traitement devraient être exclus de l’étude au moyen d’un dépistage préalable reposant sur les critères d’identification qui sont énoncés dans le protocole. Tout effet placebo important doit également être pris en considération, car les comparaisons entre des produits pharmaceutiques ne peuvent être effectuées que lorsqu’on a pris en considération a priori cet effet dans le plan de l’étude. Une phase croisée avec placebo pourrait être nécessaire à l’évaluation de l’effet placebo.
Les doses utilisées durant les études pharmacodynamiques doivent être choisies de manière à produire un éventail de valeurs de la réponse qui appuie une caractérisation minutieuse de la réponse au fil du temps, après l’administration du traitement, ainsi que l’évaluation des différences entre le produit à l’essai et le produit de référence. Ni le produit à l’essai ni le produit de référence ne devrait entraîner une réponse maximale au cours de l’étude, car il pourrait être impossible de distinguer des différences entre des formes pharmaceutiques administrées à des doses donnant un effet maximal ou quasi maximal. L’évaluation de la relation dose-réponse pourrait devoir faire partie du plan d’étude.
L’effet pharmacodynamique mesuré doit être un effet pharmacologique ou thérapeutique qui est pertinent au regard des allégations d’efficacité du produit. La réponse doit être mesurable au moyen de méthodes quantitatives qui ont fait l’objet d’une validation de la précision, de l’exactitude, de la reproductibilité et de la spécificité. Des mesures répétées de la réponse au fil du temps doivent être effectuées dans des conditions de double insu et produites ou enregistrées grâce à un instrument de manière que l’on puisse fournir un relevé des événements pharmacodynamiques qui soient des substituts des concentrations plasmatiques. Lorsqu’il est impossible d’effectuer de telles mesures, on pourrait recourir à des enregistrements à des échelles analogiques visuelles. Dans d’autres cas où les données se limitent à des mesures qualitatives (catégories), il faudra avoir recours à des méthodes spéciales d’analyse statistique.
2.3.4 Prise en compte des abandons et des retraits
Un nombre fixe de sujets, en plus du nombre estimé lors du calcul de la taille de l’échantillon, devrait être recruté dans l’étude. Cette stratégie permet de contrer les éventuels abandons. Tous les sujets qui fournissent des données évaluables concernant le produit à l’essai et le produit de référence dans une étude croisée, ou concernant un traitement dans une étude à groupes parallèles, devraient être inclus dans l’analyse statistique.
Il faudrait indiquer la raison du retrait des sujets qui ont reçu au moins une dose du médicament (par exemple réaction indésirable au médicament) et fournir les concentrations du médicament dans le plasma (ou dans le sérum ou le sang total) du sujet. Les résultats de bioanalyse de tous les échantillons des sujets qui ont été retirés de l’étude devraient être déclarés. Si un sujet se retire de l’étude pour des motifs personnels ou en est exclu en raison d’un non-respect du protocole (par exemple, dépistage positif de drogue) avant la fin d’au moins deux périodes de l’étude, il n’est pas nécessaire d’analyser les échantillons de sang de ce sujet.
Les sujets qui vomissent devraient subir une évaluation visant à déterminer, en fonction de l’impact possible des vomissements sur l’intégrité des résultats de l’étude, s’il est souhaitable qu’ils continuent de participer à l’étude. L’évaluation devrait avoir lieu le plus tôt possible après le ou les épisodes de vomissements et avant l’analyse des échantillons prélevés pendant l’étude.
Les courbes de concentration en fonction du temps des sujets dont les concentrations pré-dose dépassent de 5 % la Cmax correspondante devraient être exclues de l’analyse statistique, pourvu que la période de sevrage thérapeutique du médicament entre les doses soit adéquate. Les courbes de concentration en fonction du temps des sujets dont les concentrations pré-dose sont égales ou inférieures de moins de 5 % à la Cmax correspondante devraient être incluses dans l’analyse statistique sans correction.
2.3.5 Prise en compte des valeurs aberrantes
Les études de biodisponibilité comparatives sont de petites études comparativement à d’autres essais cliniques. Une ou deux valeurs extrêmes pourraient avoir un effet majeur sur les conclusions qui seront tirées de ces petites études. Les suppositions et estimations paramétriques usuelles ne sont pas robustes en présence de valeurs extrêmes.
Des procédures spécifiques devraient comprendre une stratégie pour identifier et prendre en compte les valeurs aberrantes. La proportion de sujets dont les valeurs sont aberrantes ne devrait pas dépasser 5 %, à moins que l’étude ne compte que 20 sujets ou moins, auquel cas un seul sujet peut être retiré. N’importe quel protocole concernant le traitement réservé aux valeurs aberrantes devrait être appliqué avant que les résultats de l’analyse ne soient traduits en intervalles de confiance (c’est-à-dire que le protocole sur les valeurs aberrantes doit être suivi que les résultats soient conformes ou non à la norme).
Le protocole concernant le traitement à réserver aux valeurs aberrantes devrait comprendre les éléments suivants :
- la valeur devrait être identifiée par un test de recherche des valeurs aberrantes. Il est recommandé d’utiliser un test simple, par exemple un résidu studentisé supérieur à 3; la valeur devrait se situer à l’extérieur de l’intervalle de toutes les autres valeurs observées, et ce, quelle que soit la forme pharmaceutique. Autrement dit, le test ne devrait permettre d’identifier que les valeurs très différentes des autres valeurs obtenues;
- Le sujet en question devrait être identifié comme une source de valeurs aberrantes pour tous les paramètres (que ce soit pour le produit à l’essai ou pour le produit de référence) sur lesquels repose la décision relative à la bioéquivalence ou à la biodisponibilité comparative. Les paramètres pertinents sont habituellement la SSC et la Cmax, mais d’autres paramètres sont requis dans certains cas.
- De nouveaux tests chez les sujets reconnus comme une source de valeurs aberrantes ne sont pas recommandés.
2.4 Conduite de l'étude
2.4.1 Normalisation
Il faut s'efforcer de normaliser les conditions de l'étude à chacun de ses stades; par exemple, l'administration du médicament à l'étude devrait se faire approximativement à la même heure chaque jour de l'étude. La posture des sujets, l'exercice, l'alimentation, l'usage du tabac et la consommation d'alcool devraient aussi être normalisés. Il est préférable de recruter des non-fumeurs; si des fumeurs sont recrutés, ils devraient être identifiés.
Les volontaires ne devraient prendre aucun autre médicament, y compris les médicaments en vente libre, les produits de santé naturels, les boissons alcoolisées et les aliments qui ont un effet sur les cytochromes P450 ou le pompe d'efflux Pgp (par exemple, jus de pamplemousse et millepertuis, respectivement). Ces restrictions devraient être appliquées dans un intervalle convenable avant et pendant l'étude. Les violations de protocole liées à la consommation d'aliments interdits ou à l'utilisation de produits de santé devraient être signalées (dose et moment de l'administration). La décision d'accepter ou de rejeter les résultats d'un sujet ayant violé le protocole établi devrait être prise avant le début de l'analyse statistique.
2.4.2 Étude en insu
Pour qu'un biais soit évité, les études de biodisponibilité comparatives devraient être menées de façon que les sujets ignorent quel produit (produit à l'essai ou produit de référence) ils reçoivent. De plus, les personnes qui vérifient la présence de réactions indésirables et celles qui effectuent la bioanalyse des échantillons ne devraient pas connaître la séquence de traitement.
2.4.3 Administration d'aliments et de liquides
Si l'administration d'une dose unique du médicament ou du produit pharmaceutique en l'absence ou en présence de nourriture comporte un risque sérieux établi pour la sécurité des sujets, une étude adéquatement conçue dans les conditions d'utilisation indiquées (à jeun ou non à jeun) pourrait être acceptable pour l'évaluation de la biodisponibilité comparative. Le promoteur devrait justifier scientifiquement et à priori une telle étude. Si des études à l'état d'équilibre sont menées, les conditions et restrictions mentionnées ci-après concernant les aliments et les liquides devraient s'appliquer le jour où les courbes plasmatiques sont obtenues ainsi que le soir précédent.
2.4.3.1 Étude chez des sujets à jeun
La prise d'aliments et de liquides devrait être surveillée étroitement. En général, les sujets devraient jeûner pendant 8 heures avant l'administration du médicament. Pour être à jeun, un sujet ne doit pas consommer d'aliments ni de solides, mais les liquides clairs ne renfermant ni alcool, ni xanthine, ni flavonoïdes sont permis la nuit précédant l'étude. L'eau peut être permise jusqu'à une heure avant l'administration du médicament. La dose devrait être prise avec de l'eau à un volume (150 à 250 mL) et à une température normalisée. Une heure après l'administration du médicament, les liquides sans xanthine ni flavonoïdes sont permis. Quatre heures après l'administration du médicament, les sujets peuvent prendre un repas normalisé. Tous les repas devraient être normalisés, et les mêmes repas devraient être servis chaque jour où le médicament est administré.
Lorsqu'on compare la performance de deux formes pharmaceutiques orales qui se désintègrent et sont conçues pour être prises sans eau, l'étude de biodisponibilité comparative devrait être conçue de façon que les formes soient soumises aux conditions les plus discriminantes qui soient. Avec de telles formes posologiques, il ne faudrait pas administrer d'eau pendant la période allant d'une heure avant à une heure après l'administration de la dose.
Dans le cas des formes pharmaceutiques orales solides dont l'étiquette prévoit d'autres modes d'administration (par exemple, saupoudrée sur un aliment mou ou dispersée dans l'eau), il est recommandé aux promoteurs de communiquer avec un représentant de Santé Canada avant le début de l'étude pour vérifier le mode d'administration du médicament le plus indiqué pour la bioétude. Il pourrait être nécessaire de fournir des données additionnelles pour démontrer que l'autre mode d'administration choisi est convenable. Par exemple, il faudrait fournir des données pour démontrer que la technologie utilisée dans la forme est robuste et que les propriétés de libération contrôlée, le cas échéant, ne sont pas altérées pendant la période proposée par suite d'une exposition aux aliments ou liquides mentionnés sur l'étiquette. Les produits devraient demeurer stables pendant la durée de l'exposition. De plus, si le produit est utilisé conjointement avec un dispositif d'administration, des tests sur le dispositif en question (par exemple, diverses seringues et sondes nasogastriques) pourraient aussi être requis pour évaluer des facteurs tels que le dépôt ou l'agrégation de granules du médicament, l'obstruction du dispositif ou la présence de résidus de médicament dans le dispositif.
2.4.3.2 Étude chez des sujets non à jeun
Le repas utilisé au cours d'une étude de biodisponibilité comparative chez des sujets non à jeun devrait perturber le plus possible la biodisponibilité générale du médicament. Il s'agit en général d'un repas hyperlipidique et hypercalorique, et on devrait employer de façon implicite ce type de repas dans les études de biodisponibilité comparatives chez des sujets non à jeun.
Dans un repas hyperlipidique (environ 50 % des calories totales sous forme de lipides) et hypercalorique (environ 800 à 1 000 kilocalories), les protéines, les glucides et les lipides devraient fournir respectivement 150, 250 et 500 à 600 kilocalories. Voici un exemple de repas (déjeuner) hyperlipidique et hypercalorique : 2 oeufs frits dans le beurre, 2 tranches de bacon, 2 tranches de pain grillé avec du beurre, 120 g de pommes de terre rissolées et 240 mL de lait entier.
Le recours à un repas qui n'est pas hyperlipidique et hypercalorique ne devrait se faire que dans des circonstances exceptionnelles et devrait être justifié scientifiquement et à priori par le promoteur. Un risque sérieux établi pour la sécurité des sujets à la suite de l'administration d'une dose unique du médicament ou du produit pharmaceutique en présence d'un repas hyperlipidique et hypercalorique pourrait justifier l'utilisation d'un autre type de repas.
Le repas devrait être pris dans les 30 minutes précédant l'administration du produit pharmaceutique.
2.4.4 Posture et activité physique
En ce qui concerne la plupart des médicaments, les sujets ne devraient pas pouvoir s'allonger pendant au moins deux heures après l'administration du médicament. L'activité physique et la posture devraient être normalisées le plus possible afin d'en limiter les effets sur le débit sanguin et la motilité gastro-intestinaux. Le même modèle de posture et d'activité physique devrait être appliqué à chaque période d'étude.
2.4.5 Intervalle entre les doses
L'intervalle entre les journées d'étude devrait être suffisamment long pour permettre à l'organisme d'éliminer pratiquement toute la dose administrée antérieurement. L'intervalle minimal entre les traitements devrait être le même pour tous les sujets et, en tenant compte de la variabilité interindividuelle de la vitesse d'élimination, il ne devrait généralement pas être inférieur à dix fois la demi-vie terminale moyenne du médicament.
En règle générale, l'intervalle entre les journées d'étude ne devrait pas dépasser trois ou quatre semaines.
2.4.6 Temps d'échantillonnage
La durée des prélèvements dans une étude devrait être suffisante pour correspondre à au moins 80 % de la SSC connue extrapolée jusqu’à l’infini (SSCI). Cette durée représente en général au moins trois fois la demi-vie terminale du médicament.
Il faudrait prélever au moins 12 échantillons par sujet par dose afin de pouvoir calculer les paramètres pharmacocinétiques pertinents. La variabilité inter-sujets de même que des facteurs tels que la possibilité de comportement erratique de certaines formes pharmaceutiques dans des conditions particulières (par exemple, des aliments peuvent influer sur la libération d’une forme pharmaceutique gastro-résistante) devraient être pris en considération au moment de déterminer le nombre total d’échantillons à prélever et le plan d’échantillonnage. L’heure exacte de prélèvement des échantillons devrait être consignée, et les prélèvements devraient être espacés de façon que les renseignements suivants puissent être estimés avec exactitude :
- la Cmax;
- la surface sous la courbe de concentration en fonction du temps au moment de la dernière concentration quantifiable (SSCT) équivalant à au moins 80 % de la SSCI;
- la constante de vitesse terminale d'élimination du médicament (λ).
Les estimations de la constante de vitesse terminale d’élimination par régression linéaire au moyen de quelques points seulement peuvent présenter des inexactitudes importantes. Pour réduire ces inexactitudes, il est préférable de déterminer au moins trois points pendant la phase terminale log-linéaire de la courbe.
2.4.7 Prélèvement des échantillons
Le sang devrait être le liquide biologique échantillonné pour le dosage du médicament. Dans la plupart des cas, les dosages peuvent être effectués dans le plasma; cependant, dans certains cas, il peut être préférable d’effectuer la bioanalyse sur du sang entier ou du sérum.
Il arrive parfois que la concentration du médicament dans un liquide autre que le sang puisse être mieux corrélée avec l’effet. Néanmoins, le médicament doit d’abord être absorbé avant d’être distribué aux autres liquides comme le liquide céphalorachidien ou les sécrétions bronchiques. Donc, pour l’estimation de la biodisponibilité, des échantillons de sang doivent quand même être prélevés et analysés.
2.4.8 Traitement des échantillons
Les échantillons devraient être prélevés, traités et conservés dans des conditions qui ne causent pas de dégradation ou d'interconversion importante des substances à analyser.
2.4.9 Détermination des incidents thérapeutiques
L'article C.05.001 du Règlement définit l'incident thérapeutique comme un « événement indésirable affectant la santé d'un sujet d'essai clinique à qui une drogue a été administrée qui peut ou non être causé par l'administration de la drogue, y compris toute réaction indésirable à une drogue ». Par conséquent, tous les signes défavorables ou inattendus (y compris des résultats de laboratoire anormaux, par exemple), symptômes ou maladies temporellement associés à l'usage d'un médicament doivent être déclarés, qu'ils soient considérés comme étant liés au médicament ou non. (Voir aussi la ligne directrice intitulée Gestion des données cliniques sur l'innocuité des médicaments : Définitions et normes relatives à la déclaration rapide - ICH thème E2A).
Dans certains cas, les incidents thérapeutiques sont attribuables à des facteurs autres que l'ingrédient actif contenu dans la forme pharmaceutique. La vitesse d'absorption et les excipients contenus dans la forme pharmaceutique peuvent influer sur la fréquence des incidents thérapeutiques, leur moment d'apparition et leur gravité. La fréquence, la gravité et la durée de tous les incidents thérapeutiques observés durant l'étude devraient être signalées. Il incombe au chercheur de juger de la probabilité qu'un incident thérapeutique soit induit par le médicament.
Il convient de faire appel au même observateur et d'utiliser le même format pour le produit à l'essai et le produit de référence afin d'obtenir et de consigner les renseignements sur les incidents thérapeutiques. L'observateur, tenu dans l'ignorance du produit reçu par les sujets, devrait interroger ces derniers sur les incidents thérapeutiques au cours de chaque période d'échantillonnage. Dans le cas des médicaments qui provoquent des incidents thérapeutiques connus (par exemple, goût de métal, hypotension orthostatique, dysrythmie cardiaque), des questions explicites au sujet de ces incidents devraient être posées. En posant les questions, l'intervieweur ne devrait pas amener le sujet à croire que les événements sont prévus ou imprévus. De plus, l'entrevue devrait être effectuée en privé. Des observations (par exemple, mesure de la pression artérielle et électrocardiogramme) devraient être faites et notées au moment où l'on sait que les incidents surviennent par rapport au moment d'administration.
2.5 Produit à l'essai et produit de référence
Les caractéristiques requises du produit à l'essai et du produit de référence qui devraient être indiquées comprennent la qualité, la posologie, la teneur, les numéros de lot et l'identité du produit de référence utilisé dans l'étude.
2.5.1 Propriétés chimiques
Le produit à l'essai et le produit de référence devraient être conformes à une norme de l'annexe B ou à une autre norme applicable acceptable pour Santé Canada. Il convient de consulter les lignes directrices en matière de propriétés chimiques et de fabrication des présentations précliniques et des présentations de drogues nouvelles pour l'interprétation des exigences techniques générales énumérées aux paragraphes C.08.005(1) et C.08.002(2), respectivement.
2.5.2 Posologie et teneur
Dans les études de biodisponibilité comparatives, il conviendrait d'utiliser la même dose de chaque produit, de préférence en une seule forme unitaire. Les lots employés pour les études de biodisponibilité comparatives devraient être représentatifs des lots de production commerciale proposés. Ils devraient provenir d'un lot équivalant à au moins 10 % de la taille d'un lot commercial, ou de 100 000 unités (le plus grand des deux), à moins qu'une justification ne soit fournie. Les lots devraient être produits à l'aide du même type d'équipement et des mêmes procédures et, dans le cas des formes à libération modifiée, au même endroit que celui proposé pour la production commerciale. La validité du biolot utilisé dans l'étude de biodisponibilité comparative pourrait être compromise s'il s'avère que le développement du produit s'est fait de façon inadéquate ou que le procédé de fabrication est inadéquat et qu'il pourrait en résulter un produit non uniforme ou de piètre qualité. Une étude de biodisponibilité comparative réalisée à l'aide d'un tel biolot ne serait pas jugée adéquate pour étayer l'innocuité et l'efficacité du produit commercial proposé.
En ce qui concerne les produits dont la proportion des excipients et les caractéristiques de dissolution sont similaires, des études de biodisponibilité comparatives pourraient ne pas être nécessaires pour toutes les teneurs. Le degré de différence entre les teneurs des formes pharmaceutiques et les résultats des études de dissolution comparatives détermineront si toutes les teneurs doivent faire l'objet d'essais. Des renseignements à ce sujet peuvent être trouvés dans la politique de la Direction des produits thérapeutiques intitulée Bioéquivalence des formulations proportionnelles - formes pharmaceutiques orales solides.
Lorsque, dans le cas d'un produit à libération modifiée sous forme de comprimé sécable, il est allégué qu'une partie du comprimé peut être administrée pour fournir une dose proportionnelle, des données devraient être présentées pour étayer cette allégation. Les comprimés séqués obtenus par fractionnement d'un comprimé sécable sont considérés comme des formes indépendantes, et il faudrait établir l'uniformité de la teneur des comprimés séqués de tous types, quel que soit le mode de libération du médicament. Dans le cas des comprimés à libération modifiée, les données fournies devraient comprendre de l'information sur la conception et la mise au point du produit indiquant que le fractionnement du comprimé n'aura pas d'effet sur son innocuité et sa performance et que l'utilisation de données sur la libération du médicament in vitro (dissolution) est justifiée. Veuillez consulter la Ligne directrice : Qualité (chimie et fabrication) : Présentations de drogue nouvelle (PDN) et présentations abrégées de drogue nouvelle (PADN) pour les exigences en matière de qualité quant à l’acceptabilité du comprimé sécable.
2.5.3 Choix du produit de référence
Dans le cas d'une nouvelle substance médicamenteuse (mise en marché initiale), le produit de référence devrait être la forme pharmaceutique utilisée dans les essais cliniques pivots.
Dans le cas des autres études de bioéquivalence pivots, le produit de référence devrait être le produit de référence canadien défini à l'article C.08.001.1 du Règlement :
- une drogue à l'égard de laquelle un avis de conformité a été délivré en application de l'article C.08.004 [du Règlement] et qui est commercialisée au Canada par son innovateur;
- une drogue jugée acceptable par le ministre qui peut être utilisée pour la détermination de la bioéquivalence d'après les caractéristiques pharmaceutiques et, le cas échéant, les caractéristiques en matière de biodisponibilité, lorsqu'une drogue pour laquelle un avis de conformité a été délivré en application de l'article C.08.004 [du Règlement] ne peut être utilisée à cette fin parce qu'elle n'est plus commercialisée au Canada;
- une drogue jugée acceptable par le ministre qui peut être utilisée pour la détermination de la bioéquivalence d'après les caractéristiques pharmaceutiques et, le cas échéant, les caractéristiques en matière de biodisponibilité, par comparaison à une drogue visée à l'alinéa a).
Le lecteur trouvera plus de renseignements à ce sujet dans la Ligne directrice de la Direction des produits thérapeutiques intitulée Utilisation d’un produit référence étranger comme produit de référence canadien.
2.6 Méthodes bioanalytiques
Pour déterminer la biodisponibilité comparative, il faut disposer de méthodes bioanalytiques bien caractérisées et validées qui permettent d'obtenir des estimations fiables de la concentration des substances à analyser.
Le laboratoire bioanalytique devrait tenir à jour une série complète de procédures opératoires normalisées qui couvre tous les aspects de la validation des méthodes et de l'analyse des échantillons des sujets. De plus, les données sur la validation des méthodes et les analyses des échantillons des sujets devraient être enregistrées et conservées.
Les principes et procédures de validation des méthodes bioanalytiques et d'analyse des échantillons d'étude décrits dans le document de l'Agence européenne des médicaments intitulé Guideline on bioanalytical method validation ICH M10 Validation des Méthodes Bioanalytiques et Analyse des Échantillons d'Étude devraient être suivis.
2.6.1 Bioanalyse des échantillons des sujets
La détermination de la biodisponibilité comparative devrait être fondée sur les données concernant le médicament mère.
Le promoteur ne peut pas se soustraire au dosage du médicament mère, sauf si la pharmacocinétique du médicament formulé ne peut pas être estimée de façon fiable, par exemple si le médicament mère n’est pas détectable en raison d’une biotransformation rapide. En de telles circonstances, l’utilisation des données sur un métabolite peut être acceptable. Le métabolite mesuré devrait être un métabolite primaire (première étape) et majeur. De plus, le promoteur devrait fournir une justification scientifique appropriée pour étayer sa décision de ne pas mesurer le médicament mère et d’utiliser les données sur un métabolite. Le recours au dosage d’un métabolite plutôt que du médicament mère doit être clairement énoncé, a priori, dans l’objectif de l’étude figurant dans le protocole d’étude.
Aux fins de la présente ligne directrice, un promédicament doit être traité comme un « médicament mère ». Autrement dit, si la substance libérée par la forme pharmaceutique pénètre intacte dans la circulation générale et qu’elle peut faire l’objet de mesures fiables dans le sang, alors cette substance devrait être utilisée pour l’évaluation de la biodisponibilité comparative.
En règle générale, on considère qu’il n’est pas nécessaire de mesurer à la fois les concentrations du médicament mère et celles d’un métabolite pour évaluer la biodisponibilité comparative. Toutefois, le dosage des métabolites peut quelquefois s’avérer utile, par exemple pour expliquer des valeurs extrêmes causées par des changements métaboliques chez un sujet.
En ce qui concerne les médicaments chiraux, rares seraient les cas où l’élimination in vivo de chaque énantiomère serait pertinente pour l’évaluation de la biodisponibilité comparative de deux formes pharmaceutiques orales solides d’un type similaire contenant un ratio défini d’énantiomères. Pour plus de renseignements à ce sujet, veuillez consulter la Ligne directrice à l’intention de l’industrie : Développement des médicaments chiraux, questions reliées à la stéréo-isomérie publiée par la Direction des produits thérapeutiques.
2.6.2 Validation des méthodes d'essai
Les méthodes bioanalytiques employées pour doser le médicament, ou le métabolite, dans le plasma, le sang total ou le sérum devraient convenir aux fins prévues. Elles devraient être reproductibles, sélectives et suffisamment sensibles, précises et exactes. Lorsque le laboratoire d’analyse aura déterminé que ces attributs sont présents, les chercheurs pourront entreprendre l’étude de biodisponibilité.
2.7 Analyse des données
La bioanalyse de tous les échantillons devrait être terminée avant le début des analyses pharmacocinétiques et des analyses statistiques.
2.7.1 Présentation des données
Les concentrations du médicament dans le plasma de chaque sujet, le temps d'échantillonnage et la forme pharmaceutique devraient être présentés sous forme de tableaux. Il faudrait fournir les concentrations mesurées, non rajustées.
Tout écart par rapport au protocole (par exemple, échantillons manquants ou prélevés tardivement) devrait être indiqué clairement dans ces tableaux.
Il faudrait établir deux graphiques pour chacun des sujets et deux graphiques pour les valeurs moyennes de tous les sujets : un graphique sur échelle linéaire et l'autre sur échelle semi-logarithmique. Ces graphiques devraient présenter les concentrations en médicament de la forme de référence et de la forme à l'essai en fonction du temps d'échantillonnage. En règle générale, les graphiques semi-logarithmiques individuels devraient présenter les lignes de régression qui ont été utilisées pour estimer la constante de vitesse terminale d'élimination (λ) des deux formes pharmaceutiques. Dans le cas des médicaments ayant une longue demi-vie pour lesquels on mesure la SSC0-72h, il ne sera peut-être pas nécessaire d'estimer la λ, la demi-vie terminale d'élimination (t½) et la SSCI ni de présenter les lignes de régression.
2.7.2 Paramètres pharmacocinétiques
Les estimations des paramètres pharmacocinétiques suivants devraient être présentées sous forme de tableaux pour chacune des combinaisons sujet-forme pharmaceutique :
- SSCT
- SSCI
- SSCT/SSCI
- Cmax
- tmax
- λ
- t½
Lorsque le moment de la manifestation de l’effet est important, le paramètre suivant devrait aussi être signalé :
h) Surface sous la courbe à la tmax du produit de référence calculée pour chaque sujet de l’étude (SSCRéftmax).
Lorsque les études portent sur des doses multiples, les paramètres suivants devraient aussi être signalés :
i) Concentration minimale observée (Cmin)
j) Concentration pré-dose déterminée immédiatement avant une dose à l’état d’équilibre (Cpd)
k) Surface sous la courbe de concentration en fonction du temps mesurée pendant l’intervalle posologique (SSCtau)
Lorsqu’on a démontré qu’un profil multiple de la concentration plasmatique d’un produit à libération modifiée faisait partie intégrante de l’effet thérapeutique, il convient de consigner les paramètres suivants :
l) La surface qui se trouve sous la courbe de la concentration en fonction du temps suivant un intervalle de temps restreint après l’administration du médicament (SSC partielle).
D’autres paramètres pharmacocinétiques peuvent aussi être présentés, mais les méthodes d’estimation utilisées pour les calculer devraient être entièrement décrites. Les moyennes et les coefficients de variation devraient être donnés pour chacun des paramètres et chacune des formes pharmaceutiques.
2.7.3 Collecte des données
Si on a recours à un plan séquentiel ou adaptatif, une description de la façon dont les changements ont été apportés à la collecte de données devrait être fournie.
2.7.4 Analyse statistique
2.7.4.1 Analyse des valeurs aberrantes
Les valeurs extrêmes devraient être identifiées. Les résultats du test de recherche des valeurs aberrantes proposé dans le protocole d'étude devraient être indiqués pour chaque paramètre. Par exemple, si on utilise le résidu studentisé, seules les valeurs supérieures à 3 pourront être considérées comme des valeurs extrêmes. Les valeurs minimales et maximales de chaque paramètre devraient aussi être identifiées. Seuls les sujets identifiés comme une source de valeurs aberrantes pour tous les paramètres pourront être retirés de l'étude.
2.7.4.2 Ajustement du modèle
Par définition, le plan croisé est un modèle à effets mixtes comportant des effets fixes et des effets aléatoires. Le plan croisé de base à deux périodes peut être analysé au moyen d'un simple modèle à effets fixes et de l'estimation des moyennes des moindres carrés. Des résultats identiques seront obtenus au moyen d'un outil d'analyse des effets mixtes tel que PROC MIXED de SAS®. Si un modèle à effets mixtes est utilisé, les contraintes imposées aux paramètres devraient être définies dans le protocole. Les modèles d'ordre supérieur, comme les plans d’étude croisés répliqués, devraient être analysés selon la démarche du modèle mixte si l’on veut estimer de façon adéquate les effets aléatoires. Dans de tel cas, on recommande d’utiliser le protocole Proc MIXED plutôt que le protocole Proc GLM lorsqu’on se sert du Système d’analyse statistique.
2.7.4.3 Analyse des effets fixes
Un résumé de l'analyse des effets liés à la séquence, à la période et à la forme pharmaceutique devrait être présenté. Les effets significatifs devraient être expliqués.
2.7.4.4 Estimation des effets aléatoires
Un résumé des estimations de la variance inter-sujets et intra-sujet devrait être fourni. Pour ce qui est des plans d’étude croisés répliqués , des estimations de la variance de l'interaction entre le sujet et la forme pharmaceutique ainsi que des estimations de la variance au sein des formes pharmaceutiques devraient être fournies.
2.7.4.5 Analyse des données
Les analyses devraient comprendre toutes les données évaluables de tous les sujets (voir la section 2.3.3, « Prise en compte des abandons et des retraits ») pour les données mesurées. Une analyse basée sur un moins grand nombre de données devrait être justifiée.
L'analyse devrait être réalisée sur les données de la SSCT et de la Cmax ayant subi une transformation logarithmique. L'analyse et les résultats pour chaque paramètre devraient être présentés sur une page distincte, comme l'explique en détail l'annexe 1, « Analyse type pour une étude de biodisponibilité comparative ». Les résultats devraient comprendre :
- les moyennes arithmétiques et les CV (inter-sujets) pour chaque produit;
- les tests et les estimations concernant les effets fixes et les effets aléatoires;
- le rapport des moyennes géométriques de la SSCT et de la Cmax du produit à l'essai et du produit de référence;
- l'intervalle de confiance adéquat au sujet du paramètre analysé.
Annexe 1 : Analyse type pour une étude de biodisponibilité comparative
Les tableaux et les figures qui suivent illustrent les données recueillies et utilisées dans une étude type de biodisponibilité. Une analyse des données est aussi présentée.
Bien qu'une étude de biodisponibilité comparative puisse inclure de nombreuses formes pharmaceutiques, l'analyse de base est la même : chaque forme pharmaceutique à l'essai est comparée à une forme de référence.
L'analyse de toute étude de biodisponibilité comparative devrait comporter les volets qui suivent :
- un schéma de randomisation pour le plan, dans lequel tous les sujets répartis au hasard dans l'étude sont inclus et identifiés par code, séquence et dates des périodes d'administration de la forme pharmaceutique à l'essai et de la forme de référence (voir la section A1.1);
- un sommaire (graphique et quantitatif) de la concentration du médicament à chaque moment d'échantillonnage et chez chaque sujet pour la forme pharmaceutique à l'essai et la forme de référence (voir la section A1.2);
- un sommaire de l'estimation des paramètres tel que définis à la section A1.3 pour la forme à l'essai et la forme de référence, incluant les moyennes, les écarts-types et les CV (voir la section A1.4);
- une analyse statistique formelle des paramètres pertinents avec comparaison entre la forme pharmaceutique à l'essai et la forme de référence (voir les sections A1.5 à A1.9);
Toutes les analyses statistiques types qui suivent comportent au minimum les deux formes minimales (forme à l'essai et forme de référence) administrées sur deux périodes ou jours.
A1.1 Schéma de randomisation du plan
Le tableau A1-A présente le schéma de randomisation du plan croisé utilisé dans l'étude. Tous les sujets ayant fait l'objet d'une randomisation dans l'étude devraient y être inclus, y compris ceux qui n'ont pas terminé l'étude (ces derniers devraient alors être identifiés comme tels). Les numéros attribués aux sujets et qui figurent sur les formulaires de consentement éclairé et sur les formulaires de présentation des données devraient être indiqués dans le tableau. De plus, il faudrait indiquer tout autre code d'identification des sujets qui a été utilisé. Il faudrait aussi donner la séquence dans laquelle le sujet a été randomisé. Enfin, toutes les périodes et dates d'administration du médicament devraient être présentées.
A1.2 Sommaire des concentrations des médicaments
Les tableaux A1.B et A1.C donnent les concentrations à chaque moment d'échantillonnage pour chacun des sujets, pour la forme pharmaceutique à l'essai et la forme de référence, respectivement. Toute concentration manquante doit être indiquée et motivée (par exemple échantillon perdu, échantillon non prélevé).
Bien qu'une analyse statistique formelle ne soit pas nécessaire pour chacun des moments d'échantillonnage, il est recommandé de présenter des statistiques sommaires pour chaque moment d'échantillonnage et chaque forme pharmaceutique. Il serait également utile d'indiquer dans le tableau la limite inférieure de quantification de la méthode bioanalytique utilisée.
| Sujet | Période | |||
|---|---|---|---|---|
| Numéro | ID | Séquence | 14 mai 2008 | 21 mai 2008 |
| 001 | A | ER | E | R |
| 002 | B | RE | R | E |
| 003 | C | RE | R | E |
| 004Note de bas de page * | D | ER | E | Aucune données |
| 005 | E | ER | E | R |
| 006 | F | RE | R | E |
| 007 | G | ER | E | R |
| 008 | H | RE | R | E |
| 009 | I | ER | E | R |
| 010Note de bas de page ** | J | RE | Aucune données | Aucune données |
| 011 | K | RE | R | E |
| 012 | L | ER | E | R |
| 013 | M | ER | E | R |
| 014 | N | RE | R | E |
| 015 | O | RE | R | E |
| 016 | P | ER | E | R |
| 017 | Q | RE | R | E |
| 018 | R | ER | E | R |
|
||||
|
ID
|
Séq
|
Période
|
Moments d'échantillonnage (heures)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
0,0
|
0,33
|
0,66
|
1,0
|
1,5
|
2,0
|
3,0
|
4,0
|
6,0
|
8,0
|
12,0
|
16,0
|
|||
| A | ER | 14 mai | 0,00 | SLQ |
52,01 | 95,03 | 122,20 | 77,88 | 65,15 | 46,24 | 19,20 | 14,99 | SLQ |
SLQ |
| B | RE | 21 mai | 0,00 | SLQ |
56,66 | 80,85 | 102,00 | 86,41 | 63,81 | 49,20 | 24,00 | 11,37 | 8,24 | SLQ |
| C | RE | 21 mai | 0,00 | 28,63 | 201,50 | 189,80 | 188,70 | 136,20 | 97,64 | 64,53 | 32,08 | 20,63 | 14,59 | SLQ |
| E | ER | 14 mai | 0,00 | SLQ |
9,04 | 34,32 | 47,70 | 52,79 | 59,47 | 32,61 | 17,61 | 8,76 | SLQ |
SLQ |
| F | RE | 21 mai | 0,00 | SLQ |
55,33 | 66,40 | 58,97 | 48,29 | 43,19 | 34,23 | 17,30 | 6,15 | SLQ |
SLQ |
| G | ER | 14 mai | 0,00 | SLQ |
33,15 | 45,64 | 54,19 | 34,13 | 32,78 | 21,55 | 10,75 | 8,35 | SLQ |
SLQ |
| H | RE | 21 mai | 0,00 | 35,38 | 79,14 | 100,90 | 70,71 | 48,43 | 30,73 | 26,19 | 8,65 | 6,83 | SLQ |
SLQ |
| I | ER | 14 mai | 0,00 | SLQ |
64,57 | 76,52 | 89,51 | 86,21 | 69,04 | 50,96 | 21,55 | 13,71 | 7,55 | SLQ |
| K | RE | 21 mai | 0,00 | SLQ |
79,34 | 99,41 | 154,80 | 58,60 | 57,12 | 32,57 | 19,82 | SLQ |
SLQ |
SLQ |
| L | ER | 14 mai | 0,00 | 14,78 | 55,54 | 56,88 | 46,87 | 37,29 | 28,75 | 25,20 | SLQ |
SLQ |
SLQ |
SLQ |
| M | ER | 14 mai | 0,00 | SLQ |
SLQ |
SLQ |
SLQ |
SLQ |
8,37 | 23,15 | 19,74 | 16,49 | 5,74 | 5,18 |
| N | RE | 21 mai | 0,00 | SLQ |
37,76 | 28,58 | 21,56 | 19,02 | 13,25 | 12,44 | 6,38 | SLQ |
SLQ |
SLQ |
| O | RE | 21 mai | 0,00 | SLQ |
27,85 | 43,30 | 43,30 | 32,57 | 29,59 | 25,42 | 16,89 | 7,68 | SLQ |
SLQ |
| P | ER | 14 mai | 0,00 | SLQ |
68,25 | 52,57 | 51,97 | 28,64 | 23,70 | 12,74 | SLQ |
SLQ |
SLQ |
SLQ |
| Q | RE | 21 mai | 0,00 | SLQ |
5,90 | 13,00 | 27,54 | 13,32 | 12,34 | 9,81 | 9,73 | SLQ |
SLQ |
SLQ |
| R | ER | 14 mai | 0,00 | SLQ |
18,92 | 35,77 | 53,93 | 60,43 | 47,44 | 41,72 | 16,66 | 8,87 | 5,49 | SLQ |
| MOY. | Aucune données | Aucune données | 0,00 | 4,92 | 52,81 | 63,69 | 70,87 | 51,26 | 42,62 | 31,80 | 15,04 | 7,73 | 2,60 | 0,32 |
| É.Aucune données T. | Aucune données | Aucune données | 0,00 | 11,26 | 47,05 | 45,04 | 49,76 | 33,66 | 24,64 | 15,42 | 8,60 | 6,57 | 4,42 | 1,29 |
| CV | Aucune données | Aucune données | Aucune données | 228,66 | 89,09 | 70,22 | 70,22 | 65,66 | 57,79 | 48,51 | 57,18 | 84,94 | 169,84 | 400 |
|
||||||||||||||
|
ID
|
Séq
|
Période
|
Moments d'échantillonnage (heures)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
0,0
|
0,33
|
0,66
|
1,0
|
1,5
|
2,0
|
3,0
|
4,0
|
6,0
|
8,0
|
12,0
|
16,0
|
|||
| A | ER | 14 mai | 0,00 | SLQ |
116,40 | 124,60 | 126,20 | 107,60 | 45,65 | 33,22 | 16,11 | 12,60 | SLQ |
SLQ |
| B | RE | 21 mai | 0,00 | SLQ |
88,45 | 121,40 | 206,90 | 179,00 | 84,53 | 40,02 | 38,01 | 15,12 | 5,39 | SLQ |
| C | RE | 14 mai | 0,00 | SLQ |
SLQ |
95,57 | 122,80 | 103,20 | 101,70 | 57,65 | 23,85 | 14,59 | 6,29 | SLQ |
| E | ER | 21 mai | 0,00 | SLQ |
3,23 | 37,26 | 35,90 | 28,87 | 28,48 | 25,10 | 24,91 | 6,72 | SLQ |
SLQ |
| F | RE | 14 mai | 0,00 | SLQ |
29,25 | 62,88 | 64,26 | 84,67 | 45,21 | 25,05 | 17,18 | 8,47 | SLQ |
SLQ |
| G | ER | 21 mai | 0,00 | SLQ |
6,89 | 50,04 | 55,27 | 51,68 | 38,58 | 26,19 | 7,79 | SLQ |
SLQ |
SLQ |
| H | RE | 14 mai | 0,00 | SLQ |
113,50 | 218,70 | 125,80 | 69,77 | 45,03 | 32,78 | 18,55 | 5,42 | SLQ |
SLQ |
| I | ER | 21 mai | 0,00 | SLQ |
181,90 | 135,80 | 96,51 | 90,50 | 62,58 | 30,43 | 18,50 | SLQ |
SLQ |
SLQ |
| K | RE | 14 mai | 0,00 | SLQ |
42,71 | 58,75 | 59,68 | 54,37 | 44,35 | 22,94 | 11,58 | 6,95 | SLQ |
SLQ |
| L | ER | 21 mai | 0,00 | SLQ |
14,29 | 21,32 | 24,32 | 25,56 | 25,51 | 10,49 | 5,49 | SLQ |
SLQ |
SLQ |
| M | ER | 21 mai | 0,00 | SLQ |
8,21 | 48,87 | 57,05 | 56,32 | 42,08 | 24,79 | 16,54 | 15,81 | 7,60 | SLQ |
| N | RE | 14 mai | 0,00 | SLQ |
47,20 | 34,90 | 34,90 | 24,19 | 20,11 | 8,08 | 7,27 | SLQ |
SLQ |
SLQ |
| O | RE | 14 mai | 0,00 | SLQ |
SLQ |
20,35 | 70,88 | 70,60 | 70,38 | 40,51 | 26,93 | 8,20 | SLQ |
SLQ |
| P | ER | 21 mai | 0,00 | SLQ |
39,23 | 86,29 | 97,46 | 52,26 | 40,53 | 26,74 | 12,54 | SLQ |
SLQ |
SLQ |
| Q | RE | 14 mai | 0,00 | SLQ |
SLQ |
30,86 | 88,38 | 37,67 | 29,28 | 14,99 | 6,38 | SLQ |
SLQ |
SLQ |
| R | ER | 21 mai | 0,00 | SLQ |
SLQ |
24,84 | 59,27 | 98,82 | 69,98 | 46,50 | 23,46 | 9,91 | 6,96 | SLQ |
| MOY. | Aucunes données | Aucunes données | 0,00 | Aucunes données | 45,33 | 73,28 | 82,85 | 70,94 | 49,62 | 29,09 | 17,19 | 6,49 | 1,64 | - |
| É.-T. | Aucunes données | Aucunes données | 0,00 | Aucunes données | 53,30 | 54,49 | 46,24 | 39,78 | 22,51 | 12,88 | 8,83 | 5,98 | 2,96 | - |
| CV | Aucunes données | Aucunes données | Aucunes données | Aucunes données | 117,59 | 74,37 | 55,82 | 56,08 | 45,37 | 44,28 | 51,38 | 92,23 | 180,73 | - |
|
||||||||||||||
A1.3 Liste des paramètres et définitions
Le tableau A1-D énumère les paramètres utilisés au cours de l'analyse et en donne la définition. Tout autre paramètre utilisé devrait également être défini clairement.
Tableau A1-D: Définitions des paramètres
|
Paramètres |
Définitions |
|---|---|
| Cmax | Concentration maximale observée. |
| tmax | Moment d'échantillonnage où la Cmax est atteinte. |
| SSCT | Surface sous la courbe au moment de la dernière concentration quantifiable, calculée au moyen de données observées à des moments précis. |
| SSCI | Surface sous la courbe à l'infini = SSCT + CT/λ, où CT est la concentration estimée au TDCQ. |
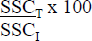 |
Pourcentage de la surface mesurée par la SSCT par rapport à la SSCI extrapolée. |
| SSCRéfTmax | Surface sous la courbe du produit à l'essai au tmax du produit de référence, calculée pour chaque sujet de l'étude. |
| SSC partielle | Surface sous la courbe du produit à l’essai au tmax du produit de référence, calculée pour chaque sujet de l’étude. Surface partielle qui se trouve sous la courbe suivant un intervalle de temps restreint après l’administration du médicament. |
| λ | Constante de vitesse terminale d'élimination calculée à partir des points dans le segment terminal log-linéaire de la courbe de concentration en fonction du temps. |
| TLIN | Point dans le temps où l'élimination log-linéaire commence. |
| TDCQ | Temps de la dernière concentration quantifiable. Moment où l'on observe la dernière concentration supérieure à la limite inférieure de quantification. |
| t½ | Demi-vie du médicament = ln(2)/λ = 0,693/λ. |
A1.4 Sommaire des paramètres estimés
Les tableaux A1-E et A1-F énumèrent, pour chaque sujet, les estimations des paramètres définis au tableau A1-D pour la forme à l'essai et la forme de référence, respectivement. Il faudrait présenter pour chaque forme pharmaceutique des statistiques sommaires (moyennes arithmétiques, écarts-types et CV, ou médianes et intervalles).
|
ID
|
Séq
|
Période
|
Forme à l'essai
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Cmax
(ng/mL) |
tmax
(h) |
SSC
T
(ng·h/mL) |
SSC
I
(ng·h/mL) |
SSC
T
(%) |
λ
(h Aucunes données 1) |
TLIN
(h) |
TDCQ
(h) |
t½
(h) |
|||
| A | ER | 14 mai | 122 | 1,50 | 365 | 409 | 89 | 0,3002 | 2,0 | 8,0 | 2,3 |
| B | RE | 21 mai | 102 | 1,50 | 405 | 432 | 94 | 0,2384 | 3,0 | 12,0 | 2,9 |
| C | RE | 21 mai | 202 | 0,66 | 703 | 774 | 91 | 0,1776 | 4,0 | 12,0 | 3,9 |
| E | ER | 14 mai | 59 | 3,00 | 233 | 256 | 91 | 0,3680 | 3,0 | 8,0 | 1,9 |
| F | RE | 21 mai | 66 | 1,00 | 247 | 265 | 93 | 0,3902 | 3,0 | 8,0 | 1,8 |
| G | ER | 14 mai | 54 | 1,50 | 178 | 205 | 87 | 0,2768 | 3,0 | 8,0 | 2,5 |
| H | RE | 21 mai | 101 | 1,00 | 246 | 263 | 94 | 0,3437 | 3,0 | 8,0 | 2,0 |
| I | ER | 14 mai | 90 | 1,50 | 408 | 433 | 94 | 0,2486 | 3,0 | 12,0 | 2,8 |
| K | RE | 21 mai | 155 | 1,50 | 315 | 372 | 85 | 0,3379 | 3,0 | 6,0 | 2,1 |
| L | ER | 14 mai | 57 | 1,00 | 140 | 331 | 42 | 0,1318 | 3,0 | 4,0 | 5,3 |
| M | ER | 14 mai | 23 | 4,00 | 165 | 195 | 85 | 0,1485 | 6,0 | 16,0 | 4,7 |
| N | RE | 21 mai | 38 | 0,66 | 88 | 113 | 78 | 0,2620 | 2,0 | 6,0 | 2,6 |
| O | RE | 21 mai | 43 | 1,00 | 183 | 215 | 85 | 0,2671 | 3,0 | 8,0 | 2,6 |
| P | ER | 14 mai | 68 | 0,66 | 122 | 148 | 83 | 0,5031 | 1,5 | 4,0 | 1,4 |
| Q | RE | 21 mai | 28 | 1,50 | 68 | 113 | 60 | 0,1833 | 1,5 | 6,0 | 3,8 |
| R | ER | 14 mai | 60 | 2,00 | 275 | 292 | 94 | 0,2546 | 3,0 | 12,0 | 2,7 |
| MOY. |
Aucunes données | Aucunes données | 79 | 1,50 | 259 | 301 | 84 | 0,2770 | 3,0 | 8,0 | 2,8 |
| É.Aucunes données T. | Aucunes données | Aucunes données | 48 | 0,89 | 158 | 164 | 14 | 0,0967 | 1,1 | 3,3 | 1,1 |
| CV | Aucunes données | Aucunes données | 61 | 59,35 | 61 | 54 | 17 | 34,92 | 37,3 | 38,5 | 37,9 |
|
|||||||||||
|
ID
|
Séq
|
Période
|
Forme de référence
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Cmax
(ng/mL) |
tmax
(h) |
SSC
T
(ng·h/mL) |
SSC
I
(ng·h/mL) |
SSC
T
(%) |
λ
(h -1) |
TLIN
(h) |
TDCQ
(h) |
t½
(h) |
|||
| A | ER | 21 mai | 126 | 1,50 | 375 | 418 | 90 | 0,2660 | 3,0 | 8,0 | 2,6 |
| B | RE | 14 mai | 207 | 1,50 | 595 | 613 | 97 | 0,2900 | 3,0 | 12,0 | 2,4 |
| C | RE | 14 mai | 123 | 1,50 | 471 | 492 | 96 | 0,2666 | 4,0 | 12,0 | 2,6 |
| E | ER | 21 mai | 37 | 1,00 | 190 | 224 | 85 | 0,2653 | 3,0 | 8,0 | 2,6 |
| F | RE | 14 mai | 85 | 2,00 | 257 | 285 | 90 | 0,3114 | 3,0 | 8,0 | 2,2 |
| G | ER | 21 mai | 55 | 1,50 | 175 | 190 | 92 | 0,5437 | 3,0 | 6,0 | 1,3 |
| H | RE | 14 mai | 219 | 1,00 | 382 | 398 | 96 | 0,4047 | 2,0 | 8,0 | 1,7 |
| I | ER | 21 mai | 182 | 0,66 | 361 | 406 | 89 | 0,3837 | 3,0 | 6,0 | 1,8 |
| K | RE | 14 mai | 60 | 1,50 | 218 | 236 | 83 | 0,3580 | 3,0 | 8,0 | 1,9 |
| L | ER | 21 mai | 26 | 2,00 | 92 | 105 | 88 | 0,4208 | 2,0 | 6,0 | 1,6 |
| M | ER | 21 mai | 57 | 1,50 | 269 | 327 | 82 | 0,1373 | 6,0 | 12,0 | 5,1 |
| N | RE | 14 mai | 47 | 0,66 | 106 | 125 | 85 | 0,3246 | 2,0 | 6,0 | 2,1 |
| O | RE | 14 mai | 71 | 1,50 | 290 | 313 | 93 | 0,4028 | 3,0 | 8,0 | 1,7 |
| P | ER | 21 mai | 97 | 1,50 | 230 | 266 | 87 | 0,3644 | 2,0 | 6,0 | 1,9 |
| Q | RE | 14 mai | 88 | 1,50 | 144 | 156 | 92 | 0,4964 | 3,0 | 6,0 | 1,4 |
| R | ER | 21 mai | 99 | 2,00 | 344 | 369 | 93 | 0,2370 | 4,0 | 12,0 | 2,9 |
| MOY. |
Aucunes données | Aucunes données | 99 | 1,50 | 281 | 308 | 90 | 0,3420 | 3,0 | 8,0 | 2,2 |
| É.-T. | Aucunes données | Aucunes données | 59 | 0,41 | 136 | 138 | 4 | 0,1017 | 1,0 | 2,0 | 0,9 |
| CV | Aucunes données | Aucunes données | 60 | 29,05 | 48 | 45 | 5 | 29,7262 | 32,6 | 29,2 | 39,4 |
|
|||||||||||
A1.5 Analyse de la SSCT
Les tableaux A1-G, A1-H et A1-I fournissent l'analyse complète requise pour la SSCT. Le tableau A1-G comprend les estimations de la SSCT à l'échelle normale et à l'échelle logarithmique. Dans cette partie, on trouve également la valeur de la SSCT de la forme pharmaceutique à l'essai en pourcentage de la SSCT de la forme de référence. Des statistiques sommaires sont calculées pour chaque variable.
ID
|
Échelle normale
|
Échelle logarithmique
|
|||
|---|---|---|---|---|---|
SSC T
essai |
SSC T
référence |
SSC T
relative (%) |
ln(SSC T)
essai |
ln(SSC T)
référence |
|
| A | 365 | 375 | 97 | 5,8998 | 5,9269 |
| B | 405 | 595 | 68 | 6,0038 | 6,3885 |
| C | 703 | 471 | 149 | 6,5553 | 6,1548 |
| E | 233 | 190 | 123 | 5,4510 | 5,2470 |
| F | 247 | 257 | 96 | 5,5093 | 5,5490 |
| G | 178 | 175 | 102 | 5,1817 | 5,1647 |
| H | 246 | 382 | 65 | 5,5053 | 5,9454 |
| I | 408 | 361 | 113 | 6,0112 | 5,8888 |
| K | 315 | 218 | 144 | 5,7525 | 5,3844 |
| L | 140 | 92 | 153 | 4,9416 | 4,5217 |
| M | 165 | 269 | 61 | 5,1059 | 5,5947 |
| N | 88 | 106 | 83 | 4,4773 | 4,6634 |
| O | 183 | 290 | 63 | 5,2094 | 5,6698 |
| P | 122 | 230 | 53 | 4,8040 | 5,4380 |
| Q | 68 | 144 | 47 | 4,2195 | 4,9698 |
| R | 275 | 344 | 80 | 5,6167 | 5,8406 |
| MOY. | 259 | 281 | 94 | 5,3903 | 5,5217 |
| É.-T. | 158 | 136 | 35 | 0,61 | 0,52 |
| CV | 61 | 48 | 37 | Aucunes données | Aucunes données |
Le tableau A1-H présente l'analyse de variance (ANOVA) pour le ln(SSCT) avec le modèle de plan croisé. Cette analyse donne une estimation adéquate de la variance intra-sujet, le CM (résiduel), pour le calcul de l'intervalle de confiance à 90 %. Tout effet significatif dans le modèle, autre que l'effet sujet(séq), devrait faire l'objet d'une enquête. Les CV intra-sujet et inter-sujets devraient également être calculés.
| Effets | Num dfNote de bas de page * | Den dfNote de bas de page ** | Valeur F | Prob > FNote de bas de page *** |
|---|---|---|---|---|
| Séq | 1 | 14 | 0,99 | 0,7699 |
| Période | 1 | 14 | 0,33 | 0,5751 |
| Forme | 1 | 14 | 1,88 | 0,1916 |
|
||||
|
Paramètre
|
Variance
|
|---|---|
|
Sujet(séq)
|
0,2648 |
|
Résiduel
|
0,0729 |

L'estimation du rapport des SSC et son intervalle de confiance à 90 % sont issus des calculs du tableau A1-J. Comme la présente étude est fondée sur un plan équilibré (c'est-à-dire un nombre égal de sujets par séquence), la différence équivaut simplement à la différence dans les moyennes arithmétiques des ln(SSC). Si le plan n'était pas équilibré, il faudrait alors utiliser une estimation de la moyenne des moindres carrés pour chaque forme afin d'établir cette différence et estimer l'erreur type appropriée.
Tableau A1-J : Analyse de la SSCT (ng·h/mL) - Calculs

A1.6 Analyse de la Cmax
L'information et le sommaire nécessaires pour l'analyse de la Cmax figurent aux tableaux A1-K à A1-N.
ID
|
Échelle normale
|
Échelle logarithmique
|
|||
|---|---|---|---|---|---|
C max
essai |
C max
référence |
C max
relative (%) |
ln(C max)
essai |
ln(C max)
référence |
|
| A | 122 | 126 | 97 | 4,8040 | 4,8362 |
| B | 102 | 207 | 49 | 4,6249 | 5,3327 |
| C | 202 | 123 | 164 | 5,3082 | 4,8121 |
| E | 59 | 37 | 160 | 4,0775 | 3,6109 |
| F | 66 | 85 | 78 | 4,1896 | 4,4426 |
| G | 54 | 55 | 98 | 3,9889 | 4,0073 |
| H | 101 | 219 | 46 | 4,6151 | 5,3890 |
| I | 90 | 182 | 49 | 4,4998 | 5,2040 |
| K | 155 | 60 | 259 | 5,0434 | 4,0943 |
| L | 57 | 26 | 223 | 4,0430 | 3,2580 |
| M | 23 | 57 | 41 | 3,1354 | 4,0430 |
| N | 38 | 47 | 80 | 3,6375 | 3,8501 |
| O | 43 | 71 | 61 | 3,7612 | 4,2626 |
| P | 68 | 97 | 70 | 4,2195 | 4,5747 |
| Q | 28 | 88 | 31 | 3,3322 | 4,4773 |
| R | 60 | 99 | 61 | 4,0943 | 4,5951 |
| MOY. | 79 | 99 | 98 | 4,2109 | 4,4244 |
| É.-T. | 48 | 59 | 68 | 0,59 | 0,61 |
| CV | 61 | 60 | 69 | Aucunes données | Aucunes données |
| Effets | Num dfNote de bas de page * | Den dfNote de bas de page ** | Valeur F | Prob > FNote de bas de page *** |
|---|---|---|---|---|
| Séq | 1 | 14 | 1,02 | 0,3306 |
| Période | 1 | 14 | 0,13 | 0,7264 |
| Forme | 1 | 14 | 1,77 | 0,2052 |
|
||||
|
Paramètre
|
Variance
|
|---|---|
|
Sujet(séq)
|
0,161 |
|
Résiduel
|
0,2048 |

Tableau A1-N : Analyse de la Cmax - Calculs

A1.7 Courbes de concentration en fonction du temps (sujet A)
La figure 1 présente un graphique de la concentration en fonction du temps pour le sujet A. Chaque graphique devrait inclure les courbes de toutes les formes pharmaceutiques administrées à ce sujet. Des courbes similaires doivent être fournies pour chacun des sujets.

La figure 2 présente un graphique du logarithme naturel (ln) de la concentration en fonction du temps pour le sujet A. Ce graphique devrait contenir les lignes de régression à partir desquelles les constantes de vitesse terminale d'élimination (λ) ont été estimées. La ligne devrait commencer et se terminer à des points dans le temps considérés comme faisant partie de la phase d'élimination log-linéaire. Tout point qui n'a pas été utilisé pour estimer la ligne de régression devrait être identifié comme tel.

La figure 3 présente la courbe des moyennes arithmétiques de tous les sujets pour chaque forme pharmaceutique et moment d'échantillonnage.

La figure 4 présente la courbe du logarithme naturel des moyennes arithmétiques de tous les sujets pour chaque forme pharmaceutique et moment d'échantillonnage.

Annexe 2 : Glossaire
- Biodisponibilité
- Vitesse et degré d'absorption d'un médicament dans la circulation générale.
- Bioéquivalence
- Forte similitude de la biodisponibilité de deux produits pharmaceutiques (de même forme pharmaceutique) provenant de la même dose molaire et qui sont peu susceptibles de produire des différences cliniques pertinentes en ce qui concerne les effets thérapeutiques, les effets indésirables ou les deux à la fois.
- Bioéquivalent
- Le produit à l'essai et le produit de référence sont dits bioéquivalents lorsqu'ils contiennent un ou des médicaments identiques et que, après comparaison dans le cadre d'une étude de biodisponibilité appropriée, ils répondent aux normes de vitesse et de degré d'absorption précisées dans la ligne directrice intitulée Normes en matière d'études de biodisponibilité comparatives : Formes pharmaceutiques de médicaments à effets systémiques.
- C max (concentration maximale observée)
- Concentration maximale (ou pic de concentration) observée.
- C min (concentration minimale observée)
- Concentration minimale observée à l'état d'équilibre.
- Concentration maximale observée (C max)
- Voir C max
- Constante de vitesse terminale d'élimination (λ)
- Constante de vitesse estimée à partir de la partie terminale de la courbe du log naturel (ln) de la concentration du médicament en fonction du temps. La demi-vie terminale (t ½) est calculée à partir de cette constante ( t ½=ln 2/λ).
- C pd (concentration pré-dose)
- Concentration pré-dose déterminée immédiatement avant l'administration d'une dose à l'état d'équilibre.
- Étiquette
- Inscriptions, mots ou marques accompagnant les drogues ou emballages. (article 2 de la Loi sur les aliments et drogues).
- Excipient
- Tout ingrédient, à l'exclusion des substances médicamenteuses, incorporé dans une forme pharmaceutique en vue d'en améliorer la stabilité, l'utilité ou l'aspect ou d'en faciliter la préparation (par exemple, base, base, vecteur, enrobage, colorant, saveur, agent de conservation, stabilisant et véhicule).
- Forme pharmaceutique
- Ingrédient ou mélange d'ingrédients particuliers, c'est-à-dire de substances médicamenteuses et d'excipients en quantités précises, définissant un produit donné.
- Forme pharmaceutique à libération modifiée
- Forme pharmaceutique pour laquelle des caractéristiques de libération du médicament dans le temps ou le lieu de libération ont été choisis pour réaliser des objectifs thérapeutiques ou pratiques qui ne sont pas à la portée des formes pharmaceutiques conventionnelles.
Les formes pharmaceutiques à libération modifiée sont des formes pharmaceutiques qui diffèrent des formes pharmaceutiques conventionnelles sur le plan de la vitesse de libération. Aux fins de la présente ligne directrice, les formes à libération modifiée englobent les formes pharmaceutiques conçues pour atteindre un ou plusieurs des objectifs suivants :- retarder la désintégration, la désagrégation ou la dissolution de manière à modifier la vitesse de dégradation
- retarder ou ralentir la vitesse d’absorption de façon à réduire la probabilité d’effets gastro-intestinaux ou d’autres effets indésirables (par exemple formes pharmaceutiques gastro-résistantes)
- fournir des concentrations efficaces de médicament pendant une période plus longue après l’administration d’une dose unique
- libérer le médicament au départ à une vitesse similaire à celle obtenue avec la forme conventionnelle, et fournir des concentrations efficaces de médicament pendant une période plus longue
- réduire au minimum les variations dans les concentrations de médicament pendant l’intervalle posologique
- faire en sorte qu’après l’administration d’une dose unique, les courbes de concentration en fonction du temps présentent de multiples pics et creux (c’est-à-dire, forme pharmaceutique multiphasique à libération modifiée)
- Intervalle de confiance à 90 %
- Intervalle autour de la valeur estimative qui garantit à 90 % qu'il contient la valeur véritable.
- Promédicament
- Précurseur inactif (ou beaucoup moins actif) qui est transformé en un médicament actif dans l'organisme.
- Rapport des C max
- Rapport des moyennes géométriques de la C max du produit à l'essai et du produit de référence. Il s'agit de l'antilogarithme de la différence entre les moyennes des logarithmes (ln) de la C max du produit à l'essai et de la C max du produit de référence.
- Rapport des SSC
- Rapport des moyennes géométriques de la SSC du produit à l'essai et du produit de référence. Il s'agit de l'antilogarithme de la différence entre les moyennes des logarithmes (ln) de la SSC du produit à l'essai et de la SSC du produit de référence.
- Repas normalisé
- Repas dont la composition en glucides, protéines, lipides et liquides est connue.
- SSC (surface sous la courbe)
- Surface sous la courbe de concentration en fonction du temps.
- SSC I (SSC à l'infini)
- Surface obtenue en extrapolant jusqu'à l'infini la SSC T. On peut calculer cette valeur en ajoutant la quantité C T/λ à la SSC T, où C T est la dernière concentration quantifiable estimée et λ est la constante de vitesse terminale d'élimination.
- SSC Réftmax
- Surface sous la courbe du produit à l'essai au moment de la concentration maximale du produit de référence, calculée pour chaque sujet de l'étude.
- SSC T (SSC au moment de la dernière concentration quantifiable)
- SSC au moment de la dernière concentration quantifiable. Le calcul de la SSC T se fonde sur des données observées à des moments précis.
- SSC Jau (SSC pendant l'intervalle posologique)
- Surface sous la courbe de la concentration en fonction du temps à l'état d'équilibre pendant l'intervalle posologique dans une étude à doses multiples
- SSC partielle
- La surface partielle qui se trouve sous la courbe de la concentration en fonction du temps suivant un intervalle de temps restreint après l’administration du médicament.
- SSC partielle0-72h (SSC jusqu’à 72 h)
- La surface quie se trouve sous la courbe de la concentration en fonction du temps pour un intervalle de temps s’échelonnant entre 0 et 72 heures.
- Temps pour atteindre la concentration maximale observée (t max)
- Temps qui s'écoule entre l'administration du médicament et l'observation de la C max.
- Vitesse d'absorption
- Vitesse à laquelle un médicament atteint la circulation générale après une administration par voie orale.