
Nouveau test diagnostique de la maladie de Creutzfeldt-Jakob : la conversion provoquée par tremblement en temps réel

 Téléchargez cet article en format PDF (309 Ko - 5 pages)
Téléchargez cet article en format PDF (309 Ko - 5 pages) Publié par : L’Agence de la santé publique du Canada
Numéro : Volume 41-8 : Troubles liés aux protéines mal repliées
Date de publication : 6 août 2015
ISSN : 1719-3109
Soumettre un article
À propos du RMTC
Naviguer
Volume 41-8, le 6 août 2015 : Troubles liés aux protéines mal repliées
Innovation
Un nouveau test diagnostique de la maladie de Creutzfeldt-Jakob : la conversion provoquée par tremblement en temps réel (RT-QulC)
Godal D1, Simon SLR1, Cheng K1,2, Knox JD1,3*
Affiliations
1 Laboratoire national de microbiologie, Agence de la santé publique du Canada, Winnipeg (Manitoba)
2 Département d'anatomie et des sciences cellulaires, Université du Manitoba, Winnipeg (Manitoba)
3 Département de microbiologie médicale, Université du Manitoba, Winnipeg (Manitoba)
Correspondance
DOI
https://doi.org/10.14745/ccdr.v41i08a02f
Résumé
À l'heure actuelle, pour diagnostiquer de manière définitive la maladie de Creutzfeldt-Jakob (MCJ), il faut procéder à l'examen du cerveau des patients décédés. En collaboration avec un consortium international, le Laboratoire national de microbiologie du Canada est à valider un nouveau test permettant de diagnostiquer la maladie de Creutzfeldt-Jakob chez des patients vivants. Ce test, qu'on appelle « conversion provoquée par tremblement en temps réel » (RT-QulC), détecte directement la forme de la protéine prion associée à la maladie dans le liquide céphalorachidien. Advenant que la RT-QuIC soit bel et bien validée, ce test représentera une avancée majeure dans le domaine du diagnostic des patients qu'on présume être atteints de la maladie de Creutzfeldt-Jakob.
Introduction
Trouble cérébral mortel dans tous les cas, la maladie de Creutzfeldt-Jakob (MCJ) est une maladie rare et non traitable. La maladie touche le plus souvent les personnes âgées à raison d'un cas annuel de maladie de Creutzfeldt-Jakob par million de personnesNote de bas de page 1. Les premiers signes comprennent souvent l'anxiété, la confusion et la perte de mémoire, qui s'apparentent à ceux d'autres troubles neurologiques tels que les maladies d'Alzheimer et de Huntington. Il existe plusieurs sous-types de la maladie de Creutzfeldt-Jakob, et les signes de l'évolution de la maladie sont très variables. Toutefois, il est courant d'observer des troubles de l'équilibre, de coordination des mains, de déambulation, de vision ainsi que des difficultés à avaler et la présence de mouvements involontaires saccadésNote de bas de page 2. À la fin, les personnes atteintes perdent la mobilité et la parole; le décès est souvent causé par une pneumonie ou d'autres infections secondaires.
Ces maladies neurologiques mortelles sont causées par de nouveaux agents transmissibles qu'on appelle « prions » et qui ne sont pas des bactéries ou des virus classiques, mais se composent plutôt d'une forme anormale d'une protéine hôte. Ce fait biologique clé exclut donc l'utilisation des technologies actuellement utilisées pour détecter directement d'autres agents infectieux. Actuellement, le diagnostic définitif de la maladie de Creutzfeldt-Jakob nécessite l'examen du cerveau des patients décédés afin de détecter la présence de dépôts de la protéine prion et la perte de cellules nerveuses associée qui entraîne l'aspect spongiforme du cerveau.
L'objectif de cet article consiste à résumer les trois types de la maladie de Creutzfeldt-Jakob, à décrire une nouvelle méthode récemment mise au point pour diagnostiquer cette maladie ainsi qu'à réfléchir aux répercussions éventuelles du nouveau test sur les patients et la pratique clinique ou en laboratoire.
Définition de la maladie de Creutzfeldt-Jakob
La maladie de Creutzfeldt-Jakob correspond à trois grandes catégories : la maladie de Creutzfeldt-Jakob sporadique (MCJs), la maladie de Creutzfeldt-Jakob génétique (MCJg) et la maladie de Creutzfeldt-Jakob acquise (MCJa). La forme sporadique, qui représente jusqu'à 85 % des cas observés et survient sans aucune raison apparente, touche généralement les gens dans la soixantaine. La dépression est un symptôme précoce courant, et la plupart des gens atteints de la MCJs meurent dans les six mois suivant leur diagnostic.
L'évolution de la MCJg peut s'apparenter à celle de la MCJs ou se manifester en tant que démence à progression lente au fil des ans. Évaluées auprès d'un nombre important de familles multigénérationnelles, certaines des mutations responsables de la maladie de Creutzfeldt-Jakob parmi les plus courantes sont transmises selon un mode autosomique dominant. Nonobstant, dans 50 % des diagnostics de la maladie de Creutzfeldt-Jakob génétique, il n'y a aucun antécédent familial de la maladie ou d'autres troubles neurologiques, ce qui tend à indiquer une faible pénétranceNote de bas de page 3.
La maladie de Creutzfeldt-Jakob acquise, qui représente moins de 1 % des cas, est causée par une exposition à l'agent infectieux. La MCJ iatrogène décrit les cas de maladie de Creutzfeldt-Jakob acquise associés à des interventions médicales. La nouvelle variante de la maladie de Creutzfeldt-Jakob (vMCJ) est acquise dans le cadre d'une exposition alimentaire à la viande de bœuf infecté par l'encéphalopathie bovine spongiforme (ESB), le terme scientifique correspondant à la « maladie de la vache folle ». À la suite de la mise en place efficace de mesures de santé publiqueNote de bas de page 4 le risque de contracter la variante de la maladie de Creutzfeldt-Jakob au Canada est presque inexistante. Par ailleurs, aucun cas domestique de la nouvelle variante de la maladie de Creutzfeldt-Jakob acquise n’a été reporté au CanadaNote de bas de page 1.
Pratique actuelle
Le diagnostic de toute forme de la maladie de Creutzfeldt-Jakob établi avant la mort repose sur le tableau clinique de la maladie ainsi que les manifestations caractéristiques de celle-ci qu'on peut observer à l'électroencéphalographie (EEG) et à l'examen d'imagerie par résonance magnétique (IRM). L'apparition de biomarqueurs de substitution, qui correspond habituellement à un changement dans la quantité de protéines spécifiques dans le liquide céphalorachidien (LCR), fournit des données supplémentaires qui appuient le diagnostic.
À l'heure actuelle, la section des maladies à prions du Laboratoire national de microbiologie mesure le taux de trois protéines (14-3-3, tau et S100) dans le LCR, afin d'aider les cliniciens qui composent avec les cas présumés de maladie de Creutzfeldt-Jakob. Les taux accrus de ces trois marqueurs de substitution concordent avec le diagnostic de la maladie de Creutzfeldt-Jakob, mais leur sensibilité et leur spécificité sont variables, ce qui fait qu'ils ne peuvent servir à diagnostiquer la maladie de Creutzfeldt-Jakob de manière définitiveNote de bas de page 5.
Le fondement moléculaire du nouveau test
Au niveau moléculaire, l'évolution de la maladie de Creutzfeldt-Jakob se caractérise par la conversion de la protéine prion exprimée normalement dans les cellules, PrPc, vers une isoforme de la protéine mal repliée qui est associée à la maladie, PRPd (Figure 1). La forme infectieuse de la protéine prion, PrPd, agit comme une « graine » et interagit avec la forme cellulaire normale de la protéine prion, PrPc, puis favorise la conversion de celle-ci en une forme infectieuse. Au fil de l'évolution de la maladie, la quantité de PrPd disponible pour convertir la forme PrPC augmente, ce qui accélère la conversion de PrPC en PrPd. Cela finit par mener à la production de grandes quantités de PrPd dans le cerveau des patients atteints de la maladie de Creutzfeldt-Jakob, qu'on peut observer après leur mort.
Contrairement à ce qu'on observe dans le cerveau, seules de petites quantités de PrPd sont présentes dans d'autres tissus, ce qui rend problématique la détection dans les échantillons facilement accessibles. La mise au point de la technique appelée « conversion provoquée par tremblement en temps réel » (RT-QulC) représente une solution à ce problème. La RT-QuIC utilise la capacité de la PrPd pour favoriser la conversion de sa configuration en celle de la PrPcNote de bas de page 6Note de bas de page 7Note de bas de page 8. Lors d'une RT-QuIC, une PrP (PrPr) recombinante fabriquée en laboratoire sert de substrat, et les échantillons de LCR des patients, qui renferment éventuellement la PrPd, sont analysés afin de vérifier s'ils amorceront le processus de conversion (Figure 1. Des cycles d'agitation et d'incubation provoqueront le LCR infecté, qui renferme la PrPd, à produire des quantités considérablement plus grandes de PrPrd, ce qui en facilitera la détection ultérieure au moyen des méthodes classiques.
Figure 1 : La protéine prion infectieuse favorise la conversion de la configuration de la protéine prion cellulaire
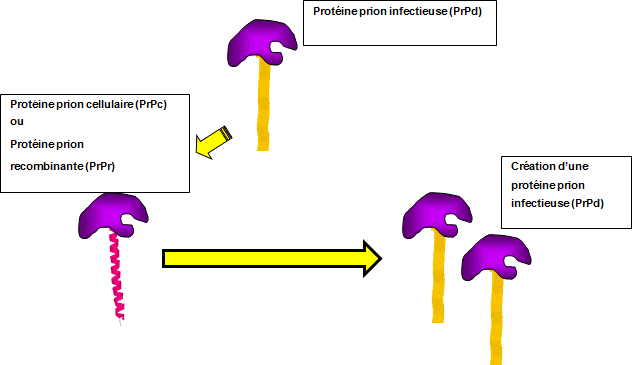
Description textuelle : Figure 1
Figure 1 : La protéine prion infectieuse favorise la conversion de la configuration de la protéine prion cellulaire
Cette figure démontre le fonctionnement du nouveau test de conversion provoquée par tremblement en temps réel (RT-QuIC). Ce test exploite le fait que la protéine prion infectieuse a la capacité de convertir la configuration de la protéine prion cellulaire afin de la rendre semblable. Si on introduit une protéine prion recombinante dans un échantillon qui contient la protéine prion infectieuse, la protéine recombinante sera convertie en protéine prion infectieuse. Ce test peut être mesuré en laboratoire.
Sommaire des résultats
La RT-QuIC s'est révélée être une méthode hautement sensible et spécifique pour détecter la présence de l'isoforme PrPd de la protéine prion associée à la maladie dans le LCR des patients atteints de maladie de Creutzfeldt-Jakob sporadiqueNote de bas de page 9Note de bas de page 10Note de bas de page 11Note de bas de page 12Note de bas de page 13. La détection directe de l'agent pathogène constitue une avancée majeure dans la mise au point d'un test diagnostique de la maladie de Creutzfeldt-Jakob sporadique, car celui-ci peut servir à poser un diagnostic définitif pendant que le patient est toujours en vie. Cependant, des études de validation en cours ont montré que les résultats des réactions de la RT-QuIC dépendent des conditions strictes liées à leur emploi (paramètres biochimiques chimiques et physiques) de même qu'à la nature de l'échantillon et du substrat recombinant. Le protocole évalué par le Laboratoire national de microbiologie a fait l'objet d'un nombre important de tests en vue de détecter la maladie de Creutzfeldt-Jakob sporadiqueNote de bas de page 10Note de bas de page 11Note de bas de page 15Note de bas de page 16. À l'aide de ce protocole, la section des maladies à prions du Laboratoire est parvenue à atteindre une sensibilité et une spécificité de 100 % dans le cadre d'une évaluation effectuée par un panel externe de vérification de la compétence relativement au LCR infecté de personnes atteints de la maladie de Creutzfeldt-Jakob sporadique. Ces résultats sont comparables à une sensibilité de 91 % et à une spécificité de 98 % déclarées par d'autres laboratoiresNote de bas de page 8Note de bas de page 9. Le fait que le consortium international utilise le même protocole normalisé devrait permettre l'analyse d'un nombre suffisant d'échantillons de vMCJ et de MCJg, qui sont des formes plus rares de la maladie, en vue de déterminer l'efficacité de notre version de la RT-QuIC dans ces cas particuliers.
Discussion
La mise au point d'un test spécifique et sensible aidera les travailleurs de la santé ainsi que les patients et leurs familles à prendre en charge les cas présumés de la maladie de Creutzfeldt-Jakob. Un test diagnostique de la maladie de Creutzfeldt-Jakob plus spécifique et plus sensible permettra aussi de se pencher sur des enjeux de santé publique concernant la transmission de la maladie de Creutzfeldt-Jakob d'un patient à lors de l'utilisation d'instruments chirurgicaux et de tissus contaminés, y compris le sang. Une fois la validation terminée, la RT-QuIC intégrera la gamme de services offerts par la section des maladies à prions du Laboratoire national de microbiologie. Ainsi, les professionnels de la santé canadiens auront accès à un test de dépistage plus sensible et le plus spécifique de la maladie de Creutzfeldt-Jakob pendant la vie des patients.
Remerciements
Nous tenons à remercier les membres du consortium du programme conjoint de recherche de l'Union européenne sur les maladies neurodégénératives (JPND), qui nous ont fourni des d'échantillons et de l'expertise, de même que le Bristol Institute for Transfusion Sciences (BITS), qui nous a fourni des PrPr.
Conflit d’intérêts
Aucun
Financement
Ce travail a été appuyé par l'Agence de la santé publique du Canada.
Détails de la page
- Date de modification :