
Intelligence artificielle et évaluation in silico de la RAM

 Téléchargez cet article en format PDF
Téléchargez cet article en format PDFPublié par : L’Agence de la santé publique du Canada
Numéro : Volume 46–6 : Intelligence artificielle en santé publique
Date de publication : 4 juin 2020
ISSN : 1719-3109
Soumettre un article
À propos du RMTC
Naviguer
Volume 46–6, le 4 juin 2020 : Intelligence artificielle en santé publique
Aperçu
Application de l’intelligence artificielle à l’évaluation in silico de la résistance aux antimicrobiens et des risques pour la santé humaine et animale présentés par les pathogènes bactériens entériques prioritaires
Rylan Steinkey1, Janice Moat2,3, Victor Gannon1, Athanasios Zovoilis3,4,5, Chad Laing2
Affiliations
1 Laboratoire national de microbiologie à Lethbridge, Agence de la santé publique du Canada, Lethbridge, AB
2 Centre national des maladies animales, Agence canadienne d’inspection des aliments, Lethbridge, AB
3 Département de chimie et de biochimie, Université de Lethbridge, Lethbridge, AB
4 Southern Alberta Genome Sciences Centre, Lethbridge, AB
5 Centre canadien de neurosciences comportementales, Lethbridge, AB
Correspondance
Citation proposée
Steinkey R, Moat J, Gannon V, Zoviolis A, Laing C. Application de l’intelligence artificielle à l’évaluation in silico de la résistance aux antimicrobiens et des risques pour la santé humaine et animale présentés par les pathogènes bactériens entériques prioritaires. Relevé des maladies transmissibles au Canada 2020;46(6):204–10. https://doi.org/10.14745/ccdr.v46i06a05f
Mots-clés : apprentissage automatique, pathogènes bactériens, séquence génomique complète, génomique prédictive, résistance aux antimicrobiens
Résumé
Chaque année, environ un Canadien sur huit est touché par des maladies d’origine alimentaire, soit par des éclosions, soit par des maladies sporadiques, les animaux constituant le principal réservoir des pathogènes. Les laboratoires de santé publique et animale procèdent désormais régulièrement à des analyses de séquençage du génome complet afin de définir des grappes de cas de maladie épidémiologique et de déterminer les sources potentielles d’infection. De même, un certain nombre d’outils bio-informatiques peuvent être utilisés pour identifier les facteurs déterminant la virulence et la résistance aux antimicrobiens (RAM) dans les génomes des souches pathogènes.
De nombreuses caractéristiques cliniques et phénotypiques importantes de ces agents pathogènes peuvent maintenant être prédites à l’aide d’algorithmes d’apprentissage automatique appliqués aux données de séquence génomique complète. Dans cet aperçu, nous comparons la capacité des machines à vecteurs de support, des arbres de décision à croissance par gradient et des réseaux neuronaux artificiels de prédire les niveaux de RAM à l’intérieur de Salmonella enterica et de la bêta-lactamase à spectre étendu (BLSE) produisant l’Escherichia coli. Nous montrons que les concentrations minimales d’inhibitrices (CMI) pour chacun des 13 antimicrobiens pour les souches de S. enterica peuvent être déterminées avec précision et ces souches E. coli produisant la BLSE peuvent être classifiées avec précision comme susceptible, intermédiaire ou résistante pour chacun des sept antimicrobiens.
En plus de la RAM et des populations bactériennes présentant les plus grands risques pour la santé humaine, les algorithmes d’intelligence artificielle offrent des outils prometteurs pour prédire d’autres phénotypes cliniquement et épidémiologiquement importants des pathogènes entériques.
Introduction
Chaque année, environ un Canadien sur huit sera touché par une maladie d’origine alimentaire, ce qui se traduira par une moyenne de 11 600 hospitalisations et 238 décès dans l’ensemble du paysNote de bas de page 1. Les animaux sont souvent le réservoir de grands pathogènes bactériens comme la Salmonella enterica et l’Escherichia coli. Ces agents pathogènes sont associés à des cas sporadiques et à des éclosions de maladies d’origine alimentaire. La résistance aux antimicrobiens (RAM) chez ces organismes est une préoccupation croissante, le traitement étant plus difficile et plus coûteux. Par exemple, la bêta-lactamase à spectre étendu (BLSE) produisant l’E. coli est multirésistante, avec des coûts de traitement jusqu’à trois fois supérieurs à ceux d’E. coli ne produisant pas la BLSENote de bas de page 2.
Les organismes nationaux et provinciaux de santé publique sont très efficaces pour identifier les sources et arrêter l’exposition aux agents pathogènes. Historiquement, la détermination de la RAM a été effectuée dans un milieu de laboratoire de travaux pratiquesNote de bas de page 3Note de bas de page 4. Deux des méthodes diagnostiques les plus couramment utilisées sont les tests de diffusion et de dilution. Les méthodes de diffusion, telles que la méthode Kirby-Bauer, exigent la culture d’un tapis bactérien dans un disque de concentration connue d’antimicrobiens ou dans une bande présentant un gradient de concentration d’antimicrobiens; la zone d’inhibition de la croissance autour de l’antimicrobien est comparée à une norme pour déterminer la résistance des bactériesNote de bas de page 3. Les méthodes de dilution impliquent des cultures liquides dans la dilution en série de chaque antimicrobien, où la croissance de l’organisme est utilisée pour déterminer la concentration minimale inhibitrice (CMI)Note de bas de page 3Note de bas de page 4.
Ces méthodes prennent beaucoup de temps parce qu’elles reposent sur la croissance des bactéries et sont coûteuses parce qu’elles nécessitent du personnel qualifié et de l’équipement spécialisé pour être appliquées.
Les analyses de la séquence génomique complète (SGC) sont devenues une partie intégrante des flux de travail en santé publique. Les tests in silico ont en grande partie remplacé de nombreux tests en laboratoire coûteux et longs en réponse aux éclosions et en surveillance de routineNote de bas de page 5Note de bas de page 6Note de bas de page 7. L’intelligence artificielle est de plus en plus utilisée pour analyser ces ensembles de données.
L’intelligence artificielle comprend la formation de machines pour faire des prédictions basées sur de grandes quantités de données. Elle a été utilisée dans des domaines aussi disparates que la reconnaissance de l’écriture manuscriteNote de bas de page 8 et les systèmes d’armes autonomesNote de bas de page 9.
L’apprentissage automatique (AA) surveillé décrit mieux l’application de l’intelligence artificielle à la prédiction des phénotypes bactériens à partir des données de SGC. Les algorithmes d’AA sont formés sur des données connues (« caractéristiques ») et prédisent ou classifient ensuite des données inconnues à l’aide des modèles formés. En général, les données utilisées pour la formation à l’AA sont particulières aux applications et peuvent inclure des images ou des renseignements sur les conditions météorologiques ou les éclosions de maladies infectieuses. Les données biologiques, et en particulier les données de SGC provenant de populations d’organismes, fournissent un très grand nombre de caractéristiques pour la formation de modèles d’AA et la prédiction des phénotypes d’intérêt. L’utilisation de ces algorithmes dans la recherche sur les maladies infectieuses n’a pas encore été pleinement exploitée, mais elle est très prometteuse.
Des algorithmes d’AA ont été utilisés pour prédire des phénotypes importants comme la RAMNote de bas de page 10Note de bas de page 11 et pour déterminer si différents groupes d’agents pathogènes de la même espèce présentent des risques différents pour la santé humaineNote de bas de page 12Note de bas de page 13Note de bas de page 14. La capacité de prédire d’importants phénotypes bactériens fondés uniquement sur les données du SGC serait d’un grand avantage pour la santé publique canadienne et pour l’industrie de l’agriculture animale.
Dans cette étude, nous avons formé trois modèles d’AA sur les données de SGC pour prédire les niveaux de résistance à 13 antimicrobiens dans les isolats de la S. enterica et pour classifier les souches d’E. coli produisant la BLSE comme susceptible, intermédiaires ou résistantes à sept antimicrobiens.
Méthodes
Les SGC de S. enterica ont été recueillis auprès de la GenBank du National Centre for Biotechnology Information. Ces 5 853 séquences ont été principalement isolées en Amérique du Nord entre 2002 et 2017; les données comprenaient 63 sérotypes avec au moins cinq membres, ainsi que des CMI phénotypiques pour 13 antimicrobiensNote de bas de page 15. Les SGC ont été décomposés en sous-chaînes séquentielles de 11 k-mers de longueur et leurs occurrences ont été comptées à l’aide du logiciel JellyfishNote de bas de page 16. Afin de limiter la sélection des caractéristiques à celles qui sont le plus associées au phénotype examiné, nous avons utilisé une analyse de la variance de la valeur F, ce qui nous a permis de maintenir les 1 000 k-mers les plus associés à chaque agent antimicrobien avant la formation au modèle. Cette sélection de fonctions permet au modèle de se concentrer sur des k-mers statistiquement importants, lesquels peuvent améliorer la précision et économiser beaucoup de temps et de ressources informatiques.
Nous avons mis en œuvre des arbres de décision à croissance par gradient en utilisant XGBoostNote de bas de page 17 et des machines à vecteurs de support en utilisant SciKit-learnNote de bas de page 18. Les analyses de données ont été effectuées au moyen d’une validation croisée quintuple, où 80 % des données ont été utilisées pour former un modèle et les 20 % restants ont été retenus pour évaluer le rendement du modèle. Les analyses ont été répétées cinq fois, avec chaque 20 % étant utilisé une fois pour évaluer le rendement. Une moyenne de l’exactitude des cinq évaluations a été calculée pour chaque répétition expérimentale. Dix répétitions expérimentales distinctes ont été effectuées avec l’attribution aléatoire de génomes à chaque fois, avec la précision totale du modèle et l’écart type calculés à partir de ces résultats.
Des réseaux neuronaux artificiels ont été mis en œuvre à l’aide de KerasNote de bas de page 19 avec un TensorFlowNote de bas de page 20 en arrière-plan et des optimisations d’hyperparamètres réalisées avec HyperasNote de bas de page 21. La validation croisée quintuple pour le réseau neuronal a consisté en un partage de 60-20-20 pour la formation, l’optimisation d’hyperparamètres et les tests, respectivement, pour chaque fois. Des mécanismes d’arrêt précoce ont été utilisés pour prévenir la suradaptation en surveillant les rendements décroissants ou négatifs à l’occasion des périodes de formation successives. En outre, une sélection aléatoire de nœuds dans le réseau et de leurs connexions a été supprimée par l’abandon afin d’empêcher la suradaptation ou la coadaptationNote de bas de page 22.
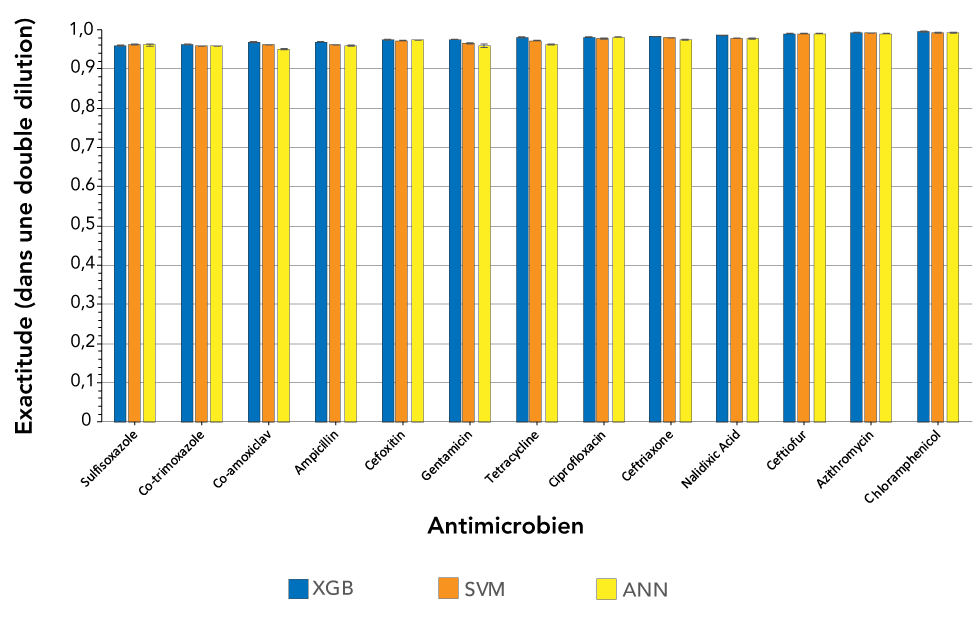
Comme le montre la figure 1, les CMI ont été prédits dans une dilution avec une précision de 97,88 % (±1,13) en utilisant XGBoost, 97,48 % (±1,20) en utilisant des machines à vecteurs de support et 97,16 % (±1,48) en utilisant des réseaux neuronaux artificiels. Les classificateurs XGBoost ont enregistré en moyenne une erreur majeure et un taux d’erreur majeur de 0,19 % (±0,19) et 0,71 % (±0,60), respectivement. Afin d’éviter l’inflation de l’exactitude des modèles, on a supprimé de ces moyennes le co-trimoxazole, la ciprofloxacine et la ceftriaxone, qui présentaient une faible diversité des classes de CMI. Les classificateurs XGBoost formés à prédire les CMI pour un seul antimicrobien utilisaient huit cœurs (processeur Intel Xeon Gold 6154), avaient un temps de formation moyen de 15 minutes et 12 secondes et ont atteint un maximum de 84,74 Go de mémoire vive.
Figure 1 : Exactitude dans une dilution double pour trois modèles d’apprentissage automatique formés sur les 1 000 principaux 11-mers et utilisés pour prédire les concentrations minimales d’inhibiteurs pour 13 antimicrobiens de Salmonella enterica
Description textuelle : Figure 1
Figure 1 : Exactitude dans une dilution double pour trois modèles d’apprentissage automatique formés sur les 1 000 principaux 11-mers et utilisés pour prédire les concentrations minimales d’inhibiteurs pour 13 antimicrobiens de Salmonella enterica
| Antimicrobien | XGB | SVM (MVS) | ANN (RNA) | |||
|---|---|---|---|---|---|---|
| Moyenne | Écart-type | Moyenne | Écart-type | Moyenne | Écart-type | |
| Sulfisoxazole | 0,958750 | 0,001282 | 0,963074 | 0,001540 | 0,961830 | 0,003175 |
| Co-trimoxazole | 0,960856 | 0,001481 | 0,958127 | 0,000286 | 0,958422 | 0,000454 |
| Co-amoxiclav | 0,967965 | 0,001001 | 0,961338 | 0,000107 | 0,949378 | 0,001805 |
| Ampicillin | 0,969231 | 0,000529 | 0,961956 | 0,000174 | 0,958878 | 0,001462 |
| Cefoxitin | 0,974201 | 0,000552 | 0,971978 | 0,000238 | 0,973140 | 0,000301 |
| Gentamicin | 0,974912 | 0,000606 | 0,965006 | 0,000680 | 0,958700 | 0,003745 |
| Tetracycline | 0,980020 | 0,000493 | 0,972590 | 0,000489 | 0,963184 | 0,001169 |
| Ciprofloxacin | 0,981727 | 0,000413 | 0,977709 | 0,000998 | 0,981252 | 0,000296 |
| Ceftriaxone | 0,981937 | 0,000698 | 0,979729 | 0,000237 | 0,975334 | 0,001893 |
| Nalidixic Acid | 0,985288 | 0,000446 | 0,978057 | 0,000142 | 0,977490 | 0,001583 |
| Ceftiofur | 0,990266 | 0,000442 | 0,990364 | 0,000092 | 0,989752 | 0,000444 |
| Azithromycin | 0,992052 | 0,000933 | 0,991365 | 0,000206 | 0,990768 | 0,000691 |
| Chloramphenicol | 0,995203 | 0,000311 | 0,992302 | 0,000056 | 0,992674 | 0,000458 |
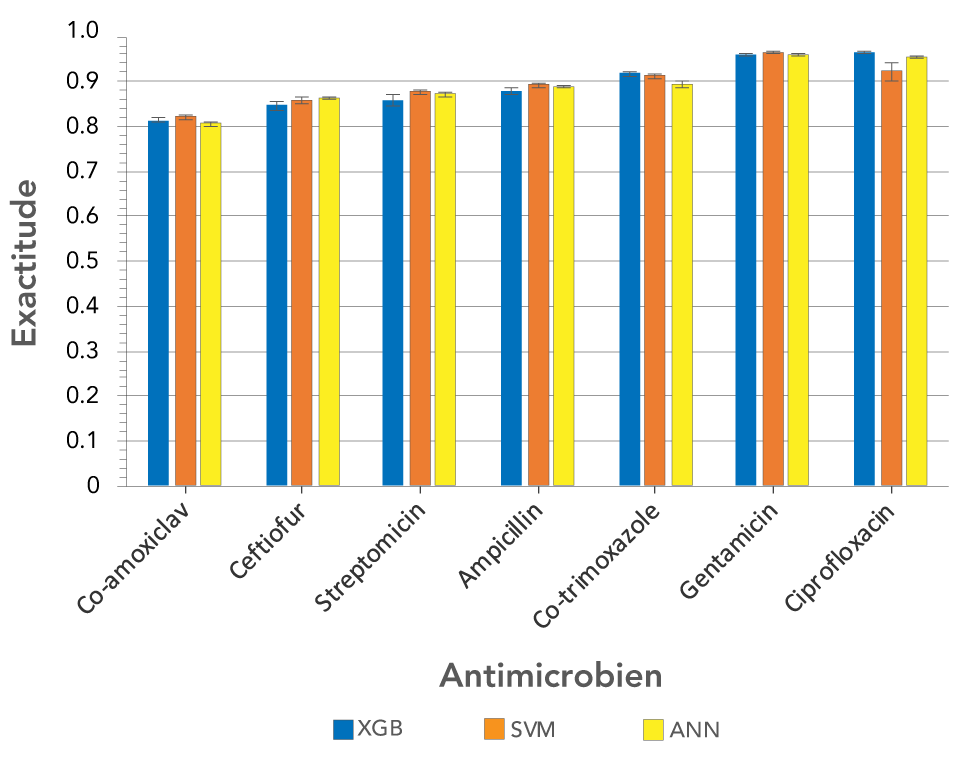
Nous avons également examiné un ensemble de 2 413 séquences d’E. coli contenant des producteurs de la BLSE, mais aucune donnée de CMI n’était disponible pour ces souches. Au lieu de cela, ils ont été classés comme susceptibles, intermédiaires ou résistants pour sept antimicrobiens. L’ensemble comprenait des échantillons de bovins, de cliniques et d’environnement isolés entre 1970 et 2017 au Canada, en Thaïlande et au Royaume-UniNote de bas de page 11Note de bas de page 23Note de bas de page 24. Nous avons analysé les séquences en utilisant l’approche k-mer décrite ci-dessus et nous les avons utilisées pour former des modèles pour classer les isolats comme susceptibles, intermédiaires ou résistants pour chaque antimicrobien. La précision moyenne des modèles dans les sept antimicrobiens était de 89,18 % (±5,44) pour XGBoost, 89,25 % (±4,43) pour les machines à vecteurs de support et 89,18 % (±5,20) pour les réseaux neuronaux artificiels (figure 2).
Figure 2 : Exactitude de trois modèles d’apprentissage automatique formés sur les 1 000 principaux 11-mers et utilisés pour prédire les classifications susceptibles, intermédiaires et résistantes de sept antimicrobiens d’Escherichia coli
Description textuelle : Figure 2
Figure 2 : Exactitude de trois modèles d’apprentissage automatique formés sur les 1 000 principaux 11-mers et utilisés pour prédire les classifications susceptibles, intermédiaires et résistantes de sept antimicrobiens d’Escherichia coli
| Antimicrobien | XGB | SVM (MVS) | ANN (RNA) | |||
|---|---|---|---|---|---|---|
| Moyenne | Écart-type | Moyenne | Écart-type | Moyenne | Écart-type | |
| Co-amoxiclav | 0,814763316 | 0,005384161 | 0,821363594 | 0,004471698 | 0,806992567 | 0,005409766 |
| Ceftiofur | 0,847536534 | 0,010266626 | 0,857223382 | 0,008464752 | 0,863486840 | 0,001456679 |
| Streptomicin | 0,857430063 | 0,011451457 | 0,876519833 | 0,006231367 | 0,872638158 | 0,005196798 |
| Ampicillin | 0,879085671 | 0,006961795 | 0,892516300 | 0,004019401 | 0,889839555 | 0,004015798 |
| Co-trimoxazole | 0,917826722 | 0,005429594 | 0,912897704 | 0,005756562 | 0,892096490 | 0,006880167 |
| Gentamicin | 0,961769581 | 0,002661451 | 0,964425197 | 0,000710687 | 0,960550072 | 0,001277735 |
| Ciprofloxacin | 0,964520099 | 0,001878239 | 0,922696229 | 0,018860678 | 0,955697705 | 0,002089745 |
Discussion
Comme nous l’avons montré, les méthodes d’AA que nous utilisons ne reposaient pas sur des génomes de référence spécifiques, ni sur une connaissance a priori des mécanismes de résistance, mais sur la classification des organismes en grands groupes phénotypiques. Ce sont les modèles d’AA qui identifient les différences génomiques sous-jacentes qui sont le plus associées au phénotype. Cela présente le double avantage de ne pas exiger de connaissances mécanistes et peut permettre d’identifier de nouveaux déterminants génomiques du phénotype à l’étude. Ces nouvelles caractéristiques extraites des modèles présentent d’énormes avantages potentiels : comme dans le cas de la résistance aux antibiotiques, ils peuvent être utilisés pour développer des bases de données publiques établies sur les mécanismes de résistance et ils peuvent être utilisés comme cibles potentielles pour des diagnostics rapides lors d’essais ultérieurs in silico ou en laboratoire de travaux pratiques.
Les modèles d’AA peuvent prédire rapidement et précisément la RAM à l’aide de données de SGC, de la classification susceptible, intermédiaire ou résistante aux valeurs quantitatives CMI. Pour les prévisions de RAM, il a été démontré que les modèles XGBoost se forment plus rapidement, utilisent moins de mémoire et sont plus précis que les méthodes d’apprentissage en profondeur. De plus, les modèles XGBoost et les modèles à vecteurs de support peuvent être utilisés pour déterminer les régions spécifiques du génome qui sont les plus prédictives d’un phénotype. C’est très difficile avec la mise en place d’un réseau neuronal dans la « boîte noire »; cependant, les réseaux neuronaux artificiels excellent encore dans la modélisation complexe des réseaux et ne devraient donc pas être exclus des études futures en génomique.
Les données de RAM souffrent généralement d’un déséquilibre de classe important, ce qui peut donner lieu à des modèles de haute précision sans valeur, comme le cas du co-trimoxazole dans nos données sur la Salmonella, où plus de 95 % des échantillons se trouvaient dans une dilution l’un de l’autre, ce qui donne un modèle capable d’une précision de 95 % sans apprendre quoi que ce soit des données sous-jacentes.
Nguyen et coll.Note de bas de page 10 ont formé des régresseurs XGBoost sur un ensemble de données contenant 4 500 séquences de génomes complets de S. enterica non typhoïdes (provenant d’un ensemble de données plus vaste de 5 278 échantillons, dont 4 595 étaient également dans notre ensemble de données). Ces modèles avaient une précision de validation croisée de 95 % pour les 10 mêmes antimicrobiens inclus dans notre étude actuelle. Nguyen et coll.Note de bas de page 10 ont utilisé un seul régresseur formé sur les 15 antimicrobiens en même temps, ce qui a pris 51 heures à former et a atteint un maximum de 1 184 Go de mémoire sur 170 cœurs (processeur Intel Xeon E5-4669v4)Note de bas de page 10. Les classificateurs XGBoost formés dans notre étude actuelle se sont améliorés sur ces temps de formation ainsi que sur l’utilisation et la précision de la mémoire. Les classificateurs XGBoost y sont arrivés en créant des modèles par antimicrobien et en sélectionnant initialement uniquement les 1 000 caractéristiques les plus importantes statistiquement. Pour mieux comparer les précisions de ces modèles, il faut utiliser un ensemble de données indépendant au lieu de s’appuyer sur les exactitudes de validation croisée rapportées.
L’ensemble de données sur l’E. coli comprenait 1 935 isolats provenant d’une étude antérieure de Moradigaravand et coll.Note de bas de page 11. Leurs méthodes exigeaient l’année d’isolement pour chaque séquence et le prétraitement des données sous forme de détermination du pangénome et de calcul de la structure de la populationNote de bas de page 11. En revanche, nos méthodes n’exigeaient que la séquence du génome associée au phénotype de résistance déterminé en laboratoire, ce qui permet la classification ainsi que l’identification de nouvelles régions qui ne sont pas actuellement associées à la RAM. Les régions pourraient être utilisées pour des tests de diagnostic ultérieurs in silico ou en laboratoire.
Bien que des classifications plus larges, comme susceptibles, intermédiaires et résistantes, soient courantes pour les diagnostics de laboratoire et utiles pour établir des lignes directrices de traitement pour une infection bactérienne, les critères de base de ces catégories sont établis par des comités, avec une certaine disparité entre les régions. La prédiction des valeurs quantifiées sous forme de CMI sera la plus utile à l’avenir, même si elles sont utilisées par la suite pour classer les bactéries dans des catégories plus larges comme la susceptible, intermédiaire et résistant.
Bien que les résultats de ces études soient encourageants, la surinterprétation des résultats pose un problème avec les données génomiques en raison du nombre élevé de caractéristiques utilisées pour faire des prédictions par rapport à la taille plus petite de l’échantillon du nombre de génomes. Cela peut conduire à une surexploitation des données et à un piètre rendement des modèles, que nous avons tenté de traiter dans les méthodes de cette étudeNote de bas de page 25.
L’utilisation d’AA a permis de prédire la RAM dans d’autres pathogènes, y compris Mycobacterium tuberculosis, où de nouvelles signatures génétiques résistantes ont été identifiéesNote de bas de page 26. L’AA s’est également révélée utile dans l’identification de nouveaux composés antimicrobiens, qui ont historiquement été marqués par des taux élevés d’échec dans les sociétés pharmaceutiquesNote de bas de page 27.
La recherche par AA sur la S. typhimurium a découvert que plus de 80 % de la source hôte pouvait être attribuée à l’aide de variantes protéiques. Ce résultat a été obtenu à l’aide d’une machine à vecteurs de support, de réseaux neuronaux artificiels et de modèles de « prévisions au hasard »Note de bas de page 28. Ce qui est particulièrement intéressant dans cette étude, c’est le chevauchement entre le réservoir animal et les cas humains. Cela indique que tous les isolats d’un agent pathogène particulier ne représentent pas le même risque de maladie et suggère que des points de contrôle plus spécifiques pourraient limiter l’infection humaine. De plus, comme plus de 60 % des agents pathogènes humains sont d’origine zoonotique, l’AA promet d’identifier les agents pathogènes émergents en analysant l’adaptation de l’hôte aux agents pathogènes animaux actuelsNote de bas de page 29.
Malgré l’utilité avérée de l’AA, les bactéries sont en constante évolution, et nos modèles aussi doivent évoluer, car ils ne sont aussi bons que les données sur lesquelles ils sont formés. La puissance de ces techniques doit être tempérée par leur utilisation judicieuse. De plus, des modèles spécifiques à la classe et à l’espèce sont toujours nécessaires pour produire des résultats significatifs, par exemple, un modèle par médicament par espèce pour prédire la RAMNote de bas de page 30.
Il convient de noter que l’AA ne saisit pas toujours avec précision les interactions complexes et que l’amélioration de la modélisation ne peut à elle seule compenser le biais d’échantillonnage ou un ensemble de données incomplet ou sujet à erreur.
Conclusion
Comme le montre cet aperçu, l’intelligence artificielle a déjà amélioré l’identification et la caractérisation des maladies infectieuses, dont les avantages toucheront les laboratoires de santé publique et de santé animale au niveau mondial. Par exemple, les régions génomiques identifiées comme prédictives pour des classes spécifiques de RAM pourraient être utilisées pour l’identification et la classification rapides en aval, y compris dans les pipelines in silico et les applications en laboratoire comme la réaction en chaîne de la polymérase.
Le futur proche promet des développements passionnants, comme l’utilisation de l’AA pour identifier les bactériophages qui lysent des groupes spécifiques de bactéries pathogènes, permettant la phagothérapie à la place des antimicrobiens traditionnelsNote de bas de page 31. Enfin, la caractérisation du « phénotype complet », avec la capacité de prédire l’expression intégrale des protéines membranaires, est de plus en plus probableNote de bas de page 32; et la formation de biofilmsNote de bas de page 33.
Malgré cela, la taille des ensembles de données requis pour former efficacement les modèles d’AA signifie que les ordinateurs de bureau sont souvent incapables d’analyser les données. Ceux qui n’ont pas accès aux ressources nécessaires doivent plutôt utiliser des méthodes analytiques qui réduisent la charge de calculNote de bas de page 34. À juste titre, l’utilisation d’AA lui-même a entraîné une augmentation de la vitesse des modèles mécanistes, dans certains cas plus de quatre ordres de grandeurNote de bas de page 35.
Nous ne sommes qu’au début du couplage de grandes quantités de données génomiques et d’intelligence artificielle, avec la promesse de nouvelles découvertes qui amélioreront la plupart des aspects de la santé animale et humaine du fardeau des pathogènes bactériens entériques.
Déclaration des auteurs
- R. J. S. — Traitement des données, analyse formelle, méthodologie, logiciel, validation, visualisation, ébauche originale, édition
- J. M. — Traitement des données, analyse formelle, méthodologie, logiciel, validation, visualisation, ébauche originale, édition
- V. P. J. G. — Conceptualisation, acquisition du financement, méthodologie, administration de projet, ressources, supervision, validation, ébauche originale, édition
- A. Z. — Conceptualisation, acquisition du financement, méthodologie, administration de projet, ressources, supervision, ébauche originale, édition
- C. R. L. — Conceptualisation, acquisition du financement, méthodologie, administration de projet, ressources, supervision, validation, ébauche originale, édition
Conflit d’intérêts
Aucun.
Financement
J. M., A. Z., C. R. L. : Ce travail a été appuyé par la subvention Antimicrobial Resistance - One Health Consortium accordée à A. Z. et C. R. L. par le ministère du Développement économique, du Commerce et du Tourisme de l’Alberta.
R. J. S., V. P. J. G., C. R. L. : Ce travail a été appuyé par le projet de l’Initiative de recherche et développement en génomique sur la résistance aux antimicrobiens. Ces travaux ont également été financés par l’Agence de la santé publique du Canada, l’Agence canadienne d’inspection des aliments et l’Université de Lethbridge.

Cette œuvre est mise à disposition selon les termes de la Licence Creative Commons Attribution 4.0 International