
Artificial intelligence and in silico assessment of AMR

 Download this article as a PDF
Download this article as a PDFPublished by: The Public Health Agency of Canada
Issue: Volume 46–6: Artificial intelligence in public health
Date published: June 4, 2020
ISSN: 1481-8531
Submit a manuscript
About CCDR
Browse
Volume 46–6, June 4, 2020: Artificial intelligence in public health
Overview
Application of artificial intelligence to the in silico assessment of antimicrobial resistance and risks to human and animal health presented by priority enteric bacterial pathogens
Rylan Steinkey1, Janice Moat2,3, Victor Gannon1, Athanasios Zovoilis3,4,5, Chad Laing2
Affiliations
1 National Microbiology Laboratory at Lethbridge, Public Health Agency of Canada, Lethbridge, AB
2 National Centre for Animal Diseases, Canadian Food Inspection Agency, Lethbridge, AB
3 Department of Chemistry and Biochemistry, University of Lethbridge, Lethbridge, AB
4 Southern Alberta Genome Sciences Centre, Lethbridge, AB
5 Canadian Centre for Behavioural Neuroscience, Lethbridge, AB
Correspondence
Suggested citation
Steinkey R, Moat J, Gannon V, Zovoilis A, Laing C. Application of artificial intelligence to the in silico assessment of antimicrobial resistance and risks to human and animal health presented by priority enteric bacterial pathogens. Can Commun Dis Rep 2020;46(6):180–5. https://doi.org/10.14745/ccdr.v46i06a05
Keywords: machine learning, bacterial pathogens, whole genome sequence, predictive genomics, antimicrobial resistance
Abstract
Each year, approximately one in eight Canadians are affected by foodborne illness, either through outbreaks or sporadic illness, with animals being the major reservoir for the pathogens. Whole genome sequence analyses are now routinely implemented by public and animal health laboratories to define epidemiological disease clusters and to identify potential sources of infection. Similarly, a number of bioinformatics tools can be used to identify virulence and antimicrobial resistance (AMR) determinants in the genomes of pathogenic strains.
Many important clinical and phenotypic characteristics of these pathogens can now be predicted using machine learning algorithms applied to whole genome sequence data. In this overview, we compare the ability of support vector machines, gradient-boosted decision trees and artificial neural networks to predict the levels of AMR within Salmonella enterica and extended-spectrum β-lactamase (ESBL) producing Escherichia coli. We show that minimum inhibitory concentrations (MIC) for each of 13 antimicrobials for S. enterica strains can be accurately determined, and that ESBL-producing E. coli strains can be accurately classified as susceptible, intermediate or resistant for each of seven antimicrobials.
In addition to AMR and bacterial populations of greatest risk to human health, artificial intelligence algorithms hold promise as tools to predict other clinically and epidemiologically important phenotypes of enteric pathogens.
Introduction
Every year, about one in eight Canadians will be affected by a foodborne illness, resulting in an average of 11,600 hospitalizations and 238 deaths nationwideFootnote 1. Animals are often the reservoir for major bacterial pathogens such as Salmonella enterica and Escherichia coli. These pathogens are associated with both sporadic cases and outbreaks of foodborne disease. Antimicrobial resistance (AMR) among these organisms is a growing concern, with treatment being more difficult and expensive. For example, extended-spectrum β-lactamase (ESBL) producing E. coli are multidrug resistant, with treatment costs up to three times that of non-ESBL-producing E. coliFootnote 2.
National and provincial public health agencies are very effective at identifying sources and halting exposure to pathogens. Historically, AMR determination has been performed in a wet lab settingFootnote 3Footnote 4. Two of the most commonly used diagnostic methods are diffusion and dilution tests. Diffusion methods, such as the Kirby–Bauer method, require growing a bacterial lawn in either a disk of known concentration of antimicrobials or a strip with a gradient of concentrations of antimicrobials; the zone of growth inhibition around the antimicrobial is compared with a standard to determine the resistance of the bacteriaFootnote 3. Dilution methods involve liquid cultures in serial dilution of each antimicrobial, where growth of the organism is used to determine the minimum inhibitory concentration (MIC)Footnote 3Footnote 4.
These methods are time consuming because they rely on the growth of bacteria, and expensive because they require trained personnel and specialized equipment to carry out.
Whole genome sequence (WGS) analyses have become integral to public health work flows. In silico tests have largely replaced many costly and time-consuming wet lab tests in outbreak response and routine surveillanceFootnote 5Footnote 6Footnote 7. Artificial intelligence is being increasingly used to analyse these datasets.
Artificial intelligence involves training machines to make predictions based on large amounts of data. It has been used in fields as disparate as handwriting recognitionFootnote 8 and autonomous weapons systemsFootnote 9.
Supervised machine learning (ML) better describes the application of artificial intelligence to the prediction of bacterial phenotypes based on WGS data. ML algorithms are trained on known data (“features”) and subsequently predict or classify unknown data using the trained models. In general, data used for ML training are application specific and can include images or information about weather or outbreaks of infectious disease. Biological data, and in particular WGS data from populations of organisms, provide an extremely large number of features for training ML models and predicting phenotypes of interest. Use of these algorithms in infectious disease research has not yet been fully exploited but holds significant promise.
ML algorithms have been used to predict important phenotypes such as AMRFootnote 10Footnote 11 and to determine if different groups of pathogens from the same species pose different risks to human healthFootnote 12Footnote 13Footnote 14. The ability to predict important bacterial phenotypes based solely on WGS data would be of enormous benefit to both Canadian public health and the animal agriculture industry.
In this study, we trained three ML models on WGS data to predict the levels of resistance to 13 antimicrobials in S. enterica isolates and to classify ESBL-producing E. coli strains as susceptible, intermediate or resistant (SIR) to seven antimicrobials.
Methods
S. enterica WGS was collected from the National Center for Biotechnology Information GenBank. These 5,853 sequences were primarily isolated within North America between 2002 and 2017; the data included 63 serotypes with at least five members, along with phenotypic MICs for 13 antimicrobialsFootnote 15. WGSs were decomposed into sequence substrings 11 k-mers in length, and their occurrences were counted using JellyfishFootnote 16. To limit the selection of features to those most associated with the phenotype being examined, we used an ANOVA F-value, keeping the top 1,000 k-mers most associated with each antimicrobial agent prior to model training. This feature selection allows the model to focus on statistically important k-mers, which can improve accuracy and saves substantial amounts of time and computing resources.
We implemented gradient-boosted decision trees using XGBoostFootnote 17 and support vector machines using SciKit-learnFootnote 18. Data analyses were conducted using five-fold cross-validation where 80% of the data was used to train a model and the remaining 20% was withheld to evaluate model performance. This was repeated five times, with each 20% being used once for evaluating performance. An average of the accuracy for the five evaluations was calculated for each experimental replicate. Ten separate experimental replicates with random assignment of genomes to each fold were performed, with the total model accuracy and standard deviation calculated from these.
Artificial neural networks were implemented using KerasFootnote 19 with a TensorFlowFootnote 20 backend and hyperparameter optimizations conducted with HyperasFootnote 21. The five-fold cross-validation for the neural network consisted of a 60-20-20 split for training, hyperparameter optimization and testing, respectively, for each fold. Early stopping mechanisms were used to prevent over-fitting by monitoring diminishing or negative returns with successive training epochs. In addition, a random selection of nodes in the network and their connections were removed via dropout to prevent over-fitting or co-adaptationFootnote 22.
As shown in Figure 1, MICs were predicted within one dilution with an accuracy of 97.88% (± 1.13) using XGBoost, 97.48% (± 1.20) using support vector machines and 97.16% (± 1.48) using artificial neural networks. XGBoost classifiers averaged a major error and major error rate of 0.19% (± 0.19) and 0.71% (± 0.60), respectively. To prevent inflating model accuracies, co-trimoxazole, ciprofloxacin and ceftriaxone, which had low MIC class diversity, were removed from these averages. XGBoost classifiers trained to predict MICs for a single antimicrobial used eight cores (Intel Xeon Gold 6154 CPU), had a mean training time of 15 minutes and 12 seconds, and peaked at 84.74 GB of random access memory (RAM).
Figure 1: Accuracies within one two-fold dilution for three machine learning models trained on the top 1,000 11-mers and used to predict minimum inhibitory concentrations for 13 Salmonella enterica antimicrobials
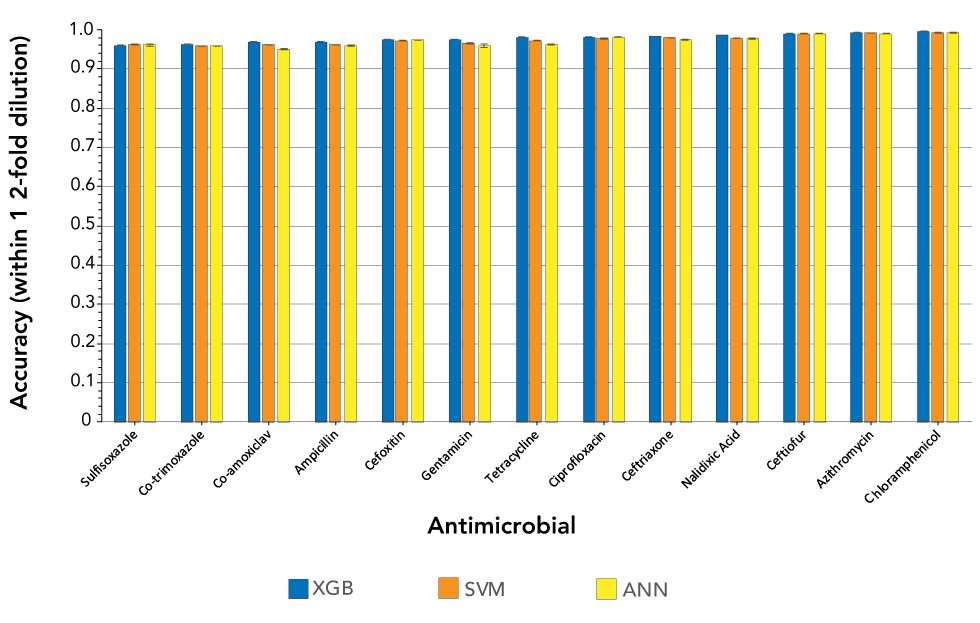
Text description: Figure 1
Figure 1: Accuracies within one two-fold dilution for three machine learning models trained on the top 1,000 11-mers and used to predict minimum inhibitory concentrations for 13 Salmonella enterica antimicrobials
| Antimicrobial | XGB | SVM | ANN | |||
|---|---|---|---|---|---|---|
| Mean | Standard deviation | Mean | Standard deviation | Mean | Standard deviation | |
| Sulfisoxazole | 0.958750 | 0.001282 | 0.963074 | 0.001540 | 0.961830 | 0.003175 |
| Co-trimoxazole | 0.960856 | 0.001481 | 0.958127 | 0.000286 | 0.958422 | 0.000454 |
| Co-amoxiclav | 0.967965 | 0.001001 | 0.961338 | 0.000107 | 0.949378 | 0.001805 |
| Ampicillin | 0.969231 | 0.000529 | 0.961956 | 0.000174 | 0.958878 | 0.001462 |
| Cefoxitin | 0.974201 | 0.000552 | 0.971978 | 0.000238 | 0.973140 | 0.000301 |
| Gentamicin | 0.974912 | 0.000606 | 0.965006 | 0.000680 | 0.958700 | 0.003745 |
| Tetracycline | 0.980020 | 0.000493 | 0.972590 | 0.000489 | 0.963184 | 0.001169 |
| Ciprofloxacin | 0.981727 | 0.000413 | 0.977709 | 0.000998 | 0.981252 | 0.000296 |
| Ceftriaxone | 0.981937 | 0.000698 | 0.979729 | 0.000237 | 0.975334 | 0.001893 |
| Nalidixic acid | 0.985288 | 0.000446 | 0.978057 | 0.000142 | 0.977490 | 0.001583 |
| Ceftiofur | 0.990266 | 0.000442 | 0.990364 | 0.000092 | 0.989752 | 0.000444 |
| Azithromycin | 0.992052 | 0.000933 | 0.991365 | 0.000206 | 0.990768 | 0.000691 |
| Chloramphenicol | 0.995203 | 0.000311 | 0.992302 | 0.000056 | 0.992674 | 0.000458 |
We also examined a set of 2,413 E. coli sequences containing ESBL producers, but no MIC data were available for these strains. Instead, they were classified as SIR for seven antimicrobials. The set included bovine, clinical and environmental samples isolated between 1970 and 2017 in Canada, Thailand and the United KingdomFootnote 11Footnote 23Footnote 24. We analyzed the sequences with the k-mer approach described above and used them to train models to classify isolates as SIR for each antimicrobial. The average accuracies of the models across the seven antimicrobials were 89.18% (± 5.44) for XGBoost, 89.25% (± 4.43) for support vector machines and 89.18% (± 5.20) for artificial neural networks (Figure 2).
Figure 2: Accuracies of three machine learning models trained on the top 1,000 11-mers, and used to predict susceptible, intermediate and resistant classifications for seven Escherichia coli antimicrobials
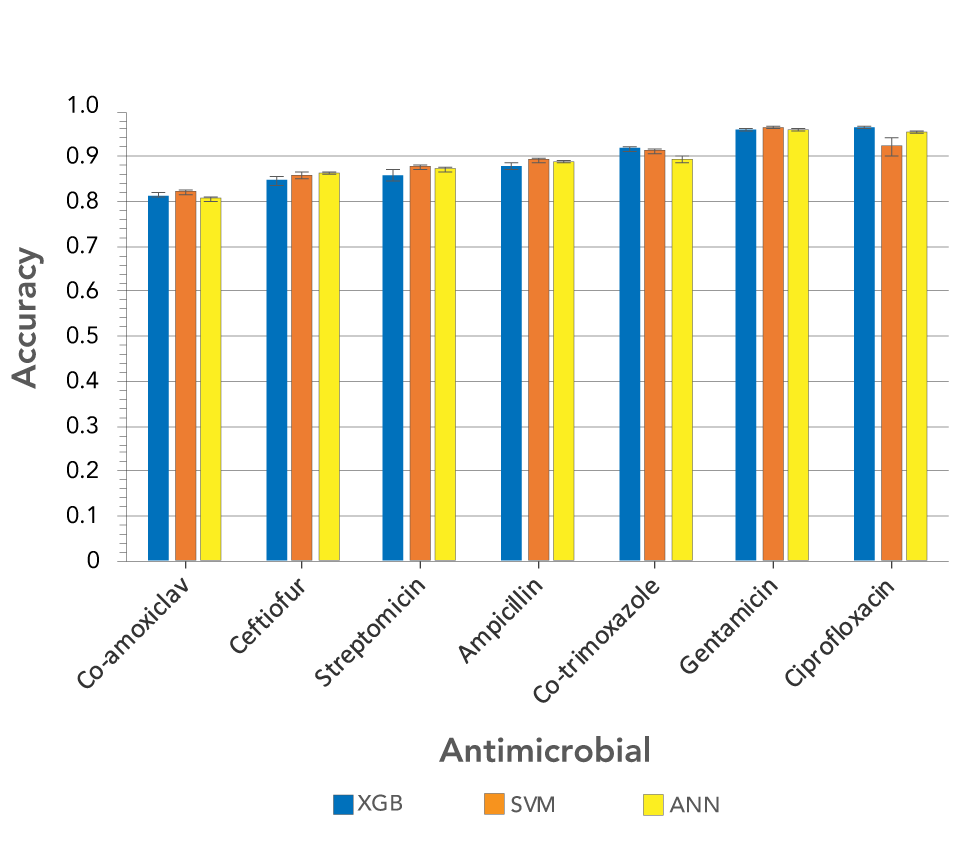
Text description: Figure 2
Figure 2: Accuracies of three machine learning models trained on the top 1,000 11-mers, and used to predict susceptible, intermediate and resistant classifications for seven Escherichia coli antimicrobials
| Antimicrobial | XGB | SVM | ANN | |||
|---|---|---|---|---|---|---|
| Mean | Standard deviation | Mean | Standard deviation | Mean | Standard deviation | |
| Co-amoxiclav | 0.814763316 | 0.005384161 | 0.821363594 | 0.004471698 | 0.806992567 | 0.005409766 |
| Ceftiofur | 0.847536534 | 0.010266626 | 0.857223382 | 0.008464752 | 0.863486840 | 0.001456679 |
| Streptomicin | 0.857430063 | 0.011451457 | 0.876519833 | 0.006231367 | 0.872638158 | 0.005196798 |
| Ampicillin | 0.879085671 | 0.006961795 | 0.892516300 | 0.004019401 | 0.889839555 | 0.004015798 |
| Co-trimoxazole | 0.917826722 | 0.005429594 | 0.912897704 | 0.005756562 | 0.892096490 | 0.006880167 |
| Gentamicin | 0.961769581 | 0.002661451 | 0.964425197 | 0.000710687 | 0.960550072 | 0.001277735 |
| Ciprofloxacin | 0.964520099 | 0.001878239 | 0.922696229 | 0.018860678 | 0.955697705 | 0.002089745 |
Discussion
As we have shown, the ML methods we employed did not rely on specific reference genomes, or a priori knowledge of the mechanisms of resistance, but on the classification of organisms into broad phenotypic groups. It is the ML models that identify the underlying genomic differences that are most associated with the phenotype. This has the double benefit of not requiring mechanistic knowledge and has the potential for identifying novel genomic determinants of the phenotype under study. These novel features extracted from the models have enormous potential benefit: as in the case of AMR, they can be used to grow established public databases of resistance mechanisms, and they can be used as potential targets for rapid diagnostics in subsequent in silico or wet lab assays.
ML models can rapidly and accurately predict AMR using WGS data, from SIR classification to quantitative MIC values. For AMR predictions, XGBoost models were shown to train faster, use less memory and be more accurate than deep-learning methods. In addition, XGBoost and support vector machine models can be used to determine the specific regions of the genome that are most predictive of a phenotype. This is very difficult with the “black box” implementation of a neural network; however, artificial neural networks still excel in complicated network modelling and therefore should not be excluded from future studies in genomics.
AMR data typically suffer from substantial class imbalance, which can result in high accuracy models that are of no value, such as the case of co-trimoxazole in our Salmonella data, where more than 95% of the samples were within one dilution of each other, resulting in a model capable of 95% accuracy without learning anything from the underlying data.
Nguyen et al.Footnote 10 trained XGBoost regressors on a dataset containing 4,500 non-typhoidal S. enterica whole genome sequences (from a larger dataset of 5,278 samples, of which 4,595 were also in our dataset). These models had a cross-validation accuracy of 95% for the same 10 antimicrobials included in our current study. Nguyen et al.Footnote 10 used a single regressor trained on all 15 antimicrobials at once, which took 51 hours to train and peaked at 1,184 GB of memory on 170 cores (Intel Xeon E5-4669v4 CPU)Footnote 10. The XGBoost classifiers trained in our current study improved upon these training times as well as memory usage and accuracy. The XGBoost classifiers did this by creating per-antimicrobial models and initially selecting only the 1,000 most statistically important features. To better compare the accuracies of these models, an independent dataset should be used instead of relying on the reported cross-validation accuracies.
The E. coli dataset included 1,935 isolates from a previous study by Moradigaravand et al.Footnote 11. Their methods required the isolation year for each sequence and data preprocessing in the form of pan-genome determination and population structure calculationFootnote 11. In contrast, our methods required only the genome sequence paired with laboratory-determined resistance phenotype, which allows classification as well as identification of novel regions not currently known to be associated with AMR. The regions could be used for subsequent in silico or wet lab diagnostic tests.
While broader classifications, such as SIR, are common for laboratory diagnostics, and useful for establishing treatment guidelines for a bacterial infection, the breakpoint criteria for these categories are established by committees, with some disparity between regions. The prediction of quantified values in the form of MICs will be of most use in future, even if they are subsequently used for classifying bacteria into broader categories such as SIR.
Though the results of these studies are encouraging, over-interpretation of results is a problem with genomic data due to the high number of features used to make predictions relative to the smaller sample size of the number of genomes. This can lead to over-fitting of data and poor performance of models, both of which we have tried to address in the methods of this studyFootnote 25.
Use of ML has proved successful for AMR prediction in other pathogens, including Mycobacterium tuberculosis, where new resistant genetic signatures were identifiedFootnote 26. ML has also proved useful in the identification of novel antimicrobial compounds, which has historically been fraught with high failure rates in pharmaceutical companiesFootnote 27.
ML research on S. typhimurium found that more than 80% of host source could be attributed using protein variants. This result was obtained using support vector machine (SVM), artificial neural networks and Random Forest modelsFootnote 28. What is particularly interesting from this study is the overlap between the animal reservoir and human cases. This indicates that not all isolates of a particular pathogen represent the same disease risk and suggests that more specific points of control could limit human infection. In addition, as more than 60% of human pathogens are of zoonotic origin, ML holds promise for identifying emerging pathogens by analyses of host adaptation of current animal pathogensFootnote 29.
Despite the proven usefulness of ML, bacteria are constantly evolving, and so our models, as they are only as good as the data they are trained on. The power of these techniques must be tempered by their judicious use. In addition, class and species-specific models are still required to generate meaningful results, for example, one model per drug per species for predicting AMRFootnote 30.
It should be noted that ML does not always accurately capture complex interactions and that improved modelling alone cannot compensate for sampling bias or an incomplete or error-prone dataset.
Conclusion
As demonstrated in this overview, artificial intelligence has already improved infectious disease identification and characterization, the benefits of which will affect public health and animal health laboratories around the world. For example, genomic regions identified as predictive for specific AMR classes could be used for rapid downstream identification and classification, including in silico pipelines and wet lab applications such as polymerase chain reaction.
The near-future promises exciting developments, such as using ML to identify bacteriophages that lyse specific groups of pathogenic bacteria, enabling phage therapy in place of traditional antimicrobialsFootnote 31. Lastly, “whole phenotype” characterization, with the ability to predict integral membrane protein expression, is becoming more likelyFootnote 32; and biofilm formationFootnote 33.
Despite this, the size of the datasets required to effectively train ML models mean that desktop computers are often incapable of analyzing the data. Those without access to the necessary resources must instead use analytical approaches that reduce the computational burdenFootnote 34. Fittingly, the use of ML itself has led to an increase in speed of mechanistic models, in some cases over four orders of magnitudeFootnote 35.
We are just at the beginning of the coupling of vast amounts of genomic data and artificial intelligence, with the promise of new discoveries that will improve most aspects of animal and human health from the burden of enteric bacterial pathogens.
Authors’ statement
- RJS — Data curation, formal analysis, methodology, software, validation, visualization, original draft, editing
- JM — Data curation, formal analysis, methodology, software, validation, visualization, original draft, editing
- VPJG — Conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, original draft, editing
- AZ — Conceptualization, funding acquisition, methodology, project administration, resources, supervision, original draft, editing
- CRL — Conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, original draft, editing
Conflict of interest
None.
Funding
JM, AZ, CRL: This work has been supported by the Antimicrobial Resistance – One Health Consortium grant to AZ and CRL from the Alberta Ministry of Economic Development, Trade, and Tourism.
RJS, VPJG, CRL: This work has been supported by the Genomics Research and Development Initiative project on antimicrobial resistance. This work was additionally funded by the Public Health Agency of Canada, the Canadian Food Inspection Agency and the University of Lethbridge.
