
Archived - Joint Audit and Evaluation of the Medical Devices Program 2013-14 to 2019-20
Table of Contents
- List of Figures
- List of Acronyms
- Executive Summary
- 1.0 Introduction and Background
- 2.0 Safety, Effectiveness and Quality of Medical Devices in Canada
- 3.0 Providing Access to Medical Devices in Canada
- 4.0 Communicating with Stakeholders and Canadians
- 5.0 Program Organization and Governance
- 6.0 Program Resources
- 7.0 Conclusions and Recommendations
- Management Response and Action Plan
- Appendix A – Joint Engagement Scope and Methodology
- Appendix B - Internal Audit Summary
- Appendix C - Summary of Evaluation Findings
- Appendix D – Logic Model
- Appendix E – MDP Key Roles and Responsibilities at the Time of the Engagement
- Endnotes
List of figures
- Figure 1: Example of Health Canada’s Involvement in the Medical Device Life cycle
- Figure 2: Organizational structure of the Medical Devices Program at the time of the engagement
- Figure 3: Proportion of medical device incident reports entered within service standards
- Figure 4: Targeted risk communications were disseminated within service standards for 3 of 5 years
- Figure 5: Proportion of DEVICE licence applications in backlog
- Figure 6: Proportion of DEVICE licence applications that complied with regulatory requirements at first review and after additional information was requested and provided
- Figure 7: Organizational structure of the Medical Device Programs in the United States and Ireland
- Figure 8: Resources and workload of the medical devices and pharmaceutical surveillance functions (2015-16)
List of Acronyms
- CMDSNET
- Canadian Medical Devices Sentinel Network
- HPFB
- Health Products and Food Branch
- HPSEB
- Health Products Surveillance and Epidemiology Bureau
- IMDRF
- International Medical Device Regulators Forum
- IT
- Information Technology
- MDB
- Medical Devices Bureau
- MDCCD
- Medical Devices and Clinical Compliance Directorate
- MDCP
- Medical Device Compliance Program
- MDP
- Medical Devices Program
- MDSAP
- Medical Device Single Audit Program
- MHPD
- Marketed Health Products Directorate
- ROEB
- Regulatory Operations and Enforcement Branch
- RTOF
- Regulatory Transparency and Openness Framework
Executive Summary
This report presents findings from a Joint Audit and Evaluation of Health Canada’s Medical Devices Program (MDP). The engagement examined MDP’s activities from April 2013 to August 2019. The MDP aims to ensure that Canadians have access to safe, effective, and quality medical devices through:
- approval of establishment and medical device licences;
- surveillance of devices available in Canada;
- inspections of manufacturers, importers, and distributors; and
- communication to Canadians about risks related to devices.
In 2018-19, MDP planned expenditures were about $33M, $15M of which was funded by Health Canada. The remaining program funds were recovered through fees for regulatory services (e.g., licences to sell devices) Footnote 1 1.
Key Findings
Ensuring the Safety, Quality and Effectiveness of Medical Devices in Canada
The MDP had sufficient controls in place for its pre-market activities to ensure the safety, effectiveness, and quality of medical devices. It also completed most of its post-market activities within service standards.However, delays in processing medical device incident reports affected the Program’s ability to proactively detect such incidents. The MDP also experienced delays in completing the assessment of medical devices incidents.
While these challenges represent a risk for the Department, there is no evidence that they affected Health Canada’s ability to ensure the safety of medical devices. The Department was perceived as a trusted regulator by the majority of external key informants and available data suggests that the safety of medical devices in Canada was comparable to other countries.
The medical device environment is complex and evolving rapidly, with the development of novel devices using innovative technologies, such as digital technology, artificial intelligence, and 3D printing. Health Canada made significant strides to adapt its regulatory framework to remain effective at regulating such rapidly changing and complex devices. The Program also progressed in its efforts to integrate sex- and gender-based analysis considerations into the delivery of its activities. However, there was room to integrate these considerations more systematically across all program functions.
Providing Access to Medical Devices in Canada
The MDP made progress in balancing safety with access to medical devices. It processed the majority of device licence applications within services standards, but most of those applications did not meet regulatory requirements when initially submitted to Health Canada. This may have increased their processing time. The reasons why the majority of device licence applications did not meet program requirements when first submitted are not fully understood. Publicly available guidance documents outlining these requirements were difficult to find and were sometimes outdated. At the same time, the Program implemented various measures to improve the industry’s understanding of the regulatory process (e.g., an e-learning course) and aligned its requirements with those of other countries, in an effort to improve access to devices in Canada. It has also put in place various initiatives to streamline the access to medical devices, such as the Special Access Program, which allows health care professionals to gain access to medical devices that are not approved for sale in Canada for use in emergencies or when conventional therapies have failed, are unavailable, or are unsuitable to treat a patient.
Communicating with Stakeholders and Canadians
The MDP increased its engagement with stakeholders through meetings with industry representatives, discussions with health care professionals, targeted outreach to patient groups, and opportunities for stakeholders and Canadians to participate in online consultations. While these efforts were well received by industry, improvements are still required for Health Canada to become a go-to source of information for non-industry stakeholders and Canadians.
Program Organization and Governance
Various factors affected the Program’s ability to function efficiently. Roles and responsibilities across the pre-market and post-market surveillance and compliance and enforcement functions were not always clearly defined, particularly at the working level. However, the Program delineated clear roles and responsibilities among scientific evaluators and medical officers.
In addition, with the creation of the new Medical Devices Directorate, the roles and responsibilities between the merged pre and post market functions are intended to be further clarified through the development of revised operating procedures and processes. The Program is based on the Medical Devices Regulations which are based on the level of risk for various classes of medical devices. All the Medical Devices Program’s core activities centre on a risk-based approach for regulating medical devices through their life cycle.The MDP, however, did not have a comprehensive framework defining roles and responsibilities, nor an integrated risk management and information-sharing strategy. This lack of clarity regarding roles and responsibilities may have contributed to the development of parallel processes across MDP functions.
Additionally, the MDP’s information was stored across several repositories. Available information had to be consolidated from various sources in order to understand the full history of Health Canada activities in relation to a medical device.
Program Resources
A lack of resources challenged the post-market surveillance function’s ability to process incident reports and complete signal assessments within service standards. Overall, post-market surveillance activities received about only 7% of MDP’s budget in 2018-19. Additionally, in recent years, the number of staff working in medical device surveillance decreased. In comparison, the number of staff increased in the other two MDP functions.
With the introduction of mandatory incident reporting by hospitals in December 2019, the workload of the post-market surveillance function and the compliance and enforcement function may significantly increase. At the time of the engagement, the workload implications of mandatory reporting were not fully analyzed. However, it should be noted that recent investments have been made through various projects. This is expected to result in an increased intake of reports that are processed through new semi-automated systems. A risk still exists, as the challenges regarding the timely assessment and completion of some surveillance activities may be exacerbated if capacity issues are not addressed in these areas.
Recommendations
-
The Health Products and Food Branch should address current resource gaps in the post-market functions responsible for processing incident reports and assessing incidents. The Regulatory Operations and Enforcement Branch should consider the subsequent impacts on the resource needs of the compliance and enforcement function. As well, both Branches should ensure that the MDP has the capacity and tools to address the mandatory reporting by hospitals introduced in December 2019, and associated enforcement of hospital vigilance practices.
MDP’s surveillance activities were challenged by a lack of resources and could not always be completed within service standards. The introduction of mandatory reporting of medical device incidents by approximately 775 Canadian hospitals in December 2019 will likely further affect the post-market surveillance function’s ability to manage its workload, as the number of incident reports submitted to Health Canada may double. Mandatory reporting may also affect the resources and activities of the compliance and enforcement function, as inspectors will be required to monitor potential under-reporting and inspect hospitals. However, at the time of the engagement, Health Canada had not fully analyzed the implications of mandatory reporting on program workload.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should continue their efforts to adapt the MDP, through both domestic and international initiatives, to address challenges emerging from the rapidly changing life cycle of medical devices and the use of novel technologies.
While Health Canada made significant strides to adapt its regulatory framework, other challenges need to be addressed to ensure that the MDP remains effective moving forward. For instance, at the time of the engagement, there was no mechanism to address the growing issue around medical devices offered through a subscription or service (e.g., online diagnostic tools), or to track individuals using implanted devices so they can be notified directly by Health Canada if there is an incident.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should consider and document how to further integrate sex- and gender-based analysis plus (SGBA+) into the life cycle of medical devices, including providing guidance to applicants and increasing awareness of MDP staff.
Evidence shows that men and women are affected differently by medical devices due to sex and gender disparities. While Health Canada progressed in its efforts to integrate sex and gender considerations into the MDP, there were additional opportunities to further integrate these considerations in all program activities, where relevant.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should maintain and scale up communication and engagement efforts with industry stakeholders, health care professionals and organizations, and patient groups. It is also recommended that the Health Product and Food Branch improve the timeliness and accessibility of program information available online. This includes ensuring that guidance documentation is reviewed and updated as required across all of the MDP's operations, including on the Government of Canada website.
Health Canada’s efforts to increase communication and engagement with stakeholders were well received by industry representatives. However, Health Canada was not a go-to source of information for health care representatives, patient groups, and the population in general. There was also a need to improve the timeliness and accessibility of information online. In particular, outdated guidance documents may have contributed to inaccuracies in application submissions for medical device licences. Also, industry key informants mentioned having difficulty finding the information and guidance required to support the development of their applications.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should ensure that roles and responsibilities at the operational level are clearly defined and distributed, and that program risks are identified and managed.
Roles and responsibilities of operational-level staff were not clearly defined across the three MDP functions. The MDP also did not have a comprehensive framework defining roles and responsibilities, nor an integrated risk management and information-sharing strategy. Challenges with roles and responsibilities may be addressed, in part, with the implementation of the new Medical Devices Directorate announced in late 2019, which will integrate the pre-market function and the function responsible for the assessments of medical device incidents. However, the compliance and enforcement function and the Health Products Surveillance and Epidemiology Bureau (i.e., the group responsible for processing medical device incident reports) will remain with their original directorates.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should ensure that a data management approach supports the monitoring of medical devices throughout their life cycle.
The MDP’s information was fragmented across several repositories. This challenged the Program’s efficiency and ability to take a life cycle approach to regulating and monitoring medical devices.
1.0 Introduction and Background
This report presents key findings and recommendations of the Joint Audit and evaluation of Health Canada’s Medical Devices Program (MDP). The engagement examined MDP’s activities from April, 2013 to August, 2019.
Areas examined include the adequacy of program controls, risk management, and the integration of sex- and gender-based analysis plus (SGBA+), as well as the MDP’s impacts, how it was positioned to adapt to an increasingly complex environment, and how the organizational structure at the time of the engagementFootnote ii supported an efficient delivery of MDP’s activities.
Since March 2020, the Medical Devices Program has been focused on meeting the immense needs of health professionals, patients and industry in the fight against COVID-19. The central role of the Program in protecting the health and safety of Canadians has been placed in the spotlight, resulting in the fast tracking of a number of significant regulatory activities, including the drafting and implementation of various Interim Orders, all targeted at providing expedited access to COVID-19 medical devices (e.g., testing devices, ventilators, gloves) at this critical time. In addition, processes have been adapted to expedite the issuance of a significant increase of Medical Device Establishment Licences in order to ensure that critical personal protective equipment (PPE) is available to combat COVID-19. Lastly, the intake of incident reports, post-market monitoring, health risk assessments, and compliance and enforcement activities have required significant resources.
Although the finalization of the Office of Audit and Evaluation’s (OAE) report and Management Response Action Plan has been delayed due to COVID-19 priorities, the Program has continued to be mindful of the OAE’s recommendations. The pandemic has provided an opportunity to make progress in addressing some of these recommendations. Specifically, the Program has continued to engage domestic and international stakeholders in relation to COVID-19. Furthermore, the Program has been cognizant of historical challenges with the existing website when developing new content and webpages to share information in relation to COVID-19.
Data was collected from a review of the literature and program documents, performance measurement data and financial information as well as from case studies, a comparative analysis, and key informant interviews with MDP staff and stakeholders. Analyses undertaken also included the testing of a sample of operational transactions from April 1st 2018 to September 2019 (see Appendix A for further details).
1.1 Benefits and Risks of Medical Devices
Medical devices, as defined in the Food and Drugs ActFootnote 2 2, cover a wide range of medical instruments, apparatus, contrivance, or similar articles, as well as in vitro reagents used in the treatment, mitigation, diagnosis, or prevention of a disease, disorder, or abnormal physical condition. Without medical devices, many common medical procedures would not be possible, such as bandaging a sprained ankle, diagnosing infectious diseases, or implanting artificial hips.
The Medical Devices Regulations Footnote 3 3 group medical devices into one of four risk classes: Class I represent the lowest risk (e.g.,
tongue depressors) and Class IV represents the highest risk (e.g., artificial heart valves).
1.3 million different types of medical devices are available for sale in Canada. These devices range from bandages, hospital beds, pacemakers, implants, and MRI machines, to smart watches.
Many Canadians are dependent on medical devices to improve their health and quality of life. In some cases, medical devices like pacemakers are also credited with saving lives. According to several external key informants who are using medical devices, having access to a device improved their health and allowed them to engage in everyday activities such as work, sports, and travel.
Medical devices also provide economic benefits for the health care system. For instance, interventional cardiology and the use of drug-secreting stents, instead of coronary bypass surgeries, was linked to substantial health system savings and improved cardiac disease management, which in turn enhanced patient outcomes, including reduced recovery times and increased capacity for managing cardiac diseaseFootnote 4 4. According to the Canadian Institute for Health Information Patient Cost Estimator, implanting a stent costs approximately $6,000, compared to $26,680 on average for coronary bypass surgeryFootnote 5 5. Additionally, a Diabetes Canada study found that specialized devices to treat diabetic foot ulcers can help prevent amputations, which could result in direct cost savings ranging from $48 to $75 million a year for the province of Ontario Footnote 6 6.
Although medical devices are valuable to Canadians and the health care system, they also involve a certain level of risk. Incidents can occur as a result of a device malfunction, application or user error, the inherent risk of a device, as well as the negative interaction between the device and another device or medical procedureFootnote 7 7. Such incidents may result in health issues or illnesses for the user, and in some cases death. For instance, in 2014, the Paradigm Insulin Pump was recalled by the manufacturer because inadvertent misuse could have resulted in the delivery of an insulin dose exceeding the amount intended by the user and potentially cause hypoglycaemiaFootnote 8 8.
The MDP aims to mitigate the risks associated with medical devices by ensuring that Canadians have access to safe, effective, and quality devicesFootnote 9 9. The MDP’s activities cover the life cycle of a medical device, from providing regulatory guidance to manufacturers during their research and product development, through to the review of device licence applications, as well as monitoring, prevention, and intervention for devices that are licensed for sale in CanadaFootnote 10 10. The MDP's compliance and enforcement function also monitors the extent to which manufacturers, distributors, and importers are compliant with the Food and Drugs Act and Medical Devices Regulations. It delivers Medical Device Establishment Licences, conducts proactive inspections for manufacturers of Class I devices, and for importers and distributors of all medical devices, as well as responsive inspections of manufacturers, importers, and distributors of all classes of devices. Figure 1 below provides examples of some of the MDP’s activities that cover the various stages of the medical device life cycle. Of note, Health Canada does not licence Class I medical devices, but monitors them through the establishment licensing of manufacturers, importers, and distributors of medical devices in CanadaFootnote 11 11.
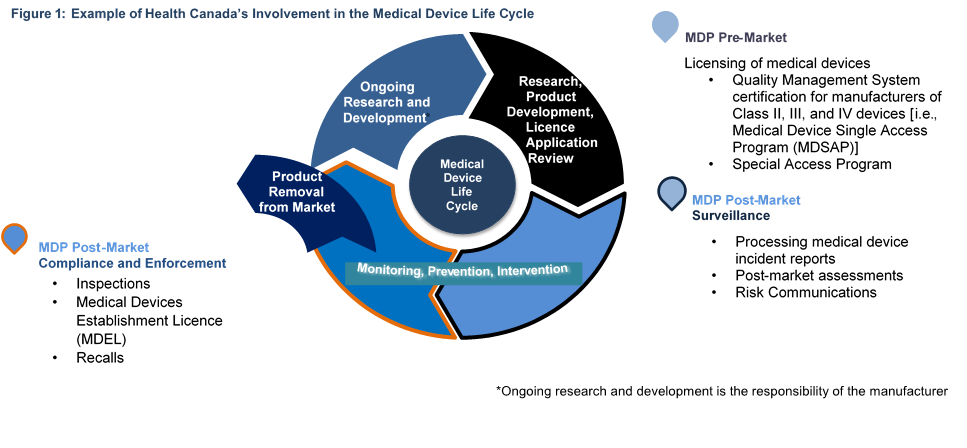
Figure 1 - Text description
This figure illustrates some of the Health Canada Medical Device Program functions and examples of activities to regulate the life cycle of medical devices. The figure is a 4-part circular model. Each section represents a particular part of the medical device life cycle. For each section, there is also an accompanying statement providing examples of activities of the Medical Device Program.
The first section of the life cycle model is labeled: Research, Product Development, Licence Application Review. The corresponding function is MDP Pre-Market Activities, with a bullet list of the following activities:
- Licensing of medical devices;
- Quality Management System certification for manufacturers of Class II and IV devices (i.e. Medical Device Single Access Program (MDSAP); and,
- Special Access Program.
The second and third sections of the model share the label: Monitoring, Prevention & Intervention. The second sections shows the MDP Post-Market Surveillance with the following examples of activities:
- Processing medical device incident reports;
- Post-market assessments; and,
- Risk Communications.
The third section shows the function: MDP Post-Market Compliance & Enforcement whit the following examples of activities:
- Inspections
- Medical Devices Establishment Licence (MDEL)
- Recalls
The third section of the model also has a smaller arrow labeled: Product Removal from Market. This arrows indicates that when a device reaches this point, it is no longer part of the model or the work of Health Canada.
The final section of the model is labeled: Ongoing Research & Development with a note indicating that ongoing research and development is the responsibility of the manufacturer. This section is included to show the entire life cycle of a medical device. It also points back to the first section of the model, which is MDP Pre-Market Activities
1.2 Health Canada’s Medical Devices Program
At the time of the engagement, the MDP was organized along separate pre-market (i.e., licence approval) and post-market functions (i.e., surveillance, as well as compliance and enforcement), each responsible for different program activities (see Figure 2).
As of November 2019, part of the Program was restructured into a new Medical Devices Directorate. This new directorate is comprised of the pre-market function and the Marketed Pharmaceuticals and Medical Devices Bureau, from the post-market surveillance function. The other half of the surveillance function (i.e., the Health Products Surveillance and Epidemiology Bureau) and the compliance and enforcement function remain with their original directoratesFootnote 12 12.
In 2018-19, MDP planned expenditures were about $33M, $15M of which was funded by Health Canada. The remaining expenditures were funded through the Program’s licensing fees.
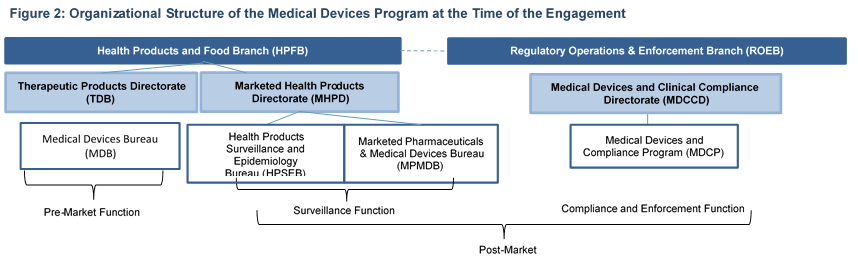
Figure 2 - Text description
This figure illustrates the organizational structure within Health Canada that comprises the primary functions of the Medical Device Program.
Two Branches deliver program activities: the Health Products and Food Branch (HPFB) and the Regulatory Operations & Enforcement Branch (ROEB).
Under HPFB, the Therapeutic Products Directorate (TPD) and Marketed Health Products Directorate (MHPD) have different divisions involved in the program:
- Under the Therapeutic Products Directorate (TPD), the Medical Devices Bureau (MDB) is responsible for the pre-market function.
- Under the Marketed Health Products Directorate (MHPD), the Health Products Surveillance and Epidemiology Bureau (HPSEB) and Marketed Pharmaceuticals and Medical Devices Bureau (MPMDB) is responsible for the post-market surveillance function.
Under the Regulatory Operations & Enforcement Branch (ROEB), the Medical Devices and Clinical Compliance Directorate (MDCCD) has a division named the Medical Device Compliance Program (MDCP). This division delivers the compliance and enforcement function, which is also a post-market function.
Overall, at the time of the engagement, the MDP’s activities were similar in scope to other Health Canada regulatory programs (e.g., pharmaceuticals)Footnote 13 13. They were also aligned with the Department’s mandate and stewardship role of protecting Canadians and facilitating access to products that are vital to their health and well-beingFootnote 14 14.
The engagement focused on the three main functions of the MDP; however, it should be noted that program activities are supported by the Health Products and Food Branch’s Resource Management and Operations Directorate and Policy Planning and International Affairs Directorate, as well as the Regulatory Operations and Enforcement Branch’s Planning and Operations Directorate and Policy and Regulatory Strategies Directorate. Moreover, the Program’s risk communications are developed in collaboration with the Marketed Health Products Directorate’s Office of Policy, Risk Advisory and Advertising.
2.0 Safety, Effectiveness and Quality of Medical Devices in Canada
2.1 Pre-market Process Controls
The MDP implemented adequate controls within its pre-market function, and the majority of medical device licence applications were processed consistently. However, guidance documents available to staff were outdated, which could represent a risk, especially for new employees who are unfamiliar with the application process.
The pre-market function established controls and provided guidance documents to MDP staff outlining the intake, screening, evaluation, and approval of medical device license applications. Transactional testing shows that approximately 94% of device licence applications were processed by the pre-market function in accordance with all of the required controls. The processing errors observed in the remaining 6% of applications were minor deficiencies, including administrative errors, formatting inconsistencies, and partially completed internal documents.
Some of the controls implemented by the pre-market function included:
- For combination products (see definition below), the MDP developed a Combination Products Policy, as well as several standard operating procedures for reviewing device licence applications for these products. The majority of device licence applications for combination products received by Health Canada in 2017-18 and 2018-19 were processed consistently, in accordance with the MDP’s standard operating procedures. Because combination products involve more than one Health Canada directorate, consultations held between the directorates when reviewing license applications for these products, were recorded, and stored in a document repository named docuBridge.
- The MDP required all staff in the pre-market function to sign conflict of interest disclaimers when they were hired and on an annual basis. These disclaimers were tracked in an electronic database. Moreover, any contractors hired by the pre-market function to review applications had to answer an extensive list of questions on conflicts of interest related to their contracts. They were not assigned to applications in which they may have had a potential vested interest.
- The pre-market function also established a control mechanism consisting of issuing licences with conditions for certain Class III and IV devices. These conditions aim to address any safety or effectiveness concerns when additional information is required from manufacturers during the application process. These licences with conditions helped ensure that medical devices with potential safety and effectiveness concerns were monitored following license approval.
Key Definition: Combination products contain multiple different components, such as a drug and a medical device, or a biological component and a medical device. These types of products typically require joint reviews between the MDP and other programs or directorates within Health Canada (e.g., the Biologics and Genetic Therapies Directorate). The primary component of the product determined which program or directorate takes the lead on reviews.
Although the pre-market function implemented processes and controls that were followed almost all of the time, many of the function’s internal guidance documents that support the review process for device licence applications were significantly outdated. For instance, some of the guidance documents for intake and application administration did not reflect the MDP’s transition to docuBridge in 2017. This represents a risk as outdated information has the potential to mislead program staff when reviewing device licence applications. This risk is likely greater for newly hired staff who may be unfamiliar with undocumented processes.
2.2 Post-market process controls
The MDP experienced delays in processing mandatory incident reports. This represents a risk to the MDP’s ability to ensure the continued safety, effectiveness, and risk-benefit profile of medical devices.
The MDP detected, monitored and followed up on medical device incidents through the collection of:
- mandatory incident reports from manufacturers or importers;
- foreign incident reports from international regulatory agenciesFootnote 15 15;
- voluntary incident reports, through the Canadian Medical Devices Sentinel Network (CMDSNet)Footnote iiiiFootnote 16 16; and
- voluntary incident reports from other sources (e.g., private clinics, industry, individuals using a medical device)Footnote 17 17.
Once received, incident reports were manually processed by MDP staff.
In addition to receiving incident reports, the MDP also conducted active surveillance of incidents through literature and media scans, monitored actions taken by international regulatory agencies, followed-up with manufacturers to ensure reporting, and implemented compliance actions in cases of underreporting. It also had several working groups and committees, with representatives from each program function that were responsible for identifying and screening these incidents.
From 2013 to 2017, the MDP received between 11,070 and 14,931 incident reports per yearFootnote 1818. The majority were mandatory reports, for which there are two categories:
- “10-day reports”, meaning that the manufacturer has 10 days to report incidents that have led to the death or serious deterioration of an individual. The MDP received 1,814 of these reports on average per year between 2013 and 2017. The processing of 10-day reports was prioritized over other reports. MDP staff had to complete the data entry for these report within a 15-day service standard.
- “30-day reports”, meaning that the manufacturer has 30 days to report all other type of incidents. The MDP received 10,317 of these reports on average per year between 2013 and 2017. The service standard for the data entry of those reports was 84 daysFootnote 19 19.
An examination of reports by fiscal year showed that the entry of 10-day reports within service standards improved over the last few years, as illustrated in Figure 3Footnote iii iii. During the same period, the number of 30-day reports entered within service standards, decreased significantly. Additional testing showed that 10-day reports exceeded service standards by up to 319 and 30-day reports exceeded service standards by up to 370 days. This included delays of up to 153 days for high-risk Class IV devices.
Internal documentation suggests that the delays experienced at the beginning of fiscal year 2019-20 were attributed to the receipt
of approximately 1,600 incident reports from a manufacturer early in 2019. Such a situation can occur when the compliance and
enforcement function discovers that a manufacturer has under-reported incidents, and is then required to send all those reports
to Health Canada. According to a few internal key informants, at the time of the engagement, the surveillance function did not have the capacity to process such a large surge of reports within program service standards.
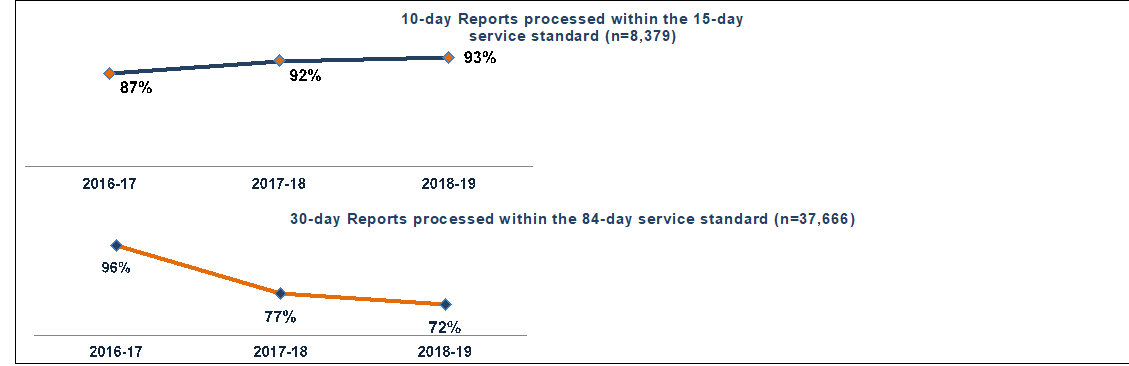
Figure 3 - Text description
This figure contains two separate line graphs. The first graph shows the proportion of 10-day reports processed within the 15-day service standard (n=8,379) over time:
- 87% in 2016-17
- 92% in 2017-18
- 93% 2018-19
The second graph is for 30-day reports processed within the 84-day service standard (n=37,666). The line graph shows the results by fiscal year starting in 2016-17 through 2018-19. The results are as follows:
- 96% in 2016-17:
- 77% in 2017-18:
- 72% in 2018-19:
The source of information for this graph is results from the audit transactional testing.
One control to ensure that 10-day reports were dealt with promptly was to verify that incidents were properly labelled as either a 10-day or 30-day report when submitted to Health Canada. However, at the time of the engagement, the triaging of reports was omitted from the intake process for medical device incidents. As a result, 10-day reports were miscategorized as 30-day reports when they were received by Health Canada, and remained in the processing queue for a longer period than would normally be expected for a 10-day report. This is a potential risk to the Canadian public, as emerging serious incidents may not be caught and addressed in a timely manner.
Some internal key informants explained that delays in completing the data entry for incident reports, lack of triaging incoming reports, and inconsistencies in the labelling of reports challenged the Program’s ability to conduct trend analysis, proactively address emerging and suspected safety issues, and conduct compliance and enforcement activities.
That being said, there were delays entering mandatory reports, as evidence shows that the voluntary reports received by the CMDSNet, which were considered to be at the same priority level as 10-day reports, were all entered within the required service standard (i.e., 15 days).
The MDP experienced delays in the completion of signal assessments, but most risk communications continued to be disseminated within service standards.
When an incident is signalled through reporting or monitoring, staff in the post-market surveillance function assess and review such signals in order to determine the right course of action to address the incident. MDP performance data shows that the majority of signal assessments and reviews were completed within the 130-day service standard for signal assessments and 60 days for reviews.
The proportion of these activities meeting service standards steadily declined from 100% in 2014-15 and 2015-16 to 89% in 2018-19. This downward trend continued in the first part of fiscal year 2019-20, as only 50% of signal assessments were completed within service standards between April and June, 2019. However, while operational data shows that service standards were met in the majority of cases before April 2019, they did not account for the time required to collect additional information about the incident from the manufacturer or other sources. If this time, excluding industry delays, is taken into consideration, approximately 30% of signals from fiscal year 2018-19 did met the required service standards.
Key Definitions
Signal Assessments aim to gather safety information on a priority issue in order to characterize it, come up with strategies to deal with it, and recommend solutions to prevent or mitigate identified risks.
Summary Safety Reviews provide information collected from monitoring licensed devices, based on several potential sources of information (e.g., domestic and foreign incident reports, medical and scientific literature) to identify potential safety issues. Each summary outlines what was assessed, what was found, and what action was taken by Health Canada, if any.
According to some internal key informants and program documents, activities pertaining to signal assessments were challenged by a lack of resources, including the redirection of staff towards other activities, such as addressing increased media attention regarding implanted devices and implementing the Medical Devices Action Plan.
The delays in conducting signal assessment did not affect the MDP’s ability to issue risk communications (e.g., recalls, warnings and safety reviews) for devices that had an incidentFootnote 20 20. As shown in Figure 4, since 2014-15, most risk communications regarding medical devices were disseminated within service standards (i.e., 25 days for standard communications, 10 days for non-standard communications, five days for expedited communications, and two days for urgent communications).
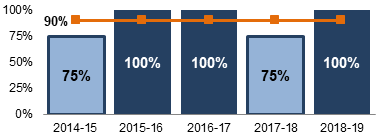
Figure 4 - Text description
This figure is a vertical bar graph showing results by fiscal year from 2014-15 through 2018-19. A target line across the top of the bar graph provides a visual representation of how close results are to the target of 90% of risk communications disseminated within services standards. The following are the results:
- 75% in 2014-15
- 100% in 2015-16
- 100% in 2016-17
- 75% in 2017-18
- 100% in 2018-19
The source of information for this graph is MDP Performance Data.
Compliance and enforcement activities were conducted within service standards.
Program performance data shows that compliance and enforcement activities for medical devices were generally conducted within performance targets:
- The target of 95% of manufacturers inspected who were in compliance with the Food and Drug Act and related regulations was exceeded every year during the 2013-14 to 2018-19 period;
- In 2018-19, the proportion of high-risk cases initially identified as non-compliant, but that eventually resulted in compliance, met the target of 95%; and
- • The target of 100% of Medical Device Establishment Licences reviewed within the 120-day service standards was achieved twice between 2014-15 and 2018-19. In other years, the proportion of licences reviewed within these service standards was higher than 99%.
Key Definition: Medical Device Establishment Licences are issued to importers and distributors of medical devices in Canada, to manufacturers selling medical devices for which they are not the licence holder, as well as manufacturers of Class I devices who distribute their own devices.
Most post-market processes were documented, but there were gaps in the documentation of service standards. In addition, inconsistencies in data entry limited the Program’s ability to analyze trends from incident report data and proactively detect incidents.
The post-market surveillance and the compliance and enforcement functions documented the operating processes and assessment criteria for the prioritization of medical device incidents, in order to ensure that high-priority incidents (i.e., 10-day and CMDSNet reports) were examined promptly. However, the post-market surveillance function did not document its service standards in its internal guidance documents, and the compliance and enforcement function did not establish service standards for the voluntary reports it received. There was no comprehensive review done by the Program to determine if the timelines for data entry, assessment, and coding of incident reports had considered all current processes. In addition, the service standards for data entry have not been reviewed since they were adopted in 2010.
The engagement team expected to find that the processes followed by post-market functions would be reflected in the Program’s guidance documents. However, all the medical device incident reports examined in the Medical Devices System database were missing one or more pieces of the scientific evaluation information that was outlined in standard operation procedures and work instructions. This information was documented in external tracking sheets instead of the database. There were also inconsistencies in data entry for the Program’s Medical Devices System database that affected data integrity. For example, Special Access Program and investigational testing reports (see section 3 for a definition), which did not have a specific label in the database, were labelled as 10-day reports. This limited the ability to interpret statistical analyses of data extracted from the Medical Devices System.
2.3. Safety of Medical Devices Available in Canada
Despite challenges in accomplishing some post-market surveillance and compliance and enforcement activities on time, Health Canada was generally effective at ensuring the safety of medical devices in Canada.
The regulatory process was seen by several external key informants as rigorous because of the requirements to be addressed. These informant’s perceptions echo results from the 2016 Regulatory Transparency and Openness Framework (RTOF) survey, which included stakeholders and members of the public. Among stakeholderFootnote iv iv respondents involved with medical devices (n=20), the majority (95%) agreed that products regulated by Health Canada were safe. Additionally, the majority (75%) of members of the Canadian public with an interest in medical devices (n=40) were confident in the products regulated by Health CanadaFootnote v v.
Several external key informants perceived Health Canada as effective at ensuring the safety of medical devices. Moreover, some experts interviewed perceived the safety of medical devices sold in Canada to be comparable to other countries. Between 2013-14 and 2018-19, Health Canada issued approximately 900 recalls, on average, per year. In comparison, Australia, which received a similar number of Class II to IV licence applications per year (i.e., approximately 2,500 applications in 2017-18, compared to 2,100 in Canada), issued an average of approximately 600 recalls annually.
A comparison of recalls in Canada and the United States suggests that the safety of devices in Canada is similar to the United States. A sample of 60 recallsFootnote vi vi issued by Health Canada in 2018 was assessed to determine whether these recalls were also issued in the United States. If the recall was not issued in the United States, the team further examined whether the device subject to the Canadian recall was approved for sale by the United States Food and Drug Administration. The analysis shows that 70% (42 out of 60) of the recalls analyzed occurred in both countries. Of these:
- The time when the recall was initiated was comparable in both countries. Of the 42 recalls, 16 were initiated in the United States, 14 were initiated in Canada and 12 were initiated in both countries at the same timeFootnote vii vii.
- In most cases (30 out of 42 recalls), the recall was posted on Health Canada’s website before it was posted on the Food and Drug Administration’s website.
- The devices most frequently subject to recalls in both countries were in the diagnostic category (e.g., immunodiagnostic products) with 10 recalls, followed by surgical instruments with eight recalls, and imaging systems (e.g., ultrasound equipment) with seven recalls. Two of the recalls sampled and found in both countries were for implantable devices.
Of the 60 recalls analyzed, 18 were only issued in Canada. Of these:
- Most of the devices were approved for sale in the United States (15 out of 18).
- Three devices were not approved in the United States. These were subject to a recall in Canada due to a: 1) licensing issue, 2) labelling issue, and 3) a reminder from the manufacturer providing some technical information about the device.
- Certain recalls issued in Canada were not relevant for the United States. For instance, a recall pertained to a packaging issue for a certain lot of devices, or the French translation was missing from the product information.
- None of the 18 recalls issued only in Canada were due to a situation where there was a reasonable probability that the use of, or exposure to, the recalled device would cause serious adverse health consequences or death. In this regard, six out of the 18 recalls were in the Health Canada’s hazard classification II, which is related to a situation where the use of, or exposure to, a recalled device could cause temporary adverse health consequences, or where there is not a significant probability of serious adverse health consequences. The other 12 recalls were in the hazard classification III, which is related to a situation where the use of, or exposure to, a recalled device is not likely to cause any adverse health consequences.
- The recalls were primarily related to diagnostic devices (eight out of 18), and two were for implantable devices.
In addition to Canada’s comparability with the United States in terms of recalls, Health Canada was sometimes at the forefront internationally when issuing warnings and recalls. This was the case with the Essure Permanent Birth Control System. Canada was among the first countries to issue a Summary Safety Review and warning to health care professionals, advising them of potentially serious complications with this device. Canada was also one of the first countries to suspend the licence for Biocell breast implants.
Key Definition: Recalls include any action taken by a manufacturer, importer, or distributor of a device to recall or correct the device, or to notify users of its potential defectiveness.
2.4 Adapting to the Evolution of Medical Devices
The medical device environment is increasingly complex and driven by the use of novel technologies.
Ensuring the safety of medical devices is complicated by the fact that the medical device environment is increasingly complex, as novel devices are being developed using new and innovative technologies, such as digital health technology, artificial intelligence, and 3D printing. The life cycle of devices is becoming shorter as they are developed and updated more rapidly.
According to a few key informants and Health Canada documents, the pace of innovation challenged the Department’s regulatory framework, which had requirements and timelines built around traditional models of manufacturing, pre-market approval, and sales, among other thingsFootnote 21 21. For instance, during the period examined, there was a growing trend towards licensing software as a medical device (e.g., online diagnostic tools) and there was no mechanism in the current regulations to address this change.
With the Medical Devices Action Plan and the Regulatory Review of Drugs and Devices, Health Canada made progress in adapting to the changing environment.
During the engagement period, Health Canada implemented several initiatives and tools to adapt its approach to regulating and monitoring medical devices. Many of these initiatives were driven by the 2018 Medical Devices Action Plan and the 2017 Regulatory Review of Drugs and DevicesFootnote 22 22. These initiatives had concrete milestones and their progress was tracked by program staff. A detailed examination of the Medical Devices Action Plan tracking tool shows that its initiatives were generally on track or completed, and that progress updates were accurate and supported by evidence (e.g., publications).
Key Definitions
The 2018 Medical Devices Action Plan focuses on 1) improving how devices get on the market; 2) strengthening the monitoring and follow-up of devices once used by Canadians; and 3) providing more information to Canadians about medical devices.
The 2017 Regulatory Review of Drugs and Devices aims to provide more timely access to drugs and devices for Canadians through collaboration with other health care organizations (e.g., Canadian Agency for Drugs and Technologies in Health).
As part of these initiatives, the MDP built internal expertise and expanded the use of outside experts and stakeholders to strengthen its competencies in key areas of growth for medical devices. This resulted in:
- The development and implementation of a Digital Health Division in 2018 to support a more targeted and comprehensive pre-market review of medical device licence applications related to digital health technologies;
- The creation of two new standing scientific advisory committees to obtain scientific, technical, and clinical advice on Digital Health Technologies (2018) and Health Products for Women (2019); and;
- The development of guidance documents on novel medical devices that use new and innovative technologies (e.g., 2019 guidance document on Supporting Evidence for Implantable Medical Devices Manufactured by 3D Printing ).
Health Canada also amended section 23 of the Food and Drugs Act in 2019 to increase the power of compliance inspectors. The amendment provided them with the ability to engage in more activities on the ground (i.e., testing, taking photographs and recordings) and to examine electronic data. The Department also implemented regulatory changes, including the mandatory reporting of medical device incidents by hospitals (see further details in section 6).
In addition, the Compliance and Enforcement Policy for Health Products (Policy 0001) was revised in December 2018. This revised version was perceived by a few internal key informants to be a more appropriate risk-based approach to enforcement, as it allows Health Canada to be more assertive in response to non-compliance. At the time of the engagement, the Department was also reviewing the scope and responsibilities of some of its compliance and enforcement activities to ensure that these activities focused on the right level of risk, and used resources effectively.
Additionally, Health Canada increased the number of annual foreign inspections to 90 (including 15 foreign on–site inspections) a year to help address challenges related to the globalization of the medical devices environment. This increase was expected to improve the compliance and enforcement of manufacturers in other counties, as well as promote international alignment and cooperation.
Engagement with international partners helps with sharing expertise and harmonizing practices.
Health Canada engaged in several international initiatives and partnerships to share expertise and harmonize practices, policies, and regulations with other jurisdictions. In particular, Health Canada was an active participant in the International Medical Device Regulators Forum (IMDRF). MDP staff participated in several working groups, including one dedicated to developing a harmonized terminology for reporting medical device incidents.
The MDP considered sex and gender differences, as they relate to medical devices.
Through the Health Portfolio Sex- and Gender-Based Analysis PolicyFootnote 23 23, Health Canada can integrate sex, gender, and diversity considerations into its programs, when appropriate, in an effort to better meet the needs of different patient groups. This is relevant for the MDP, as evidence shows that men and women are affected differently by medical devices due to sex and gender disparitiesFootnote 24 24.
In this context, the MDP implemented measures to account for these disparities in several of its activities. In addition to creating the Scientific Advisory Committee on Health Products for Women in 2019, the MDP proposed amendments to the Medical Devices Regulations to provide Health Canada with greater authority to require information from manufacturers when there is evidence of a problem, including identified risks or uncertainties for specific groups (e.g., women, people with disabilities, children)Footnote 25 25. Also, in 2013, the MDP published a guidance document, Considerations for Inclusion of Women in Clinical Trials and Analysis of Sex Differences , which outlined key considerations for including women in pre-market studies for therapeutic productFootnote 26. Health Canada also sponsored a research project that focused on applying a sex- and gender-based analysis lens to the medical device life cycle.
The pre-market function examined whether clinical studies data provided with medical device licence applications had considered specific population groups that might use the device. However, the MDP did not have structured guidance for the industry on when and how to include this information. Manufacturers were also not required to include this data in their device licence applications. As such, there was no available evidence that the MDP had conducted systematic data or trends analysis to provide information regarding sex and gender considerations related to medical devices.
While Health Canada made strides to adapt to a changing environment, some challenges remain to be addressed.
Several internal and external key informants noted that further progress was required to adapt the regulatory framework. For instance, there was no mechanism to address the growing issue of medical devices offered through a subscription or as a service (e.g., online diagnostic tools).
Although controls were in place within the compliance and enforcement function to inspect and mitigate risks, should an incident report be submitted for an unlicensed medical device, Health Canada did not have the means to directly contact individuals living with or using a medical device when there was a safety issue. The regulations required manufacturers to monitor the use of their devices, submit mandatory incident reports, and contact individuals when there was new information concerning device safety, effectiveness, or performance. However, there was a lack of clarity with respect to responsibilities for contacting individuals using devices that were no longer licensed in Canada, or that were sold by a manufacturer that did not have an active licence in Canada (e.g., their medical device licence was suspended or not renewed).
Some external key informants, including experts, health care professionals, and individuals using a medical device, felt there should be a central registry of individuals with an implantable device, in order to allow Health Canada to inform the individuals directly when there is an incident. A similar suggestion was also made by the Scientific Advisory Committee on Health Products for WomenFootnote 27 27.
Although there were ongoing mechanisms for collaboration with international regulatory agencies, some internal and international key informants noted that more could be done to communicate and collaborate with other jurisdictions. In particular, the compliance and enforcement function was often unable to engage in international collaboration due to workload pressures emerging from domestic priorities.
3.0 Providing Access to Medical Devices in Canada
3.1 Processing of Device Licence Applications
The MDP made progress in finding a balance between ensuring the safety of the devices and providing access in a timely manner.
Between 2013-14 and 2018-19, the MDP received an annual average of 1,499 new applications for Class II medical devices, 424 for Class III, and 78 for Class IV. During the same period, the Program also received an annual average of 1,382 Class II, 424 Class III, and 311 Class IV amendment applications.
During the period, the MDP improved its performance at providing decisions on medical device licence applications within service standards (i.e., 15 days for Class II licence applications, 60 days for Class III and 75 days for Class IV). Calculations undertaken for the study show that, between April 1, 2018 and June 30, 2019, almost all device licence applications (96%) were processed within service standards for all application types.
Additionally, according to program performance data, the MDP’s ability to process device licence applications within service standards improved over the engagement period. As illustrated in Figure 5, the backlog of applications was reduced for Class III and IV devices from a high of approximately 18% in 2016-17, to a low of 5% in 2018-19.
At the time of the engagement, the MDP’s service standards for reviewing device licence applications did not include the time required for administrative processing, nor for technical and regulatory screening of applications once received by Health Canada. They also did not account for the 45 days added to the review process once manufacturers respond to any requests for additional information.
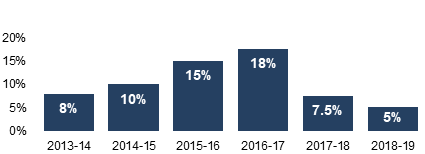
Figure 5 - Text description
This figure is a vertical bar graph that shows the proportion of licence applications in backlog by fiscal year from 2013-14 through 2018-19. The following are the results shown:
- 8% in 2013-14
- 10% in 2014-15
- 15% in 2015-16
- 18% in 2016-17
- 7.5% in 2017-18
- 5% in 2018-19
Source of information for this graph are MDB Annual Report 2018-19 and MDB Annual Report 2013-14
Several health care professionals, as well as some industry representatives and experts, perceive the regulatory process to be lengthy. However, when compared to the United States, for instance, Health Canada’s service standards for reviewing device licence applications were shorter. The United States had a 180-day service standard for the scientific review of device licence applications with a possible extension for an additional 180 days if the application is amended (e.g., more information is provided) Footnote 28 28.
A significant proportion of device licence applications did not meet regulatory requirements when first submitted to Health Canada
According to program performance data, from 2013-14 to 2018-19, between 39% and 59% of the device licence applications received by the MDP did not comply with regulatory requirements at the time of their first review decision. According to several internal and external key informants, incomplete device licence applications often increased the time required for the review process as program staff had to go back to the applicant to request additional information. Transactional testing demonstrates that 66% of device licence applications were the object of at least one additional information request or deficiency letter from the MDP to the applicant.
As shown Figure 6, once the applicant provided the additional information requested by Health Canada, the percentage of applications that remained non-compliant with regulatory requirements dropped to 8% or less between 2013-14 and 2018-19.
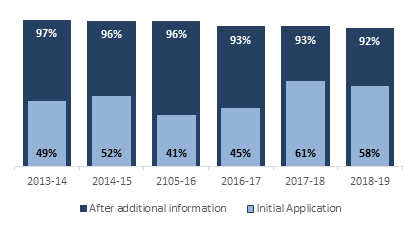
Figure 6 - Text description
This figure is an overlapping vertical bar graph showing results by fiscal year of both the proportion of initial licence application that comply with regulatory requirements at first review and the proportion of applications that became compliant after MDB requested and received additional information. The proportion of initial application that were compliant is as follows:
- 49% in 2013-14
- 52% in 2014-15
- 41% in 2015-16
- 45% in 2016-17
- 61% in 2017-18
- 58% in 2018-19
The proportion of applications that became compliant following the additional request and receipt of information is as follows:
- 97% in 2013-14
- 96% in 2014-15
- 96% in 2015-16
- 93% in 2016-17
- 93% in 2017-18
- 92% in 2018-19
Source of information for this graph is MDP Performance Data
Guidance documents on the application process were challenging to find and often not up-to-date.
The MDP published guidance documents on Health Canada’s website outlining requirements for all classes of medical devices, certain novel technologies (e.g., 3D printed devices), and combination products. These aimed to help applicants determine the classification of their product and navigate the application process.
The majority of industry key informants mentioned having difficulty finding the right information and guidance to support the development of their applications. They explained that navigating Health Canada’s website was difficult, and that the information available was not integrated in a way that helps industry understand the different steps of the application process (e.g., there is no roadmap to follow). Several industry and a few internal key informants explained that small and new manufacturers were more likely to experience challenges with the device licence application process, as they were less likely to be familiar with the regulatory process or had fewer resources with which to prepare their application materials. These perceptions align with results from the 2016 RTOF survey, where the majority of stakeholders (80%) reported that Health Canada’s information on regulatory requirements was not easy to find.
A review of guidance documents available online shows that, while the majority of guidance documents were drafted or updated within the last few years, a couple have not been updated in at least seven years. For instance, the document, Guidance on Supporting Evidence to be provided for New and Amended Licence Applications for Class III and Class IV Medical Devices, not including In Vitro Diagnostic Devices (IVDDs) , was last updated in 2012. Outdated documents may have contributed to inaccuracies in application submissions, leading to more back-and-forth between applicants and the program. This could also have affected the timeliness of device licensing decisions. There is also a risk that outdated internal guidance documentation may have caused confusion and errors when processing submissions, which could, in turn, have had impacts on the efficiency of pre-market processes.
Also, while broad information and evidence requirements for medical device licence applications were established for each class of device and available to industry stakeholders on Health Canada’s website, evidence of a defined process to periodically update these requirements was not found. This means that revisions to available guidance documents were made on an ad hoc basis.
3.2 Measures to Improve Access to Devices
The MDP’s Special Access Program provided a mechanism for Canadians to access medical devices.
According to program documents and a few external and internal key informants, challenges in accessing medical devices may also be mitigated through the MDP’s Special Access Program. This program allows health care professionals to gain access to medical devices that are not approved for sale in Canada, for use in emergencies or when conventional therapies have failed, are unavailable, or are unsuitable to treat a patient. Between 2013-14 and 2018-19, the MDP received an annual average of 4,714 applications through the Special Access Program, involving approximately 1,800 devices. On average, 95% of these applications were reviewed within the service standard of 72 hoursFootnote 29 29.
A few external and internal key informants raised concerns that accessing the Special Access Program could result in the circumvention of the full regulatory process. In this regard, the engagement team expected to find adequate controls in place to ensure that applications satisfied the criteria established by the Medical Devices Regulations, including the stipulation that manufacturers or importers must not use Special Access Program authorizations to circumvent regular licensing requirements.
The MDP implemented several controls to prevent the circumvention of licensing requirements. For example, in 2016, Health Canada published a guide informing health care professionals that the Special Access Program is not intended to provide early market access, and that manufacturers and importers are expected to pursue licensing for devices that were repeatedly authorized through the Special Access Program.
MDP staff also verified the credentials of health care professionals and restricted the number of units authorized per application. However, several controls implemented by the MDP relied on whistleblowing or self-disclosure (e.g., Health Canada prohibits applications completed by manufacturers or importers, and asks manufacturers or importers about their intentions to licence devices).
Transactional testing shows that approximately 80% of devices requested through the Special Access Program from 2013 to 2018 did not generate a subsequent device licence application. This proportion of devices also accounted for 73% of frequently-requested devices. A minority of applicants and devices accounted for the large majority of Special Access Program applications. This suggests that further incorporation of data analytics would strengthen Special Access Program controls for identifying possible attempts to circumvent regular licence requirements by facilitating the identification of risks, trends, and high-volume applications. In particular, data analytics may provide a supplemental control to help ensure that authorizations granted through the Special Access Program comply with the criteria established by the regulations, including identifying use of the Special Access Program to circumvent licensing regulations.
The MDP recently implemented several measures to mitigate challenges with the device licence application process.
In recent years, the MDP has focused on engaging with industry representatives to increase their awareness of available information and guidance documents regarding the medical device licence application process. The Program also developed and implemented several initiatives and tools in an attempt to reduce the number of applications that did not fulfill regulatory requirements, including:
- Pre-submission meetingswith industry stakeholders to discuss upcoming device licence applications. The impact of these meetings was unknown at the time of the engagement, as only a small number of industry representatives requested pre-submission meetings. The majority of industry key informants interviewed who participated in a pre-submission meeting found them to be helpful, particularly for new and novel technologies and devices;
- The launch of an e-Learning tool, Understanding how Medical Devices are Regulated in Canada – Premarket Regulation, to provide guidance on the regulatory requirements for new or amended device licence applications. While data was not collected on the reach of training, nor on feedback from participants, some industry and internal key informants noted that the course was well received; and
- The creation of the Digital Health Technology Division to facilitate the review process for devices involving new technologies. Some industry key informants recognized the Division’s efforts to facilitate and support the application process, including informational road shows and participation at industry conferences.
Harmonizing the MDP’s requirements with those of other countries can help improve access to devices .
According to program documents and interviews with several internal key informants, Health Canada was pursuing various initiatives to harmonize its requirements for medical device licence applications with those of other countries. Mainly organized through the International Medical Device Regulators Forum (IMDRF), these initiatives aimed to streamline the processes for manufacturers submitting applications in multiple jurisdictions. Examples include:
- A Table of Contents format to harmonize the format of device licence applications across IMDRF member countries. This format was perceived by a few internal key informants as a potential way to clarify the requirements of a medical device licence application. As of April 1 st, 2019 manufacturers can choose to use the Table of Content format for their Class III and IV licence applications; and
- As of January 1st, 2019, Canada implemented the Medical Device Single Audit Program (MDSAP) as a mandatory requirement for manufacturers submitting device licence applications to Health Canada. At the time of this engagement, Canada was the only IMDRF member to mandate the MDSAP. The United States and AustraliaFootnote 30 30 also accepted the MDSAP, but these countries did not implement it as a mandatory requirement.
Key Definition: The Medical Device Single Audit Program allows manufacturers to complete and submit one certificate of their Quality Management System in fulfillment of the regulatory requirements of multiple countries.
Concerns were raised by several industry key informants that the introduction of the MDSAP may have hindered access to certain medical devices. They explained that the increased costs for obtaining the MDSAP, as compared to the previous audit certification required by Health Canada, may have deterred some manufacturers from renewing their establishment licence, or from submitting device licence applications for new or amended medical devices. At the time of the engagement, it was too early to determine the impact of the MDSAP. According to program performance data, between July, 2018 and July, 2019, about 8% of the total medical device licences approved by Health Canada were cancelled, not renewed or discontinued, which was an increase compared to the annual average of 4% between July, 2013 and July, 2018 .
Transaction testing demonstrates that the MDP’s staff reviewed the certificates of manufacturers’ Quality Management System in a consistent manner when processing medical device licence applications. If deficiencies with the certificate were found during the application review, they were communicated to applicants via e-mail for Class II devices, or in a formal deficiency letter for Classes III and IV devices. However, there was a lack of integration between MDSAP audit report information and compliance and enforcement activities. Observations of post-market non-compliance were received by the pre-market function, but not flagged to the compliance and enforcement function for action, thereby limiting the Program’s ability to adopt a life cycle approach.
The MDP was exploring additional measures to further facilitate access to needed medical devices .
The MDP continued to explore measures to ensure both safety and access to medical devices in the midst of a changing environment. It is too early to assess the impacts of these measures, as they were still being implemented at the time of the engagement. These measures include:
- A regulatory pathway for advanced therapeutic products announced in Budget 2019. This proposed pathway is designed to support patient safety, while increasing the Program’s flexibility to approve novel devices that do not clearly align with the current regulatory framework; and
- The expansion of the Priority Pathway mechanism. This would allow the MDP to move certain device licence applications to the head of the queue for processing, in order to be more responsive to health care system needs and expand the agility of the regulatory system.
Key informants suggested potential opportunities for Health Canada to continue improving access to medical devices.
Several external key informants suggested examining the possibility of implementing an expedited process for approving amendment applications for medical devices that are already licensed for sale in the Canadian market, if the amendments are not expected to significantly increase the risks related to the device. This suggestion was made based on the expectation that the number of amendment applications would increase as the pace of technology continues to accelerate and updates to devices that are already licensed become more frequent (e.g., updates to software used as medical devices). A similar process for expediting amendment applications already exists in the United States [i.e., the 510(k)].
Some external key informants, including representatives from industry and health care, also suggested an expedited approval process for devices already approved for sale in countries that have regulatory processes similar to Canada. These informants further noted that, should Health Canada consider this option, it would also have to maintain its regulatory autonomy and its accountability to Canadians.
Additionally, some external key informants noted a desire for more transparency on the status of medical device licence applications submitted to Health Canada. This could be in line with the online ‘Submissions Under Review’ database for Health Canada’s pharmaceutical program. A similar desire was noted by the majority of respondents (76%) from the 2016 Health Products and Food Branch Transparency survey, who agreed that the Submissions Under Review list should be expanded to include medical devices.
4.0 Communicating with Stakeholders and Canadians
Health Canada improved its communications regarding medical devices and increased its engagement of stakeholders.
Health Canada disseminated a number of different types of communications products to help industry stakeholders navigate the Program, and help health care professionals and Canadians make informed decisions about the use of a device (e.g., Dear Health Care Professional Letter, recall notices). Communication products produced by the Department also included guidance documents (see section 3.3 for further details).
Over the last five years, Health Canada has improved its communications on medical devices through initiatives such as:
- Publishing Regulatory Decision Summaries for new and amended Class III and Class IV devices. Approximately 125 new summaries were published between March and August 2019;
- Increasing public access to medical device clinical data through a searchable web portal and to a regularly updated online database of medical device incidents, complaints, and recalls;
- Providing greater clarity and additional information on medical device inspections; and,
- Undertaking various engagement activities, including bi-annual meetings with key industry representatives, a webinar with health care professionals to discuss Health Canada's safety review of breast implant-associated anaplastic large cell lymphoma (BIA-ALCL), and targeted outreach to patient groups, including meetings with women living with breast implants to learn about their experiences.
Key Definition: Regulatory Decision Summaries explain Health Canada's decision for certain health products seeking market authorization.
Several industry representatives and a few health care professionals recognized the MDP for its engagement initiatives. Industry representatives appreciated the opportunity to engage with Health Canada representatives and hoped these types of opportunities will continue in the future. These perceptions echo findings from the 2016 RTOF survey, where the majority of stakeholder respondents (85%) agreed that Health Canada did a better job at communicating to Canadians at that time than it did ten years ago. Additionally, all stakeholders who answered the survey felt that Health Canada was a trustworthy and reliable source of health and safety information, while 95% reported using Health Canada information to inform their activities and decisions.
In addition to various engagement initiatives, Health Canada offered stakeholders and Canadians the opportunity to participate in online consultations on proposed regulatory changes, as well as the development of new or amended policies and guidance documents. In 2017-18 and 2018-19, it completed 100% of its planned consultation projects on medical devices. While industry representatives were generally happy with the opportunity to participate in these consultations, a few noted that they would have appreciated more opportunities to follow up with the MDP after their participation. This would have allowed them to provide additional details, if required, and get a better understanding of how their feedback was used by the Program. In this regard, program documents show that the MDP reviewed feedback collected through online consultations and used it, where relevant.
To date, the majority of engagement and consultation efforts by the MDP were carried out by the pre-market function. As such, there remain opportunities to further engage stakeholders in discussions related to post-market activities. The compliance and enforcement function developed a stakeholder engagement plan, but were unable to fully implement it due to a lack of resources.
Non-industry key informants, including members of the Canadian public, had a limited awareness of Health Canada’s communications related to medical devices.
The majority of key informants who were health care professionals, patient associations, or individuals using a medical device stated that they were unaware of Health Canada’s communications products on medical devices. Those who were aware indicated that they rarely used this information to inform their decisions. They relied instead on other sources of information, including manufacturers, hospital procurement departments, medical journals and publications, media, as well as colleagues or other individuals using a medical device. Nevertheless, some health care professionals and several individuals using a medical device noted that they would prefer to have accessed information from Health Canada, as they expected it to be more objective and trustworthy.
In addition to being generally unaware of Health Canada’s communications products on medical devices, the majority of non-industry key informants were also unaware of Health Canada’s online consultations on medical devices, or were not involved in any of the MDP’s engagement activities. Several external key informants indicated that more engagement is essential in order for Health Canada to learn about the impacts of medical devices and the needs of non-industry stakeholder groups.
Information posted online is often out-of-date and difficult to access.
In addition to raising concerns about the lack of timeliness of guidance documents (see section 3.1 for further details), some industry representatives and a few internal key informants noted that some Health Canada guidance documents did not necessarily reflect the realities of the current medical device environment (e.g., digital health technology, artificial intelligence). This could lead to situations where devices with more advanced technologies would not be assessed in a manner that reflects how quickly they change.
The lack of timeliness of information from Health Canada was also cited by a few external key informants as a reason why they rely on other sources of information about medical devices.
Additionally, from 2016-17 to 2018-19, only 33% to 50% of the MDP’s Summary Basis of Decisions and 7% to 33% of Regulatory Decision Summaries were posted online within program service standards. These documents were intended to provide health care professionals and Canadians with information that would lead to more informed decision making and treatmentFootnote 31 31.
Key Definition: Summary Basis of Decisions: explains why Health Canada authorized certain medical devices for sale in Canada.
Some external key informants also indicated a need for more plain language in Health Canada’s information products to make them more accessible to the general public. In this regard, a 2013 study found that most of Health Canada’s risk communications products were written at a graduate student level, which is well above the average health literacy level of the general publicFootnote 32 32.
Additionally, some external and a few internal key informants indicated that greater clarity and context around risk communications would be helpful, particularly for incidents and recalls. For instance, they explained that having more information on the causes of incidents for a particular device (e.g., device malfunction as opposed to a user error) would help inform decisions about using a device.
5.0 Program Organization and Governance
5.1 Distribution of Roles and Responsibilities and Governance
Roles and responsibilities across the three program functions were not always clearly defined, nor aligned with the nature of their mandate.
The high-level roles and responsibilities of the MDP were well defined. In particular, the Food and Drugs Act and Medical Devices Regulations clearly outline the roles and responsibilities of the Minister of Health in relation to medical devices. However, there was a lack of clarity on roles and responsibilities at the operational level across the three MDP functions. Documents and internal key informants confirmed that the MDP did not have a comprehensive framework defining roles and responsibilities, nor an integrated risk management and information-sharing strategy.
Having clearly defined roles and responsibilities is important, as it enables management to coordinate program activities, assume leadership, provide accountability, and manage risks. A lack of defined roles and responsibilities may create confusion and operational inefficiency, such as duplication of efforts across program functions and inconsistent processes.
For instance, a few internal key informants noted a challenge with the division of responsibilities for processing voluntary incident reports between the post-market surveillance function and the compliance and enforcement function. While voluntary reports received from the CMDSNet were entered, coded, and scientifically evaluated by the surveillance function, voluntary reports from other sources were entered by the compliance and enforcement function, but not coded or scientifically evaluated. This created inconsistencies in data entry, coding, and evaluation for voluntary reports. It also led to the creation of parallel processes. Of note, when the compliance and enforcement function received a voluntary report and there was no subsequent mandatory report from the manufacturer, the MDP followed up with the manufacturer, triggering the required mandatory report.
There was no evidence found that the roles and responsibilities of the compliance and enforcement function were defined for processing voluntary incident reports. There was also no evidence found that roles and responsibilities were defined in regard to sharing voluntary report information with the surveillance function. Additionally, there was no requirement for the compliance and enforcement function and the post-market surveillance functions to consult each other on process changes. It was not possible to determine whether both functions fulfilled their roles and responsibilities for the processing of incident reports.
In addition to a lack of clarity, the distribution of roles and responsibilities across the three MDP functions was not always aligned with the nature of their mandate. For instance, the pre-market function assumed several responsibilities that were predominantly perceived as post-market activities. Under section 39 of the Medical Devices Regulations, Health Canada has the power to request additional information from a manufacturer if there are reasonable grounds to believe that a licensed device is not meeting safety and effectiveness requirementsFootnote 33 33. At the time of the engagement, the pre-market function requested this additional information, even though the surveillance and compliance and enforcement functions used it to evaluate an incident. A few internal key informants explained that having to go through the pre-market function to obtain required information was not an efficient practice. To address this challenge, the surveillance function established an informal parallel process (i.e., Voluntary Request Letter) to contact manufacturer’s directly.
At the time of the engagement, the MDSAP certificate (see section 3.2) was requested, managed, and retained by the pre-market function. Several internal key informants from the compliance and enforcement function noted that their work would benefit from accessing the certificate, as it would help identify potential areas of risk and guide their inspection work. It was further noted that moving the MDSAP to the compliance and enforcement function would help reduce duplication of activities between the MDSAP audit and the inspection of manufacturers. It would also help ensure that the compliance and enforcement function is aware of any cases of non-compliance as soon as possible. Moreover, MDP inspectors had more authority (e.g., the ability to examine any articles or electronic data to which the Food and Drugs Act or Medical Devices Regulations apply) and conducted more thorough investigations of the systems and activities of manufacturers, as compared to the third party auditor’s responsible for the MDSAP reviews. At the time of the engagement, discussions between the pre-market and compliance and enforcement functions were underway to determine where the MDSAP should reside.
The MDP implemented various governance committees to foster information sharing across the three functions.
Various governance committees were implemented to oversee program operations and foster the exchange of information across the three program functions. In particular, a monthly Medical Devices Tri-Lateral Committee brought together program managers and directorsFootnote viii viii, and provided a forum to discuss issues of mutual interest or responsibility. The Tri-Lateral Committee adopted a two-fold approach where meetings including managers and directors were followed by a directors-only meeting. The MDP also implemented a number of operational-level committees, as well as a senior management
committee that provides leadership and focus.
Several internal key informants explained that communication at the management level and above generally worked well. They also noted that the various committees and meetings that took place between the MDP functions (e.g., trilateral meetings, biweekly risk meetings, monthly “hot topics” meetings, Director General and director bilateral meetings) contributed to the Program’s efficiency, including the prioritization of medical device incidents and the identification of problems before they happened. A few internal key informants explained that there remained opportunities to make the trilateral meetings more strategic and to improve the dissemination of information discussed at those meetings among a wider group of program staff.
Despite the governance structure put in place, concerns were raised over the quality of communications at the working level.
Several internal key informants raised concerns regarding the level and quality of communications at the working level across the three MDP functions. In particular, they identified a lack of communication as one of the main challenges affecting the efficiency of the Program.
Some internal key informants saw these communication challenges as negatively affecting the coordination of program activities, resulting in missed opportunities for program functions to work together or creating delays in program activities. For instance, a lack of communication between the pre-market and post-market surveillance functions regarding new and novel licensed devices limited opportunities for proactive post-market monitoring. Additionally, communication challenges between the two post-market functions regarding ongoing signal assessments resulted in situations where compliance and enforcement inspectors were unaware of upcoming signal assessments that could benefit compliance verification activities.
There was no evidence found that the MDP had a horizontal risk management framework.
The MDP had a Program Risk Management Committee that discussed emerging issues. In addition to the committee, emerging issues were managed within every program function on an ad-hoc basis. However, there was no evidence found of a horizontal risk management process. While the Health Products and Food Branch has a draft Branch Risk Register for 2018-19, an approved version was not available at the time of the engagement, nor were any previously approved versions of this document.
5.2 Creation of a new Medical Devices Directorate
Health Canada implemented a new Medical Devices Directorate bringing together pre-market activities with those targeted at reviewing incidents involving devices. This new structure is similar to the new organizational approaches implemented in the United States and Ireland.
As of November 2019, part of the MDP was restructured into a new Medical Devices Directorate, which integrated the pre-market function and the post-market surveillance functions of the Marketed Pharmaceuticals and Medical Devices Bureau (see section 1.2 and Appendix E for details on those divisions). The surveillance function’s Health Products Surveillance and Epidemiology Bureau and the compliance and enforcement function remained with their original directorates.
The new organizational structure was similar to the approach taken by IrelandFootnote 34 34 in 2018, and the United States’ Food and Drug Administration in 2019. According to international key informants and documents from the Food and Drug Administration’s website, bringing pre-market and post-market functions together was expected to enable staff to leverage each other’s knowledge and optimize decision making across the device life cycleFootnote 35 35. Figure 7 provides a high-level overview of new organizational structures implemented in these two countries.
Key informant interviews and documents from the Food and Drug Administration’s website suggested that early impacts of their reorganization included:
- Improved communication between program functions and clarity of roles and responsibilities;
- More discussions, decisions, and enhanced evidence generation throughout the device life cycle; and
- Reduced delays in getting safer and more effective innovative technologies into the market.
At the time of the engagement, it was anticipated that Health Canada’s new Medical Devices Directorate would achieve similar impacts and help foster a life cycle approach for the regulation and monitoring of medical devices. This is to be accomplished by enhancing the MDP’s scientific and policy capacity, modernizing processes to respond to rapid innovation cycles, and increasing resources to support transparency and stakeholder engagement activities across the medical device life cycleFootnote 36 36.
5.3 Management of Information
The MDP’s information technology (IT) infrastructure and its organization of information challenged program efficiency and the ability to take a life cycle approach to regulating and monitoring medical devices .
At the time of the engagement, there was no single repository for collecting all the information available on a device. However, the tracking methods, when combined, provided information about the entirety of activities relating to a device or signal. These methods included several tracking sheets within both post-market functions, docuBridge, each team’s respective shared folders, various dashboards, various forms of internal and external communications, and the Medical Devices System.
For instance, the post-market surveillance function documented its activities regarding medical device signal assessments in Excel tracking sheets, instead of the Medical Device System. It tracked the timelines for signal assessments in relation to service standards in a tool entitled the Regulatory Activity Tracking Sheet. Additionally, compliance and enforcement-related documents and surveillance-related documents, including those on CMDSNet, were saved in their respective function’s shared drives, and the information was not integrated into docuBridge.
Because not all of the data regarding a medical device was entered into a single database or repository, a consolidated life cycle report could not be generated for specific devices. As explained by some internal key informants, program staff had to connect information from various sources to piece together the full history of Health Canada’s actions related to a device.
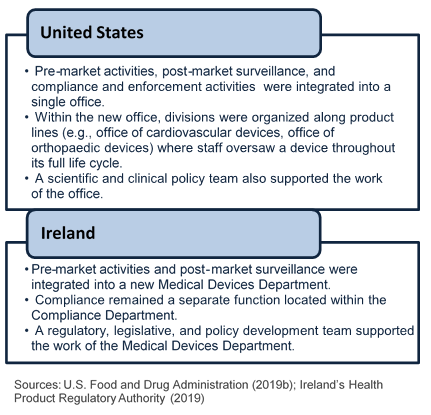
Figure 7 - Text description
This figure contains two sections, representing information gathered about the medical devices programs in the United States and Ireland.
In the first section, information about the structure in the United States is shown in a bullet list as follows:
- Pre-market activities, post-market surveillance, and compliance and enforcement activities were integrated into a single office.
- Within the new office, divisions were organised along product lines (e.g., Office of Cardiovascular Devices, Office of Orthopaedic Devices) where staff oversaw a device throughout its full life cycle.
- A scientific and clinical policy team also supported the work of the office.
- Pre-market activities and post-market surveillance were integrated into a new Medical Devices Department.
- Compliance remained a separate function located in the Compliance Department.
- A regulatory, legislative, and policy development team supported the work of the Medical Devices Department.
Sources of information for this visual are U.S. Food and Drug Administration (2019b) and Ireland’s Health Product Regulatory Authority (2019).
Fragmented information also made it difficult for the Program to conduct trend analyses and may have hindered access to some information in a timely manner. For instance, when the pre-market or compliance and enforcement functions required post-market data to inform their activities, they had to spend additional time compiling that information from various tracking sheets.
Additionally, a few key informants explained that information on the classification of a device was housed in a database held by the pre-market function. As a result, this information was not accessed in a timely fashion by inspectors when they were inspecting manufacturers. Of note, at the time of the engagement, discussions were underway between the pre-market and compliance and enforcement functions to improve the sharing of this information.
The 2016-21 Health Products and Food Branch Strategic Plan noted a commitment to investing in IT initiatives for the MDP Footnote 37 37.
Several internal key informants noted that some of the MDP’s IT systems (e.g., Medical Devices System and docuBridge) were outdated, difficult to use, and not fundamentally designed for the purposes they were used for. Moreover, a significant proportion of guidance documents were not updated to reflect changes related to IT systems, including the establishment of the Regulatory Operations and Enforcement Branch, the implementation of docuBridge, and the modernization of the Medical Devices System.
A few internal key informants also raised concerns that the IT infrastructure used to deliver program activities was not on par with today’s technology. For instance, the MDP did not have electronic interfaces to intake medical device licence applications or mandatory incident reports. Both were submitted to Health Canada using PDF forms sent via mail, email, or fax. The use of such methods did not align with the high technology world of the medical device industry. It also means that information sent to Health Canada (e.g., mandatory or voluntary incident reports) had to be processed manually by a team that was not adequately staffed to perform these tasks within targeted service standards (see section 6).
6.0 Program Resources
Delays completing certain post-market activities were primarily attributed to the level of available resources relative to workload.
In 2018-19, the MDP’s budget was allocated in the following way:
- 46% on compliance and enforcement activities;
- 37% on pre-market activities;
- 10% on program administration and other program support activities; and
- 7% on post-market surveillance activities.
Overall, the medical devices post-market surveillance function received a relatively small share of the program budget.
In the last six years, the number of non-management full-time equivalent (FTE) staff working in medical device surveillance decreased by approximately eight. This accounted for approximately 30% of the post-market surveillance functions non-management FTEs. Part of the current staff was also temporarily reassigned to other responsibilities (e.g., implementation of initiatives under the Medical Action Plan).
It also appears that the resources allocated were relatively low compared to post-market surveillance for Health Canada’s pharmaceuticals program. As illustrated in Figure 8, the number of FTEs assigned to medical device surveillance was less than a third of the number of FTEs for pharmaceutical surveillance, even though the volume of incidents to process was greater for medical devices. The difference in resources available for the surveillance of medical devices, as opposed to pharmaceuticals surveillance was a trend that continued at the time of this engagement, according to a few internal key informants.
Overall, the insufficient capacity in the post-market surveillance function was noted by several internal key informants as a reason for the backlog in processing incident reports, the inability to process a large volume of reports, the elimination of triaging for incoming incident reports, as well as the decreased number of signal assessments and reviews completed within service standards (see section 2.2).
| Medical Device Surveillance | Pharmaceutical Surveillance | |
|---|---|---|
| FTEs | 9 | 30 |
| Medical device incidents and adverse drug reactions screened | 17,133 | 4,450 |
| Potential signals for assessment | 263 | 173 |
| Source: Health Canada Internal Documents. |
In comparison, the number of FTEs in the pre-market and the compliance and enforcement functions increased during the engagement period (e.g., eight more inspectors were hired in 2019). However, several internal key informants noted that a lack of resources still challenged the pre-market, and compliance and enforcement functions. They indicated that they were facing increased workload pressures as a result of the greater scrutiny of medical devices in the Canadian market, program changes (e.g., implementation of the Medical Devices Action Plan), and the evolution of the medical device environment (e.g., novel technologies). Some internal key informants further raised concerns that, without the required resources to mitigate these challenges, the Program might not be able to efficiently and effectively review medical device licence applications for new technologies and devices, nor inspect manufacturers of these devices moving forward.
Mandatory incident reporting introduced in December 2019 will likely exacerbate the challenges faced by the post-market surveillance functions.
The Protecting Canadians from Unsafe Drugs Act, also known as Vanessa's Law, introduced amendments to the Food and Drugs ActFootnote 38 38. Among other things, it introduced the mandatory reporting of adverse drug reactions and medical device incidents for approximately 775 Canadian hospitals regulated through provincial or territorial legislation, or operated by the Government of Canada, as of December 2019Footnote 39.
Vanessa’s Law: Vanessa Young died in 2000, at the age of 15, of a cardiac arrhythmia after taking cisapride (Prepulsid®) as prescribed. A campaign for increased regulation of therapeutic products subsequently led to greater powers for Health Canada to request safety data from hospitals and industry about drugs and medical devices. Vanessa’s Law was enacted in 2014, and the mandatory reporting requirements come into effect December 16th, 2019.
According to an internal cost-benefit analysis, the number of incident reports received by the MDP may double as a result of the mandatory reporting implementationFootnote 40 40. A few internal key informants noted concerns that the surveillance function will be unable to meet the demand. The backlog of incident reports observed at the time of the engagement could continue to grow in the future. Additionally, mandatory reporting may have subsequent impacts on the resources and activities of the post-market compliance and enforcement function, as inspectors will be required to monitor potential under-reporting and inspect hospitals.
At the time of the engagement, with the exception of the cost-benefit analysis, Health Canada had not fully analyzed the implications of mandatory reporting on program workload. The Department planned on addressing the increased volume of incidents reports through resource reallocation and workload redistribution in the post-market functionFootnote 41 41. The compliance and enforcement function will need to continuously assess impact on resources and strategy should the number of reports increase significantly.
7.0 Conclusions and Recommendations
The medical device environment is complex and evolving, due to the development of novel devices using innovative technologies such as digital technology, artificial intelligence, and 3D printing. Within this complex environment, Health Canada has generally been effective at ensuring the safety, effectiveness, and quality of medical devices in the Canadian market. It also has generally implemented sufficient controls to support the rigor of its work. Overall, the safety of medical devices in Canada was found to be comparable to similar countries. At times, Canada was among the first countries to initiate recalls to address incidents with those devices, as in the cases of Essure and the Biocell breast implants.
There were, however, delays in the completion of certain post-market activities. While there was no evidence that such delays affected the safety of medical devices in the Canadian market, they did affect the MDP’s ability to proactively detect and address medical device incidents. These delays also represented a risk to the Department, as they appear to be mainly caused by a lack of capacity and resources within the post-market surveillance functions. Overall, post-market surveillance activities received about only 7% of MDP’s budget in 2018-19. As well, in recent years, the number of staff working in medical device surveillance decreased, while it increased in the two other program functions.
With the introduction of mandatory incident reporting by hospitals in December 2019, it is likely that the workload of the post-market surveillance function and the compliance and enforcement function will significantly increase. However, at the time of the engagement, the workload implications of mandatory reporting were not fully analyzed. This represents a risk, as the current challenges faced with the timely completion of some surveillance activities will likely be exacerbated if capacity issues are not addressed moving forward.
Over the last few years, the MDP implemented several initiatives to remain effective in the changing environment of medical devices. The Department has made significant strides to adapt both its regulatory framework and its post-market activities through initiatives implemented via the Medical Devices Action Plan, and the Regulatory Review of Drugs and Devices. There remained additional opportunities for Health Canada to continue adapting. This included opportunities for the MDP to increase its engagement with international counterparts, in order to align practices, particularly around compliance and enforcement activities. Also, there were opportunities for a more systematic consideration of sex- and gender-based differences in the review of device licence applications, risk communications, and compliance and enforcement activities.
During the engagement period, the MDP improved its communications regarding medical devices and its engagement with stakeholders. While these efforts were well received by industry key informants, improvements are still required for Health Canada to become a go-to source of information for health care professionals, patient groups, and the Canadian public in general. Furthermore, accessing Health Canada’s online communications products was challenging. Information available online, including guidance documents for industry, was often not up-to-date.
As for program efficiency, available evidence shows that the distribution of roles and responsibilities across the three functions was not always aligned with the nature of their core activities. This led to inefficiency, including the duplication of efforts across program functions. Moreover, the MDP did not have a comprehensive framework for defining roles and responsibilities. Also, the post-market compliance and enforcement function and the group responsible for processing medical device incident reports remain with their initial organizational structure. It was anticipated that the creation of the new Medical Devices Directorate, with the integration of the pre-market function and the Marketed Pharmaceuticals and Medical Devices Bureau (post-market function), will likely address some of these challenges and help foster a life cycle approach to regulating and monitoring medical devices.
The MDP’s IT infrastructure and organization of information did not support an efficient delivery of activities. Current program data and information was recorded in multiples different files and databases held by different program functions. This challenged the ability of program staff to access information in a timely manner for their program activities. Staff had to put together information from various files and databases in order to obtain the full history of a medical device and actions taken by the MDP related to this device.
Findings discussed in this report led to the identification of the following recommendations:
-
The Health Products and Food Branch should address current resource gaps in post-market functions responsible for processing incident reports and assessing incidents. The Regulatory Operations and Enforcement Branch should consider the subsequent impacts on the resource needs of the compliance and enforcement function. As well, both Branches should ensure that the MDP has the capacity and tools to address the mandatory reporting by hospitals introduced in December 2019, and associated enforcement of hospital vigilance practices.
MDP’s surveillance activities were challenged by a lack of resources and could not always be completed within service standards. The introduction of mandatory reporting of medical device incidents by approximately 775 Canadian hospitals in December 2019 will likely further affect the post-market surveillance function’s ability to manage its workload, as the number of incident reports submitted to Health Canada may double. Mandatory reporting may also affect the resources and activities of the compliance and enforcement function, as inspectors will be required to monitor potential under-reporting and inspect hospitals. However, at the time of the engagement, Health Canada had not fully analyzed the implications of mandatory reporting on program workload.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should continue their efforts to adapt the MDP, through both domestic and international initiatives, to address challenges emerging from the rapidly changing life cycle of medical devices and the use of novel technologies.
While Health Canada made significant strides to adapt its regulatory framework, other challenges remained to be addressed to ensure that the MDP remains effective moving forward. For instance, at the time of the engagement, there was no mechanism to address the growing issue around medical devices offered through a subscription or service (e.g., online diagnostic tools), or to track individuals using implanted devices so they can be notified directly by Health Canada if there is an incident.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should consider and document how to further integrate sex- and gender-based analysis plus (SGBA+) into the life cycle of medical devices, including providing guidance to applicants and increasing awareness of MDP staff.
Evidence shows that men and women are affected differently by medical devices due to sex and gender disparities. While Health Canada had progressed in its efforts to integrate sex and gender considerations into the MDP, there were additional opportunities to further integrate these considerations in all of program activities, where relevant.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should maintain and scale up communication and engagement efforts with industry stakeholders, health care professionals and organizations, and patient groups. It is also recommended that the Health Products and Food Branch improve the timeliness and accessibility of program information available online. This includes ensuring that guidance documentation is reviewed and updated as required across all of the MDP's operations, including on the Government of Canada website.
Health Canada’s efforts to increase communication and engagement with stakeholders were well received by industry representatives. However, Health Canada was not a go-to source of information for health care representatives, patient groups, and the population in general. There was also a need to improve the timeliness and accessibility of online information. In particular, outdated guidance documents may have contributed to inaccuracies in application submissions for medical device licences. Industry key informants also mentioned having difficulty finding required information and guidance to support the development of their applications.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should ensure that roles and responsibilities at the operational level are clearly defined and distributed and that program risks are identified and managed.
Roles and responsibilities of operational-level staff were not clearly defined across the three MDP functions. The MDP also did not have a comprehensive framework defining roles and responsibilities and an integrated risk management and information-sharing strategy. Challenges with roles and responsibilities may be addressed, in part, with the implementation of the new Medical Devices Directorate announced in late 2019, which will integrate the pre-market function and the function responsible for the assessments of medical device incidents. However, the compliance and enforcement function and the Health Products Surveillance and Epidemiology Bureau will remain with their original directorates.
-
The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should ensure that a data management approach supports the monitoring of medical devices throughout their life cycle.
The MDP’s information was fragmented across several repositories. This challenged the Program’s efficiency and ability to take a life cycle approach to regulating and monitoring medical devices.
| Recommendations | Management Response and Planned Actions | Deliverable | Completion Date | Accountability/ Responsibility |
|---|---|---|---|---|
Recommendation 1 |
Management agrees with the recommendation* *All timelines subject to change as a result of the COVID-19 pandemic |
|||
Medical device incident reporting is a key element of Health Canada’s ability to monitor, identify, assess, and respond to safety issues identified by Canadians, hospitals, medical practitioners, and manufacturers. Investments have been made in response to the new mandatory reporting requirements by hospitals. We will continue to evaluate the return on these investments as well as gaps that remain to address the post-market Medical Devices Program (MDP), including data intake and data management functions, scientific assessments, coding, evaluating, and case management to ensure that they have both the capacity and tools to deliver on program functions. |
1.1 HPFB and ROEB will continue to assess the capacity and tools in the post-market functions required to deliver effectively on mandated program activities. Examples of achievements to date:
|
|||
| a) A TOR for the team leading this work will be developed. | October 2020 COMPLETED |
Executive Directors, Bureaus of ITA, SAP, PM, MDD (HPFB) | ||
b) An implementation plan will be developed, including a plan to update guidance documents. |
December 2020 |
|||
c) A capacity gap analysis will be completed. |
March 2021 |
|||
d) An interim progress report will be completed. |
December 2021 |
|||
e) A final report will be completed. |
June 2022 |
|||
Recommendation 2 The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should continue their efforts to adapt the MDP, through both domestic and international initiatives to address challenges emerging from the fast changing life cycle of medical devices and the use of novel technologies. |
Management agrees with the recommendation* | |||
Health Canada continuously works with domestic and international partners to: (a) assess, manage and communicate the health and safety risks and benefits associated with challenges emerging from the fast-changing life cycle of medical devices and the use of novel technologies. (b) optimize the use of real-world evidence across the product life cycle with the aim of improving access to, and appropriate use of, medical devices. These relationships are critical as we develop and implement innovative approaches that improve the efficiency and sustainability of Canadian health care systems. The Program will continue to leverage and build on its solid foundation for cooperative domestic and international partnerships to address emerging challenges and issues such as International Medical Device Regulators Forum (IMDRF), Medical Device Single Audit Program (MDSAP) and Regulatory Cooperation Council (RCC). The MDP will continuously be adapted to address emerging issues and technologies so that the health and safety of Canadians is protected. |
2.1 HPFB and ROEB will continue to strengthen domestic and international outreach and will produce bi-annual progress reports Examples of achievements in the context of the pandemic include:
|
Biannual (will submit last report June 2021) |
Director, Bureau of Policy and International Programs, MDD (HPFB) |
|
2.2 HPFB and ROEB will continue to implement the Health and Biosciences Roadmap commitments that pertain to medical devices, including the items below. |
||||
2.2.1 Advanced Therapeutic Pathways (ATP), which is currently under implementation, to adapt to the fast-changing life cycle of medical devices and, in particular, develop an efficient regulatory framework for AI-related medical devices. Note – the overall project lead for ATP is not within the Medical Devices Program. |
||||
a) Biannual progress reports on MDP’s contributions will be provided. |
Biannual (will submit last report June 2021) |
Executive Director, Bureau of Evaluation, MDD (HPFB) |
||
b) Pathway to be implemented - MDP to contribute to ATP Pathway Guidance documents, including use of regulatory sandboxes. |
September 2021 |
Director, Compliance Policy and Regulatory Affairs (ROEB) |
||
2.2.2 Clinical Trial Modernization (CTM), which will include new provisions to facilitate research in Canada Note – the overall project lead for CTM is not within the Medical Devices Program. Example of an achievement to date: In October 2020, ROEB’s clinical trial inspection program conducted the first inspection of a medical device clinical trial as part of a pilot program. Two more inspections are scheduled to be conducted this year. The purpose of piloting these inspections is to gather data and information, which will lay the foundation for the implementation of a formal approach to these inspections. |
||||
a) Interim reports on progress to be provided bi-annually. |
Biannual (will submit last report December 2021) |
Director, Bureau of Policy and International Programs, MDD (HPFB) |
||
b) New or Revised Regulations to be developed and approved. |
March 2022 |
|||
2.2.3 Agile Regulations for Medical Device and Establishments licensing, which will include an analysis of the classification regime for lower-class devices, terms and conditions, and the use of foreign reviews and decisions. |
||||
a) Interim reports on progress to be provided bi-annually. |
Biannual (will submit last report December 2022) |
Director, Bureau of Policy and International Programs, MDD (HPFB) |
||
b) New or Revised Regulations to be developed and approved. |
March 2023 |
|||
Recommendation 3 The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should consider how Sex- and Gender-Based Analysis Plus (SGBA+) could be applied to program activities. |
Management agrees with the recommendation* |
|||
The Department is committed to ensuring the integration of a sex- and gender-based perspective in developing and delivering on programs including the MDP. In the context of the Medical Devices Action Plan (MDAP), the Department created a Scientific Advisory Committee on Health Products for Women to assist with identifying concrete SGBA+ solutions applicable to the Program’s activities. The Department will continue to engage with this Committee to help inform on SGBA+ considerations in program activities. |
3.1 HPFB and ROEB will integrate SGBA+ into training programs. |
|||
a) ROEB to integrate a SGBA+ lens into the “Foundations of Inspection and Regulations” course during the next review cycle, and provide training to staff, specifically to facilitators, developers, and working group members who are involved in inspector training. |
June 2021 |
Director, Program Development and Training (ROEB) |
||
b) To increase awareness of SGBA+ and its importance throughout the device life cycle, HPFB will require that staff members (e.g., Evaluators and Policy Analysts) complete mandatory SGBA+ training, either online or through face-to-face presentations. A completion report will be provided. |
June 2021 |
Director, Bureau of Planning and Operations, MDD (HPFB) |
||
3.2 HPFB and ROEB will support the researcher selected through the CIHR funding opportunity “SGBA+ Health Policy-Research Partnership Grant”. This researcher will be conducting various analyses to assess how SGBA is integrated throughout the medical device life cycle. A report will be submitted to HPFB and ROEB detailing recommendations for how SGBA+ could be better integrated throughout the medical device life cycle. |
||||
a) ROEB will provide an analysis of whether SGBA+ related risk factors exist in the context of inspection activities and assess the need to develop mitigation and monitoring strategies. |
June 2021 |
Director, Compliance Policy and Regulatory Affairs (ROEB) |
||
b) HPFB will determine recommendations that will be implemented, and will develop an implementation plan. |
December 2021 |
Director, Bureau of Policy and International Programs, MDD (HPFB) |
||
3.3 As recommended by the SAC-HPW committee members, the Program will address how manufacturers should consider SGBA+ factors when providing evidence for medical device licence applications. In particular, the guidance documents in the following sub-sections will reflect SGBA+ considerations. |
||||
a) Guidance document on Clinical Evidence Requirements |
March 2021 |
Director, Bureau of Policy and International Programs, MDD (HPFB) |
||
b) Guidance documents on Artificial Intelligence. |
December 2021 |
|||
Recommendation 4 The Health Product and Food Branch and the Regulatory Operations and Enforcement Branch should maintain and/or scale up communication and engagement efforts with industry stakeholders, health care professionals/ organisations, and patient groups. It is also recommended that the Health Product and Food Branch improve the timeliness and accessibility of program information available online. This includes ensuring that guidance documentation is reviewed and updated as required across all of the MDP's operations, including on the Government of Canada website. |
Management agrees with the recommendation* |
|||
Canadians rely on medical devices to maintain and improve their health and well-being. As reported by the International Consortium of Journalists, following the launch of the Medical Devices Action Plan, Canada has one of the best regulatory systems in the world for medical devices, with some of the most stringent requirements. The Department has taken numerous steps in the area of communication and engagement, including:
On December 16, 2019, Health Canada became the first regulator worldwide to issue regulations that require hospitals to report all serious adverse drug reactions (ADRs) and medical device incidents (MDIs). Health Canada led a significant national outreach effort throughout 2019 related to the new requirements. This involved 250 educational events with health care professionals, including face-to-face presentations and 12 webinars in both official languages to hospital-based health care professionals and administrators. Health Canada published a guidance document, new reporting forms, web site reporting page and online educational modules to support hospitals’ interpretation and understanding of the new requirements. Because of this focused engagement effort with stakeholders and over 800 hospitals across Canada, Health Canada has observed the number of reports from hospitals increase from month-to-month. The Department also developed and implemented a client strategy and consulted with health care professionals and patient groups to optimize communications, including the web sites and online incident reporting tools. The feedback received will help inform the Department’s ongoing efforts to effectively communicate and engage on program activities. |
4.1 The Department has received the results of the TBS Medical Devices Web Optimization project, including a project prototype. Examples of achievements in the context of the pandemic include: The prototype’s experimental designs have already been applied to create a Health Canada landing page for COVID-19 medical device information. In addition to guidance documents and notices, key information that is available online for Canadians includes:
Additional planned updates include publication of a COVID-19 application wizard, a review of the internet architecture of drugs and health products, creation of service initiation pages for the active licence listings (MDALL and MDEL). HPFB and ROEB will continue to implement the proposed changes and engage with stakeholders to inform of changes. Interim reports on progress to be provided quarterly |
Quarterly (will submit last report December 2022) |
Director, Bureau of Planning and Operations, MDD (HPFB) |
|
4.2.1 HPFB & ROEB will also assess the total number of guidance documents posted on its web site to determine the number of documents requiring updates, and which ones have been completed. |
March 2021 |
Director, Bureau of Policy and International Programs, MDD (HPFB) |
||
4.2.2 Based on the assessment, HPFB and ROEB will develop a plan to update the documents for approval by the MDP Trilat. |
September 2021 |
|||
4.3 HPFB and ROEB will continue to engage, create and implement an annual stakeholder engagement and compliance promotion plan, including bilateral meetings with industry associations. Here is an example of achievement in the context of the pandemic:
|
||||
4.3.1 MDD’s 2021 Stakeholder Engagement Plan will be updated and approved by the MDD-DMC |
December 2020 |
Director, Bureau of Planning and Operations, MDD (HPFB) |
||
4.3.2 MDCP 2021 stakeholder engagement plan to be updated and approved by MDCP-DMC |
December 2020 |
Director, Medical Devices Compliance , MDCCD (ROEB) |
||
4.3.3 Report on engagement activities issued quarterly |
Quarterly (will submit last report September 2021) |
Director, Bureau of Planning and Operations, MDD (HPFB) |
||
4.4 HPFB and ROEB will continue engaging with stakeholders through Scientific and Patient advisory committees (engagement with stakeholders):
Meeting minutes will be provided. |
Last minutes provided by September 2021 |
Director, Bureau of Policy and International Programs, MDD (HPFB) |
||
Recommendation 5 The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should ensure that roles and responsibilities at the operational level are clearly defined and distributed and that Program risks are identified and managed. |
Management agrees with the recommendation* |
|||
HPFB and ROEB will continue to work to build on the existing operating frameworks to clarify roles and responsibilities further at the operational level for the Program. HPFB responded to the many challenges and opportunities of medical devices development by creating a new, stand-alone Medical Devices Directorate (MDD) in January 2020. The new Directorate takes a life cycle approach to regulating medical devices, by bringing together specific post-market functions with the pre-market functions. This created an organization that is dedicated to the regulation of medical devices and facilitates the integration of risk management. |
5.1 HPFB and ROEB will collaborate to update the existing inter-branch RACI (Responsible, Accountable, Consulted and Informed) and MOU documents. Both branches will collaborate to define roles and responsibilities at the operational level within the Medical Devices Program and update SOPs accordingly. Documents will be approved by the MDP Trilat. |
|||
a) An updated RACI matrix will be developed. |
June 2021 |
Director, Bureau of Planning and Operations, MDD (HPFB) |
||
b) An updated HPBF-ROEB MOU will be developed. |
December 2021 |
|||
c) Related SOPs will be updated. |
June 2022 |
|||
Recommendation 6 The Health Products and Food Branch and the Regulatory Operations and Enforcement Branch should ensure that a data management approach supports the monitoring of medical devices throughout their life cycle. |
Management agrees with the recommendation* |
|||
Health Canada is working to strengthen further the data management approach, which would support the monitoring of medical devices throughout their life cycle. This data management approach includes different components including: IT plans, transparency principles, service standards, and records management, all to address client expectations. Investments have been made in IT projects as part of the Branch IT strategy, including a new and modernized medical device incident reporting system. These investments are intended to enhance efficiency, but will require ongoing stakeholder engagement and systems stabilization to achieve this goal. In addition, HPFB and ROEB will work towards a fully integrated and coordinated data management approach, across the medical device life cycle, which builds on the Branch IT investments. |
6.1 In light of the creation of the new Medical Devices Directorate (MDD), HPFB and ROEB will leverage and improve existing processes to ensure full integration and coordination of medical devices data across the life cycle. |
|||
a) Robotic Process Automation implemented to streamline processes, including the medical device licence renewal. |
August 2021 |
Director, Bureau of Planning and Operations, MDD (HPFB) |
||
b) The MDP will complete the conversion from Impromptu 7.5 to Cognos 11 to harmonize reporting across the device life cycle. An example of an achievement to date:
|
March 2022 |
|||
Appendix A – Joint Engagement Scope and Methodology
This joint audit and evaluation engagement was carried out by Health Canada’s Office of Audit and Evaluation. The purpose was to examine the MDP’s activities from April, 2013 to August, 2019, and the engagement applied a sex- and gender-based analysis plus (SGBA+) lens where relevant. The Internal Audit division’s testing of MDP transactions focused on the period from April, 2018 to September, 2019.
The MDP’s cost recovery model was out of scope for the engagement because a revised model will be implemented on April 1st, 2020. The Internal Audit division also did not examine Medical Device Establishment Licence inspection activities under the Regulatory Operations and Enforcement Branch, as the division recently completed an Audit of Inspection Activities. However, these inspection activities were examined in the evaluation to generate a comprehensive assessment of all the MDP’s activities.
This was the first internal audit of the MDP, which was conducted in conformance with the International Standards for the Professional Practice of Internal Auditing. The MDP was previously evaluated in 2013-14.
The audit component of the engagement examined the following criteria:
Governance, Controls and Risks
- Roles and responsibilities related to the MDP are established and followed.
- Departmental pre-market process controls are adequate.
- Departmental post-market controls are in place for medical device incidents so that high risks to the health of Canadians are prioritized and mitigated.
- Sex- and gender-based analysis is considered and implemented in the management of the MDP.
- The Department is tracking progress towards commitments made in Health Canada’s Action Plan on Medical Devices.
The evaluation component of the engagement examined the following themes and questions:
Program Impacts and Scope of Activities
- How effective has the MDP been in supporting Canadians in making informed decisions about medical devices and that Canadians have access to safe, effective, and quality medical devices?
- Is the scope of those activities in line with Health Canada’s mandate?
- How do the benefits to society achieved by the MDP compare to its costs?
Adapting to Change
- Is the MDP positioned to address both current demands, as well as those emerging from a fast-changing and increasingly complex environment?
- Are there other regulatory tools that Health Canada needs to adapt to a changing environment?
Efficiency of Program Organization
- To what extent have the organizational structure, activities, processes, and resource allocations allowed the Program to run efficiently?
- What alternatives exist to the current organizational model?
- Is the Program optimizing resources through international and domestic collaborations?
Methodology
Data for this engagement was collected using the following methods:
INTERVIEWS
- With 73 key informants, including:
- 26 Health Canada staff involved with the MDP;
- 8 experts, academics, and Scientific Advisory Committee members;
- 14 healthcare professionals or health associations;
- 10 industry members;
- 11 individuals, or family members, using a medical device; and
- 4 representatives of other countries.
DOCUMENT REVIEW
Included program files, policies, regulations, guidelines, records of decision, etc. Results from the Regulatory Transparency and Openness Framework survey and the Health Products and Food Branch Transparency survey were also included as part of the document review.
COMPARATIVE ANALYSIS
Reviewed the organizational structure of the medical device programs of six other regulatory agencies (e.g., the United States FDA, France’s National Agency for the Safety of Medicines and Health Products) and interviewed representatives from the FDA, Ireland’s Health Product Regulatory Authority, and Australia’s Therapeutic Goods Administration.
DATA ANALYSIS AND TRANSACTIONAL TESTING
Examined a sample of transactions from each of the MDP functions, as well as performance data collected by the Program.
FINANCIAL DATA REVIEW
Examined financial data for each MDP function and across the Program.
CASE STUDIES
Examined the cases of two medical devices to better understand the process for approving, monitoring, and recalling devices in Canada: (1) the Essure Permanent Birth Control device, and (2) the Paradigm Insulin Pump. The evaluation also conducted a case study of the R2D2 Project – Digital Health Technologies, in an effort to assess its early impacts on the MDP.
LITERATURE REVIEW
Examination of literature regarding risk communications, the evolution of the medical devices sector in Canada, and stakeholder engagement.
Limitations and Mitigation Strategies
The following table outlines the limitations encountered during the implementation of the data collection methods selected for this engagement. Also noted are the mitigation strategies implemented to ensure that engagement findings could be used with confidence in guiding program planning and decision making.
| Limitation | Impact | Mitigation Strategy |
|---|---|---|
There was only one data point available from the 2016 Regulatory Transparency and Openness Framework (RTOF) regarding Health Canada’s efforts to improve access to timely, useful and relevant health and safety information. As well, the survey had very low response rate |
Inability to consistently compare past and present stakeholder perceptions and inability to generalize these perceptions to the entire group of program stakeholders or the Canadian public. |
Forty-seven key informant interviews with a variety of the MDP’s stakeholders using questions validated by the Program to ensure the interviews addressed their information needs. These interviews validated some of the findings from the 2016 RTOF survey, and whether perceptions changed over time. |
External key informant interviews were only possible with a small number of individuals from different stakeholder groups. |
Findings from the interviews are not generalizable to the broader stakeholder groups or the Canadian public. |
Data from the key informant interviews were triangulated with other sources of data (e.g., documentation or program data) where possible. |
Information on the social and financial benefits, as well as the costs related to medical devices was often not publicly available. |
A roll-up of the social and financial impacts of the MDP’s activities was not possible due to a lack of available data. |
Key informant interviews and case studies were used to collect lived experience perspectives on the positive and negative impacts of medical devices, as well as perceived impacts on the health care system. A few databanks and documents were also used to draw examples of the health and financial impacts of select medical devices on the Canadian health care system. |
Appendix B- Internal Audit Summary
AUDIT OBJECTIVE: To provide assurance that the Program had sufficient controls to ensure that its mandate was fulfilled, as well as to assess sex- and gender-based analysis (SGBA+) integration into the lifecycle of the Program.
AUDIT SCOPE: This engagement will examine both pre- and post-market activities from April 2013 to August 2019, and will apply a SGBA+ lens where relevant. The testing of business transactions will cover FY 2018-19 onwards.
AUDIT CONCLUSION: Overall, we found that the Program had implemented sufficient controls in most areas to ensure that its mandate of ensuring medical devices approved for the Canadian market were safe, effective, and of high quality was fulfilled. While there were efforts demonstrating consideration of SGBA+, the Program will benefit from continuing its work to integrate SGBA+ in the life cycle of medical devices.
Controls were generally effective for pre-market side activities and active post-market surveillance was taking place. We found that defined roles and responsibilities were unclear at the operational levels, and insufficient capacity for post-market activities, outdated guidance documentation, and a lack of horizontal data analytics methodology hindered the Program’s overall efficiency and created backlogs.
| CRITERIA | CONCLUSION | RISK | RESIDUAL RISK | RECOMMENDATION |
|---|---|---|---|---|
1. Roles and responsibilities related to the Medical Devices Program were established, followed, and aligned with operational requirements. |
We found that senior management committees were in place and the broad roles and responsibilities of the Program were defined; but there was not a clear framework for roles and responsibilities that would provide more precision at the operational level between the directorates. There was also a lack of an integrated risk management approach across the Program. |
2 Minor Risk |
Unclear roles and responsibilities and a lack of an integrated risk management strategy is affecting the Program’s efficiency. With the rapid changes in this environment, the impact on the Program’s efficiency may increase in the future. |
Recommendation 5 |
2. Departmental pre-market processes controls were adequate. |
We found processes and controls were adequate. Guidance documentation was available to guide Program staff in the intake, screening, evaluation, and approval of medical device licences and applications for medical device licences. Many internal and external guiding documents, however, were significantly outdated and did not reflect current operational processes. Further incorporation of data analytics could strengthen Special Access Program controls. |
2 Minor Risk |
Outdated documentation and difficulties for stakeholders in accessing information can lead to inaccuracies in application submissions and, therefore, affect the timeliness of licensing decisions. Outdated guidance documents have the potential to mislead Program staff when reviewing submissions, particularly new hires who are unfamiliar with the process. |
Recommendation 4 |
| 2 Minor Risk |
Lack of continued and consistent use of data analytics and monitoring of the Special Access Program applications impedes identification and monitoring of higher-risk applications, such as most frequently requested devices, most frequent applicants or companies, or most frequent unique combinations of device and applicant. This, in turn, may create a risk of circumvention of the medical devices regulations. |
Recommendation 6 | ||
3. Departmental post-market controls were in place for medical device incidents so that high risks to the health of Canadians were prioritized and mitigated. |
We found the Program had established adequate post-market controls to monitor medical devices after they were licensed, to process incidents based on the risk level, and to ensure their continued safety and effectiveness. However, there were data entry delays exceeding service standards that were attributed to insufficient capacity. We found that data within the Program was fragmented into several databases, document repositories, and tracking sheets, which limited directorates’ ability to work collaboratively, and posed a risk to data integrity. A significant proportion of guidance documents were not up-to-date. |
3 Moderate Risk |
Fragmentation of data between directorates creates an obstacle to effective and efficient inter-team communication, collaboration, and access to information. The fragmentation affects the Program as a whole. |
Recommendation 6 |
| 3 Moderate Risk |
Lack of workload analysis, workforce needs analysis, and a staffing plan is negatively affecting the Program’s ability to fulfill its mandate. | Recommendation 1 | ||
| 2 Minor Risk |
Outdated guidance documentation hinders the efficiency of post-market operations, as they do not reflect Program changes, including the establishment of ROEB, the implementation of DocuBridge, and the modernization of the Medical Devices System. | Recommendation 4 | ||
| 4. Sex- and gender-based analysis is considered and implemented in the management of the Medical Devices. | We found that the Program considered and incorporated sex- and gender-based analysis + into various activities. However, it had not been implemented consistently for Program management to allow integration of this analysis into the life cycle of medical devices. | 2 Minor Risk |
Without the implementation of consistent Sex- and Gender-Based Analysis + analysis into the life cycle of medical devices, it is difficult to assess risk factors and how diverse groups of women, men, and non-binary people are affected by a variety of medical devices on the Canadian market. | Recommendation 3 |
Appendix C- Summary of Evaluation Findings
| Evaluation Questions | Indicators | Summary of Findings | Recommendations |
|---|---|---|---|
Program Impacts and Scope of Activities |
|||
How effective has the MDP been in supporting Canadians in making informed decisions about medical devices and that Canadians have access to safe, effective, and quality medical devices? |
|
Health Canada was generally effective at ensuring the safety, effectiveness, and quality of medical devices in the Canadian market. The Department was perceived to be a trusted regulator and available data suggests that the safety of medical devices in Canada is comparable to that of other countries. However, delays in processing medical device incident reports affected the Program’s ability to proactively detect such incidents. The MDP also experienced delays in completing the assessment of medical device incidents. While, these challenges represent a risk for the Department, there was no evidence that they affected Health Canada’s ability to ensure the safety of medical devices. The MDP made progress in balancing safety with access to medical devices. While the Program processed the majority of device licence applications within services standards, most of those applications did not meet regulatory requirements when submitted to Health Canada. They required additional information, which may have increased their processing time. The reasons why the majority of device licence applications did not meet program requirements when first submitted are unclear. Publicly available guidance documents outlining these requirements are difficult to find and are sometimes outdated. At the same time, the Program implemented various measures to improve industry’s understanding of the regulatory process (e.g., an e-learning course). In addition to guidance documents for industry, Health Canada disseminates a number of different types of communications products to help health care professionals and Canadians make informed decisions about the use of a device. Most of the MDP’s risk communications were disseminated within service standards. The MDP improved its communications regarding medical devices and its engagement with stakeholders. The Department also completed 100% of their planned consultations regarding medical devices in 2017-18 and 2018-19. While these efforts were well perceived by industry, improvements are still required for Health Canada to become a go-to source of information for health care professionals, patient groups, and the Canadian public in general. Accessing Health Canada’s online communications products is often challenging, and information available online is not always up-to-date. |
Recommendation 1 Recommendation 4 |
Is the scope of those activities in line with Health Canada’s mandate? |
|
The MDP’s activities cover the life cycle of a medical device from providing regulatory guidance to manufacturers during their research and product development, through to the review of device licence applications, as well as monitoring, prevention, and intervention for devices that are licensed for sale in Canada. Overall, the MDP’s activities are similar in scope to other Health Canada regulatory programs (e.g., pharmaceuticals). They also align with the Department’s mandate and stewardship role of protecting Canadians and facilitating access to products that are vital to their health and well-being. |
|
How do the benefits to society achieved by the MDP compare to its costs? |
|
Many Canadians are dependent on medical devices to improve their health and quality of life, as these devices allow them to engage in everyday activities. Medical devices also provide economic benefits for the health care system, as the cost of devices may be less than the cost of surgery or other forms of treatment (e.g. the cost of a drug secreting stent compared to coronary bypass surgery). (Note: evaluation conclusions in this area are limited due to a lack of data). |
|
Adapting to Change |
|||
Is the MDP positioned to address both current demands as well as those emerging from a fast-changing and increasingly complex environment? |
|
Health Canada made significant strides in adapting its regulatory framework to remain effective at regulating such rapidly changing and increasingly complex devices. For instance, the MDP developed and implemented a new Digital Health Division and created two new scientific advisory committees, amended the Food and Drugs Act to increase the power of compliance inspectors, and proposed regulatory amendments that would strengthen the Program’s ability to evaluate potential issues with medical devices. The MDP also developed several new guidance documents for industry, increased engagement with its stakeholders including patients and progressed in its efforts to integrate sex and gender considerations. As many of these initiatives were recently implemented it was too early to determine their impact on the Program. |
Recommendation 2 Recommendation 3 |
Are there other regulatory tools that Health Canada needs to adapt to a changing environment? |
|
While Health Canada made significant strides to adapt its regulatory framework, other challenges remain to be addressed to ensure the MDP continues to meet the demands of the changing environment. For instance, there is no mechanism to address the growing issue around medical devices offered through a subscription or service (e.g. online diagnostic tools, or to track individuals using implanted devices so they can be notified directly by Health Canada if there is an incident. There are also opportunities to further integrate sex and gender considerations in all of the program activities, where relevant. |
|
Efficiency of Program Organization |
|||
To what extent have the organizational structure, activities, processes, and resource allocations allowed the Program to run efficiently? |
|
Various factors impacted the MDP’s ability to function efficiently including a lack of clarity regarding the roles and responsibilities across the three MDP functions, as well as communications at the operational level. At times this led to inefficiency, including the duplication of efforts across program functions. Additionally, the MDP’s information is stored across several repositories, which means that it has to be consolidated from various sources in order to understand the full history of Health Canada activities in relation to a device. There was also a lack of integration between MDSAP audit information and compliance and enforcement activities, which further limited the program’s ability to adopt a life cycle approach. An insufficient capacity also challenged the post-market surveillance function’s ability to process incident reports and complete signal assessments within service standards. The implementation of mandatory reporting of medical device incidents by approximately 775 Canadian hospitals will likely further affect the post-market surveillance function’s ability to manage its workload, as the number of incident reports received may double. Mandatory reporting may also impact the resources and activities of the compliance and enforcement function, because inspectors will be required to monitor potential under-reporting and inspect hospitals. At the time of the engagement Health Canada had not fully analyzed these implications for the MDP’s workload. |
Recommendation 1 |
What alternatives exist to the current organizational model? |
|
As of November 2019, part of the MDP was restructured into a new Medical Devices Directorate, which integrates the pre-market function and the post-market surveillance function’s Marketed Pharmaceuticals and Medical Devices Bureau. This new directorate aligns with approaches taken by Ireland and the United States. The early impacts of the new medical devices programs in these two countries include improved communications, reduced delays getting devices into the market, and a greater ability to employ a life cycle approach. It is anticipated that Health Canada’s new Medical Devices Directorate will have a similar impact and help foster a life cycle approach for the regulation and monitoring of medical devices. |
Recommendation 5 |
Is the Program optimizing resources through international and domestic collaborations? |
|
Health Canada engages in several international initiatives and partnerships, including the International Medical Device Regulators Forum (IMDRF), to share expertise and harmonize practices, policies, and regulations with other jurisdictions. It is also harmonizing its requirements for medical device licence applications with other countries with the implementation of a Table of Contents and the MDSAP. |
Recommendation 2 |
Logic Model - Text description
This figure, read from bottom to top, is a logic model that describes the organization of the program to meet the outcomes.
Target Population
Industry/private sector; market authorization holders; registered establishments licence holders; health professionals and practitioners; hospitals; and, Canadian public
Activities: Actions Undertaken include:
- Review, amend, develop and/or implement regulatory framework and agreements;
- Conduct outreach and engagement activities with targeted industry partners and stakeholders; respond to inquires and provide information to Canadians;
- Conduct scientific and regulatory-based assessments of Medical Device Industry submissions and site-licence applications;
- Conduct surveillance and monitoring of Medical Device products safety on the Canadian market; and,
- Conduct compliance and enforcement activities (industry inspections, compliance verifications, investigations, etc.) and compliance promotions.
These activities then lead to the following Outputs, Products and Services:
- Legislations; regulations, proposals/amendments; standards; policies; guidelines; memoranda agreements;
- Information (e.g. data, publications, notifications, responses to public inquiries, etc.) and consultations;
- Applications review; risk-benefit assessment; investigational testing authorizations; market authorizations; special access program authorizations; establishment licences;
- Data analysis and sourcing; signal prioritization; safety reviews; regulatory and non-regulatory recommendations; post-market plans; risk communications; health risk assessments; and,
- Compliance, monitoring and enforcement report; risk management and mitigation plans; responses and decisions to identified risks (stop sale, recalls, licence & registration suspension/cancellation, seizure, prosecution).
The outputs then lead Short-term Outcome(s), Change of Awareness/Capacity:
- Industry is informed of regulatory requirements for medical devices;
- Program is delivered taking a coordinated approach with partners and stakeholders; and
- Partners, stakeholders and Canadians are informed of the risks and benefits of medical devices.
These short-term outcomes then lead to Medium-term Outcome(s),Change in Behaviour:
- Industry is compliant with pre-market and post-market regulatory requirements; and,
- Partners, stakeholders and Canadians use the information provided by the program to inform their decisions.
These medium-term outcomes then lead to Long-term Outcome(s), Change of State:
Canadians have access to safe, effective and quality medical devices
These long-term outcomes then lead to Impact(s), Result(s) for Canadians:
- Protecting Canadians from unsafe products, food and public health threats
The source for this impact statement is Draft Healthier Canadians Charter – Health Protection Outcome.
Appendix E – MDP Key Roles and Responsibilities at the Time of the Engagement
Health Products and Food Branch (HPFB):
- Therapeutic Product Directorate (TPD): responsible for regulating medical devices for human use under the authority of the Food and Drugs Act and Medical Devices Regulations.
- Medical Device Bureau (MDB): develops policy related to medical devices, reviews applications for Class II to IV medical devices to permit their importation and sale in Canada, authorizes investigational testing, gives access to non-marketed devices through the Special Access Program when needed, and supports the post-market monitoring and surveillance activities of MHPD by conducting risk assessments of post-market incidents.
- Marketed Health Products Directorate (MHPD): develops post-market surveillance policy, conducts clinical and epidemiology post-market safety surveillance, carries out risk management activities, including the issuance of risk communications, and conducts regulatory oversight of advertising.
- Health Product Safety and Effectiveness Information Bureau (HPSEB): responsible for verification reviews of mandatory problem reports and voluntary reports through the CMDSNet.
- Marketed Pharmaceuticals and Medical Devices Bureau (MPMDB): responsible for signal detection, signal assessment, and other types of safety assessment, as well as the development of recommendations in response to identified safety issues, including deciding on the need for risk communications and coordinating related content development.
Regulatory Operations and Enforcement Branch (ROEB):
- Medical Devices and Clinical Compliance Directorate (MDCCD): manages national compliance and enforcement programs to ensure the safety and quality of many therapeutic products, including medical devices.
- Medical Device Compliance Program (MDCP): focuses on the safety and quality of medical devices on the market and on manufacturers of Class I devices, as well as importers and distributers of Class I-IV devices. This program includes the Medical Device Compliance and Licensing Unit, (MDCLU) the Medical Device Inspection Program (MDIP), and the Medical Device Compliance and Enforcement Program (MDCEP).
- Policy, Planning and Analytics Unit (PPAU): provides strategic and operational policy, planning and data analytics support to help guide MDCCD compliance and enforcement activities and decision making.
Endnotes
- Footnote 1
-
Health Canada (2019). Final Report: Fees for Drugs and Medical Devices. Retrieved from: https://www.canada.ca/en/health-canada/services/publications/drugs-health-products/fees-drugs-medical-devices.html#exec
- Footnote 2
-
Food and Drugs Act, RSC 1985, c F-27. Retrieved from: https://laws-lois.justice.gc.ca/eng/acts/f-27/
- Footnote 3
-
Medical Devices Regulations, SOR/98-282. Retrieved from: https://laws-lois.justice.gc.ca/eng/regulations/sor-98-282/
- Footnote 4
-
Snowdon, A, Zur, R, Shell,J. Transforming Canada Into a Global Centre for Medical Device Innovation and Adoption. London, ON: The Ivey Centre for Health Innovation and Leadership. 2011. Retrieved from: ..\..\..\J-Evaluation Data Collection and Analysis\Preliminary findings\Analysis Files\2.2. IVEY_Transforming CAN into a Global Centre.pdf
- Footnote 5
-
Canadian Institute for Health Information(.n.d.). Patient Cost Estimator. Retrieved from: https://www.cihi.ca/en/patient-cost-estimator
- Footnote 6
-
Diabetes Canada (n.d.). Why action is needed to prevent amputations in Ontario. Retrieved from: https://diabetes.ca/DiabetesCanadaWebsite/media/Advocacy-and-Policy/Advocacy%20Reports/Amputation_Prevention_Infographic_ON_8-5x11_3.pdf
- Footnote 7
-
Health Canada (2011a). Guidance Document for Mandatory Problem Reporting for Medical Devices. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/reports-publications/medeffect-canada/guidance-document-mandatory-problem-reporting-medical-devices-health-canada-2011.html
- Footnote 8
-
Government of Canada (2014). Paradigm Insulin Pumps and Paradigm Insulin Infusion Pump (2014-01-17). Retrieved from: https://www.healthycanadians.gc.ca/recall-alert-rappel-avis/hc-sc/2014/39013r-eng.php
- Footnote 9
-
Health Canada (2019a). Medical Devices. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices.html
- Footnote 10
-
Health Canada (2018a).Drug and medical device highlights 2018: Helping you maintain and improve your health. Retrieved from: https://www.canada.ca/en/health-canada/services/publications/drugs-health-products/drug-medical-device-highlights-2018.html
- Footnote 11
-
Health Canada (2016a). Medical Device Active Licence Listing (MDALL). Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices/licences/medical-devices-active-licence-listing.html
- Footnote 12
-
Health Canada (2019b). Medical Devices Directorate. Retrieved from: https://www.canada.ca/en/health-canada/corporate/about-health-canada/branches-agencies/health-products-food-branch/medical-devices-directorate.html#_Bureau_of_Investigational
- Footnote 13
-
Health Canada (2015). How Drugs are Reviewed in Canada. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/fact-sheets/drugs-reviewed-canada.html. Health Canada (2019c). Biologics, Radiopharmaceuticals and Genetic Therapies. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies.html
- Footnote 14
-
Health Canada (2011). About Mission, Values, Activities. Retrieved from: https://www.canada.ca/en/health-canada/corporate/about-health-canada/activities-responsibilities/mission-values-activities.html
- Footnote 15
-
Health Canada. (2018b). Annual trends for the adverse reaction case reports of health products and medical device problem incidents to Health Canada (2008-2017). Retrieved from: https://www.canada.ca/en/health-canada/services/publications/drugs-health-products/annual-trends-adverse-reaction-case-reports-health-products-medical-device-problem-incidents.html
- Footnote 16
-
Health Canada (2019d). Canadian Medical Devices Sentinel Network. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/medeffect-canada/medeffect-canada-canadian-medical-devices-sentinel-network-pilot-project.html
- Footnote 17
-
Patient safety Institute (2019). Mandatory Reporting of Serious Adverse Drug Reactions and Medical Device Incidents by Hospitals. Retrieved from: ..\..\..\J-Evaluation Data Collection and Analysis\Preliminary findings\Analysis Files\Patient Safety Insitute_MDI reporting.ppt. Health Canada (2019e). Report a medical device problem: reporter. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/medeffect-canada/adverse-reaction-reporting/medical-device.html
- Footnote 18
-
Health Canada. (2018b). Annual trends for the adverse reaction case reports of health products and medical device problem incidents to Health Canada (2008-2017). Retrieved from: https://www.canada.ca/en/health-canada/services/publications/drugs-health-products/annual-trends-adverse-reaction-case-reports-health-products-medical-device-problem-incidents.html
- Footnote 19
-
Health Canada. (2018b). Annual trends for the adverse reaction case reports of health products and medical device problem incidents to Health Canada (2008-2017). Retrieved from: https://www.canada.ca/en/health-canada/services/publications/drugs-health-products/annual-trends-adverse-reaction-case-reports-health-products-medical-device-problem-incidents.html
- Footnote 20
-
Health Canada. (2011b). Risk communication - Protecting Canadians through information. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/reports-publications/medeffect-canada/risk-communication-protecting-canadians-through-information-health-canada-2011.html
- Footnote 21
-
Government of Canada. (2019c). Health and biosciences sector regulatory review roadmap. Retrieved from: https://www.canada.ca/en/health-canada/corporate/about-health-canada/legislation-guidelines/acts-regulations/targeted-regulatory-reviews/health-biosciences-sector-regulatory-review-roadmap.html#agile
- Footnote 22
-
Government of Canada. (2019c). Health and biosciences sector regulatory review roadmap. Retrieved from : https://www.canada.ca/en/health-canada/corporate/about-health-canada/legislation-guidelines/acts-regulations/targeted-regulatory-reviews/health-biosciences-sector-regulatory-review-roadmap.html#agile
- Footnote 23
-
Health Canada (2017). Health Portfolio Sex and Gender-Based Analysis Policy. Retrieved from: https://www.canada.ca/en/health-canada/corporate/transparency/corporate-management-reporting/heath-portfolio-sex-gender-based-analysis-policy.html
- Footnote 24
-
Government of Canada (2019d). Regulations Amending Certain Regulations Made Under the Food and Drugs Act (Post-market Surveillance of Medical Devices), Canada Gazette, Part 1, vol.153, no.24, p.2731, June 15, 2019. Retrieved from: http://gazette.gc.ca/rp-pr/p1/2019/2019-06-15/html/index-eng.html
- Footnote 25
-
Government of Canada (2019d). Regulations Amending Certain Regulations Made Under the Food and Drugs Act (Post-market Surveillance of Medical Devices), Canada Gazette, Part 1, vol.153, no.24, p.2731, June 15, 2019. Retrieved from: http://gazette.gc.ca/rp-pr/p1/2019/2019-06-15/html/index-eng.html
- Footnote 26
-
Health Canada (2013). Guidance Document: Considerations for Inclusion of Women in Clinical Trials and Analysis of Sex Differences. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/clinical-trials/considerations-inclusion-women-clinical-trials-analysis-data-sex-differences.html
- Footnote 27
-
Health Canada (2019g). Scientific Advisory on Health Product’s for Women Summary: Findings, Advice, 2019-05-16-17. Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/scientific-expert-advisory-committees/health-products-women/summary-findings-advice-2015-05-16-17.html
- Footnote 28
-
U.S. Food and Drug Administration (2019a). PMA Review Process. Retrieved from: https://www.fda.gov/medical-devices/premarket-approval-pma/pma-review-process
- Footnote 29
-
Health Canada (2016c). Medical Devices Bureau Annual Performance Report April 1, 2015 through March 31, 2016. ..\..\..\J-Evaluation Data Collection and Analysis\Preliminary findings\Analysis Files\254.1 MDB Annual Performance Report 2015-2016.pdf. Health Canada (2019j). Medical Devices Bureau Annual Performance Report April 1, 2018 through March 31, 2018 ..\..\..\J-Evaluation Data Collection and Analysis\Preliminary findings\Analysis Files\254.4 MDB YEARLY REPORT - 2018-19.pdf
- Footnote 30
-
Emergo by UL (2019). Australia: The Regulatory Process for Medical Devices. Retrieved from: https://www.emergobyul.com/resources/australia-process-chart
- Footnote 31
-
Health Canada (2018). Summary Basis of Decision (SBD). Retrieved from: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/summary-basis-decision.html
- Footnote 32
-
LeBrun, M., DiMuzion, J., Beauchamp, B., Reid, S., & Hogan, V. (2013). Evaluating the health literacy burden of Canada’s public advisories: A comparative effectiveness study on clarity and readability. Drug Safety, 36(12):1179-87. doi: 10.1007/s40264-013-0117-8.
- Footnote 33
-
Medical Devices Regulations, SOR/98-282. Retrieved from: https://laws-lois.justice.gc.ca/eng/regulations/sor-98-282/
- Footnote 34
-
HPRA (2018). Annual Report 2018. Retrieved from: https://www.hpra.ie/docs/default-source/publications-forms/corporate-policy-documents/annual-report-2018.pdf?sfvrsn=5
- Footnote 35
-
U.S. Food & Drug Administration. (2019b). Reorganization of the Center for Devices and Radiological Health. Retrieved from: https://www.fda.gov/about-fda/center-devices-and-radiological-health/reorganization-center-devices-and-radiological-health
- Footnote 36
-
Health Canada (2019k). The Medical Devices Directorate: Focused – Agile – World Class. Retrieved from: https://www.raps.org/RAPS/media/news-images/regulators/Executive-Summary-MDD-EN-(1).PDF
- Footnote 37
-
Health Canada (2016d). Health Products and Food Branch 2016-2021 Strategic Plan. Retrieved from: https://www.canada.ca/en/health-canada/corporate/about-health-canada/reports-publications/health-products-food-branch/health-products-food-branch-strategic-plan-2016-2021.html
- Footnote 38
-
Government of Canada (2019a). Health Canada launches consultations to improve the safety of medical devices. Retrieved from: https://www.canada.ca/en/health-canada/news/2019/06/health-canada-launches-consultations-to-improve-the-safety-of-medical-devices.html
- Footnote 39
-
Government of Canada (2019b). Regulations Amending the Food and Drug Regulations (Serious Adverse Drug Reaction Reporting — Hospitals), P.C. 2019-746, June 9, 2019, SOR/19-190, s.30(1.3), Canada Gazette, Part II, vol.153, no.13, p.3137, June 26, 2019. Retrieved from: http://www.gazette.gc.ca/rp-pr/p2/2019/2019-06-26/html/sor-dors190-eng.html
- Footnote 40
-
Government of Canada (2019b). Regulations Amending the Food and Drug Regulations (Serious Adverse Drug Reaction Reporting — Hospitals), P.C. 2019-746, June 9, 2019, SOR/19-190, s.30(1.3), Canada Gazette, Part II, vol.153, no.13, p.3137, June 26, 2019. Retrieved from: http://www.gazette.gc.ca/rp-pr/p2/2019/2019-06-26/html/sor-dors190-eng.html
- Footnote 41
-
Government of Canada (2019b). Regulations Amending the Food and Drug Regulations (Serious Adverse Drug Reaction Reporting — Hospitals), P.C. 2019-746, June 9, 2019, SOR/19-190, s.30(1.3), Canada Gazette, Part II, vol.153, no.13, p.3137, June 26, 2019. Retrieved from: http://www.gazette.gc.ca/rp-pr/p2/2019/2019-06-26/html/sor-dors190-eng.html
Footnotes
- Footnote i
-
In November, 2019, part of the MDP was reorganized into a new Medical Devices Directorate.
- Footnote ii
-
The CMDSNet is a network representing more than 260 hospitals and facilities across Canada. Although reporting is voluntary, some internal key informants noted that the CMDSNet helped to fill the gaps in surveillance and was instrumental in getting incident reports from the health care sector.
- Footnote iii
-
The receipt date and processing date were compared for each incident report to assess whether it was processed within service standards.
- Footnote iv
-
Stakeholders are defined as individuals, groups, or organizations, who are external to the Government of Canada and have a specific interest in, have some influence on, or are affected by a given policy, program, regulatory initiative, or service from Health Canada.
- Footnote v
-
The RTOF Survey was conducted in 2016 by Health Canada. The questionnaire was sent to all individuals or organizations that self-registered with Health Canada to receive information or invitations for consultations on various health-related topics. Results presented in this report pertain only to the individuals and organizations that reported an interest in medical devices. The results should be interpreted with caution due to the small number of respondents. Considering that the respondents self-registered with Health Canada, the survey results cannot be generalized the entire population or a larger group of medical device stakeholders.T
- Footnote vi
-
The sample was constructed with the first five recalls made for the months of January, March, May, July, September, and November, and the last five recalls made for the months of February, April, June, August, October, and December.
- Footnote vii
-
Difference between the “start date” reported on Health Canada’s website and the “date initiated by firm” reported on the U.S. Food and Drug Administration Web site.
- Footnote viii
-
At the time of the engagement, Membership of the Medical Devices Tri-Lateral Committee included the directors of the Medical Device Bureau, the Health Products Surveillance and Epidemiology Bureau (HPSEB), the Marketed Pharmaceutical and Medical Devices Bureau (MPMDB), and the Medical Device Compliance Program (MDCP). A representative from Health Canada’s Office of Policy, Risk Advisory and Advertising also attended the committee to provide a policy lens when required.