
Archivée - Audit et évaluation conjoints du Programme des instruments médicaux de 2013-2014 à 2019-2020
Table des matières
- Liste des figures
- Liste des acronymes
- Résumé
- 1.0 Présentation et contexte
- 2.0 Sécurité, efficacité et qualité des instruments médicaux au Canada
- 3.0 Donner accès aux instruments médicaux au Canada
- 4.0 Communication avec les intervenants et les Canadiens
- 5.0 Organisation et gouvernance du Programme
- 6.0 Ressources du Programme
- 7.0 Conclusions et recommandations
- Management Response and Action Plan
- Annexe A – Portée et méthodologie du projet conjoint
- Annexe B - Résumé de l’audit interne
- Annexe C – Résumé des résultats de l’évaluation
- Annexe D – Modèle logique
- Annexe E – Rôles et responsabilités clés de la MDP au moment du projet
- Notes de fin de document
Liste des figures
- Figure 1 : Exemple de participation de Santé Canada au cycle de vie des instruments médicaux
- Figure 2: Structure organisationnelle du Programme des instruments médicaux dans le cadre du projet
- Figure 3 : Proportion de rapports d’incidents liés aux instruments médicaux saisis en respectant les normes de service de saisie de données
- Figure 4 : Diffusion ciblée des communications sur les risques dans le cadre des normes de service
- Figure 5 : Proportion des demandes d’homologation des instruments médicaux en attente
- Figure 6 : Proportion des demandes d’homologation des instruments médicaux qui étaient conformes aux exigences réglementaires lors du premier examen et après que des renseignements supplémentaires aient été demandés et fournis
- Figure 7 : Structure organisationnelle des programmes d’instruments médicaux aux États-Unis et en Irlande
- Figure 8 : Comparaison des ressources consacrées à l’évaluation des signaux et de la charge de travail correspondante pour les instruments médicaux et les produits pharmaceutiques (2015-2016)
Liste des acronymes
- BIM
- Bureau des instruments médicaux
- BSPSE
- Bureau de la surveillance des produits de santé et de l’épidémiologie
- CRTO
- Cadre réglementaire de transparence et d’ouverture
- DCIMMC
- Direction de la conformité des instruments médicaux et en milieux cliniques
- DGORAL
- Direction générale des opérations réglementaires et de l’application de la loi
- DGPSA
- Direction générale des produits de santé et des aliments
- DPSC
- Direction des produits de santé commercialisés
- IMDRF
- Forum international des organismes de réglementation des instruments médicaux
- PCIM
- Programme de conformité des instruments médicaux
- PIM
- Programme des instruments médicaux
- PUAIM
- Programme unique d’audit des instruments médicaux
- ResSCIM
- Réseau sentinelle canadien pour les instruments médicaux
- TI
- Technologie de l’information
Résumé
Ce rapport porte sur les résultats d’un audit et d’une évaluation conjoints du Programme des instruments médicaux (PIM) de Santé Canada. Le projet a permis d’examiner les activités du PIM d’avril 2013 à août 2019. Le PIM vise à s’assurer que les Canadiens ont accès à des instruments médicaux sûrs, efficaces et de qualité de la manière suivante :
- l’approbation des licences d’établissements pour les instruments médicaux et de l’homologation des instruments médicaux;
- la surveillance des instruments disponibles au Canada;
- les inspections des fabricants, des importateurs et des distributeurs;
- la communication avec les Canadiens à propos des risques liés aux instruments.
En 2018-2019, les dépenses prévues du PIM s’élevaient à environ 33 millions de dollars, dont 15 millions de dollars ont été financés par Santé Canada. Les fonds restants de programme ont été récupérés grâce aux frais pour les services de réglementation (p. ex., licenses à vendre des instruments médicaux)Note de bas de page 1
Principales constatations
Assurer la sûreté, la qualité et l’efficacité des instruments médicaux au Canada
Le PIM avait mis en place des contrôles suffisants pour ses activités de précommerclialisation afin de garantir la sécurité, l’efficacité et la qualité des instruments médicaux. Il a également réalisé la plupart de ses activités après la mise en marché dans le respect des normes de service.Cependant, les retards dans le traitement des rapports d’incidents liés aux instruments médicaux ont affecté la capacité du Programme à détecter de tels incidents de manière proactive. Le PIM a également connu des retards dans l’évaluation des incidents liés aux instruments médicaux.
Bien que ces défis représentent un risque pour le Ministère, rien n’indique qu’ils ont nui à la capacité de Santé Canada d’assurer la sécurité des instruments médicaux. Le Ministère était perçu comme un organisme de réglementation de confiance par la majorité des informateurs clés externes et les données disponibles suggèrent que la sécurité des instruments médicaux au Canada était comparable à celle d’autres pays.
L’environnement des instruments médicaux est complexe et évolue rapidement avec le développement de nouveaux instruments utilisant des technologies innovantes, telles que la technologie numérique, l’intelligence artificielle et l’impression 3D. Santé Canada a fait d’importants progrès pour adapter son cadre réglementaire afin de demeurer efficace dans la réglementation de ces instruments complexes et en évolution rapide. Le Programme a également progressé dans ses efforts pour intégrer les considérations de l’analyse comparative fondée sur le sexe et le genre dans l’exécution des activités de programme. Toutefois, il était possible d’intégrer ces considérations de manière plus systématique dans l’ensemble des fonctions de programme.
Donner accès aux instruments médicaux au Canada
Le PIM a progressé dans la recherche d’un équilibre entre la sécurité et l’accès aux instruments médicaux. Il a traité la majorité des demandes d’homologation des instruments médicaux tout en respectant les normes de service, mais la plupart de ces demandes ne répondaient pas aux exigences réglementaires lors de leur présentation initiale à Santé Canada. Cela peut avoir augmenté leur temps de traitement. Les raisons pour lesquelles la majorité des demandes d’homologation pour un instrument médical ne répondaient pas aux exigences du Programme lors de leur présentation initiale ne sont pas entièrement comprises. Les documents d’orientation accessibles au public et décrivant ces exigences étaient difficiles à trouver et étaient parfois obsolètes. Parallèlement, le Programme a mis en œuvre diverses mesures visant à améliorer la compréhension du processus réglementaire par l’industrie (p. ex., un cours d’apprentissage en ligne) et a aligné ses exigences à celles d’autres pays, dans un effort pour améliorer l’accès aux instruments médicaux au Canada. Il a également mis en place diverses initiatives visant à rationaliser l’accès aux instruments médicaux, comme le Programme d’accès spécial, qui permet aux professionnels de la santé d’avoir accès à des instruments médicaux dont la vente n’est pas autorisée au Canada pour une utilisation en cas d’urgence ou lorsque les thérapies conventionnelles ont échoué, qui ne sont pas disponibles ou qui ne sont pas convenables pour traiter un patient.
Communiquer avec les intervenants et les Canadiens
Le PIM a renforcé sa mobilisation auprès des intervenants en organisant des réunions avec des représentants de l’industrie, des discussions avec des professionnels de la santé, des mesures de sensibilisation ciblées auprès de groupes de patients et en offrant aux intervenants et aux Canadiens la possibilité de participer à des consultations en ligne. Bien que ces efforts aient été bien accueillis par l’industrie, des améliorations sont encore nécessaires pour que Santé Canada devienne une source d’information incontournable pour les intervenants autres que de l’industrie et les Canadiens.
Organisation et gouvernance du programme
Divers facteurs ont nuits à la capacité du programme à fonctionner efficacement. Les rôles et les responsabilités dans le cadre des fonctions de surveillance et de conformité avant et après la mise en marché et d’application de la loi n’ont pas toujours été clairement définis, en particulier au niveau opérationnel. Toutefois, le Programme a défini des rôles et responsabilités clairs parmi les évaluateurs scientifiques et les agents médicaux.
De plus, avec la création de la nouvelle Direction des instruments médicaux, on s’attend que les rôles et responsabilités entre les fonctions regroupées de précommercialisation et après la mise en marché soient précisés davantage par moyen de procédures et processus normalisés révisés. Le Programme est basé sur le Règlement sur les instruments médicaux, qui est fondé sur le niveau de risque de diverses classes d’instruments médicaux. Toutes les activités de base du Programme des instruments médicaux sont axées sur une approche basée sur les risques pour la réglementation des instruments médicaux au long de leur cycle de vie. Toutefois, le PIM ne disposait pas d’un cadre global définissant les rôles et les responsabilités ni d’une stratégie intégrée de gestion des risques et de partage de l’information. Ce manque de clarté concernant les rôles et les responsabilités peut avoir contribué au développement de processus parallèles entre les fonctions du PIM.
De plus, les renseignements du PIM étaient gardés dans plusieurs dépôts. Les renseignements disponibles ont dû être consolidés à partir de diverses sources afin de comprendre l’historique complet des activités de Santé Canada concernant un instrument médical.
Ressources du Programme
Le manque de capacité a posé un défi à la capacité de la fonction de surveillance après la mise en marché de traiter les rapports d’incidents et a effectué des évaluations des signaux dans le respect des normes de service. Dans l’ensemble, les activités de surveillance après la mise en marché n’ont reçu qu’environ 7 % du budget du PIM en 2018-2019. De plus, ces dernières années, le nombre de personnes travaillant dans le domaine de la surveillance des instruments médicaux a diminué. En comparaison, le nombre d’employés a augmenté dans les deux autres fonctions du PIM.
Avec l’introduction de la déclaration obligatoire des incidents par les hôpitaux en décembre 2019, la charge de travail de la fonction de surveillance après la mise en marché et de la fonction de conformité et d’application de la loi pourrait augmenter considérablement. Au moment du projet, l’incidence en termes de charge de travail de la déclaration obligatoire n’a pas été entièrement analysée. Toutefois, il convient de noter que les investissements récents ont été par moyen de plusieurs programmes. On s’attend que ceci va mener à une hausse dans la réception de rapports qui sont traités par des systèmes semi-automatisés. Cela représente un risque, car les difficultés liées à l’achèvement en temps opportun de certaines activités de surveillance seront probablement exacerbées si les problèmes de capacité ne sont pas résolus dans ces domaines.
Recommandations
-
La Direction générale des produits de santé et des aliments devrait combler les lacunes actuelles en matière de ressources dans les fonctions suivant la mise en marché auxquelles il incombe le traitement des rapports d’incidents et l’évaluation des incidents. La Direction générale des opérations réglementaires et de l’application de la loi devrait examiner l’incidence qui suit sur les besoins en matière de ressources de la fonction de conformité et d’application de la loi. De plus, les deux directions générales devraient s’assurer que le PIM dispose des capacités et des outils nécessaires pour faire face à l’obligation de déclaration par les hôpitaux introduite en décembre 2019, et à l’application connexe des pratiques de vigilance des hôpitaux.
Les activités de surveillance du PIM ont souffert d’un manque de capacité et n’ont pas toujours pu être menées à bien dans le respect des normes de service. L’introduction de la déclaration obligatoire des incidents liés aux instruments médicaux par environ 775 hôpitaux canadiens en décembre 2019 aura probablement une incidence supplémentaire sur la capacité de la fonction de surveillance suivant la mise en marché de gérer son fardeau de travail, car le nombre de déclarations d’incidents soumises à Santé Canada pourrait doubler. La déclaration obligatoire peut également avoir un effet sur les ressources et les activités de la fonction de conformité et d’application de la loi, car les inspecteurs devront surveiller les éventuelles sous-déclarations et inspecter les hôpitaux. Toutefois, au moment du projet, Santé Canada n’avait pas encore pleinement analysé les conséquences de la déclaration obligatoire sur la charge de travail du Programme.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient poursuivre leurs efforts pour adapter le PIM au moyen d’initiatives nationales et internationales afin de relever les défis provenant du cycle de vie en évolution rapide des instruments médicaux et de l’utilisation des nouvelles technologies.
Bien que Santé Canada ait fait des progrès considérables pour adapter son cadre réglementaire, d’autres défis doivent être relevés pour que le PIM reste efficace à l’avenir. Par exemple, au moment du projet, il n’existait pas de mécanisme pour traiter le problème croissant des instruments médicaux offerts par abonnement ou par service (p. ex. les outils de diagnostic en ligne), ou pour suivre les personnes utilisant des instruments implantés afin qu’elles puissent être informées directement par Santé Canada en cas d’incident.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient examiner et documenter la manière d’intégrer davantage l’analyse comparative fondée sur le sexe et le genre plus (ACSG+) dans le cycle de vie des instruments médicaux, notamment en fournissant des conseils aux demandeurs et en sensibilisant le personnel du PIM.
Il est prouvé que les hommes et les femmes sont touchés différemment par les instruments médicaux en raison des disparités entre les sexes et les genres. Si des progrès ont été réalisés pour intégrer les considérations de sexe et de genre dans le PIM, il existe d’autres occasions d’intégrer davantage ces considérations au sein de toutes les activités du Programme, le cas échéant.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient maintenir ou intensifier leurs efforts de communication et de dialogue avec les intervenants de l’industrie, les professionnels et les organismes de soins de santé, et les groupes de patients. Il est également recommandé que la Direction générale des produits de santé et des aliments améliore l’actualité et l’accessibilité renseignements de programme accessibles en ligne. Cela comprend assurer que les des documents d’orientation sont examinés et mis à jour, au besoin, pour toutes les activités du PIM, y compris sur le site Web du gouvernement du Canada.
Les efforts de Santé Canada pour accroître la communication et la mobilisation avec les intervenants ont été bien accueillis par les représentants de l’industrie. Cependant, Santé Canada ne constituait pas une source incontournable de renseignements pour les représentants des soins de santé, les groupes de patients et la population en général. Il était également nécessaire d’améliorer l’actualité et l’accessibilité de l’information en ligne. En particulier, des documents d’orientation obsolètes peuvent avoir contribué à des inexactitudes dans les demandes d’homologation des instruments médicaux. De plus, les informateurs clés de l’industrie ont mentionné avoir eu des difficultés à trouver les renseignements et les conseils nécessaires pour appuyer l’élaboration de leurs demandes.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient s’assurer que les rôles et responsabilités au niveau opérationnel sont clairement définis et répartis, et que les risques liés au Programme sont déterminés et gérés.
Les rôles et les responsabilités du personnel opérationnel n’ont pas été clairement définis pour les trois fonctions du PIM. Le PIM ne disposait pas d’un cadre global définissant les rôles et les responsabilités ni d’une stratégie intégrée de gestion des risques et de partage de renseignements. Les défis liés aux rôles et aux responsabilités peuvent être relevés, en partie, avec la mise en œuvre de la nouvelle Direction des instruments médicaux annoncée fin 2019, qui intégrera la fonction de précommercialisation et la fonction responsable de l’évaluation des incidents liés aux instruments médicaux. Toutefois, la fonction de conformité et d’application de la loi et le Bureau de la surveillance des produits de santé et de l’épidémiologie (c.-à-d., le groupe responsable du traitement des rapports d’incidents relatifs aux instruments médicaux) relèveront toujours de leurs directions générales d’origine.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient veiller à ce qu’une approche de gestion des données permette de surveiller les instruments médicaux pendant tout leur cycle de vie.
Les renseignements du PIM étaient fragmentés dans plusieurs dépôts. Cette situation a nui à l’efficacité et à la capacité du Programme à adopter une approche axée sur le cycle de vie pour la réglementation et la surveillance des instruments médicaux.
1. Présentation et contexte
Ce rapport présente les résultats d’un audit et d’une évaluation conjoints du Programme des instruments médicaux (PIM) de Santé Canada. Le projet a permis d’examiner les activités du PIM d’avril 2013 à août 2019.
Les domaines examinés comprennent l’adéquation des contrôles de programme, la gestion des risques et l’intégration de l’analyse comparative fondée sur le sexe et le genre plus (ACSG+), ainsi que les effets du PIM, la façon dont il a été positionné pour s’adapter à un environnement de plus en plus complexe et la façon dont la structure organisationnelle au moment du projetNote de bas de page i a soutenu une exécution efficace des activités du PIM.
Depuis mars 2020, la Direction des instruments médicaux s’est efforcée de satisfaire aux besoins immenses des professionnels de santé, des patients et des industries quant à la lutte contre la COVID-19. Le rôle principal du Programme dans la protection de la santé et la sécurité des Canadiens a été mis à la lumière, ce qui a mené à l’accélération de plusieurs activités réglementaires importantes, y compris l’élaboration et la mise en œuvre de plusieurs arrêtés d'urgence, tous visant à fournir un accès rapide aux instruments médicaux liés à la COVID-19 (p. ex., instruments de dépistage, ventilateurs, gants) dans cette période critique. De plus, des processus ont été modifiés afin d’accélérer l’octroi d’une hausse significative de Licences d’établissement d’instruments médicaux afin d’assurer que l’équipement de protection individuelle (EPI) critique est disponible pour lutter contre la COVID-19. Dernièrement, la réception de rapports d’incidents, la surveillance après la mise en marché, les évaluations des risques à la santé et les activités de conformité et de l’application de la loi ont toutes demandé des ressources importantes.
Bien que la finalisation du rapport et de la réponse et plan d’action de la direction du Bureau de l’audit et de l’évaluation (BAE) a été reporté en raison des priorités imposées par la COVID-19, le Programme a continué à être attentif aux recommandations du BAE. En particulier, le Programme a continué à collaborer avec les intervenants au pays et à travers le monde en relation à la COVID-19. De plus, le Programme est reconnaissant des difficultés avec le site Web au fil du temps concernant l’élaboration de nouvelles pages et contenu pour partager des renseignements sur la COVID-19.
Les données ont été collectées à partir d’un examen de la littérature et des documents de programme, des données de mesure du rendement et de l’information financière, ainsi qu’à l’aide d’études de cas, d’une analyse comparative et d’entrevues avec les informateurs clés parmi le personnel du PIM et les intervenants. Les analyses effectuées comprenaient également la mise à l’essai d’un échantillon de transactions opérationnelles entre le 1er avril 2018 et septembre 2019 (voir l’annexe A pour plus de détails).
1.1 Avantages et risques des instruments médicaux
Le terme « instrument médical », tel qu’il est défini dans la Loi sur les aliments et droguesNote de bas de page 2, couvre un large éventail d’instruments médicaux, d’appareils, de dispositifs ou d’articles semblables ou tout réactifs in vitro utilisés dans le traitement, l’atténuation, le diagnostic ou la prévention d’une maladie, d’un trouble ou d’un état physique anormal. Sans les instruments médicaux, de nombreuses procédures médicales courantes ne seraient pas possibles, comme le bandage d’une cheville foulée, le diagnostic de maladies infectieuses ou l’implantation de hanches artificielles.
Le Règlement sur les instruments médicauxNote de bas de page 3 regroupe les instruments médicaux dans l’une des quatre classes de risque suivantes : la classe I représente le risque le plus faible (p. ex., les abaisseurs de langue) et la classe IV représente le risque le plus élevé (p. ex., les valves cardiaques artificielles).
1,3 million de types différents d’instruments médicaux peuvent être vendus au Canada. Ces instruments vont des bandages, des lits d’hôpitaux, des stimulateurs cardiaques, des implants et des appareils d’IRM, aux montres intelligentes.
De nombreux Canadiens dépendent des instruments médicaux pour améliorer leur santé et leur qualité de vie. Dans certains cas, les instruments médicaux comme les stimulateurs cardiaques ont permis de sauver des vies. Selon plusieurs informateurs clés externes qui utilisent des instruments médicaux, l’accès à un instrument a amélioré leur santé et leur a permis de participer à des activités quotidiennes telles que le travail, le sport et les voyages.
Les instruments médicaux présentent également des avantages économiques pour le système de santé. Par exemple, la cardiologie interventionnelle et le recours aux endoprothèses à élution médicamenteuse, au lieu de la réalisation de pontages coronariens, ont permis de réaliser des économies substantielles dans le système de santé et d’améliorer la gestion des maladies cardiaques, ce qui a permis d’améliorer les résultats pour les patients, notamment en réduisant le temps de récupération et en augmentant la capacité de gestion des maladies cardiaquesNote de bas de page 4. Selon l’estimateur des coûts par patient de l’Institut canadien d’information sur la santé, l’implantation d’une endoprothèse coûte environ 6 000 $, comparé à 26 680 $ en moyenne pour un pontage coronarienNote de bas de page 5.
De plus, une étude de Diabète Canada a révélé que des instruments spécialisés pour traiter les ulcères diabétiques du pied peuvent aider à prévenir les amputations, ce qui pourrait entraîner des économies directes allant de 48 à 75 millions de dollars par an pour la province de l’OntarioNote de bas de page 6.
Bien que les instruments médicaux soient précieux pour les Canadiens et le système de soins de santé, ils comportent également un certain niveau de risque. Des incidents peuvent se produire à la suite d’une défaillance d’un instrument, d’une erreur d’application ou d’utilisation, du risque inhérent à un instrument, ainsi que de l’interaction négative entre l’instrument et un autre instrument ou une technique médicaleNote de bas de page 7. De tels incidents peuvent entraîner des problèmes de santé ou des maladies pour l’utilisateur, et dans certains cas, la mort. Par exemple, en 2014, la pompe à insuline Paradigm a été rappelée par le fabricant parce qu’une mauvaise utilisation par inadvertance aurait pu entraîner l’administration d’une dose d’insuline dépassant la quantité prévue par l’utilisateur et causer une hypoglycémieNote de bas de page 8.
Le PIM vise à atténuer les risques associés aux instruments médicaux en veillant à ce que les Canadiens aient accès à des instruments sûrs, efficaces et de qualitéNote de bas de page 9. Les activités du PIM couvrent le cycle de vie d’un instrument médical, de la fourniture de conseils réglementaires aux fabricants au cours de leurs recherches et du développement de leurs produits, jusqu’à l’examen des demandes d’homologation des instruments médicaux, ainsi que la surveillance, la prévention et l’intervention pour les instruments qui sont homologués pour vente au CanadaNote de bas de page 10. La fonction de conformité et d’application de la loi du PIM permet également de contrôler dans quelle mesure les fabricants, distributeurs et importateurs respectent la Loi sur les aliments et drogues et le Règlement sur les instruments médicaux. Il octroie des licences d’établissement d’instruments médicaux, mène des inspections proactives pour les fabricants d’instruments de classe I et pour les importateurs et distributeurs de tous les instruments médicaux, ainsi que des inspections proactives des fabricants, importateurs et distributeurs de toutes les classes d’instruments. La figure 1 ci-dessous donne des exemples de certaines des activités du PIM qui couvrent les différentes étapes du cycle de vie des instruments médicaux. Il est à noter que Santé Canada n’homologue pas les instruments médicaux de classe I, mais les surveille grâce à l’octroi de licences d’établissement aux fabricants, importateurs et distributeurs d’instruments médicaux au CanadaNote de bas de page 11.
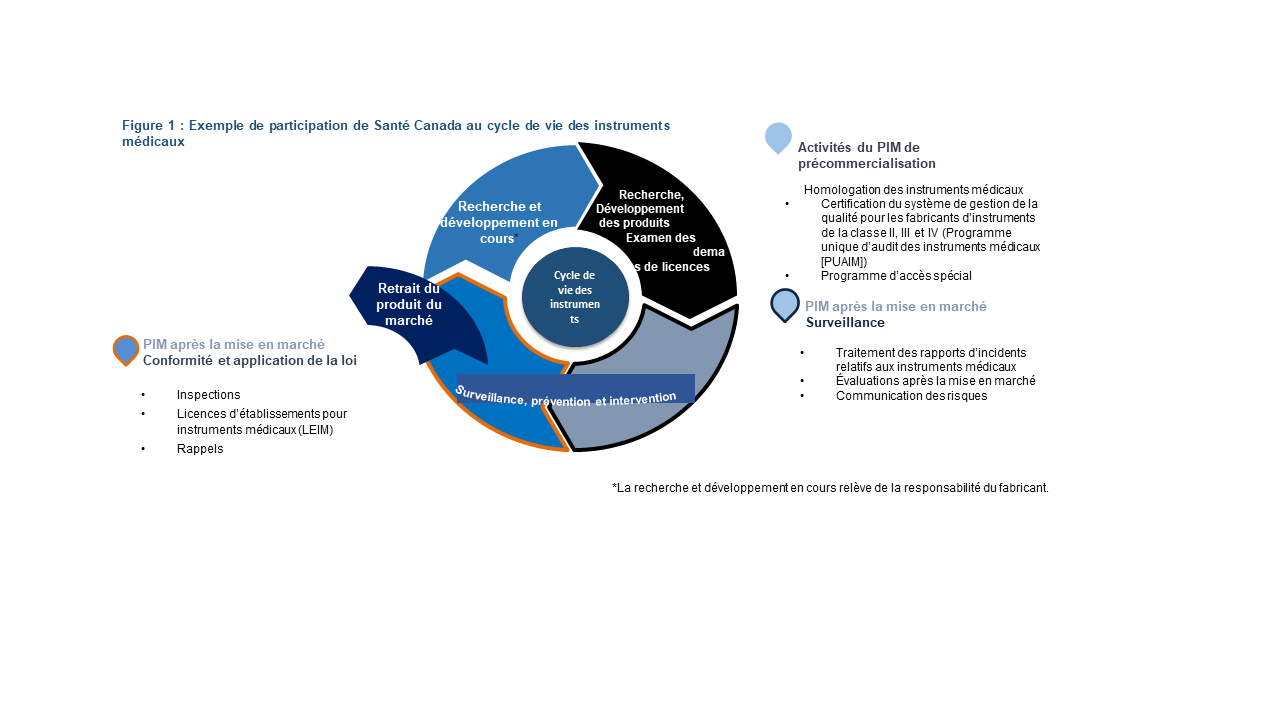
Figure 1 - Équivalent textuel
Cette figure illustre certains des rôles du Programme des matériels médicaux de Santé Canada et donne des exemples d’activités visant à réglementer le cycle de vie des matériels médicaux. La figure est un modèle circulaire en quatre parties. Chaque section représente une partie précise du cycle de vie d’un matériel médical. Chaque section est accompagnée d’un énoncé donnant des exemples d’activités du Programme des matériels médicaux.
La première section du modèle de cycle de vie est intitulée : Recherche, développement de produits, examen des demandes de licence. Cette section présente les activités du PMM avant la mise en marché, soit :
- l’homologation des matériels médicaux;
- la certification des systèmes de gestion de la qualité pour fabricants de matériels médicaux de classe II et de classe IV (c.-à-d. le Programme unique d’audit des matériels médicaux [PUAMM]);
- le Programme d’accès spécial.
Les deuxième et troisième sections du modèle se nomment : Surveillance, prévention et intervention. La deuxième section présente la surveillance après la mise en marché du PMM, soit :
- le traitement des rapports d’incidents relatifs aux matériels médicaux;
- l’évaluation après la mise en marché;
- la communication des risques.
La troisième section présente les activités en lien avec la surveillance de la conformité et l’application de la loi après la mise en marché du PMM, dont :
- les inspections;
- les licences d’établissement pour les instruments médicaux (LEIM);
- les rappels.
La troisième section du modèle comporte également une flèche plus petite intitulée : Retrait du produit du marché. Cette flèche indique que lorsqu’un dispositif atteint ce point, il ne fait plus partie du modèle ou du travail de Santé Canada.
La dernière section du modèle est intitulée : Recherche et développement en cours. Une note indique que la recherche et le développement en cours relèvent de la responsabilité du fabricant. Cette section est comprise dans la figure pour illustrer l’ensemble du cycle de vie complet d’un dispositif médical. Elle renvoie également à la première section du modèle, à savoir les activités avant la mise en marché du PMM.
1.2 Programme des instruments médicaux de Santé Canada
Au moment du projet, le PIM était organisé selon des fonctions distinctes avant la mise en marché (c.-à-d., l’approbation de la licence) et après la mise en marché (c.-à-d., la surveillance, ainsi que la conformité et l’application de la loi), chacune étant responsable de différentes activités du Programme (voir figure 2).
Depuis novembre 2019, une partie du Programme a été restructurée au sein d’une nouvelle Direction des instruments médicaux. Cette nouvelle direction assure la fonction de précommercialisation comprend le Bureau des produits pharmaceutiques et des instruments médicaux commercialisés, provenant de la fonction de surveillance après la mise en marché. L’autre moitié de la fonction de surveillance (c.-à-d., le Bureau de la surveillance des produits de santé et de l’épidémiologie) et la fonction de conformité et d’application de la loi restent sous la responsabilité de leurs directions d’origineNote de bas de page 12.
En 2018-2019, les dépenses prévues du PIM étaient d’environ 33 millions de dollars, dont 15 millions de dollars ont été financés par Santé Canada. Le reste des dépenses a été financé par les droits de licence du Programme.
Dans l’ensemble, au moment du projet, les activités du PIM avaient une portée semblable à celle d’autres programmes de réglementation de Santé Canada (p. ex., les produits pharmaceutiques)Note de bas de page 13. Elles étaient également conformes au mandat et au rôle de gérance du Ministère, qui consiste à protéger les Canadiens et à faciliter l’accès aux produits qui sont essentiels à leur santé et à leur bien-êtreNote de bas de page 14.
Le projet était concentré sur les trois principales fonctions du PIM; toutefois, il convient de noter que les activités de programme sont soutenues par la Direction de la gestion des ressources et des opérations et la Direction des politiques, de la planification et des affaires internationales de la Direction générale des produits de santé et des aliments, ainsi que par la Direction de la planification et des opérations et la Direction des politiques et des stratégies réglementaires de la Direction générale des opérations réglementaires et de l’application de la loi. De plus, les communications sur les risques du Programme sont élaborées en collaboration avec le Bureau des politiques, des conseils sur les risques et de la publicité de la Direction des produits de santé commercialisés.
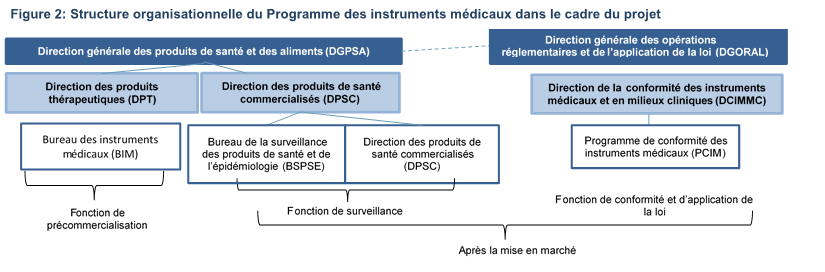
Figure 2 - Équivalent textuel
Cette figure illustre la structure organisationnelle de Santé Canada et les principaux rôles du Programme des matériels médicaux.
Deux directions générales mènent les activités du programme, soit la Direction générale des produits de santé et des aliments (DGPSA) et la Direction générale des opérations réglementaires et de l’application de la loi (DGORAL) de Santé Canada.
La Direction des produits thérapeutiques (DPT) et la Direction des produits de santé commercialisés (DPSC), qui relèvent toutes deux de la DGPSA, ont différentes divisions impliquées dans le programme :
- Pour la Direction des produits thérapeutiques (DPT), c’est le Bureau des matériels médicaux (BMM) qui assure la fonction de précommercialisation.
- Pour la Direction des produits de santé commercialisés (DPSC), c’est le Bureau de la surveillance des produits de santé et de l’épidémiologie (BSPSE) et le Bureau des produits pharmaceutiques et des instruments médicaux commercialisés (BPPIMC) qui assurent la fonction de la surveillance après la mise en marché.
La Direction de la conformité des matériels médicaux et en milieux cliniques (DCMMMC), qui relève de la Direction générale des opérations réglementaires et de l’application de la loi (DGORAL), a une division appelée le Programme de conformité des matériels médicaux (PCIM). Cette division assure la fonction de conformité et de l’application de la loi, fonction qui fait aussi partie de la surveillance après la mise en marché.
2. Sécurité, efficacité et qualité des instruments médicaux au
2.1 Contrôles des processus de précommercialisation
Le PIM a mis en place des contrôles adéquats dans le cadre de sa fonction de précommercialisation, et la majorité des demandes d’homologation pour des instruments médicaux ont été traitées de manière cohérente. Toutefois, les documents d’orientation mis à la disposition du personnel étaient obsolètes, ce qui pouvait représenter un risque, notamment pour les nouveaux employés qui ne sont pas familiarisés avec le processus de demande.
La fonction de précommercialisation a permis d’établir des contrôles et de fournir au personnel du PIM des documents d’orientation décrivant la réception, l’examen, l’évaluation et l’approbation des demandes d’homologation d’instruments médicaux. La vérification des transactions montre qu’environ 94 % des demandes d’homologation d’instruments médicaux ont été traitées par la fonction de précommercialisation, conformément à tous les contrôles requis. Les erreurs de traitement observées dans les 6 % des demandes restantes correspondaient à des déficiences mineures, notamment des erreurs administratives, des incohérences de formatage et des documents internes partiellement complétés.
Les contrôles mis en œuvre par la fonction de précommercialisation comprennent les suivantes, parmi d’autres :
- Pour les produits mixtes (voir définition ci-dessous), le PIM a élaboré une Politique sur les produits mixtes, ainsi que plusieurs procédures opérationnelles normalisées pour l’examen des demandes d’homologation de ces produits. La majorité des demandes d’homologation de produits mixtes reçus par Santé Canada en 2017-2018 et 2018-2019 ont été traitées de façon uniforme, conformément aux procédures opérationnelles normalisées du PIM. Comme les produits mixtes impliquent plus d’une direction de Santé Canada, les consultations tenues entre les directions lors de l’examen des demandes d’homologation pour ces produits ont été enregistrées et conservées dans un dépôt de documents appelé docuBridge.
- Le PIM a exigé que tous les membres du personnel de la fonction de précommercialisation signent une déclaration de conflit d’intérêts lors de leur embauche et à chaque année. Ces déclarations ont été enregistrées dans une base de données électronique. En outre, tout entrepreneur engagé par la fonction de précommercialisation pour examiner les demandes devait répondre à une longue liste de questions sur les conflits d’intérêts liés à ses contrats. Les entrepreneurs n’ont pas été affectés à des demandes dans lesquelles ils auraient pu avoir un intérêt direct potentiel.
- La fonction de précommercialisation a également établi un mécanisme de contrôle consistant à délivrer des licences assorties de conditions pour certains instruments de classe III et IV. Ces conditions visent à répondre à toute préoccupation en matière de sécurité ou d’efficacité lorsque des renseignements supplémentaires sont demandés aux fabricants au cours de la procédure de demande. Ces licences assorties de conditions ont permis de garantir que les instruments médicaux présentant des risques possibles pour la sécurité et l’efficacité sont contrôlés après l’approbation de la demande d’homologation.
Définition clé : Les produits mixtes contiennent plusieurs composantes différentes, tels qu’un médicament et un instrument médical, ou une composante biologique et un instrument médical. Ces types de produits nécessitent généralement des examens conjoints entre le PIM et d’autres programmes ou directions de Santé Canada (p. ex., la Direction des produits biologiques et des thérapies génétiques). La composante principale du produit détermine quel programme ou quelle direction prend la tête des examens.
Bien que la fonction de précommercialisation ait mis en place des processus et des contrôles qui ont été suivis presque tout le temps, de nombreux documents d’orientation internes de la fonction qui soutiennent le processus d’examen des demandes d’homologation des instruments médicaux étaient largement obsolètes. Par exemple, certains documents d’orientation pour la réception et l’administration des demandes n’ont pas tenu compte de la transition du PIM vers docuBridge en 2017. Cela représente un risque, car des renseignements obsolètes peuvent induire en erreur le personnel du Programme lors de l’examen des demandes d’homologation des instruments médicaux. Ce risque est probablement plus grand pour le personnel nouvellement embauché qui peut ne pas être familier avec les processus non documentés.
2.2 Contrôles des processus après la mise en marché
Le PIM a connu des retards dans le traitement des rapports d’incidents obligatoires. Cela représente un risque pour la capacité du PIM à assurer la sécurité, l’efficacité et le profil avantages-risques des instruments médicaux.
Le PIM a détecté, surveillé et suivi les incidents liés aux instruments médicaux grâce à la collecte des rapports suivants :
- rapports d’incident obligatoires des fabricants ou des importateurs;
- rapports d’incidents à l’étranger des organismes de réglementation internationauxNote de bas de page 15;
- rapports d’incident volontaires, par l’intermédiaire du Réseau sentinelle canadien pour les instruments médicaux (ResSCIM)Note de bas de page iiNote de bas de page 16;
- rapports d’incidents volontaires provenant d’autres sources (p. ex., les cliniques privées, l’industrie, les personnes utilisant un instrument médical)Note de bas de page 17.
Une fois reçus, les rapports d’incidents étaient traités manuellement par le personnel du PIM.
En plus de recevoir des rapports d’incidents, le PIM a également effectué une surveillance active des incidents par l’entremise d’analyses de la littérature et des médias, a suivi les mesures prises par les organismes de réglementation internationaux, a assuré un suivi auprès des fabricants pour garantir la déclaration et a mis en œuvre des mesures de conformité en cas de sous-déclaration. Il a également mis en place plusieurs groupes de travail et comités, avec des représentants de chaque fonction du Programme
qui étaient chargés d’identifier et de faire un examen préalable des incidents.
Entre 2013 et 2017, le PIM a reçu entre 11 070 et 14 931 rapports d’incidents par anNote de bas de page 18. La majorité d’entre eux étaient des rapports obligatoires, pour lesquels il existe deux catégories :
- Les « déclarations dans les 10 jours », ce qui signifie que le fabricant dispose de 10 jours pour signaler les incidents ayant entraîné le décès ou la détérioration grave de l’état de santé d’une personne. Le PIM a reçu 1 814 de ces rapports en moyenne par an entre 2013 et 2017. Le traitement des déclarations dans les 10 jours a été privilégié par rapport aux autres rapports. Le personnel du PIM a dû terminer la saisie des données pour ces rapports dans un délai respectant une norme de service de 15 jours.
- Les « déclarations dans les 30 jours » signifient que le fabricant dispose de 30 jours pour signaler tout autre type d’incident. Le PIM a reçu 10 317 de ces rapports en moyenne par an entre 2013 et 2017. La norme de service pour la saisie des données de ces rapports était de 84 joursNote de bas de page 19.
Un examen des rapports par exercice financier a montré que la prise en compte des déclarations dans les 10 jours en respectant les normes de service s’est améliorée au cours des dernières années, comme le montre la figure 3Note de bas de page iii. Au cours de la même période, le nombre de déclarations dans les 30 jours saisis en respectant les normes de service a considérablement diminué. Des examens supplémentaires ont montré que les déclarations dans les 10 jours dépassaient les normes de service jusqu’à 319 jours, tandis que les déclarations dans les 30 jours les dépassaient jusqu’à 370 jours. Cela comprenait des délais allant jusqu’à 153 jours pour les instruments médicaux à haut risque de classe IV.
Des documents internes suggèrent que les retards subis au début de l’exercice financier 2019-2020 ont été attribués à la réception d’environ 1 600 rapports d’incidents d’un fabricant au début de 2019. Une telle situation peut se produire lorsque la fonction de conformité et d’application de la loi permet de découvrir qu’un fabricant a sous-déclaré des incidents, et qu’il est alors obligé d’envoyer tous ces rapports à Santé Canada. Selon quelques informateurs internes clés, au moment du projet, la fonction de surveillance n’avait pas la capacité de traiter une aussi grande quantité de rapports en respectant les normes de service du Programme.
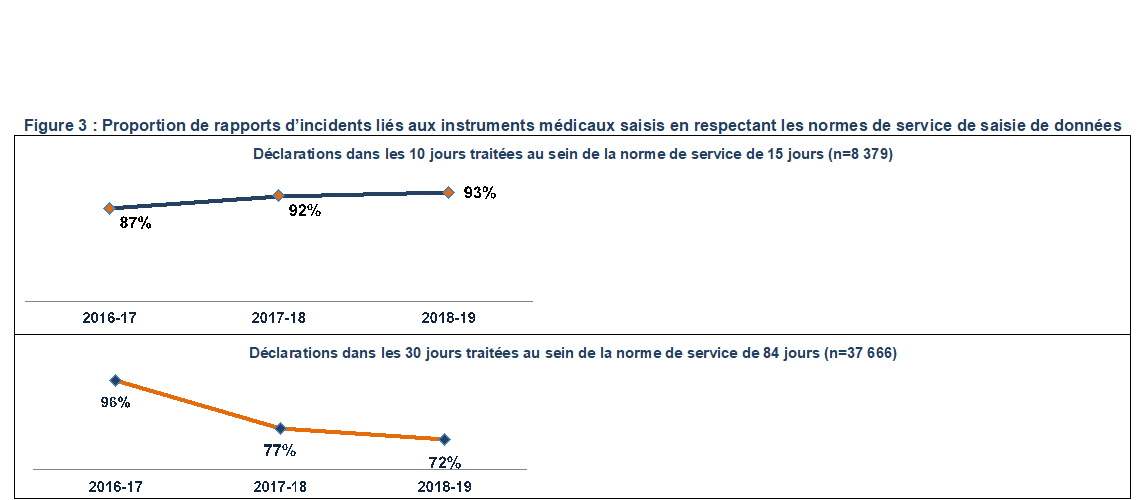
Figure 3 - Équivalent textuel
Cette figure contient deux graphiques linéaires distincts. Le premier graphique montre la proportion de déclarations dans les 10 jours traitées au sein de la norme de service de 15 jours (n = 8 379) au fils du temps :
- 87 % en 2016-2017
- 92 % en 2017-2018
- 93 % en 2018-2019
Le second graphique montre la proportion de déclarations dans les 30 jours traitées au sein de la norme de service de 84 jours (n = 37 666). Le graphique linéaire montre les résultats par exercice financier à partir de 2016-2017 jusqu’à 2018-2019, qui vont comme suit :
- 96 % en 2016-2017 :
- 77 % en 2017-2018 :
- 72 % en 2018-2019 :
Les données de ce graphique proviennent du Contrôle des transactions de l’audit.
L’un des contrôles permettant de s’assurer que les déclarations dans les 10 jours sont traitées rapidement consiste à vérifier que les incidents sont correctement étiquetés en tant que déclarations dans les 10 ou 30 jours lorsqu’ils sont présentés à Santé Canada. Cependant, au moment du projet, le triage des déclarations a été omis du processus de réception des incidents liés aux instruments médicaux. Par conséquent, les déclarations dans les 10 jours ont été classées par erreur comme des déclarations dans les 30 jours lorsqu’elles ont été reçues par Santé Canada, et sont restées dans la file d’attente de traitement pendant une période plus longue que celle à laquelle on s’attendrait normalement pour une déclaration dans les 10 jours. Il s’agit d’un risque potentiel pour le public canadien, car les nouveaux incidents graves peuvent ne pas être détectés et traités en temps opportun.
Certains informateurs internes clés ont expliqué que les retards dans la saisie des données des rapports d’incident, le manque de triage des rapports entrants et les incohérences dans l’étiquetage des rapports ont nui à la capacité du Programme à effectuer une analyse des tendances, à traiter de manière proactive les problèmes de sécurité émergents et présumés et à mener des activités de conformité et d’application de la loi.
Cela dit, il y a eu des retards dans la saisie des rapports obligatoires, car il est prouvé que les rapports volontaires reçus par le ResSCIM, qui étaient considérés comme étant au même niveau de priorité que les déclarations dans les 10 jours, ont tous été saisis dans le respect de la norme de service requise (c.-à-d., 15 jours).
Le PIM a connu des retards dans l’achèvement des évaluations des signaux, mais la plupart des communications sur les risques ont continué à être diffusées dans le respect des normes de service.
Lorsqu’un incident est signalé par l’entremise d’un rapport ou d’une surveillance, le personnel de la fonction de surveillance après la mise en marché évalue et examine ces signaux afin de déterminer la bonne marche à suivre pour répondre à l’incident. Les données sur le rendement du PIM montrent que la majorité des évaluations et des examens des signaux ont été réalisés en respectant la norme de service de 130 jours pour les évaluations des signaux et de 60 jours pour les examens.
Définitions clés
Les évaluations des signaux visent à recueillir des renseignements de sécurité sur un problème prioritaire afin de le caractériser, d’élaborer des stratégies pour y faire face et de recommander des solutions pour prévenir ou atténuer les risques déterminés.
Les résumés des examens de l’innocuité fournissent des renseignements recueillis lors de la surveillance des instruments homologués, en se fondant sur plusieurs sources d’information possibles (p. ex., les rapports d’incidents nationaux et étrangers, la littérature médicale et scientifique) pour déterminer les problèmes de sécurité éventuels. Chaque résumé présente ce qui a été évalué, ce qui a été trouvé et les mesures qui ont été prises par Santé Canada, le cas échéant.
La proportion de ces activités répondant aux normes de service a régulièrement diminué, passant de 100 % en 2014-2015 et 2015-2016, à 89 % en 2018-2019. Cette tendance à la baisse s’est poursuivie au cours de la première partie de l’année fiscale 2019-2020, puisque seulement 50 % des évaluations de signaux ont été réalisées dans le respect des normes de service entre avril et juin 2019. Toutefois, si les données opérationnelles montrent que les normes de service ont été respectées dans la majorité des cas avant avril 2019, elles ne tiennent pas compte du temps nécessaire pour recueillir des renseignements supplémentaires sur l’incident auprès du fabricant ou d’autres sources. Si l’on prend en considération ce temps, en excluant les retards de l’industrie, environ 30 % des signaux de l’année fiscale 2018-2019 ont respecté les normes de service requises.
Selon certains informateurs internes clés et documents sur le Programme, les activités relatives aux évaluations des signaux ont subi des difficultés en raison d’un manque de capacité, notamment la réorientation du personnel vers d’autres activités, comme la prise en compte de l’attention accrue des médias concernant les instruments implantés et la mise en œuvre du Plan d’action sur les instruments médicaux.
Les retards dans l’évaluation des signaux n’ont pas influé sur la capacité du PIM à émettre des communications sur les risques (p. ex., rappels, avertissements et examens de l’innocuité) pour les instruments ayant subi un incidentNote de bas de page 20. Comme le montre la figure 4, depuis 2014-2015, la plupart des communications liées au risque concernant les instruments médicaux ont été diffusées dans le respect des normes de service (c.-à-d., 25 jours pour les communications normalisées, 10 jours pour les communications non normalisées, cinq jours pour les communications accélérées et deux jours pour les communications urgentes).
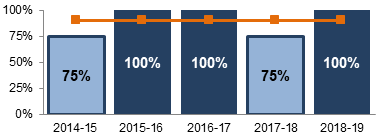
Figure 4 - Équivalent textuel
Ce graphique à colonnes montre les résultats des exercices financiers à partir de 2014-2015 jusqu’à 2018-2019. Une ligne cible, dans le haut du graphique, donne un aperçu de la proximité des résultats par rapport à l’objectif de 90 % des communications sur les risques diffusées dans le cadre des normes de service. Les résultats vont comme suit :
- 75 % en 2014-2015
- 100 % en 2015-2016
- 100 % en 2016-2017
- 75 % en 2017-2018
- 100 % en 2018-2019
Les données de ce graphique proviennent des Données sur le rendement du PMM.
Les activités de conformité et d’application de la loi ont été menées dans le respect des normes de service.
Les données sur le rendement du Programme montrent que les activités de conformité et d’application de la loi pour les instruments médicaux ont généralement été menées dans le respect des objectifs de rendement :
- L’objectif de 95 % des fabricants inspectés qui étaient en conformité avec la Loi sur les aliments et drogues et les règlements connexes a été dépassé chaque année pendant la période de 2013-2014 à 2018-2019.
- En 2018-2019, la proportion de cas à haut risque initialement déterminé comme non conforme, mais qui ont finalement été mis en conformité, a atteint l’objectif de 95 %.
- L’objectif de 100 % des licences d’établissement pour les instruments médicaux examinés dans le cadre des normes de service de 120 jours a été atteint à deux reprises entre 2014-2015 et 2018-2019. Les autres années, la proportion de licences examinées dans le cadre de ces normes de service était supérieure à 99 %.
Définition clé : Les licences d’établissement pour les instruments médicaux sont délivrées aux importateurs et aux distributeurs de instruments médicaux au Canada, aux fabricants qui vendent des instruments médicaux pour lesquels ils ne sont pas titulaires d’une licence, ainsi qu’aux fabricants d’instruments médicaux de classe I qui distribuent leurs propres instruments.
La plupart des processus après la mise en marché étaient documentés, mais il y avait des lacunes dans la documentation des normes de service. De plus, les incohérences dans la saisie des données ont limité la capacité du Programme à analyser les tendances à partir des données des rapports d’incidents et à détecter de manière proactive les incidents.
La surveillance après la mise en marché et les fonctions de conformité et d’application de la loi ont permis de documenter les processus opérationnels et les critères d’évaluation pour la priorisation des incidents liés aux instrumentsmédicaux, afin de garantir que les incidents hautement prioritaires (c.-à-d., les déclarations dans les 10 jours et les rapports du ResSCIM) soient examinés rapidement. Toutefois, la fonction de surveillance après la mise en marché n’a pas consigné ses normes de service dans ses documents d’orientation internes, et la fonction de conformité et d’application de la loi n’a pas établi de normes de service pour les déclarations volontaires qu’elle a reçues. Le Programme n’a pas procédé à un examen complet pour déterminer si les délais de saisie des données, d’évaluation et de codification des rapports d’incident avaient pris en compte tous les processus actuels. De plus, les normes de service pour la saisie des données n’ont pas été revues depuis leur adoption en 2010.
L’équipe de projet s’attendait à ce que les processus suivis par les fonctions après la mise en marché soient reflétés dans les documents d’orientation du Programme. Cependant, tous les rapports d’incidents relatifs aux instruments médicaux examinés dans la base de données du Système des instruments médicaux manquaient un ou plusieurs éléments d’information sur l’évaluation scientifique qui étaient décrits dans les procédures opérationnelles normalisées et les instructions de travail. Ces renseignements ont été documentés dans des fiches de suivi externes, plutôt que dans la base de données. Il y avait également des incohérences dans la saisie des données de la base de données du Système des instruments médicaux du programme qui ont affecté l’intégrité des données. Par exemple, les rapports du Programme d’accès spécial et les rapports d’essais expérimentaux (voir la section 3 pour obtenir une définition), qui n’avaient pas d’étiquette spécifique dans la base de données, étaient étiquetés comme des déclarations dans les 10 jours. Cela a limité la capacité à interpréter les analyses statistiques des données extraites du Système des instruments médicaux.
2.3. Sécurité des instruments médicaux disponibles au Canada
Malgré les difficultés rencontrées en effectuant certaines activités de surveillance après la mise en marché et de conformité et d’application de la loi dans les délais impartis, Santé Canada a généralement réussi à assurer la sécurité des instruments médicaux au Canada.
Le processus réglementaire a été considéré par plusieurs informateurs clés externes comme rigoureux en raison des exigences à respecter. Les perceptions de ces informateurs font écho aux résultats de l’enquête sur le Cadre réglementaire de transparence et d’ouverture (CRTO) de 2016, qui a été menée auprès des intervenants et du public. Parmi lesNote de bas de page iv répondants des parties prenantes concernées par les instruments médicaux (n=20), la majorité (95 %) ont convenu que les produits réglementés par Santé Canada étaient sûrs. De plus, la majorité (75 %) des membres du public canadien ayant un intérêt pour les instruments médicaux (n=40) avaient confiance dans les produits réglementés par Santé CanadaNote de bas de page v.
Plusieurs informateurs clés externes sont de l’avis que Santé Canada est efficace en assurant la sécurité des instruments médicaux. En outre, certains experts interrogés ont estimé que la sécurité des instruments médicaux vendus au Canada était comparable à celle d’autres pays. Entre 2013-2014 et 2018-2019, Santé Canada a procédé à environ 900 rappels, en moyenne, par année. En comparaison, l’Australie, qui reçoit un nombre similaire de demandes d’homologation des instruments médicaux de classe II à IV par année (soit environ 2 500 demandes en 2017-2018, contre 2 100 au Canada), a procédé à environ 600 rappels en moyenne par année.
Une comparaison des rappels au Canada et aux États-Unis suggère que la sécurité des instruments médicaux au Canada est similaire à celle des États-Unis. Un échantillon de 60 rappelsNote de bas de page vi émis par Santé Canada en 2018 a été évalué afin de déterminer si ces rappels ont également été émis aux États-Unis. Si le rappel n’a pas été émis aux États-Unis, l’équipe a ensuite examiné si l’instrument faisant l’objet du rappel au Canada était approuvé pour la vente par la Food and Drug Administration des États-Unis. L’analyse montre que 70 % (42 sur 60) des rappels analysés ont eu lieu dans les deux pays. De ce nombre :
- Le moment où le rappel a été lancé était comparable dans les deux pays. Sur les 42 rappels, 16 ont été lancés aux États-Unis, 14 au Canada et 12 dans les deux pays en même tempsNote de bas de page vii.
- Dans la plupart des cas (30 sur 42 rappels), le rappel a été publié sur le site Web de Santé Canada avant d’être publié sur le site Web de la Food and Drug Administration.
- Les instruments les plus fréquemment visés par des rappels dans les deux pays se trouvaient dans la catégorie des instruments de diagnostic (p. ex., les instruments d’immunodiagnostic) avec dix rappels, suivis des instruments chirurgicaux avec huit rappels et des systèmes d’imagerie (p. ex., équipement d’échographie) avec sept rappels. Deux des rappels échantillonnés et trouvés dans les deux pays concernaient des instruments implantables.
Sur les 60 rappels analysés, 18 n’ont été émis qu’au Canada. De ce nombre :
- La plupart des instruments ont été approuvés pour vente aux États-Unis (15 sur 18).
- Trois instruments n’étaient pas homologués aux États-Unis. Ils ont fait l’objet d’un rappel au Canada en raison : 1) d’un problème de licence, 2) d’un problème d’étiquetage et 3) d’un rappel du fabricant fournissant certains renseignements techniques sur l’instrument.
- Certains rappels émis au Canada n’étaient pas pertinents pour les États-Unis. Par exemple, un rappel concernait un problème d’emballage pour un certain lot d’instruments, ou la traduction française était manquante dans les renseignements sur le produit.
- Aucun des 18 rappels émis uniquement au Canada n’était attribuable à une situation où il y avait une probabilité raisonnable que l’utilisation de l’instrument faisant l’objet du rappel ou que l’exposition à celui-ci entraîne des conséquences néfastes pour la santé ou la mort. À cet égard, six des 18 rappels faisaient partie de la classification de danger II de Santé Canada, qui est liée à une situation où l’utilisation d’un instrument faisant l’objet d’un rappel ou l’exposition à celui-ci pourrait entraîner des conséquences néfastes temporaires pour la santé, ou lorsqu’il n’y a pas de probabilité importante de conséquences néfastes graves pour la santé. Les 12 autres rappels faisaient partie de la classification de danger III, qui est liée à une situation où l’utilisation d’un instrument faisant l’objet d’un rappel ou l’exposition à celui-ci n’est pas susceptible d’entraîner des conséquences néfastes pour la santé.
- Les rappels concernaient principalement des instruments de diagnostic (huit sur 18), et deux concernaient des instruments implantables.
En plus de la comparabilité du Canada avec les États-Unis en termes de rappels, Santé Canada a parfois été à l’avant-garde sur le plan international lors de l’émission d’avertissements et de rappels. Ce fut le cas avec le système de contraception permanent Essure. Le Canada a été l’un des premiers pays à publier un résumé d’examen de l’innocuité et un avertissement aux professionnels de la santé, les alertant de complications possiblement graves avec cet instrument. Le Canada a également été l’un des premiers pays à suspendre la licence pour les implants mammaires Biocell.
Définition clé : Les rappels comprennent toute mesure prise par un fabricant, un importateur ou un distributeur d’un instrument pour rappeler ou corriger l’instrument ou pour informer les utilisateurs de son éventuelle défectuosité.
2.4 S’adapter à l’évolution des instruments médicaux
L’environnement des instruments médicaux est de plus en plus complexe et déterminé par l’utilisation de nouvelles technologies.
Garantir la sécurité des instruments médicaux est compliqué par le fait que l’environnement de ces derniers est de plus en plus complexe, car de nouveaux instruments sont produits en utilisant des technologies nouvelles et innovantes, telles que la technologie de santé numérique, l’intelligence artificielle et l’impression en 3D. Le cycle de vie des instruments est de plus en plus court, car ils sont développés et mis à jour plus rapidement.
Selon quelques informateurs clés et des documents de Santé Canada, le rythme de l’innovation a entraîné la remise en question du cadre réglementaire du Ministère, dont les exigences et les échéanciers sont basés sur des modèles traditionnels de fabrication, d’approbation avant la mise en marché et de vente, entre autresNote de bas de page 21. Par exemple, au cours de la période examinée, on a constaté une tendance croissante à l’homologation des logiciels en tant qu’instruments médicaux (p. ex., les outils de diagnostic en ligne) et il n’existait aucun mécanisme dans la réglementation actuelle pour faire face à ce changement.
Avec le Plan d’action sur les instruments médicaux et l’Examen réglementaire des médicaments et des instruments, Santé Canada a fait des progrès pour s’adapter à l’environnement changeant.
Au cours de la période du projet, Santé Canada a mis en œuvre plusieurs initiatives et outils pour adapter son approche en matière de réglementation et de surveillance des instruments médicaux. Bon nombre de ces initiatives proviennent du Plan d’action sur les instruments médicaux de 2018 et de l’Examen réglementaire des médicaments et des instruments de 2017Note de bas de page 22. Ces initiatives comportaient des jalons concrets et leur progression a fait l’objet d’un suivi par le personnel de programme. Un examen détaillé de l’outil de suivi du Plan d’action sur les instruments médicaux montre que ses initiatives étaient généralement sur la bonne voie ou achevées, et que les mises à jour sur les progrès étaient précises et soutenues par des preuves (p. ex., des publications).
Définitions clés
Le Plan d’action sur les instruments médicaux de 2018 se concentre sur 1) l’amélioration de la mise en marché des instruments, 2) le renforcement de la surveillance et du suivi des instruments utilisés par les Canadiens et 3) l’offre aux Canadiens de plus amples renseignements sur les instruments médicaux.
L’Examen réglementaire des médicaments et des instruments de 2017 vise à fournir aux Canadiens un accès plus rapide aux médicaments et aux instruments médicaux grâce à une collaboration avec d’autres organismes de soins de santé (p. ex., l’Agence canadienne des médicaments et des technologies de la santé).
Dans le cadre de ces initiatives, le PIM a développé une expertise interne et a élargi le recours à des experts et à des intervenants externes pour renforcer ses compétences dans des domaines clés de croissance pour les instruments médicaux. En voici les résultats :
- L’élaboration et la mise en œuvre d’une Division de la santé numérique en 2018 pour assurer un examen plus ciblé et plus exhaustif des demandes d’homologation des instruments médicaux liés aux technologies de santé numérique avant leur mise en marché.
- La création de deux nouveaux comités consultatifs scientifiques permanents pour obtenir des avis scientifiques, techniques et cliniques sur les technologies numériques de la santé (2018) et les produits de santé destinés aux femmes (2019);
- L’élaboration de documents d’orientation sur les nouveaux instruments médicaux qui utilisent des technologies nouvelles et innovantes (p. ex., le document d’orientation 2019 intitulé Lignes directrices - Données sur les instruments médicaux implantables fabriqués par impression 3D ).
Santé Canada a également modifié l’article 23 de la Loi sur les aliments et drogues en 2019 afin d’accroître le pouvoir des inspecteurs de la conformité. La modification leur a donné la possibilité de réaliser davantage d’activités sur le terrain (c.-à-d., des essais, des prises de photos, des enregistrements) et d’examiner les données électroniques. Le Ministère a également mis en œuvre des changements réglementaires, notamment la déclaration obligatoire des incidents liés aux instruments médicaux par les hôpitaux (voir les détails dans la section 6).
De plus, la Politique de conformité et d’application de la loi pour les produits de santé (Politique 0001) a été révisée en décembre 2018. Cette version révisée a été perçue par quelques informateurs internes clés comme une approche plus appropriée d’application de la loi, fondée sur le risque, car elle permet à Santé Canada d’être plus ferme en cas de non-conformité. Au moment du projet, le Ministère examinait également la portée et les responsabilités de certaines de ses activités de conformité et d’application de la loi pour s’assurer que ces activités se concentraient sur le bon niveau de risque et utilisaient les ressources efficacement.
De plus, Santé Canada a augmenté le nombre d’inspections annuelles à l’étranger à 90 (y compris 15 inspections sur place à l’étranger) par année pour aider à relever les défis liés à la mondialisation de l’environnement des instruments médicaux. Cette augmentation devait permettre d’améliorer la conformité et l’application de la loi par les fabricants d’autres pays, ainsi que de promouvoir l’harmonisation et la coopération au niveau international.
La mobilisation des partenaires internationaux permet de partager l’expertise et d’harmoniser les pratiques.
Santé Canada a participé à plusieurs initiatives et partenariats internationaux afin de partager son expertise et d’harmoniser les pratiques, les politiques et les règlements avec d’autres administrations. En particulier, Santé Canada a participé activement au Forum international des organismes de réglementation des instruments médicaux (IMDRF). Le personnel du PIM a participé à plusieurs groupes de travail, dont un consacré à l’élaboration d’une terminologie harmonisée pour le signalement des incidents liés aux instruments médicaux.
Le PIM a pris en compte les différences de sexe et de genre en ce qui concerne les instruments médicaux.
Grâce à la Politique en matière d’analyse comparative fondée sur le sexe et le genre du portefeuille de la SantéNote de bas de page 23, Santé Canada peut intégrer les considérations de sexe, de genre et de diversité dans ses programmes, le cas échéant, afin de mieux répondre aux besoins des différents groupes de patients. Cela est pertinent pour le PIM, car il est prouvé que les hommes et les femmes sont touchés différemment par les instruments médicaux en raison des disparités entre les sexes et les genresNote de bas de page 24.
Dans ce contexte, le PIM a mis en place des mesures pour tenir compte de ces disparités dans plusieurs de ses activités. En plus de créer le Comité consultatif scientifique sur les produits de santé destinés aux femmes en 2019, le PIM a proposé des modifications au Règlement sur les instruments médicaux, afin de donner à Santé Canada un plus grand pouvoir pour exiger des renseignements de la part des fabricants lorsque les données probantes indiquent un problème, y compris des risques déterminés ou des incertitudes pour des groupes spécifiques (p. ex., les femmes, les personnes handicapées, les enfants)Note de bas de page 25. En 2013, le PIM a également publié un document d’orientation intitulé « Considérations relatives à l’inclusion des femmes dans les essais cliniques et à l’analyse des données selon le sexe », qui exposait les principales considérations relatives à l’inclusion des femmes dans les études de précommercialisation des produits thérapeutiquesNote de bas de page 26. Santé Canada a également parrainé un projet de recherche qui visait à appliquer une lentille d’analyse fondée sur le sexe et le genre au cycle de vie des instruments médicaux.
La fonction de précommercialisation a permis d’examiner si les données des études cliniques fournies avec les demandes d’homologation d’instruments médicaux avaient pris en compte des groupes de population spécifiques susceptibles d’utiliser l’instrument. Toutefois, le PIM ne comportait pas de directives structurées pour l’industrie sur le moment et la manière d’inclure ces renseignements. De plus, les fabricants n’étaient pas tenus d’inclure ces données dans leurs demandes d’homologation d’instruments médicaux. En tant que tel, il n’y avait pas de preuve que le PIM a effectué une analyse systématique des données ou des tendances pour fournir des renseignements concernant les considérations liées au sexe et au genre en ce qui concerne les instruments médicaux.
Bien que Santé Canada ait fait des progrès pour s’adapter à un environnement changeant, certains défis restent à relever.
Plusieurs informateurs internes et externes clés ont noté que des progrès supplémentaires étaient nécessaires pour adapter le cadre réglementaire. Par exemple, il n’existait pas de mécanisme permettant de répondre à la question croissante des instruments médicaux proposés par abonnement ou sous forme de service (p. ex., les outils de diagnostic en ligne).
Bien que des contrôles aient été mis en place au sein de la fonction de conformité et d’application de la loi pour inspecter et atténuer les risques, si un rapport d’incident devait être soumis pour un instrument médical non homologué, Santé Canada n’avait pas les moyens de communiquer directement avec les personnes vivant avec ou utilisant un instrument médical lorsqu’il y avait un problème de sécurité. La réglementation exige que les fabricants surveillent l’utilisation de leurs instruments, soumettent des rapports d’incidents obligatoires et contactent des personnes lorsqu’il y a de nouveaux renseignements concernant la sécurité, l’efficacité ou le rendement des instruments. Cependant, il y avait un manque de clarté en ce qui concerne les responsabilités pour contacter les personnes utilisant des instruments médicaux qui n’étaient plus homologués au Canada, ou qui étaient vendus par un fabricant qui n’avait pas d’homologation active au Canada (p. ex., leur homologation pour l’instrument médical a été suspendue ou n’a pas été renouvelée).
Certains informateurs externes clés, notamment des experts, des professionnels de la santé et des personnes utilisant un instrument médical, estiment qu’il devrait y avoir un registre central des personnes ayant un instrument implantable, afin de permettre à Santé Canada d’informer directement ces personnes en cas d’incident. Une suggestion similaire a également été faite par le Comité scientifique consultatif sur les produits de santé destinés aux femmesNote de bas de page 27.
Bien qu’il existe des mécanismes permanents de collaboration avec les organismes de réglementation internationaux, certains informateurs internes et internationaux clés ont noté que l’on pourrait faire davantage pour communiquer et collaborer avec d’autres administrations. En particulier, la fonction de conformité et d’exécution de la loi a souvent été incapable de s’engager dans des collaborations internationales en raison de pressions au travail provenant des priorités au Canada.
3.0 Donner accès aux instruments médicaux au Canada
3.1 Traitement des demandes d’homologation d’instruments médicaux
Le PIM a progressé dans la recherche d’un équilibre entre la garantie de la sécurité des instruments médicaux et un accès à ces derniers en temps opportun.
Entre 2013-2014 et 2018-2019, le PIM a reçu une moyenne annuelle de 1 499 nouvelles demandes pour des instruments médicaux de classe II, 424 pour la classe III et 78 pour la classe IV. Au cours de la même période, le programme a également reçu une moyenne annuelle de 1 382 demandes de modification d’homologation pour les instruments médicaux de classe II, 424 pour la classe III et 311 pour la classe IV.
Au cours de la période, le MDP a amélioré son rendement en rendant des décisions sur les demandes d’homologation d’instruments médicaux dans le respect des normes de service (c.-à-d., 15 jours pour les demandes d’homologation de classe II, 60 jours pour la classe III et 75 jours pour la classe IV). Les calculs effectués pour l’étude montrent qu’entre le 1er avril 2018 et le 30 juin 2019, presque toutes les demandes d’homologation d’instruments médicaux (96 %) ont été traitées dans le respect des normes de service pour tous les types de demandes.
De plus, selon les données sur le rendement du Programme, la capacité du PIM à traiter les demandes d’homologation des instruments médicaux dans le respect des normes de service s’est améliorée au cours de la période du projet. Comme l’illustre la figure 5, l’arriéré des demandes a été réduit pour les instruments des classes III et IV, passant d’un maximum d’environ 18 % en 2016-2017 à un minimum de 5 % en 2018-2019.
Au moment du projet, les normes de service du PIM pour l’examen des demandes d’homologation des instruments médicaux ne comprenaient pas le temps nécessaire au traitement administratif, ni à l’examen technique et réglementaire des demandes une fois qu’elles étaient reçues par Santé Canada. Elles n’ont pas compris non plus les 45 jours ajoutés au processus d’examen une fois que les fabricants ont répondu à toute demande de renseignements supplémentaires.
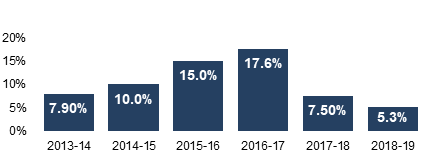
Figure 5 - Équivalent textuel
Cette figure est un graphique à colonnes qui montre la proportion de demandes d’homologation en attente par exercice financier, de 2013-2014 jusqu’à 2018-2019. Les résultats vont comme suit :
- 8 % en 2013-2014
- 10 % en 2014-2015
- 15 % en 2015-2016
- 18 % en 2016-2017
- 7,5 % en 2017-2018
- 5 % en 2018-2019
Les données de ce graphique proviennent du Rapport annuel du BMM 2018-2019 et du Rapport annuel du BMM 2013-2014
Plusieurs professionnels de la santé, ainsi que certains représentants et experts de l’industrie, perçoivent le processus réglementaire comme étant long. Toutefois, par rapport aux États-Unis, par exemple, les normes de service de Santé Canada pour l’examen des demandes d’homologation des instruments médicaux étaient plus courtes. Les États-Unis ont adopté une norme de service de 180 jours pour l’examen scientifique des demandes d’homologation des instruments médicaux, avec une possibilité de prolongation de 180 jours supplémentaires si la demande est modifiée (p. ex., des renseignements supplémentaires sont fournis)Note de bas de page 28.
Une proportion importante des demandes d’homologation des instruments médicaux ne répondaient pas aux exigences réglementaires lorsqu’elles ont été présentées pour la première fois à Santé Canada.
Selon les données sur le rendement du Programme, de 2013-2014 à 2018-2019, entre 39 et 59 % des demandes d’homologation des instruments médicaux reçues par le PIM n’étaient pas conformes aux exigences réglementaires au moment de leur première décision d’examen. Selon plusieurs informateurs internes et externes clés, les demandes d’homologation des instruments médicaux incomplètes augmentaient souvent le temps requis pour le processus d’examen, car le personnel du Programme devait consulter le demandeur pour lui demander des renseignements supplémentaires. Le contrôle des transactions montre que 66 % des demandes d’homologation des instruments médicaux ont fait l’objet d’au moins une demande de renseignements complémentaires ou d’une lettre d’avis de demande incomplète de la part du PIM adressée au demandeur.
Comme le montre la figure 6, une fois que le demandeur a fourni les renseignements supplémentaires demandés par Santé Canada, le pourcentage de demandes qui sont restées non conformes aux exigences réglementaires est tombé à 8 % ou moins entre 2013-2014 et 2018-2019.
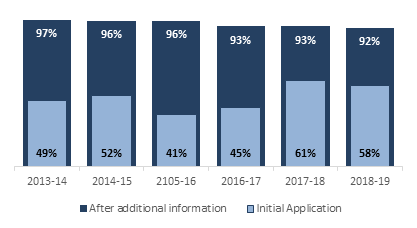
Figure 6 - Équivalent textuel
Cette figure est un graphique à colonnes qui se chevauchent et qui montre les résultats, par exercice financier, de la proportion de demandes d’homologation initiales qui sont conformes aux exigences réglementaires lors du premier examen et de la proportion de demandes devenues conformes après que le PMM a demandé et reçu des renseignements supplémentaires. Les résultats de la proportion de demandes initiales qui étaient conformes vont comme suit :
- 49 % en 2013-2014
- 52 % en 2014-2015
- 41 % en 2015-2016
- 45 % en 2016-2017
- 61 % en 2017-2018
- 58 % en 2018-2019
Les résultats de la proportion de demandes qui sont devenues conformes à la suite d’une demande de renseignements supplémentaire et de la réception de ces renseignements vont comme suit :
- 97 % en 2013-2014
- 96 % en 2014-2015
- 96 % en 2015-2016
- 93 % en 2016-2017
- 93 % en 2017-2018
- 92 % en 2018-2019
Les données de ce graphique proviennent des Données sur le rendement du PMM.
Les documents d’orientation sur la procédure de demande étaient difficiles à trouver et souvent non à jour.
Le PIM a publié sur le site Web de Santé Canada des documents d’orientation décrivant les exigences relatives à toutes les classes d’instruments médicaux, à certaines technologies nouvelles (p. ex., les instruments imprimés en 3D) et aux produits mixtes. Ces documents visent à aider les demandeurs à déterminer la classification de leur produit et à s’orienter dans le processus de demande.
La majorité des informateurs clés de l’industrie ont mentionné qu’ils avaient de la difficulté à trouver les renseignements ou les instructions appropriées pour les aider à préparer leur demande. Ils ont expliqué qu’il est difficile de naviguer sur le site Web de Santé Canada et que l’information disponible n’est pas intégrée de façon à aider l’industrie à comprendre les différentes étapes du processus de demande (p. ex., il n’y a pas de processus étape par étape à suivre). Plusieurs représentants de l’industrie et quelques informateurs internes clés ont expliqué que les petits et nouveaux fabricants étaient plus susceptibles de rencontrer des difficultés dans le cadre du processus de demande d’homologation des instruments médicaux, car ils étaient moins susceptibles de connaître le processus réglementaire ou disposaient de moins de ressources pour préparer leur trousse de demande. Ces perceptions concordent avec les résultats de l’enquête de 2016 sur le CRTO, selon laquelle la majorité des intervenants (80 %) ont indiqué que l’information de Santé Canada sur les exigences réglementaires n’était pas facile à trouver.
Un examen des documents d’orientation disponibles en ligne montre que, si la majorité des documents d’orientation ont été rédigés ou mis à jour au cours des dernières années, quelques-uns n’ont pas été mis à jour depuis au moins sept ans. Par exemple, le document Ligne directrice sur les données à fournir pour étayer les demandes d’homologation des instruments médicaux de classe III et classe IV et les demandes de modification, à l’exception des instruments de diagnostic in vitro (IDIV) a été mis à jour en 2012. Des documents obsolètes peuvent avoir contribué à des inexactitudes dans la présentation des demandes, entraînant un plus grand va-et-vient entre les demandeurs et le Programme. Cela pourrait également avoir affecté la rapidité des décisions d’homologation des instruments médicaux. Il existe également un risque que des documents d’orientation internes obsolètes aient causé des confusions et des erreurs lors du traitement des demandes, ce qui aurait pu, à son tour, avoir des répercussions sur l’efficacité des processus de précommercialisation.
De plus, bien que des exigences générales en matière d’information et de données probantes pour les demandes d’homologation des instruments médicaux aient été établies pour chaque classe d’instruments et mises à la disposition des intervenants de l’industrie sur le site Web de Santé Canada, on n’a pas trouvé de preuve d’un processus défini pour mettre à jour périodiquement ces exigences. Cela signifie que les révisions des documents d’orientation disponibles ont été effectuées selon les besoins du moment.
3.2 Mesures visant à améliorer l’accès aux instruments médicaux
Le Programme d’accès spécial du PIM fournit un mécanisme permettant aux Canadiens d’accéder aux instruments médicaux.
Selon les documents du Programme et quelques informateurs externes et internes clés, les difficultés d’accès aux instruments médicaux peuvent également être atténuées grâce au Programme d’accès spécial du PIM. Ce programme permet aux professionnels de la santé d’avoir accès à des instruments médicaux qui ne sont pas homologués pour vente au Canada, pour une utilisation en cas d’urgence ou lorsque les thérapies conventionnelles ont échoué, ne sont pas disponibles ou ne conviennent pas pour traiter un patient. Entre 2013-2014 et 2018-2019, le PIM a reçu une moyenne annuelle de 4 714 demandes par l’entremise du Programme d’accès spécial, concernant environ 1 800 instruments médicaux. En moyenne, 95 % de ces demandes ont été examinées en respectant la norme de service de 72 heuresNote de bas de page 29.
Quelques informateurs externes et internes clés ont fait part de leurs préoccupations quant au fait que l’accès au Programme d’accès spécial pourrait entraîner le contournement de l’ensemble du processus réglementaire. À cet égard, l’équipe de projet s’attendait à trouver des contrôles adéquats en place pour garantir que les demandes répondent aux critères établis par le Règlement sur les instruments médicaux, y compris la stipulation selon laquelle les fabricants ou les importateurs ne doivent pas utiliser les autorisations du Programme d’accès spécial pour contourner les exigences régulières en matière d’homologation.
Le PIM a mis en place plusieurs contrôles pour empêcher le contournement des exigences en matière d’homologation. Par exemple, en 2016, Santé Canada a publié un guide informant les professionnels de la santé que le Programme d’accès spécial n’est pas destiné à fournir un accès rapide au marché, et que les fabricants et les importateurs doivent chercher à obtenir une homologation pour les instruments médicaux qui ont été autorisés à plusieurs reprises par l’entremise du Programme d’accès spécial.
Le personnel du PIM a également vérifié les qualifications des professionnels de la santé et a limité le nombre d’unités autorisées par demande. Cependant, plusieurs contrôles mis en place par le PIM reposaient sur la dénonciation ou l’autodivulgation (p. ex., Santé Canada interdit les demandes remplies par les fabricants ou les importateurs, et demande aux fabricants ou aux importateurs s’ils ont l’intention d’homologuer les instruments médicaux).
Des vérifications des transactions montrent qu’environ 80 % des instruments médicaux demandés dans le cadre du Programme d’accès spécial de 2013 à 2018 n’ont pas donné lieu à une demande ultérieure d’homologation de l’instrument médical. Cette proportion d’instruments médicaux représente également 73 % des instruments médicaux fréquemment demandés. Une minorité de demandeurs et d’instruments médicaux représentaient la grande majorité des demandes du Programme d’accès spécial. Cela suggère qu’une intégration plus poussée de l’analyse des données renforcerait les contrôles du Programme d’accès spécial pour déterminer les éventuelles tentatives de contournement des exigences d’homologation régulières en facilitant l’identification des risques, des tendances et des demandes de grand volume. En particulier, l’analyse des données peut fournir un contrôle supplémentaire pour aider à garantir que les autorisations accordées par l’entremise du Programme d’accès spécial sont conformes aux critères établis par la réglementation, y compris en repérant le recours au Programme d’accès spécial pour contourner la réglementation sur l’homologation.
Le PIM a récemment mis en œuvre plusieurs mesures pour atténuer les difficultés liées au processus de demande d’homologation des instruments médicaux.
Ces dernières années, le PIM s’est concentré sur la mobilisation des représentants de l’industrie afin de les sensibiliser aux renseignements et aux documents d’orientation disponibles concernant le processus de demande d’homologation des instruments médicaux. Le Programme a également élaboré et mis en œuvre plusieurs initiatives et outils pour tenter de réduire le nombre de demandes ne répondant pas aux exigences réglementaires, notamment :
- Des réunions préalables à la présentation des demandes avec les parties prenantes de l’industrie pour discuter des prochaines demandes d’homologation des instruments médicaux. L’incidence de ces réunions était inconnue au moment du projet, car seul un petit nombre de représentants de l’industrie ont demandé des réunions préalables à la présentation des demandes. La majorité des informateurs clés de l’industrie interrogés qui ont participé à une réunion préalable à la présentation d’une demande ont trouvé cette réunion utile, en particulier pour les technologies et les instruments nouveaux et innovants.
- Le lancement d’un outil d’apprentissage en ligne, Comprendre comment les instruments médicaux sont réglementés au Canada avant la mise en marché, pour fournir des conseils sur les exigences réglementaires relatives aux demandes d’homologation des instruments médicaux nouveaux ou modifiés. Bien que des données n’aient pas été recueillies sur la portée de la formation ni sur les commentaires des participants, certains informateurs de l’industrie et internes clés ont noté que le cours avait été bien accueilli.
- La création de la Division de la santé numérique pour faciliter le processus d’examen des instruments médicaux impliquant de nouvelles technologies. Certains informateurs clés de l’industrie ont reconnu les efforts déployés par la Division pour faciliter et soutenir le processus de demande, notamment les tournées d’information et la participation aux conférences de l’industrie.
L’harmonisation des exigences du PIM avec celles d’autres pays peut contribuer à améliorer l’accès aux instruments médicaux .
Selon les documents du programme et les entrevues avec plusieurs informateurs internes clés, Santé Canada poursuivait diverses initiatives visant à harmoniser ses exigences en matière de demandes d’homologation d’instruments médicaux avec celles d’autres pays. Principalement organisées par le biais du Forum international des organismes de réglementation des instruments médicaux (IMDRF), ces initiatives visaient à rationaliser les processus pour les fabricants qui soumettent des demandes dans plusieurs administrations. Voici quelques exemples :
- Un format de table des matières pour harmoniser le format des demandes d’homologation d’instruments médicaux dans les pays membres de l’IMDRF. Ce format a été perçu par quelques informateurs internes clés comme un moyen potentiel de clarifier les exigences d’une demande d’homologation d’instrument médical. Depuis le 1er avril 2019, les fabricants peuvent choisir d’utiliser le format de la table des matières pour leurs demandes d’homologation d’instruments médicaux de classe III et IV.
- Depuis le1er janvier 2019, le Canada a mis en place le Programme unique d’audit pour le matériel médical (PUAIM) comme exigence obligatoire pour les fabricants qui présentent des demandes d’homologation des instruments médicaux à Santé Canada. Au moment de cette mission, le Canada était le seul membre de l’IMDRF à avoir rendu obligatoire le PUAIM. Les États-Unis et l’Australie ontNote de bas de page 30 également accepté le PUAIM, mais ces pays ne l’ont pas mis en œuvre comme une exigence obligatoire.
Définition clé : Le Programme unique d’audit pour le matériel médical permet aux fabricants de remplir et de présenter un seul certificat de leur système de gestion de la qualité pour satisfaire aux exigences réglementaires de plusieurs pays.
Plusieurs informateurs clés de l’industrie ont fait part de leurs préoccupations quant au fait que l’introduction du PUAIM pourrait avoir nui à l’accès à certains instruments médicaux. Ils ont expliqué que l’augmentation des coûts d’obtention du PUAIM, par rapport à la certification précédente exigée par Santé Canada, a pu dissuader certains fabricants de renouveler leur licence d’établissement ou de présenter des demandes d’homologation pour des instruments médicaux nouveaux ou modifiés. Au moment de la mission, il était trop tôt pour déterminer l’effet du PUAIM. Selon les données sur le rendement du Programme, entre juillet 2018 et juillet 2019, environ 8 % de l’ensemble des homologations des instruments médicaux approuvées par Santé Canada ont été annulées, non renouvelées ou abandonnées, ce qui représente une augmentation par rapport à la moyenne annuelle de 4 % entre juillet 2013 et juillet 2018.
La vérification des transactions montre que le personnel du PIM a examiné les certificats du système de gestion de la qualité des fabricants de manière cohérente lors du traitement des demandes d’homologation des instruments médicaux. Si des lacunes dans le certificat étaient cernées lors de l’examen de la demande, elles étaient communiquées aux demandeurs par courrier électronique pour les instruments médicaux de classe II, ou dans une lettre de déficiences officielle pour les instruments de classes III et IV. Toutefois, il y avait un manque d’intégration entre les renseignements des rapports d’audit du PUAIM et les activités de conformité et d’application de la loi. Les observations de non-conformité après la mise en marché ont été reçues par la fonction de précommercialisation, mais n’ont pas été signalées à la fonction de conformité et d’application de la loi pour qu’elle prenne des mesures, ce qui limite la capacité du programme à adopter une approche fondée sur le cycle de vie.
Le PIM examine des mesures supplémentaires pour faciliter davantage l’accès aux instruments médicaux nécessaires .
Le PIM a continué d’examiner des mesures visant à garantir à la fois la sécurité et l’accès aux instruments médicaux dans un environnement évoluant. Il est trop tôt pour évaluer l’effet de ces mesures, car elles étaient encore en cours d’application au moment de la mission. Ces mesures comprennent :
- Une voie réglementaire pour les produits thérapeutiques avancés annoncée dans le budget 2019. Cette voie proposée a été conçue pour assurer la sécurité des patients, tout en augmentant la flexibilité du programme pour approuver les nouveaux instruments qui ne correspondent pas clairement au cadre réglementaire actuel.
- L’extension du mécanisme d’évaluation prioritaire. Cela permettrait au PIM de placer certaines demandes d’homologation d’instruments médicaux en tête de file pour leur traitement, afin de mieux répondre aux besoins du système de santé et d’accroître la flexibilité du système réglementaire.
Les informateurs clés ont suggéré des possibilités pour que Santé Canada puisse continuer à améliorer l’accès aux instruments médicaux.
Plusieurs informateurs externes clés ont suggéré d’examiner la possibilité de mettre en œuvre un processus accéléré d’approbation des demandes de modification pour les instruments médicaux déjà homologués pour la vente sur le marché canadien, dans les cas où les modifications ne devraient pas augmenter de manière significative les risques liés à l’instrument. Cette suggestion a été faite en partant du principe que le nombre de demandes de modification augmenterait à mesure que le rythme d’évolution de la technologie continue de s’accélérer et que les mises à jour des instruments médicaux déjà homologués deviennent plus fréquentes (p. ex., les mises à jour des logiciels utilisés comme instruments médicaux). Une procédure similaire pour accélérer les demandes de modification existe déjà aux États-Unis (c.-à-d., le 510[k]).
Certains informateurs externes clés, dont des représentants de l’industrie et des soins de santé, ont également suggéré un processus d’approbation accéléré pour les instruments médicaux dont la vente est déjà autorisée dans les pays qui ont des processus réglementaires similaires à ceux du Canada. Ces informateurs ont aussi fait remarquer que si Santé Canada envisageait cette option, le Ministère devrait également maintenir son autonomie réglementaire et sa responsabilité envers les Canadiens.
De plus, certains informateurs clés externes ont noté un désir de plus grande transparence sur le statut des demandes d’homologation des instruments médicaux présentées à Santé Canada. Cela pourrait être conforme à la base de données en ligne « Présentations en cours d’examen » du programme pharmaceutique de Santé Canada. La majorité des répondants (76 %) à l’enquête de 2016 sur la transparence de la Direction générale des produits de santé et des aliments ont exprimé le même souhait et ont convenu que la liste des demandes en cours d’examen devrait être élargie pour inclure les instruments médicaux.
4.0 Communication avec les intervenants et les Canadiens
Santé Canada a amélioré ses communications concernant les instruments médicaux et a renforcé la mobilisation des intervenants.
Santé Canada a diffusé différents types de produits de communication pour aider les intervenants de l’industrie à s’y retrouver dans le Programme et pour aider les professionnels de la santé et les Canadiens à prendre des décisions éclairées sur l’utilisation d’un instrument médical (p. ex., lettre aux professionnels de la santé, avis de rappel). Les produits de communication élaborés par le Ministère comprenaient également des documents d’orientation (voir la section 3.3 pour plus de détails).
Au cours des cinq dernières années, Santé Canada a amélioré ses communications sur les instruments médicaux grâce à des initiatives telles que les suivantes :
- La publication de sommaires des décisions réglementaires pour les instruments médicaux nouveaux et modifiés de classe III et de classe IV. Environ 125 nouveaux sommaires ont été publiés entre mars et août 2019.
- L’amélioration de l’accès du public aux données cliniques relatives aux instruments médicaux grâce à un portail Web interrogeable et à une base de données en ligne régulièrement mise à jour sur les incidents, les plaintes et les rappels liés aux instruments médicaux.
- Une plus grande clarté et des renseignements supplémentaires sur les inspections des instruments médicaux.
- La réalisation de diverses activités de mobilisation, notamment des réunions semestrielles avec des représentants clés de l’industrie, un webinaire avec des professionnels de la santé pour discuter de l’examen de la sécurité du lymphome anaplasique à grandes cellules associé aux implants mammaires (LAGC AIM) par Santé Canada, et des activités de sensibilisation ciblées auprès de groupes de patients, notamment des réunions avec des femmes vivant avec des implants mammaires pour connaître leurs expériences.
Définition clé : Les sommaires des décisions réglementaires expliquent la décision prise par Santé Canada concernant certains produits de santé demandant une autorisation de mise en marché.
Plusieurs représentants de l’industrie et quelques professionnels de la santé ont souligné les initiatives de mobilisation du PIM. Les représentants de l’industrie ont apprécié l’occasion qui leur a été donnée d’engager avec les représentants de Santé Canada et ils espèrent que ce genre d’occasions se représentera à l’avenir. Ces perceptions font écho aux résultats du sondage de 2016 sur le CRTO, selon laquelle la majorité des intervenants interrogés (85 %) ont convenu que Santé Canada avait mieux communiqué avec les Canadiens à cette occasion qu’il y a dix ans. De plus, tous les intervenants qui ont répondu au sondage ont estimé que Santé Canada était une source fiable et digne de confiance d’information sur la santé et la sécurité, tandis que 95 % ont déclaré utiliser les renseignements de Santé Canada pour éclairer leurs activités et leurs décisions.
En plus de diverses initiatives de mobilisation, Santé Canada a offert aux intervenants et aux Canadiens la possibilité de participer à des consultations en ligne sur les changements réglementaires proposés, ainsi qu’à l’élaboration de politiques et de documents d’orientation nouveaux ou modifiés. En 2017-2018 et 2018-2019, le Ministère a réalisé 100 % de ses projets de consultation prévus sur les instruments médicaux. Si les représentants de l’industrie étaient généralement satisfaits de l’occasion de participer à ces consultations, quelques-uns ont fait remarquer qu’ils auraient apprécié avoir davantage de possibilités d’effectuer un suivi avec le PIM après leur participation. Cela leur aurait permis de fournir des détails supplémentaires, si nécessaire, et de mieux comprendre comment leurs commentaires ont été utilisés par le programme. À cet égard, les documents du Programme montrent que le PIM a examiné les commentaires recueillis lors des consultations en ligne et les ont utilisés, le cas échéant.
Jusqu’à présent, la majorité des efforts de participation et de consultation du PIM ont été réalisés par la fonction de précommercialisation. Il reste donc des possibilités supplémentaires pour que les intervents soient engagés dans les discussions relatives aux activités réalisées après la mise en marché. La fonction de conformité et d’application de la loi a élaboré un plan de mobilisation des intervenants, mais n’a pas pu le mettre en œuvre complètement en raison d’un manque de ressources.
Les informateurs clés non issus de l’industrie, y compris les membres du public canadien, avaient une connaissance limitée des communications de Santé Canada relatives aux instruments médicaux.
La majorité des informateurs clés qui étaient des professionnels de la santé, des associations de patients ou des personnes utilisant un instrument médical ont déclaré qu’ils n’étaient pas au courant des produits de communication de Santé Canada sur les instruments médicaux. Ceux qui étaient au courant ont indiqué qu’ils utilisaient rarement ces renseignements pour informer leurs décisions. Ils se fiaient plutôt à d’autres sources d’information, y compris les fabricants, les services d’approvisionnement des hôpitaux, les revues et publications médicales, les médias, ainsi que des collègues ou d’autres personnes utilisant un instrument médical. Néanmoins, certains professionnels de la santé et plusieurs personnes utilisant un instrument médical ont indiqué qu’ils auraient préféré avoir accès aux renseignements de Santé Canada, car ils s’attendaient à ce que celles-ci soient plus objectives et plus fiables.
En plus d’être généralement ignorants des produits de communication de Santé Canada sur les instruments médicaux, la majorité des informateurs clés non issus de l’industrie ignoraient également les consultations en ligne de Santé Canada sur les instruments médicaux, ou ne participaient à aucune des activités de mobilisation du PIM. Plusieurs informateurs externes clés ont indiqué qu’un plus grand nombre d’activités de mobilisation sont essentielles pour que Santé Canada puisse connaître l’incidence des instruments médicaux et les besoins des groupes d’intervenants non issus de l’industrie.
Les renseignements mis en ligne sont souvent obsolètes et difficiles d’accès.
En plus de soulever des préoccupations concernant le manque d’actualité des documents d’orientation (voir la section 3.1 pour plus de détails), certains représentants de l’industrie et quelques informateurs internes clés ont fait remarquer que certains documents d’orientation de Santé Canada ne reflétaient pas nécessairement les réalités de l’environnement actuel des instruments médicaux (p. ex., technologie de santé numérique, intelligence artificielle). Cela pourrait mener à des situations dans lesquelles les instruments médicaux dotés de technologies plus avancées ne seraient pas évalués d’une manière qui reflète la rapidité avec laquelle ils évoluent.
Le manque d’actualité des renseignements de Santé Canada a également été cité par quelques informateurs externes clés comme une raison pour laquelle ils se fient à d’autres sources de renseignements sur les instruments médicaux.
De plus, de 2016-2017 à 2018-2019, de 33 à 50 % des sommaires des motifs de décision du PIM et de 7 à 33 % des sommaires des décisions réglementaires ont été mis en ligne dans le cadre des normes de service du Programme. Ces documents avaient pour but de fournir aux professionnels de la santé et aux Canadiens des renseignements qui mèneront à des prises de décisions et à des traitements plus bien informésNote de bas de page 31.
Définition clé : Les sommaires des motifs de décisions expliquent pourquoi Santé Canada a autorisé la vente de certains instruments médicaux au Canada.
Certains informateurs clés externes ont également indiqué qu’il était nécessaire d’utiliser un langage plus clair dans les produits d’information de Santé Canada afin de les rendre plus accessibles au grand public. À cet égard, une étude réalisée en 2013 a révélé que la plupart des produits de communication des risques de Santé Canada étaient rédigés à niveau de connaissances ciblant les étudiants de deuxième cycle, ce qui est bien supérieur au niveau moyen de connaissances en matière de santé du grand publicNote de bas de page 32.
De plus, certains informateurs externes clés et quelques informateurs internes clés ont indiqué qu’une plus grande clarté et un meilleur contexte autour de la communication des risques seraient utiles, en particulier pour les incidents et les rappels. Par exemple, ils ont expliqué que le fait de disposer de plus d’information sur les causes des incidents pour un instrument médical particulier (p. ex., un dysfonctionnement de l’instrument par opposition à une erreur de l’utilisateur) aiderait à prendre des décisions éclairées sur l’utilisation d’un instrument médical.
5.0 Organisation et gouvernance du Programme
5.1 Répartition des rôles et des responsabilités et gouvernance
Les rôles et les responsabilités des trois fonctions du Programme n’ont pas toujours été clairement définis ni alignés sur la nature de leur mandat.
Les rôles et responsabilités de haut niveau du PIM ont été bien définis. En particulier, la Loi sur les aliments et drogues et le Règlement sur les instruments médicaux définissent clairement les rôles et les responsabilités du ministre de la Santé en ce qui concerne les instruments médicaux. Toutefois, les rôles et les responsabilités au niveau opérationnel des trois fonctions du PIM n’ont pas été clairement définis. Des documents et des informateurs internes clés ont confirmé que le PIM ne disposait pas d’un cadre global définissant les rôles et les responsabilités ni d’une stratégie intégrée de gestion des risques et de partage de l’information.
Il est important de définir clairement les rôles et les responsabilités, car cela permet à la direction de coordonner les activités du Programme, d’assumer le leadership, d’assurer la responsabilisation et de gérer les risques. Un manque de définition des rôles et des responsabilités peut être source de confusion et d’inefficacité opérationnelle, comme la duplication des efforts entre les fonctions du Programme et des processus incohérents.
Par exemple, quelques informateurs internes clés ont noté un problème de répartition des responsabilités pour le traitement des rapports d’incidents volontaires entre la fonction de surveillance après la mise en marché et la fonction de conformité et d’application de la loi. Alors que les rapports volontaires reçus du ResSCIM étaient saisis, codés et évalués scientifiquement par la fonction de surveillance, les rapports volontaires provenant d’autres sources étaient saisis par la fonction de conformité et d’application de loi, mais pas codés ni évalués scientifiquement. Ceci a créé des incohérences dans la saisie, le codage et l’évaluation des données pour les rapports volontaires. Il a également mené à la création de processus parallèles. Il est à noter que lorsque la fonction de conformité et d’application de la loi a reçu un rapport volontaire et qu’il n’y a pas eu de rapport obligatoire ultérieur de la part du fabricant, le PIM a effectué un suivi avec le fabricant, déclenchant le rapport obligatoire requis.
Il n’ya a pas de preuves que les rôles et les responsabilités de la fonction de conformité et d’application de la loi ont été définis pour le traitement des rapports d’incidents volontaires. Il n’a pas non plus été constaté que les rôles et les responsabilités étaient définis en ce qui concerne le partage de l’information des rapports volontaires avec la fonction de surveillance. De plus, il n’était pas exigé que la fonction de conformité et d’application de la loi et les fonctions de surveillance après la mise en marché se consultent sur les changements de processus. Il n’a pas été possible de déterminer si les deux fonctions ont rempli leurs rôles et responsabilités dans le traitement des rapports d’incidents.
En plus d’un manque de clarté, la répartition des rôles et des responsabilités entre les trois fonctions du PIM n’a pas toujours été conforme à la nature de leur mandat. Par exemple, la fonction de précommercialisation a assumé plusieurs responsabilités qui étaient principalement perçues comme des activités se déroulant après la mise en marché. En vertu de l’article 39 du Règlement sur les instruments médicaux, Santé Canada a le pouvoir de demander des renseignements supplémentaires à un fabricant s’il y a des motifs raisonnables de croire qu’un instrument homologué ne répond pas aux exigences de sécurité et d’efficacitéNote de bas de page 33. Au moment du projet, la fonction de précommercialisation est chargée de demander ces renseignements supplémentaires, même si ce sont les fonctions de surveillance et de conformité et d’application de la loi qui les utilise pour évaluer un incident. Quelques informateurs internes clés ont expliqué que le fait de devoir passer par la fonction de précommercialisation pour obtenir les renseignements requis n’était pas une pratique efficace. Pour répondre à ce défi, la fonction de surveillance a établi un processus parallèle informel (c.-à-d.,une lettre de demande volontaire) pour contacter directement les fabricants.
Au moment du projet, le certificat du PUAIM (voir section 3.2) était demandé, géré et conservé par la fonction de précommercialisation. Plusieurs informateurs internes clés de la fonction de conformité et d’application de la loi ont indiqué que l’accès au certificat pourrait faciliter leur travail, car il permettrait de cerner les domaines possibles de risque et d’orienter leur travail d’inspection. Il a également été noté que la transition du PUAIM vers la fonction de conformité et d’application de la loi contribuerait à réduire la duplication des activités entre l’audit du PUAIM et l’inspection des fabricants. Cela contribuerait également à garantir que la fonction de conformité et d’application de la loi soit informée de tout cas de non-conformité dans les meilleurs délais. En outre, les inspecteurs du PIM avaient plus de pouvoir (p. ex., la capacité d’examiner tout article ou toute donnée électronique visée par laLoi sur les aliments et drogues ou le Règlement sur les instruments médicaux) et menaient des enquêtes plus approfondies sur les systèmes et les activités des fabricants, par rapport aux auditeurs tiers responsables des examens du PUAIM. Au moment du projet, des discussions entre les fonctions de précommercialisation et de conformité et d’application de la loi étaient en cours pour déterminer de quelle fonction le PUAIM devait relever.
Le PIM a mis en œuvre divers comités de gouvernance pour favoriser le partage d’information entre les trois fonctions.
Divers comités de gouvernance ont été mis en œuvre pour surveiller les opérations du Programme et favoriser l’échange d’information entre les trois fonctions du Programme. En particulier, un comité trilatéral mensuel sur les instruments médicaux a rassemblé les gestionnaires et les directeurs Note de bas de page viii du Programme et a fourni un forum pour discuter des questions d’intérêt ou de responsabilité mutuelle. Le comité trilatéral a adopté une double approche, dans laquelle les réunions incluant les directeurs et les gestionnaires étaient suivies d’une réunion réservée aux directeurs. Le PIM a également mis en œuvre plusieurs comités de niveau opérationnel, ainsi qu’un comité de direction qui assure le leadership et une orientation.
Des informateurs internes clés ont expliqué que la communication au niveau de la direction et au-delà fonctionnait généralement bien. Ils ont également noté que les divers comités et réunions qui ont eu lieu entre les fonctions du PIM (p. ex., les réunions trilatérales, les réunions bihebdomadaires sur les risques, les réunions mensuelles sur les « sujets brûlants », les réunions bilatérales entre le directeur général et les directeurs) ont contribué à l’efficacité du Programme, notamment en ce qui concerne la priorisation des incidents liés aux instruments médicaux et l’identification des problèmes avant qu’ils ne se produisent. Quelques informateurs internes clés ont expliqué qu’il restait des possibilités de rendre les réunions trilatérales plus stratégiques et d’améliorer la diffusion de l’information discutée lors de ces réunions parmi un groupe plus large de personnel du Programme.
Malgré la structure de gouvernance mise en place, des inquiétudes ont été exprimées quant à la qualité des communications au niveau opérationnel.
Plusieurs informateurs internes clés ont exprimé leurs préoccupations concernant le niveau et la qualité des communications au niveau opérationnel entre les trois fonctions du PIM. En particulier, ils ont cerné un manque de communication comme l’un des principaux défis minant l’efficacité du Programme.
Certains informateurs internes clés ont estimé que ces difficultés de communication avaient un effet négatif sur la coordination des activités du Programme, ce qui a eu pour conséquence de faire manquer des occasions de collaboration entre les fonctions du Programme ou de créer des retards dans les activités du Programme. Par exemple, un manque de communication entre les fonctions de surveillance avant et après la mise en marché concernant les nouveaux instruments médicaux homologués a limité les possibilités de surveillance proactive après la mise en marché. De plus, les difficultés de communication entre les deux fonctions après la mise en marché concernant les évaluations de signaux en cours ont entraîné des situations où les inspecteurs chargés de la conformité et de l’application de la loi n’étaient pas au courant des évaluations de signaux à venir qui pourraient profiter aux activités de vérification de la conformité.
Rien n’indique que le PIM disposait d’un cadre horizontal de gestion des risques.
Le PIM disposait d’un comité de gestion des risques du Programme qui discutait des questions émergentes. En plus du comité, les questions émergentes ont été gérées au sein de chaque fonction de programme selon les besoins du moment. Toutefois, aucun élément n’a été trouvé indiquant l’existence d’un processus horizontal de gestion des risques. Bien que la Direction générale des produits de santé et des aliments dispose d’une ébauche d’un registre des risques de la Direction générale pour 2018-2019, une version approuvée n’était pas disponible au moment du projet, ni une version antérieurement approuvée de ce document.
5.2 Création d’une nouvelle Direction des instruments médicaux
Santé Canada a mis en œuvre une nouvelle Direction des instruments médicaux qui regroupe les activités de précommercialisation et celles visant à examiner les incidents impliquant des instruments médicaux. Cette nouvelle structure est similaire aux nouvelles approches organisationnelles mises en œuvre aux États-Unis et en Irlande.
En novembre 2019, une partie du PIM a été restructurée en une nouvelle Direction des instruments médicaux, qui a intégré la fonction de précommercialisation et les fonctions de surveillance après la mise en marché du Bureau des produits pharmaceutiques et des instruments médicaux commercialisés (voir la section 1.2 et l’annexe E pour plus de détails sur ces divisions). La fonction de surveillance du Bureau de la surveillance des produits de santé et de l’épidémiologie et la fonction de conformité et d’application de la loi relèvent toujours de leurs directions d’origine.
La nouvelle structure organisationnelle était similaire à l’approche adoptée par l’IrlandeNote de bas de page 34 en 2018 et par la Food and Drug Administration des États-Unis en 2019. Selon des informateurs internationaux clés et des documents du site Web de la Food and Drug Administration, le regroupement des fonctions de précommercialisation et après la mise en marché devait permettre au personnel de profiter des connaissances de chacun et d’optimiser la prise de décision tout au long du cycle de vie des instruments médicauxNote de bas de page 35. La figure 7 donne un aperçu de haut niveau des nouvelles structures organisationnelles mises en place dans ces deux pays.
Les entrevues avec des informateurs clés et les documents du site Web de la Food and Drug Administration laissent entendre que les premiers effets de leur réorganisation se sont fait sentir :
- Communications améliorées entre les fonctions du programme et clarté des rôles et des responsabilités
- Plus de discussions, de décisions et de production de preuves tout au long du cycle de vie des instruments médicaux
- Réduction des délais de mise en marché de technologies innovantes plus sûres et plus efficaces.
Au moment du projet, il était prévu que la nouvelle Direction des instruments médicaux de Santé Canada aurait des effets similaires et contribuerait à favoriser une approche fondée sur le cycle de vie pour la réglementation et la surveillance des instruments médicaux. Pour ce faire, il convient de renforcer les capacités scientifiques et politiques du PIM, de moderniser les processus pour répondre aux cycles d’innovation rapides et d’augmenter les ressources pour soutenir les activités de transparence et de mobilisation des intervenants tout au long du cycle de vie des instruments médicauxNote de bas de page 36.
5.3 Gestion de l’information
L’infrastructure de la technologie de l’information (TI) du PIM et son organisation de l’information ont mis au défi l’efficacité du Programme et la capacité d’adopter une approche de cycle de vie pour la réglementation et la surveillance des instruments médicaux .
Au moment du projet, il n’existait pas de dépôt unique pour recueillir tous les renseignements disponibles sur un instrument médical. Toutefois, les méthodes de suivi, lorsqu’elles sont combinées, fournissent des renseignements sur l’ensemble des activités liées à un instrument médical ou à un signal. Ces méthodes comprenaient plusieurs fiches de suivi au sein des deux fonctions après la mise en marché, docuBridge, les dossiers partagés respectifs de chaque équipe, divers tableaux de bord, diverses formes de communication interne et externe, et le Système des instruments médicaux.

Figure 7 - Équivalent textuel
Cette figure contient deux sections qui montrent les renseignements recueillis sur les programmes de matériels médicaux aux États-Unis et en Irlande.
Dans la première section, les renseignements sur la structure organisationnelle aux États-Unis sont présentés sous forme de liste à puces, comme suit :
- Les activités avant mise en marché, la surveillance après mise en marché et les activités de conformité et d’application de la loi ont été regroupées dans un même bureau.
- Au sein du nouveau bureau, les divisions ont été organisées selon des gammes de produits (p. ex. le Bureau des instruments cardiovasculaires, le Bureau des instruments orthopédiques) au sein desquelles le personnel supervise un instrument médical tout au long de son cycle de vie.
- Une équipe de politique scientifique et clinique a également soutenu le travail du bureau.
Dans la seconde section, les renseignements sur la structure organisationnelle en Irlande sont présentés sous forme de liste à puces, comme suit :
- Les activités avant la mise en marché et la surveillance après la mise en marché ont été regroupées dans un nouveau département des matériels médicaux.
- La conformité est restée une fonction distincte au sein du département de la conformité.
- Une équipe chargée de la réglementation, de la législation et de l’élaboration des politiques a soutenu le travail du département des matériels médicaux.
Les sources des renseignements pour cette figure proviennent de la Food and Drug Administration des États-Unis (2019 b) et de Health Product Regulatory Authority d’Irlande (2019).
Par exemple, la fonction de surveillance après la mise en marché a documenté ses activités concernant l’évaluation des signaux des instruments médicaux dans des feuilles de suivi Excel, au lieu d’utiliser le Système des instruments médicaux. Elle a suivi les délais d’évaluation des signaux par rapport aux normes de service dans un outil intitulé Fiche de suivi des activités réglementaires. De plus, les documents relatifs à la conformité et à l’application de la loi et les documents relatifs à la surveillance, y compris ceux du ResSCIM, ont été sauvegardés dans les lecteurs partagés de leurs fonctions respectives, et l’information n’a pas été intégrée dans docuBridge.
Comme toutes les données relatives à un instrument médical n’ont pas été saisies dans une base de données ou un dépôt unique, un rapport consolidé sur le cycle de vie n’a pas pu être généré pour des instruments particuliers. Comme l’ont expliqué certains informateurs internes clés, le personnel du Programme a dû regrouper l’information provenant de diverses sources pour reconstituer l’historique complet des mesures prises par Santé Canada relativement à un instrument médical.
La fragmentation de l’information a également rendu difficile la réalisation d’analyses de tendances par le Programme et a pu nuire à l’accès à certains renseignements en temps utile. Par exemple, lorsque les fonctions de précommercialisation ou de conformité et d’application de la loi ont eu besoin de données après la mise en marché pour éclairer leurs activités, elles ont dû passer des heures supplémentaires à compiler ces renseignements à partir de diverses fiches de suivi.
De plus, quelques informateurs clés ont expliqué que les renseignements sur la classification d’un instrument étaient stockés dans une base de données détenue par la fonction de précommercialisation. Par conséquent, les inspecteurs n’ont pas eu accès à ces renseignements en temps opportun lorsqu’ils ont inspecté les fabricants. Il est à noter qu’au moment du projet, des discussions étaient en cours entre les fonctions de précommercialisation et de conformité et d’application de la loi pour améliorer l’échange de ces renseignements. Le Plan stratégique 2016-2021 de la Direction générale des produits de santé et des aliments a fait état d’un engagement à investir dans des initiatives de TI pour le PIMNote de bas de page 37.
Plusieurs informateurs internes clés ont fait remarquer que certains des systèmes informatiques du PIM (p. ex., le Système sur les instruments médicaux et docuBridge) étaient désuets, difficiles à utiliser et n’étaient pas fondamentalement conçus pour les fins auxquelles ils étaient destinés. En outre, une proportion importante des documents d’orientation n’a pas été mise à jour pour refléter les changements liés aux systèmes de TI, notamment la création de la Direction générale des opérations réglementaires et de l’application de la loi, la mise en œuvre de docuBridge et la modernisation du Système des instruments médicaux.
Quelques informateurs internes clés ont également fait part de leurs préoccupations quant au fait que l’infrastructure informatique utilisée pour exécuter les activités de programme n’était pas à la hauteur de la technologie actuelle. Par exemple, le PIM ne disposait pas d’interfaces électroniques pour la saisie des demandes d’homologation de instruments médicaux ni des rapports d’incidents obligatoires. Les deux éléments sont soumis à Santé Canada au moyen de formulaires PDF envoyés par courrier, par courriel ou par télécopie. L’utilisation de ces méthodes n’était pas conforme au monde de la haute technologie de l’industrie des instruments médicaux. Cela signifie également que les renseignements envoyés à Santé Canada (p. ex., les rapports d’incidents obligatoires ou volontaires) ont dû être traités manuellement par une équipe qui n’avait pas le personnel adéquat pour effectuer ces tâches dans le cadre des normes de service ciblées (voir section 6).
6.0 Ressources du Programme
Les retards dans l’achèvement de certaines activités après la mise en marché ont été principalement attribués au niveau des ressources disponibles par rapport à la charge de travail.
En 2018-2019, le budget du PIM a été réparti de la manière suivante :
- 46 % pour les activités de conformité et d’application de la loi;
- 37 % pour les activités de précommercialisation;
- 10 % pour l’administration du Programme et les autres activités de soutien du Programme;
- 7 % pour les activités de surveillance après la mise en marché.
Dans l’ensemble, la fonction de surveillance après la mise en marché des instruments médicaux a reçu une part relativement faible du budget de programme.
Au cours des six dernières années, environ huit (8) équivalents temps plein (ETP) non gestionnaires ont quitté le domaine de la surveillance des instruments médicaux, soit environ 30 % des ETP non gestionnaires des fonctions de surveillance après la mise en marché. Une partie du personnel actuel a également été temporairement réaffectée à d’autres responsabilités (p. ex., la mise en œuvre d’initiatives dans le cadre du Plan d’action sur les instruments médicaux).
Il semble également que les ressources allouées étaient relativement faibles par rapport à la surveillance après la mise en marché du Programme des produits pharmaceutiques de Santé Canada. Comme l’illustre la figure 8, le nombre d’ETP affecté à l’évaluation des incidents liés aux instruments médicaux était inférieur à un tiers du nombre d’ETP affecté à des activités similaires pour le programme pharmaceutique, même si le volume d’incidents à traiter était plus important pour les instruments médicaux. Selon quelques informateurs internes clés, la différence entre les ressources disponibles pour la surveillance des instruments médicaux, par opposition à la surveillance des produits pharmaceutiques, correspondait à une tendance qui se poursuivait au moment du projet.
Dans l’ensemble, plusieurs informateurs internes clés ont indiqué que la capacité insuffisante de la fonction de surveillance après la mise en marché constituait l’une des raisons du retard dans le traitement des rapports d’incident, de l’incapacité à traiter un grand nombre de rapports, de l’élimination du triage des rapports d’incident entrants, ainsi que de la diminution du nombre d’évaluations et d’examens des signaux effectués dans le respect des normes de service (voir section 2.2).
| Instruments médicaux | Produits pharmaceutiques | |
|---|---|---|
| ETP | 9 | 30 |
| Incidents liés aux instruments médicaux et effets indésirables des médicaments examinés | 17 133 | 4 450 |
| Signaux potentiels pour l’évaluation | 263 | 173 |
| Source : Documents internes de Santé Canada. |
En comparaison, le nombre d’ETP dans les fonctions de précommercialisation et de conformité et d’application de la loi a augmenté pendant la période du projet (p. ex., huit inspecteurs supplémentaires ont été embauchés en 2019). Toutefois, plusieurs informateurs internes clés ont noté que les fonctions de précommercialisation et de conformité et d’application de loi étaient également mises à mal par des capacités insuffisantes. Ils ont indiqué qu’ils étaient confrontés à une charge de travail accrue en raison de l’examen plus minutieux des instruments médicaux sur le marché canadien, des changements apportés au Programme (p. ex., la mise en œuvre du Plan d’action sur les instruments médicaux) et de l’évolution de l’environnement des instruments médicaux (p. ex., les nouvelles technologies). Certains informateurs internes clés ont en outre soulevé des préoccupations selon lesquelles, sans les ressources nécessaires pour atténuer ces difficultés, le programme pourrait ne pas être en mesure d’examiner de façon efficiente et efficace les demandes d’homologation de nouvelles technologies et de nouveaux instruments médicaux ni d’inspecter les fabricants de ces instruments à l’avenir.
La déclaration obligatoire des incidents introduite en décembre 2019 va probablement exacerber les difficultés rencontrées par les fonctions de surveillance après la mise en marché.
La Loi visant à protéger les Canadiens contre les drogues dangereuses , également connue sous le nom de Loi de Vanessa, a introduit des modifications à la Loi sur les aliments et droguesNote de bas de page 38. Elle a notamment introduit la déclaration obligatoire des effets indésirables des médicaments et des incidents liés aux instruments médicaux pour environ 775 hôpitaux canadiens régis par une législation provinciale ou territoriale, ou exploités par le gouvernement du Canada, à partir de décembre 2019Note de bas de page 39.
Loi de Vanessa : Vanessa Young est décédée en 2000, à l’âge de 15 ans, d’une arythmie cardiaque après avoir pris du cisapride (Prepulsid®) conformément à la prescription de son médecin. Une campagne visant à renforcer la réglementation des produits thérapeutiques a par la suite amené Santé Canada à demander aux hôpitaux et à l’industrie des données sur l’innocuité des médicaments et la sécurité des instruments médicaux. La Loi de Vanessa a été promulguée en 2014, et les exigences de déclaration obligatoire sont entrées en vigueur le 16 décembre 2019.
Selon une analyse interne coûts-avantages, le nombre de rapports d’incidents reçus par le PIM pourrait doubler à la suite de la mise en œuvre de la déclaration obligatoireNote de bas de page 40. Quelques informateurs internes clés ont fait part de leurs préoccupations quant à l’incapacité de la fonction de surveillance à répondre à la demande. L’arriéré des rapports d’incidents observé au moment de la mission pourrait continuer à s’accroître à l’avenir. En outre, la déclaration obligatoire peut avoir des répercussions ultérieures sur les ressources et les activités de la fonction de conformité et d’application de la loi après la mise en marché, car les inspecteurs seront tenus de surveiller les éventuelles sous-déclarations et d’inspecter les hôpitaux.
Au moment du projet, à l’exception de l’analyse coûts-avantages, Santé Canada n’avait pas entièrement analysé les répercussions de la déclaration obligatoire sur la charge de travail de programme. Le Ministère a prévu de faire face au volume accru de rapports d’incidents à l’aide d’une réaffectation des ressources et d’une redistribution de la charge de travail dans la fonction après la mise en marchéNote de bas de page 41 . La fonction de conformité et d’application de la loi doit évaluer de façon constante l’incidence sur les ressources et la stratégie, particulièrement si le nombre de rapports devait augmenter de manière significative.
7.0 Conclusions et recommandations
L’environnement des instruments médicaux est complexe et en pleine évolution, en raison du développement de nouveaux instruments utilisant des technologies innovantes telles que la technologie numérique, l’intelligence artificielle et l’impression 3D. Dans cet environnement complexe, Santé Canada a généralement réussi à assurer la sécurité, l’efficacité et la qualité des instruments médicaux sur le marché canadien. Il a également mis en place des contrôles suffisants pour assurer la rigueur de son travail. Dans l’ensemble, la sécurité des instruments médicaux au Canada a été jugée comparable à celle de pays similaires. Le Canada a parfois été l’un des premiers pays à lancer des rappels pour faire face aux incidents liés à ces instruments, comme dans les cas d’Essure et des implants mammaires Biocell.
Il y a eu, cependant, des retards dans l’achèvement de certaines activités après la mise en marché. Bien que rien ne prouve que ces retards aient affecté la sécurité des instruments médicaux sur le marché canadien, ils ont affecté la capacité du PIM à détecter et à traiter de manière proactive les incidents liés aux instruments médicaux. Ces retards représentaient également un risque pour le Ministère, car ils semblent être principalement attribuables à un manque de capacité dans les fonctions de surveillance après la mise en marché. Dans l’ensemble, les activités de surveillance après la mise en marché n’ont reçu qu’environ 7 % du budget du PIM en 2018-2019. De plus, ces dernières années, le nombre d’employés travaillant au sein de la fonction de surveillance des instruments médicaux a diminué, alors qu’il a augmenté dans les deux autres fonctions du Programme.
Avec l’introduction de la déclaration obligatoire des incidents par les hôpitaux en décembre 2019, il est probable que la charge de travail de la fonction de surveillance après la mise en marché et de la fonction de conformité et d’application de la loi augmentera considérablement. Au moment du projet, les répercussions de la déclaration obligatoire, en termes de charge de travail, n’ont cependant pas été entièrement analysées. Cela représente un risque, car les défis actuels liés à l’achèvement en temps voulu de certaines activités de surveillance seront probablement exacerbés si les problèmes de capacité ne sont pas résolus à l’avenir.
Au cours des dernières années, le PIM a mis en œuvre plusieurs initiatives afin de demeurer efficace dans l’environnement changeant des instruments médicaux. Le Ministère a fait d’importants progrès pour adapter à la fois son cadre réglementaire et ses activités après la mise en marché grâce à des initiatives mises en œuvre dans le cadre du Plan d’action sur les instruments médicaux et de l’Examen réglementaire des médicaments et des instruments. Il restait à Santé Canada d’autres possibilités de continuer à s’adapter. Le PIM a notamment eu la possibilité d’accroître la mobilisation de ses homologues internationaux, afin d’harmoniser les pratiques, notamment en ce qui concerne les activités de conformité et d’application de la loi. Il a également été possible de prendre en compte de manière plus systématique les différences entre les sexes et les genres dans l’examen des demandes d’homologation des instruments médicaux, la communication des risques et les activités de conformité et d’application de la loi.
Au cours de la période du projet, le PIM a amélioré ses communications concernant les instruments médicaux et la mobilisation des intervenants. Bien que ces efforts aient été bien accueillis par les informateurs clés de l’industrie, des améliorations sont encore nécessaires pour que Santé Canada devienne une source d’information incontournable pour les professionnels de la santé, les groupes de patients et le public canadien en général. De plus, l’accès aux produits de communication en ligne de Santé Canada était difficile. Les renseignements disponibles en ligne, y compris les documents d’orientation pour l’industrie, n’étaient souvent pas à jour.
Quant à l’efficacité du programme, les éléments disponibles montrent que la répartition des rôles et des responsabilités entre les trois fonctions n’était pas toujours conforme à la nature de leurs activités principales. Cela a conduit à une inefficacité, notamment la duplication des efforts entre les fonctions du programme. En outre, le PIM ne disposait pas d’un cadre exhaustif pour définir les rôles et les responsabilités. En outre, la fonction de conformité et d’application de la loi après la mise en marché et le groupe chargé de traiter les rapports d’incidents relatifs aux instruments médicaux demeurent dans leur structure organisationnelle initiale. Il était prévu que la création de la nouvelle Direction des instruments médicaux, avec l’intégration de la fonction de précommercialisation et du Bureau des produits pharmaceutiques et des instruments médicaux commercialisés (fonction après la mise en marché), permettrait probablement de relever certains de ces défis et de favoriser une approche fondée sur le cycle de vie pour la réglementation et la surveillance des instruments médicaux.
L’infrastructure de TI et l’organisation de l’information du PIM ne permettaient pas une exécution efficace des activités. Les données et les renseignements actuels sur le programme ont été enregistrés dans de multiples fichiers et bases de données différents, détenus par différentes fonctions du programme. Cela a mis au défi la capacité du personnel du programme d’accéder à l’information en temps utile pour les activités du Programme. Le personnel a dû rassembler de l’information provenant de divers dossiers et bases de données afin d’obtenir l’historique complet d’un instrument médical et des mesures prises par le PIM liées à cet instrument médical.
Les constatations mentionnées dans le présent rapport ont conduit à la formulation des recommandations suivantes :
-
La Direction générale des produits de santé et des aliments devrait combler les lacunes actuelles en matière de ressources dans les fonctions suivant la mise en marché auxquelles il incombe le traitement des rapports d’incidents et l’évaluation des incidents. La Direction générale des opérations réglementaires et de l’application de la loi devrait examiner l’incidence qui suit sur les besoins en matière de ressources de la fonction de conformité et d’application de la loi. De plus, les deux directions générales devraient s’assurer que le PIM dispose des capacités et des outils nécessaires pour faire face à l’obligation de déclaration par les hôpitaux introduite en décembre 2019, et à l’application connexe des pratiques de vigilance des hôpitaux.
Les activités de surveillance du PIM ont été mises à mal par un manque de capacité et n’ont pas toujours pu être menées à bien dans le respect des normes de service. L’introduction, en décembre 2019, de la déclaration obligatoire des incidents liés aux instruments médicaux par environ 775 hôpitaux canadiens influera probablement davantage sur la capacité de la fonction de surveillance après la mise en marché à gérer sa charge de travail, car le nombre de déclarations d’incidents soumises à Santé Canada pourrait doubler. La déclaration obligatoire peut également toucher les ressources et les activités de la fonction de conformité et d’application de la loi, car les inspecteurs devront surveiller les éventuelles sous-déclarations et inspecter les hôpitaux. Toutefois, au moment du projet, Santé Canada n’avait pas encore pleinement analysé les répercussions de la déclaration obligatoire sur la charge de travail du Programme.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient poursuivre leurs efforts pour adapter le PIM au moyen d’initiatives nationales et internationales afin de relever les défis provenant du cycle de vie en évolution rapide des instruments médicaux et de l’utilisation des nouvelles technologies.
Bien que Santé Canada ait fait des progrès considérables pour adapter son cadre réglementaire, il reste d’autres défis à relever pour que le PIM demeure efficace à l’avenir. Par exemple, au moment de la mission, il n’existait pas de mécanisme pour traiter le problème croissant des instruments médicaux offerts par abonnement ou par service (p. ex. les outils de diagnostic en ligne), ou pour suivre les personnes utilisant des instruments implantés afin qu’elles puissent être informées directement par Santé Canada en cas d’incident.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient examiner et documenter la manière d’intégrer davantage l’analyse comparative fondée sur le sexe et le genre plus (ACSG+) dans le cycle de vie des instruments médicaux, notamment en fournissant des conseils aux demandeurs et en sensibilisant le personnel du PIM.
Il est prouvé que les hommes et les femmes sont affectés différemment par les instruments médicaux en raison des disparités entre les sexes et les genres. Bien que Santé Canada ait progressé dans ses efforts pour intégrer les considérations liées au sexe et au genre dans le PIM, il y a eu d’autres occasions d’intégrer davantage ces considérations dans toutes les activités du programme, le cas échéant.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient maintenir ou intensifier leurs efforts de communication et de dialogue avec les intervenants de l’industrie, les professionnels et les organismes de soins de santé, et les groupes de patients. Il est également recommandé que la Direction générale des produits de santé et des aliments améliore l’actualité et l’accessibilité renseignements de programme accessibles en ligne. Cela comprend assurer que les des documents d’orientation sont examinés et mis à jour, au besoin, pour toutes les activités du PIM, y compris sur le site Web du gouvernement du Canada.
Les efforts de Santé Canada pour accroître la communication et la mobilisation avec les intervenants ont été bien accueillis par les représentants de l’industrie. Cependant, Santé Canada ne constituait pas une source d’information incontournable pour les représentants des soins de santé, les groupes de patients et la population en général. Il était également nécessaire d’améliorer l’actualité et l’accessibilité de l’information en ligne. En particulier, des documents d’orientation obsolètes peuvent avoir contribué à des inexactitudes dans les demandes d’homologation de instruments médicaux. Les informateurs clés de l’industrie ont également mentionné avoir eu des difficultés à trouver les renseignements et les conseils nécessaires pour soutenir l’élaboration de leurs demandes.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient s’assurer que les rôles et responsabilités au niveau opérationnel sont clairement définis et répartis, et que les risques liés au Programme sont déterminés et gérés.
Les rôles et les responsabilités du personnel opérationnel n’ont pas été clairement définis pour les trois fonctions du PIM. Le PIM ne disposait pas non plus d’un cadre exhaustif définissant les rôles et les responsabilités ni d’une stratégie intégrée de gestion des risques et de partage de l’information. Les défis liés aux rôles et aux responsabilités peuvent être relevés, en partie, avec la mise en place de la nouvelle Direction des instruments médicaux annoncée à la fin 2019, qui intégrera la fonction de précommercialisation et la fonction responsable de l’évaluation des incidents liés aux instruments médicaux. Toutefois, la fonction de conformité et d’application de la loi et le Bureau de la surveillance des produits de santé et de l’épidémiologie resteront au sein de leurs directions d’origine.
-
La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient veiller à ce qu’une approche de gestion des données permette de surveiller les instruments médicaux pendant tout leur cycle de vie.
L’information du PIM était fragmentée dans plusieurs dépôts. Cette situation a nui à l’efficacité et à la capacité du Programme à adopter une approche fondée sur le cycle de vie pour la réglementation et la surveillance des instruments médicaux.
| Recommandations | Réponse de la direction et mesures prévues | Produit livrable | Date d’achèvement | Responsabilité |
|---|---|---|---|---|
Recommandation 1 La Direction générale des produits de santé et des aliments devrait combler les lacunes actuelles en matière de ressources dans les fonctions suivant la mise en marché auxquelles il incombe le traitement des rapports d’incidents et l’évaluation des incidents. La Direction générale des opérations réglementaires et de l’application de la loi devrait examiner l’incidence qui suit sur les besoins en matière de ressources de la fonction de conformité et d’application de la loi. De plus, les deux directions générales devraient s’assurer que le PIM dispose des capacités et des outils nécessaires pour faire face à l’obligation de déclaration par les hôpitaux introduite en décembre 2019, et à l’application connexe des pratiques de vigilance des hôpitaux. |
La direction accepte cette recommandation * * Tous les délais peuvent être modifiés en raison de la pandémie de la COVID-19. |
|||
Le signalisation des incidents concernant un instrument médical est un élément de base de la capacité de Santé Canada à surveiller, identifier, évaluer et traiter les problèmes de sécurité identifiés par les Canadiens, les hôpitaux, les médecins et les fabricants. Des investissements ont été faits pour répondre aux obligations de déclaration par les hôpitaux. Nous allons continuer à évaluer le retour sur ces investissements ainsi que les lacunes à combler dans le Programme des instruments médicaux (PIM), après la mise en marché, y compris les fonctions de la prise de données et de la gestion des données, les évaluations scientifiques, le codage, les évaluations et la gestion des cas, afin d’assurer qu’ils avaient tous les deux la capacité et les outils de livrer les fonctions programmatiques. |
1.1 La DGPSA et la DGORAL vont continuer à évaluer la capacité et les outils que la fonction après la mise en marché exige pour livrer effectivement les activités de programme relevant de son mandat. Exemples des accomplissements à date :
|
|||
a) Un cadre de référence pour l’équipe qui mène ce projet sera élaboré. |
octobre 2020 COMPLÉTÉ |
Directeurs exécutifs, bureaux de AAE, PAS, SP, DIM (DGSPA) |
||
b) UN plan de mise en œuvre sera élaboré, y compris un plan pour mettre les documents d’orientation à jour. |
décembre 2020 |
|||
c) Une analyse des lacunes dans les capacités sera complétée. |
mars 2021 |
|||
d) Un rapport d’étape intérimaire sera achevé. |
décembre 2021 |
|||
e) Un rapport définitif sera achevé. |
juin 2022 |
|||
Recommandation 2 La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient poursuivre leurs efforts pour adapter le PIM au moyen d’initiatives nationales et internationales afin de relever les défis provenant du cycle de vie en évolution rapide des instruments médicaux et de l’utilisation des nouvelles technologies. |
La direction accepte cette recommandation |
|||
Santé Canada collabore avec des partenaires nationaux et internationaux afin d’effectuer les mesures suivantes : (a) évaluer, gérer et communiquer les risques et les avantages pour la santé et la sécurité liés aux défis provenant du cycle de vie en évolution rapide des instruments médicaux et de l’utilisation des nouvelles technologies. (b) optimiser l’utilisation de données concrètes dans l’ensemble du cycle de vie dans le but d’améliorer l’accès aux instruments médicaux, ainsi que leur utilisation appropriée. Ces relations sont essentielles dans le développement et la mise en œuvre d’approches novatrices qui vont améliorer l’efficacité et la durabilité des systèmes canadiens de soins de santé. Le Programme va continuer à tirer profit et bâtir d’une fondation solide pour des partenariats canadiens et internationaux qui visent à traiter des défis et enjeux émergents, tels que le Forum international des organismes de réglementation des instruments médicaux (IMDRF), Programme unique d’audit des instruments médicaux (PUAIM) et le Conseil de coopération en matière de réglementation (CCMR). Le PIM sera toujours adapté pour répondre aux enjeux et technologies émergents afin que la santé et la sécurité des Canadiens soient protégées. |
2.1 La DGPSA et la DGORAL vont continuer à renforcer la collaboration nationale et internationale et à produire des rapports d’étapes semestriels. Voici quelques exemples des accomplissements dans le contexte de la pandémie :
|
Semestriel (le dernier rapport sera présenté en juin 2021) |
Directeur, Bureau des politiques et des programmes internationaux, DIM (DGPSA) |
|
2.2 La DGPSA et le DGORAL vont continuer à mettre en œuvre les engagements provenant de la Feuille de route du secteur de la santé et des sciences biologiques qui concernent les instruments médicaux, y compris les éléments ci-dessous. |
||||
2.2.1 Les voies thérapeutiques de pointe (VTP), qui sont présentement en train d’être mises en œuvre, afin d’adapter à l’évolution rapide du cycle de vis des instruments médicaux et, en particulier, élaborer un cadre réglementaire efficace pour les instruments médicaux liés à l’IA. À noter – la responsabilité globale du projet VPT ne relève pas du Programme des instruments médicaux. |
||||
a) Des rapports d’étape semestriels sur les contributions du PIM seront présentés |
Semestriel (le dernier rapport sera présenté en juin 2021) |
Directeur exécutif, Bureau de l’évaluation, DIM (DGPSA) |
||
b) La Voie sera mise en œuvre – le PIM va contribuer à la documentation d’orientation pour la Voie TP, y compris l’utilisation d’une réglementation évolutive. |
septembre 2021 |
Directrice, Politiques de conformité et affaires réglementaires (DGORAL) |
||
2.2.2 Modernisation des essais cliniques (MEC), qui va comprendre de nouvelles provisions pour faciliter la recherche au Canada À noter - la responsabilité globale du projet MEC ne relève pas duy Programme des instruments médicaux. Exemple d’un accomplissement à ce jour : En octobre 2020, le programme d’inspection des essais cliniques a effectué la première inspection d’un essai clinique d’un instrument médical dans le cadre d’un programme pilote. Deux inspections de plus sont prévues à être effectués cette année. L’objectif de ces pilotes liées aux inspections est de recueillir des données et des renseignements, ce qui jettera les bases pour la mise en œuvre d’une approche officielle pour ces inspections. |
||||
a) Des rapports d’étape intérimaires seront présentés de façon semestrielle. |
Semestriel (le dernier rapport sera présenté en juin 2021) |
Directeur, Bureau des politiques et des programmes internationaux, DIM (DGPSA) |
||
b) Des règlements nouveaux ou révisés seront élaborés et approuvés. |
mars 2022 |
|||
2.2.3 Règlements agiles pour les licences des instruments médicaux et les établissements, qui vont comprendre une analyse du régime de classification des instruments des classes plus bas, les modalités et l’utilisation des examens et décisions provenant de l’extérieur du pays. |
||||
a) Des rapports d’étape intérimaires seront présentés de façon semestrielle. |
Semestriel (le dernier rapport sera présenté en décembre 2022) |
Directeur, Bureau des politiques et des programmes internationaux, DIM (DGPSA) |
||
b) Des règlements nouveaux ou révisés seront élaborés et approuvés. |
mars 2023 |
|||
Recommandation 3 La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient examiner et documenter la manière d’intégrer davantage l’analyse comparative fondée sur le sexe et le genre plus (ACSG+) dans le cycle de vie des instruments médicaux, notamment en fournissant des conseils aux demandeurs et en sensibilisant le personnel du PIM. |
La direction accepte cette recommandation |
|||
Le Ministère est déterminé à assurer l’intégration d’une perspective basée sur le sexe et le genre dans l’élaboration et la prestation de programmes, y compris le PIM. Dans le contexte du Plan d’action relatif aux instruments médicaux (PARIM), le Ministère a créé un Comité consultatif scientifique sur les produits de santé destinés aux femmes afin d’aider à identifier des solutions concrètes en matière de l’ACSG+ qui s’appliquent aux activités du Programme. Le Ministère va continuer à collaborer avec ce Comité afin d’aider à éclairer les considérations de l’ACSG+ dans les activités programmatiques. |
3.1 La DGPSA et la DGORAL vont intégrer l’ACSG+ dans leurs programmes de formation. |
|||
a) La DGORAL va intégrer une perspective de l’ACSG+
dans le cours |
juin 2021 |
Directeur, Développement des programmes et formation (DGORAL) |
||
b) Afin d’accroître la sensibilisation à l’ACSG+ et son importance au long du cycle de vie de l’instrument, la DGPSA exigera que les membres de son personnel (p. ex., les évaluateurs et les analystes des politiques) complètent une formation obligatoire sur l’ACSG+, soit en ligne ou par moyen de présentations en personne. Un rapport touchant la complétion sera fourni. |
juin 2021 |
Directeur, Bureau de la planification et des opérations, DIM (DGPSA) |
||
3.2 La DGPSA et la DGORAL vont appuyer le chercheur choisi au sein de la subvention des IRSC « Partenariats entre la recherche et les politiques de santé en matière d’ACSG + ». Ce chercheur va effectuer plusieurs analyses afin d’évaluer la façon dont l’ACSG est intégrée au long du cycle de vie de l’instrument médical. |
||||
a) La DGORAL va fournir une analyse pour déterminer si des facteurs de risque liés à l’ACSG + sont présents dans le contexte des activités d’inspection et évaluer le besoin d’élaborer des stratégies d’atténuation et de surveillance. |
juin 2021 |
Directrice, Politiques de conformité et affaires réglementaires (DGORAL) |
||
b) La DGPSA va déterminer les recommandations qui seront mises en œuvre et va élaborer un plan de mise en œuvre. |
décembre 2021 |
Directeur, Bureau des politiques et des programmes internationaux, DIM (DGPSA) |
||
3.3 Comme recommandé par les membres du comité CCS-PSDF, le Programme va traiter de la question de la façon dont les fabricants devront prendre les facteurs de l’ACSG + en considération quand ils présentent les preuves pour une demande de permis pour un instrument médical. En particulier, les documents d’orientation dans les sous-sections qui suivent vont refléter des considérations de l’ACSG+. |
||||
a) Document d’orientation sur les Exigences en matière de preuves cliniques |
mars 2021 |
Directeur, Bureau des politiques et des programmes internationaux, DIM (DGPSA) |
||
b) Documents d’orientation sur l’intelligence artificielle |
décembre 2021 |
|||
Recommandation 4 La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient maintenir ou intensifier leurs efforts de communication et de dialogue avec les intervenants de l’industrie, les professionnels et les organismes de soins de santé, et les groupes de patients. Il est également recommandé que la Direction générale des produits de santé et des aliments améliore l’actualité et l’accessibilité renseignements de programme accessibles en ligne. Cela comprend assurer que les des documents d’orientation sont examinés et mis à jour, au besoin, pour toutes les activités du PIM, y compris sur le site Web du gouvernement du Canada. |
La direction accepte cette recommandation |
|||
Les Canadiens se fient aux instruments médicaux afin de maintenir et améliorer leur santé et bien-être. Comme a déclaré le Consortium international des journalistes, suite au lancement du Plan d’action sur les instruments médicaux, le Canada détient un des meilleurs systèmes réglementaires au monde quant aux instruments médicaux, avec des exigences qui sont parmi les plus rigoureux. Le Ministère a pris plusieurs étapes dans le domaine des communications et de la mobilisation, y compris :
Le 16 décembre 2019, Santé Canada est devenu le premier organe de réglementation au monde à émettre des règlements qui exigent que les hôpitaux de signaler tous les effets indésirables des médicaments (EIM) et les incidents concernant les instruments médicaux (ICIM). Santé Canada a mené un important effort national de sensibilisation au long de 2019 lié aux nouvelles exigences. Ceci a impliqué 250 événements éducatifs avec des professionnels de santé publique, y compris des présentations en personne et 12 webinaires dans les deux langues officielles destinés aux professionnels de soins de santé dans les milieux hospitaliers et les administrateurs. Santé Canada a publié un document d’orientation, de nouveaux formulaires de déclaration, une pour faire des déclarations sur le site Web et des modules pour appuyer l’interprétation et la compréhension des hôpitaux quant aux nouvelles exigences. En raison de cet effort de mobilisation avec les intervenants et plus de 800 hôpitaux partout au Canada, Santé Canada a vu que le nombre de déclarations provenant des hôpitaux a augmenté d’un mois à l’autre. Le Ministère a aussi élaboré et mis en œuvre une stratégie pour les clients et a consulté avec les professionnels des soins de santé et les groupes de patients afin d’optimiser les communications, y compris les sites Web et les outils en ligne de déclaration des incidents. La rétroaction reçue va informer les efforts continus du Ministère de communiquer de façon efficace et de mobiliser sur les activités de programme. |
4.1 Le Ministère a reçu les résultats du projet d’Optimisation du Web pour les instruments médicaux du SCT, y compris un prototype de projet. Voici quelques exemples d’accomplissements dans le contexte de la pandémie : Les modèles expérimentaux du prototype ont déjà été utilisés pour créer une page d’accueil pour les renseignements sur les instruments médicaux liés à la COVID-19. En plus de documents d’orientation et d’avis, des renseignements clés disponibles en ligne pour les Canadiens comprennent les suivants:
Les mises à jour additionnelles planifiées comprennent la publication d’un guide intelligent pour les demandes liées à la COVID-19, un examen de l’architecture des médicaments et des produits de santé, la création de Pages de lancement d’un service pour les instruments médicaux homologués en vigueur (MDALL et MDEL). La DGPSA et la DGORAL vont continuer à mettre en œuvre les changements et engager avec les intervenants pour les informer des changements. Des rapports d’étape intérimaires seront présentés chaque trimestre. |
trimestriel ((e dernier rapport sera présenté en décembre 2022) |
Directeur, Bureau de la planification et des opérations, DIM (DGPSA) |
|
4.2.1 La DGPSA et la DGORAL vont évaluer le nombre total de documents d’orientation affichés dans son site Web afin de déterminer le nombre de documents qui exigent des mises à jour, et ceux qui ont été complétés. |
mars 2021 |
Directeur, Bureau des politiques et des programmes internationaux, DIM (DGPSA) |
||
4.2.2 Selon l’évaluation, la DGPSA et la DGORAL vont élaborer un plan pour mettre à jour les documents pour être approuvées par le groupe trilatéral du PIM. |
septembre 2021 |
|||
4.3 La DGPSA et la DGORAL vont continuer à engager, créer et mettre en œuvre un plan annuel de mobilisation des intervenants et de promotion de la conformité, y compris les réunions bilatérales avec les associations industrielles. Voici quelques exemples des accomplissements dans le contexte de la pandémie :
|
||||
4.3.1 Le plan du DIM de mobilisation des intervenants sera mis à jour et approuvé par le DIM-CSM. |
décembre 2020 |
Directeur, Bureau de la planification et des opérations, DIM (DGPSA) |
||
4.3.2 Le plan du PCIM de 2021 de mobilisation des intervenants sera mis à jour et approuvé par le PCIM-CSM. |
décembre 2020 |
Director, Medical Devices Compliance , MDCCD (ROEB) |
||
4.3.3 Un rapport sur les activités de mobilisation sera produit chaque trimestre. |
trimestriel ((e dernier rapport sera présenté en septembre 2021 |
Directeur, Bureau de la planification et des opérations, DIM (DGPSA) |
||
4.4 La DGPSA et la DGORAL vont continuer à engager avec les intervenants par moyen de comités consultatifs scientifiques et des patients (engagement avec les intervenants) :
Des comptes rendus seront fournis. |
derniers comptes rendus seront fournis d’ici septembre 2021 |
Directeur, Bureau des politiques et des programmes internationaux, DIM (DGPSA) |
||
Recommandation 5 La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient s’assurer que les rôles et responsabilités au niveau opérationnel sont clairement définis et répartis, et que les risques liés au Programme sont déterminés et gérés. |
La direction accepte cette recommandation |
|||
La DGPSA et la DGORAL vont continuer à améliorer les cadres opérationnels afin de préciser davantage les rôles et les responsabilités au niveau opérationnel du Programme. La DGPSA a traité des divers défis et possibilités en créant une nouvelle Direction des instruments médicaux (PIM) en janvier 2020. Cette nouvelle direction prend une approche fixée sur le cycle de vie pour la réglementation des instruments médicaux en fusionnant des fonctions particuliers de précommercialisation avec les fonctions après la mise en marché. Ceci a créé une organisation dédiée à la réglementation des instruments médicaux et facilite l’intégration de la gestion des risques. |
5.1 La DGPSA et la DGORAL vont collaborer afin de mettre à jour les documents actuels interministériels de RACI (Responsable, Agent comptable, Consulté, Informé) et des PE. Les deux directions générales vont collaborer afin de définir les rôles et les responsabilités au niveau opérationnel au sein du Programme des instruments médicaux et de mettre à jour les PON au besoin. Les documents seront approuvés par le groupe trilatéral du PIM. |
|||
a) Une matrice mise à jour de RACI sera élaborée. |
juin 2021 |
Directeur, Bureau de la planification et des opérations, DIM (DGPSA) |
||
b) Un PE mis à jour entre la DGPSA et la DGORAL sera élaboré. |
décembre 2021 |
|||
c) Les PON connexes seront mises à jour. |
juin 2022 |
|||
Recommandation 6 La Direction générale des produits de santé et des aliments et la Direction générale des opérations réglementaires et de l’application de la loi devraient veiller à ce qu’une approche de gestion des données permette de surveiller les instruments médicaux pendant tout leur cycle de vie. |
La direction accepte cette recommandation |
|||
Santé Canada travaille pour renforcer davantage l’approche à la gestion des données, qui va soutenir la surveillance des instruments médicaux pendant leur cycle de vie. Cette approche à la gestion des données comprend des composants différents, y compris : plans de la TI, les principes de transparence, les normes de service et la gestion des documents; le tout pour répondre aux attentes des clients. Les investissements qui ont été faits dans les projets de TI comme partie de la stratégie de la TI de la direction générale, y compris un système nouveau et moderne de déclaration d’incidents des instruments médicaux. Ces investissements sont axés sur le renforcement de l’efficacité, mais vont nécessiter la mobilisation continue et la stabilisation des systèmes afin d’atteindre cet objectif. De plus, la DGPSA et la DGORAL vont collaborer pour l’établissement d’une approche pleinement intégrée et coordonnée, au long du cycle de vie de l’instrument médical, qui ajoute aux investissements de la direction générale dans la TI. |
6.1 Compte tenu la création de la nouvelle Direction des instruments médicaux (DIM), la DGPSA et la DGORAL vont tirer profit des processus actuels et améliorer ceux-ci afin d’assurer l’intégration et coordination complète des données sur les instruments médicaux au long du cycle de vie. |
|||
a) L’automatisation robotisée des processus mise en œuvre afin de rationalise les processus, y compris le renouvellement des permis d’instruments médicaux. |
août 2021 |
Directeur, Bureau de la planification et des opérations, DIM (DGPSA) |
||
b) La PIM va compléter la conversion d’Impromptu 7.5 à Cognos 11 afin d’harmoniser les déclarations au long du cycle de vie de l’instrument. Voici un exemple d’un accomplissement jusqu’à maintenant :
|
mars 2022 |
|||
Annexe A – Portée et méthodologie du projet conjoint
Ce projet conjoint a été réalisé par le Bureau de l’audit et de l’évaluation de Santé Canada. L’objectif consistait à examiner les activités du PIM d’avril 2013 à août 2019, et le projet a permis de réaliser une analyse comparative fondée sur le sexe et le genre plus (ACSG+) lorsque cela était pertinent. Le contrôle des transactions du PIM par la Division de l’audit interne a porté sur la période allant d’avril 2018 à septembre 2019.
Le modèle de recouvrement des coûts du PIM dépassait la portée du projet, car un modèle révisé sera mis en œuvre le 1er avril 2020. La Division de l’audit interne n’a pas non plus examiné les activités d’inspection des licences d’établissement pour les instruments médicaux relevant de la Direction générale des opérations réglementaires et de l’application de la loi, car la Division a récemment terminé un audit des activités d’inspection. Toutefois, ces activités ont été examinées dans le cadre de l’évaluation afin de produire une évaluation exhaustive de toutes les activités du PIM.
Il s’agissait du premier audit interne du PIM, qui a été réalisé conformément aux Normes internationales pour la pratique professionnelle de l’audit interne. Le PIM a été évalué précédemment en 2013-2014.
Le volet audit du projet a examiné les critères suivants :
Gouvernance, contrôles et risques
- Les rôles et les responsabilités liés au PIM sont établis et suivis.
- Les contrôles ministériels du processus de précommercialisation sont adéquats.
- Les contrôles ministériels après la mise en marché sont en place pour les incidents liés aux instruments médicaux afin que les risques élevés pour la santé des Canadiens soient priorisés et atténués.
- L’analyse comparative fondée sur le sexe et le genre est prise en compte et mise en œuvre dans la gestion du PIM.
- Le Ministère suit les progrès réalisés dans le cadre des engagements pris dans le Plan d’action sur les instruments médicaux de Santé Canada.
Le volet d’évaluation du projet portait sur les questions et les thèmes suivants :
Répercussions du Programme et portée des activités
- Dans quelle mesure le PIM est-il efficace pour aider les Canadiens à prendre des décisions éclairées au sujet des instruments médicaux et pour assurer l’accès à des instruments médicaux sûrs, efficaces et de qualité?
- La portée de ces activités correspond-elle au mandat de Santé Canada?
- Comment ses avantages pour la société se comparent-ils à ses coûts?
Adaptation au changement
- Le PIM peut-il répondre tant à la demande actuelle que nouvelle provenant d’un environnement en constante évolution et de plus en plus complexe?
- Santé Canada a-t-il besoin d’autres outils réglementaires pour s’adapter à un environnement en évolution?
Efficacité de l’organisation du Programme
- Dans quelle mesure la structure organisationnelle, les activités, les processus et l’affectation des ressources aident-ils le PIM à fonctionner efficacement?
- Quelles sont les solutions de rechange au modèle organisationnel actuel?
- Le PIM optimise-t-il les ressources grâce à des collaborations internationales et nationales?
Méthodologie
Les données nécessaires à la mission ont été recueillies à l’aide des méthodes suivantes :
ENTREVUES
Avec 73 informateurs clés, dont :
- 26 membres du personnel de Santé Canada participant au PIM;
- 8 experts, universitaires et membres du comité consultatif scientifique;
- 14 professionnels de la santé ou associations de santé;
- 10 membres de l’industrie;
- 11 personnes, ou membres de la famille, utilisant un instrument médical;
- 4 représentants d’autres pays.
EXAMEN DES DOCUMENTS
Inclut les dossiers de programmes, les politiques, les règlements, les lignes directrices, les comptes rendus de décisions, etc. Les résultats de l’enquête sur le cadre réglementaire de transparence et d’ouverture et de l’enquête sur la transparence de la Direction générale des produits de santé et des aliments ont également été inclus dans l’examen des documents.
ANALYSE COMPARATIVE
Examen de la structure organisationnelle des programmes des instruments médicaux de six autres organismes de réglementation (p. ex., la FDA des États-Unis, l’Agence nationale de sécurité du médicament et des produits de santé) et interrogation des représentants de la FDA, de l’autorité irlandaise de réglementation des produits de santé et de la Therapeutic Goods Administration d’Australie.
ANALYSE DES DONNÉES ET CONTRÔLE DES TRANSACTIONS
Examen d’un échantillon d’opérations de chacune des fonctions du PIM, ainsi que les données sur le rendement recueillies par le Programme.
EXAMEN DES DONNÉES FINANCIÈRES
Examen des données financières pour chaque fonction du PIM et pour l’ensemble du Programme.
ÉTUDES DE CAS
Examen de deux instruments médicaux afin de mieux comprendre le processus d’approbation, de surveillance et de rappel des instruments médicaux : 1) le dispositif de contraception permanente Essure et 2) la pompe à insuline Paradigm. L’équipe d’évaluation s’est aussi penchée sur l’Examen réglementaire des médicaments et des instruments – Technologies numériques de la santé dans le but d’évaluer sa première incidence sur le PIM.
ANALYSE DOCUMENTAIRE
Examen de la littérature concernant la communication des risques, l’évolution du secteur des instruments médicaux au Canada et la mobilisation des intervenants.
Restrictions et stratégies d’atténuation
Le tableau ci-dessous décrit les limites auxquelles se sont heurtés les évaluateurs pendant la mise en œuvre des méthodes de collecte de données choisies aux fins de la présente mission. On y précise également les stratégies d’atténuation mises en place afin que les constatations tirées de la mission puissent être utilisées en toute confiance pour orienter la planification des programmes et la prise de décisions connexes.
| Restriction | Incidence | Stratégie d’atténuation |
|---|---|---|
Le Cadre réglementaire pour la transparence et l’ouverture (CRTO) de 2016 ne contenait qu’un seul point de données concernant les efforts déployés par Santé Canada pour améliorer l’accès à des renseignements opportuns, utiles et pertinents en matière de santé et d’innocuité. De plus, le taux de réponse à l’enquête a été très faible. |
Incapacité à comparer de manière cohérente les perceptions passées et présentes des parties prenantes et incapacité à généraliser ces perceptions à l’ensemble des parties prenantes du programme ou au public canadien. |
Quarante-sept entrevues avec des informateurs clés de diverses parties prenantes du PIM à l’aide de questions validées par le programme pour s’assurer que les entrevues répondent à leurs besoins d’information. Ces entrevues ont permis de valider certaines des conclusions de l’enquête de 2016 sur le CRTO et de déterminer si les perceptions ont évolué au fil du temps. |
Les entrevues avec des informateurs clés externes n’ont été possibles qu’avec un petit nombre de personnes issues de différents groupes d’intervenants. |
Les résultats des entrevues ne sont pas généralisables à l’ensemble des groupes d’intervenants ou au public canadien. |
Les données issues des entrevues avec les informateurs clés ont été validées avec d’autres sources de données (p. ex., la documentation ou les données du Programme) dans la mesure du possible. |
Les renseignements sur les avantages sociaux et financiers, ainsi que sur les coûts liés aux instruments médicaux n’étaient souvent pas accessibles au public. |
Il n’a pas été possible d’établir un bilan des répercussions sociales et financières des activités du PIM en raison du manque de données disponibles. |
Des entrevues avec des informateurs clés et des études de cas ont été utilisées pour recueillir les points de vue des expériences vécues sur les effets positifs et négatifs des instruments médicaux, ainsi que les effets perçus sur le système de soins de santé. Quelques banques de données et documents ont également été utilisés pour tirer des exemples des effets sanitaires et financiers de certains instruments médicaux sur le système de santé canadien. |
Annexe B - Résumé de l’audit interne
OBJECTIF DE L’AUDIT : Fournir l’assurance que le Programme dispose de contrôles suffisants pour garantir que son mandat est rempli, ainsi que pour évaluer l’intégration de l’analyse comparative fondée sur le sexe et le genre (ACSG+) dans le cycle de vie du programme.
PORTÉE DE LA MISSION : La mission portera sur les activités avant et après commercialisation pour la période d’avril 2013 à août 2019 et appliquera l’ACSG+ au besoin. Le contrôle des transactions commerciales portera sur l’exercice financier 2018-2019 et au-delà.
CONCLUSION DE L’AUDIT : Dans l’ensemble, nous avons constaté que le Programme avait mis en place des contrôles suffisants dans la plupart des domaines pour garantir que son mandat, qui consiste à s’assurer que les instruments médicaux approuvés pour le marché canadien sont sûrs, efficaces et de grande qualité, était rempli. Bien que des efforts aient été déployés pour démontrer la prise en compte de l’ACSG+, le Programme bénéficiera de la poursuite de ses travaux visant à intégrer l’ACSG+ dans le cycle de vie des instruments médicaux.
Les contrôles étaient généralement efficaces pour les activités parallèles de précommercialisation et une surveillance active après la mise en marché avait lieu. Nous avons constaté que les rôles et responsabilités définis n’étaient pas clairs au niveau opérationnel, et que la capacité insuffisante pour les activités après la mise en marché, les documents d’orientation obsolètes et le manque de méthodologie d’analyse des données horizontales nuisaient à l’efficacité globale du Programme et généraient des arriérés.
| CRITÈRE | CONCLUSION | RISQUE | RISQUE RÉSIDUEL | RECOMMANDATION |
|---|---|---|---|---|
1. Les rôles et responsabilités liés au Programme des instruments médicaux ont été établis, suivis et alignés sur les exigences opérationnelles. |
Nous avons constaté que des comités de la haute direction étaient en place et que les rôles et responsabilités généraux du Programme étaient définis; mais il n’y avait pas de cadre clair pour les rôles et responsabilités qui permettrait d’apporter plus de précision au niveau opérationnel entre les directions. Il y avait également un manque d’approche intégrée de gestion des risques dans l’ensemble du Programme. |
2 Risque mineur |
Le manque de clarté des rôles et des responsabilités et l’absence d’une stratégie intégrée de gestion des risques nuisent à l’efficacité du Programme. Avec les changements rapides dans cet environnement, l’incidence sur l’efficacité du Programme pourrait s’accroître à l’avenir. |
Recommandation 5 |
2. Les contrôles ministériels des processus préalables à la mise en marché étaient adéquats. |
Nous avons constaté que les processus et les contrôles étaient adéquats. Des documents d’orientation étaient disponibles pour guider le personnel du Programme dans la réception, l’examen, l’évaluation et l’approbation des demandes d’homologation des instruments médicaux et des licences d’établissement. Toutefois, de nombreux documents d’orientation internes et externes étaient largement obsolètes et ne reflétaient pas les processus opérationnels actuels. Une intégration plus poussée de l’analyse des données pourrait renforcer les contrôles du Programme d’accès spécial. |
2 Risque mineur |
Une documentation obsolète et des difficultés d’accès à l’information pour les intervenants peuvent entraîner des inexactitudes dans les demandes d’homologation et, par conséquent, influer sur la rapidité de la prise des décisions d’homologation. Des documents d’orientation obsolètes peuvent induire en erreur le personnel du Programme lors de l’examen des demandes, en particulier les nouveaux employés qui ne connaissent pas le processus. |
Recommandation 4 |
| 2 Risque mineur |
L’absence d’utilisation continue et cohérente de l’analyse des données et du suivi des demandes au titre du Programme d’accès spécial empêche l’identification et le suivi des demandes à haut risque, tels que les instruments les plus fréquemment demandés, les demandeurs ou entreprises les plus fréquents ou les combinaisons uniques les plus fréquentes d’instrument et de demandeur. Cela peut créer un risque de contournement de la réglementation sur les instruments médicaux. |
Recommandation 6 |
||
3. Des contrôles ministériels après la mise en marché ont été mis en place pour les incidents liés aux instruments médicaux afin de prioriser et d’atténuer les risques élevés pour la santé des Canadiens. |
Nous avons constaté que le programme avait établi des contrôles après la mise en marché adéquats pour surveiller les instruments médicaux après leur homologation, pour traiter les incidents en fonction du niveau de risque et pour assurer leur innocuité et leur efficacité continues. Toutefois, des retards dans la saisie des données dépassant les normes de service ont été attribués à une capacité insuffisante. Nous avons constaté que les données au sein du programme étaient fragmentées en plusieurs bases de données, dépôts de documents et fiches de suivi, ce qui limitait la capacité des directions à travailler en collaboration et posait un risque pour l’intégrité des données. Une proportion importante des documents d’orientation n’était pas à jour. |
3 Risque modéré |
La fragmentation des données entre les directions constitue un obstacle à une communication, à une collaboration et à un accès à l’information efficaces entre les équipes. La fragmentation a une incidence sur le Programme dans son ensemble. |
Recommandation 6 |
| 3 Risque modéré |
L’absence d’analyse de la charge de travail, d’analyse des besoins en personnel et de plan de dotation en personnel nuit à la capacité du Programme à remplir son mandat. |
Recommandation 1 |
||
| 2 Risque mineur |
Des documents d’orientation obsolètes nuisent à l’efficacité des opérations après la mise en marché, car ils ne reflètent pas les changements apportés au Programme, notamment l’établissement de la DGORAL, la mise en œuvre de DocuBridge et la modernisation du Système des instruments médicaux. |
Recommandation 4 |
||
4. L’analyse comparative fondée sur le sexe et le genre est prise en compte et mise en œuvre dans la gestion des instruments médicaux. |
Nous avons constaté que le Programme a pris en compte et intégré l’analyse comparative fondée sur le sexe et le genre plus dans diverses activités. Cependant, elle n’a pas été mise en œuvre de manière cohérente pour que la direction du Programme permette l’intégration de cette analyse dans le cycle de vie des instruments médicaux. |
2 Risque mineur |
Sans la mise en œuvre d’une analyse comparative fondée sur le sexe et le genre plus cohérente dans le cycle de vie des instruments médicaux, il est difficile d’évaluer les facteurs de risque et la façon dont divers groupes de femmes, d’hommes et de personnes non binaires sont touchés par une variété d’instruments médicaux sur le marché canadien. |
Recommandation 3 |
Annexe C – Résumé des résultats de l’évaluation
| Questions d’évaluation | Indicateurs | Résumé des constatations | Recommandations |
|---|---|---|---|
Répercussions du Programme et portée des activités |
|||
Dans quelle mesure le PIM est-il efficace pour aider les Canadiens à prendre des décisions éclairées au sujet des instruments médicaux et pour assurer l’accès à des instruments médicaux sûr, efficace et de qualité? |
|
Santé Canada a généralement réussi à assurer la sécurité, l’efficacité et la qualité des instruments médicaux sur le marché canadien. Le Ministère était perçu comme un organisme de réglementation de confiance et les données disponibles suggèrent que la sécurité des instruments médicaux au Canada est comparable à celle d’autres pays. Cependant, les retards dans le traitement des rapports d’incidents liés aux instruments médicaux ont influé sur la capacité du Programme à détecter de tels incidents de manière proactive. Le PIM a également connu des retards dans l’évaluation des incidents liés aux instruments médicaux. Bien que ces défis représentent un risque pour le Ministère, rien n’indique qu’ils aient affecté la capacité de Santé Canada à assurer la sécurité des instruments médicaux. Le PIM a progressé dans la recherche d’un équilibre entre la sécurité et l’accès aux instruments médicaux. Bien que le Programme ait traité la majorité des demandes d’homologation d’instruments médicaux dans le respect des normes de service, la plupart de ces demandes ne répondaient pas aux exigences réglementaires lorsqu’elles ont été soumises à Santé Canada. Elles exigeaient des renseignements supplémentaires, ce qui a pu prolonger leur délai de traitement. Les raisons pour lesquelles la majorité des demandes d’homologation pour un instrument médical ne répondaient pas aux exigences du Programme lors de leur présentation initiale ne sont pas entièrement claires. Les documents d’orientation accessibles au public et décrivant ces exigences sont difficiles à trouver et parfois obsolètes. Parallèlement, le Programme a mis en œuvre diverses mesures visant à améliorer la compréhension du processus réglementaire par l’industrie (p. ex., un cours d’apprentissage en ligne). En plus des documents d’orientation destinés à l’industrie, Santé Canada diffuse différents types de produits de communication pour aider les professionnels de la santé et les Canadiens à prendre des décisions éclairées sur l’utilisation d’un instrument médical. La plupart des communications sur les risques du PIM ont été diffusées dans le respect des normes de service. Le PIM a amélioré ses communications concernant les instruments médicaux et la mobilisation des intervenants. Le Ministère a également mené à bien 100 % des consultations prévues concernant les instruments médicaux en 2017-2018 et en 2018-2019. Bien que ces efforts aient été bien accueillis par l’industrie, des améliorations sont encore nécessaires pour que Santé Canada devienne une source d’information incontournable pour les professionnels de la santé, les groupes de patients et le public canadien en général. L’accès aux produits de communication en ligne de Santé Canada est souvent difficile, et l’information disponible en ligne n’est pas toujours à jour. |
Recommandation 1 Recommandation 4 |
La portée de ces activités correspond-elle au mandat de Santé Canada? |
|
Les activités du PIM couvrent le cycle de vie d’un instrument médical, de la diffusion de conseils réglementaires aux fabricants au cours de leurs recherches et du développement de leurs produits, jusqu’à l’examen des demandes d’homologation des instruments médicaux, ainsi que la surveillance, la prévention et l’intervention pour les instruments qui sont homologués pour la vente au Canada. Dans l’ensemble, les activités du PIM ont une portée similaire à celle d’autres programmes réglementaires de Santé Canada (p. ex., les produits pharmaceutiques). Elles étaient également conformes au mandat et au rôle de gérance du Ministère, qui consiste à protéger les Canadiens et à faciliter l’accès aux produits qui sont essentiels à leur santé et à leur bien-être. |
|
Comment ses avantages pour la société se comparent-ils à ses coûts? |
|
De nombreux Canadiens dépendent d’instruments médicaux pour améliorer leur santé et leur qualité de vie, car ces instruments leur permettent de s’adonner à des activités quotidiennes. Les instruments médicaux présentent également des avantages économiques pour le système de soins de santé, car leur coût peut être inférieur à celui de la chirurgie ou d’autres formes de traitement (p. ex., le coût d’une endoprothèse à élution de médicaments par rapport à un pontage coronarien). (Remarque : les conclusions de l’évaluation dans ce domaine sont limitées en raison du manque de données.) |
|
Adaptation au changement |
|||
Le PIM peut-il répondre tant à la demande actuelle que nouvelle découlant d’un environnement en constante évolution et de plus en plus complexe? |
|
Santé Canada a fait d’importants progrès pour adapter son cadre réglementaire afin de demeurer efficace dans la réglementation de ces instruments complexes et en évolution rapide. Par exemple, le PIM a élaboré et mis en place une nouvelle Division de la santé numérique et a créé deux nouveaux comités consultatifs scientifiques, a modifié la Loi sur les aliments et drogues afin d’accroître le pouvoir des inspecteurs de la conformité et a proposé des modifications réglementaires qui renforceraient la capacité du Programme à évaluer les problèmes possibles liés aux instruments médicaux. Le PIM a également élaboré plusieurs nouveaux documents d’orientation pour l’industrie, a renforcé la mobilisation des intervenants, y compris les patients, et a progressé dans ses efforts pour intégrer les considérations liées au sexe et au genre. Comme beaucoup de ces initiatives ont été mises en œuvre récemment, il était trop tôt pour déterminer leur impact sur le programme. |
Recommandation 2 Recommandation 3 |
Santé Canada a-t-il besoin d’autres outils réglementaires pour s’adapter à un environnement en évolution? |
|
Bien que Santé Canada ait fait des progrès considérables pour adapter son cadre réglementaire, il reste d’autres défis à relever pour que le PIM continue de répondre aux exigences de l’environnement en évolution. Par exemple, il n’existe aucun mécanisme pour traiter le problème croissant des instruments médicaux offerts par abonnement ou par service (p. ex. les outils de diagnostic en ligne), ou pour suivre les personnes utilisant des instruments implantés afin qu’elles puissent être informées directement par Santé Canada en cas d’incident. Il est également possible d’intégrer davantage les considérations liées au sexe et au genre dans toutes les activités du Programme, le cas échéant. |
|
Efficacité de l’organisation du Programme |
|||
Dans quelle mesure la structure organisationnelle, les activités, les processus et l’affectation des ressources aident-ils le PIM à fonctionner efficacement? |
|
Divers facteurs ont eu un effet sur la capacité du PIM à fonctionner efficacement, notamment un manque de clarté concernant les rôles et les responsabilités des trois fonctions du PIM, ainsi que les communications au niveau opérationnel Cela a conduit parfois à une inefficacité, notamment la duplication des efforts entre les fonctions du Programme. En outre, les renseignements du PIM sont stockés dans plusieurs dépôts, ce qui signifie qu’ils doivent être consolidés à partir de diverses sources afin de comprendre l’historique complet des activités de Santé Canada en rapport avec un instrument. Il y avait également un manque d’intégration entre l’information d’audit du PUAIM et les activités de conformité et d’application de loi, ce qui a encore limité la capacité du Programme à adopter une approche fondée sur le cycle de vie. Le manque de capacité a également mis à mal la capacité de la fonction de surveillance après la mise en marché à traiter les rapports d’incidents et à effectuer des évaluations des signaux dans le respect des normes de service. La mise en place de la déclaration obligatoire des incidents liés aux instruments médicaux par environ 775 hôpitaux canadiens influera probablement davantage sur la capacité de la fonction de surveillance après la mise en marché à gérer sa charge de travail, car le nombre de rapports d’incidents soumis pourrait doubler. La déclaration obligatoire peut également toucher les ressources et les activités de la fonction de conformité et d’application de la loi, car les inspecteurs devront surveiller les éventuelles sous-déclarations et inspecter les hôpitaux. Au moment de la mission, Santé Canada n’avait pas entièrement analysé son incidence sur la charge de travail du PIM. |
Recommandation 1 Recommandation 5 Recommandation 6 |
Quelles sont les solutions de rechange au modèle organisationnel actuel? |
|
Depuis novembre 2019, une partie du PIM a été restructurée en une nouvelle Direction des instruments médicaux, qui intègre la fonction de précommercialisation et le Bureau des produits pharmaceutiques et des instruments médicaux commercialisés de la fonction de surveillance après la mise en marché. Cette nouvelle direction s’aligne sur les approches adoptées par l’Irlande et les États-Unis. Les premiers effets des nouveaux programmes d’instruments médicaux dans ces deux pays comprennent notamment l’amélioration des communications, la réduction des délais de mise en marché des instruments médicaux et une plus grande capacité à utiliser une approche fondée sur le cycle de vie. On s’attend à ce que la nouvelle Direction des instruments médicaux de Santé Canada ait un effet similaire et contribue à favoriser une approche fondée sur le cycle de vie pour la réglementation et la surveillance des instruments médicaux. |
Recommandation 5 |
Le PIM optimise-t-il les ressources grâce à des collaborations internationales et nationales? |
|
Santé Canada participe à plusieurs initiatives et partenariats internationaux, dont le Forum international des organismes de réglementation des instruments médicaux (IMDRF), afin de partager l’expertise et d’harmoniser les pratiques, les politiques et les règlements avec d’autres administrations. Il harmonise également ses exigences en matière de demandes d’homologation d’instruments médicaux avec celles d’autres pays grâce à la mise en place d’une table des matières et du PUAIM. |
Recommandation 2 |
Annexe D – Modèle logique
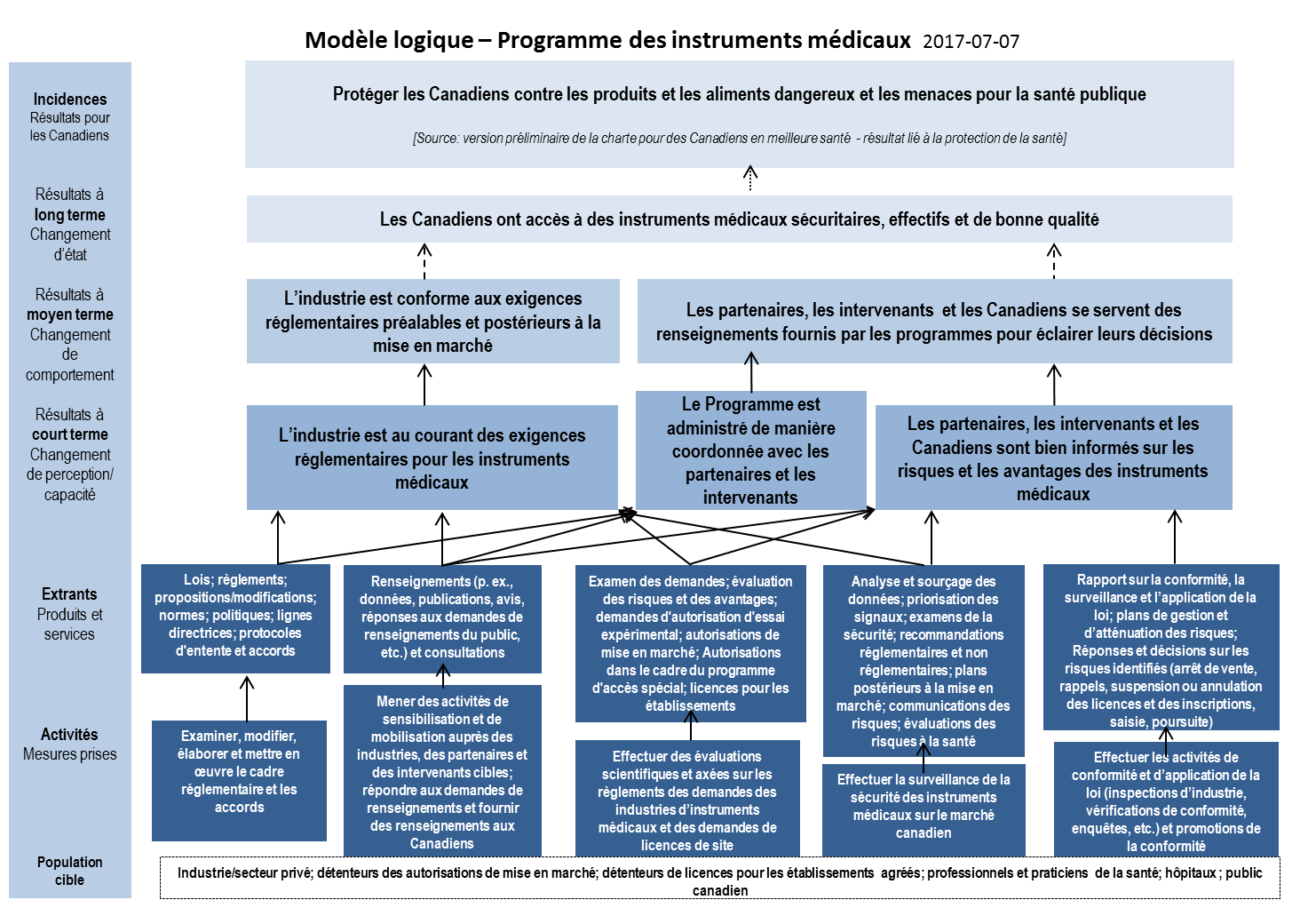
Modèle logique - Équivalent textuel
Cette figure, à lire de bas en haut, est un modèle logique qui décrit l’organisation du programme pour obtenir les résultats escomptés.
Population cible
L’industrie et le secteur privé; les titulaires d’autorisation de mise en marché; les détenteurs de licence d’un établissement enregistré; les professionnels et les praticiens de la santé; les hôpitaux et le public canadien.
Exemples de mesures prises :
- Examiner, modifier, élaborer et mettre en œuvre des cadres réglementaires et des ententes.
- Mener des activités de sensibilisation et de mobilisation avec les partenaires et les intervenants ciblés de l’industrie; répondre aux demandes de renseignements et fournir des informations aux Canadiens;
- Procéder à des évaluations scientifiques et réglementaires des demandes présentées par l’industrie des matériels médicaux et des demandes de licences d’exploitation;
- Assurer la surveillance et le suivi de la sécurité des matériels médicaux sur le marché canadien;
- Mener des activités de conformité et d’application de la loi (inspections auprès de l’industrie, vérifications de la conformité, enquêtes, etc.) et de promotion de la conformité.
Ces activités conduisent aux extrants, soit les produits et services suivants :
- Lois; propositions et modifications des règlements; normes; politiques; lignes directrices; protocoles; ententes;
- Renseignements (p. ex., données, publications, avis, réponses aux demandes de renseignements du public, etc.) et consultations;
- Examen des demandes; évaluation des risques et des avantages; autorisations d’essais expérimentaux; autorisation de mise en marché; autorisations relatives au Programme d’accès spécial; licences d’établissements;
- Analyse des données et approvisionnements; priorisation des signaux; examens de l’innocuité; recommandations réglementaires et non réglementaires; plans relatifs aux activités après la mise en marché; communication des risques; évaluations des risques pour la santé;
- Rapports de surveillance de la conformité et d’application de la loi; plan de gestion et d’atténuation des risques; interventions et décisions relatives aux risques cernés (arrêt de la vente, rappel, suspension et annulation de la licence, saisie, poursuite).
Ces extrants conduisent ensuite à des résultats à court terme, soit des changements de sensibilisation ou de capacité :
- L’industrie est informée des exigences réglementaires relatives aux matériels médicaux;
- Le programme est mis en œuvre selon une approche coordonnée avec les partenaires et les intervenants;
- Les partenaires, les intervenants et les Canadiens sont informés des risques et des avantages que comportent les matériels médicaux.
Ces résultats à court terme conduisent ensuite à des résultats à moyen terme, soit des changements de comportements :
- L’industrie se conforme aux exigences réglementaires avant et après la mise en marché;
- Les partenaires, les intervenants et les Canadiens utilisent les éclairées.
Ces résultats à moyen terme conduisent ensuite à un résultat à long terme, soit un changement d’état :
- Les Canadiens ont accès à des matériels médicaux sécuritaires, efficaces et de qualité.
- Ces résultats à long terme ont une incidence directe sur les Canadiens :
- Ils sont protégés contre les produits dangereux, les aliments insalubres et les menaces pour la santé publique.
Les renseignements de cette annexe proviennent de la version préliminaire de la charte pour des Canadiens en meilleure santé — résultats liés à la protection de la santé.
Annexe E – Rôles et responsabilités clés de la MDP au moment du projet
Direction générale des produits de santé et des aliments (DGPSA)
- Direction des produits thérapeutiques (DPT) : responsable de la
réglementation des instruments médicaux à usage humain en vertu de laLoi sur les aliments et drogues et du Règlement sur les instruments médicaux.
- Bureau des instruments médicaux (BIM) : élabore la politique relative aux instruments médicaux, examine les demandes d’instruments médicaux de classe II à IV pour en autoriser l’importation et la vente au Canada, autorise les essais expérimentaux, donne accès au besoin aux instruments non commercialisés par l’entremise du Programme d’accès spécial, et soutient les activités de contrôle et de surveillance de la DPSC après la mise en marché en effectuant des évaluations des risques d’incidents après la mise en marché.
- Direction des produits de santé commercialisés (DPSC) : élabore la
politique de surveillance après la mise en marché, effectue la
surveillance de la sécurité clinique et épidémiologique après la mise
en marché, mène des activités de gestion des risques, y compris la
publication de communications sur les risques, et assure la
surveillance réglementaire de la publicité.
- Bureau de la surveillance des produits de santé et de l’épidémiologie (BSPSE) : responsable des examens de vérification des rapports de problèmes obligatoires et des rapports volontaires par l’entremise du ResSCIM.
- Bureau des produits pharmaceutiques et des instruments médicaux commercialisés (BPPIMC) : s’occupe de la détection des signaux et de leur évaluation, de même que d’autres types d’évaluations concernant la sûreté, de l’élaboration de recommandations en réponse à des questions de sûreté cernées, ainsi que de la prise de décisions en ce qui concerne la nécessité de transmettre des communications sur les risques et de la coordination de l’élaboration du contenu connexe.
Direction générale des opérations réglementaires et de l’application de la loi (DGORAL)
- Direction de la conformité des instruments médicaux et en milieux
cliniques (DCIMMC) : gère les programmes nationaux de conformité et
d’application de la loi afin de garantir la sécurité et la qualité de
nombreux produits thérapeutiques, y compris les instruments médicaux.
- Programme de conformité des instruments médicaux (PCIM) : se concentre sur la sécurité et la qualité des instruments médicaux sur le marché et sur les fabricants d’instruments médicaux de classe I, ainsi que sur les importateurs et les distributeurs d’instruments médicaux de classe I à IV. Ce programme comprend l’Unité de la conformité et de l’homologation des instruments médicaux (UCHIM), le Programme d’inspection des instruments médicaux (PIIM) et le Programme de conformité et d’application de la loi des instruments médicaux (PCALIM).
- Unité des politiques, de la planification et de l’analyse (UPPA) : fournit un soutien stratégique et opérationnel en matière de politiques, de planification et d’analyse des données pour aider à orienter les activités et la prise de décision en matière de conformité et d’application de la loi de la DCIMMC.
Notes de fin de document
- Note de bas de page 1
-
Santé Canada (2019). Rapport final : Frais pour les médicaments et les instruments médicaux Extrait de : https://www.canada.ca/fr/services/sante/publications/medicaments-et-produits-sante/frais-medicaments-intruments-medicaux.html
- Note de bas de page 2
-
Loi sur les aliments et drogues, L.R.C.1985, ch. F-27. Accessible à l’adresse :
http://laws-lois.justice.gc.ca/fra/lois/F-27/
- Note de bas de page 3
-
Règlement sur les instruments médicaux (DORS/98-282) Accessible à l’adresse :
http://laws-lois.justice.gc.ca/fra/reglements/DORS-98-282/
- Note de bas de page 4
-
Snowdon, A, Zur, R, Shell,J. Transforming Canada Into a Global Centre for Medical Device Innovation and Adoption. London (Ontario), The Ivey Centre for Health Innovation and Leadership. 2011. Extrait de : ..\..\..\J-Evaluation Data Collection and Analysis\Preliminary findings\Analysis Files\2.2. IVEY_Transforming CAN into a Global Centre.pdf (Anglais seulement)
- Note de bas de page 5
-
Institut canadien d’information sur la santé (s.d.). Estimateur des coûts par patient. Accessible à l’adresse : https://www.cihi.ca/fr/estimateur-des-couts-par-patient
- Note de bas de page 6
-
Diabète Canada (s.d.). Why action is needed to prevent amputations in Ontario. Extrait de : https://diabetes.ca/DiabetesCanadaWebsite/media/Advocacy-and-Policy/Advocacy%20Reports/Amputation_Prevention_Infographic_ON_8-5x11_3.pdf (Anglais seulement)
- Note de bas de page 7
-
Santé Canada (2011a). Document d’orientation sur les rapports d’incident obligatoires relatifs aux instruments médicaux Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/rapports-publications/medeffet-canada/lignes-directrices-declaration-obligatoire-incidents-lies-materiels-medicaux-sante-canada-2011.html
- Note de bas de page 8
-
Gouvernement du Canada (2014). Pompe à insuline Paradigm et Pompe à perfusion d’insuline Paradigm (2014-01-17) Accessible à l’adresse : https://canadiensensante.gc.ca/recall-alert-rappel-avis/hc-sc/2014/39013r-fra.php
- Note de bas de page 9
-
Santé Canada (2019a). Instruments médicaux. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/instruments-medicaux.html
- Note de bas de page 10
-
Santé Canada (2018a). Faits saillants de 2018 sur les médicaments et les instruments médicaux : Pour maintenir et améliorer votre santé. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/publications/medicaments-et-produits-sante/faits-saillants-2018-medicaments-dispositifs-medicaux.html
- Note de bas de page 11
-
Santé Canada (2016a). Liste des instruments médicaux homologués en vigueur (MDALL).
Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/instruments-medicaux/licences/liste-instruments-medicaux-homologues-vigueur.html - Note de bas de page 12
-
Santé Canada (2019b). Direction des instruments médicaux. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/organisation/a-propos-sante-canada/directions-generales-agences/direction-generale-produits-sante-aliments/direction-instruments-medicaux.html
- Note de bas de page 13
-
Santé Canada (2015). Comment les médicaments sont examinés au Canada. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/medicaments/feuillets-information/comment-medicaments-sont-examines-canada.html. Santé Canada (2019c). Produits biologiques et radiopharmaceutiques et thérapies génétiques. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/produits-biologiques-radiopharmaceutiques-therapies-genetiques.html
- Note de bas de page 14
-
Santé Canada (2011). Mission, valeurs, activités. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/organisation/a-propos-sante-canada/activites-responsabilites/mission-valeurs-activites.html
- Note de bas de page 15
-
Santé Canada (2018b). Tendances annuelles dans les déclarations de cas d’effets indésirables des produits de santé et des incidents liés aux matériels médicaux à Santé Canada (2008-2017). Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/publications/medicaments-et-produits-sante/tendances-annuelles-declarations-cas-effets-indesirables-produits-sante-incidents-lies-materiels-medicaux.html
- Note de bas de page 16
-
Santé Canada (2019d). Réseau sentinelle canadien pour les matériels médicaux. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/medeffet-canada/medeffet-canada-projet-pilote-reseau-sentinelle-canadien-materiels-medicaux.html
- Note de bas de page 17
-
Institut canadien pour la sécurité des patients (2019). Déclaration obligatoire des réactions indésirables graves à des médicaments et des incidents liés aux instruments médicaux par les hôpitaux. Extrait de : ..\..\..\J-Evaluation Data Collection and Analysis\Preliminary findings\Analysis Files\Patient Safety Insitute_MDI reporting.ppt. Santé Canada (2019e). Déclarer un incident lié à un instrument médical : rapporteur. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/medeffet-canada/declaration-effets-indesirables/instrument-medical.html
- Note de bas de page 18
-
Santé Canada (2018b). Tendances annuelles dans les déclarations de cas d’effets indésirables des produits de santé et des incidents liés aux matériels médicaux à Santé Canada (2008-2017). Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/publications/medicaments-et-produits-sante/tendances-annuelles-declarations-cas-effets-indesirables-produits-sante-incidents-lies-materiels-medicaux.html
- Note de bas de page 19
-
Santé Canada (2018b). Tendances annuelles dans les déclarations de cas d’effets indésirables des produits de santé et des incidents liés aux matériels médicaux à Santé Canada (2008-2017). Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/publications/medicaments-et-produits-sante/tendances-annuelles-declarations-cas-effets-indesirables-produits-sante-incidents-lies-materiels-medicaux.html
- Note de bas de page 20
-
Santé Canada (2011b). Communication des risques – La protection des Canadiens par l’information. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/rapports-publications/medeffet-canada/communication-risques-protection-canadiens-information-sante-canada-2011.html
- Note de bas de page 21
-
Gouvernement du Canada (2019c). Feuille de route pour l’examen de la réglementation du secteur de la santé et des sciences biologiques. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/organisation/a-propos-sante-canada/legislation-lignes-directrices/lois-reglements/examens-reglementaires-cibles/feuille-route-examen-reglementaire-secteur-sante-sciences-biologiques.html
- Note de bas de page 22
-
Gouvernement du Canada (2019c). Feuille de route pour l’examen de la réglementation du secteur de la santé et des sciences biologiques. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/organisation/a-propos-sante-canada/legislation-lignes-directrices/lois-reglements/examens-reglementaires-cibles/feuille-route-examen-reglementaire-secteur-sante-sciences-biologiques.html
- Note de bas de page 23
-
Santé Canada (2017). Politique du portefeuille de la Santé en matière d’analyse comparative fondée sur le sexe et le genre. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/organisation/transparence/rapports-gestion/portefeuille-sante-politique-matiere-d-analyse-comparative-fondee-sur-sexe-genre.html
- Note de bas de page 24
-
Gouvernement du Canada (2019d). Règlement modifiant certains règlements pris en vertu de la Loi sur les aliments et drogues (surveillance après la mise en marché des instruments médicaux), La Gazette du Canada, Partie I, volume 153, numéro 24, p. 2731, 15 juin 2019. Accessible à l’adresse : http://gazette.gc.ca/rp-pr/p1/2019/2019-06-15/html/index-fra.html
- Note de bas de page 25
-
Gouvernement du Canada (2019d). Règlement modifiant certains règlements pris en vertu de la Loi sur les aliments et drogues (surveillance après la mise en marché des instruments médicaux), La Gazette du Canada, Partie I, volume 153, numéro 24, p. 2731, 15 juin 2019. Accessible à l’adresse : http://gazette.gc.ca/rp-pr/p1/2019/2019-06-15/html/index-fra.html
- Note de bas de page 26
-
Santé Canada (2013). Document d’orientation : Considérations relatives à l’inclusion des femmes dans les essais cliniques et à l’analyse des données selon le sexe Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/medicaments/demandes-presentations/lignes-directrices/essais-cliniques/considerations-relatives-inclusion-femmes-essais-cliniques-analyse-donnees-selon-sexe-document-orientation.html
- Note de bas de page 27
-
Santé Canada (2019g). Comité consultatif scientifique sur les produits de santé destinés aux femmes, Résumé observations, conseils, 2019-05-16-17. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/medicaments/comites-consultatifs-experts-scientifiques/produits-sante-destines-femmes/resume-observations-conseils-2019-05-16-17.html
- Note de bas de page 28
-
U.S. Food and Drug Administration (2019a). PMA Review Process. Accessible à l’adresse : https://www.fda.gov/medical-devices/premarket-approval-pma/pma-review-process
- Note de bas de page 29
-
Santé Canada (2016c). Rapport annuel sur le rendement du Bureau des matériels médicaux du 1er avril 2015 au 31 mars 2016. ..\..\..\J-Evaluation Data Collection and Analysis\Preliminary findings\Analysis Files\254.1 MDB Annual Performance Report 2015-2016.pdf. Santé Canada (2019j) (Anglais seulement). Rapport annuel sur le rendement du Bureau des matériels médicaux du 1er avril 2018 au 31 mars 2019 ..\..\..\J-Evaluation Data Collection and Analysis\Preliminary findings\Analysis Files\254.4 MDB YEARLY REPORT - 2018-19.pdf
- Note de bas de page 30
-
Emergo by UL (2019). Australia: The Regulatory Process for Medical Devices. Accessible à l’adresse : https://www.emergobyul.com/resources/australia-process-chart
- Note de bas de page 31
-
Santé Canada (2018). Sommaire des motifs de décision (SMD). Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/services/medicaments-produits-sante/medicaments/sommaire-motifs-decision.html
- Note de bas de page 32
-
LeBrun, M., DiMuzion, J., Beauchamp, B., Reid, S., et Hogan, V. (2013). Evaluating the health literacy burden of Canada’s public advisories: A comparative effectiveness study on clarity and readability. Drug Safety, 36(12):1179-87. doi: 10.1007/s40264-013-0117-8.
- Note de bas de page 33
-
Règlement sur les instruments médicaux (DORS/98-282) Accessible à l’adresse : http://laws-lois.justice.gc.ca/fra/reglements/DORS-98-282/
- Note de bas de page 34
-
HPRA (2018). Annual Report 2018. Extrait de : https://www.hpra.ie/docs/default-source/publications-forms/corporate-policy-documents/annual-report-2018.pdf?sfvrsn=5
- Note de bas de page 35
-
U.S. Food & Drug Administration (2019b). Reorganization of the Center for Devices and Radiological Health. Accessible à l’adresse : https://www.fda.gov/about-fda/center-devices-and-radiological-health/reorganization-center-devices-and-radiological-health
- Note de bas de page 36
-
Santé Canada (2019k). The Medical Devices Directorate: Focused – Agile – World Class. Extrait de : https://www.raps.org/RAPS/media/news-images/regulators/Executive-Summary-MDD-EN-(1).PDF
- Note de bas de page 37
-
Santé Canada (2016d). Plan stratégique 2016-2021 de la Direction générale des produits de santé et des aliments. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/organisation/a-propos-sante-canada/rapports-publications/direction-generale-produits-sante-aliments/plan-strategique-direction-generale-produits-sante-aliments-2016-2021.html
- Note de bas de page 38
-
Gouvernement du Canada (2019a). Santé Canada lance des consultations afin d’améliorer l’innocuité des instruments médicaux. Accessible à l’adresse : https://www.canada.ca/fr/sante-canada/nouvelles/2019/06/sante-canada-lance-des-consultations-afin-dameliorer-linnocuite-des-instruments-medicaux.html
- Note de bas de page 39
-
Gouvernement du Canada (2019b). Règlement modifiant le Règlement sur les aliments et drogues (rapports sur les réactions indésirables graves à une drogue — hôpitaux), C.P. 2019-746, 9 juin 2019, DORS/2019-190, s.30(1.3), Gazette du Canada, Partie II, volume.153, numéro 13, p. 3137, 26 juin 2019. Extrait de : http://www.gazette.gc.ca/rp-pr/p2/2019/2019-06-26/html/sor-dors190-fra.html
- Note de bas de page 40
-
Gouvernement du Canada (2019b). Règlement modifiant le Règlement sur les aliments et drogues (rapports sur les réactions indésirables graves à une drogue — hôpitaux), C.P. 2019-746, 9 juin 2019, DORS/2019-190, s.30(1.3), Gazette du Canada, Partie II, volume.153, numéro 13, p. 3137, 26 juin 2019. Extrait de : http://www.gazette.gc.ca/rp-pr/p2/2019/2019-06-26/html/sor-dors190-fra.html
- Note de bas de page 41
-
Gouvernement du Canada (2019b). Règlement modifiant le Règlement sur les aliments et drogues (rapports sur les réactions indésirables graves à une drogue — hôpitaux), C.P. 2019-746, 9 juin 2019, DORS/2019-190, s.30(1.3), Gazette du Canada, Partie II, volume.153, numéro 13, p. 3137, 26 juin 2019. Extrait de : http://www.gazette.gc.ca/rp-pr/p2/2019/2019-06-26/html/sor-dors190-fra.html
Notes de bas de page
- Note de bas de page i
-
En novembre 2019, une partie du PIM a été réorganisée au sein d’une nouvelle Direction des instruments médicaux.
- Note de bas de page ii
-
Le ResSCIM est un réseau qui représente plus de 260 hôpitaux et établissements à travers le Canada. Bien que la déclaration soit volontaire, certains informateurs internes clés ont noté que le ResSCIM a contribué à combler les lacunes en matière de surveillance et a permis d’obtenir des rapports d’incidents de la part du secteur des soins de santé.
- Note de bas de page iii
-
La date de réception et la date de traitement ont été comparées pour chaque rapport d’incident afin d’évaluer si celui-ci a été traité dans le respect des normes de service
- Note de bas de page iv
-
Les intervenants sont définis comme des personnes, des groupes ou des organisations qui ne font pas partie du gouvernement du Canada et qui ont un intérêt particulier envers une politique, un programme, une initiative réglementaire ou un service de Santé Canada, ou qui ont une certaine influence sur ceux-ci, ou qui sont touchés par ceux-ci
- Note de bas de page v
-
L’enquête sur le CRTO a été menée en 2016 par Santé Canada. Le questionnaire a été envoyé à toutes les personnes ou organisations qui se sont inscrites auprès de Santé Canada pour recevoir de l’information ou des invitations à des consultations sur divers sujets liés à la santé. Les résultats présentés dans ce rapport ne concernent que les personnes et les organisations qui ont déclaré un intérêt pour les instruments médicaux. Les résultats doivent être interprétés avec prudence en raison du petit nombre de répondants. Étant donné que les répondants se sont auto-enregistrés auprès de Santé Canada, les résultats de l’enquête ne peuvent pas être généralisés à l’ensemble de la population ou à un groupe plus large d’intervenants dans le domaine des instruments médicaux
- Note de bas de page vi
-
L’échantillon a été constitué à partir des cinq premiers rappels effectués pour les mois de janvier, mars, mai, juillet, septembre et novembre, et des cinq derniers rappels effectués pour les mois de février, avril, juin, août, octobre et décembre
- Note de bas de page vii
-
Différence entre la « date de début » indiquée sur le site Web de Santé Canada et la « date initiée par le fabricant » indiquée sur le site Web de la Food and Drug Administration des États-Unis
- Note de bas de page viii
-
Au moment de la mission, le Comité trilatéral des instruments médicaux était composé des directeurs du Bureau des instruments médicaux, du Bureau de la surveillance des produits de santé et de l’épidémiologie (BSPSE), du Bureau des produits pharmaceutiques et des instruments médicaux commercialisés (BPPIMC) et du Programme de conformité des instruments médicaux (PCIM). Un représentant du Bureau des politiques, des conseils sur les risques et de la publicité de Santé Canada a également assisté au comité pour apporter un éclairage politique au besoin