
Cleaning validation guide (GUI-0028)
Disclaimer
This document does not constitute part of the Food and Drugs Act (the Act) or its regulations and in the event of any inconsistency or conflict between the Act or regulations and this document, the Act or the regulations take precedence. This document is an administrative document that is intended to facilitate compliance by the regulated party with the Act, the regulations and the applicable administrative policies.

Download the alternative format
(PDF format, 1 MB, 37 pages)
Organization: Health Canada
Date published: 2021-06-29
Table of contents
- About this document
- Guidance
- 4. Principles
- 5. Applying QRM principles to control cross-contamination risks
- 6. Cleaning validation master plan
- 7. Cleaning validation lifecycle approach
- 8. Analytical and sampling methods
- 9. Assessment of cleaning
- 10. Establishing limits
- 11. Microbiological controls
- 12. General equipment cleaning considerations
- 13. Additional considerations for cleaning of API production equipment
- 14. Additional considerations for cleaning validation of biotechnology processes
- Appendices
The following are the two types of icons used in this document, and the way they are intended to be used.
Important: Key or cautionary information for people to know.
Important: Supplementary information like quotes and legal references.
About this document
1. Purpose
This document is for anyone involved in pharmaceutical, biological and radiopharmaceutical fabrication and packaging activities for drugs sold in Canada, including:
- regulated industry
- inspectors and evaluators
It provides guidance on cleaning validation. It will help you understand and comply with Part C, Division 2 of the Food and Drug Regulations (the Regulations).
This guide is also intended to establish inspection consistency and uniformity with respect to equipment cleaning procedures. Principles incorporated in international guidance have been taken into account when preparing this document.
2. Scope
This guide addresses special considerations and issues when validating cleaning procedures for equipment used to fabricate and package:
- active pharmaceutical ingredients (APIs)
- pharmaceuticals
- radiopharmaceuticals
- biological drugs
- veterinary drugs
It covers validation of equipment cleaning for:
- the removal of residues associated with products used in the previous production run, such as active ingredients, breakdown or by-products of concern, intermediates, residues of cleaning agents, and processing agents
- the control of potential microbial contaminants
Additional guidance on cleaning validation for certain veterinary drugs and Category IV drugs can be found in these Health Canada guidance documents:
While this document is about cleaning validation, the following references on impurities from the International Council for Harmonisation (ICH) may also be useful:
- ICH M7 - Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk
- ICH Q3A - Impurities in New Drug Substances
- ICH Q3B - Impurities in New Drug Products
- ICH Q3C - Impurities: Guideline for Residual Solvents
- ICH Q3D - Guideline for Elemental Impurities
3. Introduction
These guidelines interpret the requirements for good manufacturing practices (GMP) in Part C, Division 2 of the Regulations. They were developed by Health Canada in consultation with stakeholders.
Guidance documents like this one are meant to help industry and health care professionals understand how to comply with regulations. They also provide guidance to Health Canada staff, so that the rules are enforced in a fair, consistent and effective way across Canada.
Health Canada inspects establishments to assess their compliance with the Food and Drugs Act (the Act) and associated regulations. When Health Canada conducts an inspection, inspectors will use this document as a guide in assessing the site's compliance with GMP requirements with respect to equipment cleaning.
These guidelines are not the only way GMP regulations can be interpreted, and are not intended to cover every possible case. Other ways of complying with GMP regulations will be considered with proper scientific justification. Also, as new technologies emerge, different approaches may be called for. This document builds on other international guidance (see References)
Guidance documents are administrative and do not have the force of law. Because of this, they allow for flexibility in approach. Use this guide to help you develop specific approaches that meet your unique needs.
Guidance
4. Principles
Cleaning validation is performed to ensure that the equipment cleaning process will consistently reduce the possibility of cross contamination via carryover in a drug manufacturing process. It provides documented evidence that an approved cleaning process will reproducibly remove previous products, by-products of concern or cleaning agent residues that may remain on the equipment to below scientifically set limits. These limits are calculated based on safe threshold values, which are determined by toxicological evaluation.
All cleaning processes for product contact equipment should be validated in accordance with Quality Risk Management (QRM) principles. Consideration should also be given to non-contact parts from which product may migrate. These should be based on risk.
Remediation actions must be implemented when a cleaning process is not capable of consistently producing adequate results. Examples of remediation actions include improved cleaning procedures and equipment/facility dedication. Continued cleaning failures and/or testing until clean (i.e. continually cleaning and testing until acceptable results are achieved) are not acceptable.
It is also important to demonstrate that the facility and equipment are designed, cleaned and used in a manner that will prevent microbial contamination of products.
4.1 About safe threshold values
It is Health Canada's intention to align with guidance adopted July 1, 2018 by the Pharmaceutical Inspection Co-operation Scheme (PIC/S) on use of toxicological evaluation in setting Health Based Exposure Limits (HBEL). With this approach, an evaluation of all pharmacological and toxicological data should be undertaken by a qualified person to determine a safe daily threshold value, such as Permissible Daily Exposure (PDE) or Threshold of Toxicological Concern (TTC).
The PDE represents a substance specific dose that is unlikely to cause an adverse effect if an individual is exposed at or below this dose every day for a lifetime.
The TTC represents the genotoxic impurity exposure level associated with a theoretical cancer risk of 1 additional cancer in 100,000 patients when exposed over a lifetime.
Additional information can be found in the following question and answer document published by PIC/S.
The HBEL, such as the PDE or TTC, can then be used in risk identification and justification of maximum safe carryover limits into the next product. Other approaches to determining health based exposure limits may be considered acceptable in accordance with QRM principles and if scientifically justified.
It should be noted that the PIC/S Guideline also states that the PDE and ADE (Allowable Daily Exposure) are effectively synonymous.
5. Applying QRM principles to control cross-contamination risks
You have an obligation to prevent the cross contamination of drugs. This is achieved by developing a contamination control strategy, which will include designing and establishing appropriate controls of the premises, equipment and all associated processes. It should be recognized that equipment cleaning is only one of many measures that should be taken to control risk of cross-contamination in a multi-product facility or on equipment proposed to be shared.
- Actions should be taken on a level proportional to the identified risks e.g. greater control is required for products with lower HBELs.
- All potential sources of cross contamination should be assessed via a documented QRM process. The QRM process should evaluate risks based on scientific knowledge and assessment, and determine measures that can be taken to reduce those risks.
- The outcome of the QRM process should be the basis for determining the extent of the technical and organizational measures required to control risks for cross-contamination. Refer to Appendices of this document for a list of technical and operational measures to consider.
- Measures to prevent cross-contamination and their effectiveness should be reviewed periodically according to set procedures.
- If the QRM process confirms that the drug can safely be made on shared equipment, validate any equipment cleaning process(es) to be used.
Additional information on QRM can be found in ICH Q9 - Quality Risk Management and ASTM E3106 - 18e1 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation.
6. Cleaning validation master plan
You should maintain a Cleaning Validation Master Plan (or equivalent document) to outline the general cleaning validation policies at your site.
- Product and equipment may be grouped in accordance with QRM principles:
- You may choose to conduct cleaning validation studies on all products at the facility or on worst case products only (the product family approach). You must stipulate and justify, as required, which approach is being used in the Cleaning Validation Master Plan. If a worst case approach is being used, you should document:
- the methodology/scientific rationale used in determining the worst case products
- the actual worst case products including a listing of all products deemed to be represented by the identified worst case products
Examples of factors that can be included in the assessment of worst case products include:
- HBEL of the residue
- difficulty in cleaning or product cleanability
- solubility of residues in cleaning agents and cleaning solvents
- physical characteristics of the product, active substance or excipients
- past experience (for example during development and with similar products)
It should be noted that there may be multiple worst case products. For example, an insoluble product with a high HBEL value may be the most difficult product to clean but not necessarily worst case compared to a partially soluble product with a low HBEL value.
- You may choose to conduct cleaning validation studies for all equipment or by grouping similar equipment, such as 'like for like' equipment. A representative approach is only suitable if equipment is equivalent in terms of size, design, function, cleaning procedure and cleanability. If there are any differences in equipment, the proposal to group them should be based on data. If an equipment grouping approach is being used, you should document:
- the approach/scientific rationale by which equipment were grouped together
- the listing of all equipment in each group, identifying the equipment in each group that is considered to be worst case, with proper justification.
- You may choose to conduct cleaning validation studies on all products at the facility or on worst case products only (the product family approach). You must stipulate and justify, as required, which approach is being used in the Cleaning Validation Master Plan. If a worst case approach is being used, you should document:
- All cleaning processes must be equivalent if cleaning validation studies are to be conducted following a worst case product and/or equipment grouping approach.
7. Cleaning validation lifecycle approach
Validation, in a lifecycle approach, involves the collection and evaluation of data throughout the product's lifecycle. Learnings from each phase are critical in ensuring appropriate controls are established. Refer to section 7.2: Phase 2 - Cleaning process qualification for additional information.
For cleaning validation, the lifecycle approach normally involves the following phases:
- Phase 1 - Cleaning process design and development: Develop effective cleaning procedures in a controlled and documented manner prior to implementation.
- Phase 2 - Cleaning process qualification: Evaluate cleaning processes to ensure they are effective and reproducible. Cleaning process qualification studies involve conducting cleaning verification assessments a predetermined number of times under specified conditions.
- Phase 3 - On-going monitoring: Ensure cleaning procedures remain effective and controlled via an ongoing monitoring program.
It is important to differentiate between three important terms with respect to where they fit into the overall cleaning lifecycle approach.
Cleaning verification refers to the gathering of evidence through an appropriate analytical method after each batch/campaign to show that the residues of concern have been reduced below pre-defined carryover limits derived from scientifically set safe threshold levels.
Cleaning verification refers to an individual cleaning and sampling exercise or study to assess equipment cleanliness and is used throughout the lifecycle approach. Cleaning verification studies should be conducted in accordance with an established cleaning procedure or a protocol. Sampling requirements during a cleaning verification study should be, at a minimum, equivalent to those during the cleaning process qualification phase.
Cleaning process qualification refers to a defined phase within the cleaning validation lifecycle, which demonstrates that the cleaning process is robust and reproducible. It will normally be comprised of multiple cleaning verification runs/studies for all equipment involved in the cleaning process qualification study.
Cleaning validation refers to the overall validation program, from the development stage all the way through the ongoing monitoring stage. The cleaning validation program is comprised of appropriately controlled cleaning procedures and having sufficient data to demonstrate their effectiveness.
Figure 1 – Overview of the cleaning validation program
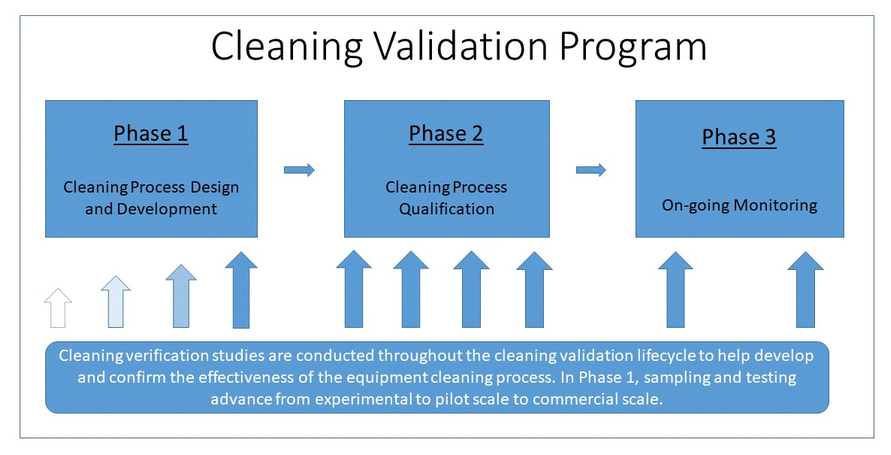
Figure 1 - Text Description
The diagram is introduced as "Figure 1 – Overview of the cleaning validation program".
The diagram consists of one large box entitled 'Cleaning Validation Program'.
Within the box are 3 smaller boxes placed horizontally with an arrow leading from the first to the second and from the second to the third. These boxes are meant to represent the 3 phases of the cleaning validation program.
The first box is described as "Phase 1 – Cleaning Process Design and Development".
The second box is described as "Phase 2 – Cleaning Process Qualification".
The third box is described as "Phase 3 – On-going Monitoring".
Below the 3 boxes are a number of arrows pointing upwards toward the boxes. These are meant to represent the formality and number of cleaning verifications performed during each phase.
There are 4 arrows below the Phase 1 box getting larger in size and increasing in colour meant to represent the increasing amount of effort and formality that the testing undergoes as the cleaning process is developed.
There are 4 arrows below the Phase 2 box all equal in large size and full colour representing the number of formal cleaning verifications to be performed during Phase 2.
There are 2 arrows below the Phase 3 box equal in large size and full colour representing a decreased frequency of cleaning verifications during the on-going monitoring phase.
There is a single box of text below the arrows meant to describe the overall process. "Cleaning verification studies are conducted throughout the cleaning verification lifecycle to help develop and confirm the effectiveness of the equipment cleaning process. In Phase 1, the sampling and testing advance from experimental to pilot scale to commercial scale".
7.1 Phase 1 - Cleaning process design and development
Cleaning procedures should be developed in a controlled manner in accordance with QRM principles and tools to ensure cleaning processes are effective and reproducible. Factors that can influence cleaning effectiveness should be identified and controlled.
- Appropriate effort and resources need to be applied when designing and developing cleaning processes. This could include laboratory, material coupon, bench top or pilot scale trials through to commercial scale trials.
- It is important that potential issues that could impact the effectiveness and reproducibility of cleaning processes be considered when developing new or revised cleaning processes. Items for consideration include:
- Understand the chemical and physical properties of the actives, excipients and by-products or degradants. This knowledge is required to help determine which cleaning agents, solvents and cleaning process parameters would be most appropriate. It should be noted that by-products can also be created through interaction with the cleaning agents and solvents.
- Review the design of the equipment. Consider engineering drawings, experience of maintenance and cleaning personnel, examination of disassembled equipment in the clean and dirty state to establish areas at risk for residue accumulation or migration. Pay attention to materials of construction for equipment parts, their smoothness, and evaluate any differences these substrates have on residue removal. All sampling sites should be appropriately justified. This is an important consideration during product scale-up, as equipment size and design should be evaluated.
- Evaluate any risk for endotoxin contamination or microbial proliferation in susceptible products through incoming materials, usage, handling, hold times and storage. Evaluate whether any additional disinfection or endotoxin control steps are required after equipment storage, where appropriate.
- Determine whether any variation in raw materials might affect cleanability.
- Examine the environment in which the cleaning is proposed to be conducted. Ensure suitable facility and environmental controls are in place to facilitate required cleaning, contact time and drying, and to prevent any potential for cross contamination.
- All cleaning process elements, parameters and controls such as cleaning agents, solvents, critical cleaning parameters (time, temperature, pressures, and action such as scrubbing, soaking, circulating or reflex) should be scientifically established. Specific challenge trials may be required. The goal is to identify critical cleaning parameters and understand the impact of variability of such parameters on cleaning performance.
- Document learnings during the cleaning development process to ensure knowledge transfer, and use them to construct a detailed cleaning procedure. Use risk assessment tools to identify any potential risks with respect to the cleaning procedure and make the necessary adjustments.
- You should have confidence that cleaning procedures will be effective and reproducible in full/commercial scale equipment. Cleaning verification(s) may be done in full-scale equipment as the last step of cleaning process design and development to confirm understanding of the effectiveness of the proposed cleaning process. See 7.2.1 Release of equipment for guidance about release of commercial equipment used for cleaning verification studies.
- The outcome of the cleaning design and development process should be a defined cleaning process that is both reproducible and effective.
7.1.1 Control of manual cleaning processes
Ensuring operator consistency is one of the biggest challenges in a manual cleaning program. The QRM evaluation should clearly identify steps required to ensure both consistency in how the procedures are conducted and the overall outcome of the cleaning validation process.
Ensure manual cleaning procedures are consistently performed by doing the following:
- Have adequately detailed instructions and establish range/value of the applicable critical process parameters:
- detailed disassembly instructions
- sequence of the cleaning steps
- cleaning agent to be used and its concentration
- cleaning agent application means (e.g., soaking or scrubbing)
- contact time
- temperature of the cleaning solutions or rinses
- rinsing techniques (i.e., pre-rinses, soaking, flushing, times and pressures)
- method of drying
- Establish and maintain operator training programs, which may include certification requirements based on risk. It is not acceptable to repeatedly justify cleaning failures on inappropriate cleaning techniques such as operator error, as this indicates inadequate control.
- Ensure that cleaning procedures and critical parameters are adequately documented and verified, where appropriate. Records should be sufficiently detailed to confirm the completion of key steps in the cleaning process.
- Verify that calibrated measuring devices (such as timers, temperature probes, dosing pumps and flow meters), if required, are used.
7.1.2 Control of automated cleaning processes
Control automated cleaning equipment (e.g. Clean-In-Place) and processes as follows:
- Have adequately detailed procedures describing the automated cleaning process, controls and requirements such as equipment preparation or disassembly, and loading patterns.
- Qualify equipment used for such cleaning and verify that all product contact surface areas are being appropriately contacted by the cleaning/rinsing agents.
- Ensure that appropriate calibration and maintenance programs are established and maintained.
- Define cleaning sequences including all temperatures, concentrations, valve openings, spray rates, pressures and volumes. It is also important to ensure that cleaning sequences controlled by automated recipes are appropriately protected against unapproved or uncontrolled changes.
- Monitor critical control points and parameters with appropriate sensors and alarms to ensure the process is highly controlled. Critical alarms should be identified and regularly checked or verified. Procedures should outline steps to be taken in response to such alarms.
- Ensure cleaning sequences and data, including alarms, are appropriately controlled and reviewed.
7.2 Phase 2 - Cleaning process qualification
- You may begin a cleaning process qualification study once you have a fully defined cleaning process. This can be before the start of commercial production if equipment, batch sizes, and formulation/operating parameters are not subject to change. The cleaning process qualification study is normally started when commercial production is initiated.
- Conduct cleaning process qualification studies for all products, or worst case products if a product family approach is used.
- Use QRM principles to determine the extent and scope of cleaning process qualification requirements.
- Determine the number of cleans to be assessed using a documented risk assessment. Although a three-clean assessment has long been the industry norm, your risk assessment may result in a recommendation to evaluate a different number of cleans.
- Although worst case challenge testing should be evaluated during the cleaning process design phase, it may be appropriate to verify critical process parameter limits during qualification studies. Examples of challenge testing may include minimum detergent contact time, minimum or maximum temperatures and minimum rinse time/volume/pressure. Worst case challenge testing is of particular importance when manual cleaning systems are employed.
- Operator variability should also be assessed, particularly when manual cleaning processes are being used.
- Document the cleaning process qualification requirements in a protocol. The protocol should include:
- objective and scope of the cleaning qualification exercise
- responsibilities for performing and approving the qualification study
- description of the equipment to be used for the process and for cleaning
- references and descriptions of the cleaning procedures and parameters to be used, with a description of all critical parameters
- any worst case challenges to be evaluated in the study
- the number of cleaning cycles to be performed
- sampling procedures
- clearly defined sampling locations, with rationale for their selection
- validated analytical methods, that are appropriate for the residue limits under consideration and data on recovery studies
- the acceptance criteria, including the rationale for setting the specific limits
Note – Rationale and data to support approaches taken may be contained in other documents to which the protocol may refer.
- Ensure you have data to demonstrate that the following variables do not impact cleaning effectiveness:
- The length of time between the completion of manufacturing and start of cleaning (dirty hold time).
- The maximum allowable number of batches of the same product manufactured prior to full cleaning, specifying maximum campaign lengths in days and/or number of batches.
- Investigate any cleaning failure as per quality system requirements.
- Prepare a final qualification report. The conclusions of this report should state if the cleaning process has been qualified successfully.
7.2.1 Release of equipment
It is understood that it may take time to assess the required number of cleaning verification runs during the qualification phase. In addition, it is anticipated that full-scale equipment used for other commercial products will sometimes need to be used for cleaning verification studies during the design and development phase and when introducing new products to a facility. QRM principles should be used to determine whether release of equipment for manufacture of other commercial products is acceptable before the cleaning qualification phase is completed. The data from the verification study(ies) should be reviewed and determined to be sufficient to support release of the equipment. The risk of subsequent failures during cleaning verification runs should be taken into consideration.
7.3 Phase 3 - Ongoing monitoring
- Establish ongoing monitoring requirements after the completion of the cleaning process qualification phase to demonstrate the process remains in a state of control.
- Ongoing monitoring can include a number of different activities such as:
- data analysis (such as data generated from automated processes)
- additional cleaning verification studies
- rinse sample analysis
- The amount and nature of ongoing monitoring requirements and the frequency at which additional cleaning verification assessments are proposed to be performed should be determined by QRM principles. Activities such as rinse sample analysis may be used to help monitor the program and provide data to indicate it remains in a state of control. Cleaning verification normally provides a more in-depth analysis of cleaning process performance. As such, activities such as rinse monitoring do not eliminate the need to conduct periodic cleaning verification assessments.
- Areas of special concern, in terms of on-going monitoring, include:
- products with low HBEL values which are generally more hazardous products
- products for which visual inspection cannot be used to estimate cleanliness of the equipment, meaning HBEL derived residue levels cannot be visually detected
- equipment and products with a history of failure or highly variable testing results during verification and qualification testing
- manual cleaning processes
- locations or surfaces that are difficult to access or clean
- equipment which cannot be appropriately visually inspected
- Appropriate and timely action must be taken if there are any signs that cleaning processes are inadequately controlled. Evidence of inadequate control can come through:
- statistical evaluation of data generated through cleaning verifications and/or any data generated from routine cleaning process itself.
- review of traditional quality indicators such as complaints, deviations and lab failures.
- Cleaning process data and quality indicators should be regularly reviewed for any trends or failures that may indicate the need for a review of technical or operational measures.
7.3.1 Change control and requalification
- Ensure a change control system is in place to assess and document all changes that might impact the cleaning process. The review should include consideration of whether the cleaning procedure should be re-developed and/or re-qualified.
- Changes that may potentially impact cleaning process qualification/validation include:
- new products
- changes to the cleaning process
- changes in the formulation and/or process of products
- raw material changes (e.g. change in impurity profile or physical properties)
- new cleaning agents and/or changes in cleaning agent formulation
- significant equipment changes or new equipment
- lot size or campaign length changes
- changes in analytical procedures or sampling method or materials
- changes to cleaning limits, which might happen upon periodic review of the data which form the basis of the HBEL
Note: The installation of used equipment such as equipment sourced from other sites, may pose special challenges in terms of ensuring the cleanliness of such equipment is appropriately evaluated prior to use. This should be considered as per QRM principles.
7.3.2 Introducing new products to a facility
All new product introductions should be reviewed through the QRM process and change control to determine whether the existing technical and organizational controls are sufficient or need to be modified. Consider the following:
- The HBEL of the new product and evaluate the suitability of the product for your facility and whether dedicated facilities/equipment or other additional controls are required.
- The ease of cleaning the equipment used to make the new product whether the new product is a new worst case product. Then determine if existing cleaning processes are adequate or if a new or revised process is required. If additional development of the cleaning process is required, see 7.1 Phase 1 - Cleaning process design and development.
- Any new equipment and/or equipment modification that may impact cleanability.
Verification studies will then normally be conducted in commercial equipment to demonstrate equipment has been adequately cleaned following production of the new product (development, technology transfer or clinical trial batches).
8. Analytical and sampling methods
- Validate analytical methods used to measure residue and contaminants on equipment (for example, product active drug or degradants and cleaning agent residue).
Refer to Q2 Validation of Analytical Procedures: Text and Methodology for general information with respect to how to validate analytical methods.
- Determine the limits of quantification and detection to ensure the sensitivity of the analytical method is appropriate for the residue levels under consideration. You may also need to evaluate whether the selectivity of the analytical method needs to be established in relation to potential degradants such as those formed during the cleaning process.
- Conduct recovery studies for all sampling methods used with analytical methods:
- Ensure the sampling method used in the laboratory is equivalent to the method used in manufacturing.
- Conduct recovery studies for all applicable product contact materials of construction to be sampled in the equipment.
- Establish percent recovery for each surface/material of construction and use this in the calculation of residual contaminants.
- Low or variable recovery of standard concentrations of residue during recovery studies may not be acceptable as it is indicative of an inadequate sampling or extraction technique.
Recovery studies demonstrate that the sampling and analytical methods can adequately measure residue that may be present on equipment surfaces. Such studies are performed by spiking material coupons with the residue under consideration at low levels representative of amounts after cleaning, then sampling the residue according to the applicable method. Testing results should then be compared with the actual quantity spiked onto the coupon.
- Non-specific analytical methods, for example total organic carbon (TOC) and conductivity, may be used with appropriate justification. In such cases, you must establish the effectiveness of your non-specific method to detect the target residue. You must assume that the testing result is entirely due to the target residue in such cases. You must still demonstrate that the method will provide adequate and reproducible recovery.
9. Assessment of cleaning
9.1 Visual inspection
Visual inspection is a qualitative method of evaluating equipment cleanliness and involves verifying that equipment is free of visible residue and foreign material at product changeover. It is also useful to detect damage or wear to equipment, which may render it more difficult to clean. This is an important element of every cleaning process, whether done during cleaning qualification studies or during routine production.
- Conduct visual inspections after all cleans and before conducting any cleaning verification/qualification/on-going monitoring sampling activities. Document the results.
- Establish procedures detailing how visual inspections are to be conducted. Include clear instructions with respect to:
- ensuring equipment is dry
- disassembly instructions
- use of an appropriate light source and lighting conditions
- how to assess difficult areas, such as the bottom of mixing blades
- Ensure visual inspection is only conducted by trained personnel. For more hazardous products, have a program in place to demonstrate the ability of visual inspection personnel to detect residues.
- Reliance of visual inspections as the sole means of routine verification and release of equipment should be based on a QRM assessment. Spiking studies may be required to determine visual inspection thresholds of different products on different surfaces. This is of particular concern for products with a lower HBEL.
- Investigate any visual inspection failures through the applicable quality system. Visual inspection failures should be rare when a cleaning process has been validated and may be indicative of an inadequately controlled cleaning process.
9.2 Equipment sampling
Equipment sampling is generally conducted via direct surface sampling (swab/wipe method), rinse sampling or a combination of the two.
9.2.1 Direct surface sampling (swab/wipe method)
Swab sampling involves wiping an equipment surface with a specified material wetted with solvent to recover residue from the surface.
- Conduct swab/wipe sampling on areas determined during the risk assessment and specifically on identified hardest to clean areas. In addition, consider taking representative samples of large surfaces. Clearly specify hardest to clean areas in relevant protocols. The choice of swabbing locations should be justified with appropriate supporting data.
- Consider the following when determining hardest to clean areas:
- accessibility
- equipment geometry
- potential for residue accumulation
- material of construction
- Specify the material to be used for swabbing and the sampling medium or solvent.
- Ensure production equipment is sampled in the same way as during recovery studies in the laboratory. Measures to ensure consistency may include:
- detailed procedures
- qualification/certification of swabbing personnel to demonstrate suitable recovery
- on the job training and supervision of swabbing personnel when swabbing manufacturing equipment
9.2.2 Rinse sampling
Rinse sampling involves rinsing the relevant equipment surfaces with a defined quantity of a specified solvent to remove residue. Measure the residue levels in the rinsing liquid. Rinse samples allow the sampling of a large surface area and of systems that are inaccessible or that cannot be routinely disassembled.
9.2.3 Placebo sampling
Placebo sampling is another alternative that can be used for assessment of cleaning effectiveness. Placebo sampling involves the processing of a placebo batch after cleaning activities have been completed and then analyzing the placebo for traces of the previous product. Such evaluations are normally conducted to complement swab and/or rinsing studies.
10. Establishing limits
The amount of residue allowed on equipment and/or a process train after cleaning is referred to as a maximum safe carry over limit. This limit is determined by calculating how much of the active substance of the first product made could safely be carried over into the second product (after the clean) such that the maximum daily dose of the second product doesn't contain more than the HBEL of the first product. When you take into account the surface area of the equipment and any other safety considerations, the preliminary swab or rinse limits can be calculated. The final cleaning limits chosen should not exceed this value.
- Use risk management principles when determining maximum allowable carryover calculations for residues of concern. Such limits should be based on toxicological evaluation and documented in the form of a risk assessment.
Further guidance on calculating limits can be found in various guidance documents such as the ISPE Baseline Guide Vol 7: Risk-Based Manufacture of Pharma Products 2nd Edition.
- Consider establishing alert limits in the event that HBEL derived cleaning limits are significantly higher than historic cleaning limits (for example, 1/1000th of a dose and 10 PPM). Cleaning procedures that are capable of achieving better limits than those derived from HBELs should continue to do so. Note that cleaning limits must also continue to meet the visually clean criteria.
- Ensure the analytical methods used are capable of detecting residues at an acceptable level below these limits. If this is not possible, improvements to the analytical methods can be explored or alternative means of risk reduction should be considered such as equipment dedication.
- Establish calculated cleaning acceptance criteria accounting for the cumulative impact of residue from multiple shared equipment (the process train effect).
- It is recommended that HBELs be periodically reevaluated and the impact of any changes on the overall cleaning validation program be assessed and documented.
11. Microbiological controls
- Ensure that equipment and facility design, operation, cleaning and maintenance will appropriately control microbiological bioburden. Focus on preventative measures rather than removal of contamination once it has occurred.
- Use QRM principles to determine:
- the need for including microbiological and/or endotoxin contamination evaluation as part of verification/qualification and on-going monitoring assessments
- sampling locations in equipment, which should consider those locations or materials that might be more prone to microbial growth
- the type, nature and scope of an ongoing environmental monitoring program.
- Areas of special concern for microbiological considerations include the following.
- Establish a maximum period of time that cleaned equipment can be held before use without re-cleaning or re-sanitization (commonly referred to as clean hold time). Demonstrate that the maximum allowable clean hold or storage time does not result in microbial proliferation.
-
Ensure that microbiological assessments are considered, as per risk management principles, when assessing maximum campaign lengths.
Note – the microbiological considerations stated above may not be applicable for some API products.
- Ensure that stagnant water is not allowed to remain in equipment after cleaning or use. Equipment should be drained/dried before use or storage.
- Ensure that procedures are established for the appropriate handling of hoses. Hoses, such as purified water hoses, are a known area of potential microbial contamination.
- Ensure that any microbiological limits are scientifically justified.
12. General equipment cleaning considerations
Ensure all processing equipment is designed to facilitate cleaning and permit visual inspection (where possible). Equipment should have smooth surfaces and be made of non-reactive materials. Piping of the equipment should be sloped continuously to ensure adequate drainability of the lines. Dead legs should be avoided.
Special consideration should be given to long transfer lines. Appropriate cleaning processes will involve flooding the entire pipe to ensure contact with all surfaces. Turbulent flow is generally preferred in terms of ensuring optimal cleaning. Consider ensuring there are removable sections, where appropriate for the process, to evaluate the efficacy of the cleaning process by visual, swab testing and/or rinse sample.
12.1 Cleaning agents:
- When selecting cleaning agents, ensure that their composition is known. Preference should be given to cleaning agents whose components have favorable toxicological profiles and limits. Ensure that you are notified of any changes in composition of the cleaning agent.
- Ensure that cleaning agents are easily removable.
Removal of cleaning agents is an important consideration in any cleaning validation program. Evidence should be available that cleaning procedures will effectively remove cleaning agents to below predetermined levels.
12.2 Last rinse:
- If water is used to perform the last rinse, ensure it is equivalent to or better than the grade and standard of water being used at that stage of the process. Water quality attributes (chemical, microbiological and endotoxin) should be appropriate for the given application.
- If the cleaning procedure requires a solvent as the last rinse, the quality of the solvent should be appropriate.
12.3 Dedicated equipment and facilities
The decision as to whether dedicated facilities or dedicated equipment are required should be based on QRM principles and toxicological evaluation. It may be possible to dedicate parts of equipment which are particularly difficult to assess or clean (e.g. filter bags, gaskets or screens), while validating the remainder of the equipment train for shared use. Refer to Good manufacturing practices guide for drug products (GUI-0001) for additional information.
Use QRM principles to determine cleaning validation requirements when using dedicated equipment or facilities. Areas of concern include:
- microbiological considerations
- cleaning agent removal
- potential product degradants and process impurities
13. Additional considerations for cleaning of API production equipment
- Cleaning validation is a requirement to minimize cross contamination risks in the production of APIs. Per risk management principles, such activities should be focused on process steps that pose the greatest risk to product quality such as later stage intermediates and final processing and handling stages.
- In general, cleaning control and evaluation requirements for the final API production processes should be equivalent to those required for finished dosage form manufacture. For example:
- relevant cleaning processes should be validated in accordance with a lifecycle approach
- equipment should be designed in accordance with the same concepts as used for finished drug products
- API cleaning processes normally involve significant use of solvents. In such cases:
- ensure the API is soluble in the agent being used for cleaning and rinse recovery studies
- ensure the solvents used for the cleaning process, including the final rinse, are of appropriate quality
- consider reflux or boil-out steps
Note: reflux or boil-out steps may be important when cleaning reactors and similar equipment to ensure appropriate solvent contact with the entire product contact equipment surface area. A reflux or boil-out step may also be included when collecting a rinse sample for qualification, verification, or monitoring activities.
14. Additional considerations for cleaning validation of biotechnology processes
- The principles outlined in this document can generally be applied to the cleaning validation of biotechnology processes as well.
- Cleaning development programs need to consider removal of a large number of substances such as media, proteins, acids, bases, salts etc.
- Biotechnology cleaning processes often involve conditions that cause protein molecules to denature or degrade so residual measurements are often performed using a non-specific test method such as TOC.
- Cleaning validation requirements for biological drugs should normally include a microbiological and endotoxin assessment.
- Validation of the cleaning of shared product-contact equipment should normally be evaluated for each product and process.
- Bracketing for similar products or equipment is acceptable, provided there is appropriate justification that is based on sound and scientific rationale. Some examples include:
- cleaning of fermenters of the same design but with different vessel capacity, used for the same type of recombinant proteins expressed in the same or similar cell lines and cultivated in closely related growth media
- for multi-antigen vaccines, use of a representative antigen (or combinations of them) when validating the same or similar equipment.
- QRM principles should be used in setting appropriate limits for carry over taking into account the manufacturing process and the stage of manufacture. Stringency of limits may increase through the purification process.
- Bulk Manufacture: Carryover calculations may not be applicable for bulk manufacture where the specific product residues may be present at low concentrations or it can be demonstrated that the cleaning conditions render the product permanently inert.
- Formulation and Final Filling: Limits are calculated based on an HBEL that has been established from a toxicological assessment.
Appendices
Appendix A – Prevention of cross-contamination
The following guidance is taken from PIC/S - Guide to Good Manufacturing Practice for Medicinal Products Part 1, Chapter 5 [PE 009-14 (Part 1)]
5.21 The outcome of the Quality Risk Management process should be the basis for determining the extent of technical and organizational measures required to control risks for cross-contamination. These could include, but are not limited to, the following:
Technical Measures
- Dedicated manufacturing facility (premises and equipment);
- Self-contained production areas having separate processing equipment and separate heating, ventilation and air-conditioning (HVAC) systems. It may also be desirable to isolate certain utilities from those used in other areas;
- Design of manufacturing process, premises and equipment to minimize risk for cross-contamination during processing, maintenance and cleaning;
- Use of "closed systems" for processing and material/product transfer between equipment;
- Use of physical barrier systems, including isolators, as containment measures;
- Controlled removal of dust close to source of the contaminant e.g. through localized extraction;
- Dedication of equipment, dedication of product contact parts or dedication of selected parts which are harder to clean (e.g. filters), dedication of maintenance tools;
- Use of single use disposable technologies;
- Use of equipment designed for ease of cleaning;
- Appropriate use of air-locks and pressure cascade to confine potential airborne contaminant within a specified area;
- Minimizing the risk of contamination caused by recirculation or re-entry of untreated or insufficiently treated air;
- Use of automatic clean in place systems of validated effectiveness;
- For common general wash areas, separation of equipment washing, drying and storage areas.
Organizational Measures
- Dedicating the whole manufacturing facility or a self-contained production area on a campaign basis (dedicated by separation in time) followed by a cleaning process of validated effectiveness;
- Keeping specific protective clothing inside areas where products with high risk of cross-contamination are processed;
- Cleaning verification after each product campaign should be considered as a detectability tool to support effectiveness of the Quality Risk Management approach for products deemed to present higher risk;
- Depending on the contamination risk, verification of cleaning of non- product contact surfaces and monitoring of air within the manufacturing area and/or adjoining areas in order to demonstrate effectiveness of control measures against airborne contamination or contamination by mechanical transfer;
- Specific measures for waste handling, contaminated rinsing water and soiled gowning;
- Recording of spills, accidental events or deviations from procedures;
- Design of cleaning processes for premises and equipment such that the cleaning processes in themselves do not present a cross-contamination risk;
- Design of detailed records for cleaning processes to assure completion of cleaning in accordance with approved procedures and use of cleaning status labels on equipment and manufacturing areas;
- Use of common general wash areas on a campaign basis;
- Supervision of working behaviour to ensure training effectiveness and compliance with the relevant procedural controls.
Appendix B - Glossary
Acronyms
- API
- Active pharmaceutical ingredient
- GMP
- Good manufacturing practices
- HBEL
- Health based exposure limit
- ICH
- International Council for Harmonisation
- PDE
- Permissible daily exposure
- PIC/S
- Pharmaceutical Inspection Co-operation Scheme
- QRM
- Quality risk management
- TOC
- Total organic carbon
- TTC
- Threshold of toxicological concern
Terms
These definitions explain how terms are used in this document, as well as in the annexes (unless otherwise specified). Definitions cited directly from other documents are noted in brackets at the end of the definition.
If there is a conflict with a definition in the Food and Drugs Act or Food and Drug Regulations, the definition in the Act/Regulations prevails.
Drug – Includes any substance or mixture of substances manufactured, sold or represented for use in:
- the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state, or its symptoms, in human beings or animals,
- restoring, correcting or modifying organic functions in human beings or animals, or
- disinfection in premises in which food is manufactured, prepared or kept;
(Section 2 of the Food and Drugs Act)
From Division 1 A of the FDR;
(2) In this Division and in Division 2, drug does not include any of the following:
- a dilute drug premix;
- a medicated feed as defined in subsection 2(1) of the Feeds Regulations, 1983;
- an active ingredient that is for veterinary use and that is not an active pharmaceutical ingredient;
- an active pharmaceutical ingredient for veterinary use that is not required to be sold pursuant to a prescription and that is also a natural health product as defined in subsection 1(1) of the Natural Health Products Regulations;
- a drug that is used only for the purposes of an experimental study in accordance with a certificate issued under section C.08.015.
Fabricate – "To prepare and preserve a drug for the purpose of sale." (C.01A.001)
Permissible daily exposure – The PDE represents a substance-specific dose that is unlikely to cause an adverse effect if an individual is exposed at or below this dose every day for a lifetime. (PIC/S PI 046-1)
Threshold of toxicological concern - The TTC represents the genotoxic impurity exposure level associated with a theoretical cancer risk of 1 additional cancer in 100,000 patients when exposed over a life time. (PIC/S PI 046-1)
Validation – A documented program that provides a high degree of assurance that a specific process, method, or system will consistently produce a result meeting pre-determined acceptance criteria. (ICH Q7)
Appendix C – References
Laws and regulations
Health Canada guidance documents
- Good manufacturing practices guide for drug products (GUI-0001)
- Annex 4 to the Current Edition of the Good Manufacturing Practices Guidelines - Veterinary Drugs (GUI-0012)
- Annex 7 to the Good Manufacturing Practices Guide for drug products - Selected non-prescription drugs (GUI-0066)
International guidance documents
- ASTM E3106 - 18e1 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation
- ICH M7 - Genotoxic Impurities - Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk
- ICH Q2 - Validation of Analytical Procedures: Text and Methodology
- ICH Q3A - Impurities in New Drug Substances
- ICH Q3B - Impurities in New Drug Products
- ICH Q3C - Impurities: Guideline for Residual Solvents
- ICH Q3D - Guideline for Elemental Impurities
- ICH Q7 - Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
- ICH Q9 - Quality Risk Management
- ISPE Baseline Guide Vol 7: Risk-Based Manufacture of Pharma Products
- Pharmaceutical Inspection Cooperation Scheme - Guideline on exposure limits - Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (PI 046-1)
- Pharmaceutical Inspection Cooperation Scheme - Questions and answers on implementation of risk-based prevention of cross-contamination in production and 'Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities' (PI 053-1).
Other related documents
- Food and Drug Administration – Validation of Cleaning Processes (7/93)
- Parenteral Drug Association - Technical Report no. 29 (Revised 2012) "Points to Consider for Cleaning Validation"
- Parenteral Drug Association - Technical Report No. 49 (Revised 2010) "Points to Consider for Biotechnology Cleaning Validation"
- Pharmaceutical Inspection Cooperation Scheme - Guide to Good Manufacturing Practice for Medicinal Product Annexes [PE 009-14(Annexes)]
- Pharmaceutical Inspection Cooperation Scheme - Guide to good Manufacturing Practice for Medicinal Products Part I, Chapter 5 [PE 009-14 (Part 1)]
- Pharmaceutical Inspection Cooperation Scheme - Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation and Cleaning Validation (PI 006-3)
- VICH GL 18 – Residual solvents in new veterinary medicinal products, active substances and excipients (Revision)