
Guide sur la validation des procédés de nettoyage (GUI-0028)
Avertissement
Le présent document ne constitue pas une partie de la Loi sur les aliments et drogues (la Loi) ou de ses règlements. En cas de contradiction ou d'incompatibilité entre la Loi ou les règlements et le présent document, la Loi ou les règlements auront préséance. Le présent document est un document administratif destiné à favoriser la conformité des parties réglementées à la Loi, aux règlements et aux politiques administratives applicables.

Télécharger le format de rechange
(Format PDF, 1,1 Mo, 42 pages)
Organisation : Santé Canada
Date publiée : 2021-06-29
Table des matières
- À propos du présent document
- Orientation
- 4. Principes
- 5. Appliquer les principes de GRQ pour contrôler les risques de contamination croisée
- 6. Plan maître de validation du nettoyage
- 7. Approche de validation du nettoyage axée sur le cycle de vie
- 8. Méthodes d'échantillonnage et d'analyse
- 9. Évaluation du nettoyage
- 10. Établir les limites
- 11. Mesures de contrôle microbiologiques
- 12. Renseignements généraux sur le nettoyage de l'équipement
- 13. Renseignements supplémentaires sur le nettoyage de l'équipement de production des IPA
- 14. Renseignements supplémentaires sur la validation du nettoyage des procédés biotechnologiques
- Annexes
Voici les deux types d'icônes utilisés dans le présent document et leur signification.
Important: Renseignement clé ou de mise en garde que les gens doivent connaître.
Renseignements : Renseignements supplémentaires, comme des citations et des références juridiques.
À propos du présent document
1. Objet
Le présent guide s'adresse à toutes les personnes qui participent à la fabrication et à la réglementation de produits pharmaceutiques, biologiques et radiopharmaceutiques vendus au Canada, notamment :
- l'industrie réglementée;
- les inspecteurs et les évaluateurs.
Il fournit des lignes directrices sur la validation des procédés de nettoyage. Il vous aidera à comprendre et à respecter le titre 2 de la partie C du Règlement sur les aliments et drogues (le Règlement).
Le présent document vise également à assurer l'uniformité et la cohérence des procédures d'inspection appliquées aux procédés de nettoyage de l'équipement. Les principes énoncés dans les directives internationales ont été pris en considération lors de la rédaction du présent document.
2. Portée
Le présent document traite de facteurs et de problèmes particuliers à prendre en compte lors de la validation des procédés de nettoyage de l'équipement utilisé pour fabriquer (manufacturer) et emballer :
- les ingrédients pharmaceutiques actifs (IPA);
- les produits pharmaceutiques;
- les produits radiopharmaceutiques;
- les produits biologiques;
- les médicaments vétérinaires.
Il couvre la validation des procédés de nettoyage de l'équipement pour :
- l'élimination des résidus associés aux produits utilisés dans le cycle de production précédent, tels que les ingrédients actifs, les produits de dégradation ou les sous-produits préoccupants, les produits intermédiaires, les résidus d'agents de nettoyage et les agents de transformation;
- le contrôle des contaminants microbiens potentiels.
Des directives supplémentaires sur la validation des procédés de nettoyage de certains médicaments vétérinaires et des médicaments de catégorie IV sont disponibles dans ces documents d'orientation de Santé Canada :
Bien que le présent guide porte sur la validation des procédés de nettoyage, les références suivantes sur les impuretés de l'International Council for Harmonisation (ICH) peuvent également être utiles :
- ICH M7 : Évaluation et contrôle des impuretés réactives de l'ADN (mutagènes) dans les produits pharmaceutiques pour limiter les risques de cancérogénicité (disponible en anglais seulement)
- ICH Q3A : Présence d'impuretés dans les nouvelles substances médicamenteuses
- ICH Q3B : Présence d'impuretés dans les nouveaux produits
- ICH Q3C : Impuretés : Directives sur les solvants résiduels
- ICH Q3D : Directive concernant les impuretés élémentaires
3. Introduction
Le présent guide décrit les exigences relatives aux bonnes pratiques de fabrication (BPF) qui énoncées au Titre 2 de la partie C du Règlement. Le document a été élaboré par Santé Canada en consultation avec des intervenants.
Les documents d'orientation comme celui-ci ont pour but d'aider l'industrie et les professionnels de la santé à comprendre comment se conformer à la réglementation. Ils guident également le personnel de Santé Canada afin que les règles soient appliquées de manière équitable, uniforme et efficace dans l'ensemble du Canada.
Santé Canada inspecte les établissements afin d'évaluer leur conformité à la Loi sur les aliments et drogues (la Loi) et aux règlements connexes. Lorsque nous effectuons une inspection, nous nous servons du présent document comme guide pour déterminer si vous vous conformez aux exigences des BPF relatives au nettoyage de l'équipement.
Le présent guide n'est pas la seule interprétation des BPF et n'aborde pas tous les cas possibles. D'autres moyens de se conformer à la réglementation des BPF seront aussi pris en considération avec les justifications scientifiques appropriées. De plus, d'autres approches deviendront peut-être nécessaires avec l'apparition de nouvelles technologies. Le présent document s'appuie sur d'autres documents d'orientation internationaux (voir la section Références).
Les documents d'orientation sont de nature administrative et n'ont pas force de loi. Pour cette raison, ils permettent une certaine souplesse sur le plan de l'approche. Ce guide vous aidera à définir des approches particulières qui répondent à vos besoins uniques.
Orientation
4. Principes
La validation des procédés de nettoyage vise à s'assurer que le procédé de nettoyage de l'équipement réduira de manière systématique les risques de contamination croisée dans un processus de fabrication de médicaments. Elle fournit des documents attestant qu'un procédé de nettoyage approuvé permet d'éliminer de manière reproductible les produits antérieurs, les sous-produits préoccupants ou les résidus d'agents de nettoyage pouvant demeurer sur l'équipement, de sorte que leurs teneurs soient inférieures aux limites scientifiquement établies. Ces limites sont calculées en fonction de valeurs correspondant au seuil de sûreté, qui sont déterminées par une évaluation toxicologique.
Tous les procédés de nettoyage de l'équipement venant en contact avec le produit doivent être validés conformément aux principes de la gestion des risques liés à la qualité (GRQ). Il faut également accorder une attention aux parties de l'équipement qui ne sont pas en contact direct avec le produit, mais dans lesquelles les produits peuvent se loger. Cette démarche devrait être fondée sur le risque.
Il convient de mettre en œuvre des mesures correctives si un procédé de nettoyage ne produit pas de façon constante des résultats adéquats. Des exemples de mesures correctives comprennent des procédés de nettoyage améliorés et l'utilisation d'équipement et d'installations dédiés. Des défaillances persistantes dans les procédés de nettoyage, ou le fait de poursuivre les nettoyages et les analyses jusqu'à l'obtention du degré voulu de propreté, ne sont pas acceptables.
Il est également important de démontrer que l'installation et l'équipement sont conçus, nettoyés et utilisés de manière à prévenir la contamination microbienne des produits.
4.1 À propos des valeurs de seuil de sureté
Santé Canada a l'intention de s'harmoniser avec les lignes directrices adoptées le 1er juillet 2018 par le Pharmaceutical Inspection Co-operation Scheme (PIC/S) sur l'utilisation de l'évaluation toxicologique dans l'établissement de limites d'exposition fondées sur la santé (LEFS). Cette approche permettra à une personne qualifiée d'entreprendre une évaluation de toutes les données pharmacologiques et toxicologiques afin de déterminer une valeur de seuil de sûreté quotidienne, comme l'exposition journalière admissible (EJA) ou le seuil de préoccupation toxicologique (SPT).
EJA représente une dose à laquelle il est peu probable qu'une substance particulière cause un effet néfaste si une personne est exposée à cette dose ou à une dose inférieure chaque jour pendant toute sa vie.
Le SPT représente le niveau d'exposition aux impuretés génotoxiques qui est associé à un risque théorique de cancer correspondant à 1 cancer supplémentaire pour 100 000 patients exposés au cours d'une vie.
Les définitions sont tirées de PIC/S Guideline on exposure limits – Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (PI 046-1) (disponible seulement en anglais).
Vous trouverez des renseignements supplémentaires dans le document de questions et réponses suivant, publié par le PIC/S.
Questions and answers on implementation of risk-based prevention of cross-contamination in production and 'Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities' (PI 053-1) (disponible seulement en anglais).
Les LEFS, comme l'EJA ou le SPT, peuvent ensuite servir à la détermination des risques et à la justification des limites maximales d'innocuité du transfert dans le produit suivant. D'autres approches pour déterminer les limites d'exposition fondées sur la santé peuvent être jugées acceptables à la lumière d'une justification scientifique et des principes de GRQ.
Il convient de noter que la directive du PIC/S établit que les termes anglais PDE (Permissible Daily Exposure) et ADE (Allowable Daily Exposure), utilisés pour désigner l'exposition journalière admissible, sont synonymes.
5. Appliquer les principes de GRQ pour contrôler les risques de contamination croisée
Vous avez l'obligation de prévenir la contamination croisée des drogues, par l'élaboration d'une stratégie de contrôle de la contamination. Cette stratégie comprendra la conception et l'établissement de contrôles appropriés des locaux, de l'équipement et de tous les processus connexes. Il convient de reconnaître que le nettoyage de l'équipement n'est qu'une des nombreuses mesures à prendre pour limiter le risque de contamination croisée dans une installation multi-produits ou sur l'équipement devant être partagé.
- Les mesures prises devraient être proportionnelles aux risques définis; par exemple, un contrôle accru est nécessaire pour les produits présentant des LEFS plus faibles.
- Toutes les sources éventuelles de contamination croisée devraient être évaluées au moyen d'un processus de GRQ documenté. Le processus de GRQ devrait inclure une évaluation des risques en fonction des connaissances et des justifications scientifiques, ainsi que la détermination des mesures qui permettraient de réduire ces risques.
- Le résultat du processus de GRQ devrait servir de base pour déterminer l'étendue des mesures techniques et organisationnelles nécessaires pour contrôler les risques de contamination croisée. Voir l'annexe A du présent document pour obtenir une liste des mesures techniques et opérationnelles possibles.
- Les mesures visant à prévenir la contamination croisée et leur efficacité doivent être examinées périodiquement selon les procédures établies.
- Si le processus de GRQ confirme que la drogue peut être fabriquée en toute sécurité sur un équipement partagé, valider tout procédé de nettoyage de l'équipement à utiliser.
Pour obtenir de plus amples renseignements sur la GRQ, consultez les documents ICH thème Q9 – Gestion des risques liés à la qualité et ASTM E3106 - 18e1 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation (disponible seulement en anglais).
6. Plan maître de validation du nettoyage
Vous devez dresser un plan maître de validation du nettoyage (ou un document équivalent) pour décrire les politiques générales de validation du nettoyage de votre site.
- Les produits et l'équipement peuvent être regroupés selon les principes de GRQ :
- Vous pouvez choisir d'effectuer des études de validation du nettoyage sur tous les produits dans l'installation ou uniquement sur les produits correspondant à la pire éventualité (l'approche par famille de produits). Précisez (et justifiez au besoin) l'approche utilisée dans le plan maître de validation du nettoyage. Documentez les éléments suivants si vous utilisez une approche de la pire éventualité :
- la méthodologie/les justifications scientifiques utilisées pour déterminer les produits correspondant à la pire éventualité;
- les produits correspondant à la pire éventualité, y compris une liste de tous les produits dont ils sont représentatifs.
Voici des exemples de facteurs pouvant être inclus dans l'évaluation des produits correspondant à la pire éventualité :
- la valeur LEFS du résidu;
- la difficulté du nettoyage ou la nettoyabilité du produit;
- la solubilité des résidus dans les agents de nettoyage et dans les solvants de nettoyage;
- les caractéristiques physiques du produit, des ingrédients pharmaceutiques actifs ou des excipients;
- l'expérience passée (par exemple, lors du développement et avec des produits similaires).
Il convient de noter qu'il peut y avoir plusieurs produits correspondant à la pire éventualité. Par exemple, un produit insoluble dont la valeur LEFS est élevée peut être le produit le plus difficile à nettoyer, mais il ne s'agit pas nécessairement de la pire éventualité si on le compare à un produit partiellement soluble dont la valeur LEFS est faible.
- Vous pouvez choisir d'effectuer des études de validation du nettoyage sur tout l'équipement ou en regroupant l'équipement semblable, par exemple l'équipement équivalent sur le plan fonctionnel. Une approche fondée sur l'équipement représentatif convient seulement si la taille, la conception, les fonctions, la procédure de nettoyage et la nettoyabilité des différents équipements sont équivalentes. S'il y a des différences dans l'équipement, la proposition de les regrouper devrait être fondée sur des données. Documentez ce qui suit si une méthode de regroupement d'équipement est utilisée :
- L'approche/les justification scientifique en vertu desquelles l'équipement a été regroupé.
- La liste de l'ensemble de l'équipement dans chaque groupe. Indiquez dans chaque groupe l'équipement qui correspond à la pire éventualité ainsi que la justification appropriée.
- Vous pouvez choisir d'effectuer des études de validation du nettoyage sur tous les produits dans l'installation ou uniquement sur les produits correspondant à la pire éventualité (l'approche par famille de produits). Précisez (et justifiez au besoin) l'approche utilisée dans le plan maître de validation du nettoyage. Documentez les éléments suivants si vous utilisez une approche de la pire éventualité :
- Tous les procédés de nettoyage doivent être équivalents si les études de validation du nettoyage doivent être effectuées selon une approche de regroupement d'équipement ou en fonction des produits constituant la pire éventualité.
7. Approche de validation du nettoyage axée sur le cycle de vie
La validation, dans le cadre d'une approche axée sur le cycle de vie, nécessite de collecter et d'évaluer des données tout au long du cycle de vie du produit. Les leçons tirées à chaque étape sont fondamentales pour s'assurer que les mesures de contrôle appropriées sont mises en place. Veuillez consulter la section 7.2 : Étape 2 – Qualification des procédés de nettoyage pour obtenir des renseignements supplémentaires.
Pour la validation du nettoyage, l'approche axée sur le cycle de vie comprend normalement les étapes suivantes :
- Étape 1 – Élaboration du procédé de nettoyage : Élaborer des procédures de nettoyage efficaces, documentées et contrôlées avant de les mettre en œuvre.
- Étape 2 – Qualification des procédés de nettoyage : Évaluer les procédés de nettoyage pour s'assurer qu'ils sont efficaces et reproductibles. Les études de qualification des procédés de nettoyage consistent à évaluer la vérification du nettoyage un nombre prédéterminé de fois dans des conditions précises.
- Étape 3 – Surveillance continue : S'assurer que les procédés de nettoyage demeurent efficaces et sont contrôlés au moyen d'un programme de surveillance continue.
Il convient de faire la distinction entre trois termes importants en fonction d'où ils s'inscrivent dans l'approche de nettoyage générale axée sur le cycle de vie.
La vérification du nettoyage renvoie à la collecte de données, au moyen d'une méthode d'analytique appropriée après chaque lot ou campagne, pour montrer que les résidus préoccupants ont été réduits en dessous des limites de transfert prédéfinies, établies selon les seuils jugés sécuritaires sur le plan scientifique.
La vérification du nettoyage renvoie à une étude ou à un exercice individuel de nettoyage et d'échantillonnage visant à évaluer la propreté de l'équipement. La vérification du nettoyage est utilisée tout au long de l'approche axée sur le cycle de vie. Les études de vérification du nettoyage devraient être menées conformément à une procédure ou à un protocole de nettoyage établi. Les exigences d'échantillonnage au cours d'une étude de vérification du nettoyage devraient être, au minimum, équivalentes à celles de l'étape de qualification des procédés de nettoyage.
La qualification des procédés de nettoyage renvoie à une étape définie du cycle de vie de la validation du nettoyage, où l'on démontre que le procédé de nettoyage est robuste et reproductible. Elle comprend normalement plusieurs essais ou études de vérification du nettoyage pour l'ensemble de l'équipement inclus dans l'étude de qualification des procédés de nettoyage.
La validation du nettoyage renvoie au programme de validation général, depuis l'étape de développement jusqu'à l'étape de surveillance continue. Le programme de validation du nettoyage comprend les procédures de nettoyage adéquatement contrôlées et la possession de données suffisantes pour prouver leur efficacité.
Figure 1 – Aperçu du programme de validation du nettoyage
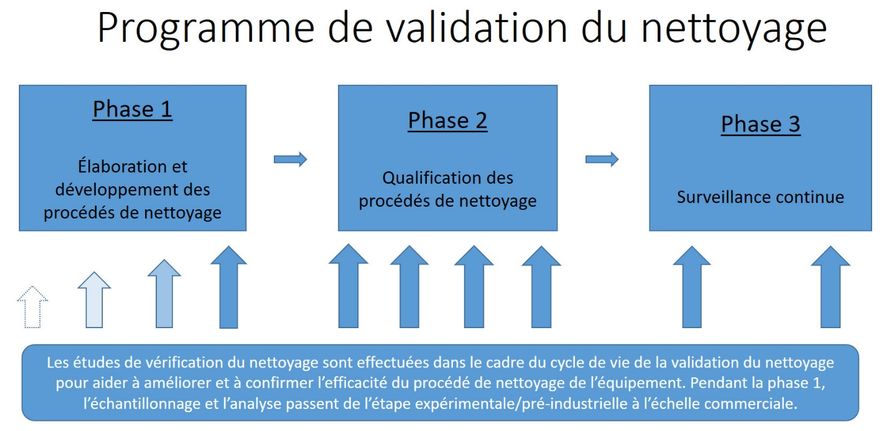
Figure 1 - Équivalent textuel
Le diagramme s'intitule « Figure 1 – Aperçu du programme de validation du nettoyage ».
Le diagramme se compose d'un grand encadré intitulé « Programme de validation du nettoyage ».
Cet encadré contient trois encadrés plus petits placés à l'horizontale, séparés par une flèche menant du premier encadré au deuxième encadré, puis du deuxième encadré au troisième encadré. Ces trois encadrés représentent les trois phases du programme de validation du nettoyage.
Le premier encadré contient la mention « Phase 1 – Élaboration et développement des procédés de nettoyage ».
Le deuxième encadré contient la mention « Phase 2 – Qualification des procédés de nettoyage ».
Le troisième encadré contient la mention « Phase 3 – Surveillance continue ».
Plusieurs flèches pointant vers le haut se trouvent sous les trois encadrés. Ces flèches représentent la formalité des vérifications de nettoyage ainsi que le nombre de vérifications effectuées pendant chaque phase.
Les quatre flèches figurant sous le premier encadré sont de plus en plus grandes et leur couleur s'accentue afin de représenter la quantité accrue d'effort et le plus grand degré de formalité auquel est assujetti l'essai pendant l'élaboration du procédé.
Les quatre flèches sous le deuxième encadré sont toutes de la même taille et de la même couleur pour représenter le nombre de vérifications de nettoyage officielles devant être effectuées pendant la phase 2.
Les deux flèches figurant sous le troisième encadré sont de même grandeur et couleur et représentent la diminution de la fréquence des vérifications de nettoyage pendant la phase de surveillance continue.
Un dernier encadré, qui figure sous les flèches, contient une description du processus général. « Les études de vérification du nettoyage sont effectuées dans le cadre du cycle de vie de la validation du nettoyage pour aider à améliorer et à confirmer l'efficacité de l'équipement. Pendant la phase 1, l'échantillonnage et l'analyse passent de l'étape expérimentale/pré-industrielle à l'échelle commerciale. »
7.1 Première étape – Élaboration et développement des procédés de nettoyage
Les procédures de nettoyage devraient être élaborées de manière contrôlée, conformément aux principes et aux outils de la GRQ, afin de s'assurer que les procédés de nettoyage sont efficaces et reproductibles. Les facteurs qui peuvent influer sur l'efficacité du nettoyage doivent être définis et contrôlés.
- Des efforts et des ressources appropriées doivent être déployés pour concevoir et mettre au point les procédés de nettoyage. Il pourrait s'agir d'essais en laboratoire, d'essais sur des coupons de matériaux, d'essais à petite échelle, d'essais à l'échelle pilote jusqu'aux d'essais à l'échelle commerciale.
- Il est important de tenir compte des problèmes potentielles qui pourraient avoir une incidence sur l'efficacité et la reproductibilité des procédés de nettoyage lors de l'élaboration de procédés de nettoyage nouveaux ou révisés. Les éléments à prendre en considération sont les suivants :
- Comprendre les propriétés chimiques et physiques des principes actifs, des excipients et des sous-produits ou des produits de dégradation. Ces connaissances sont nécessaires pour aider à déterminer les agents de nettoyage, les solvants et les paramètres de procédés de nettoyage les plus appropriés. Il convient de noter que les sous-produits peuvent également être créés par l'interaction avec les agents de nettoyage et les solvants.
- Examiner la conception de l'équipement. Considérer les dessins techniques, l'expérience du personnel d'entretien et de nettoyage et l'examen de l'équipement démonté, à l'état propre et sale, pour établir les zones sujettes à l'accumulation ou à la migration de résidus. Prêter attention aux matériaux de construction des pièces d'équipement, à leur aspect lisse, et évaluer l'impact que ces substrats ont sur l'enlèvement des résidus. Tous les endroits d'échantillonnage devraient être dûment justifiés. Il s'agit d'une considération importante lors de la mise à l'échelle en production, car la taille et la conception de l'équipement devraient être évaluées.
- Évaluer tout risque de contamination par des endotoxines ou de prolifération microbienne dans les produits sensibles par le biais de matériaux entrants, l'utilisation, la manipulation, les temps de rétention et l'entreposage. Évaluer si des mesures supplémentaires de désinfection ou de contrôle des endotoxines sont nécessaires après l'entreposage de l'équipement, le cas échéant.
- Déterminer si une variation des matières premières pourrait avoir une incidence sur la nettoyabilité.
- Examiner l'environnement dans lequel on propose d'effectuer le nettoyage. Veiller à ce que des mesures de contrôle appropriées soient mises en place pour l'installation et l'environnement, afin de faciliter le nettoyage, le temps de contact et le séchage requis et de prévenir tout risque de contamination croisée.
- Tous les éléments, paramètres et contrôles du procédé de nettoyage, tels que les agents de nettoyage, les solvants, les paramètres critiques de nettoyage (le temps, la température, les pressions et actions tel que le récurage, le trempage, la circulation ou le reflux]), doivent être scientifiquement établis. Des essais particuliers peuvent être nécessaires. L'objectif est de déterminer les paramètres de nettoyage critiques et de comprendre l'incidence de la variabilité de ces paramètres sur la performance du nettoyage.
- Documentez les apprentissages au cours du processus du développment du nettoyage pour assurer le transfert des connaissances et utilisez-les pour définir une procédure de nettoyage détaillée. Utilisez des outils d'évaluation des risques pour déterminer les risques éventuels liés à la procédure de nettoyage et effectuer les modifications nécessaires.
- Vous devez avoir l'assurance que les procédures de nettoyage seront efficaces et reproductibles avec l'équipement commercial ou à grande échelle. La ou les vérifications du nettoyage peuvent être effectuées sur l'équipement à grande échelle comme dernière étape de la conception et de l'élaboration du procédé de nettoyage, afin de confirmer l'efficacité du procédé de nettoyage proposé. Voir la section 7.2.1 pour obtenir des conseils sur le lancement de l'équipement commercial utilisé dans les études de vérification des procédés de nettoyage.
- Les travaux d'élaboration des procédés de nettoyage doivent aboutir à un procédé de nettoyage bien défini à la fois reproductible et efficace.
7.1.1 Contrôle des procédés de nettoyage manuel
L'uniformité des actions des opérateurs est l'un des plus grands défis d'un programme de nettoyage manuel. L'évaluation de la GRQ doit déterminer clairement les étapes à suivre pour assurer l'uniformité dans le mode d'exécution des procédures ainsi que dans le résultat général du processus de validation du nettoyage.
Assurez-vous que les procédures de nettoyage manuel sont exécutées de façon uniforme en procédant comme suit :
- Dotez-vous d'instructions suffisamment détaillées et établissez les limites ou la valeur des paramètres essentiels du processus :
- Des instructions détaillées de démontage;
- La séquence des étapes de nettoyage;
- L'agent de nettoyage à utiliser et sa concentration;
- Les modes d'application de l'agent de nettoyage (p. ex. trempage ou récurage);
- La durée du contact;
- La température des solutions de nettoyage ou des rinçages;
- Les techniques de rinçage (c'est-à-dire, prérinçage, trempage, rinçage, durées et pressions)
- La méthode de séchage.
- Mettez en place et tenez à jour des programmes de formation des opérateurs, qui peuvent comprendre des exigences de certification fondées sur le risque. Il n'est pas acceptable d'attribuer de façon répétée la responsabilité des défaillances dans les procédés de nettoyage aux techniques de nettoyage inappropriées (p. ex. erreur de l'opérateur), car cela indique des mesures de contrôle inadéquates.
- Veillez à ce que les procédures de nettoyage et les paramètres critiques soient dûment documentés et vérifiés, le cas échéant. Les dossiers doivent être suffisamment détaillés pour confirmer l'achèvement des étapes clés du procédé de nettoyage.
- Vérifiez que des dispositifs de mesure étalonnés (tels que des minuteries, des sondes de température, des pompes de dosage et des débitmètres) sont utilisés, au besoin.
7.1.2 Contrôle des procédés de nettoyage automatisé
Contrôlez l'équipement de nettoyage automatisé (par exemple, nettoyage en place) et les procédés comme suit :
- Assurez-vous que les procédures sont suffisamment détaillées et décrivent le nettoyage automatisé, les contrôles et les exigences comme la préparation ou le démontage de l'équipement et les configurations de charge.
- Qualifiez l'équipement utilisé pour ce type de nettoyage et vérifiez que toutes les surfaces en contact avec le produit sont en contact adéquat avec les agents de nettoyage et de rinçage.
- Assurez-vous que des programmes d'étalonnage et d'entretien appropriés sont établis et maintenus.
- Définissez les séquences de nettoyage y compris l'ensemble des températures, des concentrations, l'ouvertures de soupapes, des débits de pulvérisation, des pressions et des volumes. Il est également important de s'assurer que les séquences de nettoyage contrôlées par les recettes automatisées sont adéquatement protégées des changements non approuvés ou non contrôlés.
- Surveillez les points de contrôle critiques et les paramètres avec des capteurs et des alarmes appropriés pour assurer un contrôle élevé des procédés. Les alarmes critiques devraient être identifiées et régulièrement vérifiées. Les procédures doivent décrire les étapes à suivre en réponse à de telles alarmes.
- Veillez à ce que les séquences de nettoyage et les données, y compris les alarmes, soient contrôlées et examinées de manière appropriée.
7.2 Étape 2 – Qualification des procédés de nettoyage
- Vous pouvez entamer une étude de qualification des procédés de nettoyage une fois que vous disposez d'un procédé de nettoyage entièrement défini. Cette étude peut être effectuée avant le début de la production commerciale si l'équipement, la taille des lots et les paramètres de formulation et d'operation ne sont pas susceptibles de faire l'objet de modifications. L'étude de qualification des procédés de nettoyage est normalement initiée lorsque la production commerciale est amorcée.
- Menez des études de qualification des procédés de nettoyage pour tous les produits ou pour les produits correspondant à la pire éventualité si une approche par famille de produits est utilisée.
- Utilisez les principes de GRQ pour déterminer l'étendue et la portée des exigences de qualification des procédés de nettoyage :
- Déterminez le nombre de nettoyages à évaluer au moyen d'une évaluation des risques documentée. Bien qu'une évaluation de trois études de nettoyages ait longtemps été la norme de l'industrie, votre évaluation des risques peut aboutir à une recommandation qui vous demande d'évaluer un nombre différent d'etudes nettoyage.
- Bien que les tests de mise à l'épreuve des pires éventualités devraient être évalués au cours de l'étape de développement du procédé de nettoyage, il peut être approprié de vérifier les limites des paramètres critiques du procédé de nettoyage au cours des études de qualification. Ces tests pourraient inclure la durée minimale de contact avec le détergent, les températures minimales ou maximales, le temps minimal de rinçage, le volume minimal et la pression minimale. Les tests de mise à l'épreuve des pires éventualités sont particulièrement importants lorsque des systèmes de nettoyage manuel sont utilisés.
- La variabilité de l'opérateur doit également être évaluée, en particulier lorsque des procédés de nettoyage manuel sont utilisés.
- Documentez les exigences de qualification des procédés de nettoyage au sein d'un protocole. Le protocole doit comprendre les éléments suivants :
- L'objectif et la portée de l'exercice de qualification des procédés de nettoyage;
- Les responsabilités en matière d'exécution et d'approbation de l'étude de qualification;
- Une description de l'équipement à utiliser pour le procédé et le nettoyage;
- Les références et les descriptions des procédures de nettoyage et des paramètres à utiliser, incluant une description de tous les paramètres critiques;
- Les pires éventualités à évaluer dans l'étude;
- Le nombre de cycles de nettoyage à être exécutes;
- Les procédures d'échantillonnage;
- Les endroits d'échantillonnage clairement définis, avec une justification de leur sélection;
- Les méthodes d'analyse validées, appropriées pour les limites de résidus à évaluer , et les données sur les études de récupération;
- Les critères d'acceptation et les justifications pour l'établissement des certaines limites.
Remarque – La justification et les données à l'appui des approches adoptées peuvent figurer dans d'autres documents auxquels le protocole fait référence.
- Assurez-vous d'avoir des données pour démontrer que les variables suivantes n'ont pas d'incidence sur l'efficacité du nettoyage :
- La durée entre la fin de la fabrication et le début du nettoyage (durée du maintien en état de saleté);
- Le nombre de lots du même produit qui peuvent être fabriqués avant le nettoyage complet (précisez la durée maximale des campagnes en jours ou en nombre de lots).
- Enquêtez les défaillances dans les procédés de nettoyage conformément aux exigences du système qualité.
- Préparez un rapport de qualification final. Les conclusions de ce rapport doivent indiquer si le procédé de nettoyage a été qualifié avec succès.
7.2.1 Libération de l'équipement
Il est entendu que l'évaluation du nombre requis d'études de vérifications des procédés de nettoyage pendant l'étape de qualification peut prendre du temps. En outre, on prévoit que l'équipement utilisé pour d'autres produits commerciaux devra parfois être utilisé pour les études de vérification du nettoyage pendant l'étape de conception et de développement et lors de l'introduction de nouveaux produits dans une installation. Les principes de la GRQ devraient être utilisés pour déterminer si la libération de l'équipement destiné à la fabrication d'autres produits commerciaux est acceptable avant que l'étape de qualification des procédés de nettoyage ne soit terminée. Les données issues de l'étude de vérification devraient être examinées et jugées suffisantes pour appuyer la libération de l'équipement. Il convient de prendre en considération le risque de défaillances ultérieures lors des étapes de vérification des procédés de nettoyage.
7.3 Étape 3 – Surveillance continue
- Établissez des exigences de surveillance continue au terme de l'étape de qualification des procédés de nettoyage pour démontrer que le processus de nettoyage demeure sous contrôle.
- La surveillance continue peut comprendre un certain nombre d'activités différentes, notamment :
- L'analyse des données (telles que les données générées à partir de processus automatisés)
- Des études supplémentaires de vérification des procédés de nettoyage
- L'analyse de l'échantillon de rinçage
- Le nombre et la nature des exigences de surveillance continue ainsi que la fréquence à laquelle de vérification des procédés de nettoyage supplémentaires sont proposées devraient être déterminés selon les principes de GRQ. Des activités telles que l'analyse de l'échantillon de rinçage peuvent être utilisées pour aider à surveiller le programme et fournir des données pour indiquer qu'il demeure sous contrôle. La vérification du nettoyage fournit normalement une analyse plus approfondie de la performance du procédé de nettoyage. Ainsi, des activités comme la surveillance des échantillons de rinçage n'éliminent pas la nécessité de procéder à des évaluations périodiques de vérification des procédés de nettoyage.
- Les domaines préoccupants en ce qui a trait à la surveillance continue comprennent :
- les produits dont les valeurs LEFS sont faibles, lesquels sont généralement plus dangereux;
- les produits pour lesquels l'inspection visuelle ne peut pas être utilisée pour estimer la propreté de l'équipement, c'est-à-dire que les niveaux de résidus établis d'après les LEFS ne peuvent pas être détectés visuellement;
- l'équipement et les produits présentant des antécédents en matière de défaillance ou des résultats extrêmement variables aux tests de vérification et de qualification;
- les procédés de nettoyage manuel;
- les endroits ou les surfaces auxquels il est difficile d'accéder ou qui sont difficiles à nettoyer;
- l'équipement qui ne peut être inspecté visuellement de façon appropriée.
- Des mesures appropriées doivent être prises en temps utile s'il y a des signes indiquant que les procédés de nettoyage ne sont pas adéquatement contrôlés. Différentes démarches peuvent mettre en évidence un contrôle inadéquat :
- L'évaluation statistique des données générées par les vérifications des procédés de nettoyage et/ou toute donnée générée par le nettoyage de routine lui-même;
- L'examen des indicateurs de qualité traditionnels, tels que les plaintes, les écarts et les défaillances observées en laboratoire.
- Les données relatives aux procédés de nettoyage et les indicateurs de qualité devraient être régulièrement examinés pour déterminer les tendances ou les défaillances qui pourraient indiquer la nécessité de considérer l'examen des mesures techniques ou opérationnelles.
7.3.1 Contrôle des changements et requalification
- Assurez-vous qu'un système de contrôle des changements est en place pour évaluer et documenter tous les changements qui pourraient avoir une incidence sur les procédés de nettoyage. L'examen doit permettre d'établir si les procédés de nettoyage doivent être élaborés et/ou qualifiés de nouveau.
- Les changements susceptibles d'avoir une incidence sur la qualification ou la validation des procédés de nettoyage comprennent ce qui suit :
- Les nouveaux produits;
- Les changements apportés aux procédés de nettoyage;
- Les changements dans la formulation ou le traitement des produits;
- Les changements apportés aux matières premières (p. ex. changement du profil d'impureté ou des propriétés physiques);
- Les nouveaux agents de nettoyage ou les changements dans la formulation des agents de nettoyage;
- Les changements importants apportés à l'équipement ou utilisation d'un nouvel équipement;
- Les changements apportés à la taille du lot ou à la durée de la campagne;
- Les changements apportés aux méthodes d'analyse, aux matériaux ou à la méthode d'échantillonnage;
- Les changements apportés aux limites de nettoyage, notamment à la suite d'examens périodiques des données qui constituent la base des LEFS.
Remarque : L'installation d'équipement usagé (c.-à-d. équipement provenant d'autres sites) peut poser des défis particuliers pour ce qui est de s'assurer que la propreté de l'équipement est évaluée adéquatement avant son utilisation. Dans une telle situation, les principes de la GQR devraient être pris en compte.
7.3.2 Introduction de nouveaux produits dans une installation
Tous les nouveaux produits introduits dans les installations de fabrication devraient être examinées dans le cadre du processus de GRQ et du contrôle des changements afin de déterminer si les contrôles techniques et organisationnels existants sont suffisants ou doivent être modifiés. Il faut tenir compte des éléments suivants :
- Les LEFS du nouveau produit, la convenance du produit pour votre installation et la nécessité de recourir à des installations ou de l'équipement dédiés.
- La facilité de nettoyage de l'équipement utilisé pour fabriquer le nouveau produit, si le nouveau produit correspond à la pire éventualité. Déterminez ensuite si les procédés de nettoyage existants sont adéquats ou si un nouveau procédé ou un procédé révisé est nécessaire. Si un travail de développement supplémentaire du procédé de nettoyage doit être effectué, consultez la section 7.1.
- Tout nouvel équipement et/ou toute modification de l'équipement qui pourrait avoir une incidence sur la nettoyabilité.
Des études de vérification seront alors normalement menées sur l'équipement commercial pour démontrer que l'équipement a été correctement nettoyé après la production du nouveau produit (sur de lots de développement, de transfert de technologie ou d'essai clinique).
8. Méthodes d'échantillonnage et d'analyse
- Validez les méthodes analytiques utilisées pour mesurer les résidus et les contaminants sur l'équipement (par exemple, l'ingrédient actif, les produits de dégradation ou les résidus d'agent de nettoyage).
Consultez la directive Q2 Validation des méthodes d'analyse : Texte et méthodologie pour obtenir des renseignements généraux sur la validation des méthodes analytiques.
- Déterminez les limites de quantification et de détection pour vous assurer que la sensibilité de la méthode d'analyse est appropriée pour les niveaux de résidus qui font l'objet d'un examen. Vous devrez peut-être aussi évaluer si la sélectivité de la méthode d'analyse doit être établie par rapport aux produits de dégradation qui peuvent s'être formés pendant le procédé de nettoyage.
- Effectuez des études de récupération pour toutes les méthodes d'échantillonnage utilisées avec des méthodes analytiques :
- Assurez-vous que la méthode d'échantillonnage utilisée en laboratoire est équivalente à la méthode utilisée dans la fabrication.
- Effectuez des études de récupération pour tous les matériaux de construction à échantillonner dans l'équipement en contact avec les produits.
- Établissez un pourcentage de récupération pour chaque surface ou matériau de construction et utilisez-le dans le calcul des contaminants résiduels.
- Une récupération faible ou variable des concentrations standard de résidus au cours des études de récupération peut ne pas être acceptable, car elle indique une technique d'échantillonnage ou d'extraction inadéquate.
Les études de récupération démontrent que les méthodes d'échantillonnage et d'analyse peuvent mesurer adéquatement les résidus qui peuvent être présents sur les surfaces de l'équipement. Ces études sont effectuées au moyen de coupons de matériaux sur lesquels une faible concentration du résidu considéré a été déposée, correspondant aux quantités présentes après le nettoyage. Ceux-ci sont soumis à un échantillonnage selon la méthode applicable. Les résultats des analyses sont ensuite comparés avec la quantité réelle déposée sur le coupon.
- Des méthodes analytiques non spécifiques (par exemple, carbone organique total [COT] et conductivité) peuvent être utilisées si elles sont justifiées de manière appropriée. Dans de tels cas, vous devez établir l'efficacité de votre méthode non spécifique pour détecter le résidu cible. Vous devez partir de l'hypothèse que le résultat de l'analyse est entièrement dû au résidu cible dans de tels cas. Vous devez toujours démontrer que la méthode assurera une récupération adéquate et reproductible.
9. Évaluation du nettoyage
9.1 Inspection visuelle
L'inspection visuelle est une méthode qualitative visant à déterminer la propreté de l'équipement et à inspecter l'équipement pour s'assurer qu'il ne contient pas de résidus visibles et de corps étrangers au moment du changement de produit. Il est également utile de détecter les dommages ou l'usure de l'équipement, qui peuvent rendre le nettoyage plus difficile. Il s'agit d'un élément important de chaque procédé de nettoyage, que ce soit pendant les études de qualification du nettoyage ou pendant la production courante.
- Effectuez des inspections visuelles après tout nettoyage et avant de mener toute activité d'échantillonnage aux fins de vérification du nettoyage, de qualification ou de surveillance continue. Consignez les résultats.
- Établissez des procédures décrivant en détail la façon dont les inspections visuelles doivent être effectuées. Inclure des instructions claires concernant ce qui suit :
- La confirmation du fait que l'équipement est sec;
- Les instructions de démontage;
- L'utilisation d'une source lumineuse et de conditions d'éclairage appropriées;
- L'évaluation des zones critiques tel que la partie inférieure des hélices de mélange.
- Assurez-vous que l'inspection visuelle est effectuée par du personnel qualifié seulement. Pour les produits plus dangereux, mettez en place un programme pour démontrer la capacité du personnel à détecter les résidus lors de l'inspection visuelle.
- Le recours aux inspections visuelles comme seul moyen habituel de vérification et de libération de l'équipement devrait être fondé sur une évaluation de la GRQ. Il peut être nécessaire d'effectuer des études par ajout connu de différents produits sur différentes surfaces pour déterminer les seuils d'inspection visuelle. . À cet égard, les produits dont les LEFS sont faibles suscitent des préoccupations particulières.
- Enquêtez sur les défaillances de l'inspection visuelle grâce au système qualité applicable. Les défaillances de l'inspection visuelle devraient être rares lorsqu'un procédé de nettoyage a été validé, et elles peuvent indiquer que les procédés de nettoyage sont insuffisamment contrôlés.
9.2 Échantillonnage de l'équipement
L'échantillonnage de l'équipement est généralement effectué par échantillonnage direct de la surface (par écouvillonnage ou essuyage), échantillonnage par rinçage ou une combinaison des deux.
9.2.1 Échantillonnage direct de la surface (méthode par écouvillonnage ou par essuyage)
L'échantillonnage par écouvillonnage consiste à essuyer une surface d'équipement avec un matériel particulier imbibé de solvant pour récupérer les résidus de la surface.
- Procédez à l'échantillonnage par écouvillonnage ou par essuyage sur les zones déterminées au cours de l'évaluation des risques et, plus particulièrement, sur les zones les plus difficiles à nettoyer. De plus, pensez à prendre des échantillons représentatifs de grandes surfaces. Précisez clairement les zones les plus difficiles à nettoyer dans les protocoles pertinents. Le choix de l'emplacement des écouvillonnages devrait être justifié par des données appropriées à l'appui.
- Prenez en considération les éléments suivants lors de la détermination des zones les plus difficiles à nettoyer :
- L'accessibilité;
- La géométrie de l'équipement;
- La possibilité d'accumulation de résidus;
- Les matériaux de construction.
- Précisez le matériel à utiliser pour écouvillonner et le milieu d'échantillonnage ou le solvant.
- Assurez-vous que l'équipement de production est échantillonné de la même manière que lors des études de récupération en laboratoire. Les mesures visant à assurer l'uniformité peuvent comprendre ce qui suit :
- Des procédures détaillées;
- La qualification ou l'attestation du personnel en charge de l'échantillonnage pour démontrer une récupération appropriée;
- La formation et la supervision du personnel en charge de l'écouvillonnage sur le lieu de travail relativement à l'écouvillonnage de l'équipement de fabrication.
9.2.2 Échantillonnage par rinçage
L'échantillonnage par rinçage consiste à rincer les surfaces de l'équipement pertinentes à l'aide d'une quantité définie d'un solvant déterminé pour éliminer les résidus. Mesurez les niveaux de résidus dans le liquide de rinçage. Les échantillons de rinçage permettent d'échantillonner une grande surface et des systèmes inaccessibles ou qui ne peuvent être démontés souvent.
9.2.3 Échantillonnage de placebo
L'échantillonnage de placebo offre une autre solution pour évaluer l'efficacité du nettoyage. L'échantillonnage de placebo consiste à traiter un lot placebo une fois les activités de nettoyage terminées, puis à analyser le placebo pour repérer des traces du produit précédent. De telles évaluations sont normalement menées pour compléter les études par écouvillonnage ou par rinçage.
10. Établir les limites
La quantité de résidus autorisée sur l'équipement ou sur une chaîne d'équipements de traitement après le nettoyage est appelée limite maximale sécuritaire de résidus. Cette limite est déterminée en calculant la quantité de substance active du premier produit fabriqué qui pourrait être transférée en toute sécurité dans le deuxième produit (après le nettoyage), de sorte que la dose quotidienne maximale du deuxième produit ne contienne pas plus que la LEFS du premier produit. Lorsque vous prenez en compte la surface de l'équipement et tout autre facteur relatif à l'innocuité, vous pouvez calculer les limites préliminaires d'écouvillonnage ou de rinçage. Les limites de nettoyage finales ne doivent pas dépasser cette valeur.
- Utilisez les principes de gestion des risques pour déterminer les calculs de transfert maximaux admissibles pour les résidus préoccupants. Ces limites devraient s'appuyer sur une évaluation toxicologique et être documentées sous la forme d'une évaluation des risques.
Vous trouverez d'autres directives sur le calcul des limites dans divers documents d'orientation, dont ISPE Baseline Guide Vol 7: Risk-Based Manufacture of Pharma Products, 2e édition (disponible seulement en anglais).
- Envisagez d'établir des limites d'alerte dans le cas où les limites de nettoyage issues des LEFS sont considérablement plus élevées que les limites de nettoyage historiques (par exemple, 1/1000e d'une dose et 10 ppm). Les procédures de nettoyage qui sont capables d'atteindre de meilleures limites que celles issues des LEFS devraient continuer de le faire. Notez que les limites de nettoyage doivent également continuer à répondre aux critères de nettoyage visuel.
- Assurez-vous que les méthodes d'analyse utilisées sont capables de détecter les résidus à un niveau acceptable en dessous de ces limites. Si cela n'est pas possible, on peut envisager d'améliorer les méthodes d'analyse ou envisager d'autres moyens de réduire les risques, comme le recours à de l'équipement dédié.
- Les calculs des critères d'acceptation de nettoyage doivent tenir compte de l'impact cumulatif des résidus provenant de plusieurs équipements partagés (c.-à-d. l'effet de la chaîne de traitement).
- Il est recommandé de réévaluer périodiquement les LEFS et d'évaluer et de documenter l'incidence de tout changement sur le programme global de validation des procédés de nettoyage.
11. Mesures de contrôle microbiologiques
- Assurez-vous que la conception, le fonctionnement, le nettoyage et l'entretien de l'équipement et des installations permettront de contrôler de façon appropriée la charge microbiologique. Mettez l'accent sur les mesures préventives plutôt que sur l'élimination des contaminants après coup.
- Utiliser les principes de GRQ pour déterminer :
- la nécessité d'inclure l'évaluation de la contamination microbiologique et/ou par des endotoxines dans le cadre des évaluations de vérification, de qualification ou de surveillance continue;
- les lieux d'échantillonnage dans l'équipement, notamment les emplacements ou les matériaux qui pourraient être plus propices à la croissance microbienne;
- le type, la nature et la portée d'un programme de surveillance de l'environnement continue.
- Une importance particulière doit être portée à certains aspects des considérations microbiologiques :
- Déterminez une période maximale pendant laquelle l'équipement propre peut rester tel quel avant l'utilisation sans devoir être nettoyé ou désinfecté de nouveau avant d'être utilisé (communément appelé la durée de propreté). Démontrez que la durée maximale autorisée de maintien à l'état propre ou d'entreposage n'entraîne pas de prolifération microbienne.
-
Assurez-vous que les évaluations microbiologiques sont prises en considération (selon les principes de gestion des risques) pour évaluer la durée maximale des campagnes.
Remarque – Les considérations microbiologiques mentionnées ci-dessus peuvent ne pas s'appliquer à certains ingrédients pharmaceutiques actifs.
- Assurez-vous que de l'eau ne stagne pas dans l'équipement après son nettoyage ou son utilisation. L'équipement doit être drainé ou séché avant son utilisation ou son entreposage.
- Assurez-vous que des procédures sont établies pour manipuler de manière appropriée les tuyaux. Les tuyaux (p. ex. tuyaux d'eau purifiée) sont connus pour être une zone possible de contamination microbienne.
- Assurez-vous que les limites microbiologiques sont justifiées sur le plan scientifique.
12. Renseignements généraux sur le nettoyage de l'équipement
Assurez-vous que l'ensemble de l'équipement de traitement est conçu de manière à faciliter le nettoyage et à permettre une inspection visuelle (dans la mesure du possible). L'équipement doit avoir des surfaces lisses et être fabriqué de matériaux non réactifs. La tuyauterie de l'équipement doit être en pente pour assurer le drainage adéquat des conduites. Les tronçons morts devraient être évités.
Il convient d'accorder une attention particulière aux longues lignes de transfert. Les procédés de nettoyage appropriés consisteront à remplir l'ensemble du tuyau pour assurer le contact avec toutes les surfaces. Un régime d'écoulement turbulent est généralement préférable pour assurer un nettoyage optimal. Assurez-vous qu'il existe des sections amovibles, s'il y a lieu, pour évaluer l'efficacité du procédé de nettoyage au moyen d'un examen visuel, d'un écouvillonnage ou d'un échantillon de rinçage.
12.1 Agents de nettoyage:
- Lors de la sélection des agents de nettoyage, assurez-vous que leur composition est connue. La préférence devrait être accordée aux agents de nettoyage dont les composants présentent des profils et des limites toxicologiques favorables. Assurez-vous d'être informé de tout changement dans la composition de l'agent de nettoyage.
- Assurez-vous que les agents de nettoyage sont faciles à éliminer.
L'élimination des agents de nettoyage est un facteur important dans tout programme de validation du nettoyage. Des preuves que les procédures de nettoyage élimineront efficacement les agents de nettoyage à des niveaux inférieurs aux niveaux prédéterminés doivent être fournies.
12.2 Dernier rinçage:
- Si de l'eau est utilisée pour effectuer le dernier rinçage, assurez-vous qu'elle satisfait ou dépasse la qualité et la norme de l'eau utilisée à ce stade du procédé. Les paramètres de qualité de l'eau (chimiques, microbiologiques et relatifs aux endotoxines) devraient être appropriés pour l'usage en question.
- Si la procédure de nettoyage nécessite un solvant pour le dernier rinçage, la qualité du solvant doit être appropriée.
12.3 Équipement et installations dédiés
Pour déterminer si des installations ou du matériel dédiés sont nécessaires, on devrait se fonder sur les principes de la GRQ et sur l'évaluation toxicologique. Il peut être possible de consacrer des parties de l'équipement qui sont particulièrement difficiles à évaluer ou à nettoyer (par exemple, les sacs filtrants, les joints ou les écrans), tout en validant le reste de la chaîne d'équipement pour un usage partagé. Consultez les Bonnes pratiques de fabrication des drogues (GUI-0001) pour de plus amples renseignements.
Utilisez les principes de GRQ pour déterminer les exigences de validation des procédés de nettoyage lors de l'utilisation d'équipement ou d'installations dédiés. Les domaines préoccupants comprennent :
- les considérations microbiologiques;
- l'élimination de l'agent de nettoyage;
- les produits de dégradation potentiels et les impuretés du traitement.
13. Renseignements supplémentaires sur le nettoyage de l'équipement de production des IPA
- La validation du nettoyage constitue une condition pour minimiser la contamination croisée dans la production d'IPA. Selon les principes de gestion des risques, ces activités devraient être axées sur les étapes du procédé qui présentent le plus grand risque pour la qualité du produit, comme les produits intermédiaires à un stade avancé et les étapes finales de traitement et de manipulation.
- Généralement, les exigences en matière de contrôle et d'évaluation du nettoyage pour les procédés de production d'IPA finaux devraient être équivalentes à celles requises pour la fabrication des formes pharmaceutiques finies. Par exemple :
- Les processus de nettoyage pertinents doivent être validés conformément à une approche axée sur le cycle de vie;
- L'équipement doit être conçu selon les mêmes concepts que ceux utilisés pour les produits pharmaceutiques finis.
- Les procédés de nettoyage pour les IPA nécessitent habituellement une utilisation importante de solvants. Dans de tels cas :
- Assurez-vous que l'IPA est soluble dans l'agent utilisé pour les études sur la récupération de rinçage et le nettoyage;
- Assurez-vous que les solvants utilisés dans les procédés de nettoyage, y compris le rinçage final, sont de la qualité appropriée;
- Envisagez des étapes de reflux ou d'ébullition.
Remarque : Les étapes de reflux ou d'ébullition peuvent être importantes lors du nettoyage des réacteurs et de pièces d'équipement similaires pour assurer un contact approprié du solvant avec la surface totale de l'équipement en contact avec le produit. Une étape de reflux ou d'ébullition peut également être incluse lors de la collecte d'un échantillon de rinçage pour des activités de qualification, de vérification ou de surveillance.
14. Renseignements supplémentaires sur la validation du nettoyage des procédés biotechnologiques
- Les principes énoncés dans le présent document peuvent généralement être appliqués à la validation du nettoyage dans le cadre de procédés biotechnologiques.
- Les programmes de développement des procédés de nettoyage doivent prendre en compte l'élimination d'un grand nombre de substances comme les milieux, les protéines, les acides, les bases et les sels.
- Les procédés de nettoyage biotechnologiques impliquent souvent des conditions qui provoquent la dénaturation ou la dégradation des molécules de protéines. Ainsi, les mesures des résidus sont normalement effectuées en utilisant une méthode de test non spécifique, comme celle du COT.
- Les exigences de validation du nettoyage pour les médicaments biologiques devrait normalement inclure une évaluation microbiologique et des endotoxines.
- La validation du nettoyage de l'équipement partagé entrant en contact avec les produits devrait normalement être évaluée pour chaque produit et procédé.
- Vous pouvez utiliser la méthode des extrêmes pour des produits ou de l'équipement similaires, à la condition de disposer d'une justification adéquate fondée sur des motifs solides et scientifiques. Voici quelques exemples :
- nettoyage de fermenteurs de même conception, mais ayant des cuves de capacités différentes, qui sont utilisés pour le même type de protéines recombinées exprimées dans la même lignée cellulaire ou des lignées cellulaire similaire et cultivées dans des milieux de croissance très apparentés;
- pour les vaccins multi antigènes, l'utilisation d'un antigène représentatif (ou de combinaisons d'antigènes) lors de la validation du même équipement ou d'un équipement similaire.
- Les principes de la GRQ devraient être utilisés pour fixer des limites appropriées pour le transfert en tenant compte du procédé de fabrication et de l'étape de fabrication. La rigueur des limites peut augmenter à travers le processus de purification.
- Fabrication en vrac : Les calculs des résidus peuvent ne pas s'appliquer à la fabrication en vrac, lorsque les résidus spécifiques du produit peuvent être présents à de faibles concentrations ou qu'il peut être démontré que les conditions de nettoyage rendent le produit définitivement inerte.
- Formulation et remplissage terminal : Les limites sont calculées sur la base d'une LEFS établie à partir d'une évaluation toxicologique.
Annexes
Annexe A – Prévention de la contamination croisée
Les lignes directrices suivantes sont tirées du document PIC/S - Guide to Good Manufacturing Practice for Medicinal Products Part 1, Chapter 5 [PE 009-14 (Part 1)] (disponible seulement en anglais)
5.21 Le résultat du processus de gestion des risques liés à la qualité devrait servir de base pour déterminer l'ampleur des mesures techniques et organisationnelles nécessaires pour contrôler les risques de contamination croisée. Ces mesures peuvent comprendre les suivantes, sans toutefois s'y limiter :
Mesures techniques
- Installations de fabrication dédiées (locaux et équipement).
- Aires de production indépendantes dotées d'équipement de traitement et de systèmes de chauffage, de ventilation et de climatisation distincts. Il pourrait être également utile d'isoler certains services de ceux utilisés dans d'autres zones.
- Conception du processus, des locaux et de l'équipement de fabrication pour réduire au minimum les risques de contamination croisée durant le traitement, l'entretien et le nettoyage.
- Utilisation de « systèmes fermés » pour le traitement et le transfert de matériel ou de produits entre les équipements.
- Utilisation de systèmes à barrière physique, y compris des isolateurs, comme mesures de confinement.
- Élimination contrôlée de la poussière près des sources de contaminant; par exemple, au moyen d'une extraction localisée.
- Utilisation d'équipement, des pièces en contact avec les produits ou de certaines pièces plus difficiles à nettoyer (par exemple, filtres), et des outils d'entretien dédiés.
- Utilisation de technologies jetables à usage unique.
- Utilisation d'équipement conçu pour un nettoyage facile.
- Utilisation appropriée de sas d'air et d'une cascade de pression pour confiner les contaminants atmosphériques éventuels dans une zone donnée.
- Minimisation des risques de contamination causés par la recirculation ou la réintroduction d'air non traité ou traité de façon insuffisante.
- Utilisation de systèmes de nettoyage automatique automatisés (nettoyage en place) dont l'efficacité a été validée.
- Pour les zones de lavage général communes, séparation des aires de lavage, de séchage et d'entreposage de l'équipement.
Mesures organisationnelles
- Utilisation de l'ensemble des installations de fabrication dédiées ou d'une aire de production indépendante dédiée sur la base de campagne ( dédié par séparation dans le temps) suivie d'un processus de nettoyage dont l'efficacité a été validée.
- Port de vêtements de protection particuliers à l'intérieur des zones de traitement des produits présentant un risque élevé de contamination croisée.
- La vérification du nettoyage après chaque campagne de produit doit être considérée comme un outil de détection à l'appui de l'efficacité de l'approche de gestion des risques liés à la qualité pour les produits considères à risque élevé.
- Selon le risque de contamination, vérification du nettoyage des surfaces qui ne sont pas en contact avec les produits et surveillance de l'air dans les zones de fabrication ou les zones adjacentes afin de prouver l'efficacité des mesures de contrôle contre la contamination par de particules aéroportées ou la contamination par transfert mécanique.
- Mesures spécifiques pour la manipulation des déchets, de l'eau de rinçage contaminée et des vêtements souillés.
- Déclaration des déversements, des évènements accidentels ou des écarts par rapport aux procédures.
- Conception de processus de nettoyage des locaux et de l'équipement faisant en sorte que les procédés de nettoyage eux-mêmes ne présentent pas de risque de contamination croisée.
- Conception de documents détaillés des procédés de nettoyage afin d'assurer l'exécution complète du nettoyage conformément aux procédures approuvées et l'utilisation d'étiquettes de statut en matière de nettoyage pour l'équipement et les zones de fabrication.
- Utilisation de zones de lavage générales communes sur la base d’une campagne.
- Supervision des comportements professionnels pour assurer l'efficacité de la formation et la conformité aux mesures de contrôle des procédures pertinentes.
Annexe B – Glossaire
Acronymes
- BPF
- bonnes pratiques de fabrication
- COT
- carbone organique total
- EJA
- exposition journalière admissible
- GRQ
- gestion des risques liés à la qualité
- ICH
- International Council for Harmonisation (Conseil international pour l'harmonisation)
- IPA
- ingrédient pharmaceutique actif
- LEFS
- limite d'exposition fondée sur la santé
- PIC/S
- Pharmaceutical Inspection Co-operation Scheme
- SPT
- seuil de préoccupation toxicologique
Termes
Les définitions suivantes s'appliquent aux termes utilisés dans le présent document, ainsi que dans les annexes (sauf indication contraire). Dans le cas des définitions tirées d'autres documents, la source est indiquée entre parenthèses.
En cas de conflit avec une définition de la Loi sur les aliments et drogues ou du Règlement sur les aliments et drogues, la définition de la Loi ou du Règlement prévaut.
Drogue (médicament) – Sont compris parmi les drogues, les substances ou mélanges de substances fabriqués, vendus ou présentés comme pouvant servir :
- au diagnostic, au traitement, à l'atténuation ou à la prévention d'une maladie, d'un désordre, d'un état physique anormal ou de leurs symptômes, chez l'être humain ou les animaux;
- à la restauration, à la correction ou à la modification des fonctions organiques chez l'être humain ou les animaux;
- à la désinfection des locaux où des aliments sont gardés.
(article 2 de la Loi sur les aliments et drogues)
Titre 1 A du Règlement sur les aliments et drogues :
(2) Au présent titre et au titre 2, le terme drogue ne vise pas :
- le prémélange médicamenteux dilué;
- l'aliment médicamenté au sens du paragraphe 2(1) du Règlement de 1983 sur les aliments du bétail;
- l'ingrédient actif pour usage vétérinaire qui n'est pas un ingrédient actif pharmaceutique;
- l'ingrédient actif pharmaceutique pour usage vétérinaire qui peut être vendu sans ordonnance et qui est également un produit de santé naturel au sens du paragraphe 1(1) du Règlement sur les produits de santé naturels;
- la drogue utilisée uniquement pour une étude expérimentale menée conformément au certificat délivré en vertu de l'article C.08.015.
Manufacturer – Préparer et conserver une drogue en vue de la vente. (C.01A.001)
Exposition journalière admissible – L'exposition journalière admissible représente une dose à laquelle il est peu probable qu'une substance particulière cause un effet néfaste si une personne est exposée à cette dose ou à une dose inférieure chaque jour pendant toute sa vie. (PIC/S PI 046-1)
Seuil de préoccupation toxicologique – Le seuil de préoccupation toxicologique représente le niveau d'exposition aux impuretés génotoxiques qui est associé à un risque théorique de cancer correspondant à 1 cancer supplémentaire pour 100 000 patients exposés au cours d'une vie. (PIC/S PI 046-1)
Validation – Programme documenté fournissant un degré élevé d'assurance qu'un procédé, une méthode ou un système produira de façon constante un résultat répondant aux critères d'acceptation prédéterminés. (ICH Q7)
Annexe C — Références
Lois et règlements
Documents d'orientation de Santé Canada
- Lignes directrices sur les Bonnes pratiques de fabrication des drogues (GUI-0001)
- Annexe 4 à l'édition actuelle des Lignes directrices sur les Bonnes pratiques de fabrication — Médicaments vétérinaires (GUI-0012)
- Annexe 7 du guide sur les bonnes pratiques de fabrication – Lignes directrices sur certaines drogues en vente libre (GUI-0066)
Documents d'orientation internationaux
- ASTM E3106 - 18e1 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation (disponible seulement en anglais)
- ICH M7(R1) : Évaluation et contrôle des impuretés réactives de l'ADN (mutagènes) dans les produits pharmaceutiques pour limiter les risques de cancérogénicité
- ICH Q2(R1) : Validation des méthodes d'analyse : Texte et méthodologie
- ICH Q3A(R) : Présence d'impuretés dans les nouvelles substances médicamenteuses
- ICH Q3B : Présence d'impuretés dans les nouveaux produits
- ICH Q3C : Impuretés : Directives sur les solvants résiduels (disponible seulement en anglais)
- ICH Q3D : Directive concernant les impuretés élémentaires (disponible seulement en anglais)
- ICH Q7 : Ligne directrice sur les Bonnes pratiques de fabrication applicables aux ingrédients pharmaceutiques actifs (disponible seulement en anglais)
- ICH Q9 : Gestion des risques liés à la qualité
- ISPE Baseline Guide Vol 7: Risk-Based Manufacture of Pharma Products(disponible seulement en anglais)
- Pharmaceutical Inspection Co-Operation Scheme - Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities (PI 046-1).
- Pharmaceutical Inspection Cooperation Scheme - Questions and answers on implementation of risk-based prevention of cross-contamination in production and 'Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities' (PI 053-1).
Autres documents connexes
- Food and Drug Administration – Validation of Cleaning Processes (7/93)
- Parenteral Drug Association — Technical Report no. 29 (Revised 2012): Points to Consider for Cleaning Validation
- Parenteral Drug Association — Technical Report No. 49 (Revised 2010): Points to Consider for Biotechnology Cleaning Validation
- Pharmaceutical Inspection Cooperation Scheme - Guide to Good Manufacturing Practice for Medicinal Product Annexes [PE 009-14(Annexes)]
- Pharmaceutical Inspection Cooperation Scheme - Guide to good Manufacturing Practice for Medicinal Products Part I, Chapter 5 [PE 009-14 (Part 1)]
- Pharmaceutical Inspection Cooperation Scheme - Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation and Cleaning Validation (PI 006-3)
- VICH GL 18 – Directive sur les solvants dans les nouveaux produits médicamenteux vétérinaires, les substances actives et les excipients (révision)