
Guidance Document: Drug Identification Numbers for Schedule C Drugs (Radiopharmaceuticals and Kits)
Guidance Document
Date adopted: 2017/12/13
Effective date: 2017/12/13
Foreword
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent, and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant programme area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy, or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable Guidance documents.
Table of contents
- 1. Introduction
- 2. Guidance for implementation
- 3. Transition Period
1. Introduction
1.1 Purpose/Overview
On December 13, 2017, Health Canada published the Regulations Amending the Food and Drug Regulations (DIN Requirements for Drugs in Dosage Form Listed in Schedule C to the Food and Drugs Act) in Canada Gazette, Part II. The objective of this regulatory amendment is to apply to Schedule C Drugs (radiopharmaceuticals and kits), the requirement to have a Drug Identification Number (DIN) in order for a drug to be sold in Canada. Along with the new requirement for all Schedule C drugs to have a DIN, the labelling requirements have also been updated to reflect the new requirement: instead of requiring the Establishment Licence number to be on the label, there will now be a requirement for the DIN to be on the label.
1.2 Scope and application
This guidance document applies to all Schedule C Drugs.
1.3 Policy objectives
The policy objectives that guide the regulatory authority for DIN assignment and DIN to appear on the label of a drug product are as follows:
- to provide the public/Canadians, partners and stakeholders with timely, reliable and accurate information on the availability of drugs in Canada; and
- to help protect the health and safety of Canadians from the sale of unsafe drugs
These objectives should be considered when complying with the Regulations including when interpreting the regulatory requirements for specific situations.
1.4 Policy statements
Previous to these amendments, all drugs in dosage form, other than Schedule C drugs, regulated under the Food and Drug Regulations (FDR) have had a DIN assigned upon authorization by Health Canada which must appear on the product label. A number of regulatory inconsistencies have arisen by not requiring that a DIN be assigned to Schedule C drugs. Specifically, Schedule C drugs are not required to notify Health Canada once sale on the Canadian market has begun, nor are notices required to be given when sales of radiopharmaceuticals are discontinued. Furthermore, since no mechanism exists to cancel a Notice of Compliance (the type of market authorization that Schedule C drugs receive), Health Canada does not have the authority to cancel market status for these drugs.
With these amendments, Schedule C drugs will be subjected to the same requirements as all other DIN products, such as market notification, discontinuance of sales notification and 12 months without sale notification. This will bring the regulatory oversight of these products into alignment with other products that have been assigned a DIN.
Despite the addition of Schedule C drugs to the Drug Product Database online query (DPD) in 2015, the information within the database is still incomplete. Affected stakeholders may be unable to find adequate information relating to available alternatives when facing a possible drug shortage. As the DPD is a publicly available database, these amendments will make market status information for Schedule C drugs available to physicians and other healthcare stakeholders. This is an important mitigation measure during shortages and discontinuations of drugs. Additionally, a DIN for Schedule C drugs will allow for easier identification, monitoring, and recall of these products. For example, the Canada Border Services Agency (CBSA) will be able to access the DPD for complete and reliable information to aid in compliance and enforcement of drug importation. Health care institutions (hospitals and clinics) will be able to easily identify affected drugs during product recall situations.
1.5 Background
Prior to 1998, Schedule C Licences were issued to manufacturers annually for Schedule C drugs. Each Schedule C Licence listed all the products that were being marketed by a manufacturer at that time, and these were renewed on an annual basis. These licences were replaced by Establishment Licences in 1998. Since then, Schedule C drugs have been required to be labelled with the Establishment Licence number.
For other classes of drugs, a DIN was issued for each drug product that was authorized for sale in Canada and served as a unique identifier that had, historically, been primarily used for reimbursement purposes by insurance companies. As most radiopharmaceuticals are administered parenterally (i.e., intravenously or via intramuscular injection) and are commonly prepared and administered by health care professionals in a clinical or hospital setting, DINs were deemed not to be required for reimbursement purposes. As such, Schedule C drugs were exempted from the DIN requirement.
Over time, the purpose of the DIN has evolved. Today, Health Canada uses a DIN for a variety of regulatory activities such as the tracking of market status of a drug and displaying associated information within the DPD. The CBSA uses a DIN to aid in compliance and enforcement activities with respect to the importation of drugs. A manufacturer's authority to sell a product on the Canadian market is represented through the issuance of a DIN. Health Canada can similarly revoke the authority to sell a product which has been shown in the post-market context to be unsafe or for other reasons, such as non-compliance with the regulations, through the cancellation of a DIN.
Currently, once a DIN has been assigned, the manufacturer of the drug must provide Health Canada with a notification within 30 days of selling a drug, not selling for a period of 12 consecutive months or discontinuing the sale of a drug in Canada. By means of an Annual Drug Notification Form, they also confirm that all the information previously supplied with respect to that drug is correct. This information is used by Health Canada for a variety of regulatory activities, such as the tracking of market status of the drug in the DPD.
As Schedule C drugs are currently exempted from the DIN requirement, they are not required to provide market notification or an Annual Drug Notification Form. In the absence of such information on Schedule C Drugs in the DPD, it has been difficult to determine which radiopharmaceutical products are available on the Canadian market. This was particularly evident during the isotope shortages that followed the shutdowns of the Chalk River National Research Universal (NRU) nuclear reactor facility in 2007-2008 and 2009-2010. It was difficult for Health Canada to identify radioisotope alternatives during this shortage period due to the lack of an up-to-date list of Schedule C drugs on the market.
2. Guidance for implementation
2.1 Applying for a Drug Identification Number
2.1.1 Products with a Market Authorization issued prior to June 13, 2018
Products that have received a market authorization before June 13, 2018 have not been issued a DIN. To receive a DIN, you are required to submit a DIN Application for Schedule C drugs (DIN-SC) by filling a Drug Identification Number Application Form for Schedule C product with a Market Authorization.
The instructions on how to complete and submit the form are provided in the form which is available upon request. You can contact Biologics and Genetic Therapies Directorate (BGTD) by email at the following address to obtain a copy of the form: BGTD_ORA@hc-sc.gc.ca.
This DIN-SC application process is unique to Schedule C products that have already been granted a market authorization by Health Canada, and will only apply to those drug products that are currently sold, or will be sold in Canada. DIN Application Forms for Schedule C products that are no longer marketed in Canada (and will not be marketed in the future) should not be submitted. If there is a desire to market again in the future, a DIN-SC application must be submitted at that time, and a DIN assigned, with the labels updated, before the drug can be sold in Canada.
2.1.2 Drugs without a Market Authorization
As per section C.01.014.1(3) of the Food and Drug Regulations,the filing of a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission or an abbreviated extraordinary use new drug submission will be regarded as an application for a drug identification number.
2.2 Drug Notification Form and Market Notification
Once the review of your DIN-SC application or new drug submission is complete and the regulatory amendments come into force, Health Canada will issue you a Drug Notification Form (DNF) with the new DIN.
The DNF are issued by Health Canada in accordance with section C.01.014.2 of the Food and Drug Regulations. They contain the DIN assigned for a drug as well as some of the information included in the drug submission. In accordance with section C.01.014.3 of the FDR, the sponsor must, within 30 days after commencing sale of a drug, complete the DNF and return it to Health Canada along with the labelling material (when applicable).
p>You can find detailed instructions for submitting the market notification in the Notice: Instructions for filing Drug Notification Forms (DNF) and Supporting Documents Provided in Electronic Format.2.3 Reporting 12 months without sale (Dormant DIN)
In accordance with section C.01.014.71 of the Food and Drug Regulations, manufacturers of Schedule C drugs are required to submit within 30 calendar days after a period of twelve consecutive months that a product has not been sold on the Canadian market, a notification to Health Canada.
You can find detailed instructions for submitting the 12 months without sale notification in the Guidance Document: Cancellation of a Drug Identification Number (DIN) and Notification of the Discontinuation of Sales.
2.4 DIN Cancellation
In accordance with section C.01.014.7 of the Food and Drug Regulations,manufacturer are required to submit within 30 days after the day on which they discontinue sale of the drug a notification to Health Canada.
You can find detailed instructions for submitting the sale discontinuation notification in the Guidance Document: Cancellation of a Drug Identification Number (DIN) and Notification of the Discontinuation of Sales available on our Website at the address provided in section 2.3.
2.5 Annual Drug Notification
The Annual Drug Notification Form (ADNF) is intended to assist sponsors in complying with section C.01.014.5 of the Food and Drug Regulations which requires that every manufacturer of a drug confirm annually, before October, that specific information previously supplied with regard to the product is correct. Failure to comply may result in cancellation of existing DINs by Health Canada.
Sponsors of Schedule C products will be asked to complete the ADNF provided in the package and return it before October of each year.
Health Canada is responsible for preparing and distributing packages for the Annual Drug Notification to all manufacturers. The package is sent during the first week of June and includes instructions on how to complete and submit the ADNF.
2.6 Fees for the Right to Sell Drugs
The Fees in Respect of Drugs and Medical Devices Regulations grant Health Canada the authority to charge annual fees for the right or privilege to sell a drug in Canada. A fee is charged for each product assigned a Drug Identification Number (DIN), pursuant to Section C.01.014.2 of the Food and Drug Regulations, which has been subsequently notified as being marketed.
However, for Schedule C products, a non-application provision has been added to the Fees in Respect of Drugs and Medical Devices Regulations. Health Canada intends to consult with stakeholders in the coming months to discuss an update to the User Fee framework, after which time, it is Health Canada's intention to commence the application of the fees.
You can find additional information regarding the fees for the right to sell drugs in the Guidance Document - Fees for the Right to Sell Drugs
2.7 Fees for the Examination of a Drug Submission
All fees currently paid by radiopharmaceutical manufacturers, under the Fees in Respect of Drugs and Medical Devices Regulations, such as the fee for the examination of a new drug submission, an abbreviated new drug submission, a drug submission supplement (eg. new drug manufacturing site) or establishment licence fees, will continue to apply.
For DIN-SC applications, a non-application provision has been added to the Fees in Respect of Drugs and Medical Devices Regulations; therefore, there will be no fees applied for the review of these applications.
3. Transition Period
Health Canada is requesting that all applications for a DIN be submitted by December 13, 2018.
In an effort to reduce the impact of these amended regulations, there will be a transition period to allow authorization holders time to submit applications for DINs and also to update the respective product labels. This transitional provision will only apply to market authorization holders of Schedule C Drugs which have already received a market authorization.
3.1 If the application is received by December 13, 2018, and accepted into review
The authorization holder will have 12 months following the granting of the DIN to update the product's label for radiopharmaceuticals (and generators) or 24 months following the granting of the DIN to update the product's label for kits.
3.2 If the application is not received by December 13, 2018
The authorization holder will have until December 8, 2019 for radiopharmaceutical (and generators) or December 13, 2020 (for kits) to fully adhere to the Food and Drug Regulations as written as the transition period will expire.
3.3 Transition Period Diagram
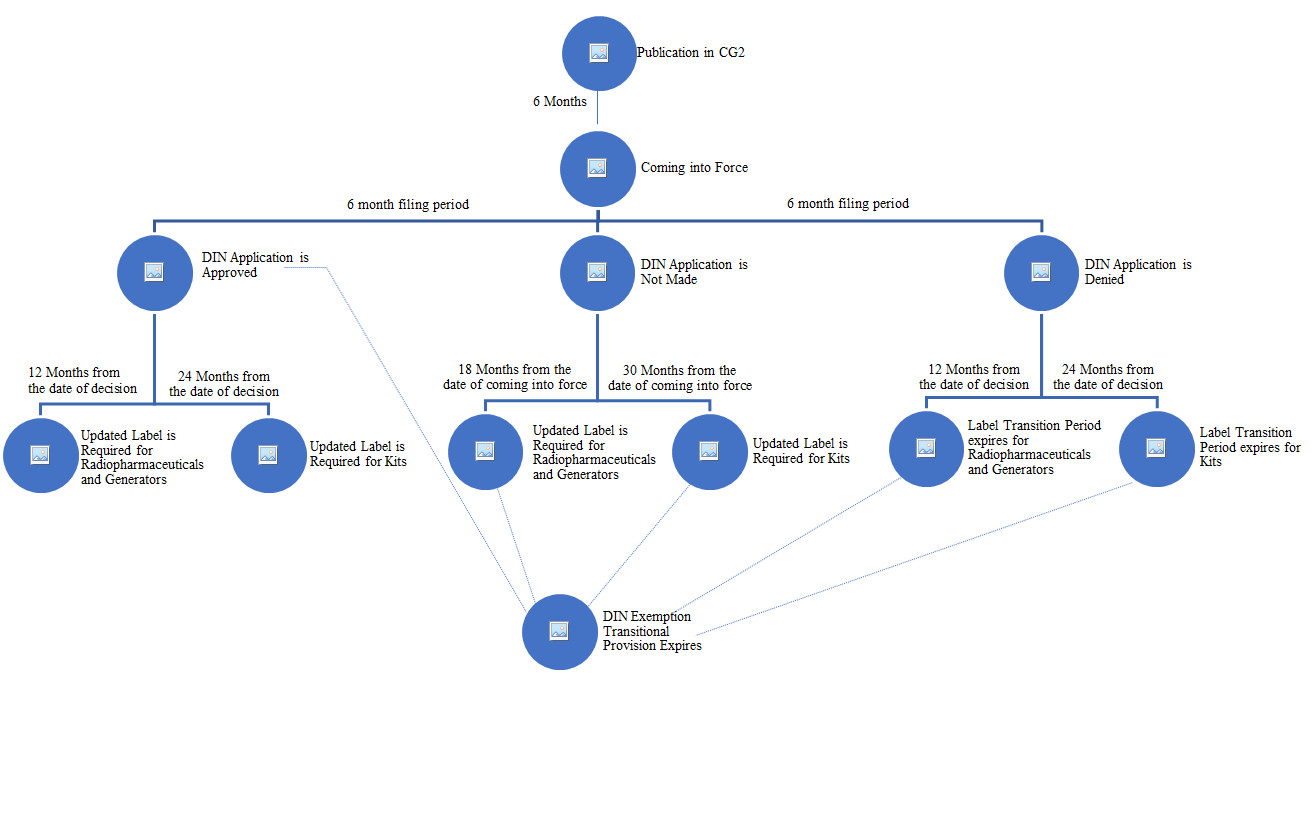
Figure 1 - Text description
This image is a flowchart describing the transition period:
- It starts at the top with the Publication in CGII.
- Then there is a 6 month period before the regulations come into force.
- Following the coming into force, there is a 6 month filing period.
The flowchart then divides into three (3) possible tracks:
- The DIN Application is approved.
- The DIN Application is not made in the 6 month filing period.
- The DIN Application is denied.
If the DIN Application is approved, the transition period for the product possessing a DIN is over.
The flowchart divides into two tracks:
- If the product is a radiopharmaceutical or a generator, the transition period for the label change will expire 12 months following the date of the decision.
- If the product is a Kit, the transition period for the label change will expire 24 months following the date of the decision.
If the DIN Application is not made:
- The transition period for the product possessing a DIN will expire 30 months following the regulations coming into force date.
The flowchart divides into two tracks:
- If the product is a radiopharmaceutical or a generator, the transition period for the label change will expire 18 months following the coming into force date.
- If the product is a Kit, the transition period for the label change will expire 30 months following the coming into force date.
If the DIN Application is denied:
- The transition period for the product possessing a DIN will expire 24 months following the date of the decision.
The flowchart divides into two tracks:
- If the product is a radiopharmaceutical or a generator, the transition period for the label change will expire 12 months following the date of the decision.
- If the product is a Kit, the transition period for the label change will expire 24 months following the date of the decision.