
Guidance for Industry - Device Licence Applications for Ultrasound Diagnostic Systems and Transducers
Notice to reader:
This guidance document is no longer current. For information on device licence applications for diagnostic ultrasound systems and transducers please consult the Device licence applications for diagnostic ultrasound systems and transducers: Notice to industry.
This HTML document is not a form. Its purpose is to display the information as found on the form for viewing purposes only. If you wish to use the form, you must use the alternate format below.
Notice
June 7, 2006
Our file number: 06-113050-982
| Change | Location | Nature of Change |
|---|---|---|
| 1 | Entire Document | original standard AIUM/NEMA UD2-1998, Revision 2 Acoustic Output Measurement Standard for Diagnostic Ultrasound Equipment. revised standard |
| 2 | Entire Document | original standard AIUM/NEMA UD3-1998, Revision 1 Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment. revised standard |
Notice
July 18, 2005
Our file number: 05-114684-13
Please find attached the finalized Guidance for Industry - Device Licence Applications for Ultrasound Diagnostic Systems and Transducers. This guidance document will supercede the previous Guidance for Manufacturers Preparing Device Licence Applications for Ultrasound Diagnostic Systems and Transducers (June 3, 2002).
The Medical Device Regulations set out the requirements governing the sale, importation and advertisement of medical devices. The goal of the Regulations is to ensure that medical devices distributed in Canada are safe and effective and meet quality standards.
This guidance document entitled Guidance for Industry - Preparing Device Licence Applications for Ultrasound Diagnostic Systems and Transducers sets out the Therapeutic Products Directorate's guidance for industry on this subject. This guidance document is to be used specifically in the preparation of new Ultrasound Diagnostic Systems and Transducers device licence applications.
The changes made to the guidance document reflect changes to the current interpretation of the materials required to demonstrate safety and effectiveness of diagnostic ultrasound systems. Specifically, the following items should be considered when preparing an application for licensing:
- Data validating Doppler sensitivity is now required.
- Global acoustic output limits for systems in conformance with the AIUM/NEMA 1998 Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment has been specified.
- It is recommended that the user's manual make reference to Health Canada's Guidelines for the Safe Use of Diagnostic Ultrasound for the application of the ALARA principle in imaging.
This final guidance does not address device-specific issues related to "significant change" and applications for device licence amendments. Manufacturers are directed to the general guidance on this matter entitled Guidance for the Interpretation of Significant Change. The Therapeutic Products Directorate may, in the future, consider the development of device-specific Significant Change guidance documents if the need arises. This guidance document is available on the website and can be accessed in the Medical Device Guidance Documents section.
Should you have any questions or comments regarding the content of the guidance, please contact :
- Device Evaluation Division
Medical Devices Bureau
Therapeutic Products Directorate
Health Products and Foods Branch
Health Canada
2934 Baseline Road, Tower B
Address Locator: 3403A
Ottawa, Ontario K1A 0K9 - Email: meddevices-instrumentsmed@hc-sc.gc.ca
- Telephone: (613) 957-7285
Fax Number: (613) 957-9969
Device Licence Applications for Ultrasound Diagnostic Systems and Transducers
- Date Adopted : 2002/06/01
- Revised Date: 2005/07/06
- Effective Date : 2005/07/06
| Change | Location | Nature of Change |
|---|---|---|
| 1 | Entire Document | Minor editorial and style changes. Updated URLs. |
| 2 | File name | File name changed from "ultrasound_e.wpd" to "draft_ultrasound_e.wpd". |
| 3 | 1.0 Background information 1.2 Section 32(3)(b): Design Philosophy, Transducer Operation 1.2.2 Operating Controls |
Sentence rewritten Original sentence Revised sentence |
| 4 | 2.0 Summary of safety and effectiveness studies 2.3 Section 32(3)(f): Summary of Studies 2.3.1 Clinical Measurement Accuracy and System Sensitivity |
Paragraph removed "For each probe/mode combination in which quantitative claims regarding Doppler sensitivity are made in the product labeling, a minimum performance specification for the Doppler sensitivity should be provided and the methodology justified. Data validating the specification shall be included in the submission and the Device Master Record. For certain special cases or claims, clinical data or special phantom testing may be more appropriate." |
| 5 | 2.0 Summary of safety and effectiveness studies 2.3 Section 32(3)(f): Summary of Studies 2.3.2 Performance Specifications |
Paragraph rewritten Original paragraph Revised paragraph |
| 6 | 2.0 Summary of safety and effectiveness studies 2.3 Section 32(3)(f): Summary of Studies 2.3.3 Accepted Acoustic Output Levels For Systems Not in Compliance with the "Output Display Standard" |
Sentence added "For Fetal Heart Rate Monitors, the maximum attainable value of spatial average, temporal average intensity at the transducer face should be less than 20 mW/cm² for continuous wave devices and the maximum attainable value of the spatial average, pulse average intensity at the transducer face should be less than 20 mW/cm² for pulsed devices." Sentence removed "Acoustic output should not exceed the following upper limits: i.e., global maximum derated ISPTA.3=720 mW/cm² and either MI=1.9 or ISPPA.3=190 W/cm²." |
| 7 | 2.0 Summary of safety and effectiveness studies 2.3 Section 32(3)(f): Summary of Studies 2.3.3 Accepted Acoustic Output Levels For Systems in Compliance with the "Output Display Standard" First paragraph, Third paragraph
|
Sentence added "Acoustic output should not exceed the following upper limits: ISPTA.3=720 mW/cm² and either MI=1.9 or ISPPA.3=190 W/cm²." Original sentence Revised sentence Sentence rewritten Original sentence Revised sentence |
| 8 | 2.0 Summary of safety and effectiveness studies 2.3 Section 32(3)(f): Summary of Studies 2.3.7 Low Output Systems |
Revised sentence Original sentence Revised sentence |
| 9 | 3.0 Section 32(3)(g):Labelling 3.1 Acoustic Output Labelling in the Operator's Manual First paragraph |
Sentence rewritten Original sentence Revised sentence |
| 10 | 3.0 Section 32(3)(g):Labelling 3.1 Acoustic Output Labelling in the Operator's Manual 3.1.1 Real Time Features First paragraph |
Sentence added "It is recommended that these include, but not be limited to, a reference to Health Canada's Guidelines for the Safe Use of Diagnostic Ultrasound, particularly sections 2 and 3. The document is available on the website. |
| 11 | 3.0 Section 32(3)(g): Labelling 3.1 Acoustic Output Labelling in the Operator's Manual 3.1.3 Low Output Systems |
Revised sentence Original sentence Revised sentence |
| 12 | 3.0 Section 32(3)(g):Labelling 3.2 Section 32(3)(g): General Labelling Information 3.2.2 Additional Labelling |
Sentence rewritten Original sentence Revised sentence |
| 13 | 4.0 Section 32(3)(j): Quality system certification | Sentence removed "This section of the Regulations will not come into force until January 1, 2003." Paragraph rewritten Original paragraph Revised paragraph |
| 14 | 5.0 Medical device licence issuance First paragraph
|
Sentence rewritten Original sentence Revised sentence Sentence rewritten Original sentence Revised sentence |
| 15 | Appendix 4: List of Standards | Sentence rewritten Original sentence
"For a complete list of TPD Recognized Medical Device Standards, refer to the Policy on Recognition and Use of Standards under the Medical Devices Regulations (dated January 2004) posted on the website. Original sentence Revised sentence |
| 16 | Appendix 5: Device Specific Definitions | Definition added "Intensity, spatial-average pulse-average: the spatial average, temporal average intensity at the face of the transducer divided by the duty factor, where the duty factor is the product of the pulse duration and the pulse repetition frequency. "Spatial-average pulse-average intensity: see intensity. |
| 17 | Appendix 5: List of Symbols | Symbol added "ISAPA=Spatial-average pulse-average intensity" |
Foreword
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with the policies and governing statutes and regulations. They also serve to provide review and compliance guidance to staff, thereby ensuring that mandates are implemented in a fair, consistent and effective manner.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate scientific justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this guidance, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable guidances.
Table of Contents
- Purpose
- Background
- Scope
- Presentation of the Review Document
- Elements of Class III Device Licence Application
- Elements of Class IV Device Licence Application
- 1.0 Background Information
- 1.1 Section 32(3)(a): Device Description
- 1.1.1 Indications for Use
- Table 1: Diagnostic Ultrasound Indications for Use
- 1.1.1 Indications for Use
- 1.2 Section 32(3)(b): Design Philosophy, Transducer Operation
- 1.2.1 Transducer Operation
- 1.2.2 Operating Controls
- 1.2.3 Unique Features
- 1.3 Section 32(3)(c): Marketing History
- 1.1 Section 32(3)(a): Device Description
- 2.0 Summary of Safety and Effectiveness Studies
- 2.1 Section 32(3)(d): List of Standards
- 2.2 Section 32(3)(e): Method of Sterilization
- 2.2.1 Components Provided Sterile
- 2.2.2 Pyrogenicity
- 2.3 Section 32(3)(f): Summary of Studies
- 2.3.1 Clinical Measurement Accuracy and System Sensitivity
- 2.3.2 Performance Specifications
- 2.3.3 Accepted Acoustic Output Levels
- For Systems Not in Compliance with the "Output Display Standard"
- Table 2-1: Accepted Acoustic Output Levels
- For Systems in Compliance with the "Output Display Standard"
- 2.3.4 Certifications
- 2.3.5 Default Settings
- 2.3.6 Thermal Index
- 2.3.7 Low Output Systems
- 2.3.8 Thermal, Mechanical and Electrical Safety
- 2.3.9 Patient Contact Material
- 2.3.9.1 Identification and Composition
- 2.3.9.2 Biocompatibility
- 2.3.10 Section 20: Software/Firmware
- 2.4 Section 32(3)(i): Bibliography
- 3.0 Section 32(3)(g): Labelling
- 3.1 Acoustic Output Labelling in the Operator's Manual
- 3.1.1 Real Time Features
- 3.1.2 Display Accuracy and Measurement Precision
- 3.1.3 Low Output Systems
- 3.1.4 Acoustic Output Reporting Tables
- 3.2 Section 32(3)(g): General Labelling Information
- 3.2.1 Re-use Instructions
- 3.2.2 Additional Labelling
- 3.1 Acoustic Output Labelling in the Operator's Manual
- 4.0 Section 32(3)(j): Quality System Certification
- 5.0 Medical Device Licence Issuance
- Appendix 1:
- Table A-1: Low Output Summary Table
- Appendix 2: Operating Modes and Acoustic Output Estimates
- Table A-2: Output Range Summary Table
- Table A-2-1: Transducer/Mode Combination Summary Table
- Table A-2: Output Range Summary Table
- Appendix 3: Acoustic Output Reporting Table
- Symbols used in the table
- Table A-3: Acoustic Output Reporting Table
- Table A-3-1: Acoustic Output Reporting Table (B-mode)
- Table A-3-2: Acoustic Output Reporting Table (Pulsed Doppler)
- Table A-3-3: Acoustic Output Reporting Table (Colour Flow M-Mode)
- Appendix 4: List of Standards
- Appendix 5:
- General Definitions
- Device Specific Definitions
- List of Symbols
Purpose
This guidance document is intended to assist manufacturers and sponsors in preparing a premarket device licence application for Class III or IV ultrasound diagnostic systems and transducers.
Background
The importation, advertising and sale of medical devices is regulated by the Food and Drugs Act and the Medical Device Regulations. The Sections 10 to 20 provide for the general safety and effectiveness requirements for all medical devices.
Section 32(3) and 32(4) of the Regulations outline specific evidence required for Class III and Class IV applications in order to meet the safety and effectiveness requirements. All Class III and Class IV medical devices require a review of submitted evidence of safety and effectiveness before the device can be recommended for licensing.
A new device licence application for a Class III or a Class IV medical device will contain a premarket review document in addition to the general requirements of Section 32(1). The premarket review document contains the evidence to support the requirements of Section 32(3) or 32(4), depending on the risk classification of the device. Portions of the review submission may reference application files previously submitted by the manufacturer.
A licence will be issued after a review of the information included in the medical device licence application has determined that the medical device conforms to the safety and effectiveness requirements.
Section 35(1) of the Medical Devices Regulations, provides a provision for additional information or documentation from the manufacturer where the evidence submitted in support of the licence application requirements of Section 32 is insufficient to determine whether the device meets the safety and effectiveness requirements of Section 10 to 20.
A copy of the Medical Devices Regulations is available on The Department of Justice Canada website.
Scope
This document is intended to be a device specific premarket guidance on the preparation of a new device licence application for Class III and Class IV ultrasound diagnostic systems and transducers. For ultrasound systems and transducers, this device-specific guidance document replaces the general premarket medical device guidance documents entitled Preparation of a Premarket Review Document for Class III and Class IV Device Licence Applications.
This guidance document does not address issues related to significant change, applications for medical device licence amendments or the general process and procedures of device licensing.
For guidance on significant change, refer to the Guidance for the Interpretation of Significant Change.
For guidance on general processes and procedures, refer to the general guidance documents such as How to Complete the Application for a New Medical Device Licence, Guidance on the Interpretation of Section 28 to 31: Licence Application Type and Guidance for the Labelling of Medical Devices under Section 21 to 23 of the Medical Devices Regulations.
Presentation of the Review Document
Manufacturers and/or device sponsors are advised to follow the structure below when submitting an application for a device licence. Sections that are not applicable must be clearly indicated and accompanied by a rationale.
Elements of Class III Device Licence Application
A licence application for a Class III medical device must contain evidence in support of the requirements of Section 32(3) of the Medical Devices Regulations. These requirements are grouped into four general sections. These sections must be easily identifiable in every licence application for a Class III medical device. The following format is advised:
- Device Licence Application Form
- Executive Summary
- Table of Contents
- Background Information
- 1.1 Device Description
- 1.2 Design Philosophy
- 1.3 Marketing History
- Summary of Safety and Effectiveness Studies
- 2.1 List of Standards
- 2.2 Method of Sterilization
- 2.3 Summary of Studies
- 2.4 Bibliography
- Labelling
- Quality System Requirements (comes into force January 1, 2003)
Health Canada will accept information as provided to another regulatory authority's prescribed format (eg. FDA 510(k) applications) provided that the information is appropriately indexed to Section 32(3) of Medical Devices Regulation requirements using the above suggested format.
Elements of Class IV Device Licence Application
Ultrasound diagnostic systems and transducers are generally classified as Class III devices. Devices, such as transducers, intended to be used in direct contact with the central nervous system or the central cardiovascular system are an exception. Application of classification Rule 1(2), these surgically invasive devices are considered to be Class IV devices.
In addition to the requirements of Class III, device licence applications for Class IV central nervous system or cardiovascular system surgically invasive ultrasound diagnostic devices would require the following information:
- Section 32(4)(d): a risk assessment comprising of an analysis and evaluation of the risks, and the risk reduction measures adopted to satisfy the safety and effectiveness requirements.
- Section 32(4)(e): a quality plan setting out the specific quality practices, resources and sequence of activities relevant to the device
- Section 32(4)(f): the specifications of the materials used in the manufacture and packaging of the device (not required if previously licensed)
- Section 32(4)(g): the manufacturing process of the device
- Section 32(4)(i): detailed information on all studies on which the manufacturer relies to ensure that the device meets the safety and effectiveness requirements, including
- pre-clinical (not required if previously licensed)
- process validation studies
- software validation studies (not required for transducers)
- literature studies (not required for established technology)
1.0 Background information
1.1 Section 32(3)(a): Device Description
Provide a general description of the subject device, including but not limited to the model, designation, design, patient contact materials, operating controls, and system operation. Components or accessories that can be sold separately and used with other medical devices, systems or units must be identified.
1.1.1 Indications for Use
Identify all indications for use of the subject device (fill out the indications for use form(s) or equivalent (refer to Table 1 below).
Table 1: Diagnostic Ultrasound Indications for Use
- System:
- Transducer:
- Intended Use: Diagnostic ultrasound imaging or fluid flow analysis of the human body as follows:
| Clinical Application | Mode of Operation | |||||||
|---|---|---|---|---|---|---|---|---|
General |
Specific |
B |
M |
PWD |
CWD |
Colour Doppler |
Combined |
OtherFootnote 1 |
| Ophthalmic | Ophthalmic |
- | - | - | - | - | - | - |
| Fetal Imaging & Other | Fetal |
- | - | - | - | - | - | - |
Abdominal |
- | - | - | - | - | - | - | |
Intra-operative (Specify) |
- | - | - | - | - | - | - | |
Intra-operative |
- | - | - | - | - | - | - | |
Laparoscopic |
- | - | - | - | - | - | - | |
Pediatric |
- | - | - | - | - | - | - | |
| Small Organ (Specify) |
- | - | - | - | - | - | - | |
Neonatal Cephalic |
- | - | - | - | - | - | - | |
Adult Cephalic |
- | - | - | - | - | - | - | |
Trans-rectal |
- | - | - | - | - | - | - | |
Trans-vaginal |
- | - | - | - | - | - | - | |
Trans-urethral |
- | - | - | - | - | - | - | |
Trans-esoph. |
- | - | - | - | - | - | - | |
Musculo-skel (conventional) |
- | - | - | - | - | - | - | |
Musculo-skel (superficial) |
- | - | - | - | - | - | - | |
Intra-luminal |
- | - | - | - | - | - | - | |
Other (Specify) |
- | - | - | - | - | - | - | |
Cardiac Adult |
- | - | - | - | - | - | - | |
| Cardiac | Cardiac Pediatric |
- | - | - | - | - | - | - |
| Trans-Esoph. (Cardiac) |
- | - | - | - | - | - | - | |
Other (Specify) |
- | - | - | - | - | - | - | |
| Peripheral Vessel |
Peripheral vessel |
- | - | - | - | - | - | - |
Other (Specify) |
- | - | - | - | - | - | - | |
|
||||||||
Additional Comments
A separate form should be filled in for the system and for each transducer. It is not necessary to fill out a form for a previously submitted/licensed transducer. In a submission for a new mode or a new indication for use on an existing system(s), a form for the system(s) and each transducer having the new mode or indication for use must be filled out.
1.2 Section 32(3)(b): Design Philosophy, Transducer Operation
1.2.1 Transducer Operation
Description of the transducer operation in each mode and mode combination including, but not limited to:
- The type of transducer (e.g. model designation, mechanical sector, rectangular phased array, curved linear array, annular phased array);
- Size and spacing of element(s), geometrical configuration, total number of elements in the array and array dimensions, as well as the maximum number of active elements for a single pulse, where applicable, and the nominal ultrasonic frequency(ies) of the transducer assembly.
1.2.2 Operating Controls
Describe the operating controls that can cause a change in the radiated field, e.g. gain, pulse repetition frequency, transmit focal length, sector angle, image rate, pulse duration, depth, and sample volume.
For devices not conforming to the AIUM/NEMA 2004 Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment (hereafter in this Guidance Document referred to as the "Output Display Standard") describe the operating controls and procedures necessary to change to an application or mode that has a higher application specific acoustic output limit.
1.2.3 Unique Features
Describe any unique features or technological characteristics of the subject device.
1.3 Section 32(3)(c): Marketing History
Provide the following:
- List of countries where device is currently being sold.
- How many units have been sold worldwide?
- Summary of reported problems, number reported and corrective action. (eg. modifications to the same device, software fixes)
- Summary of the recalls and any corrective actions as a result of the recall.
2.0 Summary of Safety and Effectiveness Studies
Section 32(3)(f) of the Medical Devices Regulations requires a summary of all studies that the manufacturer relies on to ensure that the device meets the safety and effectiveness requirements of Section 10 to 20.
For devices based on established technology, information to demonstrate that the specifications and performance are equivalent to previously licensed devices is acceptable.
2.1 Section 32(3)(d): List of Standards
Manufacturers may choose to demonstrate conformance to a recognized standard or they may elect to address the relevant safety and effectiveness issues in another manner. Conformance with recognized standards is voluntary for manufacturers. If a standard is recognized, a manufacturer applying for a device licence to which that standard applies must either:
- meet the standard; or
- meet an equivalent or a better standard; or
- provide alternate evidence of safety or efficacy.
In the case of "b" and "c", detailed information must be submitted with the device licence application.
Manufacturers are directed to the Therapeutic Products Directorate's (TPD) Guidance Document - Recognition and Use of Standards under the Medical Devices Regulations for further information on the use of standards and for the TPD list of recognized medical device standards. The guidance document may be found on the website.
The List of Recognized Standards for Medical Devices may also be found on the website. For a partial list of recognized standards that are applicable to ultrasound systems and transducers, refer to Appendix 4 of this document.
2.2 Section 32(3)(e): Method of Sterilization
In the case of a Class III device sold in a sterile condition, the applicant is requested to provide a description of the sterilization method used and the packaging used to maintain sterility. This must include the type of sterilization process, the level of sterility assurance and an attestation or certification that the process has been properly validated.
2.2.1 Components Provided Sterile
For device components provided sterile to the user (e.g. single use, disposable components), provide the following information:
- The method of sterilization and a description of the method used to validate the sterilization cycle;
- The SAL (Sterility Assurance Level) intended for the device (at least 10-6);
- A description of the packaging system used to maintain device sterility;
- If the device is sterilized using ethylene oxide, the maximum levels of residues of ethylene oxide, ethylene chlorohydrin and ethylene glycol;
- If the device is radiation sterilized, the radiation dose used to achieve sterility.
For device accessories, the above information is also applicable if the accessory is included in the same Class III or Class IV device licence application.
2.2.2 Pyrogenicity
If the device is labelled pyrogen-free, provide a description of the method (standard method) used to assess pyrogenicity. Sheaths that contact brain tissue must be pyrogen free.
2.3 Section 32(3)(f): Summary of Studies
2.3.1 Clinical Measurement Accuracy and System Sensitivity
For each transducer/mode combination, give the accuracy of any measurement (e.g. distance, volume, heart rate, Doppler frequency shift, velocity, indices etc.) that can be made in that mode, and the range over which this accuracy can be expected to be maintained. Describe and justify the test methodology (e.g. laboratory and/or electronic phantom) used to determine each accuracy.
2.3.2 Performance Specifications
For each transducer/mode combination in which quantitative claims regarding Doppler sensitivity are made in the product labelling, a minimum performance specification of the Doppler sensitivity is to be provided and the methodology justified. Data validating the specification shall be included in the submission. For certain special cases or claims, clinical data or special phantom testing may be more appropriate.
2.3.3 Accepted Acoustic Output Levels
For Systems Not in Compliance with the "Output Display Standard"
The global maximum derated acoustic output limits, given in Table 2-1, are deemed to be acceptable for devices that do not comply with the "Output Display Standard" (also known as the AIUM/NEMA 2004 Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment).
Table 2-1: Accepted Acoustic Output Levels
| Use | I SPTA.3Footnote 1 (mW/cm²) |
I SPPA.3Footnote 2 (W/cm²) |
MIFootnote 3 |
|---|---|---|---|
| Peripheral Vessel | 720 | 190 | 1.9 |
| Cardiac | 430 | 190 | 1.9 |
| Fetal Imaging & OtherFootnote 4 | 94 | 190 | 1.9 |
| Ophthalmic | 17 | 28 | 0.23 |
|
|||
Note: for purposes of acoustic output limits:
- trans-esophageal for non-cardiac use, intravascular, and musculo-skeletal applications are included in the "Fetal Imaging & Other" category;
- cardiac use includes transthoracic adult and pediatric uses as well as trans- esophageal adult and pediatric uses for visualization of the heart;
- peripheral vessel use includes vessels of the neck; and
- cephalic and transcranial are synonymous.
For Fetal Heart Rate Monitors, the maximum attainable value of spatial average, temporal average intensity at the transducer face should be less than 20 mW/cm² for continuous wave devices and the maximum attainable value of the spatial average, pulse average intensity at the transducer face should be less than 20 mW/cm² for pulsed devices.
For Systems in Compliance with the "Output Display Standard"
Acoustic output should not exceed the following upper limits: ISPTA.3=720 mW/cm² and either MI=1.9 or ISPPA .3=190 W/cm².
Devices that comply with the "Output Display Standard" must also comply with the AIUM 2004 Acoustic Output Labelling Standard for Diagnostic Ultrasound Equipment: A Standard for How Manufacturers should Specify Acoustic Output Data.
For these devices that comply with the "Output Display Standard", refer to Section 3.1.4 on Labelling of this guidance document. The related Acoustic Reporting Tables are given in Appendix 3.
For ophthalmic applications in conformance with the "Output Display Standard", the upper limits are TI=max. (TIS_as, TIC) and is not to exceed 1.0, ISPTA . 3= 50mW/cm² and MI=0.23.
Summarize the operating mode possibilities for each system/transducer combination by completing Table A-2 in Appendix 2. For each possible transducer/mode identified, specify the maximum estimated ISPTA.3, MI or ISPPA.3 and TI (TIS, TIB and TIC, each as applicable). Provide a justification of the method of estimation (e.g. preliminary or prototype measurements, theoretical calculations, estimates based on measurements of previously licensed transducers).
2.3.4 Certifications
Provide the following certifications:
- That the ultrasound system will be designed and marketed in conformance with the "Output Display Standard";
- That the measurements of acoustic output display indices - Thermal Index (TI) and the Mechanical Index (MI), will be in conformance with the requirements of Section 6 entitled, "Measurement Methodology for Mechanical and Thermal Indices" of the Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment (AIUM/NEMA 2004) or "Output Display Standard".
In all submissions, the applicant shall provide evidence that the acoustic output will be, or was measured, calculated and derated as per the most recently released revision of the Acoustic Output Measurement Standard for Diagnostic Ultrasound Equipment (AIUM/NEMA UD 2-2004) and the Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment (AIUM/NEMA UD3-2004). Any deviation from the methodologies outlined in these references shall be fully described in terms of the differing methodology used and validating data.
2.3.5 Default Settings
Specify the default setting levels (i.e. as a percentage of the maximum levels) and the rationale for selecting these default values. Refer to Section 5 of the "Output Display Standard".
2.3.6 Thermal Index
Provide a justification for any Thermal Index that exceeds a value of 6.0.
2.3.7 Low Output Systems
If no system/transducer combination is capable of exceeding either a TI of 1.0 or and MI of 1.0 in any operating mode, then completion of the Acoustic Output Reporting Tables (Appendix 3) is not necessary. However, in their place the global maximum values of the ISPTA . 3 of the TI (TIS, TIB or TIC) and MI and IPA . 3 @ MImax should be specified.
2.3.8 Thermal, Mechanical and Electrical Safety
Provide either a declaration of conformity to a recognized standard, or data showing that their system is designed to be thermally, electrically and mechanically safe. This may be provided in the form of descriptions, safety precautions, testing and data to support the electrical and mechanical safety of the device. Applicable voluntary recognized standards must be identified to which the system is specified to conform with or the applicant shall include a third party certification to demonstrate that the device meets an acceptable standard.
2.3.9 Patient Contact Material
2.3.9.1 Identification and Composition
Provide the trade name and generic material composition (polyethylene, polycarbonate, silastic, etc.) of all patient contact materials or provide previously filed device licence number that contains the material description.
2.3.9.2 Biocompatibility
Provide either a declaration of conformity to applicable ISO 10993 or equivalent standards or provide biocompatibility testing results for tests conducted according to the ISO-10993-1, Biological Evaluation of Medical Devices Part 1: Evaluation and Testing or equivalent standard for any patient contact materials.
For materials, transducers, components and accessories that have been previously approved for the same or more critical tissue contact, biocompatibility is not required if the applicant certifies that the patient contact materials are unchanged in formulation and processing from the previously licensed device. Device licence numbers shall be provided.
2.3.10 Section 20: Software/Firmware
Provide either a declaration of conformity to IEC 60601-1-4 or equivalent standard or provide a description of the software/firmware supporting the operation of the subject device as per the following guidance. This applies to new systems and to software/firmware changes made to devices with extensive marketing history and history of safe use (marketing history) in other countries. Significant changes to software must be validated and verified.
The Therapeutic Products Directorate recognizes that many of ultrasound systems have a variety of software modules controlling different functions and that the level of concern for a particular module may vary. An applicant may provide different levels of documentation for different modules, providing appropriate justification is given. Applications shall include the following:
- A summary description of new and altered algorithms and an explanation why they are considered suitable for the task;
- The software version number;
- A software structural chart;
- A system hazard analysis;
- A listing of the specific hardware/software requirements;
- A summary of the software design and development process, including the software change management process; and
- A summary of the software verification and validation processes.
2.4 Section 32(3)(i): Bibliography
To facilitate the review process, the applicant is requested to provide a bibliography of relevant reports dealing with the use of the device for new technology and new indications. Inclusion of abstracts and reprints may facilitate the review process. For established technology, a rationale for not including references must be provided.
3.0 Section 32(3)(g): Labelling
A copy of device labelling is required to be submitted for Class III and Class IV device licence application under Section 32(3)(g) and Section 32(4)(o) of the Regulations, respectively.
"Final draft device labelling" may be submitted to meet these requirements.
Labelling includes device labels, package inserts, product brochures, operator's manuals, promotional material that describe the system and associated transducers. Maintenance manuals are not a required component of the final draft device labelling.
Graphics and ink colours are not required for the "final draft device labelling". Final product labelling shall not differ in the content or context of the "final draft device labelling" submitted with the device licence application.
All final product labelling shall comply with the Labelling Requirements (Section 21 to 23) of the Regulations.
3.1 Acoustic Output Labelling in the Operator's Manual
Draft labelling of the operator's manual showing estimated global acoustic output indices for each possible system/transducer/mode combination as per Appendix 2: Tables A-2 and A-2-1, may be provided with the initial medical device licence application. However, final acoustic output indices values are required to be submitted prior to the first sale of the device as per Appendix 3: Tables A-3, A-3-1, A-3-2 and A-3-3.
The acoustic output reporting table may also be found in the AIUM labelling standard (AIUM 1998). Manufacturers are advised to use the format of the tables in the submission.
3.1.1 Real Time Features
Provide an explanation of the real-time display features and controls of the system, including default setting (refer to Section 4.2.2 of the "Output Display Standard"). Suggestions on how to use these features and controls to follow the ALARA principle should be provided. It is recommended that these include, but not be limited to, a reference to Health Canada's Guidelines for the Safe Use of Diagnostic Ultrasound, particularly sections 2 and 3.
Note that if the intended uses include neonatal cephalic, then the provisions of the "Output Display Standard" are interpreted to mean that all three thermal indices (TIS, TIB, TIC) must be available to be called up by the user, although all three indices are not required to be displayed simultaneously. In this regard, the applicant may refer to page 39 in the AIUM publication, Medical Ultrasound Safety (AIUM 1994).
3.1.2 Display Accuracy and Measurement Precision
Provide the display accuracy and measurement precision. Refer to Sections 4.2, 4.2.1, and 6.4 of the "Output Display Standard".
3.1.3 Low Output Systems
If no system/transducer combination is capable of exceeding either a TI of 1.0 or and MI of 1.0 in any operating mode, then completion of the Acoustic Output Reporting Tables (Appendix 3) is not necessary. However, in their place the global maximum values of the ISPTA . 3 of the TI (TIS, TIB or TIC) and MI and IPA . 3 @ MImax1 should be specified as shown in Appendix 1: Table A-1.
3.1.4 Acoustic Output Reporting Tables
Appendix 3 contains an example output table. Note the use of the four footnotes: a,b,c,#. The Operating Mode (B-mode, Pulsed Doppler, and/or Colour Flow (incl. M-Mode)) should be stated, and applicable data provided.
Summarize the operating mode possibilities for each system/transducer combination by completing Table A-2. For each possible transducer/mode identified specify the target range of values for the MI or ISPPA.3 and ISPTA.3 and an estimated range of TI's under the operating conditions that maximize these quantities, noting that the upper bound must not be greater than the global maximum values. Also provide the engineering basis for the range of values specified (e.g. preliminary or prototype measurements, theoretical calculations, estimates based on measurements of previously licensed transducers).
3.2 Section 32(3)(g): General Labelling Information
3.2.1 Re-use Instructions
Provide instructions for care of the device between uses, including storage, cleaning, disinfection and sterilization of all components, as appropriate. Labelling should recommend the use of sterile, when appropriate and licensed transducer sheaths, for clinical applications of a semi-critical or critical nature (i.e. intra operative, transrectal, transvaginal, trans-esophageal, biopsy procedures).
When recommending a procedure that uses a clear liquid disinfection or sterilizing agent refer the user to the labelling instructions provided by the manufacturer of that product.
When recommending a procedure other than liquid disinfection or sterilization, detailed instructions are to be provided. These procedures are validated and a summary of the validation process and representative data submitted as part of the application.
3.2.2 Additional Labelling
Additional labelling may be necessary to address safety and effectiveness concerns, depending upon the clinical application(s) of the transducer: e.g. transcranial, trans-esophageal, intra operative, transvaginal, ophthalmic or vascular diagnostic systems.
Neurological intraoperative transducers (i.e. when the transducer makes contact with the dura or any intracranial tissues) should have the following additional labelling. There should be a recommendation to use sterile, pyrogen-free sheaths. In addition, a caution should warn the user of a potential problem in using the transducer on patients with Cruetzfeld-Jacob disease. If the transducer becomes contaminated, it may have to be destroyed since it may not be adequately disinfected.
4.0 Section 32(3)(j): Quality system certification
Section 32(3)(j) requires a a copy of a quality system certificate certifying that the quality system under which the device is designed and manufactured satisfies National Standard of Canada CAN/CSA- ISO 13485-03, Medical devices -- Quality management systems -- Requirements for regulatory purposes, as amended from time to time.
5.0 Medical device licence issuance
In the case where the final global acoustic output indices (Sections 2.3.3 and 3.1) are provided in the initial medical device licence application, and it is determined that the ultrasound system/transducer meet the safety and effectiveness requirements of the Regulations, a medical device licence will be issued.
In the case where the estimated global acoustic output indices (Sections 2.3.3 and 3.1) are provided in the initial medical device licence application, and it is determined that the ultrasound system/transducer meet the safety and effectiveness requirements of the Regulations, a medical device licence with conditions will be issued. A licence with conditions sets out terms to ensure that the device continues to meet the safety and effectiveness requirements. In this case, and specifically for ultrasound systems and transducers, the licence is granted on the condition that prior to the first sale of the device, the final global acoustic output indices based on production line devices (as per Appendix 3: tables A-3. A-3-1, A-3-2, A-3-3) must be submitted. Once this information is provided, sale of the ultrasound system and/or transducer may commence. If the information is complete and contains acceptable acoustic output values (ie. the device continues to meet the safety and effectiveness requirements) then the licence will be amended to remove this condition.
Appendix 1
Table A-1: Low Output Summary Table
(for systems with no transducers having global maximum index values exceeding 1.0)
- System:
- Transducer Model
- Model A
- Model B
- Model C
- Ispta.3
- TI Type
- TI Value
- MI
- Ipa.3@MImax
- Additional Comments:
Appendix 2: Operating Modes and Acoustic Output Estimates
This approach is based upon conformance with the "Output Display Standard".
Table A-2: Output Range Summary Table
For devices that comply with the "Output Display Standard", the information submitted in this table format will provide information on the specifications and parameters to determine applicability of standards.
Table A-2-1: Transducer/Mode Combination Summary Table
Complete Table A-2-1 below for each transducer/mode combination. Indicate with a check the transducer/mode combinations for which the global maximum displayed MI or TI is greater than 1.0. For each transducer/mode combination checked, and acoustic output table should be completed (Table A-3).
- System:
- Transducer Model
- Mode of Operation
- B
- M
- PWD
- CWD
- Colour Doppler
- Combined (Specify)
- OtherFootnote 3 (Specify)
- Additional Comments:
For reporting purposes, the following mode definitions and reporting rules apply:
- B Mode
- No other modes active. Only MI (when > 1.0) need be reported for this mode.
- M Mode
- May include simultaneous B mode.
- PW Dop./ CW Dop.
- In duplex modes, report largest displayed TIS (scanned or non-scanned) if > 1.0.
- Colour Flow
- May include simultaneous Colour Flow M-mode, B-mode and M mode. In combined modes, report largest displayed TIS (scanned or non-scanned) if > 1.0.
- Combined modes
- Need only be reported as a separate mode if the largest formulation of TIS, TIB or TIC (if there is an applicable intended use; e.g. transcranial or neonatal cephalic) is greater than the corresponding value reported for all constituent modes.
TIC need not be reported if the transducer is not intended for transcranial or neonatal cephalic use.
If the acoustic output of an "other" mode is the same (within the manufacturer's stated measurement uncertainty) as that of a designated standard mode, then only one acoustic output table need be completed for both modes. However, the acoustic output table should be identified as applying to both modes.
Appendix 3: Acoustic Output Reporting Table
- Z 1
- where Z 3Z bp (centimetres)
- US
- where z Z bπ (centimetres)
- D eq(Z)
- (4/p)
All entries in Table A-3 should be obtained at the same operating conditions that give rise to the global maximum Index Value in the second row. These operating conditions should be specified. Measurement uncertainties for acoustic quantities (power, pressure, intensities, center frequency) should be provided.
Symbols used in the table are described below.
MI
TISscan
TISnon-scan
TIB
TIC
Aaprt
Pr.3
Wo
W.3(z1)
ITA.3(Z1)
Z1
Zbp
zsp
deq(Z)
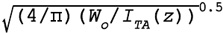
ƒc
Dim. of Aaprt
PD
PRF
Pr@PIImax
deq@PIImax
FL
IPA.3@MImax
All entries in Table A-3 should be obtained at the same operating conditions that give rise to the global maximum Index Value in the second row. These operating conditions should be specified. Measurement uncertainties for acoustic quantities (power, pressure, intensities, center frequency) should be provided.
TIS_as: The soft-tissue thermal index at surface for non-autoscanning mode;

where
- Wo1x1 is the bounded-square output power in milliwatts;
- ƒc is the center frequency in Megahertz.
- Symbol: TIS_as
- Unit: Unitless
Table A-3: Acoustic Output Reporting Table
(provide data where global maximum displayed index exceeds 1.0)
Note: See section 4.1.3.1 of the "Output Display Standard"
- Transducer Model:
- Operating Mode:
- Index Label
- Global Maximum Index Value
- Assoc Acoustic Parameter
- pr.3 (MPa)
- Wo (mW)
- min of [W.3(z1), ITA.3(z1)] (mW)
- z1 (cm)
- zbp (cm)
- zsp (cm)
- deq(zsp) (cm)
- ƒc (MHz)
- Dim of Aaprt
- X (cm)
- Y (cm)
- Other Information
- PD (µsec)
- PRF (Hz)
- pr@PIImax (MPa)
- deq@PIImax (cm)
- Focal Length
- FLx (cm)
- FLy (cm)
- IPA.3 @MImax (W/cm²)
- Operating Control Conditions
- Control 1
- Control 2
- Control 3
- Control n
- MI
- TIS
- scan
- non - scan
- Aaprt≤1
- Aaprt>1
- TIB
- non-scan
- TIC
- Notes:
- (a) This index is not required for this operating mode; see section 4.1.3.1on Output Display Standard
- (b) This probe is not inteded for transcranial or neonatal cephalic uses
- (c) This formulation for TIS is less than that for an alternate formulation in this mode.
- # No data are reported for this operating condition since the global maximum index value is not reported for the reason listed.
The next three pages contain example output tables to illustrate the use of the four footnotes (a,b,c,#). A check mark () indicates that the box should be filled in with the appropriate value; a dash (-) means that no value is required because of either scan/non-scan or aperture size considerations.
With regard to the third example, Colour flow and M-mode, the use of footnote (c) is shown. Note that if the M-mode TIS were greater than the colour flow TIS, then footnote (c) would appear under TIS(scan), and the M-mode TIS value would be listed in the appropriate TIS (non-scan) box. Therefore, it is important to list under "Operating Mode" all included modes for proper interpretation of the tabulated values.
Table A-3-1: Acoustic Output Reporting Table (B-mode)
(provide data where global maximum displayed index exceeds 1.0)
Note: See section 4.1.3.1 of the "Output Display Standard".
- Transducer Model:
- Operating Mode: B-mode
| Index Label | MI | TIS | TIB | TIC | ||||
|---|---|---|---|---|---|---|---|---|
| scan | non - scan | non-scan | ||||||
| Aaprt≤1 | Aaprt>1 | |||||||
| Global Maximum Index Value | 1.5 | Footnote a | - | - | - | Table 2 footnote a | ||
| Assoc Acoustic Parameter |
pr.3 | (MPa) | X | |||||
| Wo | (mW) | Footnote # | - | - | Footnote # | |||
| min of [W.3(z1), ITA.3(z1)] | (mW) | - | ||||||
| z1 | (cm) | - | ||||||
| zbp | (cm) | - | ||||||
| zsp | (cm) | X | - | |||||
| deq(zsp) | (cm) | - | ||||||
| ƒc | (MHz) | X | Footnote # | - | - | - | Footnote # | |
| Dim of Aaprt | X (cm) | Footnote # | - | - | - | Footnote # | ||
| Y (cm) | Footnote # | - | - | - | Footnote # | |||
| Other Information | PD | (µsec) | X | |||||
| PRF | (Hz) | X | ||||||
| pr@PIImax | (MPa) | X | ||||||
| deq@PIImax | (cm) | - | ||||||
| Focal Length |
FLx (cm) | Footnote # | - | - | Footnote # | |||
| FLy (cm) | Footnote # | - | - | Footnote # | ||||
| IPA.3 @MImax | (W/cm²) | X | ||||||
| Operating Control Conditions |
Control 1 | |||||||
| Control 2 | ||||||||
| Control 3 | ||||||||
| Control n | ||||||||
|
||||||||
Table A-3-2: Acoustic Output Reporting Table (Pulsed Doppler)
(provide data where global maximum displayed index exceeds 1.0)
Note: See section 4.1.3.1 of the Output Display Standard.
- Transducer Model:
- Operating Mode: Pulsed Doppler
| Index Label | MI | TIS | TIB | TIC | ||||
|---|---|---|---|---|---|---|---|---|
| scan | non - scan | non-scan | ||||||
| Aaprt≤1 | Aaprt>1 | |||||||
| Global Maximum Index Value | Footnote a | - | - | <1 | 2 | <1 | ||
| Assoc Acoustic Parameter |
pr.3 | (MPa) | Footnote # | |||||
| Wo | (mW) | - | - | X | Footnote # | |||
| min of [W.3(z1), ITA.3(z1)] | (mW) | Footnote # | ||||||
| z1 | (cm) | Footnote # | ||||||
| zbp | (cm) | Footnote # | ||||||
| zsp | (cm) | Footnote # | X | |||||
| deq(zsp) | (cm) | X | ||||||
| ƒc | (MHz) | Footnote # | - | - | Footnote # | X | Footnote # | |
| Dim of Aaprt | X (cm) | - | - | Footnote # | X | Footnote # | ||
| Y (cm) | - | - | Footnote # | X | Footnote # | |||
| Other Information | PD | (µsec) | Footnote # | |||||
| PRF | (Hz) | Footnote # | ||||||
| pr@PIImax | (MPa) | Footnote # | ||||||
| deq@PIImax | (cm) | X | ||||||
| Focal Length |
FLx (cm) | - | - | Footnote # | Footnote # | |||
| FLy (cm) | - | - | Footnote # | Footnote # | ||||
| IPA.3 @MImax | (W/cm²) | Footnote # | ||||||
| Operating Control Conditions |
Control 1 | |||||||
| Control 2 | ||||||||
| Control 3 | ||||||||
| Control n | ||||||||
|
||||||||
Table A-3-3: Acoustic Output Reporting Table (Colour Flow M-Mode)
(provide data where global maximum displayed index exceeds 1.0)
Note: See section 4.1.3.1 of the "Output Display Standard".
- Transducer Model:
- Operating Mode: Colour Flow (M-mode)
| Index Label | MI | TIS | TIB | TIC | ||||
|---|---|---|---|---|---|---|---|---|
| scan | non - scan | non-scan | ||||||
| Aaprt≤1 | Aaprt>1 | |||||||
| Global Maximum Index Value | Footnote a | 2 | - | Footnote c | 3 | Footnote c | ||
| Assoc Acoustic Parameter |
pr.3 | (MPa) | Footnote # | |||||
| Wo | (mW) | X | - | X | Footnote # | |||
| min of [W.3(z1), ITA.3(z1)] | (mW) | Footnote # | ||||||
| z1 | (cm) | Footnote # | ||||||
| zbp | (cm) | Footnote # | ||||||
| zsp | (cm) | Footnote # | X | |||||
| deq(zsp) | (cm) | X | ||||||
| ƒc | (MHz) | Footnote # | X | - | Footnote # | X | Footnote # | |
| Dim of Aaprt | X (cm) | X | - | Footnote # | X | Footnote # | ||
| Y (cm) | X | - | Footnote # | X | Footnote # | |||
| Other Information | PD | (µsec) | Footnote # | |||||
| PRF | (Hz) | Footnote # | ||||||
| pr@PIImax | (MPa) | Footnote # | ||||||
| deq@PIImax | (cm) | X | ||||||
| Focal Length |
FLx (cm) | X | - | Footnote # | Footnote # | |||
| FLy (cm) | X | - | Footnote # | Footnote # | ||||
| IPA.3 @MImax | (W/cm²) | Footnote # | ||||||
| Operating Control Conditions |
Control 1 | |||||||
| Control 2 | ||||||||
| Control 3 | ||||||||
| Control n | ||||||||
|
||||||||
Appendix 4: List of Standards
The following is a partial list of recognized standards that are available for use in diagnostic ultrasound submissions. For a complete list of TPD Recognized Medical Device Standards, refer to the List of Recognized Standards for Medical Devices posted on the website.
- AIUM 1998
- Acoustic Output Labelling Standard for Diagnostic Ultrasound Equipment: A Standard for How Manufacturers Should Specify Acoustic Output Data
- AIUM/NEMA UD2-2004, Revision 3
- Acoustic Output Measurement Standard for Diagnostic Ultrasound Equipment.
- AIUM/NEMA UD3-2004, Revision 2
- Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment.
- IEC 60601-1
- Medical Electrical Equipment - Part 1: General Requirements for Safety 2nd edition
- IEC 60601-1-am1
- Medical Electrical Equipment - Part 1: General Requirements for Safety Amendment No.1
- IEC 60601-1-am2
- Medical Electrical Equipment - Part 1: General Requirements for Safety Amendment No.2
- IEC 60601-1-1
- Medical Electrical Equipment - Part 1: General Requirements for Safety - Section 1: Collateral Standard: Safety Requirements for Medical Electrical Systems
- IEC 60601-1-2
- Medical Electrical Equipment - Part 1: General Requirements for Safety - Section 2: Collateral Standard: Programmable Electrical Medical Systems
- IEC 60601-1-4
- Medical Electrical Equipment - Part 1: General Requirements for Safety - Section 4: Collateral Standard: Programmable Electrical Medical Systems
- ISO-10993-1,
- Biological Evaluation of Medical Devices Part 1: Evaluation and Testing, (ISO 1993)
- IEC 60601-2-37 Ed. 1.0
- Medical Electrical Equipment - Part 2-37: Particular Requirements for the Safety of Ultrasonic Medical Diagnostic and Monitoring Equipment
- ISO 14971-1-2000
- Medical Devices - Application of Risk Management to Medical Devices
Note: This guidance makes extensive reference to the Standard for Real-Time Display of Thermal and Mechanical Acoustic Output Indices on Diagnostic Ultrasound Equipment using the generally recognized shortened name "Output Display Standard". All references to the "Output Display Standard" specifically refer to the above-mentioned standard.
Appendix 5: General Definitions
Contraindications describe situations where the device should not be used because the risk of use clearly outweighs any reasonably foreseeable benefits.
Control mechanism is a means of verifying or checking that the specifications or outputs of the device meet a standard or predetermined result. They are mechanisms put in place to maintain on-going control or regulate the output of a device.
Indications for use is the general description of the disease(s) or condition(s) the device will diagnose, treat, prevent or mitigate, including where applicable a description of the patient population for which the device is intended. The indications include all the labelled patient uses of the device, for example: the condition(s) or disease(s) to be prevented, mitigated, treated or diagnosed, part of the body or type of tissue applied to or interfaced with, frequency of use, physiological purpose and patient population. Indications for use are generally found in the indications section of the labelling, but indications may also be inferred from other parts of the labelling such as the precautions, warnings, bibliography or directions for use sections. In some instances, intended use is determined by a manufacturer's and/or distributor's statements or may be shown by the circumstances surrounding the distribution of the article.
Operating Principles are the means by which a device produces or brings about a desired or appropriate effect. They are the means whereby a device is able to have a certain influence on a person or its surroundings.
Precautions describe any special care to be exercised by practitioner or patient for the safe and effective use of a device. This definition includes limitations stated for in vitro diagnostic devices (IVDDs).
Warnings describe serious adverse reactions and potential safety hazards that can occur in the proper use, or the misuse, of a device, along with consequent restrictions in use and mitigating steps to take if problems occur.
Device Specific Definitions
Acoustic Pressure
- Symbol: p
- Unit: Pascal, Pa
ALARA
Autoscan (Autoscanning)
Bandwidth
The difference between the most widely separated frequencies ƒ1 and ƒ2 at which the transmitted acoustic pressure spectrum is 71 percent (-3 dB) of its maximum value.
- Symbol: BW
- Unit: Hertz, Hz
Beam Axis
Beam Cross-Sectional Area
- Symbol: A
- Unit: centimetre squared, cm²
Bounded-Square Output Power
- Symbol: W01X1
- Unit: milliwatt, mW
- center frequency: defined as ƒc = (ƒ1 + ƒ2)/2 where ƒ1 and ƒ2 are frequencies defined in bandwidth
- Symbol: ƒc
- Unit: Hertz, Hz
Conventional
Declaration of Conformity
- Any element of the standard that was not applicable to the device;
- If the standard is part of a family of standards which provides collateral and/or particular parts, a statement regarding the collateral and/or particular parts that were met;
- Any deviations from the standards that were applied;
- What differences exist, if any, between the tested device(s) and the device to be marketed and a justification of the test results in those areas of difference; and
- Name and address of any test laboratory or certification body involved and a reference to any accreditations of those organisations.
Derating (Derating Factor, Derated)
A factor applied to acoustic output parameters intended to account for ultrasonic attenuation of tissue between the source and a particular location in the tissue. As referred to in this document, the average ultrasonic attenuation is assumed to be a 0.3 dB/cm-MHz along the beam axis in the body. Derated parameters are denoted with a subscript ".3".
- Symbol: a
- Unit: decibel per centimetre - megahertz, dB cm-1MHz-1
Designated Standard Mode
Duty Factor
Entrance Beam Dimensions
- Symbol: EBD
- Unit: centimetre, cm
Entrance Dimensions of the Scan
- Symbol: EDS
- Unit: centimetre, cm
Envelope
Far Field
Focal Surface
- Symbol: (none)
- Unit: centimetre squared, cm²
Global Maximum
Intensity
Intensity, Instantaneous
- Symbol: I
- Unit: Watt per square-centimetre, W cm-2
Intensity, Pulse-Average
- Symbol: IPA
- Unit: Watt per square-centimetre, W cm-2
Intensity, Spatial-Average Pulse-Average
- Symbol: ISAPA
- Unit: milliwatt per square-centimetre, mW cm-2
Intensity, Spatial-Average Temporal-Average
- Symbol: ISATA
- Unit: milliwatt per square-centimetre, mW cm²
Intensity, Spatial-Peak Pulse-Average
- Symbol: ISPPA
- Unit: Watt per square-centimetre, W cm-2
Intensity, Spatial-Peak Temporal-Average
- Symbol: ISPTA
- Unit: milliwatt per square-centimetre, mW cm-2
Intensity, Temporal-Average
- Symbol: ITA
- Unit: milliwatt per square-centimetre, mW cm-2
Intensity, Temporal Peak
- Symbol: ITP
- Unit: Watt per square-centimetre, W cm-2
Invasive Transducer
Mechanical Index
The spatial-peak value of the peak rarefactional pressure, derated by 0.3 dB/cm-MHz at each point along the beam axis, divided by the square root of the center frequency, that is: = pr.3( zsp) / ƒc½ where pr.3 (Zsp) is the peak rarefactional pressure in megapascals derated by 0.3 dB/cm-MHz to the point on the beam axis, Zsp, where the pulse intensity integral (PII.3) is maximum; and ƒc is the center frequency in megahertz.
- Symbol: MI
- Unit: Unitless
Mode
Non-Autoscan(Non-Autoscanning)
Operating Condition
Output Control Settings
Output Display Standard
Peak Rarefactional Pressure; Peak Negative Pressure
- Symbol: Pr or P_
- Unit: megapascal, Mpa
Power (Ultrasonic Power)
- Symbol: Wo
- Units: Watts, W
Pressure
Pulse-Average Intensity
- Symbol: IPA
- Unit: Watt per square-centimetre, W cm-2
Pulse Duration
- Symbol: PD
- Unit: second, s
Pulse Intensity Integral
- Symbol: PII
- Unit: Joule per centimetre-squared, J cm-2
Pulse Repetition Frequency
- Symbol: PRF
- Unit: Hertz, Hz
Radiating Cross-Sectional Area (Aaprt)
- Symbol: S
- Unit: centimetre squared, cm²
Scan Cross-Sectional Area
- Symbol: (none)
- Unit: centimetre squared, cm²
Spatial-Average Pulse-Average Intensity
- Symbol: ISATA
- Unit: milliwatt per square-centimetre, mW cm-2
Spatial-Average Temporal-Average Intensity
- Symbol: ISATA
- Unit: milliwatt per square-centimetre, mW cm-2
Spatial-Peak Pulse-Average Intensity
- Symbol: ISPPA
- Unit: Watt Per Square-centimetre, W Cm-2
Spatial-Peak Temporal-Average Intensity
- Symbol: ISPTA
- Unit: milliwatt per square-centimetre, mW cm-2
Superficial
Temporal-Average Intensity
- Symbol: ITA
- Unit: milliwatt per square-centimetre, mW cm-2
Temporal-Peak Intensity
- Symbol: ITP
- Unit: Watt per square-centimetre, W cm-2
Thermal Index
- Symbol: TI
TIS_as

- Wo1x1 is the bounded-square output power in milliwatts;
- ƒc is the center frequency in Megahertz.
- Symbol: TIS_as
- Unit: Unitless
Transducer Assembly
Utrasonic Power
Waveform
Waveform Record
Wavelength
- Symbol: λ
- Unit: centimetres per cycle, cm cycle-1
Mechanical Index, MI = Pr.3 (Zsp) / (ƒc½)
Wavelength λ = V / ƒ
List of Symbols
- P
- Acoustic Pressure
- BW
- Bandwidth
- A
- Beam cross-sectional area
- ƒc
- Center frequency
- a
- Derating Factor
- I
- Instantaneous Intensity
- IPA
- Pulse-average intensity
- ISAPA
- Spatial-average pulse-average intensity
- ISATA
- Spatial-average temporal-average intensity
- ISPPA
- Spatial-peak pulse-average intensity
- ISPTA
- Spatial-peak temporal-average intensity
- ITA
- Temporal-average intensity
- ITP
- Temporal-peak intensity
- MI
- Mechanical index
- Pr
- Peak rarefactional pressure
- WO
- Power, ultrasonic power
- PD
- Pulse duration
- PII
- Pulse intensity integral
- PRF
- Pulse repetition frequency
- S
- Radiating cross-sectional area
- TI
- Thermal Index
- TIS_as
- Soft tissue thermal index at surface
- λ
- Wavelength