
Draft Guidance Document: Applications for Medical Device Investigational Testing Authorizations
Notice to the reader:
Current Status: Closed. The final guidance is available.
Draft Guidance Document
Applications for Medical Device Investigational Testing Authorizations
Published by authority of the Minister of Health
Draft Date: 2017/10/06
Health Products and Food Branch
Our mission is to help the people of Canada maintain and improve their health.
Health Canada
The Health Products and Food Branch's mandate is to take an integrated approach to the management of the risks and benefits to health related products and food by:
- minimizing health risk factors to Canadians while maximizing the safety provided by the regulatory system for health products and food; and,
- promoting conditions that enable Canadians to make healthy choices and providing information so that they can make informed decisions about their health.
Health Products and Food Branch
© Minister of Public Works and Government Services Canada 2017
Également disponible en français sous le titre : Ébauche de la ligne directrice : Demandes d'autorisation d'essai expérimental pour les instruments médicaux
Foreword
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how to implement Health Canada mandates and objectives in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable guidance documents.
Table of Contents
- 1. Introduction
- 2. Guidance for Implementation
- 2.1 Abbreviations and Definitions
- 2.2 Pre-ITA Application Meeting
- 2.3 ITA Applications
- 2.4 Responsibilities of Manufacturers and Importers
- 2.5 Requests for Revisions to an ITA
- 2.6 Cancellation of an ITA
- 2.7 Review Process for ITA Applications and Revisions to ITAs
- 2.8 Post-Authorization Requirements
- Appendix 1 - Relevant Addresses
- Appendix 2 - Useful Documents
- Appendix 3 - Determining when an ITA Application is Required
- Appendix 4 - Format for an ITA Application
- Appendix 5 - Frequently Asked Questions
1. Introduction
The Food and Drugs Act (FDA) sets out the legislative framework under which medical devices are regulated. The Medical Devices Bureau is responsible for administering the requirements within Part 3 of the Medical Device Regulations (herein referred to as the Regulations) that govern the sale and importation of a medical device for investigational testing involving human subjects. Manufacturers and importers must meet the regulatory requirements therein including requirements outlined in Subsection 83 (1) of the Regulations in order to receive authorization from Health Canada to sell a device to a qualified investigator for the purpose of conducting investigational testing.
This draft guidance document replaces the previous guidance document entitled: Preparation of an Application for Investigational Testing - Medical Devices (GD009/Rev00-MDB V3 dated 1999-02-22) and will be finalised following consideration of stakeholder comments.
1.1 Policy Objectives
To provide assistance to manufacturers and importers in preparing the documentation necessary to obtain an authorization for the sale or importation of a medical device under an Investigational Testing Authorization (ITA), while assuring the protection of research subjects, and promoting excellence in research and development in Canada.
1.2 Policy Statements
Manufacturers and importers are responsible for submitting ITA applications to Health Canada for authorization to sell or import a medical device for the purpose of conducting investigational testing in human subjects.
Health Canada's expectation is that manufacturers follow the principles of the Declaration of Helsinki and the Tri-Council Policy Statement (2nd Edition): Ethical Conduct for Research Involving Humans (2010), and conform to Good Clinical Practices (GCP) as set out by ISO 14155 - Clinical investigation of medical devices for human subjects. ISO 14155 is generally consistent with the definitions and requirements of the Regulations. Where inconsistencies exist, the Regulations take precedence.
Research Ethics Boards (REBs) have an important role in the oversight of the conduct of investigational testing. Health Canada will issue an ITA only after receipt of evidence of REB approval for Class III and IV devices. The authorization will remain valid provided that the REB approval is current, or until the device is licensed.
Manufacturers and importers are requested to submit ITA applications in the "non-eCTD electronic-only" format and follow the structure presented in Appendix 3 when applying for an ITA.
1.3 Scope and Application
This guidance document is intended to assist manufacturers and importers in organizing and submitting an ITA application to conduct investigational testing of a Class II, III or IV device, by the manufacturer, an academic institution, a health care facility or a contract research organization. It also provides details on the responsibilities of manufacturers and importers when conducting investigational testing using Class I devices. Further, it will assist investigators and institutions involved in the investigational testing of medical devices in Canada to understand their roles and responsibilities in this process.
This document is not applicable to the investigational testing of in vitro diagnostic devices (IVDDs) in Canada. Manufacturers and importers are referred to the guidance document titled Preparation of an Application for Investigational Testing - in vitro Diagnostics (GD010) available on the Government of Canada website (refer to Appendix 2).
1.4 Background
Investigational testing of medical devices in human subjects has become a growing area for research and development in Canada since the establishment of the new Medical Devices Regulations in May, 1998.
Concerns have been raised about the inconsistencies between the Food and Drug Regulations and the Medical Devices Regulations. Although these inconsistencies can only be corrected by regulatory amendment, resolution of some deficiencies can be addressed through this guidance document.
The previous guidance document, published in 1999, is no longer adequate and lacks clear and consistent guidance on a number of aspects regarding the preparation of applications for ITAs. Frequent requests for clarification by manufacturers addressed to the Medical Devices Bureau have underlined these deficiencies.
This guidance document has been updated to respond to stakeholder concerns and address topics such as the use of recognized standards under the Regulations, drug-device combination products, the use of unlicensed devices in drug studies, investigator-sponsored investigational testing, stages of product development, revisions to an investigational testing protocol, and problem reporting.
2. Guidance for Implementation
2.1 Abbreviations and Definitions
2.1.1 Abbreviations
- CTA
- Clinical Trial Application
- CTD
- Common Technical Document
- GCP
- Good Clinical Practices
- IB
- Investigator's Brochure
- ICF
- Informed Consent Form
- IFU
- Instructions for Use
- IA
- Investigator Agreement
- IT
- Investigational Testing
- ITA
- Investigational Testing Authorization
- IVDD
- In Vitro Diagnostic Devices
- ISO
- International Organization for Standardization
- MDB
- Medical Device Bureau
- MDCU
- Medical Devices Compliance Unit
- MDPR
- Medical Devices Problem Reporting
- MHPD
- Marketed Health Products Directorate
- RCT
- Randomized Controlled Trial
- REB
- Research Ethics Board
- RORB
- Regulatory Operations and Regions Branch
- SAP
- Special Access Programme
- SOP
- Standard Operating Procedure
- TPD
- Therapeutic Products Directorate
2.1.2 Terms and Definitions
Most of the definitions listed below were taken from the Regulations and ISO 14155 Clinical investigation of medical devices for human subjects - Good Clinical Practice.
Additional Information: Information requested from the device manufacturer or importer by the Medical Devices Bureau to determine whether the conditions set out in Subsection 83(1) of the Regulations have been met.
Adverse Event: Any untoward medical occurrence, unintended disease or injury, or untoward clinical signs (including abnormal laboratory findings) in subjects, users or other persons, whether or not related to the investigational medical device which includes:
- events related to the investigational medical device or the comparator; and,
- events related to the procedures involved.
For users or other persons, this definition is restricted to events related to investigational medical devices.
Bias: A systematic error in measurement during data collection.
Case Report Forms: Set of printed, optical or electronic documents for each subject on which information to be reported to the sponsor is recorded, as required by the Clinical Investigation Plan (i.e., protocol).
Clinical investigation: A systematic investigation in one or more human subjects, undertaken to assess the safety or performance of a medical deviceFootnote 1.
Clinical Investigation Report: Document describing the design, execution, statistical analysis and results of a clinical investigation.
Combination Product: Is a therapeutic product that combines a drug component and a device component (which by themselves would be classified as a drug or a device), such that the distinctive nature of the drug component and device component is integrated in a singular product.
Comparator: Medical device, therapy (e.g. active control), placebo or no treatment, used in the reference group in a clinical investigation.
Contract Research Organization: Person or organization contracted by the sponsorFootnote 2 to perform one or more of the sponsor's clinical investigation-related duties and functions.
Deviation: Instance(s) of failure to follow, intentionally or unintentionally, the requirements of the Clinical Investigation Plan.
Device: means an instrument, apparatus, contrivance or other similar article, or an in vitro reagent, including a component, part or accessory of any of them, that is manufactured, sold or represented for use in:
- diagnosing, treating, mitigating or preventing a disease, disorder or abnormal physical state, or any of their symptoms, in human beings or animals,
- restoring, modifying or correcting the body structure of human beings or animals or the functioning of any part of the bodies of human beings or animals,
- diagnosing pregnancy in human beings or animals,
- caring for human beings or animals during pregnancy or at or after the birth of the offspring, including caring for the offspring, or
- preventing conception in human beings or animals;
however, it does not include such an instrument, apparatus, contrivance or article, or a component, part or accessory of any of them, that does any of the actions referred to in paragraphs (a) to (e) solely by pharmacological, immunological or metabolic means or solely by chemical means in or on the body of a human being or animal;
Device Identifier: Means a unique series of letters or numbers or any combination of these or a bar code that is assigned to a medical device by the manufacturer and that identifies it as different from similar devices.
Device Name: In respect of a medical device, includes any information necessary for the user to identify the device and to distinguish it from similar devices.
Emergency: "An unlooked for or sudden occasion; an accident; an urgent or pressing need"Footnote 3.
Ethics Committee: Independent body whose responsibility it is to review clinical investigations in order to protect the rights, safety and well-being of human subjects participating in a clinical investigation. The regulatory requirements pertaining to ethics committees or similar institutions vary by country or regionFootnote 4.
Good Clinical Practices: Generally accepted clinical practices that are designed to ensure the protection of the rights, safety and well-being of clinical trial subjects and other persons.
Health Care Professional: Means a person who is entitled under the laws of a province to provide health services in the province.
Hypothesis: Testable statement, resulting from the objective, regarding the investigational medical device safety or performance that is used to design the clinical investigation and that can be accepted or rejected based on results of the clinical investigation and statistical calculations. The primary hypothesis is the determinant of the investigational medical device safety or performance parameters and is usually used to calculate the sample size. Secondary hypotheses concerning other points of interest can also be evaluated.
Importer: A person other than the manufacturer of a device, whose establishment is in Canada, who causes the medical device to be brought into Canada from foreign manufacturers or distributors, for sale in Canada.
Independent: Not involved in the conduct of a clinical investigation, except for their specifically assigned responsibilities, in order to avoid bias or a conflict of interest.
Informed Consent Document/Form: Process by which an individual is provided information and is asked to voluntarily participate in a clinical investigation. Informed consent is documented by means of a written, signed and dated informed consent form.
Investigation Site: Institution or site where the clinical investigation is carried outFootnote 5.
Investigational Medical Device: Medical device being assessed for safety or performance in a clinical investigation. This includes medical devices already on the market that are being evaluated for new intended uses, new populations, new materials or design changesFootnote 6.
Investigator: Individual member of the investigation site team designated and supervised by the principal investigator at an investigation site to perform critical clinical-investigation-related procedures or to make important clinical investigation-related decisionsFootnote 7.
Investigator's Brochure: Compilation of the current clinical and non-clinical information on the investigational medical device(s), relevant to the clinical investigation.
Investigator Sponsored Investigational Testing: A clinical investigation initiated by a clinician or a health care facility and not by the manufacturer of the device. The data generated is not intended to be used in support of a marketing application. The funding for such research is typically provided by a third party, such as the CIHR (Canadian Institutes of Health Research).
In these cases, the Investigator must obtain the cooperation of the manufacturer, who must be the official applicant and signatory for the ITA application. Regulatory correspondence and clinical oversight can be delegated by the manufacturer who is the legal sponsor of the clinical investigation.
Manufacturer: Means a person who sells a medical device under their own name, or under a trade-mark, design, trade name or other name or mark owned or controlled by the person, and who is responsible for designing, manufacturing, assembling, processing, labelling, packaging, refurbishing or modifying the device, or for assigning to it a purpose, whether those tasks are performed by that person or on their behalf.
Medical Device: Means a device, but does not include any device that is intended for use in relation to animals.
Monitoring: Act of overseeing the progress of a clinical investigation and to ensure that it is conducted, recorded, and reported in accordance with the Clinical Investigation Plan, written procedures, the International Standard (ISO), and the applicable regulatory requirements.
Objective: Main purpose for conducting the clinical investigation.
Principal Investigator: Qualified person responsible for conducting the clinical investigation at an investigation site. If a clinical investigation is conducted by a team of individuals at an investigation site, the principal investigator is responsible for leading the team. Whether this is the responsibility of an individual or an institution can depend on national regulations.
Product Development: Basic exploratory studies carried out to determine whether a test article has any potential utility.
Protocol: A document that describes the objectives, design, methodology, statistical considerations and organization of a clinical trial (i.e., Clinical Investigational Plan).
Qualified Investigator: Means a person who is a member in good standing of a professional association of persons entitled under the laws of a province to provide health care in the province and who is designated, by the ethics committee of the health care facility at which investigational testing is to be conducted, as the person to conduct the testing.
Randomization: Process of assigning subjects to the investigational medical device or comparator groups using an established recognized statistical methodology to determine the assignment in order to reduce bias.
Risk Classification: The classification of a medical device according to the Classification Rules set out in Schedule 1 of the Medical Devices Regulations.
Sell: includes offer for sale, expose for sale, have in possession for sale and distribute, whether or not the distribution is made for considerationFootnote 8.
Serious Adverse Event: Adverse event that:
- led to death,
- led to serious deterioration in the health of the subject, that either resulted in
- a life-threatening illness or injury, or
- a permanent impairment of a body structure or a body function, or
- in-patient or prolonged hospitalization, or
- medical or surgical intervention to prevent life-threatening illness or injury or permanent impairment to a body structure or a body function,
- led to foetal distress, foetal death or a congenital abnormality or birth defectFootnote 9.
Significant Change: Means a change that could reasonably be expected to affect the safety or effectiveness of a medical device. It includes a change to any of the following:
- the manufacturing process, facility or equipment;
- the manufacturing quality control procedures, including the methods, tests or procedures used to control the quality, purity and sterility of the device or of the materials used in its manufacture;
- the design of the device, including its performance characteristics, principles of operation and specifications of materials, energy source, software or accessories; and
- the intended use of the device, including any new or extended use, any addition or deletion of a contra-indication for the device, and any change to the period used to establish its expiry date.Footnote 10
Study Completion: A study is considered to be completed after the last subject globally completes the "end of study" visit. The "end of study visit" is the final visit for study-related tests and procedures, including the capture of any final potential study-related adverse events. A study is not considered complete if the clinical trial is suspended, cancelled or closed.
Subject: Individual who participates in a clinical investigation. A subject can be either a healthy volunteer or a patient.
2.2 Pre-ITA Application Meeting
Health Canada invites manufacturers and importers to request a pre-ITA application meeting, particularly for novel Class III and IV devices and combination products. A consultation may be useful to assist applicants in the completion of applications and to facilitate a timely regulatory decision.
The purpose of pre-ITA application meetings is to provide the manufacturer or importer an opportunity to present relevant data and discuss concerns and issues regarding product development. It also gives Health Canada an opportunity to provide guidance and highlight potential deficiencies or concerns of the proposed investigation. Manufacturers and importers may invite the qualified investigators who will be involved in the proposed investigation in Canada to attend the meeting.
2.2.1 Requesting a Pre-ITA Application Meeting
Requests for a pre-ITA application meeting should be submitted in writing by the manufacturer or importer to the Investigational Testing Division of the Medical Devices Bureau at: Investigational_Testing@hc-sc.gc.ca.
Requests should be submitted in the form of a cover letter proposing three dates and times suitable for the meeting. The cover letter should be accompanied by the following information:
- A synopsis of the proposed study;
- A list of preliminary questions to be addressed by the Bureau during the meeting;
- Sufficient information for Health Canada to assess the utility of the meeting and identify the appropriate staff necessary to discuss the proposed issues. This will assist in ensuring efficient use of Health Canada resources.
The Medical Device Bureau will acknowledge the request for a meeting in a timely manner. If the Bureau agrees to hold the meeting, the acknowledgement will indicate the date that the pre-ITA application information package is to be provided, the meeting date and location, as well as the list of attendees. Please note that the Bureau reserves the right to propose a teleconference meeting or to address the questions by email correspondence, instead of a face-to-face meeting.
2.2.2 Information Package for Pre-ITA Application Meetings
The information package should be submitted in electronic format, and should contain:
- the proposed agenda, any prepared slides, including a finalized list of questions, and a complete list of attendees [it is recognized that the slides may change prior to the meeting]
- a brief summary of all data including:
- a tabular listing of completed pre-clinical and clinical studies
- the device specifications, preclinical testing, including the results of bench tests and animal studies
- a list of the standards used in the design and manufacture of the device
- the observed adverse events and a discussion of potential safety problems
- a proposed global clinical plan for the current stage of device development including regulatory status in other countries [it is recognized that this plan is subject to change as new information becomes available]
- details of the proposed clinical investigation to be conducted in Canada, within the scope of the intended ITA, including:
- a statement of trial protocol design
- a study hypothesis and the objectives of the study
- the condition to be treated or diagnosed, and a description of the treatments available
- proposed procedures and/or criteria for patient monitoring, clinical efficacy and safety assessments
- statistical considerations
Should the pre-ITA application package be found deficient, the manufacturer or importer may be requested to reschedule or postpone the meeting to allow the manufacturer or importer to assemble a more complete package. Please note that the Medical Device Bureau reserves the right to modify or truncate the proposed agenda as it sees fit to better achieve the stated goals of the meeting.
2.2.3 Record of the Pre-ITA Application Meeting
The manufacturer or importer should prepare and send to the Bureau a written record of the discussion and conclusions of the meeting within fourteen (14) days after the meeting. All final records of this consultation will be kept on file.
A copy of the record of discussion and conclusions approved by all parties in attendance at the meeting should be included in the subsequent ITA application.
2.3 ITA Applications
Under the Regulations, only manufacturers and importers can apply for an authorization to conduct investigational testing on human subjects in Canada. In either case a senior official of the manufacturer must complete and sign the Application Form. Based on the requirements of Section 82, an editable electronic copy of the supporting records as detailed in Section 81 paragraphs (a) to (k) and Section 86 (labelling) of the Regulations must be submitted.
2.3.1 Stages of Product Development
First in Human (FIH): A first in human study is a type of study designed to evaluate a device for a specific indication for the first time in humans for the purpose of:
- gaining initial clinical insights,
- refining device design,
- addressing an unmet clinical need, or when
- additional non-clinical testing is not feasible.
Early Feasibility: An early feasibility study is a limited clinical investigation of a device early in development, typically before the device design has been finalized (e.g., innovative device for a new or established intended use or marketed device for a novel clinical application). Clinical investigation designed to obtain initial insights into the safety and performance of a new device.
Traditional Feasibility: Clinical investigation designed to capture preliminary safety and effectiveness information on a near-final or final device design to adequately plan an appropriate pivotal study.
Pivotal Study: Clinical investigation designed to collect definitive evidence of the safety and effectiveness of a device for a specified intended use, typically in a statistically justified number of subjects.
2.3.2 When to Apply for an ITA
An ITA application is required for unlicensed class II, III, and IV medical devices to be imported or sold for the purpose of investigational testing involving humans. However, if basic exploratory studies are being carried out to determine whether a product has any potential utility, and the product is not represented within the context of the definition of a "device" and "medical device", an application for ITA is not required. The classification of a product as a medical device is dependent on the manufacturer's representation of the device in the labelling and not solely on how it is being used in the study. Further information to determine the classification can be found in the Guidance on the Risk-based Classification System for Non-In Vitro Diagnostic Devices (non-IVDDs) and the Guidance for Industry - Keyword Index to Assist Manufacturers in Verifying the Class of Medical Devices (refer to Appendix 2).
An ITA is not required if there is no sale of the medical device, as defined under the Food and Drugs Act. Consequently, if the development, manufacture, and testing of the device are conducted within a single corporate entity, no sale has occurred and the requirements of the Regulations do not apply. In this case, testing must be conducted on-site and solely by the legal manufacturer.
An ITA is not required for the conduct of a study using a licensed device according to its authorized indications for use, whereas a licensed device to be used in a manufacturer sponsored study intended to generate data to support a new indication for use, is subject to an ITA Application.
Already licensed medical devices used in clinical investigations which are not sponsored by the manufacturer, and which are not intended to generate data to support a licensing application do not require an ITA. Likewise, licensed medical devices used in clinical investigations outside the scope of the authorized indications for use and not sponsored by the manufacturer do not fall under the Regulations, as this is considered off-label use.
Parallel applications for an ITA and a medical device licence should not be performed, where the IT does not gather additional safety and/or effectiveness information to support a licence, but instead is being used to gain expedited market access.
2.3.3 Filing an ITA Application
All applications must be submitted electronically in accordance with current format for the submission of ITA applications, as specified in the Notice - Applications for Investigational Testing Authorization (ITA), for Medical Devices, in the "Non-eCTD Electronics-Only" Format (refer to Appendix 4). You may also refer to the Guidance Document: Preparation of Regulatory Activities in "Non-eCTD Electronic-Only" Format for detailed guidance on filing medical device regulatory activities and subsequent transactions, in the "non-eCTD electronic-only" format. It should be noted that Health Canada no longer accepts paper copies of submissions. Applications should be submitted by e-mail to devicelicensing-homologationinstruments@hc-sc.gc.ca, or sent by mail to the bureau's mailing address (refer to Appendix 1) on a CD or DVD if it is too large to be sent by e-mail. Applications sent by email should include a subject line that clearly distinguishes it as an ITA application. Requests for revised ITA applications should be clearly marked as so and include the previously assigned application number. ITA applications are not subject to a fee.
Applications sponsored by an investigator or a third party (e.g., a funding agency, a drug manufacturer, another device manufacturer, or a health care institution), must be signed by a senior official of the manufacturer of the unlicensed device. Regulatory correspondence and clinical oversight can be delegated to the clinical investigator, manufacturer, their regulatory agent, or contract research organization. In this case, the official who has delegated authority assumes the responsibilities of the importer. For Class III or IV applications, the manufacturer must submit the non-clinical information required in support of the application and the clinical investigator or manufacturer can submit the study protocol, the patient informed consent form (ICF), the risk analysis addressing clinical concerns, the signed investigator agreement, and evidence of REB approval.
Manufacturers are encouraged to register their clinical investigations on a publicly accessible registry which accepts international clinical trial information and which is recognized by the World Health Organisation (WHO). ClinicalTrials.gov and Current Controlled Trials International Standard Randomised Controlled Trials Number Register are acceptable. Information on investigational testing of medical devices in Canada can be obtained directly from the manufacturer, or by consulting one of the registries.
2.3.3.1 Multiple Devices used in one Study
If several unlicensed devices, made by different manufacturers, are to be used under the same study protocol, each manufacturer or importer must submit a separate application to the Bureau for review. The executive summary should indicate the rationale for using the unlicensed device in conjunction with the other devices under the same study protocol as well as reference whether an application is pending or to be submitted for the other devices. To the extent possible the applications should be submitted at the same time. Authorization will not be granted until all applications have been submitted and found to satisfy the regulatory requirements.
2.3.3.2 Devices used in Drug Clinical Trials
International drug clinical trials sometimes use ancillary devices that are not licensed in Canada. For Clinical Trial Applications (CTAs) that involve the use of an unlicensed Class II, III, or IV medical device, a separate ITA application and CTA must be filed and each authorized before the trial can commence in Canada. These applications can be filed concurrently.
In this case, the manufacturer of the device must sign the ITA application, and regulatory correspondence is usually delegated to the drug sponsor. For Class III and IV devices, the pre-clinical information is submitted to the Bureau by the device manufacturer, and the study protocol and ICF is submitted by the sponsor of the drug study. Although the study protocol and the ICF are relevant to the drug study, these documents are acceptable for review under these circumstances. The CTA number should be provided in the application as well as information on when it was submitted and if a No Objection Letter (NOL) has been issued.
2.3.3.3 Combination Products and Joint Reviews
For the investigational testing of a drug-device combination product, ITA applications or CTAs must be submitted to the lead Directorate or Bureau within Health Canada, depending on the principal mechanism of action and hence the classification of the product. Refer to the guidance document entitled Drug/Medical Device Combination Products Policy (refer to Appendix 2).
Authorization for the sale and importation of all investigational products to be used within a medical device clinical investigation or a drug clinical trial must be obtained prior to the initiation of the clinical trial or implementation of the protocol amendment. Therefore, separate ITAs or CTAs must be submitted for investigational products that are not regulated as combination product.
The lead Bureau or Directorate will be responsible for communicating all regulatory decisions to the sponsor or manufacturer.
2.3.4 ITA Format
Since January 3rd, 2017, applications for ITAs for medical devices have been accepted in the "non-eCTD electronic-only" format. Refer to the Guidance Document: Preparation of Regulatory Activities in "Non-eCTD Electronic-Only" Format for detailed guidance on filing medical device regulatory activities and subsequent transactions, in the "non-eCTD electronic-only" format.
As of April 1st, 2017, Health Canada no longer accepts paper copies of regulatory activities or their related transactions, including ITA applications.
A zipped folder structure should be used by adding documents in their respective folders. Empty folders must be deleted before filing to Health Canada. Folder numbering and naming should not be changed.
2.3.5 Organization of the ITA
To facilitate the timely evaluation of ITA applications, it is recommended that the manufacturer or importer provide an executive summary, a table of contents and the distinct sections appropriate for the class of medical device. The application should be organized into folders as outlined in Appendix 4.
For cases where multiple investigations are being conducted using the same medical device, a separate application is required for each study protocol. Information submitted under a previous ITA for a separate study protocol, but for the same device, can be cross referenced if the device remains unchanged.
Information in the document may be in either French or English. Material in a foreign language must be accompanied by an English or French translation.
The manufacturer or importer is responsible for informing the Bureau of changes in regulatory contact information. Changes to the assigned regulatory contact should be submitted by the current regulatory contact or a senior official in the company and addressed to: devicelicensing-homologationinstruments@hc-sc.gc.ca.
2.3.6 The Requirements of an ITA Application
Although the information that must be submitted in an application depends on the risk class of the device, the manufacturer or importer must possess records that contain all of the information and documents required under Section 81 of the Regulations. For some Class II devices, the Medical Device Bureau may, under Section 84, request additional information normally required for a Class III or IV device.
Evidence required to demonstrate safety, effectiveness or performance for devices to be used in investigational testing is different from the evidence required for a licence application.
Applications must meet all regulatory requirements, have a favorable benefit to risk ratio, and address potential risks in a complete patient ICF.
Health Canada's expectation is that manufacturers follow the principles of the Declaration of Helsinki and the Tri-Council Policy Statement (2nd Edition): Ethical Conduct for Research Involving Humans (2010), and conform to Good Clinical Practices (GCP) as set out by ISO 14155 - Clinical investigation of medical devices for human subjects. ISO 14155 is generally consistent with the definitions and requirements of the Regulations. Where inconsistencies exist, the Regulations take precedence.
There is no requirement for Good Laboratory Practices (GLP) for the conduct of animal studies in Canada. However, conformity with GLP is strongly recommended, and animal studies should be conducted with similar scientific rigour as studies in humans.
Applications for devices which were previously authorized under a different study protocol can cross reference the previous application for device specific information. The results of the previously authorized study should be provided, if available.
The table below lists application requirements for different device classes.
Table 1
| Requirements for an Investigational Testing Authorization (ITA) | |
|---|---|
| Class I | There is no requirement to obtain an ITA for a Class I medical device. Subsection 80(3) of the Regulations permits a manufacturer or importer of a Class I medical device to sell the device to a qualified investigator for the purpose of conducting investigational testing provided that all the records and information detailed in Section 81 are on file. Institutional requirements and Research Ethics Board approval apply. |
| Class II | The information required under Subsections 81 (a), (b), (h), (i), and (j) must be submitted. The manufacturer or importer must possess records that contain all the information required by section 81 of the Regulations. It is not required that evidence of Research Ethics Board approval is provided to the Bureau, but it must be obtained prior to the initiation of the study, in accordance with institutional policies. |
| Class III and IV | The information required under Subsections 81 (a) to (k) must be submitted. Evidence of Research Ethics Board approval is required before an ITA will be issued. |
Table 2
| Contents of the Application Package | |
|---|---|
| Class II, III and IV | Cover Letter & Executive Summary Include a short Cover Letter and a one or two page Executive Summary of the application. |
| Table of Contents Include a Table of Contents listing the contents of the application and their location. |
|
| Application Form Include a copy of the completed ITA Application form (Application for Investigational Testing Authorization, refer to Appendix 2). |
|
| Pre-submission Correspondence Include copies of any decisions from pre-submission correspondence that occurred with Health Canada, including minutes from pre-ITA meetings, submission enquiries, classification decisions, as well as correspondence with other bureaux in Health Canada. |
|
Table 3
| Introduction | |
|---|---|
| Class II, III and IV Subsection 81(a) |
Manufacturer or Importer Identification Provide the complete name and address of the device manufacturer and importer, if applicable, including contact names, e-mail addresses, fax and telephone numbers. The name of the legal manufacturer must be consistent with the information found on the device labelling. |
| Class II, III and IV Subsection 81(b) |
Device Identification Specify the risk classification of the device based on the Classification Rules set out in Schedule 1 of the Medical Devices Regulations and on the manufacturer's representation in the labelling. For assistance in determining the risk class please refer to the Guidance on the Risk-based Classification System for Non-In Vitro Diagnostic Devices (non-IVDDs) and the Guidance for Industry- Keyword Index to Assist Manufacturers in Verifying the Class of Medical Devices. If, after reviewing these documents assistance is still required you may contact MDB_enquiries@hc-sc.gc.ca. This risk classification of a device under investigational testing may differ from that of the device in general sale if, for example, a new indication is being investigated. The risk class assigned to an investigational device may also be different from the classification assigned by the Bureau at the time a future licence application is filed if at the time of licensing it is deemed to present a higher risk. This section should also include the number of units of each individually sold device requested for the conduct of this study in Canada (total number and a breakdown of the number per site). |
| Class III and IV Subsection 81(c) |
Device Description Engineering diagrams of long term implantable devices aid in the determination of dimensions and relative proportions. These should be provided with an original application for investigational testing. Engineering diagrams for other device types, such as electro-medical devices may be requested as additional information if necessary to establish the safety and potential effectiveness of the device in question. If the device has previously been authorized for investigational testing or granted a medical device licence, provide the authorization number and clearly specify if changes have been made to the device. If so, provide a tabular comparison of the similarities and differences with regard to the intended use, design, key specifications and performance features, hardware, software, accessories, patient contact materials, etc. It is recommended that this comparison be provided in tabular form. |
| Class III and IV Subsection 81(d) |
Design Philosophy This section should include an overview of the purposes and principles of operation for the device and should include a summary of the method of use and operation of the device. |
| Class II, III and IV | Indications for Use, Intended Use, Contraindications Include the proposed indications for use and/or intended use as well as any contraindications for the device, as proposed by the manufacturer. |
Class II, III and IV Subsection 81(j) |
Device Labelling Health Canada expects a bilingual copy of the device label. Include a copy of the product monograph, Instructions for Use, Operator's Manual, Training Manual, and all advertising brochures intended to be used with the device. For RCTs, the required labelling (device name, manufacturer name, investigational statement) must be affixed to the outer container, and each individual device (study and control) must bear an assigned code, the manufacturer's name, and the investigational use statement in French and English. The intent of this label is to ensure that the device is not used other than under the study protocol. |
Class III and IV Subsection 81(e) |
Marketing History The marketing history should provide details of the regulatory status of the device in other jurisdictions, the volume of sales by country, a summary of reported problems with the device and details of any recalls in other jurisdictions. Include a summary of Special Access requests made to Health Canada and the outcome of these requests. |
Table 4
| Risk Assessment and Risk Reduction Measures | |
|---|---|
Class III and IV Subsection 81(f) |
Risk Assessment Include a list of all possible hazards and evaluate them against the presumed benefits of the device. Additionally, an indication of the way by which the risks have been reduced to acceptable levels must be provided. The risk analysis should be signed, and the identity of the responsible individual provided. |
Class III and IV Subsection 81(f)(i) |
Previous Studies Examples include physical and mechanical bench testing, biological safety, electrical safety, software validation and verification, biocompatibility, shelf life, sterilization validation and residuals testing, packaging validation, animal studies and clinical studies. Supporting literature and a bibliography should be provided, as applicable. Evidence of conformity to recognized standards should be provided, as described in the Guidance Document: Recognition and Use of Standards under the Medical Device Regulations (refer to Appendix 2). A Declaration of Conformity to a recognized standard can replace detailed pre-market information to satisfy the regulatory requirements. It should be noted that if conformity is declared to a standard that specifies only test methods (such as ISO 10993 for the evaluation of biocompatibility), or procedures (such as ISO 14971 for application of a risk management system for medical devices), quantitative information on test results is also required. |
Class III and IV Subsection 81(f)(ii) |
Alternate Treatments |
Class III and IV Subsection 81(f)(iii) |
Precautions |
Class II, III and IV Subsection 81(g, h) |
Institutional Information |
Class III and IV Subsection 81(k) |
Investigator Agreement(s) This agreement outlines the responsibilities of the investigator to:
A template of an investigator agreement form is available on the Government of Canada website (refer to Appendix 2). An alternate format is acceptable provided the five (5) conditions described in Subsection 81(k) are adequately addressed. |
Class III and IV Subsection 81(h) |
Research Ethics Board Approval The versions and/or dates of the protocol and ICF authorized by the REB should correspond with those provided in your application. If non-administrative changes have been made during the REB review process, clean and redlined copies of the documents highlighting the changes should be provided. Previous REB refusals (foreign and Canadian) should be reported and discussed in your application. A description of the constitution, roles, and responsibilities of an REB can be found under the TCPS-2 and ISO 14155. |
Class II, III and IV Subsection 81(i) |
Protocol Background information describing the disease or condition to be treated, prevalence, diagnostic criteria, and current treatment, study hypothesis, study objectives, study design, inclusion and exclusion criteria, number of devices and study subjects required, as well as detailed information on the duration of the investigation and the follow-up period for patients. The methods of assessing the investigational device must be fully described, including the criteria for success or failure of the performance of the device. Provide a copy of all case report forms to be used in the investigation. The proposed methods of data analysis should be described, including the identity of the person or group performing the analysis. Methods of data quality control should be specified. The control group must be fully described, and a justification provided for the type of control group chosen. Provide a full description of the subject selection, including:
Subjects should be selected to be representative of the population intended to be treated with the device, with appropriate inclusion of children, women, and ethnic groups. Specify the estimated time frame for the conduct of the study, including the duration of the enrolment phase, the duration of the treatment phase, and the duration of the follow-up phase. Also include the approximate duration from the start of enrolment to the end of follow-up period. This can be provided as part of the protocol or provided separately. Investigator's Brochure (IB) Informed Consent Form (ICF) |
Table 5
| Additional Requirements | |
|---|---|
Class II, III and IV |
Minimizing Bias Bias should be minimized by adherence to currently accepted standards of protocol design as described in the TCPS-2 and ISO 14155. |
| Inclusion of women, children, and vulnerable populations Efforts should be made to maximize compliance with the International Council for Harmonisation (ICH) Document E11 entitled Clinical Investigation of Medicinal Products in the Paediatric Population and the Health Canada Guidance Document: Considerations for Inclusion of Women in Clinical Trials and Analysis of Sex Differences (refer to Appendix 2). |
|
Clinical Trial Design and Statistical Considerations
General guidance on this topic can be found in the ICH documents E8: General Considerations for Clinical Trials, E9: Statistical Principles of Clinical Trials, and E10 Choice of Control Group and Related Issues in Clinical Trials and TCPS-2 (refer to Appendix 2). |
|
2.3.7 The Investigational Testing Authorization
The Medical Devices Bureau will issue an authorization under Section 83 of the Regulations after a review of the submitted information is deemed to satisfy the requirements of the Regulations.
This letter will be the manufacturer's legal authorization to import and sell the requisite number of devices to the investigators or institutions listed under the Authorization for use in the referenced study protocol.
The Authorization Letter will specify the name of the device, the study protocol title, date and version number, the date of the ICF, the objectives of the study, the authorized number of devices to be imported and sold and number of study subjects to be recruited in Canada, as well as the names of the investigators and institutions where testing may be conducted.
The authorization can be issued only after receipt of evidence of REB approval for Class III and IV devices, and will remain valid provided that the REB approval is current, or until the device is licensed.
2.4 Responsibilities of Manufacturers and Importers
2.4.1 Advertising
Section 87 of the Regulations prohibits advertising a medical device that is the subject of investigational testing unless an authorization has been issued, and the advertisement clearly indicates that the device is the subject of investigational testing, and the purpose of the clinical investigation.
2.4.2 Quality Management System
It is expected that manufacturers will have implemented a proper Quality Management System. However, evidence of certification to ISO13485 is not a requirement for an ITA.
2.4.3 Record Keeping
Under Section 80 of the Regulations, the manufacturer or importer of a medical device undergoing investigational testing in Canada must maintain records as detailed under Section 81 of the Regulations. The Regulations do not cover the document retention period for clinical institutions. This period should be in line with institutional policies and provincial regulating bodies for the practice of medicineFootnote 11.
2.4.4 Distribution Records
Under Section 88 of the Regulations the manufacturer, importer and distributor of a medical device undergoing investigational testing in Canada must maintain distribution records as detailed under Sections 52 to 56.
2.4.5 Mandatory Problem Reporting
The qualified investigator is required to report serious adverse events to Health Canada and to the manufacturer and importer within 72 hours of discovery. This includes cases in which the incident:
- is related to a failure of the device or a deterioration in its effectiveness, or any inadequacy in its labelling or in its directions for use; and
- has led to the death or a serious deterioration in the state of health of a patient, user or other person, or could do so were it to recur.
For an incident that occurs in Canada, the manufacturer is required to provide a preliminary and a final report in respect of the incident. The preliminary report shall be submitted:
- within 10 days after the manufacturer or importer of a medical device becomes aware of an incident, if the incident has led to the death or a serious deterioration in the state of health of a patient, user or other person, or
- within 30 days after the manufacturer or importer of a medical device becomes aware of an incident, if the incident has not led to the death or a serious deterioration in the state of health of a patient, user or other person, but could do so were it to recur;
Further details for the information required in the reports, as well as reporting for incidents outside of Canada can be found in Sections 59 to 62 of the Regulations. Reference should also be made to the Guidance Document for Mandatory Problem Reporting for Medical Devices and relevant forms (refer to Appendix 2) for the process of submitting these reports.
Additional clarification can be found in ISO 14155. It is expected that any serious untoward medical occurrence, unintended disease or injury, or untoward clinical signs (including abnormal laboratory findings) in subjects, users or other persons, whether or not related to the investigational medical device, will be reported. All non-serious and expected adverse events should be reported in the periodic and/or final study reports by the manufacturer or importer.
2.4.6 Other Obligations
Manufacturers and importers must have documented procedures in place to handle product complaints and recalls as required by Sections 57 and 58, and 63 to 65 of the Regulations. In addition, the appropriate records of these activities must be maintained.
For additional information, consult the guidance document on complaint handling and recalls (Recall Policy, refer to Appendix 2).
The manufacturer's responsibilities with regard to implant registration, as described in Sections 66 to 68, are also applicable (as appropriate) to devices authorized for investigational testing. Implant Registration cards are required for the devices listed under Schedule 2 of the Regulations.
2.5 Requests for Revisions to an ITA
Changes made to the details of the ITA, the investigation or the device require authorization. Manufacturers or importers may request a revised authorization for the below listed cases. Other changes will require a new ITA application to be submitted and the previously issued authorization to be cancelled:
- A change in the name of the manufacturer or the device;
A change to the study protocol that will not bias the data previously collected; and
A significant change to the design of the device (where the device generally maintains its original functionality and intended use) - Minor protocol of ICF revisions (e.g., clarifications, modifications to the inclusion or exclusion criteria, patient follow-up schedule), additional study subjects or additional devices;
- Minor changes to the design of the device (e.g., software upgrades);
- Changes to the list of qualified investigators; or
- Changes to the institutions where the testing is being conducted.
Examples of protocol changes that require a revised authorization are provided below to aid in determining whether a request for a revised authorization should be filed. These examples are not all inclusive. When in doubt whether an application is required, sponsors should contact the Investigational Testing Division at: Investigational_Testing@hc-sc.gc.ca.
- Protocol revisions to permit the enrolment of additional study subjects and longer term patient follow-upFootnote 12.
- Any protocol amendment that requires REB approval will also require a Health Canada authorization.
- Inclusion and exclusion criteria modifications. These include changes to eligibility criteria, tests or procedures for selecting the study population, as well as tests, procedures, or criteria for dismissing clinical trial subjects prematurely or at the end of the trial.
- Changes to the patient selection criteria, tests or procedures required for the ongoing assessment of clinical trial subjects, including assessment of safety, or evaluation of safety and efficacy. This includes protocol changes as a result of serious unexpected adverse reactions.
- Inclusion of sub-studies.
- Changes to sample size estimation or addition of interim analyses that will affect the analysis and interpretation of the study results.
- Changes to the post-treatment follow-up period that may affect the safety evaluation of the device.
- Use of ancillary medical devices for the treatment or monitoring of the study subjects that may have an impact on the analysis of efficacy or increase the risk to clinical trial subjects.
- Changes to the requirements or procedure for reporting of serious, unexpected adverse reactions.
The criteria for issuing a revised authorization are:
- the changes do not have a major impact on the risk analysis originally submitted with the application;
- the rights and safety or welfare of the study subjects are ensured;
- the integrity of the study data collected to date is secure;
- the scientific soundness of the plan is intact; and
- there are no changes to the overall study hypothesis or objectives.
2.5.1 Filing Requests for Revisions to an ITA
The requirements for filing an ITA, as discussed in section 2.3.6 of this guidance document, are applicable to ITA revision requests. All applicable review sections that have changed as a result of the modifications or are required to validate the modified device and/or support its continued safety in the investigation should be re-submitted. Include a cover letter clearly distinguishing what aspects are modified and which remain unchanged, as well as what supporting information has been provided.
If the protocol, ICF, or labelling have been revised, both clean and redlined copies of the modified documents should be provided along with a tabular list of changes made and a rationale supporting each change.
If device changes are being made or new devices added, applicable verification and validation studies should be provided.
Changes to the authorized investigators or institutions where the investigations will be conducted require the complete institutional information, investigator documentation and Research Ethics Board approval, as applicable to the device class and described in section 2.3.6 of this guidance document.
A new ITA application form is not required. However, any changes made to the device detail (e.g., name, authorized devices, catalog numbers, etc.) must be provided on a revised copy of section 11 (Device Detail) of the Application Form.
Applications for revisions to previously authorized ITAs should be submitted in electronic format following the structure under Appendix 4. All correspondence should reference the previously issued ITA number, including the email subject line and Cover Letter. Completed applications are to be submitted electronically in accordance with the current policy for the submission of ITA applications to: devicelicensing-homologationinstruments@hc-sc.gc.ca or by mail to the bureau's postal address (refer to Appendix 1).
2.5.2 Additional Information to be added to the file
Modifications made to the device or the investigation that are not deemed to be a significant change should be reported to Health Canada. These changes will be acknowledged in writing, and a revised authorization may or may not be required depending on the nature of the changes.
2.6 Cancellation of an ITA
Section 85 of the Regulations allows the Minister to cancel an ITA for Class II, III or IV devices and to stop the sale of a Class I device that has been sold for investigational purposes, as outlined in section 85(1) paragraphs (a) to (e) when:
- the testing seriously endangers the life, health or safety of patients, users or other persons;
- the testing is contrary to the best interests of patients on whom the testing is being conducted;
- the objective of the testing will not be achieved;
- the qualified investigator who is conducting the testing is not respecting the undertaking required by paragraph 81(k); or
- the information submitted in respect of the testing is false or misleading.
Prior to cancelling an ITA, Health Canada will request information from the manufacturer or importer to substantiate that the conditions set out in Subsection 83(1) are still being met. If this information is not submitted, or if it is submitted and a review of the information determines that one of the conditions set out under Section 85(1) exists, the authorization will be cancelled by written notice giving the reasons for the cancellation.
2.7 Review Process for ITA Applications and Revisions to ITAs
2.7.1 Screening
New ITA applications are screened for administrative and scientific content to ensure that the applicable regulatory requirements of Sections 81 and 86 of the Regulations have been addressed. A Screening Acceptance Letter is issued if the information is complete. If regulatory deficiencies are identified at screening, a Screening Deficiency Letter is issued which details the deficiencies. The manufacturer or importer is given fifteen (15) calendar days to provide the missing information, after which time the application is rejected and returned to the manufacturer (refer to the Policy on Management of Applications for Medical Device Licences and Investigational Testing Authorizations, Appendix 2). The response should include an Executive Summary listing the responses in question and answer format, as well as all other documents in the appropriate section of the electronic folder structure for ITA applications (refer to Appendix 4).
2.7.1.1 Screening Rejection Letter
A Screening Rejection Letter is issued for grossly deficient applications, such as when substantial information which is required under Section 81 of the Regulations is missing from the application. This may also include when a class II application is submitted for a medical device determined to be class III or IV.
A Screening Rejection Letter will also be issued if the manufacturer or importer fails to provide the records required under Section 81 within fifteen (15) days of receipt of a Screening Deficiency Letter. In the letter the sponsor will receive a list of the deficiencies. If the application is rejected, the applicant will have to file a complete new application. Cross-referencing an application rejected at screening is not permitted.
2.7.2 Review
Once the review has been initiated un-solicited information should not be provided without consent from the Investigational Testing Division Manager. The review target for new ITA applications or requests for revised ITAs, other than for combination products, is thirty (30) calendar days from when screening is initiated. If a Screening Deficiency Letter was issued, the thirty (30) days begins from when the complete application was received. For minor administrative revisions to ITAs the review target is fifteen (15) calendar days. It should be noted that these are only estimated target times and not default deadlines that result in automatic authorization.
After the review has been completed Health Canada will send a request for additional information as described in Section 84 of the Regulations for any missing information which is required under Section 81. The applicant must submit a complete response within sixty (60) calendar days. Upon receipt of the response to the additional information request from the manufacturer or importer, a new thirty (30) calendar day review period begins.
Should the applicant be unable to provide the requested information within this timeframe, the application may be withdrawn and resubmitted without prejudice within six (6) months. After this time period a new application must be filed. Requests for additional information that have not been responded to within sixty (60) days will be subject to a refusal.
If the information submitted in support of the ITA or ITA revision is deemed to be satisfactory, Health Canada will issue an authorization or revised authorization.
2.7.2.1 Refusal of an Application
The ITA application or revised application may be refused if it is determined that:
- the device cannot be used safely for investigational testing;
- the investigational testing is contrary to the best interests of patients;
- the objective of the testing cannot be achieved; or
- the records required under Section 81and Section 86 have not been provided.
A Refusal letter will be issued itemizing the deficiencies.
The manufacturer or importer may appeal a refusal to issue an ITA. Information on the appeal process for decisions made regarding investigational testing applications can be found in the document Management of Applications for Medical Device Licences and Investigational Testing Authorizations (refer to Appendix 2).
2.8 Post-Authorization Requirements
2.8.1 Discontinuation of the Investigational Testing
In the event of the premature discontinuation of a study in its entirety or at a study site for which an ITA or ITA revision has been issued in Canada, the manufacturer or importer should notify the Medical Devices Bureau as soon as possible, but no later than fifteen (15) calendar days after the date of discontinuation.
This notification should include:
- Detailed reasons for this action, including whether it is related to the safety and/or effectiveness of the medical device;
- Description of the impact on the proposed or ongoing study conducted in Canada;
- Confirmation that all qualified investigators have been notified of the discontinuation and the reasons for it and have been advised in writing of any potential risks to the health of the research subjects or other persons;
- Confirmation that the sale or importation of the device to all discontinued sites has been stopped; and
- Confirmation that the manufacturer will take reasonable measures to ensure the return of all unused devices.
Note: Notification of a premature discontinuation of international sites (due to safety reasons) for which there are ongoing studies with the medical device in Canada, should also be submitted to the Medical Devices Bureau.
2.8.2 Resumption of Investigational Testing
The sponsor may resume the investigational testing in its entirety or at a site that was previously discontinued if the sponsor submits the following information:
- The name, address and telephone number, and electronic mail address of the qualified investigator for each site and of the REB that approved the re-initiation of the investigational testing at each site;
- The name, address and telephone number and, if applicable, the fax number and electronic mail address of any REB that has previously refused to approve the re-initiation of the investigational testing , if applicable; and
- The proposed date of re-initiation of the investigational testing at each study site.
Note: The above information may be submitted as Additional Information and the bureau will provide acknowledgement of the resumption. When there has been a change to the device, the study protocol or the manufacturing process, the information should be submitted as a request for a revised ITA (refer to Section 2.5). The study may resume only when a revised Authorization Letter has been issued.
2.8.3 Study Completion and Site Closures
Manufacturers and importers are urged to report the completion of the study to the Bureau. Inclusion of a final study report is encouraged. Subsequent to this notification, Health Canada will issue a letter to confirm closure of the study. ISO 14155 provides additional guidance.
After completion of the investigational testing, re-usable devices, such as capital equipment, which are not yet licensed, should be returned to the manufacturer or importer. If the qualified investigator intends to use these devices under a different study protocol, a new application for investigational testing signed by a senior official of the manufacturer must be submitted. Clinical use of unlicensed medical devices other than under an authorised clinical protocol is prohibited.
2.8.4 Investigational Testing Report
After closure of the investigational testing, the manufacturer or importer should submit a report of the study to Health Canada even if the investigation was terminated prematurely.
- The report should include identification of the device, a description of the methodology and design of the investigation, any deviations from the protocol, data analysis together with any statistics and a critical appraisal of the aims of the investigation.
- The report should take into account the data from each investigation site and for all subjects. No subjects should be identifiable either from the investigation report or the published results.
- Where applicable, the investigation testing report should be made available to the coordinating investigator and all principal investigators for review and comment. The sponsor shall maintain records confirming that the report has been provided for review. If a reviewer does not agree with all or part of the report, their comments should be recorded and communicated to the other principal investigators.
- The sponsor and coordinating investigator should provide their signatures, indicating their agreement with the content of the report. If no coordinating investigator is appointed, the signature of the principal investigators should be obtained.
- The report should be provided to the REB and other regulatory authorities.
2.8.5 Public Release of Information about ITAs
Upon request, Health Canada will publically release certain information about issued ITAs. The primary goal is to provide transparency and improve public access to information on Canadian studies involving medical devices. For ITAs authorized since November 14, 2013 the following information will be released:
- protocol title;
- device name;
- medical condition;
- study population;
- authorization date;
- end date of the study; and
- name of the manufacturer or importer.
For all other information enquirers will be redirected to the manufacturer or importer, or referred to their health care provider.
Appendix 1 - Relevant Addresses
Medical Devices
Medical Devices Bureau
2nd Floor, 11 Holland Avenue, Tower A
Address Locator: 3002A
Ottawa, Ontario
Canada
K1A 0K9
General Enquiries:
E-mail: MDB_Enquiries@hc-sc.gc.ca
Telephone: 613-957-4786
Fax: 613-957-6345
Pharmaceutical Drugs
Office of Clinical Trials
Therapeutic Products Directorate
5th Floor, Holland Cross, Tower B
Address Locator: 3105A
1600 Scott Street
Ottawa, Ontario
Canada
K1A 0K9
General Enquiries:
E-mail: OCT_BEC_Enquiries@hc-sc.gc.ca
Telephone: 613-941-2132
Fax: 613-946-7996
Appendix 2 - Useful Documents
The following documents may be useful in the preparation of the application:
- Application for Investigational Testing Authorization [1999-02-22],
- Drug/Medical Device Combination Products Policy [2005-11-30],
- General Considerations for Clinical Trials ICH Topic E8 [1998-05-01],
- Guidance Document: Considerations for Inclusion of Women in Clinical Trials and Analysis of Sex Differences [2013-05-29],
- Guidance Document - E10: Choice of Control Group and Related Issues in Clinical Trials [2011-07-04],
- Guidance Document for Mandatory Problem Reporting for Medical Devices [2011-10-03],
- Guidance Document: Preparation of Regulatory Activities in the "Non-eCTD Electronic-Only" Format [2016-10-31],
- Guidance Document: Recognition and Use of Standards under the Medical Device Regulations [2006-09-22],
- Guidance for the Interpretation of Significant Change of a Medical Device [2011-01-20],
- Guidance for the Risk Based Classification System of In Vitro Diagnostic Devices [2016-09-23],
- Guidance on Medical Device Compliance and Enforcement (GUI-0073) [2015-06-12],
- Keyword Index to Assist Manufacturers in Verifying the Class of Medical Devices [2006-09-14],
- The List of Recognized Standards [2016-10-26],
- Management of Applications for Medical Device Licences and Investigational Testing Authorizations [2001-04-06],
- Mandatory Problem Reporting Forms For Industry [October 2011],
- Notice - Adoption of ICH Guidance: Clinical Investigation of Medicinal Products in the Pediatric Population E11 [2003-12-17],
- Notice - ICH Guidance E6: Good Clinical Practice: Consolidated guideline [1997-09-19],
- Notice - ICH Guidance E9: Statistical Principles of Clinical Trials [2003-02-10],
- Notice - Release of information about Medical Device Investigational Testing Applications authorized by Health Canada [2013-11-15],
- Preparation of an Application for Investigational Testing - In Vitro Diagnostic Devices (IVDD) V.3 [1999-02-22],
- Recall Policy (POL-0016) [2015-03-05],
- TCPS 2 (2014) - the latest edition of Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans
Appendix 3 - Determining When an ITA Application is Required
Figure 1
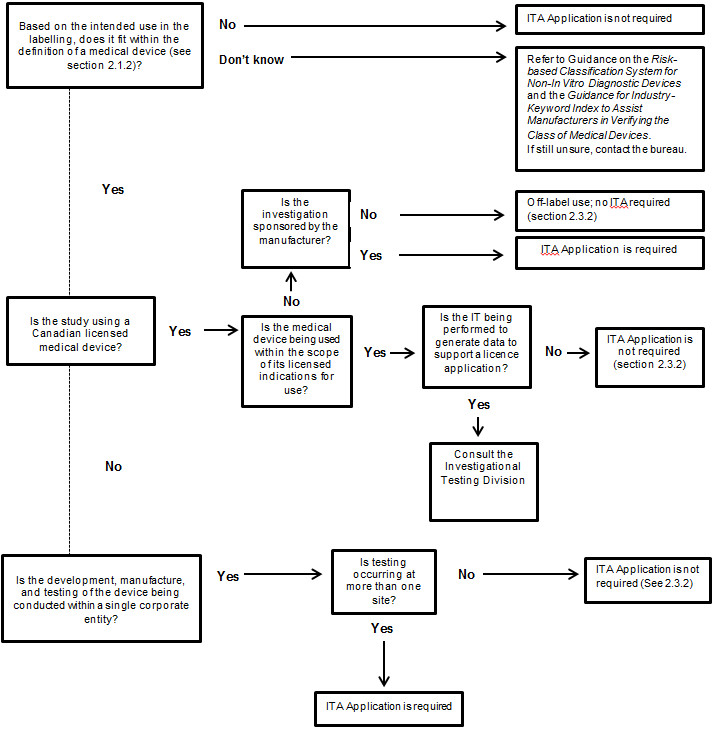 Figure 1 - Text Description
Figure 1 - Text Description
Based on the intended use in the labelling, does the device fit within the definition of a medical device (see section 2.1.2 of the ITA guidance document)? If no, an ITA Application is not required. If the manufacturer does not know, s/he should refer to the Guidance on the Risk-based Classification System for Non-In Vitro Diagnostic Devices and the Guidance for Industry- Keyword Index to Assist Manufacturers in Verifying the Class of Medical Devices. If the manufacturer is still unsure, h/she should contact the bureau.
Based on the intended use in the labelling, if the device fits within the definition of a medical device, is the study using a Canadian licensed medical device?
If yes, is the medical device being used within the scope of its licensed indications for use? If the medical device being used is within the scope of its licensed indications for use, is the IT being performed to generate data to support a licence application? If no, an ITA Application is not required (see section 2.3.2 of the ITA guidance document). If the IT being performed is to generate data to support a licence application, the manufacturer should consult the Investigational Testing Division.
If the medical device is not being used within the scope of its licensed indications for use, is the investigation sponsored by the manufacturer? If no, this activity is considered off-label use and an ITA is not required (see section 2.3.2 of the ITA guidance document). If yes, an ITA Application is required.
If the study is not using a Canadian licensed medical device, is the development, manufacture, and testing of the device being conducted within a single corporate entity? If yes, is testing occurring at more than one site? If no, an ITA Application is not required (see section 2.3.2 of the ITA guidance document). If yes, an ITA Application is required.
Appendix 4 - Format for an ITA Application
The following is the "non-eCTD electronic-only" format for ITA applications referenced in the Notice - Applications for Investigational Testing Authorization (ITA), for Medical Devices, in the "Non-eCTD Electronics-Only" Format. Some of the sections are not applicable to Class II applications. Empty folders must be deleted before filing to Health Canada. Any other supporting documentation that does not fall into one of the pre-defined folders can be provided in an appendix, but should be clearly referenced in the Executive Summary.
01- Cover Letter & Executive Summary
02- Table of Contents
03- Application Form
04- Pre-submission Correspondence
05- Introduction
05.01 Manufacturer or Importer Identification
05.02 Device Identification
05.03 Device Description
05.04 Design Philosophy
05.05 Indications For Use, Intended Use, Contraindications
05.06 Device Labelling
05.07 Market History
06- Risk Assessment and Risk Reduction Measures
06.01 Risk Assessment
06.02 Previous Studies
06.03 Alternate Treatments or Testing Options
06.04 Precautions
07- Institutional Information
07.01 Investigator Information
07.02 Curriculum Vitae
07.03 Investigator Agreements
07.04 Name and Address of Institutions
07.05 Research Ethics Board Approval
08- Study Documents
08.01 Protocol
08.02 Investigator's Brochure
08.03 Informed Consent Form
09- Appendices
Appendix 5 - Frequently Asked Questions
Investigational Testing Authorization (ITA) Applications
1. How do I submit my ITA applications?
Applications will only be received in electronic format. They can be sent by e-mail to devicelicensing-homologationinstruments@hc-sc.gc.ca. If the submission is too large to be sent by e-mail it can be sent by mail (refer to Appendix 1) on a CD or DVD. The submission must be provided in the format specified the Guidance Document: Preparation of Regulatory Activities in the "Non-eCTD Electronic-Only" Format (see Appendix 2 and section 2.3.4 of the ITA guidance document for more details).
2. Can an investigator apply for an ITA?
Applications must be submitted by the device manufacturer or the importer; however, they may delegate an investigator to serve as the regulatory contact. In the case where the importer or investigator is submitting the application, the manufacturer must sign off the application form (see section 2.3.3 of the ITA guidance document for more details).
3. Is an ITA required if a doctor intends to use an unlicensed device that s/he invented in a study to treat his own patients?
Medical devices manufactured and used in a clinical investigation within the same corporate entity are exempt from the requirements of the Medical Devices Regulations. This applies only to cases when the investigation is occurring at a single institution. In the case where the physician is the legal manufacturer of the device, as defined under the Regulations, and the device is being used on-site where the physician practices, and s/he is the only person responsible for conducting the investigational testing, there is no sale occurring as defined under the Food and Drugs Act. The requirements of the Regulations do not apply for these situations and an ITA is not required (see section 2.3.2 of the ITA guidance document for more details).
4. How do I request changes to my ITA?
Changes to the device and/or protocol that maintain the original study objectives and would not be expected to alter the risk-profile or negatively impact the integrity of the data collected can be sent in as a request for a revised ITA. Evidence to support the requirements under Section 81 and 86 of the Regulations should be provided for all requirements that are impacted by the proposed changes. Requests should be submitted electronically in accordance with the current policy for the submission of ITA applications to: devicelicensing-homologationinstruments@hc-sc.gc.ca or by mail to the bureau's postal address (see Appendix 1 and section 2.5 of the ITA guidance document for more details).
5. Is an ITA required for a Class I device?
For a class I device, there is no requirement to apply for an authorization to conduct investigational testing, but the manufacturer must keep a record of the information required by Section 81 of the Medical Devices Regulations on file and the study should be conducted in accordance with good clinical guidance (e.g., ISO 14155, Good clinical practices, etc.).
6. Do I need REB approval before I will receive an ITA?
Health Canada does not require evidence of REB approval before issuing an ITA for Class II devices. However, institutional policies will require REB approval prior to initiating the study for a class II device.
Evidence of REB approval is required before an ITA will be issued for Class III and IV devices. Conditional approval (which would state Health Canada approval is required) from an REB is acceptable. An ITA application can be submitted to Health Canada while the REB approval is pending. The review will be initiated but the ITA will not be issued until evidence of REB is received.
7. How long after the study has been completed do I have to keep the documents?
Section 55 of the Regulations requires the manufacturer, importer and distributor to retain the distribution records maintained in respect of a medical device for the longer of:
- the projected useful life of the device, and
- two years after the date the device is shipped.
The Regulations do not cover the document retention period for clinical institutions. This period should be in line with institutional policies and provincial regulating bodies for the practice of medicine.
8. Do I need to report an incident that occurred during the investigation?
The qualified investigator is required to report serious adverse events that fall within the scope of section 59 of the Regulations to Health Canada and to the manufacturer or importer within 72 hours of discovery. The manufacturer and the importer of a medical device are required to report to Health Canada incidents that fall under the scope of section 59 of the Regulations within 10 days, if the incident has led to the death or a serious deterioration in the state of health of a patient, user or other person, or within 30days the incident has not led to the death or a serious deterioration in the state of health of a patient, user or other person, but could do so were it to recur. Further details of the reporting requirements are set out under Sections 59 to 62 of the Regulations and found in section 2.4.5 of the ITA guidance document.
9. If I previously received an ITA for a re-usable device, can I continue to use it after the investigation is over or use it for a new investigation?
The ITA issued pertains specifically to the device, protocol and number of patients referenced on the authorization. After the investigation has been completed the device should be returned to the manufacturer or importer, decommissioned or disposed of appropriately. If the qualified investigator intends to use the devices under a different study protocol, a new ITA application must be submitted by the manufacturer or importer.
Footnotes
- Footnote 1
Clinical Investigation, Investigational Testing, Clinical Trial and Clinical Study are synonyms.
- Footnote 2
For medical devices, applications must be submitted by the medical device manufacturer.
- Footnote 3
Dorland's Medical Dictionary.
- Footnote 4
Ethics Committee and Institutional Review Board are synonyms.
- Footnote 5
Investigation site and investigation centre are synonyms.
- Footnote 6
The terms "investigational medical device" and "investigational device" are used interchangeably.
- Footnote 7
Although not defined in the Medical Device Regulations, an individual member of the investigation site team is commonly called “sub-investigator” or “co-investigator”.
- Footnote 8
The term “sell,” as defined in section 2 of the Food and Drugs Act, is not restricted to commercial or monetary sale and includes transactions without consideration, such as devices provided by the manufacturer to the investigator free of charge.
- Footnote 9
Planned hospitalization for a pre-existing condition, or a procedure required by the Clinical Investigation Plan, without serious deterioration in health, is not considered a serious adverse event.
- Footnote 10
Reference can be made to Guidance for the Interpretation of Significant Change of a Medical Device for general principles in determining significant changes
- Footnote 11
While this is not defined in the Medical Device Regulations for device, the Food and Drugs Regulations define a period of 25 years for the retention of records related to drug clinical studies.
- Footnote 12
If this version of the study protocol does not apply to the Canadian site, there is no need to obtain Health Canada authorization for this.