
Ebauche de la Ligne directrice : Demandes d’autorisation d’essai expérimental pour les instruments médicaux
Avis au lecteur :
État actuel : Fermé. La ligne directrice sont disponible.
Ébauche de la ligne directrice
Demandes d'autorisation d'essai expérimental pour les instruments médicaux
Publication autorisée par la ministre de la Santé
Date de l'ébauche: 2017/10/06
Direction générale des produits de santé et des aliments
Notre Mission est d'aider les Canadiens et les Canadiennes à maintenir et à améliorer leur état de santé.
Santé Canada
Le mandat de la Direction générale des produits de santé et des aliments est d'adopter une approche intégrée à la gestion des risques et des avantages pour la santé liés aux produits de santé et aux aliments :
- en réduisant les facteurs de risque pour la santé des Canadiens et des Canadiennes tout en maximisant la protection offerte par le système réglementaire des produits de la santé et des aliments;
- et en favorisant les conditions qui permettent aux Canadiens et aux Canadiennes de faire des choix sains ainsi qu'en leur donnant des renseignements afin qu'ils ou qu'elles puissent prendre des décisions éclairées en ce qui a trait à leur santé.
Direction générale des produits de santé et des aliments
© Ministre, Services publics et Approvisionnement Canada 2017
Also available in English under the following Title: Draft Guidance Document: Applications for Medical Device Investigational Testing Authorizations
Avant-propos
Les lignes directrices sont destinées à guider l'industrie et les professionnels de la santé sur la façon de se conformer aux lois et règlements qui régissent leurs activités. Elles servent également de guide au personnel sur la façon de remplir le mandat et d'atteindre les objectifs de Santé Canada d'une manière équitable, uniforme et efficace.
Les lignes directrices sont des outils administratifs n'ayant pas force de loi, ce qui permet une certaine souplesse d'approche. Les principes et les pratiques énoncés dans le présent document pourraient être remplacés par d'autres approches, à condition que celles-ci s'appuient sur une justification scientifique adéquate. Ces autres approches devraient être examinées préalablement en consultation avec le programme concerné pour s'assurer qu'elles respectent les exigences des lois et des règlements applicables.
Dans la foulée de ce qui précède, il importe également de mentionner que Santé Canada se réserve le droit de demander des renseignements ou documents complémentaires, ou de définir des conditions dont il n'est pas explicitement question dans la ligne directrice, afin que le Ministère puisse être en mesure d'évaluer adéquatement la sûreté, l'efficacité ou la qualité d'un produit thérapeutique donné. Santé Canada s'engage à justifier de telles demandes et à documenter clairement ses décisions.
Le présent document devrait être lu en parallèle avec l'avis d'accompagnement et les sections pertinentes des autres lignes directrices qui s'appliquent.
Table des matières
- 1. Introduction
- 2. Directives de mise en œuvre
- 2.1 Abréviations et définitions
- 2.2 Rencontre préalable à la demande d'AEE
- 2.3 Demandes d'AEE
- 2.4 Responsabilités des fabricants et des importateurs
- 2.5 Demande de révision d'une AEE
- 2.6 Annulation d'une AEE
- 2.7 Procédure d'examen des demandes d'AEE et des demandes de révision d'une AEE
- 2.8 Exigences suivant l'autorisation
- Annexe 1 - Adresses pertinentes
- Annexe 2 - Documents utiles
- Annexe 3 - Déterminer s'il faut demander une AEE
- Annexe 4 - Format d'une demande d'aee
- Annexe 5 - Foire aux questions
1. Introduction
La Loi sur les aliments et drogues (LAD) précise le cadre législatif auquel les instruments médicaux sous soumis. Le Bureau des matériels médicaux voit à administrer les exigences découlant de la partie 3 du Règlement sur les instruments médicaux (appelé ci-après le Règlement) qui gouverne la vente et l'importation de tout instrument médical utilisé lors d'essais expérimentaux menés sur des sujets humains. Les fabricants et les importateurs doivent respecter les exigences prévues dans le Règlement, notamment celles précisées au paragraphe 83(1), s'ils veulent obtenir l'autorisation de Santé Canada de vendre un instrument à un chercheur qualifié dans le but de mener des essais expérimentaux.
Cette présente ébauche de la ligne directrice remplace la ligne directrice intitulée Élaboration d'une demande d'essai expérimental - Instruments médicaux (GD009/Rev00-MDB V3 datée du 1999-02-22) et sera finalisée après l'examen des commentaires des parties prenantes.
1.1 Objectifs de la politique
Aider les fabricants et les importateurs à préparer la documentation nécessaire en vue d'obtenir l'autorisation de vendre ou d'importer un instrument médical en vertu d'une autorisation d'essai expérimental (AEE), tout en assurant la protection des sujets de l'étude et en favorisant l'excellence de la recherche et du développement au Canada.
1.2 Énoncés de la politique
Les fabricants et les importateurs sont responsables de présenter une demande d'AEE à Santé Canada pour avoir la permission de vendre ou d'importer un instrument médical dans le but de mener des essais expérimentaux sur des sujets humains.
Santé Canada s'attend à ce que les fabricants respectent les principes de la Déclaration d'Helsinki et de l'Énoncé de politique des trois Conseils (2e édition) : Éthique de la recherche avec des êtres humains (2010), de même que se conforment aux bonnes pratiques cliniques(BPC) décrites dans la norme ISO 14155 - Investigation clinique des dispositifs médicaux pour sujets humains. Cette norme correspond en général aux définitions et aux exigences du Règlement. En cas de divergences, le Règlement a préséance.
Les conseils d'éthique de la recherche (CER) ont un rôle de premier plan à jouer dans la surveillance de la conduite des études expérimentales. Santé Canada n'accordera une AEE qu'après avoir eu la preuve qu'un CER a approuvé l'instrument de classe III et IV. Cette autorisation demeurera valide jusqu'à l'échéance de l'approbation courante du CER ou l'homologation de l'instrument.
Les fabricants et les importateurs sont tenus de présenter une demande d'AEE en format « électronique autre que le format eCTD » et de respecter la structure présentée à l'annexe 3.
1.3 Portée et application
La présente ligne directrice a pour but d'aider les fabricants et les importateurs à organiser et à présenter une demande d'AEE afin qu'un essai expérimental puisse être mené à l'aide d'un instrument de classe II, III ou IV par le fabricant, un établissement universitaire, un établissement de soins de santé ou une organisation contractuelle de recherche. Elle précise également les responsabilités des fabricants et des importateurs au moment de mener un essai expérimental à l'aide d'un instrument de classe I. En outre, elle permet aux chercheurs et aux institutions prenant part aux essais expérimentaux d'instruments médicaux au Canada de bien comprendre leurs rôles et responsabilités dans cette démarche.
Le présent document ne concerne pas l'essai expérimental d'instruments diagnostiques in vitro (IDIV) au Canada. Les fabricants et les importateurs sont invités à consulter la ligne directrice intitulée Élaboration d'une demande d'essai expérimental - Instruments diagnostiques in vitro (GD010) disponible sur le site Web du gouvernement du Canada (voir l'annexe 2).
1.4 Contexte
Les essais expérimentaux menés à l'aide d'instruments médicaux sur des sujets humains sont devenus un domaine de recherche et de développement en croissance depuis l'établissement du nouveau Règlement sur les instruments médicaux en mai 1998.
La présence d'incohérences entre le Règlement sur les aliments et drogues et le Règlement sur les instruments médicaux a suscité des réserves chez certains intervenants. Bien que ces incohérences ne puissent être corrigées qu'en amendant les règlements, il est possible d'en atténuer quelques-unes par l'application de la présente ligne directrice.
La ligne directrice antérieure, publiée en 1999, ne convient plus, car elle ne donne pas d'indications claires et cohérentes quant à un certain nombre d'aspects concernant l'élaboration des demandes d'AEE. Les fréquentes demandes de clarification faites par les fabricants auprès du Bureau des matériels médicaux ont permis de constater ces lacunes.
La présente ligne directrice a été mise à jour pour répondre aux réserves exprimées par les intervenants et aborder certains sujets, comme l'utilisation de normes reconnues aux termes du Règlement, les produits mixtes médicaments/matériels médicaux, l'utilisation d'instruments non homologués dans les études sur les médicaments, les études cliniques parrainées par les chercheurs, les stades de développement d'un produit, les révisions apportées au protocole d'un essai expérimental et les rapports d'incident obligatoires.
2. Directives de mise en œuvre
2.1 Abréviations et définitions
2.1.1 Abréviations
- AEE
- Autorisation d'essai expérimental
- BC
- Brochure du chercheur
- BMM
- Bureau des matériels médicaux
- BPC
- Bonnes pratiques cliniques
- BPL
- Bonne pratique de laboratoire
- CER
- Conseil d'éthique de la recherche
- CTD
- Common Technical Document (dossier technique commun)
- DEC
- Demande d'essai clinique
- DGORR
- Direction générale des opérations réglementaires et des régions
- DIMM
- Déclaration des incidents liés aux matériels médicaux
- DPSC
- Direction des produits de santé commercialisés
- DPT
- Direction des produits thérapeutiques
- ECR
- Essai clinique randomisé
- EE
- Essai expérimental
- FCE
- Formulaire de consentement éclairé
- IDIV
- Instrument diagnostique in vitro
- IRSC
- Instituts de recherche en santé du Canada
- ISO
- Organisation internationale de normalisation
- MON
- Mode opératoire normalisé
- OMS
- Organisation mondiale de la Santé
- PAS
- Programme d'accès spécial
2.1.2 Termes et définitions
La plupart des définitions qui suivent sont tirées du Règlement ou de la norme ISO 14155 - Investigation clinique des dispositifs médicaux pour sujets humains - Bonnes pratiques cliniquesNote de bas de page 1.
Renseignements complémentaires : Renseignements demandés au fabricant ou à l'importateur de l'instrument par le Bureau des matériels médicaux afin de déterminer si les conditions précisées au paragraphe 83(1) du Règlement ont été respectées.
Événement indésirable : Toute manifestation clinique indésirable, maladie ou blessure non intentionnelle, ou tout signe clinique indésirable (y compris un résultat anormal de laboratoire) chez un sujet, utilisateur ou autre personne, en relation ou non avec l'instrument médical sous investigation, ce qui comprend :
- les événements liés à l'instrument médical sous investigation ou au comparateur;
- les événements liés aux procédures suivies.
Pour les utilisateurs ou autres personnes, la définition concerne uniquement les événements liés à l'instrument sous investigation.
Biais : Erreur de mesure systématique commise au cours de la collecte des données.
Cahier d'observations : Ensemble de documents imprimés, optiques ou électroniques concernant chaque sujet sur lesquels les informations à communiquer au promoteur sont enregistrées, comme l'exige le plan d'investigation clinique (soit le protocole).
Investigation cliniqueNote de bas de page 2 : Investigation systématique portant sur un ou plusieurs sujets humains, entreprise pour vérifier la sécurité ou les performances d'un instrument médical.
Rapport d'investigation clinique : Document décrivant la conception, l'exécution, l'analyse statistique et les résultats d'une investigation clinique.
Produit mixte : Produit thérapeutique réunissant une composante « médicament » et une composante « matériel médical » (qui seraient séparément classifiées comme étant soit un médicament soit un matériel médical), de manière telle que les natures distinctes de la composante « médicament » et une composante « matériel médical » sont intégrées en un seul produit.
Comparateur : Instrument médical, thérapie (par exemple, contrôle ayant une action), placebo ou absence de traitement, utilisé dans le groupe de référence d'une investigation clinique.
Organisation contractuelle de recherche : Personne ou organisation engagée par le promoteurNote de bas de page 3 pour exercer une ou plusieurs responsabilités ou tâches liées à l'investigation clinique incombant au promoteur.
Écart : Cas de non-respect, délibéré ou non, des exigences du plan d'investigation clinique.
Instrument : Tout instrument, appareil, dispositif ou article semblable ou tout réactif in vitro, y compris tout composant, partie ou accessoire de l'un ou l'autre de ceux-ci, fabriqué ou vendu pour servir à l'une ou l'autre des fins ci-après ou présenté comme pouvant y servir :
- le diagnostic, le traitement, l'atténuation ou la prévention d'une maladie, d'un désordre ou d'un état physique anormal ou de leurs symptômes, chez l'être humain ou les animaux;
- la restauration, la correction ou la modification de la structure corporelle d'un être humain ou d'un animal, ou du fonctionnement des parties du corps d'un être humain ou d'un animal;
- le diagnostic de la gestation chez l'être humain ou les animaux;
- les soins de l'être humain ou des animaux pendant la gestation ou à la naissance ou les soins postnatals, notamment les soins de leur progéniture;
- la prévention de la conception chez l'être humain ou les animaux;
Est exclu de la présente définition un tel instrument, appareil, dispositif ou article, y compris tout composant, partie ou accessoire de l'un ou l'autre de ceux-ci, servant à l'une ou l'autre des fins visées aux alinéas a) à e) uniquement par des moyens pharmacologiques, immunologiques ou métaboliques ou uniquement par des moyens chimiques à l'intérieur ou à la surface du corps d'un être humain ou d'un animal.
Identificateur de l'instrument : Série unique de lettres ou de chiffres, ou toute combinaison de ceux-ci, ou code à barres, qui est assigné à l'instrument médical par le fabricant et qui permet d'identifier l'instrument et de le distinguer d'instruments similaires.
Nom de l'instrument : Vise également tout renseignement nécessaire à l'utilisateur pour identifier l'instrument médical et le distinguer d'instruments similaires.
Urgence : Caractère de ce qui est urgent, de ce qui ne souffre aucun retard. Situation pathologique dans laquelle un diagnostic et un traitement doivent être réalisés très rapidementNote de bas de page 4.
Comité d'éthique : Organisme indépendant dont la responsabilité consiste à examiner les investigations cliniques pour protéger les droits, la sécurité et le bien-être des sujets participant à l'investigation clinique. Les exigences réglementaires relatives aux comités d'éthique ou aux institutions similaires varient d'un pays à l'autreNote de bas de page 5.
Bonnes pratiques cliniques : Pratiques cliniques généralement reconnues qui visent à assurer la protection des droits, la sécurité et le bien-être des sujets qui participent à des essais cliniques et des autres personnes.
Professionnel de la santé : Personne autorisée en vertu des lois d'une province à y fournir des services de santé.
Hypothèse : Postulat à vérifier, issu de l'objectif, concernant la sûreté ou les performances de l'instrument médical utilisé pour concevoir l'investigation clinique et pouvant être accepté ou rejeté en fonction des résultats de l'investigation et des calculs statistiques. L'hypothèse principale est le facteur déterminant des paramètres de sûreté ou de performance de l'instrument médical sous investigation et sert généralement à calculer la taille de l'échantillon. Les hypothèses secondaires concernant d'autres points d'intérêt peuvent également être évaluées.
Importateur : Personne autre que le fabricant de l'instrument, dont l'établissement est au Canada, et qui se charge de faire introduire l'instrument au Canada par des fabricants ou des distributeurs de l'étranger, en vue de sa vente au Canada.
Indépendant : Non impliqué dans la conduite d'une investigation clinique à l'exception des responsabilités affectées spécifiquement afin d'éviter les biais ou un conflit d'intérêts.
Document ou formulaire de consentement éclairé : Processus par lequel une personne est informée et invitée à participer volontairement à une investigation clinique. Un consentement éclairé est consigné sous la forme d'un formulaire de consentement éclairé écrit, signé et daté.
Site d'investigation : Institution ou lieu où est effectuée une investigation cliniqueNote de bas de page 6.
Instrument médical sous investigation : Instrument médical dont la sûreté ou les performances sont évaluées dans le cadre d'une investigation clinique. Cette définition inclut les instruments médicaux déjà commercialisés pour lesquels de nouvelles indications, de nouvelles populations, de nouveaux matériaux ou des modifications de conception sont évaluésNote de bas de page 7.
Chercheur : Membre de l'équipe du site d'investigation nommé et encadré par le chercheur principal de ce site pour réaliser des procédures essentielles liées à l'investigation clinique ou pour prendre des décisions importantes concernant celle-ciNote de bas de page 8.
Brochure du chercheur : Ensemble des informations cliniques et non cliniques actualisées concernant le ou les instruments médicaux sous investigation et qui sont pertinentes pour l'étude clinique.
Essai expérimental à l'initiative du chercheur : Essai clinique entrepris par un clinicien ou un établissement de santé, et non par le fabricant de l'instrument. Les données ainsi recueillies ne sont pas destinées à servir à des fins de commercialisation. Le financement de ce type de recherche provient généralement d'une tierce partie, comme les Instituts de recherche en santé du Canada (IRSC). Dans ce cas, le chercheur doit obtenir la coopération du fabricant, qui doit être le demandeur et signataire officiel de la demande d'AEE. La correspondance réglementaire et la surveillance clinique peuvent être déléguées par le fabricant, qui est le promoteur légal de l'investigation clinique.
Fabricant : Personne qui vend l'instrument médical sous son propre nom ou sous un nom commercial, une marque de commerce, un dessin ou un autre nom ou marque qu'elle contrôle ou dont elle est propriétaire et qui est responsable de la conception, de la fabrication, de l'assemblage, du traitement, de l'étiquetage, de l'emballage, de la remise à neuf ou de la modification de l'instrument, ou de l'assignation d'une utilisation à cet instrument, que ces opérations soient effectuées par elle ou pour son compte.
Instrument médical : S'entend d'un instrument, au sens de la Loi, à l'exclusion des instruments destinés à être utilisés à l'égard des animaux.
Surveillance : Action de superviser l'avancement d'une investigation clinique pour s'assurer que celle-ci est conduite, enregistrée et communiquée conformément au plan d'investigation clinique, aux procédures écrites, à la Norme internationale de l'ISO et aux exigences réglementaires applicables.
Objectif : Raison principale de la conduite de l'investigation clinique.
Chercheur principal : Personne qualifiée responsable de la conduite de l'investigation clinique sur un site d'investigation. Si une investigation clinique est conduite par une équipe sur un site d'investigation, le chercheur principal est le responsable de l'équipe. En fonction des réglementations nationales, la responsabilité incombe soit à une personne soit à une institution.
Développement de produit : Études exploratoires de base menées pour déterminer si un article mis à l'essai a une quelconque utilité.
Protocole : Document établissant les objectifs, la conception, la méthodologie, les considérations statistiques et l'organisation de l'essai clinique (aussi appelé « plan d'investigation clinique »).
Chercheur compétent : Personne qui est membre en règle d'une association professionnelle de personnes habilitées en vertu des lois d'une province à y offrir des soins de santé et qui a été désignée par le comité de déontologie d'un établissement de santé pour y effectuer un essai expérimental.
Randomisation : Processus d'affectation des sujets à l'instrument médical sous investigation ou aux groupes comparateurs utilisant une méthodologie statistique reconnue pour déterminer l'affectation afin de réduire les biais.
Classification du risque : Classification d'un instrument médical conformément aux Règles de classification des instruments médicaux précisées à l'annexe 1 du Règlement sur les instruments médicaux.
Vente : Est assimilé à l'acte de vendre le fait de mettre en vente, ou d'exposer ou d'avoir en sa possession pour la vente, ou de distribuer, que la distribution soit faite ou non pour une contrepartieNote de bas de page 9.
Événement indésirable grave : Événement indésirable ayant entraîné :
- la mort,
- une détérioration grave de la santé du sujet qui :
- a provoqué une maladie ou une blessure mettant en danger la vie du sujet,
- a provoqué une infirmité permanente du corps ou des fonctions corporelles,
- a nécessité l'hospitalisation du patient ou la prolongation de son séjour à l'hôpital,
- a provoqué une intervention médicale ou chirurgicale pour éviter une maladie ou une blessure mettant en danger la vie du sujet ou une infirmité permanente du corps ou des fonctions corporelles;
- une souffrance fœtale, une mort fœtale, une anormalité ou une anomalie congénitaleNote de bas de page 10.
Modification importante : Toute modification qui pourrait vraisemblablement influer sur la sûreté ou l'efficacité de l'instrument médical. Est également visée toute modification d'un des éléments suivants :
- les procédés, les installations ou l'équipement de fabrication;
- les procédures de contrôle de la qualité de la fabrication, notamment les méthodes, essais ou procédures utilisés pour contrôler la qualité, la pureté et la stérilité de l'instrument ou de ses matériaux de fabrication;
- la conception de l'instrument, notamment les principes de fonctionnement, les caractéristiques de rendement et les spécifications des matériaux, de la source d'énergie, du logiciel ou des accessoires;
- l'utilisation à laquelle l'instrument est destiné, notamment toute utilisation nouvelle ou supplémentaire, tout ajout ou suppression de contre-indications et toute modification de la période servant à fixer la date de péremptionNote de bas de page 11.
Fin de l'étude : L'étude est considérée comme terminée après que le dernier sujet s'est soumis à une visite de « fin d'étude ». La « visite de fin d'étude » est la toute dernière visite au cours de laquelle des examens et des procédures liés à l'étude ont été menés, y compris la consignation de tout événement indésirable lié à l'étude. L'étude n'est pas considérée comme terminée si l'essai clinique est suspendu, annulé ou clos.
Sujet : Personne qui prend part à une investigation clinique. Il peut s'agir d'une personne en santé se portant volontaire ou d'un patient.
2.2 Rencontre préalable à la demande d'AEE
Santé Canada invite les fabricants et les importateurs à demander une rencontre préalable à la demande d'AEE, surtout pour les instruments nouveaux de classe III et IV et les produits mixtes. Une telle consultation peut s'avérer utile pour les aider à rédiger leur demande et favoriser une décision réglementaire en temps opportun.
Cette rencontre préalable à la demande d'AEE vise à permettre au fabricant ou à l'importateur de présenter des données pertinentes et de discuter des préoccupations ou questions concernant le développement de produit. Elle offre également à Santé Canada l'occasion de donner des conseils, ainsi que de souligner les lacunes possibles ou d'exprimer des réserves quant à l'investigation proposée. Les fabricants et les importateurs peuvent inviter les chercheurs compétents qui prendront part à l'investigation proposée au Canada à assister à cette rencontre.
2.2.1 Demander une rencontre préalable à la demande d'AEE
Toute demande de rencontre préalable à la demande d'AEE doit être faite par écrit par le fabricant ou l'importateur à la Division des essais expérimentaux du Bureau des matériels médicaux à l'adresse : EssaisExperimentaux@hc-sc.gc.ca.
La demande doit être présentée sous forme de lettre d'accompagnement proposant trois choix de date et heure qui conviennent en vue de la rencontre. La lettre d'accompagnement doit contenir les renseignements suivants :
- un résumé de l'étude proposée;
- une liste des questions préliminaires auxquelles le Bureau doit répondre pendant la rencontre;
- suffisamment d'information pour que Santé Canada puisse évaluer l'utilité de la rencontre et déterminer le personnel compétent à y convier pour discuter des questions soumises. Cela permettra de veiller à ce que les ressources de Santé Canada soient utilisées à bon escient.
Le Bureau des matériels médicaux accusera réception de la demande de rencontre en temps opportun. S'il accepte de tenir une rencontre, l'accusé de réception indiquera la date à laquelle une trousse d'information préalable à la demande d'AEE doit être fournie, la date et le lieu de la rencontre, ainsi que la liste des personnes qui y seront présentes. Il convient de noter que le Bureau se réserve le droit de proposer une rencontre via téléconférence ou de répondre aux questions pour courriel au lieu de tenir une rencontre en face à face.
2.2.2 Trousse d'information en vue de la rencontre préalable à la demande d'AEE
La trousse d'information doit être présentée en format électronique et contenir :
- l'ordre du jour proposé, le jeu de diapositives utilisé, notamment la liste définitive des questions, ainsi qu'une liste complète des personnes présentes [il est convenu que les diapositives peuvent être modifiées avant la rencontre];
- un résumé de toutes les données, notamment :
- un tableau énumérant les études précliniques et cliniques terminées,
- les spécifications de l'instrument et les résultats des essais précliniques, y compris les résultats des bancs d'essai et des études expérimentales sur animal,
- une liste des normes utilisées lors de la conception et de la fabrication de l'instrument,
- les événements indésirables observés et des précisions sur les problèmes de sécurité possibles;
- le plan clinique global proposé pour le stade de développement actuel de l'instrument, notamment, ses usages homologués dans d'autres pays [il est convenu que ce plan est sujet à changement à mesure que de nouvelles données sont disponibles];
- les détails de l'investigation clinique qu'on se propose de mener au Canada, à l'intérieur du cadre de l'AEE souhaitée, y compris :
- un exposé du protocole d'essai,
- une hypothèse et les objectifs de l'étude,
- l'affection à traiter ou à diagnostiquer et une description des traitements disponibles,
- les procédures et/ou les critères envisagés pour le suivi des patients, ainsi que les évaluations de l'efficacité et de la sûreté cliniques,
- les considérations statistiques.
En présence d'une trousse d'information insatisfaisante, le fabricant ou l'importateur peut être invité à repousser ou à remettre la rencontre à plus tard pour lui permettre de réunir une trousse plus complète. Il convient de noter que le Bureau des matériels médicaux se réserve le droit de modifier ou d'abréger l'ordre du jour proposé comme bon lui semble afin d'atteindre les objectifs de la rencontre.
2.2.3 Compte rendu de la rencontre préalable à la demande d'AEE
Le fabricant ou l'importateur doit rédiger puis envoyer au Bureau un compte rendu écrit des discussions et des conclusions de la rencontre dans les quatorze (14) jours suivant la rencontre. Tous les comptes rendus définitifs de cette consultation seront versés au dossier.
Une copie du compte rendu des discussions et des conclusions approuvé par toutes les parties présentes à la rencontre doit être jointe à la demande d'AEE subséquente.
2.3 Demandes d'AEE
Aux termes du Règlement, seuls les fabricants et les importateurs peuvent demander l'autorisation de mener un essai expérimental sur des sujets humains au Canada. Dans les deux cas, un cadre supérieur du fabricant doit remplir et signer le formulaire de demande. Conformément aux exigences précisées à l'article 82, une copie électronique modifiable des documents à l'appui précisés aux alinéas 81a) à k) et à l'article 86 (Étiquetage) du Règlement doit être présentée.
2.3.1 Stades de développement du produit
Premier essai sur des sujets humains : Il s'agit du type d'étude conçu pour évaluer un instrument en fonction d'une indication précise chez l'humain pour la toute première fois :
- pour obtenir des données cliniques initiales,
- pour peaufiner la conception de l'instrument,
- pour répondre à un besoin clinique non satisfait,
- lorsque d'autres essais non cliniques ne peuvent pas être menés.
Étude de faisabilité hâtive : Il s'agit d'une investigation clinique limitée portant sur un instrument aux premiers stades de son développement, habituellement avant que sa conception ne soit définitive (p. ex. un instrument novateur en vue d'une nouvelle utilisation ou d'une utilisation prévue établie ou un instrument mis en marché pour une application clinique nouvelle). Elle est conçue pour obtenir des données initiales sur la sûreté et le rendement du nouvel instrument.
Étude de faisabilité traditionnelle : Il s'agit de l'investigation clinique conçue pour recueillir des données préliminaires sur la sûreté et l'efficacité de l'instrument dans sa forme définitive ou presque définitive afin de bien planifier une étude pivot appropriée.
Étude pivot : Il s'agit de l'investigation clinique conçue pour recueillir les preuves définitives de la sûreté et de l'efficacité d'un instrument en vue d'une utilisation prévue bien précise, habituellement chez un nombre statistiquement justifié de sujets.
2.3.2 Quand présenter une demande d'AEE
Une demande d'AEE est nécessaire pour que les instruments médicaux non homologués de classe II, III et IV puissent être importés ou vendus aux fins d'essais expérimentaux sur des sujets humains. Cependant, lorsque des études exploratoires de base sont menées pour déterminer si le produit a une utilité potentielle, et que le produit n'est pas représenté dans le cadre de la définition d'« instrument » ou d'« instrument médical », une demande d'AEE n'est pas requise. La classification d'un produit dans la catégorie des instruments médicaux repose sur la représentation que fait le fabricant de l'instrument sur l'étiquette, et non seulement sur son utilisation dans le cadre de l'étude. De plus amples renseignements pour déterminer la classification d'un instrument se trouvent dans la Ligne directrice - Orientation sur le système de classification fondé sur le risque des instruments autres que les instruments diagnostiques in vitro (IDIV) et l'Index de mots-clés pour aider les fabricants à vérifier la classification des matériels médicaux (voir l'annexe 2).
Il est inutile de demander une AEE si l'instrument médical ne fait pas l'objet d'une vente, selon la définition de la Loi sur les aliments et drogues. C'est donc dire que si le développement, la fabrication et la mise à l'essai de l'instrument s'effectuent au sein d'une seule et même entité juridique, il n'y a aucune vente; les exigences du Règlement ne s'appliquent donc pas. Dans ce cas, les essais doivent être menés sur place et uniquement par le fabricant légal.
En outre, il est tout autant inutile de demander une AEE pour mener une étude à l'aide d'un instrument homologué selon les indications d'utilisation autorisées. En revanche, un instrument homologué utilisé dans le cadre d'une étude financée par le fabricant en vue de générer des données à l'appui d'une nouvelle indication d'utilisation est assujetti à l'obligation de demander une AEE.
Les instruments médicaux déjà homologués utilisés lors d'investigations cliniques qui ne sont pas financées par le fabricant et qui ne visent pas à générer des données à l'appui d'une nouvelle demande d'homologation n'ont pas besoin d'être soumis à une AEE. Dans le même ordre d'idées, les instruments médicaux homologués utilisés lors d'investigations cliniques qui dépassent la portée des indications d'utilisation autorisées et qui ne sont pas financées par le fabricant ne relèvent pas du Règlement, puisqu'il s'agit là d'une utilisation non indiquée.
Une demande d'AEE et une demande d'homologation de l'instrument médical ne doivent pas être faites en parallèle, puisque l'EE ne permet pas de recueillir des données supplémentaires sur la sûreté ou l'efficacité permettant d'appuyer une homologation; l'EE ne sert qu'à accélérer l'accès au marché.
2.3.3 Comment remplir une demande d'AEE
Toutes les demandes doivent être présentées par voie électronique, dans le format actuellement en vigueur pour soumettre des demandes d'AEE, comme précisé dans l'Avis - Demandes d'autorisation d'essais expérimentaux (AEE) pour les instruments médicaux, en format « électronique autre que le format eCTD » (voir l'annexe 4). Il est aussi possible de consulter la Ligne directrice : Préparation des activités de réglementation en format « Électronique autre que le format eCTD » qui décrit en détail la marche à suivre pour présenter les activités réglementaires liées à l'instrument médical et les transactions subséquentes dans un format « électronique autre que le format eCTD ». Il convient de noter que Santé Canada n'accepte plus de copie papier des demandes. Les demandes doivent donc être acheminées par courriel à l'adresse : homologation_instruments@hc-sc.gc.ca ou par la poste à l'adresse postale du Bureau (voir l'annexe 1) sur CD ou DVD si leur taille ne permet pas l'envoi par courriel. Les demandes faites par courriel doivent afficher une ligne d'objet qui indique clairement qu'il s'agit d'une demande d'AEE. Toute demande de révision d'une demande d'AEE doit être clairement indiquée comme telle et s'accompagner du numéro de demande précédemment attribué. Les demandes d'AEE ne sont pas assujetties à des frais.
Les demandes présentées par un chercheur ou une tierce partie (p. ex. organisme de financement, fabricant d'un médicament, fabricant d'un autre instrument ou établissement de santé) doivent être signées par un cadre supérieur du fabricant de l'instrument non homologué. La correspondance réglementaire et la surveillance clinique peuvent être déléguées au chercheur clinique, au fabricant, à leur responsable de la réglementation ou à une organisation contractuelle de recherche. Dans ce cas, le cadre qui dispose du pouvoir délégué assume les responsabilités de l'importateur. Pour les instruments des classes III ou IV, le fabricant peut présenter les données non cliniques requises à l'appui de la demande, et le chercheur clinique ou le fabricant peut présenter le protocole d'étude, le formulaire de consentement éclairé (FCE) des patients, l'analyse des risques abordant les préoccupations cliniques, l'engagement écrit des chercheurs et la preuve de l'approbation du CER.
Les fabricants sont invités à inscrire leurs investigations cliniques dans un registre public qui accepte les données d'essais cliniques internationaux et qui est reconnu par l'Organisation mondiale de la Santé (OMS). Les registres ClinicalTrials.gov et Current Controlled Trials International Standard Randomised Controlled Trials Number Register sont acceptables. Les données liées aux essais expérimentaux d'instruments médicaux menés au Canada peuvent être obtenues directement du fabricant ou en consultant l'un des registres.
2.3.3.1 Plusieurs instruments utilisés dans la même étude
Si on se propose d'utiliser plusieurs instruments non homologués, provenant de différents fabricants, dans le même protocole d'étude, chaque fabricant ou importateur doit soumettre une demande distincte à l'examen du Bureau. Le sommaire de la demande doit justifier l'utilisation de l'instrument non homologué en même temps que les autres instruments dans le même protocole d'étude, ainsi qu'indiquer si une demande a été ou sera présentée pour les autres instruments. Dans la mesure du possible, les demandes devraient être présentées simultanément. Aucune autorisation ne sera accordée avant que toutes les demandes n'aient été présentées et jugées conformes aux exigences réglementaires.
2.3.3.2 Instruments utilisés dans des essais cliniques de médicaments
Les essais cliniques internationaux portant sur un médicament ont parfois recours à des instruments auxiliaires qui ne sont pas homologués au Canada. Pour toute demande d'essai clinique (DEC) qui nécessite l'utilisation d'un instrument médical de la classe II, III ou IV non homologué, il faut présenter une demande d'AEE distincte de la DEC, et chaque demande doit avoir été autorisée avant que l'essai ne puisse commencer au Canada. Ces demandes peuvent être présentées simultanément.
Dans ce cas, le fabricant de l'instrument signe la demande d'AEE et délègue généralement la correspondance réglementaire au promoteur du médicament. Le fabricant d'un instrument de la classe III ou IV fournit les données précliniques au Bureau, alors que le promoteur du médicament à l'étude s'occupe de présenter le protocole d'étude et les FCE. Même si ces deux documents se rapportent à l'étude d'un médicament, ils sont acceptables pour les besoins d'examen dans ces circonstances. Le numéro de la DEC, ainsi que la date de sa présentation et l'existence de toute lettre de non-objection (LNO) doivent être mentionnés dans les documents présentés.
2.3.3.3 Produits mixtes et examens conjoints
Si l'essai expérimental porte sur un produit mixte médicament/instrument médical, les demandes d'AEE ou les DEC doivent être présentées à la Direction générale ou au Bureau responsable au sein de Santé Canada, selon le principal mécanisme d'action et, de ce fait, de la classification du produit. Il faut dans ce cas consulter la politique intitulée Produits mixtes : Médicaments et matériels médicaux (voir l'annexe 2).
Il faut avoir obtenu l'autorisation de vendre et d'importer l'ensemble des produits expérimentaux qui seront utilisés dans l'investigation clinique de l'instrument médical ou l'essai clinique du médicament avant d'entreprendre l'essai expérimental ou de mettre en œuvre le protocole modifié. C'est donc dire qu'il faut présenter une demande d'AEE et une DEC distinctes pour tout produit expérimental qui n'est pas réglementé en tant que produit mixte.
La Direction générale ou le Bureau responsable verra à transmettre toutes les décisions réglementaires au promoteur ou au fabricant.
2.3.4 Format de la demande d'AEE
Depuis le 3 janvier 2017, les demandes d'AEE liées aux instruments médicaux sont acceptées en format « électronique autre que le format eCTD ». Il faut consulter la Ligne directrice : Préparation des activités de réglementation en format « Électronique autre que le format eCTD » pour connaître en détail la façon de présenter les activités réglementaires liées à l'instrument médical et les transactions subséquentes dans un format « électronique autre que le format eCTD ».
Depuis le 1er avril 2017, Santé Canada n'accepte plus de copie papier des activités réglementaires ou des transactions qui s'y rattachent, y compris les demandes d'AEE.
Il faut donc utiliser une structure de dossiers compressés et verser les documents dans le dossier pertinent. Les dossiers vides doivent être supprimés avant d'envoyer le tout à Santé Canada. Les numéros et noms de dossier ne doivent pas être modifiés.
2.3.5 Structure de la demande d'AEE
Pour accélérer l'évaluation en temps opportun des demandes d'AEE, il est recommandé au fabricant ou à l'importateur de rédiger un sommaire, d'établir une table des matières et de prévoir des sections distinctes selon la classe de l'instrument médical. La demande doit être structurée par dossier, tel que précisé à l'annexe 4.
Lorsque plusieurs investigations sont menées à partir du même instrument médical, il faut présenter une demande distincte pour chaque protocole d'étude. Il est possible de faire un renvoi aux renseignements déjà soumis dans le cadre d'une demande d'AEE concernant un autre protocole d'étude, mais portant sur le même instrument, si l'instrument n'a pas été modifié.
Les renseignements contenus dans la demande peuvent être en français ou en anglais. Les documents écrits dans une langue étrangère devront être accompagnés d'une traduction en français ou en anglais.
Le fabricant ou l'importateur voit à informer le Bureau de tout changement apporté aux coordonnées de la personne-ressource responsable de la réglementation. Tout changement de ce type doit être transmis par la personne-ressource actuelle ou un cadre supérieur de l'entreprise à l'adresse suivante : homologation_instruments@hc-sc.gc.ca.
2.3.6 Exigences d'une demande d'AEE
Bien que les renseignements à transmettre dans une demande soient déterminés par la classe de risque de l'instrument, le fabricant ou l'importateur doit conserver des registres de tous les renseignements et documents exigés aux termes de l'article 81 du Règlement. Pour certains instruments de la classe II, le Bureau des matériels médicaux peut, conformément à l'article 84, demander des renseignements complémentaires exigés normalement pour les instruments des classes III et IV.
Les données à soumettre pour prouver la sûreté, l'efficacité ou le rendement de l'instrument qui servira à mener l'essai expérimental diffèrent de celles exigées dans le cadre d'une demande d'homologation.
Les demandeurs doivent satisfaire à toutes les exigences réglementaires, démontrer qu'il y a plus d'avantages que de risques d'utiliser l'instrument et aborder tous les risques possibles dans le FCE à faire signer aux patients.
Santé Canada s'attend à ce que les fabricants respectent les principes de la Déclaration d'Helsinki et de l'Énoncé de politique des trois Conseils (2e édition) : Éthique de la recherche avec des êtres humains (2010), de même que se conforment aux bonnes pratiques cliniques(BPC) décrites dans la norme ISO 14155 - Investigation clinique des dispositifs médicaux pour sujets humains. Cette norme correspond en général aux définitions et aux exigences du Règlement. En cas de divergences, le Règlement a préséance.
Il n'est pas obligatoire de faire preuve de bonnes pratiques de laboratoire (BPL) pour mener des études expérimentales sur les animaux au Canada. Il est cependant fortement conseillé d'adopter ces BPL et de mener toute étude expérimentale sur les animaux avec la même rigueur scientifique que s'il s'agissait d'une étude chez l'humain.
Les demandes concernant des instruments qui ont déjà été autorisés dans le cadre d'un autre protocole d'étude peuvent faire des renvois à cette demande précédente en ce qui concerne les renseignements liés à l'instrument. Les résultats de l'étude autorisée antérieurement doivent être fournis, s'ils sont disponibles.
Le tableau suivant énumère les exigences à respecter dans la demande en fonction des diverses classes d'instrument.
Tableau 1
| Exigences d'une demande d'autorisation relative à un essai expérimental (AEE) | |
|---|---|
| Classe I | Il n'est pas nécessaire de faire une demande d'AEE pour un instrument médical de la classe I. Le paragraphe 80(3) du Règlement sur les instruments médicaux permet à un fabricant ou à un importateur d'un instrument médical de la classe I de vendre un tel instrument à un chercheur compétent afin d'effectuer un essai expérimental, à condition que le vendeur ait en sa possession tous les documents et tous les renseignements décrits en détail à l'article 81 du Règlement. Les exigences institutionnelles et l'approbation du Conseil d'éthique de la recherche s'appliquent. |
| Classe II | Les renseignements exigés aux termes des alinéas 81a), b), h), i) et j) doivent être soumis. Le fabricant ou l'importateur doit détenir des registres contenant tous les renseignements exigés à l'article 81 du Règlement. Il n'est pas nécessaire de soumettre la preuve de l'approbation du Conseil d'éthique de la recherche au Bureau, mais elle doit être obtenue avant d'entreprendre l'étude, conformément aux politiques institutionnelles. |
| Classes III and IV | Les renseignements exigés aux termes des alinéas 81a) à k) doivent être soumis. La preuve de l'approbation du Conseil d'éthique de la recherche est nécessaire pour qu'une AEE puisse être accordée. |
Tableau 2
| Contenu du dossier de la demande | |
|---|---|
| Classes II, III et IV | Lettre d'accompagnement et sommaire Inclure une courte lettre d'accompagnement et un sommaire d'une ou deux pages de la demande. |
| Table des matières Inclure une table des matières énumérant les éléments contenus dans la demande et leur emplacement. |
|
| Formulaire de demande Inclure une copie dûment remplie du formulaire de demande d'AEE (Demande d'autorisation pour la vente d'un instrument médical pour essai expérimental, voir l'annexe 2). |
|
| Correspondance préalable à la demande Inclure une copie de toute décision découlant de la correspondance préalable à la demande avec Santé Canada, notamment le compte rendu des rencontres préalables à la demande d'AEE, les demandes de renseignements sur la présentation d'une demande, les décisions relatives à la classification, ainsi que la correspondance avec d'autres bureaux de Santé Canada. |
|
Tableau 3
| Introduction | |
|---|---|
| Classes II, III et IV Alinéa 81a) |
Identification du fabricant ou de l'importateur Il faut donner le nom et l'adresse en entier du fabricant et de l'importateur (le cas échéant) de l'instrument, y compris le nom des personnes-ressources, courriel et les numéros de télécopieur et de téléphone. Le nom du fabricant légal doit correspondre aux renseignements figurant sur l'étiquette de l'instrument. |
| Classes II, III et IV Alinéa 81b) |
Identification de l'instrument Préciser la classification de l'instrument fondée sur le risque, telle que précisée à l'annexe 1 du Règlement sur les instruments médicaux et représentée sur l'étiquette du fabricant. Pour obtenir de l'aide dans la détermination de la classe du risque, il faut consulter la Ligne directrice - Orientation sur le système de classification fondé sur le risque des instruments autres que les instruments diagnostiques in vitro (IDIV) et l'Index de mots-clés pour aider les fabricants à vérifier la classification des matériels médicaux. Si, après avoir consulté ces documents, une aide est toujours nécessaire, écrire à l'adresse enquetes_bmm@hc-sc.gc.ca. Cette classification du risque d'un instrument faisant l'objet d'un essai expérimental peut différer de l'instrument en vente si, par exemple, une nouvelle indication est examinée. La classe de risque attribuée à un instrument expérimental peut également différer de la classification qu'attribuera le Bureau lors d'une demande d'homologation à venir si, au moment de l'homologation, l'instrument est considéré comme présentant un risque plus élevé. Cette section devrait également préciser le nombre d'unités de chaque instrument vendu séparément qui est demandé pour mener cette étude au Canada (nombre total et répartition du nombre par emplacement). |
| Classes III et IV Alinéa 81c) |
Description de l'instrument Des diagrammes techniques des instruments implantés à long terme aident à en déterminer les dimensions et les proportions relatives. Ces documents doivent être fournis avec la demande originale d'essai expérimental. Des diagrammes techniques des autres types d'instruments médicaux, tels que des instruments électromédicaux, pourraient être sollicités, au besoin, en tant que renseignements additionnels, pour établir la sûreté et l'efficacité éventuelle de ces instruments. Si l'instrument a déjà été homologué ou autorisé en vue d'un essai expérimental, il faut fournir le numéro de l'autorisation et indiquer clairement si des modifications ont été apportées à l'instrument. Si c'est le cas, fournir un tableau comparatif des similitudes et des différences en ce qui concerne l'utilisation prévue, la conception, les principales spécifications et les caractéristiques de rendement, les composantes matérielles et logicielles, les accessoires, les matériaux en contact avec le patient, etc. Il est recommandé de faire cette comparaison sous la forme d'un tableau. |
| Classes III et IV Alinéa 81d) |
Philosophie de la conception Cette section devrait comprendre un aperçu des buts et des principes de fonctionnement de l'instrument, en plus d'un résumé de la méthode d'utilisation et de fonctionnement de l'instrument. |
| Classes II, III et IV | Indications d'utilisation, utilisation prévue et contre-indications Inclure les indications d'utilisations proposées et/ou l'utilisation prévue, ainsi que toute contre-indication, pour l'instrument proposé par le fabricant. |
Classes II, III et IV Alinéa 81j) |
Étiquette d'instrument Santé Canada s'attend à recevoir une étiquette bilingue de l'instrument. Inclure une copie de la monographie du produit, des instructions d'utilisation, du manuel d'utilisation, du manuel de formation et de toute brochure promotionnelle devant accompagner l'instrument. Pour les essais cliniques randomisés, l'étiquette requise (nom de l'instrument, nom du fabricant, énoncé Instrument de recherche) doit être apposée sur le contenant extérieur, et chacun des instruments (étude et contrôle) doit porter le code qui lui a été attribué, le nom du fabricant et l'énoncé Instrument de recherche en français et en anglais. Le but de cette étiquette est de veiller à ce que l'instrument ne soit pas utilisé en dehors du cadre du protocole d'étude. |
Classes III et IV Alinéa 81e) |
Historique de la commercialisation Cette section doit fournir des détails sur la situation réglementaire de l'instrument sur les autres grands marchés mondiaux, le volume actuel des ventes par pays et les rapports de rappel dans les autres territoires de compétence, ainsi qu'un résumé des problèmes signalés relativement à l'instrument. Inclure un résumé des demandes d'accès spécial faites à Santé Canada et les résultats de celles-ci. |
Tableau 4
| Mesures d'évaluation et de réduction des risques | |
|---|---|
Classes III et IV Alinéa 81f) |
Évaluation des risques Inclure une liste de tous les risques possibles et les évaluer par rapport aux avantages prévus de l'instrument. De plus, préciser la façon dont les risques ont été réduits à un niveau acceptable. L'évaluation des risques doit être signée, et l'identité de la personne responsable doit être divulguée. |
Classes III et IV Sous-alinéa 81f)(i) |
Précédentes études Voici des exemples de précédentes études : bancs d'essai physiques et mécaniques, sécurité biologique, sécurité électrique, validation et vérification de logiciels, biocompatibilité, durée utile, validation de stérilisation et analyse des résidus, validation de l'emballage, études expérimentales sur les animaux et études cliniques. Fournir la documentation à l'appui et une bibliographie, le cas échéant. Fournir une preuve de conformité à des normes reconnues, tel que décrit dans la Ligne directrice : La Reconnaissance et l'utilisation des normes en vertu du Règlement sur les instruments médicaux (voir l'annexe 2). La présence d'une « déclaration de conformité » à une norme reconnue dans une demande éliminera, la plupart du temps, la nécessité de fournir des données de précommercialisation détaillées pour répondre aux exigences réglementaires. Il convient de noter qu'en cas de déclaration de conformité à une norme qui ne précise que les méthodes d'essai (comme ISO 10993 pour l'évaluation de la biocompatibilité), ou les procédures (comme ISO 14971 pour le recours à un système de gestion des risques des instruments médicaux), il faut aussi fournir des renseignements quantitatifs sur les résultats d'essai. |
Classes III et IV Sous-alinéa 81f)(ii) |
Traitements de remplacement |
Classes III et IV Sous-alinéa 81f)(iii) |
Précautions |
Classes II, III et IV Alinéas 81g) et h) |
Renseignements institutionnels |
Classes III et IV Alinéa 81k) |
Engagement du ou des chercheurs Cette lettre d'engagement précise les responsabilités du chercheur, soit :
Un modèle de formulaire d'engagement est disponible sur le site Web du gouvernement du Canada (voir l'annexe 2). Une version de rechange à celle-ci est acceptable, pourvu que les cinq (5) conditions décrites au paragraphe 81k) soient respectées comme il se doit, de manière adéquate. |
Classes III et IV Alinéa 81h) |
Approbation du Conseil d'éthique de la recherche Les versions et/ou dates du protocole et du FCE autorisés par le CER doivent correspondre aux renseignements fournis dans la demande. Si des modifications non administratives ont été apportées pendant le processus d'examen du CER, il faut remettre un exemplaire propre soulignant en rouge les modifications apportées au document. Tout refus émis par un CER (canadien ou étranger) précédemment doit être signalé et expliqué dans la demande. Pour une description de la constitution, des rôles et des responsabilités d'un CER, il faut consulter l'EPTC 2 et la norme ISO 14155. |
Classes II, III et IV Alinéa 81i) |
Protocole Renseignements de base décrivant la maladie ou la condition à traiter, la prévalence, les critères diagnostiques, le traitement en cours; l'hypothèse, les objectifs, la conception et les critères d'inclusion et d'exclusion de l'étude; le nombre d'instruments et de sujets requis, ainsi que des renseignements détaillés sur la durée de l'investigation et la période de suivi des patients. Les méthodes d'évaluation de l'instrument expérimental doivent faire l'objet d'une description complète, y compris les critères de succès ou d'échec en ce qui concerne le rendement de l'instrument. Il faut fournir un exemplaire de toutes les formules de rapport de cas qu'il faut utiliser au cours de l'investigation. Il faut expliquer les méthodes d'analyse des données proposées, en précisant l'identité de la personne ou du groupe qui exécute l'analyse. On doit également préciser les méthodes de contrôle de la qualité des données qui ont été choisies. Il faut décrire en détail le groupe de contrôle et justifier ce qui a amené à le choisir. Fournir une description détaillée de la sélection des sujets, c'est-à-dire :
Les sujets sélectionnés doivent être représentatifs de la population à traiter à l'aide de l'instrument, en assurant l'inclusion appropriée d'enfants, de femmes et de membres de groupes ethniques. Il faut aussi préciser l'échéancier planifié pour mener l'étude, notamment la durée de l'étape de recrutement, la durée de l'étape de traitement et la durée de l'étape de suivi. Indiquer également la durée approximative de l'investigation, du début du recrutement à la fin de la période de suivi. Cette information peut être incluse dans le protocole ou faire l'objet d'un document distinct. Brochure du chercheur (BC) Formulaire de consentement éclairé (FCE) |
Tableau 5
| Exigences supplémentaires | |
|---|---|
Classes II, III et IV |
Biais Il faut réduire au minimum les biais en respectant les normes acceptées en matière de conception de protocole précisées dans l'EPTC 2 et la norme ISO 14155. |
| Inclusion des femmes, des enfants et des populations vulnérables Il faut faire des efforts pour se conformer le plus possible au document du thème E11 de l'International Council for Harmonisation (l'ICH) intitulé Recherche clinique sur les produits médicinaux dans la population pédiatrique et au document de Santé Canada intitulé Document d'orientation : Considérations relatives à l'inclusion des femmes dans les essais cliniques et à l'analyse des données selon le sexe (voir l'annexe 2). |
|
Conception de l'essai clinique et considérations statistiques
Des directives générales sur ce sujet se trouvent dans les lignes directrices de l'ICH E8 : Considérations générales relatives aux études cliniques, E9 : Principes statistiques pour les essais cliniques et E10 : Choix d'un groupe témoin et questions connexes dans le cadre des essais cliniques, ainsi que dans l'EPTC 2 (voir l'annexe 2). |
|
2.3.7 Autorisation d'essai expérimental
Le Bureau des matériels médicaux accordera une autorisation aux termes de l'article 83 du Règlement après avoir examiné les renseignements fournis et déterminé que la demande satisfait aux exigences du Règlement.
La lettre qui sera envoyée au fabricant constituera l'autorisation légale d'importer et de vendre le nombre d'instruments requis aux chercheurs ou aux institutions énumérés dans l'autorisation pour qu'ils utilisent l'instrument dans le cadre du protocole d'étude mentionné.
La lettre d'autorisation précisera le nom de l'instrument, le titre du protocole d'étude, la date et le numéro de version, la date du FCE, les objectifs de l'étude, le nombre d'instruments qu'il est permis d'importer ou de vendre et le nombre de sujets à recruter pour mener l'étude au Canada, ainsi que le nom des chercheurs et des institutions où l'essai expérimental peut être mené.
L'autorisation ne sera donnée qu'après réception d'une preuve de l'approbation du CER pour les instruments des classes III et IV, et elle demeurera valide jusqu'à l'échéance de l'approbation courante du CER ou l'homologation de l'instrument.
2.4 Responsabilités des fabricants et des importateurs
2.4.1 Publicité
L'article 87 du Règlement interdit la publicité d'un instrument médical faisant l'objet d'un essai expérimental, sauf si une autorisation a été accordée et si la publicité précise clairement le fait que l'instrument fait l'objet d'un essai expérimental et le but de celui-ci.
2.4.2 Système de gestion de la qualité
On s'attend à ce que les fabricants disposent d'un système de gestion de la qualité adéquat. Toutefois, la preuve d'une certification à la norme ISO13485 n'est aucunement exigée dans le cadre d'une demande d'AEE.
2.4.3 Tenue de registres
Aux termes de l'article 80 du Règlement, le fabricant ou le promoteur d'un instrument médical soumis à un essai expérimental au Canada doit tenir à jour des registres décrits à l'article 81 du Règlement. Le Règlement ne précise toutefois pas la durée de conservation de ces registres par les institutions cliniques. Cette période devrait correspondre aux politiques institutionnelles et aux règles des organismes de réglementation provinciaux concernant la pratique de la médecineNote de bas de page 12.
2.4.4 Registres de distribution
Aux termes de l'article 88 du Règlement, le fabricant, l'importateur et le distributeur d'un instrument médical soumis à un essai expérimental au Canada doivent conserver des registres de distribution décrits aux articles 52 à 56 du Règlement.
2.4.5 Rapports d'incident obligatoires
Le chercheur compétent est tenu de signaler tout événement indésirable grave à Santé Canada et au fabricant ou à l'importateur dans les 72 heures suivant sa constatation. Il s'agit notamment des cas où l'incident :
- est lié à une défaillance de l'instrument, à une dégradation de son efficacité ou à un étiquetage ou mode d'emploi défectueux;
- a entraîné la mort ou une détérioration grave de l'état de santé d'un patient, utilisateur ou autre personne, ou serait susceptible de le faire s'il se reproduisait.
Pour tout incident qui se produit au Canada, le fabricant est tenu de présenter un rapport préliminaire et un rapport final sur l'incident. Le rapport préliminaire doit être présenté :
- dans les 10 jours suivant le moment où le fabricant ou l'importateur a eu connaissance de l'incident, dans le cas d'un incident qui a entraîné la mort ou une détérioration grave de l'état de santé d'un patient, utilisateur ou autre personne, ou
- dans les 30 jours suivant le moment où le fabricant ou l'importateur a eu connaissance de l'incident, dans le cas d'un incident qui n'a pas entraîné la mort ou une détérioration grave de l'état de santé d'un patient, utilisateur ou autre personne, mais qui serait susceptible de le faire s'il se reproduisait.
Les renseignements que doit contenir un tel rapport sont décrits en détail aux articles 59 à 62 du Règlement. Ces articles portent également sur la déclaration des incidents qui se produisent à l'extérieur du Canada. Il faut également consulter la Ligne directrice sur la déclaration obligatoire des incidents liés aux matériels médicaux (voir l'annexe 2) pour connaître la marche à suivre pour présenter ces rapports.
La norme ISO 14155 donne de plus amples clarifications à cet égard. On s'attend à ce que tout problème médical grave indésirable, toute maladie ou blessure non intentionnelle ou tout signe clinique indésirable (y compris un résultat anormal de laboratoire) chez un sujet, utilisateur ou autre personne, en relation ou non avec l'instrument médical expérimental soit déclaré. Tous les événements indésirables prévus et bénins doivent être déclarés dans les rapports d'étude périodiques et finaux par le fabricant ou l'importateur.
2.4.6 Autres obligations
Les fabricants et les importateurs doivent s'être dotés de procédures documentées afin de traiter les plaintes et les rappels concernant le produit, comme l'exigent les articles 57 et 58, ainsi que 63 à 65 du Règlement. En outre, des registres appropriés de ces activités doivent être tenus à jour.
Pour de plus amples renseignements, il faut consulter le document d'orientation sur la gestion des plaintes et les rappels (Politique sur les retraits/rappels, voir l'annexe 2).
Les responsabilités du fabricant en ce qui concerne l'enregistrement des implants, telles que décrites aux articles 66 à 68 du Règlement, s'appliquent également (le cas échéant) aux instruments autorisés à servir dans un essai expérimental. Une fiche d'enregistrement des implants est nécessaire pour les instruments énumérés à l'annexe 2 du Règlement.
2.5 Demande de révision d'une AEE
Les modifications apportées aux détails de l'AEE, de l'essai expérimental ou de l'instrument médical nécessitent une autorisation. Les fabricants ou les importateurs peuvent demander une autorisation révisée pour les cas ci-dessous. D'autres modifications nécessiteront la présentation d'une nouvelle demande d'AEE et l'annulation de l'autorisation précédemment émise:
- une modification apportée au nom du fabricant ou de l'instrument,
une modification apportée au protocole d'étude qui ne biaisera pas les données déjà recueillies,
une modification importante à la conception de l'instrument (lorsque l'instrument conserve généralement sa fonctionnalité première et son utilisation prévue); - correctifs mineurs apportés au protocole de FCE (p. ex. clarifications, modifications aux critères d'inclusion ou d'exclusion, modification du calendrier de suivi du patient), d'un ajout au nombre de sujets d'étude ou d'instruments étudiés;
- modifications mineures à la conception de l'instrument (p. ex. mise à niveau logicielle);
- modifications apportées à la liste des chercheurs compétents;
- changements d'institutions où l'essai sera mené.
Voici des exemples de modifications au protocole qui nécessitent la révision de l'AEE. Ces exemples sont fournis pour vous aider à déterminer si une demande d'autorisation révisée doit être présentée. Ils ne représentent pas tous les cas de figure. En cas de doute sur la nécessité de présenter une demande, les promoteurs doivent communiquer avec la Division des essais expérimentaux à l'adresse suivante : Essaisexperimentaux@hc-sc.gc.ca.
- Révisions apportées au protocole pour permettre le recrutement d'autres sujets d'étude et un suivi des patients à plus long termeNote de bas de page 13.
- Toute modification apportée au protocole qui nécessite l'approbation du CER nécessitera également l'autorisation de Santé Canada.
- Modifications apportées aux critères d'inclusion et d'exclusion. Il peut s'agir de modifications apportées aux critères d'admissibilité, aux épreuves ou aux procédures permettant de sélectionner la population de l'étude, ainsi qu'aux épreuves, procédures ou critères permettant de rejeter des sujets d'un essai clinique en cours d'étude ou à la fin de l'étude.
- Modifications apportées aux critères de sélection des patients, ainsi qu'aux épreuves ou procédures nécessaires pour assurer l'évaluation continue des sujets de l'essai clinique, notamment l'évaluation de la sécurité de l'instrument ou l'évaluation de sa sûreté et de son efficacité. Il s'agit notamment des modifications apportées au protocole à la suite de graves effets indésirables imprévus.
- Inclusion de sous-études.
- Modifications apportées à l'estimation de la taille de l'échantillon ou ajout d'analyses intérimaires qui auront une incidence sur l'analyse et l'interprétation des résultats de l'étude.
- Modifications apportées à la période de suivi post-traitement qui peuvent avoir une incidence sur l'évaluation de la sûreté de l'instrument.
- Utilisation d'instruments médicaux auxiliaires pour le traitement ou la surveillance des sujets de l'étude qui peuvent avoir une incidence sur l'analyse de l'efficacité de l'instrument ou accroître le risque de l'essai clinique pour les sujets.
- Modifications apportées aux exigences ou à la procédure liée à la déclaration de graves effets indésirables imprévus.
Voici les critères à respecter pour qu'une autorisation révisée soit donnée :
- les modifications n'auront aucune incidence importante sur l'analyse des risques originale présentée avec la demande;
- les droits et la sécurité ou le bien-être des sujets d'étude sont assurés;
- l'intégrité des données d'étude recueillies jusqu'à présent est assurée;
- le bien-fondé scientifique du plan est intact;
- aucune modification n'est apportée à l'hypothèse ou aux objectifs globaux de l'étude.
2.5.1 Comment présenter une demande de révision de l'AEE
Les exigences relatives à la présentation d'une demande d'AEE, telles que décrites à la section 2.3.6 de la présente ligne directrice, s'appliquent également à la présentation d'une demande de révision de l'AEE. Toutes les sections concernées par la révision, soit celles qui ont été touchées par les modifications apportées ou celles qui sont nécessaires pour valider l'instrument modifié ou assurer sa sécurité continue lors de l'essai, doivent être présentées de nouveau. Il faut également inclure une lettre d'accompagnement pour préciser clairement les aspects qui sont modifiés et ceux qui demeurent inchangés, ainsi que les données à l'appui qui sont fournies.
Si le protocole, le FCE ou l'étiquetage a été révisé, il faut soumettre une copie propre et annotée en rouge des documents modifiés, ainsi qu'un tableau énumérant les modifications apportées et les raisons qui justifient chacune d'elles.
Si l'instrument fait l'objet de modifications ou que de nouveaux instruments sont ajoutés, il faut soumettre les études de vérification et de validation applicables.
Si des modifications sont apportées aux chercheurs compétents ou aux établissements où auront lieu les essais, il faut fournir tous les renseignements institutionnels, les documents du chercheur et l'approbation du CER, selon la classe de l'instrument, comme décrits à la section 2.3.6 de la présente ligne directrice.
Un nouveau formulaire de demande d'AEE n'est pas nécessaire. Cependant, toute modification apportée aux détails liés à l'instrument (p. ex. nom, instruments homologués, numéro de catalogue, etc.) nécessite de présenter une copie révisée de la section 11 (Détails au sujet de l'instrument) du formulaire de demande.
Les demandes de révision d'une AEE déjà accordée doivent être présentées en format électronique et suivre la structure précisée à l'annexe 4. Toute la correspondance doit mentionner le numéro de l'AEE accordée, y compris dans l'objet du courriel et de la lettre d'accompagnement. Les demandes dûment remplies doivent être transmises par voie électronique, conformément à la politique en vigueur concernant la présentation des demandes d'AEE, à l'adresse suivante : homologation_instruments@hc-sc.gc.ca ou par la poste à l'adresse postale du bureau (voir l'annexe 1).
2.5.2 Autres renseignements à ajouter au dossier
Les modifications apportées à l'instrument ou à l'essai qui ne sont pas considérées comme une modification importante doivent être signalées à Santé Canada. Un accusé de réception des modifications sera envoyé par écrit, et une autorisation révisée pourra ou non être nécessaire, selon la nature des modifications.
2.6 Annulation d'une AEE
L'article 85 du Règlement permet au ministre d'annuler une AEE pour les instruments des classes II, III ou IV, ainsi que d'arrêter la vente des instruments de la classe I à des fins expérimentales, tel que précisé aux alinéas 85(1)a) à e), lorsque :
- l'essai présente un risque grave pour la vie, la santé ou la sécurité des patients, utilisateurs ou autres personnes;
- l'essai va à l'encontre de l'intérêt des patients en cause;
- les objectifs de l'essai ne seront pas atteints;
- le chercheur compétent qui effectue l'essai ne respecte pas l'engagement visé à l'alinéa 81k);
- les renseignements soumis concernant l'essai sont faux ou trompeurs.
Avant d'annuler une AEE, Santé Canada demandera des renseignements complémentaires au fabricant ou à l'importateur afin de vérifier que les conditions visées au paragraphe 83(1) sont toujours respectées. Si les renseignements demandés ne sont pas fournis ou, dans le cas où ceux-ci sont fournis, si un examen de ces renseignements permet de déterminer que l'une des situations visées au paragraphe 85(1) existe, l'autorisation sera annulée par avis écrit précisant les raisons de cette annulation.
2.7 Procédure d'examen des demandes d'AEE et des demandes de révision d'une AEE
2.7.1 Examen préliminaire
Les nouvelles demandes d'AEE font l'objet d'un examen préliminaire pour veiller à ce que le contenu administratif et scientifique respecte les exigences réglementaires applicables visées aux articles 81 et 86 du Règlement. Un avis d'acceptation (examen préliminaire) est expédié si les renseignements sont complets. Lorsque des lacunes sont relevées à l'examen préliminaire, un avis d'insuffisances constatées à l'examen préliminaire précisant les lacunes décelées est envoyé. Le fabricant ou l'importateur dispose de quinze (15) jours civils à partir de la date de cet avis pour fournir les renseignements manquants, à défaut de quoi la demande sera rejetée et retournée au fabricant (consulter la politique Gestion des demandes d'homologation d'instruments médicaux et d'autorisation d'essais expérimentaux, à l'annexe 2). Les renseignements à fournir doivent être présentés dans un Sommaire sous la forme de questions et réponses, ainsi que dans tout autre document versé à la section appropriée du dossier électronique, en respectant la structure d'une demande d'AEE (voir l'annexe 4).
2.7.1.1 Avis de refus (examen préliminaire)
Un avis de refus (examen préliminaire) est envoyé lorsque la demande comporte trop de lacunes, notamment lorsque les renseignements importants exigés à l'article 81 du Règlement sont absents de la demande. Un tel refus peut également survenir lorsque la demande concerne un instrument médical de la classe II, alors que l'examen préliminaire détermine qu'il s'agit plutôt d'un instrument de la classe III ou IV.
Un avis de refus (examen préliminaire) est également envoyé lorsque le fabricant ou l'importateur ne fournit pas les registres visés à l'article 81 dans les quinze (15) jours suivant la réception de l'avis d'insuffisances constatées à l'examen préliminaire. Cet avis renferme une liste des lacunes. Si la demande est rejetée, le fabricant ou l'importateur devra présenter une toute nouvelle demande. Aucune référence à une demande rejetée à l'examen préliminaire ne sera permise.
2.7.2 Examen
Une fois l'examen enclenché, aucun renseignement non sollicité ne doit être fourni sans le consentement du gestionnaire de la Division des essais expérimentaux. L'examen des nouvelles demandes d'AEE ou des demandes de révision d'une AEE, autres que celles concernant des produits mixtes, devrait prendre trente (30) jours civils à partir de la date où l'examen préliminaire a été entrepris. Si un avis d'insuffisances constatées à l'examen préliminaire a été envoyé, le délai de trente (30) jours commence à la date de réception de la demande complète. L'examen des demandes de révisions administratives mineures à une AEE devrait prendre quinze (15) jours civils. Il convient de noter que ces délais ne sont que des cibles établies en fonction d'une estimation du temps requis, et non des échéances fermes entraînant automatiquement l'autorisation si elles ne sont pas respectées.
Une fois l'examen terminé, Santé Canada enverra une demande de renseignements complémentaires, tel que décrit à l'article 84 du Règlement, si certains renseignements exigés à l'article 81 sont absents. Le fabricant ou l'importateur doit présenter une réponse détaillée dans les soixante (60) jours civils. La réception de cette réponse donne lieu à un nouveau délai de trente (30) jours civils pour l'examen de la demande.
Si le fabricant ou l'importateur ne parvient pas à fournir les renseignements demandés dans les délais impartis, il peut retirer sa demande et la présenter à nouveau, sans préjudice, dans les six (6) mois suivants. Après ce délai, une nouvelle demande doit être présentée. Les demandes de renseignements complémentaires qui demeurent sans réponse après le délai de soixante (60) jours pourraient être rejetées.
Lorsque les renseignements fournis à l'appui de l'AEE ou de la révision d'une AEE sont jugés satisfaisants, Santé Canada accordera une autorisation ou une autorisation révisée.
2.7.2.1 Refus d'une demande
Les demandes d'AEE ou de révision d'une AEE peuvent être rejetées si l'on détermine que :
- l'instrument ne peut être utilisé en toute sécurité à des fins d'essai expérimental;
- l'essai expérimental est contraire aux intérêts supérieurs des patients;
- l'objectif de l'essai n'est pas atteignable;
- les registres exigés aux articles 81 et 86 n'ont pas été fournis.
Un avis de refus décrivant les lacunes sera envoyé.
Le fabricant ou le promoteur de l'instrument pourra en appeler de la décision de refuser l'AEE. Les renseignements ayant trait à la procédure d'appel des décisions relatives aux demandes d'AEE se trouvent dans le document Gestion des demandes d'homologation d'instruments médicaux et d'autorisation d'essais expérimentaux (voir l'annexe 2).
2.8 Exigences suivant l'autorisation
2.8.1 Interruption de l'essai expérimental
En cas d'interruption d'une étude dans son ensemble ou de l'étude à un emplacement donné pour laquelle une AEE ou une AEE révisée a été accordée au Canada, le fabricant ou l'importateur doit en aviser le Bureau des matériels médicaux dès que possible, mais au plus tard quinze (15) jours civils suivant la date de l'interruption.
Cet avis doit contenir :
- les raisons détaillées de cette mesure, notamment si elles sont liées à la sûreté ou à l'efficacité de l'instrument médical;
- la description de l'incidence de cette interruption sur l'étude proposée ou en cours menée au Canada;
- la confirmation que tous les chercheurs compétents ont été informés de l'interruption et des raisons qui la sous-tendent, et qu'ils ont été avisés par écrit des risques possibles pour la santé des sujets de recherche ou d'autres personnes;
- la confirmation que la vente ou l'importation de l'instrument à tous les emplacements où l'étude a été interrompue a été arrêtée;
- la confirmation que le fabricant prendra des mesures raisonnables pour assurer le retour de tous les instruments non utilisés.
Remarque : L'avis d'interruption précoce d'une étude à des emplacements internationaux (pour des raisons de sûreté) alors que des essais expérimentaux avec l'instrument médical sont menés au Canada doit également être transmis au Bureau des matériels médicaux.
2.8.2 Reprise d'un essai expérimental
Le promoteur peut reprendre l'essai expérimental dans son ensemble ou à un emplacement à la suite d'une interruption s'il fournit les renseignements suivants :
- les nom, adresse, numéro de téléphone et courriel du chercheur compétent de chaque emplacement et du CER qui a approuvé la reprise de l'essai expérimental à chaque emplacement;
- les nom, adresse et numéro de téléphone et, le cas échéant, le numéro de télécopieur et le courriel de tout CER qui a refusé d'approuver la reprise de l'essai expérimental, le cas échéant;
- la date proposée pour la reprise de l'essai expérimental à chaque emplacement de l'étude.
Remarque : Les renseignements susmentionnés peuvent être transmis en tant que renseignements complémentaires. Le Bureau accusera réception des renseignements, ce qui autorisera la reprise. En cas de modification à l'instrument, au protocole d'étude ou au procédé de fabrication, les renseignements doivent être transmis dans une demande de révision de l'AAE (voir la section 2.5). L'étude peut reprendre seulement lorsqu'un avis d'autorisation révisée est envoyé.
2.8.3 Achèvement de l'étude et fermeture des emplacements
Les fabricants et les importateurs sont vivement invités à aviser le Bureau de l'achèvement de l'étude. Ils sont aussi encouragés à transmettre un rapport d'étude final. À la réception d'un tel avis, Santé Canada enverra une lettre confirmant la clôture de l'étude. La norme ISO 14155 donne d'autres lignes directrices à ce chapitre.
Une fois l'essai expérimental terminé, les instruments réutilisables, comme les biens d'équipement, qui ne sont pas encore homologués doivent être retournés au fabricant ou à l'importateur. Si le chercheur compétent a l'intention d'utiliser ces instruments dans le cadre d'un autre protocole d'étude, une nouvelle demande d'AEE signée par un cadre supérieur du fabricant doit être présentée. Il est interdit de faire une quelconque utilisation clinique d'instruments médicaux non homologués en dehors du cadre d'un protocole clinique autorisé.
2.8.4 Rapport d'essai expérimental
Après la clôture de l'essai expérimental, le fabricant ou l'importateur devrait remettre un rapport de l'étude à Santé Canada, même si l'essai s'est achevé prématurément.
- Le rapport devrait indiquer le nom de l'instrument, décrire la méthodologie et la conception de l'étude et mentionner tout écart par rapport au protocole, de même que présenter une analyse, ainsi que des statistiques et une évaluation critique des objectifs de l'investigation.
- Le rapport devrait tenir compte des données de chaque emplacement de l'étude et sur tous les sujets. Aucun sujet ne doit pouvoir être identifié, que ce soit dans le rapport d'étude ou les résultats publiés.
- Le cas échéant, le rapport de l'essai expérimental devrait être soumis à l'examen et aux commentaires du chercheur coordonnateur et de tous les chercheurs principaux. Le promoteur de l'étude doit tenir des registres confirmant que le rapport a été remis en vue de son examen. Si un examinateur n'est pas d'accord avec une partie ou l'ensemble du rapport, ses commentaires doivent être consignés et communiqués aux autres chercheurs principaux.
- Le promoteur de l'étude et le chercheur coordonnateur doivent signer le rapport pour montrer qu'ils sont d'accord avec son contenu. En l'absence d'un chercheur coordonnateur, la signature des chercheurs principaux doit être obtenue.
- Le rapport doit être remis au CER et aux autres autorités réglementaires.
2.8.5 Diffusion publique des renseignements concernant les AEE
Sur demande, Santé Canada diffusera publiquement certains renseignements sur les AEE accordées. Le but premier est d'assurer la transparence et d'améliorer l'accès du public aux renseignements portant sur les études canadiennes menées à l'aide d'instruments médicaux. Ainsi, pour les AEE accordées depuis le 14 novembre 2013, les renseignements suivants seront diffusés :
- titre du protocole;
- nom de l'instrument;
- problème médical;
- population visée par l'étude;
- date de l'autorisation;
- date de clôture de l'étude;
- nom du fabricant ou de l'importateur.
Pour tous les autres renseignements, les demandeurs seront invités à s'adresser au fabricant ou à l'importateur ou dirigés vers leur professionnel de la santé.
Annexe 1 - Adresses Pertinentes
Instruments médicaux
Bureau des matériels médicaux
11, avenue Holland, Tour A, 2e étage
Indice de l'adresse 3002A
Ottawa (Ontario)
Canada K1A 0K9
Demandes de renseignements généraux
Courriel : enquetes_bmm@hc-sc.gc.ca
Téléphone : 613-957-4786
Télécopieur : 613-957-6345
Médicaments pharmaceutiques
Bureau des essais cliniques
Direction des produits thérapeutiques
Holland Cross, Tour B, 5e étage
Indice de l'adresse 3105A
1600, rue Scott
Ottawa (Ontario)
Canada K1A 0K9
Demandes de renseignements généraux
Courriel : BEC_enquetes@hc-sc.gc.ca
Téléphone : 613-941-2132
Télécopieur : 613-946-7996
Annexe 2 - Documents Utiles
Les documents suivants peuvent se révéler utiles au moment de rédiger la demande.
- Avis - Adoption de la ligne directrice de l'ICH, thème E11 : Recherche clinique sur les produits médicinaux dans la population pédiatrique [2003-12-17],
- Avis - ICH thème E6 : Bonnes pratiques cliniques : directives consolidées [1997-09-19],
- Avis - ICH thème E9 : Principes statistiques pour les essais cliniques [2003-02-10],
- Avis - Divulgation de renseignements au sujet des demandes d'essais expérimentaux visant des instruments médicaux autorisés par Santé Canada [2013-11-15],
- Considérations générales relatives aux études cliniques - ICH thème E8 [1998-05-01],
- Demande d'autorisation pour la vente d'un instrument médical pour essai expérimental [1999-02-22],
- Directive sur l'interprétation d'une modification importante d'un instrument médical [2011-01-20],
- Document d'orientation : Considérations relatives à l'inclusion des femmes dans les essais cliniques et à l'analyse des données selon le sexe [2013-05-29],
- Élaboration d'une demande d'essai expérimental - Instruments diagnostiques in vitro [1999-02-22],
- EPTC 2 (2014) - dernière édition de l'Énoncé de politique des trois Conseils : Éthique de la recherche avec des êtres humains,
- Formulaire de déclaration obligatoire des incidents liés aux matériels médicaux à l'intention de l'industrie [octobre 2011],
- Gestion des demandes d'homologation d'instruments médicaux et d'autorisation d'essais expérimentaux [2001-04-06],
- Index de mots-clés pour aider les fabricants à vérifier la classification des matériels médicaux [2006-09-14],
- Ligne directrice - E10 : Choix d'un groupe témoin et questions connexes dans le cadre des essais cliniques [2011-07-04],
- Ligne directrice : Préparation des activités de réglementation en format « Électronique autre que le format eCTD » [2016/10/31],
- Ligne directrice sur la déclaration obligatoire des incidents liés aux matériels médicaux [2011-10-03],
- Ligne directrice : La reconnaissance et l'utilisation des normes en vertu du Règlement sur les instruments médicaux [2006-09-22],
- Ligne directrice : Orientation pour le système de classification fondé sur le risque des instruments diagnostiques in vitro [2016-09-23],
- Lignes directrices sur la conformité et l'application de la loi à l'égard des instruments médicaux (GUI-0073) [2015-06-12],
- Normes applicables aux instruments médicaux [2016-10-26],
- Politique - Produits mixtes : Médicaments et matériels médicaux [2005-11-30],
- Politique sur les retraits/rappels (POL-0016) [2015-03-05],
Annexe 3 - Déterminer s'il faut demander une AEE
Figure 1
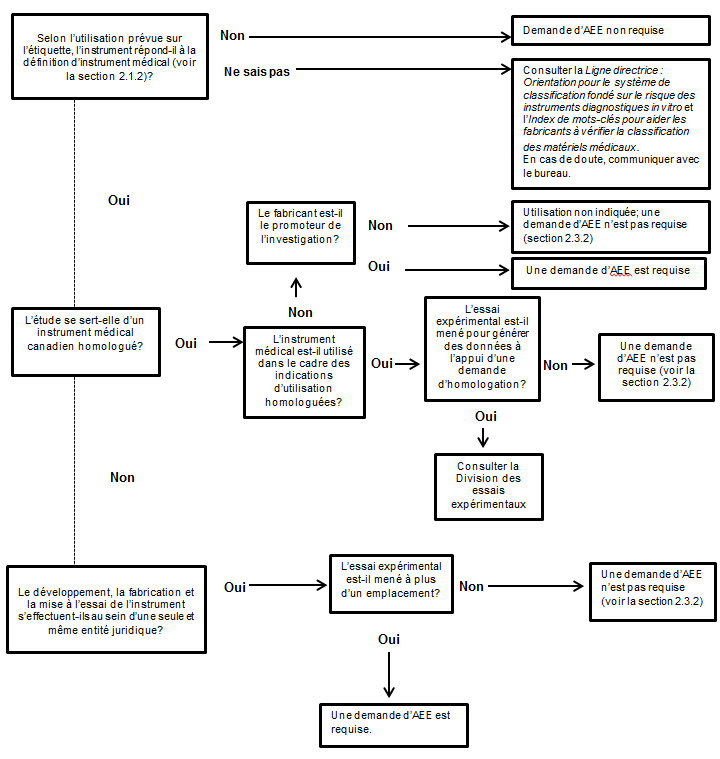 Figure 1 - Équivalent textuel
Figure 1 - Équivalent textuel
Selon l'utilisation prévue dans l'étiquette, l'instrument répond-il à la définition d'un instrument médical (voir la section 2.1.2 de la ligne directrice de demande d'AEE)? Si non, une demande d'AEE n'est pas requise. Si le fabricant ne le sait pas, il devrait consulter la Ligne directrice : Orientation pour le système de classification fondé sur le risque des instruments diagnostiques in vitro et l'Index de mots-clés pour aider les fabricants à vérifier la classification des matériels médicaux. En cas de doute, le fabricant doit communiquer avec le bureau.
Selon l'utilisation prévue dans l'étiquette, si l'instrument répond à la définition d'un instrument médical, l'étude se sert-elle d'un instrument médical canadien homologué?
Si oui, l'instrument médical est-il utilisé dans le cadre des indications d'utilisation homologuées? Si l'instrument médical est utilisé dans le cadre des indications d'utilisation homologuées, l'essai expérimental est-il mené pour générer des données à l'appui d'une demande d'homologation? Si non, une demande d'AEE n'est pas requise (voir la section 2.3.2 de la ligne directrice de demande d'AEE). Si l'essai expérimental est mené pour générer des données à l'appui d'une demande d'homologation, le fabricant doit consulter la division des essais expérimentaux.
Si l'instrument médical n'est pas utilisé dans le cadre des indications d'utilisation homologuées, le fabricant est-il le promoteur de l'investigation? Si non, cette activité est considérée comme une utilisation non indiquée et une demande d'AEE n'est pas requise (voir la section 2.3.2 de la ligne directrice de demande d'AEE). Si oui, une demande d'ITA est requise.
Si l'étude ne se sert pas d'un instrument médical canadien homologué, le développement, la fabrication et la mise à l'essai de l'instrument s'effectuent-ils au sein d'une seule et même entité juridique? Si oui, l'essai expérimental est-il mené à plus d'un emplacement? Si non, une demande d'AEE n'est pas requise (voir la section 2.3.2 de la ligne directrice de demande d'AEE). Si oui, une demande d'AEE est requise.
Annexe 4 - Format d'une demande d'aee
Ce qui suit est le format « électronique autre que le format eCTD » de toute demande d'AEE, tel que précisé dans l'Avis - Demandes d'autorisation d'essais expérimentaux (AEE) pour les instruments médicaux, en format « électronique autre que le format eCTD ». Certaines sections ne s'appliquent pas aux instruments de classe II. Les dossiers vides doivent être supprimés avant de soumettre la demande à Santé Canada. Tout document à l'appui qui ne relève d'aucun dossier préétabli peut être joint en annexe, mais il doit être clairement mentionné dans le Sommaire.
01- Lettre d'accompagnement et sommaire
02- Table des matières
03- Formulaire de demande
04- Correspondance préalable à la demande
05- Introduction
05.01 Identification du fabricant ou de l'importateur
05.02 Identification de l'instrument
05.03 Description de l'instrument
05.04 Philosophie de la conception
05.05 Indications d'utilisation, utilisation prévue, contre-indications
05.06 Étiquette d'instrument
05.07 Historique de la commercialisation
06- Mesures d'évaluation et de réduction des risques
06.01 Évaluation des risques
06.02 Précédentes études
06.03 Traitements de remplacement
06.04 Précautions
07- Renseignements institutionnels
07.01 Nom des chercheurs
07.02 Curriculum vitae
07.03 Ententes avec les chercheurs
07.04 Nom et adresse des institutions
07.05 Approbation du Conseil d'éthique de la recherche
08- Documents d'étude
08.01 Protocole
08.02 Brochure du chercheur
08.03 Formulaire de consentement éclairé
09- Annexes
Annexe 5 - Foire aux questions
Demandes d'autorisation d'essai expérimental (AEE)
1. Comment puis-je présenter une demande d'AEE?
Les demandes ne peuvent être présentées qu'en format électronique. Elles peuvent être envoyées par courriel à l'adresse homologation_instruments@hc-sc.gc.ca. Si le fichier de la demande est trop volumineux pour être envoyé par courriel, il peut être envoyé par la poste (voir l'annexe 1) sur un CD ou un DVD. La demande doit être présentée dans le format précisé dans la Ligne directrice : Préparation des activités de réglementation en format « Électronique autre que le format eCTD » (voir l'annexe 2 et la section 2.3.4 de la ligne directrice sur les demandes d'AEE pour de plus amples détails).
2. Un chercheur peut-il demander une AEE?
Les demandes d'AEE doivent être présentées par le fabricant ou l'importateur de l'instrument; cependant, ce dernier peut déléguer ce pouvoir à un chercheur, qui agira à titre de personne-ressource réglementaire. Lorsque l'importateur ou le chercheur présente la demande, le fabricant doit signer le formulaire de demande (voir la section 2.3.3 de la ligne directrice sur les demandes d'AEE pour de plus amples renseignements).
3. Une AEE est-elle nécessaire si un médecin à l'intention d'utiliser un instrument non homologué qu'il a inventé dans une étude pour traiter ses propres patients?
Les instruments médicaux qui sont fabriqués et utilisés dans le cadre d'un essai clinique au sein d'une seule et même entité juridique ne sont pas assujettis aux exigences du Règlement sur les instruments médicaux. Cette exception ne s'applique que dans les cas où l'essai est mené dans un seul établissement. Lorsque le médecin est le fabricant légal de l'instrument, tel que défini dans le Règlement, que l'instrument est utilisé dans l'établissement où exerce le médecin et que ce dernier est la seule personne responsable de mener l'essai expérimental, il n'y a aucune vente aux termes de la Loi sur les aliments et drogues. Les exigences du Règlement sur les instruments médicaux ne s'appliquent donc pas à ces situations, et une AEE n'est pas nécessaire (voir la section 2.3.2 de la ligne directrice sur les demandes d'AEE pour de plus amples renseignements).
4. Comment puis-je apporter des modifications à mon AEE?
Toute modification à l'instrument et/ou au protocole qui ne change pas les objectifs originaux de l'étude et qui ne devrait pas altérer le profil de risque ou compromettre l'intégrité des données recueillies peut faire l'objet d'une demande de révision de l'AEE. Des preuves à l'appui des exigences visées aux articles 81 et 86 du Règlement sur les instruments médicaux doivent être fournies pour toutes les exigences qui sont touchées par les modifications proposées. Les demandes doivent être présentées par voie électronique, conformément à la politique en vigueur concernant la présentation des demandes d'AEE à l'adresse : homologation_instruments@hc-sc.gc.ca ou par la poste à l'adresse postale du Bureau (voir l'annexe 1 et la section 2.5 de la ligne directrice sur les demandes d'AEE pour de plus amples renseignements).
5. Une AEE est-elle nécessaire pour un instrument de la classe I?
Pour un instrument de la classe I, il n'est pas nécessaire de demander une autorisation pour mener un essai expérimental, mais le fabricant doit garder un registre des renseignements requis à l'article 81 du Règlement sur les instruments médicaux, et l'étude doit être menée en respectant les lignes directrices de bonne pratique clinique (p. ex. la norme ISO 14155, Bonnes pratiques cliniques, etc.).
6. Ai-je besoin de l'approbation d'un comité d'éthique de la recherche (CER) avant de recevoir une AEE?
Santé Canada n'exige pas la preuve de l'approbation d'un CER avant d'accorder une AEE pour les instruments de la classe II. Cependant, les politiques institutionnelles exigent l'approbation d'un CER avant d'entreprendre l'étude d'un instrument de la classe II.
La preuve de l'approbation d'un CER est nécessaire pour qu'une AEE soit accordée lorsqu'il agit d'un instrument des classes III et IV. L'approbation conditionnelle (qui déclare que l'autorisation de Santé Canada est nécessaire) d'un CER est acceptable. Une demande d'AEE peut être présentée à Santé Canada en attendant l'approbation du CER. L'examen de la demande sera entrepris, mais aucune AEE ne sera accordée avant que la preuve de l'approbation d'un CER ne soit reçue.
7. Pendant combien de temps après la fin de l'étude dois-je conserver les documents?
L'article 55 du Règlement sur les instruments médicaux exige que le fabricant, l'importateur et le distributeur conservent leur registre de distribution concernant l'instrument médical pendant la plus longue des périodes suivantes :
- la durée de vie utile projetée de l'instrument médical,
- deux ans suivant la date d'expédition de l'instrument.
Le Règlement ne précise aucune période de conservation pour les établissements cliniques. Cette période devrait correspondre aux politiques institutionnelles et aux règles des organismes de réglementation provinciaux concernant la pratique de la médecine.
8. Dois-je déclarer un incident qui survient durant l'étude?
Le chercheur compétent est tenu de déclarer tout événement indésirable grave qui relève de la portée de l'article 59 du Règlement sur les instruments médicaux à Santé Canada, ainsi qu'au fabricant ou à l'importateur dans les 72 heures suivant sa constatation. Le fabricant ou l'importateur d'un instrument médical est tenu de signaler à Santé Canada les incidents qui relèvent de la portée de l'article 59 du Règlement dans les 10 jours, si l'incident a entraîné la mort ou une détérioration grave de l'état de santé d'un patient, utilisateur ou autre personne, ou dans les 30 jours si l'incident n'a pas entraîné la mort ou une détérioration grave de l'état de santé d'un patient, utilisateur ou autre personne, mais qui serait susceptible de le faire s'il se reproduisait. De plus amples renseignements sur les exigences de déclaration sont précisés aux articles 59 à 62 du Règlement et à la section 2.4.5 de la ligne directrice sur les demandes d'AEE.
9. Si j'ai déjà obtenu une AEE pour un instrument réutilisable, puis-je continuer de l'utiliser après l'essai expérimental ou dans le cadre d'une nouvelle investigation?
L'AEE accordée concerne précisément l'instrument, le protocole et le nombre de patients mentionnés sur l'autorisation. Une fois l'étude terminée, l'instrument devrait être retourné au fabricant ou à l'importateur, mis hors service ou éliminé de manière appropriée. Si le chercheur compétent a l'intention d'utiliser l'instrument dans le cadre d'un autre protocole d'étude, une nouvelle demande d'AEE doit être présentée par le fabricant ou l'importateur.
Notes de bas de page
- Note de bas de page 1
Les termes « investigation clinique », « essai expérimental », « essai clinique » et « étude clinique » sont synonymes.
- Note de bas de page 2
Dans le cas des instruments médicaux, les demandes doivent être présentées par leur fabricant.
- Note de bas de page 3
Dictionnaire de français Larousse.
- Note de bas de page 4
Les termes « comité d'éthique », « conseil d'éthique de la recherche », « comité d'éthique indépendant » ou « comité de révision institutionnel » sont synonymes.
- Note de bas de page 5
Les termes « site d'investigation » et « emplacement d'étude » sont synonymes.
- Note de bas de page 6
Les termes « instrument médical sous investigation » et « instrument sous investigation » sont utilisés indifféremment.
- Note de bas de page 7
Malgré l'absence de définition dans le Règlement sur les instruments médicaux, un membre de l'équipe du site d'investigation peut également être désigné par « chercheur secondaire » ou « co-chercheur ».
- Note de bas de page 8
Le terme « vente », comme défini à l'article 2 de la Loi sur les aliments et drogues, ne se limite pas à la vente commerciale ou monétaire et englobe également toute transaction sans distinction, notamment la remise sans frais d'instruments par le fabricant au chercheur.
- Note de bas de page 9
Une hospitalisation prévue en raison d'une affection préexistante ou une procédure requise par le plan d'investigation clinique, sans détérioration grave de la santé, n'est pas considérée comme un événement indésirable grave.
- Note de bas de page 10
Il faut consulter la Directive sur l'interprétation d'une modification importante d'un instrument médical pour connaître les principes généraux de la détermination de ce qui constitue une modification importante.
- Note de bas de page 11
Le Règlement sur les instruments médicaux est silencieux à ce sujet, mais le Règlement sur les aliments et drogues stipule que les registres liés aux études cliniques des médicaments doivent être conservés pendant 25 ans.
- Note de bas de page 12
Si cette version du protocole d'étude ne s'applique pas à l'emplacement canadien, il n'est pas nécessaire d'obtenir une autorisation de Santé Canada dans ce cas.