
Operational procedures for the Generic Medicines Work-Sharing Initiative (GMWSI)
Download the alternative format (PDF format, 610 KB, 9 pages)
Document change log
| Version | Description of Change | Author | Effective Date |
|---|---|---|---|
| v 1.0 | Original publication | ACSS Generic Medicines WG | 2016-05-20 |
| v 2.0 | Updated following the first application with the GMWST | ACSS Generic Medicines WG | 2017-10-05 |
| v 3.0 | Updated to include process enhancements based on experiences with the work sharing model, recent developments (e.g., addition of UK's MHRA to the Access Consortium) and feedback received from stakeholders. | Access Generic Medicines WG | 2022-10-30 |
Également disponible en français sous le titre :
Consortium Access : Procédures opérationnelles pour l'Initiative de partage du travail concernant les médicaments génériques (IPTMG)
This publication may be reproduced for personal or internal use only without permission provided the source is fully acknowledged.
Cat.: H164-344/2022E-PDF
ISBN: 978-0-660-45703-1
Pub.: 220461
Table of Contents
- Introduction
- Scope
- Application considerations
- Operational approach
- Round 1 assessment
- Round 2 assessment
- Round 3 assessment (if applicable)
- National steps
- Related links
Introduction
This document outlines operational procedures and recommendations for planning and implementing the Generic Medicines Work-sharing Initiative (GMWSI) for the regulatory agencies within the Access Consortium. The agencies are:
- Therapeutic Goods Administration (TGA), Australia
- Health Canada (HC), Canada
- Health Sciences Authority (HSA), Singapore
- Swissmedic (SMC), Switzerland
- Medicines and Healthcare products Regulatory Agency (MHRA), United Kingdom
The initiative is based on Europe's decentralized procedure, where 1 agency acts as a reference regulatory agency (RRA) and will evaluate Modules 2 to 5. Each participating agency acts as a concerned regulatory agency (CRA) and, with the RRA, evaluates their respective module 1. The CRAs peer review the assessment reports (ARs) and the proposed List of Questions (LoQ) provided by the RRA on Modules 2 to 5, consult the modules where necessary and provide supplementary comments as needed.
Each agency makes its own decision based on the recommendations made in the ARs. If, during the process, the participating agencies are unable to reconcile issues with the data, the agencies may seek additional information and undertake further independent review.
Scope
To be considered for this initiative, the proposed product should be regarded as a generic product by all the participating agencies. All pharmaceutical (dosage) forms are eligible.
Application considerations
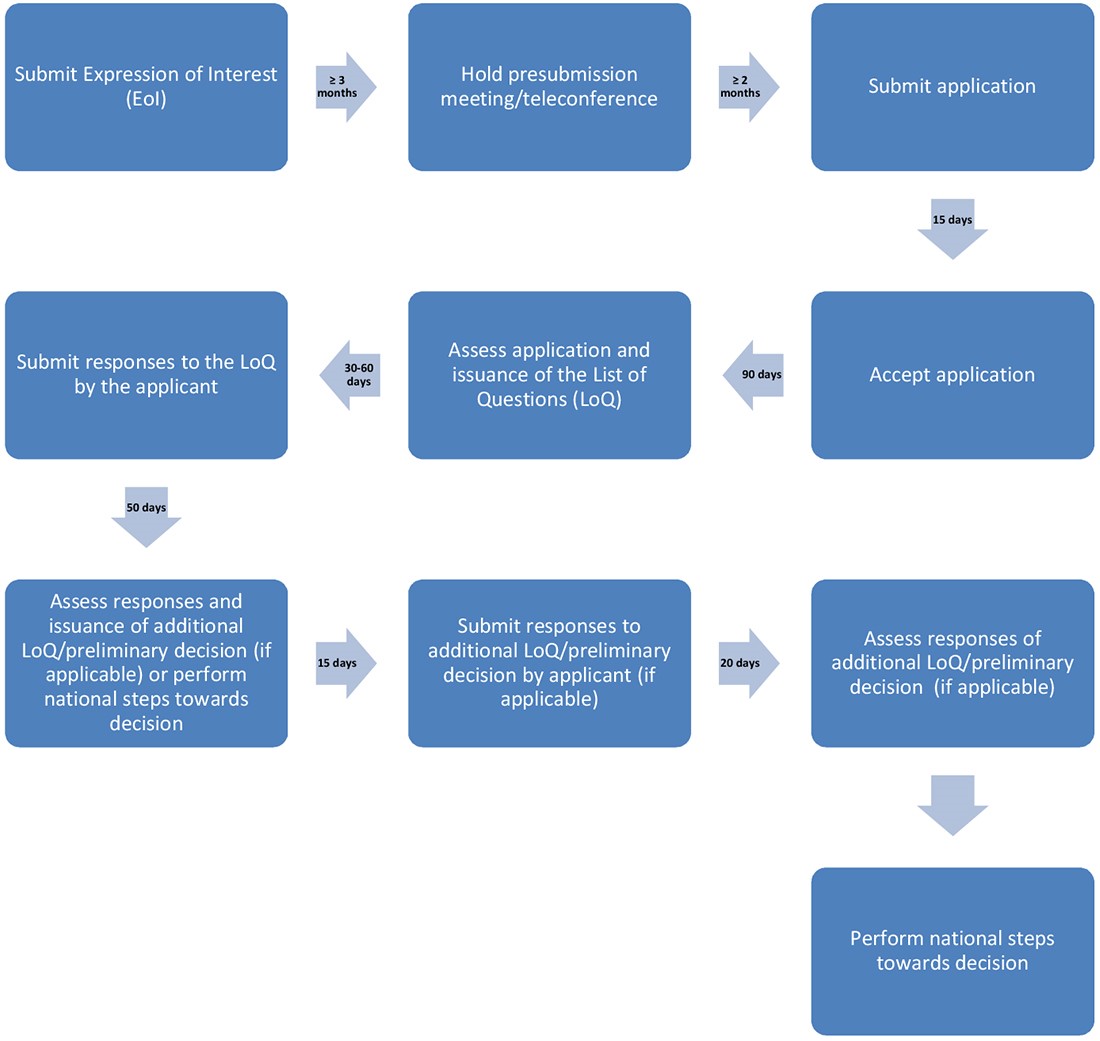
An applicant wishing to participate in this innovative work-sharing initiative must submit an Expression of Interest (EoI) form to each agency proposed for this initiative. The applicant should submit this form at least 3 months before the planned submission date.
Applications for this initiative should be submitted at the same time to at least 2 of the Access Consortium members.
The applicant should submit the same Modules 2 to 5 to all of the agencies involved in the initiative. However, there may be minor differences in the applications submitted to the various agencies participating in the initiative. For example, there may be differences in packaging formats such as bottles versus unit dose blisters. Major differences between applications may make the work-sharing process more complicated, which may delay the assessment process. For example, submitting multiple studies using different comparator products would be considered a major difference.
If there are minor differences between datasets, the applicant should provide a 'Summary of Differences' table. This table is part of the EoI form. The table should outline the differences in the quality and bioequivalence study information provided to each participating agency. The Access agencies will discuss these differences and determine if the application is suitable for GMWSI.
Please note that all Access agencies accept the use of foreign comparator products in bioequivalence studies. Therefore, for submissions requiring clinical equivalence (whether pharmacokinetic bioequivalence or therapeutic equivalence), it may be possible to use a single comparator product, along with country-specific justifications for using the foreign comparator product. Please see the related links section for references.
Also note that Module 1 is specific to each country. Thus, it will continue to be different for the applications filed in the different Access jurisdictions (as per national requirements).
Although an applicant may propose a preferred RRA, the Access agencies will ultimately determine the RRA and CRAs for any submission. The agencies base their decision on factors such as the Access Consortium's operational needs.
In general, 1 agency (the RRA) will perform the evaluation and the other participating agencies will act as CRAs. However, in some cases, the application may be split up, with multiple agencies doing the initial assessment. The roles of each participating agency will be determined in line with operational requirements.
In the EoI, the applicant should indicate their preferred timeframe for submitting their responses to the LoQs provided by the agencies. The timeframe should be either 30 or 60 calendar days.
Applications should fully address the requirements of all jurisdictions to be included in the procedure. Applicants should also acknowledge that they will need to work collaboratively with the agencies. While a single application that covers broader issues for more than 1 agency may seem more onerous, it will ultimately reduce the overall regulatory burden.
Operational approach
The process needs to work concurrently within the regulatory systems of the participating agencies. This section outlines the steps and issues that need to be considered when implementing the procedure.
All timelines/days are based on calendar days. If a milestone falls on a weekend or a national holiday, the milestone is the preceding business day. Please note that the following timeframes are target timeframes and may be adjusted depending on the complexity of the application.
Pre-submission meeting/teleconference (minimum 2 months in advance)
Once the participating agencies receive an EoI form, they will work together to discuss the application, its suitability for inclusion in the work-sharing initiative, the RRA and CRAs, and next steps.
A pre-submission meeting/teleconference between the applicant and their local Access agency, or all participating agencies if possible (may not be granted due to operational and resource challenges), is strongly recommended. This meeting is used to discuss the technical aspects of the submission, and to confirm the logistics and expectations related to requirements, timelines for assessment and the process. The meeting also gives agencies a chance to respond to any additional questions the applicant may have.
The teleconference should take place at least 2 months before the agreed-upon submission date of the application. The applicant must follow the usual procedures of their local agency when requesting a pre-submission meeting.
The applicant will be asked to provide their questions at least 2 weeks before the pre-submission teleconference. Within 2 weeks of this teleconference, the applicant must provide a record of the meeting, summarizing the points that have been agreed to.
Submitting application (less than 15 days)
Applications should be submitted to each participating agency at the same time or as agreed with the participating agencies. The process begins as soon as all agencies have received the applications. This is "Day -15" of the process.
If applicable, the Active Substance Master File/Drug Master File must be submitted to each participating agency before the application is filed, with the appropriate local forms.
Accepting application (15 days)
Once the participating agencies receive the application, the RRA and CRAs screen and validate the technical and administrative information. They check that their national legislation and data requirements (for example, application forms, user fees) have been met and that the application can be accepted for assessment.
The RRA and CRAs then inform the applicant if their application has been accepted for assessment. If accepted, the applicant is also given a summary of the target timeframes for each step in the process. The day of acceptance of the application for assessment by the RRA is "Day 0" of the process. The CRAs will make efforts to accept the application on the same day as the RRA.
Round 1 assessment
Initial assessment by the RRA (60 days)
The RRA evaluates Modules 2 to 5 and prepares an AR and an LoQ. At the same time, the RRA and CRAs evaluate their national Module 1 and prepare an LoQ for this module. The RRA then shares the AR and LOQ on Modules 2 to 5 with the CRAs.
While a consolidated LoQ is the preferred option, the RRA may instead send rolling questions to the local applicant to seek clarification during the assessment process. If applicable, the responses to these clarification questions have a short timeframe (for example, 5 days). The RRA then shares these responses with the CRAs.
Peer review by CRAs (25 days)
As part of the peer review process, the CRAs:
- conduct a peer review of the AR and LoQ
- consult the modules (as needed)
- share comments and additional questions on Modules 2 to 5 with the RRA
Finalising ARs and LoQs (5 days)
The RRA and CRAs discuss the LoQ and any additional questions. The RRA prepares the consolidated LoQ on Modules 2 to 5. The RRA and each CRA forward the consolidated LoQ, as well as their questions on Module 1 (includes questions on product information and labelling), to their local applicant.
Submitting responses to the LoQ by applicant (30 or 60 days)
The applicant prepares and sends the same responses to the LoQ on Modules 2 to 5 to the RRA and CRAs by way of the respective local applicants. At the same time, the local applicant sends responses to the Module 1 questions to their respective agencies.
As stated, the applicant should indicate in the EoI their preferred timeframe for submitting responses to the LoQ (either 30 or 60 days). However, the applicant will be able to respond any time after 30 days and before 60 days.
Round 2 assessment
Assessing responses to LoQ (30 days)
The RRA prepares an AR of the responses on the consolidated LoQ for Modules 2 to 5 and shares it with the CRAs. At the same time, the RRA and CRAs prepare an AR of the responses to their respective country-specific questions on Module 1.
Peer review by CRAs (15 days)
The CRAs conduct a peer review of the AR of the responses to Modules 2 to 5 and provide feedback. If necessary, the RRA prepares an additional LoQ, which each agency sends to the local applicant. The 15-day timeframe includes the time for peer review and for any coordination between the RRA and the CRAs.
Finalising ARs and additional LoQ (5 days)
If an additional LoQ (in general, this corresponds to the preliminary decision in Switzerland) is not necessary, each agency makes a final decision. The agencies also undertake the necessary administrative steps to finalise the process for their country.
Submitting responses to the additional LoQ (if applicable) (15 days)
The applicant prepares and sends responses to the additional LoQ (if applicable) to the RRA and CRAs.
Round 3 assessment (if applicable)
Assessing responses to the additional LoQ (15 days)
The RRA prepares an AR of the responses to the additional LoQ following the process described for round 2.
Peer review of responses by the CRAs, finalising the ARs and additional LoQ (if applicable) (5 days)
The CRAs conduct a peer review of the responses and provide feedback in order for the RRA to finalise the AR.
National steps
Each agency makes a final decision (or seeks further clarification on issues separately before making a final decision) and undertakes the necessary administrative steps to finalise the process nationally. Depending on each agency's assessment outcome, an authorisation letter or additional questions is issued.
These communications may not necessarily happen at the same time.
Total maximum elapsed timeframe from when the application is accepted (day 0) to the start of the national steps:
- 170 to 200 calendar days (including the applicant's response time and if only 1 list of questions is required) or
- 205 to 235 calendar days (if an additional list of questions is required)
Any further questions about this initiative can be directed to the local regulatory authority:
- Australia: PMABinternationalevaluations@health.gov.au
- Canada: collaboration@hc-sc.gc.ca
- Singapore: HSA_TP_Enquiry@hsa.gov.sg
- Switzerland: Networking@swissmedic.ch
- United Kingdom: Access-MHRA@mhra.gov.uk
Communications by e-mail should include "Access Consortium - GMWSI" in the subject line.
Related links
- A survey of the regulatory requirements for the acceptance of foreign comparator products by participating regulators and organizations of the International Generic Drug Regulators Programme
- Biopharmaceutic studies (TGA guidance)
- Comparator products in bioequivalence/therapeutic equivalence studies (MHRA guidance)
- Guidance document: Use of a foreign-sourced reference product as a Canadian reference product (HC guidance)
- Guidance on therapeutic product registration in Singapore: Product interchangeability and biowaiver request for chemical generic drugs application (HSA guidance)
- Guidance document: Authorisation of human medicinal product with known active pharmaceutical substances HMV4 (SMC guidance)
Page details
- Date modified: