
ARCHIVED - Good Manufacturing Practices (GMP) Guidelines - 2009 Edition (GUI-0001)
Health Products and Food Branch Inspectorate
Cover Letter
Supersedes:
2002 Edition, Version 2
Date issued:
May 8, 2009
Date of implementation:
November 8, 2009
Disclaimer
This document does not constitute part of the Food and Drugs Act (Act) or the Food and Drugs Regulations (Regulations) and in the event of any inconsistency or conflict between that Act or Regulations and this document, the Act or the Regulations take precedence. This document is an administrative document that is intended to facilitate compliance by the regulated party with the Act, the Regulations and the applicable administrative policies. This document is not intended to provide legal advice regarding the interpretation of the Act or Regulations. If a regulated party has questions about their legal obligations or responsibilities under the Act or Regulations, they should seek the advice of legal counsel.
Ce document est aussi disponible en français.
Table of Contents
- 1.0 Introduction
- 2.0 Purpose
- 3.0 Scope
- 4.0 Quality Management
- 5.0 Regulation
- Appendix A Internationally Harmonized Requirements for Batch Certification
- Appendix A1 Content of the Fabricator’s/Manufacturer’s Batch Certificate for Drug/Medicinal Products Exported to Countries under the Scope of a Mutual Recognition Agreement (MRA)
- Appendix B
- Appendix C Annexes to the Current Edition of the Good Manufacturing Practices (GMP) Guidelines
- References
- Health Products and Food Branch Inspectorate - Operational Centres
1.0 Introduction
These guidelines on Good Manufacturing Practices (GMP) pertain to Division 2, Part C of the Food and Drug Regulations. The guidelines apply to pharmaceutical, radiopharmaceutical, biological, and veterinary drugs and were developed by Health Canada in consultation with stakeholders. These guidelines are designed to facilitate compliance by the regulated industry and to enhance consistency in the application of the regulatory requirements.
Division 1A, Part C of the Food and Drug Regulations defines activities for which GMP compliance is to be demonstrated prior to the issuance of a drug establishment licence. In addition to these guidelines, further guidance in specific areas is provided in Appendix C to this document or in separate documents. The guidance regarding the fabrication, packaging, labelling, testing, distribution, and importation of medical gases is described in the guideline "Good Manufacturing Practices for Medical Gases (GUI-0031)".
The content of this document should not be regarded as the only interpretation of the GMP Regulations, nor does it intend to cover every conceivable case. Alternative means of complying with these Regulations can be considered with the appropriate scientific justification. Different approaches may be called for as new technologies emerge.
The guidance given in this document has been written with a view to harmonize with GMP standards from other countries and with those of the World Health Organization (WHO), the Pharmaceutical Inspection Cooperation/Scheme (PIC/S), and the International Conference on Harmonisation (ICH).
This document takes into account the implementation of the current Mutual Recognition Agreements (MRA). The MRA establish mutual recognition of GMP compliance certification between Regulatory Authorities that are designated as equivalent. Exemptions from requirements under C.02.012 (2) and C.02.019 (1) and (2) are provided for importers of drugs where all activities (fabrication, packaging/labelling and testing) are carried out in MRA countries. All other regulatory requirements described in the Food and Drug Regulations apply.
The present edition of this document includes modified and/or new terminology, the incorporation of most GMP questions and answers, additional requirements such as annual product quality review, additional interpretations, and an updated table of requirements.
2.0 Purpose
To provide interpretive guidance for Part C, Division 2, of the Food and Drug Regulations. These guidelines are designed to facilitate compliance by the regulated industry and to enhance consistency in the application of the regulatory requirements.
3.0 Scope
The guidelines apply to pharmaceutical, radiopharmaceutical, biological, and veterinary drugs and were developed by Health Canada in consultation with stakeholders.
Chart 1.0: GMP Regulations Applicable to Licensable Activities
| 1. Premises | C.02.004 | X | X | X | X | X | |
| 2. Equipment | C.02.005 | X | X | X | |||
| 3. Personnel | C.02.006 | X | X | X | X | X | X |
| C.02.007 | X | X | |||||
| C.02.008 | X | X | |||||
| C.02.009 | X | * | |||||
| C.02.010 | X | * | |||||
| C.02.011 | X | X | X | X | |||
| C.02.012 | X | X | X | X | X | ||
| C.02.013 | X | X | X | X | X | ||
| C.02.014 | X | X | X | X | X | ||
| C.02.015 | X | X | X | X | X | X | |
| C.02.016 | X | X | * | ||||
| C.02.017 | X | X | * | ||||
| C.02.018 | X | X | X | X | * | ||
| C.02.019 | X | X | X | * | |||
| C.02.020 | X | X | X | X | X | ||
| C.02.021 | X | X | X | X | X | X | |
| C.02.022 | X | X | X | ||||
| C.02.023 | X | X | X | X | X | ||
| C.02.024 | X | X | X | X | X | ||
| C.02.025 | X | X | X | ||||
| C.02.026 | X | X | X | ||||
| C.02.027 | X | X | * | ||||
| C.02.028 | X | X | * | ||||
| 13. Sterile Products | C.02.029 | X | X | * |
* - Where applicable depending on the nature of the activities
4.0 Quality Management
4.1 Guiding Principle
The holder of an establishment licence, or any operation to which the requirements of Division 2 Part C of the Food and Drug Regulations are applicable, must ensure that the fabrication, packaging, labelling, distribution, testing and wholesaling of drugs comply with these requirements and the marketing authorization, and do not place consumers at risk due to inadequate safety and quality.
The attainment of this quality objective is the responsibility of senior management and requires the participation and commitment of personnel in many different departments and at all levels within the establishment and its suppliers. To ensure compliance, there must be a comprehensively designed and correctly implemented quality management system that incorporates GMP and quality control. The system should be fully documented and its effectiveness monitored. All parts of the quality management system should be adequately resourced with qualified personnel, suitable premises, equipment, and facilities.
4.2 Relationship among Quality Elements
The basic concepts of quality assurance, GMP, and quality control are inter-related. They are described here in order to emphasize their relationships and their fundamental importance to the production and control of drugs.
4.2.1 Quality Assurance
Quality assurance is a wide-ranging concept that covers all matters that individually or collectively influence the quality of a drug. It is the total of the organized arrangements made with the objective of ensuring that drugs are of the quality required for their intended use. Quality assurance therefore incorporates GMP, along with other factors that are outside the scope of these guidelines.
A system of quality assurance appropriate for the fabrication, packaging, labelling, testing, distribution, importation, and wholesale of drugs should ensure that:
- Drugs are designed and developed in a way that takes into account the GMP requirements;
- Managerial responsibilities are clearly specified;
- Systems, facilities and procedures are adequate and qualified;
- Production and control operations are clearly specified;
- Analytical methods and critical processes are validated;
- Arrangements are made for the supply and use of the correct raw and packaging materials;
- All necessary control on intermediates, and any other in-process monitoring is carried out;
- Outsourced activities are subject to appropriate controls and meet GMP requirements;
- Fabrication, packaging/labelling, testing, distribution, importation, and wholesaling are performed in accordance with established procedures;
- Drugs are not sold or supplied before the quality control department has certified that each lot has been produced and controlled in accordance with the marketing authorization and of any other regulations relevant to the production, control and release of drugs;
- Satisfactory arrangements exist for ensuring that the drugs are stored, distributed, and subsequently handled in such a way that quality is maintained throughout their shelf life;
- The quality risk management system should ensure that:
- the evaluation of the risk to quality is based on scientific knowledge, experience with the process and ultimately links to the protection of the patient
- the level of effort, formality and documentation of the quality risk management process is commensurate with the level of risk.
- The effectiveness, applicability, and continuous improvement of the quality management system is ensured through regular management review and self-inspection;
- An annual product quality review of all drugs should be conducted with the objective of verifying the consistency of the existing process, the appropriateness of current specifications for both raw materials and finished product to highlight any trends and to identify product and process improvements.
4.2.2 Good Manufacturing Practices (GMP) for Drugs
GMP are the part of quality assurance that ensures that drugs are consistently produced and controlled in such a way to meet the quality standards appropriate to their intended use, as required by the marketing authorization.
GMP basic requirements are as follows:
- Manufacturing processes are clearly defined and controlled to ensure consistency and compliance with approved specifications;
- Critical steps of manufacturing processes and significant changes to the process are validated;
- All necessary key elements for GMP are provided, including the following:
- qualified and trained personnel,
- adequate premises and space,
- suitable equipment and services,
- correct materials, containers and labels,
- approved procedures and instructions,
- suitable storage and transport.
- Instructions and procedures are written in clear and unambiguous language;
- Operators are trained to carry out and document procedures;
- Records are made during manufacture that demonstrate that all the steps required by the defined procedures and instructions were in fact taken and that the quantity and quality of the drug was as expected. Deviations are investigated and documented;
- Records of fabrication, packaging, labelling, testing, distribution, importation, and wholesaling that enable the complete history of a lot to be traced are retained in a comprehensible and accessible form;
- Control of storage, handling, and transportation of the drugs minimizes any risk to their quality;
- A system is available for recalling of drugs from sale;
- Complaints about drugs are examined, the causes of quality defects are investigated, and appropriate measures are taken with respect to the defective drugs and to prevent recurrence
4.2.3 Quality Control
Quality control is the part of GMP that is concerned with sampling, specifications, testing, documentation, and release procedures. Quality control ensures that the necessary and relevant tests are carried out and that raw materials, packaging materials, and products are released for use or sale, only if their quality is satisfactory. Quality control is not confined to laboratory operations but must be incorporated into all activities and decisions concerning the quality of the product.
The basic requirements of quality control are as follows:
- Adequate facilities, trained personnel, and approved procedures are available for sampling, inspecting and testing of raw materials, packaging materials, intermediate bulk and finished products, and, where appropriate monitoring environmental conditions for GMP purposes;
- 1.1 Samples of raw materials, packaging materials, and intermediate, bulk, and finished products are taken according to procedures approved by the quality control department;
- 1.2 Test methods are validated;
- 1.3 Records demonstrate that all the required sampling, inspecting, and testing procedures were carried out, and any deviations are recorded and investigated;
- 1.4 Records are made of the results of the self-inspection program;
- 1.5 The procedures for product release include a review and evaluation of relevant production documentation and an assessment of deviations from specified procedures;
- 1.6 No drug is released for sale or supply prior to approval by the quality control department;
- 1.7 Sufficient samples of raw material and finished product are retained to permit future examination if necessary.
5.0 Regulation
In this Division,
- - "medical gas" means any gas or mixture of gases manufactured, sold, or represented for use as a drug; (gaz médical)
- - "packaging material" includes a label; (matériel d'emballage)
- - "quality control department" means a quality control department referred to in section C.02.013; (service du contrôle de la qualité)
- - "specifications" means a detailed description of a drug, the raw material used in a drug, or the packaging material for a drug and includes:
- (a) statement of all properties and qualities of the drug, raw material or packaging material that are relevant to the manufacture, packaging, and use of the drug, including the identity, potency, and purity of the drug, raw material, or packaging material,
- (b) a detailed description of the methods used for testing and examining the drug, raw material, or packaging material, and
- (c) a statement of tolerances for the properties and qualities of the drug, raw material, or packaging material. (spécifications)
C.02.002.1
This Division does not apply to fabricating, packaging/labelling, testing, storing and importing of antimicrobial agents.
Sale
No distributor referred to in paragraph C.01A.003(b) and no importer shall sell a drug unless it has been fabricated, packaged/labelled, tested, and stored in accordance with the requirements of this Division.
Premises
Regulation
The premises in which a lot or batch of a drug is fabricated or packaged/labelled shall be designed, constructed and maintained in a manner that
- (a) permits the operations therein to be performed under clean, sanitary and orderly conditions;
- (b) permits the effective cleaning of all surfaces therein; and
- (c) prevents the contamination of the drug and the addition of extraneous material to the drug.
Rationale
The pharmaceutical establishment should be designed and constructed in a manner that permits cleanliness and orderliness while preventing contamination. Regular maintenance is required to prevent deterioration of the premises. The ultimate objective of all endeavours is product quality.
Interpretation
- Buildings in which drugs are fabricated or packaged are located in an environment that, when considered together with measures being taken to protect the manufacturing processes, presents a minimal risk of causing any contamination of materials or drugs.
- The premises are designed, constructed, and maintained such that they prevent the entry of pests into the building and also prevent the migration of extraneous material from the outside into the building and from one area to another.
- 2.1 Doors, windows, walls, ceilings, and floors are such that no holes or cracks are evident (other than those intended by design).
- 2.2 Doors giving direct access to the exterior from manufacturing and packaging areas are used for emergency purposes only. These doors are adequately sealed. Receiving and shipping area(s) do not allow direct access to production areas.
- 2.3 Production areas are segregated from all non-production areas. Individual manufacturing, packaging, and testing areas are clearly defined and if necessary segregated. Areas where biological, microbiological or radioisotope testing is carried out require special design and containment considerations.
- 2.4 Laboratory animals quarters are segregated.
- 2.5 Engineering, boiler rooms, generators, etc. are isolated from production areas.
- In all areas where raw materials, primary packaging materials, in-process drugs, or drugs are exposed, the following considerations apply to the extent necessary to prevent contamination.
- 3.1 Floors, walls, and ceilings permit cleaning. Brick, cement blocks, and other porous materials are sealed. Surface materials that shed particles are avoided.
- 3.2 Floors, walls, ceilings, and other surfaces are hard, smooth and free of sharp corners where extraneous material can collect.
- 3.3 Joints between walls, ceilings and floors are sealed.
- 3.4 Pipes, light fittings, ventilation points and other services do not create surfaces that cannot be cleaned.
- 3.5 Floor drains are screened and trapped.
- 3.6 Air quality is maintained through dust control, monitoring of pressure differentials between production areas and periodic verification and replacement of air filters. The air handling system is well defined, taking into consideration airflow volume, direction, and velocity. Air handling systems are subject to periodic verification to ensure compliance with their design specifications. Records are kept.
- Temperature and humidity are controlled to the extent necessary to safeguard materials.
- Rest, change, wash-up, and toilet facilities are well separated from production areas and are sufficiently spacious, well ventilated, and of a type that permits good sanitary practices.
- Premises layout is designed to avoid mix-ups and generally optimize the flow of personnel and materials.
- 6.1 There is sufficient space for receiving and all production activities.
- 6.2 Working spaces allow the orderly and logical placement of equipment (including parts and tools) and materials.
- 6.3 Where physical quarantine areas are used, they are well marked, with access restricted to designated personnel. Where electronic quarantine is used, electronic access is restricted to designated personnel.
- 6.4 A separate sampling area is provided for raw materials. If sampling is performed in the storage area, it is conducted in such a way as to prevent contamination or cross-contamination.
- 6.5 Working areas are well lit.
- Utilities and support systems (e.g., HVAC, dust collection, and supplies of purified water, steam, compressed air, nitrogen, etc.) for buildings in which drugs are fabricated or packaged/labelled are qualified and are subject to periodic verification. Further guidance is provided in Health Canadas document entitled "Validation Guidelines for Pharmaceutical Dosage Forms (GUI-0029)".
- Outlets for liquids and gases used in the production of drugs are clearly identified as to their content.
- Premises are maintained in a good state of repair. Repair and maintenance operations do not affect drug quality.
- Where necessary, separate rooms are provided and maintained to protect equipment and associated control systems sensitive to vibration, electrical interference, and contact with excessive moisture or other external factors.
- Fabricators and packagers must demonstrate that the premises are designed in such a manner that the risk of cross-contamination between products is minimized.
- 11.1 Campaign production can be accepted where, on a product by product basis, proper justification is provided, validation is conducted and rigorous validated controls and monitoring are in place and demonstrate the minimization of any risk of cross-contamination.
- 11.2 Self-contained facilities are required for:
- 11.2.1 certain classes of highly sensitizing drugs such as penicillins and cephalosporins.
- 11.2.2 other classes of highly potent drugs such as potent steroids, cytotoxics, or potentially pathogenic drugs (e.g., live vaccines), for which validated cleaning or inactivation procedures cannot be established (e.g., the acceptable level of residue is below the limit of detection by the best available analytical methods).
- 11.3 For the types of products listed in interpretations 11.2.1 and 11.2.2, external contamination with drug product residues of the final container and primary packaging does not exceed established limits.
- 11.3.1 Storage in common areas is allowed once the products are enclosed in their immediate final containers and controls are in place to minimize risks of cross-contamination.
- 11.4 No production activities of highly toxic non-pharmaceutical materials, such as pesticides and herbicides, are conducted in premises used for the production of drugs.
Equipment
Regulation
The equipment with which a lot or batch of a drug is fabricated, packaged/labelled or tested shall be designed, constructed, maintained, operated, and arranged in a manner that
- (a) permits the effective cleaning of its surfaces;
- (b) prevents the contamination of the drug and the addition of extraneous material to the drug; and
- (c) permits it to function in accordance with its intended use.
Rationale
The purpose of these requirements is to prevent the contamination of drugs by other drugs, by dust, and by foreign materials such as rust, lubricant and particles coming from the equipment. Contamination problems may arise from poor maintenance, the misuse of equipment, exceeding the capacity of the equipment and the use of worn-out equipment. Equipment arranged in an orderly manner permits cleaning of adjacent areas and does not interfere with other processing operations. It also minimizes the circulation of personnel and optimizes the flow of materials. The fabrication of drugs of consistent quality requires that equipment perform in accordance with its intended use.
Interpretation
- The design, construction and location of equipment permit cleaning, sanitizing, and inspection of the equipment.
- 1.1 Equipment parts that come in contact with raw materials, in-process intermediates or drugs are accessible to cleaning or are removable.
- 1.2 Tanks used in processing liquids and ointments are equipped with fittings that can be dismantled and cleaned. Validated Clean-In-Place (CIP) equipment can be dismantled for periodic verification.
- 1.3 Filter assemblies are designed for easy dismantling.
- 1.4 Equipment is located at a sufficient distance from other equipment and walls to permit cleaning of the equipment and adjacent area.
- 1.5 The base of immovable equipment is adequately sealed along points of contact with the floor.
- 1.6 Equipment is kept clean, dry and protected from contamination when stored.
- equipment does not add extraneous material to the drug.
- 2.1 Surfaces that come in contact with raw materials, in-process intermediates or drugs are smooth and are made of material that is non-toxic, corrosion resistant, non-reactive to the drug being fabricated or packaged and capable of withstanding repeated cleaning or sanitizing.
- 2.2 The design is such that the possibility of a lubricant or other maintenance material contaminating the drug is minimized.
- 2.3 Equipment made of material that is prone to shed particles or to harbour microorganisms does not come in contact with or contaminate raw materials, in-process drugs or drugs.
- 2.4 Chain drives and transmission gears are enclosed or properly covered.
- 2.5 Tanks, hoppers and other similar fabricating equipment are equipped with covers.
- Equipment is operated in a manner that prevents contamination.
- 3.1 Ovens, autoclaves and similar equipment contain only one raw material, in-process drug or drug at a time, unless precautions are taken to prevent contamination and mix-ups.
- 3.2 The location of equipment precludes contamination from extraneous materials.
- 3.3 The placement of equipment optimizes the flow of material and minimizes the movement of personnel.
- 3.4 Equipment is located so that production operations undertaken in a common area are compatible and cross-contamination between such operations is prevented.
- 3.5 Fixed pipework is clearly labelled to indicate the contents and, where applicable, the direction of flow.
- 3.6 Dedicated production equipment is provided where appropriate.
- 3.7 Water purification, storage, and distribution equipment is operated in a manner that will ensure a reliable source of water of the appropriate chemical and microbial purity.
- Equipment is maintained in a good state of repair.
- 4.1 Where a potential for contamination during fabrication or packaging of a drug exists, surfaces are free from cracks, peeling paint and other defects.
- 4.2 Gaskets are functional.
- 4.3 The use of temporary devices (e.g., tape) is avoided.
- 4.4 Equipment parts that come in contact with drugs are maintained in such a manner that drugs are fabricated or packaged within specifications. Equipment used for significant processing or testing operations is maintained in accordance with a written preventative maintenance program.. Maintenance records are kept.
- Equipment is designed, located, and maintained to serve its intended purpose.
- 5.1 Measuring devices are of an appropriate range, precision and accuracy. Such equipment is calibrated on a scheduled basis, and corresponding records are kept.
- 5.2 Equipment that is unsuitable for its intended use is removed from fabrication, packaging/labelling, and testing areas. When removal is not feasible unsuitable equipment is clearly labelled as such.
- 5.3 Equipment used during the critical steps of fabrication, packaging/labelling, and testing, including computerized systems, is subject to installation and operational qualification. Equipment qualification is documented. Further guidance is provided in Health Canadas document entitled "Validation Guidelines for Pharmaceutical Dosage Forms" and "Annex 11 to the Current Edition of the Good Manufacturing Practices Guidelines - Computerised Systems (GUI-0050)".
- 5.4 Equipment used for significant processing and testing operations is calibrated, inspected or checked in accordance with a written program. Records are kept.
- 5.5 For equipment used for significant processing or testing operations, usage logs are maintained. These logs should include identification of products, dates of operation, and downtime due to frequent or serious malfunctions or breakdowns. The information should be collected to facilitate the identification of negative performance trends.
Personnel
Regulation
Every lot or batch of a drug shall be fabricated, packaged/labelled, tested and stored under the supervision of personnel who, having regard to the duties and responsibilities involved, have had such technical, academic, and other training as the Director considers satisfactory in the interests of the health of the consumer or purchaser.
Rationale
People are the most important element in any pharmaceutical operation, without the proper personnel with the appropriate attitude and sufficient training, it is almost impossible to fabricate, package/label, test, or store good quality drugs.
It is essential that qualified personnel be employed to supervise the fabrication of drugs. The operations involved in the fabrication of drugs are highly technical in nature and require constant vigilance, attention to details and a high degree of competence on the part of employees. Inadequate training of personnel or the absence of an appreciation of the importance of production control, often accounts for the failure of a product to meet the required standards.
Interpretation
- The individual in charge of the quality control department of a fabricator, packager/labeller, tester, importer, and distributor; and the individual in charge of the manufacturing department of a fabricator or packager/labeller;
- 1.1 holds a Canadian university degree or a degree recognized as equivalent by a Canadian university or Canadian accreditation body in a science related to the work being carried out;
- 1.2 has practical experience in their responsibility area;
- 1.3 directly controls and personally supervises on site, each working shift during which activities under their control are being conducted; and
- 1.4 may delegate duties and responsibility (e.g., to cover all shifts) to a person in possession of a diploma, certificate or other evidence of formal qualifications awarded on completion of a course of study at a university, college or technical institute in a science related to the work being carried out combined with at least two years of relevant practical experience, while remaining accountable for those duties and responsibility.
- The individual in charge of the quality control department of a wholesaler;
- 2.1 is qualified by pertinent academic training and experience; and
- 2.2 may delegate duties and responsibility to a person who meets the requirements defined under interpretation 2.1.
- The individual responsible for packaging operations, including control over printed packaging materials and withdrawal of bulk drugs;
- 3.1 is qualified by training and experience; and
- 3.2 is directly responsible to the person in charge of the manufacturing department or a person having the same qualifications.
- For secondary labellers, individuals in charge of labelling operations and individuals in charge of the quality control department;
- 4.1 are qualified by pertinent academic training and experience; and
- 4.2 can delegate their duties and responsibilities to a person who meets the requirements defined under 4.1.
- An adequate number of personnel with the necessary qualifications and practical experience appropriate to their responsibilities are available on site.
- 5.1 The responsibilities placed on any one individual are not so extensive as to present any risk to quality.
- 5.2 All responsible personnel have their specific duties recorded in a written description and have adequate authority to carry out their responsibilities.
- 5.3 When key personnel are absent, qualified personnel are appointed to carry out their duties and functions.
- All personnel are aware of the principles of GMP that affect them, and all personnel receive initial and continuing training relevant to their job responsibilities.
- 6.1 Training is provided by qualified personnel having regard to the function and in accordance with a written program for all personnel involved in the fabrication of a drug, including technical, maintenance, and cleaning personnel.
- 6.2 The effectiveness of continuing training is periodically assessed.
- 6.3 Training is provided prior to implementation of new or revised SOPs.
- 6.4 Records of training are maintained.
- 6.5 Personnel working in areas where highly active, toxic, infectious, or sensitizing materials are handled are given specific training.
- 6.6 The performance of all personnel is periodically reviewed.
- Consultants and contractors have the necessary qualifications, training, and experience to advise on the subjects for which they are retained.
Sanitation
Regulation
- (1) Every person who fabricates or packages/labels a drug shall have a written sanitation program that shall be implemented under the supervision of qualified personnel.
- (2) The sanitation program referred to in subsection (1) shall include:
- (a) cleaning procedures for the premises where the drug is fabricated or packaged/labelled and for the equipment used in the fabrication or packaging/labelling of the drug; and
- (b) instructions on the sanitary fabrication and packaging/labelling of drugs and the handling of materials used in the fabrication and packaging/labelling of drugs.
Rationale
Sanitation in a pharmaceutical plant, as well as employee attitude, influences the quality of drug products. The quality requirement for drug products demand that such products be fabricated and packaged in areas that are free from environmental contamination and free from contamination by another drug.
A written sanitation program provides some assurance that levels of cleanliness in the plant are maintained and that the provisions of Sections 8 and 11 of the Food and Drugs Act are satisfied.
Interpretation
- Every person who fabricates or packages/labels a drug shall have a written sanitation program available on the premises.
- The sanitation program contains procedures that describe the following:
- 2.1 cleaning requirements applicable to all production areas of the plant with emphasis on manufacturing areas that require special attention;
- 2.2 cleaning requirements applicable to processing equipment;
- 2.3 cleaning intervals;
- 2.4 products for cleaning and disinfection, along with their dilution and the equipment to be used;
- 2.5 the responsibilities of any outside contractor;
- 2.6 disposal procedures for waste material and debris;
- 2.7 pest control measures;
- 2.8 precautions required to prevent contamination of a drug when rodenticides, insecticides, and fumigation agents are used;
- 2.9 microbial and environmental monitoring procedures with alert and action limits in areas where susceptible products are fabricated or packaged; and
- 2.10 the personnel responsible for carrying out cleaning procedures.
- The sanitation program is implemented and is effective in preventing unsanitary conditions.
- 3.1 Cleaning procedures for manufacturing equipment are validated based on the Health Canada document entitled "Cleaning Validation Guidelines (GUI-0028)".
- 3.2 Residues from the cleaning process itself (e.g., detergents, solvents, etc.) are also removed from equipment.
- 3.3 Evidence is available demonstrating that routine cleaning and storage does not allow microbial proliferation; Where necessary, sanitisers and disinfectants are filtered to remove spores (e.g., isopropyl alcohol).
- 3.4 Analytical methods used to detect residues or contaminants are validated. Guidance on analytical method validation can be obtained from publications such as the International Conference on Harmonisation (ICH) document entitled "ICH Q2(R1): Validation of Analytical Procedures: Text and Methodology", or in any standard listed in Schedule B to the Food and Drugs Act .
- 3.5 A cleaning procedure requiring complete product removal may not be necessary between batches of the same drug provided it meets the requirements of interpretation 3.1.
- Individuals who supervise the implementation of the sanitation program;
- 4.1 are qualified by training or experience; and
- 4.2 are directly responsible to a person who has the qualifications described under Regulation C.02.006, interpretation 1.
- Dusty operations are contained. The use of unit or portable dust collectors is avoided in fabrication areas especially in dispensing, unless the effectiveness of their exhaust filtration is demonstrated and the units are regularly maintained in accordance with written approved procedures.
Regulation
- (1) Every person who fabricates or packages/labels a drug shall have, in writing, minimum requirements for the health and the hygienic behaviour and clothing of personnel to ensure the clean and sanitary fabrication and packaging/labelling of the drug.
- (2) No person shall have access to any area where a drug is exposed during its fabrication or packaging/labelling if the person
- (a) is affected with or is a carrier of a disease in a communicable form, or
- (b) has an open lesion on any exposed surface of the body.
Rationale
Employees health, behaviour, and clothing may contribute to the contamination of the product. Poor personal hygiene will nullify the best sanitation program and greatly increase the risk of product contamination.
Interpretation
- Minimum health requirements are available in writing.
- 1.1 Personnel who have access to any area where a drug is exposed during its fabrication or packaging/labelling must undergo health examinations prior to employment. Medical re-examinations, based on job requirements take place periodically.
Note: A person who is a known carrier of a disease in a communicable form should not have access to any area where a drug is exposed. The likelihood of disease transmission by means of a drug product would depend on the nature of the disease and the type of work the person carries out. Certain diseases could be transmitted through a drug product if proper hygiene procedures are not followed by an infected person handling the product. However, a person may also be a carrier of a communicable disease and not be aware of it. Therefore, in addition to strict personal hygiene procedures, systems should be in place to provide an effective barrier that would preclude contamination of the product. These procedures must be followed at all times by all personnel. - 1.2 Employees are instructed to report to their supervisor any health conditions they have that could adversely affect drug products.
- 1.3 Supervisory checks are conducted to prevent any person who has an apparent illness or open lesions that may adversely affect the quality of drugs from handling exposed raw materials, primary packaging materials, in-process drugs or drugs until the condition is no longer judged to be a risk.
- 1.4 When an employee has been absent from the workplace due to an illness that may adversely affect the quality of products, that employees health is assessed before he or she is allowed to return to the workplace.
- 1.5 A procedure in place which describes the actions to be taken in the event that a person who has been handling exposed raw materials, primary packaging materials, in-process drugs or drugs is identified as having a communicable disease.
- 1.6 Periodic eye examinations and/or periodic requalification are required for personnel who conduct visual inspections.
- 1.1 Personnel who have access to any area where a drug is exposed during its fabrication or packaging/labelling must undergo health examinations prior to employment. Medical re-examinations, based on job requirements take place periodically.
- The written hygiene program clearly defines clothing requirements and hygiene procedures for personnel and visitors.
- 2.1 Where a potential for the contamination of a raw material, in-process material or drug exists, individuals wear clean clothing and protective covering.
- 2.2 Direct skin contact is avoided between the operator and raw materials, primary packaging materials, in-process drugs or drugs.
- 2.3 Unsanitary practices such as smoking, eating, drinking, chewing, and keeping plants, food, drink, smoking material and personal medicines are not permitted in production areas or in any other areas where they might adversely affect product quality.
- 2.4 Requirements concerning personal hygiene, with an emphasis on hand hygiene, are outlined and are followed by employees.
- 2.5 Requirements concerning cosmetics and jewellery worn by employees are outlined and are observed by employees.
- 2.6 Soiled protective garments, if reusable, are stored in separate containers until properly laundered and, if necessary, disinfected or sterilized. A formalized procedure for the washing of protective garments under the control of the company is in place. Washing garments in a domestic setting is unacceptable.
- 2.7 Personal hygiene procedures including the use of protective clothing, apply to all persons entering production areas.
Raw Material Testing
Regulation
- (1) Each lot or batch of raw material shall be tested against the specifications for the raw material prior to its use in the fabrication of a drug.
- (2) No lot or batch of raw material shall be used in the fabrication of a drug unless that lot or batch of raw material complies with the specifications for that raw material.
- (3) Notwithstanding subsection (1), water may, prior to the completion of its tests under that subsection, be used in the fabrication of a drug.
- (4) Where any property of a raw material is subject to change on storage, no lot or batch of that raw material shall be used in the fabrication of a drug after its storage unless the raw material is retested after an appropriate interval and complies with its specifications for that property.
- (5) Where the specifications referred to in subsections (1), (2) and (4) are not prescribed, they shall
- (a) be in writing;
- (b) be acceptable to the Director who shall take into account the specifications contained in any publication mentioned in Schedule B to the Act; and
- (c) be approved by the person in charge of the quality control department.
Rationale
The testing of raw materials before their use has three objectives: to confirm the identity of the raw materials, to provide assurance that the quality of the drug in dosage form will not be altered by raw material defects, and to obtain assurance that the raw materials have the characteristics that will provide the desired quantity or yield in a given manufacturing process.
Interpretation
- Each raw material used in the production of a drug is covered by specifications (see regulation C.02.002) that are approved and dated by the person in charge of the quality control department or by a designated alternate who meets the requirements described under Regulation C.02.006, interpretation 1.4.
- Specifications are of pharmacopoeial or equivalent status and are in compliance with the current marketing authorization. Where appropriate, additional properties or qualities not addressed by the pharmacopoeia (e.g., particle size, etc.) are included in the specifications.
- Where a recognized pharmacopoeia (Schedule B of the Food and Drugs Act ) contains a specification for microbial content, that requirement is included.
- Purified water that meets any standard listed in Schedule B of the Food and Drugs Act is used in the formulation of a non-sterile drug product, unless otherwise required in one of these standards or as stated in the marketing authorization.
- 4.1 Specifications should include requirements for total microbial count, which should not exceed 100 colony forming units (cfu)/ml.
- 4.2 Purified water should be monitored on a routine basis for the purpose intended to ensure the absence of objectionable microorganisms (e.g., Escherichia coli and Salmonella for water used for oral preparations, Staphylococcus aureus and Pseudomonas aeruginosa for water used for topical preparations).
- Test methods are validated, and the results of such validation studies are documented. Full validation is not required for methods included in any standard listed in Schedule B to the Food and Drugs Act , but the user of such a method establishes its suitability under actual conditions of use. Method transfer studies are conducted when applicable.
Note: Guidance for the validation of particular types of methods can be obtained in publications such as the ICH document entitled "ICH Q2(R1): Validation of Analytical Procedures: Text and Methodology" or in any standard listed in Schedule B to the Food and Drugs Act .
- A sample of each lot of raw material is fully tested against specifications. Sampling is conducted according to a suitable statistically valid plan.
- 6.1 In addition, each container of a lot of a raw material is tested for the identity of its contents using a specifically discriminating identity test.
- 6.2 In lieu of testing each container for identity, testing a composite sample derived from sampling each container is acceptable, as long as the following conditions are met:
- 6.2.1 the number of individual containers for each composite sample does not exceed 10; and
- 6.2.2 a potency test is performed on each composite sample to establish the mass balance of the composite sample.
- 6.3 In lieu of testing each container for identity, testing only a proportion of the containers is acceptable where a validated procedure has been established to ensure that no single container of raw material has been incorrectly labelled.
- 6.3.1 Interpretation 6.3 applies to raw material coming from a single product manufacturer or plant or coming directly from a manufacturer or in the manufacturers sealed container where there is a history of reliability and regular audits of the manufacturers Quality Assurance system are conducted by or on behalf of the purchaser (drug fabricator).
- 6.3.2 Interpretation 6.3 does not apply when the raw material is used in parenterals or supplied by intermediaries such as brokers where the source of manufacture is unknown or not audited.
- 6.3.3 The validation should take into account at least the following aspects;
- 6.3.3.1 the nature and status of the manufacturer and the supplier and their understanding of the GMP requirements of the pharmaceutical industry;
- 6.3.3.2 the Quality Assurance system of the manufacturer of the raw material; and
- 6.3.3.3 the manufacturing conditions under which the raw material is produced and controlled.
- 6.4 Where a batch of any raw material, after leaving the site of its fabrication is handled in any substantial way (e.g., repackaged by a third party) prior to its receipt on the premises of the person who formulates the raw material into dosage forms, each container in that batch is sampled and its contents positively identified.
- Only raw materials that have been released by the quality control department and that are not past their established re-test date or expiry date are used in fabrication.
- 7.1 If any raw material is held in storage after the established re-test date, that raw material is quarantined, evaluated, and tested prior to use. The re-test date or expiry date is based on acceptable stability data developed under predefined storage conditions or on any other acceptable evidence. A batch of raw material can be re-tested and used immediately (i.e., within 30 days) after the re-test as long as it continues to comply with the specifications and has not exceeded its expiry date. A raw material held in storage after the established expiry date should not be used in fabrication.
Regulation
- (1) The testing referred to in section C.02.009 shall be performed on a sample taken
- (a) after receipt of each lot or batch of raw material on the premises of the fabricator; or
- (b) subject to subsection (2), before receipt of each lot or batch of raw material on the premises of the fabricator, if
- (i) the fabricator
- (A) has evidence satisfactory to the Director to demonstrate that raw materials sold to him by the vendor of that lot or batch of raw material are consistently manufactured in accordance with and consistently comply with the specifications for those raw materials, and
- (B) undertakes periodic complete confirmatory testing with a frequency satisfactory to the Director, and
- (ii) the raw material has not been transported or stored under conditions that may affect its compliance with the specifications for that raw material.
- (i) the fabricator
- (2) After a lot or batch of raw material is received on the premises of the fabricator, the lot or batch of raw material shall be tested for identity.
Rationale
Section C.02.010 outlines options as to when the testing prescribed by Section C.02.009 is carried out. The purchase of raw materials is an important operation that requires a particular and thorough knowledge of the raw materials and their fabricator. To maintain consistency in the fabrication of drug products, raw materials should originate from reliable fabricators.
Interpretation
- Testing other than identity testing:
The testing is performed on a sample taken after receipt of the raw material on the premises of the person who formulates the raw material into dosage form, unless the vendor is certified. A raw material vendor certification program, if employed, is documented in a standard operating procedure. At a minimum, such a program includes the following:- 1.1 A written agreement outlining the specific responsibilities of each party involved. The agreement specifies:
- 1.1.1 the content and the format of the certificate of analysis, which exhibits actual numerical results and makes reference to the raw material specifications and validated test methods used;
- 1.1.2 that the raw material vendor must inform the drug fabricator of any changes in the processing or specifications of the raw material; and
- 1.1.3 that the raw material vendor must inform the drug fabricator in case of any critical deviation during the manufacturing of a particular batch of a raw material.
- 1.2 An audit report is available.
- 1.2.1 For medicinal ingredients/active pharmaceutical ingredient (API), the audit report is issued by a qualified authority demonstrating that the API vendor complies with the ICH document entitled "ICH Q7: Good Manufacturing Practices Guide for Active Pharmaceutical Ingredients" or with any standard or system of equivalent quality. This report should be less than 3 years old, but is valid for 4 years from the date of the inspection. If such an audit report is unavailable or is more than 4 years old, an on-site audit of the API vendor, against the same standard or its equivalent, by a person who meets the requirements of interpretation 1 under Section C.02.006, is acceptable.
- 1.2.2 For other raw materials, an audit report issued by a person who meets the requirements of interpretation 1 under section C.02.006 is acceptable.
- 1.3 Complete confirmatory testing is performed on the first three lots of any each raw material received from a vendor and after significant change to the manufacturing process. A copy of the residual solvent profile is obtained. Additionally, for medicinal ingredients, a copy of the impurity profile is also obtained.
- 1.4 Identification of how re-testing failures and any subsequent re-qualification of the vendor are to be addressed.
- 1.5 The list of raw materials not subject to the reduced testing program (e.g., reprocessed lots).
- 1.6 Complete confirmatory testing is conducted on a minimum of one lot per year of a raw material received from each vendor, with the raw material being selected on a rotational basis.
- 1.6.1 In addition, where multiple raw materials are received from the same vendor, confirmatory testing is carried out for each raw material at least once every five years.
- 1.7 A document is issued for each vendor verifying that the vendor meets the criteria for certification. The document is approved by the quality control department and is updated at an appropriate frequency.
- 1.8 Generally, due to the nature of its operations, a broker or wholesaler of raw materials cannot be directly certified. However, when a broker or wholesaler supplies materials received from the original vendor without changing the existing labels, packaging, certificate of analysis, and general information, then certification of the original source is still acceptable.
- 1.1 A written agreement outlining the specific responsibilities of each party involved. The agreement specifies:
- Identity testing:
Specific identity testing is conducted on all lots of any raw material received on the premises of the person who formulates the raw material into dosage forms. This identity testing is performed in accordance with Regulation C.02.009, interpretation 6. - Provided that the identity test referred to in interpretation 2 is performed, the lot of raw material selected for confirmatory testing may be used in fabrication prior to completion of all tests with the approval of the quality control department.
- Conditions of transportation and storage are such that they prevent alterations to the potency, purity, or physical characteristics of the raw material. In order to demonstrate that these conditions have been met, standard operating procedures and records for shipping and receiving are available and contain:
- 4.1 the type of immediate packaging for the raw material;
- 4.2 the labelling requirements including storage conditions and special precautions or warnings, for the packaged raw material;
- 4.3 the mode(s) of transportation approved for shipping the packaged raw material;
- 4.4 a description of how the packaged raw material is sealed;
- 4.5 the verification required to ensure that each package has not been tampered with and that there are no damaged containers; and
- 4.6 evidence that special shipping requirements (e.g., refrigeration) have been met if required.
- If a delivery or shipment of raw material is made up of different batches, each batch is considered as separate for the purposes of sampling, testing, and release.
- If the same batch of raw material is subsequently received, this batch is also considered as separate for the purpose of sampling, testing, and release.
However, full testing to specifications may not be necessary on such a batch provided that all the following conditions are met:- 6. 1 a specifically discriminating identity test is conducted;
- 6. 2 the raw material has not been repackaged or re-labelled;
- 6. 3 the raw material is within the re-test date assigned by its vendor; and
- 6. 4 evidence is available to demonstrate that all pre-established transportation and storage conditions have been maintained.
Manufacturing Control
Regulation
- (1) Every fabricator, packager/labeller, distributor referred to in paragraph C.01A.003(b) and importer of a drug shall have written procedures prepared by qualified personnel in respect of the drug to ensure that the drug meets the specifications for that drug.
- (2) Every person required to have written procedures referred to in subsection (1) shall ensure that each lot or batch of the drug is fabricated, packaged/labelled and tested in compliance with those procedures.
Rationale
This Regulation requires that measures be taken to maintain the integrity of a drug product from the moment the various raw materials enter the plant to the time the finished dosage form is released for sale and distributed. These measures ensure that all manufacturing processes are clearly defined, systematically reviewed in light of experience, and demonstrated to be capable of consistently manufacturing pharmaceutical products of the required quality that comply with their established specifications.
Interpretation
- All handling of raw materials, products, and packaging materials such as receipt, quarantine, sampling, storage, tracking, labelling, dispensing, processing, packaging and distribution is done in accordance with pre-approved written procedures or instructions and recorded.
- All critical production processes are validated. Detailed information is provided in various Health Canada validation guidelines.
- Validation studies are conducted in accordance with predefined protocols. A written report summarizing recorded results and conclusions is prepared, evaluated, approved, and maintained.
- Changes to production processes, systems, equipment, or materials that may affect product quality and/or process reproducibility are validated prior to implementation.
- Any deviation from instructions or procedures is avoided. If deviations occur, qualified personnel investigate, and write a report that describes the deviation, the investigation, the rationale for disposition, and any follow-up activities required. The report is approved by the quality control department and records maintained.
- Checks on yields and reconciliation of quantities are carried out at appropriate stages of the process to ensure that yields are within acceptable limits.
- Deviations from the expected yield are recorded and investigated.
- Access to production areas is restricted to designated personnel.
- Provided that validated changeover procedures are implemented, non-medicinal products may be fabricated or packaged/labelled in areas or with equipment that is also used for the production of pharmaceutical products.
- Before any processing operation is started, steps are taken and documented to ensure that the work area and equipment are clean and free from any raw materials, products, product residues, labels, or documents not required for the current operation.
- 10.1 Operations on different products should not be carried out simultaneously or consecutively in the same room unless there is no risk of mix-up or cross-contamination.
- 10.2 Checks should be carried out to ensure that transfer lines and other pieces of equipment used for the transfer of products from one area to another are correctly connected.
- At every stage of processing, products and materials are appropriately protected from microbial and other contamination.
- In-process control activities that are performed within the production areas do not pose any risk to the quality of the product.
- Measuring devices are regularly checked for accuracy and precision, and records of such checks are maintained.
- At all times during processing, all materials, bulk containers, major items of equipment and the rooms used are labelled or otherwise identified with an indication of the product or material being processed, its strength, and the batch number and, if appropriate, the stage of manufacturing.
- Rejected materials and products are clearly marked as such and are either stored separately in restricted areas or controlled by a system that ensures that they are either returned to their vendors or, where appropriate, reprocessed or destroyed. Actions taken are recorded.
- Upon receipt, raw materials, packaging materials, in-process (intermediate) drugs, and bulk drugs, are accounted for, documented, labelled and held in quarantine until released by the quality control department.
- Procedures are in place to ensure the identity of the contents of each container. Containers from which samples have been drawn are identified.
- For each consignment, all containers are checked for integrity of package and seal and to verify that the information on the order, the delivery note and the vendor's labels is in agreement.
- Damage to containers, along with any other problem that might adversely affect the quality of a material, is recorded, reported to the quality control department, and investigated.
- Upon receipt, containers are cleaned where necessary and labelled with the prescribed data.
- Labels for bulk drugs, in-process drugs, raw materials, and packaging materials bear the following information:
- 21.1 the designated name and, if applicable, the code or reference number of the material;
- 21.2 the specific batch number(s) given by the vendor and on receipt by the fabricator or packager/labeller;
- 21.3 the status of the contents (e.g., in quarantine, on test, released, rejected, to be returned or recalled) appears on the label when a manual system is used;
- 21.4 an expiry date or a date beyond which re-testing is necessary; and
- 21.5 the stage of manufacturing of in-process material, if applicable.
Note: When fully computerized storage systems are used, backup systems are available in case of system failure to satisfy the requirements of interpretation 21.
- Raw materials are dispensed and verified by qualified personnel, following a written procedure, to ensure that the correct materials are accurately weighed or measured into clean and properly labelled containers. Raw materials which are being staged are properly sealed and stored under conditions consistent with the accepted storage conditions for that material.
Manufacturing Master Formula
- Processing operations are covered by master formula, that are prepared by, and are subject to independent checks by, persons who have the qualifications described under Regulation C.02.006 interpretation 1, including the quality control department.
- Master formula are written to provide not less than 100% of label claim. Overages may be allowed to compensate for processing losses with documented justification and approval if appropriate. In exceptional instances, overages to compensate for losses due to degradation during manufacturing or shelf-life must be scientifically justified and in accordance with the marketing authorization. Master formula also include the following:
- 24.1 the name of the product, with a reference code relating to its specifications;
- 24.2 a description of the dosage form, strength of the product, and batch size;
- 24.3 a list of all raw materials to be used, along with the amount of each, described using the designated name and a reference that is unique to that material (mention is made of any processing aids that may not be present in the final product);
- 24.4 a statement of the expected final yield, along with the acceptable limits, and of relevant intermediate yields, where applicable;
- 24.5 Identification of the principal equipment to be used, and if applicable internal codes;
- 24.6 the procedures, or reference to the procedures, to be used for preparing the critical equipment, (e.g., cleaning, assembling, calibrating, sterilizing, etc.);
- 24.7 detailed stepwise processing instructions (e.g., checks on materials, pretreatment, sequence for adding materials, mixing times or temperatures, etc.);
- 24.8 the instructions for any in-process controls, along with their limits; and
- 24.9 where necessary, the requirements for storage of the products and in-process materials, including the container-closure system, labelling storage conditions, maximum validated hold time, and any special precautions to be observed.
Packaging Master Formula
- In the case of a packaged product, the master formula also includes for each product, package size and type, the following:
- 25.1 the package size, expressed in terms of the number, weight, or volume of the product in the final container;
- 25.2 a complete list of all the packaging materials required for a standard batch size, including quantities, sizes and types with the code or reference number relating to the specifications for each packaging material;
- 25.3 where appropriate, an example or reproduction of the relevant printed packaging materials and specimens, indicating where the batch number and expiry date of the product are to be positioned;
- 25.4 special precautions to be observed, including a careful examination of the packaging area and equipment in order to ascertain the line clearance before operations begin. These verifications are recorded;
- 25.5 a description of the packaging operations, including any significant subsidiary operations and the equipment to be used; and
- 25.6 details of in-process controls, with instructions for sampling and acceptance limits.
Manufacturing Operations
- Each batch processed is effectively governed by an individually numbered manufacturing order prepared by qualified personnel from the master formula by such means as to prevent errors in copying or calculation and verified by qualified personnel.
- As it becomes available during the process, the following information is included on or with the manufacturing batch record:
- 27.1 the name of the product;
- 27.2 the number of the batch being manufactured;
- 27.3 dates and times of commencement and completion of significant intermediate stages, such as blending, heating, etc., and of production;
- 27.4 the batch number and/or analytical control number, as well as the quantity of each raw material actually weighed and dispensed (for active raw material, the quantity is to be adjusted if the assay value is less than 98% calculated on "as is" basis and on which the master formula was based);
- 27.5 confirmation by qualified personnel of each ingredient added to a batch;
- 27.6 the identification of personnel performing each step of the process; and of the person who checked each of these steps;
- 27.7 the actual results of the in-process quality checks performed at appropriate stages of the process and the identification of the person carrying them out;
- 27.8 the actual yield of the batch at appropriate stages of processing and the actual final yields, together with explanations for any deviations from the expected yield;
- 27.9 detailed notes on special problems with written approval for any deviation from the master formula; and
- 27.10 after completion, the signature of the person responsible for the processing operations.
- Batches are combined only with the approval of the quality control department and according to pre-established written procedures.
- 28.1 The introduction of part of a previous batch, conforming to the required quality, into the next batch of the same product at a defined stage of fabrication is approved beforehand. This recovery is carried out in accordance with a validated procedure and is recorded.
Packaging Operations
- Packaging operations are performed according to comprehensive and detailed written operating procedures or specifications, which include the identification of equipment and packaging lines used to package the drug, the adequate separation and if necessary, the dedication of packaging lines that are packaging different drugs and disposal procedures for unused printed packaging materials. Packaging orders are individually numbered.
- The method of preparing packaging orders is designed to avoid transcription errors.
- Before any packaging operation begins, checks are made that the equipment and work station are clear of previous products, documents, and materials that are not required for the planned packaging operations and that equipment is clean and suitable for use. These checks are recorded.
- All products and packaging materials to be used are checked on receipt by the packaging department for quantity, identity and conformity with the packaging instructions.
- Precautions are taken to ensure that containers to be filled are free from contamination with extraneous material.
- The name and batch number of the product being handled is displayed at each packaging station or line.
- Packaging orders include the following information (recorded at the time each action is taken):
- 35.1 the date(s) and time(s) of the packaging operations;
- 35.2 the name of the product, the batch number, packaging line used, and the quantity of bulk product to be packaged, as well as the batch number and the planned quantity of finished product that will be obtained, the quantity actually obtained and the reconciliation;
- 35.3 the identification of the personnel who are supervising packaging operations and the withdrawal of bulks;
- 35.4 the identification of the operators of the different significant steps;
- 35.5 the checks made for identity and conformity with the packaging instructions, including the results of in-process controls;
- 35.6 the general appearance of the packages;
- 35.7 whether the packages are complete;
- 35.8 whether the correct products and packaging materials are used;
- 35.9 whether any on-line printing is correct;
- 35.10 the correct functioning of line monitors;
- 35.11 handling precautions applied to a partly packaged product;
- 35.12 notes on any special problems, including details of any deviation from the packaging instructions with written approval by qualified personnel;
- 35.13 the quantity, lot number, and/or analytical control number of each packaging material and bulk drug issued for use; and
- 35.14 a reconciliation of the quantity of printed packaging material and bulk drug used, destroyed or returned to stock.
- To prevent mix-ups, samples taken away from the packaging line are not returned.
- Whenever possible, samples of the printed packaging materials used, including specimens bearing the batch number, expiry date, and any additional overprinting, are attached to packaging orders.
- Filling and sealing are followed as quickly as possible by labelling. If labelling is delayed, procedures are applied to ensure that no mix-ups or mislabelling can occur.
- Upon completion of the packaging operation, any unused batch-coded packaging materials are destroyed, and their destruction is recorded. A procedure is followed if non-coded printed materials are returned to stock.
- Outdated or obsolete packaging materials are destroyed and their disposal is recorded.
- Products that have been involved in non-standard occurrences during packaging are subject to inspection and investigation by qualified personnel. A detailed record is kept of this operation.
- Any significant or unusual discrepancy observed during reconciliation of the amount of bulk product and printed packaging materials and the number of units packaged is investigated and satisfactorily accounted for before release. Validated electronic verification of all printed packaging materials on the packaging line may obviate the need for their full reconciliation.
- Printed packaging materials are:
- 43.1 stored in an area to which access is restricted to designated personnel who are supervised by persons who have the qualifications outlined under Regulation C.02.006;
- 43.2 withdrawn against a packaging order;
- 43.3 issued and checked by persons who have the qualifications outlined under Regulation C.02.006 interpretation 3; and
- 43.4 identified in such a way as to be distinguishable during the packaging operations.
- To prevent mix-ups, roll-fed labels are preferred to cut labels. Gang printing (printing more than one item of labelling on a sheet of material) is avoided.
- Cut labels, cartons, and other loose printed materials are stored and transported in separate closed containers.
- Special care is taken when cut labels are used, when overprinting is carried out off-line and in hand-packaging operations. On line verification of all labels by automated electronic means can be helpful in preventing mix-ups. Checks are made to ensure that any electronic code readers, label counters or similar devices are operating correctly.
- The correct performance of any printing (e.g., of code numbers or expiry dates) done separately or in the course of the packaging is checked and recorded.
- Raw materials, packaging materials, intermediates, bulk drugs and finished products are (a) stored in locations that are separate and removed from immediate manufacturing areas, and (b) transported under conditions designated by the quality control department to preserve their quality and safety.
- All intermediate and finished products are held in quarantine and are so identified in accordance with interpretation 21, until released by the quality control department.
- Every package of a drug is identified by a lot number.
Annual Product Quality Review
- Regular periodic or rolling quality reviews of all drugs, should be conducted with the objective of verifying the consistency of the existing process, the appropriateness of current specifications for both raw materials and finished product to highlight any trends and to identify product and process improvements. Such reviews should normally be conducted and documented annually, taking into account previous reviews, and should include at least:
- 51.1 A review of critical in-process controls, finished product testing results and specifications.
- 51.2 A review of all batches that failed to meet established specification(s) and their investigation.
- 51.3 A review of all significant deviations or non-conformances, their related investigations, and the effectiveness of resultant corrective and preventative actions taken.
- 51.4 A review of all changes carried out to the processes, analytical methods, raw materials, packaging materials, or critical suppliers.
- 51.5 A review of the results of the continuing stability program and any adverse trends.
- 51.6 A review of all quality-related returns, complaints and recalls and the investigations performed at the time.
- 51.7 A review of adequacy of any other previous product process or equipment corrective actions.
- 51.8 The qualification status of relevant equipment and systems (e.g., HVAC, water, compressed gases, etc.); and
- 51.9 A review of agreements to ensure that they are up to date.
- Quality reviews may be grouped by product type (e.g., solid dosage forms, liquid dosage forms, sterile products, etc. where scientifically justified).
- The quality control department of the importer or distributor should ensure that the annual product quality review is performed in a timely manner.
- Where required, there should be an agreement in place between the various parties involved (e.g., importer and fabricator) that defines their respective responsibilities in producing and assessing the quality review and taking any subsequent corrective and preventative actions.
- The quality control department should evaluate the results of this review and an assessment should be made whether corrective and preventative action or any revalidation should be undertaken. Reasons for such corrective actions should be documented. Agreed corrective and preventative actions should be completed in a timely and effective manner. There should be procedures for the ongoing management and review of these actions and the effectiveness of these procedures verified during self-inspection.
Regulation
- (1) Every fabricator, packager/labeller, distributor referred to in section C.01A.003, importer and wholesaler of a drug shall maintain
- (a) a system of control that permits complete and rapid recall of any lot or batch of the drug that is on the market; and
- (b) a program of self-inspection.
- (2) Every fabricator and packager/labeller and subject to subsections (3) and (4), every distributor referred to in paragraph C.01A.003(b) and importer of a drug shall maintain a system designed to ensure that any lot or batch of the drug fabricated and packaged/labelled on premises other than their own is fabricated and packaged/labelled in accordance with the requirements of this Division.
- (3) The distributor referred to in paragraph C.01A.003(b) of a drug that is fabricated, packaged/labelled, and tested in Canada by a person who holds an establishment licence that authorizes those activities is not required to comply with the requirements of subsection (2) in respect of that drug.
- (4) If a drug is fabricated or packaged/labelled in an MRA country at a recognized building, the distributor referred to in paragraph C.01A.003(b) or importer of the drug is not required to comply with the requirements of subsection (2) in respect of that activity for that drug if
- (a) the address of the building is set out in that persons establishment licence; and
- (b) that person retains a copy of the batch certificate for each lot or batch of the drug received by that person.
Rationale
The purpose of a recall is to remove from the market, a drug that represents an undue health risk.
Drugs that have left the premises of a fabricator, packager/labeller, distributor, wholesaler and importer can be found in a variety of locations. Depending on the severity of the health risk, it may be necessary to recall a product to one level or another. Fabricators, packagers/labellers, distributors, wholesalers, and importers are expected to be able to recall to the consumer level if necessary. Additional guidance on recalls can be found in Health Canadas document entitled "Product Recall Procedures".
This Regulation also requires fabricators, packagers/labellers, distributors, wholesalers, and importers to maintain a program of self-inspection. The purpose of self-inspection is to evaluate the compliance with GMP in all aspects of production and quality control. The self-inspection program is designed to detect any shortcomings in the implementation of GMP and to recommend the necessary corrective actions.
Drugs offered for sale in Canada, regardless of whether they are domestically produced or imported, must meet the requirements of Part C, Division 2 of the Food and Drug Regulations. Contract production and analysis must be correctly defined, agreed on, and controlled in order to avoid misunderstandings that could result in a product, work, or analysis of unsatisfactory quality. Normally, a written agreement exists between the parties involved, and that document clearly establishes the duties of each party.
Interpretation
- A written recall system is in place to ensure compliance with Section C.01.051 of the Food and Drug Regulations and requires the following:
- 1.1 Health Canada is to be notified of the recall;
- 1.2 Action that is taken to recall a product suspected or known to be in violation is prompt and in accordance with a pre-determined plan; the procedures to be followed are in writing and are known to all concerned;
- 1.3 The person(s) responsible for initiating and co-ordinating all recall activities are identified;
- 1.4 The recall procedure is capable of being put into operation at any time, during and outside normal working hours;
- 1.5 The recall procedure outlines the means of notifying and implementing a recall and of deciding its extent;
- 1.6 Distribution records enable tracing of each drug product, and account is taken of any products that are in transit, any samples that have been removed by the quality control department, and any professional samples that have been distributed;
- 1.7 Wholesalers must obtain drug products from companies that hold an establishment licence as required in Part C, Division 1A of the Food and Drug Regulations in order to facilitate a system of control that permits complete and rapid recall;
- 1.8 A written agreement clearly describes respective responsibilities when the importer or distributor assumes some or all of the wholesalers responsibilities with respect to recalls;
- 1.9 Recalled products are identified and are stored separately in a secure area until their disposition is determined;
- 1.10 The progress and efficacy of the recall is assessed and recorded at intervals, and a final report is issued (including a final reconciliation); and
- 1.11 All Canadian and foreign establishments involved in the fabrication, distribution, or importation of the recalled product are notified.
- A self-inspection program appropriate to the type of operations of the company, in respect to drugs, ensures compliance with Division 2, Part C of the Food and Drug Regulations.
- 2.1 A comprehensive written procedure that describes the functions of the self-inspection program is available.
- 2.2 The program of a fabricator engaged in processing a drug from raw material through to the drug in dosage form addresses itself to all aspects of the operation. For packagers/labellers, distributors, importers, and wholesalers engaged only in packaging and/or distributing drugs fabricated by another fabricator, the written program covers only those aspects of the operations over which they exercise control on their premises.
- 2.3 The self-inspection team includes personnel who are suitably trained and qualified in GMP.
- 2.4 Periodic self-inspections are carried out.
- 2.5 Reports on the findings of the inspections and on corrective actions are reviewed by appropriate senior company management. Corrective actions are implemented in a timely manner.
- To ensure compliance of contractors performing fabrication and packaging/labelling:
- 3.1 All arrangements for contract fabrication or packaging/labelling are in accordance with the marketing authorization for the drug product concerned.
- 3.2 There is a written agreement covering the fabrication or packaging/labelling arranged among the parties involved. The agreement specifies their respective responsibilities relating to the fabrication or packaging/labelling and control of the product.
- 3.2.1 Technical aspects of the agreement are drawn up by qualified personnel suitably knowledgeable in pharmaceutical technology, and GMP.
- 3.2.2 The agreement permits the distributor or importer to audit the facilities of the contractor.
- 3.2.3 The agreement clearly describes as a minimum who is responsible for:
- 3.2.3.1 purchasing, sampling, testing, and releasing materials;
- 3.2.3.2 undertaking production, quality, and in-process controls; and
- 3.2.3.3 process validation.
- 3.2.4 No subcontracting of any work should occur without written authorization.
- 3.2.5 The agreement specifies the way in which the quality control department of the distributor or importer releasing the lot or batch for sale, ensures that each lot or batch has been fabricated and packaged/labelled in compliance with the requirements of the marketing authorization.
- 3.2.6 The agreement describes the handling of raw materials, packaging materials, in-process drug, bulk drug and finished products if they are rejected.
- 3.3 The contractors complaint/recall procedures specify that any records relevant to assessing the quality of a drug product in the event of complaints or a suspected defect are accessible to the distributor or importer.
- 3.4 The fabricator, packager/labeller, distributor, or importer provides the contractor with all the information necessary to carry out the contracted operations correctly in accordance with the marketing authorization and any other legal requirements. The fabricator, packager/labeller, distributor, or importer ensures that the contractor is fully aware of any problems associated with the product, work or tests that might pose a hazard to premises, equipment, personnel, other materials or other products.
- 3.5 The fabricator, packager/labeller, distributor, or importer is responsible for assessing the contractors continuing competence to carry out the work or tests required in accordance with the principles of GMP described in these guidelines.
- 3.5.1 Distributors of drugs fabricated, packaged/labelled and tested at Canadian sites are required only to have a copy of the relevant valid Canadian establishment licence held by the Canadian fabricator or packager/labeller or tester.
- 3.5.2 Importers of drugs fabricated, packaged/labelled, or tested at a foreign site must meet the requirements described in Health Canadas policy document entitled "Guidance on Evidence to Demonstrate Drug GMP Compliance of Foreign Sites (GUI-0080)".
Quality Control Department
Regulation
- (1) Every fabricator, packager/labeller, distributor referred to in paragraph C.01A.003(b), and importer shall have on their premises in Canada a quality control department that is supervised by personnel described in section C.02.006.
- (2) The quality control department shall be a distinct organizational unit that functions and reports to management independently of any other functional unit, including the manufacturing, processing, packaging or sales unit.
Rationale
Quality control is the part of GMP concerned with sampling, specifications, and testing and with the organization, documentation, and release procedures. This Regulation ensures that the necessary and relevant tests are actually carried out and that raw materials and packaging materials are not released for use, nor products released for sale, until their quality has been judged to be satisfactory. Quality control is not confined to laboratory operations but must be incorporated into all activities and decisions concerning the quality of the product.
Although manufacturing and quality control personnel share the common goal of assuring that high-quality drugs are fabricated, their interests may sometimes conflict in the short run as decisions are made that will affect a company's output. For this reason, an objective and accountable quality control process can be achieved most effectively by establishing an independent quality control department. The independence of quality control from manufacturing is considered fundamental. The rationale for the requirement that the quality control department be supervised by qualified personnel is outlined under Regulation C.02.006.
Interpretation
- A person responsible for making decisions concerning quality control requirements of the fabricator, packager/labeller, distributor, and importer is on site or fully accessible to the quality control department and has adequate knowledge of on-site operations to fulfill the responsibilities of the position.
- The quality control department has access to adequate facilities, trained personnel, and equipment in order to fulfill its duties and responsibilities.
- Approved written procedures are available for sampling, inspecting, and testing raw materials, packaging materials, in-process drugs, bulk drugs, and finished products.
- Quality control personnel have access to production areas for sampling and investigations as appropriate.
Regulation
- (1) No lot or batch of drug shall be made available for sale unless the sale of that lot or batch is approved by the person in charge of the quality control department.
- (2) A drug that is returned to the fabricator, packager/labeller, distributor referred to in paragraph C.01A.003(b) or importer thereof shall not be made available for further sale unless the sale of that drug is approved by the person in charge of the quality control department.
- (3) No lot or batch of raw material or of packaging/labelling material shall be used in the fabrication or packaging/labelling of a drug unless that material is approved for that use by the person in charge of the quality control department.
- (4) No lot or batch of a drug shall be reprocessed without the approval of the person in charge of the quality control department.
Rationale
The responsibility for the approval of all raw materials, packaging materials and finished products is vested in the quality control department. It is very important that adequate controls be exercised by this department in order to guarantee the quality of the end product.
To maintain this level of quality, it is also important to examine all returned drugs and to give special attention to reprocessed drugs.
Interpretation
- All decisions made by the quality control department pursuant to Regulation C.02.014 are signed and dated by the person in charge of the quality control department or by a designated alternate meeting the requirements described under Section C.02.006.
- The assessment for the release of finished products embraces all relevant factors, including the production conditions, the results of in-process testing, the fabrication and packaging documentation, compliance with the finished product specifications, an examination of the finished package, and if applicable, a review of the storage and transportation conditions.
- 2.1 Deviations and borderline conformances are evaluated in accordance with a written procedure. The decision and rationale are documented. Where appropriate, batch deviations are subject to trend analysis.
- 2.2 Any non-conformances, malfunctions or errors including those pertaining to premises, equipment, sanitation, and testing, that may have an impact on the quality and safety of batches pending release or released, should be assessed and the rationale documented.
- 2.3 The quality control department of the importer/distributor should assure compliance to the current master production documents and the marketing authorization.
- The quality control department ensures that raw materials and packaging materials are quarantined, sampled, tested, and released prior to their use in the fabrication or packaging/labelling of a drug.
- Finished products returned from the market are destroyed unless it has been ascertained that their quality is satisfactory. Returned goods may be considered for resale only after they have been assessed in accordance with a written procedure. The reason for the return, the nature of the product, the storage and transportation conditions, the products condition and history, and the time elapsed since it was originally sold are to be taken into consideration in this assessment. Records of any action taken are maintained.
- 4.1 Documentation is available to support the rationale to place returned goods into inventory for further resale. Wholesalers should obtain guidance from importers/distributors to make an informed decision pertaining to the restock of the product.
- Rejected materials and products are identified as such and quarantined. They are either returned to the vendors, reprocessed, or destroyed. Actions taken are recorded.
- The reworking of any lot or batch of drug is given prior approval by the quality control department. Approval of a reworked lot or batch of a drug by the quality control department is based on documented scientific data, which may include validation. The reworking of products that fail to meet their specifications is undertaken only in exceptional cases. Reworking is permitted only when the following conditions are met:
- 6.1 The quality of the finished product is not affected;
- 6.2 The reworked lot meets specifications;
- 6.3 If it is done in accordance with a defined procedure approved by the quality control department;
- 6.4 All risks have been evaluated;
- 6.5 Complete records of the reworking are kept;
- 6.6 A new batch number is assigned; and
- 6.7 The reworked lot is included in the ongoing stability program.
- The reprocessing of any lot or batch of drug is given prior approval by the quality control department. Approval of a reprocessed lot or batch of a drug by the quality control department is based on documented scientific data, which may include validation. The reprocessing of products that fail to meet their specifications is undertaken only in exceptional cases. Reprocessing is permitted only when the following conditions are met:
- 7.1 The quality of the finished product is not affected;
- 7.2 The reprocessed lot meets specifications;
- 7.3 The reprocessing is done in accordance with a defined procedure approved by the quality control department;
- 7.4 All risks have been evaluated;
- 7.5 Complete records of the reprocessing are kept;
- 7.6 A new batch number is assigned; and
- 7.7 Validation demonstrates that the quality of the finished product is not affected.
- Recovery is not considered to be either a reprocessing or a reworking operation. Guidance regarding recovery is found under Regulation C.02.011, interpretation 28.1.
- The need for additional testing of any finished product that has been reprocessed, or reworked, or into which a recovered product has been incorporated, is evaluated and acted on by the quality control department. A record is maintained.
Regulation
- (1) All fabrication, packaging/labelling, testing, storage, and transportation methods and procedures that may affect the quality of a drug shall be examined and approved by the person in charge of the quality control department before their implementation.
- (2) The person in charge of the control department shall cause to be investigated every complaint on quality that is received respecting and cause corrective action to be taken where necessary.
- (3) The person in charge of the quality control department shall cause all tests or examinations required pursuant to this Division to be performed by a competent laboratory.
Rationale
Pharmaceutical processes and products must be designed and developed taking GMP requirements into account. Production procedures and other control operations are independently examined by the quality control department. Proper storage, transportation, and distribution of materials and products minimize any risk to their quality. Complaints may indicate problems related to quality. By tracing their causes, one can determine which corrective measures should be taken to prevent recurrence. Having tests carried out by a competent laboratory provides assurance that test results are genuine and accurate.
Written agreements for consultants and contract laboratories describe the education, training, and experience of their personnel and the type of services provided and are available for examination and inspection. Records of the activities contracted are maintained.
Interpretation
The quality control department is responsible for the following:
- All decisions made pursuant to Regulation C.02.015 are signed and dated by the person in charge of the quality control department or by a designated alternate who meets the requirements described under Regulation C.02.006 as applicable to the activity.
- Ensuring that guidelines and procedures are in place and implemented for storage and transportation conditions, such as: temperature, humidity, lighting controls, stock rotation, sanitation, and any other precautions necessary to maintain the quality and safe distribution of the drug. Further guidance relating to storage and transportation are detailed in Health Canadas document entitled "Guidelines for Temperature Control of Drug Products during Storage and Transportation (GUI-0069)". Standard operating procedures and records for shipping and receiving are available and contain the following:
- 2.1 a description of the shipping configuration and the type of packaging to be employed for shipping the finished product;
- 2.2 the labelling requirements, including storage conditions and special precautions or warnings, for shipments of the finished product;
- 2.3 mode(s) of transportation approved for shipping the finished product;
- 2.4 the verifications required to ensure that no finished product in the shipment has been tampered with and that there are no damaged containers;
- 2.5 evidence that shipping requirements (e.g., temperature control) have been met if required; and
- 2.6 a written agreement clearly describes the respective responsibilities between the fabricator, the packager/labeller, the distributor, the importer, the wholesaler and the transportation provider relative to the storage and transportation of the drug.
- The sampling of raw materials, packaging materials, in-process drugs, bulk drugs, and finished products is carried out in accordance with detailed written procedures. Samples are representative of the batches of material from which they are taken.
- All complaints and other information concerning potentially defective products are reviewed according to written procedures. The complaint is recorded with all the original details and thoroughly investigated. Appropriate follow-up action is taken after investigation and evaluation of the complaint. All decisions and measures taken as a result of a complaint are recorded and referenced to the corresponding batch records. Complaint records are regularly reviewed for any indication of specific or recurring problems that require attention.
- Establishing a change control system to provide the mechanisms for ongoing process optimization and for assuring a continuing state of control. All changes are properly documented, evaluated, and approved by the quality control department and are identified with the appropriate effective date. Any significant change may necessitate re-validation.
- The tests are performed by a laboratory that meets all relevant GMP requirements.
- 6.1 Laboratory facilities are designed, equipped, and maintained to conduct the required testing.
- 6.1.1 In the microbiology laboratory, environmental monitoring is performed periodically. Microbiological cultures and sample testing are handled in an environment that minimizes contamination.
- 6.1.2 The facility used to perform the sterility testing should comply with the microbial limits of an aseptic production facility which should conform to a Grade A within a Grade B background or in an isolator of a Grade A within an appropriate background and limited access to non-essential personnel.
- 6.2 The individual in charge of the laboratory either (a) is an experienced university graduate who holds a degree in a science related to the work being carried out and has practical experience in his or her responsibility area or (b) reports to a person who has these qualifications C.02.006, interpretation 1).
- 6.3 Laboratory personnel are sufficient in number and are qualified to carry out the work they undertake.
- 6.4 Laboratory control equipment and instruments are suited to the testing procedures undertaken. Equipment and records are maintained as per the interpretations under C.02.005.
- 6.5 Computerized systems are validated, and spreadsheets are qualified.
- 6.6 Water used for microbial and analytical tests meets the requirements of the test or assay in which it is used.
- 6.7 All reagents and culture media are recorded upon receipt or preparation. Reagents made up in the laboratory are prepared according to written procedures and are properly labelled.
- 6.7.1 Prepared media are sterilized using validated procedures and stored under controlled temperatures.
- 6.7.2 Prepared media are properly labelled with the lot numbers, expiration date and media identification. The expiration date of media is supported by growth-promotion testing results that show the performance of the media still meets acceptance criteria up to the expiration date.
- 6.7.3 Sterility and growth-promotion testing are performed to verify the suitability of culture media.
- 6.7.4 All purchased ready to use media received are accompanied by a certificate of analysis with expiry date and recommended storage conditions as well as the quality control organisms used in growth-promotion and selectivity testing of that media.
- 6.7.4.1 Procedures are in place to ensure that media are transported under conditions that minimize the loss of moisture and control the temperature.
- 6.7.4.2 Media are stored according to the vendors instructions.
- 6.7.4.3 Sterility and growth-promotion testing are performed on lots received, unless the vendor is certified. Periodic confirmatory testing is performed for ready to use media received from each certified vendor.
- 6.7.4.4 Records are maintained.
- 6.8 Reference standards are available in the form of the current reference standards listed in Schedule B to the Food and Drugs Act . When such standards have not been established or are unavailable, primary standards can be used. Secondary standards are verified against a Schedule B reference standard or against the primary standard and are subject to complete confirmatory testing at predetermined intervals. All reference standards are stored and used in a manner that will not adversely affect their quality. Records relating to their testing, storage, and use are maintained.
- 6.9 Out of Specification (OOS) test results are investigated to determine the cause of the OOS.
- 6.9.1 Procedures are in place to describe the steps to be taken as part of the investigation.
- 6.9.2 In the case of a clearly identified laboratory or statistical error, the original results may be invalidated, and the test repeated. The original results should be retained and an explanation recorded.
- 6.9.3 When there is no clearly identified laboratory or statistical error and retesting is performed, the number of retests to be performed on the original sample and/or a new sample, and the statistical treatment of the resultant data, are specified in advance in the procedure.
- 6.9.4 All valid test results, both passing and suspect, should be reported and considered in batch release decisions.
- 6.9.5 If the original OOS result is found to be valid, a deviation is raised against the batch and a complete investigation is conducted. The investigation is performed in accordance to written procedures and should include an assessment of root cause, description of corrective actions and preventive actions carried out and conclusions.
- 6.10 To ensure compliance of contractors conducting testing required under Part C, Division 2 of the Food and Drug Regulations:
- 6.10.1 A Canadian contract laboratory must have a relevant valid current establishment licence. A foreign testing site must be listed on a Canadian establishment licence, as described in Health Canadas policy document entitled "Guidance on Evidence to Demonstrate Drug GMP Compliance of Foreign Sites (GUI-0080)";
- 6.10.2 All arrangements for external testing are in accordance with the marketing authorization for the drug product concerned, including the testing of in-process drugs, intermediates, raw materials, packaging materials and all other necessary testing required by Part C, Division 2 of the Food and Drug Regulations;
- 6.10.3 There is a written agreement covering all activities of testing between the contract laboratory and the parties involved. The agreement specifies their respective responsibilities relating to all aspects of testing;
- 6.10.3.1 Technical aspects of the agreement are drawn up by qualified personnel suitably knowledgeable in analysis and GMP;
- 6.10.3.2 The agreement permits audit of the facilities and operations of the external laboratory;
- 6.10.3.3 The agreement clearly describes as a minimum who is responsible for:
- 6.10.3.3.1 collection, transportation and storage conditions of samples before testing;
- 6.10.3.3.2 keeping stability samples at predetermined temperatures and humidity, if applicable;
- 6.10.3.3.3 testing methods to be used, limits and test method validation; and
- 6.10.3.3.4 retention of analytical results and supporting documentation (additional guidance under interpretations of C.02.021).
- 6.10.3.4 No subcontracting of any work should occur without written authorization.
- 6.1 Laboratory facilities are designed, equipped, and maintained to conduct the required testing.
Packaging Material Testing
Regulation
- (1) Each lot or batch of packaging material shall, prior to its use in the packaging of a drug, be examined or tested against the specifications for that packaging material.
- (2) No lot or batch of packaging material shall be used in the packaging of a drug unless the lot or batch of packaging material complies with the specifications for that packaging material.
- (3) The specifications referred to in subsections (1) and (2) shall
- (a) be in writing;
- (b) be acceptable to the Director who shall take into account the specifications contained in any publication mentioned in Schedule B to the Act; and
- (c) be approved by the person in charge of the quality control department.
Rationale
Where a drug product is presented in an inadequate package, the entire effort put into the initial research, product development and manufacturing control is wasted. Drug quality is directly dependent on packaging quality. In many cases (e.g., metered-dose aerosols), packaging quality is critical to the overall performance and effectiveness of the drug product. Faults in the packaging and labelling of a drug product continue to be a cause of drug recalls. Packaging materials are required to be tested or examined prior to their use in a packaging operation to ensure that materials of acceptable quality are used in the packaging of drugs.
Interpretation
- Each packaging material used in the packaging/labelling of a drug is covered by specifications (as defined under C.02.002) that are approved and dated by the person in charge of the quality control department or by a designated alternate who meets the requirements described under Regulation C.02.006, interpretation 1.4. The use of recycled or reprocessed primary packaging components is permitted only after a full evaluation of the risks involved, including any possible deleterious effects on product integrity. Specific provision is made for such a situation in the specifications.
- Where applicable, specifications are of pharmacopeial or equivalent status and are in compliance with the marketing authorization.
- The adequacy of test or examination methods that are not of pharmacopeial or equivalent status is established and documented.
- Only packaging materials released by the quality control department are used in packaging/labelling.
- Outdated or obsolete packaging material is adequately segregated until its disposition.
- The sampling plan for packaging materials should take into account: the quantity received, the level of quality required, the nature of the material (e.g., primary packaging materials and/or printed packaging materials), the production methods, and knowledge of the Quality Assurance system of the packaging materials manufacturer. The number of samples taken should be determined statistically and specified in a sampling plan.
- 6.1 Because of the higher risk of using cut labels, these labels are inspected upon receipt for absence of foreign labels using appropriate methods.
- Sampling should take place in an appropriate environment and with precautions to prevent contamination where necessary.
Regulation
- (1) The examination or testing referred to in section C.02.016 shall be performed on a sample taken
- (a) after receipt of each lot or batch of packaging material on the premises of the person who packages a drug; or
- (b) subject to subsection (2), before receipt of each lot or batch of packaging material on the premises of the person who packages a drug, if
- (i) that person
- (A) has evidence satisfactory to the Director to demonstrate that packaging materials sold to him by the vendor of that lot or batch of packaging material are consistently manufactured in accordance with and consistently comply with the specifications for those packaging materials; and
- (B) undertakes periodic complete confirmatory examination or testing with a frequency satisfactory to the Director,
- (ii) the packaging material has not been transported or stored under conditions that may affect its compliance with the specifications for that packaging material.
- (i) that person
- (2) After a lot or batch of packaging material is received on the premises of the person who packages a drug,
- (a) the lot or batch of the packaging material shall be examined or tested for identity; and
- (b) the labels shall be examined or tested in order to ensure that they comply with the specifications for those labels.
Rationale
Regulation C.02.017 outlines options as to when the testing or examination prescribed by Regulation C.02.016 is carried out. As with raw materials, the purchase of packaging materials is an important operation that involves personnel who have thorough knowledge of the packaging materials and vendor.
Packaging materials originate only from vendors named in the relevant specifications. It is of benefit that all aspects of the production and control of packaging materials be discussed between the manufacturer and the vendor. Particular attention is paid to printed packaging materials; labels are examined or tested after receipt on the premises of the person who packages a drug.
Interpretation
- The testing or examination of the packaging material is performed on a sample taken after their receipt on the premises of the person that packages the drug unless the vendor is certified. A packaging material vendor certification program, if employed, is documented in a standard operating procedure. At a minimum, such a program includes the following:
- 1.1 A written agreement outlines the specific responsibilities of each party involved. The agreement specifies:
- 1.1.1 all the tests to be performed by the vendor, along with the content and format of the certificate of analysis, which exhibits actual numerical results, if applicable, and makes reference to product specifications;
- 1.1.2 that the vendor must inform the drug packager/labeller of any changes in the processing or specifications of the packaging material; and
- 1.1.3 that the vendor must inform the drug packager/labeller of any critical deviations during the manufacturing of a particular batch of a packaging material.
- 1.2 In lieu of a written agreement, an on-site audit of the vendors facilities and controls by qualified personnel is acceptable. The audit ensures that all criteria described under interpretation 1.1 are verified. These audits are performed at an appropriate frequency, and the results are documented;
- 1.3 The certification procedure also outlines how re-testing failures and any subsequent re-qualification is to be addressed;
- 1.4 A document is issued for each vendor verifying that the certification criteria have been met. The document is approved by the quality control department and is updated at an appropriate frequency;
- 1.5 When a certification program is implemented, complete confirmatory examination or testing of a minimum of one lot per year per vendor is required for non-printed packaging material; and
- 1.6 Generally, due to the nature of its operations, a broker or wholesaler of packaging materials cannot be directly certified. However, when a broker or wholesaler supplies materials received from the original vendor without changing the existing labels, packaging, certificate of analysis, and general information, then certification of the original source is still acceptable.
- 1.1 A written agreement outlines the specific responsibilities of each party involved. The agreement specifies:
- Provided that the material is properly identified, the lot of packaging material selected for confirmatory testing may, with the approval of the quality control department, be used in packaging prior to completion of that testing.
- Conditions of transportation and storage are such that they prevent alterations of the characteristics of the packaging material. In order to demonstrate that these conditions have been met, standard operating procedures and records are available and contain the following:
- 3.1 the type of packaging to be employed;
- 3.2 labelling requirements;
- 3.3 mode of transportation;
- 3.4 the type of seal used on the package; and
- 3.5 the verification required to ensure that the package has not been tampered with and that there are no damaged containers.
- Positive identification of all packaging materials, along with examination of all labels and other printed packaging materials, is conducted following their receipt on the premises of the person who packages the drug.
- If a delivery or shipment of packaging material is made up of different batches, each batch is considered as separate for the purposes of sampling, testing, and release.
Finished Product Testing
Regulation
- (1) Each lot or batch of a drug shall, prior to its availability for sale, be tested against the specifications for that drug.
- (2) No lot or batch of a drug shall be available for sale unless it complies with the specifications for that drug.
- (3) The specifications referred to in subsections (1) and (2) shall
- (a) be in writing;
- (b) be approved by the person in charge of the quality control department; and
- (c) comply with the Act and these Regulations.
Rationale
Finished product tests complement the controls employed during the manufacturing process. It is the responsibility of each fabricator, packager/labeller, distributor, and importer to have adequate specifications and test methods that will help ensure that each drug sold is safe and meets the standard under which it is represented.
Interpretation
- Written specifications are approved by the person in charge of the quality control department or by a designated alternate who meets the requirements described under Regulation C.02.006 as applicable to the activity.
- 1.1. The written specifications contain a description of the drug in dosage form. This description includes all properties and qualities, including physical characteristics, identity, purity, and potency. The specifications also include tolerances and a description of all tests used to measure compliance with the established tolerances, in sufficient detail to permit performance by qualified personnel. When a unique identifier is used for identity testing, it is described in the specifications.
- 1.2 Specifications are equal to or exceed a recognized standard as listed in Schedule B to the Food and Drugs Act and are in compliance with the marketing authorization.
- 1.3 Where a recognized pharmacopoeia (Schedule B to the Food and Drugs Act ) contains a specification for microbial content, that requirement is included.
- Test methods are validated, and the results of such validation studies are documented. Method transfer studies are conducted when applicable.
Note: Guidance for the validation of particular types of methods can be obtained in publications such as the ICH document entitled "ICH Q2(R1): Validation of Analytical procedures: Text and Methodology" or any standard listed in Schedule B to the Food and Drugs Act. - All tests are performed according to the approved specifications. These tests may be carried out by the distributor or by their contracted testing laboratory when a written agreement specifically excludes the fabricator from this obligation.
- Any lot or batch of a drug that does not comply with specifications is quarantined pending final disposition and is not made available for sale.
Regulation
- (1) Subject to subsections (3) and (4), in the case of a packager/labeller, distributor referred to in paragraph C.01A.003(b) or importer, the testing referred to in section C.02.018 shall be performed on a sample taken
- (a) after receipt of each lot or batch of the drug on the premises in Canada of the packager/labeller, distributor referred to in paragraph C.01A.003(b) or importer of the drug; or
- (b) subject to subsection (2), before receipt of each lot or batch of the drug on the premises described in paragraph (a), if;
- (i) the packager/labeller, distributor referred to in paragraph C.01A.003(b) or importer
- (A) has evidence satisfactory to the Director to demonstrate that drugs sold to him by the vendor of that lot or batch of the drug are consistently manufactured in accordance with and consistently comply with the specifications for those drugs, and
- (B) undertakes periodic complete confirmatory testing with a frequency satisfactory to the Director, and
- (ii) the drug has not been transported or stored under conditions that may affect its compliance with the specifications for that drug.
- (i) the packager/labeller, distributor referred to in paragraph C.01A.003(b) or importer
- (2) Where the packager/labeller, distributor referred to in paragraph C.01A.003(b) or importer of a drug receives a lot or batch of a drug on the premises in Canada, and the useful life of the drug is more than 30 days, the lot or batch of the drug shall be tested for identity, and the packager/labeller shall confirm the identity after the lot or batch is packaged/labelled.
- (3) The distributor referred to in paragraph C.01A.003(b) of a drug that is fabricated, packaged/labelled and tested in Canada by a person who holds an establishment licence that authorizes those activities is not required to comply with the requirements of subsections (1) and (2) in respect of that drug.
- (4) If a drug is fabricated, packaged/labelled and tested in an MRA country at a recognized building, the distributor referred to in paragraph C.01A.003(b) or importer of that drug is not required to comply with the requirements of subsections (1) and (2) in respect of that drug if
- (a) the address of the building is set out in that persons establishment licence; and
- (b) that person retains a copy of the batch certificate for each lot or batch of the drug received by that person.
Rationale
C.02.019 outlines conditions and exemptions as to when finished product testing is to be performed. Paragraph C.02.019(1) (b) outlines requirements that are to be met if the finished product testing is done before receipt on the premises of the packager/labeller, distributor, or importer of the drug. Paragraphs C.02.019(3) and C.02.019(4) outline exemptions to finished product testing.
Interpretation
- Identity is confirmed by the packager/labeller after the lot or batch is packaged.
Sites Holding a Canadian Establishment Licence
- To demonstrate compliance with finished product specifications, distributors of drugs fabricated, packaged/labelled and tested at Canadian sites are required only to have a copy of the authentic certificate of analysis from the licensed Canadian fabricator. This certificate shows actual numerical results and refers to the product specifications and validated test methods used. Re-testing, including identity testing, is not required.
Recognized Buildings by a Regulatory Authority in a MRA Country
- To demonstrate compliance with finished product specifications, importers of drugs fabricated, packaged/labelled, and tested at recognized buildings authorized by a Regulatory Authority as listed by virtue of Regulation C.01A.019 and identified on their establishment licence are required only to have a batch certificate in the format agreed on by the MRA partners for each lot or batch of the drug received. Re-testing, including identity testing, is not required when the drug is fabricated, packaged/labelled, and tested in an MRA country.
Sites in Non-mra Countries
- For testing other than identity testing, the following conditions are to be met if the packager/labeller or importer chooses to rely on the test results provided by an establishment located in a non-MRA country:
- 4.1 Evidence of ongoing GMP compliance is provided according to a system described in the interpretation of Regulation C.02.012 as demonstrated by listing on the packager/labellers or importers establishment licence;
- 4.2 Each lot is accompanied by an authentic certificate of analysis or by a copy thereof (an electronic copy with an electronic signature is acceptable). The certificate of analysis exhibits actual numerical results and makes reference to the product specifications and validated test methods used;
- 4.2.1 For terminally sterilized products, documented evidence is available from the fabricator to demonstrate that each sterilizer load was individually tested; and
- 4.2.2 For aseptically filled products, evidence demonstrates that samples tested for sterility included the first container filled, the last container filled, and those filled after any significant interruption of work.
- 4.3 Evidence is available to demonstrate that each lot or batch received has been transported and stored in a manner that maintains the quality of the drug. Further requirements are described in interpretation 2 of section C.02.015;
- 4.4 Periodic complete confirmatory testing is performed on at least one lot per year per dosage form per fabricator. For each dosage form, products are selected on a rotational basis;
- 4.4.1 In addition, where multiple drugs are received from the same fabricator, confirmatory testing is carried out for each drug at least once every five years;
- 4.4.2 Confirmatory testing should be performed by an alternate laboratory. In exceptional circumstances (e.g., biologic) the original laboratory may perform confirmatory testing when justified; and
- 4.4.3 No confirmatory testing for sterility, pyrogen, bacterial endotoxin, particulate matter, or general safety is required.
- 4.5 Provided that a specific identity test is performed, a lot or batch of the finished product selected for periodic confirmatory testing may, with the approval of the quality control department, be released for sale prior to completion of all tests.
- Should any failure to conform to finished product testing requirements be identified, an investigation of the extent of the non-compliance is to be conducted. This investigation may lead to reassessment and re-testing of all dosage forms from the fabricator. This procedure may include:
- 5.1 re-evaluation of GMP compliance; and
- 5.2 additional complete confirmatory testing, based on the risk associated with the non-compliance.
- Positive identification of each lot or batch in a shipment of a drug is carried out on a sample taken after receipt on the premises of the packager/labeller or the importer. This identity testing requirement applies to lots received from any non-MRA site. Laboratory chemical/biological testing is required unless the dosage form has unique physical characteristics. Acceptable identity testing methods include the following:
- 6.1 chemical testing;
- 6.2 biological testing; and
- 6.3 physical verification in cases where the product has unique identifiers.
- 6.3.1 The unique identifier principle must be applied before the final chemical or biological identity testing is performed by the fabricator. Where only a portion of a lot is packaged/labelled for Canada, the identity testing must be performed after the unique identifier is applied on the Canadian labelled product.
- 6.3.2 For each product and each strength, uniqueness must be confirmed in writing by the fabricator to the importer at least once a year, as well as whenever a change occurs. When no such confirmation can be obtained, chemical or biological identity testing will be required from the importer.
- 6.3.3 The unique identifier must be confirmed on the certificate of analysis for each lot received from the fabricator.
Note: Label review or examination of the shape and size of the container is not generally considered adequate identity testing. - 6.3.4 The following unique identifiers are considered acceptable:
- 6.3.4.1 Tablets and capsules that are engraved, embossed, or printed with a unique logo;
- 6.3.4.2 Permanent identification on the drugs closure system that indicates the name and strength of the contents. This marking must be applied as part of a continuous filling process and only where the closure cannot be removed without being destroyed;
- 6.3.4.3 Colour closure systems as part of a continuous filling process when the fabricator uses a uniquely coloured cap or closure for only one product and strength;
- 6.3.4.4 A coloured vial, sometimes used for light-sensitive drugs, if it is unique to one product, strength, and fabricator;
- 6.3.4.5 A dedicated facility fabricating only one product;
- 6.3.4.6 Labelling, where preprinted containers are issued to the filling line and where the lot number either is pre-printed or is printed or crimped onto the package in a continuous process; and
- 6.3.4.7 Group 2 products subject to Health Canadas lot release program.
Records
Regulation
- (1) Every fabricator, packager/labeller, distributor referred to in paragraph C.01A.003(b) and importer shall maintain on their premises in Canada, for each drug sold
- (a) master production documents for the drug;
- (b) evidence that each lot or batch of the drug has been fabricated, packaged/labelled, tested and stored in accordance with the procedures described in the master production documents;
- (c) evidence that the conditions under which the drug was fabricated, packaged/labelled, tested and stored are in compliance with the requirements of this Division;
- (d) evidence establishing the period of time during which the drug in the container in which it is sold will meet the specifications for that drug; and
- (e) adequate evidence of the testing referred to in section C.02.018.
- (2) Every distributor referred to in paragraph C.01A.003(b) and importer shall make available to the Director, on request, the results of testing performed on raw materials and packaging/labelling materials for each lot or batch of a drug sold.
- (3) Every fabricator shall maintain on his premises
- (a) the written specifications for the raw material; and
- (b) adequate evidence of the raw materials testing referred to in section C.02.009.
- (4) Every person who packages a drug shall maintain on his premises
- (a) the written specifications for the packaging materials; and
- (b) adequate evidence of the packaging material examination or testing referred to in section C.02.016.
- (5) Every fabricator shall maintain on their premises in Canada
- (a) detailed plans and specifications of each building in Canada at which they fabricate, package/label or test; and
- (b) a description of the design and construction of those buildings.
- (6) Every fabricator, packager/labeller and tester shall maintain on their premises in Canada details of the personnel employed to supervise the fabrication, packaging/labelling and testing, including each person's title, responsibilities, qualifications, experience and training.
- (1) Subject to subsection (2), all records and evidence on the fabrication, packaging/labelling, testing and storage of a drug that are required to be maintained under this Division shall be retained for a period of at least one year after the expiration date on the label of the drug, unless otherwise specified in the person's establishment licence.
- (2) All records and evidence on the testing of raw materials and packaging/labelling materials that are required to be maintained under this Division shall be retained for a period of at least five years after the materials were last used in the fabrication or packaging/labelling of a drug unless otherwise specified in the person's establishment licence.
Every distributor referred to in section C.01A.003, wholesaler and importer of a drug shall retain records of the sale of each lot or batch of the drug, which enable them to recall the lot or batch from the market for a period of at least one year after the expiration date of the lot or batch, unless otherwise specified in their establishment licence.
- (1) On receipt of a complaint respecting the quality of a drug, every distributor referred to in paragraph C.01A.003(b), and importer of the drug shall make a record of the complaint and of its investigation and retain the record for a period of at least one year after the expiration date of the lot or batch of the drug, unless otherwise specified in their establishment licence.
- (2) On receipt of any information respecting the quality or hazards of a drug, every distributor referred to in paragraph C.01A.003(b) and importer of the drug shall make a record of the information and retain it for a period of at least one year after the expiration date of the lot or batch of the drug unless otherwise specified in their establishment licence.
- (1) Every fabricator, packager/labeller, distributor referred to in section C.01A.003 importer and wholesaler shall
- (a) maintain records of the results of the self-inspection program required by section C.02.012 and of any action taken in connection with that program; and
- (b) retain those records for a period of at least three years.
- (2) Every person who fabricates or packages/labels a drug shall
- (a) maintain records on the operation of the sanitation program required to be implemented under section C.02.007; and
- (b) retain those records for a period of at least three years.
Rationale
Good documentation is an essential part of the quality assurance system and should therefore be related to all aspects of GMP. Its aims are to define the specifications for all materials and methods of fabrication, packaging/labelling, and control; to ensure that the quality control department has all the information necessary to decide whether or not to release a batch of a drug for sale; and to provide an audit trail that will permit investigation of the history of any batch that is suspected to be defective.
Evidence that drugs have been fabricated and packaged/labelled under prescribed conditions can be maintained only after developing adequate record systems. The information and evidence should provide assurance that imported drugs are fabricated and packaged/labelled in a like manner to those produced in Canada.
Interpretation
For all sections of Good Manufacturing Practices guidelines, standard operating procedures (SOPs) are retained for reference and inspection. These SOPs are regularly reviewed and kept up to date by qualified personnel. The reasons for any revisions are documented. A system is in place to ensure that only current SOPs are in use. Records of SOPs for all computer and automated systems are retained where appropriate.
All relevant GMP documents (such as associated records of actions taken or conclusions reached) and SOPs are approved, signed, and dated by the quality control department. Documents are not altered without the approval of the quality control department. Any alteration made to a document is signed and dated; the alteration permits the reading of the original information. Where appropriate, the reason for the change is recorded.
Records may be maintained in electronic format provided that backup copies are also maintained. Electronic data must be readily retrievable in a printed format. During the retention period, such records must be secured and accessible within 48 hours to the fabricator, packager/labeller, distributor, or importer.
An electronic signature is an acceptable alternative to a handwritten signature. When used, such a system must be evaluated and tested for security, validity, and reliability, and records of those evaluations and tests must be maintained. The validation of electronic signature identification systems is documented.
Any documentation requested for evaluation by Health Canada is provided in one of the official languages.
- The following documents are maintained by the fabricator, packager/labeller, distributor referred to in paragraph C.01A.003(b) and importer of a drug.
- 1.1 Master production documents as defined in the Glossary of Terms.
- 1.1.1 When the fabricator is located in Canada, specific parts of a master production document considered to be a trade secret or confidential may be held by the fabricator rather than the distributor. When the fabricator is located outside Canada, specific parts of a master production document considered to be a trade secret or confidential may be held on behalf of the distributor or importer by an independent party in Canada. In either case, the distributor or importer must ensure that Health Canada has access to the data in a timely manner.
- 1.1.2 Regardless of whether the fabricator is Canadian or foreign, the master production documents retained by the distributor or importer describe in general terms whatever information has been deleted as a trade secret or confidential.
- 1.2 Evidence that each lot or batch of the drug has been fabricated, packaged/labelled, tested and stored in accordance with the procedures described in the master production documents.
- 1.2.1 This evidence includes manufacturing orders, packaging orders, and test results for raw materials, packaging materials, and drugs in dosage form. However, when the drug is fabricated or packaged outside the premises of the distributor or importer, test results for raw materials and packaging materials need only be made available on request in a timely manner.
- 1.2.2 A certificate of manufacture is considered an acceptable alternative to complete batch documentation, provided that complete documentation is made available on request in a timely manner.
- 1.2.3 Where an importer of drugs from non-MRA countries employs a system involving a "certificate of manufacture", complete batch documentation is obtained at least once per year per drug.
- 1.2.4 A certificate of manufacture alone cannot be employed where reworking has taken place. Should there be changes to the production documents, the complete documentation is provided to the importer or distributor, and any changes that have been made are indicated.
- 1.3 Evidence that the conditions under which the drug was fabricated, packaged/labelled, tested, and stored are in compliance with the requirements of Part C, Division 2 of the Food and Drug Regulations.
- 1.3.1 This evidence includes records generated under subsection C.02.012(2) and evidence of validation. For additional guidance, refer to Health Canadas document entitled "Validation Documentation Requirements and Responsibilities for Drug Fabricators, Packagers/Labellers, Testers, Distributors and Importers (GUI-0042)".
- 1.3.2 Records are maintained detailing the qualifications/experience of any consultant employed for GMP purposes, along with the services that each consultant provides.
- 1.4 Evidence establishing the period of time during which the drug in the container in which it is sold will meet the specifications for that drug.
- 1.4.1 The documentation to be maintained includes the written stability program, the data generated in accordance with that program, and the conclusions leading to the establishment of the period of time during which each drug in the package in which it is sold complies with the specifications for that drug. Also included are data generated as part of the continuing stability program.
- 1.5 For each lot of drug in dosage form, there is adequate evidence of compliance with finished product specifications.
- 1.1 Master production documents as defined in the Glossary of Terms.
- The following documents are maintained by the fabricator, packager/labeller, distributor, wholesaler, and importer of a drug as they relate to all operations in Canada.
- 2.1 Distribution records of all sales of drugs, including those of professional samples.
- 2.1.1 Records of all sales are retained or are kept readily accessible in a manner that will permit a complete and rapid recall of any lot or batch of a drug. This requirement need not necessarily involve tracking by lot number.
- 2.1.2 Records to indicate that all customers who have received a recalled drug have been notified.
- 2.2 Records of the results of the self-inspection program, evaluation, and conclusions, and corrective measures implemented.
- 2.1 Distribution records of all sales of drugs, including those of professional samples.
- The following documents are maintained by every fabricator, packager/labeller, distributor, and importer of a drug:
- 3.1 Records of complaints relating to quality and of subsequent investigations of complaints, including corrective actions taken.
- 3.2 Records concerning information received respecting the quality or hazards of a drug.
- The following documents are maintained by the fabricator:
- 4.1 the written specifications for the raw materials;
- 4.2 the results of the raw material testing;
- 4.3 the sources of the raw materials supplied;
- 4.4 records on the operation of the sanitation program required by Regulation C.02.007; and
- 4.5 detailed plans and specifications of each building where fabrication occurs, including a description of the design and construction.
- The following documents are maintained by the person who packages or labels a drug:
- 5.1 the written specifications for the packaging materials;
- 5.2 the results of the packaging material examinations or testing;
- 5.3 the sources of the packaging materials supplied; and
- 5.4 records on the operation of the sanitation program required by Regulation C.02.007.
- Every fabricator, packager/labeller, and tester maintains:
- 6.1 Details of the personnel employed to supervise the fabrication, packaging/labelling, and testing, including organization charts; each persons title, job description, responsibilities, qualifications, experience, and training; and the name(s) of each persons designated alternate(s).
- Records required under Regulations C.02.021(1), C.02.022, and C.02.023 are retained for a period of at least one year past the expiration date of the drug to which the records apply.
Samples
Regulation
- (1) Every distributor referred to in paragraph C.01A.003(b) and importer of a drug shall retain in Canada a sample of each lot or batch of the packaged/labelled drug for a period of at least one year after the expiration date on the label of the drug, unless otherwise specified in the distributor's or importer's establishment licence.
- (2) The fabricator shall retain a sample of each lot or batch of raw materials used in the fabrication of a drug for a period of at least two years after the materials were last used in the fabrication of the drug, unless otherwise specified in the fabricator's establishment licence.
The samples referred to in section C.02.025 shall be in an amount that is sufficient to determine whether the drug or raw material complies with the specifications for that drug or raw material.
Rationale
These requirements help ensure that responsible officials at the establishment and Health Canada have ready access to those samples that are essential for re-examination should a product quality concern arise.
Interpretation
- A sample of each lot or batch of a finished product is retained in Canada by the distributor referred to in paragraph C.01A.003(b) and by the importer of the drug.
- 1.1 Retention samples are kept in their trade package, or in a container that is equivalent with respect to stability. In the case of large containers of finished products, a smaller representative sample may be retained, as supported by stability data. This allowance does not apply to sterile products.
- 1.2 Retention samples are stored under the conditions indicated on the label.
- 1.3 Retention samples are maintained in accordance with a written procedure.
- 1.4 Retention samples may be stored at another site holding a Canadian Drug Establishment Licence pursuant to a written agreement clearly describing the respective responsibilities of each party.
- A sample of each lot or batch of a raw material (including both active and inactive ingredients), is retained by the fabricator of the drug.
- 2.1 The sample is stored in the same packaging system in which the raw material is stored or in one that is equivalent to or more protective than the vendors packaging system of the raw material.
- 2.2 The sample is stored under the conditions recommended by the vendor.
- 2.3 Retention samples are maintained in accordance with a written procedure.
- In determining the size of sample to be maintained, it is to be kept in mind that Health Canada needs at least enough of the material to carry out tests to determine whether the drug or the raw material complies with its specifications. The fabricator, distributor, or importer may also wish to test the material in the event of a complaint; the sample should therefore be at least double the amount needed to complete all required tests.
- This requirement is not considered to be applicable to the number of units normally required for sterility and pyrogen testing, or to water, solvents, and medical gases.
- Health Canada will consider alternate sample retention sites outside of Canada for distributors and importers of pharmaceutical, radiopharmaceutical, biological, and veterinary drugs, as referred to in sub-section C.02.025(1) if a product specific request is submitted. Further guidance is available in "Alternate Sample Retention Site Guidelines (GUI-0014)".
Stability
Regulation
Every distributor referred to in paragraph C.01A.003(b) and importer shall establish the period of time during which each drug in the package in which it is sold will comply with the specifications.
Rationale
The purpose of the written stability program is to ascertain the normal shelf life of the products that is to determine how long the products can be expected to remain within specifications under recommended storage conditions. The requirements for the stability studies (primary and commitment batches) are outlined in the various Health Canada, ICH, and Veterinary International Conference on Harmonisation (VICH) Guidelines. Each packaged dosage form must be covered by a sufficient amount of data to support its shelf life in its trade package.
Interpretation
- The stability of the drug is determined prior to marketing and prior to adoption of significant changes in formulation, fabrication procedures, or packaging materials that may affect the shelf life of the drug. This determination is made in accordance with Health Canada and ICH guidelines, which include conditions for storage of stability samples.
- 1.1 Accelerated stability data are considered to be preliminary information only. The accelerated data are supported by long term testing. When the shelf-life is assigned based on accelerated data and extrapolated long-term data, it should be verified by additional long term stability data as these data become available.
- 1.2 Stability studies are carried out on the drug in each package type in which it is to be sold in Canada.
- 1.3 For new chemical entities, at least three commercial-scale batches of each strength are sampled to verify or confirm shelf life post-approval, unless such data are submitted as a part of the application for marketing approval. For existing chemical entities (e.g., generic drugs), two commercial-scale batches of each strength are sampled. The principle of bracketing and matrixing designs may be applied if justified.
- 1.4 For imported products, stability studies originating from foreign sites are acceptable provided that the data meet the requirements of the various Health Canada and ICH guidelines regarding stability and that the site can demonstrate GMP compliance.
- 1.5 The shelf life is established based on the date of fabrication.
- 1.6 Stability data are available for drugs before and after constitution, reconstitution or dilution, if applicable.
- 1.7 Analytical test procedures used in stability evaluation are validated in accordance with the ICH document entitled "ICH Q2(R1): Validation of Analytical Procedures: Text and Methodology". Assays are to be stability-indicating, (e.g., sufficiently specific to detect and quantify degradation products and to distinguish between degraded and non-degraded materials). Limits for individual specified, unspecified, and total degradation products are included.
Regulation
Every distributor referred to in paragraph C.01A.003(b) and importer shall monitor, by means of a continuing program, the stability of the drug in the package in which it is sold.
Rationale
The purpose of the written continuing stability program is to monitor the validity of the product shelf life on an on-going basis. It also serves to determine how long the product can be expected to remain within specifications under recommended storage conditions. Each packaged dosage form must be covered by a sufficient amount of data to support its labelled expiry date in its trade package.
Interpretation
- A continuing stability program is implemented to ensure compliance with the approved shelf life specifications. A protocol is available and is implemented for each drug marketed in Canada. A summary of all the data generated, including the evaluation and the conclusions of the study, is prepared. This program includes but is not limited to the following parameters:
- number of batch(es) per strength, packaging, and batch sizes,
- relevant physical, chemical, microbiological or biological test methods,
- acceptance criteria,
- container closure system(s),
- testing frequency,
- storage conditions (and tolerances) of samples, and
- other applicable parameters specific to the drug.
- 1.1 Any differences in the protocol for the continuing stability program and the protocol for the formal stability studies are scientifically justified.
- 1.2 A minimum of one batch of every strength and container closure system of the drug is enrolled into the continuing stability program each year the drug is produced. The principle of bracketing and matrixing designs may be applied if justified in accordance with the ICH document entitled "ICH Q1A (R): Stability Testing of New Drug Substances and Products".
- 1.3 Worst case situations should be addressed by the continuing stability program (e.g., inclusion of reworked or reprocessed lots).
- 1.4 Any confirmed out of specification result, or significant negative trend that may have an impact on the quality of the product should be assessed and may require further stability studies.
- 1.5 For imported products, stability studies originating from foreign sites are acceptable, provided that the data meet the requirements of the various Health Canada and ICH guidelines regarding stability and that the site can demonstrate GMP compliance. It is the importers responsibility to obtain and maintain up to date records associated with the ongoing stability program.
Note: Chart 2.0 Stability is a guide for selecting parameters to be studied in the stability program. Each product must be examined separately.- The inclusion of a sterility test in the stability study of a sterile drug may not be necessary if the container closure system has been proven to be hermetic.
- In addition to preservative content testing, a single regular production batch of the drug is to be tested for antimicrobial preservative effectiveness at the end of the proposed shelf life.
Chart 2.0: Stability
(Information to be used as guide only for selecting parameters to be studied in the stability program. Each product must be examined separately.)
POTENCY
- Tablets: Assay all active ingredients.
- Capsules: Assay all active ingredients.
- Liquids and gels: Assay all active ingredients, plus preservatives, antioxidants, and bacteriostats if effectiveness not checked under Purity section.
- Ointments and creams: Assay all active ingredients, plus preservatives, antioxidants, and bacteriostats if effectiveness not checked under Purity section.
- Powders: Assay all active ingredients, plus complete testing data on reconstituted forms.
- Injectables: Assay all active ingredients, plus preservatives, antioxidants, and bacteriostats if effectiveness not checked under Purity section.
- Suppositories: Assay all active ingredients,
- Aerosols: Assay all active ingredients, plus quantity delivered per spray for metered dose products.
PHYSICAL CHARACTERISTICS
Containers (applicable to all listed dosage forms): (1) Appearance of inner walls and cap interiors colour (2) Integrity of seals (3) Appearance and adhesion of label.
- Tablets: Dissolution, disintegration, odour, hardness, color, appearance.
- Capsules: Dissolution, disintegration, condition of shells, color, appearance
- Liquids and gels: Odour, viscosity, specific gravity, pH, clarity of solution, precipitation of ingredients, non-homogeneity of suspensions, homogeneity (gels)
- Ointments and Creams: Odour, texture, pH, homogeneity, precipitation of ingredients
- Powders: Odour, texture, clarity of solution, homogeneity, pH (after reconstitution), particle size, flow characteristics (inhalation powders)
- Injectables: Clarity, particulate matter, pH, precipitation of ingredients, optical rotation, multiple dose vials: product integrity after initial use
- Suppositories: melting point, homogeneity
- Aerosols: net weight, delivery weight, delivery pressure, pH, delivery effectiveness (e.g. spray pattern and droplet size), number of doses or sprays per package
PURITY
Containers (applicable to all listed dosage forms): (1) Migration of drug into plastic (2) Migration of plasticisers into drug (3) Corrosion
- Tablets: moisture content, microbial test, degradation products
- Capsules: moisture content, microbial test, degradation products
- Liquids and gels: sterility for ophthalmics, particulate matter for ophthalmics, microbial test, degradation products
- Ointments and Creams: sterility for ophthalmics, particulate matter for ophthalmics, microbial test, degradation products
- Powders: moisture content, microbial test, degradation products
- Injectables: sterility, microbial test, degradation products
- Suppositories: microbial test, degradation products
- Aerosols: microbial test, degradation products
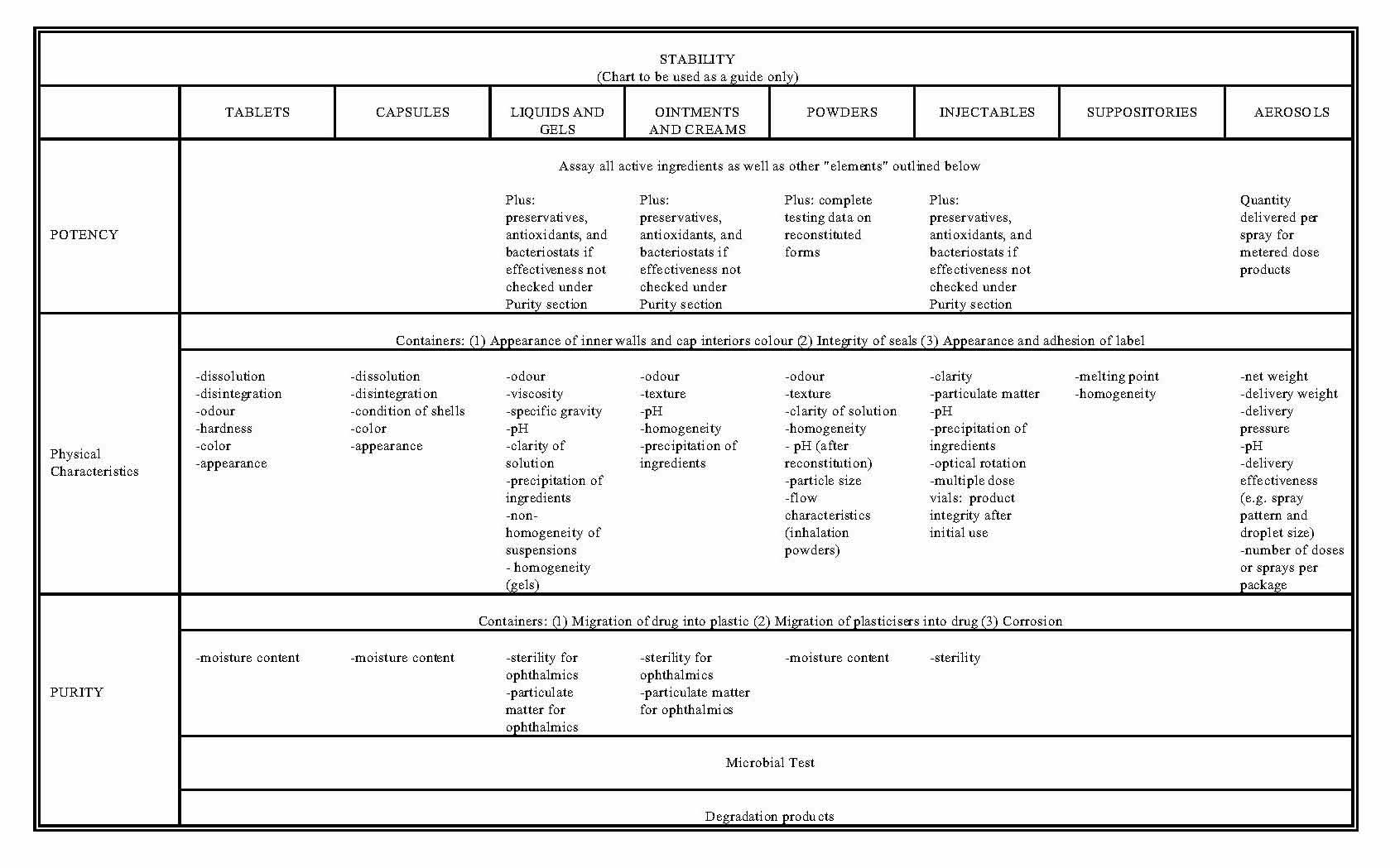{kind=link}
Sterile Products
Regulation
In addition to the other requirements of this Division, a drug that is intended to be sterile shall be fabricated and packaged/labelled
- (a) in separate and enclosed areas;
- (b) under the supervision of personnel trained in microbiology; and
- (c) by a method scientifically proven to ensure sterility.
Rationale
Sterile drugs are susceptible to particulate, pyrogenic and microbiological contamination. Due to the health hazard associated with the use of contaminated sterile products, special precautions are required in the production of these products. The skill, training, and competency of all personnel involved are critical. Quality assurance is important and the production must follow carefully established and validated methods of preparation and sterilization.
Interpretation
General
- Separate packaging and labelling operations of hermetically sealed containers are not subject to Regulation C.02.029 but are covered under Regulation C.02.011.
- When designing procedures for achieving sterility, a number of factors must be considered, particularly airborne microorganisms, particulate matter, the size of the opening of the container, the length of time contents are exposed, and assurance that all the material is exposed to the sterilization condition or process.
- All aqueous-based sterile products must be subjected to terminal steam sterilization, with the following exceptions:
- 3.1 Instances where terminal steam sterilization is not practicable (e.g., where the sterilization process would cause product or packaging degradation). The rationale for the departure from the standard is fully evaluated and documented; and
- 3.2 Aseptic processes that exclude human intervention (e.g., robotics, form-fill-seal, and barrier systems) may be employed in lieu of terminal sterilization, provided that the data developed demonstrate an acceptable level of sterility assurance. Any such methods introduced are fully validated, taking into account all critical factors of the technology used as well as the routine monitoring to be carried out.
- Environmental Grade requirements and monitoring:
Drugs subject to terminal sterilization
- 4.1 Formulation takes place in an environment with a minimum classification of Grade C, provided that the formulated bulk is immediately subjected to its subsequent processing step, (e.g., filtration, sterilization), in order to minimize bio-burden and particulates.
- 4.2 Formulation may take place in a Grade D environment if additional measures (e.g., the use of closed systems of manufacture) are taken to minimize contamination.
- 4.3 Parenterals are filled in an aseptic area with at least a Grade B environment or in a Grade A zone with at least a Grade C background, before terminal sterilization.
- 4.3.1 Parenterals that are to be terminally sterilized may be filled in a Grade C area if the process or product does not pose a high-risk of microbial contamination. Examples of high-risk situations include slow filling operations, the use of wide-necked containers, or the exposure of filled containers to the environment for more than a few seconds before sealing.
- 4.4 Non-parenterals may be filled in a Grade C environment before terminal sterilization.
Drugs not subject to terminal sterilization
- 4.5 Parenterals sterilized by filtration, are formulated in an environment with a minimum classification of a Grade C.
- 4.6 Non-parenteral products may be formulated in a Grade D environment if additional measures are taken to minimize contamination, such as the use of closed systems.
- 4.7 Sterile filtration requires a minimum filter rating of 0.2 µm. The integrity of the filter is verified before and after use by an appropriate method such as a bubble point, diffusion or pressure hold tests.
- 4.8 Filling operations are performed under local Grade A conditions within a Grade B background environment. However a lower-grade background environment may be acceptable if specialized automated or barrier techniques are employed and if those techniques are validated to demonstrate that their use has no negative impact on the quality of the drug.
Drugs not subject to filtration or terminal sterilization
- 4.9 Sterile products subject to neither filtration nor terminal sterilization, are produced from sterile raw materials and packaging components in an aseptic area under local Grade A conditions with a Grade B background. Additional information pertaining to blow-fill-seal and isolator technology is provided in interpretations 82 and 83 under the Sterile Products section.
- The air standards described in the following tables are to be achieved throughout the area when it is occupied and in operation. In the operational condition for Grade A zone, the air standards apply in the zone immediately surrounding the drug whenever it is exposed, and with at least a Grade B background. It may not always be possible to demonstrate conformity with air standards for non-viable particulates at the point of fill when filling is in progress, owing to the generation of particles or droplets by the product itself.
- 5.1 The "at rest" state is the condition where the installation is complete, including fabrication equipment installed and present in an operational condition but not in use and with operating personnel absent. The "in operation" state should not commence until all the predefined criteria have been met with the equipment and personnel in place.
- The classification of aseptic and clean areas is based on environmental results obtained using acceptable standardized air sampling methods. Such methods take into account the volume and number of samples taken at each location and the total number of sampling locations. The number of sampling locations is based on room volume and on the nature of the operations being undertaken. Sampling methods used during the operational state do not interfere with zone protection.
- Radiation sterilization is used mainly for heat-sensitive materials. Since drugs and packaging materials are radiation-sensitive, this method is permissible only when, prior to use, evidence has confirmed the absence of any damaging effects on the material.
- Ethylene Oxide sterilization is used only when other methods are not practicable. Evidence must be available to show the absence of any damaging effect on the drug when this method is used. The conditions and time allowed for degassing the drug are such that residual gas and reaction products are reduced to clearly defined acceptable limits.
- Ultraviolet irradiation is not an acceptable method of sterilization.
Basic Environmental Standards for the Manufacture of Sterile Products
| at rest (Note 5) | in operation | |||
|---|---|---|---|---|
| Grade | Maximum permitted number of particles / m3 equal to or above (Note 3) | |||
| 0.5 µm | 5 µm | 0.5µm | 5 µm | |
| A (Note 1) | 3,520 | 20 | 3,520 | 20 |
| B (Note 2) | 3,520 | 29 | 352,000 | 2,900 |
| C (Note 2) | 352,000 | 2,900 | 3,520,000 | 29,000 |
| D (Note 2) | 3,520,000 | 29,000 | not defined (Note 4) | not defined (Note 4) |
Notes :
1. Unidirectional airflow systems provide a homogeneous air speed of 0.45 meters/ second +/- 20% (guidance value) at the working position in open clean room applications. The maintenance of unidirectional air flow should be demonstrated and validated. A unidirectional air flow and lower velocities may be used in closed isolators and glove boxes.
2. In order to attain air Grades B, C, and D, the number of air changes will be related to the size of the area and to the equipment and personnel present in the area.
3. Low values for contaminants are reliable only when a large number of air samples are taken. Adequate data is available to generate confidence that the required conditions are met throughout the duration of the operations.
4. The requirement and limits for this area will depend on the nature of the operations carried out.
5. The particulate conditions given in the at rest column are to be achieved after a short clean-up period (15 to 20 minutes) in an unmanned state after completion of operations.
| Recommended limits for microbial contamination (refer to Note a and e) | ||||
|---|---|---|---|---|
| GRADE | air sample cfu/m3 | settle plates (diameter 90mm), colony-forming units (cfu)/4 hours (refer to Note b) | contact plates (diameter 55mm), colony-forming units (cfu)/plate (refer to Note c) | glove print (5 fingers) colony-forming units (cfu)/glove (refer to Note d) |
| A | < 1 | < 1 | < 1 | < 1 |
| B | 10 | 5 | 5 | 5 |
| C | 100 | 50 | 25 | - |
| D | 200 | 100 | 50 | - |
Notes :
(a) These are average values; however, averaging of results can mask unacceptable localized conditions, therefore individual excursions should be treated with caution. Appropriate alert and action limits should be set for microbial monitoring. If the limits are exceeded, operating procedures should prescribe investigation and corrective action. Samples from Grade A critical area environments should normally yield no microbial contaminants.
(b) Individual settle plates may be exposed for less than 4 hours.
(c) The surface sampled with a contact plate is subject to appropriate cleaning immediately after use.
(d) Monitoring is conducted after critical operations are complete.
(e) All indicated sampling methods are required unless alternative methods demonstrate equivalency.
Premises
- To the extent possible, premises are designed to avoid the unnecessary entry of supervisory or control personnel. Grade B areas are designed so that all critical operations can be observed from outside.
- To prevent the shedding or accumulation of dust and other particulate matter, ceilings, floors, and walls in aseptic areas, and floors and walls in clean areas, have smooth impervious surfaces that permit the repeated application of cleaning and disinfecting agents.
- To reduce the accumulation of dust and to facilitate cleaning, projecting ledges or shelves and electrical and mechanical equipment are kept to a minimum. Covings are required where walls meet floors or ceilings. Walls, floors, and ceilings form an effective seal around any traversing pipe or duct.
- False ceilings are sealed to prevent contamination from the space above them.
- Uncleanable devices, such as certain sliding-door rails, are avoided.
- Where required, sinks and drains are designed, located, and maintained so as to minimize risks of microbial contamination. Sinks and drains are excluded from areas where aseptic operations are carried out.
- Hand-washing facilities are provided only in changing rooms.
- Changing rooms are designed as airlocks and are used to separate the different stages of changing, thus minimizing microbial and particulate contamination of protective clothing. They are effectively flushed with filtered air. In the final stage, they are, at rest, the same grade as the area into which they lead.
- Access to clean and aseptic areas is provided only through air-locks. Doors to airlocks are arranged so that, either by design or by procedure, only one side or door may be opened at one time (except for emergencies).
- The air for clean and aseptic areas is supplied through filters of suitable efficiency. Unidirectional air flow systems are of appropriate design.
- The filtered air supply for clean and aseptic areas is designed to provide a fabrication environment that meets the required grade classifications. Under all operational conditions, a positive pressure of filtered airflow is maintained in relation to surrounding areas of a lower grade. Particular attention is paid to protecting critical areas, that is, the immediate environment in which the sterilized drug product, containers, and closures are exposed.
- 20.1 The air system should be provided with appropriate terminal filters such as HEPA for Grades A, B and C. An intact HEPA filter should be capable of retaining at least 99.97% of particulates greater than 0.3 µm in diameter.
- 20.2 In Grade A areas the air velocity should be sufficient to protect exposed product, product contact components and product contact surfaces from environmental contamination, by sweeping particles away from the filling/closing area and maintain a unidirectional airflow during operations. Air velocity measurements should be taken at locations where meaningful and reproducible results can be obtained. Such locations should normally be not more than 30 cm away from the work site, within the air flow.
- 20.3 In "critical areas" HEPA filters should be leak tested at least twice a year. The purpose of performing regularly scheduled leak tests is to detect leaks from the filter media, filter frame or seal. The aerosol selected for HEPA leak testing should not support microbial growth and should be composed of a sufficient number of particles at approximately 0.3 µm.
- 20.4 HEPA filtered air should be supplied in critical areas at a velocity sufficient to sweep particles away from the filling/ closing area and maintaining a unidirectional airflow. In situ air pattern analysis should be conducted at the critical area to demonstrate unidirectional air flow, sweeping action over and away from the product, and the absence of turbulence or eddy currents.
- 20.5 For aseptically filled products, the transportation and loading of partially sealed containers, such as between filling and lyophilization, should be under Grade A conditions.
- Warning systems alert personnel when air pressure or airflow falls below established limits. Pressure differentials between areas are monitored and recorded where such differences are of importance.
- 21.1 Pressure differentials between 10 and 15 Pa (0.10 cm and 0.15 cm or 0.04" and 0.06" of water) are considered effective between zones of different environmental classifications.
- 21.2 Pressure differentials between aseptic areas and adjacent areas should be monitored continuously and documented. All alarms should be documented and deviations from established limits should be investigated. When doors are open, outward air flow should be sufficient to minimize ingress of contamination. It is critical that the time the door can remain ajar be strictly controlled.
- Airflow patterns do not present a contamination risk. For example, care is taken to ensure that airflows do not distribute particles from a particle-generating person, operation, or machine to a zone of higher product risk.
- All work with microorganisms and other infectious agents known to require special precautions in manipulation is safely segregated.
Equipment
- Equipment is designed in such a way as to facilitate cleaning, disinfection, or sterilization. Electronic accessories and those parts of large equipment that are not readily amenable to such treatment are appropriately and adequately sealed or effectively isolated.
- To the extent possible, equipment fittings and services are designed and installed so that operations, maintenance, and repairs can take place outside clean or aseptic areas.
- When equipment maintenance is carried out within aseptic areas during operations, sterilized instruments and tools are used. If the required standards of cleanliness and/or asepsis are not maintained during the maintenance work, the area is cleaned and disinfected before processing recommences.
- All equipment, including sterilizers, air-filtration systems, and water-treatment systems, are subject to planned maintenance, validation, and monitoring. Following maintenance/validation, the approval for use of the equipment is documented.
- For aseptically filled products, conveyor belts do not pass through a partition from a Grade A or Grade B area to an area of lower cleanliness unless the belts are continuously sterilized (e.g., they pass through a sterilizing tunnel).
- Vent filters used on equipment directly involved in aseptic filling such as receiving tanks, transfer lines, and surge vessels should be integrity tested upon installation where practical or prior to installation and after batch completion.
- Vent filters used on stationary equipment such as Water for Injection (WFI) storage tanks and sterilizers, and membrane filters used to filter compressed gases, should be integrity tested prior to installation and periodically there after.
- Filter integrity test failures should be investigated. Filters should be replaced according to written criteria at appropriate, predefined intervals, and documented.
- All critical surfaces that come in direct contact with sterile materials should be sterile.
Water Treatment Systems
- Water treatment facilities are designed, constructed, and maintained so as to ensure the reliable production of water of an appropriate quality. They are not operated beyond their designed capacity. Water is produced, stored, and distributed in a manner that minimizes microbial growth and prevents other types of contamination.
- The quality of the raw feed water is established by specification and is periodically monitored for compliance. The sampling plan takes seasonal variations into account. Records are maintained.
- Purified water is used as feed water for WFI systems and for clean steam generators. WFI is produced either by distillation or by reverse osmosis.
- WFI is used in the formulation of parenteral, irrigation, and intra-ocular products. Purified water may be used in the formulation of ophthalmic products.
- Purified water and WFI systems are validated that is, the ability of the systems and its procedures to maintain the appropriate level of chemical and microbial control, taking seasonal variations into account, is demonstrated and documented.
- Alert and action limits should be established for bacterial endotoxins and microbial load. These limits should meet any standard listed under Schedule B to the Food and Drugs Act .
- WFI storage tanks are equipped with hydrophobic bacterial-retentive vent filters.
- Sanitization or regeneration of water systems is carried out according to a predetermined schedule and also whenever established microbial counts are exceeded within any of the system's components. Consideration is given to controlling biofilm formation.
- The WFI system is maintained at an elevated temperature and kept in continuous movement. Water velocity through pipes is sufficient to prevent microbial attachment.
- Piping is sloped to provide for complete drainage of the system. The system is free of dead legs.
- All metal surfaces in contact with WFI should be, as a minimum, 316 stainless steel, and should be passivated upon or prior to installation and after changes.
- While in use during processing, WFI is sampled daily from at least two points on a rotating basis so as to cover all outlets.
- Revalidation of water systems is required if any of the following situations arise:
- 45.1 Unscheduled or extensive maintenance is performed on the system;
- 45.2 New or revised sections or components are added to or removed from the system; and
- 45.3 The system exhibits an out-of-control trend in either chemical or microbiological parameters.
- The extent of the re-validation work necessary is determined jointly by the personnel from the quality control, engineering, production, and any other appropriate departments. A pre-approved protocol is signed and dated by the parties involved.
Note: Refer to interpretations 74 and 75 in the "Sterile Products" section for further requirements regarding water to be used in fabrication.
Personnel
- In addition to the requirements outlined under Regulation C.02.006, the personnel responsible for the fabrication and testing of sterile products have had training in microbiology.
- High standards of personal hygiene and cleanliness are maintained. Personnel involved in the fabrication of sterile preparations are instructed to report any condition that may cause the shedding of abnormal numbers or types of contaminants. Periodic health checks for such conditions are conducted, and appropriate action (e.g., deciding whether to allow an individual to be involved in a particular operation) is taken by designated qualified personnel when necessary.
- All personnel (including those whose duties involve cleaning and maintenance) employed in such areas receive regular training in disciplines relevant to the correct fabrication of sterile products, including reference to hygiene and to the basic elements of microbiology. When outside personnel who have not received such training (e.g., building or maintenance contractors) need to be brought in, particular care is taken with regard to their supervision.
- Personnel who have been engaged in the processing of animal-tissue materials or of cultures of microorganisms other than those used in the current fabrication process do not enter areas where sterile products are fabricated unless rigorous and clearly defined decontamination procedures have been followed.
- Only the minimum number of personnel required are present in areas where sterile products are fabricated; this is particularly important during aseptic processes. Inspections and controls are conducted from outside such areas to the extent that such an approach is possible.
- Outdoor clothing is not brought into these areas. Personnel entering the changing rooms are already clad in standard protective garments designed for factory facilities. Changing and washing follow written procedures.
- The clothing worn by personnel and its quality are adapted to the particular process and workplace, and the clothing is worn in such a way as to protect the product from contamination.
- Clothing is appropriate to the environmental grade of the area where the personnel will be working. Written gowning procedures must be established for each environmental grade. Personnel must be trained according to these procedures prior to entry. Such training must be documented. Descriptions of the clothing required for each grade are given below.
For Grades A and B areas: Headgear totally encloses the persons hair, as well as any beard or mustache, the headgear is tucked into the neck of the suit; a face mask is worn to prevent the shedding of droplets; sterilized non-powdered rubber or plastic gloves and sterilized or disinfected footwear are worn; trouser-bottoms are tucked inside the footwear and garment sleeves are tucked into the gloves. The protective clothing sheds virtually no fibres or particulate matter and retains particles shed by the body.
For Grade C areas: The persons hair, as well as any beard or mustache, is covered. A one- or two-piece trouser suit, gathered at the wrists and with a high neck, and appropriate shoes or overshoes are worn. The protective clothing sheds virtually no fibres or particulate matter.
For Grade D areas: The persons hair, as well as any beard or mustache, is covered. Protective clothing and appropriate shoes or overshoes are worn.
- For every worker in an aseptic (Grades A and B) area, clean sterilized protective garments are provided at each re-entry. Gloves are regularly disinfected during operations. Masks and gloves are changed prior to every new working session.
- Clothing used in clean and aseptic areas is laundered or cleaned in such a way that it does not gather additional particulate contaminants that can later be shed. Separate laundry facilities for such clothing are desirable. If fibres are damaged by inappropriate cleaning or sterilization, there may be an increased risk of shedding particles. Washing and sterilization operations follow standard operating procedures. Repair of clothing is carried out using appropriate materials (e.g., non-shedding thread).
- Behavioural techniques aimed at maintaining sterility should be employed by personnel working in aseptic areas. These include:
- 57.1 moving slowly and deliberately;
- 57.2 keeping the entire body out of the path of unidirectional airflow;
- 57.3 approaching any manipulation in a manner that does not compromise sterility of the product; and
- 57.4 maintaining proper gown control.
Sanitation
- Walls, floors, ceilings, and equipment in clean areas are cleaned and, when required, disinfected in accordance with a written procedure. This procedure differentiates between procedures that are followed daily and those that are undertaken whenever fabrication of a different drug is about to begin.
- Walls, floors, ceilings, and equipment in aseptic areas are cleaned and, when required disinfected in accordance with a written procedure. This procedure differentiates between the cleaning and disinfection procedures that are followed daily and those that are undertaken whenever fabrication of a different drug is about to begin.
- Disinfectants and cleaning agents to be used in aseptic processing areas should be sterile.
- 60.1 A disinfectant program should also include a sporicidal agent since many common disinfectants are ineffective against spores.
- Disinfectants and cleaning agents are monitored for microbial contamination and are sterile when used in Grades A or Grade B areas. Dilutions are kept in previously cleaned and sterilized containers and are not stored for long periods unless sterilized. Partly emptied containers are not topped up.
- Fumigation of clean and aseptic areas may be useful for reducing microbiological contamination in inaccessible places.
- The cleaning procedures are validated, and the disinfection procedures are monitored.
- 63.1 The suitability, efficacy and limitations of disinfecting agents and procedures should be assessed including their ability to adequately remove potential contaminants from surfaces.
Manufacturing Control
- During all processing stages, precautions are taken to minimize contamination.
- Preparations containing live microorganisms are neither made nor transferred into containers in areas used for the processing of other pharmaceutical products. Preparations containing only dead organisms or bacterial extracts may be dispensed into containers, in the same premises as other sterile pharmaceutical products, provided that validated inactivation procedures and validated cleaning procedures are followed.
- Activities in these areas are kept to a minimum, especially when aseptic operations are performed. The movement of personnel is controlled and methodical in order to avoid excessive shedding of particles and organisms. The ambient temperature and humidity are controlled and monitored to ensure the comfort of personnel.
- Prior to sterilization, possibilities for microbiological contamination of raw materials and packaging materials are kept to a minimum. Specifications include requirements for microbiological quality when monitoring has indicated the need for such requirements.
- Articles are sterilized and passed into the aseptic areas by the use of doubled-ended sterilizers equipped with interlocking doors or by another validated method.
- Written standards are available specifying the air quality, including viable and non viable counts, to be maintained in clean and aseptic areas. Viable and non viable counts are taken at least once per shift in aseptic areas, while aseptic filling and aseptic fabrication operations are carried out, and at appropriate intervals in areas where other fabrication takes place.
- 69.1 Air monitoring of "critical areas", Grade A environments should normally yield no microbiological contaminants. Contamination in a critical area should be investigated and corrective actions implemented.
- 69.2 In Grades A and B areas regular monitoring for particulates and viables should be performed during each operating shift. The total sample volume should not be less than 1 cubic meter per sample for Grades A and B areas and preferably also in Grade C. For Grade C areas, monitoring frequency is justified based on the criticality of the operations and historical data for the specified area. Where product is exposed in a Grade C area, monitoring should be conducted at least once per week. Sampling locations should be based on a formal risk analysis study considering historical results and those obtained during the classification of rooms.
- 69.3 Personnel working in aseptic processing areas should be microbiologically monitored once per shift. Typical monitoring sites should include operators gloves and one gown site. Manual aseptic production processes require more aggressive personnel monitoring than automated aseptic production processes.
- The presence of containers and materials liable to generate fibres is minimized in clean and aseptic areas.
- Following cleaning and sterilization, components, bulk-product containers, and equipment are handled in such a way that they are not re-contaminated. The stage of processing of components, bulk-product containers, and equipment is properly identified.
- The interval between cleaning and sterilization of components, bulk-product containers, and equipment, as well as between their sterilization and use, is as short as possible and subject to a time-limit appropriate to the validated storage conditions.
- The time between the start of the preparation of a solution and its sterilization or filtration through a bacteria-retentive filter is as short as possible. A maximum permissible time is validated for each product, taking into account its composition and the prescribed method of storage.
- Water used in the preparation of parenterals is tested for endotoxins and complies with its approved specifications.
- Water used for the final rinsing of container components that are used for parenteral drugs is tested for endotoxins unless such components are depyrogenated subsequently.
- Compressed air and gases that come in direct contact with the product/container primary surfaces must be of appropriate chemical, particulate and microbiological purity, free from oil, and must be filtered through a sterilizing filter at the point of use.
- The microbial contamination of products (bioburden) is minimal prior to sterilization. The maximum acceptable bioburden is established on a product by product basis, and should be founded on adequate product/process design and control. Acceptable bioburden levels are further demonstrated through the execution of validation studies. This limit is related to the efficiency of the method to be used and to the risk of pyrogens and bacterial endotoxins, which are not removed by sterilization. The bioburden should be monitored before sterilization. All solutions, particularly large-volume parenterals, are passed through a bacteria-retentive filter; if possible, this filtering occurs immediately before the filling process. Where aqueous solutions are held in sealed vessels, any pressure-release outlets are protected (e.g., by hydrophobic microbial air filters).
- Water, gas, or any heating or cooling fluid in contact with filled drug product containers (e.g., for the cool down cycle of sterilization loads) should present a low risk of microbial contamination.
- Documented evidence that establishes the validation and validity of each sterilization process is available. The validation and validity of the sterilization process are verified at scheduled intervals, at least annually, and also whenever significant modifications or changes are made to the equipment. Loading patterns for all sterilization processes are established and validated.
- 79.1 Sterilization by heat
Chemical or biological indicators may also be used, but should not take the place of physical measurements.- 79.1.1 Sufficient time is allowed for the whole load to reach the required temperature before measurement of the sterilizing time-period begins. This time is determined for each type of load to be processed.
- 79.1.2 After the high-temperature phase of a heat sterilization cycle, precautions are taken to prevent contamination of a sterilized load during cooling.
- 79.2 Sterilization by moist heat
- 79.2.1 Both temperature and pressure controls are used to monitor the process. Control instrumentation is independent from both monitoring instrumentation and recording charts. Where automated controls and monitoring systems are used for these applications, they are fully validated to ensure that the critical process requirements are met. System and cycle faults are registered by the system and observed by the operator. The reading of the independent temperature indicator is periodically monitored. For sterilizers fitted with a drain at the bottom of the chamber, it may also be necessary to record the temperature at this position throughout the sterilization period. There are frequent leak tests on the chamber when a vacuum phase is part of the cycle.
- 79.2.2 The items to be sterilized, other than products in sealed containers, are wrapped, if necessary, in a material that allows the removal of air and the penetration of steam but that prevents re-contamination after sterilization. All parts of the load are in contact with the sterilizing agent at the required temperature and pressure for the required time.
- 79.2.3 Clean steam is used for sterilization and does not contain additives at a level that could cause contamination of product or equipment.
- 79.3 Sterilization by dry heat
- 79.3.1 The process used includes air circulation within the chamber and the maintenance of a positive pressure to prevent the entry of non-sterile air. Any air admitted passes through a HEPA filter.
- 79.4 Sterilization by radiation
- 79.4.1 The radiation dose is measured during the sterilization procedure. For this purpose, dosimetry indicators that are independent of dose rate are to be used, giving a quantitative measurement of the dose received by the product itself. Dosimeters are inserted into the load in sufficient number and close enough together to ensure that there is always a dosimeter in the irradiator. Where plastic dosimeters are used, they are within the time limit of their calibration. Dosimeter absorbencies are read within a specified time period after exposure to radiation.
- 79.4.2 Biological indicators may be used as an additional control.
- 79.4.3 Materials handling procedures are designed so as to prevent mix-up between irradiated and non-irradiated materials. Radiation-sensitive colour disks are used on each package to differentiate between packages that have been subjected to irradiation and those that have not.
- 79.4.4 The total radiation dose is administered within a predetermined time span.
- 79.5 Sterilization with Ethylene Oxide (EtO)
- 79.5.1 Direct contact between gas and microbial cells is essential; precautions are taken to avoid the presence of organisms likely to be enclosed in such material as crystals or dried protein. The nature and quality of packaging materials can significantly affect the process.
- 79.5.2 Before exposure to gas, materials are brought into equilibrium with the humidity and temperature required by the process. The time required for this is balanced against the opposing need to minimize the time before sterilization.
- 79.5.3 Each sterilization cycle is monitored with suitable biological indicators, using the appropriate number of test pieces distributed throughout the load. The information so obtained is part of the batch record.
- 79.5.4 For each sterilization cycle, records are made of the time taken to complete the cycle, the pressure, the temperature and the humidity within the chamber during the process, the gas concentration, and total amount of gas used. The pressure and temperature are recorded throughout the cycle on a chart. The readings are part of the batch record.
- 79.5.5 After sterilization, the load is stored in a controlled manner under ventilated conditions to allow residual gas and reaction products to reduce to the defined level. This process is validated.
- 79.6 Biological indicators are considered only as an additional method for monitoring the sterilization, except in the case of ethylene oxide sterilization, where they are a normal part of the monitoring criteria. If they are used, strict precautions are taken to avoid transferring microbial contamination from them.
- 79.7 Records are available indicating that the requirements for each sterilization cycle have been met. These records include all recording charts (e.g., time/temperature).
- 79.8 A clear visual means is used for differentiating products that have not been sterilized from those that have been sterilized. Each basket, tray, or other carrier of products or components should be clearly labelled with the name of the material, its batch number, and an indication of whether or not it has been sterilized. Such indicators as autoclave tape or radiation sensitive colour disks may be used, where appropriate, to indicate whether or not a batch (or sub-batch) has passed through a sterilization process. These visual means are not intended to give an indication that the lot is sterile.
Note: Refer to Health Canada process validation guidelines for further guidance on these processes.
- 79.1 Sterilization by heat
- Aseptic Filling Operations
A written standard designed to test the efficiency of the overall aseptic filling operation is maintained. This standard includes a requirement to perform normal aseptic filling operations using sterile media.
- 80.1 The use of nutrient media that support microbial growth in trials to simulate aseptic operations (i.e., sterile media fills, "broth fills") is a valuable part of the overall validation of an aseptic process. Such trials have the following characteristics:
- 80.1.1 The trials simulate actual operations as closely as possible and also take into consideration worst case conditions;
- 80.1.2 The medium or media selected are capable of growing a wide spectrum of microorganisms, including those that would be expected to be found in the filling environment. Each batch of media used for process simulation must be tested for its growth promotion capabilities; and
- 80.1.3 The trials include a sufficient number of units of production to give a high degree of assurance that low levels of contamination, if present, would be detected.
- 80.2 The number of containers used for a media fill should be sufficient to allow a valid evaluation. If batches smaller than 5,000 units are filled, the minimum number of containers used for process simulation with sterile nutrient media should be equal to the maximum commercial batch size. The target is zero positives. Any positive unit indicates a potential problem regardless of the run size. All positives should be identified, and should result in a thorough, documented investigation and any identified corrective action implemented.
- 80.2.1 Recommended criteria for assessing state of aseptic line control are as follows:
- 80.2.1.1 When filling fewer than 5000 units, no contaminated units should be detected. One (1) contaminated unit is considered cause for re-validation, following an investigation.
- 80.2.1.2 When filling from 5,000 to 10,000 units, one (1) contaminated unit should result in an investigation, including consideration of a repeat media fill. Two (2) contaminated units are considered cause for re-validation, following investigation.
- 80.2.1.3 When filling more than 10,000 units, one (1) contaminated unit should result in an investigation. Two (2) contaminated units are considered cause for re-validation, following investigation.
- 80.2.2 Investigations of gross media fill failures should include an assessment of the potential impact on sterility assurance of batches filled since the last successful media fill.
- 80.2.1 Recommended criteria for assessing state of aseptic line control are as follows:
- 80.3 A matrix approach to process simulation may be developed for each filling line, and should include elements such as the type of products filled, size of lots, container and closure configuration, fill volume, line speed, filling line configuration and components, and sterile hold times.
- 80.4 A process simulation run should be performed of sufficient duration to cover all routine manipulations and operations, various interventions known to occur during normal production, as well as worst case situations.
- 80.5 The process simulation test should simulate all the specific manufacturing steps, such as product transfer, sterile filtration, filling, transfer of semi-stoppered vials to the lyophilizer, the lyophilization process, and stoppering and crimping of vials.
- 80.6 The process simulation test program incorporates a representative number, type, and complexity of normal interventions that occur with each run, as well as non-routine interventions, and events (e.g., maintenance, stoppages, equipment, adjustments). A pre-defined list of all permitted interventions should be documented and incorporated into process simulation on a periodic basis.
- 80.7 The fill volume should be sufficient to assess potential microbial contamination, and to ensure the complete contact with all sterile surfaces inside the container when the container is inverted and swirled. Consideration should be given to incubation of filled vials with media contacting the closure system (e.g., inverted storage).
- 80.8 Incubation conditions should be suitable for recovery of bioburden and environmental isolates. Following the aseptic processing of the medium, the filled containers are incubated at 22.5 °C ± 2.5 °C or at 32.5 °C ± 2.5 °C. All media filled containers should be incubated for a minimum of 14 consecutive days. If two temperatures are used for incubation of media filled samples, then these filled containers should be incubated for at least 7 consecutive days at each temperature starting with the lower temperature.
- 80.9 Initial validation or re-validation requires three successful process simulation tests. In the absence of observed issues with respect to environmental monitoring or sterility testing, re-validation should take place at least semi-annually with a minimum of a single process simulation. Whenever a significant alteration in the product, premises, equipment, or process occurs or failure of process simulation occurs, re-validation is required.
- 80.10 Every person who is normally allowed to be in the filling room during aseptic filling operations must participate in the process simulation test, during which they must perform their normal assigned duties. Only trained and qualified personnel who have successfully participated in a process simulation test should be permitted to participate in aseptic processing. Records should be maintained.
- 80.1 The use of nutrient media that support microbial growth in trials to simulate aseptic operations (i.e., sterile media fills, "broth fills") is a valuable part of the overall validation of an aseptic process. Such trials have the following characteristics:
- Sterilization by Filtration
- 81.1 The sterilizing filtration should be validated to reproducibly remove viable microorganisms from the process stream, producing a sterile effluent. Validation studies should consider factors that can affect filter performance, which generally include viscosity and surface tension of the solution to be filtered, pH, compatibility of the material or formulation components with the filter, pressures, flow rates, batch volume, maximum use time, temperature, osmolality, and the effects of hydraulic shock.
- 81.2 The microorganism Brevundimonas diminuta (ATCC 19146) when properly harvested, grown and used, is a common challenge microorganism for sterilizing filters because of its size (0.3 µm mean diameter). A challenge concentration of at least 107 organisms per cm2 of effective filtration area should generally be used, and should result in no passage of the challenge microorganism. Direct inoculation into the drug formulation is the preferred method because it provides an assessment of the effect of the drug product on the filter matrix and on the challenge organism (except for products with inherent bactericidal activity against this microbe, or oil-based formulations).
- 81.3 Use of redundant sterilizing filters should be considered. This second filtration via a further sterilized micro-organism retaining filter, immediately prior to filling, should be carried out as close as possible to the filling point.
- 81.4 When the use of one sterilizing filter has been validated to achieve sterilization of a specific product, then the sterilizing filter must satisfactorily pass integrity testing before and after use.
When more than one sterilizing filters are used in the filter train, all filters must be tested before use. The secondary sterilizing filter does not require post-use integrity testing unless the primary sterilizing filter fails. In that case, the secondary sterilizing filter must satisfactorily pass integrity testing before and after use. If there are documented reasons for not being able to perform pre-filtration filter integrity testing of either filter in a series after sterilization, (e.g., if sterility downstream of the first filter may be compromised), the filters should be tested both prior to sterilization and after use.
- Blow/fill/ seal
- 82.1 Blow/ fill/ seal equipment used for aseptic production is fitted with an effective Grade A air shower and operated in at least a Grade C background environment. The background environment should comply with the viable and non viable limits at rest and viable limit when in operation.
- 82.2 Blow/ fill/ seal equipment used for the production of products that are terminally sterilized should be installed in at least a Grade D background environment.
- Isolator technology
- 83.1 The environmental cleanliness within an isolator is a Grade A located in at least a Grade D background environment.
- 83.2 Decontamination procedures should ensure full exposure of all isolator surfaces to the chemical agent. Decontamination methods that render the inner surfaces of barrier and isolator systems free of viable microorganisms should be developed and validated. Residues from the decontamination process should not negatively impact product or primary contact surfaces.
- 83.3 For sterilization of the filling line, where decontamination methods are used to render certain product contact surfaces free of viable organisms, a minimum of a six-log reduction should be demonstrated using a suitable biological indicator.
- 83.4 When using Aseptic processing in isolators the integrity and seams of gloves and half suits should receive daily attention when in use and be addressed by a comprehensive preventative maintenance program. Replacement frequencies should be established in written standard operating procedures that will ensure that parts will be changed before they breakdown or degrade. Transfer systems, gaskets, and seals should be covered by a written maintenance program.
- 83.5 Protection against potential ingress of any airborne particles from the environment surrounding the isolator must be a design feature. A breach of isolator integrity should normally lead to a decontamination cycle. Integrity can be affected by power failures, valve failure, inadequate overpressure, holes in gloves and seams, or other leaks.
- 83.6 Air quality within the isolator should be monitored for microbiological quality and particulates during each shift.
Quality Control
- Filled containers of parenteral products are inspected individually for the presence of particulates and other defects. When inspection is done visually, it takes place under suitable and controlled conditions of illumination, and background, and line speed. Operators doing the inspection pass regular eyesight checks, while wearing corrective lenses if such lenses are normally worn, and are allowed frequent breaks from inspection. Operators are subjected to routine checks for their efficiency in detecting defective units. Where other methods of inspection are used, the process is validated and the performance of the equipment is checked at intervals.
- Filled ampules are subjected to a leaker test (e.g., dye immersion test). Samples of other containers closed by appropriately validated methods are checked for integrity of seal and/or maintenance of vacuum where applicable after an appropriate predetermined period.
- Samples taken for sterility testing are representative of the whole of the batch, but in particular include samples taken from parts of the batch considered to be most at risk of contamination, for example:
- 86.1 For products that have been filled aseptically, samples include the first and last containers filled, and those filled after any significant interruption.
- 86.2 For products that have been heat-sterilized in their final containers, consideration is given to taking samples from the potentially coolest part of the load. Each sterilizer load is treated as a separate batch for sterility testing purposes.
- 86.3 The validated sterility test applied to the finished product is only one measure taken to assure sterility. It is to be interpreted in conjunction with the environmental and batch processing records.
- 86.4 Batches failing an initial sterility test are rejected unless a thorough investigation is carried out and the initial test is invalidated. The procedure for handling sterility test failures takes into account the guidance provided in official pharmacopeias (Schedule B to the Food and Drugs Act ).
- Where authorization for parametric release has been issued, after review of the submission which includes validation and monitoring of the process data pursuant to paragraph C.01.065(b)(ii), end product sterility testing is not required.
- Biological indicators used for routine monitoring of a sterilization process and when used in validation/ re-validation studies should be tested to verify the accuracy of the population count stated by the vendor.
- Media used for environmental monitoring should be tested for its growth promotion capability, in accordance with a formal written program.
- Microbial quantification must be based on scientifically sound methods. Because devices (e.g., air sampler) vary, the user should assess the suitability of their selected monitoring devices before they are placed into service. Such devices should be calibrated and used according to appropriate procedures.
- Environmental monitoring data generated in Grade A areas should be reviewed as part of product batch release. A written plan should be available that describes the actions to be taken when an environmental excursion occurs.
Medical Gases
Regulation
The provisions of C.02.025, C.02.027, and C.02.028 do not apply to medical gases.
Appendix A
Internationally Harmonized Requirements for Batch Certification
Content of the Fabricators/Manufacturers Batch Certificate
for Drug/Medicinal Products Exported to Countries
under the Scope of a Mutual Recognition Agreement (MRA)
Explanatory Note
In the framework of Mutual Recognition Agreements, the Sectoral Annex on Good Manufacturing Practices (GMP) requires a batch certification scheme for drug/medicinal products covered by the pharmaceutical Annex. The internationally harmonized requirements for the content of the batch certificate of a drug/medicinal product are attached. The importer of the batch is to receive and maintain the batch certificate issued by the fabricator/manufacturer. Upon request, the batch certificate has to be readily available to the regulatory authority of the importing country. This certification by the manufacturer regarding the conformity of each batch is essential to exempt the importer from re-control (re-analysis).
Each batch shipped between countries having an MRA in force must be accompanied by a batch certificate issued by the fabricator/manufacturer in the exporting country. This certificate will be issued after a full qualitative and quantitative analysis of all active and other relevant constituents to ensure that the quality of the products complies with the requirements of the marketing authorization of the importing country. The certificate will attest that the batch meets the specifications and has been manufactured in accordance with the marketing authorization of the importing country; will detail the specifications of the product, the analytical methods referenced, and the analytical results obtained; and will contain a statement that the batch processing and packaging quality control records were reviewed and found in conformity with GMP. The batch certificate will be signed by the person responsible for releasing the batch for sale or supply/export at the fabrication/manufacturing recognized building.
These harmonized requirements have been agreed on by the regulatory authorities of the following parties/countries: Australia, Canada, European Community, New Zealand, and Switzerland.
Appendix A1
Content of the Fabricators/Manufacturers Batch Certificate
for Drug/Medicinal Products Exported to Countries
under the Scope of a Mutual Recognition Agreement (MRA)
[Letterhead of Exporting Manufacturer]
- Name of product.
Proprietary, brand, or trade name in the importing country. - Importing country.
- Marketing authorization number.
The marketing authorization number of the product in the importing country should be provided. - Strength/Potency.
Identity (name) and amount per unit dose are required for all active ingredients/constituents. - Dosage form (pharmaceutical form).
- Package size (contents of container) and type (e.g., vials, bottles, blisters).
- Lot/batch number.
As related to the product. - Date of fabrication/manufacture.
In accordance with national (local) requirements. - Expiry date.
- Name and address of fabricator(s)/manufacturer(s) - manufacturing recognized building(s).
All recognized buildings involved in the manufacture of the batch including packaging and quality control of the batch, should be listed. The name(s) and address(es) given must correspond to the information provided on the manufacturing authorization/establishment licence. - Number(s) of manufacturing authorization(s)/licence(s) or certificate(s) of GMP compliance held by fabricator(s)/manufacturer(s).
A number should be given for each recognized building listed under Item 10. - Results of analysis.
Should include the approved specifications, describe all results obtained, and refer to the analytical methods used (May refer to a separate certificate of analysis, which must be dated, signed, and attached). - Comments/remarks.
Any additional information that might be of value to the importer and/or inspector who must verify the compliance of the batch certificate (e.g., specific storage or transportation conditions). - Certification statement.
Should cover the fabrication/manufacturing, including packaging and quality control. The following text should be used: "I hereby certify that the above information is authentic and accurate. This batch of product has been fabricated/manufactured, including packaging and quality control, at the above-mentioned recognized building(s) in full compliance with the GMP requirements of the local regulatory authority and with the specifications in the marketing authorization of the importing country. The batch processing, packaging, and analysis records were reviewed and found to be in compliance with GMP". - Name and position/title of person approving the batch release.
Must include the persons company/recognized building name and address, if more than one company is mentioned under Item 10. - Signature of person approving the batch release.
- Date of signature.
Appendix B
Acronyms
- API:
- Active Pharmaceutical Ingredient
- CoC:
- Certificate of Compliance
- DIN:
- Drug Identification Number
- GMP:
- Good Manufacturing Practices
- HPFB:
- Health Products and Food Branch
- ICH:
- International Conference on Harmonisation
- MPD:
- Master Production Documents
- MRA:
- Mutual Recognition Agreement
- NOC:
- Notice of Compliance
- OOS:
- Out of specification
- PIC/S:
- Pharmaceutical Inspection Cooperation/Scheme
- VICH:
- Veterinary International Conference on Harmonisation
- WFI:
- Water for Injection
- WHO:
- World Health Organization
Glossary of Terms
The definitions given below apply to the terms used in these guidelines, they also apply to the terms used in the annexes unless otherwise specified therein. Definitions quoted from other documents are identified in brackets at the end of the definition.
Active Pharmaceutical Ingredient - "Any substance or mixture of substances that is intended to be used in the manufacture of a drug (medicinal) product and that, when used in the production of a drug, becomes an active ingredient of the drug product. Such substances are intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease or to affect the structure and function of the body." (ICH, Q7)
Airlock - An enclosed space with two or more doors, that is interposed between two or more rooms, usually of differing classes of cleanliness, for the purpose of controlling the airflow between those rooms when either people or goods need to enter or leave them.
Alternate Sample Retention (ASR) Site - An alternate site specified on a Drug Establishment Licence for the storage of samples pursuant to section C.02.025 (1) of the Food and Drug Regulations.
Aseptic Area - A zone or zones within a clean area where Grade A or B (see table in Section C.02.029 of these guidelines) conditions are maintained.
Aseptic Process - A method of producing a sterile product in which sterile bulk drug or sterile raw materials are compounded and assembled with sterile packaging components under Grade A or B conditions (see table in Section C.02.029 of these guidelines).
Batch - A quantity of drug in dosage form, a raw material, or a packaging material, homogeneous within specified limits, produced according to a single production order and as attested by the signatories to the order. In the case of continuous manufacture, a batch corresponds to a defined fraction of the production, that is characterized by its intended homogeneity. It may sometimes be necessary to divide a batch into a number of sub-batches, which are later brought together to form a final homogeneous batch.
Batch Certificate - "A certificate issued by the fabricator of a lot or batch of a drug that is exported within the framework of a mutual recognition agreement and in which the fabricator
- identifies the master production document for the drug and certifies that the lot or batch has been fabricated, packaged/labelled and tested in accordance with the procedures described in that document;
- provides a detailed description of the drug, including
- a statement of all properties and qualities of the drug, including the identity, potency and purity of the drug, and
- a statement of tolerances for the properties and qualities of the drug;
- identifies the analytical methods used in testing the lot or batch and provides details of the analytical results obtained;
- sets out the addresses of the buildings at which the lot or batch was fabricated, packaged/labelled and tested; and
- certifies that the lot or batch was fabricated, packaged/labelled and tested in accordance with the good manufacturing practices of the regulatory authority that has recognized those buildings as meeting its good manufacturing practices standard." (C.01A.001)
(The certificates content is also described in Appendix A).
Batch Number - A distinctive combination of numbers and/or letters that specifically identifies a batch. The batch number appears on the batch records, certificates of analysis, etc.
Biological Drug - A drug that is listed in Schedule D to the Food and Drugs Act.
Bracketing - "The design of a stability schedule such that only samples on the extremes of certain design factors (e.g., strength, package size) are tested at all time points as in a full design. The design assumes that the stability of any intermediate levels is represented by the stability of the extremes tested. Where a range of strengths is to be tested, bracketing is applicable if the strengths are identical or very closely related in composition (e.g., for a tablet range made with different compression weights of a similar basic granulation, or a capsule range made by filling different plug fill weights of the same basic composition into different sized capsule shells). Bracketing can be applied to different container sizes or to different fills in the same container closure system." (ICH, Q1AR)
Bulk Drug - A drug in dosage form that is not in its final packaging, usually in quantities larger than the largest commercially available package size.
Bulk Process Intermediate - Any intermediate form of a Schedule C or D drug (e.g., final bulk intermediate, bulk material, bulk concentrate, drug substance) which must undergo further processing before it becomes a final product. They are usually characterized by a holding time, storage conditions and the application of in-process tests.
Campaign Production - Sequential processing of material, either more than one product in a multi-product facility or more than one lot of the same product in a dedicated facility, over a defined period of time. Campaign production could occur at any point in a production process where common rooms/suites and/or equipment are reused for multiple products/lots.
Certificate of Analysis (COA) - A document containing the name and address of the laboratory performing the test(s), name and specifications of the material(s), test(s) performed, test method(s) used, actual numerical results, approval date(s), signature of approver, and any other technical information deemed necessary for its proper use.
Certificate of Compliance (CoC) - A certificate issued by a Regulatory Authority attesting to the GMP compliance of a recognized building in that country. In Canada, the CoC is issued by the HPFB Inspectorate.
Certificate of Manufacture - A document issued by a vendor to a distributor or importer that attests that a specific lot or batch of drug has been produced in accordance with its master production documents. Such certificates include a detailed summary of current batch documentation, with reference to respective dates of revision, manufacture, and packaging, and are signed and dated by the vendors quality control department. For drugs that are fabricated, packaged/labelled and tested in MRA countries, the batch certificate is considered to be equivalent.
Certificate of Pharmaceutical Product (CPP) - A certificate issued by the Inspectorate establishing the regulatory status of the pharmaceutical, biological, radiopharmaceutical or veterinary product listed and the GMP status of the fabricator of the product. This certificate is in the format recommended by the WHO.
Change Control - A written procedure that describes the action to be taken if a change is proposed (a) to facilities, materials, equipment, and/or processes used in the fabrication, packaging, and testing of drugs, or (b) that may affect the operation of the quality or support system.
Changeover Procedure - A logical series of validated steps that ensures the proper cleaning of suites and equipment before the processing of a different product begins.
Clean Area - A room or suite of rooms where Grade C or D conditions (see table in Section C.02.029 of these guidelines) are required. The rooms have a defined environmental control of particulate and microbial contamination and are constructed, maintained, and used in such a way as to minimize the introduction, generation, and retention of contaminants.
Commitment Batches - "Production batches of a drug product for which the stability studies are initiated or completed post approval through a commitment made in the registration application." (ICH Q1A (R2))
Computerized Systems - Consists of all components, including but not limited to hardware, software, personnel, and documentation, necessary to capture, process, transfer, store, display, and manage information.
Containment - Total isolation of one or more steps of a manufacturing process to prevent cross-contamination of the product, or staff, from all other steps of the process.
Contractor - Legal entity carrying out activities on behalf of a company pursuant to a written agreement. This includes other sites within the same corporate structure.
Critical Area - Area in which the sterilized drug product, containers, and closures are exposed to environmental conditions that must be designed to maintain product sterility. Activities conducted in this area include manipulations, such as aseptic connections, sterile ingredient additions, filling and closing operations.
Critical Process - A process that if not properly controlled may cause significant variation in the quality of the finished product.
Date of Fabrication - Unless otherwise defined in the Food and Drug Regulations, this is the date when any active ingredient, excipient, anti-oxidant, preservative, or air/oxygen scavenger is first added to the lot being processed.
Dilute Drug Premix - "A drug for veterinary use that results from mixing a drug premix with a feed as defined in section 2 of the Feed Regulations, 1983, with the lowest approved dosage level of the drug." (C.01.A.001)
Director - "The Assistant Deputy Minister, Health Products and Food Branch, of the Department of Health." (A.01.010)
Distributor or Manufacturer - "A person, including an association or partnership, who under their own name, or under a trade, design or word mark, trade name or other name, word, or mark controlled by them, sells a food or drug." (A.01.010)
Divisions 1A and 2 to 4 apply to the following distributors (C.01A.003):
- a distributor of a drug listed in Schedule C or D to the Act or in Schedule F to these Regulations, a controlled drug as defined in subsection G.01.001 (1) or a narcotic as defined in the Narcotic Control Regulations who does not hold the drug identification number for the drug or narcotic; and
- a distributor of a drug for which that distributor holds the drug identification number.
Dosage Form - A drug product that has been processed to the point where it is now in a form in which it may be administered in individual doses.
Drug - "Any substance or mixture of substances manufactured, sold, or represented for use in (a) the diagnosis, treatment, mitigation, or prevention of a disease, a disorder, an abnormal physical state, or the symptoms thereof, in humans or animals, (b) restoring, correcting, or modifying organic functions in humans or animals, or (c) "disinfection" in premises in which food is manufactured, prepared, or kept." (Section 2 of the Food and Drugs Act)
In Division 1A and Division 2 of the Food and Drug Regulations, "drug" means a drug in dosage form, or a drug that is a bulk process intermediate that can be used in the preparation of a drug listed in Schedule C to the Act or in Schedule D to the Act that is of biological origin. It does not include a dilute drug premix, a medicated feed as defined in Section 2 of the Feeds Regulations, 1983, a drug that is used only for the purposes of an experimental study in accordance with a certificate issued under Section C.08.015 or a drug listed in Schedule H to the Act. (C.01A.001(2))
Drug Establishment Licence - A licence issued to a drug establishment in Canada which has been inspected and assessed as being in compliance with the applicable requirements of Divisions 2 to 4 of the Food and Drug Regulations.
Drug Identification Number - A number assigned to each drug in dosage form under the Food and Drug Regulations with the exception of blood and blood components and radiopharmaceuticals.
Drug Premix - "A drug for veterinary use to which a drug identification number has been assigned, where the directions on its label specify that it is to be mixed with feed as defined in Section 2 of the Feeds Act." (C.01A.001)
Expiry Date - "Means the earlier of (a) the date, expressed at minimum as a year and month, up to and including which a drug maintains its labelled potency, purity and physical characteristics, and (b) the date, expressed at minimum as a year and month, after which the manufacturer recommends that the drug not be used." C.01.001)
Export Certificate (under Section 37 of the Food and Drugs Act) - A certificate signed by the fabricator and a Commissioner for Taking Oaths to attest that the drug for which the certificate is prepared is not manufactured or sold for consumption in Canada and that its package and the contents do not contravene any known requirement of the law of the country to which it is or is about to be consigned.
Fabricate - "To prepare and preserve a drug for the purpose of sale." (C.01A.001)
Filling - Transferring a bulk drug into its final container and enclosing it in the container.
Finished Product - A product that has undergone all stages of production, including packaging in its final container and labelling.
Formulating - Preparing components and combining raw materials into a bulk drug.
Group 2 Products - Drugs listed in Schedule D to the Act and subject to Health Canadas lot release programme which require the highest level assessment after the notice of compliance (NOC) has been issued. This assessment includes targeted testing, protocol review, and written approval for sale of each lot in Canada in the form of a release letter.
Growth Promotion - A test in which prepared media is challenged with pre-selected organisms to assure that the media is capable of supporting growth.
Import - "To import into Canada a drug for the purpose of sale" (C.01A.001)
In-process Control - Checks performed during production in order to monitor and, if necessary, to adjust the process to ensure that the finished product conforms to its specifications. The control of the production environment or equipment may also be regarded as a part of in-process control.
In-process Drug - Any material or mixture of materials that must, to become a drug in dosage form, undergo further processing.
In-process Testing - The examination or testing of any material or mixture of materials during the manufacturing process.
Installation Qualification - The documented act of demonstrating that process equipment and ancillary systems are appropriately selected and correctly installed.
Label - "Includes any legend, word, or mark attached to, included in, belonging to, or accompanying any food, drug, cosmetic, device, or package (Section 2 of the Act). As described in package/label, the action of labelling refers to affixing the inner or outer label to the drug." (C.01A.001)
Long Term Testing - "Stability studies under the recommended storage condition, for the re-test period or shelf life proposed (or approved) for labelling." (ICH, Q1AR)
Lot - A quantity of any drug in dosage form, a raw material, or a packaging material, homogeneous within specified limits, constituting all or part of a single batch and identified by a distinctive lot number that appears on the label of the finished product.
Lot Number - "Any combination of letters, figures, or both, by which any food or drug can be traced in manufacture and identified in distribution." (A.01.010)
Manufacturer or Distributor - See definition under distributor.
Manufacturing Batch Record - Records demonstrating that the batch of a drug was fabricated in accordance with the approved master production documents.
Marketing Authorization - A legal document issued by Health Canada, authorizing the sale of a drug or a device based on the health and safety requirements of the Food and Drug Act and its associated Regulations. The marketing authorization may be in the form of a Notice of Compliance (NOC), Drug Identification Number (DIN), a device licence for classes II, III and IV medical devices, or a natural product number (NPN) or homeopathic DIN (DIN-HM).
Mass Balance - "The process of adding together the assay value and levels of degradation products to see how closely these add up to 100% of the initial value, with due consideration of the margin of analytical error." (ICH, Q1AR)
Master Formula - A document or set of documents specifying the raw materials with their quantities and the packaging materials, together with a detailed description of the procedures and precautions required to produce a specified quantity of a finished product as well as the processing instructions, including the in-process controls.
Master Production Documents (MPD) - Documents that includes specifications for raw material, for packaging material and for packaged dosage form; master formula (including composition and instructions as described in the definition above), sampling procedures, and critical processing related SOPs, whether or not these SOPs are specifically referenced in the master formula.
Matrixing - "The design of a stability schedule such that a selected subset of the total number of possible samples for all factor combinations is tested at a specified time point. At a subsequent time point, another subset of samples for all factor combinations is tested. The design assumes that the stability of each subset of samples tested represents the stability of all samples at a given time point. The differences in the samples for the same drug product should be identified as, for example, covering different batches, different strengths, different sizes of the same container closure system, and possibly in some cases, different container closure systems." (ICH, Q1A(R)) The concept of matrixing may also apply in other areas such as validation.
Medical Gas - "Any gas or mixture of gases manufactured, sold or represented for use as a drug" (C.02.002)
Medicinal Ingredient - Refer to the definition of active pharmaceutical ingredient.
MRA Country - A country that is a participant to a mutual recognition agreement with Canada. (C.01A.001).
Mutual Recognition Agreement (MRA) "An international agreement that provides for the mutual recognition of compliance certification for Good Manufacturing Practices for drugs." (C.01A.001)
Operational Qualification - The documented action of demonstrating that process equipment and ancillary systems work correctly and operate consistently in accordance with established specifications.
Package - "As described in package/label, the action of packaging refers to putting a drug in its immediate container." (C.01A.001)
Package/label - "To put a drug in its immediate container or to affix the inner or outer label to the drug." (C.01A.001)
Packaging Batch Record - Records demonstrating that the batch of a drug was packaged in accordance with the approved master production documents.
Packaging Material - Labels, printed packaging materials and those components in direct contact with the dosage form. (refer to C.02.002)
Parenteral Use - "Administration of a drug by means of hypodermic syringe, needle or other instrument through or into the skin or mucous membrane." C.01.001)
Pharmaceutical - "A drug other than a drug listed in Schedule C or D to the Act." (C.01A.001).
Potency - The activity or amount of active moiety, or any form thereof, indicated by label claim to be present.
Process Validation - Establishing documented evidence with a high degree of assurance, that a specific process will consistently produce a product meeting its predetermined specifications and quality characteristics. Process validation may take the form of Prospective, Concurrent or Retrospective Validation and Process Qualification or Re-validation.
Production - All operations involved in the preparation of a finished product, from receipt of materials, through processing and packaging, to completion of the finished product, including storage.
Purified Water - As defined in any standard listed in Schedule B to the Food and Drugs Act.
Purity - The extent to which a raw material or a drug in dosage form is free from undesirable or adulterating chemical, biological, or physical entities as defined by specifications.
Qualified Authority - A member of the Pharmaceutical Inspection Cooperation/Scheme (PIC/S) or the United States Food and Drug Administration (USFDA).
Quality Risk Management - A systematic process for the assessment, control, communication and review of risks to the quality of the medicinal product. It can be applied both proactively and retrospectively (ICH, Q9).
Quarantine - Effective restriction of the availability of material or product for use (physically or by system), until released by the quality control department.
Radiopharmaceutical - "A drug that exhibits spontaneous disintegration of unstable nuclei with the emission of nuclear particles or photons." C.03.201)
Raw Material - Any substance, other than in-process drug or packaging material, intended to be used in the manufacture of drugs, including those that appear in the master formula but that do not appear in the drug such as solvents and processing aids.
Recognized Building - "In respect of the fabrication, packaging/labelling or testing of a drug, a building that a regulatory authority that is designated under subsection C.01A.019(1) in respect of that activity has recognized as meeting its Good Manufacturing Practices standards in respect of that activity for that drug." (C.01A.001)
Reconciliation - A comparison, making due allowance for normal variation, between the amount of product or materials theoretically produced or used and the amount actually produced or used.
Recovery - The introduction of all or part of previous batches of the required quality into another batch at a defined stage of manufacture.
Regulatory Authority - A government agency or other entity in an MRA country that has a legal right to control the use or sale of drugs within that country and that may take enforcement action to ensure that drugs marketed within its jurisdiction comply with legal requirements. (C.01A.001)
Re-packaging / Re-labelling - Replacement of packaging or labelling of previously packaged and labelled products.
Reprocessing - Subjecting all or part of a batch or lot of an in-process drug, a bulk process intermediate (final biological bulk intermediate) or a bulk drug of a single batch/lot to a previous step in the validated manufacturing process due to failure to meet predetermined specifications. Reprocessing procedures are foreseen as occasionally necessary and are validated and pre-approved by the quality control department or as part of the marketing authorization.
Re-test Date - "The date when a material should be re-examined to ensure that it is still suitable for use." (ICH Q7)
Re-test Period - "The period of time during which a drug substance can be considered to remain within the specifications and therefore acceptable for use in the fabrication of a given drug product, provided that it has been stored under defined conditions; after this period, the batch is re-tested for compliance with specifications and then used immediately." (ICH, Q1AR)
Reworking - "Subjecting an in-process drug, a bulk process intermediate (final biological bulk intermediate), or final product of a single batch/lot to an alternate manufacturing process due to a failure to meet predetermined specifications. Reworking is an unexpected occurrence and is not pre-approved as part of the marketing authorization." (WHO GMP)
Secondary Labelling - The operation of affixing an inner or outer label to a previously labelled container to fulfill the regulatory requirements of Part C of the Food and Drug Regulations.
Self-contained Facility - Means a premise that provides complete and total separation of all aspects of the operation, including personnel and equipment movement, with well established procedures, controls and monitoring. This includes physical barriers and separate utilities such as air handling systems. A self-contained facility does not necessarily imply a distinct and separate building.
Sell - "Offer for sale, expose for sale, have in possession for sale, and distribute, regardless of whether the distribution is made for consideration." (Section 2 of the Food and Drugs Act)
Shelf Life - The time interval during which a drug product is expected to remain within the approved specification provided that it is stored under the conditions defined on the label and in the proposed containers and closure.
Specifications - "Means a detailed description of a drug, the raw material used in a drug, or the packaging material for a drug and includes:
- a statement of all properties and qualities of the drug, raw material or packaging material that are relevant to the manufacture, packaging, and use of the drug, including the identity, potency, and purity of the drug, raw material, or packaging material,
- a detailed description of the methods used for testing and examining the drug, raw material, or packaging material, and
- a statement of tolerances for the properties and qualities of the drug, raw material, or packaging material." C.02.002.)
Standard Operating Procedure (SOP) - A written procedure giving instructions for performing operations not necessarily specific to a given product or material but of a more general nature (e.g., equipment operation, maintenance and cleaning; validation; cleaning of premises and environmental control; sampling and inspection). Certain SOPs may be used to supplement product-specific master and batch production documents.
Sterile - Free from viable microorganisms.
Sterilizing Filter - A filter used to render a material sterile. Sterilizing filters have a rated pore size of 0.2 µm or less.
System - A regulated pattern of interacting activities and techniques that are united to form an organized whole.
Terminal Sterilization - Sterilizing a drug in its final closed container.
Test - To perform the tests, including any examinations, evaluations, and assessments, as specified in the Division 2 of the Food and Drug Regulations.
Validation - The documented act of demonstrating that any procedure, process, and activity will consistently lead to the expected results. Includes the qualification of systems and equipment.
Vendor - Person who is the fabricator of the item (raw material, packaging material, medicinal ingredients, reagents).
Veterinary Drugs - Drugs that are administered to food-producing and companion animals.
Wholesale - "To sell any of the following drugs, other than at retail sale, where the seller's name does not appear on the label of the drugs:
- a drug listed in Schedule C or D to the Act or in Schedule F to these Regulations or a controlled drug as defined in subsection G.01.001 (1); or
- a narcotic as defined in the Narcotic Control Regulations." (C.01A.001)
Appendix C
Annexes to the Current Edition of the Good Manufacturing Practices (GMP) Guidelines
The list of references in this Appendix has been updated. Annex numbers and document titles have been changed to correspond to those used by the European Union (EU) and PIC/S. This is done in order to work towards the global harmonization of technical standards and procedures related to GMP, and in preparation for Health Canada's revisions on these documents, which is anticipated to start in the near future. URLs to the documents, active at the time of this GUI-0001 posting, have been provided.
Annexes are available on Health Canadas Web site in the Compliance and Enforcement section.
- Annex 1 to the Current Edition of the Good Manufacturing Practices Guidelines - Selected Category IV Monograph Drugs (GUI-0066)
- Annex 2 to the Current Edition of the Good Manufacturing Practices Guidelines - Schedule D Drugs, Biological Drugs Including Fractionated Blood Products (GUI-0027)
- Annex 3 to the Current Edition of the Good Manufacturing Practices Guidelines - Schedule C Drugs (GUI-0026)
- Annex 4 to the Current Edition of the Good Manufacturing Practices Guidelines - Veterinary Drugs (GUI-0012)
- Annex 5 to the Current Edition of the Good Manufacturing Practices Guidelines - Positron Emitting Radiopharmaceuticals (PERs) (GUI-0071)
- Annex 11 to the Current Edition of the Good Manufacturing Practices Guidelines - Computerised Systems (GUI-0050)
- Annex 13 to the Current Edition of the Good Manufacturing Practices Guidelines - Drugs Used in Clinical Trials (GUI-0036)
- Annex 14 to the Current Edition of the Good Manufacturing Practices Guidelines - Schedule D Drugs, Human Blood and Blood Components (GUI-0032)
- Annex 17 to the Current Edition of the Good Manufacturing Practices Guidelines - Guidance on Parametric Release (GUI-0046)
References
Justice Canada
Acts and regulations of Canada are available on Justice Laws Web Site.
Health Canada
Guides (GUI) and Questions and Answers (Q&As) that relate to GMPs are available on Health Canadas Web Site in the Compliance and Enforcement section under Good Manufacturing Practices.
- Alternate Sample Retention Site Guidelines (GUI-0014)
- Cleaning Validation Guidelines (GUI-0028)
- Good Manufacturing Practices - Questions and Answers
- Good Manufacturing Practices Guidelines for Medical Gases (GUI-0031)
- Good Manufacturing Practices for Medical Gases - Questions and Answers
- Guidance on Evidence to Demonstrate Drug Good Manufacturing Practices Compliance of Foreign Sites (GUI-0080)
- Guidelines for Temperature Control of Drug Products during Storage and Transportation (GUI-0069)
- Importation and Exportation Questions and Answers
- Process Validation Guidelines:
- Recall Procedures
- Product Recall Procedures
- Recall Policy (POL-0016)
- HPFBI Guidelines for Recall of Drug and Natural Health Products (GUI-0039)
- Risk Classification of Good Manufacturing Practices Observations (GUI0023)
- Validation Documentation Requirements and Responsibilities for Drug Fabricators, Packagers/Labellers, Distributors and Importers (GUI-0042)
- Validation Guidelines for Pharmaceutical Dosage Forms (GUI-0029)
Documents that relate to stability are available on Health Canadas Web site in the Drug Products secton under Application and Submissions
- Stability Requirements for Changes to Marketed New Drugs
- Stability Testing of Existing Drug Substances and Products
Guidance documents developed by the International Conference on Harmonisation (ICH) and adopted by Health Canadas Web site in the Drug Products section under ICH (International Conference on Harmonisation)
- ICH Q1A(R2): Stability Testing of New Drug Substances and Products
- ICH Q1B: Stability Testing: Photostability Testing of New Drug Substances and Products
- ICH Q1C: Stability Testing for New Dosage Forms
- Validation of Analytical Procedures: Text and Methodology (PDF Version size K)ICH Q2(R1): Validation of Analytical Procedures: Text and Methodology
- ICH Q7A: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
Health Products and Food Branch Inspectorate - Operational Centres
Atlantic Operational Centre
Health Products and Food Branch Inspectorate
16th floor, suite 1625
1505 Barrington Street
Halifax, Nova Scotia
B3J 3Y6
Tel: 902-426-2160
Fax: 902-426-6676
E-mail: insp_aoc-coa@hc-sc.gc.ca
Inspectorate Ottawa
Health Products and Food Branch Inspectorate
Graham Spry Building, 2nd Floor
250 Lanark Avenue
Address Locator #2002B
Ottawa, Ontario
K1A 0K9
Tel: 613-957-1492
Fax: 613-957-6709
E-mail: GMP_questions_BPF@hc-sc.gc.ca
Ontario Operational Centre
Health Products and Food Branch Inspectorate
2301 Midland Avenue
Scarborough, Ontario
M1P 4R7
Tel: 416-973-1600
Fax: 416-973-1954
E-mail: insp_onoc-coon@hc-sc.gc.ca
Manitoba and Saskatchewan Operational Centre
Health Products and Food Branch Inspectorate
510 Lagimodière Blvd
Winnipeg, Manitoba
R2J 3Y1
Tel: 204-984-1341
Fax: 204-984-2155
E-mail: Insp_MSOC_COMS@hc-sc.gc.ca
Quebec Operational Centre
Health Products and Food Branch Inspectorate
1001 St-Laurent Street West
Longueuil, Quebec
J4K 1C7
Tel: 450-646-1353
Fax: 450-928-4455
E-mail: QOC-COQ@hc-sc.gc.ca
Western Operational Centre
Health Products and Food Branch Inspectorate
4th Floor
4595 Canada Way
Burnaby, British-Colombia
V5G 1J9
Tel: 604-666-3704
Fax: 604-666-3149
E-mail: insp_woc-coo@hc-sc.gc.ca
Page details
- Date modified: