
Lignes directrices du CEPMB
L’entrée en vigueur du Règlement sur les médicaments brevetés modifié (le « Règlement ») a de nouveau été reportée au-delà du 1er janvier 2022. Par conséquent, les nouvelles Lignes directrices n'entreront pas en vigueur le 1er janvier 2022.
Le Conseil a annoncé sa décision suite à la consultation sur la définition des médicaments de transition et le délai de mise en conformité le 17 mars 2021.

Table des matières
- I. Préface
- II. Interprétation
- III. Cadre juridique
- IV. Exigences en matière de présentation de renseignements dans le cadre de l’examen des prix
- V. Processus d’examen des prix
- VI. Réévaluation
- VII. Enquêtes
- VIII. Engagement de conformité volontaire (ECV)
- IX. Recommandations relatives à l’audience
- X. Processus d’audience relativement aux prix excessifs et recours
- XI. Audience pour défaut de présenter les renseignements requis
- XII. Plaintes
- XIII. Annexes
- A. Comparaison selon la catégorie thérapeutique des prix nationaux (CCTn) et comparaison selon la catégorie thérapeutique des prix internationaux (CCTi)
- B. Test de la relation raisonnable et formes posologiques comparables
- C. Évaluation de la valeur pharmacoéconomique
- D. Méthode de rajustement en fonction de la taille du marché
- E. Processus d’examen scientifique
- F. Sommaire des calendriers de mise en conformité
I. Préface
1. Le Conseil d’examen du prix des médicaments brevetés (CEPMB), créé en 1987 en tant que pilier de la protection des consommateurs dans le cadre d’importantes réformes de la Loi sur les brevets (la « Loi »), est un organisme quasi judiciaire doté d’un mandat de réglementation dont l’objectif est de veiller à ce que les titulaires de brevets (ou brevetés) pharmaceutiques ne facturent pas des prix excessifs aux consommateurs pendant la période de monopole de droit. La mise sur pied du CEPMB découle de la crainte qu’une protection accrue des brevets des médicaments puisse entraîner une hausse inacceptable de leur prix et les rendre inabordables pour les consommateurs. À titre de membre du portefeuille de la Santé, le CEPMB contribue à un système de santé moderne et viable en veillant à ce que les Canadiens continuent d’avoir accès aux médicaments brevetés à des prix non excessifs.
2. Les présentes Lignes directrices, établies conformément au paragraphe 96(4) de la Loi, visent à assurer aux brevetés la transparence et la prévisibilité du processus de triage et d’examen habituellement suivi par les fonctionnaires du CEPMB (le « personnel ») pour déterminer si le prix d’un médicament breveté semble excessif sur un des différents marchés au Canada. Les Lignes directrices présentent également un aperçu des processus que les brevetés devraient connaître en ce qui a trait à leurs obligations en matière de présentation de renseignements en vertu du Règlement sur les médicaments brevetés (le « Règlement »).
3. Si le prix d’un médicament breveté semble excessif au regard des Lignes directrices et que le breveté n’a pas soumis d’engagement de conformité volontaire (ECV) acceptable, le personnel peut recommander au président de tenir une audience sur la question. Si une telle audience est jugée dans l’intérêt public par le président et qu’un panel d’audience composé de membres du Conseil (le « panel d’audience ») confirme que le prix du médicament breveté était excessif sur tout marché, le CEPMB peut rendre une ordonnance obligeant le breveté à baisser le prix du médicament et à prendre des mesures pour compenser l’excédent des recettes que lui aurait procuré la vente du médicament au prix excessif.
II. Interprétation
4. Les Lignes directrices fournissent des renseignements sur l’approche générale du CEPMB à l’égard du processus d’examen des prix et des enquêtes. Elles remplacent l’ensemble des documents d’orientation, des communiqués de politique et des énoncés écrits ou verbaux antérieurs du CEPMB concernant l’administration du processus d’examen des prix et des enquêtes, y compris les versions antérieures du Compendium des politiques, des Lignes directrices et des procédures du CEPMB. Les Lignes directrices doivent être lues en parallèle avec la Loi, le Règlement, les annexes desdites Lignes directrices et les autres documents d’orientation connexes que le CEPMB publiera à l’avenir, dont la section d’aide de l’outil de dépôt en ligne, qui remplace l’ancien Guide du breveté.
5. Conformément au paragraphe 96(4) de la Loi, les Lignes directrices ne lient ni le personnel, ni le président, ni les panels d’audience, ni les brevetés, et elles ne visent pas à créer des droits ou des présomptions juridiques, à réaffirmer la loi ou à faire une déclaration définitive concernant l’interprétation des dispositions législatives se rapportant au CEPMB. Dans chaque cas, l’approche adoptée par le personnel en matière d’application de la loi et la résolution définitive des questions dépendront des circonstances particulières de l’affaire en cause. L’interprétation définitive de la loi relève du Conseil (qui siège en tant que panel d’audience) et est soumis à un contrôle judiciaire par les tribunaux.
6. Certains aspects des Lignes directrices peuvent être revus par le CEPMB à la lumière de l’expérience et de l’évolution des circonstances. Les Lignes directrices et les politiques émises par le CEPMB sont élaborées de façon transparente et permettent une consultation complète des parties intéressées. Lorsqu’il envisagera d’apporter des modifications aux Lignes directrices, le CEPMB consultera les intervenants conformément au processus de consultation établi en vertu du paragraphe 96(5) de la Loi.
7. Le CEPMB fera tous les efforts raisonnables pour aider les brevetés à comprendre les Lignes directrices et leur application. Par exemple, le personnel informera sur demande les brevetés des méthodes à appliquer dans le cadre de l’examen du prix des médicaments brevetés vendus au Canada. En outre, si on lui en fait la demande et s’il dispose de suffisamment de renseignements pour le convaincre que le prix auquel le breveté vend ou a l’intention de vendre un médicament breveté ne serait pas jugé excessif, le Conseil peut délivrer un certificat non contraignant en ce sens en vertu du paragraphe 98(4) de la Loi.
8. Les Lignes directrices ne décrivent pas de manière exhaustive toutes les mesures qui peuvent être prises ou tous les problèmes qui peuvent survenir dans le contexte d’un examen du prix. Dans des circonstances exceptionnelles ou lors d’une audience, le CEPMB peut utiliser des méthodes ou des tests jugés appropriés et conformes à la Loi et au Règlement, même s’il n’en est pas question dans les Lignes directrices ou s’ils diffèrent de l’approche qui y est énoncée. En aucun cas le personnel ou les membres du Conseil ne seront liés ou limités par les Lignes directrices.
9. Pour obtenir de plus amples renseignements au sujet de ces Lignes directrices ou de l’approche générale du personnel à l’égard de l’examen des prix et des enquêtes, veuillez consulter le site Web du CEPMB ou communiquer avec le personnel à l’adresse suivante :
Conseil d’examen du prix des médicaments brevetés
C.P. L40
Centre Standard Life
333, avenue Laurier Ouest
Bureau 1400
Ottawa (Ontario)
K1P 1C1
À l’attention de : Secrétaire du Conseil
III. Cadre juridique
10. Le CEPMB a été créé en 1987 après que des modifications d’envergure apportées à la Loi sur les brevets sont entrées en vigueur par l’adoption du projet de loi C-22Note de bas de page 1. Ces modifications ont permis de renforcer la protection des brevets des médicaments, tant du point de vue de la portée de l’objet brevetable que de la période d’exclusivité fondée sur les brevets, instaurant ainsi au Canada ce que les décideurs croyaient être un climat d’investissement plus propice à la recherche et au développement (R-D) pharmaceutique. En guise de concession faite aux opposants à ces modifications, qui craignaient que la protection renforcée des brevets des médicaments entraîne une hausse inacceptable des prix et rende ces produits inabordables pour les Canadiens, on a également créé le CEPMB dans la foulée du projet de loi C-22. Le mandat du CEPMB consiste à s’assurer que le prix des médicaments brevetés n’est pas excessif. Essentiellement, le projet de loi C-22 cherchait à établir un équilibre entre la nécessité de reconnaître et de récompenser l’innovation pharmaceutique par l’octroi d’une période d’exclusivité de marché aux brevetés et la nécessité de veiller à ce que les prix demandés au cours de cette période d’exclusivité restent raisonnables. Comme en témoigne le statut du CEPMB, qui est le seul organisme sectoriel de réglementation des prix plafonds en vertu de la Loi, les décideurs reconnaissent que la capacité absolue d’établir le prix des médicaments brevetés n’est pas dans l’intérêt public, étant donné le préjudice unique qui peut découler des prix excessifs de ces médicaments pour les consommateurs.
11. CEPMB est investi d’un double rôle : dans son rôle de réglementation, il protège les consommateurs en veillant à ce que le prix des médicaments brevetés ne soit pas excessif; dans son rôle de rapport, il fournit des renseignements sur les tendances des prix dans l’industrie pharmaceutique au moyen de son rapport annuel. Conformément à une directive du ministre de la Santé prise en application de l’article 90 de la Loi, le CEPMB appuie également la politique éclairée et fondée sur des données probantes en matière de santé en rendant compte des tendances relatives aux prix, à l’utilisation et aux coûts des médicaments dans le cadre de l’initiative du Système national d’information sur l’utilisation des médicaments prescrits (SNIUMP).
12. Le CEPMB est composé de cinq membres nommés par le gouverneur en conseil en vertu de l’article 91 de la Loi. La durée maximale de leur mandat est de cinq ans, renouvelable une seule fois. Le président du CEPMB agit comme premier dirigeant du Conseil et, à ce titre, il en assure la direction. Aux termes de l’article 94 de la Loi, le CEPMB emploie des fonctionnaires (le « personnel ») pour l’exercice de ses activités quotidiennes. Le directeur exécutif du CEPMB est son haut fonctionnaire, son chef de l’exploitation et son dirigeant principal des finances, et il est responsable de la gestion du personnel.
13. Le CEPMB a été créé en vertu de la Loi en tant qu’organisme indépendant quasi judiciaire. Afin d’assurer l’indépendance et l’autonomie du Conseil, la Loi ne confère aucun pouvoir exprès ou implicite à Santé Canada ou à toute autre entité gouvernementale qui permettrait de diriger le CEPMB dans l’exercice de ses fonctions de réglementation. Le CEPMB n’a aucun lien de dépendance avec le ministre de la Santé (qui est responsable des articles de la Loi applicables au CEPMB), le ministre de l’Innovation, des Sciences et du Développement économique (qui est responsable de la Loi dans son ensemble) et ses différents intervenants. De même, le CEPMB est structuré de manière à ce que les activités et les fonctions du personnel, du président et des membres du Conseil soient distinctes. En effet, le personnel s’acquitte des fonctions liées aux enquêtes, aux litiges et aux rapports, qui sont distinctes des fonctions d’arbitrage, réservées aux membres du Conseil.
14. La surveillance de la conformité des brevetés aux exigences réglementaires en matière de présentation de renseignements et l’examen administratif du prix des médicaments brevetés relèvent du personnel. Lorsque le prix d’un médicament breveté semble excessif et que la question ne peut pas être résolue au moyen d’une baisse volontaire du prix ou de mesures visant à compenser les recettes procurées au breveté par la vente du médicament à ce prix, le directeur exécutif peut porter le dossier à l’attention du président, qui déterminera si la tenue d’une audience est dans l’intérêt public. S’il décide de tenir une audience, le président émettra un avis d’audience et constituera un panel d’audience chargé de trancher la question. La décision du président d’émettre un avis d’audience est de nature purement administrative et n’exprime pas son point de vue sur le bien-fondé du dossier sous-jacent.
15. Le CEPMB examine le prix des médicaments brevetés vendus sans lien de dépendance par les brevetés. Les ventes au Canada peuvent comprendre notamment les médicaments brevetés assujettis à un avis de conformité (AC), les médicaments obtenus par le Programme d’accès spécial (PAS), les médicaments figurant sur la Liste des drogues utilisées pour des besoins urgents en matière de santé publique et les médicaments visés par des demandes d’essais cliniques. Le CEPMB n’a aucun pouvoir sur les prix demandés par des parties autres que les brevetés, comme les prix pratiqués par les grossistes ou les détaillants, ou sur les honoraires des pharmaciens.
16. En vertu de la Loi, le CEPMB a compétence pour déterminer si un médicament breveté est ou a été vendu par un titulaire de droits (le titulaire d’un brevet, l’ancien titulaire d’un brevet ou la personne ayant pour le moment droit à l’avantage d’un certificat de protection supplémentaire délivré à l’égard d’une invention liée à un médicament) à un prix excessif sur un marché au CanadaNote de bas de page 2. Le terme « titulaire de brevet » (ou breveté) est défini dans la Loi comme une personne ayant droit à l’avantage d’un brevet pour une invention pendant une période, ainsi que quiconque était titulaire d’un brevet pour une telle invention ou exerce ou a exercé les droits d’un titulaire, par exemple le titulaire d’une licence implicite ou explicite.Note de bas de page 3
17. Une invention est liée à un médicament si elle est destinée à des médicaments ou à la préparation ou la production de médicaments, ou susceptible d’être utilisée à de telles fins. L’expression « liée à un médicament » a un sens large. La Cour d’appel fédérale a déterminé que la nature de ce lien peut être « ténue »Note de bas de page 4 [traduction]. Par exemple, il ne faut parfois qu’« un lien, aussi ténu soit-il, entre l’invention brevetée et le médicament vendu au Canada »Note de bas de page 5 [traduction].Note de bas de page 6
18. Au sens de la Loi, le terme « médicament » s’entend notamment d’une drogue (c.-à-d. une substance ou un mélange de substances qui est fabriqué, vendu ou présenté comme pouvant servir à l’une des fins suivantes : (i) le diagnostic, le traitement, l’atténuation, la prévention d’une maladie, d’un désordre, d’un état physique anormal ou de leurs symptômes, chez l’être humain ou les animaux; (ii) la restauration, la correction ou la modification des fonctions organiques chez l’être humain ou les animaux) et d’un ingrédient médicinalNote de bas de page 7. Sauf indication contraire, toute mention d’un « médicament » dans les présentes Lignes directrices inclut toutes les formes posologiques et toutes les concentrations (p. ex. tous les numéros d’identification de médicament ou « DIN » des médicaments pour lesquels un DIN a été attribué) du médicament.
19. Le CEPMB reconnaît que le terme « ingrédient médicinal » signifie généralement « ingrédient pharmaceutique actif » (IPA) utilisé comme matière première au cours de la fabrication de la forme posologique finale. Les médicaments brevetés relevant de la compétence du CEPMB comprennent les vaccins, les préparations topiques, les anesthésiques et les produits diagnostiques utilisés in vivo, peu importe leur mode d’administration (c.-à-d. timbre transcutané, capsule, produit injectable, inhalateur). Toutefois, le CEPMB ne considère pas les instruments médicaux, les produits diagnostiques in vitro et les désinfectants qui ne sont pas utilisés in vivo comme étant des médicaments brevetés aux fins de l’application des dispositions de la Loi relatives à l’examen des prix.
20. Le CEPMB a compétence pendant toute la durée d’un brevet admissible et délivré, ce qui comprend la période précédant la délivrance du brevet (à partir de la date de la demande de brevet). Le CEPMB a également compétence pendant la période de protection prolongée accordée par un certificat de protection supplémentaire.Note de bas de page 8
21. Le CEPMB continue d’avoir compétence sur le prix auquel un médicament breveté est vendu sur un marché canadien après la cession du brevet et jusqu’à l’annulation ou à l’abandon du brevet conformément aux dispositions expresses de la Loi ou à la péremption du brevet. La cession de brevets n’est pas expressément reconnue dans la Loi comme un mécanisme d’extinction des droits que confère un brevet avant la péremption normale du brevet.
22. Les ordonnances rendues par le CEPMB sont exécutoires de la même manière que les ordonnances de la Cour fédérale ou de toute cour supérieure au Canada, et elles peuvent être exécutées par le Conseil ou par la Cour fédérale. Les décisions contenues dans les ordonnances rendues par le CEPMB peuvent faire l’objet d’un contrôle judiciaire par la Cour fédérale, conformément aux principes de droit administratif et à la Loi sur les Cours fédérales.
IV. Exigences en matière de présentation de renseignements dans le cadre de l’examen des prix
23. Le CEPMB doit avoir accès à des renseignements exacts et opportuns au sujet de la vente des médicaments brevetés pour pouvoir s’acquitter de son mandat de réglementation. Par conséquent, les brevetés et les anciens brevetés sont tenus de présenter ces renseignements au CEPMB.
24. Les renseignements à présenter sont énoncés à l’article 82 de la Loi et dans le Règlement. La section d’aide de l’outil de dépôt en ligne, qui remplace l’ancien Guide du breveté, vise à aider les brevetés à déterminer le contenu et la forme des renseignements à présenter ainsi que les dates limites pour ce faire conformément à la loi. Les brevetés sont responsables de la conformité avec leurs obligations en matière de présentation de renseignements. Ces obligations statutaires ne peuvent être levées ou modifiées par le personnel.
25. Les renseignements que les brevetés ou les anciens brevetés peuvent être tenus de fournir comprennent ce qui suit (liste non exhaustive) :
- Un avis décrivant l’intention du breveté de mettre en vente sur un marché canadien un médicament breveté qui n’y a jamais été vendu (c.-à-d. la première vente du médicament) et les renseignements connexes (Notification de l’intention de vendre un médicament breveté);
- Les renseignements réglementaires sur l’identification et les caractéristiques d’un médicament breveté, tels que la monographie du médicament ou les renseignements équivalents, et tout DIN octroyé à chaque forme posologique et à chaque concentration du médicament;
- Les renseignements réglementaires sur le prix d’un médicament breveté, comme le prix auquel chaque forme posologique et chaque concentration du médicament est ou a été vendu sur tout marché canadien ou dans n’importe lequel des onze pays indiqués dans le Règlement (le « CEPMB11 »);
- Les renseignements réglementaires sur les analyses coût-utilité préparées par un organisme canadien d’évaluation des technologies de la santé (ÉTS) financé par l’État, dont les résultats sont exprimés en fonction du coût par année de vie ajustée en fonction de la qualité (AVAQ), pour chaque indication faisant l’objet de l’analyse;
- Les renseignements réglementaires sur l’utilisation maximale approximative d’un médicament breveté au Canada, en fonction de la quantité prévue des ventes du médicament sous sa forme posologique finale.
26. En vertu du paragraphe 7 du Règlement, les brevetés doivent présenter les renseignements exigés par courriel en utilisant les formulaires électroniques disponibles sur le site Web du CEPMB. Les formulaires doivent porter la signature électronique d’une personne autorisée qui atteste que les renseignements sont exacts et complets.
27. Il incombe à chaque breveté de s’assurer de manière indépendante que les renseignements présentés au CEPMB (dont les prix nationaux et internationaux) sont exacts. Le personnel peut de temps à autre effectuer une vérification ponctuelle des renseignements fournis par les brevetés, ce qui comprend les renseignements sur les prix, les recettes et les brevets. Dans l’éventualité d’une telle vérification, les brevetés peuvent être appelés à fournir d’autres documents à l’appui, ou encore à corriger ou à confirmer des renseignements fournis.
28. Le défaut de soumettre les renseignements requis dans le délai imparti ou le fait de présenter des renseignements erronés ou faux peuvent avoir des conséquences graves pour les brevetés ou les anciens brevetés. Le personnel peut demander au Conseil de rendre une ordonnance accordant certains recours, dont une ordonnance ex parte qui exige que les renseignements manquants soient présentés. Le dossier peut également mener à des poursuites par procédure sommaire en vertu du paragraphe 76.1(1) de la Loi. En outre, la présentation de faux renseignements constitue un acte criminel en vertu de l’article 76 de la Loi, lequel peut entraîner une amende ou un emprisonnement sur déclaration de culpabilité.
29. La Loi prévoit la confidentialité des renseignements présentés au CEPMB dans certaines circonstances. En effet, les renseignements ou les documents fournis au CEPMB en application des dispositions des articles 80, 81 et 82 de la Loi qui concernent les renseignements sur les prix ou dans le cadre d’une poursuite relative aux prix excessifs intentée en vertu de l’article 83 sont protégés et ne peuvent être divulgués au public sans l’autorisation de la partie qui les a fournis, à moins que ces renseignements n’aient été divulgués lors d’une audience publique tenue aux termes de l’article 83 de la Loi ou ne soient visés par les exceptions énoncées au paragraphe 87(2) de la Loi.
30. Les renseignements fournis au CEPMB peuvent être assujettis à certaines dispositions de la Loi sur l’accès à l’information et de la Loi sur la protection des renseignements personnels.
V. Processus d’examen des prix
31. Le processus d’examen des prix comprend une série d’étapes au cours desquelles (i) les médicaments brevetés sont catégorisés en fonction de la date de leur lancement et de leurs caractéristiques commerciales et (ii) les prix plafonds sont établis et utilisés pour évaluer le prix des médicaments brevetés. En général, c’est le personnel qui effectue l’examen des prix en utilisant les méthodes et les tests énoncés dans les présentes Lignes directrices, d’après les renseignements fournis par le breveté ou obtenus par le personnel auprès de sources externes pertinentes, comme les listes de médicaments des régimes publics.
32. Dans une première étape d’examen, les médicaments brevetés sont classés dans l’une ou l’autre des catégories suivantes : (1) médicaments bénéficiant de droits acquis; (2) élargissements de gammes de médicaments bénéficiant de droits acquis; (3) médicaments de transition; (4) nouveaux médicaments.
33. Les médicaments bénéficiant de droits acquis sont toutes les formes posologiques et les concentrations de médicaments brevetés pour lesquels un DIN a été attribué au breveté avant le 21 août 2019, même si ces formes et ces concentrations ont été approuvées pour de nouvelles indications (sans changement de DIN) après le 21 août 2019.
34. Les élargissements de gammes sont de nouvelles formes posologiques et concentrations de médicaments bénéficiant de droits acquis auxquels un DIN a été attribué à partir du 21 août 2019.
35. Les médicaments de transition sont des médicaments pour lesquels un DIN a été attribué à partir du 21 août 2019 et avant le 1er juillet 2021, et qui ont été vendus pour la première fois au Canada avant le 1er juillet 2021.
36. Les nouveaux médicaments sont toutes les autres formes posologiques et concentrations de médicaments ne relevant d’aucune des catégories susmentionnées.
37. Les facteurs d’examen des prix en vertu du paragraphe 85(1) de la Loi qui s’appliquent aux quatre catégories de médicaments brevetés susmentionnées sont les suivants :
- Médicaments bénéficiant de droits acquis :
- Le prix de vente du médicament sur un marché canadien
- Le prix de vente de médicaments de la même catégorie thérapeutique sur un marché canadien
- Le prix de vente du médicament et d’autres médicaments de la même catégorie thérapeutique à l’étranger;
- Les variations de l’indice des prix à la consommation (IPC)
- Nouveaux médicaments, élargissements de gammes et médicaments de transition :
- Le prix de vente du médicament sur un marché canadien
- Le prix de vente de médicaments de la même catégorie thérapeutique sur un marché canadien
- Le prix de vente du médicament et d’autres médicaments de la même catégorie thérapeutique à l’étranger
- Les variations de l’indice des prix à la consommation (IPC)
- La valeur pharmacoéconomique du médicament au Canada
- La taille du marché du médicament au Canada
- Le produit intérieur brut au Canada et le produit intérieur brut par habitant au Canada
38. Les processus d’examen et les tests qui s’appliquent aux nouveaux médicaments, aux médicaments bénéficiant de droits acquis, aux élargissements de gammes de médicaments et aux médicaments de transition sont expliqués plus en détail ci-après.
A. Processus d’examen du prix des nouveaux médicaments
39. Le CEPMB examine les renseignements fournis par le breveté et les renseignements obtenus d’autres sources pour déterminer si le prix d’un médicament breveté lancé sur un marché canadien semble excessif. Le diagramme ci-après illustre le processus d’examen du prix des nouveaux médicaments :
Schéma des lignes directrices provisoires
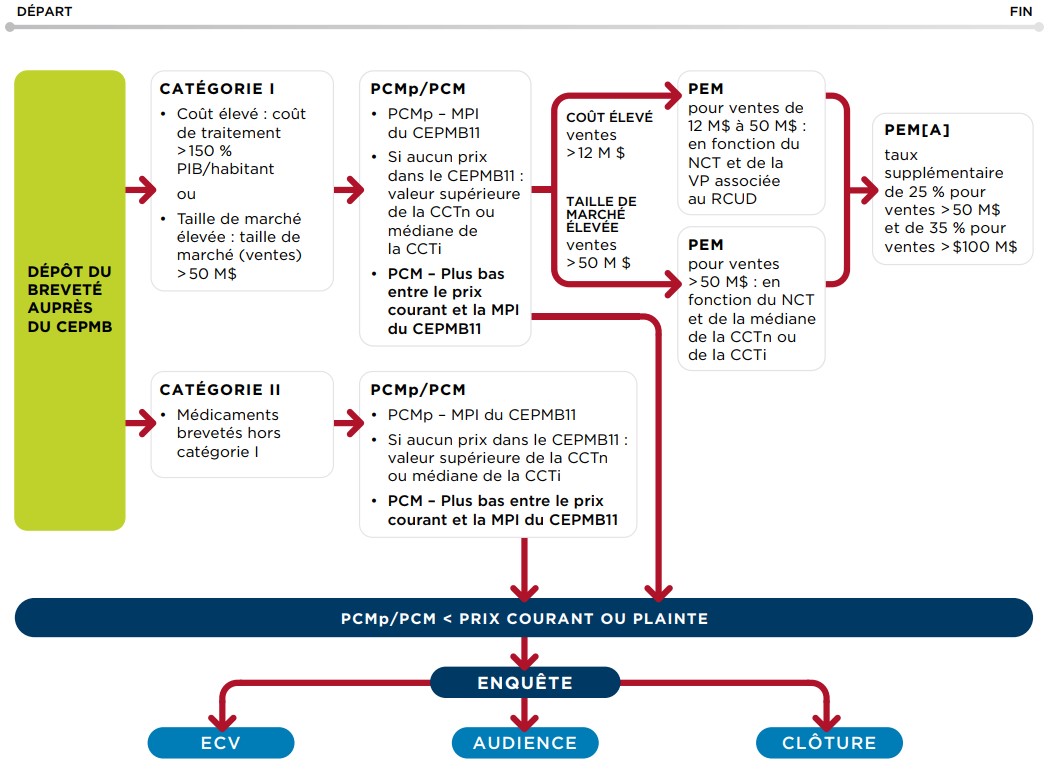
Description de la figure
Il s’agit d’un schéma décrivant de manière générale le processus d’examen des nouveaux médicaments brevetés (médicaments brevetés dont les ventes sont postérieures à janvier 2021) :
Premièrement, les brevetés présentent leurs données au CEPMB. La catégorie du nouveau médicament est établie. Le médicament breveté est classé soit dans la catégorie I (médicament dont le coût annuel du traitement est supérieur à 150 % du PIB/habitant et [ou] médicaments dont la taille maximale prévue du marché est supérieure à 50 millions de dollars), soit dans la catégorie II (tous les médicaments brevetés qui ne font pas partie de la catégorie I).
Un PCMp est déterminé en fonction de la MPI des prix du CEPMB11 disponibles. S’il n’y a pas de prix dans les pays du CEPMB11, le PCMp est basé sur le maximum de la CCTn ou de la CCTi. Le PCMp est calculé lors du lancement et maintenu jusqu’à ce que le PCM soit fixé. Si le prix courant du médicament breveté est supérieur au PCMp, une enquête peut être ouverte, qui peut conduire soit à un ECV, soit à une audience, soit à la clôture de l’enquête.
Le PCM équivaut soit au plus bas prix entre le prix courant et la MPI, une fois que des données ont été recueillies pour un minimum de 5 pays ou que 3 ans se sont écoulés depuis le lancement, soit au RR, si le médicament breveté est un élargissement de la gamme. Si, trois ans après le lancement, aucun prix n’est disponible pour les pays du CEPMB11, le PCM est basé sur le sommet de la CCTn ou la médiane de la CCTi. Le PCM est maintenu dans une marge de 10 % de la MPI jusqu’à la fin de la compétence du CEPMB. Si le prix courant du médicament breveté est supérieur au PCM, une enquête peut être ouverte, qui peut conduire soit à un ECV, soit à une audience, soit à la clôture de l’enquête.
Les médicaments de catégorie II sont uniquement soumis à un PCM.
Les médicaments de catégorie I sont également soumis un PCM. De plus, un PEM est accordé aux médicaments de catégorie I. Toutefois, seul le PCM est utilisé pour évaluer si une enquête doit être déclenchée. Le PEM ne constitue pas un élément déclencheur d’une enquête.
Pour les ventes effectuées sur un marché dont la taille annuelle est inférieure à 12 millions de dollars, le PEM est égal au PCM.
Pour les ventes effectuées après que la taille annuelle du marché ait atteint entre 12 et 50 millions de dollars, le PEM est égal à un pourcentage de réduction basé sur le NCT et le VP associés au RCUD (pour les RCUD entre 100 000 et 200 000 dollars par AVAQ) qui peut aller jusqu’à une réduction maximale de 50 % sur le PCM. S’il n’y a pas de valeur VP, la réduction maximale de 50 % sur le PCM est appliquée.
Pour les ventes effectuées après que la taille annuelle du marché ait dépassé 50 millions de dollars, le PEM est soumis à un ajustement supplémentaire de la taille du marché qui peut varier entre 25 % et 35 %.
Les tests utilisés pour le PEM pour les médicaments de la catégorie I dont la taille maximale prévue du marché est supérieure à 50 millions de dollars sont également différents selon les ventes.
Pour les ventes effectuées sur un marché dont la taille annuelle est inférieure à 50 millions de dollars, le PEM est égal au PCM.
Pour les ventes réalisées après que la taille annuelle du marché ait dépassé 50 millions de dollars, le PEM est basé sur une réduction du PCM qui elle-même est basée sur le NCT et la CCTn médiane et peut aller jusqu’à une réduction maximale de 50 %. Un ajustement supplémentaire de 25 à 35 % de la taille du marché peut également s’appliquer.
Chacune des étapes est décrite plus en détail ci-dessous.
Étape 1 : PCMp
40. Au moment du lancement (c’est-à-dire lors de la première vente sur un marché canadien), un prix courant maximum provisoire (le « PCMp ») est fixé pour la vente du médicament. Le PCMp est établi en fonction de la médiane des prix courants internationaux départ usine accessibles au public (MPI) des pays du CEPMB11 pour lesquels le breveté a fourni des renseignements pendant la période provisoire (définie ci-dessous). Le prix courantNote de bas de page 9 d’un médicament ne peut dépasser le PCMp pour la période provisoire, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
41. Si le breveté n’a pas soumis de renseignements sur les prix internationaux pour les pays du CEPMB11 après deux périodes de référence (c.-à-d. la période de lancement durant laquelle le médicament est vendu au Canada et une période subséquente), le PCMp est fixé par la valeur supérieure de la comparaison selon la catégorie thérapeutique des prix nationaux (« CCTn »). La CCTn sera calculée sur la base du coût de traitement le plus élevé parmi les médicaments de comparaison, obtenu en tenant compte du prix le plus bas des médicaments de comparaison (voir l’annexe A pour plus de détails). S’il n’existe pas de comparateur de classe thérapeutique au niveau national, le PCMp peut être fixé selon la médiane de la comparaison selon la catégorie thérapeutique des prix internationaux (« CCTi »).
42. Le PCMp n’est calculé qu’une seule fois et ne s’applique que pendant la période provisoire, qui dure jusqu’à la première des deux dates suivantes :
- trois (3) ans à compter de la date de lancement du médicament au Canada;
- lorsque le breveté fournit des renseignements sur les prix internationaux d’au moins cinq pays du CEPMB11.
À la fin de la période provisoire, on établit le PCM (voir l’étape 2), et le PCMp cesse de s’appliquer.
43. Si, au moment du lancement du médicament, le breveté fournit de l’information sur les prix internationaux pour au moins cinq des pays du CEPMB11, le PCMp ne s’applique pas et le PCM est fixé immédiatement (voir la section ci-dessous).
Étape 2 : PCM
44. Sous réserve de la procédure décrite ci-dessus, le PCMp est remplacé par un prix courant maximum ou « PCM ».
45. Si le breveté a déposé des prix internationaux avant la fin de la période provisoire, le PCM est fixé par le prix le plus bas entre le prix courant et la MPI. Sinon, le PCM est fixé par le prix le plus bas entre le prix courant et la valeur supérieure de la CCTn, laquelle est calculée sur la base du coût de traitement le plus élevé parmi les médicaments de comparaison, obtenu en tenant compte du prix le plus bas des médicaments de comparaison (voir l’annexe A pour plus de détails).
46. Si le breveté n’a pas soumis de prix international à la fin de la période provisoire, et qu’il n’existe pas de comparateur de classe thérapeutique au niveau national, le PCM peut être fixé selon la médiane de la CCTi.
47. Il peut arriver que le PCM soit inférieur au PCMp. Le cas échéant, le breveté doit se conformer au PCM dans un délai d’une (1) période de référence après l’établissement du PCM .
48. Comme il est expliqué plus en détail dans la section VI (« Réévaluation »), si, dans les périodes ultérieures, la MPI dépasse le PCM de plus de 10 %, le PCM peut être ajusté sur la base de la variation retardée de l’indice des prix à la consommation (« IPC »)Note de bas de page 10. Pour les médicaments ayant plusieurs concentrations et formes posologiques (p. ex. les médicaments pour lesquels plusieurs DIN ont été attribués), la comparaison du PCM avec la MPI n’est effectuée que pour la concentration ou la forme posologique pour laquelle le PCM a initialement été fixé par la MPI, et non pour les concentrations et formes posologiques pour lesquelles les PCM ont été fixées par le test de la relation raisonnable (« RR ») (tel que décrit à l’annexe B). Toutefois, si le PCM d’une concentration ou d’une forme posologique est ajusté en fonction de l’IPC selon le processus décrit, les PCM supplémentaires pour les concentrations et les formes posologiques fixés par le test de la RR en référence à ce PCM seront ajustés conformément à la méthodologie de la RR.
49. Le prix courant d’un médicament ne peut dépasser le PCM applicable ou le plus élevé des prix internationaux (« PEPI »), sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
a) Prix courant du médicament au Canada
50. Conformément au Règlement, le breveté doit présenter des renseignements sur le prix courant de chaque forme posologique, de chaque concentration et de chaque format d’emballage (p. ex. pour chaque DIN) du médicament dans chaque province et territoire.
51. Les prix courants peuvent varier à l’intérieur des marchés géographiques canadiens (c. à d. entre les provinces ou les territoires). Lorsqu’il y a plusieurs prix courants sur différents marchés, le prix courant le plus élevé est utilisé pour évaluer la conformité au PCMp et au PCM.
Prix courants du médicament dans les pays du CEPMB11
52. Les tests des prix internationaux sont fondés sur les renseignements fournis par les brevetés. Lorsqu’il y a plusieurs prix courants dans le même pays, le prix courant le plus bas est généralement utilisé pour établir les prix plafonds en fonction des prix internationaux.
53. Le site Web du CEPMB contient des conseils à l’intention des brevetés sur les sources potentielles de renseignements sur les prix.
54. Pour comparer les prix d’un médicament au Canada et les prix dans les pays du CEPMB11, on convertit la monnaie locale en dollars canadiens en utilisant les taux de change calculés comme la moyenne simple des trente-six (36) taux de change mensuels moyens au comptant à midi pour chaque pays (ramenés à huit [8] décimales) comme publiés par la Banque du Canada. Durant la période de lancement d’un médicament, les trente-six (36) mois se terminant au deuxième mois de la période de référence précédente (c’est-à-dire février ou août) seront utilisés. Par la suite, les trente-six (36) mois se terminant au cours du deuxième mois de la période de référence qui est examinée seront utilisés.
Étape 3 : PEM/PEM[A] (Nouveaux médicaments brevetés de la catégorie I seulement)
55. Les nouveaux médicaments sont classés comme étant des médicaments de catégorie I ou de catégorie II en fonction de certains critères, lesquels sont décrits ci-dessous. En plus du PCMp/PCM, les médicaments de la catégorie I sont soumis à un plafond de « prix escompté maximum » (« PEM ») qui peut être ajusté en fonction de la taille du marché (PEM [ajusté] (« PEM[A] »)). Le calcul du PEM et du PEM[A] est expliqué ci-dessous et dans les annexes C et D.
56. Le PEM tient compte du niveau de critères thérapeutiques (NCT) (les niveaux I à IV et le processus d’examen scientifique sont décrits à l’Annexe E), de la valeur pharmacoéconomique et de la taille du marché du médicament (voir l’annexe C). Même si le personnel calculera le PEM ou le PEM[A] et le transmettra aux brevetés aux fins d’information, une enquête ne sera déclenchée que par le défaut du breveté de se conformer au PCMp ou au PCM. Toutefois, si une enquête est déclenchée, le personnel peut examiner le prix netNote de bas de page 11 et le PEM ou le PEM[A] pour décider de la marche à suivre.
Classification d’un médicament de catégorie I
57. A New medicine will be classified as Category I if it meets either of the following criteria:
- Le coût de traitement de 12 mois est supérieur à 150 % du PIB par habitant (« médicaments à coût élevé ») : Cette valeur est basée sur les renseignements sur les prix de la période de lancement fournis par le breveté; le coût de traitement sur 12 mois du médicament calculé relativement à la dose maximale par traitement indiquée dans la monographie du produit; le nombre maximal de traitements par période de 12 mois, en tenant compte de la nature du problème de santé, des pratiques cliniques et d’autres critères pertinents; et le prix courant canadien le plus élevé. Si aucun prix courant n’est disponible, le prix net est utilisé.
- La taille estimée ou réelle du marché (ventes) dépasse 50 millions de dollars par année (« médicaments avec taille de marché élevée ») : Cette valeur peut être basée sur soit (i) une estimation déposée en vertu du Règlement ou (ii) les recettes réelles déposées en vertu du Règlement.Note de bas de page 12
58. Tous les autres nouveaux médicaments sont des médicaments de catégorie II.
59. Nonobstant ce qui précède, les médicaments biosimilairesNote de bas de page 13 et les médicaments génériquesNote de bas de page 14 seront toujours classés dans la catégorie II.
b) Calcul du PEM et du PEM[A]
60. Le PEM est calculé en fonction des critères pertinents qui ont conduit à la classification du médicament dans la catégorie I et de la disponibilité et du contenu d’une analyse coût-utilité.
61. Pour les médicaments (1) qui sont des médicaments à coût élevé qui ont des ventes au PCM supérieures à 12 millions de dollars par an et (2) qui disposent d’une analyse coût-utilité disponible, le PEM est calculé comme suit :
- Le rapport coût-utilité différentiel (« RCUD ») mesuré en années de vie ajustées en fonction de la qualité (« AVAQ ») pour chaque indication du médicament est calculé à partir des analyses coût-utilité présentées par le breveté.
- Le RCUD est comparé au seuil de la valeur pharmacoéconomique (« SVP ») et au seuil de réduction applicables, en fonction de son niveau de critères thérapeutiques (voir l’annexe C, « Évaluation de la valeur pharmacoéconomique » et le tableau reproduit ci-dessous).
- Le prix auquel le RCUD du médicament serait équivalent au SVP est déterminé (le « prix pharmacoéconomique » ou « PP »).
- Le PEM sera déterminé en supposant que les 12 premiers millions de dollars sont réalisés pour les quantités vendues au PCM, tandis que les ventes restantes jusqu’à 50 millions de dollars sont réalisées pour les quantités vendues au PP sous réserve du seuil de réduction. Le PEM peut être ajusté davantage en fonction de la taille du marché (PEM[A]) si le médicament réalise des ventes unitaires annuelles telles que, si son prix est fixé au PCM, les recettes seraient supérieures à 50 millions de dollars (voir l’annexe D « Méthode de rajustement en fonction de la taille du marché »). Comme ce rajustement a lieu à des niveaux de vente prévisibles, le breveté sera informé de son PEM actuel et de son PEM[A] futur prévu.
Rajustement du prix selon le niveau de critères thérapeutiques pour le calcul du PEM Niveau de critères thérapeutiques
(voir annexe E – Processus d’examen scientifique)SVP Seuil de réduction du PCM Niveau I 200 k$/ AVAQ 20 % Niveau II 150 k$/ AVAQ 30 % Niveau III 150 k$/ AVAQ 40 % Niveau IV 100 k$/ AVAQ 50 % Aucun RCUD déclaré dans l’analyse pharmacoéconomique Médiane de la CCTn soumise à un seuil de 50 % Aucune analyse pharmacoéconomique déposée 50 % du PCM
62. En l’absence d’un RCUD disponible à partir des renseignements déposés par le breveté, le PEM sera établi à la médiane de la CCTn, sous réserve d’un seuil de réduction de 50 % du PCM et sous réserve d’ajustements de la taille du marché.
63. Le PEM ou PEM[A] ne peut jamais dépasser le PCM.
64. Pour les médicaments qui sont des médicaments à coût élevé et qui ont un revenu maximal estimé supérieur à 12 millions de dollars par an, mais pour lesquels une analyse pharmacoéconomique n’a pas été déposée, le PEM sera fixé en fonction de 50 % du PCM (voir l’annexe C).
65. Pour les médicaments avec taille de marché élevée qui ne sont pas des médicaments à coût élevé, le PEM est calculé comme suit :
- Si la taille réelle du marché est inférieure à 50 millions de dollars, le PEM est fixé en fonction du PCM.
- Si la taille réelle du marché est supérieure à 50 millions de dollars, le PEM est fixé en fonction de la valeur la plus faible entre le PCM et la médiane de la CCTn.
66. Le PEM[A] est recalculé annuellement.
Étape 4 : Détermination de l’indication pertinente
67. Pour les médicaments ayant plus d’une indication approuvéeNote de bas de page 15, le personnel déterminera l’indication pour laquelle le PEM (et le PCM, le cas échéant) sera évalué (l’« indication pertinente »). Cela pourrait se produire lors du lancement, ou dans le cadre d’une réévaluation si des indications supplémentaires sont approuvées pendant le cycle de vie du médicament (voir la section VI).
68. S’il s’agit d’un médicament de catégorie I, l’indication pertinente sera l’indication qui remplit le critère relatif à la classification de catégorie I. Si le médicament de catégorie I possède plus d’une indication qui remplit ce critère ou n’en possède aucune, ou s’il s’agit d’un médicament de catégorie II, l’indication pertinente sera l’indication associée au problème de santé ayant la prévalence la plus élevée (c.-à-d. la plus grande population de patients) ou l’utilisation estimée.
Étape 5 : Calendrier de l’examen de la conformité
69. Le calendrier de mise en conformité avec les plafonds de PCMp/PCM décrits dans cette section est le suivant :
- PCMp : Le breveté doit se conformer au PCMp lors de l’entrée sur le marché si celui-ci a été établi à ce moment-là, ou au cours d’une (1) période de référence suivant l’établissement du PCMp.
- PCM : Le breveté doit se conformer au PCM lors de l’entrée sur le marché si celui-ci a été établi à ce moment-là, ou au cours d’une (1) période de référence suivant l’établissement du PCM.
B. Processus d’examen du prix pour les médicaments bénéficiant de droits acquis, les élargissements de gammes et les médicaments de transition
70. Les médicaments brevetés bénéficiant de droits acquis et les élargissements de gammes sont assujettis à un PCM, mais pas à un PEM. Les médicaments de transition seront évalués conformément aux procédures prévues aux paragraphes 40 à 43.
71. Le PCM pour les médicaments bénéficiant de droits acquis et les élargissements de gammes est fixé en fonction de la plus faible des deux valeurs suivantes :
- le PEPI pour les pays du CEPMB11 pour lesquels le breveté a fourni des renseignements;
- le plafond du médicament (p. ex. le « PMNE ») en vertu des Lignes directrices telles qu’elles étaient applicables avant la publication des présentes Lignes directrices.
72. La méthode de calcul du PEPI et du prix courant des médicaments bénéficiant de droits acquis et des élargissements de gammes est la même que la méthode prévue dans les présentes Lignes directrices pour les nouveaux médicaments (voir la section V[A], « Processus d’examen du prix des nouveaux médicaments »).
73. Le PCM pour les médicaments de transition est fixé en fonction de la plus faible des deux valeurs suivantes :
- la MPI pour les pays du CEPMB11 pour lesquels le breveté a fourni des informations;
- le plafond du médicament (p. ex. le « PMNE » ou le « PMMP ») en vertu des Lignes directrices telles qu’elles étaient applicables avant la publication des présentes Lignes directrices.
74. prix courant du médicament ne peut dépasser le PCM applicable, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
75. Pour les médicaments bénéficiant de droits acquis, les élargissements de gammes et les médicaments de transition, si le PCM est fixé par le PMNE du médicament et s’il est prouvé que son calcul a été fortement influencé par la déclaration d’avantages, le breveté peut soumettre une demande afin d’établir un PCM plus élevé. Toute demande de ce type doit inclure des détails et des documents justificatifs des avantages déclarés et, si possible, un historique des changements des prix courants du médicament, ainsi que toute autre information pertinente demandée par le personnel. Si les renseignements requis justifient une réévaluation, le PCM sera ajusté au prix le plus bas entre (i) les tests des prix internationaux applicables pour les pays du CEPMB11 pour lesquels le breveté a fourni des renseignements et (ii) le prix courant conforme le plus élevé du médicament selon les Lignes directrices telles qu’elles étaient applicables avant la publication des présentes Lignes directrices.
76. Le breveté est tenu de se conformer au PCM dans un délai d’une (1) période de référence suivant l’établissement du PCM pour un élargissement de gamme d’un médicament et pour un médicament bénéficiant de droits acquis ou un médicament de transition.
VI. Réévaluation
77. Il est possible que l’on réévalue la catégorie ou le prix plafond d’un médicament pour s’assurer qu’ils restent pertinents à la suite de changements importants dans les conditions du marché.
Réévaluation des nouveaux médicaments et des médicaments de transition
78. En ce qui concerne les nouveaux médicaments, il peut y avoir une réévaluation si l’une ou l’autre des situations suivantes survient :
- Une nouvelle indication d’un médicament (de catégorie I ou II) est approuvéeNote de bas de page 16;
- Les ventes d’un médicament de catégorie II ont dépassé le seuil de taille du marché (voir l’annexe D), contrairement à l’estimation initiale présentée par le breveté;
- L’analyse coût-utilité d’un médicament de catégorie I est mise à jour;
- Si, au cours de deux périodes consécutives, la MPI en vigueur est inférieure au PCM de plus de 10 %.
79. Un médicament qui est approuvé pour une nouvelle indication peut voir son indication pertinente modifiée conformément aux procédures décrites dans la section V. L’attribution d’une nouvelle indication peut avoir une incidence sur la taille du marché du médicament, les médicaments de comparaison appartenant à la même catégorie thérapeutique et le coût-efficacité. Il peut donc y avoir une augmentation ou une diminution correspondante du PEM.
80. Un médicament de catégorie II qui est approuvé pour une nouvelle indication (à l’exception des biosimilaires et des médicaments génériques) peut être réévalué en catégorie I s’il répond aux critères de sélection pertinents. Par exemple, si les ventes au PCM du médicament augmentent au-delà du seuil annuel de la taille du marché, contrairement à l’estimation initiale de la taille du marché déposée par le breveté.
81. Une réévaluation de la catégorie II à la catégorie I entraînera l’attribution d’un PEM et d’un nouveau PCM au médicament.
82. Pour les nouveaux médicaments et les médicaments de transition, si la MPI en vigueur est inférieure au PCM de plus de 10 % pendant deux périodes de référence consécutives, le PCM sera redéfini par la MPI en vigueur. Si le médicament a des formes posologiques et des concentrations supplémentaires avec des PCM déterminés par le test de la RR (voir l’Annexe B), ces PCM seront ajustés en conséquence.
Réévaluation des médicaments bénéficiant de droits acquis et des élargissements de gammes
83. Dans le cas des médicaments brevetés bénéficiant de droits acquis et de leurs élargissements de gammes, si, pendant deux périodes de référence consécutives, le PEPI en vigueur est inférieur au PCM, le PCM sera redéfini par le PEPI en vigueur.
Respect des plafonds réévalués
84. Le breveté sera informé en cas de déclenchement des critères de réévaluation et sera ensuite informé du PCM/PEM ajusté. Le breveté doit se conformer au nouveau PCM dans un délai d’une (1) période de référence suivant l’établissement du nouveau PCM.
VII. Enquêtes
85. Une enquête est un examen approfondi du prix d’un médicament breveté réalisé par le personnel. Dans le cadre d’une enquête, le personnel examine les renseignements fournis par le breveté et les renseignements pouvant être obtenus d’autres sources. Les enquêtes sont de nature purement administrative, et les membres du Conseil ne participent pas au processus. Si une enquête entraîne la tenue d’une audience, le panel d’audience doit effectuer un examen de novo indépendant du prix du médicament breveté afin de déterminer s’il est excessif au sens de l’article 83 de la Loi. Par conséquent, les positions prises par le personnel ou par le breveté au cours de l’enquête peuvent différer de celles prises au cours d’une audience.
86. Les critères justifiant la tenue d’une enquête ont été élaborés dans le but d’assurer l’utilisation la plus efficace des ressources humaines et financières du CEPMB. Le fait que le prix d’un médicament breveté ne soit pas visé par une enquête ne signifie pas nécessairement que son prix ne soit pas excessif, et inversement. Cela signifie uniquement que les critères d’enquête prévus dans les Lignes directrices n’ont pas été remplis dans les circonstances particulières.
A. Critères d’enquête
87. En règle générale, le personnel lancera une enquête si l’une ou autre des situations suivantes survient :
- Le prix courant de toute forme posologique ou concentration d’un médicament semble être supérieur de plus de 5 % au PCMp ou au PCM applicable correspondant;
- Les recettes cumulatives potentielles tirées de la vente à un prix supérieur PCMp ou au PCM (« excédent des recettes potentielles ») semblent dépasser 50 000 dollars pour le médicament (c.-à-d. pour toutes ses formes posologiques et concentrations) au cours d’une année civile;
- Une plainte est reçue.
88. Nonobstant ce qui précède, dans le cas des biosimilaires, des médicaments à usage vétérinaire, des médicaments en vente libre et des vaccinsNote de bas de page 17, une enquête ne sera ouverte par le personnel que si une plainte est reçue.
89. En ce qui concerne les médicaments génériques, le personnel n’examinera les renseignements relatifs à l’identité du médicament et au prix qu’au début d’une enquête menée par le personnel, ce qui ne peut se produire que lorsque les trois conditions suivantes sont remplies :
- une plainte a été reçue concernant le médicament générique;
- le breveté du médicament générique est la seule entreprise au Canada qui vend une version générique du médicament dans ce pays;
- le médicament générique ne fait pas l’objet d’une entente de prix avec l’Alliance pancanadienne pharmaceutique (APP) de qui il relève. Le fardeau de prouver au personnel qu’un médicament générique est assujetti à une entente d’établissement des prix avec l’APP et qu’elle y est conforme incombe au breveté pour ce médicament générique breveté.
90. Tout médicament générique faisant l’objet d’une enquête sera examiné par le personnel conformément aux lignes directrices applicables. En ce qui concerne les comparaisons des prix nationaux et internationaux des médicaments génériques, le personnel examinera, sans toutefois s’y limiter, l’utilisation des prix nationaux et internationaux du produit de référence de marque comme comparateurs. Si le produit de référence de marque n’est plus vendu sur le marché canadien ou international, le personnel envisagera, sans toutefois s’y limiter, d’utiliser l’ancien prix du produit de référence de marque comme comparateur (tel qu’ajusté par l’IPC, le cas échéant).
91. Nonobstant les paragraphes 87 et 88, le prix de tout médicament breveté figurant sur la Liste des drogues destinées aux importations et aux ventes exceptionnelles en vertu de l’Arrêté d’urgence concernant les drogues, les instruments médicaux et les aliments à des fins diététiques spéciales dans le cadre de la COVID-19 du 30 mars 2020 et dans toute liste publiée par Santé Canda conformément à l’Arrêté d’urgence concernant l’importation, la vente et la publicité de drogues à utiliser relativement à la COVID-19 du 16 septembre 2020 ne fera l’objet d’un examen ou d’une enquête par le personnel du CEPMB que si de la ministre fédérale de la Santé ou l’un de ses homologues provinciaux ou territoriaux formule une plainte concernant son prix. Ce sera le cas tant que les décrets provisoires seront en vigueur et que le médicament breveté restera inscrit sur la liste. En l’absence d’une plainte préexistante, Lorsque les décrets provisoires seront expirés ou abrogés, ou qu’un médicament breveté sera retiré de la liste applicable, le personnel réexaminera les prix des médicaments assujettis à cette mesure en fonction des prix de liste internationaux et nationaux en vigueur, et non en fonction des prix de lancement réduits au Canada.
92. Lorsque le prix courant d’un médicament est supérieur au PCMp ou au PCM en vertu des Lignes directrices, mais que les critères justifiant la tenue d’une enquête ne sont pas remplis, le breveté en est avisé, et le médicament se voit attribuer la mention « Ne justifie pas une enquête » dans le rapport annuel du CEPMB. Le cas échant, le personnel ne prend aucune mesure immédiate; toutefois, afin d’éviter une enquête à une date ultérieure, le prix courant du médicament ne doit pas dépasser le prix plafond applicable et les recettes qui peuvent avoir été tirées doivent être compensées, sans quoi il pourrait faire l’objet d’une enquête ou d’un examen supplémentaire par le personnel.
B. Processus d’enquête
93. Lorsque le personnel lance une enquête, le breveté en est avisé, et le médicament se voit attribuer la mention « Sous enquête » dans le rapport annuel du CEPMB. Le processus d’examen des prix et le processus d’enquête ne peuvent pas donner lieu à une décision judiciaire établissant que le prix d’un médicament est excessif au sens de la Loi. Une telle décision ne peut être prise qu’une fois que le breveté ou l’ancien breveté a eu la possibilité raisonnable de présenter ses observations, conformément à l’article 83 de la Loi.
1. Examen supplémentaire des renseignements présentés
94. Lorsqu’un médicament fait l’objet d’une enquête, le personnel examine l’historique des prix du médicament depuis son lancement. Tous les renseignements fournis par le breveté sont analysés, et des éclaircissements peuvent être demandés. Par exemple, si un prix semble erroné ou inattendu, ou s’il y a des divergences dans les renseignements présentés par le breveté, celui-ci pourrait devoir fournir une explication ou d’autres documents à l’appui. Le personnel pourrait également prendre en considération tous les renseignements pertinents qui n’ont pas été soumis par le breveté, comme les prix obtenus avec la CCTn et la CCTi. En outre, le personnel analyse la pertinence de la catégorie et des tests applicables en fonction des faits du dossier et des particularités des marchés pertinents sur lesquels le médicament est vendu. Par exemple, le personnel peut examiner si la taille réelle du marché est sensiblement inférieure à la taille estimée du marché, ou si le médicament est un vaccin, un produit sanguin ou un autre produit soumis à un processus d’appel d’offres.
95. Le personnel peut utiliser tout test décrit dans les Lignes directrices et toute modification ou variation de ces tests (par exemple, la MPI au lieu du PEPI ou la médiane par opposition au sommet de la CCTn, ou bien la comparaison du prix net par rapport au PEM) selon ce qu’il juge le plus approprié au vu des circonstances factuelles entourant le prix du médicament faisant l’objet de l’enquête.
2. Calcul de l’excédent des recettes potentielles
96. Lorsque le prix d’un médicament est supérieur aux prix plafonds prévus dans les Lignes directrices, le breveté est informé que le prix du médicament breveté « dépasse les seuils établis dans les Lignes directrices », et les prix plafonds ou tests applicables sont identifiés. Le personnel commence également à calculer l’excédent cumulatif des recettes potentielles en fonction des prix nets présentés par le breveté, quel que soit le prix plafond ou test utilisé. Comme le CEPMB a compétence sur le médicament breveté au cours de la période précédant la délivrance du brevet, l’excédent des recettes qu’aurait procuré la vente du médicament pendant cette période est inclus dans les calculs.
97. Dans certains cas, les tests et les plafonds utilisés au cours de l’enquête peuvent différer des seuils initiaux qui ont mené au déclenchement de l’enquête. Dans de tels cas, les plafonds de l’enquête (par opposition aux plafonds de déclenchement) seront utilisés pour calculer les recettes excédentaires potentielles.
98. Enfin, si une audience a lieu, le personnel peut soutenir que des plafonds autres que ceux qui sont prévus dans les Lignes directrices ou qui ont été utilisés lors de l’enquête d’appliquent, ou exercer un recours dans le cadre duquel l’excédent des recettes diffère de l’excédent cumulatif des recettes potentielles calculé lors de l’enquête. De plus, lorsque le personnel estime que le breveté ou l’ancien breveté s’est livré à une politique de vente d’un médicament à un prix excessif, il peut, par ordonnance, lui enjoindre de compenser jusqu’au double de l’excédent des recettes.
3. Résultats de l’enquête
99. Les résultats possibles d’une enquête sont les suivants :.
- Un engagement de conformité volontaire (« ECV »), tel qu’il est décrit dans la section VIII;
- L’émission d’un avis d’audience si, sur recommandation du personnel, le président estime que la tenue d’une audience est dans l’intérêt public;
- La clôture de l’enquête.
100. La clôture d’une enquête peut découler d’un ECV ou d’une décision selon laquelle un examen approfondi du prix d’un médicament breveté n’est pas justifié à ce moment-là, compte tenu des faits et des considérations qui ont été mis en lumière au cours de l’enquête.
101. La clôture d’une enquête est une mesure administrative; il ne s’agit pas d’une décision judiciaire établissant que le prix du médicament breveté n’est pas excessif ni d’un aveu du CEPMB en ce sens. La clôture d’une enquête n’exclut pas la possibilité que l’on rouvre une enquête, que l’on en lance une nouvelle ou que l’on tienne une audience dans l’avenir.
VIII. Engagement de conformité volontaire (ECV)
102. Le breveté peut présenter un ECV au personnel en tout temps avant l’émission d’un avis d’audience. Un ECV est une promesse par laquelle le breveté s’engage à baisser les prix du médicament visé par l’enquête ou à compenser l’excédent des recettes que lui aurait procuré la vente de ce médicament à ce prix. Une proposition d’ECV ne constitue pas un aveu du breveté quant au caractère excessif du prix du médicament.
103. Le personnel est chargé de mener les négociations relatives à l’ECV avec le breveté et, selon la politique du CEPMB, le président ne peut pas y participer. Si les négociations aboutissent à une proposition d’ECV qui, de l’avis du personnel, serait acceptable pour le président, la proposition sera soumise au président par le directeur exécutif aux fins d’examen. Le personnel ne peut pas déterminer de façon indépendante si une proposition d’ECV est acceptable, et il ne peut pas donner de garantie au breveté quant à la probabilité que le président accepte la proposition.
104. L’examen d’un ECV est une procédure administrative; il ne s’agit pas d’une décision du CEPMB établissant que le prix du médicament présenté par le breveté ou utilisé pour le calcul du montant de la compensation n’est pas excessif ni d’un aveu du CEPMB en ce sens. Toutefois, l’acceptation d’un ECV par le président donnera lieu à la clôture de l’enquête.
105. Le CEPMB rend compte publiquement de tous les ECV acceptés par le président. Lorsqu’il présente un ECV signé, le breveté doit consentir à sa publication en version entière ou expurgée. Les renseignements publiés peuvent comprendre le contenu de l’ECV, dont les conditions qui y sont énoncées. Ces renseignements peuvent figurer dans le rapport annuel du CEPMB, sur le site Web du CEPMB, dans les publications du CEPMB, comme La Nouvelle, et sur les plateformes de médias sociaux.
106. Le personnel ne peut pas se pencher sur les demandes de négociations ou de propositions d’ECV « sans préjudice ». Les ECV sont des promesses unilatérales faites par les brevetés et ne sont pas des ententes entre le CEPMB et les brevetés. Néanmoins, certains aspects des discussions entre les brevetés et le personnel qui ont trait au contenu des présentations du breveté peuvent être soumis aux protections énoncées aux articles 87 et 88 de la Loi. En outre, les dispositions dans la Loi sur l’accès à l’information peuvent s’appliquer.
107. Lorsqu’un avis d’audience est émis, le breveté peut négocier un règlement sous forme d’entente, laquelle doit être approuvée par le panel d’audience. Le personnel examine les demandes d’accord ou de proposition de règlement « sans préjudice ».
IX. Recommandations relatives à l’audience
108. Lorsqu’une enquête sur le prix d’un médicament breveté est terminée et que l’affaire n’est pas réglée, le directeur exécutif peut présenter un rapport au président. Le président peut décider d’émettre un avis d’audience s’il estime que la tenue d’une audience est dans l’intérêt public. La décision d’émettre un avis d’audience n’est pas judiciaire ou quasi judiciaire, et le président n’effectue pas d’analyse pour déterminer si les faits allégués par le personnel sont ou seront prouvés. Aucun autre membre du Conseil n’est informé de l’enquête ni des résultats de l’examen du personnel sur le prix d’un médicament breveté tant que l’affaire n’est pas soumise à un panel d’audience au cours d’une audience publique.
109. La décision quant au caractère excessif du prix d’un médicament breveté est prise par le panel d’audience seulement, après la tenue de l’audience publique.
X. Processus d’audience relativement aux prix excessifs et recours
110. Les audiences du CEPMB sont publiques. Lors d’une audience, un panel d’audience, composé d’au moins deux membres du Conseil, entend les observations et les éléments de preuve des parties. Le panel détermine si un médicament breveté est ou a été vendu à un prix excessif sur n’importe quel marché canadien en tenant compte des renseignements disponibles concernant les facteurs énoncés à l’article 85 de la Loi.
111. Pour obtenir de plus amples renseignements sur les audiences, veuillez consulter les Règles de pratique et de procédure du CEPMB, l’ensemble normalisé et publié de procédures que tous les participants à des audiences devant le CEPMB doivent suivre. Les Règles établissent les procédures du CEPMB conformément à l’exigence prévue dans la Loi selon laquelle le CEPMB doit résoudre les questions soumises à son attention sans formalisme, en procédure expéditive, dans la mesure où les circonstances et l’équité le permettent. Les instructions relatives à la pratique et d’autres renseignements au sujet des audiences en cours ou passées sont à la disposition du public sur le site Web du CEPMB.
112. En vertu de la Loi, le CEPMB est habilité à rendre des ordonnances correctives lorsqu’il détermine, à la suite d’une audience, qu’un breveté (ou un ancien breveté) vend ou a vendu un médicament breveté à un prix excessif sur un marché canadienNote de bas de page 18.
113. En termes généraux, le CEPMB a le pouvoir d’exercer des deux grandes voies de recours après une audience: i) des ordonnances enjoignant au breveté de faire baisser le prix maximum auquel il vend le médicament breveté sur un marché à un niveau que le CEPMB ne juge pas excessif ; et ii) des ordonnances enjoignant au breveté de compenser le montant des recettes excessives qu’il estime avoir tirées de la vente du médicament breveté à un prix excessif soit a) en réduisant le prix auquel il vend le médicament breveté ; (b) en réduisant le prix auquel le breveté vend un autre médicament auquel se rattache une de ses inventions brevetées ; ou (c) en versant à Sa Majesté du chef du Canada un montant spécifié dans l’ordonnance.
114. Si le panel d’audience conclut que le breveté ou l’ancien breveté s’est livré à une politique de vente du médicament breveté à un prix excessif, il peut, par ordonnance, lui enjoindre de compenser, selon lui, jusqu’au double de l’excédent des recettes procuré par la vente du médicament breveté au prix excessif. Le CEPMB examinera l’étendue et la durée des ventes du médicament breveté au prix excessif lorsqu’il arrivera à cette conclusion et rendra l’ordonnance.
XI. Audience pour défaut de présenter les renseignements requis
115. Lorsque le personnel estime qu’un breveté a omis ou a refusé de fournir au CEPMB les renseignements sur les prix, les ventes ou les recettes, ou d’autres renseignements semblables exigés par la loi, le directeur exécutif peut présenter un rapport au président. Le président peut décider d’émettre un avis d’audience s’il juge que l’intérêt public justifie la tenue d’une audience publique pour déterminer si le breveté a bel et bien manqué aux exigences en matière de présentation de renseignements prévues par la Loi et le Règlement.
116. Comme dans le cadre d’une audience sur un prix excessif, la décision d’émettre un avis d’audience n’est d’aucune façon judiciaire ou quasi judiciaire, et le président n’effectue pas d’analyse pour déterminer si les faits allégués par le personnel sont ou seront prouvés. Aucun membre du CEPMB n’est informé des résultats de l’examen ou l’enquête du personnel sur le prix d’un médicament breveté tant que l’affaire n’est pas soumise à un panel d’audience au cours d’une audience publique. Après la tenue d’une audience publique, le panel d’audience décide si l’information présentée par le breveté est suffisante. Si, à la suite d’une audience, le panel d’audience conclut que le breveté a manqué à ces exigences, il peut, par ordonnance, lui enjoindre de fournir au CEPMB les renseignements et les documents requis en vertu de l’article 81 ou 88 de la Loi.
117. En outre, aux termes du paragraphe 76.1(1) de la Loi, quiconque contrevient aux exigences en matière de présentation de renseignements énoncées aux articles 80, 81, 82 ou 88 ou à une ordonnance prise sous le régime de l’un ou l’autre de ces articles commet une infraction et encourt, sur déclaration de culpabilité par procédure sommaire, une amende ou un emprisonnement.
XII. Plaintes
118. Une personne ou un groupe qui estime que le prix d’un médicament breveté est excessif sur tout marché canadien peut déposer une plainte au CEPMB par téléphone, par écrit ou par voie électronique. Les coordonnées à utiliser se trouvent sur la page « Comment déposer une plainte » du site Web du CEPMB.
119. En règle générale, toute plainte déclenche une enquête sur le prix du médicament breveté visé par la plainte. Le plaignant ne participe pas à l’enquête ni aux audiences qui en résultent (à moins qu’il ne présente une demande pour participer aux audiences à titre d’intervenant). Le plaignant n’est pas tenu de fournir des documents ou des éléments de preuve au CEPMB. Les enquêtes sont fondées sur les documents fournis par le breveté ou obtenus autrement par le personnel.
120. En raison des limites en matière de communication énoncées aux articles 87 et 88 de la Loi sur les brevets et dans la Loi sur l’accès à l’information, le plaignant est informé du résultat d’une enquête uniquement si le processus mène à un ECV ou à un avis d’audience.
XIII. Annexes
A. Comparaison selon la catégorie thérapeutique des prix nationaux (CCTn) et comparaison selon la catégorie thérapeutique des prix internationaux (CCTi)
Test de la CCTn
Tel qu’il est décrit dans la section V des présentes Lignes directrices, le test de la comparaison selon la catégorie thérapeutique des prix nationaux (« CCTn ») sert à calculer le seuil maximum du prix d’un médicament breveté dans certains cas. Le test de la CCTn permet de comparer le prix courant d’un médicament breveté avec le prix courant d’autres médicaments sélectionnés au terme d’un examen scientifique.
Pour les médicaments à coût élevé, le PEM est établi en fonction de la valeur la plus faible entre le PCM et la médiane de la CCTn, en tenant compte des seuils de réduction applicables indiqués dans le tableau ci-dessous, et de l’application du rajustement selon la taille du marché décrit à l’annexe D.
| Niveau de critères thérapeutiques (Voir annexe E – Processus d’examen scientifique) |
Seuil de réduction de la CCTn |
|---|---|
| Niveau I | 20 % |
| Niveau II | 30 % |
| Niveau III | 40 % |
| Niveau IV | 50 % |
Sélection des médicaments aux fins de comparaison
La sélection des médicaments à utiliser aux fins de comparaison est fondée sur le Système de classification anatomique thérapeutique chimique (ATC) du Centre collaborateur de l’Organisation mondiale de la Santé (OMS) pour la méthodologie sur l’établissement des statistiques concernant les produits médicamenteux.
Les médicaments utilisés aux fins de comparaison appartiennent généralement à la souscatégorie du système ATC située immédiatement au-dessus de la substance chimique simple. Il s’agit habituellement du quatrième niveau de sous-catégorie, mais il peut également s’agir de la sous-catégorie supérieure suivante ou d’une autre sous-catégorie. Dans certains cas, la sélection peut devoir être faite au cinquième niveau, soit au niveau de la substance chimique simple.
Un médicament de la même catégorie thérapeutique du système ATC que le médicament breveté à l’étude peut être omis s’il ne se prête pas à la comparaison. Par exemple, un médicament comportant une indication ou un usage principal autre que l’indication ou l’usage principal du médicament breveté à l’étude peut être omis de la comparaison.
Tous les médicaments sélectionnés aux fins de comparaison qui comportent la même indication approuvée ou le même usage que l’indication pertinente du médicament breveté à l’étude sont inclus dans l’examen. Cet examen repose sur les recherches effectuées par le personnel et peut comprendre d’autres recherches menées par un centre d’information sur les médicaments ou une analyse des données probantes présentées par les brevetés.
Lorsqu’il y a plusieurs vendeurs d’un médicament identifié en tant que médicament comparable, le prix le plus bas des médicaments sera identifié. Le prix final de la comparaison selon la catégorie thérapeutique est le prix le plus élevé de l’ensemble des médicaments comparables pour le PCM et la médiane pour le PEM.
Lorsque le médicament breveté est une nouvelle forme posologique ou concentration du même ingrédient médicinal contenu dans un ou plusieurs médicaments existants, les médicaments existants qui sont offerts dans la même forme posologique ou dans une forme posologique comparable et qui comportent la même indication ou le même usage servent de médicaments de comparaison, et ce, que les régimes posologiques soient identiques ou diffèrent sensiblement.
Lorsque le produit est une combinaison de médicaments qui sont chacun vendus au Canada et qui comportent tous la même indication ou le même usage, les médicaments de comparaison sont limités aux médicaments composants.
Régimes posologiques comparables
Le régime posologique comparable utilisé aux fins de comparaison correspond normalement au régime posologique maximal habituellement recommandé dans la monographie de produit (ou dans des renseignements semblables), et il tient compte des variables cliniques pertinentes. La concentration est déterminée en fonction du régime posologique du médicament à l’étude. En règle générale, un régime posologique fondé sur un traitement s’appliquera aux indications aiguës, tandis qu’un régime quotidien (selon la dose d’entretien) sera utilisé pour les indications chroniques.
Sources des prix
Les brevetés ne présentent pas de renseignements sur le prix des médicaments sélectionnés aux fins de comparaison avec un médicament breveté. On consulte d’abord des sources publiques pour obtenir le prix d’un médicament de comparaison dans le cadre du test de la CCTn. Les listes de médicaments des régimes provinciaux constituent le point de départ du personnel aux fins de l’identification des prix. Pour chaque médicament de comparaison sélectionné, on utilise le prix le plus bas. Le personnel peut décider d’exclure du test de la CCTn un médicament (breveté ou non) sélectionné aux fins de comparaison s’il a des raisons de croire que le médicament est vendu à un prix excessif.
Test de la CCTn
Après avoir sélectionné les médicaments qui se prêtent à la comparaison et déterminé le prix le plus bas de chaque médicament, on calcule le coût d’un traitement comparable pour chaque médicament. Les différents coûts de traitement sont triés, et le plafond ou la médiane, établi(e). Lorsqu’un nombre pair de médicaments est utilisé aux fins de comparaison, la médiane correspond à la moyenne simple des deux coûts de traitement du milieu. Le coût médian de traitement est ensuite divisé par le nombre d’unités constituantes du traitement comparable pour le médicament à l’étude, ce qui permet d’établir un prix unitaire.
Test de la CCTI
Tel qu’il est décrit dans la section V des présentes Lignes directrices, le test de la comparaison selon la catégorie thérapeutique des prix internationaux (« CCTi ») peut être utilisé dans certains cas. Le test de la CCTi permet de comparer le prix d’un médicament breveté avec le prix courant d’autres médicaments sélectionnés au terme d’un examen scientifique aux fins de comparaison dans les onze pays de comparaison énumérés dans le Règlement.
Sélection des médicaments aux fins de comparaison et régimes posologiques comparables
Le test de la CCTi se fonde sur les mêmes médicaments sélectionnés aux fins de comparaison et les mêmes régimes posologiques comparables que ceux utilisés pour le test de la CCTn, conformément aux procédures établies précédemment. Lorsqu’aucun médicament de comparaison n’a été trouvé au pays, le personnel peut déterminer si d’autres médicaments comportent la même indication approuvée ou le même usage que le médicament à l’étude dans l’un des pays de comparaison.
Sources des prix
Les brevetés ne présentent pas de renseignements sur les prix internationaux des médicaments sélectionnés à l’étranger aux fins de comparaison avec un médicament breveté. Les listes de médicaments nationales constituent le point de départ du personnel aux fins de l’identification de prix. Pour chaque médicament de comparaison sélectionné, on utilise le prix public le plus bas.
Le personnel peut décider d’exclure du test de la CCTi un médicament (breveté ou non) sélectionné aux fins de comparaison s’il a des raisons de croire que le médicament est vendu à un prix excessif.
Test de la CCTi
Après avoir sélectionné les médicaments qui se prêtent à la comparaison et déterminé le prix le plus bas de chaque médicament, on calcule le coût d’un traitement comparable pour chaque médicament dans chaque pays de comparaison. Les différents coûts de traitement sont triés, et la médiane de chaque pays est établie. Lorsqu’un nombre pair de médicaments est utilisé aux fins de comparaison, la médiane correspond à la moyenne simple des deux coûts de traitement du milieu. Ces médianes sont triées, et une « médiane des médianes » est définie. Le coût médian de traitement est ensuite divisé par le nombre d’unités constituantes du traitement comparable pour le médicament à l’étude, ce qui permet d’établir un prix unitaire. Les devises étrangères sont converties en dollars canadiens selon la méthode décrite à la section V des présentes Lignes directrices.
B. Test de la relation raisonnable et formes posologiques comparables
Test de la relation raisonnable
Le test de la relation raisonnable (RR) peut servir à déterminer le PCM d’une nouvelle concentration supplémentaire d’un médicament breveté qui est déjà offert dans une ou plusieurs autres concentrations, pourvu que la nouvelle concentration comporte le même ingrédient médicinal, la même indication, le même régime posologique et la même forme posologique (ou une forme posologique comparable) que la ou les concentrations existantes. Il n’y a pas de test de la RR tant qu’un PCM n’est pas fixé pour la concentration de référence.
PCM
Lorsqu’un médicament breveté n’est pas ou n’est plus dans la période provisoire, la première concentration qui a satisfait aux critères de transition du PCMp au PCM deviendra la concentration de référence. S’il existe plusieurs concentrations qui répondent à ce critère au cours d’une période de référence, la concentration de référence sera choisie parmi celles-ci sur la base de considérations scientifiques. Une fois la concentration de référence définie, le plafond pour les nouvelles concentrations supplémentaires sera établi sur la base de la relation proportionnelle entre les concentrations, sous réserve d’un plafond du PEPI pour la nouvelle concentration supplémentaire. Plus précisément, le PCM de la nouvelle concentration supplémentaire plus élevée sera fixé de manière à être équivalent au prix par unité standard de la concentration de référence. Toutefois, le PCM pour la nouvelle concentration supplémentaire inférieure ne sera pas fixé à un niveau supérieur au prix de la concentration de référence, à condition que cela n’entraîne pas un prix pour la nouvelle concentration supplémentaire supérieur au PEPI.
PEM
Le PEM est calculé comme une réduction par rapport au PCM. Par conséquent, la demande de test de la RR pour une nouvelle concentration supplémentaire d’un médicament breveté soumis au PEM sera faite au niveau du PCM pour la concentration de référence et le pourcentage de réduction de prix correspondant sera reporté sur les nouvelles concentrations supplémentaires du médicament breveté.
PEM[A]
Le PEM[A] est un ajustement basé sur le PEM en fonction de la taille du marché et changera donc en conséquence.
Formes posologiques comparables
Les formes posologiques ci-après sont considérées comme étant des formes posologiques comparables pour les besoins du test de la RR. Les formulations d’un même groupe sont considérées comme étant comparables, mais les formes posologiques de groupes différents ne le sont pas.
Topique (T)
- Aérosol
- Aérosol (mousse)
- Crème
- Disque (à libération prolongée)
- Disque
- Pansement
- Gel
- Gel (à libération contrôlée)
- Liposomes
- Liquide
- Lotion
- Onguent
- Tampon
- Peinture
- Pâte
- Timbre
- Timbre (à libération prolongée)
- Crayon
- Plâtre
- Poudre
- Shampoing
- Savon en pain
- Solution
- Éponge
- Vaporisateur
- Vaporisateur (valve à poche)
- Vaporisateur (à dose mesurée)
- Bâtonnet
- Bandelette
- Écouvillon
- Teinture
Nasal (N) ou pulmonaire (P)
- Aérosol
- Aérosol à dose mesurée
- Gouttes
- Gaz
- Préparation à dose mesurée
- Poudre
- Poudre (à dose mesurée)
- Solution
- Solution (à libération prolongée)
- Vaporisateur
- Vaporisateur (à dose mesurée)
- Bâtonnet
Solide administré par voie orale (S)
- Barre à croquer
- Caplet
- Capsule
- Granules effervescents
- Poudre effervescente
- Comprimé effervescent
- Pellicule (soluble)
- Globules
- Granules
- Gomme à mâcher
- Pastille
- Caplet à libération modifiée
- Capsule à libération modifiée
- Comprimé à libération modifiée
- Granulés
- Morceau à croquer
- Poudre (à libération prolongée)
- Bandelette
- Comprimé
- Comprimé (à croquer)
- Comprimé (à dissolution orale)
- Comprimé pour suspension
- Cachet
Liquide administré par voie orale (L)
- Gouttes
- Solution buvable
- Émulsion
- Gel
- Granules pour solution
- Granules pour suspension
- Granules pour suspension (à libération retardée)
- Granules pour suspension (à libération prolongée)
- Liquide
- Liquide à libération modifiée
- Poudre (à libération prolongée)
- Poudre pour solution
- Poudre pour suspension
- Solution
- Solution (à libération prolongée)
- Vaporisateur
- Suspension
- Suspension (à libération prolongée)
- Sirop
- Sirop (à libération prolongée)
- Tisane
- Teinture
Vaginal (V)
- Cône
- Crème
- Douche
- Mousse
- Gel
- Gel (à libération contrôlée)
- Implant
- Insert
- Insert (à libération prolongée)
- Suppositoire
- Granulés
- Anneau (à libération lente)
- Éponge
- Suppositoire
- Suppositoire (à libération soutenue)
- Tampon
- Comprimé vaginal
- Comprimé vaginal (effervescent)
Parentéral (J)
- Bolus
- Implant
- Trousse
- Liposomes
- Injection à libération modifiée
- Pellet (implantable)
- Poudre pour solution
- Poudre pour suspension (à libération soutenue)
- Solution
- Solution (à libération prolongée)
- Suspension pour émulsion
- Suspension (à libération prolongée)
Otique (E) ou ophtalmique (Y)
- Gouttes
- Gel
- Gel (à libération contrôlée)
- Implant
- Insert
- Insert (à libération prolongée)
- Liquide
- Dispositif oculaire à libération modifiée
- Onguent
- Poudre pour solution
- Solution
- Solution (à libération prolongée)
- Suspension
Rectal (R)
- Crème
- Lavement
- Mousse
- Insert
- Onguent
- Suppositoire
- Bâtonnet
- Suppositoire
- Suppositoire (à libération soutenue)
- Suspension
- Suspension (à libération prolongée)
Dentaire, sublingual ou buccal (M)
- Émulsion
- Pellicule (soluble)
- Soie dentaire
- Gel
- Gel (à libération contrôlée)
- Gomme à mâcher
- Pastille
- Pompe à dose mesurée
- Comprimé buccal à libération modifiée
- Rince-bouche (gargarisme)
- Pâte
- Poudre effervescente
- Poudre pour suspension
- Solution
- Solution (à libération prolongée)
- Vaporisateur – buccal
- Vaporisateur – sublingual
- Bâtonnet
- Bandelette
- Comprimé sublingual
- Suspension
- Suspension (à libération prolongée)
- Écouvillon
- Comprimé (dissolution orale)
- Comprimé
- Dentifrice
- Poudre dentifrice
- Cachet
C. Évaluation de la valeur pharmacoéconomique
Comme il est décrit dans la section V des présentes Lignes directrices, l’évaluation de la valeur pharmacoéconomique sert à calculer le PEM d’un médicament. Le Règlement exige que les brevetés présentent des renseignements relatifs à toutes les analyses pharmacoéconomiques d’un médicament breveté préparées par des organismes canadiens financés par l’État si le coût de traitement calculé au prorata du médicament au cours d’une période de 12 mois est supérieur ou égal à 50 % du PIB par habitant au moment de la publication de l’analyse.
Les rapports pharmacoéconomiques du Programme commun d’évaluation des médicaments (PCEM) et les rapports finaux d’orientation économique du Programme pancanadien d’évaluation des anticancéreux (PPEA) de l’Agence canadienne des médicaments et des technologies de la santé (ACMTS) sont les sources habituelles d’analyses pharmacoéconomiques utilisées pour les besoins de l’évaluation de la valeur pharmacoéconomique. Les analyses préparées par l’Institut national d’excellence en santé et services sociaux (INESSS) dans ses Évaluations aux fins d’inscription ou par le Comité consultatif national de l’immunisation (CCNI) sont également étudiées.
Dans le cadre de l’évaluation de la valeur pharmacoéconomique, les calculs du personnel sont fondés sur la nouvelle analyse du scénario de référence menée par l’organisme public, par opposition à l’analyse effectuée à l’aide du modèle de scénario de référence présenté par le breveté. Il s’agit d’un modèle d’analyse coût-utilité dans lequel les résultats en matière de santé sont exprimés sous forme d’AVAQ. Le rapport coût-utilité différentiel (« RCUD ») mesuré en années de vie ajustées en fonction de la qualité (« AVAQ ») pour chaque indication du médicament sera calculé à partir des analyses coût-utilité présentées par le breveté.
Si le rapport de l’organisme ne déclare aucun RCUD, une CCTn sera effectuée. Dans ce cas, le PEM sera établi en fonction de la médiane de la CCTn, sous réserve d’un seuil de réduction du PCM de 50 % et d’ajustements en fonction de la taille du marché, comme indiqué à l’annexe D. Pour les médicaments à indications multiples, l’évaluation pharmacoéconomique pour l’indication pertinente, comme déterminée par la procédure de la section V des présentes Lignes directrices, sera utilisée.
Le RCUD sera comparé au seuil de la valeur pharmacoéconomiqueNote de bas de page 19 (« SVP ») applicable, sur la base des niveaux de critères thérapeutiques indiqués dans le tableau ci-dessous. Le PEM est basé sur le PP (le prix auquel le RCUD du médicament serait équivalent au SVP), mais il est assujetti au seuil de réduction maximal indiqué dans le tableau.
Le CEPMB compte sur des organismes comme l’ACMTS, l’INESSS et le CCNI pour fournir dans leurs rapports des estimations du PP correspondant aux SVP établis, ou bien des renseignements qui lui permettent de calculer le PP.
| Niveau de critères thérapeutiques (voir annexe E – Processus d’examen scientifique) |
SVP | Seuil de réduction du PCM |
|---|---|---|
| Niveau I | 200 k$/ AVAQ | 20 % |
| Niveau II | 150 k$/ AVAQ | 30 % |
| Niveau III | 150 k$/ AVAQ | 40 % |
| Niveau IV | 100 k$/ AVAQ | 50 % |
| Aucun RCUD déclaré dans l’analyse pharmacoéconomique | Médiane de la CCTn soumise à un seuil de 50 % | |
| Aucune analyse pharmacoéconomique déposée | 50 % du PCM | |
D. Méthode de rajustement en fonction de la taille du marché
Comme il est décrit dans la section V des Lignes directrices, un rajustement de la taille du marché est appliqué aux médicaments de la catégorie I lorsque les ventes au PEM[A] dépassent 50 millions de dollars pour toutes les formes posologiques et toutes les concentrations du médicament (p. ex. tous les DIN combinés). On applique ce rajustement chaque année conformément au tableau suivant :
| Ventes annuelles‡ | PEM | Facteur de rajustement du PEM supplémentaire |
Facteur de rajustement du PCM |
|---|---|---|---|
| < 12 M$ | PCM | 0 % | 1 |
| 12 M$ à 50 M$ | Le plus grand entre le PP et le seuil | (12 M$ - 12 M$ * (PEM/PCM)) / Ventes§ + (PEM/PCM) | |
| 50 M$ à 100 M$ | -25 % | (21,5 M$ - 9 M$ * (PEM/PCM)) / Ventes§ + (1 - 25 %) * (PEM/PCM) | |
| >100 M$ | -35 % | (32 M$ - 7,8 M$ * (PEM/PCM)) / Ventes§ + (1 - 35 %) * (PEM/PCM) |
‡ Les ventes annuelles sont calculées au PEM[A].
§ Les ventes sont calculées au PCM
| Ventes annuelles‡ | PEM | Facteur de rajustement du PEM supplémentaire |
Facteur de rajustement du PCM |
|---|---|---|---|
| < 50 M$ | PCM | 0 % | 1 |
| 50 M$ à 100 M$ |
Le plus bas entre le PCM et la médiane de la CCTn |
-25 % | (50 M$ - 37,5 M$ * (PEM/PCM)) / Ventes§ + (1 - 25 %) * (PEM/PCM) |
| > 100 M$ | -35 % | (56,7 M$ - 32,5 M$ * (PEM/PCM)) / Ventes§ + (1 - 35 %) * (PEM/PCM) |
‡ Les ventes annuelles sont calculées au PEM[A].
§ Les ventes sont calculées au PCM
Par exemple, un médicament breveté nécessitant un RCUD et réalisant entre 50 et 100 millions de dollars de ventes sur la base des unités vendues au PEM aura son PEM[A] fixé comme suit :
PEM[A] = PCM * (21,5 M$ - 9 M$ * (PEM/PCM)) / Ventes§ + (1 - 25 %) * (PEM/PCM)
PEM[A] = PCM * facteur de rajustement du PCM.
Le facteur de rajustement du PCM est calculé pour chaque fourchette de ventes annuelles, selon les formules correspondantes. Chaque facteur de rajustement du PCM est ensuite multiplié par les ventes au PCM. Le facteur de rajustement du PCM qui s’applique est celui qui, multiplié par les ventes calculées au PCM, donne un montant qui se situe dans la fourchette des ventes annuelles correspondantes. Le PEM[A] est déterminé en multipliant le PCM par ce facteur de rajustement.
E. Processus d’examen scientifique
L’examen scientifique du CEPMB est un processus qui tient compte des données cliniques, pharmacoéconomiques, et d’autres renseignements pertinents sur le médicament breveté afin de déterminer son niveau de critères thérapeutiques.
L’examen scientifique d’un médicament breveté est basé sur des renseignements provenant de diverses sources telles que celles qui sont présentées ci-dessous.
- Présentation du breveté – Le breveté peut fournir au personnel un bref document de présentation qui explique clairement la raison d’être de sa proposition quant aux critères thérapeutiques, aux médicaments retenus pour la comparaison et aux régimes posologiques comparables. Des renseignements sur les procédures de présentation sont disponibles sur l’outil de soumission en ligne du CEPMB.
- Recherches menées par un centre d’information sur les médicaments (CIM) – Le personnel peut faire appel aux services de divers centres d’information sur les médicaments pour obtenir des renseignements scientifiques, tels que des renseignements sur les essais cliniques, les directives de pratique clinique, etc. La base de l’examen par le CIM est la monographie du produit ou des renseignements similaires à ceux contenus dans une monographie de produit si un AC n’a pas été accordé.
- Recherches menées par le personnel – Le personnel peut également mettre à jour les recherches et compléter les données et les données probantes du breveté et du CIM en utilisant d’autres sources.
- Groupe consultatif sur les médicaments pour usage humain (GCMUH) – Le personnel peut, de façon ponctuelle, consulter le GCMUH quant aux aspects cliniques des renseignements scientifiques à l’étude.
Critères thérapeutiques de niveau I
Le médicament breveté est le premier médicament de sa catégorie disponible sur le marché canadien qui est efficace sur le plan clinique pour le traitement d’une maladie ou pour une indication donnée. Les améliorations qui ont une incidence sur le plan clinique sont définies comme étant celles qui contribuent à augmenter la qualité de vie, à diminuer la mortalité ou à réduire de façon marquée la gravité de la maladie ou l’utilisation des soins de santé, de préférence sur la base de données probantes de haute qualité utilisant des paramètres cliniques concrets plutôt que des résultats dévaluation de substitution. Un gain élevé des AVAQ est normalement associé aux médicaments à ce niveau.
Critères thérapeutiques de niveau II
Le médicament breveté apporte une amélioration considérable des bienfaits thérapeutiques, par rapport à d’autres médicaments vendus au Canada, d’une manière efficace sur le plan clinique. Les améliorations qui ont une incidence sur le plan clinique sont définies comme étant celles qui contribuent à augmenter la qualité de vie, à diminuer la mortalité ou à réduire de façon marquée la gravité de la maladie ou l’utilisation des soins de santé, de préférence sur la base de données probantes de haute qualité fondées sur un comparateur actif utilisant des paramètres cliniques concrets plutôt que des résultats dévaluation de substitution. Une méta-analyse de réseau de haute qualité peut également être envisagée. Un gain élevé des AVAQ est normalement associé aux médicaments à ce niveau.
Critères thérapeutiques de niveau III
Le médicament breveté apporte une amélioration absolue modérée des bienfaits thérapeutiques, par rapport à d’autres médicaments vendus au Canada. Le médicament peut a) entraîner une augmentation de l’efficacité cliniquement pertinente; b) être associé à une réduction de l’incidence ou du degré des effets indésirables importants; c) être associé à des caractéristiques de facilité d’utilisation accrue pertinentes sur le plan clinique (p. ex. voie d’administration, commodité, conformité accrue, etc.), cependant, ces améliorations peuvent avoir une incidence clinique valable limitée ou être basées sur des données probantes cliniques de moindre qualité. Les médicaments brevetés à ce niveau sont normalement associés à des gains d’AVAQ graduels modérés avec un degré de certitude relativement élevé ou à des gains d’AVAQ élevés avec un degré de certitude relativement faible.
Critères thérapeutiques de niveau IV
Le médicament breveté apporte une légère amélioration ou aucune amélioration par rapport aux autres médicaments vendus au Canada. Par ailleurs, ou en plus, le médicament dispose de très peu (ou pas) de données probantes cliniques solides pour établir clairement le degré d’amélioration thérapeutique pertinente sur le plan clinique par rapport à d’autres médicaments vendus au Canada. À ce niveau, les médicaments sont associés à des gains d’AVAQ limités ou inexistants, ou à une incertitude importante due à des données probantes de mauvaise qualité.
Sélection des médicaments aux fins de comparaison
Le Système de classification anatomique thérapeutique chimique (ATC) du Centre collaborateur de l’Organisation mondiale de la Santé (OMS) pour la méthodologie sur l’établissement des statistiques concernant les produits médicamenteux peut être utilisé pour la sélection des médicaments à utiliser aux fins de comparaison. Les médicaments utilisés aux fins de comparaison appartiennent généralement à la sous-catégorie du système ATC située immédiatement au-dessus de la substance chimique simple. Il s’agit habituellement du quatrième niveau de sous-catégorie, mais il peut également s’agir de la sous-catégorie supérieure suivante ou d’une autre sous-catégorie. Dans certains cas, la sélection peut devoir être faite au cinquième niveau, soit au niveau de la substance chimique simple. Un médicament de la même catégorie thérapeutique du système ATC que le médicament à l’étude peut être omis s’il ne se prête pas à la comparaison. Par exemple, un médicament comportant une indication ou un usage principal autre que l’indication ou l’usage principal du médicament breveté à l’étude peut être omis de la comparaison.
Tous les médicaments utilisés à des fins de comparaison auront généralement la même indication ou utilisation approuvée que le médicament breveté qui est examiné.
Si la CCTi inclut des médicaments ayant la même indication ou utilisation approuvée que le médicament breveté examiné, cette information peut être prise en compte.
Lorsque le médicament breveté est une nouvelle forme posologique ou concentration du même ingrédient médicinal contenu dans un ou plusieurs médicaments existants, les médicaments existants qui sont offerts dans la même forme posologique ou dans une forme posologique comparable et qui comportent la même indication ou le même usage servent de médicaments de comparaison. Cela s’appliquera indépendamment du fait que le régime posologique soit le même ou diffère sensiblement.
Lorsque le produit est une combinaison de médicaments qui sont chacun vendus au Canada et qui comportent tous la même indication ou le même usage, les produits de comparaison sont limités aux médicaments de la composition.
Détermination des régimes posologiques comparables
Le régime posologique comparable utilisé aux fins de comparaison correspond normalement au régime posologique maximum habituellement recommandé dans la monographie de produit (ou dans des renseignements semblables), et il tient compte des variables cliniques pertinentes. La concentration est déterminée en fonction du régime posologique du médicament à l’étude. En règle générale, un régime posologique fondé sur un traitement s’appliquera aux indications aiguës, tandis qu’un régime quotidien (selon la dose d’entretien) sera utilisé pour les indications chroniques.
F. Sommaire des calendriers de mise en conformité
| Catégorie du médicament breveté | Test utilisé pour établir le PCMp | Test utilisé pour établir le PCM (au moins 3 ans ou 5 pays) | PEM | Critères de déclenchement d’une réévaluation | Examen de la conformité |
|---|---|---|---|---|---|
| Médicament bénéficiant de droits acquis | S. O. | Plus bas entre le PMNE et le PEPI | S. O. | Si PCM > PEPI au cours de deux périodes de référence consécutives | 1 période de référence |
| Élargissement de gamme d’un médicament bénéficiant de droits acquis | S. O. | Plus bas entre le prix courant et le test de la RR ou prix échelonné à concurrence du PEPI | S. O. | Si PCM > PEPI au cours de deux périodes de référence consécutives | 1 période de référence |
| Médicament de transition | Plus bas entre le prix courant et la MPI | Plus bas entre le prix courant, le PMNE/ PMMP et la MPI | S. O. | Si PCM > MPI de 10 % au cours de deux périodes de référence consécutives | 1 période de référence |
| Nouveau médicament | MPI | Plus bas entre le prix courant et la MPI (ou CCTn/CCTi si aucun PI*) | S. O. | Si PCM > MPI de 10 % au cours de deux périodes de référence consécutives. Déclenchement dû à la taille du marché Preuves thérapeutique/ ÉTS mises à jour Nouvelle indication pertinente |
1 période de référence |
| Nouvelle forme posologique et concentration d’un nouveau médicament | MPI | Plus bas entre le prix courant et le test de la RR ou prix échelonné à concurrence du PEPI | S. O. | Comme pour les nouveaux médicaments pour toutes les formes posologiques et concentrations | 1 période de référence |
* PI : prix international